25 ALTERATIONS OF PULMONARY FUNCTION ACROSS THE LIFE SPAN
INTRODUCTION
At some stage everyone experiences an alteration to the pulmonary system. This may range from a minor respiratory illness through to chronic lung diseases and cancers of the pulmonary system. The common cold, a mild upper respiratory tract infection arising from several different viruses, is one of the most familiar pulmonary infections and most people experience one or two infections each year.1 Often more serious is the impact of influenza, commonly referred to as the ‘flu’, with an estimated 5–20% of the Australian and New Zealand populations infected each year and up to half a million deaths worldwide.2,3 Pulmonary diseases and disorders can severely limit an individual’s ability to perform activities of daily living and result in frequent hospitalisations. Moreover, alterations to the pulmonary system contribute significantly to mortality rates in Australia and New Zealand.
The lungs, with their large surface area, are constantly exposed to the external environment. Therefore, lung disease is greatly influenced by conditions of the environment, occupation and personal and social habits. For instance, individuals who smoke are known to be at a greater risk of lung conditions compared to non-smokers. Symptoms of lung disease are common and associated not only with primary lung disorders but also with diseases of other organ systems.
Alterations of respiratory function in children are influenced by physiological maturation as a function of age, genetics and environmental conditions. A variety of upper and lower airway infections can cause respiratory problems or play a role in the pathogenesis of more chronic pulmonary disease. Infants, especially premature infants, may present special problems because of the immaturity of their lung, airway and chest wall structures, as well as the immaturity of pulmonary homeostasis (for example, a lack of surfactant production) and immunological immaturity. Immunisation and attentive healthcare can greatly reduce the incidence and severity of pulmonary disorders in children.
Pulmonary disease is often classified as acute or chronic, obstructive or restrictive, or infectious or non-infectious, and may be caused by alterations in the lung or heart. Because skilful and knowledgeable clinical practice plays a major role in decreasing mortality and morbidity from pulmonary disorders, healthcare professionals who have a clear understanding of the pathophysiology of common pulmonary disorders can greatly affect the outcomes for affected individuals.
This chapter examines the more common alterations to the pulmonary system and then provides detailed information about the signs and symptoms that arise from these conditions. In this way, you can learn about the pathophysiology of the pulmonary conditions and so understand how these alterations manifest in individuals.
DISORDERS OF THE PULMONARY SYSTEM
Obstructive lung diseases
Obstructive lung diseases are characterised by airway obstruction that is worse with expiration. More force — that is, the use of the accessory muscles of expiration — is required to expire a given volume of air or emptying of the lungs is slowed, or both. In adults and children, the major obstructive lung disease is asthma, with chronic bronchitis and emphysema also prevalent in the adult population. Because many individuals have both chronic bronchitis and emphysema, these diseases together are often called chronic obstructive pulmonary disease. Asthma is more acute and intermittent than chronic obstructive pulmonary disease, even though it can be chronic. The unifying symptom of obstructive lung diseases is dyspnoea (difficulty breathing) and the unifying sign is wheezing. Individuals who have an obstructive lung disease have an increased work of breathing, ventilation/perfusion mismatching and a decreased forced expiratory volume in one second (FEV1).
Obstructive lung diseases are prevalent in the Australian and New Zealand populations and are the third leading cause of disease burden.4,5 The most prevalent is asthma,6,7 which affects both children and adults; however, fortunately the mortality rate is low. In the following section we examine the pathophysiology of asthma in both children and adults, to provide a more thorough understanding of the disease.
Asthma
It is estimated that approximately 300 million people worldwide have asthma and there is likely to be a marked increase in this number over the next two decades as modern lifestyles and urbanisation occur in developing countries.8 Asthma occurs at all ages, with approximately half of all cases developing during childhood and another third occurring before age of 40 years. Rates of asthma are higher in English-speaking countries than other countries, and the prevalence of asthma in Australia and New Zealand is high by international standards.9 The exact reasons for this are unknown.
In Australia, more than two million people have asthma (10.2% of the population), with slightly higher rates in children compared to adults. In childhood more males than females have asthma; however, this trend is reversed in adulthood, with more females than males having asthma. Overall, the rates of asthma in New Zealand are comparable with those in Australia.6,7,10 Unfortunately, as with so many chronic diseases, the Indigenous populations in both Australia and New Zealand have higher rates of asthma compared to the non-Indigenous populations: In New Zealand the prevalence of asthma in Maori and Pacific Islander adults is greater than in the non-Indigenous population,7 and in Australia asthma is the second most common illness, affecting 26% of the Indigenous population.11
Despite the high prevalence of asthma, the mortality rate is low. Asthma accounts for less than 0.5% of all deaths. Rates of hospitalisation are also low, with less than 5% of adult and child sufferers hospitalised for asthmatic episodes. Nonetheless, the economic costs of asthma are high — more than $600 million per year in Australia and $125 million per year in New Zealand.6,7
Asthma is likely to result from a complex interaction of genetic and environmental components. It can be defined as:
A chronic inflammatory disorder of the airways in which many cells and cellular elements play a role, in particular, mast cells, eosinophils, T lymphocytes, macrophages, neutrophils and epithelial cells. In susceptible individuals, this inflammation causes recurrent episodes of wheezing, breathlessness, chest tightness and coughing, particularly at night or in the early morning. These episodes are usually associated with widespread but variable airflow obstruction that is often reversible, either spontaneously or with treatment.12
Asthma is a familial disorder and many genes have been identified that may play a role in the susceptibility and pathogenesis of asthma, including those that influence the production of interleukins 4 and 5, immunoglobin E (IgE), eosinophils, mast cells, β (beta)-adrenergic receptors (for the stress response using adrenaline) and bronchial hyperresponsiveness (meaning the bronchioles have increased responses/constriction).10,13
Risk factors for asthma, in addition to family history, include allergen exposure, living in urban areas, exposure to air pollution and cigarette smoke, recurrent respiratory viral infections and other allergic diseases, such as allergic rhinitis.14 There is considerable evidence that exposure to high levels of certain allergens during childhood increases the risk for asthma. Furthermore, decreased exposure to certain infectious organisms appears to create an immunological imbalance that favours the development of allergy and asthma. This complex relationship has been called the hygiene hypothesis, where it is thought that living with low levels of infectious organisms can make the immune system particularly sensitive.15,16 Urban environments have been shown to have a higher disposition of asthma compared to rural areas. The likely exposure to air pollution combined with decreased exercise also play a role in the increasing prevalence of asthma.15,16
PATHOPHYSIOLOGY
Inflammation resulting in hyperresponsiveness of the airways is the major pathological feature of asthma. This means that the airways respond in an abnormal, exaggerated way to inflammatory mediators, such as an allergen or irritants like exercise or cold air. It is initiated by a type I hypersensitivity reaction (see Chapter 15). Exposure to allergens or irritants results in a cascade of events, beginning with mast cell degranulation and the release of multiple inflammatory mediators (see Figure 25-1). Some of the most important mediators that are released during an acute asthmatic episode are histamine, interleukins, prostaglandins, leukotrienes and nitric oxide. The vasoactive effects of these cytokines include vasodilation and increased capillary permeability. This causes an increase in blood flow to the area, and inflammatory cells and chemicals move through the cells into the interstitial tissue. Chemotactic factors (chemicals that attract inflammatory cells to the site of inflammation) are produced, which result in bronchial infiltration by neutrophils, eosinophils and lymphocytes (different types of white blood cells). Eosinophils release a variety of chemicals that contribute to inflammation and tissue damage. The resulting inflammatory process produces bronchial smooth muscle spasm, vascular congestion, oedema formation, production of thick mucus, impaired mucociliary function (see Figure 25-2), thickening of airway walls and increased bronchial hyperresponsiveness. In addition, there is alteration to the normal autonomic control of bronchial smooth muscle because the production of neuropeptides (small protein-like substances that are released by neurons to communicate with other neurons) leads to acetylcholine-mediated bronchospasm. These changes, combined with epithelial cell damage caused by eosinophil infiltration, produce airway hyperresponsiveness and obstruction and, untreated, can lead to long-term airway damage that is irreversible.15–18 16 17 18
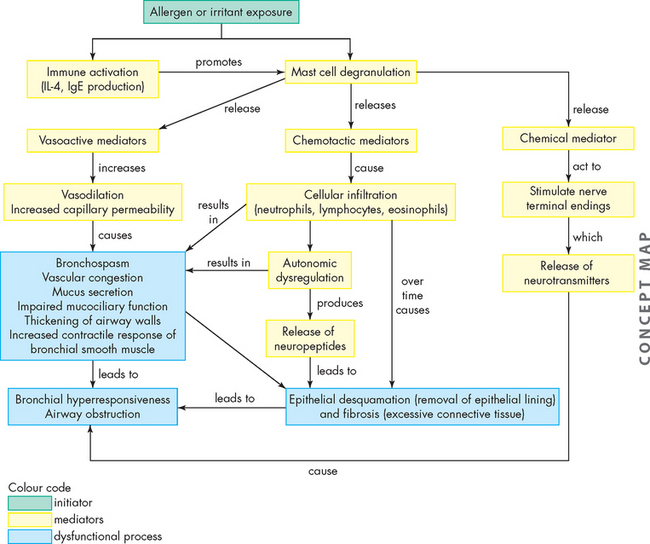
FIGURE 25-1 The pathophysiology of asthma.
A concept map outlining the effects of exposure to an allergen or irritant, which causes an inflammatory cascade leading to acute and chronic airway dysfunction.
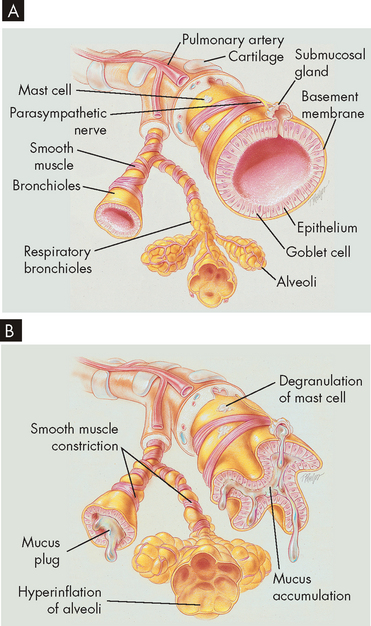
FIGURE 25-2 Changes in airways due to asthma.
A Normal lung with clear airways. B Thick mucus, mucosal oedema and smooth muscle spasm causing obstruction of small airways occurs in asthma, breathing becomes laboured and expiration is difficult due to the airway restrictions.
Source: Des Jardins T, Burton GG. Clinical manifestations and assessment of respiratory disease. 5th edn. St Louis: Mosby; 2006.
Examination of postmortem lung specimens of individuals who have died from asthma reveals abnormalities consistent with both acute and chronic changes in the airways. These include extensive mucus plugging, mucosal oedema and denudation of bronchial and bronchiolar epithelium (loss of epithelium). Eosinophilia (an increased amount of eosinophils) is present in the submucosa and a multicellular inflammatory infiltrate accumulates in the airways. Thickening of the basement membrane, airway smooth muscle hypertrophy and mucous gland hypertrophy are often noted, sometimes even in pathology specimens from mild asthmatics, providing evidence that there may be long-term airway structural changes associated with asthma.
For acute allergen-induced asthma, the paradigm of the early asthmatic response remains useful (see Figure 25-3A). This begins immediately after exposure and lasts up to 2 hours. The allergen binds to preformed immunoglobin E (IgE) on the surface of mucosal mast cells, and cross-linking of these IgE molecules triggers degranulation of the mast cells, releasing mediators such as histamine, leukotrienes, prostaglandin D2, platelet-activating factor and certain cytokines. These mediators cause airway smooth muscle constriction (bronchospasm), increased vascular permeability (mucosal oedema) and mucus secretion.
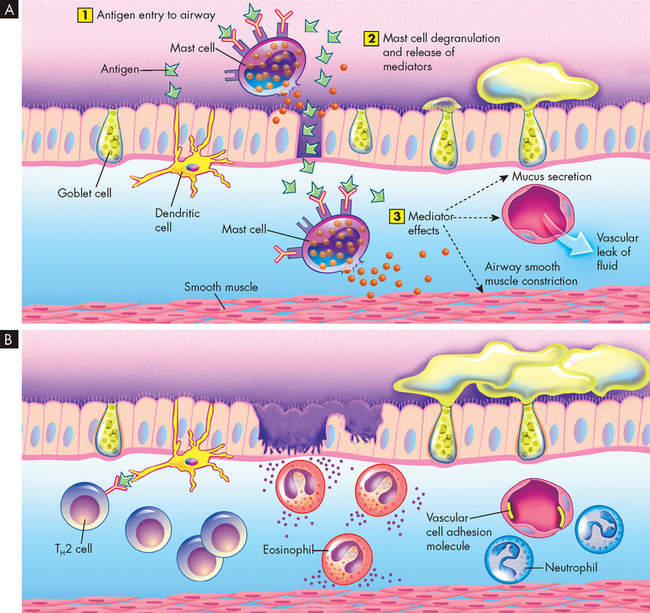
FIGURE 25-3 Asthmatic responses at a cellular level.
A Early asthmatic response. (1) Inhaled antigen enters the airway and binds to IgE on mast cells. (2) Mast cells degranulate and release mediators such as histamine, prostaglandin D2 and platelet-activating factor, which promotes inflammation. (3) These chemicals open junctions between cells, allowing the allergen to penetrate below the epithelial surface, which induces active bronchospasm, oedema and mucus secretion. At the same time, as shown on the left, antigen may be received by dendritic cells and later present to T helper (TH) lymphocytes in the airway mucosa (see B). B Late asthmatic response. There are areas of epithelial damage caused at least in part by toxicity of eosinophil. Local T lymphocytes produce IL-4 and IL-13, which promote switching of B cells to favour IgE production, and IL-3, IL-5 and granulocyte-macrophage colony-stimulating factor, which encourage eosinophil differentiation and survival.
The late asthmatic response starts 4–8 hours after the initial exposure and may persist for up to 24 hours (see Figure 25-3B). The response is characterised by inflammatory cell recruitment (neutrophils, eosinophils, basophils, lymphocytes) that was triggered earlier by chemotactic factors and endothelial adhesion molecules (molecules that attach to the endothelial). Another wave of mediator release occurs, again causing bronchospasm, oedema and mucus secretion. Epithelial damage and impaired mucociliary function (the sweeping ability of the cilia lining the airways) may be seen because of the direct toxic effects of products such as proteins released from eosinophils. This local injury stimulates local nerve endings, which may aggravate bronchoconstriction and mucus secretion through autonomic pathways.
In chronic asthma, some of these mechanisms may be operational on an ongoing basis. There are increased numbers of inflammatory cells, which may lead to long-term changes such as goblet cell hyperplasia (abnormally increased number of cells) and airway wall remodelling (subepithelial fibrosis, smooth muscle hypertrophy).
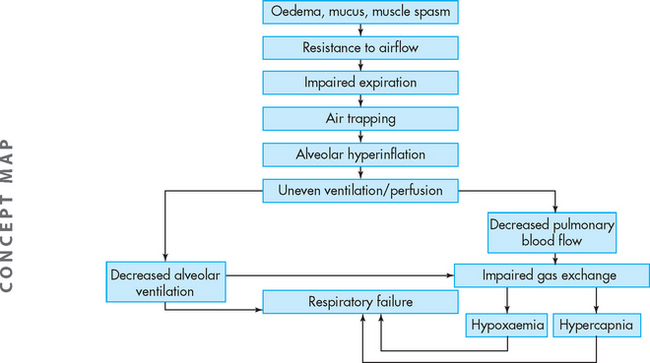
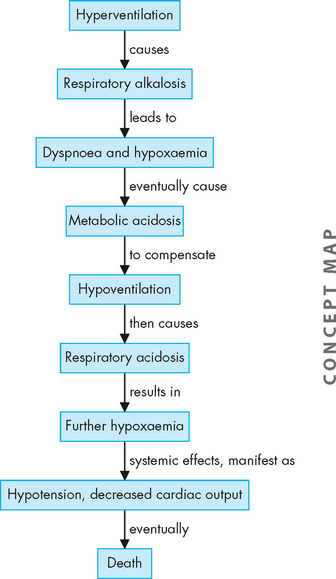
Airway obstruction increases resistance to airflow and decreases flow rates, primarily expiratory flow. For instance, a 10% reduction in airway calibre leads to a 2% increase in resistance. Impaired exhalation causes air trapping and hyperinflation distal to obstructions and increases the work of breathing. Intrapleural and alveolar gas pressures rise and cause decreased perfusion of the alveoli. These changes lead to uneven ventilation–perfusion relationships causing hypoxaemia (a reduction in oxygen levels in the blood). Hyperventilation is triggered by lung receptors responding to the hyperinflation and causes a decrease in carbon dioxide levels in the blood (PaCO2) and increased pH (which results in respiratory alkalosis). As the obstruction becomes more severe, the number of alveoli being adequately ventilated and perfused decreases. Air trapping continues to worsen and the work of breathing increases further, leading to hypoventilation (decreased tidal volume), carbon dioxide retention and respiratory acidosis (from the reduction in carbon dioxide removal). Respiratory acidosis signals respiratory failure (Figure 25-4 summarises these steps).
CLINICAL MANIFESTATIONS
When individuals with asthma are asymptomatic, pulmonary function tests are normal. However, at the beginning of an acute asthmatic episode (commonly referred to as an asthma attack), the individual experiences chest constriction, expiratory wheezing, dyspnoea, non-productive coughing, prolonged expiration, tachycardia and tachypnoea (increased ventilatory rate). Severe episodes involve the accessory muscles of ventilation and wheezing is heard during both inspiration and expiration. Pulsus paradoxus (an exaggerated decrease in systolic blood pressure during inspiration) may be noted. Peak flow measurements (the rate of maximal air flow out of the lungs) are usually reduced. Because the severity of blood gas alterations is difficult to evaluate by clinical signs alone, arterial blood gas levels should be measured if oxygen saturation falls below 90%. The usual findings are hypoxaemia with an associated respiratory alkalosis. The severity of acute asthmatic episodes is outlined in Table 25-1.
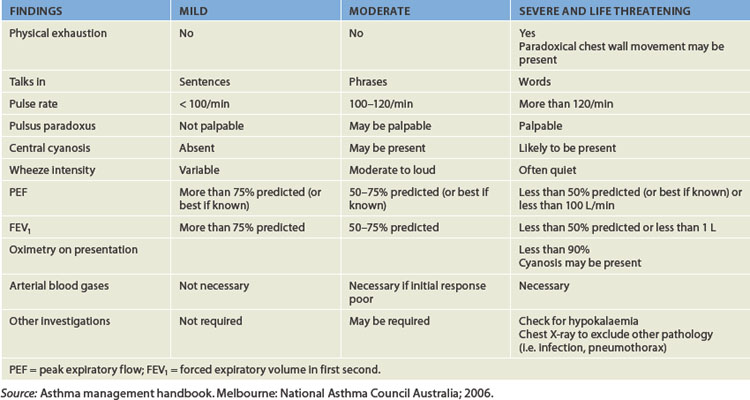
The typical arterial blood gas abnormalities seen in acute asthma are hypoxaemia, hypocapnia (low blood carbon dioxide levels) and respiratory alkalosis (a pH level above 7.45). As bronchial obstruction is non-uniform, ventilation becomes uneven, causing ventilation–perfusion mismatch and further hypoxaemia. The degree of hypoxaemia is usually mild; however, arterial saturations of less than 90% indicate severe airway obstruction. Pulmonary circulation may be altered by regional hypoxic vasoconstriction, as well as the effect of increased intra-alveolar pressure (caused by hyperinflation) to decrease perfusion of the alveolar capillaries. Typically, the ventilatory rate (commonly referred to as the respiratory rate in clinical environments) is elevated to compensate for hypoxaemia, with reduced minute ventilation because of increased airway resistance and lung hyperinflation. Thus, the carbon dioxide level is low (30–35 mmHg compared to the normal level of 35–45 mmHg) or can be normal. Retention of carbon dioxide is a late finding and reflects inadequate alveolar ventilation and increased functional dead space as little air is being moved. Alterations of pH homeostasis usually start with respiratory alkalosis (pH greater than 7.45) caused by hyperventilation, which literally ‘blows off’ carbon dioxide. With severe airway obstruction, the end result of the pathophysiological processes may be respiratory failure, with acute carbon dioxide retention and respiratory acidosis (pH less than 7.35).
When bronchospasm worsens during a severe asthmatic episode the individual may progress to a condition known as status asthmaticus. This is defined as a severe asthmatic episode that does not respond to pharmacological control. Acute airway inflammation causes bronchospasm to worsen. Mucus plugging, oedema and cellular infiltration lead to further airway narrowing. Partial obstruction leads to segmental hyperinflation, which may become extreme and compromise effective tidal volume. Expiratory flow rates such as FEV1 and peak flow are also markedly reduced. If status asthmaticus continues, hypoxaemia worsens, expiratory flows and volumes decrease further, and effective ventilation decreases. Metabolic acidosis may accompany status asthmaticus as the carbon dioxide level in the blood begins to rise. Asthma becomes a life-threatening condition at this point, often with impending respiratory or cardiac arrest if treatment does not reverse this process quickly. A silent chest (no audible air movement) and a carbon dioxide level over 70 mmHg are ominous signs of impending death.
EVALUATION AND TREATMENT
For objective evaluation of asthma, the best indicators are measures of pulmonary function using spirometry. Spirometry may document reversible decreases in FEV1 during an induced asthmatic episode. An example of a reduction in FEV1 compared to normal spirometry is provided in Figure 25-5. However, performing spirometry is not always possible, especially in children less than 4 years of age. In such cases, a history from parents is crucial. Between episodes, the diagnosis of asthma is supported by a history of allergies and recurrent episodes of breathlessness or exercise intolerance. For home management of asthma, peak flow meters are often used to help guide treatment in the face of increased symptoms or illness.
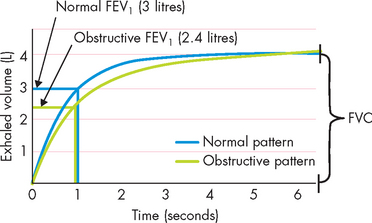
FIGURE 25-5 Changes in expiratory flow associated with obstructive lung disease such as asthma.
The defining characteristic is a reduction in FEV1 with a normal forced vital capacity (FVC) with obstructive lung disease. In the above example the FVC in both cases was identical yet FEV1 was reduced. The FEV1/ FVC ratio was therefore below the normal 80% in healthy individuals.
Source: Chu MW, Han JK. Introduction to pulmonary function. Otolaryngol Clin North Am 2008; 41(2):387–396.
Management of asthma begins with avoidance of allergens and irritants and patient education, which have been shown to reduce morbidity and mortality.10 In Australia and New Zealand, asthmatics are encouraged to employ an asthma action plan outlining the changes in the pulmonary system that may trigger an asthmatic episode and importantly what types and dose of drugs are effective. Long-term management is based on the stage of asthma and includes the use of three different types of pharmacological agents:
There are also combination medications that contain a symptom controller and preventer medication, enabling the individual to administer medication in a single dose. Examples include fluticasone and salmeterol (Seretide), and budesonide and eformoterol (Symbicort). More recently, the use of anti-immunoglobulin therapy has shown promise in some asthmatic individuals. Omalizumab is a recombinant humanised monoclonal antibody to IgE. This means that the medication acts more on the start of inflammation, rather than its effects (see the box ‘Health alert: monoclonal antibodies to IgE for treatment of asthma’).10,12,19–21 4 6
There are a growing number of options for management of chronic asthma depending on the duration of the condition and the severity of the symptoms, as well as individual adherence issues. Guidelines have been outlined and widely distributed by the National Asthma Council Australia (www.nationalasthma.org.au) and the New Zealand Guidelines Group (www.nzgg.org.nz). The most important element of chronic asthma management appears to be reduction of inflammation.
Acute asthma episodes can be life-threatening and therapy should be directed at maintaining a patent (open) airway and providing rapid bronchodilation and effective ventilation to maintain adequate gas exchange (see Table 25-1 for details of the severity of asthmatic episodes).10,12,20 Administration of oxygen and rapid-acting bronchodilators such as salbutamol (β2-agonist — that is, one that will interact with a group of adrenergic receptors in the lungs; see Chapter 6) are typically used for management of acute asthma, as well as systemic steroids for moderate to severe attacks to decrease inflammatory responses in the lungs.10,22
Monoclonal antibodies to IgE for treatment of asthma
In genetically predisposed individuals, exposure to inhaled allergens can lead to an allergic response that is manifested clinically as asthma. This immunological response is a type I hypersensitivity reaction (see Chapter 15) and results from a complex interaction of macrophages, T lymphocytes, antibodies, mast cells and eosinophils. The immune response leads to a profound inflammatory response, with resultant bronchospasm, mucus formation and mucosal oedema in the airways. Allergen avoidance and immunotherapy can be effective in preventing this type I hypersensitivity reaction. Omalizumab (Xolair) is a recombinant humanised monoclonal antibody to IgE that is indicated for individuals with moderate or severe asthma who do not respond to standard therapy. Omalizumab can improve symptoms, reduce hospitalisations and limit the need for systemic corticosteroid therapy.
Source: Mathur SK, Busse WW. Asthma: diagnosis and management. Med Clin North Am 2006; 90(1):39–60; Davydov L. Omalizumab (Xolair) for treatment of asthma. Am Family Phys 2005; 71:341–342; Marcus P. Practice Management Committee, American College of Chest Physicians: Incorporating anti-IgE (omalizumab) therapy into pulmonary medicine practice: practice management implications. Chest 2006; 129(2):466–474. Holgate ST et al. The use of omalizumab in the treatment of severe allergic asthma: a clinical experience update. Respir Med 2009; 103:109–113.
 PAEDIATRICS
PAEDIATRICSAsthma in children
Asthma affects more children than adults in Australia and New Zealand. There appear to be certain types of wheezing that are associated with asthma during childhood that are not carried through to adulthood, hence the differences in adult and child asthma prevalence.23 Interestingly, while the prevalence of childhood asthma in Australia and New Zealand is among the highest in the world, unlike asthma prevalence in adults it is not higher in Indigenous populations compared with non-Indigenous populations.7,10 However, the incidence of severe asthmatic episodes and the rate of hospitalisations is greater in Indigenous populations compared with non-Indigenous populations.6,7,10,11,24
While the pathophysiology of asthma in children is similar to that in adults, several pertinent points need to be highlighted. Asthma is clinically different in children due to the pattern of asthma, natural history and anatomical features.10 For instance, wheezing in childhood can be both associated and not associated with asthma. The classification of asthma is based on the clinical patterns, rather than objective evaluation using spirometry. There are currently many theories regarding the mechanisms of the disease in childhood. There is not one single gene responsible for the manifestation of asthma.25 The wide spectrum of clinical disease probably reflects a complex interaction between genetic susceptibility and environmental factors, including early exposure to allergens and infections, particularly viral respiratory infections26 (see the box ‘Health alert: asthma genes and tailored therapies’).
CLINICAL MANIFESTATIONS
Although the genetic expression of asthma is difficult to identify, many discrete clinical presentations have been demonstrated. There are at least three different manifestations for childhood asthma.23 These are:
Atopy (an allergic reaction when IgE increases due to environmental allergens — associated with a strong family history of allergies) is strongly associated with classical asthma that persists into adulthood. However, wheezing illnesses in childhood usually resolve by about 6 years of age, especially when the wheezing is intermittent.6,10,12
The classification of childhood asthma is divided into three levels: infrequent intermittent, frequent intermittent and persistent (see Figure 25-6). Broadly speaking, these classifications include the following:
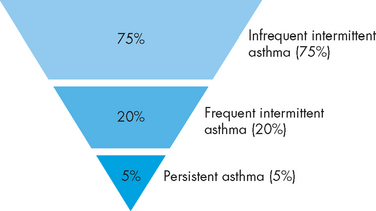
FIGURE 25-6 Classification of childhood asthma and their distribution.
Source: Asthma management handbook. Melbourne: National Asthma Council Australia; 2006.
In a typical asthmatic episode, the major complaints are cough, wheeze and shortness of breath. There may or may not have been signs of a preceding upper respiratory infection, such as rhinorrhoea (discharge of nasal mucus, often termed ‘runny’ nose) or low-grade fever. In children, about 70–80% of acute wheezing episodes are associated with viral respiratory infections. In infants and toddlers under 2 years old, the most common of these is respiratory syncytial virus. In older children and adults, the major viral trigger is rhinovirus (commonly referred to as the ‘common cold’ virus).27
On physical examination, there is an expiratory wheeze that is often described as high-pitched and musical, and exhalation is unusually longer than inhalation. Breath sounds may become faint when air movement is poor. The child may speak in short sentences or not at all because of dyspnoea (difficulty breathing). Ventilatory rate and heart rate are elevated to compensate for the low oxygen levels and increased work of breathing. Nasal flaring and use of accessory muscles with retractions in the substernal, subcostal, intercostal, suprasternal or sternocleidomastoid muscle areas are evident. The child may appear anxious or be diaphoretic (excessive sweating), which are often important signs of respiratory compromise.
Asthma genes and tailored therapies
Genomic screening of populations suggests that there is no single ‘asthma gene’ but rather numerous genes that may be associated with asthma. It may ultimately be possible to associate certain gene variants with specific clinical patterns of asthma and with responsiveness to specific asthma treatments. For example, one variant (or polymorphism) is the gene for the β2-adrenergic receptor, which has been shown to be associated with a poor or even adverse response to salbutamol in one study. If findings such as these can be corroborated and expanded in larger studies, ultimately it may be possible to develop individual profiles to optimise asthma therapy.
Source: Steinke JW, Borish L. Genetics of allergic disease. Med Clin N Am 2006; 90(1):1–15; Meurer JR, Lustig JV, Jacob HJ. Genetic aspects of the etiology and treatment of asthma. Pediatr Clin North Am 2006; 53(4):715–725.
Chronic obstructive pulmonary disease
Unlike asthma, chronic obstructive pulmonary disease (COPD) is associated with a high mortality rate. The exact prevalence of COPD is difficult to determine; however, it is estimated that approximately 200,000 New Zealanders (about 15% of the adult population over the age of 45 years) and approximately 600,000 Australians are affected.4,5,28 COPD is the fourth leading cause of death after cardiovascular disease, cancer and stroke, and forms a large percentage of all respiratory deaths (45%). It is estimated to cost up to $900 million annually in Australia4,28 and is a major cause of disability. Individuals with COPD often have a reduced quality of life and require frequent hospitalisations as the disease progresses.28
COPD is a syndrome that includes the pathological lung changes consistent with emphysema or chronic bronchitis. It is characterised by abnormal tests of expiratory airflow that do not change markedly over time or exhibit major reversibility in response to pharmacological agents. There are similarities with asthma; however, individuals with totally irreversible airflow obstruction following administration of a bronchodilator do not have COPD. The syndrome has been defined as:
a preventable and treatable disease with some significant extrapulmonary effects that may contribute to the severity in individual patients. Its pulmonary component is characterized by airflow limitation that is not fully reversible. The airflow limitation is usually progressive and associated with an abnormal inflammatory response of the lung to noxious particles or gases.29
Chronic obstructive pulmonary disease is diagnosed in clinical practice based on a history of smoking (or exposure to other noxious inhalational agents) and with a FEV1/FVC ratio of less than 0.7 (70%) following administration of a bronchodilator.4 The ratio should be at about 0.8 (80%) or higher in individuals without lung pathology:
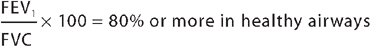
The disease is primarily caused by cigarette smoke and both active and passive smoking have been implicated. This is the most important cause of COPD and smokers demonstrate a steady decline in pulmonary function, irrespective of the development of COPD (see Figure 25-7). The risk of developing COPD with continued long-term smoking, irrespective of cigarette type, is high.30 Other risks include occupational exposure and air pollution. Genetic susceptibilities have also been identified.4,31 In the following sections we take a closer look at the two main conditions that result in COPD.
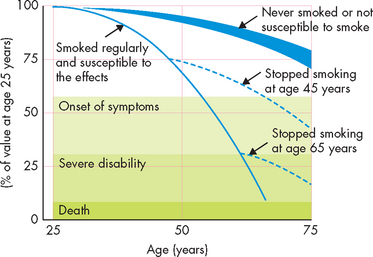
FIGURE 25-7 Time course of smoking and the changes with smoking cessation at 45 and 65 years of age.
Notice that for the smoker who quit at age 45, the serious progression of COPD is much slower than that for the smoker who quit at age 65. In both cases, the disease progression is slower than for those who continue smoking.
Source: Modified from Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J 1977; 1:1645–1648.
PATHOPHYSIOLOGY
Inspired irritants result in airway inflammation with infiltration of neutrophils, macrophages and lymphocytes into the bronchial wall. Continual bronchial inflammation causes bronchial oedema and increases the size and number of mucous glands and goblet cells in the airway epithelium.32 Thick, tenacious mucus is produced and cannot be cleared because of impaired ciliary function. The defence mechanisms of the pulmonary system are compromised, increasing susceptibility to pulmonary infection and injury. Frequent infectious exacerbations are complicated by bronchospasm with dyspnoea and a productive cough.33 The pathophysiology of chronic bronchitis is shown in Figure 25-8.34
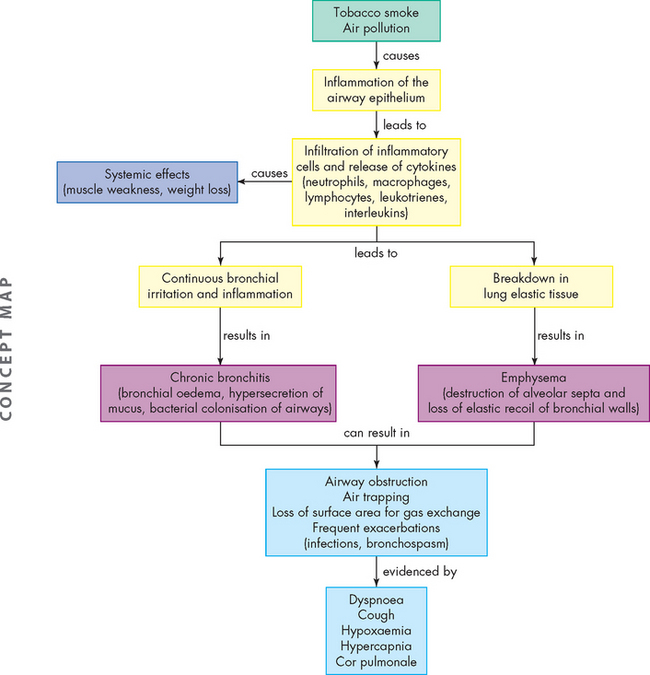
Initially this process affects only the larger bronchi, but eventually all airways are involved. As the airways become increasingly narrowed, expiratory airway obstruction results (see Figures 25-9 and 25-10). The airways collapse early in expiration, trapping gas in the distal portions of the lung. Eventually ventilation–perfusion mismatching (see Chapter 24) and hypoxaemia occurs. Extensive air trapping puts the respiratory muscles at a mechanical disadvantage, resulting in hypoventilation and hypercapnia.
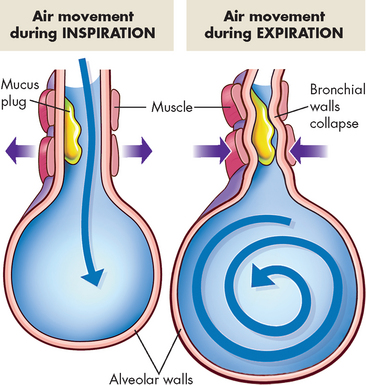
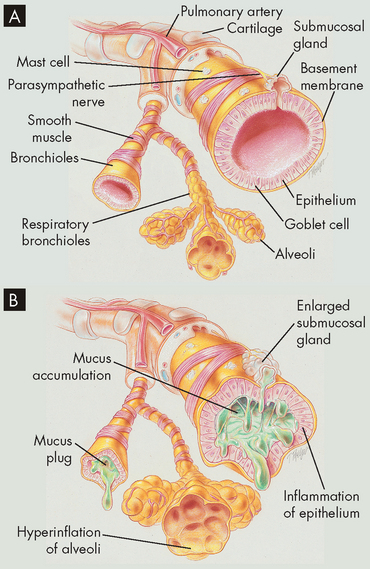
FIGURE 25-10 Airway obstruction resulting from chronic bronchitis.
A Normal lungs with clear airways. B Inflammation and airway thickening of mucous membrane with accumulation of mucus and pus leading to obstruction and characterised by a cough.
Source: Des Jardins T, Burton GG. Clinical manifestations and assessment of respiratory disease. 5th edn. St Louis: Mosby; 2006.
CLINICAL MANIFESTATIONS
Table 25-2 lists the common clinical manifestations of COPD including chronic bronchitis.
Table 25-2 CLINICAL MANIFESTATIONS OF COPD
| VARIABLES | BRONCHITIS | EMPHYSEMA |
|---|---|---|
| Age (years) | 40–45 | 50–75 |
| Infections | Common | Occasional |
| Dyspnoea | Mild, late in course | Severe, early in course |
| Productive cough | Classical sign | Late in course with infection |
| Wheezing | Intermittent | Common |
| History of smoking | Common | Common |
| Prolonged expiration | Always present | Always present |
| Cyanosis | Common | Uncommon |
| Chronic hypoventilation | Common | Late in course |
| Chest X-ray findings | Prominent vessels | Hyperinflation |
| General appearance | ‘Blue bloater’ | ‘Pink puffer’ |
| Barrel chest | Occasionally | Classic |
Source: Based on Kumar V. Robbins & Cotran pathologic basis of disease. 7th edn. Philadelphia: Saunders; 2004.
EVALUATION AND TREATMENT
Diagnosis is based on physical examination, chest X-ray, pulmonary function tests and blood gas analyses; these tests reflect the progressive nature of the disease. The best ‘treatment’ for chronic bronchitis is prevention, because once pathological changes have occurred they are not reversible. Unfortunately, by the time an individual seeks medical assistance for symptoms, considerable airway damage is present. If the individual stops smoking, disease progression can be halted but importantly not reversed — this means that although quitting smoking will not allow for repair of the airway damage, it is still important as it will stop the disease worsening. If smoking is stopped before symptoms occur, the risk of chronic bronchitis decreases considerably.
Bronchodilators, expectorants and chest physiotherapy are employed as needed to control cough and reduce dyspnoea.35 Education includes nutritional counselling (see the box ‘Health alert: nutrition and COPD’), respiratory hygiene (such as deep breathing and coughing), recognition of the early signs of infection and techniques that relieve dyspnoea, such as pursed-lip breathing. During acute exacerbations (infection and bronchospasm), individuals require treatment with antibiotics and steroids.36 Chronic oral steroids may be needed late in the course of the disease but should be considered a last resort. Individuals with severe hypoxaemia will require home oxygen therapy.
Emphysema
Emphysema is abnormal permanent enlargement of gas-exchange airways accompanied by destruction of alveolar walls. Obstruction results from changes in lung tissues, rather than mucus production and inflammation as in chronic bronchitis. The major mechanism of airflow limitation is loss of elastic recoil (see Figure 25-9). The major cause of emphysema is cigarette smoking, although air pollution and childhood respiratory infections are known to be contributing factors.
PATHOPHYSIOLOGY
Emphysema begins with destruction of alveolar septa, which eliminates portions of the pulmonary capillary bed and increases the volume of air in the alveoli (see Figure 25-11). It is postulated that inhaled oxidants in tobacco smoke and air pollution stimulate inflammation, which over time causes alveolar destruction and loss of the normal elastic recoil of the bronchi (see Figure 25-8). Alveolar destruction produces large air spaces within the lung tissue and air spaces adjacent to pleurae. These areas are not effective in gas exchange. The loss of alveolar tissue means a loss of the respiratory membrane where gases cross between air and the blood, resulting in a significant ventilation–perfusion mismatching and hypoxaemia. Expiration becomes difficult because loss of elastic recoil reduces the volume of air that can be expired passively and air is trapped in the lungs (see Figure 25-8). Air trapping causes an increase in expansion of the chest, which puts the muscles of ventilation at a mechanical disadvantage. This results in increased workload of breathing, so that late in the course of disease, many individuals will develop hypoventilation and hypercapnia.
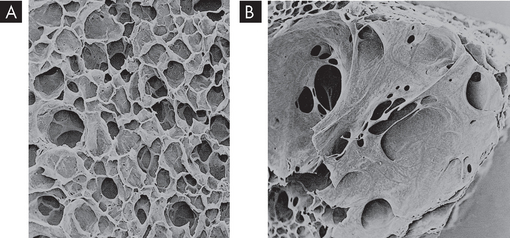
FIGURE 25-11 The effects of emphysema on the gas exchange units.
A Normal lung with many small alveoli. B Lung tissue affected by emphysema. Notice that the alveoli have merged into larger air spaces, reducing the surface area for gas exchange.
Source: Thibodeau GA. The human body in health & disease. 5th edn. St Louis: Mosby; 2010.
EVALUATION AND TREATMENT
Pulmonary function testing, chest X-ray, high-resolution CT scanning and arterial blood gas measurement are used to diagnose emphysema.4,29 Pulmonary function measurements are helpful in determining the stage of disease, appropriate treatment and prognosis. Treatment for emphysema is similar to that for chronic bronchitis and includes smoking cessation, bronchodilators, nutritional support, chest physiotherapy, anti-inflammatory medications and antibiotics for acute infections. The most recent recommendations for the management of chronic symptoms of COPD have been shown in two large studies, which demonstrated that long-acting bronchodilators, inhaled corticosteroids or a combination of these two can reduce mortality and the decline in FEV1 and augment quality of life.37,38 However, it should be noted that in these studies mortality was not reduced, despite halting the decline of pulmonary function.
Nutrition and COPD
Malnutrition is a major concern for individuals with COPD because they have increased energy expenditure, decreased energy intake and impaired oxygenation. The disproportionate muscle wasting is similar to that which occurs with other chronic diseases, such as cancer, heart failure and AIDS. Systemic inflammatory mediators may impair appetite and contribute to hypermetabolism. Malnutrition: (1) adversely affects exercise tolerance by limiting skeletal and respiratory muscle strength and aerobic capacity; (2) limits surfactant production; (3) reduces cell-mediated immune responses; (4) reduces protein synthesis; and (5) increases morbidity and mortality. The goal of medical nutrition therapy is to maintain an acceptable and stable weight for the individual. This can be accomplished by including foods of high energy density, snacking frequently, choosing soft foods, having an adequate intake of fluids and providing assistance with shopping and meal preparation. Increasing omega-3 fatty acids and antioxidant intake may modulate the effects of systemic inflammation. Protein intake should be maintained at 1.0–1.5 g/kg of body weight, and a daily vitamin C supplement should be added to the diet if the individual is still smoking.
Source: Schols AM et al. Weight loss is a reversible factor in the prognosis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157(6):1791–1797; Ferreira IM et al. Nutritional supplementation for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2005; 2:CD000998; MacNee W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2(1):50–60.
Restrictive lung diseases
Restrictive lung diseases are not as prevalent as obstructive lung diseases in the Australian and New Zealand populations. They are fundamentally different from obstructive diseases, but many of the clinical manifestations are similar. Therefore, it is essential that you can differentiate between these two groups of disorders, such that clinical management can be directed appropriately.
Restrictive lung diseases are characterised by decreased compliance (stretchiness) of the lung tissue, resulting in an increased work of breathing. Individuals with lung restriction complain of dyspnoea and have an increased ventilatory rate and decreased tidal volume — that is, they breathe fast but with smaller breath size. Pulmonary function testing reveals a decrease in FVC often accompanied with a reduction in FEV1. Therefore, the ratio of FEV1/FVC can be normal — that is, about 80% of the forced expired air is expelled from the lungs in the first second — yet the overall amount of air forcibly exhaled is less than normal (see Figure 25-12). Restrictive lung diseases commonly affect the alveolar–capillary membrane and cause decreased diffusion of oxygen from the alveoli into the blood, resulting in hypoxaemia. Some of the most common restrictive lung diseases in adults are acute respiratory distress syndrome and inhalational disorders.
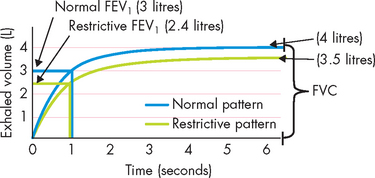
FIGURE 25-12 Changes in expiratory flow associated with restrictive lung disease.
Note that there is a reduction in both FEV1 and FVC. Therefore, when calculated, the FEV1/ FVC ratio is not different from that in healthy individuals; however, there is restriction throughout the entire expiratory phase.
Source: Based on Chu MW, Han JK. Introduction to pulmonary function. Otolaryngol Clin North Am 2008; 41(2):387–396.
Acute respiratory distress syndrome
Acute respiratory distress syndrome is a dramatic life-threatening condition characterised by acute lung inflammation and diffuse alveolar capillary injury. It can affect all age groups. Individuals who progress to acute respiratory distress syndrome typically are critically ill and require intensive care treatment. The mortality rate is high; however, advances in therapy have decreased mortality in people younger than 60 years. The most common predisposing factors are sepsis and multiple trauma; however, there are many other causes, including pneumonia, burns, aspiration, cardiopulmonary bypass surgery, pancreatitis, blood transfusions, drug overdose, high concentrations of supplemental oxygen and disseminated intravascular coagulation.
PATHOPHYSIOLOGY
The hallmark of acute respiratory distress syndrome is lung inflammation. There is activation of the inflammatory response (see Figure 25-13), including complement, cytokines, arachidonic acid metabolites and platelet-activating factor.
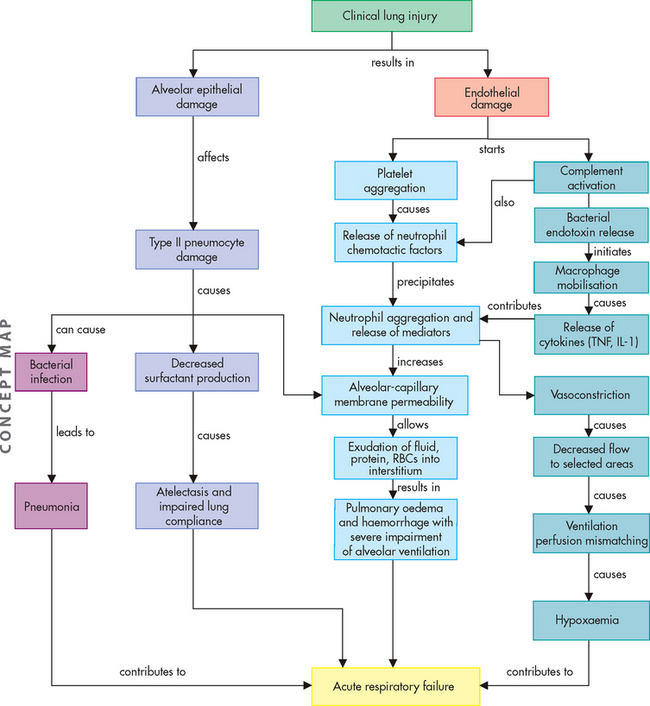
FIGURE 25-13 The proposed mechanism for the pathophysiological changes associated with acute respiratory distress syndrome.
TNF = tumour necrosis factor; IL-1 = interleukin-1; RBCs = red blood cells.
Source: Based on McCance KL, Huether SE. Pathophysiology: the biologic basis for disease in adults and children. 6th edn. St Louis: Mosby; 2010.
All disorders causing acute respiratory distress syndrome cause massive pulmonary inflammation that injures the alveolar–capillary membrane and produces severe pulmonary oedema and hypoxaemia (Figure 25-13). The damage can occur directly, as with the aspiration of highly acidic gastric contents or the inhalation of toxic gases, or indirectly from chemical mediators released in response to systemic disorders such as sepsis. Injury to the pulmonary capillary endothelium stimulates platelet aggregation (platelets sticking together) and intravascular thrombus formation. Endothelial damage also initiates the complement cascade, stimulating neutrophil and macrophage activity and the inflammatory response.39
Once activated, macrophages produce toxic mediators such as tumour necrosis factor-alpha (TNF-α) and interleukin-1 (IL-1) (see Chapter 12). The role of neutrophils is central to the development of acute respiratory distress syndrome. Activated neutrophils release a battery of inflammatory mediators, including proteolytic enzymes (enzymes that break down proteins), toxic oxygen products, arachidonic acid metabolites (prostaglandins, thromboxanes, leukotrienes) and platelet-activating factor. These mediators extensively damage the alveolar–capillary membrane and greatly increase capillary membrane permeability. This allows fluids, proteins and various blood cells to leak from the capillary bed into the pulmonary interstitium and alveoli. The resulting pulmonary oedema severely reduces lung compliance and impairs alveolar ventilation. Mediators released by neutrophils and macrophages also cause pulmonary vasoconstriction, which leads to worsening of ventilation–perfusion mismatching and hypoxaemia. This vicious cycle continues and is difficult to halt.
The initial lung injury also damages the alveolar epithelium. This cell injury increases alveolar capillary permeability, increases susceptibility to bacterial infection and pneumonia, and decreases surfactant production. Alveoli and respiratory bronchioles fill with fluid or collapse. The lungs become less compliant, ventilation of alveoli decreases and pulmonary blood flow is shunted right to left. The work of breathing increases. The end result is acute respiratory failure (see ‘Clinical manifestations of pulmonary alterations’ below).
The chemical mediators responsible for the alveolar capillary damage of acute respiratory distress syndrome often cause widespread inflammation, endothelial damage and capillary permeability throughout the body, resulting in the systemic inflammatory response syndrome, which then leads to multiple organ dysfunction syndrome. In fact, death may not be caused by respiratory failure alone but by multiple organ dysfunction syndrome associated with acute respiratory distress syndrome. (Multiple organ dysfunction syndrome is discussed in Chapter 23.)
CLINICAL MANIFESTATIONS
Acute respiratory distress syndrome develops acutely after the initial insult, usually within 24 hours, though occasionally it is delayed up to a few days. The classic signs and symptoms of acute respiratory distress syndrome are marked dyspnoea, rapid shallow breathing, inspiratory crackles, respiratory alkalosis, decreased lung compliance, hypoxaemia unresponsive to oxygen therapy (called refractory hypoxaemia) and diffuse alveolar infiltrates seen on chest X-rays, without evidence of cardiac disease. Symptoms develop progressively, as shown in Figure 25-14.
EVALUATION AND TREATMENT
Diagnosis is based on physical examination, analysis of blood gases and radiological examination. Treatment for acute respiratory distress syndrome remains supportive in nature and the goals are to maintain adequate tissue oxygenation, minimise acute lung injury and avoid further pulmonary complications. Most individuals with acute respiratory distress syndrome require mechanical ventilation and often relatively high levels of positive end-expiratory pressure to promote alveolar ventilation and stabilisation and redistribution of alveolar oedema fluid into the interstitium.
Inhalation disorders
Exposure to toxic gases
Inhalation of gaseous irritants can cause significant respiratory dysfunction. Gases that are toxic to the pulmonary system include smoke, ammonia, hydrogen chloride, sulfur dioxide, chlorine and nitrogen dioxide. Inhalation of a toxic gas results in severe inflammation of the airways, alveolar and capillary damage and pulmonary oedema. Initial symptoms include burning of the eyes, nose and throat; coughing, chest tightness and dyspnoea. Hypoxaemia is common. Treatment includes supplemental oxygen, mechanical ventilation and support of the cardiovascular system due to hypotension. Most individuals respond quickly to therapy. Some, however, may improve initially and then deteriorate as a result of bronchiectasis (persistent dilation of the bronchioles) or bronchiolitis (inflammation of the bronchioles).
Pneumoconiosis
Pneumoconiosis represents any change in the lung caused by inhalation of inorganic dust particles, usually in the workplace. As in all cases of environmentally acquired lung disease, the individual’s history of exposure is important in determining the diagnosis. Pneumoconiosis often occurs after years of exposure to the offending dust, with progressive fibrosis of lung tissue. Asbestosis, silicosis and coal worker’s pneumoconiosis are among the three most important dust-related diseases from occupational exposure in Australia.40 Asbestosis has the highest mortality of these three; however, the risk of environmental exposure has been recognised for decades and exposure to asbestos hopefully will be limited in the future.40
Deposition of dusts from silica, asbestos and coal leads to chronic inflammation. In addition, scarring of the alveolar–capillary membrane leads to a build-up of connective tissue in the lung (termed pulmonary fibrosis).41 These dust deposits are permanent and leads to progressive pulmonary deterioration. Clinical manifestations with advancement of disease include cough, chronic sputum production, dyspnoea, decreased lung volumes and hypoxaemia. Diagnosis is confirmed by chest X-ray and CT scans. Treatment is usually palliative (to reduce symptoms of the disease, such as pain control) and focuses on preventing further exposure, particularly in the workplace.
INFECTIONS OF THE PULMONARY SYSTEM
Infections of the pulmonary system are some of the most common infections in humans. On the whole, they cause only minor problems for those affected, such as increased sputum, cough, sore throat and fever, and do not require medical intervention. Most of these infections — the common cold, pharyngitis (sore throat) and laryngitis — involve only the upper airways (that is, the top part of the conducting airways). Although the lungs have direct contact with the atmosphere, they usually remain sterile as the upper airways filter and clear the inspired air of contaminants and thus more serious infections are prevented. Infections of the lower respiratory tract occur most often in individuals whose normal defence mechanisms are impaired and often provide more serious alterations to the pulmonary system, which can also have profound systemic effects, such as changes in cellular metabolism, affecting homeostasis. Of all the pulmonary infections in adults, pneumonia is the most serious and a leading cause of death in both males and females in Australia and New Zealand, especially in people older than 65 years of age.28,42 We now examine the pathophysiology of this serious infection.
Pneumonia
Pneumonia is infection of the lower respiratory tract caused by bacteria, viruses, fungi, protozoa or parasites. Risk factors for pneumonia include advanced age, individuals who are immunocompromised, underlying lung disease, alcoholism, altered consciousness, smoking, malnutrition and immobilisation. The causative microorganism influences how the individual presents clinically, how the pneumonia should be treated and the prognosis. Community-acquired pneumonia tends to be caused by different microorganisms compared to healthcare-acquired infections (healthcare-acquired infections are discussed in Chapter 14). In addition, the characteristics of the individual are important in determining which microorganism is likely; for example, immunocompromised individuals tend to be susceptible to opportunistic infections (pathogens that cause infections but not in healthy individuals) that normally are uncommon in adults. In general, infections acquired within healthcare facilities and those affecting immunocompromised individuals have a higher mortality rate than community-acquired pneumonia. Some of the most common causal microorganisms are listed in Table 25-3.43–45 4 6
Table 25-3 COMMON MICROORGANISMS OF PNEUMONIA
| COMMUNITY-ACQUIRED PNEUMONIA | HOSPITAL-ACQUIRED (NOSOCOMIAL) PNEUMONIA | IMMUNOCOMPROMISED INDIVIDUALS |
|---|---|---|
| Streptococcus pneumoniae | Pseudomonas aeruginosa | Pneumocystis jiroveci(Pneumocystis pneumonia) |
| Mycoplasma pneumoniae | Staphylococcus aureus | Mycobacterium tuberculosis |
| Haemophilus influenzae | Klebsiella pneumoniae | Atypical mycobacteria |
| Oral anaerobic bacteria | Escherichia coli | Fungi |
| Influenza virus | Respiratory viruses | |
| Legionella pneumophilae | Protozoa | |
| Chlamydia pneumoniae | Parasites | |
| Moraxella catarrhalis |
The most common community-acquired pneumonias are caused by bacteria, particularly those caused by Streptococcus pneumoniae (also known as the pneumococcus), which has a relatively high mortality rate in the elderly.46 Mycoplasma pneumoniae is a common cause of pneumonia in young people living in close contact, such as in dormitories.47 Influenza is the most common viral community-acquired pneumonia in adults and children; respiratory syncytial virus and parainfluenza virus are common aetiological microorganisms.48 Legionella species is also an important cause of community-acquired pneumonia. Pseudomonas aeruginosa, other gram-negative microorganisms and Staphylococcus aureus are the most common aetiological agents in nosocomial pneumonia.44 Immunocompromised individuals (for example, people with HIV or individuals who have undergone organ transplantation) are especially susceptible to Pneumocystis jiroveci (formerly called Pneumocystis carinii), mycobacterial infections and fungal infections, such as aspergillus, of the respiratory tract.45,49 These infections can be difficult to treat and have a high mortality rate.
PATHOPHYSIOLOGY
Aspiration of oropharyngeal secretions is the most common route of lower respiratory tract infection; thus, the nasopharynx and oropharynx constitute the first line of defence for most infectious agents. Another route of infection is through the inhalation of microorganisms that have been released into the air when an infected individual coughs, sneezes or talks, or from aerosolised water such as that from contaminated respiratory therapy equipment. This route of infection is most important in viral and mycobacterial pneumonias and in Legionella outbreaks. Pneumonia can also occur when bacteria are spread to the lungs in the blood from bacteraemia (bacteria within the blood) that can result from infection elsewhere in the body or from intravenous drug abuse.
In healthy individuals, pathogens that reach the lungs are expelled or held in check by mechanisms of self-defence (see Chapters 12 and 13). If a microorganism gets past the upper airway defence mechanisms, such as the cough reflex and mucociliary clearance, the next line of defence is the alveolar macrophage (see Chapter 24 for details on pulmonary system defence mechanisms). This phagocyte is capable of removing most infectious agents without setting off significant inflammatory or immune responses. However, if the microorganism is virulent (small numbers can be pathogenic) or present in large enough numbers, it can overwhelm the alveolar macrophages. This results in a full-scale activation of the body’s defence mechanisms, including the release of multiple inflammatory mediators, cellular infiltration and immune activation.50,51 These inflammatory mediators and immune complexes can damage bronchial mucous membranes and alveolar–capillary membranes, causing the alveoli and terminal bronchioles to fill with infectious debris and exudate (fluid moving into a site of inflammation). In addition, some microorganisms release toxins from their cell walls that can cause further lung damage. The accumulation of exudate in the alveoli leads to dyspnoea, ventilation–perfusion mismatching and hypoxaemia.
There are many viruses that can cause pneumonia, including influenza virus, respiratory syncytial virus, adenoviruses and parainfluenza virus. Viral pneumonia is the primary cause of pneumonia in children and older adults. Although viral pneumonia can be severe, it is usually mild and self-limiting. However, it can set the stage for a secondary bacterial infection by providing an ideal environment for bacterial growth and by damaging ciliated epithelial cells, which normally prevent pathogens from reaching the lower airways. Viral pneumonia can be a primary infection or a complication of another viral illness, such as chickenpox or measles (spread from the blood). The virus not only destroys the ciliated epithelial cells but also invades the goblet cells and bronchial mucous glands. Sloughing of destroyed bronchial epithelium occurs throughout the respiratory tract, preventing mucociliary clearance. Bronchial walls become oedematous and infiltrated with leucocytes. In severe cases, the alveoli are involved, with decreased compliance and increased work of breathing.
CLINICAL MANIFESTATIONS
Many cases of pneumonia are preceded by an upper respiratory infection, which is often viral. Individuals then develop fever, chills, productive or dry cough, malaise, pleural pain and sometimes dyspnoea and haemoptysis (blood in the sputum). Physical examination may reveal signs of pulmonary consolidation, such as dullness to percussion (creation of vibrations, typically by tapping the chest) and inspiratory crackles. Individuals may also demonstrate symptoms and signs of underlying systemic disease or sepsis.
EVALUATION AND TREATMENT
Diagnosis is made on the basis of the physical examination, white blood cell count, chest X-ray, stains and cultures of respiratory secretions, and blood cultures.52 The white blood cell count is usually elevated, although it may be low if the individual is debilitated or immunocompromised. Chest X-rays show infiltrates that may involve a single lobe of the lung or may be more diffuse (see Figure 25-15). Once the diagnosis of pneumonia has been made, the pathogen is identified by means of sputum characteristics (gram stain; see Chapter 14 for details) and cultures or, if sputum is absent, blood cultures. Because many pathogens exist in the normal oropharyngeal flora, the specimen may be contaminated with pathogens from oral secretions. If sputum studies fail to identify the pathogen, the individual is immunocompromised or the individual’s condition worsens, further diagnostic studies may include bronchoscopy (in which a scope with a camera is introduced into the lungs to visualise the airways) or lung biopsy. Positive identification of viruses can be difficult. Blood cultures often help to identify the virus if systemic disease is present.
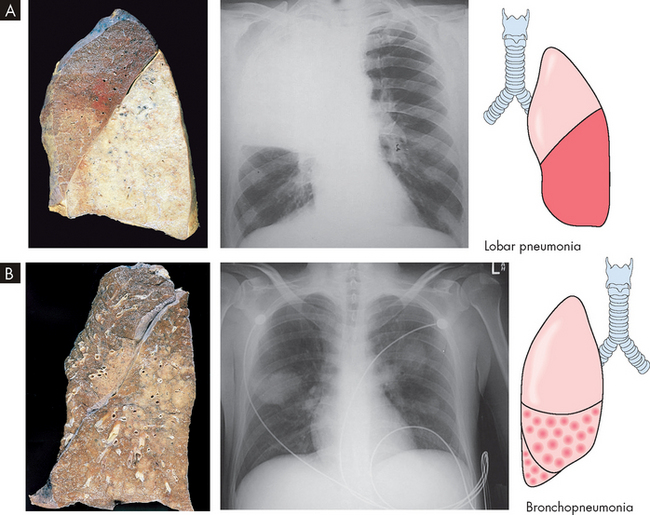
FIGURE 25-15 Bacterial pneumonia seen in gross lung, chest X-ray and illustration.
A Lobar pneumonia occurs when bacteria infection occurs in a portion of the lobe or the entire lobe. B Bronchopneumonia with patchy consolidation throughout the lung.
Source: A Based on Kumar V. Robbins & Cotran pathologic basis of disease. 8th edn. Philadelphia: Saunders; 2010. B Klatt EC. Robins & Cotran atlas of pathology. 2nd edn. Philadelphia: Saunders; 2010. Thibodeau GA. The human body in health & disease. 5th edn. St Louis: Mosby; 2010.
Antibiotics are used to treat bacterial pneumonia; however, resistant strains of pneumococcus are on the rise.53 Antibiotics are chosen based on the likely causative microorganism according to the clinical presentation and history.52 Viral pneumonia is usually treated with supportive therapy alone; however, antiviral medication may be needed in severe cases. Infections with opportunistic microorganisms may be polymicrobial (many species of microorganism) and require multiple drugs, including antifungals. Adequate hydration and good pulmonary hygiene (e.g. deep breathing, coughing, chest physiotherapy) are important aspects of treatment for all types of pneumonia.
Tuberculosis
Tuberculosis is an infection caused by Mycobacterium tuberculosis, a bacterium that usually affects the lungs but may invade other body systems. Worldwide, tuberculosis is the leading cause of death from a curable infectious disease and was responsible for an estimated 1.6 million deaths in 2005.54 There are new cases of tuberculosis each year in Australia and New Zealand although the rates are very low compared with other developed countries. However, as with many other diseases, the rates are higher in the Indigenous populations and people born overseas.28
PATHOPHYSIOLOGY
Tuberculosis is transmitted from person to person in airborne droplets. Microorganisms lodge in the lung periphery, usually in the upper lobe. Once the bacteria are inspired into the lung, they multiply and cause lung inflammation. Some bacteria migrate through the lymphatics and become lodged in the lymph nodes, where they encounter lymphocytes that initiate the immune response.
Inflammation in the lung causes activation of alveolar macrophages and neutrophils. These cells engulf the bacteria and begin the process by which the body’s defence mechanisms isolate and prevent their spread. The neutrophils and macrophages seal off the colonies of bacteria, forming granulomatous lesions called tubercles. Infected tissues within the tubercles die, forming cheese-like material that is necrotic (see Figure 25-16).55,56 Scar tissue then grows around the tubercles, completing isolation of the bacteria. The immune response is complete after about 10 days, preventing further spread of the bacteria.
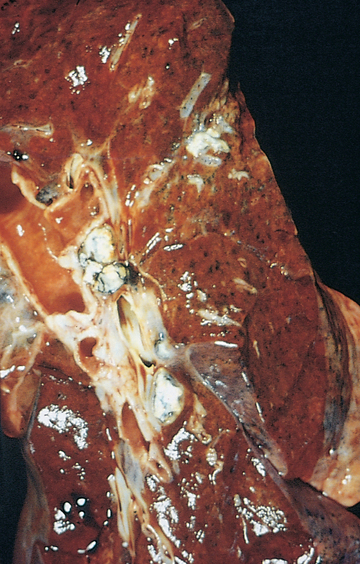
FIGURE 25-16 Tuberculosis in the lung.
The grey-white areas represent the lesions formed from the bacteria.
Source: Kumar V. Robbins & Cotran pathologic basis of disease. 8th edn. Philadelphia: Saunders; 2010.
Once immunity develops, tuberculosis may remain dormant for life.55,56 If the immune system is impaired or if live bacteria escape into the bronchi, active disease occurs and may spread through the blood and lymphatics to other organs.
CLINICAL MANIFESTATIONS
In many infected individuals, tuberculosis is asymptomatic. In others, symptoms develop so gradually that they are not noticed until the disease is advanced. Common clinical manifestations include fatigue, weight loss, lethargy, anorexia (loss of appetite) and a low-grade fever that usually occurs in the afternoon. A cough that produces purulent (containing pus) sputum develops slowly and becomes more frequent over several weeks or months. Night sweats and general anxiety are often present. Dyspnoea, chest pain and haemoptysis may occur as the disease progresses.
EVALUATION AND TREATMENT
Tuberculosis is usually diagnosed by a positive tuberculin skin test, sputum culture and chest X-rays. However, due to the high rate of false positives with the tuberculin skin test (meaning that the test reveals a positive tuberculosis result when the disease is not present), newer diagnostic tests have been developed. One such test, the interferon gamma release assay, measures interferon gamma that has been released from T cells of the immune system. When an individual becomes infected with the pathogenic bacteria, Mycobacterium tuberculosis, the bacterial antigen is recognised by the immune system and T cells are sensitised. The T cells then release the cytokine, interferon gamma, which stimulates macrophages to phagocytose bacteria (see Chapter 13 for more details on immune responses).
Treatment consists of antibiotic therapy to control active or dormant tuberculosis and prevent transmission. Today, with the increased numbers of immunosuppressed individuals and drug-resistant bacteria, treatment is never single drug therapy as resistance appears rapidly; the recommended treatment includes a combination of drugs to which the organism is susceptible, including isoniazid, rifampin, pyrazinamide and ethambutol. Combination therapy is usually continued for 6 months.57
In the past, individuals with active tuberculosis were isolated from the community. Today, individuals remain at home or, rarely, in hospital, until sputum cultures show that the active disease has been eliminated.
Acute bronchitis
Acute bronchitis is an acute infection or inflammation of the airways or bronchi and is usually self-limiting. In the vast majority of cases, it is caused by viruses.35 Many clinical manifestations are similar to those of pneumonia (fever, cough, chills and malaise — a general feeling of being unwell), but chest X-rays show no infiltrates. Individuals with viral bronchitis present with a non-productive cough that often occurs in paroxysms (sudden fits of coughing) and is aggravated by cold, dry or dusty air. In some cases, purulent sputum is produced. Chest pain often develops from the effort of coughing. Treatment consists of rest, aspirin, humidity and a cough suppressant, such as codeine.35
Bacterial bronchitis is rare in previously healthy adults except after viral infection but is common in patients with COPD. Although individuals with bronchitis do not have signs of pulmonary consolidation on physical examination (e.g. crackles), many will require chest X-ray evaluation to exclude the diagnosis of pneumonia. Bacterial bronchitis is treated with rest, antipyretics (fever-reducing drugs) and antibiotics.
Avian and swine influenza
A highly pathogenic virus known as H5N1 influenza virus (avian influenza), dubbed ‘avian flu’, has caused massive infections in poultry in Asia and was first found to infect humans in Hong Kong in 1997. Human infections occurred only in those individuals who had been in close contact with infected birds. In 2003, the H5N1 virus was found in poultry in South-East Asia, this time spreading to 8 countries with subsequent human infections. The virus has since spread to Europe, Africa and the Middle East. Despite the geographical spread of the virus, the number of human deaths has been relatively low, especially when compared to annual deaths from seasonal influenza (about 0.5 million). As of July 2009, 436 humans were infected with H5N1 virus and 262 people across 12 countries had died as a result of the virus. The primary reason for this limitation in human deaths is the lack of human-to-human transmission with H5N1.
In contrast, a new strain of type A influenza virus (H1N1) was found in April 2009. This subtype had never been found before in humans and was termed ‘swine flu’ because some of the genes were similar to those found in influenza infecting pigs. However, the virus is actually a mutation, containing two genes from pig influenza, one avian gene and one human gene. The place of origin of the virus is unknown. Unlike avian influenza, this new type A H1N1 influenza virus can be transmitted from human to human and is as contagious as seasonal influenza. Accordingly, the World Health Organization declared an influenza pandemic and guidelines were instituted to limit the spread. As of February 2010, Australia had 39,693 confirmed cases of pandemic H1N1 with 191 deaths. In 2009, New Zealand had 3200 cases and 20 deaths. The symptoms of swine flu are similar to those of seasonal influenza, with cough, fever, sore throat, malaise and myalgia most commonly reported. The incubation period for the virus is from 3 to 7 days. In Australia, the Panvax® H1N1 vaccine was made available in September 2009 and 5.1 million doses have been distributed to immunisation providers. There were limited supplies in New Zealand, with enough for 150,000 immunisations, which are directed to higher risk priority groups, such as healthcare workers.
Source: Epidemiology of WHO-confirmed human cases of avian influenza A(H5N1) infection (2006). Weekly epidemiological record no. 26, 81. Available at www.who.int/wer; Cheng A et al. ASID/TSANZ guidelines: treatment and prevention of H1N1 influenza 09 (human swine influenza) with antiviral agents. MJA 2009; 191:1–8; www.who.int/csr/disease/avian_influenza/en/index.html; www.wpro.who.int/health_topics/h1n1/; www.who.int/csr/disease/swineflu/frequently_asked_questions/about_disease/en/index.html; www.healthemergency.gov.au/internet/healthemergency/publishing.nsf/Content/healthprof#antiviral.
Influenza
Influenza is a common respiratory viral infection that affects millions of people worldwide.3 The influenza virus can infect all age groups, but the young and the elderly are at greater risk and have the highest mortality from the infection. The virus can rapidly spread worldwide and has a seasonal variation that affects Australia and New Zealand predominately from June to September.2,28
PATHOPHYSIOLOGY
There are three main strains of influenza virus: type A, type B and type C. All three can cause influenza in humans, but type A is the most prevalent and is responsible for the yearly influenza known as ‘seasonal flu’. Type A has many different subtypes, which are classified using the letters ‘H’ and ‘N’, denoting two different surface proteins. For example, the most common virus causing infection in humans is type A (H1N1), which itself has many different subtypes. However, the virus can change — called antigenic drift, which means that mutations occur in the virus antigen such that the body’s antibodies cannot recognise the virus and hence it represents a new primary immune response (refer to Chapter 12). This is the primary reason why ‘new’ types of flu circulate each year. Antigenic drift has led to major pandemics that have resulted in massive mortality worldwide.3 Worryingly, type A affects not only humans, but also horses, pigs, birds and aquatic birds. Avian and swine type A influenza have infected humans, and human-to-human transmission has occurred, leading to pandemics (see the box ‘Health alert: avian and swine influenza’).
The influenza virus enters the upper airways from airborne secretions of an infected individual. If the virus is not immobilised by the inflammatory and immune systems, it invades the respiratory tract lining and proliferates. The triggered inflammatory mediators cause mucosal hyperaemia (redness to the mucosal lining), upper airway oedema and excess mucous secretion. The incubation period (until the appearance of symptoms) is up to 72 hours.
CLINICAL MANIFESTATIONS
The classic signs and symptoms of cough and fever are usually indicative of influenza infection. They are often accompanied by generalised myalgia (muscle pain), headache and sore throat. The onset of the illness is abrupt and usually lasts between 3 and 5 days. Influenza infections can invade the lower respiratory tract and cause pneumonia, especially in children, the elderly and immunocompromised individuals (see Figure 25-17).
EVALUATION AND TREATMENT
Diagnosis of influenza is often difficult because of the rapid onset and relatively short duration. In addition, it is often hard to obtain isolation of the virus in specimens. The most effective treatment is prevention. Hand-washing combined with pulmonary hygiene lowers the risk of acquiring the virus. In Australia and New Zealand, influenza vaccines are available for those at higher risk of attaining the virus, such as healthcare workers, infants and the elderly.
Childhood pulmonary infections
Respiratory infections are common in children and are a frequent cause of hospitalisations. Clinical presentation, the age of the child and the season of the year can often provide clues to the type of microorganism, even when the agent cannot be proven.
Bronchiolitis
Bronchiolitis is a rather common, viral-induced lower respiratory tract infection that occurs almost exclusively in infants and young toddlers. It has a seasonal, yearly incidence (May–October) and is the leading cause of hospitalisations for infants during the winter season. The most common associated pathogen is respiratory syncytial virus, which accounts for 50–75% of cases,58,59 but it also may be associated with adenoviruses, influenza, parainfluenza and mycoplasma. Healthy infants usually make a full recovery from respiratory syncytial virus bronchiolitis, but infants who were born premature with a birth weight of less than 2500 grams may have a much higher risk for a more severe or even deadly course.
PATHOPHYSIOLOGY
Viral infection causes necrosis of the bronchial epithelium and destruction of ciliated epithelial cells. There is infiltration with lymphocytes around the bronchioles and a cell-mediated hypersensitivity to viral antigens with release of lymphokines causing inflammation, as well as activation of eosinophils, neutrophils and monocytes.60 The submucosa becomes oedematous and cellular debris and fibrin form plugs within the bronchioles.
Oedema of the bronchiolar wall, accumulation of mucus and cellular debris and, perhaps, bronchospasm narrow many peripheral airways. Other airways become partially or completely occluded. Atelectasis (collapse of lung tissue) occurs in some areas of the lungs and hyperinflation in others. There is air trapping and functional residual capacity is greatly increased. Compliance is decreased because the lungs are already highly inflated and because airway resistance within the lungs is uneven and increased. The decrease in compliance and the increase in airway resistance result in a substantial increase in the work of breathing. Serious alterations in gas exchange occur because of airway obstruction and patchy atelectasis. Hypoxaemia develops because of ventilation–perfusion mismatch and hypercapnia may occur in severe cases. It has been suggested that children with acquired bronchiolitis may later develop asthma, but the relationship between these two respiratory disorders is unclear.
CLINICAL MANIFESTATIONS
Symptoms usually begin with significant rhinorrhoea (runny nose) followed by a tight cough over the next few days, along with systemic signs of poor feeding, lethargy and fever. Infants typically have tachypnoea (increased ventilatory rate), variable degrees of respiratory distress and abnormal auscultatory findings of the chest. Wheezing is most common.
EVALUATION AND TREATMENT
Diagnosis of bronchiolitis is made by a review of the signs and symptoms (e.g. rhinitis, cough, wheezing, chest retractions, tachypnoea) and chest X-ray findings. Treatment is determined by the severity of the disease and the age of the child. Most cases are mild and usually require no specific treatment. Preventive treatment using pulmonary and hand hygiene combined with decreased exposure to people in the susceptible months decreases the risk of infection. Respiratory syncytial virus antibody is recommended for high-risk infants under 2 years old.58
Pertussis
Pertussis is caused by the bacterium Bordetella pertussis. The symptoms are thick secretions, a chronic cough and spasm following coughing fits, which give a characteristic ‘whoop’ sound — hence the common name ‘whooping cough’. The infection has an incubation period of 7–10 days and is highly contagious, but it is preventable with vaccination. However, despite the availability of a vaccine, Australia and New Zealand experience periodic outbreaks of pertussis, with 11,200 cases reported in Australia in 2008.28,61 Pertussis is particularly lethal in newborns and infants prior to immunisation (see Chapter 14 for immunisation schedules).
LUNG CANCER
Lung cancer arises from the epithelium of the respiratory tract. Therefore, the term lung cancer excludes other pulmonary tumours such as sarcomas, lymphomas, blastomas, haematomas and mesotheliomas.
Approximately 7500 people die from lung cancer each year in Australia.62,63 Of all the cancers, lung cancer is the leading cause of death in Australia.62,63 Lung cancer is the fourth most common cancer and those diagnosed with it have a low survival rate. For females, lung cancer deaths were higher than breast cancer deaths in 2005 and it is predicted that this trend will continue due to the high smoking rates in females in the 1970s and 1980s. However, despite lung cancer remaining the biggest killer of all cancers in men, the rate of new lung cancer cases is declining because smoking rates in men have declined.63 The incidence and mortality rates of lung cancer in the Indigenous population are approximately double those of the non-Indigenous population. Concomitantly, smoking rates in the Indigenous population are also greater.64
In New Zealand, lung cancer is the most common cause of cancer death in males and the third most common in females.42 Similar to in Australia, lung cancer incidence and mortality in females is projected to rise, while the rates for males are in decline. Unfortunately, the mortality and incidence rates for lung cancer in the Indigenous population are up to three times higher than those in the non-Indigenous population.65
The most common cause of lung cancer is cigarette smoking. It is likely that up to 90% of lung cancers are attributable to smoking.5,66 It has been shown that the amount that people smoke and the number of years they smoke are directly related to the risk of developing lung cancer. There is an increased risk of developing lung cancer with advancing age, with a three times greater risk in people aged 65 years and older compared with their younger counterparts. This is evidenced by the fact that only 1% of lung cancers occur in people less than 40 years of age.63 In addition, second-hand (passive or environmental) smoke exposure has been identified as a risk for lung cancer. Smokers with obstructive lung disease (low FEV1) are at even greater risk. Genetic predisposition to developing lung cancer also plays a role in the pathophysiology. Other risk factors include occupational exposure to certain workplace toxins, radiation, air pollution and tuberculosis.
Types of lung cancer
Primary lung cancers arise from the bronchi within the lungs and are therefore called bronchogenic carcinomas. Although there are many types of lung cancer, lung cancer is divided into two major categories: non–small cell carcinoma (75–85% of all lung cancers) and small cell carcinoma (15–25% of all lung cancers). The category non–small cell carcinoma can be subdivided into three common types of lung cancer: squamous cell carcinoma, adenocarcinoma and large cell carcinoma. Characteristics of these tumours, including the clinical manifestations, are listed in Table 25-4. Many cancers that arise in other organs of the body metastasise to the lungs; however, these are not considered as lung cancers and are categorised by their primary site of origin.
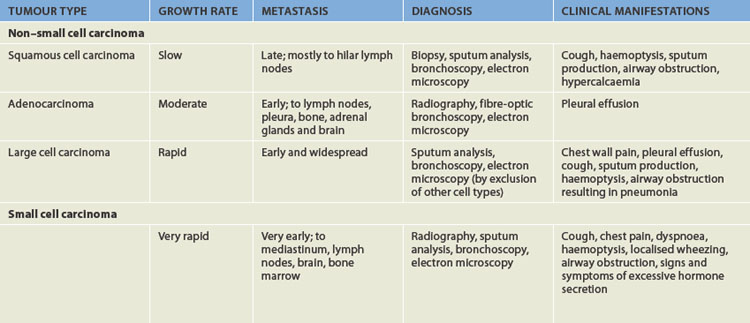
Non–small cell carcinoma
Squamous cell carcinoma accounts for about 30% of bronchogenic carcinomas. These tumours are typically located near the hilum and project into the bronchi (see Figure 25-18). Because of the location in the central bronchi, obstructive manifestations are nonspecific and include non-productive cough or haemoptysis (which is the coughing up of blood in the sputum; see ‘Clinical manifestations of pulmonary alterations’ below). Pneumonia and atelectasis are often associated with squamous cell carcinoma. Chest pain is a late symptom associated with large tumours. These tumours can remain fairly well localised and tend not to metastasise until late in the course of the disease. The preferred treatment is surgical resection, although once metastasis (spread away from the original site) has taken place, total surgical resection is more difficult and survival rates decrease dramatically.67,68 Radiation therapy and chemotherapy improve outcomes in many individuals.69
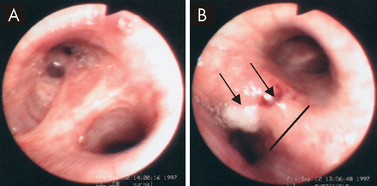
FIGURE 25-18 Squamous cell carinoma.
A Normal carina and bronchi of left upper lobe. B Carina of left lower lobe with swollen mucosa (thin dark line showing extent of swelling), white lesion (squamous cell cancer, bottom arrow) and haemorrhage on upper surface (top arrow).
Source: Goldman L. Cecil medicine. 23rd edn. Philadelphia: Saunders; 2007.
Adenocarcinoma (meaning that the tumour arises from the glands) constitutes 35–40% of all bronchogenic carcinomas (see Figure 25-19). The increase in incidence of adenocarcinoma has been ascribed to the increasing occurrence of lung cancer in females, environmental and occupational carcinogens, and changes in the histological criteria for diagnosis. These tumours, which are usually smaller than 4 cm, more commonly arise in the peripheral regions of the lung tissue. They may be asymptomatic and discovered by routine chest X-ray in the early stages or the individual may present with pleuritic chest pain and shortness of breath from pleural involvement by the tumour. Surgical resection is possible in a high proportion of cases, but because metastasis occurs early, the 5-year survival rate is low.
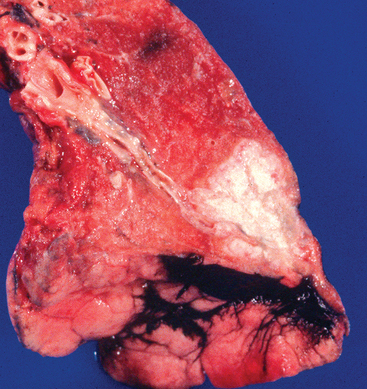
This gross lung lobe has a large white mass (carcinoma). The cancers are predominately on the peripheral aspects of the lung, not in the large airways.
Source: Zander DS, Farver CF. Pulmonary pathology. Churchill Livingstone; 2008.
Large cell carcinomas constitute 10–15% of bronchogenic carcinomas (see Figure 25-20). This cell type has lost all evidence of differentiation and therefore is sometimes referred to as undifferentiated large cell anaplastic cancer (literally meaning that the cells revert back to an immature form). Because large cell carcinomas show none of the histological findings of squamous cell carcinoma or adenocarcinoma, they are diagnosed by a process of exclusion. The cells are large and contain darkly stained nuclei. These tumours commonly arise centrally and can grow to distort the trachea and cause widening of the carina, which can result in breathing difficulties. Once metastasis has occurred, surgical therapy is limited to palliative procedures — meaning that care is for comfort only, as the individual cannot be cured.
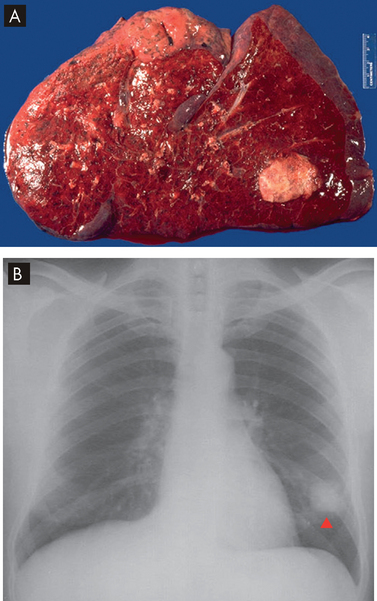
Small cell carcinoma
Small cell carcinomas constitute 15–20% of bronchogenic carcinomas. Most of these tumours are central in origin (see Figure 25-21). This cell type has the strongest correlation with cigarette smoking. Because these tumours show a rapid rate of growth and tend to metastasise early and widely, small cell carcinomas have the worst prognosis. Survival time for untreated small cell carcinoma is usually only 1–3 months. Approximately only 14% of treated individuals are alive 2 years after diagnosis.
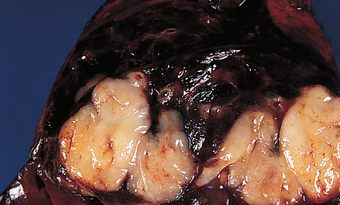
FIGURE 25-21 Small cell carcinoma.
Source: Damjanov I, Linder J (eds). Anderson’s pathology. 10th edn. St Louis: Mosby; 1996.
Small cell carcinoma is most often associated with ectopic hormone production, meaning that hormones are produced in tissues, in this case cancerous lung tissue, away from the usual glands. Neuroendocrine cells containing neurosecretory granules exist throughout the tracheobronchial tree and may be associated with small cell carcinoma.70 Ectopic hormone production is important to the clinician because resulting signs and symptoms may be the first manifestation of the underlying cancer. Small cell carcinomas most commonly produce antidiuretic hormone from associated neuroendocrine cells and develop the syndrome of inappropriate antidiuretic hormone secretion (see Chapter 11). They can also produce gastrin-releasing peptide, calcitonin, arginine, vasopressin and adrenocorticotropic hormone. As a result of adrenocorticotropic hormone secretion, individuals with lung cancer secrete large quantities of steroids, leading to the development of an atypical Cushing’s syndrome (see Chapter 11).
Signs and symptoms related to this condition include muscular weakness, facial oedema, hypokalaemia, alkalosis, hyperglycaemia, hypertension and increased pigmentation. Treatment of small cell carcinoma is usually palliative. More than 85% of tumours will have metastasised by the time of diagnosis. Chemotherapy and radiation can significantly prolong life and relieve symptoms, but relapse is inevitable in most individuals.71
PATHOPHYSIOLOGY
Tobacco smoke contains more than 30 carcinogens and is responsible for causing 80–90% of lung cancers. These carcinogens, along with probable inherited genetic predisposition to cancers, result in multiple genetic abnormalities in bronchial cells including deletions of chromosomes, activation of oncogenes and inactivation of tumour suppressor genes (see Chapter 36).72 The most common genetic abnormality associated with lung cancer is loss of the tumour suppressor gene p53; mutations in this gene have been found in 50–60% of non–small cell carcinomas and 90% of small cell carcinomas.73 Once lung cancer is initiated by these carcinogen-induced mutations, further tumour development is promoted by growth factors. Further cellular toxicity is enhanced through smoke-induced toxic free radical production.
The bronchial mucosa suffers multiple carcinogenic ‘hits’ due to repetitive exposure to cigarette smoke and eventually epithelial cell changes begin to be visible on biopsy. These changes progress from metaplasia (changing from one cell type to another cell type) to carcinoma in situ and finally to invasive carcinoma. Further tumour progression includes invasion of surrounding tissues and finally metastasis to distant sites including the brain, bone marrow and liver.
CLINICAL MANIFESTATIONS
There are many different signs and symptoms in individuals with lung cancer. It is somewhat dependent on the location of the cancer in the pulmonary system as to what clinical manifestations will arise. Table 25-4 summarises the characteristic clinical manifestations according to tumour type. By the time there are manifestations severe enough for the individual to notice them, the disease is usually advanced.
EVALUATION AND TREATMENT
Diagnostic tests for the evaluation of lung cancer include chest X-ray, sputum cytology, chest-computed tomography, fibreoptic bronchoscopy and biopsy. Biopsy determines the cell type, and the evaluation of lymph nodes and other organ systems is used to determine the stage of the cancer. The histological cell type and the stage of the disease are the major factors that influence the choice of therapy. The current accepted system for the staging of non–small cell carcinoma is the TNM classification. This system is a code in which T denotes the extent of the primary tumour, N indicates the lymph node involvement and M describes the extent of metastasis. Small cell carcinoma is so rapidly progressive that its staging system consists of only two stages: limited and extensive disease.
Genetic and immunological therapies for lung cancer
Although new chemotherapeutic agents have slightly improved outcomes in the management of lung cancer, overall survival rates remain poor and the toxicities of these regimens limit their use. New understandings of the genetic and immunological features of lung cancer cells have led to new treatments. Gene therapy is emerging as a way of restoring normal tumour suppressor gene function and increasing tumour responsiveness to chemotherapy and radiation therapy. Immunological therapies include antibodies to growth factor receptors and anti-angiogenesis drugs (those that prevent the growth of new blood vessels from the tumour). The effectiveness of these strategies is still being evaluated, but new knowledge is leading to new opportunities for treatment.
Source: González G et al. Therapeutic vaccination with epidermal growth factor (EGF) in advanced lung cancer: analysis of pooled data from three clinical trials. Hum Vaccin 2007; 3(1):8–13; Ruttinger D et al. Immunotherapy of lung cancer: an update. Onkologie 2006; 29(1–2):33–38; Sandler AB. Targeting angiogenesis in lung cancer. Sem Oncol 2005; 32(6 suppl 10):S16–S22; Toloza EM. Gene therapy for lung cancer. Thorac Surg Clin 2006; 16(4):397–419.
The only proven way of reducing the risk for lung cancer is the cessation of smoking. To date, trials evaluating the use of various early screening modalities such as chest X-ray and CT scanning have not resulted in a decrease in lung cancer mortality.74 The management of lung cancer has been outlined here under each cell type, but it is generally chosen on the basis of tumour stage and patient functional status. Current modalities include combinations of surgical resection, chemotherapy and radiation; however, new genetic and immunological therapies are being explored.75–79 76 77 78 79 (see the box ‘Health alert: genetic and immunological therapies for lung cancer’).
OBSTRUCTIVE SLEEP APNOEA
Obstructive sleep apnoea generally results from upper airway obstruction recurring during sleep, with excessive snoring and multiple apnoeic episodes (periods were there is no breathing) that last 10 seconds or longer. It is estimated that 3.1% of Australian males have obstructive sleep apnoea;80 however, a more recent estimate concluded that approximately 9–25% of the middle-aged population in Australia have obstructive sleep apnoea, with the prevalence unknown in older people.81 In New Zealand, the prevalence of obstructive sleep apnoea has been estimated at 4.1% for males and 0.7% for females.82 Within Indigenous populations, the prevalence is higher for both males and females. Childhood obstructive sleep apnoea is also quite common, with an estimated prevalence of 1–10%.83 In children, unlike in adults, obstructive sleep apnoea occurs equally among girls and boys.
There is an increased risk of death from cardiovascular disease associated with obstructive sleep apnoea.84 The exact reason for this is unknown; however, the repeated episodes of hypoxia related to apnoea during sleep are likely to impact on the cardiovascular system. It has also been shown that obstructive sleep apnoea is an independent risk factor for increased mortality from any cause.85 Individuals with moderate to severe obstructive sleep apnoea have been shown to be up to 4.5 times more at risk of mortality from any cause compared to individuals without sleep apnoea.85
PATHOPHYSIOLOGY
Anatomical and structural factors in the upper airway play a crucial role in the development of obstructive sleep apnoea. The tonsils, adenoids, tongue and soft tissue that surrounds the pharynx become enlarged, reducing the lumen (airway size). This is more pronounced in obese individuals who have a large neck circumference. Collectively, these factors predispose the individual to upper airway collapse during sleep.86,87 In children, the most pathophysiological reason for obstructive sleep apnoea is due to adenotonsillar hypertrophy; however, rates of childhood obesity are likely to contribute to the rates of childhood sleep apnoea.
When obstructive sleep apnoea is present, the pharyngeal tissue completely obstructs the airway, preventing ventilation. This causes a cycle of obstructive breathing during sleep when the airway repeatedly collapses, and this periodic breathing eventually produces arousal, which interrupts the sleep cycle, reducing total sleep time and producing sleep deprivation.88,89 The often sustained and repeated apnoeic periods result in hypoxaemia (inadequate oxygen levels in the blood), which, it has been proposed, influences neural control of the upper airway. Hypoxaemia is more pronounced during periods of rapid eye movement (REM) sleep. The reason for this is unknown, but it may explain the tiredness and insomnia reported by individuals with obstructive sleep apnoea.89
Sleep apnoea produces low oxygen saturation (see Figure 25-22) and eventually leads to polycythaemia (a blood disorder causing excessive red cell production), pulmonary hypertension, right-sided heart failure, liver congestion, cyanosis and peripheral oedema.90 Systemic hypertension may be a response to repeated episodes of apnoea and hypoxaemia.91
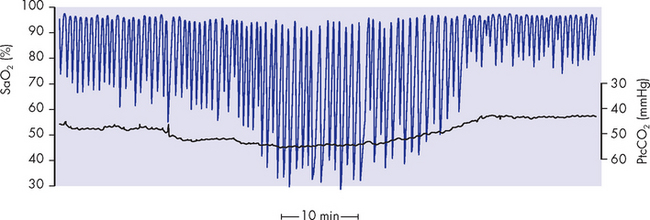
CLINICAL MANIFESTATIONS
Due to obstruction of the upper airway, snoring is the most common manifestation. There may be periods of increased ventilatory effort without an audible airflow. During the apnoeic periods, breathing can cease for 10 seconds up to 1 minute and these episodes can occur repeatedly throughout sleep. Therefore, sleep is often restless, and there is daytime tiredness and sleepiness. This chronic tiredness impacts on daytime cognitive and neurobehavioural performance. For instance, obstructive sleep apnoea has been associated with an increased mortality from traffic accidents.92 Cardiac arrhythmias during sleep apnoea are common, such as sinus pauses (temporary cessation of sinus node activity) and premature ventricular contractions.93 In children, bedwetting and chronic mouth breathing are associated with obstructive sleep apnoea.
EVALUATION AND TREATMENT
There usually is a history of snoring and laboured breathing during sleep, which may be continuous or intermittent in individuals with obstructive sleep apnoea. Associated risk factors include advancing age, obesity, gender (males are more affected than females), genetic predisposition, smoking and alcohol consumption.94 The most accurate diagnosis of obstructive sleep apnoea is an overnight polysomnogram, also known as a sleep study.94 This test records brain activity (electroencephalogram), eye movement, muscle activity (electromyogram), heart rate, air flow and oxygen haemoglobin (SpO2) levels during sleep and collectively allows classification of the amount and duration of apnoeic periods, thereby permitting diagnosis of obstructive sleep apnoea.
Treatments include nasal continuous positive airway pressure and dental devices, upper airway and jaw surgery in selected individuals, and management of obesity.95 In children, if obstructive sleep apnoea is documented or strongly suspected clinically, tonsillectomy and adenoidectomy are the treatments of choice.96 For severely affected children who do not respond to surgery or who have different problems, such as obesity, that cannot be remedied rapidly, continuous positive airway pressure, similar to adult management, may be required.
CHILDHOOD PULMONARY DISORDERS
There are some important childhood pulmonary disorders that need to be explored. In this section we examine some of the major childhood disorders, starting with croup.
Croup
Classic croup is an acute inflammation of the upper airways and almost always occurs in children between 6 months and 5 years of age.97 In 85% of cases, croup is caused by a virus, most commonly parainfluenza and in other instances by influenza A or respiratory syncytial virus. The incidence of croup is higher in males and is most common during the winter months.
PATHOPHYSIOLOGY
Airway obstruction occurs in the subglottic region of the trachea, just below the vocal cords. Contributory factors include mucosal oedema and secretions related to the viral infection. Anatomically, the subglottic region is slightly narrower than the rest of the trachea and in children the subglottic mucous membrane is more loosely attached and more vascular than in adults. These factors make the airway susceptible to compromise in children. If there is significant narrowing of the airway in this area, work of breathing will increase and the excessive negative pressure generated may even cause the airway structures higher up to collapse with inspiration (see Figure 25-23). The turbulent flow across this obstruction will cause stridor (an abnormal, harsh, high-pitched sound caused by turbulent flow in a partially obstructed upper airway) on inspiration and sometimes also on expiration (see Figure 25-24). Croup tends to affect younger children more prominently because they have smaller airways that are therefore compromised more easily (see Figure 25-25).
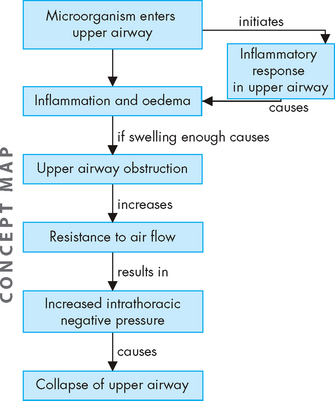
CLINICAL MANIFESTATIONS
Typically, the child experiences rhinorrhoea, sore throat and low-grade fever for a few days, then develops a seal-like barking cough. Most cases resolve spontaneously within 24–48 hours and do not warrant hospitalisation. However, the presence of inspiratory stridor or respiratory distress suggests a more severe situation.
EVALUATION AND TREATMENT
The degree of symptoms determines the level of treatment. Treatment may include injected, oral or nebulised glucocorticoids to reduce the inflammation. The presence of stridor at rest, moderate or severe retractions of the chest or agitation suggests more severe disease and requires hospitalisation for observation and treatment. Severe obstruction requires emergency care to protect the airway.
Respiratory distress syndrome of the newborn
The name respiratory distress syndrome of the newborn refers to a lung disorder that remains a significant cause of neonatal morbidity and mortality.98 It occurs almost exclusively in premature infants. Respiratory distress syndrome occurs in 60% of infants born at less than 28 weeks gestation, in 30% of those born at 28–34 weeks and in fewer than 5% of those born after 34 weeks. The incidence and death rates have declined significantly since the introduction of antenatal steroid therapy and postnatal surfactant therapy.99 Risk factors are as follows:
PATHOPHYSIOLOGY
Respiratory distress syndrome is caused by surfactant deficiency and also a deficiency in alveolar surface area for gas exchange. Surfactant is the material that lines the alveoli and is required for maintaining their inflation. Without surfactant, which lowers surface tension, alveoli would tend to collapse at the end of each exhalation. Surfactant is not normally secreted by the alveolar cells until approximately 30 weeks gestation. In addition to the functional surfactant deficiency of the premature lung, structural immaturity is a problem. Premature infants are born with many underdeveloped and small alveoli that are difficult to inflate. In the most extreme premature infants, the ‘alveoli’ have thick walls and inadequate capillary blood supply such that gas exchange is significantly impaired. In addition, the chest wall is weak and highly compliant and the rib cage tends to collapse inwards with ventilatory effort. The net effect of all these adverse factors is atelectasis (collapsed alveoli), which is difficult for the neonate to overcome because it requires a significant negative inspiratory pressure to open the alveoli with each breath. The infant uses more oxygen to sustain the work of breathing and becomes hypoxaemic and hypercapnic (low blood oxygen and high carbon dioxide, respectively). Hypoxia and atelectasis cause pulmonary vasoconstriction and increase intrapulmonary resistance. This results in hypoperfusion of the lung and a decrease in effective pulmonary blood flow. The pathogenesis of respiratory distress syndrome is summarised in Figure 25-26.
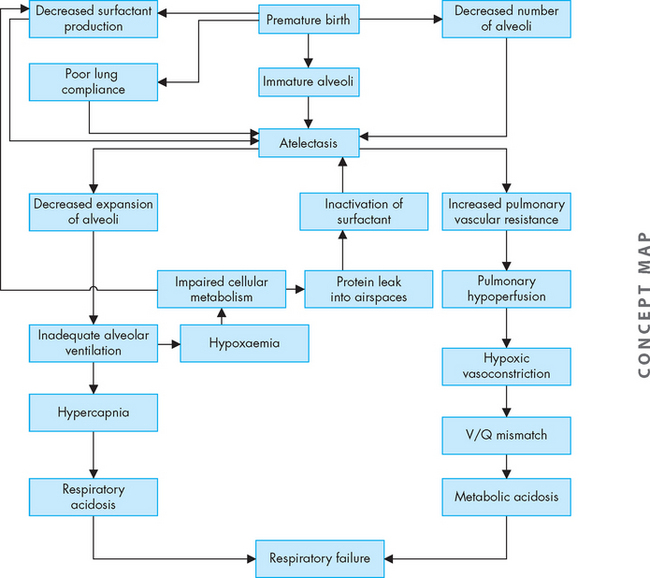
CLINICAL MANIFESTATIONS
Signs of respiratory distress syndrome appear within minutes of birth. Some neonates require immediate resuscitation because of asphyxia or severe respiratory distress. Characteristic signs are tachypnoea (respiratory rate over 60 breaths per minute), expiratory grunting, intercostal and subcostal retractions, nasal flaring and pale colour. The natural course is characterised by progressive hypoxaemia and dyspnoea. Apnoea and irregular respirations occur as the infant becomes fatigued from the difficulty of breathing. The typical chest X-ray shows diffuse, fine granular densities within the first 6 hours of life. In most cases the clinical manifestations reach a peak within 3 days, after which there is gradual improvement.
EVALUATION AND TREATMENT
Diagnosis is made on the basis of prematurity or other risk factors and chest X-rays. The ultimate treatment for respiratory distress syndrome would be prevention of premature birth; however, this is not always possible and often not foreseeable. Antenatal treatments with glucocorticoids are given to women at 24–34 weeks gestation for those at risk of premature delivery, and in preterm labour, unless delivery is imminent. Glucocorticoids induce a significant and rapid acceleration of lung maturation and there is extensive evidence that maternal steroid therapy significantly reduces the incidence of respiratory distress syndrome and death.100 Surfactant therapy should be considered complementary to antenatal corticosteroids.
Supportive care includes oxygen and often continuous positive airway pressure or mechanical ventilation. Most infants survive respiratory distress syndrome with treatment. In many cases, recovery may be complete within 10–14 days. However, the incidence of subsequent chronic lung disease is significant among very low birth weight infants.
Cystic fibrosis
Cystic fibrosis is an autosomal recessive inherited disease that results from defective epithelial chloride ion transport. The chromosomal mutation results in the abnormal expression of the protein, cystic fibrosis transmembrane conductance regulator, which is a chloride channel (it allows the diffusion of chloride out of the cell) present on the surface of many types of epithelial cells including the airways, bile ducts, pancreas, sweat ducts and vas deferens.101 In Australia and New Zealand, approximately 1 in 2800 are born with cystic fibrosis and about 1 in 25 are carriers who are not affected by the mutation.102 There are approximately 2700 children and adults in Australia and 400 in New Zealand with cystic fibrosis.103,104
PATHOPHYSIOLOGY
Although cystic fibrosis affects many organs (endocrine, gastrointestinal, renal and reproductive systems) the most important effects are on the lungs and in 90% of cases, chronic pulmonary infections eventually lead to respiratory failure and death.102 The typical features of cystic fibrosis lung disease are mucus plugging, chronic inflammation and chronic infection. The mucus plugging seen in cystic fibrosis probably results from both increased production of mucus and altered chemical properties of the mucus. Mucus-secreting airway cells (goblet cells and submucosal glands) are increased in number and size. Cystic fibrosis mucus is dehydrated and viscous because of defective chloride secretion and excess sodium absorption — as a result, the mucus secretions are thick and sticky. This decreases the fluid volume on the airway surface, impairing the mobility of the cilia and thereby allowing mucus to adhere to the airway epithelium, along with bacteria and injurious by-products from neutrophils.105,106
Chronic inflammation is believed to contribute to long-term lung damage and there is evidence that this process may even begin in infancy. Abnormal cytokine profiles promote a proinflammatory state.108
Children with cystic fibrosis have a propensity for chronic bronchial infection that remains poorly understood. It is likely that local factors in the cystic fibrosis airway microenvironment favour bacterial colonisation, because there is no systemic immune defect. Staphylococcus aureus is common and Pseudomonas aeruginosa ultimately colonises airways in at least 75% of children with cystic fibrosis. Combined with chronic bacterial infection, these lead to microabscess formation, bronchiectasis, patchy consolidation and pneumonia, peribronchial fibrosis and cyst formation (see Figure 25-27). There is a progressive decrease in the amount of available and functional lung tissue. The pathophysiology for these changes is outlined in Figure 25-28. Over time, pulmonary vascular remodelling occurs because of localised hypoxia and arteriolar vasoconstriction, and pulmonary hypertension and cor pulmonale (right ventricular enlargement) may develop in the late stages of disease.
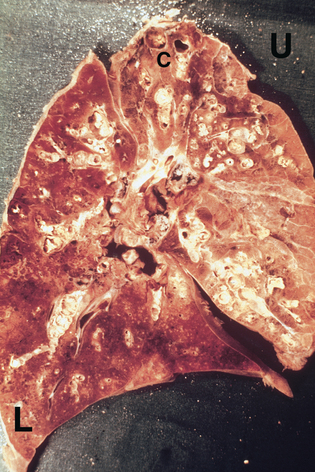
FIGURE 25-27 The pathology of the lung in cystic fibrosis.
Key features are widespread mucus impaction of airways and bronchiectasis (especially from upper lobe [U]), with haemorrhagic pneumonia in the lower lobe (L). Small cysts (C) are present at the apex of the lung.
Source: Kleinerman J, Vauthy P. Pathology of the lung in cystic fibrosis. Atlanta: Cystic Fibrosis Foundation; 1976.
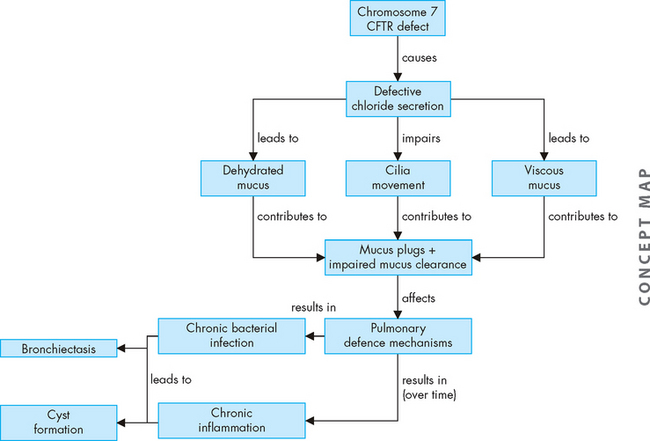
CLINICAL MANIFESTATIONS
The most common presentations are respiratory or gastrointestinal. Respiratory symptoms include persistent cough or wheeze and recurrent or severe pneumonia. Lung function decreases in individuals with cystic fibrosis with increasing age. For instance, at 18 years and older, forced expiratory volume (FEV1) is approximately 68% of predicted volumes.109 Physical signs that develop over time include barrel chest and digital clubbing. Over time, more serious pulmonary conditions may arise, such as haemoptysis and pneumothorax (see ‘Clinical manifestations of pulmonary alterations’). Classic gastrointestinal presentations include meconium ileus (meconium blocking the bowel) at birth, which is indicative of cystic fibrosis. Approximately half of all children present with failure-to-thrive and malabsorptive symptoms, such as frequent, loose and oily stools. More subtle presentations include chronic sinusitis, nasal polyps and rectal prolapse. Complications of cystic fibrosis may include liver disease (approximately 5%) and diabetes mellitus (10–25%).
EVALUATION AND TREATMENT
In Australia and New Zealand, newborn infants are screened for cystic fibrosis. The blood test measures pancreatic enzyme levels and, if the levels are abnormal, genetic testing for the cystic fibrosis mutation is undertaken. However, the testing is not definitive: there are numerous cystic fibrosis associated mutations and up to 5% of all tests will not be conclusive. Therefore, the definitive diagnosis is confirmed from a sweat test, which determines the level of sweat chloride concentration (indicative when in excess of 60 mmol/L).
Treatment is primarily focused on pulmonary health and nutrition. Common pulmonary therapies include techniques to promote mucus clearance, such as chest physical therapy and related mechanical devices, bronchodilators and aerosolised DNase, which liquefies mucus. Inhaled maintenance antibiotics can be used to suppress Pseudomonas aeruginosa when it is present and this has a beneficial clinical impact.107 Oral antibiotics are used fairly liberally for minor pulmonary exacerbations. Intravenous antibiotics are used to treat major exacerbations of pulmonary infection. Lung transplantation is an option for selected individuals with end-stage lung disease from cystic fibrosis. Complications are usually related to infection or rejection and survival at 5 years post-transplant is approximately 60%.
Approximately 90% of children with cystic fibrosis have pancreatic insufficiency and therefore need to take pancreatic enzymes (for digestion of nutrients) before meals and snacks for their entire lifetime. Fat-soluble vitamins must be supplemented. Energy needs are high, especially with advancing lung disease and high-kilojoule supplements or even gastrostomy feeding may be warranted. Nutritional care for children with cystic fibrosis has become increasingly aggressive because of the documented link to better long-term outcomes.
Fortunately, frequent hospitalisations and early debilitation are no longer common in individuals with cystic fibrosis. Life expectancy is currently in the mid-30s and in the next decade, with better treatments, it is expected to extend to 50 years of age.109
Sudden infant death syndrome
Sudden infant death syndrome (SIDS) remains a disease of unknown cause and is the most common cause of unexplained infant death in Western countries.110 It is defined as ‘sudden death of an infant under 1 year of age which remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene and review of the clinical history’.110
The incidence of SIDS is low during the first month of life but sharply increases in the second month and peaks at 3–4 months of age, then gradually declines. It is more common in males (60%) than females (40%). It almost always seems to occur during night-time sleep, when infants are least likely to be observed. A seasonal variation has been noted, with higher frequencies during the winter months. This has been related to a higher rate of respiratory tract infections during these months and, in fact, such infections are often reported to have preceded the death.
Clinical risk groups include babies who were preterm or low birth weight, who were one of simultaneous multiple births and who had siblings die of SIDS. Nevertheless, about three-quarters of all SIDS cases have no known predisposing clinical risk factor.
Additional risk factors fall into the categories of socioeconomic or maternal factors and factors in the baby’s sleeping situation. Maternal factors that predict increased risk are maternal smoking, young maternal age (under 20 years), less prenatal care, poverty and illicit drug use. Risk factors that relate to the baby’s sleeping situation are prone positioning, sleeping on a soft surface and overheating.111 Prone sleeping has been concluded to be a major and modifiable risk factor. Infants should sleep on their backs, even in preference over side sleeping. Other avoidable risk factors include sleeping on top of any soft surface and loose bedding. Overwrapping the infant or overheating the room also appears to increase risk, particularly if the infant is sleeping prone.
The aetiology of SIDS remains unknown but probably involves a combination of predisposing factors and external stressors.110
Currently, the best strategies for reducing SIDS seem to be avoidance of all the controllable risk factors. In Australia, infant mortality from SIDS fell by 83% between 1985 and 2005.112 This dramatic reduction was attributed to a successful national health education campaign that raised awareness of the risk factors and promoted safe infant sleeping practices, such as positioning the baby on their back during sleeping.
ALTERATIONS OF PULMONARY BLOOD FLOW AND PRESSURE
Blood flow through the lungs can be disrupted by disorders that occlude the vessels, increase pulmonary vascular resistance or destroy the vascular bed. The effects of altered pulmonary blood flow and pressure range from insignificant dysfunction to severe and life-threatening changes in ventilation–perfusion ratios.
Pulmonary embolism
Pulmonary embolism is occlusion of a portion of the pulmonary vascular bed by an embolus (see Figure 25-29), which can be a thrombus (blood clot), tissue fragment, lipids (fats), foreign body or an air bubble (air embolism). More than 90% of pulmonary emboli result from clots formed in the veins of the legs and pelvis.
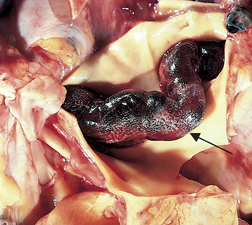
FIGURE 25-29 Large pulmonary embolus (arrow) lying in the pulmonary artery (retracted back for visualisation).
Source: Based on Kumar V. Robbins & Cotran pathologic basis of disease. 8th edn. Philadelphia: Saunders; 2010.
Risk factors for pulmonary thromboembolism, or the obstruction of a pulmonary vessel by a thrombus, include conditions and disorders that promote blood clotting as a result of venous stasis (slowing or stagnation of blood flow through the veins), hypercoagulability (increased tendency of the blood to form clots) and injuries to the endothelial cells that line the vessels. No matter the source, a blood clot becomes an embolus when all or part of it breaks away from the site of formation and begins to travel in the bloodstream. Thromboembolism or deep vein thrombus is described further in Chapter 23.
Although the overall incidence of pulmonary embolism has declined (2 per 1000 people per year), it remains an important cause of death, especially in the elderly and hospitalised individuals. Trauma, especially head injuries and fractures of the lower extremities, spine or pelvis, confers a high risk for venous thromboembolism.
PATHOPHYSIOLOGY
The impact or effect of the embolus depends on the extent of pulmonary blood flow obstruction, the size of the affected vessels, the nature of the embolus and the secondary effects. Pulmonary emboli can occur as any of the following:
The pathogenesis of pulmonary embolism caused by a thrombus is summarised in Figure 25-30.
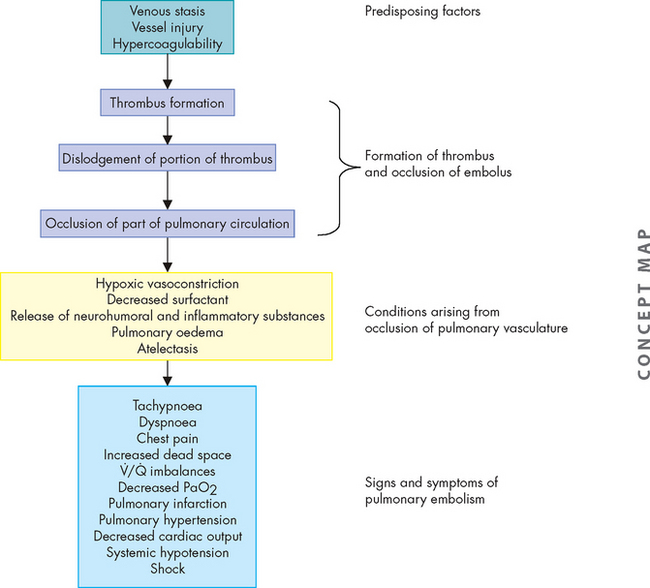
FIGURE 25-30 The pathogenesis of massive pulmonary embolism caused by a thrombus (venous thromboembolism).
If the embolus does not cause infarction in the lung tissue that is not receiving blood, the clot will be dissolved by the fibrinolytic system (see Chapter 16) and pulmonary function will return to normal. If pulmonary infarction occurs, shrinking and scarring develops in the affected area of the lung.
CLINICAL MANIFESTATIONS
In most cases, the clinical manifestations of pulmonary embolism are nonspecific, so evaluation of risk factors and predisposing factors is an important aspect of diagnosis. Although most emboli originate from clots in the lower extremities, specifically the iliac and femoral veins,113 deep vein thrombosis is often asymptomatic and clinical examination has low sensitivity for the presence of clot, especially in the thigh.
An individual with pulmonary embolism usually presents with the sudden onset of chest pain, dyspnoea, tachypnoea, tachycardia and unexplained anxiety. Occasionally syncope (fainting) or haemoptysis occurs. With large emboli, a pleural friction rub, pleural effusion, fever and leucocytosis may be noted. Recurrent small emboli may not be detected until progressive incapacitation, precordial pain, anxiety, dyspnoea and right ventricular enlargement are exhibited. Massive occlusion causes severe pulmonary hypertension, shock and sudden death.
EVALUATION AND TREATMENT
Routine chest X-rays and pulmonary function tests are not definitive for pulmonary embolism. On chest X-rays, the infarcted portion of the lung appears as a nonspecific infiltrate in a classic wedge shape bordering the pleura. Arterial blood gas analyses usually demonstrate hypoxaemia and hyperventilation (leading to respiratory alkalosis). A ventilation–perfusion scan, in which lungs are scanned after injection and inhalation of a radioactive substance, may indicate embolism (see Figure 25-31). Today, the diagnosis is made by measuring elevated levels of D-dimer in the blood (an indicator of fibrinolysis) in combination with spiral CT scanning.113,114
FIGURE 25-31 Ventilation-perfusion scan.
A Normal study with no defects visible. B Defects in scan showing lack of radioactive tracer uptake indicative of pulmonary embolism.
Source: Adam A. Grainger & Allison’s diagnostic radiology. 5th edn. Churchill Livingstone; 2008. Courtesy of Dr SE M Clarke, Guy’s Hospital, London.
The ideal treatment for pulmonary embolism is prevention through elimination of predisposing factors for individuals at risk. Venous stasis in hospital patients is minimised by leg elevation, bed exercises, position changes, early postoperative ambulation and pneumatic calf compression. Clot formation is also prevented by prophylactic low-dose anticoagulant therapy usually with low-molecular-weight heparin or warfarin.
Anticoagulant therapy is the primary treatment for pulmonary embolism. Intravenous administration of heparin is begun immediately and is followed by oral doses of warfarin. Studies suggest that low-molecular-weight heparins (e.g. enoxaparin) are as safe and effective as standard heparin but are easier to administer.113–115 4 6 If a massive life-threatening embolism occurs, a fibrinolytic agent is sometimes used and some individuals will require surgical thrombectomy.
Cor pulmonale
Cor pulmonale consists of right ventricular enlargement (hypertrophy or dilation, or both) and failure. It is most commonly caused by pulmonary hypertension.
PATHOPHYSIOLOGY
Cor pulmonale develops as pulmonary hypertension and creates chronic pressure overload in the right ventricle, similar to that created in the left ventricle by systemic hypertension. (Hypertension is discussed in Chapter 23.) Pressure overload increases the work of the right ventricle and first causes hypertrophy of the normally thin-walled heart muscle, but eventually leads to dilation and failure of the ventricle. Acute hypoxaemia, as with pneumonia, can exaggerate pulmonary hypertension and dilate the ventricle as well. The right ventricle usually fails when pulmonary artery pressure equals systemic blood pressure.
CLINICAL MANIFESTATIONS
The clinical manifestations of cor pulmonale may be obscured by primary respiratory disease and appear only during exercise testing. The heart may appear normal at rest, but with exercise, cardiac output falls. The electrocardiogram may show right ventricular hypertrophy. Increased pressures in the systemic venous circulation can cause jugular venous distension, hepatosplenomegaly (enlarged liver and spleen due to venous engorgement) and peripheral oedema.
EVALUATION AND TREATMENT
Diagnosis is based on physical examination, radiological examination, electrocardiogram and echocardiography. The goal of treatment for cor pulmonale is to decrease the workload of the right ventricle by lowering pulmonary artery pressure. Treatment success depends on reversal of the underlying lung disease.
CLINICAL MANIFESTATIONS OF PULMONARY ALTERATIONS
So far in this chapter we have explored the pathophysiology of the major pulmonary system disorders. In the following sections we look at the manifestations of these disorders — the conditions that result from pulmonary system alterations and their signs and symptoms. You should be familiar with the diseases and now need to consolidate your knowledge with an understanding of the clinical manifestations that individuals with pulmonary system alterations will exhibit.
Conditions caused by pulmonary alterations
Pulmonary oedema
One of the most serious conditions resulting from alterations to either the pulmonary system or the cardiovascular system is pulmonary oedema. Simply, pulmonary oedema is excess water in the lungs. The normal lungs are kept free from excess water by lymphatic drainage and a balance among capillary hydrostatic pressure, capillary oncotic pressure and capillary permeability (see Chapter 22). In addition, surfactant lining the alveoli repels water, keeping fluid from entering the alveoli. Predisposing factors for pulmonary oedema include heart disease, acute respiratory distress syndrome and inhalation of toxic gases. The pathogenesis of pulmonary oedema is shown in Figure 25-32.
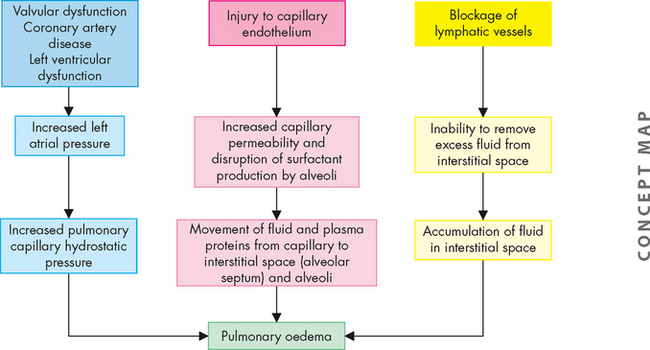
The most common cause of pulmonary oedema is heart disease. When the left ventricle fails, filling pressures on the left side of the heart increase and vascular volume redistributes into the lungs, subsequently causing an increase in pulmonary capillary hydrostatic pressure and a back-tracking of excess fluid into the lungs. When the hydrostatic pressure exceeds oncotic pressure (which holds fluid in the capillary), fluid moves out into the interstitial spaces (the spaces within the alveolar septum between the alveolus and capillary). When the flow of fluid out of the capillaries exceeds the lymphatic system’s ability to remove it, pulmonary oedema develops.
Another cause of pulmonary oedema is capillary injury that increases capillary permeability, as in cases of acute respiratory distress syndrome. Capillary injury causes water and plasma proteins to leak out of the capillary and move into the interstitial spaces, increasing interstitial oncotic pressure, which is usually very low. As the interstitial oncotic pressure begins to equal capillary oncotic pressure, water moves out of the capillary and into the lungs. (This phenomenon is discussed in Chapter 22.)
Clinical manifestations of pulmonary oedema include dyspnoea, hypoxaemia and increased work of breathing. Physical examination may reveal inspiratory crackles and dullness to percussion over the lung bases. In severe pulmonary oedema, pink frothy sputum is expectorated (coughed up) and oxygen levels decrease, while carbon dioxide levels increase, due to inadequate gas exchange.
The mainstay of therapy is supplemental oxygen. Individuals with pulmonary oedema usually require the delivery of a higher concentration of oxygen. The treatment of pulmonary oedema depends on its cause. If the oedema is caused by increased hydrostatic pressure that results from heart failure, therapy is geared towards improving cardiac output with diuretics (to reduce fluid volume), vasodilators (to redistribute blood to other areas of the body) and drugs that improve the contraction of the heart muscle (to allow normal blood flow throughout the systemic circulation). If oedema is the result of increased capillary permeability resulting from injury, the treatment is focused on removing the offending agent and supportive therapy to maintain adequate ventilation and circulation. When oxygen therapy alone is inadequate to meet metabolic demand, positive-pressure mechanical ventilation may be needed to improve ventilation and oxygenation.
Hypoxaemia
Hypoxaemia, or reduced oxygenation of arterial blood (reduced PaO2), is caused by respiratory alterations, whereas hypoxia, or reduced oxygenation of cells in tissues, may be caused by alterations of other systems as well. Although hypoxaemia can lead to tissue hypoxia, tissue hypoxia can result from other abnormalities unrelated to alterations of pulmonary function, such as low cardiac output.
Hypoxaemia results from problems with one or more of the major mechanisms of oxygenation:
The amount of oxygen in the alveoli is dependent on two factors. The first factor is the presence of adequate oxygen content in the inspired air. The amount of oxygen in inspired air is expressed as the percentage or fraction of air that is composed of oxygen. Anything that decreases the oxygen content of inspired air (such as high altitude) decreases oxygen in the alveoli. The second factor is the amount of alveolar minute volume (see Chapter 24 for alveolar ventilation). Hypoventilation results in an increased alveolar carbon dioxide and decreased oxygen such that diffusion across the alveoli is impacted. This type of hypoxaemia can be completely corrected if alveolar ventilation is improved by increasing the rate and depth of breathing. Hypoventilation causes hypoxaemia in unconscious individuals; in those with neurological, muscular or bone diseases that restrict chest expansion; and in individuals who have COPD.
Diffusion of oxygen from the alveoli into the blood is also dependent on two factors. The first is the balance between the amount of air getting into the alveoli and the amount of blood perfusing the capillaries around the alveoli. Normally, alveolar capillary lung units receive almost equal amounts of ventilation and perfusion. The normal ventilation/perfusion ratio is 0.8:0.9 because perfusion is somewhat greater than ventilation in the lung bases and because some blood is normally distributed to the bronchial circulation. An abnormal ventilation/perfusion ratio is the most common cause of hypoxaemia (see Figure 25-33). This is referred to as a ventilation–perfusion mismatch (clinically abbreviated to V˙/Q˙ mismatch). Hypoxaemia can be caused by inadequate ventilation of well-perfused areas of the lung (low ventilation–perfusion). Mismatching of this type occurs in atelectasis, in asthma as a result of bronchoconstriction and in pulmonary oedema and pneumonia when alveoli are filled with fluid. When blood passes through portions of the pulmonary capillary bed that receive no ventilation, blood is not oxygenated, resulting in hypoxaemia. Hypoxaemia can also be caused by poor perfusion of well-ventilated portions of the lung (high ventilation–perfusion), resulting in wasted ventilation. The most common cause of high ventilation–perfusion mismatching is a pulmonary embolus that impairs blood flow to a segment of the lung. An area where alveoli are ventilated but not perfused is termed alveolar dead space.
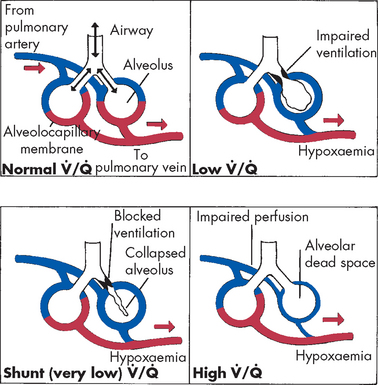
The second factor affecting diffusion of oxygen from the alveoli into the blood is the alveolar–capillary barrier. Diffusion of oxygen through the alveolar–capillary membrane is impaired if the membrane is thickened or the surface area available for diffusion is decreased. Thickened alveolar–capillary membranes, as occur with oedema (tissue swelling) and fibrosis (formation of fibrous lesions), increase the time required for oxygen to diffuse from the alveoli into the capillaries. If diffusion is slowed enough, the oxygen in the alveoli and capillary blood do not have time to equilibrate and hence oxygenation of the blood is limited.
Hypercapnia
Hypercapnia, or increased carbon dioxide in the arterial blood, is caused by hypoventilation of the alveoli. As discussed in Chapter 24, carbon dioxide is easily diffused from the blood into the alveolar space; thus, minute volume (ventilation rate × tidal volume) determines not only alveolar ventilation but also carbon dioxide levels in the blood.
There are many causes of hypercapnia. Most are a result of decreased drive to breathe or an inadequate ability to respond to ventilatory stimulation. Some of these causes include: (1) depression of the respiratory centre in the brainstem by drugs such as morphine and heroin; (2) diseases of the medulla, including infections of the central nervous system or trauma; (3) abnormalities of the spinal conducting pathways, as in spinal cord disruption; (4) diseases of the neuromuscular junction or of the respiratory muscles themselves, as in myasthenia gravis or muscular dystrophy; (5) thoracic cage abnormalities, as in chest injury or congenital deformity; (6) large airway obstruction, as in tumours or sleep apnoea; and (7) increased work of breathing or physiological dead space, as in emphysema.
Acute respiratory failure
Respiratory failure is defined as inadequate gas exchange such that arterial oxygen levels are less than 50 mmHg or arterial carbon dioxide levels are greater than 50 mmHg with pH less than 7.25. Respiratory failure can result from direct injury to the lungs, airways or chest wall, or indirectly because of injury to another body system, such as the brain or spinal cord. It can occur in individuals who have an otherwise normal pulmonary system or in those with underlying chronic pulmonary disease. Most pulmonary diseases can cause episodes of acute respiratory failure. If the respiratory failure is primarily hypercapnic (that is, due to high carbon dioxide levels), it is the result of inadequate alveolar ventilation. If the respiratory failure is primarily hypoxaemic (that is, due to low oxygen levels), it is the result of inadequate exchange of oxygen between the alveoli and the capillaries. Many individuals will have combined hypercapnic and hypoxaemic respiratory failure.
Atelectasis
Atelectasis is the collapse of lung tissue. It can occur due to lack of lung expansion, such as that experienced after surgery. There are two types of atelectasis:
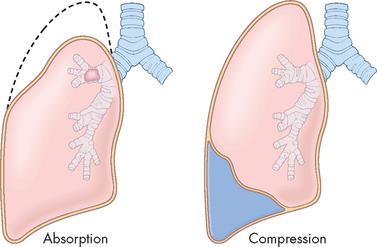
FIGURE 25-34 Different forms of atelectasis.
Source: Based on Kumar V. Robbins & Cotran pathologic basis of disease. 8th edn. Philadelphia: Saunders; 2010.
Clinical manifestations of atelectasis are similar to those of pulmonary infection including dyspnoea, cough and fever.
Atelectasis tends to occur after surgery.116 Postoperative patients may have received supplemental oxygen or inhaled anaesthetics and they are usually in pain, shallow breathe, are reluctant to change position and produce thick secretions that tend to pool in dependent portions of the lungs. Prevention and treatment of postoperative atelectasis usually include deep breathing, frequent position changes and early ambulation. Deep breathing opens connections between patent and collapsed alveoli, called pores of Kohn. This allows air to flow into the collapsed alveoli (collateral ventilation) and aids in the expulsion of intrabronchial obstructions.
Bronchiectasis
Bronchiectasis is persistent abnormal dilation of the bronchi and has a distinctive appearance on chest X-rays (see Figure 25-35). It usually occurs in conjunction with other respiratory conditions and can be caused by obstruction of an airway with mucus plugs, atelectasis, aspiration of a foreign body, infection, cystic fibrosis, tuberculosis or impaired defence mechanisms.
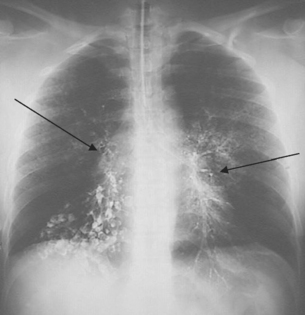
FIGURE 25-35 Bronchiectasis on a chest X-ray.
Note the dilated bronchi close to the midline (see arrows).
Source: Klatt EC. Robbins and Cotran atlas of pathology. 2nd edn. Philadelphia: Saunders; 2010.
The symptoms of bronchiectasis may date back to a childhood illness or infection. The disease is commonly associated with recurrent lower respiratory tract infections and expectoration of large amounts of purulent sputum (measured in cupfuls). If the individual is not receiving antibiotics, the sputum has a foul odour. Haemoptysis is common. Pulmonary function studies show decreased vital capacity and expiratory flow rates. Bronchiectasis is often associated with bronchitis and atelectasis.
Pneumothorax
Pneumothorax is the presence of air or gas in the pleural space caused by a rupture in the visceral pleura (which surrounds the lungs) or the parietal pleura and chest wall. As air separates the visceral and parietal pleurae, it destroys the negative pressure of the pleural space and disrupts the equilibrium between the elastic recoil forces of the lung and chest wall. The lung then tends to recoil by collapsing towards the hilum (see Figure 25-36).
Air in the pleural space causes the lung to collapse around the hilus and may push mediastinal contents (heart and great vessels) towards the other lung.
Pneumothorax can occur spontaneously or secondary to trauma. The most common presentation of spontaneous pneumothorax occurs unexpectedly in healthy males aged 20–40 years. Secondary pneumothorax can result from rib fractures, COPD or chest stabbings or shootings.
Both spontaneous and secondary pneumothorax can present as either open or tension. In open pneumothorax, air pressure in the pleural space equals barometric pressure because air that is drawn into the pleural space during inspiration (through the damaged chest wall and parietal pleura or through the lungs and damaged visceral pleura) is forced back out during expiration. In tension pneumothorax, however, the site of pleural rupture acts as a one-way valve, permitting air to enter on inspiration but preventing its escape by closing up during expiration. As more and more air enters the pleural space, air pressure in the pneumothorax begins to exceed barometric pressure. Tension pneumothorax is life-threatening. Air pressure in the pleural space pushes against the already recoiled lung, causing compression atelectasis and against the mediastinum, compressing and displacing the heart and great vessels.
Clinical manifestations of spontaneous or secondary pneumothorax begin with sudden pleural pain, tachypnoea and dyspnoea (rapid breathing and difficulty breathing, respectively). The manifestations depend on the size of the pneumothorax. Physical examination may reveal absent or decreased breath sounds. Tension pneumothorax may be complicated by severe hypoxaemia, tracheal deviation away from the affected lung and hypotension (low blood pressure). Deterioration occurs rapidly and immediate treatment is required. Diagnosis of pneumothorax is made with chest X-rays and CT scans. Pneumothorax is treated with insertion of a chest tube that is attached to a water-seal drainage system with suction, such that negative pressure is restored. After the pneumothorax re-expands and the pleural rupture is healed, the chest tube is removed.
Pleural effusion
Pleural effusion is the presence of excess fluid in the pleural space. The most common mechanism of pleural effusion is migration of fluids and other blood components through the walls of intact capillaries bordering the pleura. Pleural effusions that enter the pleural space from the intact blood vessels can be transudative (watery) or exudative (high in concentrations of white blood cells and plasma proteins). Mechanisms of pleural effusion are summarised in Table 25-5.
Table 25-5 MECHANISMS OF PLEURAL EFFUSION
| TYPE OF FLUID/EFFUSION | SOURCE OF ACCUMULATION | PRIMARY OR ASSOCIATED DISORDER |
|---|---|---|
| Transudate (hydrothorax) | Watery fluid that diffuses out of capillaries beneath the pleura (i.e. capillaries in the lungs or chest wall) | Cardiovascular disease that causes high pulmonary capillary pressures; liver or kidney disease that disrupts plasma protein production, causing hypoproteinaemia (decreased oncotic pressure in the blood vessels) |
| Exudate | Fluid rich in cells and proteins (leucocytes, plasma proteins of all kinds; see Chapter 13) that migrates out of the capillaries | Infection, inflammation or malignancy of the pleura that stimulates mast cells to release biochemical mediators that increase capillary permeability |
| Pus (empyema) | Debris of infection (microorganisms, leucocytes, cellular debris) dumped into the pleural space by blocked lymphatic vessels | Pulmonary infections, such as pneumonia; lung abscesses; infected wounds |
| Blood (haemothorax) | Haemorrhage into the pleural space | Traumatic injury, surgery, rupture or malignancy that damages blood vessels |
| Chyle (chylothorax) | Chyle (milky fluid containing lymph and fat droplets) that is dumped by lymphatic vessels into the pleural space instead of passing from the gastrointestinal tract to the thoracic duct | Traumatic injury, infection or disorder that disrupts lymphatic transport |
Small collections of fluid normally can be drained away by the lymphatics. Dyspnoea, compression atelectasis with impaired ventilation and mediastinal shift occur with large effusions. Pleural pain is present if the pleura are inflamed and cardiovascular manifestations occur in a large, rapidly developing effusion.
Diagnosis is confirmed by chest X-ray (see Figure 25-37) and thoracentesis (needle aspiration), which can determine the type of effusion and provide symptomatic relief. If the effusion is large, drainage usually requires the placement of a chest tube.
Empyema
Empyema (infected pleural effusion) is the presence of pus in the pleural space. It is thought to develop when the pulmonary lymphatics become blocked, leading to an outpouring of contaminated lymphatic fluid into the pleural space. Empyema occurs most commonly in older adults and children and usually develops as a complication of pneumonia, surgery, trauma or bronchial obstruction from a tumour.117
Individuals with empyema present clinically with cyanosis, fever, tachycardia (rapid heart rate), cough and pleural pain. Diagnosis is made by chest X-rays, thoracentesis and sputum culture.
The treatment for empyema includes the administration of appropriate antimicrobials and drainage of the pleural space with a chest tube.
Aspiration
Aspiration is the inhalation of fluid and solid particles into the lung. It tends to occur in children between the ages of 1 and 3 years or in individuals whose normal swallowing mechanism and cough reflex are impaired by central or peripheral nervous system abnormalities. Predisposing factors include an altered level of consciousness caused by substance abuse, sedation or anaesthesia; seizure disorders; cerebrovascular accident; and neuromuscular disorders that cause dysphagia (see Chapter 27). The right lung, particularly the right lower lobe, is more susceptible to aspiration than the left lung because the branching angle of the right main stem bronchus is straighter than the branching angle of the left main stem bronchus (see Chapter 24).
Foreign bodies lodged in the larynx or upper trachea cause cough, stridor, hoarseness or inability to speak, respiratory distress and agitation or panic. The presentation is often dramatic and frightening.
Aspiration of acidic gastric fluid (pH of less than 2.5) may cause lung inflammation. Bronchial damage includes inflammation, loss of ciliary function and bronchospasm. In the alveoli, acidic gastric fluid damages the alveolar–capillary membrane, allowing plasma and blood cells to move from the capillaries into the alveoli. The lung becomes stiff and non-compliant as surfactant production is disrupted, leading to further oedema and collapse.
Preventive measures for individuals at risk are more effective than treatment of known aspiration. The most important preventive measures include the semi-recumbent position, the surveillance of enteral feeding and the avoidance of excessive sedation.118 Nasogastric tubes, which are often used to remove stomach contents, are used to prevent aspiration but can also cause aspiration if fluid and particulate matter are regurgitated as the tube is being placed.
Treatment of aspiration of foreign bodies may include bronchoscopy to remove the foreign body, if sneezing and coughing does not displace the object. More serious aspirations with either stomach contents or ingestion of solids or fluids into the lungs may necessitate supplemental oxygen, mechanical ventilation, fluid restriction and steroids. Bacterial pneumonia may develop as a complication of aspiration and must be treated with broad-spectrum antimicrobials.
Signs and symptoms of pulmonary alterations
Dyspnoea
Dyspnoea is the subjective sensation of uncomfortable breathing, the feeling of not being able to get enough air. It is often described as breathlessness, air hunger, shortness of breath and laboured breathing. Everyone experiences dyspnoea at some stage. One of the most common non-pathological reasons is when you exercise heavily and become short of breath — that is dyspnoea. Our discussion here concerns dyspnoea that occurs at rest and is due to pulmonary system pathophysiology.
Dyspnoea can be caused by many pulmonary disorders. Disturbances of ventilation, gas exchange or ventilation–perfusion relationships can cause dyspnoea, as can increased work of breathing or any disease that damages lung tissue. One proposed mechanism for dyspnoea is a mismatch between sensory and motor input from the respiratory centre in the brainstem such that there is more urge to breathe than there is response by the respiratory muscles. Other causes of dyspnoea include stimulation of central and peripheral chemoreceptors and stimulation of afferent receptors in the lungs and chest wall.119,120
The signs of dyspnoea include flaring of the nostrils, use of accessory muscles of ventilation and retraction (pulling back) of the intercostal spaces. In dyspnoea caused by lung tissue disease (e.g. pneumonia), retractions of tissue between the ribs may be observed, although retractions are more common in children than in adults. Dyspnoea can be quantified using scales (such as the Borg dyspnoea scale), and is frequently associated with significant anxiety.121
Dyspnoea can occur transiently or can become chronic. One cause of dyspnoea is pulmonary congestion usually resulting from heart disease. Pulmonary congestion tends to cause dyspnoea when the individual is lying down (orthopnoea). The horizontal position redistributes body water, causes the abdominal contents to exert pressure on the diaphragm and decreases the efficiency of the respiratory muscles. Sitting up in a forward-leaning posture or supporting the upper body on several pillows generally relieves orthopnoea. Some individuals with pulmonary or cardiac disease wake up at night gasping for air and have to sit up or stand to relieve the dyspnoea (paroxysmal nocturnal dyspnoea).
Cough
A cough is a protective reflex that cleanses the lower airways by an explosive expiration. Inhaled particles, accumulated mucus, inflammation or the presence of a foreign body initiates the cough reflex by stimulating the irritant receptors in the airways. There are only few of these receptors in the most distal bronchi and the alveoli, thus it is possible for significant amounts of secretions to accumulate in the distal respiratory tree without cough being initiated. The cough consists of inspiration, closure of the glottis and vocal cords, contraction of the expiratory muscles and reopening of the glottis, causing a sudden, forceful expiration that removes the offending matter. The effectiveness of the cough depends on the depth of the inspiration and the degree to which the airways narrow, increasing the velocity of expiratory gas flow.
Acute cough is cough that resolves within 2–3 weeks of the onset of illness or resolves with treatment of the underlying condition. It is most commonly the result of upper respiratory infections, acute bronchitis, pneumonia, heart failure, pulmonary embolus or aspiration. Chronic cough is defined as cough that has persisted for more than 3 weeks. In non-smokers, chronic cough is almost always the result of postnasal drainage syndrome, asthma or gastro-oesophageal reflux disease. In smokers, chronic bronchitis is the most common cause of chronic cough, although lung cancer must always be considered.122 Approximately 20% of patients who take angiotensin-converting enzyme (ACE) inhibitors (see Chapter 23) develop a persistent dry cough, with some patients experiencing severe cough that requires the drug to be discontinued.
Hypoventilation and hyperventilation
Hypoventilation is inadequate alveolar ventilation in relation to metabolic demand. Hypoventilation occurs when minute volume (tidal volume × ventilatory rate) is reduced. It is caused by alterations in pulmonary mechanics or in the neurological control of breathing. When alveolar ventilation is normal, carbon dioxide is removed from the lungs at the same rate as that produced by cellular metabolism; therefore, arterial and alveolar carbon dioxide values remain at normal levels (between 35 and 45 mmHg). With hypoventilation, carbon dioxide removal does not keep up with carbon dioxide production and the level of carbon dioxide in the arterial blood increases, causing hypercapnia (a carbon dioxide level more than 45 mmHg). This results in respiratory acidosis (pH less than 7.35), which can affect the function of many tissues throughout the body. Blood gas analysis (i.e. measurement of the arterial carbon dioxide level) reveals hypoventilation.
Hyperventilation is alveolar ventilation exceeding metabolic demands. The lungs remove carbon dioxide faster than it is produced by cellular metabolism, resulting in decreased carbon dioxide levels in the blood, or hypocapnia (a carbon dioxide level less than 35 mmHg). Hypocapnia results in respiratory alkalosis (pH greater than 7.45), which also can interfere with tissue function. Like hypoventilation, hyperventilation can be determined by arterial blood gas analysis. Increased respiratory rate or tidal volume can occur with severe anxiety, acute head injury, pain and in response to conditions that cause insufficient oxygenation of the blood.
Abnormal breathing patterns
Normal breathing (eupnoea) is rhythmic and effortless. The resting ventilatory rate in adults is usually between 8 and 16 breaths per minute and tidal volume (the amount of air in each breath) ranges from 400–800 mL. A short expiratory pause occurs with each breath and the expiratory phase is longer than inspiration, usually in a ratio of 1:2 (for inspiration time: expiration time). Disease states can alter this ratio.
Laboured breathing occurs whenever there is an increased work of breathing, especially if the airways are obstructed. If the large airways are obstructed, a slow ventilatory rate, large tidal volume, increased effort, prolonged inspiration and expiration, and stridor or audible wheezing (depending on the site of obstruction) are typical. In small airway obstruction like that seen in asthma and COPD, a rapid ventilatory rate, small tidal volume, increased effort and prolonged expiration are often present.
Strenuous exercise or metabolic acidosis induces Kussmaul breathing (slow deep breathing), or hyperpnoea (excessive breathing), which is characterised by an increased ventilatory rate, very large tidal volumes and no expiratory pause. Another abnormal breathing pattern is Cheyne-Stokes breathing, characterised by alternating periods of deep and shallow breathing. Apnoea lasting from 15 to 60 seconds is followed by breaths that increase in volume until a peak is reached; then breathing decreases again to apnoea. Cheyne-Stokes breathing results from any condition that slows the blood flow to the brainstem, which in turn slows impulses sending information to the respiratory centres of the brainstem. Neurological impairment above the brainstem is also a contributing factor. Cheyne-Stokes breathing indicates that severe pathophysiological disturbances have occurred and often is present immediately before death.
Haemoptysis
Haemoptysis is the coughing up of blood or bloody secretions. This should not be confused with haematemesis, which is the vomiting of blood. Blood that is coughed up is usually bright red, has an alkaline pH and is mixed with frothy sputum, whereas blood that is vomited is dark, has an acidic pH and is mixed with food particles. However, both are serious conditions.
Haemoptysis indicates a localised abnormality, usually infection or inflammation that damages the bronchi, such as bronchiectasis, or the lung tissue, such as tuberculosis and cancer. Bronchoscopy, combined with chest CT scans, is used to confirm the site of bleeding.
Cyanosis
Cyanosis is a bluish discolouration of the skin and mucous membranes caused by increasing amounts of desaturated or reduced haemoglobin (which is bluish) in the blood. It generally develops when 5 g of haemoglobin is desaturated, regardless of haemoglobin concentration.
Cyanosis can be caused by decreased arterial oxygenation, ventilation–perfusion inequalities, decreased cardiac output, a cold environment or anxiety. In adults, cyanosis is not evident until severe hypoxaemia is present and therefore is an insensitive indication of respiratory failure. Severe anaemia (inadequate haemoglobin concentration) can cause inadequate oxygenation of tissues without causing cyanosis. However, individuals with polycythaemia (an abnormal increase in the numbers of red blood cells) may have cyanosis when oxygenation is adequate. Therefore, cyanosis must be interpreted in relation to the underlying pathophysiology. If cyanosis is suggested, the oxygen levels in the blood should be measured. Central cyanosis (decreased oxygen saturation of haemoglobin in arterial blood) is best seen in buccal (cheek) mucous membranes and lips. Peripheral cyanosis (slow blood circulation in fingers and toes) is best seen in nail beds.
Disorders of the pulmonary system
 Obstructive lung disease is characterised by airway obstruction that causes difficult expiration. Obstructive lung disease can be acute or chronic in nature and includes asthma, chronic bronchitis and emphysema. Asthma is the result of a type 1 hypersensitivity immune response involving the activity of lymphocytes, IgE, mast cells and eosinophils. In asthma, obstruction is caused by exacerbation episodes of bronchospasm, bronchial inflammation, mucosal oedema and increased mucus production. Asthma is a prevalent and important childhood problem. Its origins are probably multifactorial, including genetic, allergic and viral-triggered mechanisms. Effective management is aimed at decreasing chronic inflammation in the lungs, eliminating known triggers from the environment, and early recognition and treatment of acute symptoms. Chronic obstructive pulmonary disease (COPD) is the coexistence of chronic bronchitis, emphysema and sometimes asthma. Chronic bronchitis causes airway obstruction resulting from bronchial smooth muscle hypertrophy and production of thick, tenacious mucus. In emphysema, destruction of the alveolar septa and loss of passive elastic recoil lead to airway collapse and obstruct gas flow during expiration. Acute respiratory distress syndrome results from an acute diffuse injury to the alveolar–capillary membrane and decreased surfactant production, which increases membrane permeability and causes oedema and atelectasis. There is progressive respiratory distress with severe hypoxaemia and respiratory failure.
Obstructive lung disease is characterised by airway obstruction that causes difficult expiration. Obstructive lung disease can be acute or chronic in nature and includes asthma, chronic bronchitis and emphysema. Asthma is the result of a type 1 hypersensitivity immune response involving the activity of lymphocytes, IgE, mast cells and eosinophils. In asthma, obstruction is caused by exacerbation episodes of bronchospasm, bronchial inflammation, mucosal oedema and increased mucus production. Asthma is a prevalent and important childhood problem. Its origins are probably multifactorial, including genetic, allergic and viral-triggered mechanisms. Effective management is aimed at decreasing chronic inflammation in the lungs, eliminating known triggers from the environment, and early recognition and treatment of acute symptoms. Chronic obstructive pulmonary disease (COPD) is the coexistence of chronic bronchitis, emphysema and sometimes asthma. Chronic bronchitis causes airway obstruction resulting from bronchial smooth muscle hypertrophy and production of thick, tenacious mucus. In emphysema, destruction of the alveolar septa and loss of passive elastic recoil lead to airway collapse and obstruct gas flow during expiration. Acute respiratory distress syndrome results from an acute diffuse injury to the alveolar–capillary membrane and decreased surfactant production, which increases membrane permeability and causes oedema and atelectasis. There is progressive respiratory distress with severe hypoxaemia and respiratory failure.Infections of the pulmonary system
Upper respiratory tract infections are the most common cause of short-term disability in Australia and New Zealand. Serious lower respiratory tract infections occur most often in the elderly and in individuals with impaired immunity or underlying disease. Viral pneumonia can be severe, but is more often an acute self-limiting lung infection usually caused by the influenza virus. In tuberculosis, the inflammatory response proceeds to isolate colonies of bacterium by enclosing them in tubercles and surrounding the tubercles with scar tissue. These may remain dormant within the tubercles for life or, if the immune system breaks down, cause recurrence of active disease.Lung cancer
Obstructive sleep apnoea
Childhood pulmonary disorders
Croup is an acute respiratory illness of young children, usually caused by parainfluenza virus. This infection causes swelling of the upper trachea. The typical sign is a seal-like barking cough, which appears after a few days of rhinorrhoea, sore throat and low-grade fever. Respiratory distress syndrome of the newborn usually occurs in premature infants who are born before surfactant production and alveolar capillary development are complete. Atelectasis and hypoventilation cause hypoxaemia and hypercapnia. Prenatal steroids and postnatal surfactant are beneficial therapies. Cystic fibrosis is an autosomal recessive genetic disease that affects many organ systems, especially the lungs and digestive system. Airway secretions are particularly thick and tenacious and the airways develop chronic bacterial infection with pathogens such as Pseudomonas aeruginosa and Staphylococcus aureus. Chronic infection, plugged airways and severe inflammation cause long-term lung damage and ultimately death. However, the prognosis is improving and most patients with cystic fibrosis now survive to adulthood. Sudden infant death syndrome (SIDS) is the leading cause of postnatal death for infants outside of the hospital setting and is associated with low birth weight, a prone sleeping position and other environmental factors. Some risk factors are modifiable — the prime example is the profound reduction in SIDS since widespread adoption of recommendations for supine positioning of infants during sleep.Alterations of pulmonary blood flow and pressure
Clinical manifestations of pulmonary alterations
Pulmonary oedema is excess water in the lungs caused by disturbances of capillary hydrostatic pressure, capillary oncotic pressure or capillary permeability. A common cause is left heart failure, which increases the hydrostatic pressure in the pulmonary circulation. Hypoxaemia is a reduced oxygen level in the blood caused by (1) decreased oxygen content of inspired gas, (2) hypoventilation, (3) diffusion abnormality or (4) ventilation–perfusion mismatch. Atelectasis is the collapse of alveoli resulting from compression of lung tissue or absorption of gas from obstructed alveoli. Bronchiectasis is abnormal dilation of the bronchi secondary to another pulmonary disorder, usually infection or inflammation. Pneumothorax is the accumulation of air in the pleural space. It can be caused by spontaneous rupture of weakened pleural areas or it can be secondary to pleural damage caused by disease or trauma. Pneumothorax can be open, which means that the lung only partially collapses, or tension, which means that pressure builds up in the pleural space and can compress both the affected lung and the mediastinum. Pleural effusion is the accumulation of fluid in the pleural space, usually resulting from disorders that promote transudation or exudation from capillaries underlying the pleura but occasionally resulting from blockage or injury that causes lymphatic vessels to drain into the pleural space. Dyspnoea is the feeling of breathlessness and increased respiratory effort. It is a common pulmonary disorder symptom. Hypoventilation is decreased alveolar ventilation caused by airway obstruction, chest wall restriction or altered neurological control of breathing. Hypoventilation causes increased carbon dioxide levels. Hyperventilation is increased alveolar ventilation produced by anxiety, head injury or severe hypoxaemia. Hyperventilation causes decreased carbon dioxide levels. Abnormal breathing patterns are adjustments made by the body to minimise the work of the respiratory muscles. They include Kussmaul and Cheyne-Stokes breathing.John is 63 years old and has had asthma since childhood. His asthma has been well controlled with reliever, symptom controller and preventer medication. However, for the last five years his medication doses and frequency have increased. His wife died five years ago and since then he has not been taking care of himself.
He presented to hospital with exacerbation of asthma induced from an upper respiratory tract infection (influenza virus). He had not had the influenza vaccine, although he was recommended to do so by his general practitioner. Physical examination revealed decreased breath sounds throughout both lung fields, expiratory wheeze, tachypnoea (ventilatory rate: 32 breaths per minute) with accessory muscle use, small tidal volume, pursed lip breathing, tachycardia (heart rate: 116 beats per minute), oxygen saturations of 89% and anxiety. John could only speak one or two words in response to questions.
An urgent arterial blood gas was performed, which revealed respiratory alkalosis and hypoxaemia. An oxygen mask was applied at a flow of 8 L/min and nebulised salbutamol (5 mg with saline) was administered. John responded quickly to the salbutamol: his ventilatory rate decreased and he could speak in short sentences. However, he was coughing and producing sputum.