Role of the Liver in Metabolism
Learning objectives
After reading this chapter you should be able to:
 Discuss the participation of the liver in carbohydrate metabolism, and in particular its role in endogenous glucose production.
Discuss the participation of the liver in carbohydrate metabolism, and in particular its role in endogenous glucose production.
Discuss the role of the liver in lipid metabolism.
Outline changes in the hepatic protein synthesis that take place during the acute phase reaction.
Describe ubiquitin-mediated mechanisms of proteolysis.
Describe the pathway of heme synthesis.
Describe the metabolism of bilirubin and the main types of jaundice.
Comment on the mechanism of hepatotoxicity of drugs and alcohol.
Introduction
The liver has a central role in metabolism, because of both its anatomic placement and its many biochemical functions
The liver receives venous blood from the intestine: thus all of the products of digestion, ingested drugs and other xenobiotics reach it and may be further metabolized before entering the systemic circulation. The hepatic parenchymal cells, the hepatocytes, have an immensely broad range of synthetic and catabolic functions, which are summarized in Table 30.1.
Table 30.1
Functions of hepatic parenchymal cells and their disturbances in liver disease
| Function | Markers of impairment in plasma |
| Heme catabolism | bilirubin |
| Carbohydrate metabolism | ↓glucose |
| Protein synthesis | ↓albumin |
| prolonged prothrombin time | |
| Protein catabolism | ↑ammonia |
| ↓urea | |
| Lipid metabolism | ↑cholesterol |
| ↑triglycerides | |
| Drug metabolism | biological half-time of a drug |
| Bile acid metabolism | ↑bile acids |
The liver plays important roles in carbohydrate, lipid and amino acid metabolism, in the synthesis and breakdown of plasma proteins, and in the storage of vitamins and metals. It also has the ability to metabolize, and so detoxify, an infinitely wide range of xenobiotics. The liver also has an excretory function, in which metabolic waste products are secreted into a branching system of ducts known as the biliary tree, which in turn drains into the duodenum; the biliary constituents are then excreted in feces.
The liver is the largest organ in the body and has a substantial reserve metabolic capacity
Mild disease may cause no symptoms and is detected only by biochemical changes in the blood. However, the patient with severe liver disease has a yellow pigmentation of the skin (jaundice), bruises readily and may bleed profusely, has an abdomen distended with fluid (ascites), and may be confused or unconscious (hepatic encephalopathy) (Fig. 30.1). This chapter will describe the specialized metabolic functions of the liver and the abnormalities that occur in liver disease.
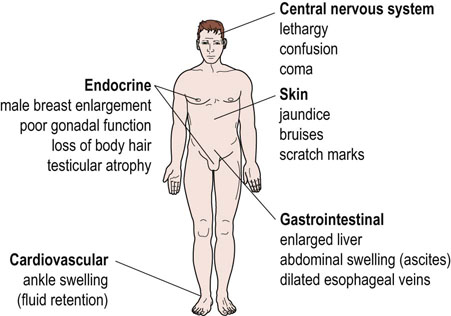
Structure of the liver
The liver is the largest solid organ in the body and, in adults, weighs about 1500 g. Approximately 75% of its blood flow is supplied by the portal vein, which arises from the intestine. The hepatic artery supplies the remainder of its blood. Blood leaving the liver enters the systemic venous system in the hepatic vein. The biliary component of the liver comprises the gallbladder and bile ducts.
Structure of the liver facilitates exchange of metabolites between hepatocytes and plasma
Under the microscope, the substance of the liver is composed of a very large number of hepatocytes arranged in polyhedral lobules (Fig. 30.2). Portal tracts at the ‘corners’ of these polyhedrons contain branches of the portal vein, hepatic artery and interlobular bile ducts. Blood sinusoids arise from the terminal branches of the portal vein and interconnect and interweave through the hepatocytes before joining the central lobular vein, which in turn eventually flows into the hepatic vein.
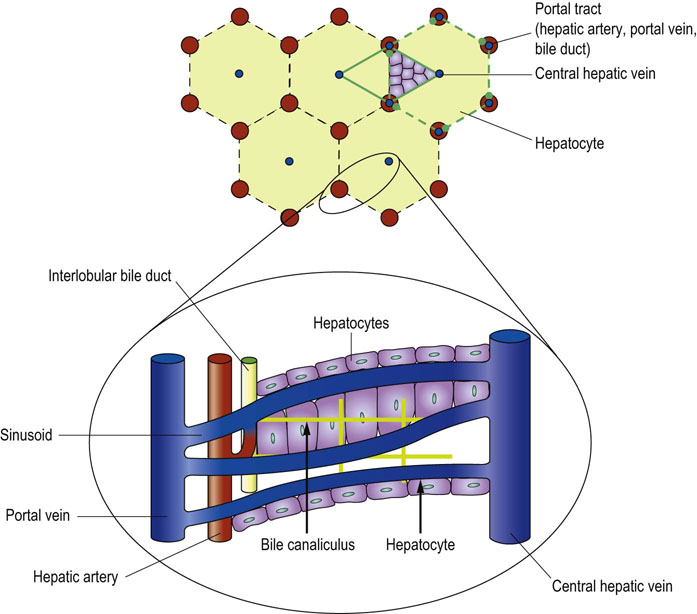
Sinusoids are lined by two cell types. The first are vascular endothelial cells, which are loosely connected one with another, leaving numerous gaps. There is no basement membrane between the endothelial cells and the hepatocytes. The second type of sinusoidal cells, known as Kupffer cells, are mononuclear phagocytes; they are generally found in the gaps between endothelial cells.
These anatomical arrangements facilitate the exchange of metabolites between hepatocytes and plasma, allow the hepatocytes to receive an arterial supply and permit the excretory products from hepatocyte metabolism destined for biliary excretion to enter the biliary ducts.
Liver and carbohydrate metabolism
The liver plays a central role in glucose metabolism, specifically in maintaining the circulating concentration of glucose (Chapters 13 and 21). This function depends upon its ability both to store a supply of glucose as glycogen, and to synthesize glucose from noncarbohydrate sources, other than lipid, through gluconeogenesis.
Depending on metabolic conditions, the liver can either take up or produce glucose
The liver possesses glucose-6-phosphatase, which permits the release of free glucose to the blood. Although muscle stores more glycogen than the liver, it has no glucose-6-phosphatase and therefore cannot directly contribute glucose to the blood. The kidney has both the ability to synthesize glucose-6-phosphate de novo by gluconeogenesis, and has glucose-6-phosphatase activity, but quantitatively it contributes much less than the liver (Chapter 23). Moreover, kidneys do not store glycogen.
Certain organs, notably the brain, are dependent upon glucose as a source of energy, and so the adult liver in the fasting state releases around 9 g of glucose each hour to the blood to maintain the blood glucose concentration. The substrates for gluconeogenesis are derived from lactate released by glycolysis in the peripheral tissues and from hepatic deamination of amino acids (mainly alanine) generated from the proteolysis of skeletal muscle (Chapter 21).
Liver and protein metabolism
Most plasma proteins are synthesized in the liver
Hepatocellular disease may alter protein synthesis both quantitatively and qualitatively. Albumin is the most abundant protein in blood and is synthesized exclusively by the liver (Chapter 4). Low plasma albumin concentration occurs commonly in liver disease. It is however, a poor index of hepatic synthetic function because in systemic illness (which often accompanies hepatic disease) there is an increased vascular endothelial permeability that allows the leakage of albumin into the interstitial space.
A better index of hepatocyte synthetic function is the production of the coagulation factors II, VII, IX, and X
All the coagulation factors undergo post-translational γ-carboxylation of specific glutamyl residues, allowing them to bind calcium. As a group, their functional concentration can be readily assessed in the hematology laboratory by measuring the prothrombin time (PT; Chapter 7).
The liver also synthesizes most of the plasma α- and β-globulins. Their plasma concentrations change in hepatic disease and in systemic illness; in the latter case, these changes form part of the acute phase response.
Response to an acute insult is associated with wide-ranging changes in liver protein synthesis
The ‘acute phase response’ is a term encompassing all the systemic changes which occur in response to infection or inflammation (Chapter 4). The liver synthesizes a number of ‘acute phase proteins’, which have been defined as those whose plasma concentrations change by more than 25% within a week of the inflammatory or infective insult. The production of these proteins is stimulated by pro-inflammatory cytokines released by macrophages, and of these interleukin-1 (IL-1), IL-6 and tumor necrosis factor (TNF) have a central role. The acute phase proteins have a number of different functions. Binding proteins, opsonins, such as C-reactive protein (CRP), bind to macromolecules released by damaged tissue or infective agents and promote their phagocytosis (Chapter 38). Complement factors also promote the phagocytosis of foreign molecules. Protease inhibitors, such as α1-antitrypsin and α1-antichymotrypsin, inhibit proteolytic enzymes. The latter two also stimulate fibroblast growth and the production of connective tissue required for the repair and resolution of the injury.
A substantial supply of amino acids is required as substrates for this increase in hepatic protein synthesis and these are derived from the proteolysis of skeletal muscle. TNF and IL-1, again, are involved by stimulating the breakdown of specific intracellular proteins by the ubiquitin–proteasome system (see below).
Protein degradation by the ubiquitin–proteasome system
Ubiquitin marks intracellular proteins for proteasomal degradation
Hepatic protein turnover is highly regulated, which allows metabolic pathways to adapt to changing physiologic circumstances. Mammalian cells possess several proteolytic systems.
Plasma proteins and membrane receptors are endocytosed and then hydrolyzed by acid proteases within the lysosomes. Intracellular proteins, on the other hand, are degraded within structures known as proteasomes by the so-called ubiquitin–proteasome system (UPS) (Chapter 33). The discoverers of protein ubiquinylation were awarded the Nobel Prize in chemistry in 2003. The UPS is important in the activation of the NFκB pro-inflammatory pathway and the function of UPS is modified by reactive oxygen species (Chapter 37; see also Fig. 34.10).
Removal of nitrogen
The urea cycle is essential for the removal of nitrogen generated by amino acid metabolism
Catabolism of amino acids generates ammonia (NH3) and ammonium ions (NH4+). Ammonia is toxic, particularly to the central nervous system (CNS). Most ammonia is detoxified at its site of formation, by amidation of glutamate to glutamine, which is mainly derived from muscle and is used as an energy source by the enterocytes. The remaining nitrogen enters the portal vein either as ammonia or as alanine, both of which are used by the liver for the synthesis of urea (Chapter 19).
Impaired clearance of ammonia causes brain damage
The urea cycle is the major route by which waste nitrogen is excreted, and is described in Chapter 19. In neonates, inherited defects of any of the enzymes of the urea cycle lead to hyperammonemia, which impairs the function of the brain, causing encephalopathy. Such problems arise within the first 48 hours of life and inevitably are made worse by protein-rich foods such as milk (see Clinical Box on p. 243).
Heme synthesis
Heme is a constituent of hemoglobin, myoglobin and cytochromes
Heme is synthesized in most cells of the body. The liver is the main nonerythrocyte source of its synthesis. Heme is a porphyrin, a cyclic compound which contains four pyrrole rings linked together by methenyl bridges. It is synthesized from glycine and succinylcoenzyme A, which condense to form 5-aminolevulinate (5-ALA). This reaction is catalyzed by 5-ALA synthase, located in mitochondria, and is rate limiting in heme synthesis. Subsequently, in the cytosol, two molecules of 5-ALA condense to form a molecule containing a pyrrole ring, porphobilinogen (PBG). Then, four PBG molecules combine to form a linear tetrapyrrole compound, which cyclizes to yield uroporphyrinogen III and then coproporphyrinogen III. Final stages of the pathway occur again in the mitochondria where a series of decarboxylation and oxidation of side chains in uroporphyrinogen III yield protoporphyrin IX. At the final stage, iron (Fe2+) is added by ferrochelatase to protoporphyrin IX to form heme. Heme controls the rate of its synthesis by feedback inhibition of 5-ALA synthase (Fig. 30.3).
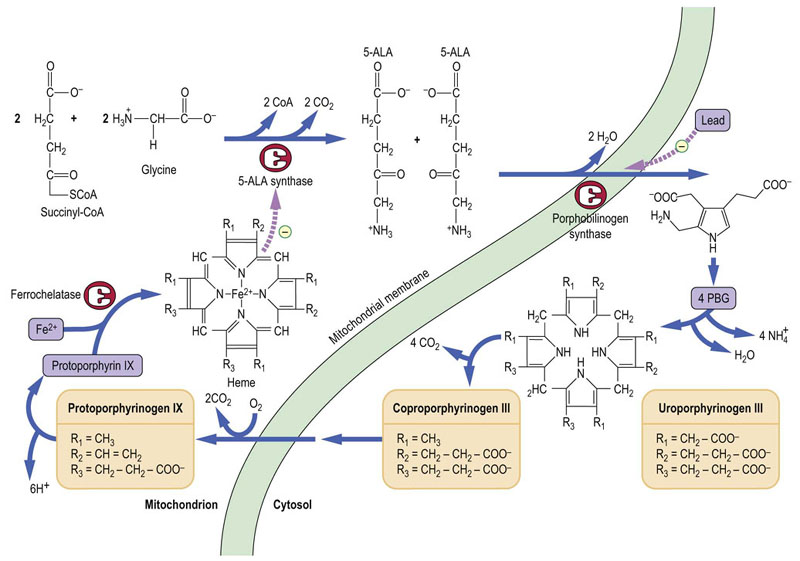
Fig. 30.3 The pathway of heme synthesis.
Part of the pathway is located in the mitochondria and part in the cytosol. ALA, 5-aminolevulinate; PBG, porphobilinogen. Hemoglobin is discussed in Chapter 5.
 Advanced concept box The porphyrias
Advanced concept box The porphyrias
Defects in the heme synthetic pathway lead to rare disorders known as porphyrias. Different porphyrias are caused by deficiencies of different enzymes in the biosynthetic pathway, starting from 5-ALA synthase and ending with ferrochelatase. Porphyrias are classified as hepatic or erythropoietic, depending on the primary organ affected.
Three porphyrias are known as acute porphyrias and can be a cause of emergency admissions with abdominal pain (which needs to be differentiated from various surgical causes). They also cause neuropsychiatric symptoms. Acute intermittent porphyria (AIC) is caused by the deficiency of hydroxymethylbilane synthase, an enzyme converting PBG to a linear tetrapyrrole; in this disorder the concentrations of 5-ALA and PBG increase in plasma and urine. Hereditary coproporphyria is due to a defect in the conversion of coproporphyrinogen III to protoporphyrinogen III (coprooxidase). The third acute porphyria is the variegate porphyria, the clinical manifestations of which are very similar to AIC.
Other porphyrias, such as porphyria cutanea tarda, present clinically as the sensitivity of skin to light (photosensitivity) which may cause disfiguration and scarring. Also, the pathway is inhibited by lead at the stage of porphobilinogen synthase.
Bilirubin metabolism
Excess bilirubin causes jaundice
Bilirubin is the catabolic product of heme. About 75% of all bilirubin is derived from the breakdown of hemoglobin from senescent red blood cells, which are phagocytosed by mononuclear cells of the spleen, bone marrow, and liver (reticulo-endothelial cells). In normal adults the daily load of bilirubin is 250–350 mg. The ring structure of heme is oxidatively cleaved to biliverdin by heme oxygenase, a P-450 cytochrome (see below). Biliverdin is, in turn, enzymatically reduced to bilirubin (Fig. 30.4). The normal plasma concentration of bilirubin is less than 21 µmol/L (1.2 mg/dL). Increased concentrations (more than 50 µmol/L or 3 mg/dL) are readily recognized clinically, because at this concentration or more bilirubin imparts a yellow color to the skin and conjunctivae known clinically as jaundice, or icterus. Abnormalities in bilirubin metabolism are clinical important pointers to the presence of liver disease.
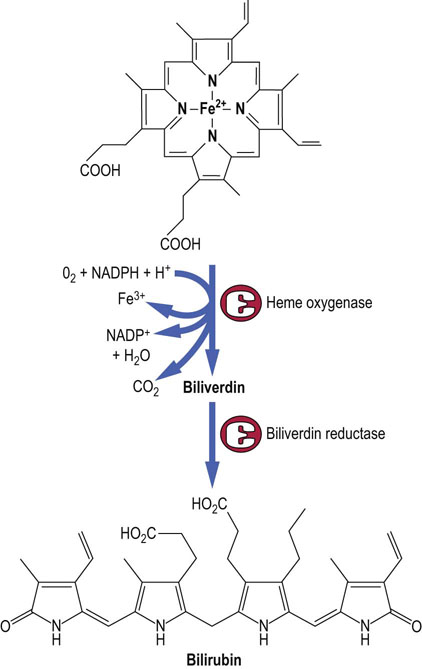
Bilirubin is metabolized by the hepatocytes and excreted in bile
Whereas biliverdin is water soluble, bilirubin, paradoxically, is not. Therefore, it must be further metabolized before excretion (Fig. 30.5). Bilirubin produced by the catabolism of heme in the reticuloendothelial cells is transported in plasma bound to albumin. The hepatic uptake of bilirubin is mediated by a membrane carrier and may be competitively inhibited by other organic anions. The hydrophilicity of bilirubin is increased by esterification, usually known as conjugation, of one or both of its carboxylic acid side chains with glucuronic acid, xylose or ribose. The glucuronide diester is the major conjugate and its formation is catalyzed by the uridine diphosphate (UDP)-glucuronyl transferase. Conjugated bilirubin is water soluble and may be secreted by the hepatocyte into the biliary canaliculi. If this excretory process is impaired and the patient becomes jaundiced as a result, some conjugated bilirubin may be lost into the urine, imparting a characteristically dark color.
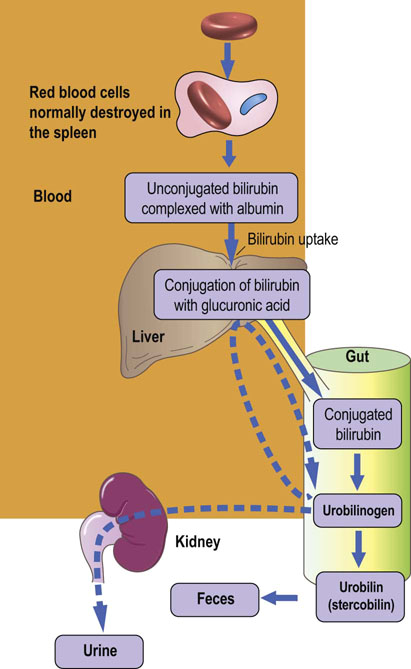
Conjugated bilirubin in the gut is catabolized by bacteria to form stercobilinogen, also known as fecal urobilinogen, which is colorless. On oxidation, however, stercobilinogen forms stercobilin (otherwise known as fecal urobilin), which is colored; most stercobilin is responsible for the color of feces. Some stercobilin may be reabsorbed from the gut and can then be re-excreted by either the liver or the kidneys.
Bile acid and cholesterol metabolism
Bile acids are key elements in fat metabolism
Bile acids are synthesized in the hepatocytes and have a detergent-like effect, solubilizing biliary lipids and emulsifying dietary fat in the gut to facilitate its digestion. Their metabolism is described in Chapters 10 and 17. Bile is also the only route of cholesterol excretion from the body (Chapters 17 and 18).
Drug metabolism
Low substrate specificity of some hepatic enzymes produces a wide-ranging capability for drug metabolism
Most drugs are metabolized in the liver. Among other effects, this hepatic metabolism usually increases the hydrophilicity of drugs and therefore their ability to be excreted through the kidneys or in bile. Generally, drug metabolites are less pharmacologically active than the parent drugs; however, some drugs are inactive when administered (prodrugs) but are converted to their active forms as a result of liver processing. The hepatic drug-metabolizing systems need to act on an infinite range of molecules that can be encountered in the environment; this is achieved by the enzymes involved having low substrate specificity.
Drug metabolism proceeds in two phases
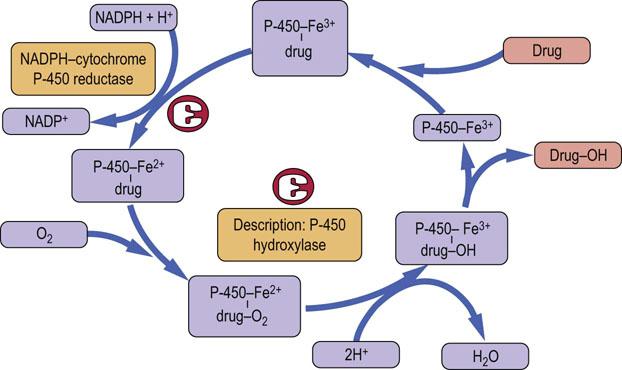
Phase I is the addition of the polar group: the polarity of the drug is increased by oxidation or hydroxylation catalyzed by a family of microsomal cytochrome P-450 oxidases.
Phase II is conjugation: cytoplasmic enzymes conjugate the functional groups introduced in the first phase reactions, most often by glucuronidation or sulfation, and also acetylation and methylation.
Three of the 12 cytochrome P-450 gene families share the responsibility for drug metabolism
The cytochrome P-450 enzymes are heme-containing proteins that co-localize with NADPH:cytochrome P-450 reductase. They are present in the endoplasmic reticulum. Most metabolism associated with the cytochrome P-450 superfamily takes place in the liver but these enzymes are also present in the epithelium of the small intestine. The reaction sequence catalyzed by these enzymes is shown in Figure 30.6. There are 12 cytochrome P-450 gene families, of which three, designated CYP1, CYP2 and CYP3, are responsible for most of the phase I drug metabolism. In fact, six enzymes, CYP1A2, CYP3A4, CYP2C9, CYP2C19, CYP2D6 and CYP2E1, are responsible for approximately 90% of drug metabolism. CYP3A4 is one of the most important cytochrome P-450 enzymes.
Induction, and competitive inhibition of cytochrome P-450 enzymes are mechanisms of drug interactions
Hepatic synthesis of cytochromes P-450 is induced by certain drugs and other xenobiotic agents: this increases the rate of phase I reactions. On the other hand, drugs that form a relatively stable complex with a particular cytochrome P-450 inhibit the metabolism of other drugs that are normally substrates for that cytochrome. For instance, CYP1A2 metabolizes, among others, caffeine and theophylline. It can be inhibited by grapefruit juice, that contains a substance known as naringin, or by the antibiotic ciprofloxacin. When a person takes any of the inhibitory substances, normal substrates for CYP1A2 are metabolized more slowly and their plasma levels increase.
During immunosuppressive therapy the dose of the immunosuppressant cyclosporine may need to be reduced by up to 75% if the patient also takes the antifungal drug ketoconazole (see Wilkinson in Further Reading). Otherwise adverse reactions may occur.
The drugs that induce induction or repression of CYP3A enzymes often act through the nuclear receptor mechanism. They combine with nuclear receptors (i.e. in case of CYP3A4, the pregnane X receptor, PXR), which then form heterodimers with retinoid X receptors (Chapter 17). Such complexes upregulate CYP3 synthesis by binding to response elements in the gene promoter.
Cytochrome P-450 gene polymorphisms determine response to many drugs
Allelic variation that affects the catalytic activity of a cytochrome P-450 also affects the pharmacologic activity of drugs. The best described example of such polymorphism is that of the P-450 cytochrome CYP2D6, which was recognized initially in the 5–10% of normal individuals who were noted to be slow to hydroxylate debrisoquine, a now little used blood pressure-lowering drug. However, CYP2D6 also metabolizes a significant number of other commonly used drugs, so that ‘debrisoquine polymorphism’ remains clinically significant.
Antiplatelet therapy with clopidogrel is the current standard of care for coronary artery disease patients undergoing revascularization. However, approximately 25% of patients experience a subtherapeutic antiplatelet response. Clopidogrel is a prodrug that undergoes hepatic biotransformation by CYP2C19 into its active metabolite. Several studies have reported that carriers of CYP2C19 variant allele exhibit a significantly lower capacity to transform clopidogrel to its active metabolite and are therefore at significantly higher risk of adverse cardiovascular events. Consequently, in the US, the FDA has recently changed clopidogrel's prescribing information to highlight the impact of CYP2C19 genotype on the clinical response to clopidogrel.
Genotyping of cytochromes P-450 to identify gene-relevant polymorphisms may become more common in an attempt to personalize an individual's response to a particular drug.
Drug hepatotoxicity
Drugs that exert their toxic effects on the liver may do so through the hepatic production of a toxic metabolite. Drug toxicity may occur in all individuals exposed to a sufficient concentration of a particular drug. However, a drug may be toxic in some individuals at concentrations normally tolerated by most other patients. This phenomenon is known as idiosyncratic drug toxicity and may be due to a genetic or immunologic cause.
The commonly prescribed drug acetaminophen (paracetamol) is hepatotoxic in excess
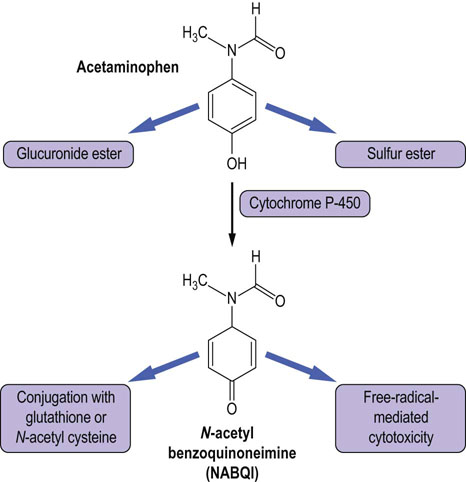
Acetaminophen is widely used as a painkiller. Taken in the usual therapeutic doses, it is conjugated with glucuronic acid or sulfate, which is then excreted by the kidneys. In overdose, however, the capacity of these conjugation pathways is overwhelmed and acetaminophen is oxidized by a liver P-450 cytochrome CYP3A4 to N-acetyl benzoquinoneimine (NABQI). NABQI can cause a free radical-mediated peroxidation of membrane lipids, and consequently hepatocellular damage. It may be detoxified by conjugation with glutathione, but in acetaminophen overdose glutathione stores also become exhausted, causing hepatotoxicity (Fig. 30.7). Therapeutically, a sulfhydryl compound, N-acetylcysteine (NAC), is routinely used as an antidote to acetaminophen poisoning. It promotes detoxification of NABQI by the glutathione pathway and also scavenges free radicals. The risk of hepatotoxicity can be reliably predicted from measurement of the plasma concentration of acetaminophen in relation to the time which has elapsed since the overdose, and NAC can be given to patients who are at risk of liver damage. Measurement of paracetamol is thus one of the emergency tests offered by clinical laboratories.
Alcohol
Alcohol excess is a major cause of liver disease
Excess intake of ethyl alcohol (ethanol) remains the most common cause of liver disease in the Western world. Ethanol may cause excessive fat deposition in the liver (alcoholic steatosis), hepatitis or finally fibrosis (known as cirrhosis), which in turn leads to liver failure. There are over 25,000 deaths associated with liver disease in the US annually, and 40% of these are linked to alcoholic cirrhosis (see Donohue in Further Reading).
Ethanol is oxidized in the liver, mainly by alcohol dehydrogenase (ADH), to form acetaldehyde, which is in turn oxidized by aldehyde dehydrogenase (ALDH) to acetate. Nicotinamide adenine dinucleotide (NAD+) is the cofactor for both these oxidations, being reduced to NADH. A P-450 cytochrome, CYP2E1, also contributes to ethanol oxidation but is quantitatively less important than the ALD–ALDH pathway. Liver damage in patients who abuse alcohol may arise from the toxicity of acetaldehyde, which forms Schiff base adducts with other macromolecules.
Ethanol oxidation alters the redox potential of the hepatocyte
Ethanol oxidation results in the increased ratio of NADH to NAD+ This inhibits oxidation of lactate to pyruvate (a step that requires NAD+ as a cofactor) and also creates potential for the development of lactic acidosis. Because pyruvate is a substrate for hepatic gluconeogenesis, there is also a risk of hypoglycemia. The risk of hypoglycemia is further increased in alcoholics, when, because of poor nutrition, they have low hepatic glycogen stores. Also, the shift in the NADH/NAD+ ratio inhibits β-oxidation of fatty acids and promotes triglyceride synthesis: excess of triglycerides is deposited in the liver and secreted into plasma as VLDL (see Clinical Box on p. 197). Hepatic steatosis can be readily diagnosed by ultrasonography of the liver when one sees a uniform increased echogenicity (Fig. 30.8). It is often associated with elevation in serum levels of the transaminase enzymes.
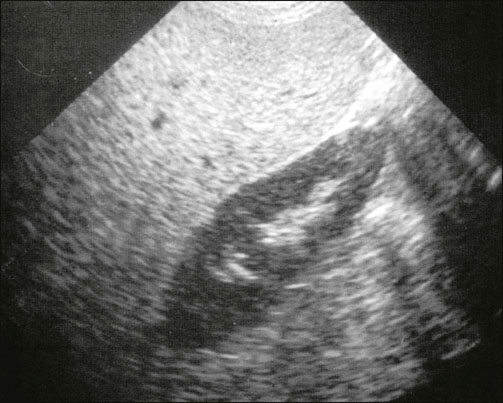
Fig. 30.8 An ultrasound scan of a liver showing steatosis. Courtesy Dr A Bannerjee, Birmingham Heartlands and Solihull NHS Trust, UK.
Ethanol consumption also affects the ubiquitin system of protein degradation. Chronic alcohol consumption decreases proteasome activity. This can deregulate the hepatocyte signaling system by inhibiting the Janus Kinase-Signal Transducer and Activator of Transcription (JAK-STAT) signalling pathway, which is involved in the acute phase response, antiviral defense and hepatic repair (Chapter 40). Inhibition of proteasome activity may also lead to increased apoptosis (Chapter 43), a feature of alcoholic liver disease (ALD). The ethanol-induced decrease in proteasome activity prevents the degradation of CYP2E1, which is involved in peroxidation reactions; this increases oxidative stress and may be another factor contributing to ALD.
Finally, alcohol-induced decrease in proteasome activity may lead to the accumulation of protein in the liver, which in turn causes liver enlargement (hepatomegaly; common in ALD). Other ethanol-induced phenomena include increased secretion of chemokines (including IL-8 and monocyte-chemoattractant protein-1 (MCP-1; Chapter 18)) by hepatocytes, leading to liver infiltration by neutrophils.
 Clinical box A 22-year-old woman who took acetaminophen overdose
Clinical box A 22-year-old woman who took acetaminophen overdose
A 22-year-old woman was admitted to hospital in a semiconscious state. She had been found with a suicide note and empty acetaminophen containers. Tests revealed aspartate aminotransferase (AST) 5500 U/L, alkaline phosphatase (ALP) 125 U/L, bilirubin 70 µmol/L (4.1 mg/dL), prothrombin time 120 s (reference range 10–15 s), creatinine 350 µmol/L (4.0 mg/dL; reference range 44–80 µmol/L, 0.50–0.90 mg/dL), glucose 2.6 mmol/L (47 mg/dL; reference range 4.0–6.0 mmol/L, 72–109 mg/dL), and blood pH 7.1 (Reference range 7.35-7.45; This equals to 80 nmol/L H+ with reference range 35-45 nmol/L). No acetaminophen was found in her plasma.
The patient had acute hepatic failure, most probably caused by acetaminophen poisoning. Blood acetaminophen may be undetectable if the patient first comes to medical attention more than 24 h after an overdose. The hepatocellular damage worsens over the first 72 h but may improve spontaneously after that, as a result of regeneration of hepatocytes. However, in patients with a metabolic acidosis (pH <7.35 or H+ above 45 mnmol/L, after fluid resuscitation), markedly increased prothrombin time (>100 s) or serum creatinine >300 µmol/L (3.4 mg/dL), mortality is of the order of 90% and liver transplantation may be necessary. For reference ranges (Chapter 40), see Table 30.2 and Appendix 1.
Table 30.2
Laboratory tests used in differential diagnosis of jaundice
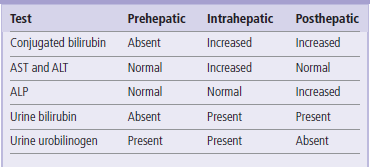
Reference ranges for liver function tests:
AST (aspartate aminotransferase), men 15–40 U/L, women 13–35 U/L; ALT (alanine aminotransferase), men 10–40 U/L, women 7–35 U/L; ALP (alkaline phosphatase), 50–140 U/L: ALP is physiologically elevated in children and adolescents; bilirubin <21 μmol/L (<1.0 mg/dL); (γ-glutamyl transpeptidase:) GGT, men <90 U/L, women <50 U/L.
Symptoms of alcohol intolerance are exploited to reinforce abstinence
Both ADH and ALDH are subject to genetic polymorphisms, which have been investigated as a potential inherited basis of susceptibility to alcoholism and ALD. Possession of the ALDH22 allele, which encodes an enzyme with reduced catalytic activity, leads to increased plasma concentrations of acetaldehyde after the ingestion of alcohol. This causes the individual to experience unpleasant flushing and sweating, which discourages alcohol abuse. Disulfiram, a drug that inhibits ALDH, also causes these symptoms when alcohol is taken, and may be given to reinforce abstinence from alcohol.
Pharmacogenomics
The response to any particular drug is influenced by drug's kinetic properties (pharmacokinetics) and its effects (pharmacodynamics)
An individual's response to a drug can be influenced by genes that code for drug-metabolizing enzymes, receptors and transporters. Any variability in these genes may lead to inter-individual differences in response to that drug.
The effectiveness and safety of drug therapy, particularly in elderly patients or those with renal or hepatic diseases as co-morbidities, and in patients whose metabolic capacity is diminished, is currently a major problem. Approximately 3% of hospital admissions in the US are linked to drug–drug interactions and a Dutch study reported values as high as 8.4%. In the US, there are 2 million cases of adverse drug reactions annually, including 100,000 deaths. Combined with the fact that most drugs are effective in only 25–60% of patients to whom they are prescribed, this makes research into individual response to drugs absolutely essential.
Pharmacogenomics studies the effects of genetic heterogeneity on drug responsiveness
Since the liver plays a central role in drug metabolism, the pharmacogenomics of some hepatic drug-metabolizing enzymes, specifically the cytochrome P-450 oxidases, is clinically very relevant. CYP2D6 is responsible for the metabolism of more than 100 pharmaceuticals, and a polymorphism of this enzyme is responsible for the long-established variation in the metabolism of debrisoquine, mentioned above. Patients are classified as ultra-rapid, extensive, intermediate and poor metabolizers of debrisoquine. There is one CYP2D6 genetic locus, and individuals may have two, one or no functional alleles, corresponding to extensive, intermediate and poor metabolizers, respectively: gene multiplication can lead to three functional alleles and the ultra-rapid metabolizer phenotype. Seventy-five CYP2D6 alleleic variants have been identified and pharmacogenetic techniques can identify the metabolizer phenotype, thereby predicting clinical response to treatment. Although debrisoquine is now obsolete, the CYP2D6 polymorphism is relevant for some drugs used in cardiac and psychiatric practice. For instance, poor metabolizers are more likely than other individuals to experience drug toxicity, and less likely to gain benefit from the analgesic codeine, a prodrug which is metabolized by CYP2D6 to morphine, the active drug. A polymorphism of CYP2C19, again leading to extensive and poor metabolizer phenotypes, affects the metabolism of the proton pump inhibitor drugs used in gastroesophageal reflux disease and the effectiveness of treatment (Chapter 8).
Biochemical tests of liver function
Clinical laboratories offer a panel of measurements on plasma or serum specimens (Table 30.2). This group of tests is usually, and incorrectly, described as liver ‘function’ tests. While plasma activities of liver enzymes are markers of liver disease, they do not exactly reflect the function of the liver. The rate of prothrombin synthesis, assessed by prothrombin time (PT), is a better indicator of liver synthetic function.
The tests commonly include the measurements of:
Transaminases
Aspartate aminotransferase (AST) and alanine amino-transaminase (ALT) are involved in the interconversion of amino and ketoacids, and are required for metabolism of proteins and carbohydrates (Chapter 19). Both are located in the mitochondria; ALT is also found in the cytoplasm. Serum activity of ALT and AST increases in liver disease (ALT is the more sensitive measurement).
Prothrombin time
In liver disease, the synthetic functions of the hepatocytes are likely to be affected, and so the patient would be expected to have a prolonged prothrombin time and low serum albumin concentration.
Alkaline phosphatase
ALP is synthesized both by the biliary tract and by bone, and in pregnancy by the placenta, but these tissues contain different ALP isoenzymes. The origin of the ALP may be determined from the isoenzyme pattern. Alternatively, the plasma activity of another enzyme, such as γ-glutamyl transpeptidase (GGT), which also originates in the biliary tract, may be measured and used to confirm the hepatic origin for a raised serum ALP activity.
Clinical box A seemingly healthy 45-year-old man with abnormal transaminases
A 45-year-old businessman had a routine medical examination, at which he was found to have a slightly enlarged liver. Tests revealed bilirubin 15 µmol/L (0.9 mg/dL), AST 434 U/L, ALT 198 U/L, ALP 300 U/L, GGT 950 U/L, and albumin 40 g/L (4 g/dL). He seemed perfectly well.
The patient has asymptomatic liver disease. The biochemical tests show evidence of hepatocellular damage. This may be due to excess alcohol intake, in which case there may also be enlarged red blood cells (macrocytosis) and an increased serum uric acid concentration. Patients may deny alcohol abuse. However nonalcoholic fatty liver disease (NAFLD) is increasingly recognized as a cause of isolated abnormalities in serum transaminase concentrations. NAFLD occurs in 40% of patients with the so-called metabolic syndrome, in which central overweight due to the accumulation of visceral fat leads to insulin resistance, hypertension, dyslipidemia and hepatic steatosis. The later may lead on to cirrhosis, as may alcohol-induced liver disease. Needle biopsy of the liver may be necessary for diagnosis. In this patient, microscopic examination of the tissue revealed characteristic changes of alcoholic steatosis, hepatitis and central fibrosis. This may be the forerunner of cirrhosis and the patient should abstain from alcohol. Other causes, such as chronic viral infection of the liver or an autoimmune active chronic hepatitis, can be detected by blood tests. For reference ranges, see Table 30.2.
Classification of liver disorders
Hepatocellular disease
Inflammatory disease of the liver is termed hepatitis and may be of short (acute) or long (chronic) duration. Viral infections, particularly hepatitis A and E, are common infectious causes of acute hepatitis, whereas alcohol and acetaminophen are the most common toxicologic causes. Chronic hepatitis, defined as inflammation persisting for more than 6 months, may also be due to the hepatitis B and C viruses, alcohol, and immunologic diseases, in which the body produces antibodies against its own tissues (autoimmune diseases, Chapter 38). Cirrhosis is the result of chronic hepatitis and is characterized microscopically by fibrosis of the hepatic lobules. The term ‘hepatic failure’ denotes a clinical condition in which the biochemical function of the liver is severely, and potentially fatally, compromised.
Cholestatic disease
Cholestasis is the clinical term for biliary obstruction, which may occur in the small bile ducts in the liver itself or in the larger extrahepatic ducts. Biochemical tests cannot distinguish between these two possibilities, which generally have radically different causes; imaging techniques such as ultrasound are more helpful.
Jaundice
Jaundice can be pre-, post- or intrahepatic
Jaundice is clinically obvious when plasma bilirubin concentrations exceed 50 µmol/L (3 mg/dL). Hyperbilirubinemia is the result of an imbalance between its production and excretion. The causes of jaundice (Table 30.3) are conventionally classified as:
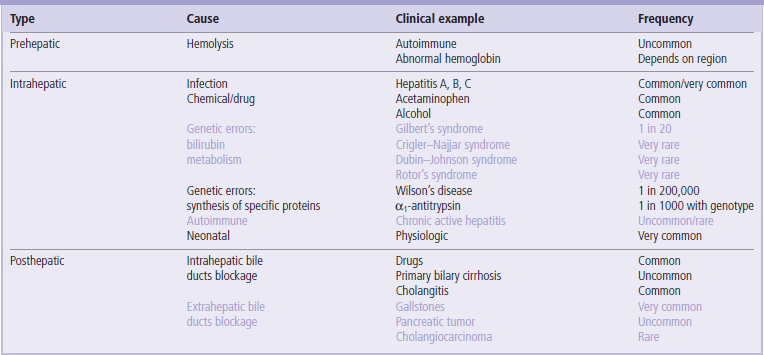
Prehepatic: increased production or impaired hepatic uptake of bilirubin (Fig. 30.9).
Fig. 30.9 Prehepatic (hemolytic) jaundice.
There is an increased concentration of plasma total bilirubin due to excess of the unconjugated fraction (see also Table 30.2).
Intrahepatic: impaired hepatic metabolism or secretion of bilirubin (Fig. 30.10).
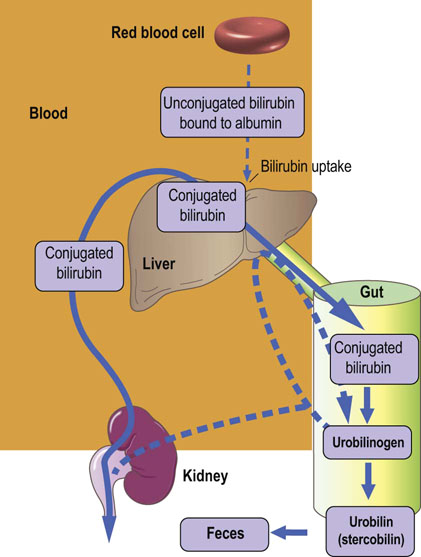
Fig. 30.10 Intrahepatic jaundice.
Bilirubin in plasma is increased due to an increase in the conjugated fraction. Increased serum enzyme activities signify hepatocyte damage (see also Table 30.2).
Posthepatic: obstruction to biliary excretion (Fig. 30.11 and Box on p. 210).
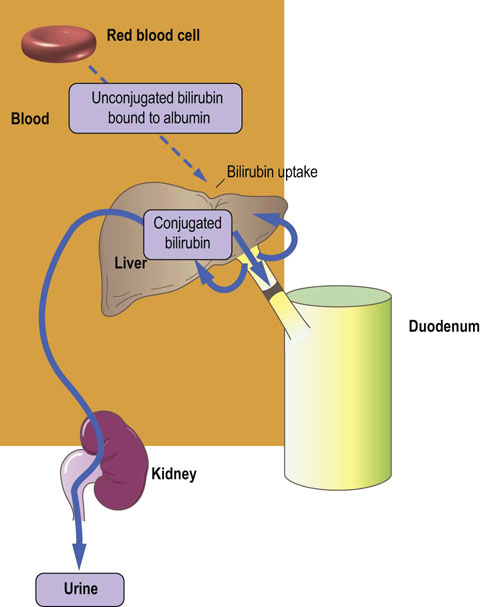
Fig. 30.11 Posthepatic jaundice.
Plasma bilirubin is elevated due to an increase in the conjugated fraction. Obstruction of the bile duct does not allow passage of bile to the gut. Stools are characteristically pale in color (see also Table 30.2). GGT, γ-glutamyl transpeptidase; ALP, alkaline phosphatase.
Prehepatic hyperbilirubinemia, results from excess production of bilirubin caused by hemolysis, or a genetic abnormality in the hepatic uptake of unconjugated bilirubin
Hemolysis is usually the result of immune disease, presence of structurally abnormal red cells or breakdown of extravasated blood. Intravascular hemolysis results in the release of hemoglobin into the plasma, where it is either oxidized to methemoglobin (Chapter 5) or complexed with haptoglobin. More commonly, red cells are hemolyzed extravascularly, within phagocytes, and hemoglobin is converted to bilirubin, which is unconjugated. Unconjugated and conjugated bilirubin can be distinguished in the laboratory as so-called indirect and direct bilirubin.
Intrahepatic jaundice reflects a generalized hepatocyte dysfunction
In this condition, hyperbilirubinemia is usually accompanied by other abnormalities in biochemical markers of hepatocellular function.
In neonates, transient jaundice is common, particularly in premature infants, and is due to immaturity of the enzymes involved in bilirubin conjugation. Unconjugated bilirubin is toxic to the immature brain and causes a condition known as kernicterus. If plasma bilirubin concentrations are judged to be too high, phototherapy with blue-white light, which isomerizes bilirubin to more soluble pigments that might be excreted with bile, or exchange blood transfusion to remove the excess bilirubin, is necessary to avoid kernicterus.
Posthepatic jaundice is caused by obstruction of the biliary tree
In this condition the plasma bilirubin is conjugated and other biliary metabolites, such as bile acids, accumulate in the plasma. The clinical features are pale-colored stools, caused by the absence of fecal bilirubin and urobilin, and dark urine as a result of the presence of water-soluble conjugated bilirubin. In complete obstruction, urobilinogen and urobilin are absent from urine, as there can be no intestinal conversion of bilirubin to urobilinogen/urobilin, and hence no renal excretion of reabsorbed urobilinogen/urobilin.
Clinical box A 3-day-old neonate who developed jaundice
the significance of neonatal jaundice
A normal term baby developed jaundice on the third day of life, with a bilirubin concentration of 150 µmol/L (8.8 mg/dL), predominantly of the indirect form. The baby was otherwise well.
About 50% of normal babies become jaundiced 48 h after birth. This physiologic jaundice is caused by temporary inefficiency in bilirubin conjugation, and resolves in the first 10 days. The hyperbilirubinemia is unconjugated in nature; if severe, it may require phototherapy (ultraviolet light to photoisomerize bilirubin into a nontoxic form) or exchange blood transfusion to prevent damage to the brain (kernicterus). Bruising from delivery, infection or poor fluid intake may exaggerate the hyperbilirubinemia. Jaundice in the first 24 h of life is abnormal and requires investigation to exclude hemolysis, as does jaundice that presents later or persists, after 10 days. This is always abnormal and is likely to indicate an inborn error of metabolism or structural defects of the bile ducts.
Clinical box A 65-year-old man with jaundice and no abdominal symptoms
the significance of adult jaundice
A 65-year-old man was admitted to hospital because of jaundice. There was no abdominal pain, but he had noticed dark urine and pale stools. The gallbladder appeared palpable but not tender. Liver function tests showed bilirubin 230 µmol/L (13.5 mg/dL), AST 32 U/L, and ALP 550 U/L. Dipstick urine testing revealed the presence of bilirubin, but no urobilin.
The patient had a history typical of obstructive jaundice. The increased ALP and normal AST concentrations were consistent with this, and the absence of urobilin in the urine indicated that the biliary tract was obstructed. It was important to carry out liver imaging tests to find the site of the obstruction; the absence of pain suggested that gall-stones were not the cause. Ultrasound showed dilatation of the bile duct and a computed tomography scan showed a mass in the head of the pancreas. During surgery, cancer of the pancreas was confirmed. For reference ranges, see Table 30.2.
Genomics of liver disease
Several hepatic diseases arise due to single gene disorders. Genetic techniques can identify individuals with a propensity to develop a disease or confirm the diagnosis in affected persons.
Hereditary hemochromatosis is a genetically determined disorder of iron metabolism
It is common in northern Europeans, with a population prevalence of 1 in 200 to 1 in 300. Patients absorb excessive amounts of iron from the gut, and tissue iron overload leads to multiorgan dysfunction, including cirrhosis of the liver. A transmembrane glycoprotein, known as HFE, modulates iron uptake. Mutations in the genetic locus which encodes this protein, the HFE gene, underlie hereditary hemochromatosis. Two point mutations, C282Y and H63D, are found in the majority of those with hereditary hemochromatosis and can be identified easily by PCR-based assays.
Wilson's disease is a condition associated with liver and CNS damage, results from genetic deficiency of ceruloplasmin
Ceruloplasmin is the major copper-containing protein of the liver and plasma, and functions as an iron oxidizing enzyme (ferroxidase): oxidation of Fe2+ to Fe3+ is necessary for the mobilization of stored iron, and nutritional copper deficiency produces anemia. Wilson's disease is a condition associated with damage to both the liver and the CNS.
Cirrhosis in this condition is due to excess copper deposition in the liver. Wilson's disease is also caused by a single gene defect but multiple mutations have been found in affected patients; no routine assay is available for diagnosis. The liver also synthesizes proteins responsible for storage (ferritin) and transport (transferrin) of iron (Chapters 11 and 22).
Deficiency of α1-antitrypsin presents in infancy as liver disease, or in adulthood as lung disease
α1-Antitrypsin is a member of the serpin family of serine protease inhibitors, and, contrary to its name, its predominant target is macrophage-derived elastase. Genetic deficiency of α1-antitrypsin presents in infancy as liver disease, or in adulthood as lung disease caused by elastase-mediated tissue destruction – early-onset lung disease and liver cirrhosis.
Several isoforms of α1-antitrypsin exist as a result of allelic variation of the AA1T gene: the normal isoform is known as M and the two common defective isoforms as S and Z; the null allele produces no α1-antitrypsin.
More than 90 allelic variants of the AA1T gene, at the so-called proteinase inhibitor (Pi) locus, have been described, the majority of which do not affect plasma levels or activity of AA1T. Phenotypic variants in AA1T were initially described by their relative mobility on electrophoresis, with the most common variant, M, having medium mobility. The Z and S variants are most frequently associated with AA1T deficiency, and are both due to point mutations which can also be detected by PCR assays.
Liver cancer is associated with particularly high plasma concentrations of α-fetoprotein
α-Fetoprotein (AFP) and albumin have considerable sequence homology, and appear to have evolved by reduplication of a single ancestral gene. In the fetus, AFP appears to serve physiologic functions similar to those performed by albumin in the adult; furthermore, by the end of the first year of life, AFP in the plasma is entirely replaced by albumin. During hepatic regeneration and proliferation, AFP is again synthesized; thus high plasma concentrations of AFP are observed in liver cancer.
There are a number of genetic disorders that impair bilirubin conjugation or secretion
Gilbert's syndrome affects up to 5% of the population, and causes a mild unconjugated hyperbilirubinemia that is harmless and asymptomatic. It is caused by a dinucleotide polymorphism in the TATA box promoter (Fig. 34.1) of the bilirubin UDP-glucuronyl transferase gene which impairs the hepatic uptake of unconjugated bilirubin.
Other inherited diseases of bilirubin metabolism are rare. Crigler–Najjar syndrome, which is the result of a complete absence or marked reduction in bilirubin conjugation, causes severe unconjugated hyperbilirubinemia that presents at birth; when the enzyme is completely absent, the condition is fatal. The Dubin–Johnson and Rotor's syndromes impair biliary secretion of conjugated bilirubin and therefore cause conjugated hyperbilirubinemia, which is usually mild.
Summary
The liver plays a central role in human metabolism.
It is extensively involved in the synthesis and catabolism of carbohydrates, lipids and proteins.
It synthesizes an array of acute phase proteins in response to inflammation and infection, and laboratory measurements of such proteins are clinically useful in monitoring disease progress.
It is involved in the metabolism of bilirubin derived from the catabolism of heme.
Disease processes often cause the patient to present with jaundice due to hyperbilirubinemia.
The liver has a central role in the detoxification of drugs.
Its biochemical function is assessed in clinical practice using a panel of blood tests, called liver function tests, abnormalities of which can point to disease affecting the hepatocellular or biliary systems.
Bernal, W, Auzinger, G, Dhawan, A, et al. Acute liver failure. Lancet. 2010; 376:190–201.
Donohue, TM, Cederbaum, AI, French, SW. Role of the proteasome in ethanol-induced liver pathology. Alcohol Clin Exp Res. 2007; 31:1446–1459.
Hall, P, Cash, J, What is the real function of the liver ‘function’ tests? Ulster Med J. 2012;81(1):30–3. www.ums.ac.uk/umj081/081(1)030.pdf
Puy, H, Gouya, L, Deybach, J-C. Porphyrias. Lancet. 2010; 375:924–937.
Schuckit, MA. Alcohol-use disorders. Lancet. 2009; 373:492–501.
Wijnen, PAHM, Op den Buijsch, RAM, Drent, M, et al. Review article: the prevalence and clinical relevance of cytochrome P450 polymorphisms. Aliment Pharmacol Ther. 2007; 26(suppl2):211–219.
Wilkinson, GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005; 352:2211–2221.
Liver diseases. MedlinePlus. www.nlm.nih.gov/medlineplus/liverdiseasesgeneral.html.
Liver disease. Lab Tests Online www.labtestsonline.org/understanding/conditions/liver_disease.html.
Liver function tests. Lab Tests Online www.labtestsonline.org.uk/understanding/analytes/liver-panel.
PharmGKB. The Pharmacogenomics Knowledgebase. www.pharmgkb.org/index.jsp.