Chapter 42 Antineoplastic Agents and Adjuncts
Antineoplastic agents are used, along with surgery and radiation therapy, in treatment of cancer. As described in the previous chapter, this is a rapidly expanding field of pharmacology, in which understanding of molecular biology is particularly important. As the mechanisms and pathways underlying development of cancers are gradually being elucidated, so many new antineoplastic agents are being developed and trialled.
It is more difficult to generalise about these drugs, as their mechanisms of action vary greatly. The actions and clinical uses of the cytotoxic agents (alkylating agents, antimetabolites, antibiotic antitumour agents and mitotic inhibitors) and other miscellaneous antineoplastic agents will be described. Newer agents are the tyrosine kinase inhibitors, various monoclonal antibodies and miscellaneous drugs. Hormones, antihormones and inhibitors of hormone synthesis and release are the other main type of drugs used in the treatment of cancer.
Antineoplastic drugs (in particular the cytotoxic agents) have many significant adverse effects; common adverse effects are reviewed, and methods of treating them are described. Cancer chemotherapy requires supportive adjunctive therapy with various drugs and other modalities, including palliative care, immunostimulatory agents and drugs used to protect bone, kidneys and blood cells during cancer therapy.
BCG Bacille Calmette–Guérin, or the Mycobacterium bovis bacillus
G-CSF granulocyte colony-stimulating factor
GnRH gonadotrophin-releasing hormone
mTOR mammalian target of rapamycin
Key background: treatment of neoplasia
ANTINEOPLASTIC agents, i.e. drugs that are used to treat neoplasia, include cytotoxic drugs, hormones and antihormones, monoclonal antibodies, tyrosine kinase inhibitors and various other agents that impair tumour growth by miscellaneous mechanisms. The general mechanisms by which these drugs affect neoplastic cells, particularly their actions in the cell cycle and on macromolecular synthesis, are discussed in Chapter 41; in this chapter, the drug groups and their clinical uses are described in more detail. Drugs used as supportive or adjunctive therapies, and to treat adverse effects of antineoplastic drugs, are also considered.
Because oncology is a highly specialised area of medicine, antineoplastic drugs are usually administered in specialist cancer hospitals or oncology units of major hospitals. Many new antineoplastic agents are available only under special access schemes or on a clinical trial basis; some new drugs are still too expensive for general use. Consequently, we deal with the anticancer drugs in rather less detail than for other areas of pharmacology where the drugs are more widely available, prescribed and used.
This is a rapidly changing area of pharmacology, with new drugs and treatment regimens being introduced continually. Individual drug information and treatment protocols from specialist oncology units should be consulted for the latest information on indications, doses, administration techniques, combination regimens, adverse drug reactions, precautions and contraindications.
Cytotoxic agents
Cytotoxic agents do not directly kill tumour cells: they act by interfering with cell proliferation or replication, processes described in the previous chapter. The cytotoxic agents are divided into various classes based on their probable major mechanisms of action (see Table 42-1 for classifications, indications and major adverse effects); many drugs act by more than one mechanism, and for some drugs the precise mechanisms have not yet been clarified. Consequently, adverse effects tend to be specific for individual drugs, rather than common to a class of drugs—with the exception that all antiproliferative agents show similar adverse effects on rapidly-dividing cells.
| Drug | Primary indications | Major toxicities* |
| Alkylating agents | ||
| 1 Nitrogen mustards | ||
| Chlorambucil | CLL, Hodgkin’s and non-Hodgkin’s lymphomas | Bone marrow suppression, GIT disorders |
| Cyclophosphamide | Various cancers & autoimmune disorders (See Drug Monograph 42-1) | Bone marrow suppression (severe), haemorrhagic cystitis |
| Ifosfamide | Lung, ovary, testicular tumours; sarcomas, lymphomas | Bone marrow suppression, encephalopathy, nephrotoxicity |
| Melphalan | Multiple myeloma, malignant melanoma | Bone marrow suppression, allergic reactions |
| 2 Nitrosoureas | ||
| Carmustine (BCNU) | Primary brain tumours, multiple myeloma, lymphomas | Severe bone marrow suppression, pulmonary fibrosis, nephrotoxicity |
| Fotemustine | Melanoma | Bone marrow suppression, reversible neuro- and hepatic toxicity |
| Lomustine | Glioma, Hodgkin’s lymphoma | Bone marrow suppression, anorexia, nausea, vomiting |
| 3 Other | ||
| Amsacrine | Acute leukaemia | (May be available through SAS) |
| Busulfan | CML | Bone marrow suppression, diarrhoea, hyperpigmentation, anorexia |
| Dacarbazine (DTIC), procarbazine | Melanoma, sarcomas, glioma, lymphomas | Severe bone marrow suppression, GIT disorders, flu-like syndrome, neurotoxicity |
| Temozolomide | Gliomas, melanoma | Bone marrow suppression, neurological disorders |
| Thiotepa (rarely used) | Bladder cancers, lymphomas, malignant effusions | Bone marrow suppression, CNS depression |
| Antimetabolites | ||
| 1 Folate antagonists | ||
| Methotrexate | Many cancers & autoimmune disorders (See Drug Monograph 42-2) | Bone marrow suppression, diarrhoea, stomatitis, liver & lung toxicity; photosensitivity |
| Pemetrexed | Some lung cancers | Bone marrow suppression, rashes, GIT disorders |
| Raltitrexed | Colorectal | Fever, GIT disorders, flu-like symptoms |
| 2 Purine analogues | ||
| Cladribine | Leukaemias | Bone marrow suppression, fever |
| Fludarabine | CLL | Bone marrow suppression, fever, chills, infection, CNS disturbances |
| Mercaptopurine | ALL, AML, CML | Bone marrow suppression, cholestasis |
| Thioguanine | AML, CML | Bone marrow suppression |
| 3 Pyrimidine analogues | ||
| Capecitabine | Breast, colorectal | Bone marrow suppression; GIT, skin & nail disorders; PPE |
| Cytarabine | AML, ALL, CML, lymphomas | Bone marrow suppression, anorexia, oral & GI ulceration, bone & muscle pain, PPE |
| Fluorouracil | Many solid tumours including GIT, breast, pancreas | Diarrhoea, stomatitis, bone marrow suppression; PPE |
| Gemcitabine | Pancreas, lung, bladder | Bone marrow suppression, oedema, flu-like syndrome |
| 4 Other | ||
| Hydroxyurea | CML, melanoma, ovary | Bone marrow suppression, rash, itch |
| Antibiotics | ||
| Bleomycin | Squamous cell carcinoma, lymphomas, testicular cancer | Chills, fever, pneumonitis, mucositis, lung fibrosis, skin reactions |
| Dactinomycin | Wilms’ tumour, Ewing’s sarcoma, choriocarcinoma, testes, rhabdomyosarcoma | Severe bone marrow suppression, myalgia |
| Daunorubicin | HIV–Kaposi’s sarcoma, leukaemias, neuroblastoma | Bone marrow suppression, cardiomyopathy, severe mucositis, allergies |
| Doxorubicin | Sarcomas, breast, endometrium, carcinoid | Allergies, PPE, infusion reactions |
| Epirubicin | Breast, sarcomas, gastric | Bone marrow suppression, GIT, skin & urinary tract disorders |
| Idarubicin | AML, ALL, breast cancer | Severe bone marrow suppression, diarrhoea |
| Mitomycin | Bladder; palliation of many cancers | Bone marrow suppression |
| Mitozantrone | Breast, lymphoma, leukaemias | Bone marrow suppression, GIT disturbances |
| Mitotic inhibitors | ||
| 1 Taxanes | ||
| Docetaxel, paclitaxel | Ovary, breast, lung, prostate | CV disorders, hypersensitivity reactions |
| 2 Vinca alkaloids | ||
| Vinblastine | Bladder, testis, Kaposi’s sarcoma, lymphomas | GIT bleeding |
| Vincristine | Wide range of solid and haematological malignancies | Mild to severe paraesthesias, ataxia, anaphylactic reactions (fatal if given intrathecally) |
| Vinorelbine | Non-small-cell lung cancer, breast | Nausea, vomiting, chest pain |
| Topoisomerase I inhibitors | ||
| Irinotecan | Colorectal | Diarrhoea, respiratory & CV disorders |
| Topotecan | Ovary, lung, cervix | Respiratory disorders, diarrhoea, dizziness |
| Topoisomerase II inhibitors (podophyllotoxins) | ||
| Etoposide | Testicular tumours, small-cell lung cancer, leukaemias, lymphomas | GIT disorders, weakness |
| Teniposide | ALL, lymphomas, glioma, bladder | CNS depression |
| Other cytotoxic agents | ||
| 1 Platinum compounds | ||
| Carboplatin | Head and neck carcinomas, lung, ovary, testes | Bone marrow suppression, nausea, vomiting, neurotoxicity, neuropathies, ototoxicity, hypersensitivity reactions |
| Cisplatin | Head and neck, cervix, lung, bladder, ovary | Nephrotoxicity, severe nausea and vomiting, bone marrow suppression, electrolyte disturbances, neuropathies |
| Oxaliplatin | Colorectal | Bone marrow suppression, neurotoxicity, liver disorders, anaemia |
| 2 Other | ||
| Bortezomib | Multiple myeloma | Weakness, GIT disturbances, thrombocytopenia, neuropathy |
ALL = acute lymphoblastic leukaemia; AML = acute myelogenous leukaemia; CLL = chronic lymphocytic leukaemia; CML = chronic myelocytic leukaemia; CNS = central nervous system; CV = cardiovascular; GIT = gastrointestinal tract; PPE = palmar–plantar erythrodysaesthesia (hand–foot syndrome); SAS = Special Access Scheme.
* Note that most cytotoxic agents can cause nausea and vomiting (see Table 41-2 for emetogenic comparisons), gastrointestinal tract and reproductive disturbances and alopecia, so these are omitted from this Table.
Alkylating agents
Alkylating agents are commonly used in anticancer chemotherapy and were the first class of drugs applied clinically in the modern era of antineoplastic drug therapy (see Clinical Interest Box 42-1).
Clinical interest Box 42-1 History of antineoplastic chemotherapy
While natural products have been used for thousands of years in medicine, the era of scientific anticancer chemotherapy can perhaps be dated to 1865, when a patient with leukaemia was administered potassium arsenite solution, with some positive results.
A group of chemical warfare agents developed as blistering agents and used during World War I included the sulfur mustards, such as ‘mustard gas’, also known as Kampfstoff. Related compounds, the nitrogen mustards, were also synthesised and studied during later military campaigns, and antidotes were developed, but neither side dared to use them during WW II because of their toxicity. After WW II, a clinical trial was published by Goodman, Gilman and colleagues with results showing the effective use of nitrogen mustards in cancer chemotherapy against lymphomas. Patients were treated cautiously with these agents, and with considerable success. These are powerful alkylating agents and resulted in ‘spin-offs’ of effective anticancer drugs such as lomustine and cyclophosphamide (Drug Monograph 42-1).
The next lines of research were into agents that impair the co-factor functions of folic acid, used in the formation of new blood cells; the analogue aminopterin was shown in the 1940s to cause striking remissions in childhood leu kae mia. This led to the development of methotrexate, still an important anticancer drug (see Drug Monograph 42-2).
Another approach was to study the pathways whereby nucleic acids are synthesised. This led to the cytotoxic purine and pyrimidine analogues, such as 6-mercaptopurine and 5-fluorouracil, thought to act by being incorporated into false nucleotides or by inhibiting enzymes in the pathways.
Along the way, some natural compounds from plants and fungi have been found with useful anticancer actions (see Clinical Interest Box 42-2). More recent research is targeting growth factors and their receptors; oncogenes and their product proteins; regulatory processes in cell biology, including signal transduction, apoptosis, cell cycle checkpoints and tumoursuppressor genes; and pharmaceutical and pharmacokinetic techniques to deliver agents directly to tumour cells or activate them there.
Mechanisms and groups
Alkylating agents contain alkyl groups (e.g. methyl, ethyl) and form highly reactive chemical structures that react rapidly with an electron donor group, such as a nitrogen atom in a guanine base of DNA, forming strong bonds as the alkyl group is ‘donated’ to the DNA base (see Figure 42-1). The alkylating agents usually are bifunctional, i.e. they have two active ‘arms’ that can link across or along the strands of DNA chains. This effectively ties the double helix together (like a zipper that has become stuck by having thread caught in it and cannot be unzipped), and interferes with the unwinding of the DNA strands in the processes of transcription to RNA and replication of DNA. Thus the cell cycle (Figure 41-1) is blocked mainly at the S phase, before the G2 phase, and cell proliferation is slowed or stopped. Some alkylating agents are considered cell-cycle-specific, others are cell-cycle-non-specific.
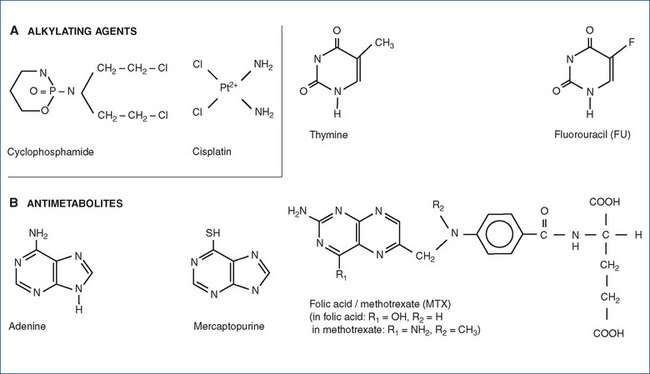
Figure 42-1 Chemical structures of representative antineoplastic agents. A Alkylating agent (cyclophosphamide) and cisplatin. B Antimetabolites: mercaptopurine, fluorouracil (FU) and methotrexate (MTX); compare with endogenous compounds adenine, thymine and folate and see also Figure 41-3.
Various types of alkylating agents are available, such as the nitrogen mustards (including chlorambucil, cyclophosphamide [Drug Monograph 42-1 and Figure 42-1], melphalan and ifosfamide) and nitrosoureas (carmustine, fotemustine and lomustine). Nitrosoureas are highly lipophilic alkylating agents that readily cross the blood–brain barrier and are thus useful for treating primary brain tumours. Other alkylating agents include busulfan, dacarbazine (and its orally active analogue temozolomide) and thiotepa.
Drug monograph 42-1 Cyclophosphamide
Cyclophosphamide is a cell-cycle-non-specific agent that crosslinks DNA strands and inhibits protein synthesis and DNA replication.
INDICATIONS Cyclophosphamide is indicated for acute and chronic leukaemias, lymphomas, multiple myeloma, carcinoma of the ovary, neuroblastomas, retinoblastoma and mycosis fungoides. It is also used as an immunosuppressant in autoimmune disorders resistant to milder therapy and to prevent transplant rejection.
PHARMACOKINETICS The drug is well absorbed after oral or parenteral administration, and the drug and its metabolites cross the blood–brain barrier. Cyclophosphamide undergoes hepatic metabolism via mixed-function oxidases and CYP2B6 and 3A4 to active (alkylating) and inactive metabolites. Excretion is primarily via the kidneys; accumulation of active metabolites in the bladder can cause nephrotoxicity and cystitis. The elimination half-life is about 4 hours.
DRUG INTERACTIONS See Drug Interactions 42-1.
ADVERSE REACTIONS Severe bone marrow suppression occurs 1–2 weeks after treatment, with dose-limiting neutropenia and leucopenia. Other adverse reactions include nephrotoxicity with haemorrhagic cystitis, severe nausea and vomiting, gastrointestinal (GI) tract dysfunction, hair loss and impaired wound healing, darkening of the skin, cardiotoxicity, hyperuricaemia and pneumonitis or interstitial pulmonary fibrosis.
The nephrotoxic effects in the renal tubular epithelium may be minimised by pre-treatment with mesna (see later discussion).
WARNINGS AND CONTRAINDICATIONS Baseline assessment before administration should include full blood cell count, tests of liver and kidney function and evaluation of disease course and progress. Patients must be closely monitored during therapy. Patients need to be hydrated before and during therapy, with 2–3 L fluid PO or infused. Staff are warned to observe safe handling procedures for cytotoxics.
Use with caution in patients with a history of cystitis, gout or urate kidney stones or cardiovascular disease. There is an increased risk of secondary neoplasia, especially of the bladder.
Avoid use in people with cyclophosphamide hypersensitivity, bone marrow suppression, chicken pox, herpes zoster or other untreated infections, urinary tract damage or infection. Contraindicated in pregnancy or breastfeeding. Dose should be reduced in severe kidney impairment
DOSAGE AND ADMINISTRATION The usual adult antineoplastic loading dose is 40–42 mg/kg IV in divided doses over 2–5 days; a typical maintenance dosage is 1–5 mg/kg orally daily. Oncology units have developed chemotherapy regimens depending on indication, other antineoplastics being administered concurrently, kidney function etc (see Table 41-1). The maximum tolerated dose is given, using depression in white cell count as a guide to toxicity. The immunosuppressive dose is markedly lower, starting at 1–3 mg/kg orally once daily, then reducing.
Drug interactions 42-1 Cyclophosphamide
The following effects can occur when cyclophosphamide is given with other drugs
| Drug | Possible effects and management |
| Bone marrow depressants or radiation; immunosuppressant drugs | Increased bone marrow depression may occur, with risk of infections and secondary neoplasia |
| If necessary to use concurrently, a decrease in drug dosage may be indicated | |
| Drugs that induce CYP2B6 or 3A4 microsomal enzymes, including phenytoin & rifampicin | Increase metabolism of cyclophosphamide to active metabolites, hence may enhance cytotoxic effects and toxic reactions |
| Suxamethonium | Cyclophosphamide has significant anticholinesterase activity, hence potentiates depolarising neuromuscular blockade |
| Vaccines, live viral | Cyclophosphamide-induced immunosuppression can lead to enhanced effects of live viral vaccines |
Antimetabolite drugs
The antimetabolites group contains drugs that are analogues of folic acid or of the purine and pyrimidine bases. These agents act by inhibiting enzymes involved in the pathways for macromolecular synthesis and/or as false ‘building blocks’, causing damaged polymers of nucleic acids to be built up into impaired DNA and RNA.
Groups of antimetabolites
The chemical structures of three typical antimetabolites (*) are shown in Figure 42-1. The antimetabolites fall into three main groups:
Folic acid antagonists
Folic acid is an essential co-factor in many biochemical reactions, particularly in one-carbon transfers, and is essential for the synthesis of purines and the methylation of uracil. An enzyme essential for the activation of folate to tetrahydrofolate is dihydrofolate reductase. This enzyme is inhibited by the folic acid antagonist methotrexate (MTX; see Drug Monograph 42-2). MTX has a structure very similar to folic acid (as shown in Figure 42-1B) so it competes with dihydrofolate for the active site on the enzyme. The blockade of biochemical reactions dependent on the co-factor activities of folic acid impairs synthesis of macromolecules required for cell proliferation, and halts cell cycling. (The sulfonamide antibacterial drugs have a similar mechanism of action and are also classed as antifolate drugs, but are more specific for bacterial metabolic pathways than those in neoplastic cells.) Pemetrexed, a new antifolate drug, also inhibits folatedependent enzymes.
Drug monograph 42-2 Methotrexate
Methotrexate (MTX) is an antimetabolite that competitively inhibits dihydrofolate reductase, impairing the synthesis of DNA and RNA, hence is cell-cycle-specific for the S phase in rapidlydividing cells, thus inhibiting cancer growth. The drug is also used for some other conditions marked by excessive cell proliferation, such as psoriasis, and its immunosuppressant actions make it useful in autoimmune conditions such as rheumatoid arthritis. MTX is a toxic drug with many serious adverse effects, drug interactions and contraindications, of which only a brief summary is given below.
INDICATIONS MTX is indicated for:
PHARMACOKINETICS MTX is administered orally or parenterally (IM, IV or intrathecal routes). The oral preparation produces peak plasma concentrations within 1–2 hours. Only limited amounts of MTX can cross the blood–brain barrier; however, after intrathecal drug administration significant quantities may pass into the systemic circulation. It is bound about 50% to plasma proteins and excreted largely unchanged by the kidneys.
DRUG INTERACTIONS There are many clinically significant drug interactions with MTX. Some of the more important are listed in Drug Interactions 42-2; see also Australian Medicines Handbook, Appendix B.
ADVERSE REACTIONS Toxic effects are possible even with low doses, and fatalities have occurred. Major adverse effects are bone marrow suppression (leucopenia, thrombocytopenia, anaemia), immunosuppression (infections, acne, boils) and loss of hair. GI tract effects include nausea, vomiting, anorexia, GI ulcers and stomatitis. With prolonged daily therapy, liver toxicity, pneumonitis or pulmonary fibrosis can occur, and carcinogenic effects and impaired fertility. With high-dose therapy, renal failure, photosensitivity and severe skin reactions are possible; these may be minimised with leucovorin rescue (see Drug Monograph 42-5), effective hydration and urine alkalinisation to enhance excretion of MTX.
WARNINGS AND CONTRAINDICATIONS Because of its very low therapeutic index, MTX should be prescribed only by specialist physicians with experience in its use. Patients should be warned of the high risk of dangerous adverse reactions and monitored closely (blood count, renal and liver function tests).
Use with caution in patients with aciduria (urine pH below 7), gout, GI obstruction, a history of kidney stone formation, nausea or vomiting (which may exacerbate dehydration) and in dehydrated patients. Health-care professionals dealing with MTX should observe safe handling guidelines.
Avoid use in people with MTX hypersensitivity, ascites, pleural effusions, liver or kidney impairment, bone marrow suppression, chickenpox (recent or current), herpes zoster, other infections, peptic ulcer or ulcerative colitis or oral mucositis. MTX is contraindicated in pregnancy (Australian Category D) and breastfeeding.
DOSAGE AND ADMINISTRATION MTX dosages vary according to the indication and course of treatment. The antineoplastic adult dosage orally is generally 15–30 mg daily for 5 days, repeated 3–5 times with a 7–14-day interval between courses. The paediatric oral dosage is 20–40 mg/m2 once a week.
The doses for immunosuppression are much lower, e.g. oral adult dose 7.5 mg once weekly, on a nominated day (e.g. on Tuesdays) to avoid risk of inadvertent daily dosing.
Drug interactions 42-2 Methotrexate
The following effects can occur when methotrexate is given with other drugs.
| Drug | Possible effects and management |
| Alcohol, leflunomide, etretinate & other hepatotoxic drugs | Increased risk of hepatotoxicity; avoid and/or monitor liver enzymes |
| Calcium folinate | NB: Intentional drug interaction, given after MTX to ‘rescue’ cells by restoring intracellular folate levels |
| Bone marrow depressants or radiation | Bone marrow-depressant effects may be increased; avoid or reduce drug dosage |
| Aspirin and other NSAIDs | Additive platelet inhibition; avoid. Low-dose aspirin may be used concurrently |
| Probenecid or salicylates | Can reduce excretion of MTX, resulting in elevated plasma concentrations & increased toxicity |
| Sulfonamides & trimethoprim | Additive antifolate actions, hence enhanced toxicity |
Purine and pyrimidine antagonists
The purine and pyrimidine base analogues can be incorporated into DNA strands in place of the true bases, forming permanently modified DNA and leading to improper base pairing during replication of DNA and improper transcription to RNA. They may also act as specific inhibitors of enzymes involved in DNA synthesis. Thus macromolecular synthesis and cell duplication are impaired. The antimetabolites are considered to be phasespecific agents, as they act particularly at the S phase of the cell cycle.
A cunning technique for making anticancer drugs more specific for neoplastic cells is exemplified in the new drug capecitabine, which is a prodrug for fluorouracil (FU). It has been rationally designed to be ‘tumour-activated’: after oral administration it is metabolised in three stages, the last of which involves the enzyme thymidine phosphorylase, which is more active in the liver and tumour cells than in normal cells. Hence 2.5-fold higher levels of active FU are reached in tumour cells than in adjacent tissue, which optimises therapeutic effects and minimises adverse effects. Capecitabine is proving particularly effective in breast and colorectal cancers.
Antibiotic antitumour drugs
The third main group of cytotoxic agents is the antibiotic antitumour agents, such as the anthracyclines and bleomycins. These drugs are defined as antibiotics because they are compounds that are isolated from one type of organism (usually fungi) and act against another type of organism. In this context the antibiotic activity is against neoplastic cells, rather than bacterial cells as for traditional antibiotics such as penicillins. The early agents in this class (daunorubicin, doxorubicin) caused the clinically limiting adverse effect of irreversible cardiomyopathy; newer agents are being sought that lack this cardiac toxicity. (See Table 42-1 for the drugs in this classification, primary indications and major toxicities.)
Mechanisms of action
Anthracyclines
The anthracyclines—daunorubicin, doxorubicin (previously called Adriamycin), idarubicin, epirubicin, valrubicin and the related compound mitozantrone—are complex polycyclic molecules derived from Streptomyces bacteria. They act by various mechanisms:
As the anthracyclines have actions at many sites in the cell cycle, and may also affect non-cycling cells, they are said to be cell-cycle-non-specific agents. They are effective against a wide range of cancers, including leukaemias and lymphomas, sarcomas and breast, gastric and bladder cancers. Potential cardiac toxicity is increased by other cardiotoxic agents and in patients with reduced cardiac reserve or cardiac disease.
Dactinomycin
The actinomycins are complex polypeptide antibiotics isolated from Streptomyces species of soil bacteria.
Dactinomycin (formerly known as actinomycin-D) is an intercalating agent and topoisomerase II inhibitor. It interferes with DNA-dependent RNA synthesis and also acts as an immunosuppressant. It is mainly indicated for treatment of sarcomas, Wilms’ tumour, choriocarcinoma and testicular cancer.
Bleomycins
The bleomycins are antibiotics first isolated from a Streptomyces species. They have complex glycopeptide structures, and the family members differ in some substituents on the tail of the molecule. ‘Bleomycin’ is a mixture mainly of bleomycins A2 and B2. They have cytotoxic and mutagenic actions: they chelate metal ions, generate reactive radicals, degrade preformed DNA into fragmented chains and block incorporation of thymidine into DNA. They are cell-cycle-non-specific agents, active against cells in G0, G2 and M phases of the cell cycle. They cause little bone marrow suppression or immunosuppression, but can cause pulmonary fibrosis in up to 50% of patients administered the drug (see review by Chen and Stubbe [2005]). Mitomycin has actions similar to those of both bleomycin and the alkylating agents.
Mitotic inhibitors
Important antineoplastic agents that are natural products isolated from plants include the vinca alkaloids, the podophyllotoxin derivatives and the taxanes (see Clinical Interest Box 42-2). These agents are mitotic inhibitors. (See Table 42-1 for primary indications and major toxicities.)
Clinical interest Box 42-2 Antineoplastic agents from natural sources
About two-thirds of commercially available anticancer drugs are derived from, or related to, natural products, including not only enzymes, hormones, interferons, oncogene proteins and antimetabolites but also plant and fungal extracts (see Figure 42-2).
The autumn crocus plant, Colchicum autumnale, was prized in Roman times as a treatment for gout, and later for rheumatism also. Its major active ingredient, colchicine, is effective as an antiinflammatory agent because of its ability to bind to tubulin, a structural protein in the microtubules in cells, and hence to interfere with the migration of neutrophils into a joint. This mechanism also accounts for the antitumour activity of colchicine, as inhibition of tubulin action inhibits mitosis and has cytotoxic effects. Colchicine is, however, too toxic for regular use.
An extract of wild chervil was mentioned in a medical book written in about 950 AD as being useful as a salve (ointment) against tumours. This may have been due to a cytotoxic agent now known as podophyllotoxin, also present in Podophyllum species plants. Native Americans used the root extracts as a purgative, emetic, poison and treatment for warts. Because of the similarity between viral warts and viral tumours, the extract was tested for antitumour activity and found to be effective but toxic. Synthesis of compounds related to the natural lignans produced two potent cytotoxic agents effective against leukaemia and lung cancers: etoposide and teniposide. These act by inhibition of topoisomerase II.
Extracts of the rosy periwinkle plant, Catharanthus roseus (formerly called Vinca rosea), were used in several cultures to treat diabetes. When tested in animals (in the 1950s), the extracts showed not antidiabetic effects but immunosuppressant effects, with severe depletion of white cells. As this is a common adverse effect of cytotoxic agents, the extracts were tested against animal tumours, with excellent results, leading to the development of the drugs vincristine and vinblastine. The 5-year survival rate for patients with Hodgkin’s disease rose from 5% in 1970 to more than 98% with the combination chemotherapy regimen of vincristine, MTX, mercaptopurine and prednisolone.
Antibiotics with antitumour activity include the anthracyclines, such as doxorubicin and daunorubicin; dactinomycin; and the bleomycins, mainly extracted from Streptomyces species (see earlier section on antitumour antibiotics).
More recent natural products with anticancer activities include the taxanes (paclitaxel and docetaxel), agents that have antimitotic and immunostimulatory effects and are derived from bark of the yew tree Taxus baccata; and the camptothecins (topotecan and irinotecan), a group of drugs with topoisomerase inhibitory actions derived from Camptotheca accuminata.
It is recognised by scientists in research institutions and drug companies that the millions of diverse species in tropical rainforests and marine environments potentially contain novel compounds with important antineoplastic (and other useful medical) actions. Screening programs with fast throughputs test millions of new compounds annually for potentially useful actions against human neoplastic cell lines (see Clinical Interest Box 4-2). Compounds isolated from marine molluscs and sponges are currently being trialled for their anticancer properties. However, deforestation of rainforests and overfishing and blanching of coral reefs may cause loss of potentially useful products before their discovery.

Figure 42-2 Plants from which some antineoplastic drugs are sourced. APodophyllum spp, source of podophyllotoxins; BCatharanthus roseus (formerly known as Vinca rosea), source of vinca alklaloids; CTaxus baccata, the Pacific yew tree, source of taxanes; DCamptotheca accuminata, source of camptothecins.
Mechanisms of action
During the metaphase stage of mitotic division (see Figure 41-1), the replicated chromosomes line up on a spindle formed from microtubules. Mitotic inhibitors bind to the protein tubulin, a constituent of microtubules, which inhibits its polymerisation into microtubules, disrupts spindle formation and arrests mitosis in metaphase. Thus they are said to be cell-cycle-phase-specific agents, inhibiting cell cycling during the late G2 and M phases. (Inhibition of microtubule actions also impairs other processes in cells, including chemotaxis, phagocytosis and axonal transport of neurotransmitters.) The mitotic inhibitors may also have other actions that contribute to their cytotoxic effects; e.g. the vinca alkaloids impair uridine incorporation into mRNA.
Drug groups
Vinca alkaloids
The vinca alkaloids vinblastine and vincristine (from the plant Catharanthus or Vinca rosea; Clinical Interest Box 42-2) and the related semisynthetic alkaloid vinorelbine have different therapeutic indications and different adverse effects. They have been used in the treatment of various lymphomas, carcinoma of the breast and testes, leukaemias and Hodgkin’s disease, and for non-small-cell lung cancer. They are relatively non-toxic compared with other cytotoxic agents: they have mild bone marrow-suppressant and neurotoxic effects and can cause hypersensitivity reactions. Vincristine is an irritant and is potentially fatal if administered intrathecally.
Taxanes
The taxanes (paclitaxel and docetaxel, from yew trees; Clinical Interest Box 42-2) are agents with antimitotic and immunostimulatory effects; they bind to microtubules with high affinity, stabilise microtubule bundles and thus inhibit mitosis and initiate apoptosis. Their effects in stimulating immune responses and regulating lymphocyte activation are also useful in cancer chemotherapy. They are used particularly in breast, lung, ovarian and prostate cancers. Dexamethasone is usually given before the taxane drug to prevent oedema and allergic reactions.
Paclitaxel is marketed for the treatment of metastatic ovarian cancer refractory to other drug treatments. It is also used for the treatment of metastatic breast cancer, and some studies indicate that it should be used earlier, such as immediately after surgery, as it then produces better effects than hormone therapy alone. Adverse reactions include severe allergic reactions, bone marrow suppression, peripheral neuropathy, muscle pain, alopecia and gastric distress.
Epothilones
This is a novel group of drugs, some natural (from the myxobacterium Sorangium cellulosum) and some synthetic. They have a similar mechanism of action to the taxanes, with affinity for the same binding site on tubulin, causing stabilisation of microtubules and thus antimitotic and pro-apoptotic effects. The first in clinical use is ixabepilone, in multi-drug resistant metastatic breast cancer; others are patupilone and sagopilone.
Other cytotoxic agents
Platinum compounds
The platinum-containing compounds cisplatin, carboplatin and oxaliplatin are sometimes considered as alkylating agents, as they have a rather similar mechanism of action. In cisplatin, the platinum atom is bonded to two amine groups, which cross-link between DNA strands (see Figure 42-1). Cell division is inhibited, leading to apoptosis; these compounds are cell-cycle-non-specific. Cisplatin is particularly emetogenic, and most likely to cause nephroand neurotoxicities. The three platinum compounds have different indications, contraindications, adverse drug reactions, and dosage and administration guidelines, so individual specialist oncology protocols should be consulted for these details.
Podophyllotoxins
The podophyllin-type compounds etoposide and teniposide are sometimes included with mitotic inhibitors as they can cause metaphase arrest; however, their mechanisms of action are not simple, as they also kill cells in the S and G2 phases of the cell cycle and may inhibit topoisomerase II (see Clinical Interest Box 42-2). They are used mainly in leukaemias and lymphomas.
Topoisomerase I inhibitors
The topoisomerase type I enzymes are involved in the untwisting, nicking and resealing of DNA strands during the processes of DNA duplication. Agents that inhibit these enzymes cause breaks in double-stranded DNA, blocking macromolecular synthesis in the cell cycle and leading to tumour cell death. Two new antineoplastic agents with this mechanism of action are derivatives of the plant Camptotheca accuminata and have been given the generic name camptothecins. They are S phase-specific.
Topotecan, a topoisomerase I inhibitor, is indicated for the treatment of relapsed or refractory metastatic carcinoma of the ovary after failure of other therapies, and also for small-cell lung cancer. Adverse reactions include neutropenia (a dose-limiting toxicity), leucopenia, thrombocytopenia, anaemia, headache, GI tract disturbances, alopecia, tiredness, dyspnoea and neuromuscular pain. The usual dose is 1.5 mg/m2 by IV infusion over 30 minutes daily for 5 days.
Irinotecan, another topoisomerase I inhibitor, is indicated for the treatment of metastatic colorectal cancer that has occurred or progressed after FU chemotherapy. Irinotecan can cause severe diarrhoea, which requires immediate treatment with atropine or loperamide. Severe myelosuppression, nausea and vomiting may also occur.
Colaspase (formerly known as asparaginase)
The enzyme asparaginase hydrolyses the amino acid L-asparagine to L-aspartic acid and ammonia. Asparagine is necessary for cell survival and, because normal body cells are capable of synthesising adequate supplies of asparagine, they are not affected by an asparagine deficiency. Certain cancer cells, however, are unable to synthesise asparagine and depend on a circulating supply of asparagine within the blood; administration of asparaginase enhances the breakdown of asparagine, so the cancer cells will die. Colaspase is sometimes classed as an antimetabolite, as it exploits differences between metabolic pathways in normal and neoplastic cells.
Colaspase is a form of asparaginase produced from cultures of Escherichia coli; as it is a protein, it cannot be given orally, but is administered by IM injection or IV infusion to treat leukaemias and lymphoma. Adverse reactions include allergic reactions (including anaphylaxis), a decrease in the blood clotting factors, hyperammonaemia (headache, anorexia, nausea, vomiting and abdominal cramps), liver toxicity and nervous system dysfunction. The drug should be administered only in hospital settings because of the risk of anaphylaxis.
Altretamine
Altretamine is a cytotoxic agent for the palliative treatment of persistent or recurrent ovarian cancer. Its mechanism of action is unknown, although chemically it resembles the alkylating agents. Clinically, however, it is effective for ovarian tumours that are resistant to the previously marketed alkylating agents. Its main adverse reactions are bone marrow suppression, GIT dysfunction and neurotoxicity.
Hydroxyurea
Hydroxyurea inhibits DNA synthesis by interfering with the conversion of ribonucleotides to deoxyribonucleotides; it is sometimes classified with the antimetabolites. It is indicated for the treatment of ovarian carcinoma, chronic myelocytic leukaemia and malignant melanoma. Adverse reactions include bone marrow suppression and GI tract dysfunctions.
Hormones
Treatment of hormone-dependent cancers
Hormonal agents are used in the treatment of neoplasia that are sensitive to hormonal growth controls in the body. Growth of prostate cancer, for example, is stimulated by the male sex hormones (androgens), breast cancers by oestrogens and thyroid cancer by thyrotrophin. Hormones used in cancer chemotherapy are not specifically antiproliferative or cytotoxic in neoplastic cells, but have their usual hormonal actions, thus they are more selective and less toxic than other antineoplastics. Hormonal agents used include corticosteroids, androgens and antiandrogens, oestrogens and anti-oestrogens, progestogens, and analogues of GnRH (see Table 42-2 for their indications and major adverse effects). Thus there are several options for treatment of these cancers.
Table 42-2 Summary of hormonal agents used in neoplastic conditions
| Hormonal agents | Clinical indications | Common adverse effects |
| Antiandrogens Bicalutamide, cyproterone, flutamide, nilutamide |
Advanced prostatic cancer, metastatic prostatic cancer, suppression of GnRH ‘flare’ | Impotence, impaired libido, decreased spermatogenesis, gynaecomastia, nausea, dizziness, alcohol intolerance, dyspnoea, hepatotoxicity, impaired dark–light adaptation |
| Anti-oestrogens & SERMs Fulvestrant, raloxifene, tamoxifen, toremifene |
Breast cancer | Hot flushes, dizziness, nausea and vomiting, oedema, vaginal bleeding, musculoskeletal pain |
| Aromatase inhibitors Anastrozole, exemestane, letrozole |
Postmenopausal breast cancer (ER receptor-positive) | Hot flushes, vaginal bleeding, hair thinning, nausea, GIT disturbances, joint pain, rash, fatigue, oedema, headache |
| Progestogens Megestrol, medroxyprogesterone acetate |
Palliative therapy of metastatic breast, endometrial and renal cell cancers | Hypersensitivity, nausea and vomiting, CNS disturbances, breast tenderness, menstrual irregularities, weight gain |
| GnRH analogues (= LHRH agonists) Goserelin, leuprorelin, triptorelin |
Palliative treatment of prostate cancer, advanced breast cancer (premenopausal) | Males: ‘flare-up’ of prostate cancer (bone pain, ureter obstruction, spinal cord compression), oedema, hot flushes, testicular atrophy, GIT disorders, reduced libido. Females: decreased libido, hot flushes, headache, abdominal pain, hypertension, dysmenorrhoea |
| Somatostatin analogues Lanreotide, octreotide |
Carcinoid tumours of GIT and pancreas secreting VIP or 5-hydroxytryptamine (serotonin) | GIT disorders (pain, bloating, diarrhoea) |
Note: In addition, oestrogens can be used in treatment of prostate cancers, and androgens in breast cancers.
ER = (o)estrogen receptors; GIT = gastrointestinal tract; GnRH = gonadotrophin-releasing hormone; LHRH = luteinising hormone-releasing hormone; SERM = selective (o)estrogen receptor modulator; VIP = vasoactive intestinal polypeptide.
Prostate cancer
For example, prostate cancer may be treated by:
In the early stages, ‘watchful waiting’ is also an option, while monitoring levels of prostate-specific antigen (PSA) as an indicator of disease progression, and considering benefits and risks of the various treatment modalities. The decision as to which type of therapy will be used is usually based on the preference of the patient as well as the clinical expertise of the oncologist, and judgement as to the progress and prognosis of the cancer and the relative adverse effects of the treatments. If resistance develops (hormone-refractory prostate cancer) docetaxel is the drug of choice. Trials of various new chemotherapeutic agents before surgery and in metastatic disease are in progress (see review by Michael et al [2009]).
Breast cancer
Breast cancer, the commonest cancer in Australian women (27% of all diagnoses) may be genetic, but more commonly occurs sporadically and the risk is related to length of oestrogen exposure. The risk of developing breast cancer can be reduced by adopting a healthy lifestyle (low alcohol consumption, low-fat and high-fibre diet, exercise and low weight) and by early detection of cancer (regular breast selfexamination and mammography). Women at high genetic risk for breast cancer can reduce the risk by prophylactic use of antioestrogens (tamoxifen or raloxifene), or even by pre-emptive surgery to remove the breasts.
If breast cancer develops, the initial treatment is usually surgery and radiation therapy, then chemotherapy with an antioestrogen or SERM and possibly cytotoxic agents. Biopsies can be taken and tested for levels of oestrogen and progesterone receptors and for levels of the proto-oncogene c-her2/neu, which encodes the protein EGF receptor 2; if the latter levels are high, the drug trastuzumab (Herceptin), a monoclonal antibody against the her2 protein, can be effective.
Corticosteroids
Glucocorticoids retard lymphocytic proliferation by their effects in suppressing white cell production, hence are used in the treatment of lymphocytic leukaemias and lymphomas. Prednisone and dexamethasone are also used in conjunction with radiation therapy to decrease the occurrence of radiation oedema in critical areas such as the superior mediastinum, brain and spinal cord. In addition, they are often used in conjunction with antiemetic drugs and as supportive therapy for their general metabolic, anti-inflammatory and euphoric effects (see Chapters 35 and 47).
Androgens and antiandrogens
Androgens
Androgens such as testosterone (Drug Monograph 39-1) and fluoxymesterone are used to treat advanced breast carcinoma if surgery, radiation and other therapies are inappropriate or ineffective.
Antiandrogens
The group of antiandrogenic agents includes flutamide, bicalutamide, nilutamide and cyproterone (a partial agonist). These drugs inhibit the uptake or the binding of androgens at their target cells or receptors. The result is suppression of ovarian and testicular steroidogenesis, thus inducing a ‘medical castration’. They are indicated in combination with surgery and a GnRH analogue (see below) for treatment of advanced prostate cancer. This combination has been reported to prolong survival by at least 25% compared with GnRH therapy alone. Adverse reactions include diarrhoea, impotence and other symptoms of low testosterone levels, and hepato-toxicity.
In many men with prostate cancer, the tumour becomes resistant to the actions of antineoplastics, so-called ‘castration-resistant prostate cancer’, due to mutations in or increased expression of the androgen receptor, increased synthesis of androgens or alteration or upregulation of some TK pathways. Second-generation antiandrogens attempt to overcome this resistance; one new analogue under clinical trial in advanced prostate cancer, the drug MDV3100, has greater relative affinity for the androgen receptor than bicalutamide and shows sustained declines in levels of PSA, a marker for prostate overactivity. Other drugs being tested in resistant prostate cancer include drugs that reduce androgen synthesis, inhibit transcription of androgen receptors or enhance androgen receptor degradation.
Gonadotrophin-releasing hormone analogues
GnRH (also known as LHRH) synthetic peptide analogues such as goserelin and leuprorelin, when administered on a continuous basis, effectively suppress production of gonadotrophins from the pituitary gland, and thus have indirect antiandrogenic and anti-oestrogenic effects (see Figure 38-2). They are used for gonadal suppression in precocious puberty, endometriosis, polycystic ovary syndrome and prostatic and premenopausal breast cancer (see Table 38-1). In prostate cancer, GnRH agonists can cause ‘chemical castration’.
Goserelin is used as a palliative agent in the treatment of advanced prostate carcinoma. With chronic administration, there is an initial surge in gonadotrophin release (causing a ‘flare’ in prostatic cancer growth), then the plasma concentrations of testosterone usually drop to the range seen in surgically castrated men within 2–4 weeks after initiation of drug therapy. A 3.6-mg dose as a prolonged-release formulation is implanted subcutaneously in the upper abdominal wall every 28 days, or 10.8 mg into the anterior abdominal wall every 3 months. Adverse reactions reported generally are related to the lowered testosterone levels and may include sexual dysfunction, hot flushes and decreased erections, and cardiovascular dysfunction.
A new analogue, triptorelin, acts by the same mechanism in prostate cancer; there is a similar initial surge in LH concentration, which may exacerbate symptoms such as bone pain and obstruction to bladder outflow. Triptorelin can also be given by monthly or 3-monthly IM injections; adverse effects include decreased libido, impotence, breast pain, hot flushes, skeletal pain and hypertension.
Oestrogens and anti-oestrogens
Oestrogens
Oestrogens may be used to treat androgen-sensitive prostatic carcinomas or advanced breast carcinoma in postmenopausal women. Oestrogens such as diethylstilboestrol and ethinyloestradiol, for example, have been used to treat advanced prostatic carcinoma; however, they are rarely used for this indication now due to adverse cardiovascular effects (see Chapter 38 and Drug Monograph 38-2 for typical oestrogenic actions).
Oestrogens may occasionally be used in breast cancer to ‘recruit’ resting cells from the G0 phase into active cell cycling again (G1 phase), so the cells will be sensitive to cytotoxic agents.
Anti-oestrogens
Anti-oestrogens are useful in treatment of postmenopausal breast cancers that are oestrogen receptor (ER)-positive, i.e. tumours that contain high concentrations of ERs. They have replaced both androgens and oestrogens as the initial approach in breast cancer therapy. (Anti-oestrogens are less useful in premenopausal women because their effects would be swamped by the high levels of oestrogens produced by the ovaries.)
Tamoxifen
Tamoxifen (Drug Monograph 42-3) is a synthetic non-steroidal anti-oestrogen preparation with both agonist and antagonist effects; it can be considered a partial agonist at ERs. It is believed to bind to ERs in breast cancer cells, where it acts as a competitive inhibitor of oestrogen. It is an oestrogen agonist in the liver, which has desirable effects on plasma lipids in postmenopausal women. It also helps to preserve bone mineral density, which may decrease the osteoporosis risk in these women. The adverse effects are mainly those of low oestrogen levels, i.e. hot flushes, vaginal disorders and nausea and vomiting. Unfortunately, resistance to the actions of tamoxifen frequently develops, due partly to the expression of various TK enzymes including HER-2 and EGFR.
Tamoxifen is a non-steroidal compound with a range of both agonist and antagonist activities at oestrogen receptors (ER) in various tissues. In patients with ER-positive breast tumours, it acts primarily as an anti-oestrogen, binding to the receptors and inhibiting growth of the tumour.
INDICATIONS Tamoxifen is indicated for treatment of breast cancer, especially postmenopausal ER-positive cancers. Taken prophylactically for at least 5 years, it reduces the risk of ERpositive breast cancer in women at high risk.
PHARMACOKINETICS Tamoxifen is administered PO and absorbed from the GIT; peak plasma levels are reached 3–6 hours after a single dose, and concen tration at steady state is achieved about 4 weeks after commencement of once-daily dosing. The drug is distributed widely to many organs and tissues, including uterus and ovary. It is highly bound to plasma albumin (>99%), also to ERs in target tissues. Tamoxifen is extensively metabolised in the liver; the major metabolite has actions similar to those of the parent drug. Poor metabolisers may show reduced responses and may suffer relapses. Slow elimination via the faeces accounts for the accumu lation and long half-life (5–7 days) of the drug and its major metabolite.
DRUG INTERACTIONS Tamoxifen can be considered a prodrug; it is metabolised by CYP3A4, 2D6 and 2C9 to some active metabolites, so its level and activities can be affected by the many other drugs that affect or are affected by these enzymes. CYP3A4 inducers that can reduce tamoxifen effects include rifampicin, corticosteroids, pioglitazone, many anticonvulsants and St John’s wort; while CYP3A4 inhibitors that can enhance the effects of tamoxifen include some TK inhibitors, ciprofloxacin, clarithromycin, erythromycin, many -conazole antifungals, many antivirals and grapefruit juice (see Australian Medicines Handbook, Appendix Table B-1). Combinations with 2D6 inhibitors such as the SSRIs, cimetidine and terbinafine should be avoided. Tamoxifen increases the anticoagulant effects of warfarin, and bleeding is likely; bleeding time should be monitored by INR and the dose of warfarin may need to be reduced.
ADVERSE REACTIONS Abnormal gynaecological reactions are common, such as vaginal bleeding and hot flushes; endometrial changes such as polyps and cancer can also develop. Other adverse reactions include leg cramps, pneumonitis, visual disturbances and leucopenia.
WARNINGS AND CONTRAINDICATIONS Tamoxifen is contraindicated in pregnancy, breastfeeding women and children; also in those who have shown hypersensitivity reactions to it. If a woman patient is of childbearing age and sexually active, effective non-hormonal contraception must be practised.
DOSAGE AND ADMINISTRATION The usual daily dose is 20 mg; in advanced stages of breast cancer, the dose may be raised to 40 mg/day.
Toremifene is a newer anti-oestrogen product for the treatment of metastatic breast cancer in postmenopausal women; the actions and adverse effects profile are similar to those of tamoxifen. Fulvestrant, another anti-oestrogen, is used in treatment of hormone receptor-positive metastatic breast cancer in postmenopausal women with disease progression following therapy with an anti-estrogen or aromatase inhibitor. It has no agonist effects, and works both by downregulating and by degrading the estrogen receptor. It is administered as a once-monthly IM injection.
Selective ER modulators
Selective ER modulators (SERMs) have been designed to block ERs in breast cancers but maintain ER agonist actions in other tissues such as bone and cardiovascular system where oestrogen has protective actions. Tamoxifen and toremifene can in fact be classed as SERMs.
Raloxifene is a SERM currently used to prevent postmenopausal osteoporosis, as it has oestrogenic effects in bone, but anti-oestrogenic effects in uterus and breast tissues (see Chapter 38). It is also being studied for the treatment of breast cancer, with results showing a significant decrease in invasive breast cancer in women at increased risk of breast cancer (see Vogel et al [2006]; Sengupta and Jordan [2008]).
Aromatase inhibitors
In the biochemical pathways for the synthesis of oestrogens, a critical stage is the ‘aromatisation’ of the steroid A ring, from testosterone to oestradiol (see Figures 33-3 and 35-1); this step was difficult in the early attempts to synthesise oestrogens in the laboratory. In postmenopausal women, the main source of oestrogens in the body is from androgens via the aromatase enzyme actions in peripheral (non-ovary) tissues. Various compounds that inhibit the aromatase enzyme have been synthesised, including anastrozole, letrozole and exemestane. They are indicated for use in women with natural or induced postmenopausal status, whose breast cancer has progressed despite anti-oestrogen therapy. Aromatase inhibitors do not block the synthesis of glucocorticoids or mineralocorticoids.
Progestogens
Progestogens such as medroxyprogesterone (Drug Monograph 38-3) and megestrol are used to treat advanced endometrial cancer, and breast cancer unresponsive to anti-oestrogens or aromatase inhibitors, because they suppress gonadotrophin release. This is primarily a palliative approach that seeks tumour regression and an increase in the patient’s survival time. Megestrol is also indicated for advanced carcinoma of the breast, and medroxyprogesterone is used in patients with advanced renal carcinoma.
Somatostatin analogues
Analogues of somatostatin (= growth hormone releaseinhibiting factor; see Table 33-2 and Drug Monograph 33-1) are used to treat cancers of the pituitary gland that produce excess growth hormone (causing acromegaly) and in carcinoid tumours of the GIT that secrete excess 5-hydroxytryptamine. The analogues are octreotide and lanreotide; adverse reactions in the GIT are common (see Table 42-2).
Other antineoplastic agents
Miscellaneous agents include those that cannot readily be classified by their mechanism of action into any of the previous groups. As these drugs are not cytotoxic agents, they are unlikely to cause severe bone marrow depression, nausea and vomiting, or hair loss. Some of these new drugs have been designed specifically to target enzymes or growth factors in neoplastic cells.
Antineoplastic monoclonal antibodies
Monoclonal antibodies are immunoglobulins produced synthetically from a single clone (genetically identical cells) of B lymphocytes; each clone will produce identical proteins with specificity against the same antigen. The binding of the antibody to the antigen usually inactivates the antigen and/or causes cell lysis. The suffix -mab has been adopted to indicate a monoclonal antibody. Monoclonal antibodies are most effective when administered in combination with chemotherapeutic regimens. As they are large proteins, antibodies must be administered parenterally (see Ward [2003]).
Trastuzumab
Trastuzumab targets a growth factor receptor. It is a monoclonal antibody against the receptor for epidermal growth factor (EGF), a protein encoded by an oncogene (known as her-2) overexpressed in about 30% of women with breast cancer.1 By blocking the EGF receptor, trastuzumab slows breast cancer progression and increases tumour reduction in women with this altered gene. It is used as monotherapy or has synergistic effects when used in combination with paclitaxel or with an antitumour antibiotic and cyclophosphamide. The antibody is administered by IV infusion. It has a very long half-life (about 6 days), but steady state is not reached for 4–8 months as the pharmacokinetics are non-linear. Hypersensitivity reactions and cardiotoxicity can occur.
Panitumumab is also an antibody against the EGF receptor, hence it inhibits cell growth and production of some cytokines and growth factors and induces apoptosis. It is indicated for use in patients with EGFR-positive metastatic colorectal cancer whose tumours have progressed despite treatment with cytotoxic chemotherapy; it is most effective in tumours expressing the wild-type KRAS gene. Skin-related toxicity is common, due to impairment of epidermal growth factor actions; other serious adverse effects include thromboembolism and hypomagnesaemia.
Rituximab
Rituximab is another genetically engineered monoclonal antibody, this one specific against an antigen (CD20) located on the surface of both normal and malignant B lymphocytes. The CD20 antigen governs the early steps in cell cycle initiation and differentiation, and it is found on more than 90% of B-cell non-Hodgkin’s lymphomas. Rituximab is indicated in treatment of B-cell non-Hodgkin’s lymphoma and is usually administered with the CHOP regimen: cyclophosphamide/hydroxydaunomycin/Oncovin (vincristine)/prednisone.
Others
Other new monoclonal antibodies include bevacizumab, specific against a vascular EGF, hence useful in slowing growth of new blood vessels supplying a tumour (angiogenesis); it is proving useful in treatment of renal cell carcinoma, which has previously been resistant to treatment with the usual antineoplastic drugs. Cetuximab is useful as an antibody against an EGF receptor overexpressed in many patients with colorectal cancer. Gemtuzumab is an antibody against CD33, a transmembrane receptor associated with myeloid cells; the antibody has antineoplastic activity in CD33-positive acute myeloid leukaemias and may be administered attached to a cytotoxic antibiotic (ozogamicin) to enhance the effects. Alemtuzumab, an antilymphocyte antibody, is used in treatment of chronic lymphocytic leukaemia; it binds to the CD52 glycoprotein and induces cell lysis. Infusion reactions are common, so dosage starts low, and premedication with steroids, an analgesic and an antihistamine is recommended. Most patients develop cytopenia and may require transfusion of blood or platelets; fatal infections have occurred.
Tyrosine kinase inhibitors
As described in Chapter 41 and Figure 41-2, the cell cycle is regulated by many growth factors, some of which act by binding to specific transmembrane receptors, which ‘switches on’ the receptor’s kinase (phosphorylating) enzyme activity to cause phosphorylation of the tyrosine residues in proteins that induce cell growth or differentiation. Deregulation of TK activity is a major mechanism by which cancer cells evade normal regulation of cell growth and cycling. Drugs designed to inhibit specific protein kinase enzymes (tyrosine kinase inhibitors) can thus act as inhibitors of particular metabolic reactions in cell growth or differentiation pathways, and hence act as antineoplastic agents. If the enzyme inhibitors are small molecules this is an advantage as they can be administered orally. Over 500 kinases are encoded in the human genome, with at least 30 different TKs currently being investigated as targets for anticancer drugs (see review by Zhang et al [2009]).
Imatinib and gefitinib
Two such tyrosine kinase inhibitors (-tinibs) are imatinib and gefitinib. They are relatively new drugs, so clinical experience with their use is still accumulating. Imatinib (Drug Monograph 42-4) inhibits the TK in the receptor for Philadelphia chromosomal platelet-derived growth factor, and was the first anticancer drug targeted to a signal transduction protein unique to cancer cells. It is indicated for use in chronic myelocytic leukaemia (CML), various other leukaemias and gastrointestinal tumours. Adverse reactions including bone marrow suppression, gastrointestinal disorders and fluid retention are com mon. Gefitinib inhibits the TK associated with the receptor for EGF, which occurs particularly in solid tumours derived from epithelial tissues, such as non-small-cell lung cancers. Its major adverse reactions are interstitial pneumonitis and gastrointestinal and skin disorders. Drug interactions common to TK inhibitors are summarised in Drug Interactions 42-3.
Imatinib was designed specifically to inhibit the tyrosine kinase Bcr-Abl in the receptor for Philadelphia chromosomal plateletderived growth factor; it also inhibits platelet-derived growth factor receptors.
INDICATIONS It is indicated for use in chronic myeloid leukaemia, Philadelphia chromosome-positive acute lymphoblastic leukaemia, various other myeloproliferative disorders and gastrointestinal stromal tumours.
PHARMACOKINETICS Imatinib is well absorbed PO, independent of dosage or food; maximum levels are reached after 2–4 hours. It is widely distributed into tissues, with a volume of distribution of approximately 435 L. It is metabolised by CYP3A4 and other CYP enzymes (one metabolite has equal TK inhibitor activity), and competitively inhibits metabolism of drugs that are substrates for these enzymes. Metabolites are primarily eliminated via bile. Hepatic and renal dysfunction, and presence of liver metastases, may increase bioavailability; however, dosage is only reduced in severe liver disease (see below). The terminal elimination half-life is approximately 18 hours; once daily dosage is effective.
DRUG INTERACTIONS See Drug Interactions 42-3 for interactions typical of TK inhibitors. Others specific for imatinib include interactions with simvastatin (metabolism of simvastatin inhibited with risk of rhabdomyolysis; dose of statin should be halved) and with thyroid hormones (metabolism of thyroxine increased, thyroid function should be monitored; dose of thyroxine may need increasing).
ADVERSE REACTIONS These include bone marrow suppression (especially neutropenia and thrombocytopenia); gastrointestinal disorders and fluid retention (in eye, lower limb, lung and pericardium and ascites); muscle and joint disorders; and liver, cardiovascular and respiratory dysfunction; fever and photosensitivity.
WARNINGS AND CONTRAINDICATIONS Imatinib should be used with caution in the elderly and in patients with cardiac disease (fluid retention more common), thyroid disorders (risk of hypothyroidism) and in those with hepatic impairment (lower initial dose required). Complete blood count, liver function tests and oedema should be monitored before and regularly during treatment. Contraindicated in pregnancy and breastfeeding.
DOSAGE AND ADMINISTRATION Specialist oncology units have protocols for usage; typical adult doses are 400 mg 1–2 times daily (in severe liver impairment: 300 mg once daily), taken with food and a large glass of water or juice to avoid stomach upset. Patients should be advised to weigh themselves regularly (to monitor fluid retention) and avoid sun exposure and use sunscreen.
Drug interactions 42-3 Tyrosine kinase inhibitors
| Drug | Possible effects and management |
| Other antineoplastic agents | Enhanced antiproliferative effects, especially causing myelosuppression; combination protocols should take these into account |
| Rifampicin | Metabolism of TK inhibitors increased, possibly reducing their efficacy; dose may need to be increased |
| Drugs affecting platelet function (aspirin, other NSAIDs) | Avoid combination if possible |
| Drugs that induce CYP3A4 (e.g. corticosteroids, many anticonvulsants & antivirals, St John’s wort) | Metabolism may be increased and efficacy reduced; dose may need increasing |
| Drugs that inhibit CYP3A4 (e.g. clari- and erythromycin, cimetidine, -azole antifungals, other TK inhibitors, some antivirals) and grapefruit juice | Metabolism may be decreased and efficacy & toxicity enhanced; dose may need reducing |
| H2-antagonists & proton pump inhibitors | May reduce absorption of TK inhibitors due to reduced acidity; avoid using if possible |
Newer TK inhibitors
Several other new TK inhibitors are becoming available, such as lapatinib, nilotinib, sorafenib, erlotinib and sunitinib; each targets a particular TK and thus has specific indications for use in particular cancers, and specific adverse effects, depending on the TK inhibited. Sorafenib and sunitinib are oral TK inhibitors effective in some cases of renal cell carcinoma. Lapatinib is active against breast cancers overexpressing the HER-2 growth factor receptor and, when used in combination chemotherapy with capecitabine, it prolongs the time to disease progression. Nilotinib has been designed to bind to a specific TK in CML cells with the mutant Philadelphia chromosome and shows benefits in patients who have developed resistance to imatinib; sudden cardiovascular deaths have occurred due to prolongation of the QT interval. Dasatinib binds to a broader range of TKs than does imatinib, so is proving effective in some leukaemias that have developed resistance to the latter drug. Myelosuppression is common, so blood counts should be monitored regularly. Erlotinib is indicated in specialist protocols for non-small-cell lung cancer and pancreatic cancer. It has many serious adverse effects, including rashes, abnormal hair and nail growth, diarrhoea, GIT and liver disorders and, rarely, corneal or GIT perforation and interstitial lung diseases.
Other miscellaneous antineoplastics
mTOR inhibitors
The mTOR (mammalian target of rapamycin) protein complex functions as an integration centre for various intracellular signalling pathways. For example, inhibition of the mTOR complex by sirolimus (aka rapamycin) is an important target for anticancer drugs (see review by Figlin et al [2008]).
Temsirolimus has an indirect mechanism of action, binding to an intracellular protein and blocking a target of a kinase and inhibiting the mTOR pathway; cell cycling is arrested and angiogenesis inhibited, leading to tumour suppression. It is used in treatment of renal cell carcinoma. Serious adverse effects include infusion reactions and hypersensitivity reactions, delayed wound healing and infections due to leucopenia. A similar drug, everolimus, has been approved in the USA for use in advanced kidney cancer. (Other -limus drugs such as tacrolimus are more selective for immunosuppressant actions, via blocking the action of calcineurin in activated T-cells, and are used in immune disorders and to prevent transplant rejection; see Chapter 47.)
BRAF inhibitors
The BRAF gene in humans encodes a protein B-RAF, belonging to a family of serine/threonine protein kinases. This protein is involved in regulating a pathway affecting cell division and differentiation and secretion; mutations in the gene are associated with various cancers, including colorectal, thyroid and lung and malignant melanoma. In late 2009, a breakthrough in the treatment of melanoma was announced when results from the first phase I clinical trial with a BRAF inhibitor were reported: whereas traditional chemotherapy often has little effect on melanomas, the BRAF inhibitor caused marked shrinkage of melanomas; larger scale trials are to be carried out in New York, Sydney and Melbourne.
Tumour vaccines
Immune surveillance2 by T-cells against non-self cells implies that the specific defence systems in the body can mount an immune response against antigens expressed on the surface of tumour cells and destroy the cells. Only when the immune system is depressed and/or the load of mutated tumour cells becomes too great for the immune system to dispose of them, do cancers continue to develop (see Figure 41-4). This raises the hope that vaccines can be developed against cancers (see again Clinical Interest Box 41-7). The field of cancer immunology is beyond the scope of this chapter; however, it is worth noting that many anti-tumour vaccines are being developed, including against some oncogenic viruses. A notable recent vaccine against the human papilloma virus (HPV) is the non-live vaccine (Gardasil or Cervarix) developed in Australia. It is already in large-scale use in teenage girls and young women in many countries and provides effective protection against various strains of HPV.
Other strategies in cancer immunotherapy include the following (see review by Mastini et al [2009]):
Proteasome inhibitors
Proteasomes are large protease-containing complexes in cells, in which ubiquitinated proteins are degraded. There are hundreds of ubiquitin-protein ligases by which chains of poly-ubiquitinated proteins can be formed and thus proteins can be targeted for destruction by proteolysis in proteasomes. Complete inhibition of proteasome functions kills cells, so researchers have sought small-molecule specific inhibitors for regulatory proteins in the proteasome complex.
Bortezomib
Bortezomib, a new anticancer drug, is a proteasome inhibitor; it can be classed with cytotoxic agents. By blocking the targeted proteolysis normally performed by the proteasome, bortezomib disrupts various cell signalling pathways, leading to cell cycle arrest, apoptosis and inhibition of angiogenesis. Specifically, the agent inhibits nuclear factor (NF)-kappa B, a protein that is constitutively activated in some cancers. The drug thus interferes with NF-kappa B-mediated cell survival, tumor growth and angiogenesis. In vivo, bortezomib delays tumour growth and enhances the cytotoxic effects of radiation and chemotherapy.
It is being used in treatment of multiple myeloma, a cancer of the plasma cells (a type of B-lymphocyte) in which large amounts of useless immunoglobulins are formed. Multiple myeloma is characterised by bone lesions and pain, renal failure, anaemia and elevated calcium levels, and has been resistant to treatment by usual chemotherapeutic agents (previously melphalan and prednisolone, plus autologous stem cell transplantation). The relative roles of bortezomib, thalidomide and lenalidomide in treatment of multiple myeloma are still being established.
Radioactive isotopes
As described in Chapter 41 under ‘Treatment modalities’, radioactive isotopes may be used in cancer therapy. Radiopharmaceuticals used include iodine-131 (see Drug Monograph 34-3 and Clinical Interest Box 41-3), phophorus-32 and strontium-89. Preparing and administering these isotopes is a highly specialised area of hospital pharmacy practice.
Anagrelide
Anagrelide is a different type of agent; it has specific actions in reducing the platelet count and so is useful in essen tial thrombocytopenia, a condition in which platelets proliferate.
Methyl aminolevulinate
This unusual drug is a photosensitising agent that is applied as a cream to skin lesions (actinic keratoses or basal cell carcinomas) for 3 hours, then the area is exposed to red light, which activates the chemical to produce reactive oxygen radicals that selectively destroy tumour cells. Common adverse reactions include burning pain and ulceration at the site of application. The drug is a porphyrin precursor, and is related to verteporfin, used in a similar way as a photosensitiser to treat ocular macular degeneration (see Chapter 31).
Potentially toxic chemicals
Tretinoin
Retinoids (vitamin A derivatives) such as tretinoin have wide-ranging actions in many tissues and at high concentrations are toxic and teratogenic. They have useful effects in cancer cells because of their antiproliferative and prodifferentiation actions, especially in leukaemic cells, and are being used to treat acute promyelocytic leukaemia (as well as acne; see Drug Monograph 48-2 and Clinical Interest Box 48-4). Adverse effects occur particularly in the skin and mucous membranes. Retinoids are known to be teratogenic (Pregnancy Category D or X) and are contraindicated in pregnancy or in women of childbearing age unless using effective contraception.
Arsenic trioxide
Arsenic has long been known as a toxic chemical, blocking the tricarboxylic acid cycle and causing kidney damage, psychotomimetic effects and skin cancers. It has been used to treat psoriasis and various infections including syphilis, and as a weed-killer, insecticide and rodenticide, and was found in a traditional Chinese medicine used for leukaemia. Recent research has shown that arsenic trioxide activates cysteine proteases, and thus promotes cell differentiation and enhances apoptosis; hence it may have useful antineoplastic actions, and can be made available in Australia through the Special Access Scheme. It is administered by daily IV infusions for up to 60 days in treatment of acute promyelocytic leukaemia refractory to or relapsed after other treatments. Cardiovascular adverse effects are common, also bone marrow suppression; arsenic itself is a carcinogen.
Rose Bengal
Rose Bengal is a dye (traditionally used by Bengali women as a symbol for marriage) used in ophthalmic medicine as a stain. It is now being trialled in treatment of melanoma, a rapidly-advancing highly malignant skin cancer. Injected into the tumours, Rose Bengal can cause shrinkage and slowed growth, and appears to activate the immune system against other nearby melanomas.
Adjunctive treatments
Treatment of adverse drug reactions
As discussed in Chapter 41, adverse drug reactions to cytotoxic drugs most commonly impair rapidly dividing cells, such as those in the hair, skin, GIT mucosa and bone marrow. As the drugs are usually excreted via the kidneys, the kidney tubules are also very vulnerable. These adverse effects may be sufficiently severe as to require acute or prophylactic treatment. In addition, particular drugs may cause other specific adverse reactions, e.g. anthracycline antibiotics cause cardiomyopthy, and sex hormone inhibitors cause reduced libido. (Other general adverse reactions may be treated symptomatically, e.g. infections due to myelosuppression are treated with specific antimicrobial agents and fever with antipyretic drugs. Pain relief is discussed under ‘Palliative care’.)
Treatment of nausea and vomiting
The commonest and most distressing adverse reactions to cytotoxic drugs include nausea and vomiting. The chemoreceptor trigger zone in the medulla oblongata is sensitive to chemical stimuli, including emetogenic substances produced by cytotoxics, and to endogenous substances produced in radiation sickness and in the tumour lysis syndrome. Cytotoxics can be compared in terms of their emetogenic potential (see Table 41-2).
Nausea and vomiting are treated with antiemetic drugs, preferably prophylactically (see Chapter 29, and Drug Monographs 29-3 [metoclopramide] and 29-4 [ondansetron]); for low-risk agents, metoclopramide is given when necessary; for intermediate-risk agents, metoclopramide 20 mg IV or orally plus dexamethasone 20 mg IV or orally. For drugs with high emetic risk, a stronger antiemetic is required, such as an oral 5-hydroxytryptamine (5-HT3) antagonist (e.g. ondansetron) plus dexamethasone before chemotherapy. Other similar antiemetics are dolasetron, tropisetron and granisetron (also usefully available in a transdermal formulation). Phenothiazine antiemetic drugs (e.g. prochlorperazine) and antianxiety agents may also be helpful. For severe delayed emesis (>24 hours after chemotherapy), regular administration of IV antiemetics for 2–4 days may be necessary.
Aprepitant is a new antiemetic acting by a different mechanism: it inhibits substance P-mediated vomiting by selectively antagonising the neurokinin-1 receptor. Currently, it is recommended for use only with highly emetogenic cytotoxics such as high-dose cisplatin or cyclophosphamide plus an anthracycline. Fosaprepitant, a prodrug, is an IV formulation producing higher concentrations of aprepitant; it is effective in stopping delayed emesis.
Treatment of myelosuppression (bone marrow depression)
Bone marrow depression is usually the limiting factor in the clinical use of cytotoxics, causing treatment delays and dose reductions. White cells and platelets are the first affected, leading to immunosuppression, infections, bruising and bleeding. Neutrophil counts and platelet counts are monitored to indicate when, after cessation of chemotherapy, levels have risen high enough for another cycle of chemotherapy to commence. The main risk from thrombocytopenia (low platelet count) is haemorrhage; this may be treated or prevented with infusions of platelets or with haemostatic agents.
Folinic acid rescue
Myelosuppression from the antifolate agent MTX may be minimised with folinic acid rescue, in which calcium folinate (an analogue of tetrahydrofolate; see Drug Monograph 42-5) is administered a few hours after high-dose MTX. In normal cells, calcium folinate bypasses the enzyme blocked by MTX and helps prevent much of the bone marrow toxicity without reversing the antineoplastic effects in cancer cells. It also enhances the cytotoxicity of FU by further inhibiting thymidylate synthetase.
Drug monograph 42-5 Calcium folinate
Calcium folinate (aka folinic acid, or Leucovorin) is a precursor to tetrahydrofolate, the conversion to which does not require dihydrofolate reductase. It enters and ‘rescues’ normal cells preferentially to cancer cells owing to differences in the uptake mechanisms. It is therefore used to prevent or treat toxicity induced by folic acid antagonists, especially methotrexate (MTX), and to enhance the cytotoxicity and therapeutic efficacy of fluorouracil.
INDICATIONS Calcium folinate is indicated as an antidote (prophylaxis and treatment) for folic acid antagonists, such as MTX, pyrimethamine or sulfadiazine, and in treating megaloblastic anaemias caused by folic acid deficiencies, sprue, or pregnancy, and whenever oral folic acid therapy is not appropriate.
PHARMACOKINETICS Calcium folinate is rapidly absorbed orally and converted by the intestinal mucous membrane and liver to 5-methyltetrahydrofolate, an active metabolite. The onset of action is 20–30 minutes orally, 10–20 minutes IM and <5 minutes IV. The duration of action by all routes is 3–6 hours. Metabolites are primarily excreted by the kidneys. Doses >25 mg should be administered IV as oral absorption is saturable.
DRUG INTERACTIONS Calcium folinate may enhance the toxicity of fluorouracil. The antagonism between calcium folinate and folic acid antagonists such as MTX or raltitrexed is intentional.
ADVERSE REACTIONS These include allergic reactions (rash, itching and wheezing) and convulsions.
WARNINGS AND CONTRAINDICATIONS Use with caution in patients with aciduria (urine pH < 7), ascites, gastrointestinal obstruction, pleural effusion or nausea and vomiting (decreased hydration may result in an increase in MTX toxicity) and in dehydrated patients. Avoid use in persons with calcium folinate hypersensitivity and kidney function impairment. Calcium folinate is not effective in treating anaemias associated with a deficiency of vitamin B12.
DOSAGE AND ADMINISTRATION The dosage and duration of ‘rescue’ with calcium folinate depends on the MTX dose, concentration of MTX in serum and serum creatinine levels. A typical regimen is 15 mg orally, IM or IV every 6 hours for 60 hours (10 doses starting 24 hours after start of MTX infusion), plus hydration and urinary alkanisation. For treatment of acute MTX overdose, calcium folinate dosage starts at 10 mg/m2 IV.
Granulocyte colony-stimulating factors
Bone marrow suppression may also be overcome by SC administration of recombinant versions of granulocyte colony-stimulating factor (G-CSF: filgrastim or lenograstim; see below under ‘Immunomodulatory agents’). These cytokines stimulate neutrophil precursor cells, reducing the duration of neutropenia and risk of infections after cytotoxic chemotherapy or bone marrow transplant. A related growth factor for haemopoietic stem cells is ancestim. They should not be administered within 24 hours before or after chemotherapy, as rapidly dividing cells (i.e. those stimulated by the GCSF) are most sensitive to cytotoxics.
Hydration therapy and treatment of tumour lysis syndrome
Rehydration, to prevent renal tubular damage or hyperuricaemia, requires vigorous IV fluid infusion. During therapy with high-dose cisplatin, for example, about 2.5 litres of fluids (saline, mannitol and/or glucose) are infused before chemotherapy over 2–3 hours, with frusemide to encourage diuresis if necessary; then, after the chemotherapy, 2 litres of fluids are given over the next 10 hours.
Mesna and amifostine
The phosphamide alkylating agents (cyclophosphamide, ifosfamide) are specifically toxic in the kidney epithelium, causing haemorrhagic cystitis. This toxicity may be prevented or reduced by prior administration of a sulfurdonating compound such as mesna or amifostine. These release thiol (sulfhydryl) groups in the kidneys, which detoxify metabolites of the alkylating agents and protect against toxicity from alkylating agents or platinumcontaining cytotoxic agents. Two specific examples of effective cytoprotective combinations are described here.
Ifosfamide, an alkylating agent, is used for the treatment of germ cell testicular tumours, but its adverse effect of haemorrhagic cystitis (urotoxicity) has limited its usefulness. Mesna, a thiol donor in the kidneys, acts as a specific antidote for this type of toxicity. Using the drugs in combination therefore allows more aggressive therapy while reducing the dose-limiting adverse effects of ifosfa-mide.
Amifostine is a cytoprotective agent administered before platinum compounds or alkylating agents to reduce their potential for renal toxicity. It is a prodrug activated to free thiol groups that protect normal cells against radiation and DNA-binding agents. It is also used to reduce xerostomia (excessively dry mouth) caused by radiotherapy to the head and neck.
Tumour lysis syndrome
This condition is due to massive release of cell breakdown products from large tumour masses (especially leukaemias and lymphomas) treated with chemotherapy; it is mani fested by excessive levels of potassium, phosphate and uric acid in the bloodstream (see description in Chapter 41).
Allopurinol is a drug primarily used in treatment of gout. It is a xanthine oxidase inhibitor that reduces the synthesis of uric acid and hence the load of urate to be excreted, thus relieving nephropathy, kidney stone formation and gouty arthritis. In neoplastic disease and myeloproliferative disease, there is also a build-up of urates in the body, due both to the high turnover rates of neoplastic cells and to the cell death induced by cytotoxic agents. Hyperuricaemia is therefore often a clinical feature of cancers, so allopurinol is given to reduce the manifestations of urate deposition.
Rasburicase is a new drug, a recombinant form of an enzyme that catalyses the conversion of uric acid to a more soluble metabolite. It is given for prophylaxis and treatment of hyperuricaemia associated with cytotoxic chemotherapy of haematological malignancies.
Treatment of mucositis
Mouth ulcers from chemotherapy have traditionally been treated with mouthwashes of antiseptics and zinc.
Palifermin
Palifermin is an unusual drug being used to treat mucositis by rescuing keratinocytes; it is a recombinant form of the endogenous human keratinocyte growth factor. It binds to epithelial cell surface receptors in the lining of the mouth and gastrointestinal tract, resulting in stimulation of epithelial cell proliferation, differentiation and upregulation of cytoprotective mechanisms. In a clinical trial in patients with advanced colorectal cancer treated with FU, palifermin reduced the frequency and severity of mouth sores, and patients who took palifermin were more likely than those taking a placebo to receive their full dose of chemotherapy. It is given by IV bolus injection for 3 consecutive days finishing 1–2 days before myelotoxic therapy, then again for 4 days afterwards.
Preservation of fertility
By far the majority of cancers develop in people over the age of 70 years, for whom adverse effects of cytotoxic agents on germ cells (ova, spermatozoa) are usually not an issue. However, with the increased survival rate of children from childhood cancers such as leukaemias and lymphomas, it is estimated now that one in every 250 adults is a childhood cancer survivor, and may have impaired fertility from earlier treatments. Also, with women in many societies delaying childbearing into their 30s, there are more women with early-onset cancers (breast, melanoma, cervical, leukaemias and lymphomas) anxious to preserve their fertility. Assisted reproduction technologies are helpful in preserving fertility, but require interdisciplinary cooperation between the various medical specialists involved in a woman’s treatment.
Immunomodulatory agents
Immunodeficiency is a serious problem in many cancer patients, partly because cancer itself is immunosuppressant and especially because cytotoxic antineoplastic agents cause T-cell depletion. Alkylating agents, purine antimetabolites and corticosteroids all have major immunosuppressant actions. Some subsets of T cells recover relatively quickly post-chemotherapy, especially in children, but there can be prolonged T-cell depletion in adults.
Immunosuppression commonly predisposes to increased incidence of infections. When white cell counts become very low, patients may require antibiotics, anti fungal agents and/or antiviral therapy. If a patient has an acute infection, chemotherapy is usually deferred until the patient has recovered from the infection.
Clinical management of patients with iatrogenic T-cell depletion involves monitoring and effective antimi crobial treatment of opportunistic infections, irradiation of blood products for infusion, prophylaxis and possibly re-immunisation against viral infections, and administration of immunostimulatory agents.
Interferons
Interferons are naturally occurring small protein molecules that have antiviral, antiproliferative and immunostimulating actions. They can be produced by recombinant technology, resulting in highly purified proteins that have effects similar to the interferon subtypes produced by human leucocytes. Interferons are expensive, however, and the cost of treatment courses is often prohibitive.
Immunomodulatory properties include enhanced phagocyte activity and increased cytotoxic properties of lymphocytes for target cells. Their mechanism of action as antineoplastic agents is unknown, but may be the result of one or more of the three properties identified above. In some types of cancer, for example, interferon appears to have a dual effect of both cytotoxicity and immune stimulation. Some patients demonstrate an increase in haematological factors, granulocytes, platelets and haemoglobin plasma levels.
Interferons alfa-2a and alfa-2b are indicated for the treatment of some leukaemias and lymphomas, genital warts, AIDS-related Kaposi’s sarcoma, bladder cancer, osteosarcoma and chronic active hepatitis. Toxicities reported include a flu-like syndrome with fever, chills, muscle pain, loss of appetite and lethargy. At higher doses, myelosuppression, nausea, vomiting, neurotoxicity and cardiotoxicity can occur. (Other interferons are more selective in actions against multiple sclerosis or viral infections.)
Levamisole
Levamisole was initially used clinically as a treatment for intestinal worm infestations. It was also found to have useful immunostimulant effects, enhancing T-cellmediated immunity and macrophage actions. Levamisole is used in combination with FU to treat colorectal carcinoma. This combination has resulted in a lengthened survival time (decreased mortality) and lowered risk of cancer recurrence. Levamisole can be administered orally, and adverse effects are usually mild; however, reversible bone marrow sup pression and a flu-like syndrome can occur.
Aldesleukin
Aldesleukin is another immunoregulatory lymphokine that stimulates immune function and is a recombinant version of human interleukin-2 (IL-2). This substance appears to stimulate T-cell proliferation and is a co-factor in enhancing growth of the body’s natural killer cells and the lymphokine-activated killer cells; it also increases production of interferons. Aldesleukin is indicated for the treatment of metastatic renal cell carcinoma, melanoma and thymoma, and after bone marrow transplant. Adverse effects of aldesleukin include oedema, anaemia, thrombocytopenia and hypotension. It is not generally available in Australia, but may be via the Special Access Scheme.
Colony-stimulating factors
Granulocyte colony-stimulating factors (G-CSFs: filgrastim, lenograstim, pegfilgrastim) are recombinant versions of bone marrow-stimulating factors with immunomodulatory actions. G-CSFs are used to mobilise stem cells to stimulate the production of phagocytes and thus decrease the potential for infection in people receiving myelosuppressant agents (such as cytotoxics) that are associated with severe neutropenia and fever. Neutrophil counts are closely monitored after the nadir (lowest level) induced by bone marrow transplant or chemotherapy. G-CSFs should be discontinued when the absolute neutrophil count reaches 10,000/mm3 or higher. The major adverse reactions include splenomegaly, hair loss, diarrhoea, fevers, mucositis, anorexia and fatigue. Bone pain has been reported about 2–3 days before the increase in neutrophil count. This pain is usually controlled with non-opioid analgesics. The usual doses are in the range 1–20 mcg/kg per day by SC injection.
BCG (non-vaccine)
BCG stands for bacille Calmette–Guérin, or the Mycobacterium bovis bacillus. The BCG vaccine is a live bacterial vaccine for immunisation against tuberculosis; when commonly used in the early 20th century it was found that BCG vaccine reduced risk of some cancers including bladder cancer (see Clinical Interest Box 41-7). The non-vaccine formulation is also a preparation of an attenuated strain of the bacterium; however, it is administered for its immunostimulatory effects rather than to produce immunity against the particular organism. The mechanism whereby BCG reduces cancerous lesions of the urinary bladder is unknown; it promotes a local inflammatory response and white cell infiltration of tissue.
It is administered not intradermally (as is the vaccine) but instilled by urethral catheter into the urinary bladder of patients with bladder carcinoma, to reduce tumour recurrence. Patients who are immunocompromised are at risk of systemic tuberculosis infection; other adverse effects include urinary tract pain and dysfunction and fever and malaise.
Other immunostimulating therapy
Imiquimod
Imiquimod is a new drug that enhances the immune response to tumours and viruses. It is indicated in treatment of some warts, and of basal cell carcinomas where surgery is inappropriate.
Thalidomide and lenalidomide
Thalidomide is the drug infamous for having caused thousands of congenital malformations during the late 1950s and 1960s in babies whose mothers took it as a supposedly safe sedative in early pregnancy. After having been banned from use for many years, it is undergoing re-evaluation for limited specific uses, such as in treatment of leprosy and as adjunctive therapy in some AIDSassociated infections and tumours (see Clinical Interest Box 4-1). It has immunostimulatory actions and inhibits tumour necrosis factor (TNF), and is being trialled as an antiangiogenic agent in several cancers, including multiple myeloma, renal cell carcinoma and glioblastoma. In Australia, it is tightly controlled because of its teratogenicity (pregnancy category X); patients must give written informed consent before treatment. Thalidomide was listed on the Australian Pharmaceutical Benefits Scheme (PBS) in February 2006, with strong warnings that even one dose can cause birth defects.
A new analogue, lenalidomide, is indicated for treatment of multiple myeloma. It has direct cytotoxic effects via induction of apoptosis; stimulation of production of T-cells, IL-2 and interferon-gamma; inhibition of TNF-alfa and IL-6; and reduced angiogenesis. It enhances the survival rate when added to dexamethasone therapy. Serious adverse effects include venous thromboembolism, leucopenias and GIT disturbances. Like thalidomide, it is a potential teratogen.
Treatment of problems due to bony metastases
Bisphosphonates
The bisphosphonates (including pamidronate, zoledronic acid, ibandronic acid and sodium clodronate; see Chapter 37) bind to hydroxyapatite in bone and specifically inhibit osteoclast-mediated bone resorption. They are used to improve bone mineral density in osteoporosis and Paget’s disease of bone, and to prevent corticosteroid-induced osteoporosis (see Drug Monograph 37-5). In cancers, they are used to reduce the skeletal morbidity that occurs with bony metastases and to reduce tumour-induced hypercalcaemia.
Palliative care
Palliative care provides integrated and comprehensive care for all the medical and nursing needs to improve the quality of life of a patient for whom cure is not possible. Palliative care at the end of life aims to treat all aspects of a patient’s suffering—physical, psychological, social, cultural and spiritual—and to include the patient’s family and close friends in the care. Traditionally, palliative care was employed when anticancer treatment had failed and active medical treatment of related medical problems had ceased. It is now recognised that palliative care is most successful when introduced at an earlier stage, in conjunction with other therapeutic modalities. It is also usefully employed in the end-stage of neurological diseases, such as multiple sclerosis, dementia and motor neuron disease, and in severe renal or hepatic failure and AIDS.
Optimal care acknowledges that suffering includes many aspects and that a multidisciplinary approach is needed. Aspects to be considered include the patient’s responses to diagnosis of a life-threatening illness; the patient’s choice to transfer from curative to palliative care; the deterioration to a terminal stage; discussion (or not) of death and dying; optimal ways to answer questions put by patients and their families (see Clinical Interest Box 42-3); ethical issues related to withdrawal of treatment and lifesupport; support for completing essential life-tasks; and the later bereavement and grief of family and friends.
Clinical interest Box 42-3 Frequently asked questions about palliative care (relevant to pharmacology)
Questions asked by health care providers:
When is it appropriate to introduce palliative care?
When the goal of care is comfort rather than cure; disease-modifying treatments may be continued if they benefit the patient.
How can I introduce the idea of palliative care?
With the idea that a team of medical, nursing, allied health and support staff can work together with the general practitioner to improve the quality of life for the patient and family.
Do I have to take morphine/opioids?
These medications provide the best pain relief, and are safe and predictable; however, there is a range of pain relief medicines and other techniques available.
Does taking morphine/opioids mean that I am going to die soon?
No, these medications can be taken for long periods to improve quality of life and allow more activities.
Will I become addicted to them? Should I ‘save them for later’?
No, people taking opioids for pain relief rarely become addicted.
Controlling pain early is important.
Will opioids make me ‘woozy’ or unsafe to drive?They will help you sleep better at night. They may make you drowsy, especially on long trips; be especially careful if you have taken ‘top-up’ doses.
Can I keep taking my herbal medicines (or using other CAM)?
If they are not dangerous or too expensive for you; please tell us what you are taking so we can check for any interactions with prescribed medicines.
Questions asked by family and carers:
Why does (s)he need painkillers if (s)he is unconscious?
The patient may regain consciousness, and deserves ongoing pain relief.
Shouldn’t the patient be ‘on a drip’ (IV fluids)?
Only if dehydrated; as the body ‘closes down’, less fluid is required or tolerated.
Adapted from: Palliative Care Expert Group 2010; Lorenz et al 2010.
Palliative care may be carried out in a variety of settings, including general practice, the home, aged-care facilities, hospitals and hospices. Large hospitals with oncology units usually have a palliative care service and a multidisciplinary team of health-care professionals to whom patients can be referred (see Palliative Care Expert Group 2010). The team aims to provide specialist palliative care services coordinated by one member; the team may include the patient’s general practitioner, a palliative care nurse, palliative medicine specialist, other specialist physicians (e.g. oncologist, radiologist, neurologist), clinical pharmacist, social worker, counsellor, chaplain, dietician, other allied health professionals, volunteer carer and patient support group representative. Team members themselves need support and care, as this is a very demanding area of medicine.
Pharmacological aspects of palliative care
As well as the drugs used to treat the disease for which the patient is receiving care, many other drugs may be useful in providing improved quality of life and comfort, such as analgesics for pain and dyspnoea and drugs to treat side effects of the primary drugs (e.g. antiemetics, immunostimulants, GCSFs). Decisions may also need to be made as to whether or for how long to continue with drugs being administered for other concurrent chronic conditions, such as diabetes mellitus, hypertension, arthritis or asthma.
Drugs commonly administered in palliative care situations include:
Analgesics in palliative care
Pain is one of the commonest symptoms in advanced cancer and the most feared; it is often not adequately treated. Apart from physical signs and symptoms, pain may exacerbate anxiety and depression, social problems and cultural and spiritual problems. Patients need reassurance that there are effective analgesic drugs to treat pain, that they are unlikely to become dependent on opioid analgesics and that satisfactory pain control can be achieved for more than 90% of cancer patients. Analgesics are used in the stepwise ‘ladder’ approach: non-opioids first (aspirin), then mild opioids (codeine), then strong opioids (morphine or fentanyl); see Chapter 15 and Figure 15-5. Sufentanil, a fentanyl analogue, may be available through the Special Access Scheme.
Analgesic drugs need to be chosen appropriately and given in adequate doses and frequently enough to keep the patient pain-free, with instructions for extra dosing for ‘breakthrough’ pain, and warning of and treatments for adverse effects such as constipation.
Breakthrough pain (BTP)
Adequate control of breakthrough pain is an important issue in palliative care, for example in patients with bony metastases from cancer. It is defined as pain that occurs between regular doses of an analgesic that normally controls the patient’s pain. It is important for the patient’s quality of life that this pain be controlled effectively, usually with an extra dose of the patient’s regular opioid. The breakthrough dose is determined by consideration of the current 24-hour analgesic dose equivalent, one-sixth of which is given every 4 hours (for immediate-release formulations). The breakthrough dose may be 50%–100% of the regular 4-hourly dose, at intervals not less than 30 minutes, for up to three doses. The normal dose should then be given at the regular time. The number of breakthrough doses required over a 24-hour period is an indication of the need to consider increasing the regular dose. Fentanyl lozenges (‘lollipops’) which allow absorption through the buccal membranes are especially useful for breakthrough pain (see Table 15-3).
Particularly careful dosing of opioids is required in elderly patients, those with renal or severe liver impairment and when changing between formulations (oral to parenteral or transdermal, or immediate- to sustainedrelease) or between opioids (morphine to fentanyl).
Treatment of opioid-induced constipation
An unfortunate side effect of opioid use is constipation, which may be so severe as to deter the patient from using opioids. If possible, this should be pre-empted by prophylactic use of laxatives. A new use for methylnaltrexone, a peripherally-acting μ-opioid receptor antagonist, is for treatment of refractory opioid-induced constipation in patients with advanced illness such as cancer or AIDS. It can be administered SC once daily, and stimulates bowel movement in most patients within 4 hours. Pain relief is generally undiminished, as the drug preferentially blocks opioid receptors in the periphery due to poor transfer across the blood–brain barrier.
Cancer chemotherapy research
Research into the cell biology and pathology of cancer and into new agents for cancer chemotherapy is a priority area of study in medical science. Many new drugs are being developed and trialled with the hope of improving the treatment and survival of cancer patients. Some of the new ‘targets’ for anticancer drugs are discussed in Chapter 41; new drug types include TK inhibitors, inhib itors of oncogenes and their protein products, inhibitors of growth factors, activators of apoptosis, inhibitors of angiogenesis, immunostimulators and antitumour vaccines. There is also ongoing research and clinical trials on potentially protective agents including NSAIDs and COX-2 inhibitors, polyphenols, 5α-reductase inhibitors and selective oestrogen or testosterone receptor modulators.
Clinical trials of anticancer drugs
Because of their potential toxicity, antineoplastic agents are usually not tested on healthy volunteers but are fasttracked through to phase II of clinical trials. Their first use in humans may be in a small number of patients with the condition to be treated, starting with low doses, and with regular monitoring for safety and efficacy. Patients are often invited to participate in multicentre clinical trials, as cooperation between medical scientists and physicians in several oncology units will increase the numbers of patients with a particular condition able to be recruited and thus will reduce the time taken for trials to be completed and results published and implemented.
Special Access Scheme
The Special Access Scheme (SAS) is a process administered in Australia by the Therapeutic Goods Administration of the Commonwealth Department of Health and Ageing (see www.tga.gov.au/index.htm). It refers to arrangements whereby an unapproved therapeutic good (e.g. drug or device) can be imported or supplied for a single patient on a case-by-case basis. The medical practitioner must provide details of the patient, the product and the prescriber, and give clinical justification for the requirement for the product. The SAS may be applied in the case of seriously ill patients who may benefit from a new anticancer agent that is being used overseas but has not yet been approved for general use in Australia. Some drugs included in the SAS in 2010 are the antineoplastic agents aldesleukin, amsacrine and gemtuzumab; sufentanil for pain relief and cyclizine for nausea and vomiting.
Targeting drugs
The aim of all cancer treatments is for ‘selective toxicity’—i.e. killing of cancer cells but not normal cells. (This concept is discussed in Chapter 41; see review by Fuchs and Bachran [2009].) Some new ways to target cancer cells include:
Drugs targeting hypoxic cells
Tumour cells that have become hypoxic are resistant to killing by radiation therapy or by most antineoplastic drugs; this limits the success of therapy of most large solid tumours. Tirapazamine is a novel anticancer drug that targets hypoxic cells. Its mechanism of action is to be reduced in hypoxic cells to a toxic free radical that decays to oxidising free radicals that damage DNA or damage topoisomerase II enzymes. Early clinical trials of tirapazamine in lung or head and neck cancers looked promising; however, later trials showed little benefit in survival times when added to chemo- or radiation therapy. Trials are ongoing (see review by Reddy and Williamson [2009]).
Similar ‘hypoxic cytotoxins’ that are activated in hypoxic cells from prodrugs are banoxantrone and a new class of benzamide mustards. Other techniques targeting hypoxic cells include using drugs that sensitise hypoxic cells to radiation therapy (metronidazole and related compound nimorazole are being trialled), and targeting hypoxia-associated genes and proteins (see review by Bache et al [2008]).
Smart drug delivery systems
Liposomes
Another promising way of targeting drugs is the use of liposomes as a drug delivery system for lipid-soluble drugs. Liposomes are synthetic spherical vesicles consisting of one or more lipid bilayers. They are used as artificial membrane models and diagnostic agents, and as carriers to deliver drugs, vaccines, genes or imaging agents. Drugs encapsulated in a liposome capsule can be distributed differently in the body from free drugs. Liposomes accumulate at sites of inflammation and infection, as well as in some solid tumours, and can be ‘triggered’ to release their drug contents in specific cell types, by light, heat or enzymes.
Liposomes are under study for the drugs used in treatment of systemic fungal infections (amphotericin B) and for the treatment of specific cancers. Doxorubicin, cisplatin and MTX are antineoplastic agents undergoing clinical testing in liposome formulations. Doxorubicin in liposomes has been reported to deliver the drug more directly to the site of action, resulting in fewer cardiac and other adverse reactions. Cisplatin in liposomes has been reported to cause much less kidney damage than other formulations. The potential exists for liposomes to be an exciting avenue of drug delivery.
Nanoparticles
Tiny particles of liposomes, polymers, micelles or viruses at the nanometre level are being developed to direct molecules to very specific sites in the body (see Clinical Interest Box 42-4). This technology is particularly exciting in drug delivery in cancer patients, as it could reduce adverse effects in normal tissues. For example, liposomal nanoparticles small enough to be ‘internalised’ into cells (by phagocytosis or endocytosis) can be targeted to deliver nucleic acids and synthetic oligonucleotides, proteins and drugs to specific organs or to the lymphatic system. Depending on the synthetic method, the stability, half-life and size of the particle can be adjusted. Nanoparticle carriers can be made non-toxic and non-immunogenic, and even target specific cell surface receptors or carry interfering RNAs into cells to impair transcription of genes coding for ribonucleotide reductase. Clinical trials of IV-injected antineoplastic drug-nanoparticle complexes have been carried out in lung cancers, and further trials are underway (see review by Templeton [2009]).
Clinical interest Box 42-4 Nanopharmacology
The term ‘nanotechnology’ refers to the study of controlling matter at the nanometre (nm) level; a nanometre is one-billionth of a metre, or 10–9 m. One nm is about 7 or 8 carbon–carbon bond lengths, and a DNA double-helix has a diameter of about 2 nm. Thus nanotech is about matter on the scale of atoms and molecules. Very different physical properties can be expected of particles of nano-scale size, where surface tension is more important than gravity and the particles are small enough to pass readily into cells.
Nanotech has become very trendy (especially when scientists are applying for funding for research), and many nano- terms are bandied around. Nanotechnology can be applied in diverse fields, including device physics, molecular self-assembly, microscopy, development of new materials, production of energy in solar cells, machines, robots and switches operating on the molecular scale and control of atomic matter.
In the field of medicine, nanotechnology is being applied in development of microsensors, in-vivo imaging techniques, in targeting drugs to specific sites in the body (as described in text) and in preparing formulations of sunscreens with nanoparticlesize UV screening agents that do not make the skin look white.
In the future there may be nanosurgery to repair damaged tissues, neuro-electronic interfaces to allow external computer control of neuromuscular function, nano-scale artificial kidneys and even cell repair machines.
However, there are potential drawbacks in delving into this nano-realm: the toxicities of such minute particles may be very different from those of larger particles made of similar chemicals. Studies in rats have shown that inhaled nanoparticles such as carbon nanotubes can reach the brain and lungs, raising the spectre of health issues analogous to mesothelioma from inhaled minute asbestos particles; and nanoparticles in micronised sunscreens may generate reactive free radicals.
‘Nano-doomsday prophets’ warn that nanotechnology could lead to development of untraceable weapons of mass destruction and of cameras used for control by governments. Potentially damaging environmental effects are also being studied: silver nanoparticles used in socks to reduce odour can be released in the wash, with potential undesirable antimicrobial actions when the water is spread to treatment plants and oceans. There have been calls for specific regulation of nanotechnology, due to the potential harmful effects.
Complementary and alternative medicines modalities
Many people search for relief from symptoms of cancer in treatment modalities other than scientific medicine (although, as described earlier, a large proportion of anticancer agents do in fact come from or are based on natural products from plants, fungi and mammalian cells; see Clinical Interest Box 42-2). This is a worldwide phenomenon, especially in people in developed countries who are wealthy, well educated and under 50 years old. The types of complementary and alternative medicine (CAM) modalities that are popular include ‘metabolic’ therapies, diets, mega-doses of vitamins, acupuncture, electrotherapy, herbal remedies, imagery, homeopathy, spiritual methods and ‘immune’ treatments. Although some of the methods may be useful, inexpensive or harmless, many are costly and potentially toxic and have never been subjected to the rigorous scientific testing that is required of drugs before approval and marketing.
CAM in cancer
Sadly, many people with cancer rely on unproven ‘natural remedies’ until a tumour has grown so large that it is too late for conventional anticancer treatments (surgery, radiation, drugs) to be effective.
With respect to prevention of cancer, it is worth recalling that up to 80% of cancers are initiated by or exacerbated by natural causes, including carcinogens in the environment (e.g. tars and other hydrocarbons, UV radiation) or dietary or lifestyle factors (high-fat/low-fibre diets, nitrosamines, aflatoxins, cigarettes, alcohol). We cannot alter our genotype, which may endow us with oncogenes or predisposition to particular cancers, but some of the environmental factors are avoidable.
Patients often use both prescribed medical therapies and complementary and alternative therapies concurrently. It is becoming apparent that drug interactions between conventional medical treatments and CAM do occur and can have adverse effects, so it is important that patients discuss with their physicians what other remedies they are using. Some unorthodox methods tried in treatment of cancer are listed in Clinical Interest Box 42-5. Complementary and alternative techniques have also been used to treat the symptoms of cancer and the adverse effects of conventional treatments, such as pain, breathlessness, nausea and vomiting and mucositis. Relaxation training and hypnosis have been shown to improve patients’ coping skills.
Clinical interest Box 42-5 Complementary and alternative therapies in cancer
Many CAM modalities have been tried in prevention and treatment of cancer.
Other non-pharmacological or -dietary techniques tried include physical techniques (chiropractic, massage, acupuncture, yoga, exercise) and mind–body techniques (hypnotherapy, meditation, biofeedback, art therapy, music therapy, prayer).
Adapted from: Spencer & Jacobs 1999; Braun & Cohen 2007; Cassileth et al 2008.
 Key points
Key points
 Drugs used to treat cancers, i.e. antineoplastic agents, include cytotoxic agents, hormones and antihormones, immunostimulating agents and agents acting by miscellaneous mechanisms. The use of cytotoxic agents in the treatment of cancer is based on the individual drug’s effects in inhibiting macromolecular synthesis and thus interfering with cell division or replication at some point in the cell cycle. These drugs are classified according to their potential mechanisms of action: alkylating agents, antimetabolites, antibiotic antitumour agents and mitotic inhibitors. Alkylating agents such as cyclophosphamide contain reactive alkyl groups that can bind strongly to bases in DNA, thus impairing DNA replication and transcription of RNA. Antimetabolites are chemically related to folic acid (e.g. methotrexate) or to a purine or pyrimidine base (mercaptopurine, fluorouracil). They inhibit macromolecular synthesis by causing copying errors during DNA synthesis, or by inhibiting enzymes in pathways to the macromolecules DNA, RNA and proteins. Antibiotic antitumour agents include the anthracycline, bleomycin and actinomycin groups. These may bind to DNA, intercalate between DNA strands or inhibit topoisomerase II enzymes. Mitotic inhibitors such as the vinca alkaloids and taxanes are phase-specific agents, inhibiting the cell cycle during the mitosis stage. Cytotoxic drugs are usually non-selective, with antiproliferative actions on all rapidly dividing cells. They can therefore impair normal body cells that have high rates of growth, such as those in the GI tract, bone marrow and hair follicles, commonly causing mouth ulceration and GI tract dysfunction, bone marrow suppression and alopecia. Growth of hormone-dependent tumours can be inhibited by depriving the tumour of its hormone (by surgery, radiation or suppression of synthesis or release) or by use of an antagonistic hormone. Prostate cancers may be treated pharmacologically with antiandrogens (e.g. flutamide), and breast cancers with anti-oestrogens (tamoxifen), SERMs or aromatase inhibitors (exemestane). Other antineoplastic agents include platinum compounds, podophyllotoxins, topoisomerase I inhibitors (the camptothecins), the enzyme colaspase, specific tyrosine kinase inhibitors, monoclonal antibodies against specific oncogene proteins or receptors, mTOR inhibitors, vaccines and radioactive isotopes. Serious adverse drug reactions to antineoplastic agents are treated with specific methods or drugs, e.g. with powerful antiemetics or bone marrow-supportive drugs. Immune responses against neoplastic cells can be enhanced by administration of immunostimulating agents such as interferons, other cytokines, colony-stimulating factors and BCG (non-vaccine). Other adjunctive and supportive therapies include drugs to reduce uric acid load or minimise bone resorption in metastatic cancers. Palliative care involves attention to all the medical and nursing needs of the cancer patient, including physical, psychological, social and spiritual aspects; this usually requires a multidisciplinary approach.
Drugs used to treat cancers, i.e. antineoplastic agents, include cytotoxic agents, hormones and antihormones, immunostimulating agents and agents acting by miscellaneous mechanisms. The use of cytotoxic agents in the treatment of cancer is based on the individual drug’s effects in inhibiting macromolecular synthesis and thus interfering with cell division or replication at some point in the cell cycle. These drugs are classified according to their potential mechanisms of action: alkylating agents, antimetabolites, antibiotic antitumour agents and mitotic inhibitors. Alkylating agents such as cyclophosphamide contain reactive alkyl groups that can bind strongly to bases in DNA, thus impairing DNA replication and transcription of RNA. Antimetabolites are chemically related to folic acid (e.g. methotrexate) or to a purine or pyrimidine base (mercaptopurine, fluorouracil). They inhibit macromolecular synthesis by causing copying errors during DNA synthesis, or by inhibiting enzymes in pathways to the macromolecules DNA, RNA and proteins. Antibiotic antitumour agents include the anthracycline, bleomycin and actinomycin groups. These may bind to DNA, intercalate between DNA strands or inhibit topoisomerase II enzymes. Mitotic inhibitors such as the vinca alkaloids and taxanes are phase-specific agents, inhibiting the cell cycle during the mitosis stage. Cytotoxic drugs are usually non-selective, with antiproliferative actions on all rapidly dividing cells. They can therefore impair normal body cells that have high rates of growth, such as those in the GI tract, bone marrow and hair follicles, commonly causing mouth ulceration and GI tract dysfunction, bone marrow suppression and alopecia. Growth of hormone-dependent tumours can be inhibited by depriving the tumour of its hormone (by surgery, radiation or suppression of synthesis or release) or by use of an antagonistic hormone. Prostate cancers may be treated pharmacologically with antiandrogens (e.g. flutamide), and breast cancers with anti-oestrogens (tamoxifen), SERMs or aromatase inhibitors (exemestane). Other antineoplastic agents include platinum compounds, podophyllotoxins, topoisomerase I inhibitors (the camptothecins), the enzyme colaspase, specific tyrosine kinase inhibitors, monoclonal antibodies against specific oncogene proteins or receptors, mTOR inhibitors, vaccines and radioactive isotopes. Serious adverse drug reactions to antineoplastic agents are treated with specific methods or drugs, e.g. with powerful antiemetics or bone marrow-supportive drugs. Immune responses against neoplastic cells can be enhanced by administration of immunostimulating agents such as interferons, other cytokines, colony-stimulating factors and BCG (non-vaccine). Other adjunctive and supportive therapies include drugs to reduce uric acid load or minimise bone resorption in metastatic cancers. Palliative care involves attention to all the medical and nursing needs of the cancer patient, including physical, psychological, social and spiritual aspects; this usually requires a multidisciplinary approach.Review exercises
References and further reading
Australian Drug Evaluation Committee. Prescribing Medicines in Pregnancy: An Australian Categorisation of Risk of Drug Use in Pregnancy, 4th edn. Canberra: Therapeutic Goods Administration; 1999.
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Bache M., Kappler M., Said H.M., et al. Detection and specific targeting of hypoxic regions within solid tumors: current preclinical and clinical strategies. Current Medicinal Chemistry. 2008;15:322-338.
Balducci L. Supportive care in elderly cancer patients. Current Opinion in Oncology. 2009;21(4):310-317.
Braun L., Cohen M. Herbs and Natural Supplements: An Evidence-Based Guide, 2nd edn. Sydney: Elsevier Mosby; 2007.
Cassileth B., Heitzer M., Gubili J. Integrative oncology: complementary therapies in cancer care. Cancer and Chemotherapy Reviews. 2008;3(4):204-211.
Chen J., Stubbe J. Bleomycins: towards better therapeutics. Nature Reviews: Cancer. 2005;5:102-112.
Clarke S., Sharma R. Angiogenesis inhibitors in cancer: mechanisms of action. Australian Prescriber. 2006;29(1):9-12.
Dagher R., Johnson J., Williams G., Keegan P., Pazdur R. Accelerated approval of oncology products: a decade of experience. Journal of the National Cancer Institute. 2004;96(20):1500-1509.
Figlin R.A., Brown E., Armstrong A.J., et al. NCCN Task Force Report: mTOR inhibition in solid tumors. Journal of the National Comprehensive Cancer Network. 2008;6(Suppl 5):S1-S20.
Fournier P., Schirrmacher V. Randomized clinical studies of anti-tumour vaccination: state of the art in 2008. Expert Review of Vaccines. 2009;8(1):51-66.
Fuchs H., Bachran C. Targeted tumor therapies at a glance. Current Drug Targets. 2009;10(2):89-93.
Lorenz K.A., Lynn J., Dy S.M., et al. Evidence for improving palliative care at the end of life: a systematic review. Annals of Internal Medicine. 2008;148(2):147-159.
Mainwaring P. Angiogenesis inhibitors in cancer: clinical applications. Australian Prescriber. 2006;29(1):13-15.
Mann J. Murder Magic and Medicine. Oxford: Oxford University Press; 1992.
Mastini C., Martinengo C., Inghirami G., Chiarle R. Anaplastic lymphoma kinase: an oncogene for tumor vaccination. Journal of Molecular Medicine. 2009;87(7):669-677.
Medical Journal of Australia 2001; 175 [whole issue devoted to death and dying].
Michael A., Syrigos K., Pandha H. Prostate cancer chemotherapy in the era of targeted therapy. Prostate Cancer and Prostatic Diseases. 2009;12(1):13-16.
Newell D.R. How to develop a successful cancer drug: molecules to medicines or targets to treatments? European Journal of Cancer. 2005;41(5):676-682.
Osborn M., Horvath N., To L.B. New drugs for multiple myeloma. Australian Prescriber. 2009;32(4):95-98.
Palliative Care Expert Group. Therapeutic Guidelines: Palliative Care version 3. Melbourne: Therapeutic Guidelines; 2010.
Pavlakis N. Drug treatment of renal cancer. Australian Prescriber. 2006;29(6):151-153.
Peter MacCallum Cancer Institute. Standard Treatment Guidelines (Draft). Melbourne: Medical Oncology Unit, Peter MacCallum Cancer Institute; 2000.
Reddy S.B., Williamson S.K. Tirapazamine: a novel agent targeting hypoxic tumor cells. Expert Opinion on Investigational Drugs. 2009;18(1):77-87.
Segelov E. The emperor’s new clothes: can chemotherapy survive? Australian Prescriber. 2006;29(1):2-3.
Sengupta S., Jordan V.C. Selective estrogen receptor modulators as an anticancer tool: mechanisms of efficiency and resistance. Advances in Experimental Medicine and Biology. 2008;630:206-219.
Spencer J.W., Jacobs J.J. Complementary/Alternative Medicine: An Evidence-Based Approach. St Louis: Mosby; 1999.
Templeton N.S. Nonviral delivery for genomic therapy of cancer. World Journal of Surgery. 2009;33(4):685-697.
Torchilin V.P. Recent advances with liposomes as pharmaceutical carriers. Nature Reviews. Drug Discovery. 2005;4(2):145-160.
Vogel V.G., Costantino J.P., Wickerham D.L., et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. Journal of the American Medical Association. 2006;295:2727-2741.
Ward R. Antineoplastic antibodies: clinical applications. Australian Prescriber. 2003;26(6):141-143.
Watson M.S., Lucas C.F., Hoy A.M., Back I.N. Oxford Handbook of Palliative Care. Oxford: Oxford University Press; 2005.
Zhang J., Yang P.L., Gray N.S. Targeting cancer with small molecule kinase inhibitors. Nature Reviews: Cancer. 2009;9(1):28-39.
Cancer Council Australia: www.cancer.org.au/Home.htm.
New Zealand Medicines and Medical Devices Safety Authority: www.medsafe.govt.nz
More weblinks at: http://evolve.elsevier.com/AU/Bryant/pharmacology