Carbohydrate Storage and Synthesis in Liver and Muscle
Learning objectives
After reading this chapter you should be able to:
 Describe the structure of glycogen.
Describe the structure of glycogen.
Identify the primary sites of glycogen storage in the body and the function of glycogen in these tissues.
Outline the metabolic pathways for synthesis and degradation of glycogen.
Describe the mechanism by which glycogen is mobilized in liver in response to glucagon, in muscle during exercise, and in both tissues in response to epinephrine.
Explain the origin and consequences of glycogen storage diseases in liver and muscle.
Describe the mechanism for counterregulation of glycogenolysis and glycogenesis in liver.
Outline the pathway of gluconeogenesis, including substrates, unique enzymes and regulatory mechanisms.
Describe the complementary roles of glycogenolysis and gluconeogenesis in maintenance of blood glucose concentration.
Introduction
The red cell and the brain have an absolute requirement for blood glucose for energy metabolism. Together, they consume about 80% of the 200 g of glucose consumed in the body per day. There are only about 10 g of glucose in the plasma and extracellular fluid volume, so that blood glucose must be replenished constantly. Otherwise, hypoglycemia develops and compromises brain function, leading to confusion and disorientation, and possibly life-threatening coma at blood glucose concentrations below 2.5 mmol/L (45 mg/dL). We absorb glucose from our intestines for only 2–3 h following a carbohydrate-containing meal, so there must be a mechanism for maintenance of blood glucose between meals.
Glycogen, a polysaccharide storage form of glucose, is our first line of defense against declining blood glucose concentration. During and immediately following a meal, glucose is converted into glycogen, a process known as glycogenesis, in both liver and muscle. The tissue concentration of glycogen is higher in liver than in muscle but because of the relative masses of muscle and liver, the majority of glycogen in the body is stored in muscle (Table 13.1).
Hepatic glycogenolysis and gluconeogenesis are required for maintenance of normal blood glucose concentration
Hepatic glycogen is gradually degraded between meals, by the pathway of glycogenolysis, releasing glucose to maintain blood glucose concentration. However, total hepatic glycogen stores are barely sufficient for maintenance of blood glucose concentration during a 12-h fast.
During sleep, when we are not eating, there is a gradual shift from glycogenolysis to de novo synthesis of glucose, also an hepatic pathway, known as gluconeogenesis (Fig. 13.1). Gluconeogenesis is essential for survival during fasting or starvation, when glycogen stores are depleted. The liver uses amino acids from muscle protein as the primary precursor of glucose, but also makes use of lactate from glycolysis and glycerol from fat catabolism. Fatty acids, mobilized from adipose tissue triglyceride stores, provide the energy for gluconeogenesis.
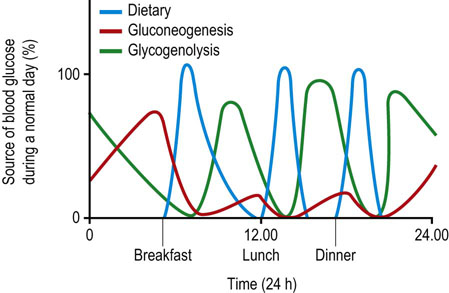
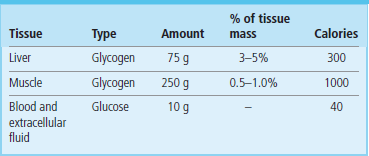
Fig. 13.1 Sources of blood glucose during a normal day.
Between meals, blood glucose is derived primarily from hepatic glycogen. Depending on the frequency of snacking, glycogenolysis and gluconeogenesis may be more or less active during the day. Late in the night or in early morning, following depletion of a major fraction of hepatic glycogen, gluconeogenesis becomes the primary source of blood glucose.
Glycogen is stored in muscle for use in energy metabolism
Muscle glycogen is not available for maintenance of blood glucose. Glucose obtained from blood and glycogen is used exclusively for energy metabolism in muscle, especially during bursts of physical activity. Although cardiac and skeletal muscles rely on fats as their primary source of energy, some glucose metabolism is essential for efficient fat metabolism in these tissues.
This chapter describes the pathways of glycogenesis and glycogenolysis in liver and muscle, and the pathway of gluconeogenesis in liver.
Structure of glycogen
Glycogen, a highly branched glucan, is the storage form of glucose in tissues
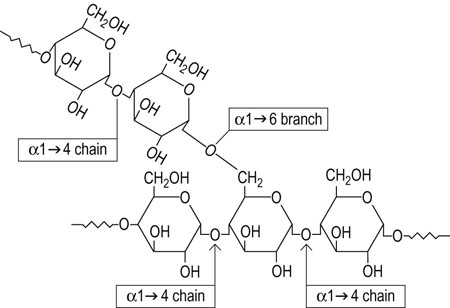
Glycogen is a branched polysaccharide of glucose. It contains only two types of glycosidic linkages, chains of α1→4-linked glucose residues with α1→6 branches spaced about every 4–6 residues along the α1→4 chain (Fig. 13.2). Glycogen is closely related to starch, the storage polysaccharide of plants, but starch consists of a mixture of amylose and amylopectin. The amylose component contains only linear α1→4 chains; the amylopectin component is more glycogen-like in structure but with fewer α1→6 branches, about one per 12 α1→4-linked glucose residues. The gross structure of glycogen is dendritic in nature, expanding from a core sequence bound to a tyrosine residue in the protein glycogenin and developing into a final structure resembling a head of cauliflower. The enzymes of glycogen metabolism are bound to the surface of the glycogen particle; many terminal glucose molecules on the surface of the molecule provide ready access for rapid release of glucose from the glycogen polymer.
Pathway of glycogenesis from blood glucose in liver
Glycogenesis is activated in liver and muscle following a meal
The liver is rich in the high-capacity, low-affinity (Km >10 mmol/L) glucose transporter GLUT-2, making it freely permeable to glucose delivered at high concentration in portal blood during and following a meal (see Table 8.2). The liver is also rich in glucokinase, an enzyme that is specific for glucose and converts it into glucose 6-phosphate (Glc-6-P). Glucokinase (GK) is inducible by continued consumption of a high-carbohydrate diet. It has a high Km, about 5–7 mmol/L, so that it is poised to increase in activity as portal glucose increases above the normal 5 mmol/L (100 mg/dL) blood glucose concentration. Unlike hexokinase, GK is not inhibited by Glc-6-P, so that the concentration of Glc-6-P increases rapidly in liver following a carbohydrate-rich meal, forcing glucose into all the major pathways of glucose metabolism: glycolysis, the pentose phosphate pathway, and glycogenesis (see Fig. 12.2). Glucose is channeled into glycogen, providing a carbohydrate reserve for maintenance of blood glucose during the postabsorptive state. Excess Glc-6-P in liver, beyond that needed to replenish glycogen reserves, is then funneled into glycolysis, in part for energy production but primarily for conversion into fatty acids and triglycerides, which are exported for storage in adipose tissue. Glucose that passes through the liver causes an increase in peripheral blood glucose concentration following carbohydrate-rich meals. This glucose is used in muscle for synthesis and storage of glycogen and in adipose tissue as a source of glycerol for triglyceride biosynthesis.
The pathway of glycogenesis from glucose (Fig. 13.3A) involves four steps:
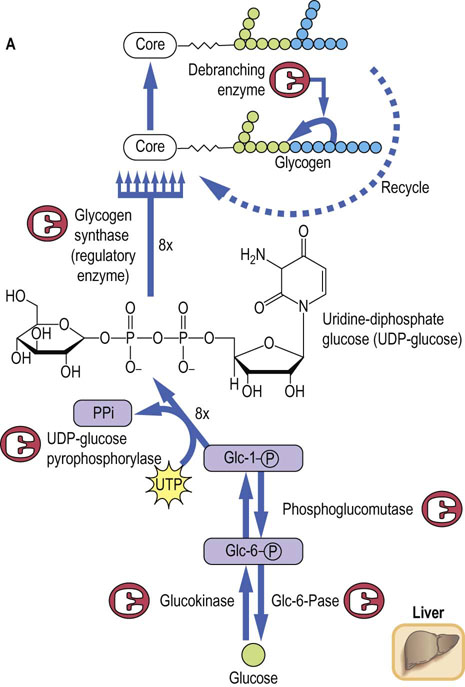
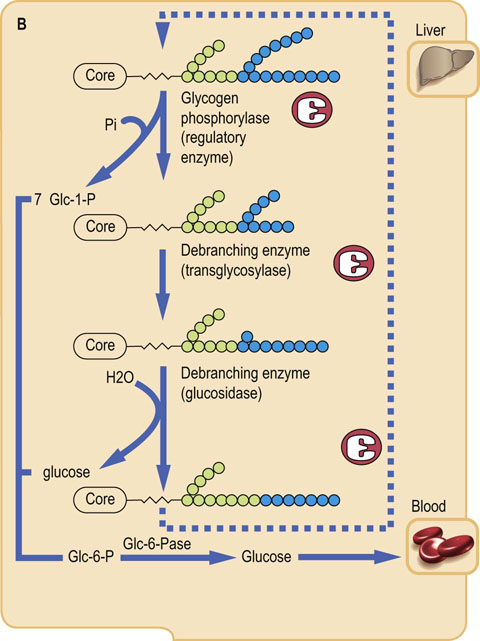
Conversion of Glc-6-P into glucose-1-phosphate (Glc-1-P) by phosphoglucomutase.
Activation of Glc-1-P to the sugar nucleotide uridine diphosphate (UDP)-glucose by the enzyme UDP-glucose pyrophosphorylase.
Transfer of glucose from UDP-Glc to glycogen in α1→4 linkage by glycogen synthase, a member of the class of enzymes known as glycosyl transferases.
When the α1→4 chain exceeds eight residues in length, glycogen branching enzyme, a transglycosylase, transfers some of the α1→4-linked sugars to an α1→6 branch, setting the stage for continued elongation of both α1→4 chains until they, in turn, become long enough for transfer by branching enzyme.
Glycogen synthase is the regulatory enzyme for glycogenesis, rather than UDP-glucose pyrophosphorylase, because UDP-glucose is also used for synthesis of other sugars, and as a glycosyl donor for synthesis of glycoproteins, glycolipids and proteoglycans (Chapters 27–29). Pyrophosphate (PPi), the other product of the pyrophosphorylase reaction, is a high energy phosphate anhydride. It is rapidly hydrolyzed to inorganic phosphate by pyrophosphatase, providing the thermodynamic driving force for biosynthesis of glycogen.
Pathway of glycogenolysis in liver
Hepatic glycogen phosphorylase provides for rapid release of glucose into blood during the postabsorptive state
As with most metabolic pathways, separate enzymes, sometimes in separate subcellular compartments, are required for the forward and reverse pathways. The pathway of glycogenolysis (Fig. 13.3B) begins with removal of the abundant, external α1→4-linked glucose residues in glycogen. This is accomplished not by a hydrolase but by glycogen phosphorylase, an enzyme that uses cytosolic phosphate and releases glucose from glycogen in the form of Glc-1-P. The Glc-1-P is isomerized by phosphoglucomutase to Glc-6-P, placing it at the top of the glycolytic pathway; the phosphorylase reaction, in effect, bypasses the requirement for ATP in the hexokinase or glucokinase reactions. In liver, the glucose is released from Glc-6-P by glucose-6-phosphatase (Glc-6-Pase), and the glucose exits via the GLUT-2 transporter into blood. The rate-limiting, regulatory step in glycogenolysis is catalyzed by phosphorylase, the first enzyme in the pathway.
Phosphorylase is specific for α1→4 glycosidic linkages; it cannot cleave α1→6 linkages. Further, this large enzyme cannot approach the branching glucose residues efficiently. Thus, as shown in Figure 13.3B, phosphorylase cleaves the external glucose residues until the branches are three or four residues long, then debranching enzyme, which has both transglycosylase and glucosidase activity, moves a short segment of glucose residues bound to the α1→6 branch to the end of an adjacent α1→4 chain, leaving a single glucose residue at the branch point. This glucose is then removed by the exo-1,6-glucosidase activity of debranching enzyme, allowing glycogen phosphorylase to proceed with degradation of the extended α1→4 chain until another branch point is approached, setting the stage for a repeat of the transglycosylase and glucosidase reactions. About 90% of the glucose is released from glycogen as Glc-1-P, and the remainder, derived from the α1→6 branching residues, as free glucose.
 Clinical box Von gierke's disease: glycogen storage disease caused by glucose-6-phosphatase deficiency
Clinical box Von gierke's disease: glycogen storage disease caused by glucose-6-phosphatase deficiency
A baby girl was chronically cranky, irritable, sweaty, and lethargic, and demanded food frequently. Physical evaluation indicated an extended abdomen, resulting from an enlarged liver. Blood glucose, measured 1 h after feeding, was 3.5 mmol/L (70 mg/dL); normal value <5 mmol/L (100 mg/dL). After 4 h, when the child was exhibiting irritability and sweating, her heart rate was increased (pulse = 110), and blood glucose had declined to 2 mmol/L (40 mg/dL). These symptoms were corrected by feeding. A liver biopsy showed massive deposition of glycogen particles in the liver cytosol.
This child cannot mobilize glycogen. Because of the severity of hypoglycemia, the most likely mutation is in hepatic Glc-6-Pase, which is required for glucose production by both glycogenolysis and gluconeogenesis. Treatment involves frequent feeding with slowly digested carbohydrate, e.g. uncooked starch, and nasogastric drip-feeding during the night.
Hormonal regulation of hepatic glycogenolysis
Three hormones (insulin, glucagon and cortisol) counter-regulate glycogenolysis and glycogenesis
Glycogenolysis is activated in liver in response to a demand for blood glucose, either because of its utilization during the postabsorptive state or in preparation for increased glucose utilization in response to stress. There are three major hormonal activators of glycogenolysis: glucagon, epinephrine (adrenaline), and cortisol (Table 13.2).
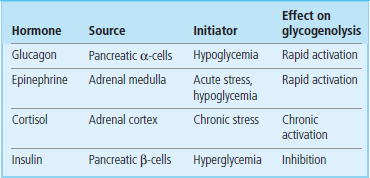
Glucagon is a peptide hormone (3500 Da), secreted from the α-cells of the endocrine pancreas. Its primary function is to activate hepatic glycogenolysis for maintenance of normal blood glucose concentration (normoglycemia). Glucagon has a short half-life in plasma, about 5 min, as a result of receptor binding, renal filtration, and proteolytic inactivation in liver. Glucagon concentration in plasma therefore changes rapidly in response to the need for blood glucose. Blood glucagon increases between meals, decreases during a meal, and is chronically increased during fasting or on a low-carbohydrate diet (Chapter 21).
Glycogenolysis is also activated in response to both acute and chronic stress. The stress may be:
physiologic, e.g. in response to increased blood glucose utilization during exercise;
pathologic, e.g. as a result of blood loss (shock);
psychological, e.g. in response to acute or chronic threats.
Acute stress, regardless of its source, causes an activation of glycogenolysis through the action of the catecholamine hormone epinephrine, released from the adrenal medulla. During prolonged exercise, both glucagon and epinephrine contribute to the stimulation of glycogenolysis and maintenance of blood glucose concentration.
Increased blood concentrations of the adrenocortical steroid hormone cortisol also induce glycogenolysis. Levels of the glucocorticoid cortisol vary diurnally in plasma, but may be chronically elevated under continuously stressful conditions, including psychological and environmental (e.g. cold) stress.
Glucagon serves as a general model for the mechanism of action of hormones that act by way of cell surface receptors. Cortisol, which acts at the level of gene expression, will be discussed later in Chapters 35 and 39
Clinical box
McArdle's diseasea glycogen storage disease that reduces capacity for exercise
A 30-year-old man consulted his physician because of chronic arm and leg muscle pains and cramps during exercise. He indicated that he had always had some muscle weakness and, for this reason, was never active in scholastic sports, but the problem did not become severe until he recently enrolled in an exercise program to improve his health. He also noted that the pain generally disappeared after about 15–30 min, and then he could continue his exercise without discomfort. His blood glucose concentration was normal during exercise, but serum creatine kinase (MM isoform from skeletal muscle) was elevated, suggesting muscle damage. Blood glucose declined slightly during 15 min of exercise, but unexpectedly blood lactate also declined, rather than increased, even when he was experien cing muscle cramps. A biopsy indicated an unusually high level of glycogen in muscle, suggesting a glycogen storage disease.
This patient suffers from McArdle's disease, a rare deficiency of muscle phosphorylase activity. The actual enzyme deficiency must be confirmed by enzyme assay, since a number of other mutations could also affect muscle glycogen metabolism. During the early periods of intense exercise, the muscle obtains most of its energy by metabolism of glucose, derived from glycogen. During cramps, which normally occur during oxygen debt, most of the pyruvate produced by glycolysis is excreted into blood as lactate, leading to an increase in blood lactate concentration. In this case, however, the patient had cramps but did not excrete lactate, suggesting a failure to mobilize muscle glycogen to produce glucose. His recovery after 15–30 min results from epinephrine-mediated activation of hepatic glycogenolysis, which provides glucose to blood and relieves the deficit in muscle glycogenolysis. Treatment of McArdle's disease usually involves exercise avoidance or carbohydrate consumption prior to exercise. Otherwise, the course of the disease is uneventful.
Mechanism of action of glucagon
Glucagon activates glycogenolysis during the post-absorptive state
Glucagon binds to an hepatic plasma membrane receptor and initiates a cascade of reactions that lead to mobilization of hepatic glycogen (Fig. 13.4) during the postabsorptive state. On the inside of the plasma membrane there is a class of signal transduction proteins, known as G-proteins, that bind guanosine triphosphate (GTP) and guanosine diphosphate (GDP), nucleotide analogues of ATP and ADP. GDP is bound in the resting state. Binding of glucagon to the plasma membrane receptor stimulates exchange of GDP for GTP on the G-protein, and the G-protein then undergoes a conformational change that leads to dissociation of its α-subunit, which then binds to and activates the plasma membrane enzyme adenylate cyclase. This enzyme converts cytoplasmic ATP into cyclic-3′,5′-AMP (cAMP), a soluble mediator that is described as the ‘second messenger’ for action of glucagon (and other hormones). Cyclic AMP binds to the cytoplasmic enzyme protein kinase A (PKA), causing dissociation of inhibitory (regulatory) subunits from the catalytic subunits of the heterodimeric enzyme, relieving inhibition of PKA (see Chapters 21 and 40), which then phosphorylates serine and threonine residues on target proteins and enzymes.
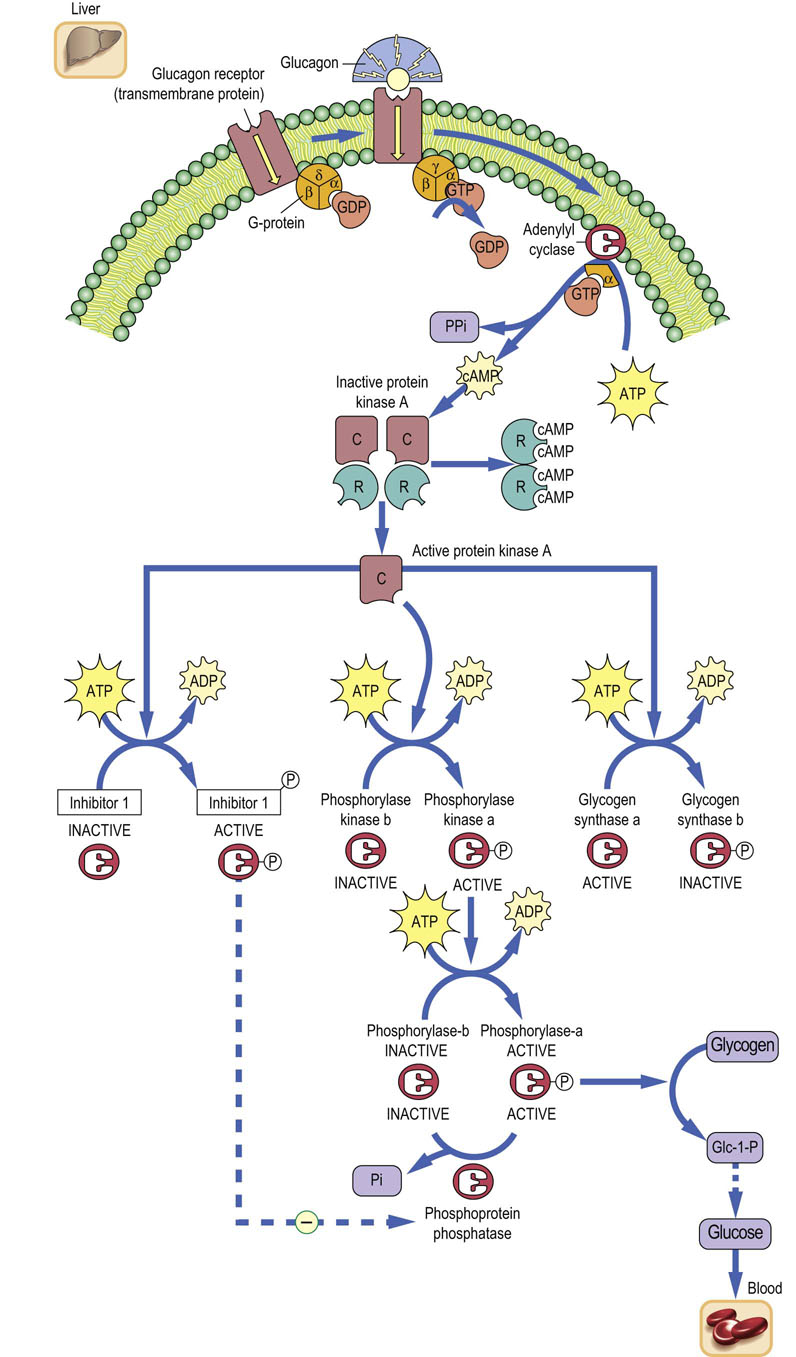
Fig. 13.4 Cascade amplification system.
Mobilization of hepatic glycogen by glucagon. A cascade of reactions amplifies the hepatic response to glucagon binding to its plasma membrane receptor. cAMP is known as the second messenger of glucagon action. PKA indirectly activates phosphorylase via phosphorylase kinase and directly inactivates glycogen synthase. C, catalytic subunits; R, regulatory (inhibitory) subunits; PKA, protein kinase A.
 Advanced concepts box G-proteins
Advanced concepts box G-proteins
G-proteins are plasma-membrane, guanosine-nucleotide-binding proteins that are involved in signal transduction for a wide variety of hormones (Fig. 13.4; see also Chapter 39). In some cases they stimulate (Gs) and in other cases they inhibit (Gi) protein kinases and protein phosphorylation. G-proteins are closely associated with hormone receptors in plasma membranes and consist of α, β, and γ subunits. The Gα-subunit binds GDP in the resting state. Following hormone binding (ligation), the receptor recruits G-proteins, stimulating exchange of GDP for GTP on the Gα-subunit. GTP binding leads to release of the β- and γ-subunits, and the α-subunit is then free to bind to and activate adenylate cyclase. The hormonal response is amplified following receptor binding, because a single receptor can activate many α-subunits. Hormonal responses are also turned off at the level of receptors and G-proteins by two mechanisms:
The Gα-subunit has a sluggish guanosine triphosphate phosphatase (GTPase) activity that hydrolyzes GTP, with a half-time measured in minutes, so that it gradually dissociates from, and thereby ceases to activate, adenylate cyclase.
Phosphorylation of the hormone receptor by protein kinase A decreases its affinity for the hormone, a process described as desensitization or hormone resistance.
Advanced concepts box Protein kinase a is very sensitive to small changes in camp concentration
As illustrated in Figure 13.4, cAMP-dependent PKA is a tetrameric enzyme with two different types of subunits (R2C2); the catalytic C-subunit has protein kinase activity, and the regulatory R-subunit inhibits the protein kinase activity. The R-subunit has a sequence of amino acids that would normally be recognized and phosphorylated by the C-subunit, except that this sequence in R contains an alanine, rather than a serine or threonine, residue. Binding of two molecules of cAMP to each R-subunit results in conformational changes that lead to dissociation of a (cAMP2-R2) dimer from the C-subunits. The monomeric, active C-subunits then proceed to phosphorylate serine and threonine residues in target enzymes. PKA is not a typical allosteric enzyme, in that the binding of the allosteric effector (cAMP) causes subunit dissociation; however, the complete activation of PKA involves cooperative binding of four molecules of cAMP to two R-subunits. PKA is fully activated at submicromolar concentrations of cAMP, so that it is exquisitely sensitive to small changes in adenylate cyclase activity in response to glucagon.
The pathway for activation of glycogen phosphorylase (see Fig. 13.4) involves phosphorylation of many molecules of phosphorylase kinase by PKA, which then phosphorylates and activates many molecules of glycogen phosphorylase. The net effect of these sequential steps, beginning with activation of many molecules of adenylate cyclase by G-proteins, is a ‘cascade amplification’ system, not unlike that of a series of amplifiers in a radio or stereo set, resulting in a massive increase in signal strength within seconds after glucagon binding to the hepatocyte plasma membrane. Phosphorylation of phosphorylase activates glycogenolysis, leading to production of Glc-6-P in liver, which is then hydrolyzed to glucose and exported into blood. Another target of PKA is inhibitor-1, a phosphoprotein phosphatase inhibitor protein, which is activated by phosphorylation. Phosphorylated inhibitor-1 inhibits cytoplasmic phosphoprotein phosphatases, which would otherwise reverse the phosphorylation of enzymes and quench the response to glucagon (see Fig. 13.4).
Glyocogenolysis and glycogenesis are counter-regulated by protein kinase A, which activates phosphorylase and inhibits glycogen synthase
Glycogenolysis and glycogenesis are opposing pathways. Theoretically, Glc-1-P produced by phosphorylase could be rapidly activated to UDP-glucose and reincorporated into glycogen. To prevent this wasteful or futile cycle, PKA also acts directly on glycogen synthase, in this case inactivating the enzyme. Thus, the activation of phosphorylase (glycogenolysis) is coordinated with inactivation of glycogen synthase (glycogenesis). Other hepatic biosynthetic pathways, including protein, cholesterol, fatty acid, and triglyceride synthesis, as well as glycolysis, are also regulated by phosphorylation of key regulatory enzymes, generally limiting biosynthetic reactions and focusing liver metabolism in response to glucagon on the provision of glucose to blood for maintenance of vital body functions (see Chapter 21).
Perhaps in order to balance the cascade of events amplifying the response to glucagon, there are multiple, redundant mechanisms to insure rapid termination of the hormonal response (Table 13.3). In addition to the slow GTPase activity of the Gα-subunit, there is also a phosphodiesterase in the cell that hydrolyzes cAMP to AMP, permitting reassociation of the inhibitory and catalytic subunits of PKA, decreasing its protein kinase activity. There are also phosphoprotein phosphatases that remove the phosphate groups from the active, phosphorylated forms of phosphorylase kinase and phosphorylase. The decrease in cAMP concentration and PKA activity also leads to decreased phosphorylation of inhibitor-1, permitting increased activity of phosphoprotein phosphatases. Thus, an array of mechanisms act in concert to insure that hepatic glycogenolysis declines rapidly in response to increasing blood glucose and decreasing blood glucagon concentrations following a meal.

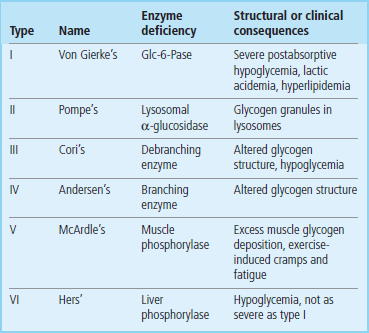
There are a number of autosomal recessive genetic diseases affecting glycogen metabolism (Table 13.4). These diseases, known as glycogen storage diseases, are characterized by accumulation of glycogen granules in tissues, which eventually compromises tissue function. Predictably, glycogen storage diseases affecting hepatic glycogen metabolism are characterized by fasting hypoglycemia and may be life-threatening, while defects in muscle glycogen metabolism are characterized by rapid muscle fatigue during exercise.
Mobilization of hepatic glycogen by epinephrine
Epinephrine activates glycogenolysis during stress, increasing blood glucose concentration
Epinephrine works through several distinct receptors on different cells. The best studied of these receptors are the α- and β-adrenergic receptors; they recognize different features of the epinephrine molecule, bind epinephrine with different affinities, work by different mechanisms, and are inhibited by different classes of drugs. During severe hypoglycemia, glucagon and epinephrine work together to magnify the glycogenolytic response in liver. However, even when blood glucose is normal, epinephrine is released in response to real or perceived threats, causing an increase in blood glucose to support a ‘fight or flight’ response. Caffeine in coffee and theophylline in tea are inhibitors of phosphodiesterase and also cause an increase in hepatic cAMP and blood glucose. Like epinephrine, caffeine, administered in the form of a few strong cups of coffee, can also make us alert and responsive – and aggressive.
Epinephrine action on hepatic glycogenolysis proceeds by two pathways. One of these, through the epinephrine β-adrenergic receptor, is similar to that for glucagon, involving a plasma membrane epinephrine-specific receptor, G-proteins, and cAMP. The epinephrine response augments the effects of glucagon during severe hypoglycemia (metabolic stress) and also explains, in part, the rapid heartbeat, sweating, tremors, and anxiety associated with hypoglycemia. Epinephrine also works simultaneously through an α-adrenergic receptor, but by a different mechanism. Binding to α-receptors also involves G-proteins, common elements in hormone signal transduction, but in this case, the G-protein is specific for activation of a membrane isozyme of phospholipase C (PLC), which is specific for cleavage of a membrane phospholipid, phosphatidylinositol bisphosphate (PIP2) (Fig. 13.5). Both products of PLC action, diacylglycerol (DAG) and inositol trisphosphate (IP3), act as second messengers of epinephrine action. DAG activates protein kinase C (PKC) which, like PKA, initiates phosphorylation of serine and threonine residues on target proteins. IP3 promotes the transport of Ca2+ into the cytosol. Ca2+ then binds to the cytoplasmic protein calmodulin, which binds to and activates phosphorylase kinase, leading to cAMP-independent phosphorylation and activation of phosphorylase. A Ca2+–calmodulin-dependent protein kinase and other enzymes are also activated, either by phosphorylation or by association with the Ca2+–calmodulin complex (Fig. 13.5). Thus, a range of metabolic pathways is activated in response to stress, especially those involved in the mobilization of energy reserves.
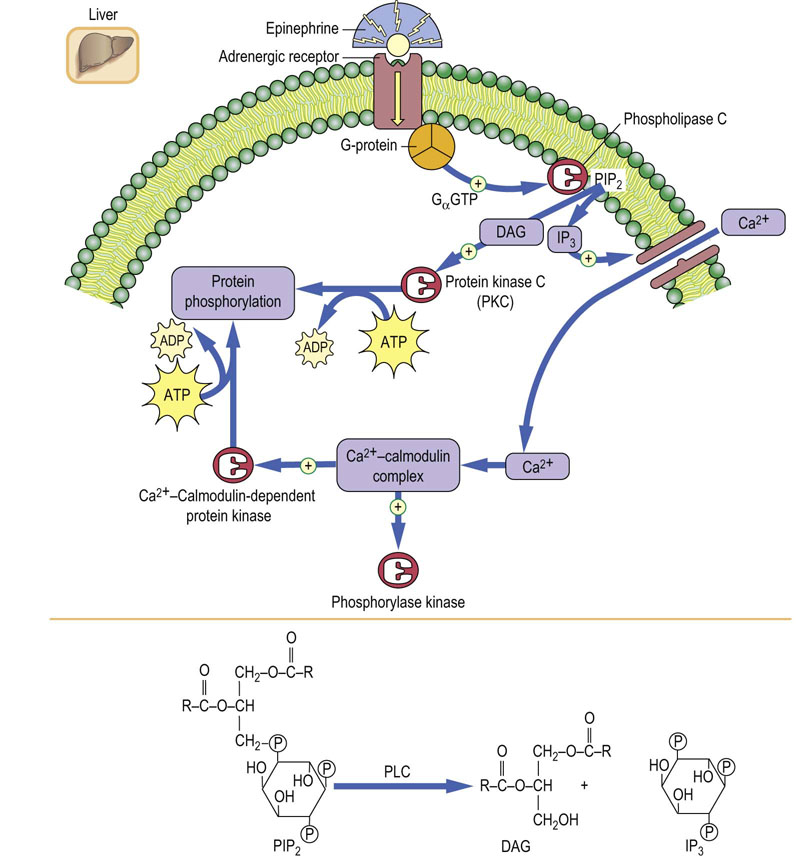
Fig. 13.5 Mechanism of activation of glycogenolysis in liver via the α-adrenergic receptor.
Diacylglycerol (DAG) and inositol trisphosphate (IP3) are second messengers mediating the α-adrenergic response. PIP2, phosphatidylinositol bisphosphate; PKC, protein kinase C. See also Chapter 40.
Glycogenolysis in muscle
Muscle lacks a glucagon receptor and glucose-6-phosphatase, and retains glucose for energy metabolism, even during hypoglycemia
The tissue localization of hormone receptors provides tissue specificity to hormone action. Thus, only those tissues with glucagon receptors respond to glucagon. Muscle may be rich in glycogen, even during hypoglycemia, but it lacks both the glucagon receptor and Glc-6-Pase. Therefore muscle glycogen cannot be mobilized to replenish blood glucose. Muscle glycogenolysis is activated in response to epinephrine through the cAMP-dependent β-adrenergic receptor, but the glucose is metabolized through glycolysis for energy production. This occurs not only during ‘fight or flight’ situations but also in response to metabolic demands during prolonged exercise. There are also two important hormone-independent mechanisms for activation of glycogenolysis in muscle (Fig. 13.6). First, the influx of Ca2+ into the muscle cytoplasm in response to nerve stimulation activates the basal, unphosphorylated form of phosphorylase kinase by action of the Ca2+–calmodulin complex. This hormone-independent activation of phosphorylase provides for rapid activation of glycogenolysis during short bursts of exercise, even in the absence of epinephrine action. A second mechanism for activation of muscle glycogenolysis involves direct allosteric activation of phosphorylase by AMP. Increased usage of ATP during a rapid burst of muscle activity leads to rapid accumulation of ADP, which is converted in part into AMP by action of the enzyme myokinase (adenylate kinase), which catalyzes the reaction:
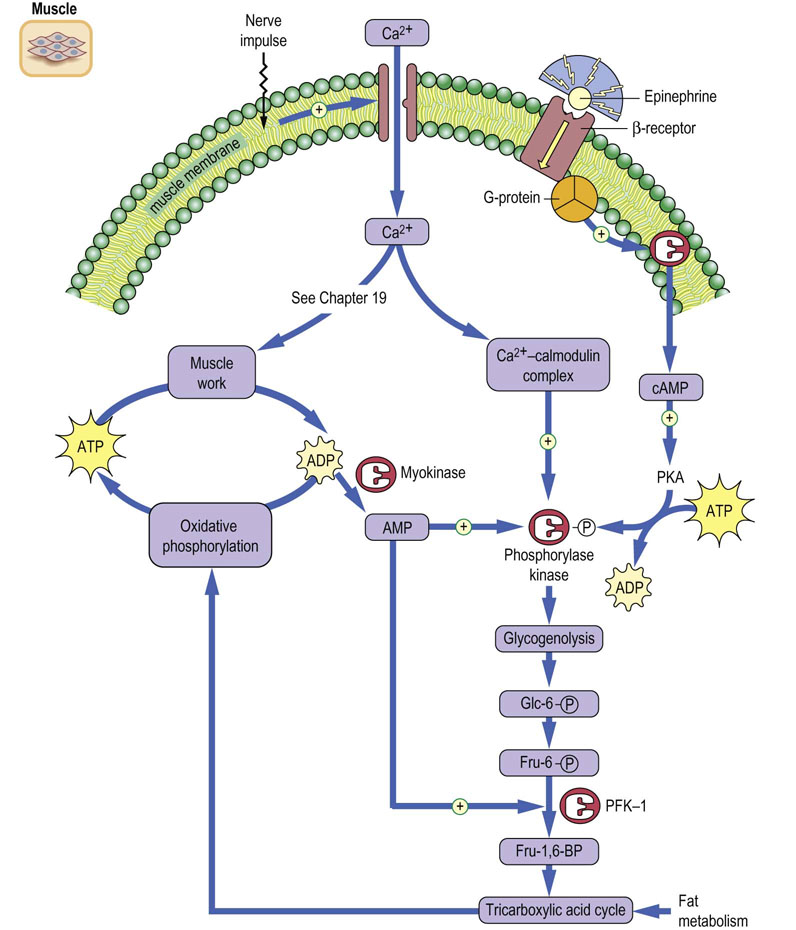
Fig. 13.6 Regulation of protein kinase A (PKA) in muscle.
Activation of glycogenolysis and glycolysis in muscle during exercise. PFK-1, phosphofructokinase-1. Compare Figure 8.4.
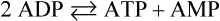
Advanced concepts box Maximal inhibition of glycogen synthase is achieved only through sequential action of several kinases
When both glucagon and epinephrine are acting on liver, the activation of glycogenolysis and inhibition of glycogenesis is mediated by at least three kinases: protein kinase A (PKA), protein kinase C (PKC), and Ca2+–calmodulin activated protein kinase. All three of these protein kinases phosphorylate key serine and threonine residues in regulatory enzymes. These and other protein kinases work in concert with one another in a process known as sequential or hierarchical phosphorylation, leading to phosphorylation of up to nine amino acid residues on glycogen synthase. Maximal inhibition of glycogen synthase is achieved only through the sequential activity of several kinases. In some cases, certain serine or threonine residues must be phosphorylated in a specific sequence by cooperative action of different kinases, i.e. phosphorylation of one site by one enzyme requires prior phosphorylation of another site by a separate enzyme.
AMP activates both the basal and phosphorylated forms of phosphorylase, enhancing glycogenolysis in either the absence or presence of hormonal stimulation. AMP also relieves inhibition of phosphofructokinase-1 (PFK-1) by ATP (see Chapter 12), stimulating the utilization of glucose through glycolysis for energy production. The stimulatory effects of Ca2+ and AMP insure that the muscle can respond to its energy needs, even in the absence of hormonal input.
Regulation of glycogenesis
Insulin opposes the action of glucagon and stimulates gluconeogenesis
Glycogenesis, and energy storage in general, occurs during and immediately following meals. Glucose and other carbohydrates, rushing into the liver from the intestines via the portal circulation, are efficiently trapped to make glycogen. Excess glucose proceeds to the peripheral circulation, where it is taken up into muscle and adipose tissue for energy reserves or storage. We normally eat sitting down, rather than during exercise, so that the opposing pathways of uptake and storage versus mobilization and utilization of energy supplies are temporally compartmentalized functions in our lives.
Energy storage is under the control of the polypeptide hormone insulin, which is stored in β-cells in the pancreatic islets of Langerhans (Chapter 21). Insulin is secreted into blood following a meal, tracking blood glucose concentration. It has two primary functions in carbohydrate metabolism: first, insulin reverses the actions of glucagon in phosphorylation of proteins, turning off glycogen phosphorylase and activating glycogen synthase, promoting glucose storage; second, it stimulates the uptake of glucose into peripheral tissues (muscle and adipose tissue), facilitating synthesis and storage of glycogen and triglycerides. Insulin also acts at the level of gene expression, stimulating the synthesis of enzymes involved in carbohydrate metabolism and storage and conversion of glucose into triglycerides. It also acts as a growth hormone, stimulating protein turnover, both synthesis and degradation.
Protein tyrosine phosphorylation, rather than serine and threonine phosphorylation, is a characteristic feature of insulin and growth factor activity. Insulin binding to its transmembrane receptor (Fig. 13.7) stimulates aggregation of receptors and promotes tyrosine kinase activity in the intracellular domain of the receptor. The insulin receptor autophosphorylates its tyrosine residues, enhancing its protein tyrosine kinase activity, and phosphorylates tyrosine residues in other intracellular effector proteins, which then activate secondary pathways. Among these are kinases that phosphorylate serine and threonine residues on proteins, but at sites and on proteins distinct from those phosphorylated by PKA and PKC. Insulin-dependent activation of GTPase, phosphodiesterase and phosphoprotein phosphatases also checks the action of glucagon, which is typically present at high concentration in the blood at mealtimes, i.e. several hours since the last meal.
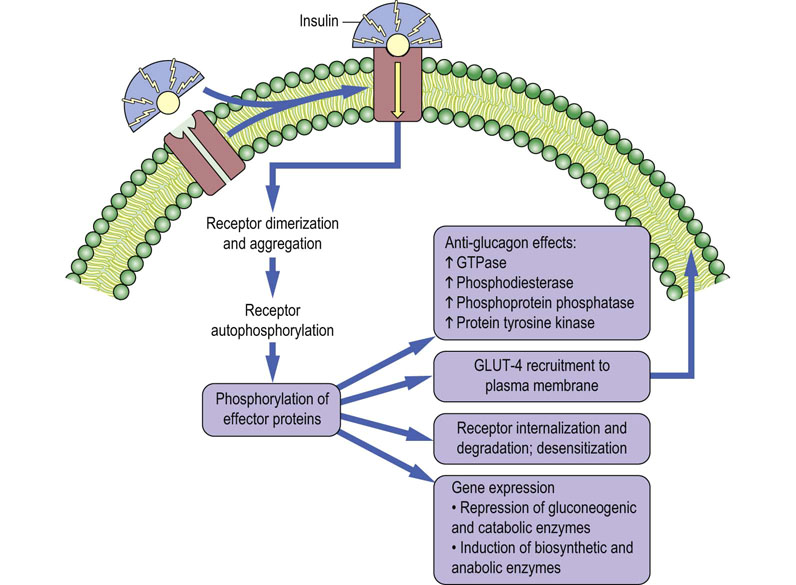
Fig. 13.7 Mechanisms of insulin action.
Regulatory effects of insulin on hepatic and muscle carbohydrate metabolism. (See also Chapter 21.)
The liver also appears to be directly responsive to ambient blood glucose concentration, since hepatic glycogen synthesis increases following a meal, even in the absence of hormonal input. Thus, the increase in hepatic glycogenesis begins more rapidly than the increase in insulin concentration in blood. Perfusion of liver with glucose solutions in vitro, in the absence of insulin, also leads to inhibition of glycogenolysis and activation of glycogenesis. This appears to occur by direct allosteric inhibition of phosphorylase by glucose and secondary stimulation of protein phosphatase activity.
Most, if not all, cells in the body are responsive to insulin in some way, but the major sites of insulin action, on a mass basis, are muscle and adipose tissue. These tissues normally have low levels of cell surface glucose transporters, restricting the entry of glucose – they rely mostly on lipids for energy metabolism. In muscle and adipose tissue, insulin receptor tyrosine kinase activity induces movement of glucose transporter-4 (GLUT-4; see Table 8.2) from intracellular vacuoles to the cell surface, increasing glucose transport into the cell. The glucose is then used in muscle for synthesis of glycogen, and in adipose tissue to produce glyceraldehyde-3-phosphate which is converted to glycerol-3-phosphate for synthesis of triglycerides (Chapter 16). The insulin-stimulated, GLUT-4-mediated uptake of glucose into muscle and adipose tissue is the primary mechanism limiting the increase in blood glucose following a meal.
Clinical box large child born of a diabetic mother
A baby boy, born of a poorly controlled, chronically hyperglycemic, diabetic mother, was large and chubby (macrosomic) at birth (5 kg) but appeared otherwise normal. He declined rapidly, however, and within 1 h showed all the symptoms of hypoglycemia, similar to the case of the baby girl born of a malnourished mother. The difference, in this case, was that the boy was obviously on the heavy side, rather than thin and malnourished.
This child has experienced a chronically hyperglycemic environment during uterine development. He adapted by increasing endogenous insulin production, which has a growth hormone-like activity, resulting in macrosomia. At birth, when placental delivery of glucose ceases, he has a normal blood glucose concentration and a substantial supply of hepatic glycogen. However, chronic hyperinsulinemia prior to birth probably represses gluconeogenic enzymes, and his high blood insulin concentration at birth promotes glucose uptake into muscle and adipose tissue. In the absence of a maternal source of glucose, insulin-induced hypoglycemia leads to a stress response, which was corrected by glucose infusion. After 1–2 days, his ample body mass will provide a good reservoir for synthesis of blood glucose from muscle protein.
Gluconeogenesis
Gluconeogenesis is required to maintain blood glucose during fasting and starvation
Unlike glycogenolysis, which can be turned on rapidly in response to hormonal stimulation, gluconeogenesis increases more slowly, depending on changes in gene expression, and reaches maximal activity over a period of hours (see Fig. 13.1); it becomes the primary source of our blood glucose concentration about 8 hours into the postabsorptive state (Chapter 21). Gluconeogenesis requires both a source of energy for biosynthesis and a source of carbons for formation of the backbone of the glucose molecule. The energy is provided by metabolism of fatty acids released from adipose tissue. The carbon skeletons are provided from three primary sources:
Lactate produced in tissues such as the red cell and muscle.
Amino acids derived from muscle protein.
Glycerol released from triglycerides during lipolysis in adipose tissue.
Among these, muscle protein is the major precursor of blood glucose during fasting and starvation – the rate of gluconeogenesis is often limited by the availability of substrate, including the rate of proteolysis in muscle or, in some cases, muscle mass. During prolonged fasting, malnutrition or starvation, we lose both adipose and muscle mass. The fat is used both for the general energy needs of the body and to support gluconeogenesis, while most of the amino acids in protein are converted into glucose; urinary nitrogen (urea) excretion is also increased.
Gluconeogenesis from lactate
Gluconeogenesis uses lactate, amino acids and glycerol as substrates for synthesis of glucose; fatty acids provide the energy
Gluconeogenesis from lactate is conceptually the opposite of anaerobic glycolysis but proceeds by a slightly different pathway, involving both mitochondrial and cytosolic enzymes (Fig. 13.8). During hepatic gluconeogenesis lactate is converted back into glucose, using, in part, the same glycolytic enzymes involved in conversion of glucose into lactate. The lactate cycle involving the liver, red cells, and muscle, known as the Cori cycle, is discussed in detail in Chapter 21. At this point, we focus on the metabolic pathway for conversion of lactate to glucose.
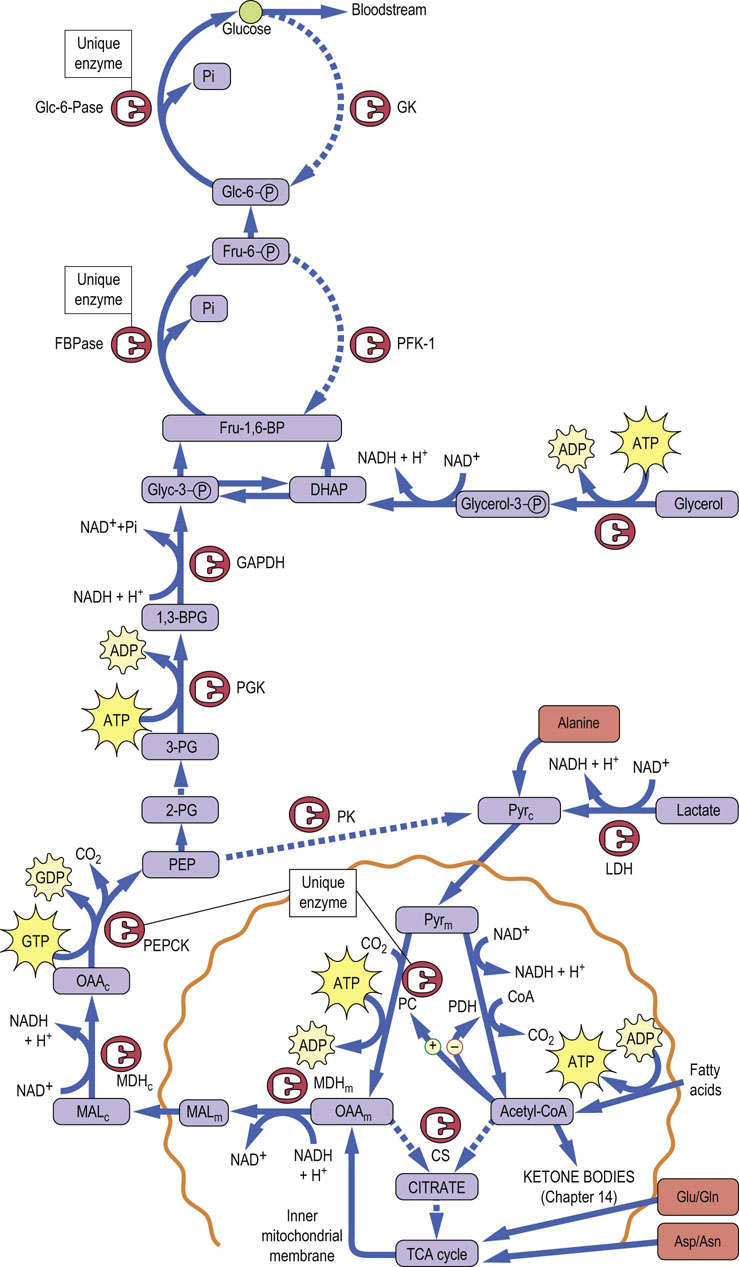
Fig. 13.8 Pathway of gluconeogenesis.
Gluconeogenesis is the reverse of glycolysis. Unique enzymes overcome the irreversible kinase reactions of glycolysis. Compartments: c, cytoplasmic; imm, inner mitochondrial membrane; m, mitochondrial. Enzymes: CS, citrate synthase; Fru-1,6-BPase, fructose-1,6-bisphosphatase; GAPDH, glyceraldehyde-3-P dehydrogenase; Glc-6-Pase, glucose-6-phosphatase; GK, glucokinase; MDH, malate dehydrogenase; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PGK, phosphoglycerate kinase. Substrates: 2,3-BPG, bisphosphoglycerate; DHAP, dihydroxyacetone phosphate; Fru-1,6-BP, fructose-1,6-bisphosphate; Glyc-3-P, glyceraldehyde 3-phosphate; MAL, malate; OAA, oxaloacetate; PEP, phosphoenolpyruvate; PEPCK, PEP carboxykinase; Pyr, pyruvate; 3-PG, 3-phosphoglycerate. Solid lines: active during gluconeogenesis. Dotted lines: inactive during gluconeogenesis.
A critical problem in the reversal of glycolysis is overcoming the irreversibility of three kinase reactions: glucokinase (GK), phosphofructokinase-1 (PFK-1), and pyruvate kinase (PK). The fourth kinase in glycolysis, phosphoglycerate kinase (PGK), catalyzes a freely reversible, equilibrium reaction: a substrate-level phosphorylation reaction, transferring a high-energy acyl phosphate in 1,3-bisphosphoglycerate to an energetically similar pyrophosphate bond in ATP. To circumvent the three irreversible reactions in glycolysis, the liver uses four unique enzymes: pyruvate carboxylase (PC) in the mitochondrion and phosphoenolpyruvate carboxykinase (PEPCK) in the cytoplasm to bypass PK, fructose-1,6-bisphosphatase (Fru-1,6-BPase) to bypass PFK-1, and Glc-6-Pase to bypass GK (see Fig. 13.8). Gluconeogenesis from lactate involves, first, its conversion into phosphoenolpyruvate (PEP), a process requiring investment of two ATP equivalents because of the high energy of the enol–phosphate bond in PEP. Lactate is first converted into pyruvate by lactate dehydrogenase (LDH), and then enters the mitochondrion, where it is converted to oxaloacetate by PC, using biotin and ATP. Oxaloacetate is reduced to malate by the TCA cycle enzyme, malate dehydrogenase, exits the mitochondrion, and is then reoxidized to oxaloacetate by cytosolic malate dehydrogenase. The cytosolic oxaloacetate is then decarboxylated by PEPCK, using GTP as a co-substrate, yielding PEP. The energy for synthesis of PEP from oxaloacetate is derived from both the hydrolysis of GTP and the decarboxylation of oxaloacetate.
Glycolysis may now proceed backwards from PEP until it reaches the next irreversible reaction, PFK-1. This enzyme is bypassed by a simple hydrolysis reaction, catalyzed by Fru-1,6-BPase without production of ATP, reversing the PFK-1 reaction and producing Fru-6-P. Similarly, the bypass of GK is accomplished by hydrolysis of Glc-6-P by Glc-6-Pase, without production of ATP. The free glucose is then released into blood.
Gluconeogenesis is fairly efficient – the liver can make a kilogram of glucose per day by gluconeogenesis, and actually does so in poorly controlled, hyperglycemic diabetic patients. Normal glucose production, in the absence of dietary carbohydrate, is ~200 g/day, almost a half-pound of glucose. Gluconeogenesis from pyruvate is moderately expensive, requiring a net expenditure of the equivalent of 4 moles of ATP per mole of pyruvate converted into glucose, i.e. 2 mole ATP at the PC reaction and 2 mole of GTP at the PEPCK reaction. The ATP and GTP are provided by oxidation of fatty acids (Chapter 15).
Gluconeogenesis from amino acids and glycerol
Most amino acids are glucogenic (Chapter 19): i.e. following deamination, their carbon skeletons can be converted into glucose. Alanine and glutamine are the major amino acids exported from muscle for gluconeogenesis. Their relative concentrations in venous blood from muscle exceed their relative concentration in muscle protein, indicating considerable reshuffling of muscle amino acids to provide gluconeogenic substrates. As discussed in more detail in Chapter 19, alanine is converted directly into pyruvate by the enzyme alanine aminotransferase (alanine transaminase, ALT), and then gluconeogenesis proceeds as described for lactate. Other amino acids are converted into tricarboxylic acid cycle (TCA cycle) intermediates, then to malate for gluconeogenesis. Aspartate, for example, is converted into oxaloacetate by aspartate aminotransferase (aspartate transaminase, AST), and glutamate into α-ketoglutarate by glutamate dehydrogenase. Some glucogenic amino acids are converted by less direct routes into alanine or intermediates in the tricarboxylic acid cycle for gluconeogenesis. The amino groups of these amino acids are converted into urea, via the urea cycle in hepatocytes, and the urea is excreted in urine (Chapter 19).
Glycerol enters gluconeogenesis at the level of triose phosphates (see Fig. 13.8). Following release of glycerol and fatty acids from adipose tissue into plasma, the glycerol is taken up into liver and phosphorylated by glycerol kinase. Following action of glycerol-3-phosphate dehydrogenase (see Fig. 9.7), glycerol enters the gluconeogenic pathway as dihydroxyacetone phosphate. Only the glycerol component of fats can be converted into glucose. The incorporation of glycerol into glucose requires only 2 mole of ATP per mole of glucose produced.
Clinical box child born of malnourished mother may have hypoglycemia
A baby girl was born at 39 weeks of gestation to a young, malnourished mother. The child was also thin and weak at birth and within 1 h after birth was showing signs of distress, including rapid heartbeat and respiration. Her blood glucose was 3.5 mmol/L (63 mg/dL) at birth, and declined rapidly to 1.5 mmol/L (27 mg/dL) by 1 h, when she was becoming unresponsive and comatose. Her condition was markedly improved by infusion of a glucose solution, followed by a carbohydraterich diet. She improved gradually over the next 2 weeks before discharge from the hospital.
During development in utero, the fetus obtains glucose exogenously, from the placental circulation. However, following birth, the child relies at first on mobilization of hepatic glycogen and then on gluconeogenesis for maintenance of blood glucose. Because of the malnourished state of the mother, this child was born with negligible hepatic glycogen reserves. Thus, she was unable to maintain blood glucose homeostasis postpartum and rapidly declined into hypoglycemia, initiating a stress response. After surviving the transient hypoglycemia, she probably still lacked adequate muscle mass to provide a sufficient supply of amino acids for gluconeogenesis. Infusion of glucose, followed by a carbohydrate-rich diet, would address these deficits, but may not correct more serious damage from prolonged malnutrition during fetal development.
Glucose cannot be synthesized from fatty acids!
As discussed in Chapter 15, metabolism of fatty acids involves their conversion in two carbon oxidation steps to form acetyl-CoA, which is then metabolized in the tricarboxylic acid cycle following condensation with oxaloacetate to form citrate. While the carbons of acetate are theoretically available for gluconeogenesis by conversion to malate, during the pathway from citrate to malate, two molecules of CO2 are eliminated, at the isocitrate and α-ketoglutarate dehydrogenase reactions. Thus, although energy is produced in the tricarboxylic acid cycle, the two carbons invested for gluconeogenesis from acetyl-CoA are lost as CO2. For this reason, acetyl-CoA, and therefore even-chain fatty acids, cannot serve as substrates for net gluconeogenesis. However, odd-chain and branched-chain fatty acids, which form propionyl-CoA, can serve as minor precursors for gluconeogenesis. Propionyl-CoA is first carboxylated to methylmalonyl-CoA, which undergoes racemase and mutase reactions to form succinyl-CoA, a tricarboxylic acid cycle intermediate (see Chapter 15). Succinyl-CoA is converted into malate, which exits the mitochondrion and is oxidized to oxaloacetate. Following decarboxylation by PEPCK, the three carbons of propionate are conserved in PEP and glucose.
Regulation of gluconeogenesis
Fructose-2,6-bisphosphate allosterically counter-regulates glycolysis and gluconeogenesis
Like glycogen metabolism in liver, gluconeogenesis is regulated primarily by hormonal mechanisms. In this case, the regulatory process involves counterregulation of glycolysis and gluconeogenesis, largely by phosphorylation/dephosphorylation of enzymes, under control of glucagon and insulin. The primary control point is at the regulatory enzymes PFK-1 and Fru-1,6-BPase which, in liver, are exquisitely sensitive to the allosteric effector fructose 2,6-bisphosphate (Fru-2,6-BP). Fru-2,6-BP is an activator of PFK-1 and an inhibitor of Fru-1,6-BPase, counterregulating the two opposing pathways. As shown in Figure 13.9, Fru-2,6-BP is synthesized by an unusual, bifunctional enzyme, phosphofructokinase-2/fructose-2,6-bisphosphatase (PFK-2/Fru-2,6-BPase) which has both kinase and phosphatase activities. In the phosphorylated state, effected by glucagon through protein kinase A, this enzyme displays Fru-2,6-BPase activity, which reduces the level of Fru-2,6-BP. The decrease in Fru-2,6-BP simultaneously decreases the stimulation of glycolysis at PFK-1 and relieves inhibition of gluconeogenesis at Fru-1,6-BPase. In this way, glucagon-mediated phosphorylation of PFK-2/Fru-2,6-BP places the liver cell in a gluconeogenic mode. The coordinate, allosterically mediated increase in Fru-1,6-BPase and decrease in PFK-1 activities ensure that glucose made by gluconeogenesis is not consumed by glycolysis in a futile cycle, but released into blood by Glc-6-Pase. Similarly, any flux of glucose from glycogen through glycogenolysis, also induced by glucagon, is diverted to blood, rather than to glycolysis, by inhibition of PFK-1. PK is also inhibited by phosphorylation by protein kinase A (PKA), providing an additional site for inhibition of glycolysis (Fig 13.9).
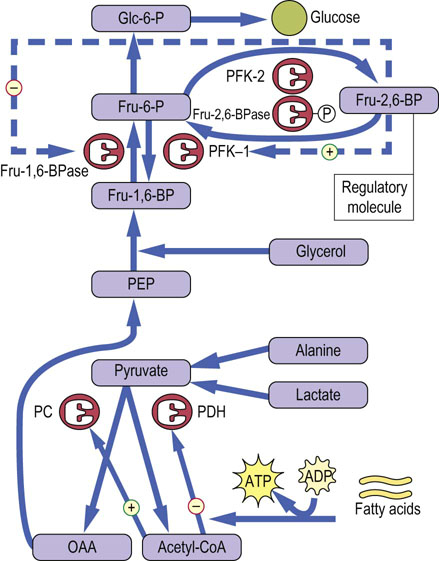
Fig. 13.9 Regulation of gluconeogenesis.
Gluconeogenesis is regulated by hepatic levels of Fru-2,6-BP and acetyl-CoA. The upper part of the diagram focuses on the reciprocal regulation of Fru-1,6-BPase and PFK-1 by Fru-2,6-BP and the lower part on the reciprocal regulation of pyruvate dehydrogenase (PDH) and pyruvate carboxylase (PC) by acetyl-CoA.
When glucose enters the liver following a meal, insulin mediates the dephosphorylation of PFK-2/Fru-2,6-BPase, turning on its PFK-2 activity. The resultant increase in Fru-2,6-BP activates PFK-1 and inhibits Fru-1,6-BPase activity. Gluconeogenesis is inhibited and glucose entering the liver is then incorporated into glycogen or routed into glycolysis for lipogenesis. Thus, liver metabolism following a meal is focused on synthesis and storage of both carbohydrate and lipid energy reserves, which are used later, in the postabsorptive state, for maintenance of blood glucose and fatty acid homeostasis.
Gluconeogenesis is also regulated in the mitochondrion by acetyl-CoA. The influx of fatty acids from adipose tissue, stimulated by glucagon to support gluconeogenesis (see Chapter 15), leads to an increase in hepatic acetyl-CoA, which is both an inhibitor of pyruvate dehydrogenase (PDH) and an essential allosteric activator of pyruvate carboxylase (PC) (see Fig. 13.8). In this way, fat metabolism inhibits the oxidation of pyruvate and favors its use for gluconeogenesis in liver. In muscle during the fasting state, glucose utilization for energy metabolism is limited both by the low level of GLUT-4 in the plasma membranes (because of the low plasma insulin concentration) and by inhibition of PDH by acetyl-CoA. Active fat metabolism and high levels of acetyl-CoA in muscle promote the excretion of a significant fraction of pyruvate as lactate, even in the resting state. The carbon skeleton of glucose is returned to the liver via the Cori cycle (Chapter 21), and recycling of pyruvate into glucose, in effect, conserves muscle protein.
Conversion of fructose and galactose to glucose
As discussed in detail in Chapter 27, fructose is metabolized almost exclusively in the liver. It enters glycolysis at the level of triose phosphates, bypassing the regulatory enzyme, PFK-1. Following consumption of fruit juices, Gatorade® or foods containing high fructose corn syrup, large amounts of pyruvate may be forced on the mitochondrion for use in energy metabolism or fat biosynthesis. During a gluconeogenic state, this fructose may also proceed toward Glc-6-P, providing a convenient source of blood glucose. Gluconeogenesis from galactose is equally efficient, since Glc-1-P, derived from galactose 1-phosphate (Chapter 27), is readily isomerized to Glc-6-P by phosphoglucomutase. Fructose and galactose are good sources of glucose, independent of glycogenolysis and gluconeogenesis.
Summary
Glycogen is stored in two tissues in the body for different reasons: in liver for short-term maintenance of blood glucose homeostasis, and in muscle as a source of energy. Glycogen metabolism in these tissues responds rapidly to both allosteric and hormonal control.
In liver, the balance between glycogenolysis and glycogenesis is regulated by the balance between concentrations of glucagon and insulin in the circulation, which controls the state of phosphorylation of enzymes. Phosphorylation of enzymes under the influence of glucagon directs glycogen mobilization and is the most common condition in the liver, e.g. during sleep and between meals.
Increases in blood insulin during and after meals promote dephosphorylation of the same enzymes, leading to glycogenesis. Insulin also promotes glucose uptake into muscle and adipose tissue for glycogen and triglyceride synthesis following a meal.
Epinephrine increases phosphorylation of liver enzymes, enabling a burst in hepatic glycogenolysis and an increase in blood glucose for stress responses.
Muscle is responsive to epinephrine, but not to glucagon; in this case the glucose produced by glycogenolysis is used for muscle energy metabolism – fight or flight. In addition, muscle glycogenolysis is responsive to intracellular Ca2+ and AMP concentrations, providing a mechanism for coupling glycogenolysis to normal energy consumption during exercise.
Gluconeogenesis takes place primarily in liver, and is designed for maintenance of blood glucose during the fasting state. It is essential after 12 h of fasting, when the majority of hepatic glycogen has been consumed.
The major substrates for gluconeogenesis are lactate, amino acids, and glycerol; fatty acid metabolism provides the energy. The major control point is at the level of phosphofructokinase-1 (PFK-1), which is activated by the allosteric effector Fru-2,6-BP.
The synthesis of Fru-2,6-BP is under control of the bifunctional enzyme, PFK-2/Fru-2,6-BPase, whose kinase and phosphatase activities are regulated by phosphorylation/dephosphorylation, under hormonal control by insulin and glucagon.
During fasting and active gluconeogenesis, glucagon mediates phosphorylation and activation of the phosphatase activity of this enzyme, leading to a decrease in the level of Fru-2,6-BP and a corresponding decrease in glycolysis; carbohydrate degradation is inhibited and fats become the primary energy source during fasting and starvation. Oxidation of pyruvate is also inhibited in the mitochondrion by inhibition of PDH by acetyl-CoA, derived from fat metabolism.
Following a meal, the decrease in phosphorylation of enzymes enhances PFK-2 activity; the increase in Fru-2,6-BP concentration activates PFK-1 and promotes glycolysis, providing pyruvate, which is converted to acetyl-CoA for lipogenesis. The actions of insulin, glucagon, and epinephrine illustrate many of the fundamental principles of hormone action (Table 13.5).
Table 13.5
General features of hormone action
Hormonal regulation of glucose metabolism illustrates fundamental principles of hormone action (see Chapter 39).
Angelini, C, Semplicini, C. Enzyme replacement therapy for Pompe disease. Curr Neurol Neurosci Rep. 2012; 12:70–75.
Hicks, J, Wartchow, E, Mierau, G. Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol. 2011; 35:183–196.
Johnson, RJ, Murray, R. Fructose, exercise, and health. Curr Sports Med Rep. 2010; 9:253–258.
Obel, LF, Müller, MS, Walls, AB, et al. Brain glycogen – new perspectives on its metabolic function and regulation at the subcellular level. Front Neuroenergetics. 2012; 4:3.
Roach, PJ, Depaoli-Roach, AA, Hurley, TD, et al. Glycogen and its metabolism: some new developments and old themes. Biochem J. 2012; 441:763–787.
Schutz, Y. Protein turnover, ureagenesis and gluconeogenesis. Int J Vitam Nutr Res. 2011; 81:101–107.
Stanhope, KL. Role of fructose-containing sugars in the epidemics of obesity and metabolic syndrome. Annu Rev Med. 2012; 63:329–343.
Stanley, CA. Hypoglycemia in the neonate. Pediatr Endocrinol Rev. 2006; 4(Suppl 1):76–81.