Membrane Receptors and Signal Transduction
Learning objectives
After reading this chapter you should be able to:
 Distinguish between steroid and polypeptide hormones, and outline their mechanisms of action.
Distinguish between steroid and polypeptide hormones, and outline their mechanisms of action.
Describe G-protein coupled receptors.
Outline the activation of downstream intracellular signaling cascades by heterotrimeric G-proteins.
Discuss the generation of second messengers such as cyclic AMP, inositol trisphosphate (IP3), diacylglycerol (DAG) and Ca2+, and explain how they activate key protein kinases.
Explain how phospholipases generate a diverse array of lipid second messengers.
Discuss how the generation of a variety of second messengers can amplify signals and lead to the generation of specific biological responses.
Introduction
Cellular signals are processed by cassettes comprising specific receptors-effector elements and regulatory proteins
Cells sense, respond to, and integrate a multiplicity of signals from their environment. These signals can be hormones produced elsewhere from their site of action (endocrine signaling); signals generated locally to the target cell (paracrine signaling); signals from cells in physical contact with the target cell (juxtacrine signaling); or signals generated by the target cell itself (autocrine signaling). Hydrophobic signals and small molecules can cross the plasma membrane and exert their effects via receptors within the cell, whereas the majority of signals are hydrophilic, are not able to cross the lipid plasma membrane and require specific cell surface membrane receptors. In either case, signals are sensed and processed by cellular signal transduction cassettes that comprise specific receptors, effector signaling elements, and regulatory proteins. These signaling cassettes serve to detect, amplify, and integrate the diverse external signals to generate the appropriate cellular response (Fig. 40.1). Ultimately, signals can rapidly alter cellular processes such as exocytosis and metabolism, or alter transcription factor activity, leading to changes in target gene expression.
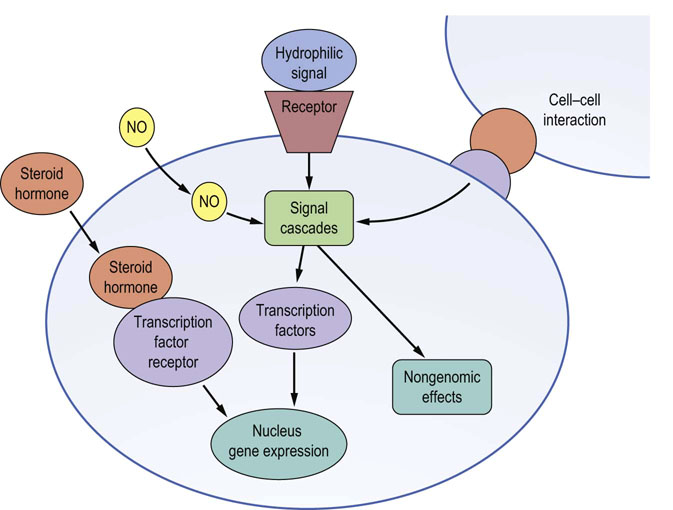
In this chapter, we first discuss how cell surface receptors sense and transduce their specific signal by transmembrane coupling to effector enzyme systems, generating low-molecular-mass molecules termed second messengers. We then discuss the diversity of these second messengers and how they influence the activity of a range of key protein kinases with distinct substrates that ultimately determine the type of obtained biological response.
Types of hormone and monoamine receptors
Hormone receptors for steroid hormones are different from those for polypeptide hormones and monoamines
Hormones are biochemical messengers that act to orchestrate the responses of different cells within a multicellular organism (Chapter 39). They are generally synthesized by specific tissues and secreted directly into the blood, which transports them to their target responsive organs. Hormone signaling can broadly be subdivided into two major classes:
Hormones achieve their biological effects by interacting with specific receptors to induce intracellular signaling cascades (Table 40.1).
Table 40.1
Classification of membrane receptors
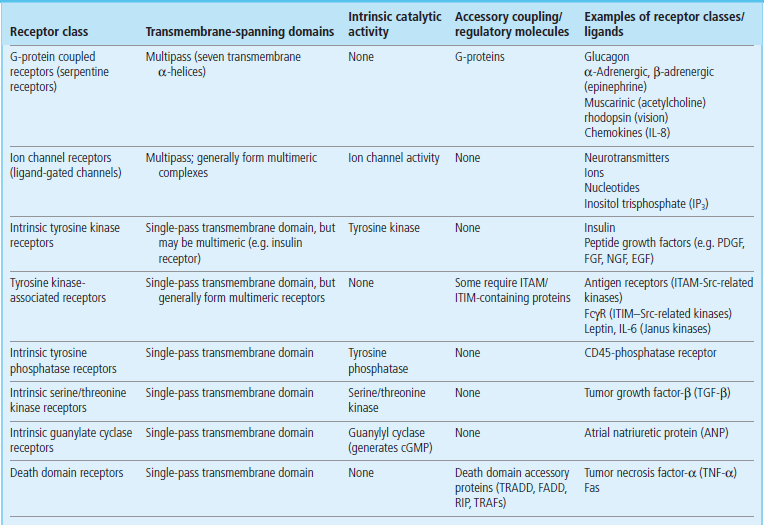
cGMP, cyclic guanosine monophosphate; FADD, Fas-associated death domain; FcγR, Fc-γ receptor (receptor for immunoglobulin G); IL, interleukin; ITAM/ITIM, immunoreceptor tyrosine activation/inhibition motif; RIP, receptor-interacting protein; Src, Src-tyrosine kinase; TRADD, TNF-receptor-associated death domain; TRAFs, TNF-receptor-associated factors. PDGF, platelet-derived growth factor; FGF, fibroblast growth factor; EGF, epidermal growth factor; NGF, nerve growth factor FGF, fibroblast growth factor; EGF, epidermal growth factor; NGF, nerve growth factor
Steroid hormones traverse cell membranes
Because of the cholesterol-based nature of their structure, steroid hormones, such as glucocorticoids, mineralocorticoids, sex hormones, and vitamin D, can traverse the plasma membrane of cells to initiate their responses via cytoplasmic located receptors called steroid hormone receptors (Fig. 40.1). These receptors belong to a superfamily of cytoplasmic receptors called the intracellular receptor superfamily, which also transduce signals from other small hydrophobic signaling molecules such as the tyrosine-derived thyroid hormones (e.g. thyroxine) and the vitamin A-derived retinoids (e.g. retinoic acid).
Intracellular receptors for steroid and thyroid hormones, and retinoids, are transcription factors
The intracellular receptors for these steroid and thyroid hormones, and retinoids, are transcription factors; they bind to regulatory regions of the DNA of genes that are responsive to the particular steroid/thyroid hormone. Such ‘ligand binding’ (ligation) induces a conformational change in the transcription factor that allows it to activate or repress gene induction. Although all the target cells have specific receptors for the individual hormones, they express distinct combinations of cell type-specific regulatory proteins that cooperate with the intracellular hormone receptor to dictate the precise repertoire of genes that are induced. Hence the hormones induce distinct sets of responses in different target cells (Chapter 35).
Polypeptide hormones act through membrane receptors
In contrast to the steroid hormones, polypeptide hormones cannot cross cell membranes and must initiate their effects on their target cells via specific cell surface receptors (Fig. 40.1). As they do not themselves enter the target cell, they are sometimes termed ‘first messengers’. Binding to the specific cell surface receptor causes a conformational change in that receptor that can engage signaling cascades in several different ways. Receptor binding can:
Regulate the production of low-molecular-weight signaling molecules, such as cyclic adenosine monophosphate (cAMP) or calcium, which are called ‘second messengers’
Alter the intrinsic catalytic activity of the receptor.
Alter the recruitment of regulatory molecules to the receptor (Table 40.1).
Other molecules that signal through membrane receptors
In addition to polypeptide hormones, a wide range of signaling molecules use transmembrane signal transduction cassettes to elicit their biological effects. Such signals include polypeptide growth factors, polypeptide signals that mediate inflammation and immunity (cytokines and chemokines) and small hydrophilic molecules (such as acetylcholine, purines, nucleotides or inositol trisphosphate) (Table 40.1).
Some low-molecular-mass signaling molecules traverse the cell membrane
Although most extracellular signals mediate their effects via receptor–ligand interaction of either cell surface or cytoplasmic receptors, some low-molecular-mass signaling molecules are able to traverse the plasma membrane and directly modulate the activity of the catalytic domains of transmembrane receptors or cytoplasmic signal transducing enzymes (Fig. 40.1). For example, nitric oxide (NO), which has a variety of functions including the relaxation of smooth muscle cells in blood vessels, can stimulate guanylyl cyclase, leading to the generation of the second messenger, cGMP. Patients with angina pectoris are treated with glyceryl trinitrate (GTN), which is converted to NO, resulting in relaxation of blood vessels delivering oxygen and nutrients to the heart. The consequent improvement in oxygen delivery to the heart muscle eases the pain that was caused by inadequate blood flow to the heart.
Receptor coupling to intracellular signal transduction
Membrane receptors couple to signaling pathways utilizing diverse mechanisms
Some membrane receptors, for example the β-adrenergic receptors or the antigen receptors on lymphocytes, have no intrinsic catalytic activity and serve simply as specific recognition units. These receptors use a variety of mechanisms, including adaptor molecules or catalytically active regulatory molecules such as G-proteins (guanosine triphosphatases, GTPases, which hydrolyze GTP) to couple them to their effector signaling elements, which are generally enzymes (often called signaling enzymes or signal transducers) or ion channels (Fig. 40.2). In contrast, other receptors – such as the intrinsic tyrosine kinase receptors for insulin and many growth factors, or the intrinsic serine kinase receptors for molecules like transforming growth factor-β (TGF-β) – have extracellular ligand-binding domains and cytoplasmic catalytic domains. After receptor ligation, these receptors can directly initiate their signaling cascades by phosphorylating and modulating the activities of target signal-transducing molecules (downstream signaling enzymes). These in turn propagate the growth factor signal by modulating the activity of further specific signal transducers or transcription factors, leading to gene induction (Chapter 35). Furthermore, sensory systems such as vision (Chapter 41.2), taste and smell use similar mechanisms of cell surface membrane receptor-coupled signal transduction (Table 40.1).

Fig. 40.2 Mechanism of G-protein signaling.
In the inactive state, G-proteins exist as heterotrimers with GDP bound tightly to the α-subunit. None of the subunits are integral membrane proteins; however, the G-protein is anchored to the plasma membrane by lipid modification of the γ-subunits (prenylation) and some of the α-subunits (myristoylation in the Giα family). Ligation of the receptor (R) drives exchange of GDP for GTP and induces a conformational change in Gα, which results in a decrease in its affinity both for the receptor and for the βγ-subunits, leading to dissociation of the receptor–G-protein complex. The activated Gα (GTP-bound) or the released βγ-subunits, or both, can then interact with one or more effectors, to generate intracellular second messengers that activate downstream signaling cascades. Signaling is terminated by the intrinsic GTPase activity of the α-subunit, which hydrolyzes GTP to GDP to allow reassociation of the inactive, heterotrimeric G-protein, Gαβγ.
Some receptors possess intrinsic protein kinase activity
Ligand binding to many growth factor receptors stimulates protein kinase activity of an intracellular domain of the receptor complex. The activated receptor subsequently phosphorylates substrate proteins, in which the γ-phosphate from ATP is transferred to the side-chain hydroxyl groups on serine, threonine or tyrosine residues. All receptor protein kinases are either specific serine/threonine kinases or tyrosine kinases, but never both. Furthermore, protein kinases phosphorylate substrates at specific serine, threonine or tyrosine residues, depending on the sequence surrounding the site of phosphorylation. Upon ligand binding, receptor protein kinases autophosphorylate (self-phosphorylate). The introduction of the bulky charged phosphate moiety during autophosphorylation or onto other substrate proteins markedly alters the conformation of the protein, leading to changes in activity or acting as docking sites for other (adapter) proteins. Adapter proteins contain specific domains that recognize and bind to the phosphorylated proteins. As receptor protein kinases often phosphorylate at several sites, this can lead to several different adapter proteins being recruited to the activated receptor complex. Subsequently these adapter proteins can engage several different signaling pathways.
The example of insulin signaling
An example of this is insulin signaling. Insulin binding to the insulin receptor (IR) causes activation and autophosphorylation of the intracellular tyrosine kinase domains. Adapter proteins including the insulin receptor substrate (IRS) proteins and Shc (Src-homology and collagen-like) proteins bind the phosphotyrosine residues on the IR. Next, IRSs are themselves tyrosine phosphorylated by the IR on tyrosine residues, generating docking sites for the lipid kinase phosphatidylinositol 3′-kinase (PI3K), which generates phosphatidylinositol 3,4,5-trisphosphate (PIP3) at the plasma membrane. Newly formed PIP3 recruits the serine/threonine protein kinase Akt to the plasma membrane, and Akt is subsequently phosphorylated and activated by other protein kinases (Fig. 40.3). Activation of Akt is the key signaling pathway by which insulin exerts the majority of its metabolic effects, including stimulation of glucose transport and suppression of gluconeogenesis (Chapter 21). Binding of Shc to the activated IR leads to recruitment of growth factor receptor-bound protein (Grb2) to Shc, which subsequently activates a guanine nucleotide exchange factor (SOS), which stimulates the small G-protein Ras, which initiates the a protein kinase cascade in which several protein kinases phosphorylate each other in turn. Signaling through this pathway is associated with the mitogenic, growth-promoting actions of insulin. Therefore ligand binding can initiate multiple signaling pathways with different cellular effects.
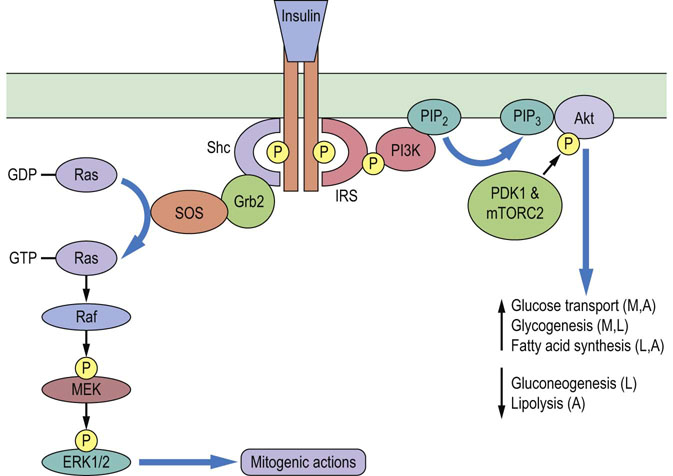
Fig. 40.3 Insulin signaling pathways.
Insulin binding to the dimeric insulin receptor tyrosine kinase stimulates autophosphorylation. The insulin receptor substrate (IRS) and Src-homology and collagen-like (Shc) adapter proteins bind to the phosphotyrosines on the insulin receptor. IRSs are subsequently phosphorylated by the insulin receptor, generating docking sites for phosphatidylinositol 3'-kinase (PI3K), which generates the phosphorylated lipid signaling molecule phosphatidylinositol 3,4,5-trisphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate (PIP2). PIP3 recruits the serine/threonine protein kinase Akt to the plasma membrane, where Akt is phosphorylated and activated by phosphoinositide-dependent kinase 1 (PDK1) and the mammalian target of rapamycin complex 2 (mTORC2). Akt is essential for the metabolic effects of insulin in muscle (M), liver (L) and adipose tissue (A). Insulin receptor-bound Shc recruits growth factor receptor-bound protein 2 (Grb2), which is bound to the guanine nucleotide exchange factor Son of Sevenless (SOS). SOS catalyzes GDP–GTP exchange on the small G-protein Ras. The GTP-bound active Ras initiates a protein kinase cascade in which the protein kinase Raf phosphorylates and activates another protein kinase MEK, which subsequently phosphorylates and activates the protein kinases ERK1 and ERK2, which mediate many of the mitogenic actions of insulin.
Some membrane receptors are coupled to G-proteins
G-protein coupled receptors comprise a superfamily of structurally related receptors for hormones, neurotransmitters, inflammatory mediators, proteinases, taste and odorant molecules, and light photons. A classic example of this class of receptors is the β-adrenergic receptor (for which the ligand is epinephrine) as its structure–function properties have been extensively studied with respect to its activation of signal transduction cascades. G-protein coupled receptors are integral membrane proteins characterized by the seven transmembrane-spanning helices within their structure. They generally comprise an extracellular N-terminus, seven transmembrane-spanning α-helices (20–28 hydrophobic amino acids each), three extracellular and intracellular loops, and an intracellular C-terminal tail. Ligands, such as epinephrine, typically bind to the G-protein coupled receptor by sitting in a pocket formed by the transmembrane helices (Fig. 13.5). G-protein coupled receptors have no intrinsic catalytic domains; upon activation they recruit G-proteins via their third cytoplasmic loop to couple to their signal transduction elements. G-protein coupled receptors are often the target of drugs; indeed it has been estimated that approximately 30% of all currently available therapeutics act on G-protein coupled receptors. Furthermore, using the information gained by sequencing the human genome, it is apparent that there are many additional members of the G-protein coupled receptor family for which the signal has not yet been identified.
G-proteins regulate a diverse range of biological processes
G-proteins constitute a group of regulatory molecules that are involved in the regulation of a diverse range of biological processes, including signal transduction, protein synthesis, intracellular trafficking (targeted delivery to the plasma membrane or intracellular organelles) and exocytosis, as well as cell movement, growth, proliferation and differentiation. The G-protein superfamily predominantly comprises two major subfamilies: the small, monomeric Ras-like G-proteins (Chapter 42) and the heterotrimeric G-proteins. Heterotrimeric G-proteins regulate the transduction of transmembrane signals from cell surface receptors to a variety of intracellular effectors, such as adenylyl cyclase, phospholipase C (PLC), cGMP-phosphodiesterase (PDE) and ion channel effector systems (Fig. 40.2). Heterotrimeric G-proteins consist of 3 subunits: α (39–46 kDa), β (37 kDa), and γ (8 kDa). In general, effector specificity is conferred by the α-subunit, which contains the GTP-binding site and an intrinsic GTPase activity. However, it is now widely accepted that βγ-complexes can also directly regulate effectors such as phospholipase A2 (PLA2), PLC-β isoforms, adenylyl cyclase and ion channels in mammalian systems, as well as cellular responses such as mating factor receptor pathways in yeast. Four major subfamilies of α-subunit genes have been identified on the basis of their cDNA homology and function: Gsα, Giα, Gq/11α, and G12/13α (Table 40.2). Many of these Gα subunits have been shown to exhibit a rather ubiquitous pattern of expression in mammalian systems, at least at the mRNA level, but it is also clear that certain α-subunits have a tissue-restricted profile of expression. Moreover, there is evidence of differential expression of α-subunits during cellular development.
Table 40.2
Properties of mammalian G-protein α-subunits
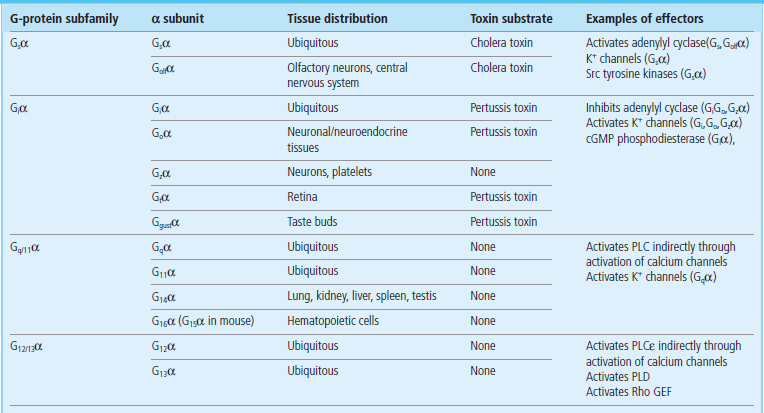
cGMP, cyclic guanosine monophosphate; PLC, phospholipase C; PLD, phospholipase D; Rho GEF, Rho GTPase guanine nucleotide exchange factor.
G-proteins act as molecular switches
Heterotrimeric G-proteins regulate transmembrane signals by acting as molecular switches, linking cell surface G-protein coupled receptors to one or more downstream signaling molecules (Fig. 40.2). Ligation of the receptor initiates an interaction with the inactive, GDP-bound heterotrimeric G-protein. This interaction drives exchange of GDP for GTP, inducing a conformational change in Gα, which results in a decrease in its affinity for both the receptor and the βγ-subunits, leading to dissociation of the receptor–G-protein complex. The activated Gα (GTP-bound) or released βγ-subunits, or both, can then interact with one or more effectors to generate intracellular second messengers, which activate downstream signaling cascades. Signaling is terminated by the intrinsic GTPase activity of the α-subunit, which hydrolyzes GTP to GDP to allow reassociation of the inactive heterotrimeric G-protein (Gαβγ).
Second messengers
 Advanced concept box Bacterial toxins which target G-proteins cause several diseases
Advanced concept box Bacterial toxins which target G-proteins cause several diseases
A variety of bacterial toxins exert their toxic effects by covalently modifying G-proteins and hence irreversibly modulating their function. For example, cholera toxin from Vibrio cholerae contains an enzyme (subunit A) that catalyzes the transfer of ADP-ribose from intracellular NAD+ to the α-subunit of Gs; this modification prevents the hydrolysis of Gs-bound GTP, resulting in a constitutively (permanently) active form of the G-protein. The resulting prolonged increase in cAMP concentrations within the intestinal epithelial cells leads to PKA-mediated phosphorylation of Cl– channels, causing a large efflux of electrolytes and water into the gut, which is responsible for the severe diarrhea that is characteristic of cholera. Enterotoxin action is initiated by specific binding of the B (binding) subunits of the cholera toxin (AB5) to the oligosaccharide moiety of the monosialoganglioside, GM1, on epithelial cells. A similar molecular mechanism has been attributed to the action of the heat-labile enterotoxin, labile toxin secreted by several strains of Escherichia coli responsible for ‘traveler’s diarrhea’.
In contrast, pertussis toxin (another AB5 toxin) from Bordetella pertussis, the causative agent of whooping cough, catalyzes the ADP-ribosylation of Giα, which prevents Giα from interacting with activated receptors. Hence, the G-protein is inactivated and cannot act to inhibit adenylyl cyclase, activate PLA2 or PLC, open K+ channels, or open and close Ca2+ channels, causing a generalized uncoupling of hormone receptors from their signaling cascades.
Cyclic AMP (cAMP) is a key molecule in signal transduction
cAMP is a small molecule that has a key role in the regulation of intracellular signal transduction cAMP is derived from ATP by the catalytic action of the signaling enzyme adenylyl cyclase (Fig. 40.4). This cyclization reaction involves the intramolecular attack of the 3′-OH group of the ribose unit on the α-phosphoryl group of ATP to form a phosphodiester bond; this is driven by the subsequent hydrolysis of the released pyrophosphate. The activity of cAMP is terminated by the hydrolysis of cAMP to 5′-AMP by specific cAMP phosphodiesterases.

Glucagon and β-adrenergic receptors are coupled to cAMP
Glucagon and β-adrenergic receptors are coupled to the generation of cAMP. The β-adrenergic hormone epinephrine induces the breakdown of glycogen to glucose-1-phosphate in muscle and, to a lesser extent, in the liver. The breakdown of glycogen in the liver is predominantly stimulated by the polypeptide hormone glucagon, which is secreted by the pancreas when plasma glucose is low (Chapters 13 and 21). One of the earliest signaling events after binding of these hormones to their receptors is the generation of cAMP. The importance of cAMP in regulating glycogen breakdown was demonstrated by a series of experiments showing not only that hormones that activate adenylyl cyclase activity in liver and muscle cells, also stimulate glycogen breakdown, but also demonstrated that cell-permeant analogues of cAMP, such as dibutyryl cAMP, can mimic the effects of these hormones in inducing glycogen breakdown.
Adenylyl cyclase is regulated by G-protein α-subunits
The β-adrenergic and glucagon receptors are coupled to adenylyl cyclase activation by the action of a specific form of the α-subunit of the G-protein, termed Gsα. Although hydrolysis of GTP by the intrinsic GTPase of the Gsα-subunit acts to switch off adenylyl cyclase activation, the hormone–receptor complex must also be deactivated to return the cell to its resting, unstimulated state. In the case of β-adrenergic receptors, this receptor desensitization, which occurs after prolonged exposure to the hormone, involves phosphorylation of the C-terminal tail of the hormone-occupied β-adrenergic receptor by a kinase known as β-adrenergic receptor kinase. Other G-protein coupled receptors, such as α2-adrenergic receptors in smooth muscle, act to inhibit adenylyl cyclase and cAMP generation. In this case, receptors are coupled to a specific inhibitory form of the α-subunit of the G-protein, termed Giα (Table 40.2), which inhibits adenylyl cyclase activity, reducing cAMP concentrations.
Signals can activate different receptor subtypes, with different consequences
Receptor subtypes are expressed in a tissue-specific manner for some signals such as epinephrine and angiotensin II. These different receptor subtypes may couple in different ways; for example, epinephrine stimulates cAMP synthesis through Gsα-coupled β-adrenergic receptors in skeletal muscle. whereas in smooth muscle, epinephrine inhibits cAMP synthesis through Giα-coupled α2-adrenergic receptors. Therefore the same signal can have differing effects on intracellular signaling cascades dependent on the tissue being examined.
Protein kinase A
cAMP transduces its effects on glycogen–glucose-1-phosphate interconversion by regulating a key signaling enzyme, protein kinase A (PKA), which phosphorylates target proteins on serine and threonine residues.
Protein kinase A binds cAMP and phosphorylates other enzymes
PKA is a multimeric enzyme comprising two regulatory (R) subunits and two catalytic (C) subunits: the R2C2 tetrameric form of PKA is inactive, but binding of four molecules of cAMP to the R-subunits leads to the release of catalytically active C-subunits, which can then phosphorylate and modulate the activity of two key enzymes, phosphorylase kinase and glycogen synthase (Fig. 40.5), which are involved in regulation of glycogen metabolism (Chapter 13).
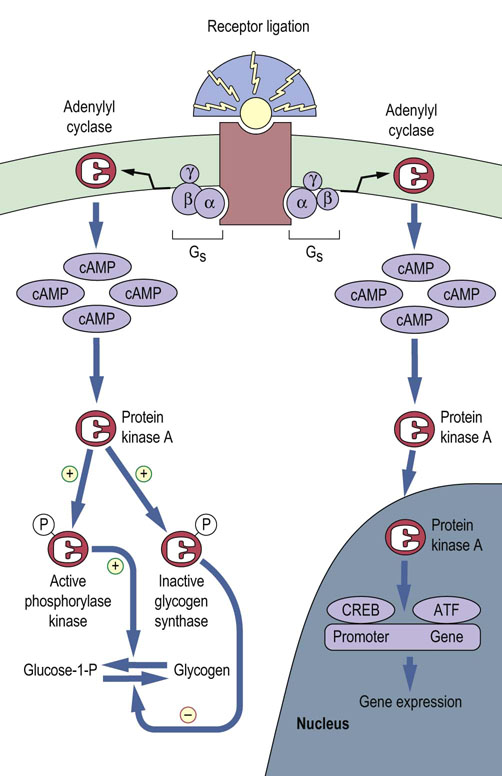
Fig. 40.5 Protein kinase A (PKA) acts as a signaling enzyme for the second messenger, cAMP.
Binding of a stimulatory G-protein (Gs) to the hormone–receptor complex activates adenylyl cyclase, which catalyzes the production of cAMP. PKA is activated by binding four molecules of cAMP. Translocation of PKA into the nucleus modulates activity of transcription factors, for example CREB and ATF (see text), leading to induction or repression of gene expression. (Compare Fig. 13.6.)
Many other cellular responses can be mediated by the cAMP–PKA signaling cassette
PKA-mediated phosphorylation can regulate the activity of a number of ion channels, such as K+, Cl– and Ca2+ channels, and that of phosphatases involved in the regulation of cell signaling. In addition, translocation of activated PKA into the nucleus allows modulation of the activity of transcription factors such as the cAMP response element-binding protein (CREB) and the activation transcription factor (ATF) families, leading to either the induction or the repression of expression of specific genes (Fig. 40.5 and Chapter 34).
cAMP can stimulate cellular signaling independent of PKA
It has become clear that not all actions of cAMP are mediated by PKA. cAMP can also bind to Epacs (exchange proteins directly activated by cAMP), which are guanine-nucleotide-exchange factors for the Rap small GTPase. Epac activation has been implicated in the anti-inflammatory action of cAMP and in neuronal growth and development.
Signal cascades amplify signals initiated by receptor binding
The concentrations of hormones and other signals are often in the nanomolar (10–9 mol/L) or picomolar (10–12 mol/L) range. As a consequence, it is important that the signal is amplified. Multilayered signal transduction cascades cause substantial amplification of the original signal at each stage of the cascade, ensuring that binding of only a few hormone molecules leads to an appropriate biological response. For example, stimulation of glycogen breakdown by glucagon or epinephrine involves amplification of the signal at the level of G-proteins, adenylyl cyclase, protein kinase A and phosphorylase, such that many glucose-1-phosphate molecules are released (Fig. 40.6).
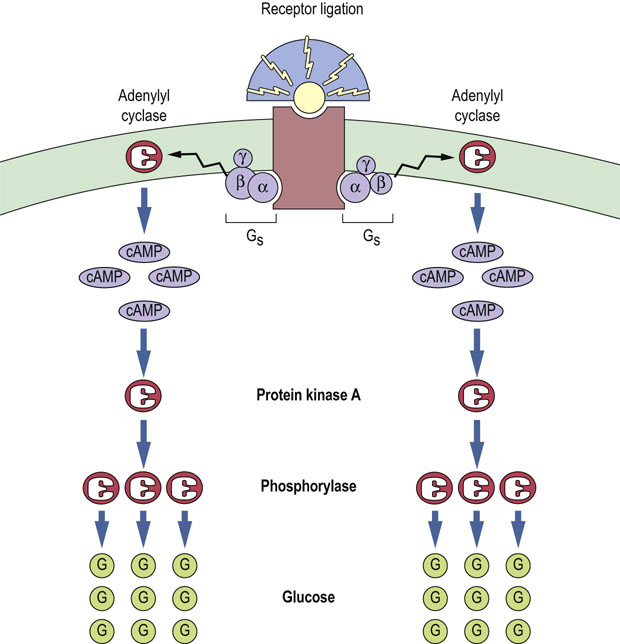
Fig. 40.6 Signal cascade induces amplification of hormone signal.
Each activated hormone–receptor complex can stimulate multiple Gs molecules. Each adenylyl cyclase can catalyze the generation of many cAMP molecules, and each protein kinase A can activate many phosphorylase molecules, leading to the breakdown of glycogen into many glucose-1-phosphate molecules as a result of glycogen degradation. (Compare Fig. 13.4.)
 Clinical box Heart failure – potential for targeting β-adrenergic receptor (βAR) signaling by gene therapy?
Clinical box Heart failure – potential for targeting β-adrenergic receptor (βAR) signaling by gene therapy?
Heart disease is a major cause of death in developed countries and despite advances with electrophysiologic therapies and pharmacologic agents including β-adrenergic receptor (βAR) blockers (commonly known as β-blockers), angiotensin-converting enzyme (ACE) inhibitors and diuretics, the intracellular signal transduction abnormalities that underlie the development and progression of heart failure are not addressed.
Normally, binding of catecholamines (epinephrine and norepinephrine) to βARs activates Gαs to stimulate adenylyl cyclase and generate cAMP, resulting in the activation of PKA and, ultimately, via calcium-mediated excitation–contraction coupling, regulation of heart rate (chronotropy), contractility (inotropy) and relaxation (lusitropy). By contrast, in heart failure, myocytes exhibit characteristic changes in βAR signaling and consequent calcium mobilization, resulting in reduction of contractile function. Specifically, βAR expression is reduced and the remaining receptors appear to be desensitized due to increased levels of the negative regulator of βAR signaling, βAR kinase (βARK). Moreover, specific polymorphisms (mutations) in βARs have been identified in humans that are associated with poor prognosis following heart failure. As a consequence, much interest has focused on the therapeutic potential of modulating signaling via this pathway by cardiac gene therapy.
Gene therapy, where genetic material is introduced directly into the cell using viral vectors is the only approach that would effectively target signaling proteins within cardiomyocytes, in contrast to drugs that also have actions in tissues other than cardiac muscle. For example, studies in animal models to date have suggested that there is potential therapeutic value in terms of contractility and myocyte survival in increasing expression of β2AR, but not β1AR. Moreover, expression of the carboxy-terminal sequence of βARK (βARKct), which inhibits βARK-mediated desensitization of βARs, has been shown to prevent heart failure in rodents. Likewise, overexpression of downstream βAR effectors such as ACVI, the major isoform of adenylyl cyclase in myocardium, has produced therapeutic effects. Finally, as βAR coupling to PKA signaling regulates the receptors and ion channels responsible for the calcium signals required for excitation–contraction coupling, downstream inotropic regulators of contractile function, such as the sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) and the EF hand-type calcium sensing protein (S100A1), which are also downregulated in heart failure, are also currently under study as potential therapeutic targets. Indeed, a clinical trial using adeno-associated virus to increase SERCA2a is currently underway in patients with advanced heart failure.
Phosphodiesterases terminate the cAMP signal
Phosphodiesterases (PDEs) terminate the cAMP signal by converting cAMP to its 5′-AMP metabolite (Fig. 40.4); thus they have the potential to play key roles in the regulation of various physiologic responses in many different cells and tissues. There are many different isoforms of PDEs that exhibit a tissue-specific pattern of expression and different selectivity for cAMP or cGMP. PDEs have been demonstrated to regulate platelet activation, vascular relaxation, cardiac muscle contraction and inflammation.
Inhibitors of phosphodiesterases are used as therapeutic agents
Selective inhibitors of PDEs have been used as therapeutic agents for asthma (methylxanthines), erectile dysfunction (sildenafil) and heart failure (milrinone). Milrinone is selective for the PDE3 isoforms, which increases the force of contraction of the heart, presumably due to increased cAMP concentrations and PKA activity, leading to phosphorylation of cardiac calcium channels and a subsequent increase in intracellular calcium concentration.
Phospholipase-derived second messengers
Phospholipase C hydrolyzes the membrane phospholipid phosphatidylinositol 4,5-bisphosphate to generate two second messengers
G-protein coupled receptors that are coupled to the Gqα subtype of the G-protein α-subunit stimulate the activity of phospholipase C (PLC). In addition, other types of membrane receptor, such as the vascular endothelial growth factor (VEGF) receptor, that has intrinsic tyrosine kinase activity, also are able to stimulate PLC. PLC catalyzes the hydrolysis of a minor phospholipid species, phosphatidylinositol 4,5-bisphosphate (PIP2). PLC-mediated hydrolysis of PIP2, which typically represents about 0.4% of total phospholipids in membranes, generates two second messengers: inositol -1,4.5- trisphosphate (IP3) and diacylglycerol (DAG) (Fig. 40.7). IP3 is a water-soluble product, which is released into the cytosol and has been shown to mobilize intracellular stores of calcium. DAG is a lipid second messenger, which is anchored in the plasma membrane by virtue of its hydrophobic fatty acid side chains and activates a key family of signaling enzymes known as protein kinase C (PKC).
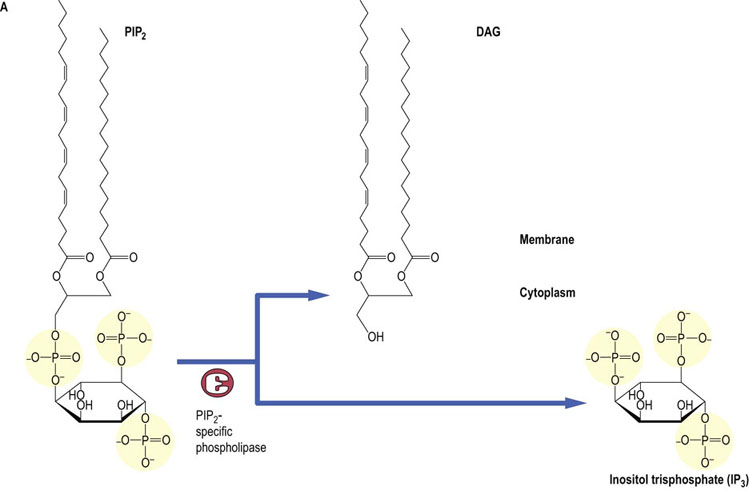
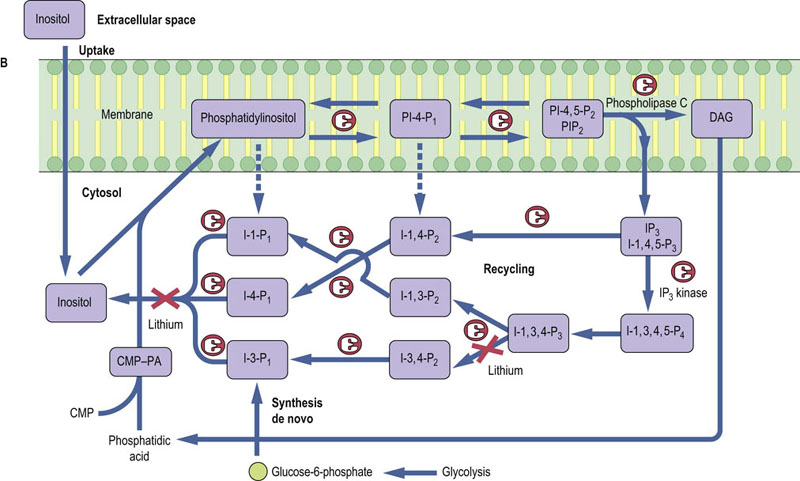
Fig. 40.7 Synthesis and metabolism of phosphatidylinositol 4,5-bisphosphate.
Phosphatidylinositol 4,5-bisphosphate (PIP2) is hydrolyzed by a PIP2-specific PLC, to generate two second messengers: Inositol-1,4,5-P3 (IP3) and DAG (A). IP3 is released into the cytosol and has been shown to mobilize intracellular stores of calcium. DAG is a lipid second messenger which is anchored in the plasma membrane and activates PKCs. PIP2 is generated from phosphatidylinositol by phosphatidylinositol 4-kinase and phosphatidylinositol 5-kinase (B). IP3 is degraded by (i) the sequential action of phosphatases converting Ins1,4,5-P3 to inositol, and (ii) an IP3 kinase, which generates Inositol-1,3,4,5-P4, which is in turn sequentially degraded to inositol by inositol phosphate-specific phosphatases, some of which can be inhibited by lithium. DAG, diacylglycerol; PA, phosphatidic acid; CMP-PA, cytosine monophosphate-phosphatidic acid.
Inositol 1,4,5-trisphosphate (IP3) stimulates intracellular calcium mobilization
Once synthesized from PIP2, IP3 binds to receptors found on the endoplasmic reticulum of all cells. IP3 receptors are a family of related glycoproteins (molecular mass 250 kDa) comprising six transmembrane-spanning domains. The active receptor is expressed as a multimer of four IP3 receptor molecules that acts as a ligand-gated Ca2+ channel. The tetrameric structure of the IP3 receptor gives rise to cooperativity in Ca2+ channel activity. It has been estimated that stimulation with IP3 causes transport of 20–30 calcium ions, revealing the amplification inherent in this signaling cascade. Consistent with the transient nature of the release of intracellular calcium that is observed after hormone receptor ligation, cellular concentrations of IP3 are rapidly returned to resting values (10 nmol/L) by more than one route of degradation (Fig. 40.7).
Calcium and calmodulin. Signal transduction by calcium ions
Calcium (Ca2+) is a ubiquitous messenger with an important role in the transduction of signals leading to diverse cellular responses such as cell motility changes, egg fertilization, neurotransmission, secretion, differentiation and proliferation. Cells expend a considerable amount of energy maintaining a Ca2+ concentration gradient such that the intracellular Ca2+ concentration in resting, unstimulated cells is of the order of 10–7 mol/L, whereas the extracellular Ca2+ concentration is approximately 10,000-fold greater, typically 10−3 mol/L. This steep gradient allows for rapid, abrupt, transient changes in Ca2+ concentration in response to signals. Ligand binding by a wide range of receptors leads to a PLC-mediated rapid (within seconds) and transient increase in intracellular Ca2+ concentration to the micromolar range (compare Fig. 8.4). The rapid changes in Ca2+ concentrations are very tightly regulated and utilize a variety of mechanisms involving cell compartmentalization. For example, intracellular Ca2+ concentrations can be lowered by sequestration of Ca2+ into the endoplasmic reticulum by Ca2+-ATPases, or into the mitochondria using the energy-driven electrochemical gradient. Alternatively, free Ca2+ can be chelated by Ca2+-binding proteins such as calsequestrin.
Many downstream signaling events mediated by Ca2+ are modulated by a Ca2+-sensing and binding protein, calmodulin
Calmodulin (CaM) is a 17 kDa protein found in all animal and plant cells (comprising up to 1% of cellular protein). It belongs to a family of proteins characterized by one or more copies of a Ca2+-binding structural motif called an EF hand motif (Fig. 40.8). CaM is composed of two similar globular domains joined by a long α-helix, each globular lobe having two EF hand motifs/Ca2+-binding sites placed 1.1 nm (11 Å) apart. Binding of three to four calcium ions occurs when the intracellular Ca2+ concentration is increased to about 500 nmol/L, inducing a major conformational change that allows CaM to bind to and modify target proteins. Binding of several calcium ions allows cooperativity in the activation of CaM, such that small changes in Ca2+ concentration cause large changes in the concentration of an active Ca2+/CaM complex, providing amplification of the original hormone signal.
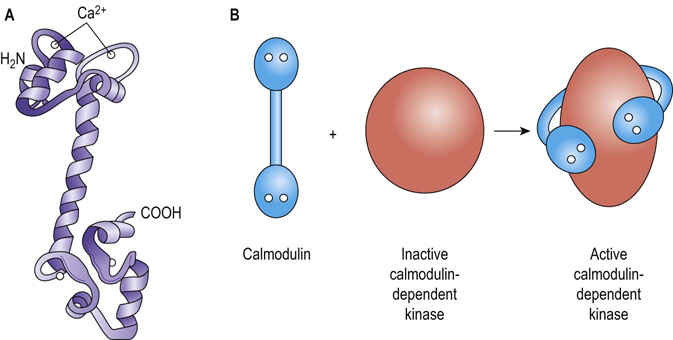
Calmodulin has a wide range of target effectors
CaM has a wide range of target effectors, including nitric oxide synthase (NOS), which stimulates NO synthesis in response to Ca2+-mobilizing signals, and Ca2+/CaM-dependent protein kinases, which phosphorylate serine-threonine residues on proteins to regulate a variety of processes. For example, the broad-specificity kinase, Ca2+/CaM-kinase II, is involved in the regulation of fuel metabolism, ion permeability, neurotransmitter biology and muscle contraction. Interestingly, CaM serves as a permanent regulatory subunit of phosphorylase kinase, and may also regulate non-kinase effectors such as certain adenylyl cyclase isoforms and also cAMP-specific PDEs, permitting ‘cross-talk’ between cAMP- and Ca2+-dependent signaling pathways.
Diacylglycerol (DAG) activates protein kinase C
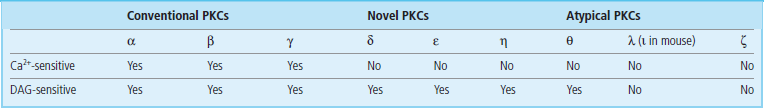
DAG fulfills its second messenger role by activating protein kinase C (PKC), which phosphorylates a wide range of target signal transduction proteins on serine or threonine residues. PKC was originally identified as a calcium- and lipid (phosphatidylserine)-dependent kinase important in the regulation of cell proliferation. However, in recent years, it has become clear that PKC is, in reality, a generic name for a superfamily of related kinases that have different activation requirements (Table 40.3). PKC family members also exhibit tissue-specific expression. Nevertheless, all these enzymes share some conserved features; most notably they comprise two major domains: an N-terminal regulatory domain and a C-terminal catalytic kinase domain. The regulatory domain contains a pseudosubstrate sequence that resembles the consensus phosphorylation site in PKC substrates. In the absence of activating cofactors (Ca2+, phospholipid, DAG), this pseudosubstrate sequence interacts with the substrate-binding pocket in the catalytic domain and represses PKC activity; binding of cofactors reduces the affinity of this interaction, induces a conformational change in the PKC, and allows stimulation of PKC activity. Consistent with the fact that the activator/cofactor, DAG, is anchored in the membranes, PKC activation is generally associated with translocation from the cytosol to the plasma mebrane or nuclear membranes.
Other phospholipases hydrolyze phosphatidylcholine or phosphatidylethanolamine, generating a range of lipid second messengers
Additional receptor-coupled lipid signaling pathways have been identified involving hydrolysis of phosphatidylcholine or phosphatidylethanolamine, which can give rise to DAG and other biologically active lipids (Fig. 40.9) in response to a wide range of growth factors and mitogens. Phosphatidylcholine comprises about 40% of total cellular phospholipids. It can be hydrolyzed by distinct phospholipases, generating a diversity of lipid second messengers, including fatty acids such as arachidonic acid (generated by PLA2) as well as different species of DAG (generated by PLC) and phosphatidic acid (generated by PLD). Hormone-stimulated phosphatidylethanolamine-PLD activities have also been reported. Some hormones or growth factors can stimulate only one or other of these phospholipases, but other ligands can stimulate all these pathways after binding to their specific receptors.
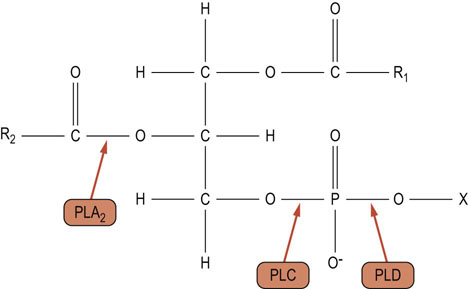
Fig. 40.9 Sites of action of phospholipases.
Hydrolysis of phosphatidylcholine or phosphatidylethanolamine by phospholipase A2 (PLA2) results in the production of lysophosphatidylcholine or lysophosphatidylethanolamine and a fatty acid. Hydrolysis by phospholipase C (PLC) results in the synthesis of DAG and phosphocholine or phosphoethanolamine. Hydrolysis of phosphatidylcholine or phosphatidylethanolamine by phospholipase D (PLD) results in the production of phosphatidic acid and choline or ethanolamine. R1, R2: fatty acyl chains; X: choline/ethanolamine.
There is growing evidence that all these distinct lipid second messengers have different targets. For example, it has recently been suggested that the saturated/monounsaturated fatty acid-containing DAGs derived from phosphatidylcholine-specific phospholipase D (PLD) activation are unable to activate PKC isoforms, and that it is only the stearoyl-arachidonyl-phosphatidic acid species that can modulate activity of the GTPase Ras (Chapter 43). Generation of these diverse but related lipid second messengers therefore provides a mechanism for initiating or terminating hormone-specific responses via particular signal transducers, including differential activation of PKC isoforms.
Arachidonic acid is a second messenger regulating phospholipases and protein kinases
Arachidonic acid is a C20 polyunsaturated fatty acid containing four double bonds. Increased arachidonic acid synthesis has been demonstrated to regulate several signaling enzymes, including PLC and PKC-α, -β and -γ isoforms. Furthermore, arachidonic acid is a key inflammatory intermediate. However, the arachidonic acid involved in these disparate functions appears to be generated by two distinct PLA2 routes. Arachidonic acid generated for signaling purposes appears to be derived from the action of a phosphatidylcholine-specific cytosolic phospholipase A2 (cPLA2), which has a molecular mass of 85 kDa and is regulated by phosphorylation of key serine residues. In contrast, inflammatory arachidonic acid is generated by the action of a family of low-molecular-mass secretory PLA2 (sPLA2) proteins (14–18 kDa), which appear to be ubiquitous and are found in high concentrations in snake venom and pancreatic juices. In addition, arachidonic acid can be generated by DAG lipase.
Arachidonic acid is the precursor of eicosanoids, which encompass prostaglandins, prostacyclins, thromboxanes, and leukotrienes
As a key inflammatory mediator, arachidonic acid is the major precursor of the group of molecules termed eicosanoids, which encompass prostaglandins, prostacyclins, thromboxanes, and leukotrienes. Eicosanoids act like hormones and signal via G-protein coupled receptors. They have a wide variety of biological activities, including modulating smooth muscle contraction (vascular tone), platelet aggregation, gastric acid secretion, and salt and water balance, as well as mediating pain and inflammatory responses. Prostaglandins, prostacyclins, and thromboxanes are synthesized in membranes from arachidonic acid by the successive actions of several enzymes, starting with cyclooxygenase (Fig. 40.10 and see also Fig 7.2).
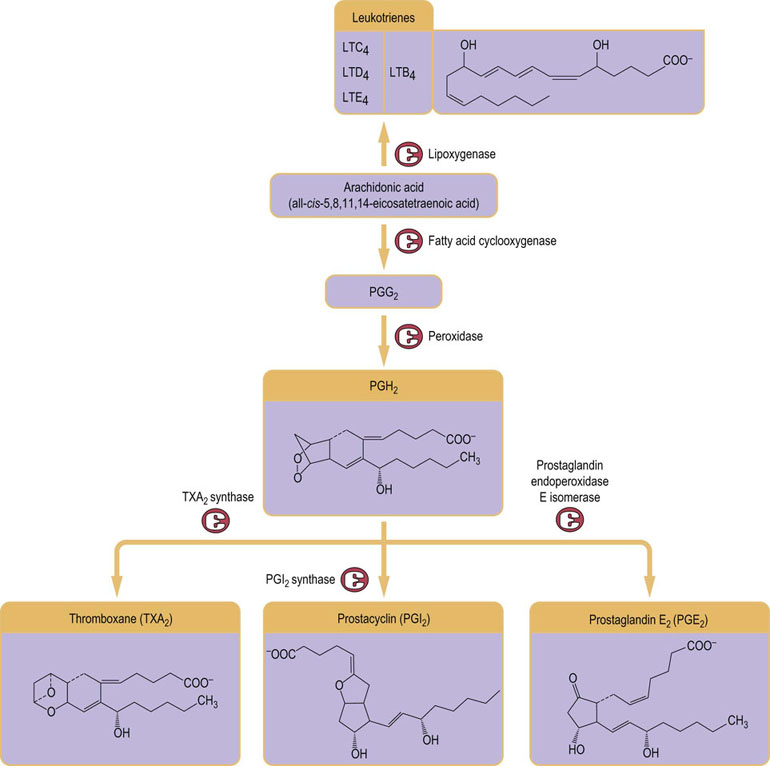
Fig. 40.10 Synthesis of eicosanoids.
Eicosanoids are primarily derived from arachidonic acid. Leukotrienes (LT) are synthesized via a lipoxygenase-dependent pathway, whereas prostaglandins (PG), prostacyclins and thromboxanes (TX) arise from cyclooxygenase-dependent routes.
Clinical box A child with premature breast development: mcCune–albright syndrome
A 3-year-old girl was brought to hospital because her mother had been concerned about apparent breast development over the last 6 months, and a spot of blood on her pants last week. On examination, she had Tanner Stage 3 breast development. On her trunk she had three areas of brown skin pigmentation with ragged edges.
This child is suffering from McCune–Albright syndrome. She is likely to develop polyostotic fibrous dysplasia, with areas of thinning and sclerosis in her long bones which may fracture. Other endocrinopathies include thyrotoxicosis, growth hormone hypersecretion, Cushing's syndrome (cortisol excess) and hyperparathyroidism. The cause is an activating missense mutation in the gene encoding the Gsα-subunit of the G-protein that stimulates cyclic AMP formation. The problem presents following a somatic cell mutation with clinical features dependent on a mosaic distribution of aberrant cells. The incidence of the syndrome is 1 in 25,000.
Clinical box Fibroblast growth factor receptor 3 and achondroplasia
Fibroblast growth factor (FGF) receptor 3 (FGFR3) is an intrinsic tyrosine kinase receptor with an important role in the regulation of bone growth. In chondrocytes (the cells that synthesize cartilage at the epiphyses of long bones), FGF binding to two FGFR3 monomers causes dimerization of the receptor, allowing the intracellular tyrosine kinase domains to transphosphorylate each other. This autophosphorylation of FGFR3 leads to activation of signaling pathways including the transcription factor, signal transducer and activators of transcription-1 (STAT1) and the small G-protein Ras, which subsequently activates the Raf-MEK-ERK protein kinase cascade. Activation of these pathways inhibits chondrocyte differentiation and proliferation. Thus, FGFR3 stimulation suppresses long bone growth as the reduced number of chondrocytes decreases cartilage deposition that would normally serve as a template for osteoblasts to form bone by ossification.
Achondroplasia, characterized by short stature and macrocephaly, has an incidence of 1 in 15,000–40,000 newborns. The majority of people with achondroplasia have mutations in FGFR3 that increase its tyrosine kinase activity in the absence of FGF. As a consequence, chondrocyte proliferation and cartilage deposition are impaired, leading to reduced length of long bones.
Summary
Cells specifically respond to a multiplicity of signals from their environment via signal transduction cassettes, which comprise specific cell surface membrane receptors, effector signaling systems (e.g. adenylyl cyclase, phospholipases, or ion channels) and regulatory proteins (e.g. G-proteins or protein kinases).
These signal transduction cassettes serve to detect, amplify and integrate diverse external signals to generate the appropriate cellular response.
The families of cell surface receptors sense and transduce their specific hormone signal by transmembrane coupling to different effector systems to generate low-molecular-weight molecules, termed second messengers, such as cAMP, IP3, DAG, and Ca2+, which mediate their signaling functions by activating key protein kinases.
The specificity of a particular hormone response can be further heightened by the variety of available phospholipase-signaling activities (PLC, PLD and PLA2). Taking into account their range of potential lipid substrates (e.g. PIP2, phosphatidylcholine, and phosphatidylethanolamine) and products (e.g. DAG, phosphatidic acid and arachidonic acid), the phospholipases can generate a diverse array of lipid second messengers.
Fredholm, BB, Hökfelt, T, Milligan, G. G-protein-coupled receptors: an update. Acta Physiol (Oxf). 2007; 190:3–7.
Halls, ML, Cooper, DM. Regulation by Ca2+-signaling pathways of adenylyl cyclases. Cold Spring Harb Perspect Biol. 2011; 3:a004143.
Houslay, MD. Underpinning compartmentalised cAMP signaling through targeted cAMP breakdown. Trends Biochem Sci. 2010; 35:91–100.
Lefkowitz, RJ. Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf). 2007; 190:9–19.
Leto, D, Saltiel, AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol. 2012; 13:383–396.
Michel, T, Vanhoutte, PM. Cellular signaling and NO production. Pflugers Arch. 2010; 459:807–816.
Osborne, JK, Zaganjor, E, Cobb, MH. Signal control through Raf: in sickness and in health. Cell Res. 2012; 22:14–22.
Parekh, AB. Decoding cytosolic Ca2+ oscillations. Trends Biochem Sci. 2011; 36:78–87.
Smith, WL. Nutritionally essential fatty acids and biologically indispensable cyclooxygenases. Trends Biochem Sci. 2008; 33:27–37.
Science magazine's signal transduction knowledge environment. www.stke.org.
Kimball's biology pages. http://users.rcn.com/jkimball.ma.ultranet/BiologyPages/C/CellSignaling.html.
The Biology Project (University of Arizona). www.biology.arizona.edu/cell_bio/problem_sets/signaling/Index.htm.
BioCarta's cell signaling pathways. www.biocarta.com/genes/CellSignaling.asp.
Cell signaling pathway maps. www.cellsignal.com/reference/pathway/index.html.