Muscle
Energy Metabolism and Contraction
Learning objectives
After reading this chapter you should be able to:
 Describe muscle structure and its function in mechanical force production, including differences among skeletal, cardiac and smooth muscle types that are related to their physiologic functions.
Describe muscle structure and its function in mechanical force production, including differences among skeletal, cardiac and smooth muscle types that are related to their physiologic functions.
Describe the structure and protein composition of the sarcomere, the sliding filament model of muscle contraction, and the source of the banding pattern in striated muscle. Describe the sequence of events in excitation–contraction coupling, including the roles of membrane depolarization, the sarcoplasmic reticulum, and calcium triggering.
Identify the key sites of energy utilization during muscle contraction, the role of creatine phosphate in skeletal muscle, and the impact of skeletal muscle fiber type on substrate utilization and muscle function.
Describe the changes in skeletal muscle mass and metabolism with age, in response to acute and prolonged exercise, and in diseases such as sarcopenia, metabolic syndrome, and wasting conditions.
Introduction
There are three types of muscle: – skeletal, cardiac, and smooth muscle – each with a unique physiologic role
All muscles function to convert chemical energy to mechanical energy, but the various types of muscle differ in their mechanism of initiation of contraction, rate of force development, duration of contraction, ability to adapt to their environment, and substrate utilization. Muscle accounts for about 40% of total body mass, and muscle metabolism is a major determinant of whole-body metabolic rate in both the basal and active state. Changes in skeletal muscle metabolism occur with physical activity and are directly related to the required force output and duration of activity. These factors also affect the muscle's relative utilization of glucose and fatty acids for fuel. Besides locomotion, skeletal muscle is also a source of body heat, provides amino acids for hepatic gluconeogenesis during fasting, and is a major site of glucose and triglyceride disposal following a meal. Because of its critical role in the regulation of systemic fuel flux and metabolism, loss of muscle mass has a profound effect on overall metabolism. Advancing age, sepsis, and wasting diseases, such as AIDS and cancer, are conditions associated with loss of muscle mass, and this loss is associated with increased morbidity and mortality.
The primary focus of this chapter will be on skeletal muscle, supplemented by discussion of similarities and differences in skeletal, cardiac and smooth muscle structure, function and metabolism. The chapter will begin with a discussion of the mechanism of muscle contraction, proceed to the signaling that initiates the contractile process, and then examine energy metabolism essential for contraction.
 Clinical box Muscular dystrophies
Clinical box Muscular dystrophies
A young boy was brought to the clinic because his mother had noticed that he walked with a waddling gait. Physical evaluation confirmed muscle weakness especially in the legs, although his calf muscles were large and firm. There was a 20-fold elevation in serum creatine (phospho) kinase (CK) activity, identified as the MM (muscle) isozyme. Histology revealed muscle loss, some necrosis, and increased connective tissue and fat volume in muscle. A tentative diagnosis of Duchenne muscular dystrophy (DMD) was confirmed by immunoelectrophoretic (Western blot) analysis showing the lack of the cytoskeletal protein dystrophin in muscle.
Though there are many forms of muscular dystrophy, some genetic and some acquired, DMD is the most common genetic dystrophy and is lethal. Dystrophin is a high-molecular-weight cytoskeletal protein that reinforces the plasma membrane of the muscle cell and mediates interactions with the extracellular matrix. In its absence, the plasma membrane of muscle cells shears during the contractile process, leading to muscle cell death.
The dystrophin gene is located on the X-chromosome and is nearly 2.5 × 106 base pairs in length. Spontaneous mutations in this gene are relatively common, the frequency of DMD being approximately 1 in 3500 male births. DMD is a progressive myodegenerative disease, commonly leading to confinement to a wheelchair by puberty, with death by age 30 years from respiratory or cardiac failure. Dystrophin is completely absent in DMD patients, though a variant of the disease, known as Becker muscular dystrophy, has milder symptoms and is characterized by expression of an altered dystrophin protein and survival into the fifth decade. Although there is currently no treatment for DMD, gene therapy still holds some promise and newer technologies that utilize ‘exon skipping’ are allowing cells to skip over mutated exons and thereby translate a slightly smaller, but still functional, protein product. The smaller dystrophin protein produces Becker-like symptoms in animal experiments and thus could translate to a doubling of human life span if the results are reproducible in humans.
Muscle structure
The sarcomere: the functional contractile unit of muscle
A common characteristic of cardiac myocytes, smooth muscle cells and skeletal myofibers is that their cytoplasm is packed full of contractile protein. The contractile protein is organized in linear arrays of sarcomere units in skeletal myofibers and cardiac myocytes, giving these muscles a striated appearance; thus, the term striated muscle. Contractile protein in smooth muscle cells is not organized into a sarcomeric structure, and this tissue is described as nonstriated muscle. Skeletal muscle's hierarchic structure (Fig. 20.1) consists of bundles (fasciculi) of elongated, multinucleated fiber cells (myofibers). The myofiber cells contain bundles of myofibrils which are, in turn, composed of myofilament proteins, primarily myosin and actin, that form the sarcomere (Table 20.1). Electron microscopic analysis of muscle reveals a repeating pattern of light- and dark-staining regions in the myofibril (Fig. 20.2). These regions are known as the I (isotropic)- and A (anisotropic)-bands, respectively. At the center of the I-band is a discrete, darker staining Z-line, while the center of the A-band has a lighter-staining H-zone with a central M-line. The contractile unit, the sarcomere, is centered on the M-line, extending from one Z-line to the next. Smooth muscle lacks a defined Z-line.
Table 20.1
The structural elements of skeletal muscle arranged in descending order of size
| Microscopic unit | Fasciculus: bundle of muscle cells |
| Cellular unit | Myofiber cell: long, multinucleated cell |
| Subcellular unit | Myofibril: composed of myofilament proteins |
| Functional unit | Sarcomere: contractile unit, repeating unit of the myofibril |
| Myofilament components | Proteins: primarily actin and myosin |
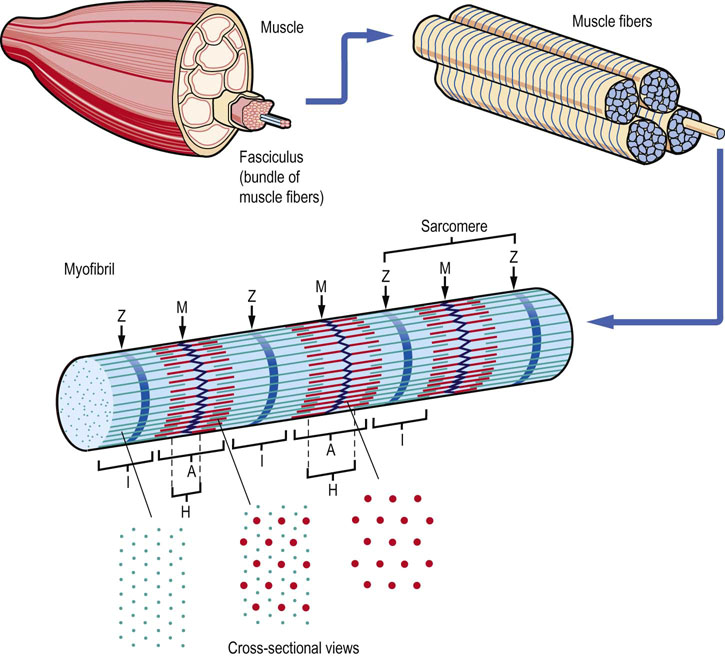
Fig. 20.1 Hierarchic structure of muscle.
Hierarchic structure of skeletal muscle, showing an exploding view of fasciculi, myofibers, myofibrils and myofilament proteins. The location of the I-band (thin, actin filaments extending from the Z-line) and the A-band (thick, myosin filaments, extending from the M-line), with darker-staining regions of the A-band corresponding to the region of overlap of actin and myosin filaments.
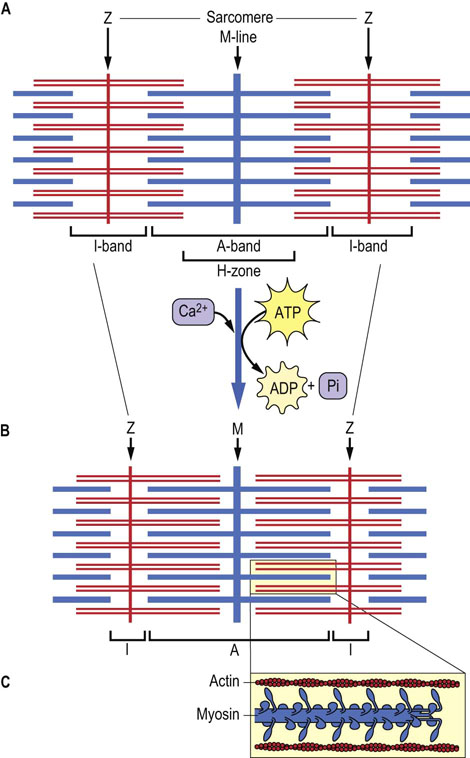
Fig. 20.2 Schematic structure of the sarcomere, indicating the distribution of actin and myosin in the A- and I-bands.
(A) Relaxed sarcomere. (B) Contracted sarcomere. (C) Magnification of contracted sarcomere, illustrating the polarity of the arrays of myosin molecules. Increased overlap of actin and myosin filaments during contraction, accompanied by a decrease in the length of the H-zones and I-bands, illustrates the sliding filament model of muscle contraction.
The thick and thin filaments
Actin and Myosin account for over 75% of muscle protein.
The sarcomere may shorten by as much as 70% in length during muscle contraction (see Fig. 20.2). The components effecting the contraction are the thick and thin filaments. The thick filament is composed of myosin and titin protein, and the thin filament is mainly made up of actin, with associated proteins, tropomyosin and troponins. The thin filament also has some interaction with titin. Thick and thin filaments extend in opposite directions from both sides of the M- and Z-lines, respectively, and overlap and slide past one another during the contractile process (see Fig. 20.2). The M- and Z-lines are, in effect, base plates for anchoring the myosin and actin filaments. In striated muscle, increased thick–thin filament intercalate during contraction, causing the H-zone (myosin only) and I-bands (actin only) to shrink. In smooth muscle, thick and thin filaments are anchored at structures called dense bodies that are further anchored by intermediate filaments. Although all three muscle types contain the same proteins, each muscle type expresses tissue-specific isoforms; the cardiac actin and troponins, for example, differ slightly from those in skeletal muscle.
Sarcomere proteins
Myosin
Interaction between actin and myosin during muscle contraction is dependent on cytoplasmic Ca++ concentration
Myosin is one of the largest proteins in the body, with a molecular mass of approximately 500 kDa, and accounts for more than half of muscle protein (Table 20.2). Under the electron microscope, myosin appears as an elongated protein with two globular heads. It is the primary component of the thick filament in muscle. Each myosin molecule is made up of two heavy chains (approx. 200 kDa) and four light chains (approx. 20 kDa). The heavy chain can be subdivided into the helical tail and globular head regions; the four light chains are bound to the globular heads. Structural analysis by limited proteolysis indicates that there are two flexible hinge regions in the myosin molecule (Fig. 20.3): one where the globular head attaches to the helical region and the other further into the helical region. The myosin filaments are associated through their helical regions and extend outward from the M-line toward the Z-line of each myofibril (see Figs 20.2 and 20.3). The hinge regions allow the myosin heads to interact with actin and provide the flexibility needed for reversible interactions and conformational changes during muscle contraction.
Table 20.2
Muscle proteins and their functions
| Protein | Function |
| Myosin | Ca2+-dependent ATPase activity |
| C-protein | assembly of myosin into thick filaments |
| M-protein | binding of myosin filaments to M-line |
| Actin | G-actin polymerizes to filamentous F-actin |
| tropomyosin | stabilization and propagation of conformational changes of F-actin |
| troponins-C, I and T | modulation of actin–myosin interactions |
| α- and β-actinins | stabilization of F-actin and anchoring to Z-line |
| nebulin | possible role in determining length of F-actin filaments |
| titin | control of resting tension and length of the sarcomere |
| desmin | organization of myofibrils in muscle cells |
| dystrophin | reinforcement of cytoskeleton and muscle cell plasma membrane |
Actin and myosin account for over 90% of muscle proteins, but several associated proteins are required for assembly and function of the actomyosin complex.
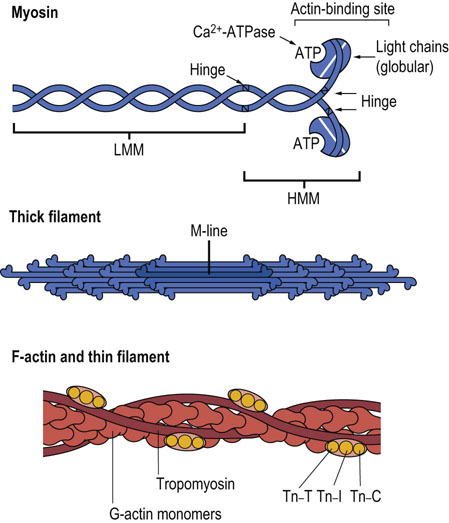
Fig. 20.3 Polymerization of myosin and actin into thick and thin filaments.
Tn-C, calcium-binding troponin; Tn-I, troponin inhibitory subunit; Tn-T, tropomyosin-binding troponin. LMM, light meromyosin; HMM, heavy meromyosin.
There are several features of myosin that are essential for muscle contraction.
The myosin globular heads have binding sites for ATP and its hydrolysis products, ADP and phosphate (Pi).
The myosin globular heads have a Ca2+-dependent ATPase activity.
Myosin binds reversibly to actin as a function of Ca2+, ATP, and ADP + Pi concentrations.
The binding of calcium and hydrolysis of ATP lead to major changes in the conformation of the myosin molecule and its interaction with actin.
Myosin-ATPase activity, myosin–actin interactions and conformational changes are integrated into the sliding filament model of muscle contraction (below). They also explain the development of rigor mortis. The increase in Ca2+ in the muscle cytoplasm (sarcoplasm) and decrease in ATP after death lead to tight binding between myosin and actin, forming rigid muscle tissue.
Actin
Actin is composed of 42 kDa subunits, known as G-actin (globular), which polymerize into a filamentous array (F-actin). Two polymer chains coil around one another to form the F-actin myofilament (see Fig. 20.3). F-actin is the major component of the thin filament and interacts with myosin in the actomyosin complex. The F-actin chains extend in opposite directions from the Z-line, overlapping with the myosin chains extending from the M-line. Each myosin-containing thick filament is surrounded by six actin molecule containing thin filaments. Each thin filament interacts with three myosin-containing thick filaments (see Fig. 20.1 for a cross-sectional view).
Tropomyosin and troponins
Troponins modulate the interaction between actin and myosin
Calcium activation of muscle contraction in striated muscle involves thin filament-associated proteins, tropomyosin and the troponins. Tropomyosin is a fibrous protein that extends along the grooves of F-actin, each molecule contacting about seven G-actin subunits. Tropomyosin has a role in stabilizing F-actin and coordinating conformational changes among actin subunits during contraction. In the absence of Ca2+, tropomyosin blocks the myosin-binding site on actin.
A complex of troponin proteins is bound to tropomyosin: Tn-T (tropomyosin-binding), Tn-C (calcium-binding) and Tn-I (inhibitory subunit). Calcium binding to Tn-C, a calmodulin-like protein, induces changes in Tn-I which shift the interaction between tropomyosin and actin, exposing the myosin-binding site on F-actin and permitting actin–myosin interactions. For a description of the diagnostic use of cardiac troponin measurements see Box at end of this chapter.
Clinical box Muscle loss during sepsis
Skeletal muscle affects both the morbidity and mortality of sepsis. Sepsis is the leading cause of death in noncoronary intensive care units (ICUs) and the 10th leading cause of death in the United States overall. The incidence of severe sepsis in the United States is approximately 700,000 cases per year and is increasing by 8% each year. Numerous factors are contributing to this increase (e.g. increasing population age, overuse of antibiotics). Sepsis can be defined as an inappropriate regulation of the immune and physiologic response to a pathogen. Normally, the body responds to an infection by eradicating the pathogen when it first comes in contact with immune cells. When this system becomes overwhelmed it disturbs whole body homeostasis and often results in multiple organ failure.
Indeed, the mortality rate of the more severe forms of sepsis is >50%. While controlling the inflammatory response and eradicating the pathogen is the primary therapeutic goal, skeletal muscle plays an important role in patient prognosis. Sepsis results in severe muscle mass loss and patients with low muscle mass are more likely to die of sepsis. The muscle degradation pathway is activated by the inflammatory cytokines such as IL-6 and TNF-α caused by the uncontrolled immune response. Muscle protein breakdown and amino acids release into the blood, are similar to starvation. However, different molecular pathways are involved; increasing nutrition or protein intake is not effective as anabolic signaling pathways are not responsive. As muscle loss is a contributing factor to morbidity and mortality of patients and affects long-term recovery outcomes, it becomes of paramount importance to consider methods of attenuating the muscle loss associated with sepsis. There are several protein degradation pathways related to muscle loss, but the ubiquitin-proteasome (UbP) pathway (see Chapter 34 for details) appears to be the primary pathway activated during sepsis. The myofibrillar proteins are most susceptible and 3-Methylhistidine, a post-translationally modified amino acid found in actin and myosin, is increased in blood and urine as a result of protein turnover. The UbP pathway is currently a target for drugs in development that might be used specifically to treat muscle loss during sepsis and thus improve mortality rates.
Titin
Titin modulates the passive tension of muscle
Titin is the largest protein in the human body, with more than 34,000 amino acids and a mass of 3800 kDa. Structurally, titin spans half the length of the sarcomere, with its N-terminus anchored to the Z-line, and its C-terminus to the thick filament at the M-line. Titin has an elastic, extensible PEVK domain (rich in Pro, Glu, Val and Lys) that contributes to passive myocardial and skeletal muscle tension, and a kinase domain that participates in intracellular signaling. Depending on the skeletal muscle, titin may account for more than half of the passive tension of the muscle, and contributes a spring-like property to the sarcomere – when a muscle is stretched, potential energy is stored in the PEVK domain, which re-coils during relaxation. Mutations in one region of titin may cause a genetic disease of the heart (e.g. hypertrophic cardiomyopathy), while a mutation elsewhere in the gene causes a disease of skeletal muscle only (e.g. limb girdle muscular dystrophy).
The contractile process
The sliding filament model of muscle contraction
The sliding filament model describes how a series of chemical and structural changes in the actomyosin complex can induce sarcomere shortening
The contractile response depends on reversible, Ca2+-dependent cross-bridge formation between the myosin head and its binding site on actin. A conformational change in the hinge regions of myosin occurs after cross-bridge formation, providing the power stroke for muscle contraction (Fig. 20.4). This conformational change, the relaxation of the high-energy form of myosin, is accompanied by dissociation of ADP and Pi. After the stroke is completed, the binding and hydrolysis of ATP restore the high-energy conformation. The stability of the contracted state is maintained by multiple and continuous Ca2+-dependent actin–myosin interactions, so that slippage is minimized until calcium is removed from the sarcoplasm, allowing dissociation of the actomyosin complex and muscle relaxation.
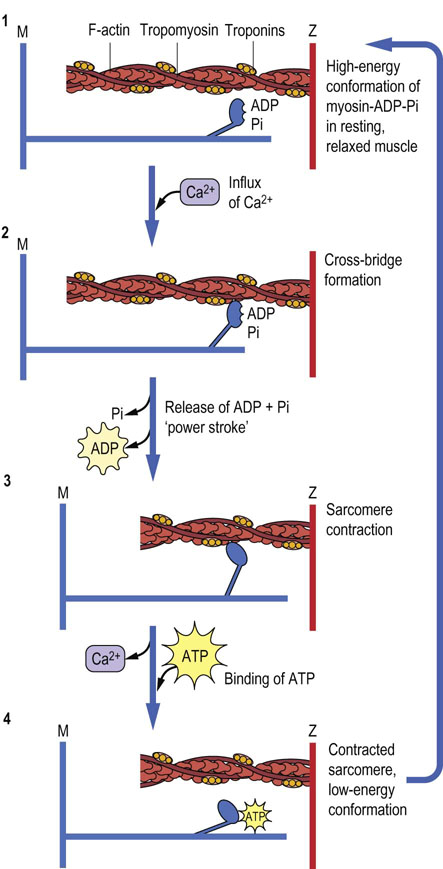
Fig. 20.4 Proposed stages in muscle contraction, according to the sliding filament model.
(1) In resting, relaxed muscle, calcium concentration is~107 mol/L. The head group of myosin chains contains bound ADP and Pi, and is extended from the axis of the myosin helix in a high-energy conformation. Although the myosin-ADP-Pi complex has a high affinity for actin, binding of myosin to actin is inhibited by tropomyosin, which blocks the myosin-binding site on actin at low calcium concentration. (2) When muscle is stimulated, calcium enters the sarcoplasm through voltage-gated calcium channels (see Chapter 8). Calcium binding to Tn-C causes a conformational change in Tn-I, which is transmitted through Tn-T to tropomyosin. Movement of tropomyosin exposes the myosin-binding site on actin. Myosin-ADP-Pi binds to actin, forming a cross-bridge. (3) Release of Pi, then ADP, from myosin during the interaction with actin, is accompanied by a major conformational change in myosin, producing the ‘power stroke’, which moves the actin chain about 10 nm (100 Å) in the direction opposite the myosin chain, increasing their overlap and causing muscle contraction. (4) The uptake of calcium from the sarcoplasm and binding of ATP to myosin leads to dissociation of the actomyosin cross-bridge. The ATP is hydrolyzed, and the free energy of hydrolysis of ATP is conserved as the high-energy conformation of myosin, setting the stage for continued muscle contraction in response to the next surge in Ca2+ concentration in the sarcoplasm.
Higher myosin-ATPase activity increases cross-bridge cycling, which allows for increased rate of contraction. Different myosin isoforms have varying levels of ATPase activity, with fast muscles having higher rates of myosin-ATPase activity. Isoforms of actin and myosin are also found in the cytoskeleton of nonmuscle cells, where they have roles in diverse processes such as cell migration, vesicle transport during endocytosis and exocytosis, maintenance or changing of cell shape, and anchorage of intracellular proteins to the plasma membrane.
Excitation–contraction coupling: muscle membrane depolarization
T-tubules transmit electrochemical signals for efficient muscle contraction
Skeletal muscle contraction is initiated by neuronal stimulation at the neuromuscular endplate. As described previously (see Fig. 8.4), this stimulus leads to depolarization of the electrochemical gradient across the muscle plasma membrane (sarcolemma). The depolarization, caused by an influx of Na+, propagates rapidly along the sarcolemma membrane and signals a voltage-gated calcium release from the sarcoplasmic reticulum (SR), a membrane-bound, calcium-sequestering compartment inside the muscle cell. The influx of Ca2+ from the SR to the sarcoplasm initiates cross-bridge formation and excitation–contraction coupling (see Fig. 20.4). In striated muscle, depolarization is transmitted into the muscle fiber by invaginations of the plasma membrane, called transverse tubules (T tubule) (Fig. 20.5). The transmission of depolarization through the highly branched T-tubule network, which interacts closely with the SR, leads to rapid, concerted release of calcium from the SR into the sarcoplasm. In order for depolarization to occur again, sodium must be actively pumped out of the cytosol, by Na+/K+-ATPase pumps located in the sarcolemma. The rate of muscle repolarization is affected by both the rate and density of these pumps. Higher levels of Na+/K+-ATPase activity are found in fast contracting muscles, and increased Na+/K+-ATPase pump density is an important adaptation to exercise.

Fig. 20.5 Side view of the transverse tubular network in skeletal muscle cells.
Transverse tubules are invaginations of the sarcolemma, which are connected to the sarcoplasmic reticulum (SR) by protein channels. The SR is a continuous, tubular compartment in close association with the myofibrils. The transverse tubules are extensions of the sarcolemma around the Z-line. They transmit the depolarizing nerve impulse to terminal regions of the SR, coordinating calcium release and contraction of the myofibril.
Skeletal, cardiac, and smooth muscle differ in their mechanism of neural stimulation, and have different structural adaptations for propagating the depolarization. Skeletal muscle contraction is volitional and fibers are innervated by motor nerve endplates that originate in the spinal cord; acetylcholine functions as the neurotransmitter (see Chapter 41). The neuromuscular junction is a special structural feature of skeletal muscle that is not found in cardiac or smooth muscle. Each individual fiber is innervated by only one motor nerve, and all the fibers innervated by one nerve are defined as a motor unit. Motor unit control and synchronization is the basis for coordinated whole-muscle contraction. Skeletal muscle cramping is a nonvoluntary muscle contraction resulting from alterations in neuromuscular control and/or from electrolyte imbalances after excessive fluid loss typically during intense exercise in hot, humid conditions.
Cardiac muscle is striated and contracts rhythmically under involuntary control. The general mechanism of contraction of heart muscle is similar to that in skeletal muscle; however, the sarcoplasmic reticulum is less developed and the transverse tubule network is more developed in the heart. The heart is more dependent on, and actually requires, extracellular calcium for its contractile response (see Fig. 8.4); the influx of extracellular calcium enhances Ca2+ release from the SR. Lacking direct neural contact, cardiac myocytes propagate depolarization from a single node, the SA node, throughout the myocardium. The depolarization is passed cell to cell along specialized membrane structures called intercalated disks. Cardiac muscle is also more responsive to hormonal regulation. For example, cAMP-dependent protein kinases phosphorylate transport proteins and Tn-I, mediating changes in the force of contraction in response to epinephrine.
Smooth muscle can respond to both neural and circulating factors. Unlike skeletal muscle, neural input to smooth muscle innervates bundles of smooth muscle cells that cause both phasic (rhythmic) and tonic (sustained) contractions of the tissue. Smooth muscle can also be induced to depolarize by ligand–receptor interactions at the sarcolemma. This is called pharmacomechanical coupling, and is the basis for many drugs that target smooth muscle contraction or relaxation. Nitric oxide donors, such as amyl nitrite and nitroglycerin, used for treatment of angina, relax vascular smooth muscles, increasing the flow of blood to cardiac muscle.
Excitation–contraction coupling: the calcium trigger
The calcium content of the sarcoplasm is normally very low, 10-7 mol/L or less, but increases rapidly by over 100-fold in response to neural stimulation. The sarcoplasmic reticulum, a specialized organelle derived from the smooth endoplasmic reticulum, is rich in a Ca2+-binding protein, calsequestrin, and serves as the site of calcium sequestration inside the cell. In striated muscle, T-tubule depolarization opens the Ca2+ channels in the SR (see Fig. 20.5). The influx of Ca2+ into the sarcoplasm triggers both actin–myosin interactions and myosin-ATPase activity, leading to muscle contraction. Troponins are not expressed in smooth muscle. In this case, calcium triggers contraction by binding to calmodulin and activating myosin light chain kinase, which then phosphorylates myosin. Myosin phosphorylation enhances myosin–actin interaction.
Increased intracellular calcium activates more cross-bridges and causes sarcomere shortening through activation of myosin-ATPase. Thus, higher calcium levels increase muscle contractile force until saturation is reached. Calcium channel blockers used for treatment of hypertension, such as nifedipine, inhibit the flow of Ca2+ into the SR, thereby limiting the force of contraction of cardiac myocytes. While muscle contraction is triggered by increased calcium, muscle relaxation is dependent on calcium being actively pumped back into the SR. The rate of muscle relaxation is directly related to SR Ca2+-ATPase activity. The SR is rich in Ca2+-ATPase, which maintains cytosolic calcium in the sarcoplasm at submicromolar (~10-7 mol/L) concentrations. As intracellular calcium levels decrease, the number of active cross-bridges also decreases, and muscle contractile force declines.
Clinical box Malignant hyperthermia
About 1 in 150,000 patients treated with halothane (gaseous halocarbon) anesthesia or muscle relaxants responds with excessive skeletal muscle rigidity and severe hyperthermia with a rapid onset, up to 2°C (4°F) within 1 hour.
Unless treated rapidly, cardiac abnormalities may be life-threatening; mortality from this condition exceeds 10%. This genetic disease results from excessive or prolonged release of Ca2+ from the SR, most commonly the result of mutations in gene(s) that code for the Ca2+-release channels within the SR. Excessive release of Ca2+ leads to a prolonged increase in sarcoplasmic Ca2+ concentration. Muscle rigidity results from Ca2+-dependent consumption of ATP, and hyperthermia results from increased metabolism to replenish the ATP.
As muscle metabolism becomes anaerobic, lacticacidemia and acidosis may develop. The cardiac abnormalities result from hyperkalemia, caused by release of potassium ions from muscle; as supplies of ATP are exhausted, muscle is unable to maintain ion gradients across its plasma membrane. Treatment of malignant hyperthermia includes use of muscle relaxants, e.g. dantrolene, an inhibitor of the ryanodine-sensitive Ca2+-channel, to inhibit Ca2+ release from the SR. Supportive therapy involves cooling, administration of oxygen, correction of blood pH and electrolyte imbalances and also treatment of cardiac abnormalities.
Muscle energy metabolism
Energy resources in the muscle cell
Muscle is the primary site of glucose disposal (uptake from the circulation) in the body and is thus a natural target for treatment of the hyperglycemia of diabetes. The glucose transporter GLUT-4 is transported to the cell surface not only in response to insulin or pharmaceuticals but also in response to cellular energy status and by muscle contractions. Thus, exercise serves as a regulator of blood glucose levels and, in effect, a treatment for diabetes – exercise, in this context, is good medicine. The muscle content and activity of hexokinase also increases with exercise, both acutely (~3 hours after one session) and chronically (after several weeks of training). Studies in animal models have shown that exercise and drugs activate both similar and distinct biochemical signaling cascades in muscle; thus, the two treatments appear to be complementary.
ATP is used for muscle contraction
Three ATPases are required for muscle contraction: Na+/K+-ATPase, Ca2+-ATPase and myosin-ATPase. Decreased ATP availability or inhibition of any of these ATPases will cause a decrease in muscle force production. However, the intracellular concentration of ATP does not change dramatically during exercise. Actively contracting muscle relies on the rapid resynthesis of ATP from ADP. Energy systems that synthesize ATP for muscle contraction include the creatine phosphate shuttle, anaerobic glycolysis from plasma glucose or glycogen, and aerobic metabolism of glucose and fatty acids via oxidative phosphorylation. The energy systems that synthesize ATP are not equivalent, and directly affect the amount and duration of power output from the contracting muscle.
Short-duration, high-power output contractions
Creatine phosphate is a high energy phosphate buffer used for rapid regeneration of ATP in muscle
A metabolic reality for skeletal muscle is that high force output can only be maintained for a short period of time. Contractions at or near maximal power levels depend on high myosin-ATPase activity and rapid ATP resynthesis by substrate-level phosphorylation using the high-energy compound creatine phosphate (creatine-P). Creatine (see Table 9.2) is synthesized from arginine and glycine and is phosphorylated reversibly to creatine-P by the enzyme creatine (phospho)kinase (CK or CPK) (Fig. 20.6). CK is a dimeric protein and exists as three isozymes: the MM (skeletal muscle), BB (brain) and MB isoforms. The MB isoform is enriched in cardiac tissue.

Fig. 20.6 Synthesis and degradation of creatine phosphate (creatine-P).
Creatine is synthesized from glycine and arginine precursors. Creatine-P is unstable and undergoes slow, spontaneous degradation to Pi and creatinine, the cyclic anhydride form of creatine, which is excreted from the muscle cell into plasma and then into urine.
The level of creatine-P in resting muscle is several-fold higher than that of ATP (Table 20.3). Thus, ATP concentration remains relatively constant during the initial stages of exercise. It is replenished not only by the action of CK but also by adenylate kinase (myokinase) as follows:
Table 20.3
Changes in energy resources in working muscle: concentrations of energy metabolites in human leg muscle during bicycle exercise
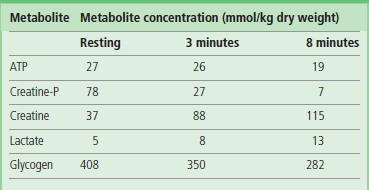
These experiments were conducted during ischemic exercise, which exacerbates the decline in ATP concentration. They illustrate the rapid decline in creatine-P and the increase in lactate from anaerobic glycolysis of muscle glycogen.
Data are adapted from Timmons JA, et al: J Clin Invest101:79–85, 1998.

Creatine phosphate stores decline rapidly during the first minute of high power output muscle contraction. As creatine phosphate stores are depleted, the muscle becomes unable to sustain the high force output, and contractile force rapidly declines. At this point, muscle glycogenolysis becomes a major source of energy. Calcium entry into muscle, in addition to its role in activation of myosin-ATPase-dependent contraction, also leads to formation of a Ca2+–calmodulin complex, which activates phosphorylase kinase, catalyzing the conversion of phosphorylase b to phosphorylase a. AMP also allosterically activates muscle phosphorylase and phosphofructokinase-1, accelerating glycolysis from muscle glycogen (see Chapter 13).
A further decline in force occurs as pyruvate and lactate gradually accumulate in the contracting muscle, resulting in a decrease in muscle pH. Force will then decline to a level that can be maintained by aerobic metabolism of fatty acids. Maximal aerobic power is about 20% of maximal power output, and about 50–60% of maximal aerobic power can be sustained for long periods of time.
Low-intensity, long-duration contractions
Fatty acids are the major source of energy in muscle during prolonged exercise
The availability and utilization of oxygen in working muscle are major limitations for maintaining continuous physical activity. Long-duration contractile activity requires adequate oxygen delivery and the capacity for the muscle to utilize the oxygen delivered. Oxygen delivery to muscle is affected by the red blood cell and hemoglobin concentrations in blood, the number of capillaries within the muscle and heart pump capacity. Highly oxidative muscle has a higher capillary density than glycolytic muscle, and muscle capillary density increases with endurance exercise training. Muscle oxygen utilization is also directly related to the number and size of muscle mitochondria. Muscles subjected to continual contractile activity, such as postural muscles, have more mitochondria than infrequently contracted muscle. A standard observation in muscle subjected to increased contractile demands is an elevation in oxidative enzyme activity.
At rest or at low intensities of physical work, oxygen is readily available and the aerobic oxidation of lipid predominates as the main source of ATP synthesis. However, at higher work intensities, oxygen availability for lipid catabolism can become limiting, and subsequently the muscle work rate decreases. During the first 15–30 minutes of exercise, there is a gradual shift from glycogenolysis and glycolysis to aerobic metabolism of fatty acids. Perhaps this is an evolutionary response to deal with the fact that lactate, produced by glycolysis, is more acidic and less diffusible than CO2. As exercise continues, epinephrine contributes to activation of hepatic gluconeogenesis, providing an exogenous source of glucose for muscle. Lipids gradually become the major source of energy in muscle during long-term, lower-intensity exercise when oxygen is not limiting.
Long-term muscle performance (stamina) depends on levels of muscle glycogen
Fats burn in the flame of carbohydrates; glycogen is required for efficient metabolism of lipids in muscle
Marathon runners typically ‘hit the wall’ when muscle glycogen reaches a critically low level. Glycogen is the storage form of glucose in skeletal muscle and its muscle concentration can be manipulated by diet, e.g. by carbohydrate loading prior to a marathon run. Fatigue, which can be defined as an inability to maintain the desired power output, occurs when the rate of ATP utilization exceeds its rate of synthesis. For efficient ATP synthesis, there is a continuing, but poorly understood, requirement for a basal level of glycogen metabolism, even when glucose is available from plasma and when fats are the primary source of muscle energy. Carbohydrate metabolism is important as a source of pyruvate, which is converted to oxaloacetate by the anaplerotic, pyruvate carboxylase reaction. Oxaloacetate is required to maintain the activity of the TCA cycle, for condensation with acetyl-CoA derived from fats. To some extent, muscle glycogen can be spared and performance time increased during long-term vigorous physical activity by increasing the availability of circulating glucose, either by gluconeogenesis or by carbohydrate ingestion, e.g. bread or Gatorade®. Increased utilization of fatty acids during early stages of exercise is an important training adaptation to regular vigorous physical activity – it serves to spare glycogen stores.
Muscle consists of two types of striated muscle cells: fast-glycolytic and slow-oxidative fibers
Striated muscle cells are generally classified by their physiologic contractile properties (fast versus slow) and primary type of metabolism (oxidative versus glycolytic). The muscle type is closely related to muscle function in skeletal muscle, and this comparison can easily be seen with muscles whose contraction is for infrequent-burst activities versus muscles used continuously for maintaining posture (antigravity). The coloring of the two striated muscle types readily distinguishes them. Fast-glycolytic muscle used for burst activity is white in appearance (like chicken breast – chickens squawk a lot, but cannot fly far!) because of less blood flow, lower mitochondrial density and decreased myoglobin content compared with slow-twitch oxidative muscle, which is red. Fast-glycolytic fibers also have increased glycogen stores and lower fat content; they rely on glycogen and anaerobic glycolysis for short bursts of contraction when additional muscle force is required such as in the ‘fight or flight’ stress response. These muscle fibers are not capable of sustaining contraction for long periods. In contrast, slow-oxidative fibers in postural muscles (and in goose breast – geese are migratory birds) are well perfused with blood, rich in mitochondria and myoglobin. This muscle type has the ability to sustain low-intensity contractions for long periods. Slow muscle uses fatty acid oxidation for ATP synthesis, which requires mitochondria. Cardiac muscle, which is continuously contracting, has many contractile and metabolic characteristics that are similar to slow-oxidative skeletal muscle. Cardiac muscle is well perfused with blood, rich in mitochondria, and relies largely on oxidative metabolism of circulating fatty acids. Goose breast, which powers long, migratory flights, is a fairly fatty and dark meat, compared to chicken breast, and has many characteristics of cardiac muscle.
Clinical box Muscle wasting syndromes
Many patients with conditions that include HIV and many cancers experience severe body weight loss, a condition known as cachexia. Patients with cachexia are frequently unable to tolerate radiation or chemotherapy and have higher morbidity and mortality. The loss of body weight is often independent of caloric intake, and not just akin to starvation. Appetite stimulants alone are often not effective. The weight loss is associated with the loss of both muscle and adipose tissue. Health problems may be magnified in cachectic individuals due to the metabolic dysregulation that can accompany the loss of adipose and muscle tissue. There also appears to be an effect of fiber type on muscle loss, which may be related to metabolism. Fast-glycolytic muscle fibers undergo more protein loss than slow-oxidative muscle fibers. This preferential loss of fast-glycolytic fibers with wasting is the opposite of what is seen in muscle with extended periods of disuse (disuse atrophy).
Slow-oxidative fibers atrophy preferentially during muscle disuse. Although the exact mechanisms inducing wasting are not certain, prime candidates with many wasting syndromes involve systemic inflammatory signaling by cytokines, such as TNF-α and IL-6. Inflammatory signaling induced by the disease process can activate muscle protein degradation, inhibit muscle protein synthesis, and induce adipose tissue lipolysis. Maintaining or preventing severe body weight loss in many disease states can improve patient treatment options, survival and quality of life. Anabolic agents, like testosterone, have proven beneficial in maintaining muscle mass in AIDS patients and are widely used clinically. With other wasting diseases, research in animal models has demonstrated that inhibition of inflammatory signaling can inhibit wasting. Further research is necessary before this approach is widely applied to human populations.
 Clinical test box Assay of creatine to assess renal function and urine dilution
Clinical test box Assay of creatine to assess renal function and urine dilution
Since creatine phosphate concentration is relatively constant per unit muscle mass, the production of creatinine (see Fig. 20.6) is relatively constant during the day. Creatinine is eliminated in urine at a relatively constant amount per hour, primarily by glomerular filtration and to a lesser extent by tubular secretion. Since its concentration in urine varies with the dilution of the urine, levels of metabolites in random urine samples are often normalized to the urinary concentration of creatinine. Otherwise, a 24 h collection would be required to assess daily excretion of a metabolite. Normal creatinine concentration in plasma is about 20–80 mmol/L (0.23–0.90 mg/dL). Increases in plasma creatinine concentration are commonly used as an indicator of renal failure. The albumin : creatinine ratio in a random urine sample, an indicator of protein filtration selectivity of the glomerulus, is used as a measure of the microalbuminuria to assess the progression of diabetic nephropathy (see also Chapter 24.).
 Advanced concept box Sarcopenia
Advanced concept box Sarcopenia
Sarcopenia, the loss of skeletal muscle mass, develops gradually in humans after the fifth decade of life and can lead to frailty and loss of functional capacity. Besides the basic erosion of quality of life, loss of skeletal muscle mass also increases the risk of mortality and morbidity. The cause of sarcopenia appears to be related to gradual decreases in physical activity and loss of regenerative capacity. Muscle fiber innervation by spinal motor neurons is critical to both development and maintenance of muscle fibers (cells). Spinal motor neurons decrease in number with advancing age, possibly because of cumulative oxidative damage to these postmitotic cells. The loss of motor neurons appears to cause the substantial (>40%) loss in the muscle fiber number, which is the primary determinant of age-dependent sarcopenia, and is accompanied by an increase in motor unit size, and decrease in fine motor skill. Sarcopenia has also been linked to age-induced systemic changes to the endocrine, cardiovascular, and immune systems, whose functions are all critical for the maintenance of skeletal muscle mass.
The scientific evidence is clear that most older individuals can increase muscle strength and mass with a regular resistance exercise program. Pharmaceutical treatments have also been examined for individuals who cannot regularly exercise. Currently there is no treatment for spinal motor neuron loss. Pharmaceutical treatments targeting muscle have had varying degrees of success and are usually limited by side effects. The treatments include endocrine interventions with male or female sex hormone replacement therapy, and growth hormone therapy. Anti-inflammatory medication is also employed to allow individuals to participate in physical activity programs. One of the best defenses against sarcopenia is regular exercise in order to maintain muscle mass during middle age.
Tissue engineering and replacement of muscle
As the field of tissue engineering advances, muscle tissue is at the forefront of experiments to grow an organ outside the human body. Muscle is derived from proliferating cells that originate in the mesenchyme germ layer in the developing embryo. These cells are ‘determined’ into the muscle lineage and then become myoblasts. Myoblasts exit the cell cycle and differentiate into a mature multinucleated muscle cell. Skeletal muscle cells (SkMC) are terminally differentiated, but skeletal muscle contains a small population (<5% of myonuclei) of undifferentiated muscle precursor cells, satellite cells. Satellite cell proliferation and differentiation are critical events for postnatal muscle growth and repair, e.g. in response to exercise, and for regeneration after damage. Skeletal muscle is one of the few human tissues that can largely regenerate itself after extensive injury. Muscle is also an ideal candidate for tissue ‘replacement’ following severe injury, as it quickly adapts to its mechanical and chemical environment. Skeletal muscles are highly specialized to their location and particular job. Muscle replacement surgeries using donor muscle for the hand have shown that morphological and biochemical differences must be taken into account when selecting muscle tissue for transplantation. Yet as muscle is highly adaptive (plastic fantastic), it is likely that skeletal muscle will be one of the first tissues (along with skin) to be completely engineered ex vivo for in vivo transplantation.
Terminally differentiated myoblasts in the heart are called cardiac myocytes; these cells remain single or binucleated throughout life. The heart has very limited regenerative capacity, so that the effects of myocardial infarction are long-lasting. Myoblasts of smooth muscle differentiate into their mature smooth muscle cells (SMC). But SMC, unlike heart and skeletal muscle, is not terminally differentiated. SMC phenotype also varies, based on the cell's location and function. SMCs are found throughout the body in the vascular wall, and retain the ability to proliferate, e.g. in response to hypertension or during angiogenesis.
Clinical test box Diagnosis of myocardial infarction
Myocardial infarction (MI) is the result of blockage of blood flow to the heart. Tissue damage results in leakage of intracellular enzymes into blood (Fig. 20.7). Among these are glycolytic enzymes, such as LDH (Chapter 12); however, measurements of myoglobin, total plasma CK and CK-MB isozymes are more commonly used for diagnosis and management of MI. Myoglobin, a small protein (17 kDa), rises rapidly in plasma, within 2 hours following MI. Although it is sensitive, it lacks specificity for heart tissue. It is cleared rapidly by renal filtration and returns to normal within 1 day. Since plasma myoglobin also increases following skeletal muscle trauma, it would not be useful for diagnosis of MI, e.g. following an automobile accident. Total plasma CK and the CK-MB isozyme begin to rise within 3–10 hours following an MI, and reach a peak value of up to 25 times normal after 12–30 hours; they may remain elevated for 3–5 days. Total CK may also increase as a result of skeletal muscle damage but the measurement of CK-MB provides specificity for cardiac damage.
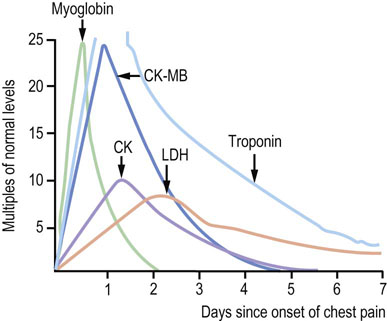
Fig. 20.7 Serum enzyme changes after myocardial infarction (MI).
Various marker enzymes increase in plasma following MI. These are still used for the diagnosis of MI, but the currently recommended test is the measurement of serum troponin concentration. CK, creatine phosphokinase; CK-MB, cardiac isozyme of CK; LDH, lactate dehydrogenase. Adapted from Pettigrew AR, Pacanis A: Diagnosis of myocardial infarction. In Dominiczak MH, editor: Seminars in clinical biochemistry, Glasgow, 1997, University of Glasgow Computer Publishing Unit.
Enzyme-linked immunosorbent assays (ELISA) for the myocardial troponins are now recommended for use in diagnosis and management of MI. These assays depend on the presence of unique isoforms of troponin subunits in the adult heart. Plasma Tn-T concentration increases within a few hours after a heart attack, peaks at up to 300 times normal plasma concentration, and may remain elevated for 1–2 weeks. An assay for a specific isoform in an adult heart, Tn-T2, is essentially 100% sensitive for diagnosis of MI and yields fewer than 5% false-positive results. Significant increases in plasma Tn-T2 are detectable even in patients with unstable angina and transient episodes of ischemia in the heart. Troponins are commonly used as a component of an algorithm to differentiate high-risk from low-risk patients in terms of need for immediate invasive intervention. The recent definition of myocardial infarction is based on observed serum troponin concentrations.
Effect of exercise
Strength or resistance training increases muscle mass
A change in daily use of skeletal muscle has a profound effect upon its functional capacity. Both an increase and a decrease in daily activity level can change muscle structure, force production capacity, and fatigability. From a biochemical point of view, these changes are primarily caused by changes in tissue perfusion and metabolic enzymes, and thus the muscle's ability to take up glucose, utilize fat as an energy source, and generate ATP. The amount and intensity of daily physical activity occurs on a continuum, and muscle adaptation to this occurs in response to the specific stress placed upon it. For the sake of simplicity, and because this is how most research studies are designed, we can separate increased use (exercise training) into two categories: strength and aerobic training. The primary purpose of strength training, also called resistance training, is to increase the ability of a specific muscle, or group of muscles, to produce force. This is typically conducted through a small number of repetitions of one exercise movement against a resistance that only allows the muscle to contract through a full range of motion a very limited number of times (e.g. 6 to 8 repetitions of a bicep curl). The primary purpose of aerobic training, also called endurance training, is to increase endurance and decrease fatigue during prolonged, lower-intensity physical activity, e.g. running or walking. This is achieved through a high number of repetitions of muscle contractions at a low resistance. Each muscle contraction in strength training might be 75–90%, while in an aerobic training session it might be 15–20%, of the maximal voluntary force production of that muscle. The biochemical changes in response to these types of exercise are distinct.
Strength training has minimal effects on muscle biochemistry. The increase in force production capacity that occurs with strength training is due to increased cell size, i.e. hypertrophy. The hypertrophy of individual muscle cells occurs as a result of an increase in structural and sarcomeric proteins. With more myofibrils and sarcomeres (the contractile units of muscle) comes an increase in force production capability. When glycolytic enzymes are examined and normalized to the increased cell size, there is no change with strength training. When mitochondrial enzyme activity is normalized to the increased cell size of strength training, there is usually a slight decrease, suggesting that while force production capacity increases, ATP production capacity (at least based on the size of the cell) has slightly decreased. In terms of contraction speed and sarcomere cross-bridge cycling, this is primarily determined by myosin-ATPase activity, which remains relatively unchanged in response to resistance training.
Endurance or aerobic training increases the oxidative metabolic capacity of muscle
In response to aerobic training, the primary biochemical change is an increase in capacity to metabolize fat, supported by increases in mitochondrial number, size and enzymes. All muscle fiber types (fast and slow) will increase their concentration and activity of citrate synthase and cytochrome-c by 2–3-fold, resulting in increased ATP production at a given workload (i.e. exercise intensity), so that muscle can then rely more on fat oxidation and less on anaerobic metabolism. The shift toward aerobic metabolism delays muscle fatigue; there are only minor effects on glycolytic enzymes in response to aerobic training, and the effects on cell size due to aerobic training are also minimal. Small shifts in myosin-ATPase composition may also occur, leading to a slower muscle phenotype (slower cross-bridge formation during contraction) due to aerobic training. Increases in glucose utilization as a result of increased expression of GLUT-4 and hexokinase also develop more in response to aerobic training, as opposed to strength training, but it is easy to see, considering the amount of skeletal muscle in the body, how blood glucose is decreased in a person with diabetes by an exercise program. It should also be noted that nearly all of these adaptations will occur in reverse in response to any form of de-training, whether that be due to cessation of an exercise program or bed rest due to injury or disease. Decreased use of muscle causes it to become much less metabolically efficient; unfortunately this de-adaptation becomes apparent within a few days following cessation of exercise. Other factors induced by endurance training include changes in cardiac output, increases in capillary density, and increases in glycogen stores. Of critical importance to health and medicine is the continuum within which these adaptations occur and the fact that small changes may impact many chronic diseases, including diabetes, atherosclerosis and cancer cachexia. Further, as changes occur relative to the original status of the muscle, older sedentary persons will see responses in muscle biochemistry comparable to those observed in younger persons. Thus, regardless of age, sedentary individuals who start even a moderate exercise program are likely to see substantial biochemical adaptations and health benefits. Much research is still ongoing in these areas in an attempt to understand the molecular genetic and signaling pathways that bring about these responses and understanding how they might be modified after injury or disease.
Summary
Muscle is the major consumer of fuels and ATP in the body. Glycogenolysis, blood glucose, glycolysis and lipid metabolism are essential for optimal muscle activity. Reliance on these energy-producing pathways varies with muscle type and its prior contractile activity.
Skeletal, cardiac, and smooth muscle have a common actomyosin contractile complex, but differ in innervation, contractile protein arrangement, calcium regulation of contraction, and propagation of depolarization from cell to cell.
The sarcomere is the fundamental contraction unit of striated muscle and is defined by Z-lines and thick and thin filament overlap.
Contraction is described by a ‘sliding filament’ model in which hydrolysis of ATP is catalyzed by an influx of Ca2+ into the sarcoplasm and is coupled to changes in the conformation of myosin. Relaxation of the high-energy conformation of myosin during interaction with actin produces a ‘power stroke’, resulting in increased overlap of the actin–myosin filaments and shortening of the sarcomere.
The ATP produced in muscle drives the maintenance of ion gradients, restoration of intracellular calcium levels, and the contractile process.
Fast-glycolytic muscle relies largely on glycogen and anaerobic glycolysis for short, high-intensity bursts of muscle activity.
Slow-oxidative muscle is an aerobic tissue; at rest, it uses fats as its primary source of energy. During the initial phases of exercise, it relies on glycogenolysis and glycolysis, but then gradually shifts to fat metabolism for long-term energy production. Enzymes and proteins are released from muscle in response to damage.
Measurements of plasma CK-MB activity and troponin concentration are used as biomarkers of damage to cardiac muscle and are commonly used in the diagnosis and treatment of myocardial infarction.
Exercise is good medicine; it increases insulin sensitivity and glucose disposal, and assists in maintenance of muscle mass and function during aging.
Cooke, R. The sliding filament model: 1972–2004. J Gen Physiol. 2004; 123:643–656.
Mayer, F, Scharhag-Rosenberger, F, Carlsohn, A, et al. The intensity and effects of strength training in the elderly. Dtsch Arztebl Int. 2011; 108:359–364.
Mitchell, WK, Williams, J, Atherton, P, et al. Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review. Front Physiol. 2012; 3:260.
Muscaritoli, M, Lucia, S, Molfino, A, et al. Muscle atrophy in aging and chronic diseases: is it sarcopenia or cachexia? Intern Emerg Med. 2013; 8:553–560.
Shave, R, Baggish, A, George, K, et al. Exercise-induced cardiac troponin elevation: evidence, mechanisms, and implications. J Am Coll Cardiol. 2010; 56:169–176.
Tiwari, RP, Jain, A, Khan, Z, et al. Cardiac troponins I and T: molecular markers for early diagnosis, prognosis, and accurate triaging of patients with acute myocardial infarction. Mol Diagn Ther. 2012; 16:371–381.
Muscular dystrophies. www.muscular-dystrophy.org/conditions.
Animations: numerous excellent videos of muscle structure and contraction a. www.youtube.com