HEPATIC DISORDERS
Etiology
Hepatitis is an acute or chronic inflammation of the liver that can result from several different causes (e.g., virus, chemical or drug reaction, or other diseases). Nonviral causes of hepatitis include autoimmune hepatitis, Wilson disease, α1-antitrypsin deficiency, and steatohepatitis. The following six viruses cause 90% of cases of viral hepatitis (Table 24-4):
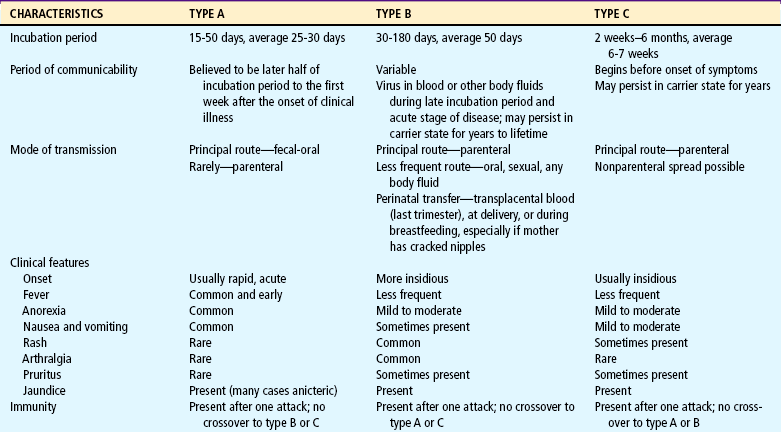
Hepatitis A.: HAV is the most common form of acute viral hepatitis in most parts of the world. It is a member of the picornavirus family. The virus produces a contagious disease transmitted primarily in contaminated stool spread via the fecal-oral route from person to person. HAV has been associated with miniepidemics in areas of poor hygiene and high population density. There is no chronic or carrier state. HAV infection affects individuals of all ages, but the highest incidence occurs among preschool- or school-age children younger than 15 years. Children may serve as the source of HAV infection in adults, such as in childcare center exposures. Usually HAV disease in children is mild. It is frequently anicteric and often subclinical. Infected children who show no symptoms may still spread the virus to others. HAV can be severe in children with immunodeficiency disorders. The incubation period is approximately 3 weeks. Although some cases may be prolonged, the prognosis is excellent. A highly effective vaccine for HAV was introduced in 1997; in 1999 the Advisory Committee on Immunization Practices recommended vaccination for all children over 24 months old living in the 11 states with the highest incidence of HAV (Amon, Darling, Fiore, and others, 2003; Balistreri, Chang, Ciocca, and others, 2002).
Hepatitis B.: HBV infection can occur as an acute or chronic infection and may range from being asymptomatic and limited to causing fatal fulminant (rapid and severe) hepatitis. HBV varies greatly throughout the world. High-prevalence areas have been identified in Africa and Asia; the United States is considered a low-prevalence area. Transmission is usually via the parenteral route through the exchange of blood or any bodily secretion or fluid. Infections from blood transfusion have been reduced as a result of blood product–screening procedures. Transplantation of organs, intimate physical contact, transmission from mother to infant, and the splashing of contaminated fluids into the mouth or eyes are other sources of infection. Adults whose occupations are associated with exposure to blood or blood products (such as health care workers) are at increased risk for infection and should receive HBV vaccination.
Most HBV infection in children is acquired perinatally. Newborns are at risk for hepatitis if the mother is infected with HBV or was a carrier of HBV during pregnancy. Possible routes of maternal-fetal or maternal-infant transmission include (1) leakage of virus across the placenta late in pregnancy or during labor and (2) ingestion of amniotic fluid or maternal blood. Infants who have HBV infection are more than 90% likely to become chronic carriers (Broderick and Jonas, 2003). The incubation period of HBV infection varies from 45 to 160 days.
HBV infection occurs in children and adolescents in the following high-risk groups:
Hepatitis C.: Approximately 1 in 50 people in the general population is positive for HCV antibodies (Centers for Disease Control and Prevention, 2001). About 0.2% to 0.4% of children younger than 12 years of age are infected with HCV. It is estimated that 4 million people in the United States are anti–HCV-positive. Approximately 7% of HCV-infected mothers transmit HCV to their newborns (Balistreri, Chang, Ciocca, and others, 2002). Another common route of infection is by percutaneous exposure, which occurs through transfusion of blood or blood products, transplantation of organs or tissues, or sharing of used needles. Transfusion-associated HCV infection is low, but a common cause of infection is injection drug use. The American Academy of Pediatrics (1998) suggests screening the following groups:
 All infants born to HCV-infected women
All infants born to HCV-infected women
Individuals who received blood products before 1992
The clinical course of HCV infection is variable. Incubation averages 6 to 7 weeks, with a range of 2 weeks to 6 months. Both acute and chronic HCV infection often produces only mild nonspecific symptoms or no symptoms at all (Bonkovsky and Mehta, 2001).
The length of time that maternal antibody is present in infants born to HCV-infected women must be considered, and screening should be done after the infant is 12 months old. However, a routine screening program, such as that for HBV, is not recommended. Current recommendations are to evaluate HCV-infected children at regular intervals to monitor for chronic hepatitis. Most children will be asymptomatic with evidence of chronic hepatitis on liver biopsy. Liver enzyme levels may fluctuate between periods of normal and elevated values.
Hepatitis D.: HDV is an important cause of acute and chronic liver disease. HDV is a defective RNA virus that requires the presence of HBV. HDV infection occurs primarily in hemophiliac patients and IV drug abusers. The incubation period is 2 to 8 weeks. Both acute and chronic forms are more severe than HBV infection and can lead to cirrhosis. Testing for HDV infection is recommended in children with chronic HBV infection or severe liver disease and in children with acute exacerbation of a previously stable liver disease.
Hepatitis E.: HEV infection is enterally transmitted. Transmission may occur through the fecal-oral route or from contaminated water. The incubation period is 2 to 9 weeks. This illness is uncommon in children, does not cause chronic liver disease, is not a chronic condition, and has no carrier state. The mortality rate resulting from submassive hepatic necrosis is low except in pregnant women in their third trimester, in whom mortality reaches 20%.
Hepatitis G.: HGV is a blood-borne virus that may also be transmitted by organ transplantation. High-risk groups include transfusion recipients, IV drug users, and individuals infected with HCV. Individuals with the virus are often asymptomatic, and most infections are chronic. The incubation period is unknown.
Diagnostic Evaluation
Diagnosis is based on the history (especially regarding possible exposure to a hepatitis virus); physical examination; and serologic markers (antibodies or antigens) indicating the presence of active infection with hepatitis A, B, or C or previous infection. Because the liver has a large functional reserve, abnormal laboratory tests may be the only indication of hepatitis. However, LFTs are not specific for the diagnosis of viral hepatitis. Although serum aspartate and alanine aminotransferase (AST and ALT) levels are markedly elevated in viral hepatitis, other diseases or conditions may cause their elevation. Serum bilirubin levels peak 5 to 10 days after clinical jaundice appears. When hepatitis is severe, albumin levels are depressed and prothrombin times are increased.
Diagnosis of viral hepatitis is based on the presence of specific viral markers. Diagnosis of acute HAV infection is based on the presence of anti-HAV immunoglobulin (immunoglobulin M [IgM]) antibody in the serum. HBV diagnosis depends on the presence of hepatitis B surface antigen (HBsAg) or anti-HBV core (anti-HBc) IgM antibody. Chronic HBV infection is associated with the persistence of HBsAg and HBV DNA markers. The diagnosis of HCV is based on the detection of anti-HCV antibodies and confirmation by polymerase chain reaction for hepatitis C RNA.
An abdominal ultrasound provides measurement of liver size, detection of cystic lesions and stones, and imaging of the gallbladder. Cholescintigraphy radionuclide imaging detects abnormalities in liver uptake, concentration, and excretory function. Finally, a liver biopsy aids in assessing the severity of the disease.
Pathophysiology
Pathologic changes occur primarily in the parenchymal cells of the liver and result in varying degrees of swelling, infiltration of liver cells by mononuclear cells, subsequent degeneration, necrosis, and fibrosis.
Hepatitis can be self-limited, and complete regeneration of liver cells without scarring may occur. However, some forms of hepatitis do not result in complete return of liver function. These include fulminant hepatitis, which is characterized by a severe, acute course and massive destruction of the liver, resulting in liver failure and death in 1 to 2 weeks. Subacute or chronic active hepatitis is characterized by progressive liver destruction, uncertain regeneration, scarring, and potential cirrhosis.
The initial anicteric (absence of jaundice) phase usually lasts 5 to 7 days and is often mistaken for influenza. Symptoms include nausea, vomiting, extreme anorexia, malaise, easy fatigability, arthralgia, skin rashes, slight to moderate fever, and epigastric or upper right quadrant abdominal pain. Dark urine is a symptom of the icteric (jaundice) phase. Pruritus may accompany jaundice and can be bothersome, but many children with acute viral hepatitis do not develop jaundice.
Therapeutic Management
Treatment options for viral hepatitis are limited. The goals of management include early detection, recognition of chronic liver disease, support and monitoring, and prevention of spread of the disease.
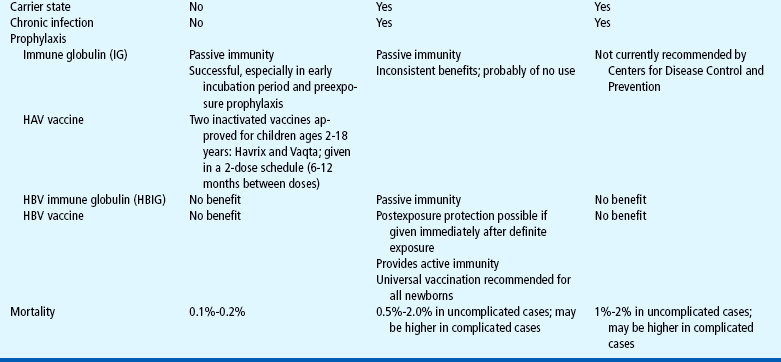
HAV infection is an acute disease that resolves with support and management of symptoms. HBV and HCV treatment is directed at managing the viral load to prevent further destruction of the liver. Currently, HBV and HCV are treated with interferons, naturally occurring proteins that exert antiviral, antiproliferative, and immunomodulatory effects. A recent interferon formulation, pegylated interferon, can be administered once a week and has been found to sustain plasma levels and enhance viral suppression (Karnam and Reddy, 2003). Lamivudine and adefovir are two other interferon analogs that suppress the replication of HBV (Yuen and Lai, 2001). A combination of α-interferon and ribavirin has resulted in a sustained response in only 50% of patients with HBV and HCV (Waters and Nelson, 2006).
Another important aspect of the therapeutic management of hepatitis involves hospitalization. Hospitalization is necessary if coagulopathy or fulminant hepatitis is present.
Prevention.: Proper hand washing and standard isolation precautions can prevent the spread of hepatitis. Prophylactic use of standard immune globulin (Ig) is effective in preventing HAV infection in situations of preexposure (e.g., anticipated travel to areas where HAV is prevalent) or in situations of postexposure during the early part of the incubation period. Hepatitis B immune globulin (HBIg) is effective in preventing HBV infection after exposure. Ig and HBIg must be administered less than 2 weeks after exposure.
Vaccines have been developed to prevent HAV and HBV infection. HBV vaccination is recommended for all newborns and for high-risk groups. HAV is also recommended for high-risk groups (see Immunizations, Chapter 10). Active immunizations are not available against HCV. It is possible to prevent HDV infection by preventing HBV infection.
Prognosis.: The prognosis for children with hepatitis is variable and depends on the type of virus. HAV usually causes a mild and brief illness with no carrier state. HBV causes a wide spectrum of acute and chronic illness. Approximately 5% of individuals develop chronic hepatitis B each year, and about half of these develop fulminant liver failure, leading to death in the absence of liver transplantation (Kim, Gores, Benson, and others, 2005). Hepatocellular carcinoma is a potentially fatal complication of HBV infection. HCV causes acute hepatitis that progresses to chronic disease in more than 85% of affected individuals, with approximately 15% to 20% developing cirrhosis or hepatocellular carcinoma (Centers for Disease Control and Prevention, 2001; Richmond, Dunning, and Desmond, 2004). Chronic HCV infection is the leading indication for liver transplantation in adults in the United States (Regev and Schiff, 2000).
Nursing Care Management
Nursing objectives depend on the severity of the hepatitis, the medical management, and factors influencing the control and transmission of the disease. Children with benign viral hepatitis are frequently cared for at home, and the clinic or office nurse must explain the medical therapy and control measures. If further assistance is needed for parents to comply with therapy, a public health nursing referral may be necessary.
A well-balanced diet and a realistic schedule of rest and activity adjusted to the child’s condition are encouraged. HAV is not infectious within a week after the onset of jaundice, and children may feel well enough to resume school. Parents are cautioned about administering any medication to the child, since normal dosages of many drugs may become dangerous because of the liver’s inability to detoxify and excrete them. Hand washing is the single most critical measure in reducing risk of transmission. The nurse should explain to parents and children the ways in which HAV (oral-fecal route) and HBV (parenteral route) are spread.
Nurses caring for young people with HBV infection and a known or suspected history of illicit drug use should help these teens realize the dangers of drug abuse. Nurses should stress the parenteral mode of transmission of hepatitis and encourage them to seek counseling through a drug program. HBV and HCV are chronic diseases that require frequent monitoring and management. Many communities have multidisciplinary clinics dedicated to the management of these diseases.
CIRRHOSIS
Cirrhosis occurs at the end stage of many chronic liver diseases, including biliary atresia (BA) and chronic hepatitis. Cirrhosis can also result from infectious, autoimmune, or toxic factors and from chronic diseases such as hemophilia and cystic fibrosis. A cirrhotic liver is irreversibly damaged.
Clinical manifestations in children are similar to those seen with all chronic liver disorders. Children exhibit jaundice, poor growth, anorexia, muscle weakness, and lethargy. Ascites, edema, GI bleeding, anemia, and abdominal pain may be present with impaired intrahepatic blood flow. Pulmonary function may be impaired because of pressure against the diaphragm from hepatosplenomegaly and ascites. Dyspnea and cyanosis may occur, especially on exertion. Intrapulmonary arteriovenous shunts may develop and cause hypoxemia. Spider angiomas and prominent blood vessels are often present on the upper torso.
Therapeutic Management
Therapy is directed toward (1) frequent assessment of liver status with physical examination and LFTs and (2) management of specific complications. The only successful treatment for end-stage liver disease and liver failure may be liver transplantation, which has improved the prognosis substantially for many children with cirrhosis. Currently, the 1-year survival rate for liver transplantation is 85%. Increasing numbers of recipients are reaching their second decade after transplant. The increasing life span after transplantation is related to advances in surgical techniques and improved preoperative, intraoperative, and postoperative care (Atkison, Ross, Williams, and others, 2002).
Prognosis.: Liver transplantation has revolutionized the approach to liver cirrhosis. Liver failure and cirrhosis are indications for transplantation. Liver transplantation reflects the failure of other medical and surgical measures to prevent or treat cirrhosis. Careful monitoring of the child’s condition and quality of life is necessary to evaluate the need for and timing of transplantation (see Family Focus box).
 FAMILY FOCUS
FAMILY FOCUSIn many cases the child and family must cope with an uncertain progression of the disease. The only hope for long-term survival may be liver transplantation. Transplantation can be successful, but the waiting period may be long, and there are many more children in need of organs than there are donors. The procedure is expensive and is performed only at designated medical centers, which are often far from the family’s home. The nurse should recognize the unique stresses of coping with end-stage liver disease and waiting for transplantation and assist the family in coping with these stressors. The assistance of social workers and support from other parents can be beneficial.
Nursing Care Management
Nursing care of the child with cirrhosis is determined by the cause of the cirrhosis, the severity of complications, and the prognosis. The prognosis for life is poor unless successful liver transplantation occurs. Nursing care of this child is similar to that for any child with a life-threatening illness (see Chapter 18). Hospitalization is usually required when complications occur.
BILIARY ATRESIA
BA is a destructive, idiopathic, inflammatory process that leads to fibrosis and obliteration of the biliary tree (Emerick and Whitington, 2006). BA has been detected in 3.7 in 10,000 live births (Chen, Chang, Du, and others, 2006). The disorder is more common in girls and preterm infants. In the United States, the incidence is twice as high in African-Americans as in Caucasian infants, and more common in Chinese than in either Japanese or Caucasian populations.
Etiology and Pathophysiology
The exact cause of BA is unknown. Because BA has two distinct forms, postnatal and fetal-embryonic, different pathogenic mechanisms are suggested. Postnatal BA represents 65% to 90% of cases and is probably the result of infection or an immune-mediated mechanism. Jaundice, manifesting with yellow discoloration of the skin or sclerae, is the most common early symptom of BA. Jaundice, indicating cholestasis (the accumulation of compounds that cannot be excreted because of occlusion or obstruction of the biliary tree), can be visible at a total serum bilirubin concentration as low as 5.0 mg/dl. An abnormal direct bilirubin has been designated as greater than 1.0 mg/dl if the total bilirubin is less than 5 mg/dl or a value of direct bilirubin that represents more than 20% of the total bilirubin if it is greater than 5 mg/dl (Emerick and Whitington, 2006). Direct hyperbilirubinemia first appears after the resolution of physiologic jaundice. Jaundice is often associated with pale stool and dark urine. Histologic study demonstrates bile duct remnants and a progressive inflammatory process. In the fetal embryonic form of BA, which represents 10% to 35% of cases, there is a congenital absence of biliary ductal patency and an absence of bile duct remnants. Many infants have associated congenital anomalies. Varying degrees of cholestasis occur, resulting in retention of irritants and toxins. Injury to the liver occurs as the result of the inflammation caused by the cholestasis.
Diagnostic Evaluation
Early diagnosis is the key to the survival of the child with BA. Infants who undergo surgery in the first 60 days of life have an 80% chance of establishing bile flow. Between 60 to 90 days of life, the chance of reestablishing flow drops to 50%, and after 90 days to 10% (Chen, Chang, Du, and others, 2006). The typical infant is thriving, appears well, and has only very mild jaundice during the first 6 to 8 weeks (Emerick and Whitington, 2006) but will soon begin failing to thrive. Several clinical signs may indicate the presence of BA (Box 24-10). Blood tests should include a CBC, electrolytes, bilirubin, and liver enzymes. Additional laboratory analyses, including α1-antitrypsin level, TORCH titers (see Maternal Infections, Chapter 9), hepatitis serology, α-fetoprotein, urine cytomegalovirus, and a sweat test, are indicated to rule out other conditions that cause persistent cholestasis and jaundice. Abdominal ultrasonography allows inspection of the liver and biliary system. Hepatobiliary scintigraphy demonstrates biliary patency but does not provide diagnostic certainty. Endoscopic retrograde cholangiopancreatography (ERCP) is performed in very young infants. This procedure, which is done using general anesthesia, has an 80% reported diagnostic accuracy. Percutaneous liver biopsy is highly reliable when the biopsy contains specimens from a number of portal areas. Definitive diagnosis of BA is obtained during surgical laparotomy and an intraoperative cholangiogram.
BOX 24-10 Clinical Manifestations of Extrahepatic Biliary Atresia
Earliest manifestation and most striking feature of disorder
May be present at birth, but usually not apparent until age 2 to 3 weeks
Stools lighter than expected or white or tan
Hepatomegaly and abdominal distention common
Therapeutic Management
The primary treatment of BA is hepatic portoenterostomy (Kasai procedure), in which a segment of intestine is anastomosed to the resected porta hepatis to attempt bile drainage. Bile drainage is achieved in approximately 80% to 90% of infants who undergo surgery when younger than 10 weeks of age (Ohi, 2001). However, progressive cirrhosis still occurs in many children, necessitating liver transplantation. Prophylactic antibiotics are given after the Kasai procedure to minimize the risk of ascending cholangitis.
Medical management is primarily supportive. It includes nutritional support with infant formulas that contain medium-chain triglycerides and essential fatty acids. Supplementation is usually required with fat-soluble vitamins; a multivitamin; and minerals, including iron, zinc, and selenium. Aggressive nutritional support with continuous tube feedings or TPN is indicated for moderate to severe failure to thrive. The enteral solution should be low in sodium. Ursodeoxycholic acid is used to treat pruritus and hypercholesterolemia.
Prognosis.: Untreated BA results in progressive cirrhosis and death in most children by 2 years of age. The Kasai procedure improves the prognosis but is not a cure. Biliary drainage can often be achieved if the surgery is done before the intrahepatic bile ducts are destroyed. Long-term survival has been reported in children who receive the Kasai procedure; however, even with successful bile drainage, many children ultimately develop liver failure.
Advances in surgical techniques and the use of immunosuppressive and antifungal drugs have improved the success of transplantation. The major obstacle continues to be a shortage of donor livers. Reduced-size, split-liver transplantation, retransplantation, and increased public awareness may improve donor organ availability in the future (see Ethical Case Study).
ETHICAL CASE STUDY: Liver Transplant

J.T. is a 3-year-old African-American boy diagnosed with biliary atresia at the age of 6 months. Although he underwent a Kasai procedure, no bile ducts were found. His condition has progressively deteriorated until he is in need of a liver transplant. There are not enough donated livers to go around. If J.T. receives a liver, his biliary atresia will be cured, and he can be expected to live a normal life. Should he and other children like him with curable diseases receive preference over adults with diseases that could recur in the new liver (such as those with hepatitis B or C)?
Treat All Involved with Respect
Representatives of all sides of the issue from various racial, ethnic, and socioeconomic groups should be included in the discussion.
The voices representing all sides of this issue should be listened to, including pediatric proponents, adult proponents, geriatric proponents, and recovered alcoholics.
After all sides have had their say, those who set the priorities regarding who gets available livers will decide whether the priorities need to be revised. They might want to consider increasing the pool of available livers by allowing relatively healthy older adults to donate to other adults whose disease is likely to return in the new liver.
Nursing Care Management
Nursing interventions for the child with BA include support of the family before, during, and after surgical procedures and education regarding the treatment plan. In the postoperative period of a portoenterostomy, nursing care is similar to that after major abdominal surgery. Family members need education relating to the proper administration of medications and nutritional therapy, including special formulas, vitamin and mineral supplements, tube feedings, or parenteral nutrition. Pruritus can often be relieved by drug therapy or comfort measures such as baths.
Children and their families also need psychosocial support. The uncertain prognosis, discomfort, and waiting for transplantation produce stress, and hospitalizations, pharmacologic therapy, and nutritional therapy impose financial burdens on the family. Families can receive help from the Children’s Liver Disease Foundation,* an organization that provides educational materials, programs, and support systems.


 answers to CRITICAL THINKING EXERCISE
answers to CRITICAL THINKING EXERCISE