Central Consequences of Peripheral Nerve Damage
Introduction
In the first Textbook of Pain, edited by Patrick Wall and Ronald Melzack and published in 1984, basic aspects of the relationship between damage to a peripheral nerve and pain were discussed by Marshall Devor (1984), as well as in several clinically oriented chapters, including one by John Scadding (1984) on peripheral neuropathies. Devor’s chapter dealt with nerve injury and focused on the pathophysiology and anatomy of the damaged nerve, in particular, on processes leading to the production of abnormal impulse discharges and pain. In the mid-1970s, Wall and Gutnick (1974) reported massive spontaneous discharges in the L4 and L5 dorsal rootlets after producing an experimental neuroma by creating a peripheral lesion of the sciatic nerve. Govrin-Lippmann and Devor (1978) and Scadding (1981) described the time course: a lack of discharges during the first few days followed by spontaneous high activity for several weeks. It was proposed that the nerve damage and neuroma lead to the establishment of ectopically generated abnormal impulses and amplification of impulse discharges and that this effect contributes to chronic neuropathic pain.
At that time, little was known about the dramatic and robust chemical changes occurring in the dorsal root ganglia (DRGs) after peripheral nerve damage, which presumably contributed to the changes in electrical phenomena monitored by Wall, Devor, and their collaborators, as well as others. In this chapter we focus on such chemical changes induced in DRG neurons by nerve injury, including changes in intracellular messengers and their receptors, enzymes, ion channels, and other molecules. We also discuss the issue of nerve injury–induced sprouting of primary afferents in the dorsal horn.
Much of the earlier work was carried out on rats after complete transection of the sciatic nerve, the model introduced by Wall and collaborators, which resulted in robust and reproducible effects. Subsequently, several other “functional” pain models were introduced (see Chapters 11 and 65). Nahin and colleagues (1994) were the first to show changes in peptide expression in DRGs in the chronic nerve constriction model (four loose ligatures) developed by Bennett and Xie; these changes are similar to those seen after complete nerve transection (see Nahin et al 1994). The purpose and consequences of these changes are still not well understood, but they may contribute to survival and regeneration of the damaged neuron and also to generation and/or attenuation of pain. It is, however, clear that these changes are not confined to neurons but affect satellite and other non-neuronal cells in the DRG as well. They also extend to the spinal cord as trans-ganglionic effects, which we will discuss with regard to functional, anatomical, and chemical consequences. However, we will not deal with events occurring at higher centers (see Chapter 7). It may be anticipated that more complete comprehension of these processes may lead to improved understanding of and ultimately novel treatment strategies for chronic pain. This topic has been dealt with in many reviews, and a few published after the fifth edition of Textbook of Pain may be mentioned (Basbaum et al 2009, Costigan et al 2009) (see also Chapters 5, 6, and 61).
Gene Expression in Dorsal Root Ganglia After Nerve Injury
Sixty years ago, Fred Lembeck in Graz suggested that substance P, an 11–amino acid peptide, could be a transmitter in sensory neurons. The presence of this and several other peptides—for example, calcitonin gene-related peptide (CGRP), somatostatin, and pituitary adenylate cyclase–activating peptide—in subpopulations of normal DRGs in rats and other species could subsequently be demonstrated.
Early evidence that nerve injury could influence the expression of messenger molecules in DRGs was presented by Jessell, Otsuka, and associates, who showed that peripheral transection of the sciatic nerve causes a decrease in substance P levels in the dorsal horn. It could later be demonstrated that this decrease in fact represented down-regulation of substance P synthesis in DRG neurons (Nielsch et al 1987). Evidence of nerve injury–induced messenger up-regulation in DRG neurons was first obtained for vasoactive intestinal polypeptide (VIP) by Shehab and Atkinson (1986). Subsequently, it has turned out that a very large number of molecules—including several neuropeptides such as substance P, CGRP, neuropeptide Y (NPY), galanin, and pituitary adenylate cyclase–activating peptide (Fig. 63-1A and B); enzymes such as nitric oxide synthase; and receptors for peptide and classic transmitters synthesized in DRG neurons—are regulated by axotomy (see Hökfelt et al 1994, Zigmond et al 1996). This is also seen in several neuropathic pain models without complete transection.
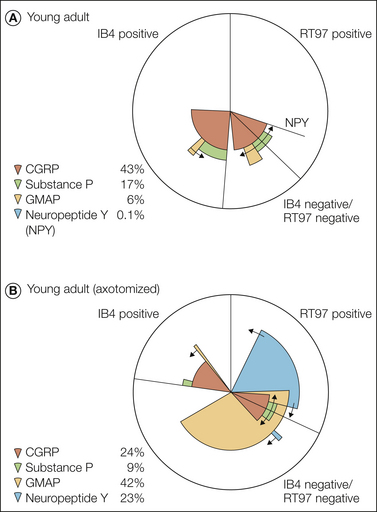
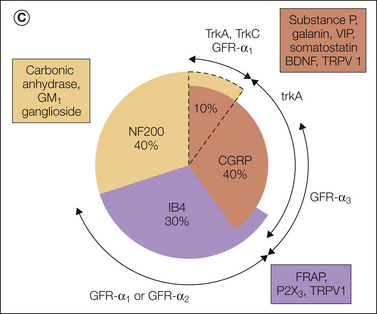
Figure 63-1 Pie charts summarizing the expression pattern of several neuropeptides, nitric oxide synthase, and GAP-43 in a subpopulation of dorsal root ganglion (DRG) neurons in non-lesioned (A) and axotomized (B) young adult rats, as well as in the main neurochemical populations of DRG neurons (C) as defined by markers for large myelinated neurons (RT97) and the small unmyelinated or thinly myelinated axons, predominantly nociceptors, that make up the two populations (isolectin B4 [IB4] positive and calcitonin gene–related peptide [CGRP] positive) and their relationship to several growth factor receptors. BDNF, brain-derived neurotrophic factor; FRAP, fluoride-resistant acid phosphatase; GFR-α2, glial cell line-neurotrophic receptor α; GMAP, galanin message-associated peptide; NF200, neurofilament 200; P2X3, purinoceptor 2X3; TRPV1, transient receptor potential vanilloid 1; VIP, vasoactive intestinal polypeptide. (A and B, From Bergman E, Johnson H, Zhang X, et al 1996 Neuropeptides and neurotrophin receptor mRNAs in primary sensory neurons of aged rats. Journal of Comparative Neurology 375:303–319; C, from Priestley JV, Michael GJ, Averill S, et al 2002 Regulation of nociceptive neurons by nerve growth factor and glial cell line derived neurotrophic factor. Canadian Journal of Physiology and Pharmacology 80:495–505.)
Importantly, in inflammatory models substance P and CGRP are up-regulated in DRGs, but no distinct effects on galanin, VIP, or NPY have been recorded. Instead, inflammation activates opioid and other peptides in the dorsal horn (see Dubner and Ruda 1992).
The DRG neurons discussed thus far are often termed the peptide (-positive) population because of their expression of several peptides. They represent 35–40% of all DRG neurons, have unmyelinated or thinly myelinated axons, and are assumed to be involved in nociception.
The non-peptide DRG neurons represent a second class (McMahon and Priestley 2005). They account for 20–30% of the DRG neurons and are characterized by several markers, such as the non-lysosomal fluoride-resistant acid phosphatase, the P2X3 purinoceptor, the vanilloid receptor 1 (VR1, now called transient receptor potential vanilloid 1 [TRPV1]), and receptor components of glial cell line–derived neurotrophic factor (GDNF), especially RET mRNA. They bind the lectin Griffonia simplicifolia isolectin B4 (IB4). Some of these markers, such as TRPV1 and P2X3, are up-regulated after peripheral nerve injury, whereas others are down-regulated. These neurons are also small with non-myelinated axons and are assumed to represent nociceptors.
The remaining 30–40% of DRG neurons are large and medium sized. These neurons have fast-conducting, myelinated Aα/β-range axons and receive input from peripheral mechanoreceptors.
It has long been assumed that glutamate is the principal transmitter in DRG neurons. However, only after the three vesicular glutamate transporters (VGLUT1–3) had been discovered was it possible to identify, with certainty, glutamatergic neurons with the microscope (see Fremeau et al 2004). In fact, with this approach it now seems that in rodents all DRG neurons express one or more VGLUTs (Landry et al 2004, Morris et al 2005, Brumovsky et al 2007b), thus confirming the general view that they use glutamate as a transmitter. Axotomy causes a reduction in VGLUT1 and -2 in DRG neurons, although a population of small neurons show increased staining (Brumovsky et al 2007b).
For a comprehensive account of neuron types, their receptors, and ion channels expressed in DRGs (and examples of their regulation), we refer readers to a review by McMahon and Priestley (2005).
Some Methodological Aspects
Many early nerve injury studies are based on complete transection of the sciatic nerve of the rat at the mid-thigh level, and in general, percentages of DRG neuron profiles expressing a particular molecule (immunohistochemistry) or a transcript (in situ hybridization) are calculated. It is important to note that this lesion will affect only approximately 70–80% of the L5 DRG neurons projecting into the sciatic nerve because the remaining ones branch off central to the transection (Devor et al 1985). Moreover, an important issue is to what extent the nerve injury causes cell loss, which has not been monitored in most studies. More recently, stereological techniques have shown that in the rat no significant loss of neurons occurs up until 4 weeks after a lesion at the mid-thigh level (Tandrup et al 2000, but see McKay Hart et al 2002) whereas a spinal nerve lesion (close to the cell bodies) causes a progressive loss of neuronal cells (Vestergaard et al 1997). In the mouse, axotomy at the mid-thigh level causes a 24% loss after 7 days and a 50% loss after 28 days (Shi et al 2001), possibly because of the short distance between the lesion and cell bodies in this small animal. To what extent neurogenesis occurs in DRGs, particularly after nerve injury, has not been established. For animal pain models, see Chapter 11.
Global Gene Expression Studies
Global gene expression has been analyzed with array techniques to monitor genes expressed in DRGs after peripheral nerve injury versus those expressed in non-lesioned ganglia (Costigan et al 2002, Wang et al 2002, Xiao et al 2002). In these analyses many different types of genes show marked changes (Table 63-1).
Table 63-1
Strongly Regulated Genes with More Than Two-Fold Changes in the cDNA Array Ratio 2, 7, 14, and 28 Days after Peripheral Axotomy
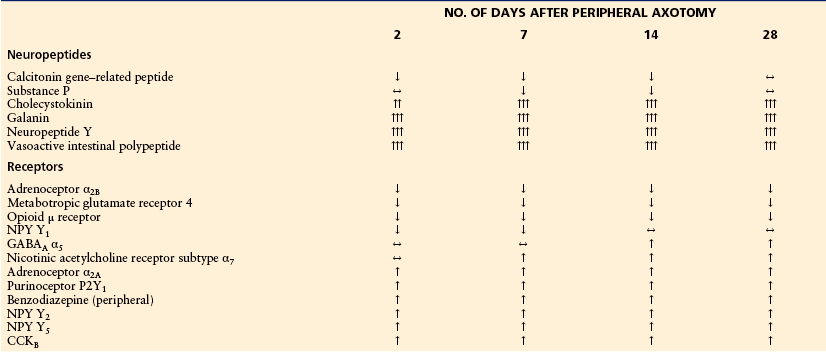

Changes are indicated by arrows: ↑↑↑ = >10-fold; ↑↑ = 10–5-fold; ↑ = 5–2-fold; ↔ = 2–0.5-fold; ↓ = 0.5–0.2-fold; ↓↓ = <0.2-fold. ATPase, adenosine triphosphatase; GABA, γ-aminobutyric acid; NPY, neuropeptide Y. Numbers indicate the cDNA array ratio of normalized volume (nVol) of axotomized dorsal root ganglia (DRG)/control DRG.
From Xiao HS, Huang QH, Zhang FX, et al 2002 Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America 99:8360–8365, with permission of the National Academy of Sciences of the United States of America.
Costigan and co-workers (2002) found changes in 178 or 240 genes (depending on the cutoff) after axotomy. Wang and associates (2002) showed greater than two-fold up-regulation of 102 genes and greater than two-fold down-regulation of 46 genes in a spinal nerve ligation neuropathic pain model. As shown in Figure 63-2, approximately 2.3 and 2.8% of the genes have been down- and up-regulated, respectively, if genes with a more than 1.5-fold regulation and P < 0.05 are considered. The genes have been categorized as indicated in Figure 63-2, and bidirectional changes are found in all classes except for translational regulation genes, where no down-regulation was observed. In many cases, approximately the same number of genes go in either direction, although up-regulation dominates in the categories apoptosis, cytoskeleton, and immunologically related genes. Down-regulation prevails in the categories ion channels, neurotransmission, vesicle trafficking, and unknown genes.
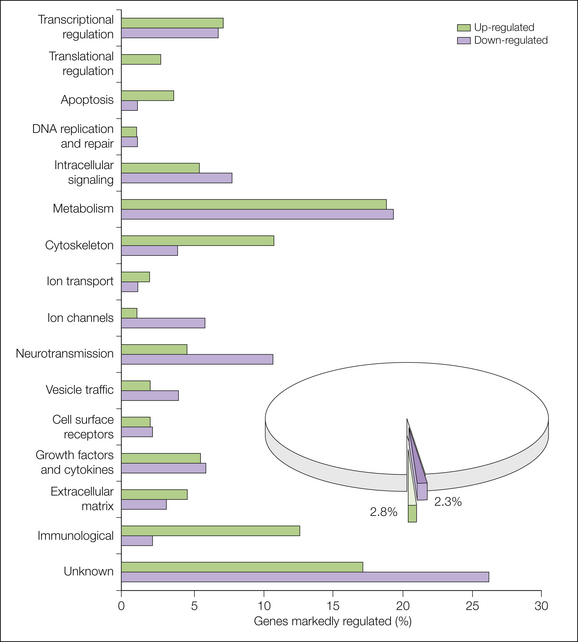
Figure 63-2 Genes regulated above 1.5-fold (P > 0.05) have been classified into functional classes, and the percentage of up- and down-regulated genes are shown for each functional class. (From Costigan M, Befort K, Karchewski L, et al 2002 Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neuroscience 3:16.)
Xiao’s group (2002) examined 7523 genes and sequence tags expressed after axotomy, of which 122 and 51, respectively, were strongly changed (see Table 63-1). In a similar manner as the Costigan study (2002), several families were identified, although under somewhat different headings and confined to 10 classes (Table 63-1).
Xiao and colleagues used a two-fold change in signal intensity as a cutoff line. They identified 122 genes and 51 expressed sequence tags, which represented 2.3% of the total genes examined. Among these the vast majority (86%) had not previously been identified in DRGs after nerve injury (Xiao et al 2002). Thus, these array studies have not only provided evidence of regulatory changes but also showed the presence of a number of genes hitherto not identified in DRGs. Interestingly, the changes are long lasting, often throughout the entire period studied (28 days) (see Table 63-1). It is known from other studies that changes in expression can last for long periods if regeneration is efficiently prevented. If not, regeneration will take approximately a month in the rat after complete mid-thigh transection, and most changes then revert to normal. The significance of some of these results has been discussed by Xiao and co-authors (2002, and references therein) and is summarized here.
Not unexpectedly, neuropeptides belonged to the group of compounds showing the most dramatic changes, particularly with regard to up-regulation (see Table 63-1). One reason is that neuropeptides, in contrast to enzymes and other proteins, are released from the neuron and have to be replaced via ribosomal synthesis in the cell soma. This is reflected by distinct changes in peptide and, especially, mRNA levels and can be conveniently recorded in single cells by in situ hybridization or immunohistochemistry. In contrast, most other messenger molecules, such as the classic transmitters (e.g., acetylcholine and noradrenaline), are synthesized by enzymes in all parts of neurons, and efficient transporter molecules allow reuptake and recycling of the transmitter. Down-regulation of excitatory peptides (substance P and CGRP) will attenuate transmission, and some up-regulated peptides (VIP, galanin, and NPY) affect transmission but in addition improve survival and regeneration (see, for example, Hobson et al 2008).
Compounds of importance for survival—such as heat shock protein 27, endoplasmic reticular stress protein, and the LIM domain protein CLP36—are transiently up-regulated. In addition, the transcription factor Jun-D and eukaryotic initiation factor 4E, a translation initiation factor, were up-regulated. Interestingly, two important growth factors, brain-derived neurotrophic factor (BDNF) and glial cell line–derived neurotrophic factor (GDNF), were increased less than twofold. However, basic fibroblast growth factor was strongly up-regulated.
With regard to synaptic transmission, most molecules were down-regulated, and only synaptotagmin IV showed an increase. However, synaptotagmin IV can form hetero-oligomers with synaptotagmin I, and this results in less efficient Ca2+ coupling. Thus, in general, the changes in synaptic vesicle proteins tend to work toward attenuation of transmission in the dorsal horn.
The expression of receptors for messenger molecules is often changed. For example, several of the neuropeptide receptors—such as the cholecystokinin B receptor (CCKB, CCK2) and the NPY Y2 and Y5 receptors, as well as a nicotinic acetylcholine receptor subtype (α7), a purinoceptor (P2Y1), the α2A-adrenergic receptor, and the benzodiazepine receptor of the peripheral type—were up-regulated, whereas others were down-regulated. Clearly, it is a difficult puzzle to understand the functional significance of these complex regulations.
Peripheral nerve injury also causes molecular modification in the dorsal horn of the spinal cord. Using a cDNA array, Yang and colleagues (2004) found that expression of the genes encoding 14 channels, 25 receptors, and 42 signal transduction–related molecules is strongly regulated in the spinal dorsal horn following peripheral axotomy. Although knowledge about these regulated genes and the related functional consequences is very limited, current findings suggest that novel signal pathways may be generated in the dorsal spinal cord after peripheral nerve injury. For example, there is a significant increase in the expression of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (Yang et al 2004), which may be accessible to the elevated levels of glutamate resulting from the decrease in glutamate uptake (Sung et al 2003). These changes may be involved in mechanisms underlying the increased neuronal sensitivity after nerve injury (Leem et al 1996).
Classic Growth Factors
Classic growth factors play a major role in the development, physiology, and pathophysiology of DRGs and sensory mechanisms. They are often target derived and therefore localized in non-neuronal cells, whereas their tyrosine kinase (Trk) receptors (TrkA, TrkB, and TrkC) are often present in neurons. Many studies have been devoted to analysis of these growth factors in non-neuronal cells (including satellite and Schwann cells) and their receptors under normal circumstances and after injury, but this topic is not discussed here (but see Del Fiacco and Priestley 2001). However, we will address BDNF and its reaction to nerve injury and inflammation because it is neuronally localized. (GDNF and nerve growth factor [NGF] have, however, been suggested to have a neuronal localization in some studies.)
Under normal circumstances, BDNF is constitutively expressed in 11–38% of DRG neurons, mainly in small TrkA-positive neurons (Wetmore and Olson 1995; Zhou and Rush 1996; Cho et al 1997, 1998; Kashiba and Senba 1999; Mannion et al 1999; Michael et al 1999). Axotomy of a peripheral nerve remote from the cell body induces up-regulation of BDNF mRNA, mainly in medium-sized and large-diameter neurons expressing TrkB and TrkC, that is, NGF-insensitive neurons (Cho et al 1998, Kashiba and Senba 1999, Michael et al 1999, Fukuoka et al 2001). NPY is up-regulated in the same neurons (Kashiba and Senba 1999, Michael et al 1999). BDNF expression in most TrkA cells was, however, unchanged (Michael et al 1997).
In contrast, spinal nerve ligation increases BDNF expression mainly in small-sized neurons (Fukuoka et al 2001), thus showing clear differential regulation depending on the type of nerve injury. Peripheral tissue inflammation increases BDNF mRNA in DRG neurons (Cho et al 1997), whereas capsaicin causes a marked down-regulation (Kashiba and Senba 1999). Interestingly, BDNF is packaged in large dense-core vesicles (the same vesicles that store neuropeptides) and then transported into the dorsal horn of the spinal cord (Michael et al 1999), which suggests that it can be released and control spinal cord excitability in a transmitter-like fashion (see Thompson et al 1999). In Figure 63-1C, the relationship of several growth factor receptors and other molecules to some major DRG neuron markers (CGRP, IB4, and neurofilament 200 [NF200]) has been summarized.
Species Differences
Most studies on the effect of nerve injury on the expression of various molecules in DRGs have been done on rats, but mice are increasingly also being analyzed and have demonstrated no obvious qualitative differences. However, in other species, differences have been encountered (see Hökfelt et al 1997). Thus, in the guinea pig, up-regulation of galanin is much more restricted than in the rat and mouse, whereas NPY regulation is similar. In addition, up-regulation of galanin is very modest in the cat. Up-regulation of galanin is strong in the monkey (Macaca mulatta), whereas NPY expression is hardly detectable. In human ganglia, galanin is expressed in some 10–15% of the DRG neuron profiles versus around 40–50% of CGRP-positive neurons (Landry et al 2003), a proportion that is similar to that found in the monkey. Thus, galaninergic mechanisms may exist at the spinal level in humans as in certain rodents and M. mulatta.
Role of Voltage-Gated Sodium Channels
Voltage-gated sodium channels in DRG neurons play a crucial role in chronic pain. Many subtypes of sodium channels are localized in DRG neurons (see McMahon and Priestley 2005). Based on their sensitivity to tetrodotoxin (TTX), these sodium channels are classified as TTX-sensitive or TTX-resistant subtypes. Both electrophysiological experiments and pain behavior tests show that the TTX-sensitive subtypes of sodium channels play important roles in generating ectopic discharges in injured sensory neurons and in maintaining allodynic behavior in animal models of neuropathic pain (Lyu et al 2000, Liu et al 2001). Moreover, following peripheral nerve injury, expression of the TTX-sensitive sodium channels Nav1.3 and Nav2 is up-regulated in primary sensory neurons, whereas Nav1.1, Nav1.2, Nav1.6, Nav1.7, Nav1.8, and Nav1.9 are down-regulated (Baker and Wood 2001, Kim et al 2002, Xiao et al 2002, Lai et al 2003). These findings suggest that TTX-sensitive sodium channels, especially Nav1.3 and some other sodium channels, are potentially important in generating and maintaining neuropathic pain.
Mechanisms Underlying Injury-Induced Phenotypic Changes
The mechanisms underlying up- and down-regulation of the various molecules in sensory ganglia are only incompletely understood. In several cases, classic neurotrophic factors play a role (see Priestley et al 2002, McMahon and Priestley 2005). A common denominator does not seem to exist, and every molecule may have its own regulatory pathway. These problems have been analyzed in vivo in DRGs for substance P, CGRP, and galanin, whereas knowledge of VIP and NPY mainly stems from in vitro work and studies on cell lines. In the case of the distinct down-regulation of the two excitatory peptides substance P and CGRP, a clear mechanism has, however, been identified. Thus it was early shown that NGF is essential for the expression of these two peptides and that axotomy-induced down-regulation can be reversed by the administration of exogenous NGF (see Del Fiacco and Priestley 2001). The nerve transection interrupts the centripetal transport of peripheral, target-derived NGF to the cell body; even if the injury increases NGF production in Schwann cells and fibroblasts around the injury site, this cannot compensate for the loss of NGF produced in the skin (see Del Fiacco and Priestley 2001).
There is some evidence of the mechanisms underlying axotomy-induced regulation of galanin. Galanin up-regulation is markedly impaired in mice lacking the gene for leukemia inhibitory factor (LIF) (Rao et al 1993). LIF is produced in DRG neurons during development and is up-regulated in Schwann cells after nerve injury (Murphy et al 1993, Del Fiacco and Priestley 2001). Also, NGF may play a role by having antagonistic action on LIF induction. The nerve injury–induced inhibition of NGF transport to the cell bodies may stimulate the synthesis of LIF and thus enhance galanin production. In addition, the cytokine interleukin-6 (IL-6) may participate, because IL-6 transcript levels increase in DRG neurons and Schwann cells after nerve injury and galanin up-regulation is impaired in IL-6 knockout mice (Murphy et al 1999). It was therefore suggested that the effect of LIF on galanin synthesis is mediated through IL-6 (Murphy et al 1999).
In the case of VIP, focus has been on ciliary neurotrophic factor, a member of the neuropoietic cytokine family just like LIF (see Pitts et al 2001 and references therein). Ciliary neurotrophic factor and fibroblast-transforming growth factor-β (FGF-β) induce transcription of VIP through a 180–base pair cytokine response element in the VIP promoter. These molecules act synergistically, with ciliary neurotrophic factor inducing signal transducer and activator of transcription (STAT)- and small (body size) mothers against decapentaplegic (Smad)-containing complexes.
With regard to the NPY gene, both NGF and BDNF stimulate the NPY promoter (Minth-Worby 1994, Wernersson et al 1998, Williams et al 1998 and references therein). In vivo studies suggest that members of the FGF family can attenuate NPY up-regulation in large DRG neurons, in agreement with the presence of these growth factors and their receptors on DRG neurons and associated cells (Oellig et al 1995, Grothe et al 2001).
Subsequently, focus turned to GDNF and related molecules, and it was demonstrated that they not only partially or completely reverse many of the nerve injury–induced morphological and neurochemical changes described earlier but also block the associated neuropathic pain state (Gardell et al 2003, Wang et al 2003). These findings open up new venues for understanding the mechanisms underlying neuropathic pain and its treatment.
Role of the DRG Neuron Cell Soma
Our view of the role of the soma of DRG neurons is at present expanding. As discussed by Devor (1984) in the first edition of this textbook, this part of the neuron was not supposed to be involved in signal generation or processing but should be responsible for the metabolic demands of neurons. However, DRG neurons may discharge spontaneously because of high levels of excitability, and such discharge is increased by peripheral nerve injury (see Devor 1984). Moreover, the DRG somata are not innervated; that is, they do not have nerve endings synapsing onto them, in contrast to sympathetic ganglion neurons and most other neurons, but evidence of chemically mediated cross-excitation in rat DRGs has been reported (Amir and Devor 1996).
There is both immunohistochemical and electrophysiological evidence of functional peptide receptors on the DRG soma membrane. For example, NPY (Abdulla and Smith 1999), substance P (Abdulla et al 2001), and galanin (Kerekes et al 2003) all distinctly modulate membrane excitability after micropipette application, thus providing further evidence of a more active role of the DRG soma. Moreover, the demonstration that neuropeptides can be released from the DRG soma (Huang and Neher 1996) and from neuronal soma and dendrites in general (see Morris et al 1993, Ludwig and Pittman 2003) strongly indicates the possibility that signaling molecules can be released intraganglionically and control membrane excitability and neuronal activity, perhaps in particular after nerve injury, when the levels of some peptides are dramatically increased. Kerekes and colleagues (2003) recorded the effects on electrical activity of DRG neurons acutely dissociated with galanin at concentrations as low as 10−14 M. It does not seem unreasonable to assume that such peptide concentrations can be achieved around cell bodies when they discharge spontaneously after nerve injury.
Functional Consequences of Altered Gene Expression in the DRG
The significance of the dramatic changes in gene expression is only very incompletely understood, not least because many of them have been identified fairly recently. However, in addition to the overview already presented (under Global Gene Expression Studies), here we would like to expand on the significance of some of the profound changes in neuropeptide expression, in particular, that of galanin and NPY, because of the large body of evidence that has been collected over the past 20 years or so.
Because peptides penetrate the blood–brain barrier only to a very limited extent and because, at least for galanin, hardly any non-peptide agonists or antagonists have been developed, these types of molecules have to be delivered intrathecally, a route originally described by Yaksh and Rudy. Woolf and Wiesenfeld-Hallin (1986) subsequently used this intrathecal approach in conjunction with the (nociceptive) flexor reflex model introduced by Wall and Woolf for studies on CGRP. The role of CGRP and substance P as excitatory messengers is generally accepted, as is the idea that their down-regulation in DRGs after nerve injury attenuates transmission in the dorsal horn.
Under normal circumstances, galanin is detectable in only a small number of DRG neurons, mainly small ones. After axotomy, up to 50% express the peptide, again mainly small neurons, but also some medium-sized and large ones; it seems likely that this up-regulation occurs in neurons that normally produce CGRP (Villar et al 1989).
Early on, intrathecal application of galanin was shown to have a biphasic effect on the flexor reflex—facilitation at low and depression at high concentrations—with enhancement of the inhibitory effect after sciatic nerve section, thus suggesting both pro- and antinociceptive effects. Meanwhile, three galanin receptors have been identified (GalR1–R3) (see Branchek et al 2000), and it is clear that GalR1 and GalR2 exhibit robust expression in rat DRGs and that GalR1 in addition is present in interneurons in the superficial dorsal horn (see O’Donnell et al 2003). Early work suggested that up-regulated galanin may serve to inhibit the increased nociceptive input after nerve injury, and further studies using a partial nerve injury model or genetically modified mice have largely supported this notion (see Wiesenfeld-Hallin and Xu 2001, Liu and Hökfelt 2002, Wiesenfeld-Hallin et al 2005, Xu et al 2008). A reasonable conclusion at the moment, based on a long series of studies in our and other laboratories, seems to be that the pro-nociceptive effect of galanin is mediated via GalR2 located on DRG neurons whereas the antinociceptive effect is exerted via GalR1 on dorsal horn interneurons (Liu and Hökfelt 2002, Alier et al 2008).
With regard to NPY, the situation appears even more complex. NPY can normally be detected only in single DRG neurons, is up-regulated by nerve injury in some 25% of DRG neurons, almost exclusively the large ones (Wakisaka et al 1991), and is present in numerous local dorsal horn neurons. Intrathecal injection of NPY in normal rats produces an antinociceptive action (Hua et al 1991), but biphasic responses have been recorded in the flexor reflex model (Xu et al 1999), and White (1997) showed a pro-nociceptive effect of NPY. In mice with a genetically deleted NPY Y1 receptor, the pain threshold is strongly reduced (Naveilhan et al 2001), thus suggesting a tonic antinociceptive role for this receptor (but note that the receptor is deleted in the entire animal). Furthermore, the NPY receptors show complex regulation after axotomy; the Y1 receptor, normally present in small DRG neurons, is markedly down-regulated, and the Y2 receptor, normally present in small DRG neurons, is strongly up-regulated in large neurons (Zhang et al 1997). Y1 receptors are also present on dorsal horn neurons. Thus, NPY at the spinal level, released either from local dorsal horn neurons, perhaps even from descending systems, or from large DRG neurons (after nerve injury), could be involved in controlling the pain threshold by acting at at least five sites (Brumovsky et al 2007a):
In terms of combating pain there are several options: (1) GalR1 and NPY Y1 agonists acting in the spinal cord and (2) GalR2 and NPY Y2 antagonists acting on somatic receptors in DRGs and possibly on presynaptic receptors in the spinal cord (Brumovsky et al 2007a, Xu et al 2008).
Discovery of the three vesicular glutamate transporters (VGLUT1–3) (see previously) has made it possible to genetically manipulate mice and to study the involvement of glutamate transmission. Thus, deletion of VGLUT2 abolishes the mechanical hypersensitivity induced by spared nerve injury (Moechars et al 2006, Lagerström et al 2010, Liu et al 2010), a neuropathic pain model (Decosterd and Woolf 2000). Similarly, loss of VGLUT3 markedly attenuates the pronounced reduction in the mechanical threshold in the same pain model (Seal et al 2009).
Cancer Pain
Pain is common with cancer and perhaps the most serious complication with regard to quality of life. Moreover, just as with neuropathic pain, present treatment alternatives (e.g., for bone pain) are not efficacious. However, comparatively little is known about the relationship between cancer pain and the nervous system. It could be guessed that an eroding tumor also damages nerves, thereby inducing cancer pain of the neuropathic type. However, studies on a murine model show that bone cancer appears to represent a unique pain state that is neurochemically distinct from the profiles existing in both inflammatory and neuropathic pain (Honore et al 2000, Mantyh 2006, Schmidt et al 2010). Thus, the dramatic phenotypic changes described earlier for inflammatory and neuropathic pain are not seen with bone pain. For example, there are no apparent changes in substance P and CGRP (up-regulated with inflammatory pain and down-regulated with neuropathic pain), and galanin and NPY are down-regulated (up-regulated with neuropathic pain) (Honore et al 2000). It remains to be analyzed to what extent this is also true for other types of cancers (see Chapter 72).
The Sprouting Paradigm
A major hypothesis in our attempts to understand neuropathic pain has been the so-called sprouting theory developed by Woolf and collaborators (see Woolf and Salter 2000). In short, it was proposed that transection of a peripheral nerve induces sprouting of large, myelinated, non-nociceptive, primary afferent fibers from deeper laminae into laminae I and II of the spinal cord, that is, where nociceptive afferents normally terminate. Thus, nerve injury may induce not only major chemical alterations in DRG neurons as already described but also, in fact, profound anatomical changes.
It is well known (see Chapter 5) that small and large DRG neurons, respectively, give rise to C fibers terminating in laminae I and II and to A fibers terminating in laminae III and IV. A third group, thin myelinated, nociceptive Aδ afferents, terminate in laminae I and V. It was shown that two retrograde and trans-ganglionic neuronal tracers, cholera toxin B (CTB) and CTB conjugated to horseradish peroxidase, which bind to cell membrane glycoconjugates (especially the monoganglioside GM1), are normally taken up mainly by large neurons and label A fibers in the deep dorsal horn (Robertson and Grant 1985). However, after nerve injury there was a marked increase in CTB-labeled afferents in laminae I and II, and this was interpreted as sprouting of Aβ afferents from deeper laminae and suggested to be an anatomical basis for the development of neuropathic pain (see Woolf and Mannion 1999, Woolf and Salter 2000).
However, Tong and associates (1999) showed that peripheral nerve injury apparently causes a phenotypic change in small DRG neurons that results in marked expansion of the capacity of DRG neurons to take up and transport CTB so that now not only large but also almost all small neurons are labeled, presumably as a result of up-regulation of GM1. These findings have now been confirmed in various ways (Bao et al 2002, Hughes et al 2003, Santha and Jancso 2003, Shehab et al 2003). For example, Bao’s group injected CTB into the peripheral nerve before nerve injury (pre-injury labeling), and as expected, this resulted in labeling mainly of large DRG neurons or Aβ afferents (Bao et al 2002). Two weeks later, after peripheral nerve injury, the distribution of CTB-labeled afferents in the dorsal horn was similar to that seen after injection of CTB into an intact nerve (Fig. 63-3A). Hughes and colleagues performed intra-axonal labeling and could not show any certain sprouting of Aβ afferents in laminae I and II 7–10 weeks after nerve injury (Hughes et al 2003). However, limited sprouting may occur because a small number of Aβ afferents may enter the inner part of lamina II after nerve injury (Bao et al 2002). Taken together, these findings strongly suggest that CTB-labeled fibers in lamina I and II after axotomy mainly represent C fibers and therefore seriously question the role of anatomical sprouting as an important basis for neuropathic pain.
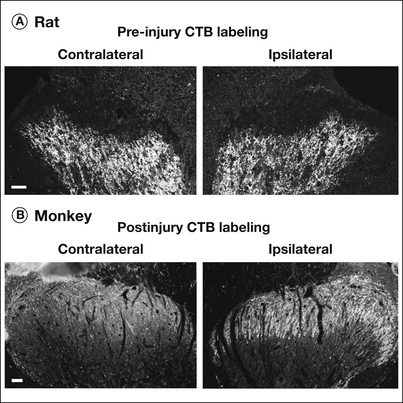
Figure 63-3 Distribution of labeled fibers 14 days after unilateral sciatic nerve transection.
A, A fibers labeled with cholera toxin B (CTB) 4 days before nerve injury (pre-injury CTB labeling) are distributed in laminae III and IV of the ipsilateral dorsal horn of rat spinal cord, which is similar to that seen in the contralateral dorsal horn. B, In monkey spinal cord, CTB-labeled afferents are distributed in the contralateral laminae I and II. After nerve injury the number of labeled afferents is increased in the superficial dorsal horn on the ipsilateral side. Bars indicate 50 μm.
It should be noted that after peripheral nerve injury, neurons in laminae I and II can be activated by selective electrical stimulation of Aβ fibers as determined by both the proto-oncogene c-fos as a functional marker (Shortland and Molander 1998) and electrophysiology (Kohama et al 2000). However, the structural and molecular mechanisms for this apparent increased responsiveness of neurons in the superficial laminae to stimulation of Aβ fibers remain to be elucidated.
In sharp contrast to the rat, in the normal monkey (M. mulatta), CTB labels both small and large DRG neurons, and CTB-labeled afferents are present in laminae I and II of the spinal cord (Tong et al 1999). After peripheral nerve injury, more than 70% of the DRG neurons (up from 11%), including both small and large neurons, are labeled by CTB, and the number of CTB-labeled afferents is increased in laminae I and II of monkey spinal cord, with a limited increase in lamina III (Fig. 63-3B). These findings suggest distinct anatomical differences with regard to termination of primary afferents in the dorsal spinal cord between the rat and monkey, at least M. mulatta. Therefore, to understand the mechanisms underlying allodynia, particularly in humans, further studies are needed on the anatomical and neurochemical network in the spinal cord and on its plasticity in response to peripheral nerve injury.
Functional Aspects of Spinal Cord Plasticity after Peripheral Nerve Damage
Injury or disease processes affecting peripheral nerves induce complex direct and secondary changes in the spinal cord; such changes eventually reach the level of the cerebral cortex, where the somatosensory map related to the injured nerve’s peripheral territory becomes reorganized. Here we concentrate on physiological changes in the spinal cord, including the terminals of sensory afferents, in response to peripheral nerve injury, but cell loss and glial responses are also briefly discussed. Physiologically, peripheral injury leads to direct uncoupling of some afferent input to dorsal horn neurons, as well as to changes in the properties of afferent terminals, synaptic efficacy, and the response characterization of dorsal horn neurons and inhibitory modulation. The result is a complex plasticity beyond a simple reduction in synaptic input, and this plasticity may participate in the development of neuropathic pain (see Chapter 6).
Alteration in the Receptive Field of Dorsal Horn Neurons
In normal animals, the skin surface of the hindlimb is mapped somatotopically in the dorsal horn of the lumbar spinal cord. Neurons in the medial region of the low lumbar dorsal horn have their receptive fields in the toe–foot region, whereas those in the lateral region correspond to the upper and lower portions of the legs (see Devor 1988). Immediately after total transection of major nerves innervating the hindpaw, such as the sciatic and saphenous nerves, there is a period of loss of input to neurons in the medial dorsal horn (Devor 1988). However, within a few days in rats and a few weeks in cats, many neurons in the medial dorsal horn start to respond to cutaneous stimulation applied to the upper part of the leg, thigh, and perineum (Devor 1988). Similar plasticity has also been found to occur in neurons receiving input from the sciatic versus the saphenous nerve (Devor 1988).
In addition to complete nerve section, animal models of partial nerve injury and neuropathic pain have been developed. Although earlier studies using the chronic nerve constriction model did not reveal major abnormalities in the receptive field characteristics of dorsal horn neurons (Laird and Bennett 1993), some more recent studies involving several partial injury models have shown an expansion of the receptive fields of dorsal horn neurons (Takaishi et al 1996, Suzuki et al 2000). These enlargements are often moderate and depend on time and the modality (Takaishi et al 1996, Suzuki et al 2000).
There is evidence that such plasticity in receptive field organization is not a result of peripheral changes, such as afferent sprouting or crosstalk between the afferents innervating the two regions, but rather a result of central changes occurring in the spinal cord (see Devor 1988), with at least two potential underlying mechanisms. One is the growth of new axons or dendrites across previous somatotopic boundaries. Another, more likely scenario, is increased effectiveness of pre-existing synapses, so-called weak or silent synapses, which have been demonstrated in dorsal horn neurons in both anatomical and electrophysiological studies (see Devor 1988). Thus, an increased excitatory drive resulting from, for example, repetitive C-fiber stimulation or reduced spinal inhibitory control can readily modify the receptive fields of dorsal horn neurons through alterations in synaptic strength (see Woolf and Salter 2000). As discussed later, peripheral nerve injury induces a cascade of changes in the dorsal horn that lead to reduced inhibition and/or increased excitation (see Chapter 6).
Response Characteristics of Dorsal Horn Neurons
It has long been known that, in humans, deafferentated dorsal horn neurons develop spontaneous activity after dorsal root injury, which has also been observed in the cat or rat after rhizotomy and peripheral nerve injury (Dalal et al 1999). Thus, the percentage of dorsal horn neurons exhibiting ongoing discharges is markedly increased in comparison to normal animals. Deafferentation, or loss of input, is not, however, a requisite for ongoing activity after axotomy because in many studies, particularly in those after partial injury, there seems to be no difference in the level of spontaneous activity in neurons with or without a measurable peripheral receptive field (Palecek et al 1992, Laird and Bennett 1993, Behbehani and Dollberg-Stolik 1994, Takaishi et al 1996, Chapman et al 1998, Dalal et al 1999, Pertovaara et al 2001, Weng et al 2003). Thus dorsal horn neurons per se can be the source of ongoing activity after nerve injury, but in addition it may result from ectopic discharges generated in the neuroma and DRG. This has also been recorded in rats with experimentally induced diabetic neuropathy despite lack of evidence of increased ongoing primary afferent activity (Pertovaara et al 2001).
Taken together, increased ongoing activity in dorsal horn neurons, especially those that normally receive nociceptive input, is likely to be an important mechanism in the development of neuropathic pain. Such activity may give rise to the spontaneous pain-like behavior observed in different pain models and may also contribute to the development of hypersensitivity when occurring in neurons with intact peripheral receptive fields. Importantly, procedures known to alleviate neuropathic pain–like behavior in animal models have also been shown to reduce such ongoing activity (Yakhnitsa et al 1999).
Response of Dorsal Horn Neurons to Mechanical and Thermal Stimulation
In contrast to the consistent observation of increased ongoing activity in dorsal horn neurons following peripheral deafferentation, the responses of these neurons to peripheral electrical, mechanical, and thermal stimulation have been found to be normal in the majority of studies. Thus after axotomy, the threshold and magnitude of the A- and C-fiber response of dorsal horn neurons to electrical stimulation of the proximal end of the cut nerve did not alter (Wall et al 1981). More remarkably, the majority of studies using a partial injury model also failed to demonstrate an increased response of dorsal horn neurons to mechanical and/or heat stimulation despite the presence of behavioral hypersensitivity in these animals. For example, the threshold and stimulus–response relationship to heat stimulation has been reported to be comparable in dorsal horn neurons after nerve injury and in neurons of sham-injured rats in almost all studies (Palecek et al 1992, Laird and Bennett 1993, Takaishi et al 1996, Chapman et al 1998, Pertovaara et al 2001). The response to mechanical stimulation is variable, with some authors reporting hypersensitivity (Yakhnitsa et al 1999, Weng et al 2003) and others reporting reduced sensitivity (Laird and Bennett 1992, Takaishi et al 1996, Chapman et al 1998).
Following partial nerve injury, responses of dorsal horn neurons to natural stimulation are very much influenced by the extent of the nerve injury, as well as the relative involvement of myelinated versus unmyelinated input, and this may contribute to the variable neuronal responses. Overall, the less robust sensory responses of dorsal horn neurons in rats with partial nerve injury suggest that additional mechanisms may account for the allodynia-type pain in animals. Such mechanisms may include expansion of the receptive field of individual dorsal horn neurons, as well as recruitment of more neurons resulting from the activation of previously inactive synapses. In agreement, Colvin and co-workers (1996) have shown that after sciatic nerve constriction, rats have an increase in dorsal horn post-synaptic currents in relation to afferent fiber input as measured by surface compound potentials. Finally, some of the behavioral changes in animals with partial nerve injury may be caused by direct stimulation of the injured nerve site, which is known to trigger marked responses in dorsal horn neurons after nerve injury (Laird and Bennett 1993).
Afterdischarges
In addition to spontaneous activity, prolonged afterdischarges represent a consistent feature of the response of dorsal horn neurons to mechanical stimulation after partial nerve injury (Palecek et al 1992, Laird and Bennett 1993, Takaishi et al 1996, Yakhnitsa et al 1999). This may also be important for pain perception in these animals because of temporal summation, and the behavioral correlate of such afterdischarges may be the prolonged paw withdrawal in neuropathic animals following noxious mechanical stimulation. Such afterdischarges could be caused by crosstalk of afferent fibers at the injury site or in the DRG, or by increased neuronal excitability in the dorsal horn.
Activity-Dependent Increase (Central Desensitization) in Spinal Excitability in Nerve-Injured Animals
An activity-dependent increase in spinal cord excitability may represent an important mechanism underlying the abnormal pain conditions after tissue injury and may share similar mechanisms with memory formation (see Ji et al 2003). In earlier studies, central sensitization after repetitive activation of C fibers was unaltered or reduced in rats after axotomy in the flexor reflex preparation (Wall and Woolf 1986). Thus, the wind-up of dorsal horn neurons after C-fiber stimulation was unchanged, and there was no Aβ fiber–induced wind-up in rats after spinal nerve ligation (Chapman et al 1998). However, more recently, Weng’s group reported that in rats with vincristine-induced neuropathy, wind-up occurs at lower-frequency stimulation than in normal animals (Weng et al 2003).
Miletic and Miletic (2000) showed that tetanic stimulation of A fibers in the sciatic nerve at low frequency triggers long-term depression of dorsal horn field potentials and that this was markedly reduced in rats with nerve ligation as compared with normal rats. Moreover, when stimulated at high frequency, A-fiber tetanic stimulation generated a prolonged increase in spinal excitability in nerve-injured rats, in contrast to normal rats, in which a brief facilitation was followed by depression. These results demonstrated that the spinal plasticity that occurs after partial nerve injury leads to an overall shift in response to A-fiber tetanic stimulation from depression to excitation, which can result from either reduced spinal inhibitory mechanisms or increased excitatory drive. At least some of the changes may be caused by an alteration in descending influences from the brain (Carlson et al 2007).
N-Methyl-D-aspartate (NMDA) receptor activation is one of the principal mechanisms in central sensitization, and its role in neuropathic pain is implicated by a large number of preclinical studies showing that NMDA receptor antagonists are effective in alleviating experimental neuropathic pain (see Woolf and Salter 2000). Following chronic constriction injury, the content of glutamate, aspartate, and intracellular calcium is increased in dorsal horn slices (Kawamata and Omote 1996), possibly related to alterations in spinal glutamate transporters (Sung et al 2003). In addition, spinal nerve ligation causes facilitation of NMDA-induced currents and calcium transients in rat substantia gelatinosa neurons (Isaev et al 2000). This may be caused by post-synaptic intracellular events subsequent to NMDA receptor activation, such as activation of protein kinase C, because peripheral nerve injury does not seem to alter the level of NMDA receptors or expression of NMDA receptor subunits in the dorsal horn (Mao et al 1995). Furthermore, increased excitatory drive in the spinal cord may also involve activation of AMPA receptors and metabotropic glutamate receptors. Finally, a role for neuropeptides such as VIP and pituitary adenylate cyclase–activating peptide, the synthesis of which is increased in sensory neurons after axotomy, has been suggested (Wiesenfeld-Hallin et al 1990; also see Dickinson and Fleetwood-Walker 1999), and they appear to take over the excitatory role of substance P in normal rats in mediating repetitive C-fiber stimulation–induced facilitation of the nociceptive flexor reflex (Wiesenfeld-Hallin et al 1990).
Reduced Inhibitory Mechanism in the Spinal Cord
Transmission of sensory information from the periphery to the central nervous system is normally subjected to either pre- or post-synaptic inhibitory control maintained by activity in sensory afferents, dorsal horn interneurons, and descending pathways. Wall and colleagues showed a marked axotomy-induced reduction in the magnitude of the dorsal root potential, a measure of presynaptic inhibition (Devor 1988), and this was later confirmed in several other studies, including models of partial nerve injury (Laird and Bennett 1992). Furthermore, the post-synaptic inhibition on dorsal horn neurons exerted by A-afferent input was also reduced after axotomy (Devor 1988). Such reduction in pre- and post-synaptic inhibition of spinal sensory input was later shown to be paralleled by a decrease in the content of the important inhibitory neurotransmitter γ-aminobutyric acid (GABA) (Castro-Lopes et al 1993), as well as GABA receptors (Castro-Lopes et al 1995). More recent work has further demonstrated that partial nerve injury (chronic constriction and spared nerve injury), but not nerve transection, induces selective loss of GABA-mediated inhibition on superficial dorsal horn neurons as a result of reduced release of GABA (Moore et al 2002).
The endogenous opioid peptides and their receptors represent another important spinal inhibitory system on nociception. Opioids are very effective in treating nociceptive or inflammatory pain. Opioids are also effective in certain types of neuropathic pain, although the mechanism of their reduced efficacy remains to be understood (Arnér and Meyerson 1988, Sindrup and Jensen 1999, Dworkin et al 2003, Rowbotham et al 2003, Finnerup et al 2010). One explanation may be injury-induced loss of μ- and δ-opioid receptors on afferent terminals and/or dorsal horn interneurons (Besse et al 1992). Interestingly, a very recent study on rats has shown that brief, high-dose opioid administration can “erase a spinal memory trace of pain,” thereby offering new treatment strategies, perhaps even “cure” for certain types of pain (Drdla-Schutting et al 2012).
It should be mentioned that opioid antinociception is also subjected to modulation by several anti-opioid systems, including activation of NMDA receptors (Mao et al 1995) and peptides such as CCK (see Wiesenfeld-Hallin and Xu 2001) and dynorphin (Gardell et al 2004). Interestingly, there is evidence of coupling between the activity-dependent insertion of δ-opioid receptors and the release of pro-nociceptive neuropeptides (Bao et al 2003), as well as evidence of dynorphin being a mediator of neuropathic pain (see Gardell et al 2004 and references therein).
Nerve Injury–Induced Cell Loss in the Dorsal Horn
More recently, attention has been paid to the possibility that peripheral nerve injury can in fact cause cell loss in the dorsal horn and that this could contribute to neuropathic pain. Thus, the Sugimoto group early on described neurons with signs of degeneration in the lumbar dorsal horn, on both sides, after unilateral constriction of the sciatic nerve, with a greater increase occurring ipsilaterally (Sugimoto et al 1990). However, after sciatic nerve transection there was no evidence of dark (degenerating) neurons. Later, Azkue and colleagues (1998), using the terminal deoxynucleotidyl transferase nick end labeling (TUNEL) technique, demonstrated nuclear fragmentation in neurons in the superficial layers and in the neck of the dorsal horn, probably reflecting apoptotic cell death. This was seen at 7 but not at 3 or 14 days after sciatic nerve transection and could be prevented with an NMDA receptor antagonist (MK-801), thus suggesting the involvement of glutamatergic mechanisms and perhaps excessive glutamate release. Coggeshall and associates (2001), using stimulation with parameters that selectively activate Aβ fibers, then provided evidence that myelinated afferents (A fibers), using glutamate as a transmitter, are responsible for cell death in the dorsal horn.
They did not see this after sciatic nerve transection alone, but activation of A fibers in a previously sectioned sciatic nerve caused cell death. The type of neuron undergoing cell death has not been identified, but GABA neurons represent a large population of dorsal horn neurons, and neuropathic injury induces a decrease in GABA content in the dorsal horn (Castro-Lopes et al 1993). Furthermore, partial nerve injury decreases dorsal horn levels of the GABA-synthesizing enzyme glutamic acid decarboxylase and induces neuronal apoptosis (Moore et al 2002).
Changes in Spinal Glia after Peripheral Nerve Injury
A role of spinal glial cell activation in chronic pain processing has increasingly been recognized, as reflected in a large number of review articles on this topic during the past decade (Tsuda et al 2005, Scholz and Woolf 2007, Suter et al 2007, Milligan and Watkins 2009, Svensson and Brodin 2010). Nerve injury induces marked gliosis of microglia and astrocytes in the spinal cord, including morphological changes (e.g., hypertrophy), up-regulation of glial markers (e.g., CD11d for microglia and glial fibrillary acidic protein [GFAP] for astrocytes), and proliferation of microglia (Suter et al 2007). Nerve injury also induces up-regulation of P2X4 receptors and activation of p38 mitogen–activated protein kinase (MAPK) in microglia and activation of c-Jun N-terminal kinase (JNK) MAPK in astrocytes, which leads to the production of pro-inflammatory cytokines and chemokines (e.g., tumor necrosis factor-α [TNF-α], IL-1β, CCL2) and growth factor (e.g., BDNF). All these mediators have been shown to induce hyperexcitability in dorsal horn nociceptive neurons. Importantly, drugs that block the action of P2X4, TNF-α, IL-1β, p38, JNK, CCL2, and BDNF, as well as glial toxins and inhibitors, can all attenuate neuropathic pain symptoms in different nerve injury models (Raghavendra et al 2003, Suter et al 2007, White et al 2007, Inoue and Tsuda 2009, Ji et al 2009, Milligan and Watkins 2009). For further information, see Chapter 4.
Conclusion
Many changes occur in DRGs and the spinal cord in response to peripheral nerve injury, and recent global expression analyses have expanded the number of genes known to be involved. Thus, in this chapter, even though we have discussed several molecules, many others have been left out. Some of the changes may play a role in the generation and maintenance of neuropathic pain (Fig. 63-4) but may also have antinociceptive effects. Taken together, exact knowledge of the role of the molecules regulated by nerve injury and of the mechanisms underlying their regulation may not only offer an understanding of neuropathic pain but may also suggest novel strategies for treatment of this type of pain. However, the very large number of injury-regulated genes makes it even more difficult to define the mechanisms underlying this type of pain.
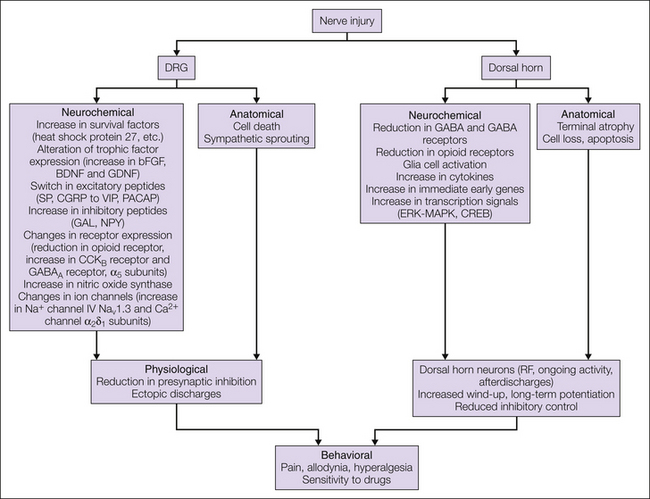
Figure 63-4 Schematic summary of changes occurring in dorsal root ganglia (DRGs) and the dorsal horn in response to peripheral nerve injury and their functional consequences.
Peripheral nerve injury induces complex anatomic, neurochemical, and functional changes in DRGs and the dorsal horn. Some of these changes may represent survival and regenerative mechanisms, some may be adaptive responses as a consequence of loss of peripheral innervation or afferent input, and some may be defensive mechanisms against excessive nociceptive stimulation. Together, such changes in plasticity are important not only in development of the pain syndrome in nerve-injured patients but also in the response of such pain to treatments. BDNF, brain-derived neurotrophic factor; bFGF, basic fibroblast growth factor; CREB, cyclic adenosine monophosphate response element–binding protein; CGRP, calcitonin gene–related peptide; ERK, extracellular signal–regulated kinase; GABA, γ-aminobutyric acid; GAL, galanin; GDNF, glial cell line–derived neurotrophic factor; MAPK, mitogen-activated protein kinase; NPY, neuropeptide Y; PACAP, pituitary adenylate cyclase–activating peptide; RF, radiofrequency; SP, substance P; VIP, vasoactive intestinal polypeptide.
The distinctly different patterns of regulation seen after peripheral inflammation (up-regulation of opioid and other peptides in dorsal horn neurons; see Dubner and Ruda 1992) and after nerve injury (regulation of molecules in DRG neurons) suggest the existence of separate defense systems associated with dorsal horn neurons and DRG neurons, respectively, for these two types of pain (see Hökfelt et al 1997).
In the spinal cord the terminals of sensory afferents undergo plasticity reflecting the changes occurring in their cell bodies in the DRG. Plastic changes in dorsal horn neurons include expanded or novel receptive fields, ongoing activity, afterdischarges, and in some cases, hypersensitivity. The overall excitatory drive in the dorsal horn may be increased, and there is strong evidence that inhibitory control in the spinal cord is compromised after nerve injury.
The unique efficacy of morphine in treating inflammatory pain has raised the question of whether there is a single molecule that can abolish neuropathic pain in a similar way. The fact that neurons produce and release multiple messengers (Hökfelt 1991)—a single primary pain afferent can, for example, probably release glutamate, adenosine triphosphate, nitric oxide, substance P, CGRP, and galanin—calls for several antagonists to block transmission. In fact, a neurokinin 1 (NK1) antagonist has been shown to be unable to antagonize pain in humans (see Rupniak and Kramer 1999). This has led to the dramatic idea of deleting entire dorsal horn projection neurons with the help of a conjugate of a toxin (saporin) coupled to substance P, which is internalized via the NK1 receptor (Mantyh et al 1997). However, injury-induced neuropathic pain can be reversed by antisense oligonucleotides against the TTX-resistant sodium channel Nav1.8 (Lai et al 2002). Moreover, investigators treated rats with neuropathic pain induced by spinal nerve ligation with systematically administered GDNF and artemin, a member of the GDNF family (Gardell et al 2003, Wang et al 2003). They observed not only pain reversal but also at least partial normalization of a number of morphological and neurochemical features, including changes in neuropeptides and the Nav1.8 sodium channel.
Beyond the conventional design of exogenous agonists and antagonists acting (mostly) on seven-transmembrane, G protein–coupled receptors, transcription factors and other molecules can be targeted to activate endogenous downstream antinociceptive systems. For example, inhibition of the enzyme enkephalinase, which inactivates the opioid peptides methionine- and leucine-enkephalin, represents one approach (Roques and Noble 1995). Furthermore, Cheng and coworkers genetically deleted the transcription factor downstream regulatory element antagonistic modulator (DREAM), which tonically suppresses the opioid peptide dynorphin, and in this way created a mouse with an increased pain threshold in a variety of pain models as a result of increased spinal dynorphin levels (Cheng et al 2002). This study demonstrates the power of mobilizing endogenous receptor ligands to counteract pain and points to future research directions. Finally, inhibition of phospholipase C, a molecule downstream of GalR2, as well as many other G protein–coupled receptors, not only attenuates inflammatory hyperalgesia (Hou et al 2004, Galeotti et al 2006, Joseph et al 2007) but also causes a long-lasting (≈48-hour) elevation in pain threshold in a neuropathic mouse pain model (Shi et al 2008).
Acknowledgments
This work was supported by the Swedish Research Council, the Marianne and Marcus Wallenberg Foundation, the Knut and Alice Wallenberg Foundation, an Unrestricted Bristol-Myers Squibb Neuroscience Grant, Funds from Karolinska Institutet, the National Science Foundation China, and the “973” Program China. We thank Professor Gunnar Grant, Department of Neuroscience, Karolinska Institutet, Stockholm, Sweden, and Dr. Ru-Rong Ji, Sensory Plasticity Laboratory, Pain Research Center, Preoperative and Pain Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, United States, for valuable comments on this manuscript. We thank Drs. Esbjörn Bergman, Stephen McMahon, John Priestly, and Clifford Woolf for generous permission to reproduce figures from their work.
The references for this chapter can be found at www.expertconsult.com.
References
Abdulla F.A., Smith P.A. Nerve injury increases an excitatory action of neuropeptide Y and Y2-agonists on dorsal root ganglion neurons. Neuroscience. 1999;89:43–60.
Abdulla F.A., Stebbing M.J., Smith P.A. Effects of substance P on excitability and ionic currents of normal and axotomized rat dorsal root ganglion neurons. European Journal of Neuroscience. 2001;13:545–552.
Alier K.A., Chen Y., Sollenberg U.E., et al. Selective stimulation of GalR1 and GalR2 in rat substantia gelatinosa reveals a cellular basis for the anti- and pro-nociceptive actions of galanin. Pain. 2008;137:138–146.
Amir R., Devor M. Chemically mediated cross-excitation in rat dorsal root ganglia. Journal of Neuroscience. 1996;16:4733–4741.
Arnér L.D., Meyerson B.A. Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain. 1988;33:11–23.
Azkue J.J., Zimmermann M., Hsieh T.F., et al. Peripheral nerve insult induces NMDA receptor–mediated, delayed degeneration in spinal neurons. European Journal of Neuroscience. 1998;10:2204–2206.
Baker M.D., Wood J.N. Involvement of Na+ channels in pain pathways. Trends in Pharmacological Sciences. 2001;22:27–31.
Bao L., Jin S.X., Zhang C., et al. Activation of delta opioid receptors induces receptor insertion and neuropeptide secretion. Neuron. 2003;37:121–133.
Bao L., Wang H.F., Cai H.J., et al. Peripheral axotomy induces only very limited sprouting of coarse myelinated afferents into inner lamina II of rat spinal cord. European Journal of Neuroscience. 2002;16:175–185.
Basbaum A.I., Bautista D.M., Scherrer G., et al. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284.
Behbehani M.M., Dollberg-Stolik O. Partial sciatic nerve ligation results in an enlargement of the receptive field and enhancement of the response of dorsal horn neurons to noxious stimulation by an adenosine agonist. Pain. 1994;58:421–428.
Bergman E., Johnson H., Zhang X., et al. Neuropeptides and neurotrophin receptor mRNAs in primary sensory neurons of aged rats. Journal of Comparative Neurology. 1996;375:303–319.
Besse D., Lombard M.C., Perrot S., et al. Regulation of opioid binding sites in the superficial dorsal horn of the rat spinal cord following loose ligation of the sciatic nerve: comparison with sciatic nerve section and lumbar dorsal rhizotomy. Neuroscience. 1992;50:921–933.
Branchek T.A., Smith K.E., Gerald C., et al. Galanin receptor subtypes. Trends in Pharmacological Sciences. 2000;21:109–116.
Brumovsky P., Shi T.S., Landry M., et al. Neuropeptide tyrosine and pain. Trends in Pharmacological Sciences. 2007;28:93–102.
Brumovsky P., Watanabe M., Hokfelt T. Expression of the vesicular glutamate transporters-1 and -2 in adult mouse dorsal root ganglia and spinal cord and their regulation by nerve injury. Neuroscience. 2007;147:469–490.
Carlson J.D., Maire J.J., Martenson M.E., et al. Sensitization of pain-modulating neurons in the rostral ventromedial medulla after peripheral nerve injury. Journal of Neuroscience. 2007;27:13222–13231.
Castro-Lopes J.M., Malcangio M., Pan B.H., et al. Complex changes of GABAA and GABAB receptor binding in the spinal cord dorsal horn following peripheral inflammation or neurectomy. Brain Research. 1995;679:289–297.
Castro-Lopes J.M., Tavares I., Coimbra A. GABA decreases in the spinal cord dorsal horn after peripheral neurectomy. Brain Research. 1993;620:287–291.
Chapman V., Suzuki R., Dickenson A.H. Electrophysiological characterization of spinal neuronal response properties in anaesthetized rats after ligation of spinal nerves L5-L6. Journal of Physiology. 1998;507:881–894.
Cheng H.-Y.M., Pitcher G.M., Laviolette S.R., et al. DREAM is a critical transcriptional repressor for pain modulation. Cell. 2002;108:31–43.
Cho H.J., Kim J.K., Park H.C., et al. Changes in brain-derived neurotrophic factor immunoreactivity in rat dorsal root ganglia, spinal cord, and gracile nuclei following cut or crush injuries. Experimental Neurology. 1998;154:224–230.
Cho H.J., Kim S.Y., Park M.J., et al. Expression of mRNA for brain-derived neurotrophic factor in the dorsal root ganglion following peripheral inflammation. Brain Research. 1997;749:358–362.
Coggeshall R.E., Lekan H.A., White F.A., et al. A-fiber sensory input induces neuronal cell death in the dorsal horn of the adult rat spinal cord. Journal of Comparative Neurology. 2001;435:276–282.
Colvin L.A., Mark M.A., Duggan A.W. Bilaterally enhanced dorsal horn postsynaptic currents in a rat model of peripheral mononeuropathy. Neuroscience Letters. 1996;207:29–32.
Costigan M., Befort K., Karchewski L., et al. Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neuroscience. 2002;3:16.
Costigan M., Scholz J., Woolf C.J. Neuropathic pain: a maladaptive response of the nervous system to damage. Annual Review of Neuroscience. 2009;32:1–32.
Dalal A., Tata M., Allegre G., et al. Spontaneous activity of rat dorsal horn cells in spinal segments of sciatic projection following transection of sciatic nerve or of corresponding dorsal roots. Neuroscience. 1999;94:217–228.
Decosterd I., Woolf C.J. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158.
Del Fiacco M., Priestley J.V. Neurotrophins and adult somatosensory neurons. In: Mocchetti I., ed. Neurobiology of the neurotrophins. Johnson City, TN: FP Graham Publishing; 2001:205–236.
Devor M. The pathophysiology and anatomy of damaged nerve. In: Wall P.D., Melzack R., eds. Textbook of pain. Edinburgh: Churchill Livingstone; 1984:49–64.
Devor M. Central changes mediating neuropathic pain. In: Dubner R., Gebhart G.F., Bond M.R., eds. Proceedings of the 5th World Congress on Pain. Amsterdam: Elsevier; 1988:114–127.
Devor M., Govrin-Lippmann R., Frank I., et al. Proliferation of primary sensory neurons in adult rat dorsal root ganglion and the kinetics of retrograde cell loss after sciatic nerve section. Somatosensory Research. 1985;3:139–167.
Dickinson T., Fleetwood-Walker S.M. VIP and PACAP: very important in pain? Trends in Pharmacological Sciences. 1999;20:324–329.
Drdla-Schutting R., Benrath J., Wunderbaldinger G., Sandkuhler J. Erasure of a spinal memory trace of pain by a brief, high-dose opioid administration. Science. 2012;335:235–238.
Dubner R., Ruda M.A. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends in Neurosciences. 1992;15:96–103.
Dworkin R.H., Backonja M., Rowbotham M.C., et al. Advances in neuropathic pain: diagnosis, mechanisms, and treatment recommendations. Archives of Neurology. 2003;60:1524–1534.
Finnerup N.B., Sindrup S.H., Jensen T.S. The evidence for pharmacological treatment of neuropathic pain. Pain. 2010;150:573–581.
Fremeau R.T., Jr., Voglmaier S., Seal R.P., et al. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends in Neurosciences. 2004;27:98–103.
Fukuoka T., Kondo E., Dai Y., et al. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. Journal of Neuroscience. 2001;21:4891–4900.
Galeotti N., Stefano G.B., Guarna M., et al. Signaling pathway of morphine induced acute thermal hyperalgesia in mice. Pain. 2006;123:294–305.
Gardell L.R., Ibrahim M., Wang R., et al. Mouse strains that lack spinal dynorphin upregulation after peripheral nerve injury do not develop neuropathic pain. Neuroscience. 2004;123:43–52.
Gardell L.R., Wang R., Ehrenfels C., et al. Multiple actions of systemic artemin in experimental neuropathy. Nature Medicine. 2003;9:1383–1389.
Govrin-Lippmann R., Devor M. Ongoing activity in severed nerves. Source and variation with time. Brain Research. 1978;159:406–410.
Grothe C., Meisinger C., Claus P. In vivo expression and localization of the fibroblast growth factor system in the intact and lesioned rat peripheral nerve and spinal ganglia. Journal of Comparative Neurology. 2001;434:342–357.
Hobson S.A., Bacon A., Elliot-Hunt C.R., et al. Galanin acts as a trophic factor to the central and peripheral nervous systems. Cellular and Molecular Life Sciences. 2008;65:1806–1812.
Hökfelt T. Neuropeptides in perspective: the last ten years. Neuron. 1991;7:867–879.
Hökfelt T., Zhang X., Wiesenfeld-Hallin Z. Messenger plasticity in primary sensory neurons following axotomy and its functional implications. Trends in Neurosciences. 1994;17:22–30.
Hökfelt T., Zhang X., Xu Z.-Q., et al. Cellular and synaptic mechanisms in transition of pain from acute to chronic. In: Jensen T.S., Turner J.A., Wiesenfeld-Hallin Z., eds. Proceedings of the 8th World Congress on Pain. Progress in Pain Research and Management. Seattle: IASP Press; 1997:133–153.
Honore P., Rogers S.D., Schwei M.J., et al. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience. 2000;98:585–598.
Hou C., Kirchner T., Singer M., et al. In vivo activity of a phospholipase C inhibitor, 1-(6-((17beta-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione (U73122), in acute and chronic inflammatory reactions. Journal of Pharmacology and Experimental Therapeutics. 2004;309:697–704.
Hua X.Y., Boublik J.H., Spicer M.A., et al. The antinociceptive effects of spinally administered neuropeptide Y in the rat: systematic studies on structure-activity relationship. Journal of Pharmacology and Experimental Therapeutics. 1991;258:243–248.
Huang L.-Y.M., Neher E. Ca2+-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron. 1996;17:135–145.
Hughes D.I., Scott D.T., Todd A.J., et al. Lack of evidence for sprouting of Abeta afferents into the superficial laminas of the spinal cord dorsal horn after nerve section. Journal of Neuroscience. 2003;23:9491–9499.
Inoue K., Tsuda M. Microglia and neuropathic pain. Glia. 2009;57:1469–1479.
Isaev D., Gerber G., Park S.K., et al. Facilitation of NMDA-induced currents and Ca2+ transients in the rat substantia gelatinosa neurons after ligation of L5-L6 spinal nerves. Neuroreport. 2000;11:4055–4061.
Ji R.R., Gereau R.W., Malcangio M., et al. MAP kinase and pain. Brain Research Reviews. 2009;60:135–148.
Ji R.R., Kohno T., Moore K.A., et al. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends in Neurosciences. 2003;26:696–705.
Joseph E.K., Bogen O., Alessandri-Haber N., et al. PLC-beta 3 signals upstream of PKC epsilon in acute and chronic inflammatory hyperalgesia. Pain. 2007;132:67–73.
Kashiba H., Senba E. Up- and down-regulation of BDNF mRNA in distinct subgroups of rat sensory neurons after axotomy. Neuroreport. 1999;10:3561–3565.
Kawamata M., Omote K. Involvement of increased excitatory amino acids and intracellular Ca2+ concentration in the spinal dorsal horn in an animal model of neuropathic pain. Pain. 1996;68:85–96.
Kerekes N., Mennicken F., O’Donnell D., et al. Galanin increases membrane excitability and enhances Ca(2+) currents in adult, acutely dissociated dorsal root ganglion neurons. European Journal of Neuroscience. 2003;18:2957–2966.
Kim C.H., Oh Y., Chung J.M., et al. Changes in three subtypes of tetrodotoxin sensitive sodium channel expression in the axotomized dorsal root ganglion in the rat. Neuroscience Letters. 2002;323:125–128.
Kohama I., Ishikawa K., Kocsis J.D. Synaptic reorganization in the substantia gelatinosa after peripheral nerve neuroma formation: aberrant innervation of lamina II neurons by Abeta afferents. Journal of Neuroscience. 2000;20:1538–1549.
Lagerström M.C., Rogoz K., Abrahamsen B., et al. VGLUT2-dependent sensory neurons in the TRPV1 population regulate pain and itch. Neuron. 2010;68:529–542.
Lai J., Gold M.S., Kim C.S., et al. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain. 2002;95:143–152.
Lai J., Hunter J.C., Porreca F. The role of voltage-gated sodium channels in neuropathic pain. Current Opinion in Neurobiology. 2003;13:291–297.
Laird J.M., Bennett G.J. Dorsal root potentials and afferent input to the spinal cord in rats with an experimental peripheral neuropathy. Brain Research. 1992;584:181–190.
Laird J.M., Bennett G.J. An electrophysiological study of dorsal horn neurons in the spinal cord of rats with an experimental peripheral neuropathy. Journal of Neurophysiology. 1993;69:2072–2085.
Landry M., Aman K., Dostrovsky J., et al. Galanin expression in adult human dorsal root ganglion neurons: initial observations. Neuroscience. 2003;117:795–809.
Landry M., Bouali-Benazzouz R., El Mestikawy S., et al. Expression of vesicular glutamate transporters in rat lumbar spinal cord, with a note on dorsal root ganglia. Journal of Comparative Neurology. 2004;468:380–394.
Leem J.W., Choi E.J., Park E.S., et al. N-Methyl-D-aspartate (NMDA) and non-NMDA glutamate receptor antagonists differentially suppress dorsal horn neuron responses to mechanical stimuli in rats with peripheral nerve injury. Neuroscience Letters. 1996;211:37–40.
Liu H.X., Hökfelt T. The participation of galanin in pain processing at the spinal level. Trends in Pharmacological Sciences. 2002;23:468–474.
Liu X., Zhou J.L., Chung K., et al. Ion channels associated with the ectopic discharges generated after segmental spinal nerve injury in the rat. Brain Research. 2001;900:119–127.
Liu Y., Abdel Samad O., Zhang L., et al. VGLUT2-dependent glutamate release from nociceptors is required to sense pain and suppress itch. Neuron. 2010;68:543–556.
Ludwig M., Pittman Q.J. Talking back: dendritic neurotransmitter release. Trends in Neurosciences. 2003;26:255–261.
Lyu Y.S., Park S.K., Chung K., et al. Low dose of tetrodotoxin reduces neuropathic pain behaviors in an animal model. Brain Research. 2000;871:98–103.
Mannion R.J., Costigan M., Decosterd I., et al. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus–induced inflammatory pain hypersensitivity. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:9385–9390.
Mantyh P.W. Cancer pain and its impact on diagnosis, survival and quality of life. Nature Reviews. Neuroscience. 2006;7:797–809.
Mantyh P.W., Rogers S.D., Honore P., et al. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278:275–279.
Mao J., Price D.D., Mayer D.J. Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain. 1995;62:259–274.
McKay Hart A., Brännström T., Wiberg M., et al. Primary sensory neurons and satellite cells after peripheral axotomy in the adult rat: time course of cell death and elimination. Experimental Brain Research. 2002;142:308–318.
McMahon S.B., Priestley J.V. Nociceptor plasticity. In: Hunt S.P., Koltzenburg M., eds. The neurobiology of pain. Oxford, UK: Oxford University Press; 2005:35–64.
Michael G.J., Averill S., Nitkunan A., et al. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. Journal of Neuroscience. 1997;17:8476–8490.
Michael G.J., Averill S., Shortland P.J., et al. Axotomy results in major changes in BDNF expression by dorsal root ganglion cells: BDNF expression in large trkB and trkC cells, in pericellular baskets, and in projections to deep dorsal horn and dorsal column nuclei. European Journal of Neuroscience. 1999;11:3539–3551.
Miletic G., Miletic V. Long-term changes in sciatic-evoked A-fiber dorsal horn field potentials accompany loose ligation of the sciatic nerve in rats. Pain. 2000;84:353–359.
Milligan E.D., Watkins L.R. Pathological and protective roles of glia in chronic pain. Nature Reviews. Neuroscience. 2009;10:23–36.
Minth-Worby C.A. Transcriptional regulation of the human neuropeptide Y gene by nerve growth factor. Journal of Biological Chemistry. 1994;269:15460–15468.
Moechars D., Weston M.C., Leo S., et al. Vesicular glutamate transporter VGLUT2 expression levels control quantal size and neuropathic pain. Journal of Neuroscience. 2006;26:12055–12066.
Moore K.A., Kohno T., Karchewski L.A., et al. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. Journal of Neuroscience. 2002;22:6724–6731.
Morris J.F., Pow D.V., Sokol H.W., et al. Dendritic release of peptides from magnocellular neurons in normal rats, Brattleboro rats and mice with hereditary nephrogenic diabetes insipidus. In: Gross P., Richter D., Robertson G.L., eds. Vasopressin. Paris: John Libbey Eurotext; 1993:171–182.
Morris J.L., Konig P., Shimizu T., et al. Most peptide-containing sensory neurons lack proteins for exocytotic release and vesicular transport of glutamate. Journal of Comparative Neurology. 2005;483:1–16.
Murphy M., Reid K., Brown M.A., et al. Involvement of leukemia inhibitory factor and nerve growth factor in the development of dorsal root ganglion neurons. Development. 1993;117:1173–1182.
Murphy P.G., Ramer M.S., Borthwick L., et al. Endogenous interleukin-6 contributes to hypersensitivity to cutaneous stimuli and changes in neuropeptides associated with chronic nerve constriction in mice. European Journal of Neuroscience. 1999;11:2243–2253.
Nahin R.L., Marino De León K.R., Ruda M. Primary sensory neurons exhibit altered gene expression in a rat model of neuropathic pain. Pain. 1994;58:95–108.
Naveilhan P., Hassani H., Lucas G., et al. Reduced antinociception and plasma extravasation in mice lacking a neuropeptide Y receptor. Nature. 2001;409:513–517.
Nielsch V., Bisby M.A., Keen P. Effect of cutting or crushing the rat sciatic nerve on synthesis of substance P by isolated L5 dorsal root ganglia. Neuropeptides. 1987;10:137–145.
O’Donnell D., Mennicken F., Hoffert C., et al. Localization of galanin receptor subtypes in the rat CNS. In: Quirion R., Björklund A., Hökfelt T., eds. Handbook of chemical neuroanatomy peptide receptors, part II. Amsterdam: Elsevier; 2003:195–244.
Oellig C., Pirvola U., Taylor L., et al. Acidic FGF and FGF receptors are specifically expressed in neurons of developing and adult rat dorsal root ganglia. European Journal of Neuroscience. 1995;7:863–874.
Palecek J., Paleckova V., Dougherty P.M., et al. Responses of spinothalamic tract cells to mechanical and thermal stimulation of skin in rats with experimental peripheral neuropathy. Journal of Neurophysiology. 1992;67:1562–1573.
Pertovaara A., Wei H., Kalmari J., et al. Pain behavior and response properties of spinal dorsal horn neurons following experimental diabetic neuropathy in the rat: modulation by nitecapone, a COMT inhibitor with antioxidant properties. Experimental Neurology. 2001;167:425–434.
Pitts R.L., Wang S., Jones E.A., et al. Transforming growth factor-beta and ciliary neurotrophic factor synergistically induce vasoactive intestinal peptide gene expression through the cooperation of Smad, STAT, and AP-1 sites. Journal of Biological Chemistry. 2001;276:19966–19973.
Priestley J.V., Michael G.J., Averill S., et al. Regulation of nociceptive neurons by nerve growth factor and glial cell line derived neurotrophic factor. Canadian Journal of Physiology and Pharmacology. 2002;80:495–505.
Raghavendra V., Tanga F., DeLeo J.A. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. Journal of Pharmacology and Experimental Therapeutics. 2003;306:624–630.
Rao M.S., Sun Y., Escary J.L., et al. Leukemia inhibitory factor mediates an injury response but not a target-directed developmental transmitter switch in sympathetic neurons. Neuron. 1993;11:1175–1185.
Robertson B., Grant G. A comparison between wheat germ agglutinin– and choleragenoid–horseradish peroxidase as anterogradely transported markers in central branches of primary sensory neurones in the rat with some observations in the cat. Neuroscience. 1985;14:895–905.
Roques B.P., Noble F. Dual inhibitors of enkephalin-degrading enzymes (neutral endopeptidase 24.11 and aminopeptidase N) as potential new medications in the management of pain and opioid addiction. NIDA Research Monograph. 1995;147:104–145.
Rowbotham M.C., Twilling L., Davies P.S., et al. Oral opioid therapy for chronic peripheral and central neuropathic pain. New England Journal of Medicine. 2003;348:1223–1232.
Rupniak N.M., Kramer M.S. Discovery of the antidepressant and anti-emetic efficacy of substance P receptor (NK1) antagonists. Trends in Pharmacological Sciences. 1999;20:485–490.
Santha P., Jancso G. Transganglionic transport of choleragenoid by capsaicin-sensitive C-fibre afferents to the substantia gelatinosa of the spinal dorsal horn after peripheral nerve section. Neuroscience. 2003;116:621–627.
Scadding J.W. Development of ongoing activity, mechanosensitivity, and adrenaline sensitivity in severed peripheral nerve axons. Experimental Neurology. 1981;73:345–364.
Scadding J.W. Peripheral neuropathies. In: Wall P.D., Melzack R., eds. Textbook of pain. Edinburgh: Churchill Livingstone; 1984:413–425.
Schmidt B.L., Hamamoto D.T., Simone D.A., et al. Mechanism of cancer pain. Molecular Interventions. 2010;10:164–178.
Scholz J., Woolf C.J. The neuropathic pain triad: neurons, immune cells and glia. Nature Neuroscience. 2007;10:1361–1368.
Seal R.P., Wang X., Guan Y., et al. Injury-induced mechanical hypersensitivity requires C–low threshold mechanoreceptors. Nature. 2009;462:651–655.
Shehab S.A., Atkinson M.E. Vasoactive intestinal polypeptide (VIP) increases in the spinal cord after peripheral axotomy of the sciatic nerve originate from primary afferent neurons. Brain Research. 1986;372:37–44.
Shehab S.A., Spike R.C., Todd A.J. Evidence against cholera toxin B subunit as a reliable tracer for sprouting of primary afferents following peripheral nerve injury. Brain Research. 2003;964:218–227.
Shi T.J., Liu S.X., Hammarberg H., et al. Phospholipase Cβ3 in mouse and human dorsal root ganglia and spinal cord is a possible target for treatment of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20004–20008.
Shi T.J., Tandrup T., Bergman E., et al. Effect of peripheral nerve injury on dorsal root ganglion neurons in the C57 BL/6J mouse: marked changes both in cell numbers and neuropeptide expression. Neuroscience. 2001;105:249–263.
Shortland P., Molander C. The time-course of A beta-evoked c-fos expression in neurons of the dorsal horn and gracile nucleus after peripheral nerve injury. Brain Research. 1998;810:288–293.
Sindrup S.H., Jensen T.S. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83:389–400.
Sugimoto T., Bennett G.J., Kajander K.C. Transsynaptic degeneration in the superficial dorsal horn after sciatic nerve injury: effects of a chronic constriction injury, transection, and strychnine. Pain. 1990;42:205–213.
Sung B., Lim G., Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. Journal of Neuroscience. 2003;23:2899–2910.
Suter M.R., Wen Y.R., Decosterd I., et al. Do glial cells control pain? Neuron Glia Biology. 2007;3:255–268.
Suzuki R., Kontinen V.K., Matthews E., et al. Enlargement of the receptive field size to low intensity mechanical stimulation in the rat spinal nerve ligation model of neuropathy. Experimental Neurology. 2000;163:408–413.
Svensson C.I., Brodin E. Spinal astrocytes in pain processing: non-neuronal cells as therapeutic targets. Molecular Interventions. 2010;10:25–38.
Takaishi K., Eisele J.H., Jr., Carstens E. Behavioral and electrophysiological assessment of hyperalgesia and changes in dorsal horn responses following partial sciatic nerve ligation in rats. Pain. 1996;66:297–306.
Tandrup T., Woolf C.J., Coggeshall R.E. Delayed loss of small dorsal root ganglion cells after transection of the rat sciatic nerve. Journal of Comparative Neurology. 2000;422:172–180.
Thompson S.W., Bennett D.L., Kerr B.J., et al. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7714–7718.
Tong Y.G., Wang H.F., Ju G., et al. Increased uptake and transport of cholera toxin B-subunit in dorsal root ganglion neurons after peripheral axotomy: possible implications for sensory sprouting. Journal of Comparative Neurology. 1999;404:143–158.
Tsuda M., Inoue K., Salter M.W. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends in Neurosciences. 2005;28:101–107.
Vestergaard S., Tandrup T., Jakobsen J. Effect of permanent axotomy on number and volume of dorsal root ganglion cell bodies. Journal of Comparative Neurology. 1997;388:307–312.
Villar M.J., Cortés R., Theodorsson E., et al. Neuropeptide expression in rat dorsal root ganglion cells and spinal cord after peripheral nerve injury with special reference to galanin. Neuroscience. 1989;33:587–604.
Wakisaka S., Kajander K.C., Bennett G.J. Increased neuropeptide Y (NPY)-like immunoreactivity in rat sensory neurons following peripheral axotomy. Neuroscience Letters. 1991;124:200–203.
Wall P.D., Fitzgerald M., Gibson S.J. The response of rat spinal cord cells to unmyelinated afferents after peripheral nerve section and after changes in substance P levels. Neuroscience. 1981;6:2205–2215.
Wall P.D., Gutnick M. Ongoing activity in peripheral nerves: the physiology and pharmacology of impulses originating from a neuroma. Experimental Neurology. 1974;43:580–593.
Wall P.D., Woolf C.J. The brief and the prolonged facilitatory effects of unmyelinated afferent input on the rat spinal cord are independently influenced by peripheral nerve section. Neuroscience. 1986;17:1199–1205.
Wang H., Sun H., Della Penna K., et al. Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience. 2002;114:529–546.
Wang R., Guo W., Ossipov M.H., et al. Glial cell line–derived neurotrophic factor normalizes neurochemical changes in injured dorsal root ganglion neurons and prevents the expression of experimental neuropathic pain. Neuroscience. 2003;121:815–824.
Weng H.R., Cordella J.V., Dougherty P.M. Changes in sensory processing in the spinal dorsal horn accompany vincristine-induced hyperalgesia and allodynia. Pain. 2003;103:131–138.
Wernersson J., Johansson I., Larsson U., et al. Activated transcription of the human neuropeptide Y gene in differentiating SH-SY5Y neuroblastoma cells is dependent on transcription factors AP-1, AP-2alpha, and NGFI. Journal of Neurochemistry. 1998;70:1887–1897.
Wetmore C., Olson L. Neuronal and nonneuronal expression of neurotrophins and their receptors in sensory and sympathetic ganglia suggest new intercellular trophic interactions. Journal of Comparative Neurology. 1995;353:143–159.
White D.M. Intrathecal neuropeptide Y exacerbates nerve injury–induced mechanical hyperalgesia. Brain Research. 1997;750:141–146.
White F.A., Jung H., Miller R.J. Chemokines and the pathophysiology of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:20151–20158.
Wiesenfeld-Hallin Z., Xu X.J. Neuropeptides in neuropathic and inflammatory pain with special emphasis on cholecystokinin and galanin. European Journal of Pharmacology. 2001;429:49–59.
Wiesenfeld-Hallin Z., Xu X.J., Crawley J.N., et al. Galanin and spinal nociceptive mechanisms: recent results from transgenic and knock-out models. Neuropeptides. 2005;39:207–210.
Wiesenfeld-Hallin Z., Xu X.J., Håkanson R., et al. Plasticity of the peptidergic mediation of spinal reflex facilitation after peripheral nerve section in the rat. Neuroscience Letters. 1990;116:293–298.
Williams A.G., Hargreaves A.C., Gunn-Moore F.J., et al. Stimulation of neuropeptide Y gene expression by brain-derived neurotrophic factor requires both the phospholipase Cgamma and Shc binding sites on its receptor, TrkB. Biochemical Journal. 1998;333:505–509.
Woolf C.J., Mannion R.J. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964.
Woolf C.J., Salter M.W. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769.
Woolf C.J., Wiesenfeld-Hallin Z. Substance P and calcitonin gene–related peptide synergistically modulate the gain of the nociceptive flexor withdrawal reflex in the rat. Neuroscience Letters. 1986;66:226–230.
Xiao H.S., Huang Q.H., Zhang F.X., et al. Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8360–8365.
Xu I.S., Hao J.X., Xu X.J., et al. The effect of intrathecal selective agonists of Y1 and Y2 neuropeptide Y receptors on the flexor reflex in normal and axotomized rats. Brain Research. 1999;833:251–257.
Xu X.J., Hokfelt T., Wiesenfeld-Hallin Z. Galanin and spinal pain mechanisms: where do we stand in 2008? Cellular and Molecular Life Sciences. 2008;65:1813–1819.
Yakhnitsa V., Linderoth B., Meyerson B.A. Spinal cord stimulation attenuates dorsal horn neuronal hyperexcitability in a rat model of mononeuropathy. Pain. 1999;79:223–233.
Yang L., Zhang F.X., Huang F., et al. Peripheral nerve injury induces trans-synaptic modification of channels, receptors and signal pathways in rat dorsal spinal cord. European Journal of Neuroscience. 2004;19:871–883.
Zhang X., Shi T., Holmberg K., et al. Expression and regulation of the neuropeptide Y Y2 receptor in sensory and autonomic ganglia. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:729–734.
Zhou X.F., Rush R.A. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience. 1996;74:945–953.
Zigmond RE, Hyatt-Sachs H, Mohney RP, et al. Changes in neuropeptide phenotype after axotomy of adult peripheral neurons and the role of leukemia inhibitor factor. Perspectives in Developmental Neurobiology. 1996;4:75–90.
Amir R., Devor M. Chemically mediated cross-excitation in rat dorsal root ganglia. Journal of Neuroscience. 1996;16:4733–4741.
Arnér L.D., Meyerson B.A. Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain. 1988;33:11–23.
Bao L., Jin S.X., Zhang C., et al. Activation of delta opioid receptors induces receptor insertion and neuropeptide secretion. Neuron. 2003;37:121–133.
Basbaum A.I., Bautista D.M., Scherrer G., et al. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284.
Besse D., Lombard M.C., Perrot S., et al. Regulation of opioid binding sites in the superficial dorsal horn of the rat spinal cord following loose ligation of the sciatic nerve: comparison with sciatic nerve section and lumbar dorsal rhizotomy. Neuroscience. 1992;50:921–933.
Castro-Lopes J.M., Malcangio M., Pan B.H., et al. Complex changes of GABAA and GABAB receptor binding in the spinal cord dorsal horn following peripheral inflammation or neurectomy. Brain Research. 1995;679:289–297.
Chapman V., Suzuki R., Dickenson A.H. Electrophysiological characterization of spinal neuronal response properties in anaesthetized rats after ligation of spinal nerves L5-L6. Journal of Physiology. 1998;507:881–894.
Coggeshall R.E., Lekan H.A., White F.A., et al. A-fiber sensory input induces neuronal cell death in the dorsal horn of the adult rat spinal cord. Journal of Comparative Neurology. 2001;435:276–282.
Colvin L.A., Mark M.A., Duggan A.W. Bilaterally enhanced dorsal horn postsynaptic currents in a rat model of peripheral mononeuropathy. Neuroscience Letters. 1996;207:29–32.
Costigan M., Befort K., Karchewski L., et al. Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neuroscience. 2002;3:16.
Costigan M., Scholz J., Woolf C.J. Neuropathic pain: a maladaptive response of the nervous system to damage. Annual Review of Neuroscience. 2009;32:1–32.
Del Fiacco M., Priestley J.V. Neurotrophins and adult somatosensory neurons. In: Mocchetti I., ed. Neurobiology of the neurotrophins. Johnson City, TN: FP Graham Publishing; 2001:205–236.
Devor M. The pathophysiology and anatomy of damaged nerve. In: Wall P.D., Melzack R., eds. Textbook of pain. Edinburgh: Churchill Livingstone; 1984:49–64.
Dubner R., Ruda M.A. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends in Neurosciences. 1992;15:96–103.
Fremeau R.T., Jr., Voglmaier S., Seal R.P., et al. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends in Neurosciences. 2004;27:98–103.
Hobson S.A., Bacon A., Elliot-Hunt C.R., et al. Galanin acts as a trophic factor to the central and peripheral nervous systems. Cellular and Molecular Life Sciences. 2008;65:1806–1812.
Hökfelt T., Zhang X., Wiesenfeld-Hallin Z. Messenger plasticity in primary sensory neurons following axotomy and its functional implications. Trends in Neurosciences. 1994;17:22–30.
Ji R.R., Gereau R.W., Malcangio M., et al. MAP kinase and pain. Brain Research Reviews. 2009;60:135–148.
Kashiba H., Senba E. Up- and down-regulation of BDNF mRNA in distinct subgroups of rat sensory neurons after axotomy. Neuroreport. 1999;10:3561–3565.
Kohama I., Ishikawa K., Kocsis J.D. Synaptic reorganization in the substantia gelatinosa after peripheral nerve neuroma formation: aberrant innervation of lamina II neurons by Abeta afferents. Journal of Neuroscience. 2000;20:1538–1549.
Lagerström M.C., Rogoz K., Abrahamsen B., et al. VGLUT2-dependent sensory neurons in the TRPV1 population regulate pain and itch. Neuron. 2010;68:529–542.
Lai J., Hunter J.C., Porreca F. The role of voltage-gated sodium channels in neuropathic pain. Current Opinion in Neurobiology. 2003;13:291–297.
Liu H.X., Hökfelt T. The participation of galanin in pain processing at the spinal level. Trends in Pharmacological Sciences. 2002;23:468–474.
Ludwig M., Pittman Q.J. Talking back: dendritic neurotransmitter release. Trends in Neurosciences. 2003;26:255–261.
Mantyh P.W. Cancer pain and its impact on diagnosis, survival and quality of life. Nature Reviews. Neuroscience. 2006;7:797–809.
Mantyh P.W., Rogers S.D., Honore P., et al. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278:275–279.
Michael G.J., Averill S., Shortland P.J., et al. Axotomy results in major changes in BDNF expression by dorsal root ganglion cells: BDNF expression in large trkB and trkC cells, in pericellular baskets, and in projections to deep dorsal horn and dorsal column nuclei. European Journal of Neuroscience. 1999;11:3539–3551.
Milligan E.D., Watkins L.R. Pathological and protective roles of glia in chronic pain. Nature Reviews. Neuroscience. 2009;10:23–36.
Nahin R.L., Marino De León K.R., Ruda M. Primary sensory neurons exhibit altered gene expression in a rat model of neuropathic pain. Pain. 1994;58:95–108.
Naveilhan P., Hassani H., Lucas G., et al. Reduced antinociception and plasma extravasation in mice lacking a neuropeptide Y receptor. Nature. 2001;409:513–517.
Schmidt B.L., Hamamoto D.T., Simone D.A., et al. Mechanism of cancer pain. Molecular Interventions. 2010;10:164–178.
Seal R.P., Wang X., Guan Y., et al. Injury-induced mechanical hypersensitivity requires C–low threshold mechanoreceptors. Nature. 2009;462:651–655.
Shehab S.A., Atkinson M.E. Vasoactive intestinal polypeptide (VIP) increases in the spinal cord after peripheral axotomy of the sciatic nerve originate from primary afferent neurons. Brain Research. 1986;372:37–44.
Shi T.J., Liu S.X., Hammarberg H., et al. Phospholipase Cβ3 in mouse and human dorsal root ganglia and spinal cord is a possible target for treatment of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20004–20008.
Sugimoto T., Bennett G.J., Kajander K.C. Transsynaptic degeneration in the superficial dorsal horn after sciatic nerve injury: effects of a chronic constriction injury, transection, and strychnine. Pain. 1990;42:205–213.
Suter M.R., Wen Y.R., Decosterd I., et al. Do glial cells control pain? Neuron Glia Biology. 2007;3:255–268.
Villar M.J., Cortés R., Theodorsson E., et al. Neuropeptide expression in rat dorsal root ganglion cells and spinal cord after peripheral nerve injury with special reference to galanin. Neuroscience. 1989;33:587–604.
Thompson S.W., Bennett D.L., Kerr B.J., et al. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7714–7718.
Tong Y.G., Wang H.F., Ju G., et al. Increased uptake and transport of cholera toxin B-subunit in dorsal root ganglion neurons after peripheral axotomy: possible implications for sensory sprouting. Journal of Comparative Neurology. 1999;404:143–158.
Tsuda M., Inoue K., Salter M.W. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends in Neurosciences. 2005;28:101–107.
Wakisaka S., Kajander K.C., Bennett G.J. Increased neuropeptide Y (NPY)-like immunoreactivity in rat sensory neurons following peripheral axotomy. Neuroscience Letters. 1991;124:200–203.
Wall P.D., Gutnick M. Ongoing activity in peripheral nerves: the physiology and pharmacology of impulses originating from a neuroma. Experimental Neurology. 1974;43:580–593.
Wiesenfeld-Hallin Z., Xu X.J., Håkanson R., et al. Plasticity of the peptidergic mediation of spinal reflex facilitation after peripheral nerve section in the rat. Neuroscience Letters. 1990;116:293–298.
Woolf C.J., Salter M.W. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769.
Xiao H.S., Huang Q.H., Zhang F.X., et al. Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8360–8365.
Xu X.J., Hokfelt T., Wiesenfeld-Hallin Z. Galanin and spinal pain mechanisms: where do we stand in 2008? Cellular and Molecular Life Sciences. 2008;65:1813–1819.
Yakhnitsa V., Linderoth B., Meyerson B.A. Spinal cord stimulation attenuates dorsal horn neuronal hyperexcitability in a rat model of mononeuropathy. Pain. 1999;79:223–233.
Zigmond R.E., Hyatt-Sachs H., Mohney R.P., et al. Changes in neuropeptide phenotype after axotomy of adult peripheral neurons and the role of leukemia inhibitor factor. Perspectives in Developmental Neurobiology. 1996;4:75–90.