Protein Synthesis and Turnover
Introduction
Translation is the process by which the information endoded in an mRNA is translated into the primary structure of a protein
Protein synthesis or translation represents the culmination of the transfer of genetic information, stored as nucleotide bases in deoxyribonucleic acid (DNA), to protein molecules that are the major structural and functional components of living cells. It is during translation that this information, expressed as a specific nucleotide sequence in a ribonucleic acid (mRNA) molecule, is used to direct the synthesis of a protein. The protein then folds into a three-dimensional structure that is defined, in large part, by its amino acid sequence. In order to translate an mRNA into protein, three main RNA components are necessary:
The ribosome, composed of rRNAs and a number of proteins, is the macromolecular machine on which all protein synthesis occurs. The information required to direct the synthesis of the primary sequence of the protein is contained in mRNA. The amino acids that are to be incorporated into the protein are attached to tRNAs. The ribosome interacts with the tRNA molecules and mRNA so that the correct amino acid is incorporated into the protein. The translation of mRNA begins near the 5′ end of the template and moves towards the 3′ end, and proteins are synthesized starting with their amino-terminal ends. Therefore, the 5′ end of the RNA encodes the amino-terminal end of the protein and the 3′ end of the RNA encodes the carboxyl-terminal end of the protein.
This chapter begins with an introduction to the genetic code and the components needed for protein synthesis. This is followed by presentation of the structure and function of the ribosome, detailing the process of translation by outlining the initiation, elongation, and termination of protein synthesis, and the mechanism by which proteins are targeted to specific locations in the cell. Following a discussion of post-translational modifications of proteins, the chapter ends with description of the role of a second macromolecular complex, the proteasome, in protein turnover.
The genetic code
The genetic code is degenerate and not quite universal
The mRNA to be used for translation has only four nucleotides – adenosine, A; cytidine, C; guanosine, G; and uridine, U – but it will encode a protein containing as many as 20 different amino acids. So there is not a one-to-one correspondence between nucleotide and amino acid sequence; instead, a series of three nucleotides in the mRNA, known as a codon, are required to specify each amino acid. When all combinations of four nucleotides are taken into account, three at a time, 64 possible codons result (Table 34.1). Three of these codons (UAA, UAG, UGA) are stop codons, used to signal the termination of protein synthesis; they do not specify an amino acid. The rest specify the 20 amino acids, which illustrates a feature of the genetic code known as degeneracy: more than one codon can specify a specific amino acid. For example, codons GUU, GUC, GUA, and GUG all code for the amino acid valine. Indeed, all the amino acids, with the exception of methionine (AUG) and tryptophan (UGG), have more than one codon. The codon AUG, which specifies only methionine, encodes methionine anywhere it appears in the RNA and it also marks the starting point for protein synthesis (see below for a few exceptions).
Table 34.1
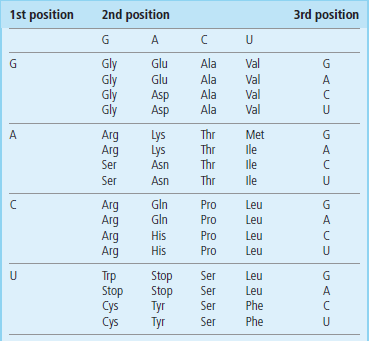
The genetic code is degenerate, meaning more than one codon can code for an amino acid, and in many cases changing the nucleotide at the third position does not change the amino acid encoded. In order to find the sequence(s) of the codons that encode a particular amino acid, one simply finds the amino acid in the table and combines the nucleotide sequence for each position. For example, methionine (Met) is encoded by the sequence AUG. To find the amino acid that matches a codon sequence, reverse this process.
The genetic code as specified by the triplet nucleotides is, for the most part, the same for bacteria and humans, and is referred to as ‘universal’. However, there are exceptions and some examples are found in bacteria and mitochondria. In bacteria, if the codons GUG and UUG occur at the beginning of protein synthesis, they can be read as a methionine codon. There are also minor differences in the genetic code in mitochondria; for instance, in vertebrate mitochondria there are additional codons that can encode methionine, UGA which is normally a stop codon, encodes tryptophan, and there are additional stop codons.
Another aspect of the genetic code is that once synthesis has started at an AUG codon for methionine, each successive triplet from that start point will be read in register without interruption until a termination codon is encountered. Thus the reading frame of the mRNA will be dictated by the start codon. Mutations that cause the addition or deletion of even single nucleotides will cause a reading frame shift, resulting in a protein with a different amino acid sequence after the mutation or a protein that is prematurely terminated if a stop codon is now in frame (nonsense mutation; Table 34.2).
Table 34.2
Effect of mutations on protein synthesis
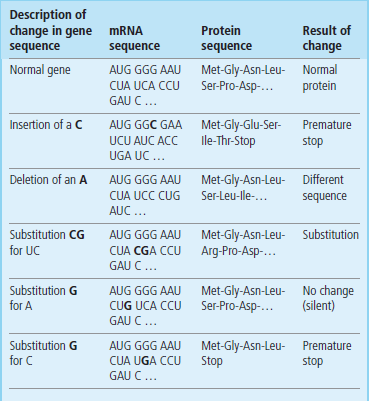
Mutations in a gene are transcribed into the mRNA and the resulting changes in the protein sequence are shown. Note that, depending on the position of the mutation, single nucleotide substitutions can result in silent changes, a change in a single amino acid (missense) or even premature termination (nonsense).
 Clinical box Sickle cell anemia: mutation of the genetic code
Clinical box Sickle cell anemia: mutation of the genetic code
Sickle cell anemia is an example of a disease in which a single nucleotide change within the coding region of the gene for the β-chain of hemoglobin A, the major form of adult hemoglobin, yields an altered protein that has impaired function (see Chapter 5). The mutation that causes this disease is a single nucleotide change in a codon that normally specifies glutamate (GAG) and which now produces a codon that specifies valine (GUG). Under conditions of low oxygen tension, this single amino acid change allows the protein to polymerize into rod-shaped structures, resulting in deformation of red blood cells and in altered flow properties of the cells in vessels and capillaries. When oxygen is not bound to hemoglobin, the conformation of the protein, deoxyhemoglobin, exposes a hydrophobic patch, allowing the mutant proteins with valine to interact more readily and form rods. This substitution of an amino acid with an acidic side chain for an amino acid with a nonpolar, hydrophobic side chain is termed a nonconservative mutation. Conservative replacement of one amino acid by another with similar physical and chemical properties may have less severe consequences, e.g. an Arg → Lys or Asp → Glu mutation.
The machinery of protein synthesis
The ribosome is multi-step assembly line for protein synthesis
Ribosomes, the molecular machines that conduct protein synthesis, consist of a small and a large subunit that, when associated with each other, possess three specific sites at which tRNAs bind. These sites are known as the aminoacyl-tRNA, or A site, the peptidyl-tRNA, or P site, and the exit site, or E site. The A site is where a donor tRNA molecule, carrying the appropriate amino acid on its acceptor stem, is positioned before that amino acid is incorporated into the protein. The P site is the location in the ribosome that contains a tRNA molecule with the amino-terminal polypeptide of the newly synthesized protein still attached through its carboxyl terminus to its acceptor stem. It is within these sites that the process of peptide bond formation takes place. This process is catalyzed by a peptidyl transferase activity, which forms the peptide bond between the amino group of the amino acid in the A site and the carboxyl terminus of the nascent peptide attached to the tRNA in the P site. The E site is where the deacylated tRNA moves once the peptide bond is formed and it will soon be exiting the ribosome. The E site, which provides a third site of interaction between tRNA and mRNA on the ribosomes, appears to be essential for maintaining the reading frame and assuring the fidelity of translation.
Each amino acid has a specific synthetase that attaches it to all the tRNAs that encode it
There is a distinct tRNA molecule for most of the codons represented in Table 34.1. The amino acid is attached to the acceptor stem of the tRNA by an enzyme called aminoacyl-tRNA synthetase. This enzyme catalyzes the formation of an ester bond linking the 3′ hydroxyl group of the adenosine nucleotide of the tRNA to the carboxyl group of the amino acid (Fig. 34.1). The attachment of an amino acid to a tRNA is a two-step reaction. The carboxyl group of the amino acid is first activated by reaction with adenosine triphosphate (ATP) to form an amino-acyladenylate intermediate, which is bound to the synthetase complex. The enzymology of activation of the carboxyl group of amino acids is similar to that for activation of fatty acids by thiokinase (Chapter 15), but, rather than transfer of the acyl group to the thiol group of coenzyme A, the aminoacyl group is transferred to the 3′-hydroxyl of the tRNA. The product is described as a charged tRNA molecule. At this point it is ready to bind to the A site of the ribosome, where it will contribute its amino acid to a growing peptide chain. There is a different synthetase specific for each of the 20 amino acids in protein. This synthetase attaches the appropriate amino acid to all the tRNAs that bind that amino acid.
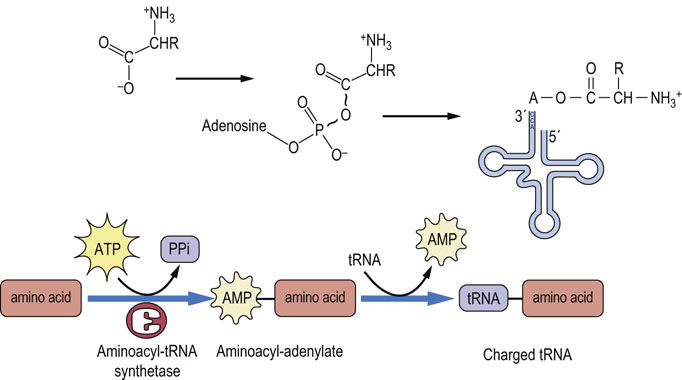
Fig. 34.1 Activation of an amino acid and attachment to its cognate tRNA.
The amino acid must be activated by an aminoacyl-tRNA synthetase to form an aminoacyl-adenylate intermediate, before its attachment to the 3′ end of the tRNA. AMP, adenosine monophosphate; PPi, inorganic pyrophosphate.
 Advanced concept box Fidelity of translation
Advanced concept box Fidelity of translation
Aminoacyl-tRNA synthetases have proofreading ability.
To guarantee the accuracy of protein synthesis, mechanisms have evolved to ensure selection of the correct amino acid for acylation and for proofreading of already charged tRNAs. One such mechanism is found in the enzymes responsible for attaching an amino acid to the correct tRNA. The aminoacyl-tRNA synthetases have the ability not only to discriminate between amino acids before they are attached to the appropriate tRNA but also to remove amino acids that are attached to the wrong tRNA. In addition, these enzymes must discriminate between the multitude of tRNAs and be able to pair the correct tRNA with the appropriate amino acid. These abilities exhibited by the synthetases are accomplished by a series of hydrogen bonding interactions between the enzyme and the amino acid and between the enzyme and the tRNA. These mechanisms combine to ensure the accurate transfer of information from mRNA to protein.
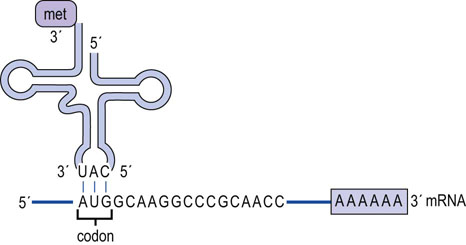
Some flexibility in base pairing occurs at the 3′ base of the mRNA codon
Interaction of the charged tRNA with its cognate codon is accomplished by association of the anticodon loop in tRNA with the codon in mRNA through hydrogen bonding of complementary base pairs (Fig. 34.2). The base-pairing rules are the same as those for DNA (Chapter 32), except at the third position, or 3′ base, of the codon. At this position, nonclassical base pairs can form between this nucleotide and the first, or 5′ base, of the anticodon. The so-called wobble hypothesis of codon–anticodon pairing allows a tRNA that has an anticodon that is not perfectly complementary to the mRNA codon to recognize the sequence and allow for the incorporation of the amino acid into the growing peptide chain. Thus, if a guanine residue is at the 5′ position of the anticodon, it can form a base pair with either a cytidine or a uridine residue in the 3′ position of the codon. If the modified adenosine residue, inosine, occurs at the 5′ position of the anticodon, it can form a base pair with uridine, adenosine, or even cytidine at the 3′ position of the codon (Table 34.3). This would allow a tRNA with the anticodon GAG to decode the codons CUU and CUC, both of which code for leucine. The wobble provides a mechanism for dealing efficiently with the degeneracy of the genetic code since the degeneracy always occurs in the third residue of the codon.
How does the ribosome know where to begin protein synthesis?
The mRNA molecule carries the information that will be used to direct the synthesis of the protein. However, not all the information carried on the mRNA is used for this purpose. Most eukaryotic mRNAs contain regions both before and after the protein-coding region, called 5′ and 3′ flanking sequences or 5′ and 3′ UTRs (untranslated regions). These sequences are involved in regulating the site and rate of protein synthesis and the stability of the mRNA. Thus, the protein coding region does not start immediately at the beginning of the mRNA, which raises the question of how the ribosome knows where to start synthesis. In the case of eukaryotic cells, the ribosome first binds to the 7-methylguanine ‘cap’ structure (Chapter 33) at the 5′ end of the mRNA, and then moves down the molecule until it encounters the first AUG codon (Fig. 34.3). This signals the ribosome to begin synthesizing the protein, beginning with a methionine residue, and to continue until it encounters one of the termination codons. On some viral and eukaryotic mRNAs, the first AUG is not used and, instead, an alternative start codon is defined by an internal ribosome entry site (IRES; see Fig. 34.3).
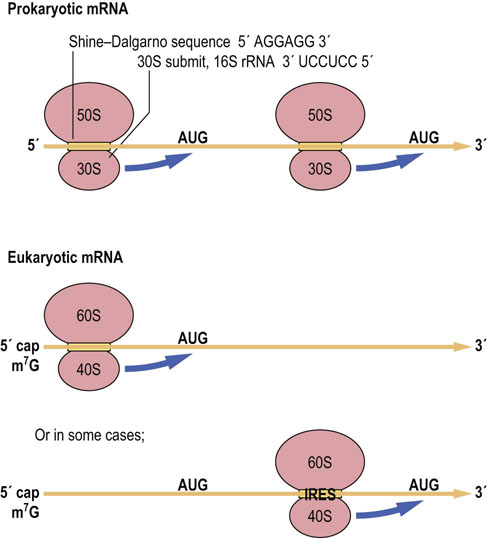
Fig. 34.3 Finding the protein-coding region.
The ribosome binds to the mRNA before locating the protein-coding region. Bacterial ribosomes (a portion of 16 S rRNA) bind to complementary sequences in the mRNA that are termed Shine–Dalgarno sequences, which are a short distance from the start of the protein-coding region. Eukaryotic ribosomes bind to the 5′ cap of mRNAs and then move down the mRNA until they encounter the first AUG codon or in a few cases they bind internally at an internal ribosome entry site (IRES) and then move to the AUG.
In the case of bacterial cells, there is no m7G cap. Knowing what portion of the mRNA is to be used to synthesize a protein is complicated by the fact that there can be several proteins encoded by a single mRNA, each out of register with one another, so that proteins of different sequence may be obtained from the same ribonucleotide sequence. This problem has been solved by the discovery of a sequence in the mRNA that helps to precisely position the ribosome at the beginning of each protein-coding region. This sequence, known as the Shine–Dalgarno sequence, is found in most bacterial mRNAs and is complementary to a portion of the 16 S rRNA in the small bacterial ribosomal subunit (Fig. 34.3). Through the formation of hydrogen bonds, the ribosome is then positioned at the start of each protein-coding region.
The process of protein synthesis
Initiation
Synthesis of a protein is initiated at the first AUG (methionine) codon in the mRNA
Initiation of protein synthesis in eukaryotes takes place when a dissociated small subunit of the ribosome forms a complex with eukaryotic initiation factors (eIF), eIF-1, eIF-1 A, and eIF-3, which binds to an eIF-2/Met-tRNA complex. This preinitiation complex is directed to the 5′ end of the mRNA by the binding of eIF-4F, and other factors, to the 5′ cap. The complex scans the mRNA until it locates the first AUG codon, using ATP to power this process. The large ribososmal subunit then binds to the small subunit/Met-tRNA/mRNA complex, and the Met-tRNA eventually is directed to the P site (Fig. 34.4), requiring eIF-5, hydrolyzing GTP and releasing initiation factors in the process. In eukaryotic cells, there are at least 12 different initiation factors. In prokaryotic cells, the process involves three initiation factors and the initiation complex first forms just 5′ to the coding region, as a result of the interaction of 16 S rRNA in the small subunit with the Shine–Dalgarno sequence on the mRNA. N-Formyl methionine (fmet), encoded by AUG, is the first amino acid in all bacterial proteins, instead of methionine.
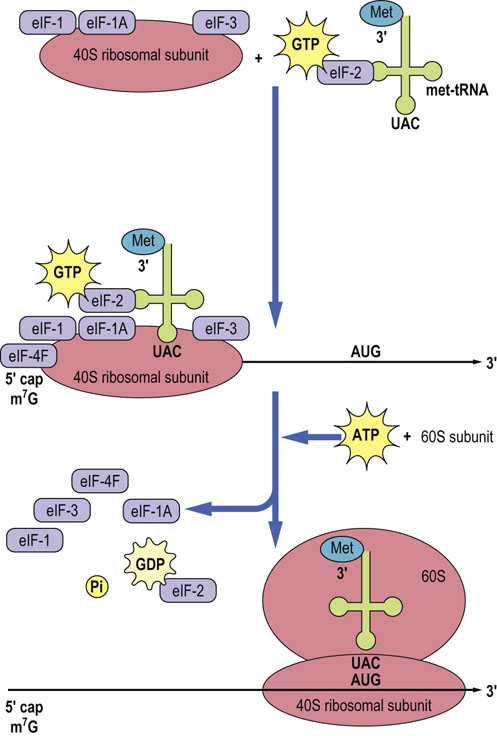
Fig. 34.4 Initiation of protein synthesis in eukaryotic cells.
The 40 S ribosomal subunit with bound initiation factors eIF-1, eIF-1A, and eIF-3, mRNA with eIF-4F bound to the 5′ cap, and met-tRNA bound to eIF-2 are brought together. Once these components are assembled, the complex translocates to the AUG, scanning the mRNA sequence and hydrolyzing ATP in the process. The 60 S ribosomal subunit completes the initiation complex and in the process the initiation factors are released. Note that, at initiation, the P site is occupied by the initiator met-tRNA, as shown in Fig. 34.5. GDP, guanosine diphosphate; eIF, eukaryotic initiation factor.
Elongation
Factors involved in the elongation stage of protein synthesis are targets of some antibiotics
After initiation is complete, the process of translating the information in mRNA into a functional protein starts. Elongation begins with the binding of a charged tRNA to the A site of the ribosome. In eukaryotic cells, the charged tRNA molecule is brought to the ribosome by the action of an elongation factor called eEF-1A (Fig. 34.5). For eEF-1A to be active, it must have a GTP molecule associated with it. If the charged tRNA is correct, one in which the anticodon of the tRNA forms base pairs with the codon on the mRNA, then GTP is hydrolyzed and eEF-1A is released. For the eEF-1A factor to bring another charged tRNA molecule to the ribosome, it must be regenerated by an elongation factor called eEF-1B which will promote the association of eEF-1A with GTP so that it may bind to another charged tRNA molecule (see Fig. 34.5). Once the correct charged tRNA molecule has been delivered to the A site of the ribosome, the peptidyl transferase activity of the ribosome catalyzes the formation of a peptide bond between the amino acid in the A site and the amino acid at the end of the growing peptide chain in the P site. The tRNA-peptide chain is now transiently bound to the A site. The ribosome is then moved one codon down the mRNA (towards the 3′ end), with the help of a factor known as eEF-2, and the tRNA in the A site, with the nascent peptide chain attached, moves to the P site. The uncharged tRNA originally in the P site moves to the E site so that there is a total of nine nucleotide pairs involved in stabilization of the ribosome–mRNA–tRNA complex. The whole process recycles for addition of the next amino acid (Fig. 34.6). The mechanics of this complex process are identical in prokaryotic cells, but the ribosomes and factors are different and this helps to explain the utility of antibiotics that preferentially inhibit protein synthesis in bacteria (Table 34.4).
Table 34.4
Selected antibiotics that affect protein synthesis
| Antibiotic | Target |
| Tetracycline | Bacterial ribosome A site |
| Streptomycin | Bacterial 30 S ribosome subunit |
| Erythromycin | Bacterial 50 S ribosome subunit |
| Chloramphenicol | Bacterial ribosomepeptidyl transferase |
| Puromycin | Causes premature termination |
| Cycloheximide | Eukaryotic 80 S ribosome |
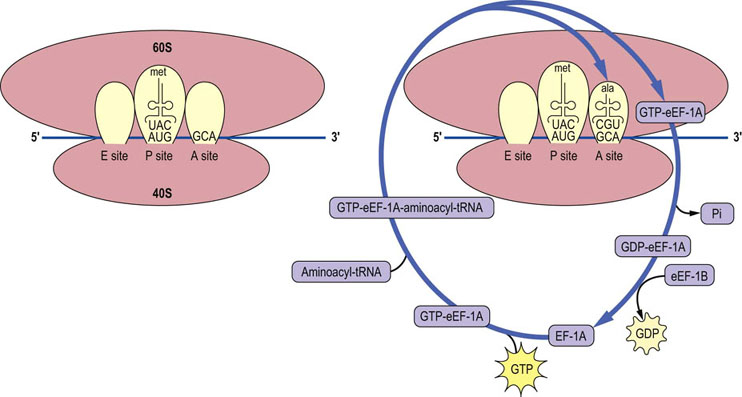
Fig. 34.5 Recycling of elongation factor eEF-1A.
A charged tRNA molecule is brought to the A site of the initiation complex, with the aid of eEF-1A with bound GTP, to begin the process of elongation. The factor is released once GTP is hydrolyzed and the process of recycling eEF-1A is aided by the exchange factor eEF-1B. Each successive amino acid addition requires that the correctly charged tRNA molecule be brought to the A site of the ribosome. ala, alanine; Pi, inorganic phosphate.
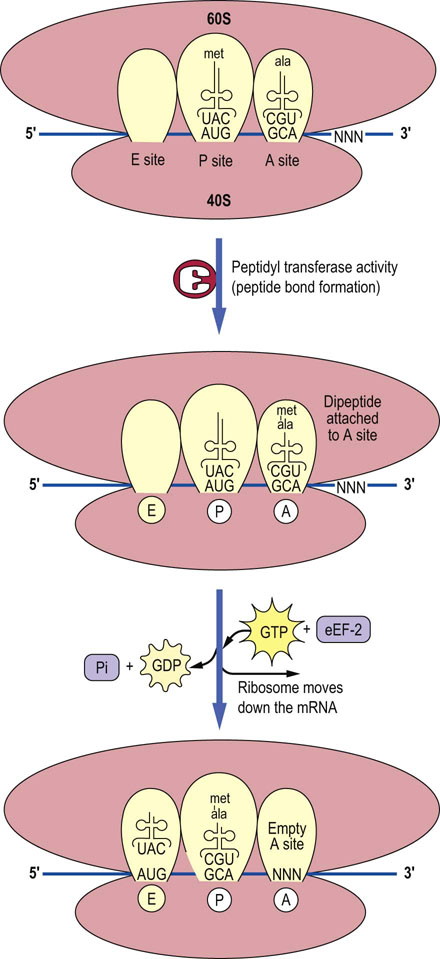
Fig. 34.6 Peptide bond formation and translocation.
The formation of the peptide bond between each successive amino acid is catalyzed by peptidyl transferase. Once the peptide bond is formed, an elongation factor (eEF-2 in this case) will move the ribosome down one codon on the mRNA, so that the A site is vacant and ready to receive the next charged tRNA. The E site is now occupied by the uncharged tRNA (met). NNN is the codon for next amino acid.
Termination
Termination of protein synthesis in both eukaryotic and bacterial cells is accomplished when the A site of the ribosome reaches one of the stop codons of the mRNA. Proteins called releasing factors recognize these codons, and cause the protein that is attached to the last tRNA molecule in the P site to be released (Fig. 34.7). This process is an energy-dependent reaction catalyzed by the hydrolysis of GTP, which transfers a water molecule to the end of the protein, thus releasing it from the tRNA. After release of the newly synthesized protein, the ribosomal subunits, tRNA, and mRNA dissociate from each other, setting the stage for the translation of another mRNA.
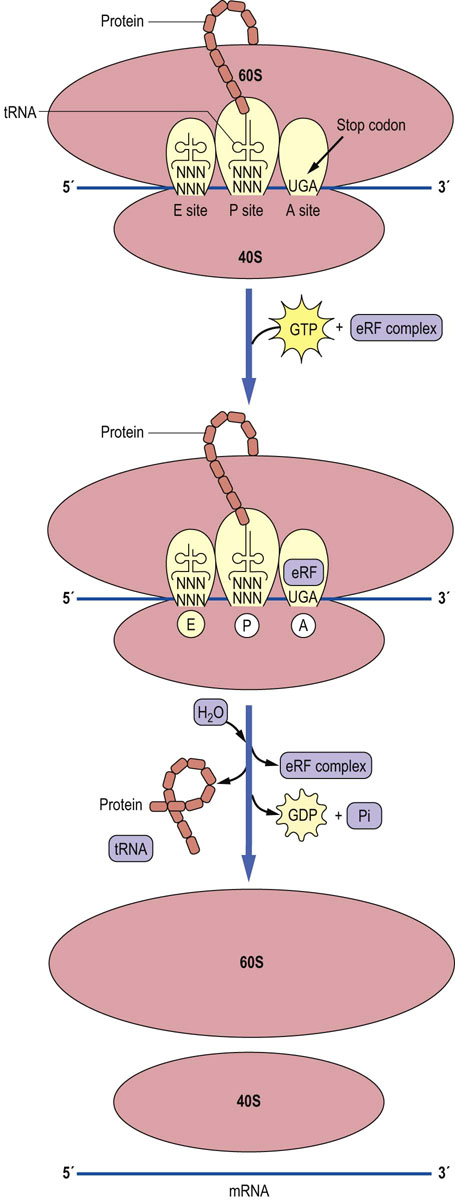
Fig. 34.7 Termination of protein synthesis.
Termination of protein synthesis occurs when the A site is located over a termination codon. A releasing factor complex (eRF), with eRF-1 in the A site, will cause the completed protein to be released and the ribosome, mRNA, and tRNA will dissociate from each other to begin another cycle of translation.
Clinical box A noncompliant patient who was prescribed an antibiotic
A young man you were treating for a sinus infection returns to your clinic after 1 week, still complaining of sinus headaches and stuffiness. He explains that he began to feel better about 3 days after starting to take the antibiotic tetracycline, which you had prescribed. You inquire whether he continued to take the full dose of the drug, even after he began to feel better. He reluctantly admits that, as soon as he felt better, he stopped taking the drug. How do you explain to your patient that it is important that he takes the drug for as long as you prescribed it, even if he feels better after only a few days?
As a physician, you know that tetracycline is a broad-spectrum antibiotic that inhibits the protein synthetic machinery of the bacterial cell by binding to the A site of the ribosome (Table 34.4). You also know that, if the drug is removed, protein synthesis can resume. If the drug is not taken for the entire period recommended, bacteria may begin to grow again, leading to the resurgence of the infection. Further, those bacteria that begin to grow after early termination of treatment are likely to be the most resistant to the drug. Because of the selection for more resistant mutant strains, the secondary infection is likely to be more difficult to control.
Advanced concept box Protein synthesis: peptidyl transferase
Peptidyl transferase is not your typical enzyme. It is a ribozyme.
Peptidyl transferase is the activity responsible for peptide bond formation during protein synthesis. This enzyme activity catalyzes the reaction between the amino group of the aminoacyl-tRNA in the A site and the carboxyl carbon of the peptidyl-tRNA in the P site, forming a peptide bond from an ester bond. The activity is located in the ribosome, but none of the ribosomal proteins has the capacity to catalyze this reaction. Crystal structures of the ribosome have shown that the peptidyl-transferase center is composed entirely of rRNA, and although proteins or specific amino acids are important for positioning the tRNAs or in stabilizing rRNA structure, the catalytic activity resides in the rRNA.
Protein folding and endoplasmic reticulum (ER) stress
ER stress, the result of errors in protein folding, develops in many chronic conditions, including obesity, diabetes and cancer
For a newly synthesized protein to become functionally active, it must be folded into a unique three-dimensional structure. Since there are too many random conformations that the newly synthesized proteins could possibly adopt, the proteins achieve their native structures with the help of a class of proteins called chaperones. Chaperones promote the correct folding, assembly and organization of proteins and macromolecular structures, including the nucleosome and electron transport complexes. In the ER they bind to exposed hydrophobic regions of unfolded proteins, preventing the misfolding of proteins and formation of nonspecific aggregates by shielding the interactive surfaces. The chaperones promote the correct folding of newly synthesized proteins by cycles of protein (substrate) binding and release regulated by an ATPase activity and by cofactor proteins. Heat shock proteins are a group of chaperone proteins that are expressed by cells in response to high temperature; they assist in the refolding of denatured proteins, not only as a result of heat but also in response to physical and chemical stresses. A protein, GRP78/BiP (glucose regulated protein 78/binding immunoglobulin protein) binds and traps misfolded proteins in the ER, preventing their further transport and secretion. The proteins are then routed to the ER-associated degradation (ERAD) pathway, which facilitates export to the cytosol and proteasomal (see below) degradation of the misfolded protein.
A condition known as ER stress develops if chaperone and ERAD activity is overwhelmed and protein aggregates accumulate in the lumen of the ER (e.g. as a result of a protein mutation, a glycosyl transferase deficiency or inhibitor, such as tunicamycin; Fig. 27.11). ER stress activates the unfolded protein response (UPR), which induces higher expression of several chaperone and ERAD proteins. In addition, the protein PERK (protein kinase RNA-like ER kinase) oligomerizes and autophosphorylates, then phosphorylates eIF-2, which inhibits initiation (Fig. 34.4) and slows down the rate of synthesis of many proteins. If homeostasis is not restored by the UPR, apoptosis is eventually initiated to eliminate the cell. A number of human diseases are characterized by ER stress and the UPR, including some forms of cystic fibrosis and retinitis pigmentosa. ER stress and the UPR also inhibit insulin signaling, causing insulin resistance, and play a role in development of pathology in obesity and type 2 diabetes mellitus (Chapter 21).
Protein targeting and post-translational modifications
Protein targeting
An mRNA can have several bound ribosomes at one time and this is known as a polyribosome or polysome (Fig. 34.8). There are two general classes of polysomes found in cells: those that are free in the cytoplasm and those that are attached to the endoplasmic reticulum. Those mRNAs encoding proteins destined for the cytoplasm or nucleus are translated primarily on polysomes free in the cytoplasm, while mRNAs encoding membrane and secreted proteins are translated on polysomes attached to the ER. Regions of the ER studded with bound ribosomes are described as the rough endoplasmic reticulum (RER).
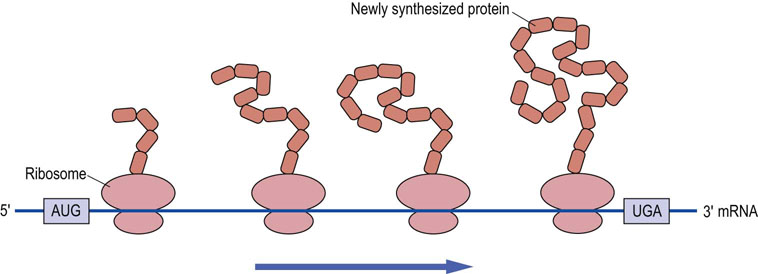
Fig. 34.8 Protein synthesis on polysomes.
Protein can be synthesized by several ribosomes bound to the same mRNA, forming a structure known as a polysome.
Cellular fate of proteins is determined by their signal peptide sequences
Proteins that are destined for export, for insertion into membranes, or for specific cellular organelles, must in some way be distinguished from proteins that reside in the cytoplasm. The distinguishing characteristic of proteins targeted for these locations is that they contain a signal sequence usually comprising the first 20–30 amino acids on the amino-terminal end of the protein. In the case of secretory or membrane proteins, shortly after the signal sequence is synthesized, it is recognized by a ribonucleoprotein complex known as the signal recognition particle (SRP), composed of a small RNA and six proteins. The SRP binds to the signal sequence and halts translation of the remainder of the protein. This complex then binds to the SRP receptor located on the membrane of the endoplasmic reticulum. After the SRP has delivered the ribosome-bound mRNA with its nascent protein to the endoplasmic reticulum, the signal sequence is inserted through the membrane, the SRP dissociates, and translation continues with the polypeptide chain being moved, as it is synthesized, across the membrane into the interstitial space of the endoplasmic reticulum (Fig. 34.9). The protein is then transferred to the Golgi apparatus and then to its final destination.
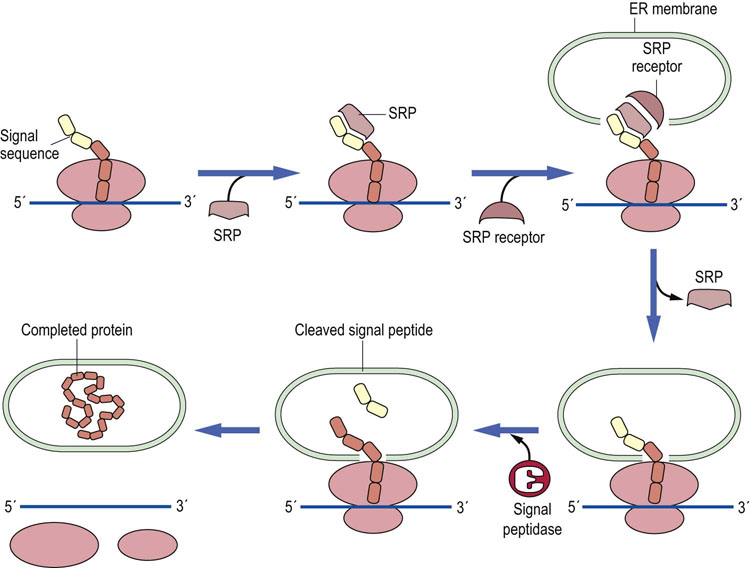
Fig. 34.9 Protein synthesis on the endoplasmic reticulum.
The signal sequence of the protein being translated is bound to an SRP and this complex is recognized by the SRP docking protein (SRP receptor) on the endoplasmic reticulum (ER), where the signal sequence is inserted through the membrane. A signal peptidase typically removes the signal sequence but not on every protein. Once synthesis of the protein is complete, the protein is inserted into the membranes or secreted.
While Man-6-P in high-mannose oligosaccharides is the signal that directs some glycoproteins to the lysosomal compartment (Fig. 27.14), targeting of proteins to other subcellular compartments is directed by a signal peptide sequence in the peptide chain. In contrast to secretory and membrane proteins, mitochondrial and nucleoplasmic proteins are transported after their translation is complete. In the case of proteins destined for the mitochondrion, they may have two signal sequences on their N-terminal end, depending on whether they are destined for the matrix or the intermembrane space. Mitochondrial proteins must be unfolded before they can be transported through transporters in the inner and outer membranes (TIM and TOM; Fig. 9.3). In contrast, nuclear proteins may have nuclear localization signals anywhere in the protein sequence, but exposed on the protein surface, and do not have to be unfolded before transport. Nuclear pores are very large complexes that can accommodate the recognition and transport of a protein in its native state.
Post-translational modification
Most proteins require post-translational modification before they become biologically active
The endoplasmic reticulum and Golgi apparatus are major sites of post-translational modification of proteins. In the endoplasmic reticulum, an enzyme called signal peptidase removes the signal sequence from the amino-terminus of the protein, resulting in a mature protein that is 20–30 amino acids shorter than that encoded by the mRNA. In the endoplasmic reticulum and Golgi apparatus, carbohydrate side chains are added and modified at specific sites on the protein (Chapter 27). One of the common amino-terminal modifications of eukaryotic cells is the removal of the amino-terminal methionine residue that initiates protein synthesis. Finally, many proteins, e.g. the hormones insulin and glucagon, are synthesized as preproproteins and proproteins that must be proteolytically cleaved for them to be active. The cleavage of a precursor to its biologically active form is usually accomplished by a specific protease, and is a regulated cellular event.
Proteasomes: cellular machinery for protein turnover
Unlike DNA, proteins are not repaired, but degraded
Protein degradation is a complex process that is critical to cellular regulation. There are a number of reasons why a protein would need to be degraded. The protein is just worn out – it has aged passively by gradual denaturation during normal environmental stress. Proteins might also be modified by reaction with reactive intracellular compounds, such as glycolytic intermediates, or by reaction with products of lipid peroxidation during oxidative stress (Chapter 37). Other proteins might be part of cellular responses to hormones or perhaps are components of the cell cycle or are transcription factors; they need to be removed, sometimes rapidly, to attenuate the response or signal. Some of these latter proteins have characteristic amino acid sequences or N-terminal residues that promote their rapid turnover. The PEST (ProGluSerThr) sequence marks some proteins for rapid turnover, while proteins with N-terminal arginine generally have short half-lives, compared to proteins with N-terminal methionine.
The proteasome is a multicatalytic, complex for designed for degradation of cytosolic proteins
Since protein degradation is a destructive process, it must be sequestered within specific cellular organelles. Lysosomes, for example, ingest and degrade damaged mitochondria and other membranous organelles. However, most soluble, cytoplasmic proteins are degraded in structures called proteasomes. The 26 S proteasome (Fig. 34.10) consists of two types of subunits: a 20 S multimeric, multicatalytic protease (MCP) and a 19 S ATPase. The proteasome is a barrel-shaped structure, formed by a stack of four rings of seven homologous monomers, α-type subunits in the outer ends and β-type subunits on the inner rings of the barrel. The proteolytic activity – three different types of threonine proteases – resides on β-subunits with active sites facing the inside of the barrel, thereby protecting cytoplasmic proteins from inappropriate degradation. The ATPase subunits are attached at either end of the barrel and act as gatekeepers, allowing only proteins destined for destruction to enter the barrel. The proteins are unfolded in a process that requires ATP and degraded by the protease activities to small peptides, 6–9 amino acids in length, that are released into the cytoplasm for further degradation.
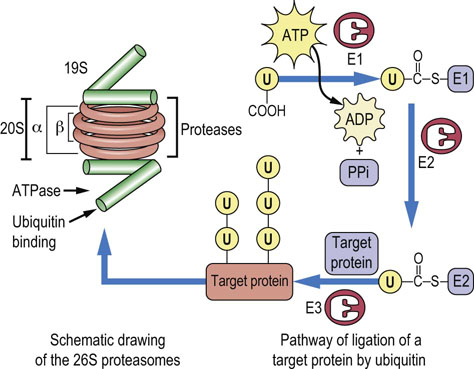
Fig. 34.10 Structure of the proteasome and the role of ubiquitin (U) in protein turnover.
The proteasome is shown at the left, a barrel-shaped structure. The 20 S multicatalytic protease in the middle rings of the barrel has protease activity on the inner face. The 19 S caps at the end of the barrel have ubiquitin binding and release and ATPase activity, and control access of proteins to the inside of the barrel for degradation. The ubiquitin cycle is involved in marking proteins for degradation by the proteasome. Ubiquitin is first activated as a thioester derivative of ubiquitin-activating enzyme E1; it is then transferred to ubiquitin-conjugating enzyme E2, then to a lysine residue on the target protein, catalyzed by ubiquitin-ligase E3. Ubiquitin is frequently copolymerized on target proteins. The more highly ubiquinated the protein, the more susceptible it is to proteasomal degradation. Note that the drawing is not to scale; the proteasome is a 26 S macromolecular complex (≥2,000,000 Da); target proteins are smaller, while ubiquitin is less than 10,000 Da.
Ubiquitin targets proteins to the proteasome for degradation
Proteins destined for destruction are directed to the proteasome primarily by covalent modification with a very highly conserved, 76-amino acid residue protein called ubiquitin, which is found in all cells. Ubiquitin must be activated to fulfill its role (see Fig. 34.10); this is accomplished by a ubiquitin-activating enzyme called E1. Activation occurs when E1 is attached via a thioester bond to the C-terminus of ubiquitin by ATP-driven formation of an ubiquitin–adenylate intermediate. The activated ubiquitin is then attached to a ubiquitin carrier protein, known as E2, by a thioester linkage. A ubiquitin protein ligase known as E3 transfers the ubiquitin from E2 to a target protein, forming an isopeptide bond between the carboxyl-terminus of ubiquitin and the ε-amino group of a lysine residue on a target protein. Denatured and oxidized proteins are commonly ubiquitinated by this mechanism. The 19 S subunit of the proteasome has a ubiquitin-binding site that allows proteins with a covalently attached ubiquitin protein to enter the barrel; the ubiquitin is then released by a ubiquitinase activity and recycled to the cytosol for reuse. Polymerization of ubiquitin on target proteins (polyubiquitination; Fig. 34.10) significantly enhances the degradation of the protein.
The ubiquitin pathway leading to proteasomal degradation of proteins is complex. Although the number of E1 enzymes is typically small, there are several E2 and E3 proteins with different target specificities, and there are six different ATPase activities associated with the 19 S proteasome subunit. The ATPases are thought to be involved in denaturation of ubiquinated proteins, opening the proteolytic core, and transporting the peptide chain into the core of the proteasome. All these variations in components of the pathway, as well as changes during the cell cycle and in response to hormonal stimulation, provide a flexible and regulated pathway for protein turnover.
Advanced concept box Inhibiting the proteasome to treat cancer
Multiple myeloma is a cancer of plasma cells (B lymphocytes), which are normally found in bone and synthesize antibodies as part of the immune system (see Chapter 2). The unchecked growth of these plasma cells results in anemia, tumors in the bones, a compromised immune response, and unfortunately usually a poor prognosis. Patients with recurring multiple myeloma have few treatment options, but a new drug that is an inhibitor of protease activity in the proteasome, bortezomib, has been added to the arsenal. It has been shown to improve the chances for survival of these patients and increase the length of time before remission, especially when combined with other therapies such as radiation or additional chemotherapy. Bortezomib inhibits the degradation of proteins involved in programmed cell death (apoptosis) of cancer cells, thereby enhancing the self-destruct signal in myeloma cells. Second-generation proteosome inhibitors targeting different parts of the complex are currently in clinical trials.
Summary
 Protein synthesis is the culmination of the transfer of genetic information from DNA to proteins. In this transfer, information must be translated from the four-nucleotide language of DNA and RNA to the 20-amino acid language of proteins.
Protein synthesis is the culmination of the transfer of genetic information from DNA to proteins. In this transfer, information must be translated from the four-nucleotide language of DNA and RNA to the 20-amino acid language of proteins.
The genetic code, in which three nucleotides in mRNA (codon) specify an amino acid, represents the translation dictionary of the two languages.
The tRNA molecule is the bridge between the sequence of the nucleotides in mRNA and the amino acids protein. It tRNA accomplishes this task by virtue of its anticodon loop, which interacts with specific codons on the mRNA and also with amino acids via its amino acid attachment site located on the 3′ end of the molecule.
The process of translation consists of three parts: initiation, elongation, and termination.
Initiation involves the assembly of the ribosome and charged tRNA at the initiation codon (AUG) of the mRNA. This assembly process is mediated by initiation factors and requires the expenditure of energy in the form of GTP.
Elongation is a stepwise addition of the individual amino acids to a growing peptide chain by the action of the ribozyme, peptidyl transferase. The charged tRNA molecules are brought to the ribosome by elongation factors at the expense of GTP hydrolysis.
Termination of protein synthesis occurs when the ribosome reaches a stop codon and releasing factors catalyze the release of a protein.
After release, the newly synthesized protein must be correctly folded with the help of ancillary proteins called chaperones, and targeted to specific subcellular compartments by signal sequences.
Many newly synthesized proteins must also be modified, by a variety of chemical and structural changes, before they are biologically active.
Back, SH., Kaufman, RJ. Endoplasmic stress and type 2 diabetes. Annu Rev Biochem. 2012; 81:767–793.
Garcia-Robles, I, Sanchez-Navarro, J, de la Pena, M. Intronic hammerhead ribozymes in mRNA biogenesis. Biol Chem. 2012; 393:1317–1326.
Grudnik, P, Bange, G, Sinning, I. Protein targeting by the signal recognition particle. Biol. Chem. 2009; 390:775–782.
Korostelev, A, Noller, HF. The ribosome in focus: new structures bring new insights. Trends Biochem Sci. 2007; 32:434–441.
Rodnina, MV, Beringer, M, Wintermeyer, W. How ribosomes make peptide bonds. Trends Biochem Sci. 2007; 32:20–26.
Schmeing, TM, Ramakrishnan, V. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009; 461:1234–1242.
Walter, P, Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011; 334:1081–1086.
Wang, S, Kaufman, RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012; 197:857–867.