Proteins With Biological Activity
Native conformation of a protein is adapted to serve specific biological functions. For example, β-conformation provides soft and flexible silk fibres, and the α-helical conformation provides tough, insoluble protective structures. In this chapter, some biologically active proteins are described with a special emphasis on structure-function relationships.
After going through this chapter, the student should be able to understand:
Distribution, primary and secondary structures, and properties of fibrous proteins, e.g. collagen and elastin: synthesis and degradation of collagen, and molecular basis of various inherited or deficiency diseases that affect collagen structure and integrity, hybrid nature, structure and function; synthesis and degradation of glycoproteins and the role of dolichol.
General structure and properties of immunoglobulin molecules; function and distinctive characteristics of different classes of antibodies; function and diagnostic significance of various plasma protein fractions; various disorders leading to over- or underproduction of plasma proteins and immunoglobulins.
I Collagen
A Overview
Multicellular organisms contain collagen, which is the major molecular component of connective tissue. Connective tissue is the principal structural and supportive element of the body—present in tendons, ligaments, cartilages, and organic matrix of bones. It also forms a structural layer under the skin, and envelops of blood vessels.
Connective tissue comprises of amorphous ground substance, in which fibrous elements like collagen and elastin are present. The ground substance comprises mainly of proteoglycans, water, minerals and proteins.
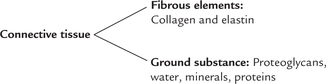
Thus, major molecular components of connective tissue are: collagen, elastin and proteoglycans. The word collagen is derived from Greek language which means to produce glue. Collagen is fibrous protein that possesses high tensile strength and cannot be stretched. Collagen is most abundant of all human proteins, constituting about one-third of total protein mass of the body. It is present in all tissues and organs where it serves as the major structural component.
In addition to collagen, the other fibrous component of connective tissue is elastin. However, unlike collagen, elastin can be readily stretched.
Proteoglycans are hybrid molecules of proteins and polysaccharides. They form the ground substance in which various fibrous elements of connective tissue are embedded. The ground substance acts as a cushion or a lubricant.
All the three molecular components of connective tissue are synthesized in specialized cells. These are fibro-blasts in connective tissue, osteoblasts in bones, and chondroblasts in embryonic cartilage.
B Basic Structure
Collagen is not a single protein but a large protein family with close to 20 members. Basic structural unit of all col-lagens is a trimer of polypeptides, called tropocollagen, that forms a characteristic triple helix. But the supermo-lecular structures of various collagen types differ, and they have different distributions and functions.
Tropocollagen is a rod-shaped molecule, about 300 nm long and 1.5 nm thick. Several tropocollagen molecules are arranged head to tail in parallel bundles to form fibrils (Fig. 5.1a ), commonly known as microfibrils. The heads of tro-pocollagen molecules are staggered along the length of the fibrils meaning that adjacent molecules are displaced approximately one-fourth of their length (67 nm). This quarter-staggered array accounts for the characteristic 67 nm spacing of the cross-striations in most collagens. The fibrils further associate to form larger fibres, which have high tensile strength: a fibres of 1 mm diameter can withstand a load of 10-40 kg before it breaks. Hence, the collagen infrastructure— a regular array of parallel, staggered tropocollagen molecules—provides a final product with a tremendous amount of strength.
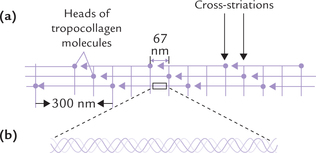
Fig. 5.1 Structure of collagen. (a) A fibril showing arrangement of tropocollagen molecules—in a regular, staggered parallel array, (b) Portion of tropocollagen molecule magnified to show the triple-helical structure of collagen—consisting of three helical polypeptide chains. Each chain is twisted into a left-handed helix and these chains wrap around each other to form a right handed triple helix.
Triple Helix
The structure of collagens is “triple-helical ”. This is because tropocollagen consists of three helical polypeptide chains, each having approximately 1050 amino acids (Fig. 5.1b). These chains, known as the α-chains (nothing to do with the α-helix), are tightly wound around one another. Col-lagens also contain non-helical regions which are relatively small. The principal features of triple helix are:
1. Individual α-chains form a tight, left-handed triple helix with 3.6 amino acids per turn and a rise per amino acid of 0.15 nm. This structure is favoured by proline residues, which are present in large amounts in these chains (proline introduces a sharp bend in a polypeptide chain).
2. The three intertwined α-chains form a superhelix with a right-handed sense. Thus, the secondary structure of tropocollagen is helical on two levels:
The opposite direction of twists of the individual chains and the triple helix means that tropocollagen is very resistant to unwinding by tension.
3. In order to form a triple-helix a polypeptide chain must contain glycine as every third residue in the sequence. This is because side chain of only glycine is small enough to permit close contact between main chains.
4. The peptide bonds are internal, and therefore, colla-gens are very resistant to digestion by proteases. The triple helix is stabilized by hydrogen bonds between the peptide bonds of different chains.
Types of Collagens
The α-chains associate in different combinations in the trimer to form various types of collagen molecules, of which about 20 types are known. The α-chains are identical in some collagen types and non-identical in others. As shown in Table 5.1, different types of α-chains are numbered according to the collagen types in which they occur. Thus, type I collagen is a heterotrimer, containing an α 1(I) chain that is present in two copies, and an α 2(I) chain in one copy, yielding the structural formula [α1(I)]2 α 2(I). The type II and type III are homotrimers composed of three identical α1(II) and α1(III) chains respectively.
Table 5.1
Composition and distribution of different types of collagen
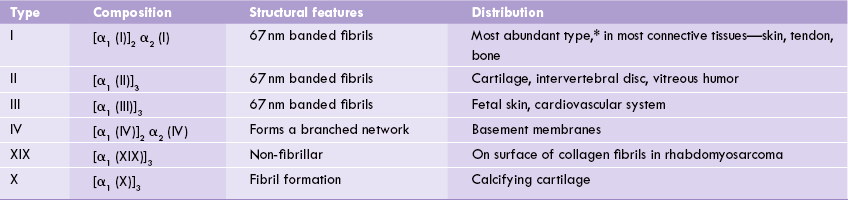
*Type I accounts for about 90% of all collagens in humans.
In most of these collagen types (notably types I, II, III, V, XI and XII), tropocollagen molecules associate in an ordered manner to form fibrils. The fibril formation depends on interactions between amino acid side chains in neighbouring molecules. Other types, such as type IV—a major constituent of basement membranes, do not form such fibrils, but are involved in the formation of an extensively branched mesh.
Amino Acid Composition and Sequence
Collagen has a unique primary structure, represented as -Gly-X-Y, which implies that glycine is present at every third position in the polypeptide chain. Thus, this amino acid constitutes about 33% of the total amino acid residues (Fig. 5.2 ). × and Y can be any amino acids, but certain ones predominate in these positions:
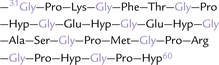
Collagens can thus be identified by their amino acid sequence, and by high proportion of certain amino acids: 33% glycine and 10% proline. Most remarkably, collagen contains 4-hydroxyproline (10%), 3-hydroxyproline (< 0.5%), and 5-hydroxylysine (1%), which do not occur in most other proteins. These hydroxylated amino acids are not represented in the genetic code, and therefore they have to be synthesized post-translationally from prolyl and lysyl residues in the polypeptides. Each of these amino acids plays a specific role in folding of the polypeptide chain, discussed as follows:
1. Proline residues introduce sharp bends in the α-chains (because R group of proline is part of a cyclic ring), which helps tight wrapping of these chains around one another. Such tight wrapping enhances strength of the triple helix.
2. Glycine residues play an indirect role in permitting extremely tight interwinding of the α-chains. The winding is so compact that there is little space available towards the interior, only the smallest amino acid, i.e. glycine, can be accommodated at that place. Importance of glycine in collagen is illustrated by the fact that substitution of this amino acid by a bulkier one results in marked reduction in the strength of collagen (Case 5.1).
3. Hydroxyproline and Hydroxylysine are modified amino acids in collagen, each containing an additional hydroxyl group (Fig. 5.3 ). Each hydroxyl group in turn can participate in the formation of an additional hydrogen bond. Formation of a large number of hydrogen bonds is thus possible, which gives enormous collective strength.
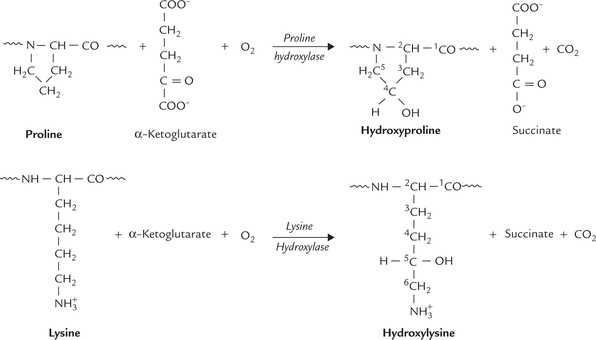
Formation of these special amino acids occurs from the corresponding primary amino acids, proline and lysine, by hydroxylation reactions. These reactions are catalyzed by the enzymes proline hydroxylase and lysine hydroxylase, respectively.
C Biosynthesis of Collagen
The collagens are synthesized and secreted by specialized cells called fibroblasts. Like other secreted proteins, collagen polypeptides are synthesized by ribosomes on the rough endoplasmic reticulum (RER; Fig. 5.4 ). The poly-peptide chain then passes through the RER and Golgi apparatus before being secreted. Along the way it is post-translationally modified by glycosylation, hydroxylation proteolytic cleavage, and cross-linking. Many of these modifications are unique to collagen.
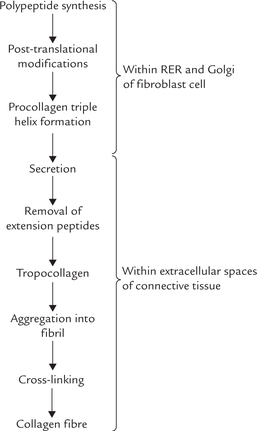
Polypeptide Synthesis
Collagen genes are some 10 times longer than the mature mRNAs that they encode, and contain more than 50 short exons separated by introns of different sizes.
The nascent chain initially synthesized is called pre-pro-α-chain, or simply pre-pro-collagen. It contains a sequence of 15-30 hydrophobic amino acids at its N-terminal end, called signal sequence. This sequence directs entry of the pre-pro-α-chain into cisternae of endoplasmic reticulum, where it is converted to proα-chain by removal of this signal sequence.
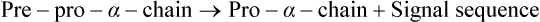
Post-translational Modifications
Biosynthesis of collagen provides an excellent example of post-translational modification of a protein. As noted earlier, it involves a number of reactions: hydroxylation and glycosylation initially (followed by chain assembly, proteolysis and cross-linking, later on).
Hydroxylation
Selected proline and lysine residues undergo hydroxyl-ation to form the corresponding hydroxylated amino acids: hydroxyproline and hydroxylysine. Action of proline hydroxylase and lysine hydroxylase, respectively catalyze these reactions. These enzymes have an Fe2+ ion at their active site and require ascorbic acid (vitamin C) for activity. The ascorbic acid acts as an anti-oxidant, keeping the Fe2+ ion in its reduced state. Both the enzymes are dioxygenases using molecular oxygen. α-Ketoglutarate, the TCA cycle intermediate, is a coreductant, which is oxidized to succinate during the reaction (Fig. 5.3). Moreover, both the enzymes hydroxylate only those proline and lysine residues that are incorporated in a poly-peptide chain.
Because the hydroxylated amino acids are important for stabilizing the structure of collagen through hydrogen bond formation, in vitamin C deficiency decreased synthesis of the hydroxylated amino acids results in weakening of the collagen fibres. This leads to the fragile blood vessels, incomplete bone formation and poor wound healing—the characteristic of the disease scurvy (Case 18.1).
Glycosylation
Collagen is unique in having covalently attached monosaccharides (galactose) or disaccharides (glucose-galactose), linked to the hydroxyl of 5-hydroxylysine. These are attached in the endoplasmic reticulum by Mn2+ containing UDP-glycosyltransferases. Like hydroxyl-ation, glycosylation occurs concomitantly with chain synthesis and is inhibited by triple helix formation. The carbohydrate content of collagens varies considerably from 0.5-1.0% in types I and III to nearly 15% in type IV, but precise significance of glycosylation is yet to be elucidated.
Procollagen Triple Helix Formation
Three pro-α-chains assemble together to form the triple helix. The process occurs while the chains are still in the endoplasmic reticulum. Note that when the collagen polypeptides are initially synthesized, they have additional amino acid residues (100-300) on both their N and C termini that are absent in the mature collagen fibre (Fig. 5.5 ). They are called extension peptides, and they help to correctly align the three polypeptides as they come together in the procollagen triple helix. This process may be aided by the formation of disulphide bonds between extension peptides on neighbouring polypep-tide chains. The extension peptides also prevent the premature aggregation of the procollagen triple helices within the cell.
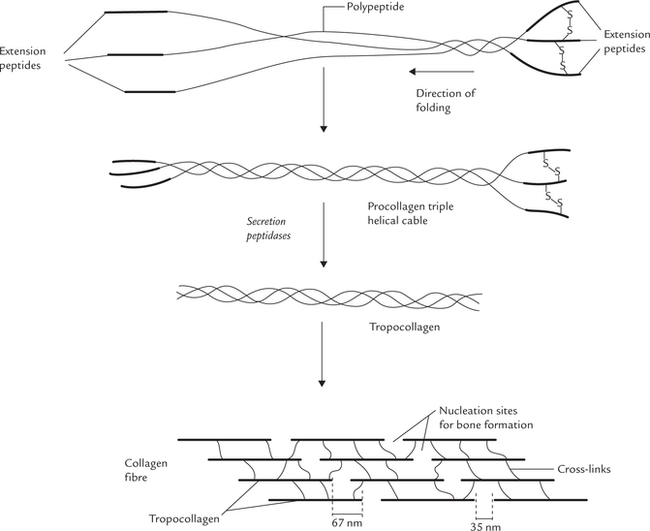
Fig. 5.5 Formation of procollagen and its processing by proteolysis to yield tropocollagen. The tropocollagen molecules aggregate to form collagen fibrils, which cross link extensively to form collagen fibres.
The procollagen is packaged into secretory vesicles within Golgi apparatus, and then secreted out of fibroblasts.
Procollagen to Tropocollagen Formation by Removal of Extension Peptides
On secretion out of the fibroblast the extension peptides are removed by the action of extracellular peptidases. Procollagen peptidases, removes them leaving behind the triple helical, mature collagen monomer, the tropo-collagen.
Collagen Fibril Formation
The newly formed tropocollagen molecules aggregate to form collagen fibrils (which would cross-link extensively to form collagen-fibres). For the formation of a fibril, the tropocollagen molecules aggregate in a staggered head-to-tail arrangement. As shown in Figure 5.5, each tropo-collagen associates itself in a parallel, overlapping staggered array with other tropocollagen molecules. A typical fibril consists of hundreds or even thousands of tropocollagen molecules in cross section.
Cross-linking and Fibre Formation
The tropocollagen molecules (which have aggregated into fibrils) now become extensively cross-linked covalently to produce mature collagen fibres. The special tensile strength of collagen fibres is provided by these covalent cross-links both between and within tropocollagen molecules (though hydrogen bonds also have a stabilizing effect).
As there are few, cysteine residues in the final mature collagen, these cross-links are not disulphide bonds as commonly found in proteins, but rather are unique crosslinks formed between lysine and its aldehyde derivative allysine. Allysine residues are formed from lysine by the action of monooxygenase lysyl oxidase (Fig. 5.6 ). It is a copper containing enzyme, also requiring pyridoxal phosphate, derived from vitamin B6. The aldehyde group on allysine then reacts spontaneously with either the side-chain amino group of Lys or with other allysine residues on other poly-peptide chains to form covalent interchain bonds.
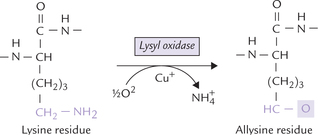
 Tropocollagen, formed after removal of extension peptide, aggregate into fibrils and are covalently cross-linked to form mature collagen fibres.
Tropocollagen, formed after removal of extension peptide, aggregate into fibrils and are covalently cross-linked to form mature collagen fibres.
Decreased maturation due to impairment of formation of various linkages alters mechanical properties of skeletomuscular system (Case 5.2).
D Degradation of Collagen
Collagen tissue undergoes constant but slow turnover, half-life being up to several months. The degradation occurs during tissue repair and during normal growth and development, and is initiated via the enzymes collagenases. These are extracellular zinc containing proteinases that initiate degradation by splitting a single Gly-Ile bond in each of the three-tropocollagen strands. Two large fragments are produced, which then spontaneously denature and become susceptible to attack by several intracellular proteases (e.g. cathepsins). Further degradation is carried out by a multipronged attack by these enzymes.
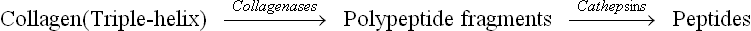
Action of collagenases is important because native collagen is resistant to attack by most proteinases, unless it is denatured. Ability to secrete large amount of collagenase is possessed by tumour cells, which helps their metastatic spread.
E Genetic Defects of Collagen Biosynthesis and Metabolism
Inherited abnormalities in the procollagen chains or deficiencies of enzymes for their post-translational processing may lead to certain genetic disorders. The underlying biochemical defects include abnormal collagen genes, abnormal post-translational modification of collagen, or deficiency of cofactors needed by the enzymes that carry out the post-translational modifications. Defective collagen formed in these diseases is unable to provide the required tensile strength.
Ehlers—Danlos Syndrome
It is heterogeneous group of genetic disorders characterized by stretchy skin and loose joints. At least 10 types are known, some of which may be traced to defective collagen synthesis or to incomplete removal of propeptides (Table 5.2 ). Type IX, for example, arises due to X-linked recessive deficiency of lysyl oxidase, resulting in impairment of cross-link formation. As a result, joint hypermobility and hyperextensible skin is seen. Autosomal recessive deficiency of lysyl hydroxylase occurs in type VI disorder. It manifests as hyperextensible skin, scoliosis and hyperex-tensible joints (Case 5.2).
Table 5.2

*AD, Autosomal dominant; AR, Autosomal recessive.
**These tissues are rich in type III collagen.
The “India rubber man ” who could bend and twist himself into incredible shapes and package himself into tiny boxes had Ehlers-Danlos syndrome probably.
Osteogenesis Imperfecta (OI)
It is a group of genetic disorders in which biosynthesis of type I collagen is defective. Point mutations in the genes for pro-α1(I) or pro-α2(I) have been identified as causing OI.
OI is characterized by fragile bones, hence also known as the brittle bone syndrome. Clinical features vary widely in severity: in mild forms occasional pathological fractures may be the only presentation, whereas in severe forms skeletal deformities and dwarfism, with even perinatal deaths are seen. Extra skeletal manifestations include a blue discolouration of sclera, hearing loss, and poor teeth development. The incidence of osteogenesis imperfecta is approximately 1 in 10,000, and the inheritance is autosomal dominant in most cases.
Epidermolysis Bullosa
It is a heritable disease in which glycosylation of collagen is impaired and the skin characteristically blisters. The type VII collagen is absent; this type of collagen forms anchoring fibrils at the dermis-epidermis junction, and they anchor the basement membrane to the underlying dermis.
Dermatosparaxis
There is a hereditary impairment in the removal of the terminal propeptide extensions. Because fibril formation can occur only when these propeptides have been removed, there is impairment in fibril formation and hence, reduced tensile strength of collagen.
Disorders of Collagen Degradation
Many collagen disorders are characterized by abnormal activities of collagen-degrading enzymes (Case 5.3). These enzymes are discussed earlier (p. xxx). Their activity is low in adult human tissues, but in some pathological states characterized by increased collagen degradation, such as Paget’s disease, rheumatoid arthritis, and tumour metastasis, higher activity of these enzymes occur.
F Effect of Aging and Disease on Collagen Metabolism
The gourmet knows that meat of young animal tastes better for being soft and tender, whereas that of old animals is tough and unpalatable. The same is true for human flesh. The reason for this age related change is that the collagen of old animals and humans have more cova-lent cross links than those of the young. Also the amount of collagen, relative to the proteins of parenchymal cells, increases with age.
Collagen synthesis is also stimulated by tissue injury. During wound healing, the fibroblasts migrate to the site of injury and produce abundant collagen. Scar tissue consist mostly of collagen. The death of parenchymal tissue is followed by deposition of a collagen-rich extracellular matrix. For example, in liver cirrhosis, which is a common outcome of a variety of hepatic disorders, most of the parenchymal cells are replaced by fibrous (scar) connective tissue. Collagen metabolism is affected in a number of other diseases, e.g. scurvy (see Case 18.3) and lathyrism. In the latter condition cross-linking of collagen is impaired due to inhibition of the enzyme lysyl oxidase by a natural product (3-amino-propionitrile, an irreversible inhibitor). Copper deficiency also leads to enzyme inhibition resulting in failure of collagens to acquire the tensile strength they need to function.
II Elastin
Elastin is a fibrous element which (unlike collagen) can be readily stretched. It is the major fibrous protein of yellow, elastic connective tissue of ligaments and large arteries.
The inextensibility of collagen suits it to forming extracellular matrices that have high tensile strength (Achilles tendon can withstand about 2000 kg cm–) or have ability to withstand compression (250 kg cm– for the intervertebral discs). In some tissues (skin, ligaments, and large arteries) there is requirement for elastic deform-ability. Such tissues have high content of elastic fibres (10% in skin to over 50% in large arteries and some ligaments). These fibres consist mainly of elastin.
Structure
The basic unit of elastin structure is tropoelastin, a polypeptide of 800-850 amino acid residues (MW 72000). Like tropocollagen, the tropoelastin has aberrant amino acid composition, with high proportions of glycine (31%), alanine (22%), and hydrophobic amino acids (40%). About 10-13% proline and 4-hydroxyproline are present, but there is no hydroxylysine. It has been proposed that tropoelastin units are present in the random coil conformation and are extensively cross-linked. This makes such a network kinetically free: free to stretch and recoil. Like in case of collagen, these cross-links are derived from allysine. Therefore, lysyl oxidase is required for the synthesis of elastin as well as that of collagen.
Cross-linking
The cross-linking among tropoelastin chains involves allysine and lysine residues. As shown in Figure. 5.7, three molecules of allysine and one molecule of lysine come together to form a new amino acids: desmosine and isodesmosine. Both contain a pyridinium nucleus, derived from the R groups of lysine and allysine residues. Thus, desmosine serves as the central connection that connects different polypeptides.
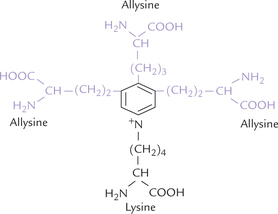
Fig. 5.7 The structure of desmosine showing 3 molecules of ally-sine and one molecule of lysine attached to pyridinium nucleus. Besides desmosine, elastin also contains some cross-links typical of collagen.
The non-specific inhibitors of collagen cross-linking, such as copper deficiency or lathyrism, inhibit cross-linking of elastin also. It is well known, for instance, that copper deficiency produces dissecting aneurysms in blood vessels of animals through incomplete elastin cross-linking. It appears, however, that elastin abnormalities observed in some inherited collagen diseases such as Marfan’s syndrome (one of the glycoproteins in microfibrils, fibrillin, is defective due to gene mutation) may be secondary to aberrations in collagen structure. Patients with this dominantly inherited condition have unusually tall stature, with long, spidery fingers; the lens is displaced; and the media of the large arteries is abnormally weak. Many patients die suddenly in mid-life after rupture of dilated aorta.
III Glycoproteins
A Overview
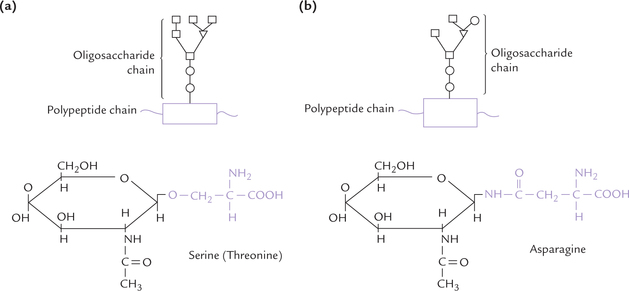
Glycoproteins are conjugated proteins. Like all other conjugated proteins, the glycoproteins contain a non-protein portion (i.e. prosthetic group) that is covalently attached with the protein portion of the molecule. The non-protein portion in case of glycoproteins are short, branched oligosaccharide units that are covalently attached to the polypeptide chain (i.e. aglycone) at several points (Fig. 5.8 ).
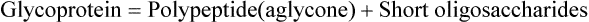
Glycosaminoglycans also consist of same two components as glycoproteins. However, these are three remarkable differences:
(a) the length of the oligosaccharide chain is relatively short (less than 10 sugar residues) in glycoproteins, whereas it is longer in glycosaminoglycans
(b) the carbohydrate chains in glycoproteins are often branched
(c) The oligosaccharide chains of glycoproteins lack a serial repeating unit; this is in contrast to the carbohydrate chains in glycosaminoglycan that consist of repeating disaccharide units (amino sugar-acid sugar; Chapter 2).
B Functions of Glycoproteins
Several cellular functions require participation of glycoproteins. Some of these are:
1. Glycoproteins are chief components of mucin of the gastrointestinal and urogenital tracts, where they act as protective lubricants.
2. The cellular phenomena, including cell surface antigenicity (e.g. the blood group antigens, refer to Box 5.1 for details); and the cell surface recognition (by other cells, hormones, viruses) require glycoproteins.
3. Several hormones are glycoprotein in nature, including the follicle-stimulating hormone (FSH), luteinizing hormone (LH), chorionic gonadotropin, etc.
4. Several proteins that are secreted from cells into extracellular fluids are glycoproteins. Almost all plasma proteins are glycoproteins. Other proteins like cerulo-plasmin, plasminogen, prothrombin, and immunoglobulins are also glycoproteins.
5. Glycoproteins are integral components of cell membranes, where they may play important role in the group behaviour of cells. They determine polarity of the biological membranes as well.
6. Several structural proteins, such as collagen, laminin, and fibronectin contain attached saccharide units.
C Structure of Glycoproteins
The Carbohydrate Component
The oligosaccharide units of glycoproteins are short and branched heteropolymers. In general, the sugar moieties are in the D-form. The principal moieties present in these oligosaccharides are: D-mannose (common in N-linked oligosaccharides), D-galactose, L-fucose (a-6-deoxy hex-ose), N-acetyl glucosamine, N-acetyl galactosamine and N-acetyl neuraminic acid (NANA). The latter is usually found in terminal positions of oligosaccharides.
Oligosaccharides of glycoproteins show great structural diversity. They range in size from 2 sugar residues in the most simple (O-linked) oligosaccharides to more than 15 in some more complex (N-linked) oligosaccharides. The carbohydrate content of different glycoproteins accordingly varies from less than 5% to more than 50%. For instance: IgG contains as little as 4% carbohydrate by weight, whereas glycophorin, a red cell glycoprotein, contains as much as 60% carbohydrates.
The heterogeneity among glycoproteins is due to difference in the sugar component. For example, pancreatic ribonuclease A and B have identical amino acid sequences, but their carbohydrate composition is significantly different. A given glycoprotein from different animal species usually has identical amino acid sequence in the polypeptide chains, but variable carbohydrate components, which accounts for their different properties. Thus the carbohydrate component is responsible for the distinct identity of a glycoprotein.
The surface of the human erythrocyte is covered with specific antigenic determinants, many of which are saccharides linked with polypeptides (i.e. glycoproteins). They determine the antigen specificity of the erythrocytes. There are about 160 blood group determinants, belonging to 21 independent human blood group systems. The most widely studied are the antigenic determinants of the ABO blood group system, and the closely related Lewis’s system. In both these systems, the antigenic determinants are glycoproteins only. A minor alteration of the saccharide units of the glycoproteins can alter the antigenicity.
There are four main groups (A, B, O, and AB), distinguished on the basis of three blood group substances on erythro-cytes. Individuals belonging to the group A have A antigen and those belonging to group B possess B antigen. In plasma, there are two naturally occurring antibodies (α and β) to these antigens. The group A persons have β-antibody and the group B persons have α-antibody. AB people have both A and B antigens but no antibody in their plasma. These people are universal recipients. The universal donors on the other hand are those that have no reactive antigens but have both α- and β-antibodies. The blood group of universal donors is denoted as O. They are said to carry H antigen, which represents the basic antigenic structure, as shown below:
Fucose—Gal—NAc—Protein Gal NAc = N-acetyl galactose
The A and B antigens differ from the H antigen in having additional sugar units, as shown below:

What is the genetic basis of different blood groups? Presence or absence of structural genes for the specific glycosyl transferase causes different blood groups. For instance, persons with blood group A have structural genes for N-acetyl galactosyl transferase. This enzyme transfers an additional sugar unit (GalNAc) to the basic antigenic structure. The people with group B have galactosyl transferase activity. Lack of both the structural genes leads to blood group O, whereas presence of both leads to blood group AB.
Role of Oligosaccharides: Oligosaccharides of glyco-proteins are important for many of these biological functions of glycoproteins. They play a role in maintenance of their higher-order structure, water solubility, antige-nicity, cell surface recognition and regulation of the metabolic fate of the protein.
Link between Carbohydrate and Protein
The saccharide unit of a glycoprotein are covalently linked to the polypeptide by specific glycosidic linkages, termed the glycopeptide bonds. Two types of such linkages are present in glycoproteins: O-glycosidic or N-glycosidic linkages (Fig. 5.8). In general, the O-glycosidic linkage is formed by attachment of the sugar unit to a hydroxyl (-OH) group of the aglycone whereas the N-glycosidic linkage is formed by attachment of the sugar unit to an amino (-NH2) group of the aglycone. In case of glycoproteins, these linkages are formed as here:
• O-Glycosidic linkage joins the sugar unit to the hydroxyl group at side chain of either serine or threo-nine.
• N-Glycosidic linkage joins the sugar unit to the amide group of an asparagine side-chain.
One or both types of linkages may be present in a given glycoprotein molecule.
D Synthesis of Glycoproteins
Synthesis of the N-linked and the O-linked glycosides differ from each other in certain important aspects, therefore, discussed separately.
Synthesis of O-linked Glycosides
The polypeptide chain of the glycoprotein is synthesized first; and later on the individual sugar units are sequentially attached to it, one at a time. The following steps are involved:
1. Synthesis of the polypeptide chain: It occurs in ribo-somes that are associated with the membrane of endoplasmic reticulum (ER). The completed chain is transported into the lumen of the ER where the glyco-sylation would occur.
2. Attachment of sugar units to polypeptide (Glycosylation): It begins in ER. The polypeptide chain is then transported to Golgi apparatus, where the glycosylation continues and oligosaccharides are constructed by stepwise addition of monosaccharides. The enzymes catalyzing glycosylation are glycosyl transferases (trans-glycosylases), which are bound to the membranes of these subcellular organelles, i.e. the ER and the Golgi apparatus.
Substrates for the glycosyl transferases are nucleo-side diphosphate sugars (NDP derivatives). These sugar nucleotides represent high energy state, and hence are capable of donating sugar to the growing oligosaccharide chain; for instance, the UDP glucose can add a glucose moiety to the growing oligosaccha-ride chain (Fig. 5.9 ), UDP galactose can add galactose moiety, and so on.
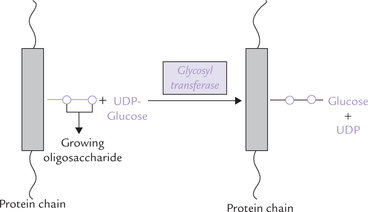
Fig. 5.9 Synthesis of O-linked glycoside by stepwise addition of individual sugar units (UDP = urine diphosphate).
3. The glycoproteins synthesized in this way are either released by the cell into extracellular fluid (ECF) or become part of the cell membrane.
Synthesis of N-linked Glycosides
Synthesis of N-linked glycosides is more complex and has additional processing steps. The characteristic feature of the synthetic process is that the polypeptide chain does not become glycosylated with individual sugar units; but rather a (mannose rich) core-oligosaccharide is constructed initially, attached to a lipid, dolichol (Fig. 5.10 ). Subsequently, the core-oligosaccharide is transferred from the dolichol to the newly synthesized polypeptide chain. The following steps are involved in the biosynthesis of the N-linked glycosides:
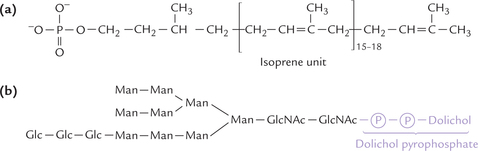
Fig. 5.10 Synthesis of N-linked oligosaccharides in glycoproteins. (a) Structure of dolichol phosphate. This lipid is present in the ER membrane, where it is used as a carrier of the core oligosaccharide, (b) Dolichol bound to the precursor oligosaccharide unit. This oligosaccharide is synthesized by the stepwise addition of the monosaccharides from activated precursors. Transfer of the precursor oligosaccharide to an asparagine side chain of the polypeptide. This transfer reaction is cotranslational.
1. Synthesis of oligosaccharide unit: It requires participation of dolichol, a polyisoprenol in the membrane of ER consisting of 15-18 isoprene units (Fig. 5.10a), which can be synthesized from acetyl CoA. Sugar units are attached to the dolichol pyrophosphate from their activated precursors in a stepwise manner. The enzyme glycosyl transferase catalyses the construction of branched oligosaccharides (Fig. 5.10b). The first sugars to be added by these membrane-bound enzymes are N-acetylglucosamine, mannose and glucose.
2. Synthesis of polypeptide chain: At the same time, a poly-peptide chain is synthesized in ribosomes associated with membrane of endoplasmic reticulum (ER). It is then transported into the lumen of the ER (Fig. 5.11 ).
Fig. 5.11 Synthesis of N-linked glycoside. Polypeptide chain is synthesized in ribosomes associated with membrane of endo-plasmic reticulum. There is a concomitant construction of oligosaccharide unit attached to dolichol phosphate. Transfer of the oligosaccharide unit to the amide group of asparagines occurs next; this transfer reaction is cotranslational.
3. Transfer of oligosaccharides from dolichol to polypeptide: The oligosaccharide unit is then transferred from the dolichol pyrophosphate to side group of an aspara-gine of the growing polypeptide chain. The enzyme catalyzing this transfer, protein-oligosaccharyl transfer-ase (E’), is present in the lumen of ER.
4. Trimming and/or addition of monosaccharides: After incorporation into the protein, the oligosaccharide is processed by removal of specific carbohydrate residues (mannose or glucose) as the glycoprotein moves through. The enzymes responsible for such processing are termed glycosidases. This process continues in the Golgi apparatus also. Finally, certain sugar units are added to complete the oligosaccharide chain synthesis.
Ultimately, the N-linked glycoproteins are either released extracellularly or become part of the cell membrane.
E Degradation of Glycoproteins
Degradation of glycoproteins requires specific lysosomal enzymes called glycosidases. Usually these enzymes are exo-glycosidases: they remove sugars in a series of reactions from the non-reducing end of the oligosaccharide chain. With each removal, the substrate is exposed for the action of the next enzyme. If any one of the exoglycosidases is absent, degradation of the glycoprotein by the next exo-glycosidase cannot occur. Action of the exoglycosidases is supplemented by that of the endoglycosidases which attack glycosidic bonds that lie towards the interior. A concerted action by both exo- and endoglycosidases is highly effective in degrading the large glycoprotein molecules.
Genetic deficiency of one or more of the lysosomal hydrolytic enzymes results in accumulation of partially degraded glycoproteins and oligosaccharides in cells. These disorders are termed the glycoprotein storage diseases.
IV Plasma Proteins
Plasma contains over 200 individual proteins that are structurally and functionally different from each other. Plasma concentration of these proteins is 6.3-8.8 g/dL. They are broadly divided into three types: albumin, globulin, and fibrinogen; their plasma concentrations are:
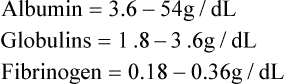
The globulin fraction is subdivided into numerous components like α, β, γ, etc.
All these proteins except the 7-globulins are synthesized in the liver. It may be noted that plasma is the soluble fraction of blood, which contains all plasma proteins, including fibrinogen and other clotting factors. If, however, a blood-clot forms before centrifugation, the supernatant no longer contains fibrinogen, and some of the other clotting factors are consumed as well. This solution is called serum. For most diagnostic tests, either plasma or serum can be used. Most of the serum proteins as well as fibrinogen have been isolated and thoroughly characterized, forthwith discussed in this section.
Concentration of plasma proteins is between 6.3 g/dL and 8.8 g/dL. With important exception of γ-globulins (the immunoglobulins), most of the plasma proteins are synthesized in liver.
Separation of Serum Proteins
Individual proteins can be isolated by an array of techniques that depend on their size, shape, net charge, and solubility. Among these techniques the common ones are:
1. Stepwise precipitation of proteins with salts is known as salting out. Different proteins precipitate at different salt concentrations; for example, 0.8 M ammonium sulphate precipitates fibrinogen, whereas a concentration of 2.4 M is needed to precipitate serum albumin.
2. Chromatography on cation and anion exchangers.
3. Fractionation with organic solvents such as ethanol or ether.
The basic principle of these techniques have been described in Chapter 3.
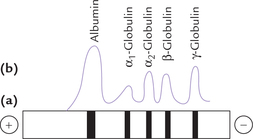
Electrophoresis: Among all the techniques enlisted above, electrophoresis is the most important for separation of serum proteins. Paper or agar gel electrophoresis with barbitone buffer at pH 8.6 yields an electrophoretic pattern shown in Figure 5.12 . The fractions in the order of increasing mobility towards anode are albumin, α1-, α2-, β- and γ-globulin.
Albumin/Globulin (A/G) Ratio
The normal serum albumin concentration is 3.6-5.4 g/dL and that of globulins is 1.8-3.6 g/dL; therefore, the normal A/G ratio is 1.36 (mean), with a normal range of 1.2-1.6. It is lowered in various conditions where serum albumin concentration falls (hypoalbuminaemia) and/ or when globulin concentration rises (hyperglobulinae-mia). Various causes of decreased A/G ratio and its importance in clinical diagnosis is discussed later in this chapter.
A Albumin
Albumin is the most abundant protein in plasma, which accounts for about 55–60% of the total plasma proteins. It is produced exclusively in liver; a healthy liver can synthesize 10–15 g of albumin per day. For this reason serum albumin levels are determined to assess a patient’s liver function (decreased synthesis and thus fall of serum albumin occurs in hepatic disorders).
Human albumin has a molecular weight of 69,000 and it consists of a single polypeptide chain of 585 amino acids with 17 disulphide bonds. Due to its low molecular weight it is one of the first proteins to appear in the urine after renal damage. Half-life of albumin is 20–25 days.
Functions
Albumin performs a variety of important functions described here.
Oncotic Pressure
Albumin plays an important role in osmotic regulation and fluid balance. As the capillary walls are relatively impermeable to plasma proteins, the latter exert an osmotic force across the capillary walls. This force helps to retain water in the intravascular compartment. The total colloidal osmotic pressure (oncotic pressure) is 25 mmHg, of which albumin accounts for about 80% (i.e. 20 mmHg). This is accounted by high concentration and low molecular weight of albumin. Hypoalbuminaemia, therefore, results in a significant reduction in osmotic pressure of blood, with consequent movement of water into interstitial spaces of the extremities and other parts of the organism, causing swelling. The condition is called oedema.
Transport
Albumin is involved in transport of several substances because of its predominantly polar nature. Nearly 40% of plasma calcium is bound-up with albumin. Bilirubin, steroid hormones, free fatty acids, and many non-polar drugs are also bound up with (and transported by) albumin.
Circulating Hormone Reservoirs
Albumin forms complexes with some of the circulating steroid hormones, thus preventing their filtration in the glomeruli. Such complexes act as circulating reservoirs of the hormones, which tissues can draw at the time of need.
Buffers
Albumin, along with other plasma proteins, is responsible for 15% of the total buffering capacity of blood. Among these proteins albumin has maximum buffering capacity.
Nutritive Function
Albumin degradation is an important source of essential amino acids during malnutrition, which are required for the maintenance of the organism’s vital processes. Serum albumin concentration, is therefore, measured for assessing a person’s protein nutritional state.
Diseases and Disorders
Concentration of serum albumin may increase (hyperal-buminaemia) or decrease (hypoalbuminaemia) in a number of conditions.
Hyperalbuminaemia
Dehydration, particularly if it develops rapidly, results in haemoconcentration. As a result, serum albumin concentration shows an apparent rise. The A : G ratio remains unaltered because serum globulin concentration also elevates proportionately. Vomiting and diarrhoea are major causes of dehydration.
In most cases, certain associated factors are present which oppose the effect of dehydration. For instance, vomiting and diarrhoea are associated with decreased protein absorption from the intestines. Consequently, serum albumin concentration may remain normal or even fall.
Hypoalbuminaemia
Certain conditions that result in hypoalbuminaemia are as follows:
Renal causes
The renal disorders in which loss of proteins in urine (i.e. proteinuria) occurs result in decreased serum albumin concentration. In nephrotic syndrome, a marked fall occurs because of a massive urinary loss of proteins (> 4 g/day) and the total serum protein concentration may fall below 4 g/dL.
Gastrointestinal causes
The gastrointestinal disorders in which the intestinal absorption of the digested molecules (including amino acids) is impaired result in hypoalbuminaemia. Some of the pathological conditions associated with malabsorption are enteritis, sprue, steat-orrhoea, and malignant diseases involving intestine or pancreas.
Loss of proteins into the alimentary tract (known as protein-losing enteropathy) can occur in a number of diseases of the stomach and intestines. Occasionally, it may develop as a primary condition. Such a loss of proteins can be demonstrated by administration of intravenous Cr-51, followed by monitoring excretion of this compound in stools.
Insufficient dietary intake of proteins, for a prolonged period, may also lead to hypoalbuminaemia. This is because the serum albumin is degraded to provide amino acids for tissue protein synthesis.
Hepatic causes
As liver is the only organ for albumin synthesis, hepatic disorders cause decrease in serum albumin concentration (Case 6.3). In most cases, hypoalbuminaemia is accompanied by hypoproteinaemia as well. In some cases, however, there is an associated rise in the globulin fraction, so that total protein concentration remains normal or may even increase.
Other causes
Hypoalbuminaemia develops following severe haemorrhage, particularly if the blood loss is rapid. Initially there is loss of both plasma and proteins, but the plasma is restored earlier than the proteins. This results in decreased plasma albumin concentration. Similar effect is observed in shock, burns, crush-injuries, and in post-operative period. In burns, the fall in albumin concentration is greater because of extravasation of proteins.
Analbuminaemia
It is a rare genetic disorder where albumin is virtually absent. However, oedema does not generally develop since globulin levels are elevated in this condition, which tend to compensate for the absence of albumin.
Prealbumin (Transthyretin)
It is called so because it moves slightly ahead of albumin during electrophoresis. It participates in the transport of retinol from its major storage site in the liver to the extra-hepatic tissues (Chapter 18): the retinol released from hepatic tissue is in tight binding to retinol binding protein (RBP). In blood circulation, the retinol-RBP forms a ternary complex with transthyretin.
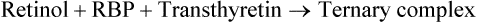
The formation of this ternary complex is important because RBP (MW 21,000) is easily filtered at glomeruli and lost in urine. The ternary complex is large enough to escape this fate.
B Globulins
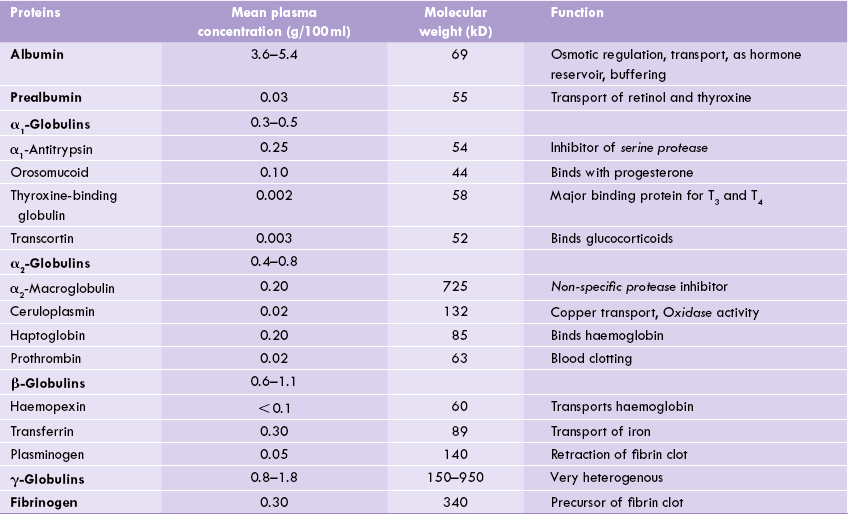
Globulins comprise a number of proteins, which are separated into four distinct bands (α1,, α2, β, and γ). Each of these bands is a mixture of proteins. Thus, the γ-globulin component consists of a variety of immunoglobulins of the IgG type. Taken together, these immunoglobulins constitute the second most abundant component of serum proteins after albumin (Table 5.3 ). Other immu-noglobulin types—IgA and IgM, have (β-globulin mobility. The β-globulin group also contains haemopexin, trans-ferrin, plasminogen, prothrombin and a lipoprotein called LDL (Chapter 12). Among α2-globulins are— α2-macroglobulin, haptoglobin and ceruloplasmin; while α1-globulins include some hormone carrying proteins (e.g. thyroxine-binding globulin and the steroid-binding proteins, transcortin and orosomucoid) and α1-antitrypsin (Table 5.3).
Table 5.3
Plasma proteins and their functions in the human body
Immunoglobulins: IgA, IgG, IgM, IgD and IgE are mostly γ-globulins (some are (β-globulins); their characteristics are outlined in Table 5.4.
Biological significance of various globulins is summarized in Table 5.3; see also Box 5.2.
Important Globulins and their Functions
α1-Antitrypsin
α1-Antitrypsin is an α 1-migrating glycoprotein. It is a serine protease inhibitor, active against a number of proteo-lytic enzymes, particularly the elastase released by neutrophils. Its role in the body is to protect lung tissue against proteolytic damage. It is implicated in two diseases: emphysema and liver disease.
Emphysema: It is a chronic disease characterized by abnormal lung distention. At least 5% of all cases of emphysema are due to impaired α 1-antitrypsin activity. Functional inactivation of α1-antitrypsin occurs in smokers, and therefore chronic smokers are more prone to lung damage by elastase, and hence develop emphysema (Case 4.2). Smoking also increases neutrophil levels in the lungs (and hence release of elastase) and decreases α1-antitrypsin levels in the bloodstream. α 1-Antitrypsin levels of emphysema patients are almost always below normal and this can also serve as a diagnostic tool for this disease.
Liver diseases: Some patients with α1-antitrypsin deficiency develop neonatal hepatitis or infantile cirrhosis. In all likelihood this is caused not by lack of protease inhibition but rather by aggregation of mutant form of α 1-antitrypsin to form polymers. These non-secretable polymers accumulate in vesicles within the hepatocytes and cause damage by unknown mechanisms.
Orosomucoid
Orosomucoid is an important constituent of α1-globulins that binds progesterone and some drugs. Its concentration rises in trauma, malignancy, or inflammatory conditions.
α2-Macroglobulin
α2-Macroglobulin is an α 2-migrating glycoprotein of high molecular weight (725,000). It inhibits activities of proteases, such as trypsin, chymotrypsin, plasmin, throm-bin and kallikrein.
Apart from α2-macroglobulin, another α2-migrating glycoprotein, α2-antichymotrypsin, is a non-specific protease inhibitor, though less important than the former.
Ceruloplasmin
Ceruloplasmin is a copper containing α 2-globulin that binds approximately 60% of the copper in the serum and is itself loosely complexed to albumin or with histi-dine. A ceruloplasmin molecule binds with six atoms of copper, and also possesses oxidase activity, as discussed under copper metabolism (Chapter 19).
Haptoglobin
Haptoglobin is an α2-migrating glycoprotein. It serves as a scavenger of the free intravascular haemoglobin. Haemolysis releases haemoglobin into plasma, from where it is at risk of being lost in urine. Haptoglobin prevents such loss by binding with haemoglobin to form a complex which is too large to be filtered in glomeruli. This complex is subsequently taken up by the reticulo-endothelial cells where the globin chain is degraded to release amino acids, and the haem is converted to bilirubin. The iron of haem is recycled for utilization. In this way, haptoglobin prevents wasteful loss of haemoglobin and its useful components (Fig. 5.13 ).
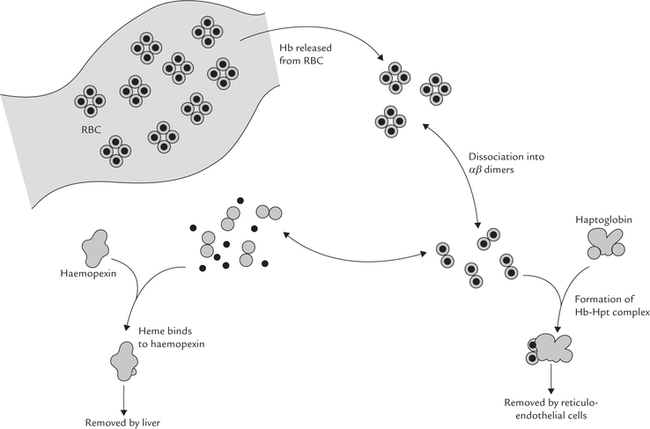
Normal plasma concentration of haptoglobin is 100300 mg/dL. In haemolytic anaemia more of this protein is used for forming a complex with haemoglobin.
Half-life of this complex is much shorter (90 minutes) compared to that of haptoglobin (5 days), and so it is rapidly cleared from the plasma. As a result plasma concentration of haptoglobin falls.
Haemopexin
Haemopexin is a β-migrating glycoprotein. It also serves as a scavenger protein for haemoglobin, but only after the haemoglobin-binding capacity of haptoglobin is saturated. Excess haemoglobin is dissociated into the globin chains and haem; the haem is dissociated into αβ dimers, carried to liver taken up where it is by haemopexin, where iron atom is released for storage and reutilization. Thus, hae-mopexin and haptoglobin have analogous functions: both help in conserving the body iron (Fig. 5.13).
When binding capacities of both haptoglobin and hae-mopexin are exceeded, a complex of haem and albumin, called methaemalbumin, is formed. The latter continues to circulate until enough haemopexin becomes available.
Transferrin
Transferrin is a liver-derived ( -globulin. It is the major iron transporter; essentially all iron in circulation binds with this glycoprotein.
Diseases and Disorders
Various pathological conditions are known to cause a generalized increase in serum globulin concentration (hyperglobulinaemia). Decreased serum globulin concentration (hypoglobulinaemia) however, never occurs.
Hyperglobulinaemia
It occurs in the following pathological conditions.
Advanced liver diseases
The serum globulin levels are typically elevated, often between 5–7 g/dL (normal = 1.8–3.6 g/dL). Since the albumin concentration is usually in the lower part of the normal range, or reduced (because of decreased synthesis), the A:G ratio is low.
Multiple myeloma
It is a group of diseases characterized by proliferation of a single plasma cell clone to cause extraordinary accumulation of immunoglobulin molecules in the patient’s serum. Being derived from a single ancestral B cell, the proliferating plasma cells all produce the same (monoclonal) immunoglobin. Such immunoglobulins are termed paraproteins, and they produce a sharp peak in the γ-globulin fraction. The paraproteins may be of the IgG, IgA, IgM, IgD or IgE type. The malignant plasma cells grow in the bone marrow, where they cause bone pain, pathological fractures and radiological abnormalities. In many cases, a multiple myeloma patient excretes protein in the urine. This protein called Bence-Jones protein, is a dimer of K or λ-chains that are formed in paraprotein.
Diagnosis
Demonstration of paraprotein upon serum electrophoresis is a reliable test for diagnosis of multiple myeloma. In addition, the classical heat test is performed. It involves the precipitation of Bence-Jones proteins when slightly acidified urine is heated to 40°-50°C and redissolve on further heating urine to boiling point. They reappear on cooling urine to about 70°C. A marked rise in serum globulin concentration is also observed in multiple myeloma, usually between 5 g/dL and 10 g/dL, but sometimes up to 14 g/dL (Case 5.5).
A/G Ratio and Electrophoretic Pattern in Some Diseases
As noted earlier, the mean A/G ratio of 1.36 decreases in various disease processes, for example in nephrotic syndrome, where albumin levels are low and/or globulin levels are high. Abnormal electrophoretic pattern, with decreased albumin and a prominent α2-globulins are seen.
Some other important conditions where A/G ratio is reversed are malnutrition and liver diseases (albumin decreases), and chronic infections and multiple myeloma (globulin rises). Abnormal electrophoretic patterns are also seen; for example, in multiple myeloma a sharp distinct M band appears in γ-globulin region, in infections the α1 and α2 bands are increased.
C Fibrinogen
Fibrinogen, the precursor of fibrin clot, appears to be formed in the liver. It is a soluble glycoprotein that consists of three pairs of identical polypeptide chains, called the α-, β- and γ-chains (MW 63,500, 56,000 and 47,000, respectively). Blood coagulation is initiated by the action of thrombin on fibrinogen, whereby two small peptides are removed from the fibrinogen. This results in formation of a fibrin monomer. Several monomers then aggregate via physical interactions to form a polymer that has properties of a soft gel. Further polymerization to form tough insoluble clots is accomplished by a specific enzyme called factor XIII: its action serves to join a number of linear fibrin polymers to form cross-linked fibrin lattices.
Diseases and Disorders
Decreased fibrinogen level in plasma is observed in severe liver diseases (e.g. acute hepatic necrosis). Low fibrinogen levels observed in antepartum haemorrhage, quickly return to normal after delivery.
Increased fibrinogen level is found in acute infections such as pneumonia, rheumatic fever, and tuberculosis. Since erythrocyte sedimentation rate (ESR) is affected by fibrinogen, it is markedly increased in these conditions. Hence, measurement of ESR is used for assessing the severity and course of these disorders. In contrast to most other acute infections, fibrinogen level is reduced in typhoid fever.
V Immunoglobulins
The immune system is remarkable for its ability to generate a variety of specific proteins that include antibodies (immunoglobulins). The antibody reacts specifically with the antigen that triggered its production. Antigens are mostly proteins, but can be nucleic acids or polysac-charides. Thus, various bacterial and viral cell components may act as antigens. Antibodies recognize the antigen on the surface of the intruding organisms. The specific antigen-antibody interaction leads to activation of the complement system which destroys the foreign cell. Antibodies play important role in providing resistance against bacterial infections. Though it is mainly the γ-fraction of plasma proteins that comprises immunoglobulins, sometimes they separate with (β- and α-globulin bands of electro-phoretogram. Plasma concentration of immunoglobu-lins is about 1.0-2.5 g/dL.
Immunoglobulins are synthesized by plasma cells. Plasma cells develop from B lymphocytes that have contacted foreign antigen. A single plasma cell can make only one type of antibody molecule. It is the accumulation of many plasma cells, each producing a distinct antibody, that gives rise to the multiple antibody specificities. Specific affinity of an antibody is not for the entire macro-molecular antigen but for a particular site on it called antigenic determinant (or epitope).
This property is widely used in purification of proteins and in affinity chromatography (Chapter 4). The affinity of antibody for an epitope is due to the complementary structure of the two. If there is a cleft on the surface of the epitope, a corresponding elevation is envisioned on the antibody. If an antigen is injected, the body generates an antibody specific to each of the epitopes on the antigen.
Immunoglobulins or antibodies, a group of plasma proteins that are secreted by plasma cells, play an important role in body’s defenses by specifically recognizing foreign molecules (antigens) and facilitating their selective elimination.
A Structure
The basic structural unit of an immunoglobulin is a hetero-tetramer, made up of two light chains (L) and two heavy chains (H), joined through non-covalent interaction and disulphide bridges. Thus, the simplest description of an antibody molecule is H2L2 (Fig. 5.14 ). The light chains are simple proteins of 212 amino acids (MW 23,000 each) and the heavy chains are glycoproteins comprising approximately 440 amino acids (MW 53,000-75,000) each.
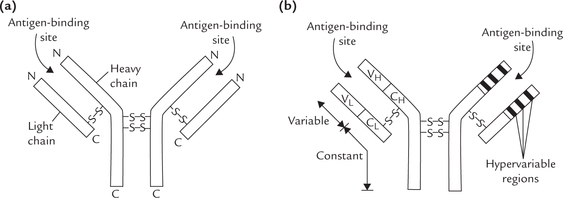
Fig. 5.14 Structure of an antibody molecule. (a) Each antibody (immunoglobulin) molecule has two identical light chains (L) and two identical heavy chains (H) consisting of 212 and 440 amino acids respectively, (b) Each chain has a variable region, where amino acid sequences shows considerable variations between individual immunoglobulin molecules and constant region where little variation occurs within one immunoglobulin class. The generic terms for these regions in the light chain are VL and CL and for heavy chain are VH and CH.
The light chains are of two types: K or λ. A given immunoglobulin contains 2K or 2X chains, but never a mixture of the two. In humans, 60% light chains are of K type.
There are five types of heavy chains: γ, μ, α, ε and δ. Depending on the heavy chain make up, the immunoglobulins are differentiated into five classes: IgA (α-chain), IgD (δ-chain), IgE (ε-chain), IgG (γ-chain), and IgM (μ-chain). All classes of immunoglobulins are present in every human being. (Within a class are found subclasses due to subtle differences in the amino acid sequences of heavy chains of the same class.)
Thus, structure of IgG can be represented as κ2 γ2 or as λ2 γ2; the IgA molecule as κ2 α2 or as λ2 α2 and so on. The heavy chains are hinged (flexible) in the vicinity of two inter-chain disulphide bridges. At these places, several prolyl residues are present, which confer a degree of flexibility on the molecule.
Human immunoglobulins exist as IgA, IgD, IgE, IgG and IgM classes, which contain α, δ, ε, γ and μ heavy chains respectively.
Each of the four chains consists of variable region and a constant region, discussed later.
Both heavy and light chains have distinct globular regions called domains that have a highly organized tertiary structure. Each of these structural domains comprise about 110 aminoacyl residues. The light chain has two such domains, one in the variable region of the chain (VL) and the other in the constant (CL) region. The heavy chain, has four or five domains. The latter are termed VH or CH depending on whether they are present in the variable region or the constant region of heavy chain. Figure 5.15 shows domain arrangement of a typical immunoglobulin molecule.
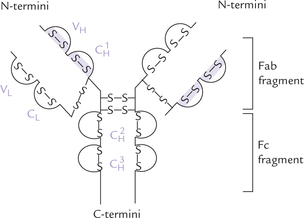
Fig. 5.15 Structure of an immunoglobulin molecule showing the disulphide-bridge limited domains. Domains on light chain are VL and CL; and the domains on heavy chain are: 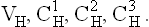
Each domain is stabilized internally by a disulphide bond and generates a globular three-dimensional structure. Thus, antibody molecule can be visualized as being made up of distinct domains, connected by relatively non-ordered polypeptide segments. These domains have well defined biological functions.
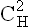 domain binds the complement.
domain binds the complement.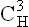 domain helps adherence to the cell surface.
domain helps adherence to the cell surface.All immunoglobulins are glycoproteins, and the degree of glycosylation in the region of the domain 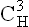 is variable.
is variable.
B Variability
The amino acid sequences at the N-termini of both light and heavy chains show considerable variation between individual immunoglobulin molecules. Half of the light chain and quarter of the heavy chain constitute the variable region. Rest of the molecule, known as constant region, shows little variation within one immunoglobulin class (Fig. 5.14b). The light chain contains one variable (VL) and one constant (CL) region, whereas the heavy chain contains one variable and three or more constant regions. The variations in the amino acid sequences of the variable region permit the body to construct a large variety of three dimensional structures.
Moreover, within these V regions, certain segments are hypervariable. The 29-32, 48-52, and 93-96 aminoacyl residues in light chain and 30-36, 50-53 and 93-98 positions in heavy chains are termed hypervariable regions. They define the actual binding site for antigen. They are also termed the complementarity-determining regions (CDRs) as they form antigen binding site complementary to the topology of the antigen structure. In the three-dimensional structure of the immunoglobulin molecule, the hypervariable parts of both the light and the heavy chains are looped together to form the antigen-binding site. Thus, they are referred to as the “hot spots ” where amino acid sequence varies from one molecule to another. Such sequence variations are specific for each of the antibody molecules and are known as idiotypic variations.
Variability in the variable regions of both light and heavy chains is mainly localized to three hypervariable regions in each chain, which are looped together to form the antigen-binding site.
The remaining parts of variable region are far less variable and are called framework regions.
Proteolytic Cleavage of Immunoglobulin
Treatment with the proteolytic enzymes, pepsin and papain, cleaves the immunoglobulin (IgG) as shown in Figure 5.16 . Papain digestion yields two univalent Fab fragments and an Fc fragment, whereas pepsin digestion yields a bivalent F(ab’)2 fragment. The cleavage occurs in the hinge region, where the antibody molecule can have limited degree of three dimensional movement. As discussed earlier, the hinge region lies in vicinity of two interchain disulphide bridges.
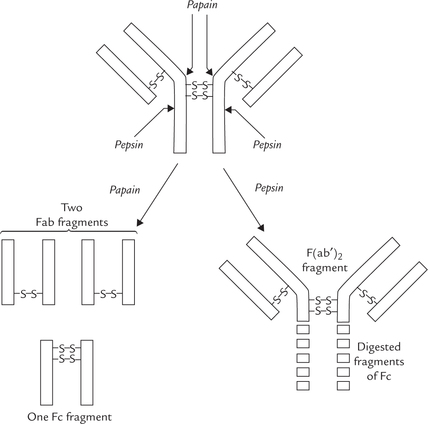
The Fab (fraction antibody) fragment binds with the corresponding antigen. It is constituted by whole of the light chain and N-terminal half of the heavy chain. Fc (fraction crystallizable) portion, constitutes the remaining part of the immunoglobulin molecule. It is concerned with activation of complement cascade and interaction with the cellular elements of the immune response. It triggers immune response that leads to the lysis of the intruding organisms.
Immunoglobulins from any class can also exist in two types: This is due to presence of either kappa (K) or lambda (X) light chain, giving rise to 10 different permutations of heavy and light chains. The K chains are more common in humans than the × chains.
While IgD, IgE, and IgG occur as a single Y-shaped antibody unit, IgM occurs as a pentamer and IgA can exist as a monomer, dimer, or trimer. Characteristics of various classes of immunoglobulins are summarized in Table 5.4 .
Table 5.4
Characteristics of the immunoglobulins
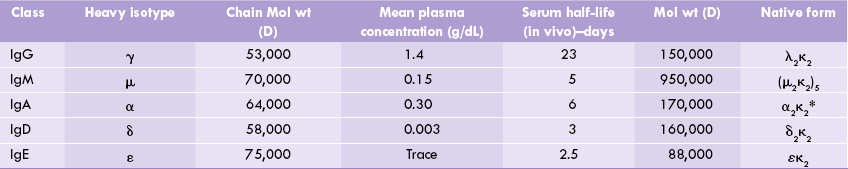
*Can form polymer structures of the basic structural unit.
Primary and Secondary Antibody Responses
Animals are capable of synthesizing specific antibodies virtually against all chemical groups. When the body system is challenged by exposure to a foreign antigen, distinct primary response (IgM dominated) and secondary antibody responses are observed. IgM is the major antibody of primary response, whereas IgG dominates the secondary response. Figure 5.17 shows kinetics of the appearance of IgM and IgG following injection of foreign antigen.
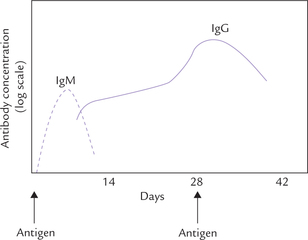
Fig. 5.17 Primary (---) and secondary (—) antibody response after the immune system is challenged by foreign antigen.
The IgM are first to appear as they are synthesized at a faster rate than IgG. Thus, they serve as first line of defense until the particular type of IgG is produced. Level of serum IgM starts rising a few days after the (first) injection of antigen. Its level declines approximately 10 days after the injection. A booster dose of antigen at this time produces a further increase in the level of IgG in the serum.
C Functions
The common function of all immunoglobulins is to serve as the first line of defense against foreign antigens. They recognize and bind with foreign antigens. In addition, Igs must interact with other cells and proteins for generating effector functions which are necessary for killing and eliminating foreign antigens. So functions of Igs can be grouped as below:
Functions Based on Antigen Recognition and Binding
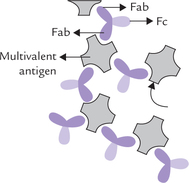
These are mediated by Fab region of the immunoglobu-lin, the region via which the immunoglobulin binds its antigen (ab stands for “antigen binding ” F for “fragment ”). Each Fab contains a binding site for an antigenic determinant. Being univalent (with a single binding site) a Fab alone cannot cross-link the antigen molecules that contain multiple determinants (i.e. multivalent antigens). Such cross-linking is possible with intact IgG molecule, which contains two identical antigen-binding sites. Each of these sites can bind to different antigen determinant on a multivalent antigen as shown in Figure 5.18 . This opens the possibility of formation of large interlinked structures in which antibodies serve as bridges between the multiva-lent antigens. This may lead to precipitation of soluble antigens by cross-linking, or to agglutination by cross-linking of particulate antigens (e.g. viruses, bacteria, or parasites). Blocking of attachment of viruses or bacterial toxins (i.e. neutralization) may also occur in this manner.
Functions Based on Effector Response
The Fc fragment, called Fc because it is crystallized rapidly and does not bind with antigen. However, it mediates certain important protective responses called effector functions which enable the immunoglobulin to destroy the target cell. The following effector functions are known:
Complement activation: The complement factors, about 20 in number, get activated by Ig molecules and cause lysis of the invading antigen. The Ig molecules that are capable of activating complement (IgG and IgM) have receptors for the complementary factors in their Fc region through which they bind these factors and activate the complement cascade. The complement activation enhances the immune response by
Opsonization: It is the process in which Ig molecules promote uptake of antigen by phagocytic cells. The phagocytes have receptors for Fc portion of Ig, and therefore when an antigen binds an Ig molecule, it is easily recognized by phagocyte (Fig. 5.19 ).
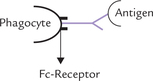
Binding to high affinity Fc receptors also leads to activation of phagocytes which now display enhanced phago-cytic activity.
The stimulation of phagocytosis by an Ig molecule bound to the surface of a foreign particle (antigen) is called opsonization; the Ig is called opsonin. Complement factors can also serve as opsonins.
Antibody-dependent cell-mediated cytolysis: It is the process in which Ig molecules mediate destruction of virus infected cells, or tumour cells, by killer cells (Fig. 5.20 ). The killer cells involved in this process are called the natural killer cells (NK cells), or ADCC effector cells. They possess Fc receptors on their cell surface, which can bind with the Fc portion of the antibody.
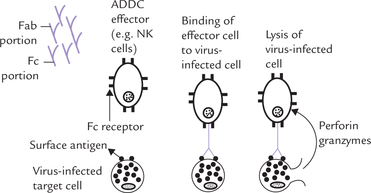
The virus-infected cells bind the antibodies through the Fab portion of the latter. The Fc portion of these antibodies bind with the Fc receptors of the NK cells. Thus antibodies serve as a bridge between the infected target cell and the effector cell. Lysis of the target cell is then brought about by the secretion of lytic enzymes (from the NK cells) such as the perforins and the powerful proteases called granzymes.
Classes of Immunoglobulins and their Effector Functions
Properties of various classes of immunoglobulins and their distinct effector functions are described here.
IgG
It is the major immunoglobulin that constitutes about 75-80% of the total immunoglobulins in circulation. The subclasses of IgG are: IgG1, IgG2, IgG3, IgG4. They differ in their degree of segmental flexibility and their capability to trigger complement fixation and other effector functions.
IgG1 makes up about 50% of the total IgG. It is the only type of immunoglobulin that can cross placenta and is localized in the vascular and extracellular compartments. IgG can enter the extravascular space and get involved in all the three effector functions (Table 5.5 ).
Table 5.5
| Class of Ig | Function |
| IgG | Crosses placenta to provide immunity to newborn. |
| Acts as opsonin, i.e. binds to Fc-receptors on macrophages, monocytes and neutrophils and enhances the phagocytosis of the bound antigen. | |
| Potent activator of complement system. | |
| Is the only class of Ig that is active in mediating antibody-dependent cell-mediated cytolysis. | |
| Easily diffuses into extracellular spaces, so plays a major role in neutralizing toxins. | |
| IgM | Most effective activator of classical complement pathway due to multiple binding sites. |
| Acts as opsonin. | |
| Present on the B-cell surface and acts as a receptor for antigen binding. | |
| Especially potent against multivalent bacterial agents, which are agglutinated. | |
| IgA | Present in breast milk, so provides immunological protection to infant’s gut. |
| Binds with antigen on epithelial cell surfaces and prevents their entry. | |
| IgE | Responsible for immediate hypersensitivity reaction or allergic reaction. |
| IgD | Involved in B-cell activation: present on surface of B-cell and (along with IgM) acts as receptor for antigen binding. |
IgM
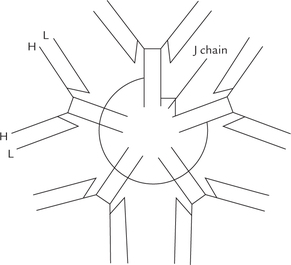
It is the antibody that is produced first when the immune system is challenged. Thus, IgM dominates the primary immune response. IgM, the largest immunoglobulin is a pentamer (Fig. 5.21 ) that is confined to vascular spaces. It cannot traverse blood vessels due to its large size. The five subunits of the IgM are joined by linkers called J pep-tides. The IgM serves as receptor on the B lymphocytes and helps in establishing humoral immunity, being the major antibody of primary immune responses. Natural antibodies are also IgM in nature. The natural antibodies are produced without any antigenic stimulation. For example, a person having blood group A will have anti-B group antibodies in his circulation.
IgA
IgA is the major immunoglobulin in seromucous secretions, e.g. saliva, tears, sweat, walls of intestine and bronchial mucous. It is abundant in colostrum, the initial secretion from the mother’s breast after childbirth. It may occur as a single (monomer) or a double unit (dimer).
IgA provides surface immunity by binding with antigen on outer epithelial surfaces, and eliminating them. In this way it prevents entry of foreign antigen into the body.
IgE
IgE is a Y-shaped monomer, plasma concentration of which is lowest among all the five immunoglobulin classes. It is a Y-shaped monomer, connected by the tail to the cell membrane of mast cells and basophils. Upon binding to an antigen, IgE stimulates degranul ation of these cells and release of inflammatory substances (i.e. histamine). Its concentration increases in response to allergic reactions. Probably it has a role in immunity to helminths and is associated with immediate hypersensitivity.
IgD
IgD is present on the surface of B-lymphocytes but its role is not clear. Probably it is involved in the antigen recognition process.
In addition to the five major classes of immunoglobulins just described, subclasses within each class also exist. They differ from one another by slight variations in the sequence of amino acids in the heavy chain. The subclasses of IgG (IgG1, IgG2, IgG3 and IgG4) correspond to four different types of heavy chains (γ1, γ2, γ3 and γ4) IgM and IgA have two subclasses each.
D Isotypes, Allotypes and Idiotypes
Isotypes are variants in molecules seen in all normal persons such as classes and subclasses of immunoglobulins (IgG, IgA, IgM, etc.). They depend on the heavy chain make up; for example, IgG possesses the 7 heavy chain isotype, IgM has μ, IgA has α, and so on (Table 5.4).
Allotypes are variations within a subclass; for example, Gma+ and Gma– are the allotypes within the IgG class. In people with Gma+, IgG1 molecule has an amino acid sequence, Asp-Glu-Leu Thr. The corresponding sequence in Gma– people is Met-Glu-Glu-Thr. These markers show Mendelian recessive hereditary character.
Idiotypes: Unique sequence of amino acids is present in hypervariable regions of an antibody, and is individually specific for each antibody molecule. Such sequence variation is known as idiotypic variation and each individual determinant is called idiotype (or idiotypic determinant).
The idiotypic determinants are located in the variable part of an antibody (VL and VH). If three classes of antibodies (IgG, IgM and IgA) are raised against the same antigen, they will have identical variable domains, both in the light chain (VL) and the heavy chain (VH). These three antibodies will share an idiotype. Idiotypic variation is specific for each antibody molecule, as mentioned earlier.
E Diseases Involving Antibodies
The diseases associated with antibodies are divided into two types. They are caused by:
• Underproduction of antibody (causing hypo- or agammaglobulinaemia).
• Overproduction of antibody (causing hypergamma-globulinaemia).
Hypogammaglobulinaemia
Various inherited disorders are known in which underproduction of immunoglobulins occurs. Most severe of these is agammaglobulinaemia a rare, X-chromosome associated disorder, in which there is total absence of immunoglobulins. The affected children are highly susceptible to the bacterial infections but react normally to the viral infections. In some other milder forms, termed hypogammaglobulinaemia, the underproduction may be restricted to a single class of immunoglobulins.
Hypogammaglobulinaemia may also be an acquired disease. It commonly occurs in hematological malignancies, such as chronic lymphatic leukemia, multiple myeloma and Hodgkin’s disease. It can arise as a complication following the use of cytotoxic drugs or as a symptom of severe protein losing state such as the nephrotic syndrome.
Hypogammaglobulinaemia may arise in some physiological states also. At the time of birth, plasma concentration of all classes of immunoglobulins, except IgG, are low. The IgG molecules, present in relatively higher concentration, are of maternal origin that crossed placenta in the last trimester of pregnancy. The physiological hypogammaglobulinaemia in infants partially accounts for their increased susceptibility to infections.
Hypergammaglobulinaemia
Increased plasma immunoglobulin levels occur in various auto-immune disorders, such as rheumatoid arthritis, systemic lupus erythematosus (SLE), and in some liver diseases that have auto-immune basis. In auto-immune diseases, body rejects its own proteins which became antigenic. Probably, buried antigen sites in endogenous proteins (epitopes) get exposed; or certain exogenous triggers bind with “self-proteins ” and make them antigenic. An immune response, involving production of antibodies against these proteins, is aroused. This leads to the destruction of self-proteins.
Markedly increased immunoglobulin levels occur in the patients suffering from myelomatosis (immunocy-toma tumours). The condition is known as multiple myeloma, as discussed earlier. The immunoglobulins are produced from a single clone of cells of β-lymphocyte series, most frequently plasma cells. Due to common origin these immunoglobulins are identical and are known as paraproteins. They are mostly monoclonal IgG, but may also be IgA or IgD. They are seen as a single discreet M band on an electrophoretogram, usually in the gamma region. In the urine of these patients, massive excretion of Bence-Jones proteins occurs. The latter are identified as low molecular weight proteins (light chains). For clinical description of an immunoglobulin disorder (see Case 5.4).
The diseases involving antibodies are discussed further in Chapter 33.
Exercises
Essay type questions
1. Write briefly on plasma proteins and their functions. Discuss methods for their separation and explain why albumin deficiency causes oedema.
2. Draw electrophoretogram of plasma proteins in a normal subject and compare it with that of a multiple myeloma patient.
3. Describe the characteristics, structural features and functions of different immunoglobulins.
4. What are glycoproteins? Discuss their structural features and biomedical importance.
5. Explain the differences between N- and O-linked oligosaccharides in glycoproteins. How do the physical properties of glycoproteins relate to their biological roles.