Chapter 5 Molecular Aspects of Drug Action and Pharmacodynamics
Drugs have been one of the mainstays of therapeutics for centuries. Belief in their ‘magical’ powers has now been supplanted by scientifi c understanding of the basis of drug action. This knowledge has enabled health-care professionals to use drugs more effectively and safely and pharmaceutical companies and research scientists to develop new drugs that produce more selective effects with diminished adverse effects. An understanding of the molecular targets for drug action and of the relation between the concentration of a drug and the pharmacological response it produces underpins many aspects of the use of drugs.
Key abbreviations
cAMP cyclic adenosine monophosphate
cGMP cyclic guanosine monophosphate
FOR centuries the curative and palliative power of medicinal products was embedded in the belief that their actions were brought about by ‘magic’. Thanks to the work of Paul Ehrlich (1845–1915) and others, the myths of magical forces were dispelled and foundations were laid for the concept that the action of a drug involves a chemical interaction between the drug and a biological target.
Drugs do not confer any new functions on a tissue or organ in the body; they modify existing physiological, biochemical or biophysical functions. Their effects can be recognised by alterations to known functions or processes. For example, when an antihypertensive drug is prescribed for a person with hypertension, the health-care professional can monitor the effectiveness of the drug by repeated measurements of the person’s blood pressure. Drugs can act by combining with a small molecule (e.g. antacids neutralise gastric acid), producing an alteration of cell membrane activity (e.g. local anaesthetics) or combining with receptors (e.g. atropine reduces the rate of salivation by interacting with receptors on salivary glands). With the exception of drugs that act on DNA, all drugs act by binding to a protein, the molecular target or site of action.
Molecular targets for drug action
An ideal drug would interact with only one molecular target, at one site, and have only one effect. Such a drug would be described as having complete specificity; unfortunately, no drugs can lay claim to that title. Most drugs show selectivity: that is, they show a preference for a molecular target. Selectivity of a drug depends on its chemical structure, molecular size and electrical charge. Changes in any of these parameters can dramatically increase or decrease the binding of a drug to its molecular target, altering its therapeutic efficacy or toxicity.
An example of a non-selective drug is isoprenaline, which interacts with β1 receptors in the heart, causing tachycardia, and β2 receptors in the lungs, causing bronchodilation. In contrast, salbutamol is a selective β2 bronchodilator, and greater site (in this case tissue) selectivity is achieved when the drug is inhaled. At higher doses, salbutamol causes muscle tremor by interacting with β2 receptors in skeletal muscle—selectivity for β2 receptors is retained but tissue selectivity is lost. Other examples of selective drugs in terms of molecular targets include raloxifene, the selective oestrogen receptor modulator, the selective serotonin reuptake inhibitors (commonly referred to as SSRIs) and celecoxib, the cyclooxygenase-2 inhibitor.
To understand how drugs act we need to understand the sites at which they bind, the molecular mechanisms by which an extracellular signal alters an intracellular pathway and causes a functional change in a cell, and why under some circumstances the response to drugs decreases with time. The binding of a drug to its molecular target can occur via multiple interactions, including simple hydrogen bonding, ionic or hydrophobic interactions, van der Waals forces (these are the forces between molecules of non-polar compounds) and covalent interactions. The latter may increase the duration of drug action, e.g. acetylcholinesterase inhibitors (see Chapter 13). The strength of the interaction between a drug and its molecular target is defined as the affinity, which is measured by the dissociation constant often referred to as the KS, when discussing enzyme kinetics.
Not all drugs act in exactly the same manner but, in general, drugs act on four main types of proteins. These are called regulatory proteins because they mediate the actions of hormones, neurotransmitters and autocoids. The four types of regulatory proteins are:
Carriers
The integrity of the cell membrane is essential for normal cell physiology. Ions and small molecules that lack sufficient lipid solubility to enable them to diffuse across biological membranes must be transported. Examples of carriers include those that transport glucose and move sodium and calcium ions out of cells. Transport of organic molecules is often coupled to transport of an ion like sodium. If the movement of molecules by a carriermediated transporter is in the same direction it is referred to as a symporter (e.g. the transport of sodium/potassium/chloride in the loop of Henle of the kidney nephron). If the transport protein moves the molecules in opposite directions it is called an antiporter (e.g. the exchange of sodium and potassium in the proximal convoluted tubule in the nephron). Other important carriers include those that are involved in the uptake of chemicals acting at nerve terminals, such as noradrenaline, 5-hydroxytryptamine (5-HT, serotonin) and glutamate. These specific carriers are often targets for drugs. The tricyclic antidepressants and cocaine are examples of drugs that inhibit carriermediated uptake of noradrenaline, an important transmitter in the sympathetic nervous system (see Chapter 12).
Enzymes
Enzymes are indispensable biological catalysts that control all the biochemical reactions of the cell. A drug can inhibit the action of a specific enzyme and so alter a physiological response; for example, neostigmine combines with the enzyme acetylcholinesterase to prevent the breakdown of acetylcholine at the neuromuscular junction. This drug is used to manage myasthenia gravis, a disease characterised by muscle weakness resulting from progressive loss of acetylcholine receptors (refer to Chapter 20).
Drugs that interact with enzymes are thought to do so by virtue of their structural resemblance to an enzyme’s substrate molecule (the substance acted on by an enzyme). A drug may resemble an enzyme’s substrate so closely that the enzyme combines with the drug instead of the substrate. An example is the lipid-lowering drug simvastatin. Simvastatin closely resembles the endogenous substrate HMG-CoA, which is normally metabolised to the cholesterol precursor mevalonate by the enzyme HMG-CoA reductase. Simvastatin is an HMG-CoA reductase inhibitor. Drugs resembling enzyme substrates are often termed ‘antimetabolites’ and can either block normal enzymatic action or result in the production of other substances with different biochemical properties. An example of an antimetabolite is the anticancer drug methotrexate.
Many commonly used drugs that target enzymes are listed in Table 5-1 along with the numbers of the chapters in which the individual drugs are discussed.
Table 5-1 Common enzyme-inhibiting drugs
| Enzyme | Drugs | Chapter(s) |
| Acetylcholinesterase | Neostigmine | 13 |
| Angiotensin-converting enzyme | Captopril | 23 |
| Lisinopril | ||
| Catechol-O-methyltransferase (COMT) | Entacapone | 20 |
| Cyclo-oxygenases (COX-1 and COX-2) | Aspirin | 47 |
| Celecoxib | ||
| Ibuprofen | ||
| Dihydrofolate reductase | Methotrexate | 42, 47 |
| Trimethoprim | 44 | |
| 3-Hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase) | Atorvastatin | 24 |
| Simvastatin | ||
| Phosphodiesterase | ||
| PDE III | Milrinone | 22 |
| PDE IV | Sildenafil | 40 |
| Thymidine kinase | Aciclovir | 45 |
| Topoisomerase IV | Ciprofloxacin | 44 |
| Xanthine oxidase | Allopurinol | 47 |
Ion channels
Cell membranes are complex lipoprotein structures that regulate the flow of ions and metabolites in a highly selective manner through ion channels, thereby maintaining an electrochemical gradient between the interior and exterior of the cell. A variety of drugs target ion channels. These include the diuretic amiloride (see Chapter 25), which blocks entry of sodium into renal tubular cells, and the very large group of calcium channel-blocking drugs, such as verapamil, nifedipine and diltiazem (see Chapter 23).
Receptors
Receptors form a large group of proteins that are targets for drugs. However, the term ‘receptor’ is widely and loosely used. Some authors when referring to receptors mean any molecular drug target, for example a peptide, a membrane component, an enzyme, or even a cell or organ. Throughout this text the term ‘receptor’ refers specifically to ‘cellular macromolecules that are concerned directly and specifically in chemical signalling between and within cells. Combination of a hormone, neurotransmitter, drug or intracellular messenger with its receptor(s) initiates a change in cell function’ (International Union of Pharmacology 1998).
Structural specificity is an essential postulate of the receptor theory of drug action. In essence, a certain portion of the drug molecule selectively combines or interacts with the receptor to produce a pharmacological effect. The relationship of a drug to its receptor has often been likened to that of the fit of a key in a lock. The drug represents the key that fits into the lock, or receptor. Thus a complementary spatial relationship exists between a certain portion of the drug molecule and the receptor site.
Families of receptors
The number of receptor families and receptor subtypes within each family continues to grow and a comprehensive listing can be found in the 2nd edition of the ‘Guide to Receptors and Channels’ (Alexander et al 2006). For simplicity there are four major families of receptors.
Clinical interest Box 5-1 Drugs and the theory of receptors
John N Langley (1852–1926) first came up with the idea that drugs act on receptors when he was studying the effects of atropine and pilocarpine on saliva flow in cats. The term ‘receptor’ or ‘receptive substance’ is generally attributed to the Nobel laureate Paul Ehrlich. When studying more than 900 compounds in search of a drug to treat syphilis, he observed that many of the compounds produced antimicrobial effects with a high degree of selectivity. These observations led in 1913 to his proposal that the interaction of a drug with its receptor is akin to that of a lock and key, and that only certain drugs would fit into the receptor and activate it.
Knowledge of cell receptors is now a mainstay of pharmacology research and drug development as numerous drugs are targeted to act on specific receptors.
receptor (Chapter 17). The response time of these transmitter-gated ion channels is rapid, usually in the order of milliseconds.
Types 1–3 are found bound in the cell membrane while Type 4, the nuclear receptors that affect gene transcription (e.g. the glucocorticoid receptor), are located in the cytosol. Specific examples of the membrane-bound receptors include the nicotinic acetylcholine receptor, which is discussed in detail in Chapter 13 and is the site of action of neuromuscular blocking drugs, and the large family of guanine nucleotide protein-coupled receptors. These receptors, more commonly referred to as GPCRs, include the receptors that are perhaps more familiar to health-care professionals as muscarinic acetylcholine receptors, the receptors for adrenaline and noradrenaline (adrenoceptors), dopamine receptors, opioid and opioid-like receptors and many more. As GPCRs are important sites exploited in pharmacotherapy, a brief summary of how they influence cell functioning follows.
G-protein-coupled receptors and second messengers
Once a drug binds to a receptor, many different cellular responses can be elicited. The response depends on the mechanism of coupling of the receptor to the intracellular system that produces the functional response. GPCRs form one of the largest families of receptors on the cell surface. They consist of an extracellular (amino) terminus that projects above the membrane, seven membrane spanning helices (designated I–VII) separated by loops of varying sizes and an intracellular (carboxyl) terminus (Figure 5-1). Interaction with the receptor occurs when a ligand (e.g. a drug such as adrenaline or the transmitter acetylcholine) binds to either a cleft within the membrane-spanning regions or a ligand-binding domain located in the extracellular amino terminus.
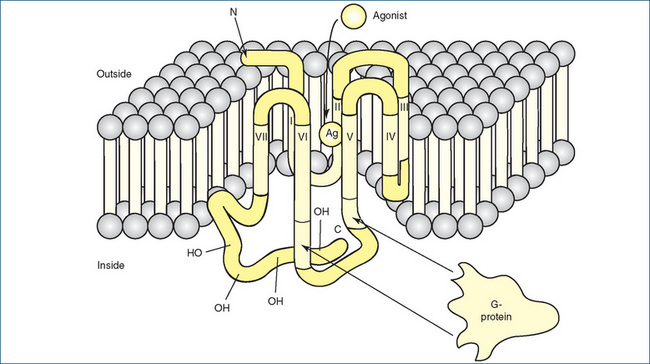
Figure 5-1 Membrane localisation of a typical G-protein-coupled receptor. The receptor’s amino (N) terminus is above the plane of the membrane and is extracellular; the carboxyl (C) terminus is located inside the membrane and is intracellular. The termini are connected by seven membrane-spanning helices (designated I–VII) that traverse the membrane and are separated by loops of varying sizes. The agonist (Ag) approaches the receptor from the extracellular fluid and binds to a site surrounded by the transmembrane regions of the receptor protein. G-proteins (G) interact with cytoplasmic regions of the receptor, especially with portions of the third cytoplasmic loop between transmembrane regions V and VI.
Source: Katzung, Basic and Clinical Pharmacology, 2007. Reproduced with permission of The McGraw-Hill Companies.
It is important to appreciate that G-proteins comprise three subunits (α, β and γ), which are essential for normal function. The guanosine nucleotides bind to the α subunit while the β and γ subunits remain together as a complex. When an agonist binds (e.g. morphine to the opiate receptor), the bound GDP dissociates from the α subunit in exchange for GTP. This leads to a change from the inactive state of a GDP-bound G-protein to an active GTP-bound G-protein. The α-GTP complex dissociates from both the receptor and the βγ complex and interacts with the effector protein, e.g. an ion channel or adenylyl cyclase. The βγ complex can also interact with a second effector protein. The G-protein remains active until GTP is hydrolysed, by the intrinsic GTPase activity of the G-protein, to GDP and the G-protein returns to its inactive GDP-bound state (Figure 5-2). Cells may express >20 GPCRs and each has a specific function. There are many different types of G-proteins and, through a series of reactions, the activated G-protein changes the activity of a second messenger specific to the type of G-protein. A simplified schema is shown in Figure 5-3.
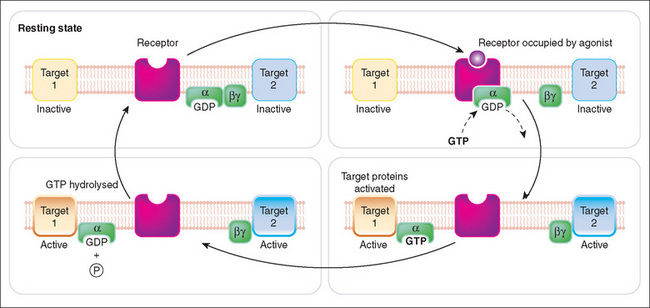
Figure 5-2 Schematic representation of activation of G-protein-coupled receptors by drugs. The second messenger systems involved include (1) cAMP, which activates various protein kinases linked to cellular functions (e.g. smooth muscle relaxation), and (2) activation of phospholipase C, which cleaves phosphatidylinositol-4,5-bisphosphate (PIP2) to form diacylglycerol (DAG), which actives protein kinase C, and inositol triphosphate (IP3), which releases intracellular calcium. ATP = adenosine triphosphate.
Source: Rang, Dale, Ritter & Flower, 2007 [Fig 3.8].

Figure 5-3 Schematic representation of activation of G-protein-coupled receptors by drugs. The second messenger systems involved include (1) cAMP, which activates various protein kinases linked to cellular functions (e.g. smooth muscle relaxation), and (2) activation of phospholipase C, which cleaves phosphatidylinositol-4, 5-bisphosphate (PIP2) to form diacylglycerol (DAG) that activates protein kinase C, and inositol triphosphate (IP3), which releases intracellular calcium. ATP = adenosine triphosphate.
Second messengers
For a cell to respond to an external stimulus (e.g. binding of a drug or hormone to a receptor), the signal has to be communicated from the exterior of the cell to the respective response elements within the cell. This mechanism of communication often involves a second messenger system, which initiates signalling within the cell through a specific biochemical pathway. The signal and the response are highly coordinated within the cell and this often involves multiple highly integrated pathways.
cAMP and cGMP
One of the most studied second messengers is cyclic adenosine monophosphate (cAMP), which is synthesised by membrane-bound adenylyl cyclase under the control of a number of GPCRs. Cyclic AMP mediates effects such as the breakdown of fat, conservation of water by the kidney and the rate and force of contraction of the heart. It exerts most of its effects through a series of protein kinases that control cell function by phosphorylating proteins (adding phosphate groups to the protein) (Figure 5-3). The breakdown of cAMP by the enzyme phosphodiesterase terminates its action. Inhibition of phosphodiesterase, which results in an increase in the intracellular con centration of cAMP and hence calcium, is one of the mechanisms by which caffeine and theophylline can produce cardiac effects. The cAMP second messenger system is linked to the action of β-adrenoceptors and many other receptors.
Another important second messenger is cyclic guanosine monophosphate (cGMP), which is involved in controlling the function of smooth muscle and nerve cells and monocytes and platelets. cGMP is formed by two distinct forms of guanylyl cyclase; the soluble form is activated to cGMP by nitric oxide (NO). NO is important in cardiovascular health and plays a role in both the autonomic and central nervous systems. The second form of guanylyl cyclase is membrane-bound and is activated by natriuretic peptides. Similar to cAMP the effects of cGMP are terminated by the phosphodiesterase enzymes. Sildenafil, a drug used to treat erectile dysfunction, inhibits phosphodiesterase 4 (PDE IV), which results in an increased concentration of NO that enhances the action of NO on penile vascular smooth muscle.
Phosphoinositides and calcium
Another well-studied second messenger system involves hydrolysis of a minor component of cell membranes, splitting it into two second messengers, diacylglycerol and inositol triphosphate (Figure 5-3). The diacylglycerol is confined to the cell membrane where it activates protein kinase C, which causes changes in the activity of other enzymes that ultimately produce the functional response (e.g. increased glandular secretions). The inositol triphosphate diffuses through the cytoplasm and causes the release of calcium from storage sites. The increased intracellular calcium then regulates the activity of other enzymes, producing a response such as increased contractility. These particular second messengers are important for producing the effects mediated by α-adrenoceptors and muscarinic receptors.
Receptor desensitisation and turnover
Receptor populations are not static and receptors may undergo several changes, including loss of responsiveness or a decrease or increase in the number of receptors. The term used clinically to describe diminished responsiveness after repeated exposure to the same concentration of the drug that stimulates the receptor is tachyphylaxis. It is rapid in onset and the individual’s initial response to the drug cannot be reproduced, even with larger doses of the drug. Transdermal glyceryl trinitrate used in the treatment of angina is an example of a drug that requires an intermittent dosing schedule (12 hours on, 12 hours off) to limit the problem of tachyphylaxis.
The term desensitisation (also referred to as adaptation or refractoriness) refers more specifically to a decrease in the response of the receptor–second messenger system and is a common feature of many receptors. The mechanisms underlying receptor desensitisation are complex and include (1) an uncoupling of the receptor from its second messenger system, (2) altered binding of the drug to the receptor and (3) a decrease in the total number of receptors.
Phosphorylation and dephosphorylation of proteins (adding or removing phosphate groups, respectively) is an important mechanism for controlling protein function. With the GPCRs uncoupling occurs when phosphorylation of the agonist-bound GPCR complex facilitates recruitment of arrestins. Arrestins, which are cytosolic proteins, uncouple the G-protein from the receptor. This can be thought of as ‘arresting’ or halting the function of the receptor. Further details on desensitisation will be provided in later chapters where relevant to specific drugs, e.g. glyceryl trinitrate (Chapter 23).
The total number of receptors in the cell membrane at any one time can change. A decrease in receptor number is called downregulation and can contribute to desensitisation and loss of response. An increase in receptor number is referred to as upregulation and can cause receptor super-sensitivity. For example, upregulation of receptors often occurs after chronic use of drugs that block receptors; when the drug is abruptly removed the person may experience increased responsiveness to stimuli (e.g. rebound hypertension).
Pharmacodynamics
Pharmacodynamics is the study of the interaction between a drug and its molecular target and of the pharmacological response: what the drug does to the body. The magnitude of a pharmacological effect depends on the concentration of a drug at its molecular target or site of action. Factors that influence this include the absorption, distribution, metabolism and excretion of the drug. These aspects are discussed in detail in Chapter 6. Binding of a drug to a receptor produces a functional response, which is governed initially by the affinity of the drug for the receptor as determined by the chemical forces that cause the drug to bind. In order to appreciate the therapeutic use of drugs, it is appropriate at this stage to consider the relationship between the concentration of the drug and the response. This relationship is commonly depicted as a dose–response curve.
Drugs that bind to a receptor are termed agonists or antagonists. An agonist binds to (occupies) and activates the receptor and produces the same response as the endogenous ligand. Examples of endogenous ligands include hormones (e.g. oestrogen), neurotransmitters such as dopamine and catecholamines (e.g. adrenaline). Some drugs are considered partial agonists as they produce less than the maximal effect even when all receptors are occupied. An antagonist (commonly called a ‘blocker’) binds to the receptor and blocks access of the endogenous ligand, thus diminishing the normal response. Drugs may act as competitive (reversible) or irreversible antagonists.
Drugs that are receptor agonists
How does knowledge of the concentration–response relationship for a drug serve a useful purpose? When an agonist drug is administered, the response usually increases in proportion to the dose until the receptors are saturated. Increasing the dose further at this stage does not produce any further increase in response. When plotted on an arithmetic scale, the relation between the concentration of the drug and the response elicited is hyperbolic (Figure 5-4). This relationship is described by the equation
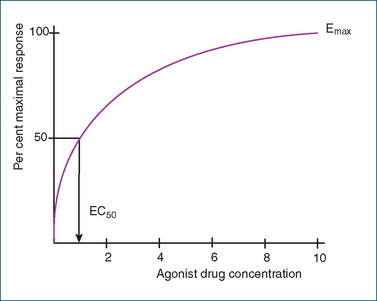
Figure 5-4 A drug concentration–response curve plotted on an arithmetic scale. The EC50 is the drug concentration at which 50% of the maximal response is observed. Emax is the maximal response when all the receptors are occupied.
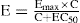
E = the effect observed at a drug concentration of C
Emax = the maximal response that the drug can produce
EC50 = the concentration at which the drug produces 50% of its maximal response.
The EC50 is an easy method for determining agonist potency and thus allows comparison of drugs in the same test system.
Drug potency
Drugs are often referred to as ‘potent’ or ‘very potent’, but what does this mean and how is it calculated? If we use the relationship described previously, then the EC50 reflects the affinity or attraction between the drug and the receptor, and is a measure of drug potency. Plotting the concentration–response data for several drugs using a semi-logarithmic scale allows us to easily determine the relative potencies of the drugs (Figure 5-5). The sigmoid shape of the curves on a logarithmic plot includes a linear portion that occurs between 20% and 80% of the maximal response. This section ‘most often applies to drugs at therapeutic concentrations and increasing drug concentration above 80% maximal response achieves very little in terms of extra therapeutic effects, but increases the risk of adverse effects’ (Birkett 2010).
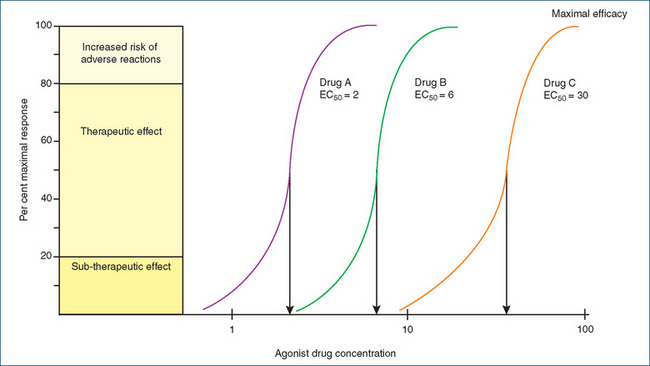
Figure 5-5 Theoretical concentration–response curves on a logarithmic scale for drugs A, B and C. The drugs are all agonists acting on the same receptor and eliciting the same response. Drug A (EC50 = 2) is three times more potent than drug B (EC50 = 6), which is five times more potent than drug C (EC50 = 30). Drugs A, B and C all differ in their potency but have the same maximal efficacy.
Maximal drug efficacy
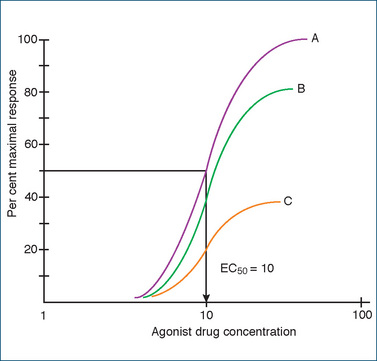
Another term that is also commonly used to describe drugs is their maximal efficacy (often simply called efficacy). Again, the concentration–response curves allow us to determine the maximal efficacy of a drug, i.e. the maximum response a drug can produce (Emax).
Several drugs may have the same potency (EC50) but differ in their efficacy (Figure 5-6). Conversely, as shown in Figure 5-5, drugs may differ in their potency but have the same maximal efficacy. This is important clinically because the effectiveness of a drug depends on its maximal efficacy and not on its potency. To illustrate this point, let us assume that the three drugs in Figure 5-6 are used as bronchodilators in the treatment of asthma. The question could be asked: Does it matter which drug is used if they are equipotent as bronchodilators? Knowing the concentration–response curves for the various drugs would provide the answer. Drugs A and B would provide a greater clinical response (bronchodilation) than drug C as they have greater efficacy. Generally drugs that are agonists possess high efficacy whereas an antagonist drug may in the simplest case have zero efficacy.
Drugs that are receptor antagonists
We also need to consider drugs that act as receptor antagonists. Drugs of this type bind to the receptor (they retain their affinity for the receptor) without eliciting a response (they have no efficacy) and prevent the binding of the endogenous agonist. An example of a receptor antagonist is propranolol, a β-adrenoceptor antagonist (commonly called a β-blocker) that blocks the action of circulating adrenaline and slows the heart rate. Antagonists can be divided into two types: those that compete with the endogenous agonist and those that bind to the receptor in an irreversible manner.
Competitive (reversible) antagonists
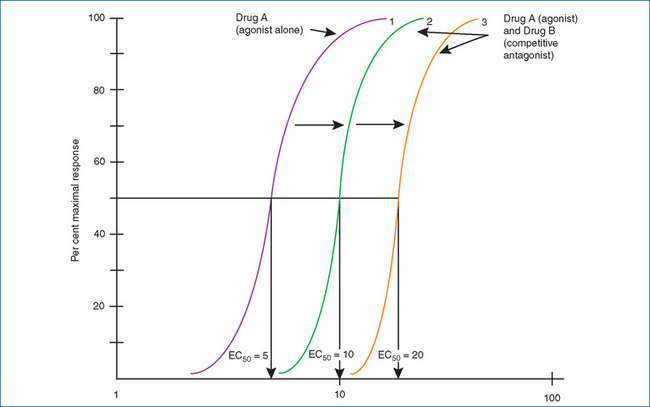
Competitive antagonists interfere with the binding of the endogenous agonist: that is, they ‘compete’. Their action can be overcome by increasing the concentration of the agonist. In essence, the agonist displaces the antagonist but the maximal response produced by the agonist does not change. On a concentration–response curve of an agonist in the presence of a competitive antagonist, the curve is shifted to the right. How far it is shifted to the right depends on the concentration of the competitive antagonist and the affinity of the antagonist for the receptor. This indicates that a much higher concentration of agonist is needed to produce 50% of the maximal response (Figure 5-7). For example, higher concentrations of adrenaline are needed to overcome the competitive blockade of β-adrenoceptors by propranolol.
Competitive (irreversible) antagonists
Competitive irreversible antagonists have limited therapeutic usefulness, as they make the target receptor permanently unavailable for binding of the endogenous agonist. This is explained by the competitive antagonist having a high affinity for the receptor and dissociating from the receptor so slowly it is in essence an ‘irreversible’ antagonist. Used experimentally to investigate receptor function, their action is usually prolonged and is not terminated until the receptors ‘die’ and are replaced by new receptors. Examples of chemicals in this class include some inhibitors of acetylcholinesterase and chemicals such as nerve gases (see Chapter 13).
Non-competitive antagonists
Non-competitive antagonists block the response to an agonist at some point within the cascade of intracellular events. In general, non-competitive antagonists reduce both the maximal response and the steepness of the slope of the dose–response curve. Examples of drugs in this category include the calcium channel blockers, which bind to the calcium channel and prevent the influx of calcium through the open channel. When another drug (agonist) binds to the calcium channel, the loss of calcium influx caused by the calcium channel blocker prevents smooth muscle contraction, which normally results from the binding of the agonist.
Another type of non-competitive antagonism can be produced when a drug binds to a second site on the target protein, which is not the main site of binding of the primary agonist. This type of drug is referred to as an allosteric antagonist. The binding of the drug at the second site produces a change in the protein either causing reduced affinity of the primary agonist or potentiating the effect of the primary agonist. An example of allosteric antagonism is the effect produced by benzodiazepines (refer to Chapter 16). Some benzodiazepines bind to the GABAA receptor causing a change that increases the affinity of the receptor for the endogenous inhibitory neurotransmitter GABA thus enhancing sedation and hypnosis.
Key points



 Drugs do not confer any new functions on a tissue or organ in the body; they modify existing physiological, biochemical or biophysical functions. With the exception of many cancer chemotherapeutic drugs that act on DNA, all drugs act by binding to proteins, which are the molecular targets or sites of action. An ideal drug would interact with only one molecular target, at one site, and have only one effect, i.e. it would be specific. Most drugs show selectivity, i.e. they show a preference for a molecular target. Selectivity of a drug for any molecular target depends on its chemical structure, molecular size and electrical charge. There are four main types of regulatory proteins that drugs act on: carriers, enzymes, ion channels and receptors. A large group of proteins that are targets for drugs are receptors, which are cellular macromolecules directly concerned with chemical signalling to initiate a change in cell function. Many receptors are coupled through G-proteins linked to second messengers that produce the functional response to agonist binding. Receptors can lose responsiveness (tachyphylaxis), become desensitised, or be downregulated or upregulated. Pharmacodynamics is the study of the interaction between a drug and its molecular target and the pharmacological response: what the drug does to the body. An agonist binds to (occupies) and activates a receptor producing the same response as the endogenous ligand. Some drugs are partial agonists as they produce less than the maximal effect even when all receptors are occupied. An antagonist binds to a receptor and blocks access of the endogenous ligand, thus diminishing the normal response. Drugs may act as competitive (reversible) or irreversible antagonists. (Drugs that are antagonists are commonly called ‘blockers’.) When a drug is administered, the response usually increases in proportion to the dose until the receptors are saturated. Increasing the dose further does not produce any further increase in response. The EC50 reflects the affinity or attraction between the drug and the receptor, and is a measure of drug potency. Competitive antagonists interfere with the binding of the endogenous agonist, i.e. they ‘compete’, and their action can be overcome by increasing the concentration of the agonist. Competitive irreversible antagonists have limited therapeutic usefulness as they bind irreversibly to the receptor, making it permanently unavailable for binding of the agonist. Their action is usually prolonged and is not terminated until the receptors are replaced by new receptors. Non-competitive antagonists block the response to an agonist at some point within the cascade of intracellular events. In general, non-competitive antagonists reduce both the maximal response and the steepness of the slope of the dose–response curve. Non-competitive antagonism can also occur when a drug binds to a second site on the target protein, which is not the main site of binding of the primary agonist. This type of drug is referred to as an allosteric antagonist. The binding of the drug at the second site produces a change in the protein either causing reduced affinity of the primary agonist or potentiating the effect of the primary agonist.
Drugs do not confer any new functions on a tissue or organ in the body; they modify existing physiological, biochemical or biophysical functions. With the exception of many cancer chemotherapeutic drugs that act on DNA, all drugs act by binding to proteins, which are the molecular targets or sites of action. An ideal drug would interact with only one molecular target, at one site, and have only one effect, i.e. it would be specific. Most drugs show selectivity, i.e. they show a preference for a molecular target. Selectivity of a drug for any molecular target depends on its chemical structure, molecular size and electrical charge. There are four main types of regulatory proteins that drugs act on: carriers, enzymes, ion channels and receptors. A large group of proteins that are targets for drugs are receptors, which are cellular macromolecules directly concerned with chemical signalling to initiate a change in cell function. Many receptors are coupled through G-proteins linked to second messengers that produce the functional response to agonist binding. Receptors can lose responsiveness (tachyphylaxis), become desensitised, or be downregulated or upregulated. Pharmacodynamics is the study of the interaction between a drug and its molecular target and the pharmacological response: what the drug does to the body. An agonist binds to (occupies) and activates a receptor producing the same response as the endogenous ligand. Some drugs are partial agonists as they produce less than the maximal effect even when all receptors are occupied. An antagonist binds to a receptor and blocks access of the endogenous ligand, thus diminishing the normal response. Drugs may act as competitive (reversible) or irreversible antagonists. (Drugs that are antagonists are commonly called ‘blockers’.) When a drug is administered, the response usually increases in proportion to the dose until the receptors are saturated. Increasing the dose further does not produce any further increase in response. The EC50 reflects the affinity or attraction between the drug and the receptor, and is a measure of drug potency. Competitive antagonists interfere with the binding of the endogenous agonist, i.e. they ‘compete’, and their action can be overcome by increasing the concentration of the agonist. Competitive irreversible antagonists have limited therapeutic usefulness as they bind irreversibly to the receptor, making it permanently unavailable for binding of the agonist. Their action is usually prolonged and is not terminated until the receptors are replaced by new receptors. Non-competitive antagonists block the response to an agonist at some point within the cascade of intracellular events. In general, non-competitive antagonists reduce both the maximal response and the steepness of the slope of the dose–response curve. Non-competitive antagonism can also occur when a drug binds to a second site on the target protein, which is not the main site of binding of the primary agonist. This type of drug is referred to as an allosteric antagonist. The binding of the drug at the second site produces a change in the protein either causing reduced affinity of the primary agonist or potentiating the effect of the primary agonist.Review exercises
References and further reading
Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels, 2nd edition. British Journal of Pharmacology 2006; 147 (Supplement 3), S1-S180.
Baillie G.S., Houslay M.D. Arrestin times for compartmentalized cAMP signaling and phosphodiesterase 4 enzymes. Current Opinion in Cell Biology. 2005;17:1-6.
Birkett D.J. Pharmacokinetics Made Easy, 2nd edn. Sydney: McGraw-Hill; 2010.
Costa T., Cotecchia S. Historical review: negative efficacy and constitutive activity of G-protein-coupled receptors. Trends in Pharmacological Sciences. 2005;26:618-624.
International Union of Pharmacology. The IUPHAR Compendium of Receptor Characterization and Classification. Foxton, UK: The International Union of Pharmacology, IUPHAR Media Ltd, Burlington Press, 1998.
Katzung B.G., editor. Basic and Clinical Pharmacology, 10th edn., New York: The McGraw-Hill Companies, Inc, 2007. [ch 2]
Lefkowitz R.J., Pitcher J., Krueger K., et al. Mechanism of β-adrenergic receptor desensitization and resensitization. Advances in Pharmacology. 1998;42:416-420.
Nahorski S.R. Pharmacology of intracellular signalling pathways. British Journal of Pharmacology. 2006;147(Suppl):38-45.
Rang H.P., Dale M.M., Ritter J.M., Flower R.J. Pharmacology, 6th edn. Edinburgh: Churchill Livingstone; 2007. [ch 2]
Rang H.P., Dale M.M., Ritter J.M., Flower R.J. Pharmacology, 6th edn. Edinburgh: Churchill Livingstone; 2007. [ch 3]
Spiegel A.M., Weinstein L.S. Inherited diseases involving G proteins and G-protein-coupled receptors. Annual Review of Medicine. 2004;55:27-39.