Water and Electrolyte Homeostasis
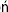 ska-Konkel
ska-KonkelLearning objectives
After reading this chapter you should be able to:
 Describe the body water compartments in the adult and the composition of the main body fluids.
Describe the body water compartments in the adult and the composition of the main body fluids.
Explain the role of albumin in the movement of water between plasma and interstitial space, including the consequences of proteinuria.
Describe how sodium influences the movement of water between the extracellular and intracellular space.
Explain why sodium/potassium ATPase is essential for normal cell hydration, and comment on the consequences of its inhibition.
Describe factors affecting plasma potassium concentration.
Discuss the links between sodium and water homeostasis.
Describe the clinical assessment of water and electrolyte status.
Introduction
Water and electrolytes are constantly exchanged with the environment, and their body content depends on the balance between intake and loss
Water is essential for survival and accounts for approximately 60% of the body weight in an adult person. This changes with age: it is about 75% in the newborn and decreases to below 50% in older individuals. Water content is greatest in the brain tissue (about 90%) and least in the adipose tissue (10%).
The stability of subcellular structures and activities of numerous enzymes are dependent on adequate cell hydration. The maintenance of ion gradients and electrical potential across membranes is also crucial for survival and underlies muscle contraction, nerve conduction and secretory processes (Chapter 8).
Both water deficiency and water excess impair function of organs and tissues. Water balance (daily intake and loss) and water distribution between cells and the surrounding fluid are subject to complex regulation.
Clinical relevance
Water and electrolyte disorders are common in clinical practice. Historically, textbooks of biochemistry (and many biochemistry courses) have treated water and electrolyte balance somewhat peripherally. And yet it is fundamental to maintaining metabolism, and underlies many essential treatment procedures.
Body water compartments
Approximately two-thirds of total body water is in the intracellular fluid (ICF), and one-third remains in the extracellular fluid (ECF). ECF consists of interstitial fluid and lymph (15% body weight), plasma (3% body weight), and the so-called transcellular fluids, which include gastrointestinal fluid, urine and cerebrospinal fluid (CSF) (Fig. 24.1
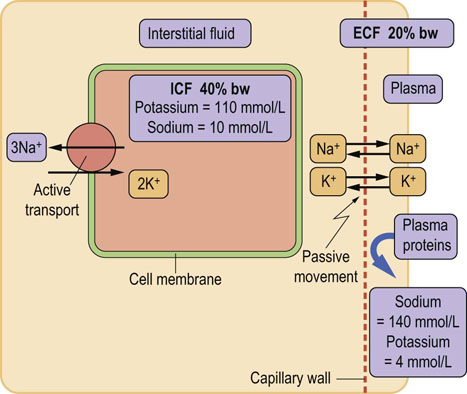
Fig. 24. 1 Distribution of body water, sodium and potassium.
The main body water compartments are the intracellular fluid (ICF) and the extracellular fluid (ECF). ECF includes interstitial fluid and plasma. The gradient of sodium and potassium concentrations is maintained across cell membranes by the Na+/K+-ATPase. Sodium is a major contributor to the osmolality of the ECF, and a determinant of the distribution of water between ECF and ICF. Distribution of water between plasma and interstitial fluid is determined by the oncotic pressure exerted by plasma proteins. bw, body weight.
 Clinical box Concentration of ions in plasma and serum
Clinical box Concentration of ions in plasma and serum
All physiologic phenomena occur in plasma – and therefore discussion of physiologic or pathologic conditions relates to plasma concentrations of ions.
However, in practice, the concentration of most ions is measured after the sampled blood has been allowed to clot (i.e using serum). Therefore, in the discussion of laboratory results we often mention serum values (Chapter 4).
). Two barriers are important for the understanding of exchanges taking place between different compartments: the cell membrane and the wall of the capillary vessel.
Capillary vessel wall separates plasma from the surrounding interstitial fluid
The capillary wall separates plasma from the interstitial fluid and is freely permeable to water and electrolytes but not to proteins. Ions and low-molecular-weight molecules are present in similar concentrations in the ECF and plasma but protein concentration is 4–5 times greater in plasma than it is in the interstitial fluid. The total plasma concentration of cations is about 150 mmol/L, of which sodium is approximately 140 mmol/L and potassium 4 mmol/L. The most abundant plasma anions are chloride and bicarbonate, with average concentrations of 100 mmol/L and 25 mmol/L, respectively (Fig. 24.2). In clinical practice the rest of the anions are, for the purposes of electrolyte balance, considered together, constituting the so-called anion gap (AG), which is calculated as follows.
Fig. 24. 2 Ions present in the plasma and in the intracellular fluid.
The most important ions in plasma are sodium, potassium, calcium, chloride, phosphate, and bicarbonate. Sodium chloride, in a concentration close to 0.9% (thus ‘physiologic saline’), is the main ionic component of the extracellular fluid. Potassium is the main intracellular cation. Glucose and urea also contribute to plasma osmolality. Their contribution is normally small, because they are present in plasma in relatively low molar concentrations (about 5 mmol/L each). However, when glucose concentration increases in diabetes, its contribution to osmolality becomes significant. Plasma urea increases in renal failure but it does not contribute to water movement between ECF and ICF because it freely crosses cell membranes. The main intracellular cation is potassium and the main anions are phosphates and proteins. There is also a substantial amount of magnesium in cells.
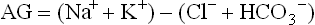
The anion gap (in a healthy person, approximately 10 mmol/L) includes phosphate, sulfate, protein, and organic anions such as lactate, citrate, pyruvate, acetoacetate, and β-hydroxybutyrate. However, it may increase several-fold in conditions where inorganic and organic anions accumulate, e.g. in renal failure or diabetic ketoacidosis. For this reason it is clinically important.
Plasma membrane separates the intracellular and extracellular fluid
In the ICF, the main cation is potassium, present in a concentration of about 110 mmol/L. This is almost 30-fold greater than its concentration in the ECF and in plasma (4 mmol/L). The main anions in the ICF are proteins and phosphate. In the ECF, the situation is reversed: the main cation is sodium, present in a concentration of about 145 mmol/L. In the ICF the concentration of sodium (and chloride) is only 10 mmol/L.
Water diffuses freely across most cell membranes but the movement of ions and neutral molecules is restricted
Small molecules are transported across cell membranes by specific transport proteins, the ion pumps. The most important is the sodium-potassium ATPase (Na+/K+-ATPase), also referred to as the sodium–potassium pump.
Na+/K+-ATPase maintains the sodium and potassium gradients across the cell membrane
Na+/K+-ATPase can be considered either as an ion transporter (sodium pump) or as an enzyme (ATPase). It maintains chemical and electrical potential gradients (it is electrogenic) across the cell membrane (Fig. 24.3). It hydrolyzes one ATP molecule, and the released energy drives the transfer of three sodium ions from the cell to the outside, and two potassium ions from the outside into the cell (Fig. 24.4).
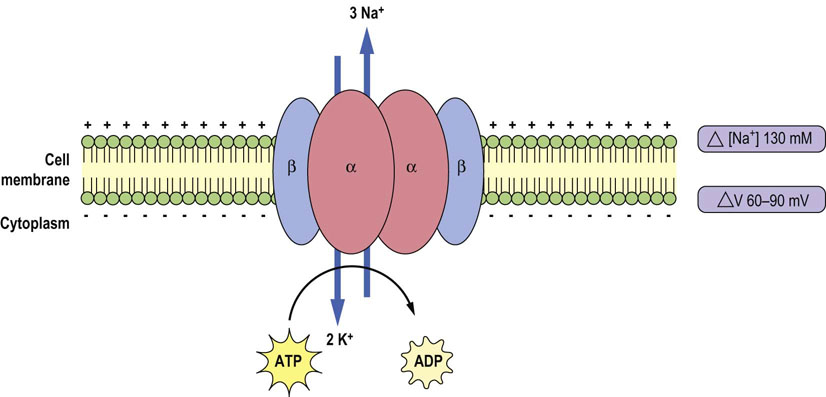
Fig. 24.3 Na+/K+-ATPase (the sodium–potassium pump) generates transmembrane potential and ion concentration gradients across the cell membrane.
The transmembrane difference in sodium concentration (ΔNa+), and transmembrane voltage difference (ΔV) are shown on the right. For each molecule of hydrolyzed ATP, it moves two potassium ions into the cell and three sodium ions out of the cell. Na+/K+-ATPase consists of two main subunits – catalytic subunit (α) and structural subunit (β).
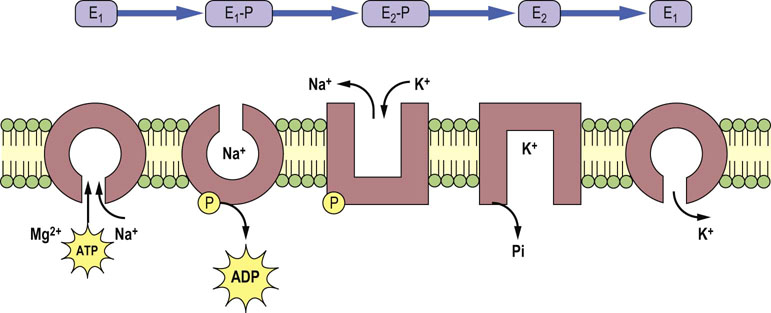
Fig. 24.4 Catalytic function of the Na+/K+-ATPase.
The catalytic subunit of the Na+/K+-ATPase can be either phosphorylated (E1-P and E2-P) or dephosphorylated (E1 and E2) and this changes its conformation and affinity towards substrates. The E1 form exhibits high affinity towards ATP, magnesium and sodium and low affinity towards potassium, whereas the E2 form exhibits high affinity for potassium and low for sodium. After the release of ADP, there is conformational change from E1-P to E2-P. This promotes extracellular delivery of sodium and the binding of extracellular potassium. The latter process induces dephosphorylation of E2-P and potassium release into the intracellular compartment.
The Na+/K+-ATPase is the major determinant of cytoplasmic sodium concentration (Chapter 8). It also has an important role in regulating cell volume, cytoplasmic pH and calcium levels through the Na+/H+ and Na+/Ca2+exchangers. One of the primary requirements for continuous sodium-pump-driven adaptation comes from changes in dietary sodium and potassium. Hormones that control the volume and ionic composition of the ECF often act directly on the sodium pump in the kidney and intestine.
Na+/K+-ATPase activity is subject to short- and long-term regulation by a number of hormones
The Na+/K+-ATPase is activated by sodium and ATP at cytoplasmic sites. The structures of the catalytic subunit of the enzyme and its phosphorylation sites are shown in Fig. 24.5. Half-maximal activation of the enzyme by intracellular sodium occurs at sodium concentration of 10–40 mM. which is often above the steady-state concentration. Accordingly, small changes in the cytoplasmic sodium concentration can have large effect on its activity. Some hormones appear to alter the Na+/K+-ATPase activity by changing its apparent affinity for sodium (for instance, angiotensin II and insulin increase the affinity).
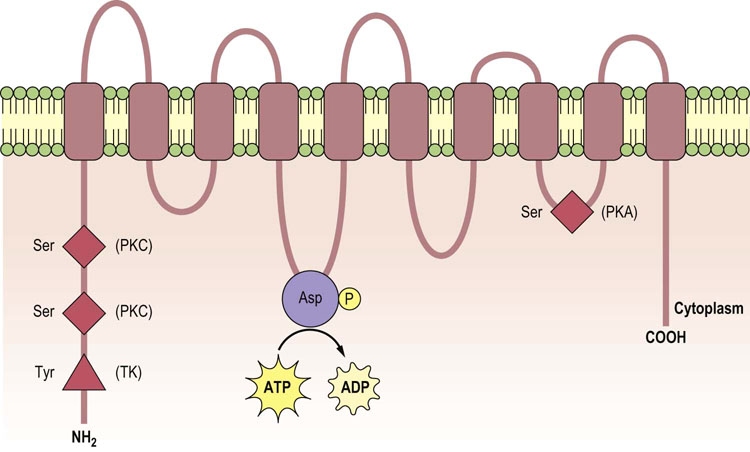
Fig. 24.5 Structure of the α-subunit of the Na+/K+-ATPase.
The α-subunit consists of 10 membrane-spanning domains (M1–M10) with intracellular amino- and carboxy-terminal domains. The ATP-binding domain and the phosphorylation site are located in the long M4–M5 cytoplasmic loop (where Asp residue undergoes phosphorylation). Other phosphorylation sites on serine and tyrosine residues are mediated by protein kinase A (PKA), protein kinase C (PKC) and tyrosine kinase (TK).
The Na+/K+-ATPase is subject to regulation by a number of hormones, including aldosterone. Short-term regulation involves either direct effects on the kinetic properties of the enzyme or its translocation between the plasma membrane and intracellular stores. Long-term regulatory mechanisms affect the enzyme's synthesis or degradation.
Peptide hormones such as vasopressin and PTH that act through G-protein-coupled receptors affect the activity of Na+/K+-ATPase. The G-proteins activate adenylyl cyclase, which generates cAMP. cAMP, in turn, activates protein kinase A (PKA). PTH, angiotensin II, norepinephrine and dopamine also trigger G-protein-mediated activation of phospholipase C, which activates protein kinase C (PKC). Both PKA and PKC affect Na+/K+-ATPase by serine phosphorylation of its α-subunit (Fig. 24.5). Insulin increases the apparent sodium affinity of Na+/K+-ATPase through the activation of the tyrosine kinase receptor and phosphorylation of the α-subunit of the enzyme.
Passive movement of electrolytes through ion channels is driven by the electrochemical gradient
For most cells, the membrane potential ranges from 50 to 90 mV, being negative inside the cell. The electrochemical gradient is a source of energy for transport of many substances such as the co-transport of the sodium ions with glucose (the SGLT transporter, Chapter 10), amino acids, and phosphate. Membrane depolarization also promotes an increase in intracellular calcium by activating voltage-dependent Ca2+channels (Chapter 8). The role of ion gradients in nerve transmission is described in Chapter 41.
Because water and sodium transport on the luminal side of the epithelial cells (in the intestine and the kidneys) is linked to the ion gradient generated by the Na+/K+-ATPase, this enzyme is critical to water absorption in the intestine and its reabsorption in the kidneys. The impairment of the sodium pump function in the kidney and small intestine is linked to pathophysiology of hypertension and chronic diarrhea, respectively.
Osmolality: osmotic and oncotic pressures
Molecules dissolved in body water contribute to the osmotic pressure
Osmolality depends on the concentration of molecules in water and osmotic pressure is proportional to the molal concentration of a solution. One millimole of a substance dissolved in 1 kg H2O at 38°C exerts an osmotic pressure of approximately 19 mmHg. Under physiologic conditions, the average concentration of all osmotically active substances in the ECF is 290 mmol/kg H2O. Normally, the ICF osmolality is identical.
Differences in osmolality cause movement of water between intracellular and extracellular fluid
A change in the concentration of osmotically active ions in either ECF or ICF creates a gradient of osmotic pressure and, consequently, causes the movement of water. Water always diffuses from lower osmolality to higher to equalize osmotic pressures (Fig. 24.6).
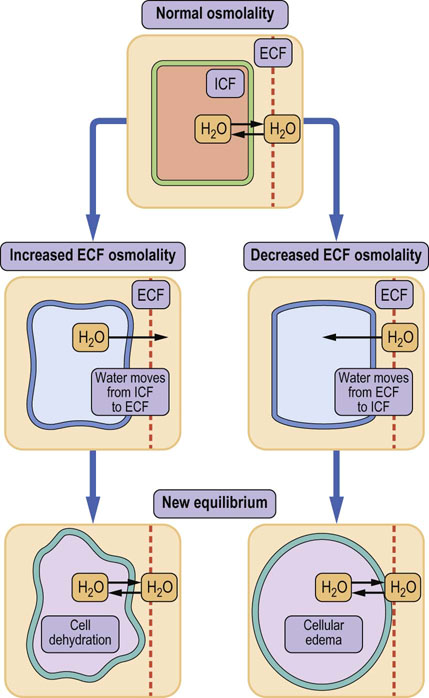
Fig. 24.6 Water redistribution caused by changes in osmolality.
Osmotic pressure controls the movement of water between compartments. An increase in ECF osmolality draws water from the cells, and leads to cellular dehydration. On the other hand, when ECF osmolality decreases, water moves into the cells and this may cause cell edema. The arrows indicate direction of water movement. ECF, extracellular fluid; ICF, intracellular fluid.
Because sodium is the most abundant ion in the ECF, it is also the most important determinant of its osmolality. Glucose at its normal plasma concentration (5 mmol/L; 90 mg/dL) does not contribute significantly to osmolality but it becomes its major determinant when its concentration increases in diabetes (remember the ‘osmotic symptoms’ in poorly controlled diabetes (Chapter 21).
Balance between the oncotic and hydrostatic pressure changes across the vascular bed and is fundamental for the circulation of substrates and nutrients
The movement of water between the plasma and interstitial fluid depends on the plasma protein concentration. Proteins, particularly albumin, exert osmotic pressure in the plasma (about 3.32 kPa; 25 mmHg). This is known as the oncotic pressure and it retains water in the vascular bed. It is balanced by the hydrostatic pressure, which forces fluid in the opposite direction, i.e. out of the capillaries. In the arterial part of the capillaries, the hydrostatic pressure prevails over the oncotic pressure, and water and low-molecular-weight compounds filter out into the extravascular space. In contrast, in the venous part of capillaries, oncotic pressure prevails over hydrostatic pressure, and fluid is drawn into the vascular lumen (Fig. 24.7). A reduction in plasma oncotic pressure, which occurs, for instance, as a consequence of a decrease in the plasma albumin concentration, results in the movement of fluid into the extravascular space and in the development of edema.
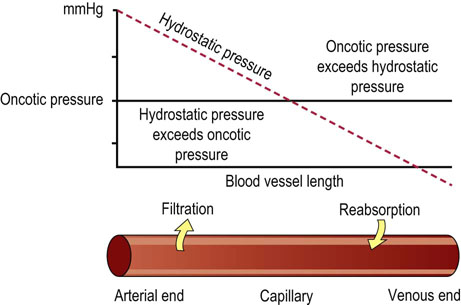
Fig. 24.7 Oncotic and hydrostatic pressures determine movements of fluid between plasma and interstitial fluid.
Clinical box Edema results from a loss of protein
An 8-year-old girl was referred to a nephrologist after it had been noticed that her face was puffy and her ankles became swollen over a period of about 2 weeks. Dipstick test for urine protein yielded a strongly positive (++++) result and measurement in a 24-hour collection showed protein excretion of 7 g/day. The reference value for urinary protein excretion is less than 0.15 g/day.
The cause of the proteinuria was the damage to the renal filtration barrier. Renal biopsy showed the so-called minimal change disease, with the urinary protein loss causing, in turn, hypoalbuminemia and a decrease in the plasma oncotic pressure. This led to edema. The condition went into remission after treatment with a glucocorticoid.
Cells protect themselves against changes of osmolality and volume
An increase in the intracellular concentration of sodium stimulates the Na+/K+-ATPase, which extrudes sodium from the cell. This is followed by the egress of water and protects the cell from volume changes. Another protective mechanism is the intracellular generation of osmotically active substances. For instance, the brain cells adapt to increased ECF osmolality by increasing their amino acid concentration, and cells in the renal medulla exposed to a hyperosmotic environment produce an osmotically active alcohol, sorbitol, and increase the concentration of the amino acid taurine.
The body constantly exchanges water with the environment
In a steady state, the intake of water equals its loss. The main source of water is oral intake and the main source of its loss is urine excretion. Water is also lost through the lungs, sweat and feces: this is called the ‘insensible’ loss and in normal circumstances amounts to approximately 500 mL daily (Fig. 24.8). Insensible loss can increase substantially in high temperatures, during intensive exercise, and also as a result of fever. Checking the patient's fluid balance is one of the essential daily routines on medical and surgical wards.
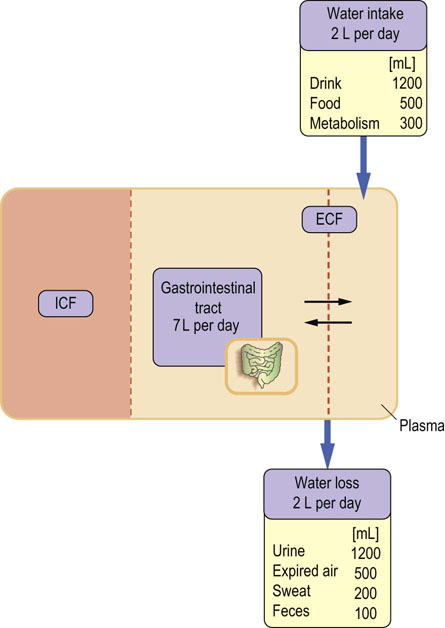
Fig. 24.8 Daily water balance in an adult person.
Water is obtained from the diet and from oxidative metabolism, and is lost through the kidneys, skin, lungs, and the intestine. Note how much water enters and leaves the gastrointestinal tract daily; this explains why severe diarrhea quickly leads to dehydration. See also Chapter 10.
Clinical box Body fluids differ in ionic composition
Clinical abnormalities that may develop after fluid loss depend on the composition of what is lost. For instance, sweat contains less sodium than extracellular fluid: therefore, excessive sweating leads to a predominant loss of water and ‘concentrates’ sodium in the extracellular fluid, causing hypernatremia. On the other hand, sodium content of the intestinal fluid is similar to that of plasma but contains considerable amounts of potassium. Thus, its loss (for instance, in severe diarrhea) would result in dehydration and hypokalemia, but may not change plasma sodium concentration (Table 24.1).
Table 24.1
Electrolyte content of the body fluids
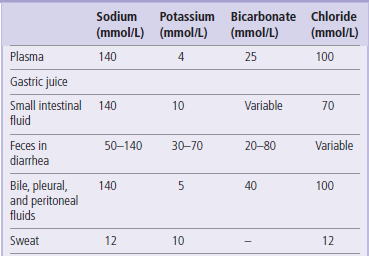
Loss of fluid that has an electrolyte content similar to that of plasma leads to dehydration with normal serum electrolyte concentrations. On the other hand, when the sodium content of the lost fluid is less than that of plasma (e.g. sweat), dehydration may be accompanied by hypernatremia. Overhydration is usually accompanied by hyponatremia.
Adapted with permission from Dominiczak MH, editor: Seminars in Clinical Biochemistry, ed. 2, Glasgow, 1997, Glasgow University.
Potassium
Monitoring serum potassium concentration is fundamentally important
Normal serum concentration of potassium is 3.5–5 mmol/L. Because its intracellular concentration is much higher than concentration in plasma, a relatively minor shift of potassium between the ECF and ICF may result in major changes in its serum concentration. Both high and low concentrations of potassium (hyperkalemia and hypokalemia, respectively) affect the cardiac muscle and can be life-threatening.
On the EKG, hyperkalemia can lead to the loss of P-wave, characteristic tall peaked T-waves, and widened QRS complexes. Hypokalemia, on the other hand, may prolong PR interval, cause peaked P-wave, flatten the T-wave and cause prominent U-waves.
Serum potassium concentration below 2.5 mmol/L or above 6.0 mmol/L is dangerous (Fig. 24.9). The most common cause of severe hyperkalemia is renal failure: in this condition potassium cannot be adequately excreted in the urine. On the other hand, low serum potassium usually results from excessive losses, either in urine or through the gastrointestinal tract. Kidneys account for more than 90% of the body potassium loss, and diuretic therapy can induce both hypo- and hyperkalemia. Changes in serum potassium concentration are also associated with acid–base disorders (Chapter 25).
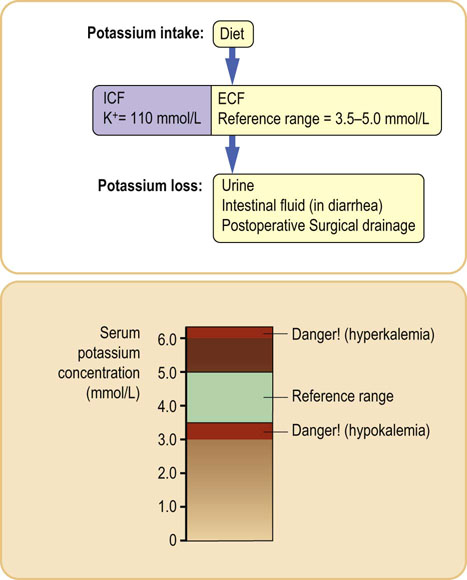
Fig. 24.9 Potassium balance.
Serum potassium concentration is maintained within narrow limits. Both low (hypokalemia) and high (hyperkalemia) concentrations are be dangerous, as potassium affects the contractility of heart muscle. Generally, serum potassium concentrations above 6.0 mmol/L and below 2.5 mmol/L are regarded as emergencies. The upper panel shows main sources of potassium loss.
Renin–angiotensin system
Renin–angiotensin system controls blood pressure and the vascular tone
Renin is an enzyme produced principally in the juxtaglomerular apparatus of the kidney; it is stored in the secretory granules and released in response to a decreased renal perfusion pressure (decreased delivery of Na+ to the macula densa) and increased sympathetic tone. Renin is a protease that uses circulating angiotensinogen as its substrate. Renin secretion is regulated by pathways involving G-protein-coupled receptors and adenylate cyclase – PKA pathway, which activates the cAMP-responsive binding protein (CREB). CREB is a transcription factor which subsequently recruits its co-activators and binds to the cAMP-responsive element in the renin gene promoter, initiating transcription. Renin secretion is also stimulated by norepinephrine and prostaglandin E2 (PGE2).
Another signaling pathway involving the G-proteins and an increase in cytosolic calcium decreases the activity of the adenylate cyclase and inhibits renin secretion (this pathway is activated by angiotensin II, and endothelins I and II).
Angiotensinogen is a glycoprotein comprising more than 400 amino acids synthesized in the liver, and its different forms have variable structures and molecular mass. Renin cleaves a 10-amino acid peptide, angiotensin I, from the angiotensinogen. Angiotensin I then becomes a substrate for peptidyl-dipeptidase A (angiotensin-converting enzyme; ACE). ACE removes two amino acids from the angiotensin I, producing angiotensin II. This reaction can also be catalyzed by enzymes such as chymase and cathepsin. Another form of angiotensin, angiotensin 1–9, is formed by an isoform of ACE (ACE2) and is subsequently degraded to angiotensin 1–7. The latter can also be formed from angiotensin II by endopeptidases. Substantial amounts of angiotensin II are formed in the kidney. Juxtaglomerular cells contain ACE, angiotensin I and angiotensin II. Angiotensin II is also synthesized in the glomerular and tubular cells, and is secreted into the tubular fluid and the interstitial space. Angiotensin II receptors are present on the tubular and renal vascular cells: therefore, locally produced angiotensin II probably influences tubular reabsorption and renal vascular tone through autocrine and paracrine action. The renin–angiotensin system is illustrated in Figure 24.10.
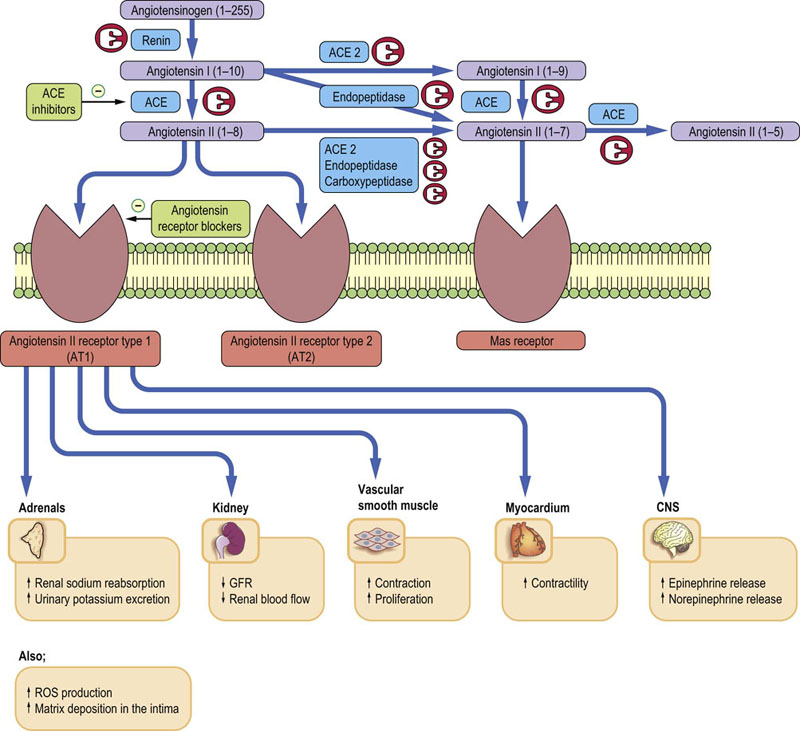
Fig. 24.10 Renin–angiotensin system.
Renin converts angiotensinogen into angiotensin I. Angiotensin I is further converted into angiotensin II by the angiotensin-converting enzyme (ACE). It also yields other angiotensin peptides. Cellular actions of angiotensins are mediated by angiotensin receptors type 1 (AT1), type 2 (AT2) and MAS receptors that bind angiotensin (1–7). The renin–angiotensin system is a target for two major classes of hypotensive drugs: ACE blockers (e.g. ramipril, enalapril) and AT1 receptor antagonists (e.g. losartan). ACE blockers are also extensively used in the treatment of heart failure. VSMC, vascular smooth muscle cells; CNS, central nervous system; ROS, reactive oxygen species; *AT1 receptor blocked by, e.g., losartan; **AT2 receptor blocked by saralasin.
Angiotensin receptors are important in the pathogenesis of cardiovascular disease
Angiotensin II constricts vascular smooth muscle, thereby increasing blood pressure and reducing renal blood flow and glomerular filtration rate. It also promotes aldosterone release and vascular smooth muscle proliferation through the activation of AT1 receptors which signal through G-proteins and phospholipase C (Fig. 24.11). Generally, AT1 receptor activation has effects that promote cardiovascular disease: the stimulation of inflammatory phenomena, extracellular matrix deposition, and generation of reactive oxygen species (ROS). It is also pro-thrombotic. These actions are counteracted by the stimulation of the AT2 receptors, which causes vasodilatation through stimulation of NO production, promotes sodium loss and inhibits vascular smooth muscle cell proliferation. The actions of angiotensin (1–7), which acts through the so-called MAS receptor (it may also bind to AT1 and AT2), also seem to be cardioprotective. Drugs that inhibit ACE are now extensively used in the treatment of hypertension and heart failure (see Fig. 24.11 and Box on p. 61).
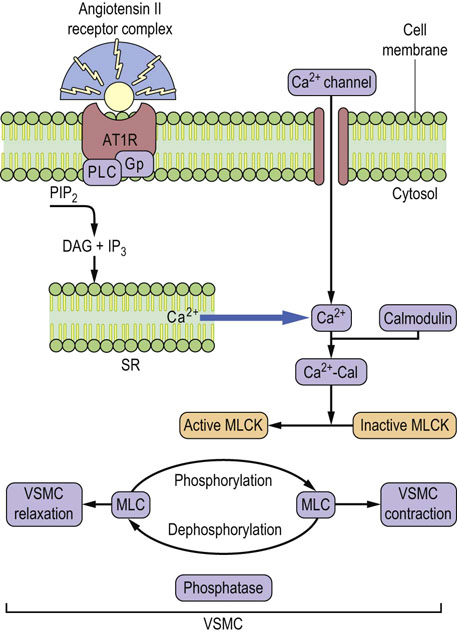
Fig. 24.11 Angiotensin II-induced vasoconstriction.
The angiotensin type 1 receptor (AT1R) is coupled to G-proteins. The binding of angiotensin II leads to phospholipase C-mediated formation of inositol 1,4,5-trisphosphate (IP3) and 1,2-diacylglycerol. This mobilizes Ca2+ from the sarcoplasmic reticulum (SR), and causes entry of the extracellular Ca2+ through activated calcium channels. An increase in the cytosolic Ca2+ initiates contractile response of the vascular smooth muscle cells. Ca2+ subsequently binds to calmodulin (Cal). The Ca2+–calmodulin complex activates myosin light-chain kinase. The kinase phosphorylates myosin light chains and elicits muscular tension. This effect is terminated by the dephosphorylation of myosin (Chapter 20). AT1 receptor antagonists, such as losartan, inhibit vasoconstrictor effects of angiotensin II and are used in the treatment of hypertension. AT1R, angiotensin type 1 receptor; DAG, 1,2,diacylglycerol; VSMC, vascular smooth muscle cell; MLCK, myosin light-chain kinase; Cal, calmodulin.
Clinical box Renin–angiotensin-aldosterone system and cardiac failure
A 65-year-old man with a previous anterior myocardial infarction presented with increasing fatigue, shortness of breath and ankle edema. Physical examination showed mild tachycardia, and a raised jugular venous pressure. An echocardiogram showed that the function of the left ventricle during systole was poor. The patient's serum measurements revealed sodium 140 mmol/L, potassium 3.5 mmol/L, protein 34 (normal 35–45) g/dL, creatinine 80 µmol/L (0.90 mg/dL), and urea 7.5 mmol/L (45 mg/dL).
This man presents with symptoms and signs of cardiac failure. The impaired function of the heart leads to a decreased blood flow through the kidney, activation of the renin–angiotensin system and stimulation of aldosterone secretion. Aldosterone causes an increased renal reabsorption of sodium and water retention, thereby increasing extracellular fluid volume and edema.
Clinical box Arterial hypertension is a common disease
Hypertension is inappropriately increased arterial blood pressure. The desirable level of systolic blood pressure is below 140 mmHg and diastolic pressure 90 mmHg (optimal values are still lower, below 120/80 mmHg). According to the World Health Organization, up to 20% of the population of the developed world may suffer from the condition. Arterial hypertension has been classified as ‘essential’ (primary) or ‘secondary’. A cause of essential hypertension has not yet been identified, although it is known to involve multiple genetic and environmental factors including neural, endocrine, and metabolic components. A sodium-rich diet is a recognized factor in the development of hypertension.
Hypertension is associated with an increased risk of stroke and myocardial infarction and causes one in every eight deaths worldwide. A range of drugs is used in the modern treatment of hypertension. These include diuretics such as bendrofluazide, drugs blocking adrenoreceptors, inhibitors of the angiotensin-converting enzyme and the antagonists of angiotensin receptors type 1 (see Box on p. 61 and Box on p. 315; pheochromocytoma is described on p. 558)
Aldosterone
Aldosterone regulates sodium and potassium homeostasis
Aldosterone is a major mineralocorticosteroid hormone in man (Chapter 17), and is produced in the adrenal cortex. It regulates extracellular volume and vascular tone, and controls renal sodium and potassium transport. It binds to the cytosolic mineralocorticoid receptor in the epithelial cells, principally in the renal collecting ducts. The receptor moves to the nucleus and binds to specific domains on targeted genes, altering their expression.
Aldosterone regulates the Na+/K+-ATPase in both the long and short term, and also regulates transporters such as the Na+/H+ exchanger type 3 in the proximal tubule, the Na+/Cl– co-transporter in the distal tubule, and the epithelial sodium channel in the renal collecting duct. The overall result is an increased sodium reabsorption, and increased potassium and hydrogen ion secretion.
Hyperaldosteronism is a common finding in hypertension
Primary hyperaldosteronism occurs as a result of abnormal adrenal activity and is rare. It may be a result of a single adrenal tumor, an adenoma (Conn's syndrome). The more common secondary hyperaldosteronism is due to an increased secretion of renin. Pheochromocytomas are catecholamine-secreting tumors that cause hypertension in about 0.1% of hypertensive patients. It is important to correctly diagnose pheochromocytoma, because it can be surgically removed (Chapter 43 and Box on p. 558).
Natriuretic peptides
Natriuretic peptides are important markers of heart failure
A family of peptides known as the natriuretic peptides are involved in the regulation of fluid volume. The two main ones are the atrial natriuretic peptide (ANP) and the brain natriuretic peptide (BNP). ANP is synthesized predominantly in the cardiac atria as a 126-amino acid propeptide (pro-ANP). It is then cleaved into a smaller 98-amino acid N-terminal peptide, and the biologically active 28-amino acid ANP. BNP is synthesized in the cardiac ventricles as a 108-amino acid propeptide, and is cleaved into a 76-amino acid N-terminal peptide and a biologically active 32-amino acid BNP. BNP 32 and another peptide, CNP (23 amino acids long), were isolated from the porcine brain, thus the name. All natriuretic peptides possess a ring-type structure due to the presence of a disulfide bond.
Natriuretic peptides promote sodium excretion and decrease the blood pressure. ANP and BNP are secreted in response to atrial stretch and to ventricular volume overload. They bind to G-protein-linked receptors: the A-type receptors are located predominantly in the endothelial cells and the B-type receptors in the brain. There is cross-reactivity between different natriuretic receptors with regard to these peptides.
The signaling pathway includes the membrane guanyl cyclase and the soluble guanyl cyclase, the latter being stimulated by NO. The generated cGMP acts on protein kinase C and phosphodiesterase 2 and 3, thus regulating cAMP synthesis.
 Clinical test box Diagnostic use of the brain natriuretic peptide (BNP) propeptides
Clinical test box Diagnostic use of the brain natriuretic peptide (BNP) propeptides
Instead of measuring the active forms of natriuretic peptides in plasma, it is more convenient to measure the propeptides which are present in plasma in equimolar amounts to the active species. Thus, proBNP (1–76) reaches higher levels in cardiac failure than BNP 32. Similarly, proANP (1–98) has a longer half-life in plasma than biologically active 1–28 ANP and therefore is present in the circulation in higher concentrations.
Clinical test box ANP and BNP as markers of heart failure
Importantly, the levels of ANP and BNP are increased in heart failure and therefore their measurements are used as early biochemical markers of this condition. These measurements are particularly useful for excluding heart failure in patients who present with nonspecific symptoms such as shortness of breath.
Vasopressin and aquaporins
Reabsorption of water in the collecting ducts of the kidney is controlled by the posterior pituitary hormone vasopressin through its control of membrane water channels, the aquaporins.
Vasopressin determines the final volume and concentration of the urine
Vasopressin (also known as the antidiuretic hormone, ADH) controls water reabsorption in the collecting ducts of the kidney. Vasopressin is synthesized in the supraoptic and paraventricular nuclei of the hypothalamus and is transported along axons to the posterior pituitary. It is stored there before being further processed and released. It binds to a receptor located on the membranes of tubular cells in the collecting ducts (Fig. 24.12). The receptor is coupled to G-proteins and activates PKA. The PKA phosphorylates aquaporin 2 (AQP2). This stimulates AQP2's translocation to the cell membrane, increasing water reabsorption in the collecting duct. On the other hand, Vasopressin secretion needs to be suppressed to allow urine dilution. Failure to maximally suppress vasopressin results in the inability to dilute urine below the osmolality of plasma.
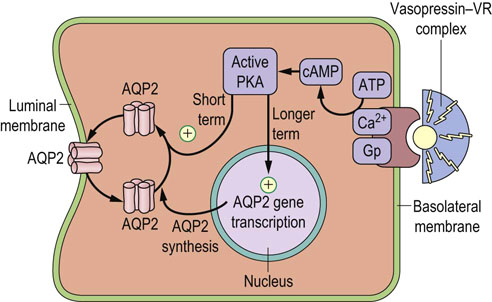
Fig. 24.12 Vasopressin regulates water reabsorption in the collecting duct.
Vasopressin controls the aquaporin 2 (AQP2) water channel. Vasopressin binds to its receptor (VR) and, through G-proteins, (Gp) stimulates production of cAMP, which, in turn, activates protein kinase A (PKA). PKA phosphorylates cytoplasmic AQP2 and induces its translocation to the cell membrane, increasing capacity for water transport. Vasopressin also regulates expression of the AQP2 gene.
Glucocorticoids stimulate vasopressin secretion primarily through their hemodynamic effects, which decrease arterial pressure. Incidentally, vasopressin is also stimulated by nicotine.
Aquaporins are membrane channel proteins which transport water
The aquaporin water channel is illustrated in Figure 24.13. AQP 2 and 3 are present in the collecting duct and are regulated by vasopressin. AQP1 is expressed on the apical and basolateral membranes of the proximal tubules and in the descending loop of Henle, and is not under vasopressin control. It is also present in erythrocytes, renal proximal tubular cells, and in the capillary endothelium.
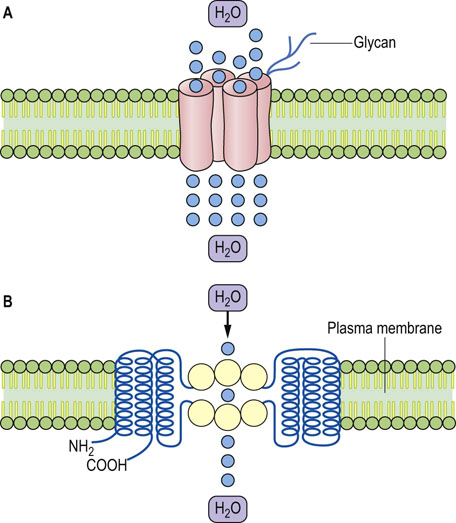
Defects in vasopressin secretion and mutations of genes coding for aquaporins cause clinical conditions
Vasopressin deficiency causes the condition known as diabetes insipidus, in which large amounts of dilute urine are excreted. On the other hand, an excessive secretion of vasopressin may occur following major trauma or surgery. This is known as the syndrome of inappropriate antidiuretic hormone secretion (SIADH), and leads to water retention. Mutations in the vasopressin receptor gene and also in the AQP2 gene lead to different types of nephrogenic diabetes insipidus, a condition associated with passing large amounts of urine and with dehydration.
Integration of water and sodium homeostasis
Aldosterone and vasopressin together control the handling of sodium and water
Normally, despite variations in fluid intake, plasma osmolality is maintained within narrow limits (280–295 mmol/kg H2O). Vasopressin contributes to the control of plasma osmolality by regulating water metabolism. It responds to both osmotic and volume signals: on the one hand, its secretion, and thirst, are stimulated by signals from osmoreceptors which respond to very small (approximately 1%) increases in plasma osmolality; on the other hand, vasopressin release is stimulated by a decrease (more than 10%) in the circulating volume.
Water excess increases plasma volume, renal blood flow, and GFR
When there is water excess, the production of renin is suppressed. Low concentration of aldosterone allows the urinary sodium loss. Because excess water ‘dilutes’ the plasma, plasma osmolality decreases. This decrease in osmolality, sensed by the hypothalamic osmoreceptors, suppresses both thirst and the secretion of vasopressin. Suppression of vasopressin leads to the urinary loss of water. Thus, the overall response to water excess is increased loss of sodium and water in urine.
Water deficit (dehydration) decreases plasma volume, renal blood flow and GFR
When there is water deficit (dehydration), the decrease in renal blood flow stimulates the renin–angiotensin–aldosterone system. Aldosterone inhibits urinary sodium excretion. In parallel, water deficit causes an increase in the plasma osmolality. This stimulates vasopressin secretion, with a consequent decrease in the urine volume. Thus, the overall response to water deficit is sodium and water retention (Fig. 24.14).
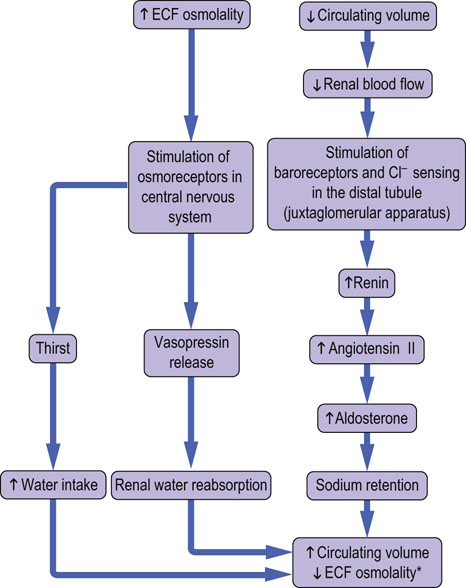
Fig. 24.14 Links between water and sodium metabolism.
Water and sodium metabolism are closely interrelated. An increase in ECF osmolality stimulates secretion of vasopressin and leads to increased renal water reabsorption. This ‘dilutes’ the ECF and the osmolality decreases. This response is reinforced by the stimulation of thirst. A decrease in the plasma volume also stimulates water retention through stimulation of the pressure-sensitive receptors (baroreceptors) in the juxtaglomerular apparatus. *Osmolality will decrease if the degree of water retention is relatively greater than that of sodium retention.
Clinical box Poor fluid intake leads to dehydration
An 80-year-old man had been admitted to hospital after lying for a prolonged period on the floor at home after suffering acute stroke. He had poor tissue turgor, dry mouth, tachycardia and he was hypotensive. Serum measurements revealed: sodium 150 mmol/L, potassium 5.2 mmol/L, bicarbonate 35 mmol/L, urea 19 mmol/L (90.3 mg/dL) and creatinine 110 µmol/L (1.13 mg/dL).
Serum sodium concentration is a marker of fluid and electrolyte disorders
Water and electrolyte disturbances result from an imbalance between the intake of fluids and electrolytes and their loss, and from the movement of water and electrolytes between body compartments. A decreased sodium concentration (hyponatremia) usually indicates that the extracellular fluid is being ‘diluted’ (due to an excess of water), whereas an increased sodium concentration (hypernatremia) means that the extracellular fluid is being ‘concentrated’ (due to water loss). Hyponatremia may also result from loss of sodium but this is rare.
Assessment of water and electrolyte status is an important part of clinical practice
The assessment of water and electrolyte balance is an important part of clinical examination. In addition to the physical examination and medical history, the following measurements are required:
Serum electrolyte concentrations: the profile commonly requested by a physician includes sodium, potassium, chloride and bicarbonate concentrations.
Serum urea (blood urea nitrogen) and creatinine.
Urine volume, osmolality and sodium concentration.
Fluid balance chart: patients who have or who are at risk of developing abnormalities of water or electrolyte balance need a daily record of fluid intake and loss.
Summary
Both deficit of body water (dehydration) and its excess (overhydration) cause potentially serious clinical problems. Therefore, the assessment of water and electrolyte balance is an important part of clinical examination.
Body water balance is closely linked to the balance of dissolved ions (electrolytes), the most important of which are sodium and potassium.
Movement of water between ECF and ICF is controlled by osmotic gradients.
Movement of water between the lumen of a blood vessel and the interstitial fluid is controlled by the osmotic and hydrostatic pressures.
The main regulators of water and electrolyte balance are vasopressin (water) and aldosterone (sodium and potassium).
The renin–angiotensin–aldosterone system is the principal regulator of the blood pressure and vascular tone.
Measurements of natriuretic peptides help to diagnose the cardiac failure.
Androgue, HJ, Madias, NE. Mechanisms of disease: sodium and potassium in the pathogenesis of hypertension. N Engl J Med. 2007; 356:1966–1978.
Bekheirnia, R, Schrier, RW. Pathophysiology of water and sodium retention: edematous states with normal kidney function. Curr Opin Pharmacol. 2006; 6:202–207.
Chobanian, A, Bakris, GL, Black, HR, et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure. JAMA. 2003; 289:2560–2572.
Ellison, DH, Berl, T. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007; 356:2064–2072.
Ernst, ME, Moser, M. Use of diuretics in patients with hypertension. N Engl J Med. 2009; 361:2153–2164.
Goetze, JP. B-type natriuretic peptide: from posttranslational processing to clinical measurement. Clin Chem. 2012; 58:83–91.
Richards, AM, Troughton, RW. Use of natriuretic peptides to guide and monitor heart failure therapy. Clin Chem. 2012; 58:62–71.
Schrier, RW. Body water homeostasis: clinical disorders of urinary dilution and concentration. J Am Soc Nephrol. 2006; 17:1820–1832.
Verkman, AS. Aquaporins in clinical medicine. Ann Rev Med. 2012; 63:303–316.
Biesalski, HK, Bischoff, SC, Boehles, HJ, et al, Water, electrolytes, vitamins and trace elements – Guidelines on Parenteral Nutrition. Ger Med Sci Ger Med Sci 2009. www.ncbi.nlm.nih.gov/pmc/articles/PMC2795367
Medline Plus. Water and electrolyte balance. www.nlm.nih.gov/medlineplus/fluidandelectrolytebalance.html.
Powell-Tuck, J, Peter Gosling, P, Dileep, N, et al. British Consensus Guidelines on Intravenous Fluid Therapy for Adult Surgical Patients. www.bapen.org.uk/pdfs/bapen_pubs/giftasup.pdf, 2011.