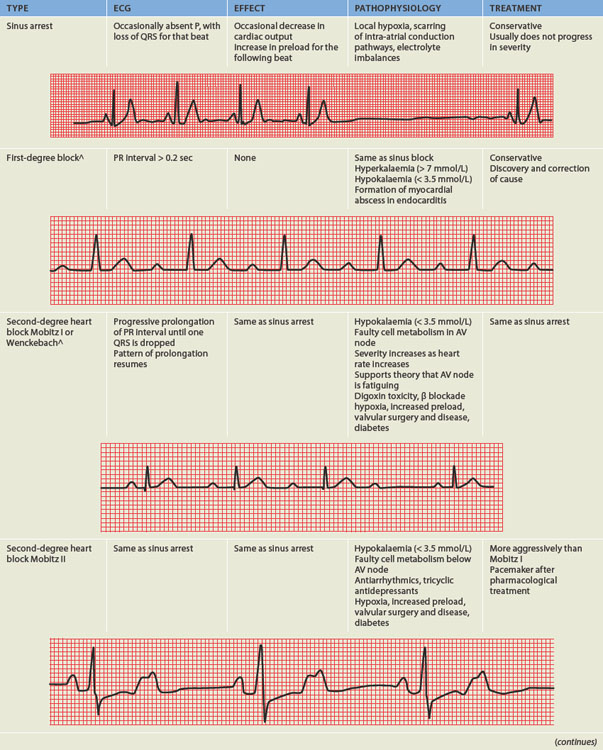
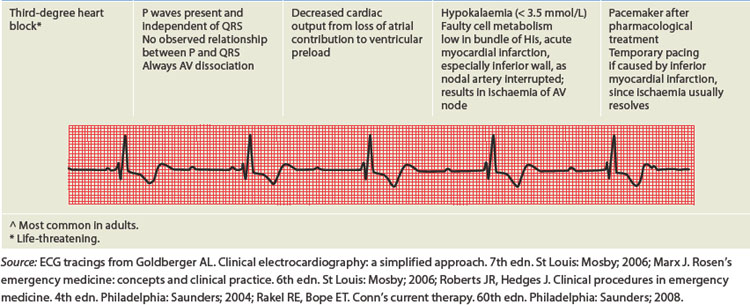
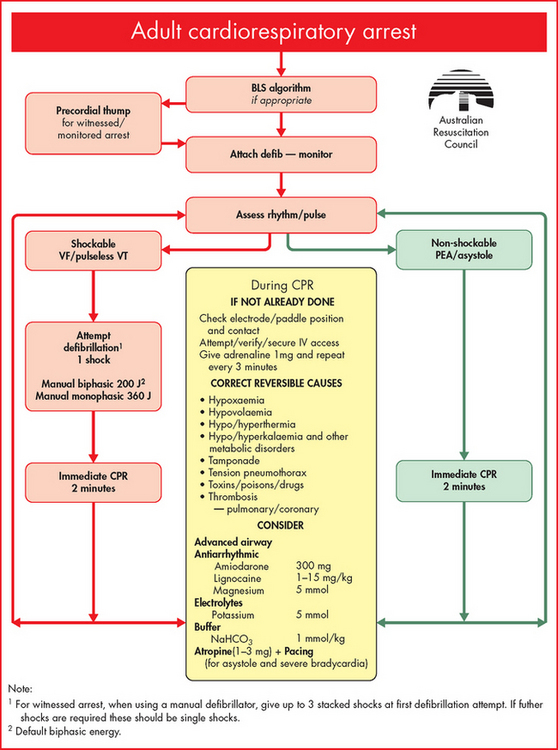
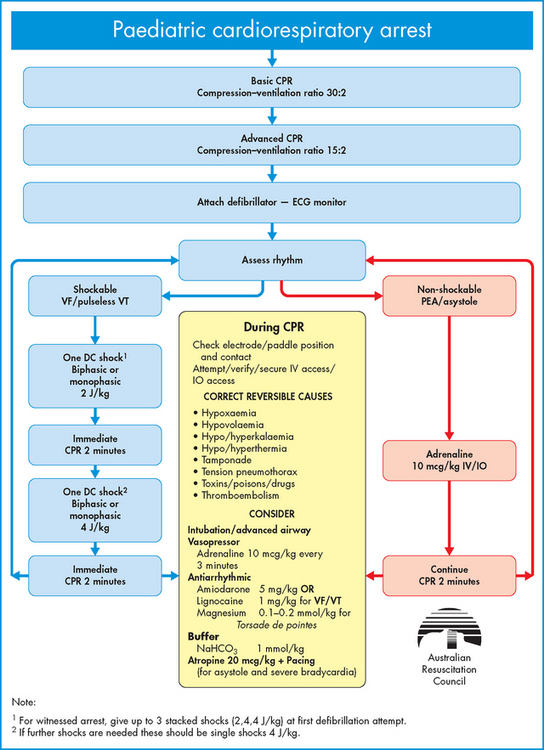
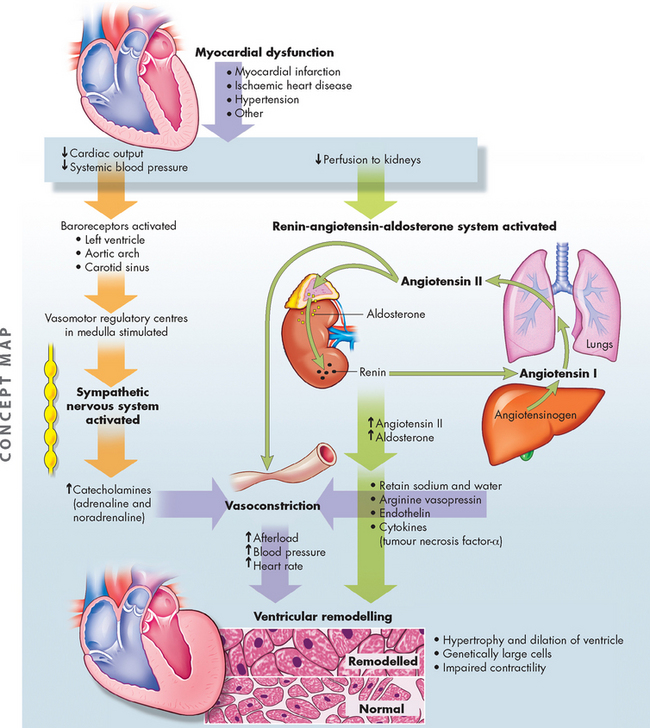
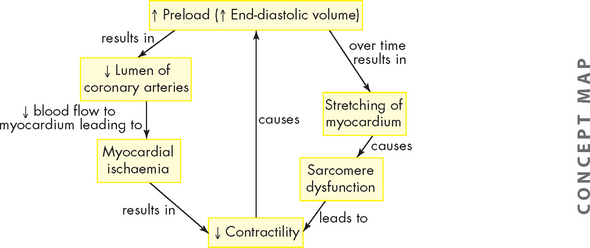
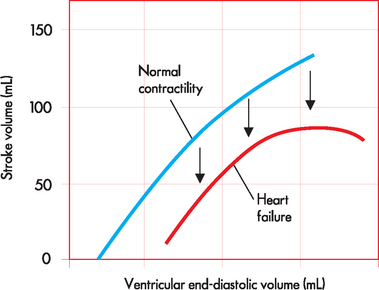
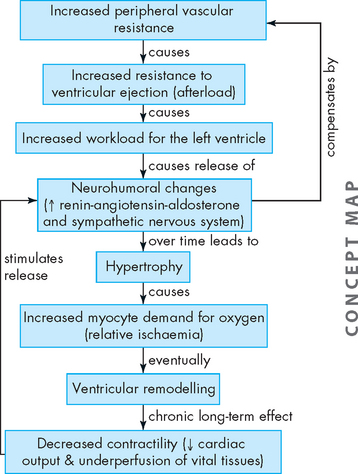
ALTERATIONS OF THE HEART WALL
Disorders of the pericardium
As you will recall, the pericardium is the outer layer of the heart, having approximately 10–30 mL of pericardial fluid to lubricate and protect the heart from infection and inflammation. Inflammation of the pericardium, known as pericarditis, is usually a response to other cardiac conditions, such as acute myocardial infarction or diseases of the thorax. The most common symptom arising from pericarditis is pain. Pericardial disease is often a localised manifestation of another disorder, such as infection (bacterial, viral, fungal or parasite); trauma or surgery; neoplasm; or a metabolic, immunological or vascular disorder (uraemia, rheumatoid arthritis, systemic lupus erythematosus).
Acute pericarditis
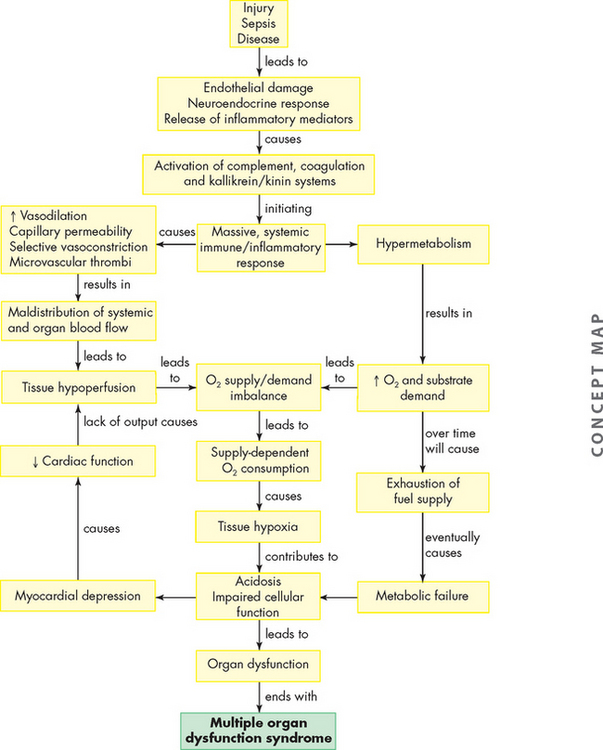
Approximately 90% of cases of acute pericarditis are caused by viruses or are idiopathic.77 Acute pericarditis can also be caused by acute myocardial infarction, uraemia, cardiac surgery, some medications and autoimmune disorders. There are many forms of acute pericarditis, including serous, fibrinous, purulent and haemorrhagic. The pericardial membranes become inflamed and roughened and an exudate may develop. Figure 23-41 shows deposits of fibrin on the pericardium possibly following an acute myocardial infarction.
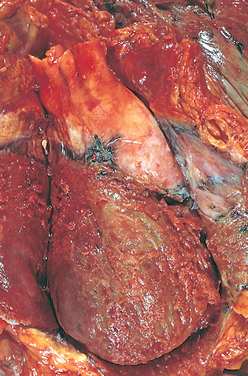
FIGURE 23-41 Fibrinous pericarditis.
Deposits of fibrin are shown on the pericardium.
Source: Kumar V. Robbins & Cotran pathologic basis of disease. 8th edn. Philadelphia: Saunders; 2010.
Symptoms include the sudden onset of severe retrosternal chest pain that worsens with respiratory movements and with lying down. The pain may radiate to the back as a result of irritation of the phrenic nerve (which innervates the trapezius muscles) as it traverses the pericardium.77 Individuals with acute pericarditis also report dysphagia, restlessness, irritability, anxiety, weakness and malaise.
Physical examination often discloses low-grade fever (< 38°C) and sinus tachycardia. Friction rub — a short, scratchy, grating sensation similar to the sound of sandpaper — may be heard at the cardiac apex and left sternal border and is diagnostic for pericarditis. The rub is caused by the roughened pericardial membranes rubbing against each other. Friction rubs are not present in approximately 15% of individuals with acute pericarditis or they may be intermittently heard and transient. Hypotension or the presence of pulsus paradoxus (an exaggerated decrease in systolic blood pressure with inspiration) is suggestive of cardiac tamponade (pericardial fluid impairing heart function; see below), which can be lethal. Electrocardiographic changes may reflect inflammatory processes through PR segment depression and diffuse ST segment elevation without Q waves, and they may remain abnormal for days or even weeks.
Treatment for uncomplicated acute pericarditis consists of relieving symptoms and includes anti-inflammatory agents. Exploration of the underlying cause is important. If pericardial effusion develops, aspiration of the excessive fluid may be necessary. Acute pericarditis is usually self-limiting.
Pericardial effusion
Pericardial effusion — the accumulation of fluid in the pericardial cavity — can occur in all forms of pericarditis. The fluid may be a transudate, such as the serous effusion that develops with left heart failure, overhydration or hypoproteinaemia. More often, however, the fluid is an exudate, which reflects pericardial inflammation like that seen with acute pericarditis, heart surgery, some chemotherapeutic agents, infections and autoimmune disorders such as systemic lupus erythematosus.
Pericardial effusion, even in large amounts, is not necessarily clinically significant, except that it indicates an underlying disorder. If an effusion develops gradually, the pericardium can stretch to accommodate large quantities of fluid without compressing the heart. If the fluid accumulates rapidly, however, even a small amount (50–100 mL) may create sufficient pressure to cause cardiac compression, a serious condition known as cardiac tamponade. The danger is that pressure exerted by the pericardial fluid eventually will equal diastolic pressure within the heart chambers, which will interfere with right atrial filling during diastole. This causes increased venous pressure, systemic venous congestion, and signs and symptoms of right heart failure (distention of the jugular veins, oedema, hepatomegaly). Decreased atrial filling leads to decreased ventricular filling, decreased stroke volume and reduced cardiac output. Life-threatening circulatory collapse may occur.
An important clinical finding is pulsus paradoxus, in which there is a substantial decrease in systolic blood pressure during inspiration. Pulsus paradoxus in the setting of a pericardial effusion indicates tamponade and reflects impairment of diastolic filling of the left ventricle plus reduction of blood volume within all four cardiac chambers.
Other clinical manifestations of pericardial effusion are distant or muffled heart sounds, poorly palpable apical pulse, dyspnoea on exertion and dull chest pain. A chest X-ray film may disclose a water-bottle configuration of the cardiac silhouette. Doppler echocardiogram can detect an effusion as small as 20 mL.
Treatment of pericardial effusion or tamponade generally consists of pericardiocentesis (aspiration of excessive pericardial fluid) and treatment of the underlying condition. Persistent pain may be treated with analgesics, anti-inflammatory medications or steroids.
Disorders of the myocardium: the cardiomyopathies
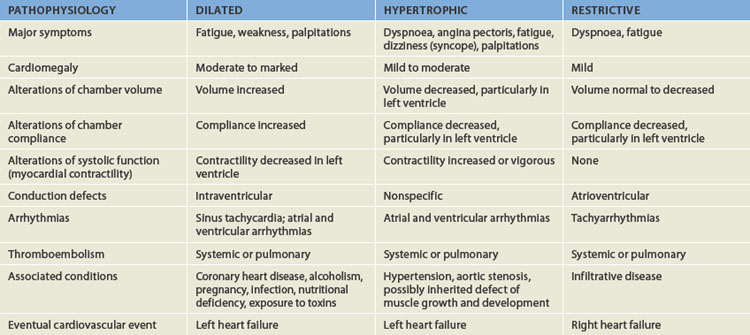
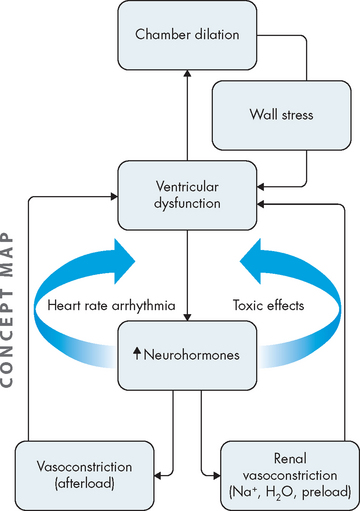
The cardiomyopathies are a diverse group of diseases that primarily affect the myocardium itself. Most are the result of remodelling caused by myocardial and neurohumoral responses (both neural and hormonal) to ischaemic events and hypertension. They may, however, be secondary to infectious disease, exposure to toxins, systemic connective tissue disease, infiltrative and proliferative disorders or nutritional deficiencies.78 However, many cases are idiopathic — that is, their cause is unknown. The cardiomyopathies are categorised as dilated (formerly, congestive), hypertrophic or restrictive, depending on their physiological effects on the heart (see Figure 23-42 and Table 23-10).
FIGURE 23-42 Diagram showing the three types of cardiomyopathy.
A Normal heart. B Dilated cardiomyopathy demonstrating enlargement of all four chambers. C Hypertrophic cardiomyopathy showing a thickened left ventricle. D Restrictive cardiomyopathy characterised by a small left ventricular volume.
Source: Copstead-Kirkhorn L-EC, Banasik JL. Pathophysiology. 4th edn. Philadelphia: Saunders; 2009.
Disorders of the endocardium
Valvular dysfunction
Disorders of the endocardium (the innermost lining of the heart wall) damage the heart valves, which are composed of endocardial tissue. Endocardial damage can be either congenital or acquired. The acquired forms cause inflammatory, ischaemic, traumatic, degenerative or infectious alterations of valvular structure and function. One of the most common causes of acquired valvular dysfunction is degeneration or inflammation of the endocardium secondary to rheumatic heart disease (see Table 23-11). Valvular stenosis occurs when the valve orifice is constricted and narrowed, so that blood cannot flow forwards and the workload of the cardiac chamber ‘behind’ the diseased valve increases (see Figure 23-43). Pressure within the ventricular or atrial chamber rises to overcome resistance to flow through the valve, causing the myocardium to work harder and producing myocardial hypertrophy.
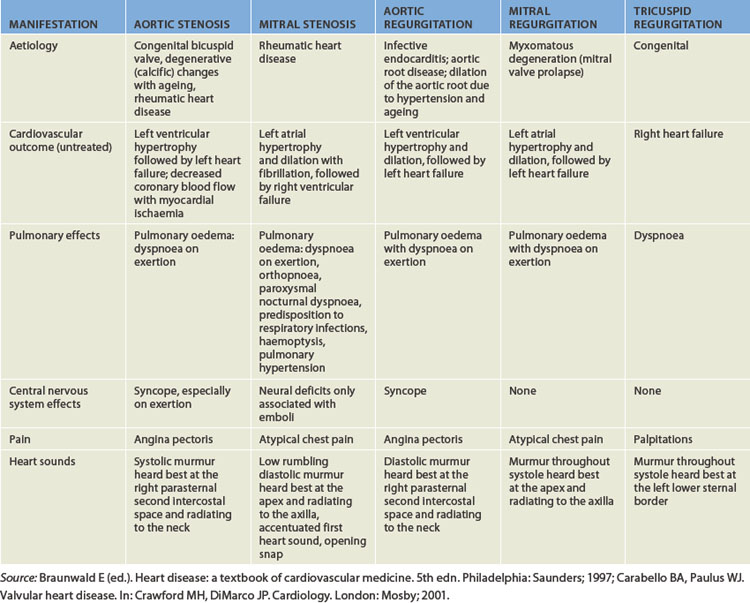
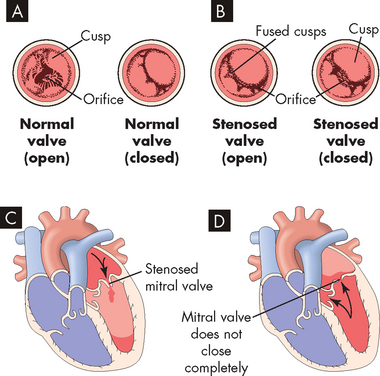
FIGURE 23-43 Valvular stenosis and regurgitation.
A Normal position of the valve leaflets, or cusps, when the valve is open and closed. B Open position of a stenosed valve (left) and open position of a closed regurgitant valve (right). C Haemodynamic effect of mitral stenosis. The stenosed valve is unable to open sufficiently during left atrial systole, inhibiting left ventricular filling. D Haemodynamic effect of mitral regurgitation. The mitral valve does not close completely during left ventricular systole, permitting blood to re-enter the left atrium.
In valvular regurgitation (also called insufficiency or incompetence), the valve leaflets, or cusps, fail to shut completely, permitting blood flow to continue even when the valve is supposed to be closed (see Figure 23-43). Blood can leak back into the chamber ‘upstream’, which increases the volume of blood the heart must pump and increases the workload of both the atrium and the ventricle. Increased volume leads to chamber dilation with cardiomegaly (see Figure 23-44) and increased workload leads to hypertrophy. Although all four heart valves may be affected, in adults those of the heart’s left side (the mitral and aortic valves) are far more commonly affected than those of the right (the tricuspid and pulmonary valves).
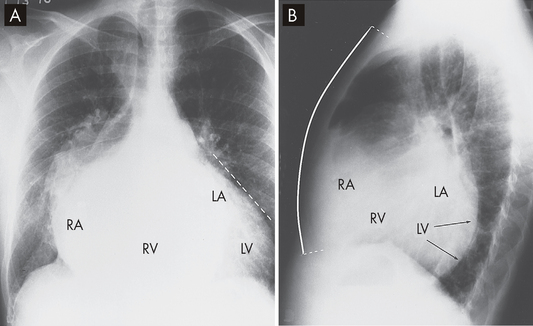
FIGURE 23-44 Cardiomegaly due to mitral and tricuspid regurgitation.
Note the enlarged heart, which is wider than normal. A On the posteroanterior chest X-ray there is marked enlargement of not only the left atrium (LA) but also the left ventricle (LV; seen as straightening of the left cardiac border), as well as right-sided enlargement, particularly of the right atrium (RA; seen by marked prominence of the right cardiac border). B On the lateral view of the chest the left ventricle can be seen overlapping the spine, and the right atrium and right ventricle (RV) have filled in the retrosternal space to more than the usual lower one-third.
Source: Metter FA. Essentials of radiology. 2nd edn. Philadelphia: Saunders; 2005.
Valvular dysfunction stimulates chamber dilation and myocardial hypertrophy, both of which are compensatory mechanisms intended to increase the pumping capability of the heart but which lead to cardiac dysfunction over time. Eventually, myocardial contractility diminishes, the ejection fraction is reduced, diastolic pressure increases and the ventricles fail from overwork. Depending on the severity of the valvular dysfunction and the capacity of the heart to compensate, valvular alterations cause a range of symptoms and some degree of incapacitation (see Table 23-11).
Stenosis
Aortic stenosis
Aortic stenosis is now the most common valvular abnormality, affecting nearly 2% of adults older than 65 years of age. It has three common causes:
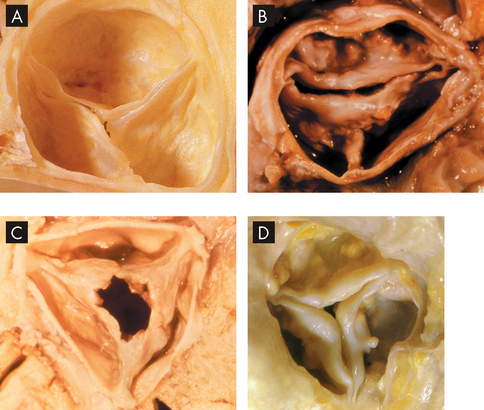
A Normal aortic valve. B Congenital bicuspid aortic stenosis. C Rheumatic aortic stenosis. D Calcified degenerative aortic stenosis.
Source: A & D Manabe H, Yutani C (eds). Atlas of valvular heart disease. Singapore: Churchill Livingstone; 1998. B & C Courtesy of William C Roberts, MD.
Aortic valve degeneration with ageing is associated with lipoprotein deposition in the tissue with chronic inflammation and leaflet calcification.80 The orifice of the aortic semilunar valve narrows, causing diminished blood flow from the left ventricle into the aorta. Outflow obstruction increases pressure within the left ventricle as it tries to eject blood through the narrowed opening.
Aortic stenosis tends to develop gradually. Clinical manifestations include decreased stroke volume, reduced systolic blood pressure and narrowed pulse pressure (the difference between systolic and diastolic pressure). Heart rate is often slow and pulses are faint. Left ventricular hypertrophy develops to compensate for the increased workload. Eventually, hypertrophy increases myocardial oxygen demand, which the coronary arteries may be unable to meet. In addition, aortic stenosis is frequently accompanied by atherosclerotic coronary disease, further contributing to inadequate coronary perfusion.80
Untreated aortic stenosis can lead to arrhythmias, myocardial infarction and heart failure. Echocardiography can be used to assess the severity of valvular obstruction before the onset of symptoms, and management almost always includes valve replacement with a prosthetic valve (see Figure 23-46), followed by long-term anticoagulation and prophylaxis for endocarditis, as needed.
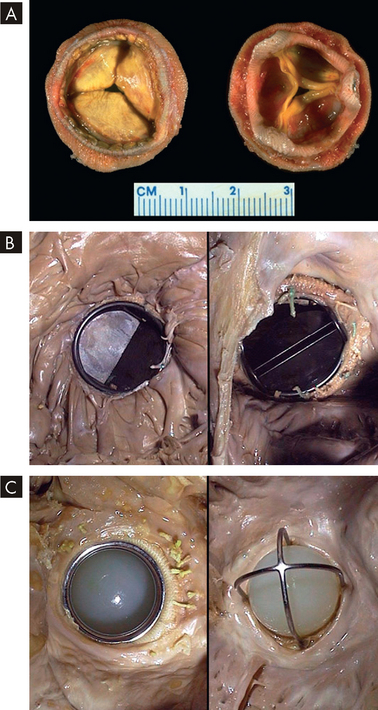
FIGURE 23-46 Various valve replacements.
A A porcine bioprosthesis. B A mechanical valve prosthesis of the tilting disc variety replacing the native mitral valve. C A mechanical valve prosthesis of the older ball-and-cage variety.
Source: Klatt EC. Robbins and Cotran atlas of pathology. 2nd edn. Philadelphia: Saunders; 2010.
Mitral stenosis
Mitral stenosis impairs the flow of blood from the left atrium to the left ventricle. Mitral stenosis is most commonly caused by rheumatic heart disease. In fact, when stenosed mitral valves are examined after removal for mitral valve replacement, almost all have evidence of rheumatic changes.81 In addition, mitral stenosis is two to three times more common in women than in men.82 Autoimmunity in response to group A β-haemolytic streptococcal protein antigens leads to inflammation and scarring of the valvular leaflets. Scarring causes the leaflets to become fibrous and fused and the chordae tendineae become shortened.
Impedance to blood flow results in incomplete emptying of the left atrium and elevated atrial pressure as the chamber tries to force blood through the stenotic valve. Continued increases in left atrial volume and pressure cause atrial dilation and hypertrophy. The risk of developing atrial arrhythmias (especially fibrillation) and arrhythmia-induced thrombi is high. As mitral stenosis progresses, symptoms of decreased cardiac output occur, especially during exertion. Continued elevation of left atrial pressure and volume causes pressure to rise in the pulmonary circulation. If untreated, chronic mitral stenosis develops into pulmonary hypertension, pulmonary oedema and right ventricular failure.
Management includes anticoagulation and endocarditis prophylaxis along with β-blockers or calcium channel blockers to slow the heart rate. Mitral stenosis can often be repaired surgically but may require valve replacement (usually porcine; see Figure 23-46A) in advanced cases.
Regurgitation
Aortic regurgitation
Aortic regurgitation results from an inability of the aortic valve leaflets to close properly during diastole resulting from abnormalities of the leaflets or the aortic root and annulus, or both.83 It can be congenital (bicuspid valve abnormalities) or acquired. Acquired aortic regurgitation can be caused by rheumatic heart disease, bacterial endocarditis, syphilis, hypertension, connective tissue disorders (e.g. Marfan’s syndrome), appetite-suppressing medications, trauma or atherosclerosis. In more than one-third of cases of aortic regurgitation there is no known cause.
The haemodynamic abnormalities depend on the size of the ‘leak’. During systole, blood is ejected from the left ventricle into the aorta. During diastole, some of the ejected blood flows back into the left ventricle. Volume overload occurs in the ventricle because it receives blood from both the left atrium and the aorta during diastole. Over time, the end-diastolic volume of the left ventricle increases and myocardial fibres stretch to accommodate the extra fluid. Compensatory dilation permits the left ventricle to increase its stroke volume and maintain cardiac output. Ventricular hypertrophy also occurs as an adaptation to the increased volume and because of increased afterload created by the high stroke volume and resultant systolic hypertension. Ventricular dilation and hypertrophy eventually cannot compensate for aortic incompetence and heart failure develops.
Clinical manifestations include widened pulse pressure resulting from increased stroke volume and diastolic backflow. Other symptoms are usually associated with heart failure, which occurs when the ventricle can no longer pump adequately. Arrhythmias and endocarditis are common complications of aortic regurgitation. The severity of regurgitation can be estimated by echocardiography, and surgical management (valve replacement; see Figure 23-46) may be delayed for many years through careful use of vasodilators and inotropic agents.
Mitral regurgitation
Mitral regurgitation has many possible causes, including mitral valve prolapse (see below), rheumatic heart disease, infective endocarditis, acute myocardial infarction, connective tissue diseases (such as Marfan’s syndrome) and congestive cardiomyopathy (myocardial disease). Mitral regurgitation permits backflow of blood from the left ventricle into the left atrium during ventricular systole. Eventually, the left ventricular volume increases, causing it to become dilated and hypertrophied to maintain adequate cardiac output. The volume of backflow re-entering the left atrium gradually increases, causing atrial dilation and associated atrial fibrillation. As the left atrium enlarges, the valve structures stretch and become deformed, leading to further backflow. As mitral valve regurgitation progresses, left ventricular function may become impaired to the point of failure. Eventually, increased atrial pressure also causes pulmonary hypertension and failure of the right ventricle. Mitral incompetence is usually well tolerated — often for years — until ventricular failure occurs. Most clinical manifestations are caused by heart failure. The severity of regurgitation can be estimated by echocardiography, and surgical repair or valve replacement may become necessary (see Figure 23-46).
Tricuspid regurgitation
Tricuspid regurgitation is more common than tricuspid stenosis (narrowing) and is usually associated with failure and dilation of the right ventricle secondary to pulmonary hypertension. Tricuspid valve incompetence leads to volume overload in the right ventricle, increased systemic venous blood pressure and right heart failure. Pulmonary semilunar valve dysfunction can have the same consequences as tricuspid valve dysfunction.
Mitral valve prolapse syndrome
In mitral valve prolapse syndrome one or both of the cusps of the mitral valve billow upwards (prolapse) into the left atrium during systole. The most common cause of mitral valve prolapse is degeneration of the leaflets in which the cusps are redundant, thickened and scalloped because of changes in tissue proteoglycans and infiltration by myofibroblasts.84 Mitral regurgitation occurs if the ballooning valve permits blood to leak into the atrium.
Many cases of mitral valve prolapse are completely asymptomatic. Symptomatic mitral valve prolapse can cause palpitations related to arrhythmias, tachycardia, light-headedness, syncope, fatigue (especially in the morning), lethargy, weakness, dyspnoea, chest tightness, hyperventilation, anxiety, depression, panic attacks and atypical chest pain. Many symptoms are vague and puzzling and are unrelated to the degree of prolapse.
Evaluation of mitral valve prolapse includes physical assessment and laboratory evaluation. Echocardiography is the procedure of choice for diagnosing the disorder. Most individuals with mitral valve prolapse have an excellent prognosis, do not develop symptoms and do not require any restriction in activity or medical management. However, some individuals have an increased risk for complications such as infective endocarditis, stroke and sudden death.
Rheumatic heart disease
Rheumatic fever is a diffuse, inflammatory disease caused by a delayed exaggerated immune response to infection by the group A β-haemolytic streptococcus in genetically predisposed individuals. In its acute form, rheumatic fever is a febrile illness characterised by inflammation of the joints, skin, nervous system and heart.85 If untreated, rheumatic fever can cause scarring and deformity of cardiac structures resulting in rheumatic heart disease.
With increases in living standards and infection control, the incidence of rheumatic heart disease has been in decline in Western countries.85 However, Australia has the unenviable position of recording the highest rates of rheumatic heart disease in the world.86 Alarmingly, the rates are highest among Aboriginal people living in the Northern Territory and have been related to poor hygiene and standards of living compared to the non-Indigenous population87 — in fact, death rates from rheumatic heart disease are 15 and 23 times higher for Aboriginal men and women, respectively.88
PATHOPHYSIOLOGY
Acute rheumatic fever can develop only as a sequel to pharyngeal infection by group A β-haemolytic streptococcus. Streptococcal skin infections do not progress to acute rheumatic fever, although both skin and pharyngeal infections can cause acute glomerulonephritis. This is because the strains of the microorganism that affect the skin do not have the same antigenic molecules in their cell membranes as those that cause pharyngitis and therefore do not elicit the same kind of immune response. Acute rheumatic fever is the result of an abnormal humoral and cell-mediated immune response to group A streptococcal cell membrane antigens called M proteins (see Figure 23-47).
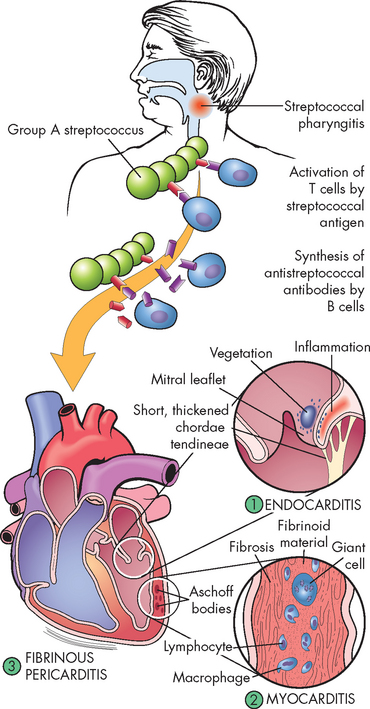
FIGURE 23-47 The pathogenesis and structural changes of rheumatic heart disease.
Beginning usually with a sore throat, rheumatic fever can develop only as a sequel to pharyngeal infection by group A β-haemolytic streptococcus. Suspected as a hypersensitivity reaction, it is proposed that antibodies directed against the M proteins of certain strains of streptococci cross-react with tissue glycoproteins in the heart, joints and other tissues. The exact nature of cross-reacting antigens has been difficult to define, but it appears that the streptococcal infection causes an autoimmune response against self-antigens. Inflammatory lesions are found in various sites; the most distinctive within the heart are called Aschoff bodies. The chronic sequelae result from progressive fibrosis because of healing of the inflammatory lesions and the changes induced by valvular deformities.
Source: Damjanov I. Pathology for the health professions. 3rd edn. St Louis: Saunders; 2006.
Diffuse, proliferative and exudative inflammatory lesions develop in the connective tissues, especially in the heart. The inflammation may subside before treatment, leaving behind damage to the heart valves and increasing the individual’s susceptibility to recurrent acute rheumatic fever after any subsequent streptococcal infections. Repeated attacks of acute rheumatic fever cause chronic proliferative changes (scarring) in the affected regions.
Approximately 10% of individuals with rheumatic fever develop rheumatic heart disease. The primary lesion usually involves the endocardium. Endocardial inflammation causes swelling of the valve leaflets, with secondary erosion along the lines of leaflet contact. Small beadlike clumps of vegetation containing platelets and fibrin are deposited on eroded valvular tissue and on the chordae tendineae (see Figure 23-48).
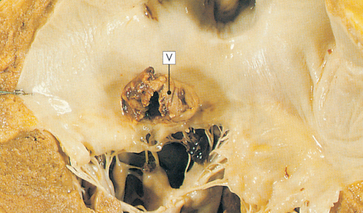
FIGURE 23-48 Mitral stenosis with vegetation.
Mitral stenosis and clumps of vegetation (V) containing platelets and fibrin are shown in this micrograph. The mitral leaflets are thickened and fused.
Source: Stevens A, Lowe J. Pathology. Edinburgh: Mosby; 2000.
Myocarditis (inflammation of the myocardium) may occur. Cardiomegaly and left heart failure may occur during episodes of untreated acute or recurrent rheumatic fever. Conduction defects and atrial fibrillation are often associated with rheumatic heart disease.
CLINICAL MANIFESTATIONS
Many common clinical manifestations of acute rheumatic fever — fever, lymphadenopathy, arthralgia (painful joints), nausea, vomiting, epistaxis (nose bleeds), abdominal pain and tachycardia — are also associated with other disorders and are therefore not diagnostic of the disease. The major clinical manifestations of acute rheumatic fever are carditis, acute migratory polyarthritis (inflammation of more than one joint), chorea (a central nervous system disorder — see Chapter 9) and erythema marginatum (truncal rash), which may occur singly or in combination 1–5 weeks after streptococcal infection of the pharynx.
EVALUATION AND TREATMENT
When combined with physical assessment findings, laboratory values lend significant support to the diagnosis of acute rheumatic fever. A positive throat culture for group A β-haemolytic streptococci can be an important finding when associated with certain physical signs. Elevated white blood cell count, erythrocyte sedimentation rate and C-reactive protein indicate inflammation. All three are usually increased at the time cardiac or joint symptoms begin to appear. They are more useful in identifying an acute inflammatory process and suggesting prognosis than in diagnosing acute rheumatic fever. These test results return towards normal levels as the inflammatory process resolves.
Therapy for acute rheumatic fever is aimed at eradicating the streptococcal infection and involves a 10-day regimen of oral penicillin or erythromycin administration. Non-steroidal anti-inflammatory drugs (NSAIDs) are used as anti-inflammatory agents for both rheumatic carditis and arthritis. Serious carditis may require adding cardiac glycosides, corticosteroids, diuretics and bed rest to the regimen. Surgical repair of damaged valves may be needed in chronic recurrent rheumatic fever and carditis leading to rheumatic heart disease.
Research suggests that a rheumatic recurrence will develop in 50–65% of children with known rheumatic fever if they have another group A streptococcal infection. To prevent a recurrence of acute rheumatic fever, continuous prophylactic antibiotic therapy may be necessary for as long as 5 years.85 Appropriate antibiotic therapy given within the first 9 days of infection usually prevents rheumatic fever.
Infective endocarditis
Infective endocarditis is a general term used to describe infection and inflammation of the endocardium, especially the cardiac valves. Bacteria are the most common cause of infective endocarditis including Streptococcus viridans, Staphylococcus aureus, Staphylococcus epidermidis and group A β-haemolytic streptococci. Infective endocarditis was once a lethal disease, but morbidity and mortality diminished significantly with the advent of antibiotics and improved diagnostic techniques (see the box ‘Risk factors for ineffective endocarditis’).89
PATHOPHYSIOLOGY
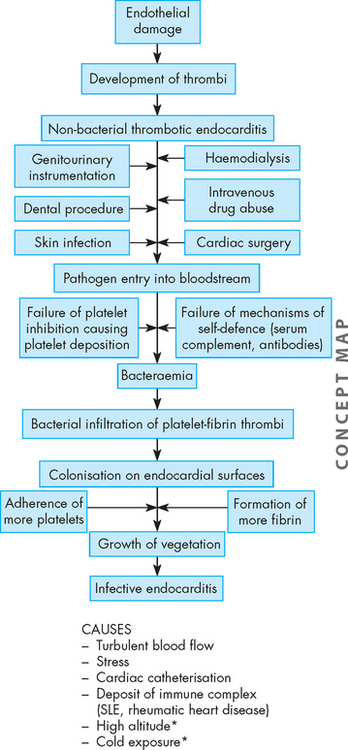
The pathogenesis of infective endocarditis requires at least three critical elements (see Figure 23-49): (1) the endocardium (e.g. heart valve) must be ‘prepared’, usually by endothelial damage, for microorganism colonisation; (2) blood-borne microorganisms must adhere to the damaged endocardial surface; and (3) the microorganisms must proliferate and promote the propagation of infective endocardial vegetation.
Endocarditis risk in children
Children with congenital heart disease are at risk for developing endocarditis. Although the risk is low, a transient bacteraemia has been noted to follow dental and surgical procedures and instrumentation involving mucosal surfaces. A blood-borne pathogen can settle in areas of the heart where there is high turbulence, an abnormal valve or vessel, or an artificial material such as a valve or homograft. Streptococcus viridans (α-haemolytic streptococci) is the most commonly found pathogen following dental or oral procedures. Enterococcus faecalis (enterococci) is the most common bacterium found following genitourinary and gastrointestinal tract surgery or instrumentation. The Infective Endocarditis Prophylaxis Expert Group convened by the Therapeutics Guidelines has provided updated guidelines for the prevention of bacterial endocarditis. The type and dose of antibiotic prophylaxis recommended depend on the procedure and the cardiac classification of risk for endocarditis.
Source: The Infective Endocarditis Prophylaxis Expert Group. Prevention of endocarditis. 2008 update from Therapeutic Guidelines: antibiotic version 13, and Therapeutic Guidelines: oral and dental version 1. Melbourne: Therapeutic Guidelines Limited; 2008.
Endocardial damage exposes the collagen within the endothelial basement membrane — this collagen attracts platelets and thereby stimulates thrombus formation on the membrane. This causes an inflammatory reaction. Infective endocarditis cannot develop unless microorganisms gain access to the bloodstream. They may enter the bloodstream during intravenous drug use, trauma or minor procedures such as dental cleaning or bladder catheterisation, or they may spread from uncomplicated upper respiratory or skin infections. A significant number of cases of infective carditis are healthcare–acquired infections in origin, especially those that result from genitourinary or gastrointestinal procedures or from surgical wound infections.
Once the endocardial surface is colonised, infected vegetations form (see Figure 23-50). Bacteria may accelerate fibrin formation by activating the coagulation cascade. Although endocardial tissue is constantly bathed in antibody-containing blood and is surrounded by scavenging monocytes and leucocytes, bacterial colonies are inaccessible to host defences because they are embedded in the protective fibrin clots. The lesions can form anywhere on the endocardium but usually occur on the endocardial surfaces of heart valves and surrounding structures.
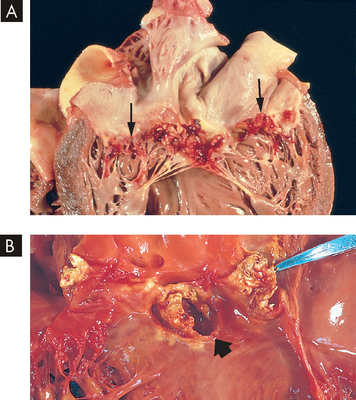
FIGURE 23-50 Infective endocarditis.
A Endocarditis of the mitral valve (subacute, caused by Streptococcus viridans). The large, friable vegetations are denoted by arrows. B Acute endocarditis of congenitally bicuspid aortic valve (caused by Staphylococcus aureus) with extensive cuspal destruction and ring abscess (arrow).
Source: Schoen FJ. Surgical pathology of removed natural and prosthetic heart valves. Human Pathol 1987; 18:558.
CLINICAL MANIFESTATIONS
Infective endocarditis may be acute, subacute or chronic. It causes varying degrees of valvular dysfunction and may be associated with manifestations involving several organ systems (lungs, eyes, kidneys, bones, joints, central nervous system), making diagnosis exceedingly difficult. The ‘classic’ findings are fever and petechial lesions of the skin, conjunctiva and oral mucosa. Other manifestations include weight loss, back pain, night sweats and heart failure.
EVALUATION AND TREATMENT
The criteria for the diagnosis of infective endocarditis include persistent bacteraemia, new heart murmurs, vascular complications and appropriate echocardiographic findings.90 Antimicrobial therapy is generally given for 4 to 6 weeks. Other drugs may be necessary to treat left heart failure secondary to valvular dysfunction. Surgery to remove infected tissue, with or without valve replacement, improves outcomes in many patients with infective endocarditis, especially those with severe heart failure or persistent bacteraemia despite antibiotic therapy.91
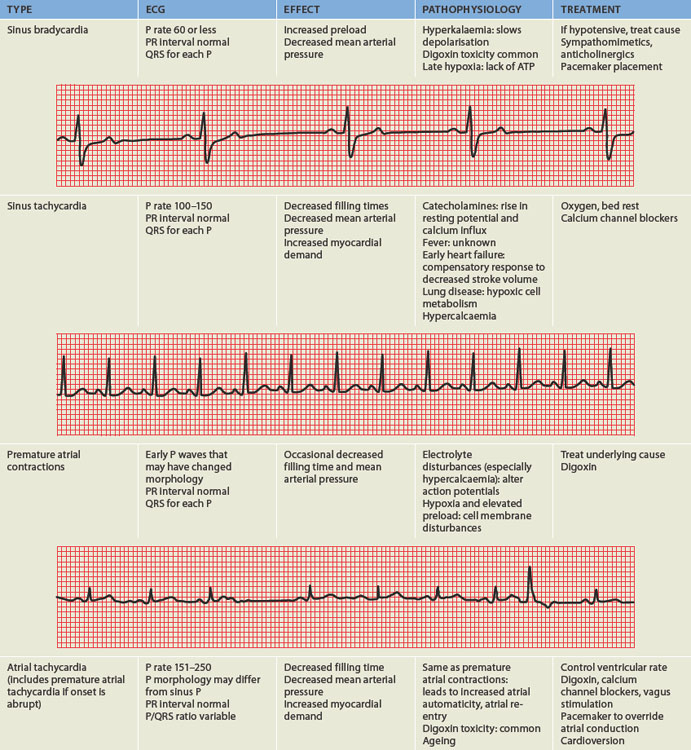
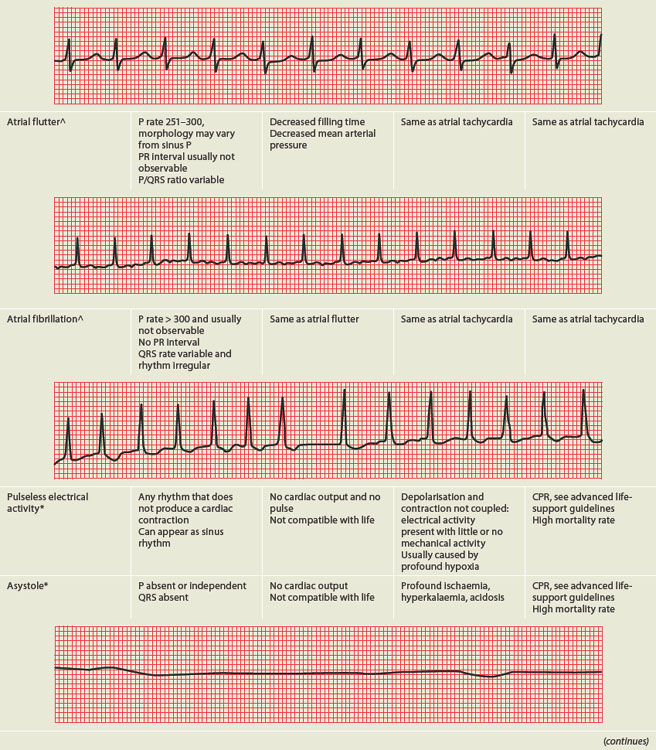
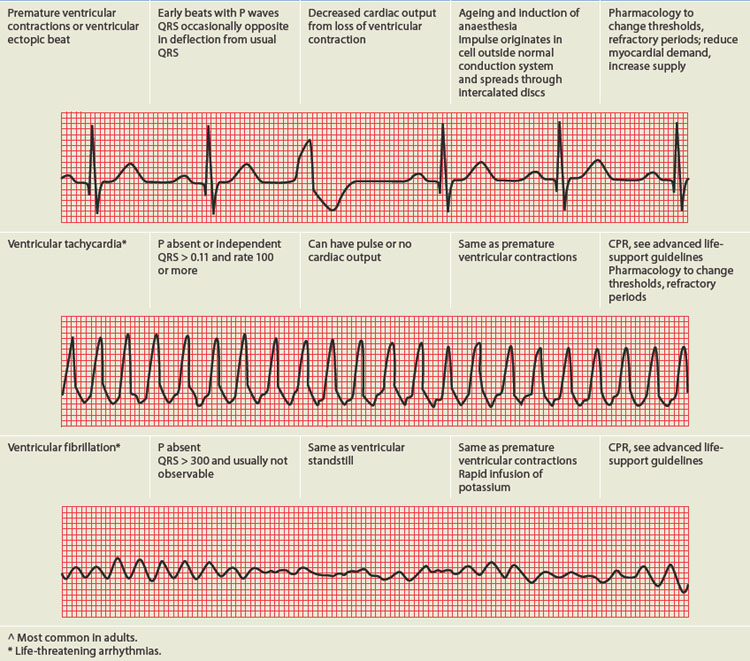
 PAEDIATRICS
PAEDIATRICS