chapter 68 Pharmacologic Management of Lower Urinary Tract Storage and Emptying Failure
The function of the lower urinary tract (LUT) is to effect efficient and low-pressure bladder filling, low-pressure urine storage with normal sensation and perfect continence, and periodic complete voluntary emptying, again at low pressure. The structures involved include the smooth musculature of the bladder and the bladder outlet and the striated muscle, both intrinsic (to the bladder outlet) and extrinsic (the striated musculature surrounding the bladder outlet and the striated musculature of the pelvic floor). These component structures are controlled by a complex interplay between the central and peripheral nervous systems and local regulatory factors. Pharmacologic alteration of LUT function can occur at any point along the afferent or efferent limb of this complex neuromuscular cascade, either by receptor activation/stimulation or blockade, by affecting the concentration of neurotransmitter at an activation site, or by stimulating or inhibiting signal transduction mechanisms (Andersson and Wein, 2004; Andersson et al, 2009; Birder et al, 2009; Fry et al, 2009; see also Chapter 60).
The focus of this chapter is the pharmacologic management of bladder filling/storage and bladder emptying/voiding dysfunction. The conceptual basis is that of the expanded functional classification seen in Table 61–5 and the division of therapies in the relatively simple-minded manner of those that facilitate urine storage/bladder filling and those that facilitate bladder emptying/voiding (see Tables 61–3 and 61–4). Although the principles expressed are generally applicable to all ages, specifics concerning usage in the elderly and pediatric age groups are considered in detail in Chapters 75, 126, and 127. Specific information regarding the pharmacologic management of LUT dysfunction secondary to obstruction by benign prostatic enlargement is considered in detail in Chapter 91, whereas drug therapy for the treatment of bladder and pelvic pain disorders is considered in detail in Chapters 11 and 12.
As an apology in explanation to significant contributors to the field whose works have not been specifically referenced by name as frequently as they could have been, please note the citations have been chosen primarily because of their comprehensive review or specific informational content and not because of originality or initial publication on a particular subject, except where noted.
Pharmacologic Therapy to Facilitate Bladder Filling and Urine Storage
Inhibiting Bladder Contractility, Decreasing Sensory Input, Increasing Bladder Capacity
Bladder Contraction and Muscarinic Receptors
The major portion of the neurohumoral stimulus for physiologic bladder contraction is acetylcholine-induced stimulation of postganglionic parasympathetic muscarinic cholinergic receptor sites in the bladder (detrusor smooth muscle and possibly other sites) (see Chapter 60). Atropine and atropine-like agents will depress normal bladder contractions and involuntary bladder contractions of any cause (Andersson, 1988; 1993; Andersson and Wein, 2004). In patients with involuntary contractions, the volume to the first involuntary contraction will generally be increased, the amplitude of the contraction decreased, and the total bladder capacity increased (Jensen, 1981).
Previously, it has been stated (Wein, 1998) that bladder compliance in normal individuals and in those with neurogenic detrusor overactivity (NDO; Abrams et al, 2002), in whom the initial slope of the filling curve on cystometry is normal before the involuntary contraction, does not seem to be significantly altered by antimuscarinic agents and that the effect of pure antimuscarinics in those patients who exhibit only decreased compliance had not been well studied. Regarding the subject of bladder tone during filling, Andersson (1999a, 1999b, 2004; Andersson and Yoshida, 2003) has pointed out that, although it is widely accepted that there is normally no sacral parasympathetic outflow to the bladder during filling, antimuscarinic drugs increase, and anticholinesterase inhibitors decrease, bladder capacity. Because antimuscarinic drugs do seem to affect the sensation of urgency during filling, this suggests an ongoing acetylcholine-mediated stimulation of detrusor tone (see later). If this is correct, agents inhibiting acetylcholine release or activity would be expected to contribute to bladder relaxation or the maintenance of low bladder tone during filling with a consequent decrease in filling/storage symptomatology unrelated to the occurrence of an involuntary contraction. Outlet resistance, at least as reflected by urethral pressure measurements, does not seem to be clinically affected.
Although the antimuscarinic agents usually produce significant clinical improvement in patients with involuntary contractions and associated symptoms, generally only partial inhibition results. In many animal models, atropine only partially antagonizes the response of the whole bladder to pelvic nerve stimulation and of bladder strips to field stimulation, although it does completely inhibit the response of bladder smooth muscle to exogenous cholinergic stimulation. This phenomenon, which is called atropine resistance, is secondary to release of a transmitter other than acetylcholine (see Andersson, 1993; Andersson et al, 1999c; Andersson and Wein, 2004; see also Chapter 60). Atropine resistance is the most common hypothesis invoked to explain the clinical difficulty in eradicating involuntary contractions with antimuscarinic agents alone, and it is also invoked to support the rationale of combined treatment of DO with agents that have different mechanisms of action (Andersson, 2006).
Andersson and Wein (2004) cite references stating that atropine resistance seems to be of little importance in normal human bladder muscle but point out that atropine-resistant (nonadrenergic, noncholinergic) contractions have been reported in human detrusor smooth muscle and in morphologically and/or functionally changed bladders in individuals with various types of voiding dysfunction. Thus, the importance or nonimportance of an atropine-resistant component to detrusor contraction in the treatment of DO in humans remains to be established.
Muscarinic Receptors
In the human bladder, where the mRNAs for all the five pharmacologically defined receptors, M1 to M5, have been demonstrated (Sigala et al, 2002; Abrams et al, 2006a; Giglio and Tobin, 2009), there is a predominance of mRNAs encoding M2 and M3 receptors (Yamaguchi et al, 1996; Sigala et al, 2002; Abrams et al, 2006a; Giglio and Tobin, 2009). This seems to be the case also in the animal species investigated (Hegde and Eglen, 1999; Chess-Williams, 2002; Andersson and Arner, 2004). Both M2 and M3 receptors can be found on detrusor muscle cells, where M2 receptors predominate at least 3 : 1 over M3 receptors, but also in other bladder structures, which may be of importance for detrusor activation. Thus, muscarinic receptors can be found on urothelial cells, on suburothelial nerves, and on other suburothelial structures, such as interstitial cells (Chess-Williams, 2002; Gillespie et al, 2003; Gillespie, 2004b; Mansfield et al, 2005; Bschleipfer et al, 2007; Giglio and Tobin, 2009).
In human as well as animal detrusor, the M3 receptors are believed to be the most important for contraction (Andersson, 1993; Chess-Williams, 2002; Abrams et al, 2006a; Giglio and Tobin, 2009). No differences between genders could be demonstrated in rat and human bladders (Kories et al, 2003). The functional role for the M2 receptors has not been clarified, and even in M3 receptor knockout mice they seem responsible for less that 5% of the carbachol-mediated detrusor contraction (Matsui et al, 2000). Stimulation of M2 receptors has been shown to oppose sympathetically (β-adrenoreceptor [β-AR]) mediated smooth muscle relaxation (Hegde et al, 1997). However, based on animal experiments, M2 receptors have been suggested to directly contribute to contraction of the bladder in certain disease states (denervation, outflow obstruction). Experiments on human detrusor muscle by Stevens and associates (2007) could not confirm this; however, Pontari and colleagues (2004) analyzed bladder muscle specimens from patients with neurogenic bladder dysfunction to determine whether the muscarinic receptor subtype mediating contraction shifts from M3 to the M2 receptor subtype, as found in the denervated, hypertrophied rat bladder. They concluded that normal detrusor contraction is mediated by the M3 receptor subtype whereas contractions can be mediated by the M2 receptors in patients with neurogenic bladder dysfunction.
Muscarinic receptors are coupled to G proteins, but the signal transduction systems may vary. Generally, M1, M3, and M5 receptors are considered to couple preferentially to Gq/11, activating phosphoinositide hydrolysis, in turn leading to mobilization of intracellular calcium (Ca2+). M2 and M4 receptors couple to pertussis toxin–sensitive Gi/o, resulting in inhibition of adenylate cyclase activity. In the human detrusor, Schneider and coworkers (2004), confirming that the muscarinic receptor subtype mediating carbachol-induced contraction is the M3 receptor, also demonstrated that the phospholipase-C inhibitor U73122 did not significantly affect carbachol-stimulated bladder contraction, despite blocking inositol 1,4,5-triphosphate generation. They concluded that carbachol-induced contraction of the human urinary bladder is mediated via M3 receptors and largely depends on Ca2+ entry through nifedipine-sensitive channels and activation of the Rho-kinase pathway. Thus, it may be that the main pathways for muscarinic receptor activation of the detrusor via M3 receptors are Ca2+ influx via L-type Ca2+ channels and increased sensitivity to Ca2+ of the contractile machinery via inhibition of myosin light-chain phosphatase through activation of Rho-kinase.
The signaling mechanisms for the M2 receptors are less clear than those for M3 receptors. As mentioned previously, M2 receptor stimulation may oppose sympathetically induced smooth muscle relaxation, mediated by β-ARs via inhibition of adenylyl cyclase (Hegde et al, 1997). In agreement with this, Matsui and colleagues (2003) suggested, based on results obtained in M2 receptor knockout mice, that a component of the contractile response to muscarinic agonists in smooth muscle involves an M2 receptor–mediated inhibition of the relaxant effects of agents that increase cyclic adenosine monophosphate (cAMP) levels. M2 receptor stimulation can also activate nonspecific cation channels and inhibit KATP channels through activation of protein kinase C (Bonev and Nelson, 1993; Kotlikoff et al, 1999).
Muscarinic receptors may also be located on the presynaptic nerve terminals and participate in the regulation of transmitter release. The inhibitory prejunctional muscarinic receptors have been classified as muscarinic M2 in the rabbit (Tobin and Sjögren, 1995) and rat (Somogyi and de Groat, 1992) and as M4 in the guinea pig (Alberts, 1995) and human bladder (D’Agostino et al, 2000). Prejunctional facilitatory muscarinic receptors appear to be of the M1 subtype in the bladders of rats, rabbits (Somogyi and de Groat, 1992; Tobin and Sjögren, 1995, 1998), and humans (Somogyi and de Groat, 1999; Giglio and Tobin, 2009). The muscarinic facilitatory mechanism seems to be upregulated in overactive bladders from chronic spinal cord–transected rats. The facilitation in these preparations is primarily mediated by M3 muscarinic receptors (Somogyi and de Groat, 1999).
The relative roles of the different presynaptic and postsynaptic receptor subtypes in normal and abnormal bladder function still require clarification, and thus speculation regarding optimal drug therapy based only on in-vitro receptor selectivity profiles represents, at the very least, a gross oversimplification of assumptions regarding the muscarinic regulation of bladder function. The muscarinic receptor functions may be changed in different urologic disorders, such as outflow obstruction, neurogenic bladders, DO without overt neurogenic cause, and diabetes (Andersson, 2000b). However, it is not always clear what the changes mean in terms of changes in detrusor function.
In general, all drug therapy for LUT dysfunction is hindered by a concept that can be expressed in one word: “uroselectivity” (Andersson, 1998). The clinical utility of available antimuscarinic agents is limited by their lack of selectivity, which is responsible for the classic peripheral antimuscarinic side effects of dry mouth, constipation, blurred vision, and increase in heart rate and for the effects on cognitive functions. Although M3 receptor selective agents have the potential to eliminate some of these side effects, it would appear that the M3 receptors in tissues of the LUT are identical to those elsewhere in the body (Caulfield and Birdsall, 1998). It may be speculated, however, that there is some heterogeneity among M3 receptors, and this has prompted many pharmaceutical companies to continue to search for the “ideal” antimuscarinic agent to treat DO, one that would be relatively selective for muscarinic receptors involved in the regulation of bladder contraction. Receptor selectivity, however, is not the only basis on which a drug may be “uroselective.” From a clinical standpoint it would seem particularly important to be able to describe in relative terms the ratio between a drug dose required for a desired therapeutic action and the dose producing side effects. A differential effect could be based not only on receptor selectivity but also on other known and as yet undefined physiologic, pharmacologic, or metabolic characteristics. Organ selectivity would thus seem to be the “holy grail” of such therapy. The same problematic set of concepts applies to virtually all drugs used for the treatment of LUT dysfunction.
Drugs Used for Treatment of Detrusor Overactivity/Overactive Bladder Symptomatology
The prevalence of the overactive bladder (OAB) syndrome varies with the criteria for diagnosis. According to Irwin and associates (2006), using the 2002 International Continence Society (ICS) definition, the overall prevalence (the EPIC study) was 11.8%. The rates were similar in men and women and increased with age. A similar study by Herschhorn and coworkers (2008) found the prevalence to be 13% in Canadian men and 14% in Canadian women. It appears that OAB/DO may be the result of several different mechanisms, both myogenic and neurologic (Morrison et al, 2002; Birder et al, 2009; Koebl et al, 2009). Most probably, both factors contribute to the genesis of the disease.
An abundance of drugs has been used for the treatment of OAB/DO. It should be stressed that in many trials on OAB/DO there has been such a high placebo response that meaningful differences between placebo and active drug cannot be demonstrated (Thüroff et al, 1998). However, drug effects in individual patients may be both distinct and useful.
The 4th International Consultation on Incontinence (ICI) (2008) assessed drugs used for treatment of incontinence (Andersson et al, 2009). The assessment criteria (Table 68–1) were based on the Oxford guidelines, and the drugs included are given in Table 68–2.
Table 68–1 International Consultation on Incontinence (ICI) Assessments 2004, 2008: Oxford Guidelines (Modified)
| Levels of Evidence |
| Grades of Recommendation |
Table 68–2 Drugs Used in the Treatment of Detrusor Overactivity (ICI, 2008; Andersson et al, 2009)
| ANTIMUSCARINIC DRUG | LEVEL OF EVIDENCE | GRADE OF RECOMMENDATION |
|---|---|---|
| Tolterodine | 1 | A |
| Trospium | 1 | A |
| Solifenacin | 1 | A |
| Darifenacin | 1 | A |
| Fesoterodine | 1 | A |
| Propantheline | 2 | B |
| Atropine, hyoscyamine | 3 | C |
| Drugs Acting on Membrane Channels | ||
| Calcium antagonists | 2 | D |
| Potassium channel openers | 2 | D |
| Drugs with Mixed Actions | ||
| Oxybutynin | 1 | A |
| Propiverine | 1 | A |
| Flavoxate | 2 | D |
| Antidepressants | ||
| Imipramine | 3 | C |
| Duloxetin | 2 | C |
| α-Adrenergic Receptor Antagonists | ||
| Alfuzosin | 3 | C |
| Doxazosin | 3 | C |
| Prazosin | 3 | C |
| Terazosin | 3 | C |
| Tamsulosin | 3 | C |
| β-Adrenergic Receptor Antagonists | ||
| Terbutaline (β2) | 3 | C |
| Salbutamol (β2) | 3 | C |
| YM178 (β3) | 2 | BPDE- |
| Phosphodiesterase-5 Inhibitors | ||
| Sildenafil, Tadalafil, Vardenafil | 2 | B |
| Cyclooxygenase Inhibitors | ||
| Indomethacin | C | |
| Flurbiprofen | 2 | C |
| Toxins | ||
| Botulinum toxin (neurogenic)* | 2 | A |
| Botulinum toxin (idiopathic)* | 3 | B |
| Capsaicin† | 2 | C |
| Resiniferatoxin† | 2 | C |
| Other Drugs | ||
| Baclofen‡ | 3 | C |
| Hormones | ||
| Estrogen | 2 | C |
| Desmopressin§ | 1 | A |
Antimuscarinic (Anticholinergic) Agents
Mechanism of Action
For many years, antimuscarinic drugs have been the gold standard for treatment of OAB. Still, the way(s) by which they exert their beneficial effect has (have) not yet been established.
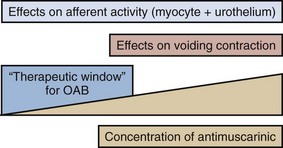
Acetylcholine stimulates both muscarinic and nicotinic receptors. Antimuscarinic agents block selectively muscarinic receptors, and they are currently the mainstay of treatment of OAB/DO (Abrams and Andersson, 2007). The traditional view is that in OAB/DO the drugs act by blocking the muscarinic receptors on the detrusor muscle, which are stimulated by acetylcholine released from activated cholinergic (parasympathetic) nerves. Thereby, they decrease the ability of the bladder to contract. However, antimuscarinic drugs act mainly during the storage phase, decreasing urgency and increasing bladder capacity; and during this phase there is normally no parasympathetic input to the LUT (Morrison et al, 2002; Andersson, 2004). Furthermore, antimuscarinic agents are usually competitive antagonists. This implies that when there is a massive release of acetylcholine, as during micturition, the effects of the drugs should be decreased, otherwise the reduced ability of the detrusor to contract would eventually lead to urinary retention. Undeniably, high doses of antimuscarinic agents can produce urinary retention in humans, but in the dose range used for beneficial effects in OAB/DO there is little evidence for a significant reduction of the voiding contraction (Finney et al, 2006) (Fig. 68–1). There is indirect clinical evidence for release of acetylcholine during bladder filling in certain abnormal conditions. Smith and colleagues (1974) found that in patients with recent spinal cord injury, inhibition of acetylcholine breakdown by use of cholinesterase inhibitors could increase resting tone and induce rhythmic contractions in the bladder. Yossepowitch and coworkers (2001) inhibited acetylcholine breakdown with edrophonium in a series of patients with disturbed voiding or urinary incontinence. They found a significant change in sensation and decreased bladder capacity, induction or amplification of involuntary detrusor contractions, or significantly decreased detrusor compliance in 78% of the patients with the symptom pattern of OAB but in no patients without specific complaints suggesting DO. Thus, during the storage phase, acetylcholine may be released from both neuronal and non-neuronal sources (e.g., the urothelium/suburothelium) and directly or indirectly (by increasing detrusor smooth muscle tone) excite afferent nerves in the suburothelium and within the detrusor. There is also good experimental evidence that antimuscarinic agents act during the storage phase by decreasing the activity in afferent nerves (both C and Aδ fibers) from the bladder (De Laet et al, 2006; Iijima et al, 2007).
Muscarinic receptors are found on bladder urothelial cells where their density can be even higher than in detrusor muscle. The role of the urothelium in bladder activation has attracted much interest (Andersson, 2002a; de Groat, 2004; Birder and de Groat, 2007; Birder et al, 2009; Giglio and Tobin, 2009), but whether the muscarinic receptors on urothelial cells can influence micturition has not yet been established. Yoshida and colleagues (2004, 2006, 2008) found that there is basal acetylcholine release in the human bladder. This release was resistant to tetrodotoxin and much diminished when the urothelium was removed; thus, the released acetylcholine was probably of non-neuronal origin and, at least partly, generated by the urothelium. Thus, during the storage phase, acetylcholine and adenosine triphosphate (ATP) may be released from both neuronal and non-neuronal sources (e.g., the urothelium) and directly or indirectly (by increasing detrusor smooth muscle tone) excite afferent nerves in the suburothelium and within the detrusor. These mechanisms may be important in the pathophysiology of OAB and represent possible targets for antimuscarinic drugs.
Pharmacologic Properties
Generally, antimuscarinic agents can be divided into tertiary and quaternary amines (Guay, 2003; Abrams and Andersson, 2007). They differ with regard to lipophilicity, molecular charge, and even molecular size, with tertiary compounds generally having higher lipophilicity and molecular charge than quaternary agents. Atropine, darifenacin, fesoterodine (and its active metabolite 5-hydroxymethyl-tolterodine), oxybutynin, propiverine, solifenacin, and tolterodine, are tertiary amines. They are generally well absorbed from the gastrointestinal tract and should theoretically be able to pass into the central nervous system (CNS), dependent on their individual physicochemical properties. High lipophilicity, small molecular size, and less charge will increase the possibilities to pass the blood-brain barrier, but for some of the drugs this is counteracted by active transport out of the CNS by P-glycoprotein. Quaternary ammonium compounds, such as propantheline and trospium, are not well absorbed, pass into the CNS to a limited extent, and have a low incidence of CNS side effects (Guay, 2003). They still produce well-known peripheral antimuscarinic side effects, such as accommodation paralysis, constipation, increase in heart rate, and dryness of mouth.
Many antimuscarinic drugs are metabolized by the cytochrome P450 enzyme system to active and/or inactive metabolites (Guay, 2003). The most commonly involved P450 enzymes are CYP2D6 and CYP3A4. The metabolic conversion creates a risk for drug-drug interactions, resulting in either reduced (enzyme induction) or increased (enzyme inhibition, substrate competition) plasma concentration/effect of the antimuscarinic and/or interacting drug. Antimuscarinic agents secreted by the renal tubules (e.g., trospium) may theoretically be able to interfere with the elimination of other drugs using this mechanism.
Antimuscarinic drugs are still the most widely used treatment for urgency and urgency incontinence (Andersson, 2004; Andersson et al, 2009). However, currently used drugs lack selectivity for the bladder, and effects on other organ systems (Table 68–3) may result in side effects that limit their usefulness. For example, all antimuscarinic drugs are contraindicated in patients with untreated narrow-angle glaucoma.
Table 68–3 Muscarinic Receptors: Distribution and Function
| REGION | SUBTYPE OF MUSCARINIC RECEPTOR | FUNCTION |
|---|---|---|
| Bladder | M2 > M3 (3 : 1) | M3: mediates human detrusor contraction |
| Salivary glands, parotid gland | M1, M3 | M1: high viscosity lubrication |
| M3: salivation | ||
| Gastrointestinal tract | M2 > M3 (4 : 1) | M3: stimulation of gastrointestinal motility |
| Brain | M1 to M5 (M3 sparse) | Involved in higher cognitive processes such as learning and memory (mostly M1) |
| Eye | M1 to M5 (M3 predominates) | Controls iris sphincter contraction |
| Heart | M2 | Modulates pacemaker activity, atrioventricular conduction, and the force of contraction |
Data from Abrams P, Andersson K-E, Buccafusco JJ, et al. Muscarinic receptors: their distribution and function in body systems, and the implications for treating overactive bladder. Br J Pharmacol 2006;148:565.
Theoretically, drugs with selectivity for the bladder could be obtained if the subtype(s) mediating bladder contraction, and those producing the main side effects of antimuscarinic drugs, were different. Unfortunately this does not seem to be the case. One way of avoiding many of the antimuscarinic side effects is to administer the drugs intravesically. However, this is practical only in a limited number of patients.
Clinical Efficacy
Several antimuscarinic drugs are and have been used for treatment of OAB/DO. For many of them documentation of effects is not based on randomized controlled trials (RCTs) satisfying currently required criteria, and some drugs can be considered as obsolete (e.g., emepronium). Information on these drugs is found elsewhere (Andersson, 1988; Andersson et al, 1999c).
The clinical relevance of efficacy of antimuscarinic drugs relative to placebo has been questioned. Herbison and associates (2003) stated in a widely discussed article: “Anticholinergics produce significant improvements in overactive bladder symptoms compared with placebo. The benefits are, however, of limited clinical significance.” Large meta-analyses of studies performed with the currently most widely used drugs (Chapple et al, 2005, 2008a; Novara et al, 2008) clearly show that antimuscarinic drugs are of significant clinical benefit. Novara and colleagues (2008) reviewed 50 RCTs and 3 pooled analyses that they considered of good methodologic quality. They concluded that still more clinical studies are needed to decide which of the drugs should be used as first-, second-, or third-line treatment. Reviewing information from more than 12,000 references, Chapple and colleagues (2008a) based their conclusions (“antimuscarinics are efficacious, safe, and well tolerated treatments”) on 73 RCTs selected for their meta-analysis. It was recommended that because the profiles of each drug and dosage differ that these factors should be considered in making treatment choices.
The consequence of this is that, in the authors’ opinion, none of the antimuscarinic drugs in common clinical use (darifenacin, fesoterodine, oxybutynin, propiverine, solifenacin, tolterodine, or trospium) is ideal as a first-line treatment for all OAB/DO patients. Optimal treatment should be individualized, implying that the patient’s comorbidities and concomitant medications, and the pharmacologic profiles of the different drugs, should be taken into consideration (Chapple et al, 2008a).
Behavioral therapy (see Chapter 69) should always be used in conjunction with drug therapy for OAB/DO because most studies show the effects of the two combined are greater than the effect of either alone (Burgio et al, 2000; Mattiasson et al, 2003; Klutke et al, 2009).
Tolerability and Safety
An extensive literature supports that antimuscarinic drugs for the treatment of OAB symptoms are generally well tolerated. The adverse effect profiles of the different drugs are determined by their organ and muscarinic receptor subtype selectivities and pharmacokinetic parameters. The most commonly reported adverse effects are dry mouth, constipation, headache, and blurred vision.
Among the more serious possibilities related to antimuscarinic use is the potential risk of cardiac adverse effects, particularly increases in heart rate and QT prolongation and induction of polymorphic ventricular tachycardia (torsades de pointes). QT prolongation and its consequences are not related to blockade of muscarinic receptors but rather linked to inhibition of the hERG potassium (K+) channel in the heart (Roden, 2004). Thus QT prolongation is not a class effect of antimuscarinic drugs. In general the cardiovascular safety for antimuscarinic drugs seems to be good. However, the potential of the different agents to increase heart rate or to prolong the QT time has not been extensively explored. Differences between the drugs cannot be excluded, but risk assessments based on available evidence are not possible.
Another concern is that antimuscarinic drugs commonly used to treat OAB can be associated with CNS side effects, including cognitive dysfunction, memory impairment, dizziness, fatigue, and headache. With the exception of oxybutynin IR, CNS-related side effects are not commonly found when investigated. The potential to cause CNS-related adverse effects may differ between drugs, but in the absence of comparative trials, relative risk assessments are not possible.
Atropine Sulfate
Atropine (dl-hyoscyamine) is rarely used for treatment of OAB/DO because of its systemic side effects, which preclude its use as an oral treatment. However, in patients with neurogenic DO, intravesical atropine may be effective for increasing bladder capacity without causing any systemic adverse effects, as shown in open pilot trials (Ekström et al, 1992; Glickman et al, 1995; Deaney et al, 1998; Enskat et al, 2001; Fader et al, 2007). It appears that intravesical atropine may be as effective as intravesical oxybutynin in patients with neurogenic DO (Fader et al, 2007). The pharmacologically active antimuscarinic component of atropine is l-hyoscyamine. Although it is still used, few clinical studies are available to evaluate the antimuscarinic activity of l-hyoscyamine sulfate (Muskat et al, 1996). For assessment, see Table 68–2.
Propantheline Bromide
Propantheline bromide (Pro-Banthine, others) is a quaternary ammonium compound, nonselective for muscarinic receptor subtypes, that has a low (5% to 10%) and individually varying biologic availability. It is metabolized (metabolites inactive) and has a short plasma half-life (<2 hr) (Beermann et al, 1972). It is usually given in a dose of 15 to 30 mg four times daily, but, to obtain an optimal effect, individual titration of the dose is necessary and often higher dosages are required. Using this approach in 26 patients with DO, Blaivas and associates (1980) in an open study obtained a complete clinical response in all patients but one, who did not tolerate more than propantheline, 15 mg, four times daily. The range of dosages varied from 7.5 to 60 mg four times daily. In contrast, Thüroff and coworkers (1991) comparing the effects or oxybutynin 5 mg three times a day, propantheline 15 mg three times a day, and placebo in a randomized, double-blind, multicenter trial on the treatment of frequency, urgency, and incontinence related to DO (154 patients) found no differences between the placebo and propantheline groups. In another randomized comparative trial with crossover design (23 women with idiopathic DO), and with dose titration, Holmes and associates (1989) found no differences in efficacy between oxybutynin and propantheline. There is a surprising lack of evaluable data on the effectiveness of propantheline for the treatment of DO. The Agency for Health Care Policy and Research (AHCPR) Clinical Practice Guidelines (U.S. Department of Health and Human Services, 1992) lists five randomized controlled trials reviewed for propantheline, with 82% female patients. Percent cures (all figures refer to percent effect on drug minus percent effect on placebo) are listed as 0% to 5%, reduction in urgency incontinence as 0% to 53%, and percent side effects and percent dropouts as 0% to 50% and 0% to 9%, respectively. RCTs (n = 6) reviewed by Thüroff and associates (1998) confirmed a positive, but varying, response to the drug.
Although the effect of propantheline on OAB/DO has not been well documented in controlled trials satisfying standards of today, it can be considered effective and may, in individually titrated doses, be clinically useful (see Table 68–2). No new studies on the use of this drug for the treatment of OAB/DO seem to have been performed during the past decade.
Trospium Chloride
Trospium is a quaternary ammonium compound with a biologic availability of less than 10% (Fusgen and Hauri, 2000; Doroshyenko et al, 2005). The drug has a plasma half-life of approximately 20 hours and is mainly (60% of the dose absorbed) eliminated unchanged in the urine. The concentration obtained in urine seems to be enough to affect the mucosal signaling system in a rat model (Kim et al, 2006). Whether it contributes to the clinical efficacy of the drug remains to be established. Trospium is not metabolized by the cytochrome P450 enzyme system (Beckmann-Knopp et al, 1999; Doroshyenko et al, 2005). It is expected to cross the blood-brain to a limited extent and seems to have no negative cognitive effects (Fusgen and Hauri, 2000; Todorova et al, 2001; Wiedemann et al, 2002).
Trospium has no selectivity for muscarinic receptor subtypes. In isolated detrusor muscle it was more potent than oxybutynin and tolterodine to antagonize carbachol-induced contractions (Uckert et al, 2000).
Several RCTs have documented positive effects of trospium both in neurogenic (Stöhrer, et al, 1991; Madersbacher et al, 1995; Menarini et al, 2006) and non-neurogenic DO (Allousi et al, 1998; Cardozo et al, 2000; Jünemann and Al-Shukri, 2000; Halaska et al, 2003; Zinner et al, 2004a; Rudy et al, 2006; Staskin et al, 2007; Dmochowski et al, 2008). In a placebo-controlled, double-blind study on patients with neurogenic DO (Stöhrer et al, 1991) the drug was given twice daily in a dose of 20 mg over a 3-week period. It increased maximum cystometric capacity, decreased maximum detrusor pressure, and increased compliance in the treatment group, whereas no effects were noted in the placebo group. Side effects were few and comparable in both groups. In another RCT including patients with spinal cord injuries and neurogenic DO, trospium and oxybutynin were equally effective; however, trospium seemed to have fewer side effects (Madersbacher et al, 1995). The effect of trospium in urgency incontinence has been documented in several RCTs. Allousi and associates (1998) compared the effects of the drug with those of placebo in 309 patients in a urodynamic study of 3 weeks’ duration. Trospium, 20 mg, was given twice daily. Significant increases were noted in volume at first involuntary contraction and in maximum bladder capacity. Cardozo and coworkers (2000) investigated 208 patients with DO who were treated with trospium, 20 mg twice daily, for 2 weeks. Also in this study, significant increases were found in mean volume at first unstable contraction (from 233 to 299 mL; placebo 254 to 255 mL) and in maximum bladder capacity (from 329 to 356 mL; placebo 345 to 335 mL) in the trospium-treated group. Trospium was well tolerated with similar frequency of adverse effects as in the placebo group. Jünemann and Al-Shukri (2000) compared trospium, 20 mg twice daily, with tolterodine, 2 mg twice daily, in a placebo-controlled double-blind study on 232 patients with urodynamically proven DO, urgency incontinence without demonstrable DO, or mixed incontinence. Trospium reduced the frequency of micturition, which was the primary end point, more than tolterodine and placebo and also reduced the number of incontinence episodes more than the comparators. Dry mouth was comparable in the trospium and tolterodine groups (7% and 9%, respectively). Halaska and colleagues (2003) studied the tolerability and efficacy of trospium chloride in doses of 20 mg twice daily for long-term therapy in patients with urgency syndrome. The trial comprised a total of 358 patients with urgency syndrome or urgency incontinence. After randomization in the ratio of 3 : 1, participants were treated continuously for 52 weeks with either trospium chloride (20 mg twice daily) or oxybutynin (5 mg twice daily). Urodynamic measurements were performed at the beginning and at 26 and 52 weeks to determine the maximum cystometric bladder capacity. Analysis of the micturition diary clearly indicated a reduction of the micturition frequency, incontinence frequency, and a reduction of the number of urgency episodes in both treatment groups. Mean maximum cystometric bladder capacity increased during treatment with trospium chloride by 92 mL after 26 weeks and 115 mL after 52 weeks (P = .001). Further comparison with oxybutynin did not reveal any statistically significant differences in urodynamic variables between the drugs. Adverse events occurred in 65% of the patients treated with trospium and 77% of those treated with oxybutynin. The main symptom encountered in both treatment groups was dryness of the mouth. An overall assessment for each of the drugs revealed a comparable efficacy level and a better benefit-risk ratio for trospium than for oxybutynin due to better tolerability. Zinner and coworkers (2004a) treated 523 patients with symptoms associated with OAB and urgency incontinence with trospium 20 mg twice daily or placebo in a 12-week, multicenter, parallel, double-blind, placebo-controlled trial. Dual primary end points were change in average number of toilet voids and change in urgency incontinent episodes per 24 hours. Secondary efficacy variables were change in average of volume per void, voiding urgency severity, urinations during day and night, time to onset of action, and change in Incontinence Impact Questionnaire. By week 12, trospium significantly decreased average frequency of toilet voids per 24 hours (−2.37; placebo −1.29) and urgency incontinent episodes (−59%; placebo −44%). It significantly increased average volume per void (32 mL; placebo: 7.7 mL) and decreased average urgency severity and daytime frequency. All effects occurred by week 1, and all were sustained throughout the study. Nocturnal frequency decreased significantly by week 4 (−0.43; placebo: 0.17), and Incontinence Impact Questionnaire scores improved at week 12. Trospium was well tolerated. The most common side effects were dry mouth (21.8%; placebo 6.5%), constipation (9.5%; placebo 3.8%). and headache (6.5%; placebo 4.6%). In a large U.S. multicenter trial with the same design, and including 658 patients with OAB, Rudy and coworkers (2006) confirmed the data by Zinner and associates (2004a), both with respect to efficacy and adverse effects. An extended-release formulation of trospium allowing once-daily dosing (60 mg) has been introduced and its effects tested in controlled trials (Staskin et al, 2007; Dmochowski et al, 2008). These RCTs demonstrated similar efficacy as found with previous formulations.
The most frequent side effects were dry mouth (12.9%; placebo 4.6%) and constipation (7.5%; placebo 1.8%) (Dmochowski et al, 2008).
Intravesical application of trospium may be an interesting alternative. Fröhlich and colleagues (1998) performed a randomized, single-blind, placebo-controlled, monocenter clinical trial in 84 patients with urgency or urgency incontinence. Compared with placebo, intravesical trospium produced a significant increase in maximum bladder capacity and a decrease of detrusor pressure accompanied by an increase of residual urine. There was an improvement in uninhibited bladder contractions. No adverse events were reported. Interestingly, intravesical trospium does not seem to be absorbed (Walter et al, 1999), thus offering an opportunity for treatment with minimal systemic antimuscarinic effects.
Trospium is a well-documented alternative for treatment of OAB/DO and seems to be well tolerated.
Tolterodine Tartrate
Tolterodine is a tertiary amine, rapidly absorbed and extensively metabolized by the cytochrome P450 system (CYP2D6). The major active 5-hydroxymethyl metabolite (5-HMT) has a similar pharmacologic profile as the mother compound (Nilvebrant et al, 1997b) and significantly contributes to the therapeutic effect of tolterodine (Brynne et al, 1997, 1998). Both tolterodine and 5-HMT have plasma half-lives of 2 to 3 hours, but the effects on the bladder seem to be more long lasting than could be expected from the pharmacokinetic data. Urinary excretion of tolterodine accounted for less than 1.0% to 2.4% of the dose; 5% to 14% of 5-HMT is eliminated in the urine (Brynne et al, 1997). Whether the total antimuscarinic activity of unchanged tolterodine and 5-HMT excreted in urine is sufficient to exert any effect on the mucosal signaling mechanisms has not been established. However, the preliminary studies by Kim and coworkers (2006) and Chuang and colleagues (2008) do not support such an effect. The relatively low lipophilicity of tolterodine and even lesser one of 5-HMT implies limited propensity to penetrate into the CNS, which may explain a low incidence of cognitive side effects (Hills et al, 1998; Clemett et al, 2001; Salvatore et al, 2008). However, tolterodine may disturb sleep in subjects unable to form the even less lipophilic 5-HMT due to a low activity of CYP2D6 (Diefenbach et al, 2008).
Tolterodine has no selectivity for muscarinic receptor subtypes but is claimed to have functional selectivity for the bladder over the salivary glands (Stahl et al, 1995; Nilvebrant et al, 1996, 1997a, 1997c). In healthy volunteers, orally given tolterodine in a high dose (6.4 mg) had a powerful inhibitory effect on micturition and also reduced stimulated salivation 1 hour after administration of the drug (Stahl et al, 1995); however, 5 hours after administration, the effects on the urinary bladder were maintained whereas no significant effects on salivation could be demonstrated. Tolterodine is available as immediate-release (TOLT-IR; 1 or 2 mg; twice-daily dosing) and extended-release (TOLT-ER) forms (2 or 4 mg; once-daily dosing). The ER form seems to have advantages over the IR form in terms of both efficacy and tolerability (Van Kerrebroeck et al, 2001).
Several randomized, double-blind, placebo-controlled studies on patients with OAB/DO (both idiopathic and neurogenic DO) have documented a significant reduction in micturition frequency and number of incontinence episodes (Hills et al, 1998; Clemett et al, 2001; Salvatore et al, 2008). Comparative RCTs such as the OBJECT (Overactive Bladder: Judging Effective Control and Treatment) and the OPERA (Overactive Bladder; Performance of Extended Release Agents) studies have further supported its effectiveness. The OBJECT trial compared oxybutynin ER (OXY-ER) 10 mg once daily with TOLT-IR 2 mg twice daily (Appell et al, 2001) in a 12-week randomized, double-blind, parallel-group study including 378 patients with OAB. Participants had between 7 and 50 episodes of urgency incontinence per week and 10 or more voids in 24 hours. The outcome measures were the number of episodes of urgency incontinence, total incontinence, and micturition frequency at 12 weeks adjusted for baseline. At the end of the study, OXY-ER was found to be significantly more effective than TOLT-IR in each of the main outcome measures adjusted for baseline (see also later discussion on oxybutynin chloride). Dry mouth, the most common adverse event, was reported by 28% and 33% of participants taking OXY-ER and TOLT-IR, respectively. Rates of CNS and other adverse events were low and similar in both groups. The authors concluded that OXY-ER was more effective than TOLT-IR and that the rates of dry mouth and other adverse events were similar in both treatment groups. In the OPERA study (Diokno et al, 2003), OXY-ER at 10 mg/day or TOLT-ER at 4 mg/day was given for 12 weeks to women with 21 to 60 urgency incontinence episodes per week and an average of 10 or more voids per 24 hours. Episodes of incontinence episodes (primary end point), total (urgency and nonurgency) incontinence, and micturition were recorded in seven 24-hour urinary diaries at baseline and at weeks 2, 4, 8, and 12 and compared. Adverse events were also evaluated. Improvements in weekly urgency incontinence episodes were similar for the 790 women who received OXY-ER (n = 391) or TOLT-ER (n = 399). OXY-ER was significantly more effective than TOLT-ER in reducing micturition frequency, and 23.0% of women taking OXY-ER reported no episodes of urinary incontinence compared with 16.8% of women taking TOLT-ER. Dry mouth (usually mild) was more common with OXY-ER. Adverse events were generally mild and occurred at low rates, with both groups having similar discontinuation of treatment due to adverse events. The conclusions were that reductions in weekly urgency incontinence and total incontinence episodes were similar with the two drugs. Dry mouth was more common with OXY-ER, but tolerability was otherwise comparable, including adverse events involving the CNS.
In the ACET (Antimuscarinic Clinical Effectiveness Trial) (Sussman and Garely, 2002) study, which consisted of two trials, patients with OAB were randomized to 8 weeks of open-label treatment with either 2 mg or 4 mg of once-daily TOLT-ER (study 1) and to 5 mg or 10 mg of OXY-ER (study 2). A total of 1289 patients were included. Fewer patients prematurely withdrew from the trial in the TOLT-ER 4-mg group (12%) than either the OXY-ER 5-mg (19%) or OXY-ER 10-mg groups (21%). More patients in the OXY-ER 10-mg group than the TOLT-ER 4-mg group withdrew because of poor tolerability (13% vs. 6%). After 8 weeks, 70% of patients in the TOLT-ER 4-mg group perceived an improved bladder condition, compared with 60% in the TOLT-ER 2-mg group, 59% in the OXY-ER 5-mg group and 60% in the OXY-ER 10-mg group. Dry mouth was dose dependent with both agents, although differences between doses reached statistical significance only in the oxybutynin trial (OXY-ER 5 mg vs. OXY-ER 10 mg; P = .05). Patients treated with TOLT-ER 4 mg reported a significantly lower severity of dry mouth compared with OXY-ER 10 mg. The conclusion that the findings suggest improved clinical efficacy of TOLT-ER (4 mg) over OXY-ER (10 mg) is weakened by the open-label design of the study.
Zinner and associates (2002) evaluated the efficacy, safety, and tolerability of TOLT-ER in older (≥65) and younger (<65) OAB patients in a 12-week RCT including 1015 patients with urgency incontinence and urinary frequency. Patients were randomized to treatment with TOLT-ER 4 mg once daily (n = 507) or placebo (n = 508) for 12 weeks. Efficacy, measured with micturition charts (incontinence episodes, micturitions, volume voided per micturition) and subjective patient assessments, safety, and tolerability end points were evaluated, relative to placebo. Compared with placebo, significant improvements in micturition chart variables with TOLT-ER showed no age-related differences. Dry mouth (of any severity) was the most common adverse event in both the TOLT-ER and placebo treatment study arms, irrespective of age (<65: TOLT-ER 22.7%, placebo 8.1%; ≥65: TOLT-ER 24.3%, placebo 7.2%). A few patients (<2%) experienced severe dry mouth. No CNS (cognitive functions were not specifically studied), visual, cardiac (per electrocardiogram), or laboratory safety concerns were noted in this study. Withdrawal rates due to adverse events on TOLT-ER 4 mg once daily were comparable in the two age cohorts (<65: 5.5%; ≥65: 5.1%).
The central symptom in the OAB syndrome is urgency. Freeman and coworkers (2003) presented a secondary analysis of a double-blind, placebo-controlled study evaluating the effect of once-daily TOLT-ER on urinary urgency in patients with OAB. Patients with urinary frequency (eight or more micturitions per 24 hours) and urgency incontinence (five or more episodes per week) were randomized to oral treatment with TOLT-ER 4 mg once daily (n = 398) or placebo (n = 374) for 12 weeks. Efficacy was assessed by use of patient perception evaluations. Of patients treated with TOLT-ER, 44% reported improved urgency symptoms (compared with 32% for placebo) and 62% reported improved bladder symptoms (placebo, 48%). The proportion of patients unable to hold urine on experiencing urgency was decreased by 58% with TOLT-ER, compared with 32% with placebo (P < .001). In the IMprovement in Patients: Assessing symptomatic Control with Tolterodine ER (IMPACT) study (Elinoff et al, 2006), the efficacy of TOLT-ER for patients’ most bothersome OAB symptom was investigated in an open-label, primary care setting. Patients with OAB symptoms for more than 3 months received TOLT-ER (4 mg once daily) for 12 weeks. By week 12 there were significant reductions in the patients’ most bothersome symptom—incontinence—urgency episodes, and nocturnal and daytime frequency. The most common adverse events were dry mouth (10%) and constipation (4%), and it was concluded that, in primary care practice, bothersome OAB symptoms can be effectively and safely treated with TOLT-ER, even in patients with comorbid conditions.
Various aspects of the efficacy and tolerability of tolterodine have been further documented in a number of RCTs (Dmochowski et al, 2007a, 2007b; Barucha et al, 2008; Choo et al, 2008; Coyne et al, 2008; Rogers et al, 2008; Rovner et al, 2008b; for further discussion see Chapple et al, 2008b; Novara et al, 2008). Importantly, the QTc effects of tolterodine were determined in a crossover-designed QT study of recommended (2 mg twice daily) and supratherapeutic (4 mg twice daily) doses of tolterodine, moxifloxacin (400 mg once daily), and placebo. No subject receiving tolterodine exceeded the clinically relevant thresholds of 500 msec absolute QTc or 60 msec change from baseline, and it was concluded that tolterodine does not have a clinically significant effect on QT interval (Malhotra et al, 2007). Olshansky and colleagues (2008) compared, in a randomized, placebo-controlled, double blind crossover study, the effects on heart rate of TOLT-ER 4 mg/day with those of darifenacin 15 mg/day and placebo in 162 healthy volunteers. They found that tolterodine, but not darifenacin and placebo, significantly increased mean heart rate per 24 hours. The treatment difference in adjusted mean change from baseline was +1.84 beats per minute (P = .0004) for tolterodine versus darifenacin and +1.42 beats per minute (P < .001) for tolterodine versus placebo. The proportion of subjects with an increase greater than 5 beats per minute was significantly greater in those receiving TOLT-ER (1 of 4) than with darifenacin (1 of 10). The clinical significance of this has yet to be established.
In a prospective, open study, Song and associates (2006) compared the effects of bladder training and/or tolterodine as first-line treatment in female patients with OAB. One hundred and thirty-nine female patients with OAB were randomized to treatment with bladder training, tolterodine (2 mg twice daily), or both for 12 weeks. All treatments were efficacious; however, combination therapy was the most effective. Mattiasson and colleagues (2003) compared the efficacy of tolterodine 2 mg twice daily plus simplified bladder training with tolterodine alone in patients with OAB in a multicenter single-blind study. At the end of the study the median percentage reduction in voiding frequency was greater with tolterodine plus bladder training than with tolterodine alone (33% vs. 25%; P < .001), whereas the median percentage increase in volume voided per void was 31% with tolterodine plus bladder training and 20% with tolterodine alone (P < .001). There was a median of 81% fewer incontinence episodes than at baseline with tolterodine alone, which was not significantly different from that with tolterodine plus bladder training (−87%). It was concluded that the effectiveness of tolterodine 2 mg twice daily can be augmented by a simplified bladder training regimen. However, Millard and colleagues (2004) investigated whether the combination of tolterodine plus a simple pelvic floor muscle exercise program would provide improved treatment benefits compared with tolterodine alone in 480 patients with OAB. Tolterodine therapy for 24 weeks resulted in significant improvement in urgency, frequency, and incontinence; however, no additional benefit was demonstrated for a simple pelvic floor muscle exercise program.
The beneficial effect of TOLT-ER in men with benign prostatic enlargement and LUTS, including OAB, has been well documented. Both as monotherapy, but particularly in combination with an α-adrenergic receptor (AR) antagonist, TOLT-ER was found effective (Kaplan et al, 2006; Höfner et al, 2007; Kaplan et al, 2008a; 2008b; Roehrborn et al, 2009; Rovner et al, 2008a). This effect was obtained irrespective of prostate size and was not associated with increased incidence of acute urinary retention (Roehrborn et al, 2009).
Tolterodine, in both the immediate- and extended-release forms, has a well-documented effect in OAB/DO.
Darifenacin Hydrobromide
Darifenacin is a tertiary amine with moderate lipophilicity, well absorbed from the gastrointestinal tract after oral administration, and extensively metabolized in the liver by the cytochrome P450 isoforms CYP3A4 and CYP2D6, the latter saturating within the therapeutic range (Skerjanec 2006). UK-148,993, UK-73,689, and UK-88862 are the three main circulating darifenacin metabolites, of which only UK-148,993 is said to have significant antimuscarinic activity. However, available information suggests that various metabolites of darifenacin contribute little to its clinical effects (Michel and Hegde, 2006). The metabolism of darifenacin by CYP3A4 suggests that coadministration of a potent inhibitor of this enzyme (e.g., ketoconazole) may lead to an increase in the circulating concentration of darifenacin (Kerbusch et al, 2003). Darifenacin is a relatively selective muscarinic M3 receptor antagonist. In vitro, it is selective for human cloned muscarinic M3 receptors relative to M1, M2, M4, or M5 receptors. Theoretically, drugs with selectivity for the M3 receptor can be expected to have clinical efficacy in OAB/DO with reduction of the adverse events related to the blockade of other muscarinic receptor subtypes (Andersson, 2002b). However, the clinical efficacy and adverse effects of a drug are dependent not only on its profile of receptor affinity but also on its pharmacokinetics and on the importance of muscarinic receptors for a given organ function.
Darifenacin has been developed as a controlled-release formulation, which allows once-daily dosing. Recommended dosages are 7.5 and 15 mg/day. The clinical effectiveness of the drug has been documented in several RCTs (Haab et al, 2004; Cardozo and Dixon 2005; Chapple et al, 2005, 2007a; Foote et al, 2005; Steers et al, 2005; Haab et al, 2006; Hill et al, 2006; Zinner et al, 2006; Abrams et al, 2008, Chancellor et al, 2008b; Dwyer et al, 2008; for reviews, see Guay, 2005; Zinner, 2007; Chapple et al, 2008; Novara et al, 2008). Haab and coworkers (2004) reported a multicenter, double-blind, placebo-controlled, parallel-group study that enrolled 561 patients (age 19 to 88; 85% female) with OAB symptoms for more than 6 months and included some patients with prior exposure to antimuscarinic agents. After washout and a 2-week placebo run-in, patients were randomized (1 : 4 : 2 : 3) to once-daily oral darifenacin controlled-release tablets: 3.75 mg (n = 53), 7.5 mg (n = 229) or 15 mg (n = 115) or matching placebo (n = 164) for 12 weeks. Patients recorded daily incontinence episodes, micturition frequency, bladder capacity (mean volume voided), frequency of urgency, severity of urgency, incontinence episodes resulting in change of clothing or pads, and nocturnal awakenings due to OAB using an electronic diary during weeks 2, 6, and 12 (directly preceding clinic visits). Tolerability data were evaluated from adverse event reports. Darifenacin 7.5 mg and 15 mg had a rapid onset of effect, with significant improvement compared with placebo being seen for most parameters at the first clinic visit (week 2). Darifenacin 7.5 mg and 15 mg were significantly superior to placebo for (median) improvements in micturition frequency (7.5 mg: −1.6; 15 mg: −1.7; placebo −0.8), frequency of urgency per day (−2.0; −2.0; −0.9), and number of incontinence episodes leading to a change in clothing or pads (−4.0; −4.7; −2.0). There was no significant reduction in nocturnal awakenings due to OAB. The most common adverse events were mild-to-moderate dry mouth and constipation with a CNS and cardiac safety profile comparable to that of placebo. No patients withdrew from the study as a result of dry mouth, and discontinuation related to constipation was rare (0.6% placebo vs. 0.9% darifenacin).
In a dose titration study on 395 OAB patients, darifenacin, allowing individualized dosing (7.5 or 15 mg), was found to be effective and well tolerated (Steers et al, 2005). A 2-year open-label extension study of these investigations (i.e., Haab et al, 2004; Steers et al, 2005) confirmed a favorable efficacy tolerability and safety profile (Haab et al, 2006). A review of the pooled darifenacin data from the three phase 3, multicenter, double-blind clinical trials in patients with OAB was reported by Chapple and coworkers (2005). After a 4-week washout/run-in period, 1059 adults (85% female) with symptoms of OAB (urgency incontinence, urgency and frequency) for at least 6 months were randomized to once-daily oral treatment with darifenacin: 7.5 mg (n = 337) or 15 mg (n = 334) or matching placebo (n = 388) for 12 weeks. Efficacy was evaluated using electronic patient diaries that recorded incontinence episodes (including those resulting in a change of clothing or pads), frequency and severity of urgency, micturition frequency, and bladder capacity (volume voided). Safety was evaluated by analysis of treatment-related adverse events, withdrawal rates, and laboratory tests. Relative to baseline, 12 weeks of treatment with darifenacin resulted in a dose-related significant reduction in median number of incontinence episodes per week: 7.5 mg, −8.8 (−68.4%; placebo −54%, P < .004); 15 mg, −10.6 (−76.8%; placebo 58%, P < .001). Significant decreases in the frequency and severity of urgency, micturition frequency, and number of incontinence episodes resulting in a change of clothing or pads were also apparent, along with an increase in bladder capacity. Darifenacin was well tolerated. The most common treatment-related adverse events were dry mouth and constipation, although together these resulted in few discontinuations (7.5 mg, 0.6% of patients; 15 mg, 2.1%; placebo, 0.3%). The incidence of CNS and cardiovascular adverse events was comparable to that of placebo. The results were confirmed in other RCTs, including also a pooled analysis of three phase 3 studies in older patients (>65 years), showing that darifenacin (7.5 and 15 mg) had an excellent efficacy, tolerability, and safety profile (Foote et al, 2005; Zinner et al, 2005; Hill et al, 2006).
One of the most noticeable clinical effects of antimuscarinic agents is their ability to reduce urgency and allow patients to postpone micturition. A study was conducted to assess the effect of darifenacin on the “warning time” associated with urinary urgency. This was a multicenter, randomized, double-blind, placebo-controlled study consisting of 2 weeks’ washout, 2 weeks’ medication-free run-in, and a 2-week treatment phase (Cardozo and Dixon, 2005). Warning time was defined as the time from the first sensation of urgency to voluntary micturition or incontinence and was recorded via an electronic event recorder at baseline (visit 3) and study end (visit 4) during a 6-hour clinic-based monitoring period, with the subject instructed to delay micturition for as long as possible. During each monitoring period, up to three urgency-void cycles were recorded. Of the 72 subjects who entered the study, 67 had warning time data recorded at both baseline and study end and were included in the primary efficacy analysis (32 on darifenacin, 35 on placebo). Darifenacin treatment resulted in a significant (P < .004) increase in mean warning time with a median increase of 4.3 minutes compared with placebo (darifenacin group, from 4.4 to 1.8 minutes; placebo, from 7.0 to −1.0 minute). Overall, 47% of darifenacin-treated subjects compared with 20% receiving placebo achieved a 30% or more increase in mean warning time. There were methodologic problems associated with this study; it utilized a dose of 30 mg (higher than the dose recommended for clinical use), the treatment period was short, it was conducted in a clinic-centered environment, the methodology carried with it a significant potential training effect, and the placebo group had higher baseline values than the treatment group. In another warning time study (Zinner et al, 2006) with 445 OAB patients, darifenacin treatment (15 mg) resulted in numerical increases in warning time; however, these were not significant compared with placebo.
Further studies have demonstrated that darifenacin treatment is associated with clinically relevant improvements on health-related quality of life in patients with OAB (Abrams et al, 2008), and such improvements were sustained as shown in a 2-year extension study (Dwyer et al, 2008). It was shown that the positive effects neither on micturition variables nor on health-related quality of life produced by darifenacin (7.5 and 15 mg) were further enhanced by a behavioral modification program including timed voiding, dietary modifications, and Kegel exercises (Chancellor et al, 2008b). Several studies have been devoted to study possible effect on cognition by darifenacin. Neither in healthy volunteers (19 to 44 years) and healthy subjects (>60 years) nor in volunteers 65 years or older could any effect of darifenacin (3.75 to 15 mg/day) be demonstrated compared with placebo (Kay and Wesnes, 2005; Lipton et al, 2005; Kay et al, 2006; Kay and Ebinger, 2008).
To study whether darifenacin had any effect on QT/QTc intervals, Serra and associates (2005) performed a 7-day, randomized, parallel-group study (n = 188) in healthy volunteers receiving once-daily darifenacin at steady-state therapeutic (15 mg) and supratherapeutic (75 mg) doses alongside control subjects receiving placebo or moxifloxacin (positive control, 400 mg) once daily. No significant increase in QTcF interval could be demonstrated compared with placebo. Mean changes from baseline at pharmacokinetic Tmax versus placebo were −0.4 and −2.2 msec in the darifenacin 15-mg and 75-mg groups, respectively, compared with +11.6 msec in the moxifloxacin group (P < .01). The conclusion was that darifenacin does not prolong the QT/QTc interval. Darifenacin 15 mg/day given to healthy volunteers did not change heart rate significantly compared with placebo (Olshansky et al, 2008).
Darifenacin has a well-documented effect in OAB/DO, and the adverse event profile seems acceptable.
Solifenacin Succinate
Solifenacin is a tertiary amine and well absorbed from the gastrointestinal tract (absolute bioavailability 90%). The mean terminal half-life is 45 to 68 hours (Kuipers et al, 2004; Smulders et al, 2004). It undergoes hepatic metabolism involving the cytochrome P450 enzyme system (CYP3A4). In subjects who received a single oral dose of 10 mg solifenacin on day 7 of a 20-day regimen of ketoconazole administration (200 mg) Cmax and AUC0-inf were increased by only approximately 40% and 56%, respectively (Swart et al, 2006). Solifenacin has a modest selectivity for M3 over M2 receptors and a marginal selectivity over M1 receptors (Ikeda et al, 2002; Abrams and Andersson, 2007).
Two large-scale phase 2 trials with parallel designs, comprising men and women, were performed (Smith et al, 2002; Chapple et al, 2004a). The first dose-ranging study evaluated solifenacin (2.5 mg, 5 mg, 10 mg, and 20 mg) and tolterodine (2 mg twice daily) in a multinational placebo-controlled study of 225 patients with urodynamically confirmed DO (Chapple et al, 2004a). Patients received treatment for 4 weeks followed by 2 weeks of follow-up. Inclusion criteria for this and subsequent phase 3 studies of patients with OAB included at least eight micturitions per 24 hours and either one episode of incontinence or one episode of urgency daily as recorded in 3-day micturition diaries. Micturition frequency, the primary efficacy variable, was statistically significantly reduced in patients taking solifenacin 5 mg (−2.21), 10 mg (−2.47), and 20 mg (−2.75) but not in patients receiving placebo (−1.03) or tolterodine (−1.79). This effect was rapid, with most of the effect observed at the earliest assessment visit, 2 weeks after treatment initiation. In addition there were numerically greater reductions in episodes of urgency and incontinence when compared with placebo. Study discontinuations due to adverse events were similar across treatment groups, albeit highest in the 20-mg solifenacin group. Because the 5-mg and 10-mg doses caused lower rates of dry mouth than tolterodine, and superior efficacy outcomes relative to placebo, these dosing strengths were selected for further evaluation in large-scale phase 3 studies. The second dose-ranging study of solifenacin 2.5 mg to 20 mg was carried out in the United States (Smith et al, 2002). This trial included 261 evaluable men and women receiving solifenacin or placebo for 4 weeks followed by a 2-week follow-up period. Micturition frequency was statistically significantly reduced relative to placebo in patients receiving 10 mg and 20 mg solifenacin. The number of micturitions per 24 hours showed reductions by day 7 and continued to decrease through day 28; day 7 was the earliest time point tested. Efficacy was demonstrated at this time. The 5-mg, 10-mg, and 20-mg dosing groups experienced significant increases in volume voided; the 10-mg solifenacin dose was associated with significant reductions in episodes of incontinence.
In one of the early RCTs, a total of 1077 patients were randomized to 5 mg solifenacin, 10 mg solifenacin, tolterodine (2 mg twice daily), or placebo (Chapple et al, 2004b). This study was powered only to compare active treatments to placebo. Compared with placebo (−8%), mean micturitions per 24 hours were significantly reduced with solifenacin 10 mg (20%), solifenacin 5 mg (17%), and tolterodine (15%). Solifenacin was well tolerated, with few patients discontinuing treatment. Incidences of dry mouth were 4.9% with placebo, 14.0% with solifenacin 5 mg, 21.3% with solifenacin 10 mg, and 18.6% with tolterodine 2 mg given twice daily.
Cardozo and colleagues (2004b) randomized 911 patients to 12-week once-daily treatment with solifenacin 5 mg, solifenacin 10 mg, or placebo. The primary efficacy variable was change from baseline to study end point in mean number of micturitions per 24 hours. Secondary efficacy variables included changes from baseline in mean number of urgency, nocturia, and incontinence episodes per 24 hours and mean volume voided per micturition. Compared with changes obtained with placebo (−1.6), the number of micturitions per 24 hours was statistically significantly decreased with solifenacin 5 mg (−2.37) and 10 mg (−2.81). A statistically significant decrease was observed in the number of all incontinence episodes with both solifenacin doses (5 mg: −1.63, 61%; 10 mg: −1.57, 52%), but not with placebo (−1.25, 28%). Of patients reporting incontinence at baseline, 50% achieved continence after treatment with solifenacin (based on a 3-day micturition diary); placebo responses were not given. Episodes of nocturia were statistically significantly decreased in patients treated with solifenacin 10 mg versus placebo. Episodes of urgency and mean volume voided per micturition were statistically significantly reduced with solifenacin 5 mg and 10 mg. Treatment with solifenacin was well tolerated. Dry mouth, mostly mild in severity, was reported in 7.7% of patients receiving solifenacin 5 mg and 23% receiving solifenacin 10 mg (vs. 2.3% with placebo). A 40-week follow-up of these studies (i.e., Chapple et al, 2004b and Cardozo et al, 2004b) demonstrated that the favorable profile, both in terms of efficacy and tolerability, was maintained over the study period (Haab et al, 2005).
The STAR trial (Chapple et al, 2005, 2007b) was a prospective, double-blind, double-dummy, two-arm, parallel-group, 12-week study conducted to compare the efficacy and safety of solifenacin 5 or 10 mg and TOLT-ER 4 mg once daily in patients with OAB. The primary effect variable was micturition frequency. After 4 weeks of treatment, patients had the option to request a dose increase but were dummied throughout, because approved product labeling only allowed an increase for those on solifenacin. The results showed that solifenacin, with a flexible dosing regimen, was “non-inferior” to tolterodine concerning the primary effect variable of micturition frequency. However, solifenacin showed significantly greater efficacy to tolterodine in decreasing urgency episodes (−2.85 vs. −2.42), incontinence (−1.60 vs. −0.83), urgency incontinence (−1.42 vs. −0.83), and pad usage (−1.72 vs. −1.19). More solifenacin-treated patients became continent by study end point (59% vs. 49%) and reported improvements in perception of bladder condition (1.51 vs. −1.33) assessments. However, this was accompanied by an adverse event incidence that was greater with solifenacin than with tolterodine. Dry mouth and constipation (mild + moderate + severe) were the most common (solifenacin 30% and 6.4%, tolterodine 23% and 2.5%). The majority of side effects were mild to moderate, and discontinuations were comparable and low (5.9 and 7.3%) in both groups.
A number of studies and reviews have further documented the effects of solifenacin (Cardozo et al, 2006; Chapple et al, 2006a, 2007b; Maniscalco et al, 2006; for further discussion see also Chapple et al, 2008; Novara et al, 2008). In a pooled analysis of four RCTs, Abrams and Swift (2005) demonstrated positive effects on urgency, frequency, and nocturia symptoms in OAB dry patients. In an analysis of four phase 3 clinical trials, Brubaker and FitzGerald (2007) confirmed a significant effect of solifenacin 5 and 10 mg on nocturia in patients with OAB (reductions of nocturia episodes with 5 mg: −0.6, P < .025; with 10 mg: −0.6, P < .001 vs. placebo: −0.4) but without nocturnal polyuria. Kelleher and colleagues (2006) and Staskin and Te (2006) presented data showing efficacy in patients with mixed incontinence. A pooled analysis of four studies confirmed the efficacy and tolerability of solifenacin 5 and 10 mg in elderly (>65 years) patients and also showed a high level of persistence in a 40-week extension trial (Wagg et al, 2006). Improvement of quality of life by solifenacin treatment has been documented in several studies (Kelleher et al, 2005; Garely et al, 2006). In female volunteers, aged 19 to 79 years, the effect of 10 mg and 30 mg solifenacin on the QT interval was evaluated at the time of peak solifenacin plasma concentration in a multiple-dose, randomized, double-blind, placebo- and positive-controlled (moxifloxacin 400 mg) trial. The QT interval prolonging effect appeared greater for the 30 mg (8 msec, 4, 13: 90% confidence interval [CI]) compared with the 10 mg (2 msec, −3, 6) dose of solifenacin. Although the effect of the highest solifenacin dose (three times the maximum therapeutic dose) studied did not appear as large as that of the positive control moxifloxacin at its therapeutic dose, the confidence intervals overlapped. This study was not designed to draw direct statistical conclusions between the drugs or the dose levels.
Michel and colleagues (2008) studied cardiovascular safety and overall tolerability of solifenacin in routine clinical use in a 12-week, open-label, post-marketing surveillance study. They concluded that “in real-life conditions, that is, with inclusion of large numbers of patients with cardiovascular comorbidities and taking comedications, therapeutically effective doses of solifenacin did not increase heart rate or blood pressure.”
Solifenacin has a well-documented effect in OAB/DO, and the adverse event profile seems acceptable.
Fesoterodine Fumarate
Fesoterodine functions as an orally active prodrug that is converted to the active metabolite 5-hydroxymethyltolterodine (5-HMT) by nonspecific esterases (Michel, 2008; McKeage and Keating, 2009). This compound, which is chemically identical to the 5-hydroxy metabolite of tolterodine, is (like fesoterodine itself) a non-subtype selective muscarinic receptor antagonist (Ney et al, 2008). All of the effects of fesoterodine in humans are thought to be mediated via 5-HMT, because the parent compound remains undetectable on oral dosing. 5-HMT is metabolized in the liver, but a significant part of 5-HMT is excreted renally without additional metabolism. Because the renal clearance of 5-HMT is about 250 mL/min, with more than 15% of the administered fesoterodine dose excreted as unchanged 5-HMT, this raises the possibility that 5-HMT also could work from the luminal side of the bladder (Michel, 2008).
Fesoterodine is indicated for use at doses of 4 and 8 mg once daily. In clinical studies, both doses of fesoterodine were consistently superior to placebo in improving the symptoms of OAB (Chapple et al, 2007c, 2008b; Nitti et al, 2007; McKeage and Keating, 2009), with 8 mg/day having significantly greater effects than 4 mg/day (Khullar et al, 2008). Analysis of pooled data on quality of life, using King’s Health Questionnaire and ICI Questionnaire-Short Form, showed that both doses of the drug caused a significant improvement in quality of life. Compared with TOLT-ER (4 mg), fesoterodine (8 mg) had statistically significant advantages for improving incontinence episodes, severe urgency with incontinence, mean voided volumes, and number of continent days a week (Chapple et al, 2008b; Khullar et al, 2008). Adverse events were characteristic for an antimuscarinic drug, with dry mouth being the most frequently reported and rated as mild to moderate in most cases. In one phase 3 study it was seen in 7%, 16%, and 36% of patients receiving placebo and 4 and 8 mg/day fesoterodine, respectively (Nitti et al, 2007), whereas in the another phase 3 study the findings were 7.1%, 21.7%, and 33.8% in the same groups (16.9% for 4 mg/day TOLT-ER) (Chapple et al, 2007c). Kelleher and colleagues (2008) evaluated the effect of fesoterodine on health-related quality of life in patients with OAB syndrome. Pooled data from two randomized placebo-controlled phase 3 studies were analyzed. Eligible patients were randomized to placebo or fesoterodine 4 or 8 mg for 12 weeks; one trial also included TOLT-ER 4 mg. By the end of treatment, all active-treatment groups had significantly improved health-related quality of life compared with those on placebo. A study on possible effects on QT intervals has been performed (Michel, 2008). This included parallel groups of 64 to 68 subjects each who were treated for 3 days with 4 mg/day fesoterodine, the highly supratherapeutic dose of 28 mg/day fesoterodine, the active control moxifloxacin 400 mg/day, or placebo. Both the standard dose of 4 mg/day and the highly supratherapeutic dose of 28 mg/day did not provide any evidence of QT interval prolongation (e.g., QTc for 28 mg/day from 404.5 ± 16.7 to 400.1 ± 14.0 msec, delta: −5.0 ± 7.9 msec).
Fesoterodine has a well-documented beneficial effect in OAB (see Table 68–2), and the adverse event profile seems acceptable.
Drugs Acting at Membrane Channels
Calcium Antagonists
Activation of detrusor muscle, both through muscarinic receptor and nonadrenergic, noncholinergic pathways, seems to require influx of extracellular Ca2+ through Ca2+ channels, as well as via mobilization of intracellular Ca2+ (Andersson, 1993; Andersson and Arner, 2004). The influx of extracellular Ca2+ can be blocked by Ca2+ antagonists, blocking L-type Ca2+ channels; and, theoretically, this would be an attractive way of inhibiting DO. However, there have been few clinical studies of the effects of Ca2+ antagonists in patients with DO and the ones that have been done suggest no beneficial effect. For example, Naglie and coworkers (2002) evaluated the efficacy of nimodipine for geriatric urgency incontinence in a randomized, double-blind, placebo-controlled crossover trial. Thirty milligrams of nimodipine was given twice daily for 3 weeks in older persons with DO and chronic urgency incontinence. A total of 86 participants with a mean age of 73.4 years were randomized. The primary outcome was the number of incontinent episodes, as measured by the self-completion of a 5-day voiding record. Secondary outcomes included the impact of urinary incontinence on quality of life measured with a modified incontinence impact questionnaire and symptoms, as measured by the American Urological Association (AUA) symptom score. In the 76 (88.4%) participants completing the study there was no significant difference in the number of incontinent episodes with nimodipine versus placebo. Scores on the incontinence impact questionnaire and the AUA symptom score were not significantly different with nimodipine versus placebo, and the authors concluded that treatment of geriatric urgency incontinence with 30 mg nimodipine twice daily was unsuccessful.
Available information does not suggest that systemic therapy with Ca2+ antagonists is an effective way to treat OAB/DO.
Potassium Channel Openers
Opening of K+ channels and subsequent efflux of K+ will produce hyperpolarization of various smooth muscles, including the detrusor (Andersson, 1992; Andersson and Arner, 2004). This leads to a decrease in Ca2+ influx by reducing the opening probability of Ca2+ channels with subsequent relaxation or inhibition of contraction. Theoretically, such drugs could be active during the filling phase of the bladder, abolishing bladder overactivity with no effect on normal bladder contraction. The K+ channel openers, such as pinacidil and cromakalim, have been effective in animal models (Andersson, 1992; Andersson and Arner, 2004), but clinically the effects have not been encouraging. The first generation of openers of ATP-sensitive K+ channels, such as cromakalim and pinacidil, were found to be more potent as inhibitors of vascular preparations than of detrusor muscle, and in clinical trials performed with these drugs no bladder effects have been found at doses already lowering blood pressure (Hedlund et al, 1991; Komersova et al, 1995). However, new drugs with K+ ATP-sensitive channel opening properties have been described, which may be useful for the treatment of OAB (Andersson and Arner, 2004). Although K+ channel openers are believed mainly to act directly on smooth muscle cells (Gopalakrishnan and Shieh, 2004), they may also at least in part affect bladder function by modulating the activity of afferent neurons (Tanaka et al, 2003).
Although the previous data demonstrate the potential of K+ channel openers to inhibit nonvoiding detrusor contractions these channels are expressed not only in bladder but also, for example, in vascular smooth muscle. Therefore, K+ channel openers may also affect cardiovascular function and in effective doses may considerably lower blood pressure (Howe et al, 1995; Shieh et al, 2007). Although some compounds of this class have a certain degree of selectivity for the bladder as compared with the cardiovascular system, it remains unclear whether the degree of selectivity offers a sufficiently large therapeutic window for clinical use. This consideration has led to a considerable hesitancy to study K+ channel openers in patients with OAB. Nevertheless, one randomized, placebo-controlled clinical study on the K+ ATP opener ZD0947 has been reported (Chapple et al, 2006b). Although ZD0947 at the chosen dose did not lower blood pressure or cause adverse events typical for a vasodilating drug, it failed to achieve superiority relative to placebo for the treatment of OAB symptoms. Therefore, despite promising preclinical efficacy data, K+ channel openers at present are not a therapeutic option and may never become one owing to a lack of selectivity for bladder over cardiovascular tissues.
At present there is no evidence from RCTs to suggest that K+ channel openers represent a treatment alternative.
Drugs with “Mixed” Actions
Some drugs used to block DO have been shown to have more than one mechanism of action. They all have a more or less pronounced antimuscarinic effect and, in addition, an often poorly defined “direct” action on bladder muscle. For several of these drugs the antimuscarinic effects can be demonstrated at much lower drug concentrations than the direct action, which may involve blockade of voltage-operated Ca2+ channels. Most probably, the clinical effects of these drugs can be explained mainly by their antimuscarinic action. Among the drugs with mixed actions was terodiline, which was withdrawn from the market because it was suspected to cause polymorphic ventricular tachycardia (torsades de pointes) in some patients (Connolly et al, 1991; Stewart et al, 1992).
Oxybutynin Chloride
Oxybutynin is a tertiary amine that is well absorbed and undergoes extensive upper gastrointestinal and first-pass hepatic metabolism via the cytochrome P450 system (CYP3A4) into multiple metabolites. The primary metabolite, N-desethyloxybutynin (DEO), has pharmacologic properties similar to those of the parent compound (Waldeck et al, 1997) but occurs in much higher concentrations after oral administration (Hughes et al, 1992). It has been implicated as the major cause of the troublesome side effect of dry mouth associated with the administration of oxybutynin. It seems reasonable to assume that the effect of oral oxybutynin to a large extent is exerted by the metabolite. The occurrence of an active metabolite may also explain the lack of correlation between plasma concentration of oxybutynin itself and side effects in geriatric patients reported by Ouslander and associates (1995). The plasma half-life of oxybutynin is approximately 2 hours, but with wide interindividual variation (Douchamps et al, 1988; Hughes et al, 1992).
Oxybutynin has several pharmacologic effects in vitro, some of which seem difficult to relate to its effectiveness in the treatment of DO. It has both an antimuscarinic and a direct muscle relaxant effect, and, in addition, local anesthetic actions. The latter effect may be of importance when the drug is administered intravesically but probably plays no role when it is given orally. In vitro, oxybutynin was 500 times weaker as a smooth muscle relaxant than as an antimuscarinic agent (Kachur et al, 1988). Most probably, when given systemically, oxybutynin acts mainly as an antimuscarinic drug. Oxybutynin has a high affinity for muscarinic receptors in human bladder tissue and effectively blocks carbachol-induced contractions (Waldeck et al, 1997). The drug was shown to have slightly higher affinity for muscarinic M1 and M3 receptors than for M2 receptors (Nilvebrant and Sparf, 1986; Norhona-Blob et al, 1991), but the clinical significance of this is unclear.
The immediate release (IR) form of oxybutynin (OXY-IR) is recognized for its efficacy, and most of the newer antimuscarinic agents have been compared with it once efficacy over placebo has been determined. In general, the new formulations of oxybutynin and other antimuscarinic agents offer patients efficacy roughly equivalent to that of OXY-IR and the advantage of the newer formulations lies in improved dosing schedules and side effect profile (Appell et al, 2001; Dmochowski et al, 2002; Diokno et al, 2003). An extended-release oxybutynin (OXY-ER) once-daily oral formulation, an oxybutynin patch transdermal delivery system (OXY-TDS), and an oxybutynin gel are available. OXY-TDS offers a twice-weekly dosing regimen and the potential for improved patient compliance and tolerability. The gel requires once-daily application. Available formulations of oxybutynin were overviewed by McCrery and Appell (2006). Clinical use of the gel was first described by Staskin and coworkers (2009).
Immediate-Release Oxybutynin (OXY-IR)
Several controlled studies have shown that OXY-IR is effective in controlling DO, including neurogenic DO (Yarker et al, 1995; Andersson and Chapple, 2001). The recommended oral dose of the IR form is 5 mg three times daily or four times daily, even if lower doses have been used. Thüroff and associates (1998) summarized 15 randomized controlled studies on a total of 476 patients treated with oxybutynin. The mean decrease in incontinence was recorded as 52%, and the mean reduction in frequency per 24 hours was 33% (data on placebo not presented). The overall “subjective improvement” rate was reported as 74% (range 61% to 100%). The mean percent of patients reporting an adverse effect was 70% (range 17% to 93%). Oxybutynin, 7.5 to 15 mg/day, significantly improved the quality of life of patients suffering from OAB in a large open multicenter trial. In this study, patient compliance was 97% and side effects, mainly dry mouth, were reported by only 8% (Amarenco et al, 1998). In nursing home residents (n = 75), Ouslander and colleagues (1995) found that oxybutynin did not add to the clinical effectiveness of prompted voiding in a placebo-controlled, double-blind, crossover trial. On the other hand, in another controlled trial in elderly subjects (n = 57), oxybutynin with bladder training was found to be superior to bladder training alone (Szonyi et al, 1995).
Several open studies in patients with spinal cord injuries have suggested that oxybutynin, given orally or intravesically, can be of therapeutic benefit (Kim et al, 1996; Szollar and Lee, 1996). The therapeutic effect of OXY-IR on DO is associated with a high incidence of side effects (up to 80% with oral administration). These are typically antimuscarinic in nature (dry mouth, constipation, drowsiness, blurred vision) and are often dose limiting (Baigrie et al, 1988; Jonville et al, 1992). The effects on the electrocardiogram of oxybutynin were studied in elderly patients with urinary incontinence (Hussain et al, 1996); no changes were found. It cannot be excluded that the commonly recommended dose of 5 mg three times a day is unnecessarily high in some patients and that a starting dose of 2.5 mg twice daily with following dose titration would reduce the number of adverse effects (Malone-Lee et al, 1992; Amarenco et al, 1998).
Extended-Release Oxybutynin (OXY-ER)
This formulation was developed to decrease liver metabolite formation of desethyloxybutynin (DEO) with the presumption that it would result in decreased side effects, especially dry mouth, and improve patient compliance with remaining on oxybutynin therapy. The formulation utilizes an osmotic system to release the drug at a controlled rate over 24 hours distally primarily into the large intestine where absorption is not subject to first-pass metabolism in the liver. This reduction in metabolism is meant to improve the rate of dry mouth complaints when compared with OXY-IR. DEO is still formed through the hepatic cytochrome P450 enzymes, but clinical trials have indeed demonstrated improved dry mouth rates compared with OXY-IR (Appell et al, 2003). Salivary output studies have also been interesting. Two hours after administration of OXY-IR or TOLT-IR, salivary production decreased markedly and then gradually returned to normal. With OXY-ER, however, salivary output was maintained at predose levels throughout the day (Chancellor et al, 2001).
The effects of OXY-ER have been well documented (Siddiqui et al, 2004). In the OBJECT study (Appell et al, 2001) the efficacy and tolerability of 10 mg OXY-ER was compared with a twice-daily 2-mg dose of TOLT-IR. OXY-ER was statistically more effective than the TOLT-IR in weekly urgency incontinence episodes (OXY-ER from 25.6% to 6.1%; TOLT-IR 24.1% to 7.8%), total incontinence (OXY-ER from 28.6% to 7.1%; TOLT-IR 27.0% to 9.3%), and frequency (OXY-ER from 91.8% to 67.1%; TOLT-IR 91.6% to 71.5%), and both medications were equally well tolerated. The basic study was repeated as the OPERA study (Diokno et al, 2003) with the difference that this study was a direct comparison of the two extended-release forms, OXY-ER (10 mg) and TOLT-ER (4 mg); and the results were quite different. In this study there was no significant difference in efficacy for the primary end point of urgency incontinence; however, TOLT-ER had a statistically lower incidence of dry mouth. OXY-ER was only statistically better at 10 mg than TOLT-ER 4 mg in the reduction of the rate of urinary frequency. These studies made it clear that in comparative studies IR entities of one drug should not be compared with ER entities of the other. Greater reductions in urgency and total incontinence have been reported in patients treated in dose escalation studies with OXY-ER. In two randomized studies, the efficacy and tolerability of OXY-ER were compared with OXY-IR. In the 1999 study (Anderson et al, 1999), 105 patients with urgency or mixed incontinence were randomized to receive 5 to 30 mg OXY-ER once daily or 5 mg of OXY-IR one to four times a day. Dose titrations began at 5 mg, and the dose was increased every 4 to 7 days until one of three end points was achieved. These were (1) the patient reported no urgency incontinence during the final 2 days of the dosing period; (2) the maximum tolerable dose was reached; and (3) the maximum allowable dose (30 mg for OXY-ER or 20 mg for OXY-IR) was reached. The mean percentage reduction in weekly urgency and total incontinence episodes was statistically similar between OXY-ER and OXY-IR, but dry mouth was reported statistically more often with OXY-IR. In the 2000 study (Versi et al, 2000), 226 patients were randomized between OXY-ER and OXY-IR with weekly increments of 5 mg/day up to 20 mg/day. As in the 1999 study, OXY-ER again achieved a greater than 80% reduction in urgency and total incontinence episodes and a significant percentage of patients became dry. A negative aspect of these studies is that there were no naive patients included, as all patients were known responders to oxybutynin. Similar efficacy results have been achieved, however, with OXY-ER in a treatment-naive population (Gleason et al, 1999). In an RCT comparing different daily doses of oxybutynin (5, 10, and 15 mg), Corcos and colleagues (2006) found a significant dose-response relationship for both urgency incontinence episodes and dry mouth. The greatest satisfaction was with 15 mg oxybutynin/day.
Transdermal Oxybutynin (OXY-TDS)
Transdermal delivery also alters oxybutynin metabolism reducing DEO production to an even greater extent than OXY-ER. A study (Davila et al, 2001) comparing OXY-TDS with OXY-IR demonstrated a statistically equivalent reduction in daily incontinence episodes (from 7.3 to 2.3: 66% for OXY-TDS, and 7.4 to 2.6: 72% for OXY-IR) but much less dry mouth (38% for OXY-TDS and 94% for OXY-IR). In another study (Dmochowski et al, 2002), the 3.9-mg daily dose patch significantly (vs. placebo) reduced the mean number of daily incontinence episodes (from 4.7 to 1.9; placebo from 5.0 to 2.9), while reducing average daily urinary frequency confirmed by an increased average voided volume (from 165 to 198 mL; placebo from 175 to 182 mL). Furthermore, the dry mouth rate was similar to that of placebo (7% vs. 8.3%). In a third study (Dmochowski et al, 2003b), OXY-TDS was compared not only with placebo but also with TOLT-ER. Both drugs equivalently and significantly reduced daily incontinence episodes and increased the average voided volume, but TOLT-ER was associated with a significantly higher rate of antimuscarinic adverse events. The primary adverse event for OXY-TDS was application site reaction pruritus in 14% and erythema in 8.3% with nearly 9% feeling that the reactions were severe enough to withdraw from the study, despite the lack of systemic problems.
The pharmacokinetics and adverse effect dynamics of OXY-TDS (3.9 mg/day) and OXY-ER (10 mg/day) were compared in healthy subjects in a randomized, two-way crossover study (Appell et al, 2003). Multiple blood and saliva samples were collected, and pharmacokinetic parameters and total salivary output were assessed. OXY-TDS administration resulted in greater systemic availability and minimal metabolism to DEO compared with OXY-ER, which resulted in greater salivary output in OXY-TDS patients and less dry mouth symptomatology than when taking OXY-ER. Dmochowski and coworkers (2005) analyzing the combined results of two RCTs concluded that OXY-TDS was shown to be efficacious and well tolerated. The most common systemic side effect was dry mouth (7.0% vs. placebo 5.3%). Application-site erythema occurred in 7% and pruritus in 16.1%. Also Cartwright and Cardozo (2007), reviewing published and presented data, concluded that OXY-TDS has a good balance between efficacy and tolerability with a rate of systemic antimuscarinic side effects lower than that with oral antimuscarinic agents; however, this benefit was offset by the rate of local skin reaction. Sahai and associates (2008) largely confirmed these conclusions.
Oxybutynin Gel
Oxybutynin topical gel was introduced in 2009 as a transdermal formulation applied once daily to the abdomen, thigh, shoulder, or upper arm area (Staskin et al, 2009). The 1-g application dose delivers approximately 4 mg of drug to the circulation with stable plasma concentration. Staskin and associates (2009) reported a randomized controlled double-blind trial of patients (89% women) with urgency-predominant urinary incontinence. The mean number of urgency-predominant urinary incontinence episodes on a 3-day voiding diary was reduced by 3 episodes per day versus 2.5 in the placebo study arm (P < .0001). Urinary frequency was decreased by 2.7 episodes per day versus 2.0 episodes (P = .0017), and voided volume increased by 21 mL versus 3.8 mL (P = .0018). Dry mouth was reported in 6.9% of the treatment group versus 2.8% in the placebo group. Skin reaction at the application site occurred in 4.5% of the treatment group versus 1% in the placebo arm. It was believed that the improved skin tolerability over the transdermal patch system was secondary to a lack of adhesive and of skin occlusion. Person-to-person transference via skin contact is a risk but largely eliminated if clothing is worn over the application site.
Other Administration Forms
Rectal administration (Collas and Malone-Lee, 1997; Winkler and Sand, 1998) was reported to have fewer adverse effects than the conventional tablets. Administered intravesically, oxybutynin has in several studies been demonstrated to increase bladder capacity and produce clinical improvement with few side effects, both in neurogenic and in other types of DO, and both in children and adults (Weese et al, 1993; Lose and Norgaard, 2001; Fader et al, 2007; George et al, 2007; Guerra et al, 2008), although adverse effects may occur (Kasabian et al, 1994; Palmer et al, 1997). Intravesical administration of oxybutynin is a conceptually attractive form of drug delivery, especially for patients who already perform clean intermittent catheterization (CIC). However, a specific intravesical formulation is not available, and, currently, the oral formulation, either liquid or crushed tablet in solution, is delivered by periodic insertion through a catheter. There does appear to be some systemic absorption of oxybutynin placed intravesically. Plasma oxybutynin but not DEO levels were measured after intravesical and oral administration by Madersbacher and Jilg (1991). It is still not entirely clear what portion of the effects of the drug are due to local versus absorbed parent drug and primary metabolite. Certainly this is a circumstance in which the non-antimuscarinic actions of oxybutynin (see earlier) might be relevant.
Effects on Cognition
Several studies have documented the possibility that oxybutynin may have negative effects on cognitive functions, particularly in the elderly population but also in children (see Kay et al, 2006; Klausner al Steers, 2007; Kay and Ebinger, 2008). This factor should be taken into consideration when prescribing the drug.
Oxybutynin has a well-documented efficacy in the treatment of OAB/DO (see Table 68–2). Despite the adverse effect profile, it is still an established therapeutic option.
Dicyclomine
Dicyclomine (Bentyl) has attributed to it both a direct relaxant effect on smooth muscle and an antimuscarinic action (Downie et al, 1977). An oral dose of 20 mg three times a day in adults was reported to increase bladder capacity in patients with neurogenic DO (Fischer et al, 1978). Beck and associates (1976) compared the use of 10 mg of dicyclomine, 15 mg of propantheline, and placebo three times a day in patients with DO. The reported cure or improved rates, respectively, were 62%, 73%, and 20%. Awad and coworkers (1977) reported that 20 mg of dicyclomine three times a day caused resolution or significant improvement in 24 of 27 patients with involuntary bladder contractions.
Even if published experiences of the effect of dicyclomine on DO are favorable, the drug is not widely used, and controlled clinical trials documenting its efficacy and side effects are scarce.
Propiverine Hydrochloride
Several aspects of the preclinical, pharmacokinetic, and clinical effects of propiverine have been reviewed by Madersbacher and Murz (2001). The drug is rapidly absorbed (Tmax 2 hr), but has a high first-pass metabolism, and its biologic availability is about 50%. Propiverine is an inducer of hepatic cytochrome P450 enzymes in rats in doses about 100 times above the therapeutic doses in man (Walter et al, 2003).
Several active metabolites are formed that quantitatively and qualitatively differ from the mother compound (Haustein et al, 1988; Muller et al, 1993; Wuest et al, 2006; Sugiyama et al, 2008; Zhu et al, 2008). Most probably these metabolites contribute to the clinical effects of the drug, but their individual contributions have not been clarified (Michel and Hegde, 2006). The half-life of propiverine is 11 to 14 hours. An extended-release preparation was shown to be effective (Jünemann et al, 2006; May et al, 2008).
Propiverine has combined antimuscarinic and Ca2+ antagonistic actions (Haruno, 1992; Tokuno et al, 1993). The importance of the Ca2+ antagonistic component for the drug’s clinical effects has not been established. Propiverine has no selectivity for muscarinic receptor subtypes. The drug is not available in the United States.
Propiverine has been shown to have beneficial effects in patients with DO in several investigations. Thüroff and coworkers (1998) collected nine randomized studies on a total of 230 patients and found a 17% reduction in micturitions per 24 hours, a 64-mL increase in bladder capacity, and a 77% (range 33% to 80%) subjective improvement. Side effects were found in 14% (range 8% to 42%). In patients with neurogenic DO, controlled clinical trials have demonstrated propiverine’s superiority over placebo (Stöhrer et al, 1999). Propiverine also increased bladder capacity and decreased maximum detrusor contractions. Controlled trials comparing propiverine, flavoxate, and placebo (Wehnert et al, 1989) and propiverine, oxybutynin, and placebo (Wehnert et al, 1992; Madersbacher et al, 1999) have confirmed the efficacy of propiverine and suggested that the drug may have equal efficacy and fewer side effects than oxybutynin. In a comparative RCT including 131 patients with neurogenic DO, propiverine and oxybutynin were compared (Stöhrer et al, 2007). The drugs were found to be equally effective in increasing bladder capacity and lowering bladder pressure. Propiverine caused a significantly lower frequency of dry mouth than oxybutynin. Also in children and adolescents with neurogenic DO, propiverine was found to be effective (Grigoleit et al, 2006; Schulte-Baukloh et al, 2006), with a low incidence rate of adverse events: less than 1.5% (Grigoleit et al, 2006).
Madersbacher and colleagues (1999) compared the tolerability and efficacy of propiverine (15 mg three times daily), oxybutynin (5 mg twice daily), and placebo in 366 patients with urgency and urgency incontinence in a randomized, double-blind placebo-controlled clinical trial. Urodynamic efficacy of propiverine was judged similar to that of oxybutynin, but the incidence of dry mouth and the severity of dry mouth were judged less with propiverine than with oxybutynin. Dorschner and coworkers (2000) investigated in a double-blind, multicenter, placebo-controlled randomized study the efficacy and cardiac safety of propiverine in 98 elderly patients (mean age 68 years) suffering from urgency, urgency incontinence, or mixed urgency stress urinary incontinence (SUI). After a 2-week placebo run-in period the patients received propiverine (15 mg three times daily) or placebo (three times daily) for 4 weeks. Propiverine caused a significant reduction of the micturition frequency (from 8.7 to 6.5) and a significant decrease in episodes of incontinence (from 0.9 to 0.3 per day). The incidence of adverse events was very low (2% dryness of the mouth under propiverine—2 of 49 patients). Resting and ambulatory electrocardiograms indicated no significant changes.
In a randomized, double-blind multicenter clinical trial, patients with idiopathic DO were treated with 15 mg propiverine twice daily or 2 mg TOLT-IR twice daily over a period of 28 days (Jünemann et al, 2005). The maximum cystometric capacity was determined at baseline and after 4 weeks of therapy. The difference of both values was used as the primary end point.
Secondary end points were voided volume per micturition, evaluation of efficacy (by the investigator), tolerability, postvoid residual urine, and quality of life. It was found that the mean maximum cystometric capacity increased significantly (P < .01) in both groups. The volume at first urgency and the frequency/volume chart parameters also showed relevant improvements during treatment. The most common adverse event, dry mouth, occurred in 20 patients in the propiverine group and in 19 patients in the tolterodine group. The scores for the quality of life improved comparably in both groups. Abrams and associates (2006b) compared the effects of propiverine and oxybutynin on ambulatory urodynamic monitoring parameters, safety, and tolerability in OAB patients. Patients (n = 77) received two of the following treatments during two 2-week periods: propiverine 20 mg once daily, propiverine 15 mg three times daily, oxybutynin 5 mg three times daily, and placebo. They found that oxybutynin 15 mg was more effective than propiverine 20 mg in reducing symptomatic and asymptomatic involuntary detrusor contractions in ambulatory patients. Oxybutynin had a higher rate of dry mouth, and propiverine had a more pronounced effect on gastrointestinal, cardiovascular, and visual function.
A randomized, double-blind, placebo-controlled trial with parallel-group design in children aged 5 to 10 years was performed by Marschall-Kehrel and coworkers (2009). Of 171 randomized children, 87 were treated with propiverine and 84 with placebo. Decrease in voiding frequency per day was the primary efficacy parameter; secondary end points included voided volume and incontinence episodes. There was a significant decrease in voiding frequency episodes for propiverine versus placebo. Superiority could also be demonstrated for voided volume and incontinence episodes per day. Propiverine was well tolerated: 23% of side effects were reported for propiverine and 20% for placebo.
Yamaguchi and colleagues (2007) performed a multicenter, 12-week, double-blind phase III trial in Japanese men and women with OAB (1593 patients were randomized and 1584 were treated), comparing solifenacin 5 or 10 mg, propiverine 20 mg, and placebo. Changes at end point in number of voids per 24 hours, urgency, incontinence, urgency incontinence and nocturia episodes, volume voided/void, restoration of continence, and quality of life were examined. It was found that at end point there were greater reductions in mean (SD) voids per 24 hours with all drug regimens than with placebo. All active treatments improved the volume voided and quality of life versus placebo; solifenacin 10 mg reduced nocturia episodes and significantly improved urgency episodes and volume voided versus propiverine 20 mg, and solifenacin 5 mg caused less dry mouth. Solifenacin 10 mg caused more dry mouth and constipation than propiverine 20 mg.
Propiverine has a documented beneficial effect in the treatment of OAB/DO (see Table 68–2) and seems to have an acceptable side effect profile.
Flavoxate Hydrochloride
Flavoxate is well absorbed, and oral bioavailability appeared to be close to 100% (Guay, 2003). The drug is extensively metabolized, and plasma half-life was found to be 3.5 hours (Sheu et al, 2001). Its main metabolite (3-methylflavone-8-carboxylic acid [MFCA]) has been shown to have low pharmacologic activity (Cazzulani et al, 1988; Caine et al, 1991). The main mechanism of flavoxate’s effect on smooth muscle has not been established. The drug has been found to possess a moderate Ca2+ antagonistic activity, to have the ability to inhibit phosphodiesterase (PDE), and to have local anesthetic properties; no antimuscarinic effect was found (Guarneri et al, 1994). Uckert and associates (2000) found that in strips of human bladder the potency of flavoxate to reverse contraction induced by muscarinic receptor stimulation and by electrical field stimulation was comparable. Tomoda and coworkers (2005) studied the effects of flavoxate on K+-induced tension using strips prepared from human urinary bladder and also investigated the effects of flavoxate on voltage-dependent nifedipine-sensitive Ba2+ currents (i.e., L-type Ca2+ currents) in single freshly dispersed human detrusor smooth muscle myocytes by use of whole-cell patch-clamp techniques. They found that flavoxate caused a concentration-dependent reduction of the K+-induced contraction of the bladder strips and that in the human detrusor myocytes, flavoxate inhibited the peak amplitude of voltage-dependent, nifedipine-sensitive inward Ba2+ currents in a voltage- and concentration-dependent manner. They concluded that flavoxate caused muscle relaxation through the inhibition of L-type Ca2+ channels in the human detrusor.
It has been suggested that pertussis toxin-sensitive G proteins in the brain are involved in the flavoxate-induced suppression of the micturition reflex, since intracerebroventricularly or intrathecally administered flavoxate abolished isovolumetric rhythmic bladder contractions in anesthetized rats (Oka et al, 1996).
The clinical effects of flavoxate in patients with DO and frequency, urgency, and incontinence have been studied in both open and controlled investigations, but with varying rates of success (Ruffmann, 1988). Stanton (1973) compared emepronium and flavoxate in a double-blind, crossover study of patients with detrusor instability and reported improvement rates of 83% and 66% after flavoxate or emepronium, respectively, both administered as 200 mg three times daily. In another double-blind, crossover study comparing flavoxate 1200 mg/day with that of oxybutynin 15 mg daily in 41 women with idiopathic motor or sensory urgency and utilizing both clinical and urodynamic criteria, Milani and associates (1993) found both drugs effective. No difference in efficacy was found between them, but flavoxate had fewer and milder side effects. Other investigators, comparing the effects flavoxate with those of placebo, have not been able to show any beneficial effect of flavoxate at dosages up to 400 mg three times daily (Briggs et al, 1980; Chapple et al, 1990; Dahm et al, 1995).
In general, few side effects have been reported during treatment with flavoxate. On the other hand its efficacy, compared with other therapeutic alternatives, is not well documented. No RCTs seem to have been performed with flavoxate during the past decade.
α-Adrenergic Receptor Antagonists
Even if it is well known that α1-AR antagonists can ameliorate lower urinary tract symptoms (LUTS) in men with benign prostatic enlargement (Andersson et al, 2002) there are no controlled clinical trials showing that they are an effective alternative in the treatment of OAB/DO in this patient category. In an open-label study, Arnold (2001) evaluated the clinical and pressure-flow effects of tamsulosin 0.4 mg once daily in patients with LUTS caused by benign prostatic obstruction. He found that tamsulosin produced a significant decrease in detrusor pressure, an increase in flow rate, and a symptomatic improvement. In a study in which tamsulosin was given alone, or together with tolterodine, to male patients with LUTS, monotherapy with the drug was not effective (Kaplan et al, 2006). An RCT, comprising 364 women with OAB, revealed no effect of tamsulosin versus placebo (Robinson et al, 2007). On the other hand, voiding symptoms in women with functional outflow obstruction, or LUTS, were reported as successfully treated with an α1-AR antagonist (Kessler et al, 2006; Low et al, 2008).
α1-AR antagonists have been used to treat patients with neurogenic DO (Abrams et al, 2003); however, the success has been moderate. Thus there is no convincing evidence that α1-AR antagonists are effective in patients with storage symptoms. Although α1-AR antagonists may be effective in selected cases, convincing effects documented in RCTs are lacking (see Table 68–2). In women, these drugs may produce or aggravate SUI (Dwyer and Teele, 1992).
β-Adrenergic Receptor Agonists
In isolated human bladder, non-subtype selective β-AR agonists such as isoprenaline have a pronounced inhibitory effect, and administration of such drugs can increase bladder capacity in humans (Andersson, 1993). However, the β-ARs of the human bladder were shown to have functional characteristics typical of neither β1- nor β2-ARs, because they could be blocked by propranolol but not by practolol or metoprolol (β1) or butoxamine (β2) (Nergardh et al, 1977; Larsen, 1979). On the other hand, early receptor binding studies using subtype selective ligands suggested that the β-ARs of the human detrusor are primarily of β2 subtype (Andersson, 1993), and favorable effects on DO were reported in open studies with selective β2-AR agonists such as terbutaline (Lindholm and Lose, 1986). In a double-blind investigation clenbuterol 0.01 mg three times daily was shown to have a good therapeutic effect in 15 of 20 women with DO (Gruneberger, 1984). Other investigators, however, have not been able to show that β-AR agonists represent an effective therapeutic principle in elderly patients with DO (Castleden and Morgan, 1980) or in young patients with myelodysplasia and DO (Naglo et al, 1981).
However, three subtypes (β1, β2, and β3) have been identified in the detrusor of most species, including humans (Andersson and Arner, 2004; Michel and Vrydag, 2006). Also the human urothelium contains all three receptor subtypes (Otsuka et al, 2008). Studies, using real-time polymerase chain reaction, have revealed a predominant expression of β3-AR mRNA in human detrusor muscle (Nomiya and Yamaguchi, 2003; Michel and Vrydag, 2006); and the functional evidence for an important role in both normal and neurogenic bladders is convincing (Fujimura et al, 1999; Igawa et al, 1999, 2001; Takeda et al, 1999; Morita et al, 2000; Biers et al, 2006; Michel and Vrydag, 2006; Badawi et al, 2007; Leon et al, 2008). The human detrusor also contains β2-ARs, and most probably both receptors are involved in the physiologic effects (relaxation) of norepinephrine in this structure (Andersson and Arner 2004; Michel and Vrydag, 2006).
The generally accepted mechanism by which β-ARs induce detrusor relaxation in most species is activation of adenylyl cyclase with the subsequent formation of cAMP. However, there is evidence suggesting that, in the bladder, K+ channels, particularly BKCa channels, may be more important in β-AR–mediated relaxation than cAMP (Hudman et al, 2000; Frazier et al, 2005; Uchida et al, 2005; Frazier et al, 2008).
Because β-ARs are present in the urothelium, their possible role in bladder relaxation has been investigated (Murakami et al, 2007; Otsuka et al, 2008). Murakami and coworkers (2007) found that the relaxation responses of the detrusor were not influenced by the urothelium. However, isoprenaline was more potent at inhibiting carbachol contractions in the presence of the urothelium than in its absence. It was suggested that this might reflect the release of an inhibitory factor from the urothelium. Further support for this hypothesis was given by Otsuka and associates (2008). However, to what extent a urothelial signaling pathway contributes in vitro and in vivo to the relaxant effects of β-AR agonists in general, and β3-AR agonists specifically, remains to be elucidated.
The in-vivo effects of β3-AR agonists on bladder function have been studied in several animal models. It has been shown that compared with other agents (including antimuscarinic drugs), β3-AR agonists increase bladder capacity with no change in micturition pressure and the residual volume (Fujimura et al, 1999; Woods et al, 2001; Kaidoh et al, 2002). For example, Hicks and associates (2007) studied the effects of the selective β3-AR agonist GW427353 in the anesthetized dog and found that the drug evoked an increase in bladder capacity under conditions of acid-evoked bladder hyperactivity, without affecting voiding.
A number of β3-AR selective agonists are currently being evaluated as potential treatment for OAB in humans, including GW427353 (solabegron) and YM178 (Colli et al, 2007). Takasu and associates (2007) found that the selective β3-AR agonist YM178 mediated muscle relaxation of human bladder strips. Chapple and coworkers (2008c) reported the results of a controlled clinical trial with this drug in patients with OAB. Tolterodine and placebo served as controls. The primary efficacy analysis showed a statistically significant reduction in mean micturition frequency, compared with placebo (−2.19 vs. −1.18). With respect to secondary variables YM178 (100 mg) was significantly superior to placebo concerning mean volume voided per micturition (26 vs. 11 mL), mean number of incontinence episodes (−2.17 vs. −1.01), and urgency episodes per 24 hour (−2.30 vs. −1.03). The drug was well tolerated, and the most commonly reported side effects were headache and gastrointestinal adverse effects. The results of this proof of concept study showed that the principle of β3-AR agonism may be useful for treatment of patients with OAB (see Table 68–2). However, to show that this class of drugs offers a viable therapeutic alternative or complement to current treatment of LUTS/OAB requires further well-designed RCTs.
Phosphodiesterase Inhibitors
Drugs stimulating the generation of cAMP are known to relax smooth muscles, including the detrusor (Andersson, 1999b; Andersson and Arner, 2004). It is also well established that drugs acting through the nitric oxide/cyclic guanosine monophosphate (NO/cGMP) system can relax the smooth muscle of the bladder outflow region (Andersson and Arner, 2004). Use of PDE inhibitors to enhance the presumed cAMP- and cGMP-mediated relaxation of LUT smooth muscles (detrusor prostate, urethra) should then be a logical approach (Andersson et al, 2007). There are presently 11 families of PDEs, some of which preferentially hydrolyze either cAMP or cGMP (Andersson et al, 2007).
As a basis for PDE inhibitor treatment of LUTS, Truss and colleagues (2001) investigated human bladder tissue, revealing mRNA for PDE1, PDE2, PDE4, and PDE5; most of these PDEs preferably inhibit the breakdown of cAMP. In vitro, human detrusor muscle responded poorly to sodium nitroprusside and to agents acting via the cGMP system (Truss et al, 2001). However, significant relaxation of human detrusor muscle, paralleled by increases in cyclic nucleotide levels, was induced by papaverine, vinpocetine (a low affinity inhibitor of PDE1), and forskolin (stimulating the generation of cAMP), suggesting that the cAMP pathway and PDE1 may be important in regulation of detrusor smooth muscle tone. Significant dose-dependent relaxations were also induced by human cAMP analogues (Truss et al, 2001). With these studies as a background, Truss and colleagues (2000) presented preliminary clinical data with vinpocetine in patients with urgency/urgency incontinence or low compliance bladders and not responding to standard antimuscarinic therapy. This initial open pilot study suggested a possible role for vinpocetine in the treatment of OAB. However, the results of a larger RCT in patients with DO showed that vinpocetine showed statistically significant results only for one parameter (Truss et al, 2001). Studies with other PDE1 inhibitors than vinpocetine (which may not be an optimal drug for elucidating the principle) do not seem to have been performed.
PDE4 (which also preferably hydrolyzes cAMP) has been implicated in the control of bladder smooth muscle tone. PDE4 inhibitors reduced the in-vitro contractile response of guinea pig (Longhurst et al, 1997) and rat (Nishiguchi et al, 2007; Kaiho et al, 2008) bladder strips and also suppressed rhythmic bladder contractions of the isolated guinea pig bladder (Gillespie, 2004a). Previous experiences with selective PDE4 inhibitors showed emesis to be a dose-limiting effect (Giembycz, 2005). If this side action can be avoided, PDE4 inhibition seems to be a promising approach.
NO has been demonstrated to be an important inhibitory neurotransmitter in the smooth muscle of the urethra, and its relaxant effect is associated with increased levels of cGMP (Andersson and Wein, 2004). However, few investigations have addressed the cAMP- and cGMP-mediated signal transduction pathways and its key enzymes in the mammalian urethra. Morita and associates (1994) examined the effects of isoproterenol, prostaglandin E1 and E2, and sodium nitroprusside on the contractile force and tissue content of cAMP and cGMP in the rabbit urethra. They concluded that both cyclic nucleotides can produce relaxation of the urethra. Werkström and associates (2006) characterized the distribution of PDE5, cGMP, and PKG1 in female pig and human urethra and evaluated the effect of pharmacologic inhibition of PDE5 in isolated smooth muscle preparations. After stimulation with the NO donor, DETA NONO-ate, the cGMP-immunoreactivity in urethral and vascular smooth muscles increased. There was a wide distribution of cGMP- and vimentin-positive interstitial cells between pig urethral smooth muscle bundles. PDE5 immunoreactivity could be demonstrated within the urethral and vascular smooth muscle cells but also in vascular endothelial cells that expressed cGMP immunoreactivity. Nerve-induced relaxations of urethral preparations were enhanced at low concentrations of sildenafil, vardenafil, and tadalafil, whereas there were direct smooth muscle relaxant actions of the PDE5 inhibitors at high concentrations.
The distribution of PDEs in the male urethral structures does not seem to have been studied.
The observation that patients treated for erectile dysfunction with PDE5 inhibitors had an improvement of their LUTS has sparked a new interest in using these drugs also for treatment of LUTS and OAB. After the report in an open study (Sairam et al, 2002) that treatment with sildenafil appeared to improve urinary symptom scores in men with erectile dysfunction and LUTS, this observation has been confirmed in several well-designed and conducted RCTs (McVary et al, 2007a, 2007b; Stief et al, 2008).
McVary and colleagues (2007a) evaluated the effects of sildenafil (50 to 100 mg daily for 12 weeks) on erectile dysfunction and LUTS in men 45 years or older who scored 25 or less on the erectile function domain of the International Index of Erectile Function (IIEF) and 12 or greater on the International Prostate Symptom Score (IPSS). In 189 men receiving sildenafil, significant improvements were observed in IPPS (−6.32 vs. −1.93, P < .0001), Benign Prostatic Hyperplasia Impact Index (−2.0 vs. −0.9, P < .0001), mean IPSS quality of life score (−0.97 vs. −0.29, P < .0001) and total Self-Esteem and Relationship questionnaire scores (24.6 vs. 4.3, P < .0001). Interestingly there was no difference in urinary flow between the groups (P = .08). Significantly more sildenafil versus placebo-treated patients were satisfied with treatment (71.2 vs. 41.7, P < .0001). Sildenafil was well tolerated.
In an RCT, treatment with tadalafil once daily, in addition to improving erectile function in men with LUTS, was demonstrated to produce a clinically meaningful and statistically significant symptomatic improvement of LUTS (McVary et al, 2007b). In another RCT, vardenafil given twice daily for 8 weeks to men with erectile dysfunction and LUTS was shown to significantly improve LUTS, erectile function, and quality of life (Stief et al, 2008).
The mechanism behind the beneficial effect of the PDE inhibitors on LUTS/OAB and their site(s) of action largely remain to be elucidated. If the site of action were the smooth muscles of the outflow region (and the effect relaxation), an increase in flow rate should be expected. In none of the trials referred to, such an effect was found. However, there are several other structures in the LUT that may be involved, including those in the urothelial signaling pathway (urothelium, interstitial cells, and suburothelial afferent nerves).
PDE5 inhibitors have a documented effect in men with LUTS/OAB (see Table 68–2). The place of PDE5 inhibitors in the treatment of OAB/DO remains to be established. Specifically, there seems to be no published information on the effects of these drugs in women with OAB/DO.
Antidepressants
Many clinicians believe that tricyclic antidepressants, particularly imipramine (Tofranil, others), are useful agents for facilitating urine storage, both by decreasing bladder contractility and by increasing outlet resistance (Wein, 1995a, 1995b). These agents have been the subject of a voluminous amount of highly sophisticated pharmacologic investigation to determine the mechanisms of action responsible for their varied effects (Maggi et al, 1989b; Richelson, 1994; Baldessarini, 2006). Most data have been accumulated as a result of trying to explain the antidepressant properties of these agents and are thus primarily from CNS tissue. The results, conclusions, and speculations inferred from the data are extremely interesting, but it should be emphasized that it is essentially unknown whether they apply to or have relevance for the LUT.
Tricyclic antidepressants possess varying degrees of at least three major pharmacologic actions: (1) they have central and peripheral antimuscarinic effects at some, but not all, sites; (2) they block the active transport system in the presynaptic nerve ending that is responsible for the reuptake of the released amine neurotransmitters norepinephrine and serotonin; and (3) they are sedatives, an action that occurs presumably on a central basis but is perhaps related to antihistaminic properties (at H1 receptors, although they also antagonize H2 receptors to some extent). There is also evidence that they desensitize at least some α2- and some β-AR agonists. Paradoxically, they also have been shown to block some α-AR agonists and 5-hydroxytryptamine type 1 receptors.
Imipramine
As indicated earlier, imipramine has complex pharmacologic effects, and its mode of action in OAB/DO has not been established (Hunsballe and Djurhuus, 2001). Even if it is generally considered that imipramine is a useful drug in the treatment of OAB/DO, no good quality RCTs that can document this have been retrieved.
It has been known for a long time that imipramine can have favorable effects in the treatment of nocturnal enuresis in children with a success rate of 10% to 70% in controlled trials (Hunsballe and Djurhuus, 2001; Glazener et al, 2003).
The risks (see later) and benefits of imipramine in the treatment of voiding disorders do not seem to have been assessed. Very few studies have been performed during the past decade (Hunsballe and Djurhuus, 2001). No good quality RCTs have documented that the drug is effective in the treatment DO. However, a beneficial effect has been documented in the treatment of nocturnal enuresis.
Doxepine
Doxepine is another tricyclic antidepressant that was found to be more potent, using in-vitro rabbit bladder strips, than other tricyclic compounds with respect to antimuscarinic and musculotropic relaxant activity (Levin and Wein, 1984). Lose and associates (1989), in a randomized, double-blind crossover study of females with involuntary bladder contractions, and either frequency, urgency, or urgency incontinence, found that this agent caused a significant decrease in urine loss (pad weighing test) and in the cystometric parameters of first sensation and maximum bladder capacity. The dosage of doxepin used was either a single 50-mg bedtime dose or this dose plus an additional 25 mg in the morning. The number of daytime incontinence episodes decreased in both doxepin and placebo groups, and the difference was not statistically significant. Doxepine treatment was preferred by 14 patients, whereas 2 preferred placebo. Three patients had no preference. Of the 14 patients who stated a preference for doxepine, 12 claimed that they became continent during treatment whereas 2 claimed improvement; the 2 patients who preferred placebo claimed improvement. The AHCPR Guidelines combine results for imipramine and doxepine, citing only three randomized controlled trials, with an unknown percent of female patients. Percent cures (all figures refer to percent drug effect minus percent effect on placebo) are listed as 31%, percent reduction in urgency incontinence as 20% to 77%, and percent side effects as 0% to 70% (U.S. Department of Health and Human Services, 1992).
Duloxetine
Duloxetine is a serotonin-norepinephrine reuptake inhibitor that has been shown to significantly increase sphincteric muscle activity during the filling/storage phase of micturition in the cat acetic acid model of irritated bladder function (Thor et al, 1995; Katofiasc et al, 2002). Bladder capacity was also increased in this model, both effects mediated centrally through both motor efferent and sensory afferent modulation (Fraser et al, 2003). The effects of duloxetine were studied in a placebo-controlled study comprising 306 women (aged 21 to 84 years) with OAB, randomly treated with placebo (153) or duloxetine (80 mg/day for 4 weeks, which was increased to 120 mg/day for 8 weeks (Steers et al, 2007). The primary efficacy analysis compared the treatment effects on mean change from baseline to end point in the mean number of voiding episodes per 24 hours. Patients randomized to duloxetine had significant improvements over those randomized to placebo for decreases in voiding and incontinence episodes (−1.81 vs. −0.62, for increases in the daytime voiding intervals (29 vs. 7 minutes), and for improvements in Incontinence Quality of Life scores at both doses of duloxetine. Urodynamic studies showed no significant increases in maximum cystometric capacity or in the volume threshold for DO. The most common treatment-emergent adverse events with duloxetine (nausea 31%, placebo 4.6%; dry mouth 16%, placebo 1.3%; dizziness 14%, placebo 0.7%; constipation 14%, placebo 3.3%; insomnia 13%, placebo 1.3%; and fatigue 11%, placebo 2.0%) were the same as those reported by women with SUI (see later) and were significantly more common with duloxetine than placebo. Also in females with mixed incontinence, improvement of the OAB component has been demonstrated (Bent et al, 2008; Schagen van Leeuwen et al, 2008). For assessment, see Table 68–2.
When used in the generally larger doses employed for antidepressant effects the most frequent side effects of the tricyclic antidepressants are those attributable to their systemic antimuscarinic activity (Richelson, 1994; Baldessarini, 2006). Allergic phenomena, including rash, hepatic dysfunction, obstructive jaundice, and agranulocytosis may also occur but rarely. CNS side effects may include weakness, fatigue, parkinsonian effect, a fine tremor noted most in the upper extremities, a manic or schizophrenic picture, and sedation, probably from an antihistaminic effect. Postural hypotension may also be seen, presumably on the basis of selective blockade (a paradoxical effect) of α1-AR agonists in some vascular smooth muscle. Tricyclic antidepressants can also cause excess sweating of obscure etiology and a delay of orgasm or orgasmic impotence, the cause of which likewise is unclear. They can also produce arrhythmias and interact in deleterious ways with other drugs, and so caution must be observed in their use in patients with cardiac disease (Baldessarini, 2006). Whether cardiotoxicity will prove to be a legitimate concern in patients receiving the smaller doses (than for treatment of depression) for LUT dysfunction remains to be seen but is a potential matter of concern. Consultation with a patient’s internist or cardiologist is always helpful before instituting such therapy in questionable situations. It is well established that therapeutic doses of tricyclic antidepressants, including imipramine, may cause serious toxic effects on the cardiovascular system (orthostatic hypotension, ventricular arrhythmias). Imipramine prolongs QTc intervals and has an antiarrhythmic (and proarrhythmic) effect similar to that of quinidine (Bigger et al, 1977; Giardina et al, 1979). Children seem particularly sensitive to the cardiotoxic action of tricyclic antidepressants (Baldessarini, 2006).
With respect to the potential cardiovascular risks of antidepressant medication, two additional points need to be made. First of all, it may be depression itself that is associated with an increased risk of myocardial infarction, cardiovascular disease, and all-cause mortality (see references in Cohen et al, 2000). Although treatments for depression, including antidepressive medications, are certainly a potential factor underlying this association, the separation from disease association and treatment association is difficult at best. The second point relates to whether there is a difference in this regard between the use of tricyclic antidepressants and selective serotonin reuptake inhibitors. Data presented by Cohen and coworkers (2000) suggest with respect to long-term adverse cardiovascular outcome there is an association between the use of tricyclic antidepressants but not selective serotonin reuptake inhibitors, a conclusion that differs from earlier data that indicated no significant differences in the safety or efficacy of these two groups of agents (AHCPR Report, cited by Cohen et al, 2000). As a closing statement on this subject, it should be noted that data in the literature refer to therapeutic doses of these medications for depression and not the smaller (in comparison) doses of imipramine used for the treatment of voiding dysfunction.
The use of imipramine is contraindicated in patients receiving monoamine oxidase inhibitors, because severe CNS toxicity can be precipitated, including hyperpyrexia, seizures, and coma. Some potential side effects of the antidepressants may be especially significant for the elderly, specifically weakness, fatigue, and postural hypotension. Psychotropic drugs in general have been shown to increase the risk of falls and hip fractures in the elderly (Liu et al, 1998). If imipramine or any of the tricyclic antidepressants is to be prescribed for the treatment of voiding dysfunction, the patient should be thoroughly informed of the fact that this is not the usual indication for this drug and that potential side effects exist. Reports of significant side effects (severe abdominal distress, nausea, vomiting, headache, lethargy, and irritability) after abrupt cessation of high doses of imipramine in children would suggest that the drug should be discontinued gradually, especially in patients receiving high doses.
Cyclooxygenase Inhibitors
Human bladder mucosa has the ability to synthesize eicosanoids (Jeremy et al, 1987), and these agents can be liberated from bladder muscle and mucosa in response to different types of trauma (Downie and Karmazyn, 1984; Leslie et al, 1984). Even if prostaglandins cause contraction of human detrusor (Andersson, 1993) it is still unclear whether prostaglandins contribute to the pathogenesis of DO. More important than direct effects on the bladder muscle may be sensitization of sensory afferent nerves, increasing the afferent input produced by a given degree of bladder filling. Involuntary bladder contractions can then be triggered at a small bladder volume. If this is an important mechanism, treatment with prostaglandin synthesis inhibitors could be expected to be effective. However, clinical evidence for this is scarce.
Cardozo and colleagues (1980) performed a double-blind controlled study of 30 women with DO using the prostaglandin synthesis inhibitor flurbiprofen at a dosage of 50 mg three times daily. The drug was shown to have favorable effects, although it did not completely abolish DO. There was a high incidence of side effects (43%) including nausea, vomiting, headache, and gastrointestinal symptoms. Palmer (1983) studied the effects of flurbiprofen 50 mg four times daily versus placebo in a double-blind, crossover trial in 37 patients with idiopathic DO (27% of the patients did not complete the trial). Active treatment significantly increased maximum contractile pressure, decreased the number of voids, and decreased the number of urgent voids compared with baseline. Indomethacin 50 to 100 mg/day was reported to give symptomatic relief in patients with DO, compared with bromocriptine in a randomized, single-blind, crossover study (Cardozo and Stanton, 1980). The incidence of side effects was high, occurring in 19 of 32 patients. However, no patient had to stop treatment because of side effects.
The few controlled clinical trials on the effects of prostaglandin synthesis inhibitors in the treatment of DO, and the limited number of drugs tested, make it difficult to evaluate their therapeutic value (see Table 68–2). No new RCTs on the effects of COX inhibitors in OAB/DO patients seem to have been published during the past decade.
Dimethyl Sulfoxide
Dimethyl sulfoxide (DMSO) is a relatively simple, naturally occurring organic compound that has been used as an industrial solvent for many years. It has multiple pharmacologic actions (membrane penetrant, anti-inflammatory, local analgesic, bacteriostatic, diuretic, cholinesterase inhibitor, collagen solvent, vasodilator) and has been used for the treatment of arthritis and other musculoskeletal disorders, generally in a 70% solution. The formulation for human intravesical use is a 50% solution. Sant (1987) has summarized the pharmacology and clinical use of DMSO and has tabulated “good to excellent” results in 50% to 90% of collected series of patients treated with intravesical instillation for interstitial cystitis. However, DMSO has not been shown to be useful in the treatment of neurogenic or idiopathic DO or in any patients with urgency/frequency but without interstitial cystitis. The subject of interstitial cystitis and its treatment is considered in Chapter 12.
Polysynaptic Inhibitors
Baclofen (Lioresal) is discussed primarily along with agents that decrease outlet resistance secondary to striated sphincter dyssynergia. Baclofen is a γ-aminobutyric acid type B (GABAB) receptor agonist that depresses monosynaptic and polysynaptic motoneurons and interneurons in the spinal cord (Andersson et al, 1999, 2000a). It has also been shown capable of depressing neurogenic DO secondary to a spinal cord lesion (Kiesswetter and Schober, 1975). Taylor and Bates (1979), in a double-blind crossover study, reported it to be very effective also in decreasing daytime and night-time urinary frequency and incontinence in patients with idiopathic DO. Cystometric changes were not recorded, however, and considerable improvement was also obtained in the placebo group. Effective intrathecal use of baclofen for treatment of neurogenic DO was reported by Kums and Delhaas (1991), Steers and associates (1992), and Bushman and colleagues (1993). Little on this subject has appeared during the past decade.
Toxins
Vanilloids
By means of capsaicin, a subpopulation of primary afferent neurons innervating the bladder and urethra, the “capsaicin-sensitive nerves,” has been identified. It is believed that capsaicin exerts its effects by acting on specific “vanilloid” receptors on these nerves (Szallasi, 1994). Capsaicin exerts a biphasic effect: initial excitation is followed by a long-lasting blockade, which renders sensitive primary afferents (C fibers) resistant to activation by natural stimuli. In sufficiently high concentrations, capsaicin is believed to cause “desensitization” initially by releasing and emptying the stores of neuropeptides and then by blocking further release (Maggi, 1993). Resiniferatoxin (RTX) is an analogue of capsaicin, approximately 1000 times more potent for desensitization than capsaicin (Ishizuka et al, 1995a) but only a few hundred times more potent for excitation (Szallasi and Blumberg, 1999). Possibly, both capsaicin and RTX can have effects on Aδ fibers. It is also possible that capsaicin at high concentrations (mM) has additional, nonspecific effects (Kuo, 1997).
The rationale for intravesical instillations of vanilloids is based on the involvement of C fibers in the pathophysiology of conditions such as bladder hypersensitivity and neurogenic DO. In the healthy human bladder C fibers carry the response to noxious stimuli, but they are not implicated in the normal voiding reflex. After spinal cord injury major neuroplasticity appears within bladder afferents in several mammalian species, including humans. C-fiber bladder afferents proliferate within the suburothelium and become sensitive to bladder distention. Those changes lead to the emergence of a new C-fiber–mediated voiding reflex, which is strongly involved in spinal neurogenic DO. Improvement of this condition by defunctionalization of C-fiber bladder afferents with intravesical vanilloids has been widely demonstrated in humans and animals.
Cystometric evidence that capsaicin-sensitive nerves may modulate the afferent branch of the micturition reflex in humans was originally presented by Maggi and colleagues (1989a), who instilled capsaicin (0.1 to 10 µM) intravesically in five patients with hypersensitivity disorders with attenuation of their symptoms a few days after administration. Intravesical capsaicin, given in considerably higher concentrations (1 to 2 mM) than those administered by Maggi and colleagues (1989a), has since been used with success in neurologic disorders such as multiple sclerosis or traumatic chronic spinal lesions (Fowler et al, 1992b, 1994; Geirsson et al, 1995; DeRidder et al, 1997; for more discussion see de Seze et al, 1999; DeRidder and Baert, 2000; Andersson et al, 2002, 2009; Cruz, 2004; de Seze et al, 2004). Side effects of intravesical capsaicin include discomfort and a burning sensation at the pubic/urethral level during instillation, an effect that can be overcome by prior instillation of lidocaine, which does not interfere with the beneficial effects of capsaicin (Chandiramani et al, 1996). No premalignant or malignant changes in the bladder have been found in biopsy specimens of patients who had repeated capsaicin instillations for up to 5 years (Dasgupta et al, 1998).
The beneficial effect of RTX has been demonstrated in several studies (see Andersson 2002a, 2009; Kim et al, 2003; Kuo et al, 2003; de Seze et al, 2004; Giannantoni et al, 2004; Watanabe, 2004).
de Seze and associates (2004) compared the efficacy and tolerability of nonalcohol capsaicin (1 mM) versus RTX (100 nM) in 10% alcohol in a randomized, double-blind, parallel-group study in 39 spinal cord–injured adult patients with neurogenic DO (hyperreflexia). Efficacy (voiding chart and cystomanometry) and tolerability were evaluated during a 3-month follow-up. On day 30, clinical and urodynamic improvement was found in 78% and 83% of patients with capsaicin versus 80% and 60% with RTX, respectively, without a significant difference between the two treated groups. The benefit remained in two thirds of the two groups on day 90. There were no significant differences in regard to the incidence, nature, or duration of side effects in capsaicin- versus RTX-treated patients. The data suggested that capsaicin and RTX are equally efficient for relieving the clinical and urodynamic symptoms of neurogenic DO and that glucidic capsaicin is as well tolerated as ethanolic RTX.
Available information (including data from RCTs) suggests that both capsaicin and RTX may have useful effects in the treatment of neurogenic DO. There may be beneficial effects also in non-neurogenic DO in selected cases refractory to antimuscarinic treatment, but further RCT-based documentation is desired. RTX is an interesting alternative to capsaicin, but the drug is currently not in clinical development owing to formulation problems.
Neither capsaicin nor RTX is approved in the United States for clinical use. It is obvious, however, that the intravesical use of such agents has the potential to significantly contribute to the treatment of DO in patients with neurogenic and other types of LUT dysfunction. Various techniques of administration of intravesical capsaicin and RTX differ slightly among experienced users and are described in detail in the articles by Chancellor and de Groat (1999), Fowler (2000), and DeRidder and Baert (2000).
Botulinum Neurotoxin (BoNT)
BoNT is produced by Clostridium botulinum. Of the seven subtypes of BoNT, subtype A (BoNT/A) is the most relevant clinically. Most of the intravesical experience reported on BoNT/A deals with Botox. However, BoNT/A toxin is also available under the trade names of Dysport and Xeomin. Bladder experience with the latter was, however, never reported. Available information indicates that Botox is roughly three times more potent than Dysport (Cruz and Silva, 2004; Nitti et al, 2006). However, this relation has never been clearly defined. Therefore in a bladder setting these equivalents should be approached with caution (for clinical reviews, see Nitti et al, 2006; Patel et al, 2006; Cruz and Dinis, 2007; Karsenty et al, 2008). In addition to subtype A, recent reports have investigated the detrusor injection of BoNT subtype B (Neurobloc, Miobloc). BoNT was shown to exert a prolonged local effect when injected directly into skeletal muscles, and the effects are dose dependent and reversible.
BoNT consists of a heavy and a light chain linked by a disulfide bond. In the synaptic cleft the toxin binds to synaptic vesicle protein or SV2 (Dong et al, 2006) through its heavy chain and is internalized by the nerve terminal. On cleavage, the light chain is released in the cytosol, where it impedes binding of neurotransmitter-containing synaptic vesicles to the plasma membrane. This is achieved through the N-ethylmaleimide-sensitive factor attachment protein (SNARE) complex made up of synaptosome-associated protein 25 kD (SNAP 25), synaptobrevin (vesicle-associated membrane protein [VAMP]), and syntaxin. BoNT/A cleaves SNAP 25 rendering the SNARE complex inactive (Humeau et al, 2000; Chancellor et al, 2008a). Subtype B acts preferentially through the inactivation of VAMP (Humeau et al, 2000).
The rationale for using BoNT to treat human DO is based on the assumption that effects of the toxin on skeletal muscle would be replicated in bladder smooth muscle (Schurch et al, 2000a, 2000b) and that detrusor muscle paralysis would reduce the symptoms of OAB (Duthie et al, 2007). BoNT has been developed as a second-line treatment option (after failure of, or intolerance to, appropriate antimuscarinic therapy) for patients with NDO with urinary incontinence or other neurogenic OAB symptoms and who are able and willing to perform CIC.
The first report of the application of BoNT/A in NDO appeared in 2000 (Schurch et al, 2000b). Injection of BoNT/A 200 to 300 units at 20 to 30 detrusor muscle sites restored continence in 17 of 19 patients (89.5%) with severe neurogenic DO and incontinence secondary to traumatic spinal cord injury at the 6-week follow-up. No side effects were reported, mean reflex volume and mean maximum cystometric bladder capacity increased significantly from baseline, and there was a significant decrease in mean maximum detrusor pressure. Since then, the efficacy of BoNT/A injection into the detrusor muscle in adult patients with NDO refractory to, and/or intolerant of, antimuscarinic agents has been confirmed in a number of studies, which have been the subject of systematic review (Karsenty et al, 2008; Apostolidis et al, 2009; see also Andersson et al, 2009). The analysis by Karsenty and colleagues (2008) evaluated 18 trials with Botox, involving a total of 698 patients, of whom 83% had NDO with urinary incontinence. Significant benefits were seen in clinical variables (micturition frequency and number of incontinence episodes) as well as urodynamic variables (maximum detrusor pressure, maximum cystometric capacity). Complete continence was achieved in 40% to 80% of patients. Efficacy has also been demonstrated in spinal cord–injured patients with detrusor–striated sphincter dyssynergia (Dykstra et al, 1990, 1998; Schurch et al, 1996). A significant response to BoNT/A is seen as early as 1 week after treatment; however, maximum effects were seen between 1 and 4 weeks. The efficacy of BoNT/A appears to persist for at least 3 to 4 months and up to 1 year but does decline over time (Grosse et al, 2005), meaning that repeat injections are required for continued therapeutic effect. Repeat injections have been shown to be effective and well tolerated (Grosse et al, 2005; Karsenty et al, 2006; Giannantoni et al, 2009), and there is no reported evidence of a reduction in response over time, after two to nine repeat injections. The beneficial clinical and urologic effects of BoNT/A in adults with NDO are accompanied by improvement in patients’ quality of life (Kalsi et al, 2006; Schurch et al, 2007).
BoNT/A is generally well tolerated. Data from a systematic review of the role of BoNT/A in NDO indicated that the most frequent adverse events are injection site pain, procedure-related urinary tract infection, and mild hematuria (Karsenty et al, 2008). A potential adverse effect resulting from the use of BoNT in patients not using CIC is an increase in postvoid residual volume that may result in de novo CIC (6% to 88% of patients), with associated impact on quality of life (Karsenty et al, 2008; Shaban et al, 2008). Systemic side effects, although rare, could be very disabling for patients with spinal cord injury (De Laet et al, 2005).
Important factors to consider in relation to the risk of adverse events during urologic use of BoNT/A are the drug dosage, the formulation used, and the injection technique. Available data indicate that Dysport may be associated with a higher risk of side effects related to drug migration (e.g., muscle weakness) than Botox (Dmochowski and Sand, 2007). The doses typically evaluated in clinical trials in urologic indications differ between the commercially available formulations of BoNT/A. Additionally, systemic adverse reactions, including respiratory compromise and death, have been reported after the use of BoNT/A and BoNT/B for both U.S. Food and Drug Administration (FDA) approved and unapproved uses (Dmochowski and Sand, 2007). The most serious cases involved treatment of children for cerebral palsy–associated limb spasticity, and the FDA is currently reviewing safety data relating to marketed BoNT products.
Accumulating evidence indicates that BoNT/A may be a useful option for treating children with NDO and urinary incontinence and whose disorders are refractory to antimuscarinic therapy. A systematic review on the use of BoNT/A (Botox) in children with NDO identified six small (10 to 26 patients) prospective studies primarily involving children with myelomeningocele (Game et al, 2009). BoNT/A treatment resulted in a reduction in urinary incontinence of 40% to 80%, and between 65% and 87% of patients became completely continent between CIC. In addition, all studies showed a significant impact of treatment on urodynamic variables, including maximum detrusor pressure and maximum cystometric capacity. Improvement was observed within 2 weeks of BoNT/A injection and persisted for up to 6 months. The amount of Botox injected ranged from 5 to 12 units/kg, with a maximum dose of 360 units. Repeat administration of BoNT/A was associated with similar therapeutic effects to the first dose. Available data therefore indicate that BoNT/A may have the ability to prevent or delay the need for surgery in children with NDO, but further studies with the appropriate design and with a longer follow-up are required to confirm this. The safety profile in children has not been established, and there is a need for studies that evaluate the effect of long-term, repeated administration on the bladder wall. Although the value of BoNT for the treatment of NDO is now recognized in European and U.S. consensus reports (Naumann et al, 2008; Apostolides et al, 2009), at the time of this writing, the only approval for BoNT/A in a urologic indication is for Botox for the treatment of OAB in Brazil. On the whole, studies on BoNT/A are small (<50 patients), short term, and are largely restricted to patients with spinal cord injury or multiple sclerosis. Indeed, several questions remain unanswered. These include the optimal dose and frequency of administration to balance efficacy with safety, the most appropriate method and location of administration, and how to identify patients who would benefit the most. Considerable systematic research is still required to answer these questions. Large phase 3 registration studies are being conducted to support the registration of this treatment for NDO, including the optimal dose recommendation.
There are limited data on the use of botulinum toxin B (BoNT/B) for incontinence, particularly in patients with NDO. In the only two published trials on the use of BoNT/B in urologic indications, the vast majority of patients had non-neurogenic OAB (Dykstra et al, 2003a, 2003b; Ghei et al, 2005). Data on the use of BoNT/B in patients with NDO come primarily from three case reports (Dykstra et al, 2003b; Pistolesi et al, 2004; Reitz and Schurch, 2004). In one report, BoNT/B 5000 units was injected into 10 different areas of the bladder wall in a patient with multiple sclerosis whose disease was refractory to oral and topical antimuscarinic agents. The patient reported a positive response within 24 hours and had no adverse effects. The beneficial effects of BoNT/B began to decline after 4 months, and a repeat BoNT/B dose of 7500 units was given that also provided therapeutic benefit for 4 months (Dykstra et al, 2003b). BoNT/A and BoNT/B interfere with different presynaptic proteins, meaning that a primary non-response to the type A toxin does not necessarily imply a non-response to the type B toxin. Indeed, data from the other two case reports indicate that BoNT/B may have a place for the management of patients with NDO resistant to therapy with the type A toxin. However, the duration of effect was only 4 to 6 weeks (Pistolesi et al, 2004; Reitz and Schurch, 2004). There are no data on the use of BoNT/B in children.
Estrogens for Urgency Urinary Incontinence and Overactive Bladder Symptoms
Estrogen has been used to treat postmenopausal urgency and urgency incontinence for many years, but there have been few controlled trials to confirm that it is of benefit (Hextall, 2000; for assessment, see Table 68–2). A double-blind multicenter study of 64 postmenopausal women with “urgency syndrome” failed to show efficacy (Cardozo et al, 1993). All women underwent pretreatment urodynamic investigation to ensure that they had either “sensory urgency” or DO. They were randomized to treatment with oral estriol 3 mg daily or placebo for 3 months. Compliance with therapy was confirmed by a significant improvement in the maturation index of vaginal epithelial cells in the active but not the placebo group. Estriol produced subjective and objective improvements in urinary symptoms but was not significantly better than placebo.
Another recent RCT from the same group using 25 mg estradiol implants confirmed the previous findings (Rufford et al, 2003) and additionally found a high complication rate in the estradiol-treated patients (i.e., vaginal bleeding).
Evidence from Recent Large Clinical Trials
The HERS study included 763 postmenopausal women younger than the age of 80 years with coronary heart disease and intact uteri (Grady et al, 2001). It was designed to evaluate the use of estrogen in secondary prevention of cardiac events. In a secondary analysis, 1525 participants who reported at least one episode of incontinence per week at baseline were included. Participants were randomly assigned to 0.625 mg of conjugated estrogens plus 2.5 mg of medroxyprogesterone acetate in one tablet (n = 768) or placebo (n = 757) and were followed for a mean of 4.1 years. Severity of incontinence was classified as improved, unchanged, or worsened. The results showed that incontinence improved in 26% of the women assigned to placebo compared with 21% assigned to hormones whereas 27% of the placebo worsened compared with 39% of the hormone group (P = .001). This difference was evident by 4 months of treatment, for both urgency incontinence and SUI. The number of incontinent episodes per week increased an average of 0.7 in the hormone group and decreased by 0.1 in the placebo group (P < .001). The authors concluded that daily oral estrogen plus progesterone therapy was associated with worsening urinary incontinence in older postmenopausal women with weekly incontinence and did not recommend this therapy for treatment of incontinence. However, it is possible that the progestogen component may have had an influence on the results of this study.
The Women’s Health Initiative (WHI) was a multicenter double-blind, placebo-controlled RCT of menopausal hormone therapy in 27,347 postmenopausal women aged 50 to 79 years enrolled between 1992 and 1998 for whom urinary incontinence symptoms were known in 23,296 participants at baseline and 1 year. The women were randomized based on hysterectomy status to active treatment or placebo. Those with a uterus were given 0.625 mg/day of conjugated equine estrogen (CEE) plus 2.5 mg/day of medroxyprogesterone acetate (CEE+MPA), whereas those who had undergone hysterectomy received estrogen alone (CEE). At 1 year hormone therapy was shown to increase the incidence of all types of urinary incontinence among women who were continent at baseline. The risk was highest for SUI, with results as follows: CEE+MPA: relative risk [RR] 1.7, 95% CI 1.61 to 2.18; CEE alone: RR 2.15, 95% CI 1.77 to 2.62; followed by mixed urinary incontinence CEE+MPA: RR 1.49, 95% CI 1.10 to 2.01. Results for CEE alone were RR 1.79 and 95% CI 1.26 to 2.53. The combination of CEE and MPA had no significant effect on developing urgency urinary incontinence: RR 1.15, 95% CI 0.99 to 1.34; but CEE alone increased the risk: RR 1.32, 95% CI 1.10 to 1.58. For those women experiencing urinary incontinence at baseline, frequency worsened in both active groups: CEE+MPA: RR 1.38, 95% CI 1.28 to 1.49; CEE alone: RR 1.47, 95% CI 1.35 to 1.61. Quantity of urinary incontinence worsened at 1 year in both active groups: CEE+MPA: RR 1.20, 95% CI 1.06 to 1.76; CEE alone: RR 1.59, 95% CI 1.39 to 1.82. Those women receiving hormone therapy were more likely to report that urinary incontinence limited their daily activities: CEE+MPA: RR 1.18, 95% CI 1.06 to 1.32; CEE alone: RR 1.29, 95% CI 1.15 to 1.45 at 1 year. Thus based on this secondary analysis of data from a huge study, conjugated equine estrogen alone or in combination with medroxyprogesterone acetate was shown to increase the risk of urinary incontinence among continent women and worsen urinary incontinence among symptomatic women after 1 year of therapy.
The Nurses Health Study (Grodstein et al, 2004) was a biennial postal questionnaire starting in 1976. In 1996, 39,436 postmenopausal women aged 50 to 75 years who reported no urinary leakage at the start of the study were followed-up for 4 years to identify incident cases of urinary incontinence: 5060 cases of occasional and 2495 cases of frequent incontinence were identified. The risk of developing urinary incontinence was increased among postmenopausal women taking hormones compared with women who had never taken hormones, respectively (oral estrogen: RR 1.54, 95% CI 1.44, 1.65; transdermal estrogen: RR1.68, 95% CI 1.41, 2.00; oral estrogen with progestin: RR 1.34, 95% CI 1.24, 1.44; transdermal estrogen with progestin: RR 1.46, 95% CI 1.16, 1.84). After cessation of hormone therapy there was a decreased risk of incontinence such that 10 years after stopping hormones the risk was identical in women who had and who never had taken hormone therapy.
Conclusions
Estrogen has an important physiologic effect on the female LUT and its deficiency is an etiologic factor in the pathogenesis of a number of conditions. However, the use of estrogen either alone or in combination with progestogen has yielded poor results. The current level 1 evidence against the use of estrogens for the treatment of urinary incontinence comes from studies powered to assess their benefit in the prevention of cardiovascular events and therefore the secondary analyses have been based only on self-reported symptoms of urinary leakage without any objective data. Despite this, all of these large randomized controlled trials show a worsening of preexisting urinary incontinence (both stress and urgency) and an increased new incidence of urinary incontinence with both estrogen and estrogen plus progesterone. However, the majority of patients in all of these studies were taking combined equine estrogen and this may not be representative of all estrogens taken by all routes of administration.
In a systematic review of the effects of estrogens for symptoms suggestive of an OAB, the conclusion was that estrogen therapy may be effective in alleviating OAB symptoms and that local administration may be the most beneficial route of administration (Cardozo et al, 2004c). It is quite possible that the reason for this is that the symptoms of urinary urgency, frequency, and urgency incontinence may be a manifestation of urogenital atrophy in older postmenopausal women rather than a direct effect on the LUT (Robinson and Cardozo, 2003). Although there is good evidence that the symptoms and cytologic changes of urogenital atrophy may be reversed by low dose (local) vaginal estrogen therapy there is currently no evidence that estrogens with or without progestogens should be used in the treatment of urinary incontinence. Of interest would be studies looking at (1) the role of prophylactic versus therapeutic estrogen and (2) a double-blind study of transvaginal estrogen in various but low doses.
Concerns about the use of estrogen are summarized by Loose and Stancel (2006). Unopposed estrogen for hormone treatment in postmenopausal women increases the risk of endometrial carcinoma by 5 to 15 fold, but this increased risk can be prevented if a progestin is coadministered with the estrogen, and this is now standard practice. In the WHI study (see earlier), an estrogen-progestin combination increased the total risk of breast cancer by 24%, translating into an absolute increase in attributable cases of disease of 6 per 1000 women requiring 3 or more years of treatment. In women without a uterus who received estrogen alone, the relative risk of breast cancer was actually decreased by 23%, and the decrease only narrowly missed reaching statistical significance. It appears to be the progestin component that plays a major role in the increased risk of breast cancer. Estrogens increase cholesterol levels in bile and cause a relative twofold to threefold increase in gallbladder disease. The HERS study and the WHI study indicate that combination estrogen-progestin does not protect against coronary heart disease, but the specific populations studied and the specific preparations utilized may prohibit generalization of these conclusions to other preparations, doses, and patient populations. It is clear, however, that the risk of thromboembolic disease is clearly elevated in women taking oral estrogen, both healthy women and women with preexisting cardiovascular disease. In the WHI study the estrogen-progestin combination was associated with an increase in eight cases of stroke per 10,000 women and a similar increase in pulmonary embolism. Loose and Stancel (2006) also mention as possibilities altered cognition, changes in carbohydrate and lipid metabolism, hypertension, nausea, migraine, and changes in mood.
Future Possibilities
Peripherally Acting Drugs
A few new peripheral targets suitable for treatment have been identified and are under evaluation.
Vitamin D3 Receptor Analogues
Rat and human bladders were shown to express receptors for vitamin D (Crescioli et al, 2005), which makes it conceivable that the bladder may also be a target for vitamin D. Analogues of vitamin D3 have also been shown to inhibit benign prostatic hyperplasia (BPH) cell proliferation and to counteract the mitogenic activity of potent growth factors for BPH cells (Crescioli et al, 2002, 2003, 2004). Experiments in rats with bladder outflow obstruction (Schröder et al, 2006) showed that one of the analogues, BXL-628, at nonhypercalcemic doses, did not prevent bladder hypertrophy but reduced the decrease in contractility of the bladder smooth muscle that occurred with increasing bladder weight (Schröder et al, 2006). The mechanism of action for the effects has not been clarified. However, elocalcitol was shown to have an inhibitory effect on the RhoA/Rho kinase pathway (Morelli et al, 2007). Upregulation of this pathway has been associated with bladder changes associated with diabetes, outflow obstruction, and DO (Peters et al, 2006; Christ and Andersson, 2007). The effect of elocalcitol on prostate volume was evaluated in patients with BPH, and it was found that BXL628 was able to arrest prostate growth within 12 weeks in men aged 50 years or older with prostatic volume of 40 mL or more (Colli et al, 2006). In an RCT enrolling 120 female patients with OAB, in which the primary end point was an increase in the mean volume voided, a significant increase versus placebo (22% vs. 11%) was demonstrated (Colli et al, 2007). Whether vitamin D receptor agonism (monotherapy or in combination) will be a useful alternative for the treatment of LUTS/OAB requires further RCTs.
Centrally Acting Drugs
Many parts of the brain seem to be activated during storage and voiding (see, Griffiths 2007; Fowler et al, 2008; Griffiths and Tadic, 2008), and there is increasing interest in drugs modulating the micturition reflex by a central action (Andersson and Pehrson, 2003). Several drugs used for pain treatment also affect micturition, with morphine and some antiepileptic drugs being a few examples. However, CNS mechanisms have so far not been preferred targets for drugs aimed to treat OAB, because selective actions may be difficult to obtain. Holstege (2005), reviewing some of the CNS mechanisms involved in micturition, including the periaqueductal gray matter and the pontine micturition center, suggested that the problem in OAB or urgency incontinence is at the level of the periaqueductal gray matter or pontine micturition center and their connections, and possible treatments for this condition, should target the micturition pathways at that level.
Gonadotropin-Releasing Hormone Antagonists
The beneficial effects of the 5α-reductase inhibitors finasteride and dutasteride in the treatment of male LUTS are well documented. The efficacy of other hormonal treatments, for example, antiandrogens or gonadotropin-releasing hormone (GNRH; also known as luteinizing hormone-releasing hormone [LHRH]) agonists is either poor or at the expense of unacceptable side effects such as medical castration associated with hot flushes, decrease of potency and libido, and negative effects on bone density and cardiac health after long-term androgen ablation (Schroeder et al, 1986; Peters et al, 1987; Bosch et al, 1989; Eri and Tveter, 1993). With LHRH antagonists, submaximal, noncastrating blockade of the androgen testosterone and consequently of dihydrotestosterone can be achieved, thus avoiding medical castration.
Debruyne and coworkers (2008) demonstrated in a phase 2 RCT that the LHRH antagonist cetrorelix given subcutaneously weekly for 20 weeks to 140 men with LUTS (IPSS ≥ 13, peak urinary flow rates 5 to 13 mL/sec) rapidly caused a significant improvement in the mean IPSS: the peak decrease was −5.4 to −5.9 versus −2.8 for placebo. All dosage regimens tested were well tolerated, and the authors concluded that the drug offered a safe and effective treatment of male LUTS. Further studies are needed to assess whether this therapeutic principle is a useful addition to the current treatment alternatives.
Gabapentin
Gabapentin is one of the new first-generation antiepileptic drugs that expanded its use into a broad range of neurologic and psychiatric disorders (Striano and Striano, 2008). It was originally designed as an anticonvulsant γ-aminobutyric acid (GABA) mimetic capable of crossing the blood-brain barrier (Maneuf et al, 2003). The effects of gabapentin, however, do not appear to be mediated through interaction with GABA receptors, and its mechanism of action remains controversial (Maneuf et al, 2003). It has been suggested that it acts by binding to a subunit of the α2δ unit of voltage-dependent Ca2+ channels (Gee et al, 1996; Striano and Striano, 2008). Gabapentin is also widely used not only for seizures and neuropathic pain but also for many other indications, such as anxiety and sleep disorders, because of its apparent lack of toxicity.
Carbone and coworkers (2006) reported on the effect of gabapentin on neurogenic DO. They found a positive effect on symptoms and significant improvement in urodynamic parameters and suggested that the effects of the drug should be explored in further controlled studies in both neurogenic and non-neurogenic DO. Kim and colleagues (2004) studied the effects of gabapentin in patients with OAB and nocturia not responding to antimuscarinic agents. They found that 14 of 31 patients experienced improvement of their symptoms with orally administered gabapentin. The drug was generally well tolerated, and the authors suggested that it can be considered in selective patients when conventional modalities have failed. It is possible that gabapentin and other α2δ ligands (e.g., pregabalin and analogues) will offer new therapeutic alternatives.
Tramadol
Tramadol is a well-known analgesic drug (Grond and Sablotzski, 2004). By itself it is a weak µ-receptor agonist, but it is metabolized to several different compounds, some of them almost as effective as morphine at the µ receptor. However, the drug (metabolites) also inhibits serotonin (5-HT) and norepinephrine reuptake (Grond and Sablotzski, 2004). This profile is of particular interest because both µ-receptor agonism and amine reuptake inhibition may be useful principles for treatment of LUTS/OAB/DO, as shown in a placebo-controlled study with duloxetine (Steers et al, 2008).
In rats, tramadol abolished experimentally induced DO caused by cerebral infarction (Pehrson et al, 2003). Tramadol also inhibited DO induced by apomorphine in rats (Pehrson and Andersson, 2003), a crude model of bladder dysfunction in Parkinson disease. Singh and associates (2008) gave tramadol epidurally and found the drug to increase bladder capacity and compliance and to delay filling sensations without adverse effects on voiding. Safarinejad and Hosseini (2006) evaluated in a double-blind, placebo-controlled randomized study the efficacy and safety of tramadol in patients with idiopathic DO. A total of 76 patients 18 years or older were given 100 mg tramadol in sustained-release form every 12 hours for 12 weeks. Clinical evaluation was performed at baseline and every 2 weeks during treatment. Tramadol significantly (P < .01) reduced the number of incontinence periods per 24 hours from 3.2 ± 3.3 to 1.6 ± 2.8 and induced improvements in urodynamic parameters. The main adverse event was nausea. It was concluded that, in patients with non-neurogenic DO, tramadol provided beneficial clinical and urodynamic effects. Even if tramadol may not be the best suitable drug for treatment of LUTS/OAB the study suggests efficacy for modulation of micturition via the µ receptor.
NK1-Receptor Antagonists
The main endogenous tachykinins substance P (SP), neurokinin A (NKA), and neurokinin B (NKB) and their preferred receptors NK1, NK2, and NK3, respectively, have been demonstrated in various CNS regions, including those involved in micturition control (Lecci and Maggi, 2001; Covenas et al, 2003; Saffroy et al, 2003). NK1 receptor–expressing neurons in the dorsal horn of the spinal cord may play an important role in DO, and tachykinin involvement via NK1 receptors in the micturition reflex induced by bladder filling has been demonstrated (Ishizuka et al, 1994) in normal and, more clearly, rats with bladder hypertrophy secondary to bladder outlet obstruction. Capsaicin-induced DO was reduced by blocking NK1 receptor–expressing neurons in the spinal cord, using intrathecally administered substance P/saponin conjugate (Seki et al, 2005). Furthermore, blockade of spinal NK1 receptor could suppress detrusor activity induced by dopamine receptor (levodopa) stimulation (Ishizuka et al, 1995b).
In conscious rats undergoing continuous cystometry, antagonists of both NK1 and NK2 receptors inhibited micturition, decreasing micturition pressure and increasing bladder capacity at low doses and inducing dribbling incontinence at high doses. This was most conspicuous in animals with outflow obstruction (Gu et al, 2000). Intracerebroventricular administration of NK1 and NK2 receptor antagonists to awake rats suppressed detrusor activity induced by dopamine receptor (levodopa) stimulation (Ishizuka et al, 2000). Taken together, available information suggests that spinal and supraspinal NK1 and NK2 receptors may be involved in micturition control.
Aprepitant, an NK-1 receptor antagonist used for treatment of chemotherapy-induced nausea and vomiting (Massaro and Lenz, 2005), was reported to improve symptoms of OAB in postmenopausal women with a history of urgency incontinence or mixed incontinence (with predominantly urgency urinary incontinence), as shown in a well-designed pilot RCT (Green et al, 2006). The primary end point was percent change from baseline in average daily micturitions assessed by a voiding diary. Secondary end points included average daily total urinary incontinence and urgency incontinence episodes and urgency episodes. Aprepitant significantly (P < .003) decreased the average daily number of micturitions (−1.3 ± 1.9) compared with placebo (−0.4 ± 1.7) at 8 weeks. The average daily number of urgency episodes was also significantly (P < .047) reduced (−23.2 ± 32%) compared with placebo (−9.3 ± 40%) and so were the average daily number of urgency incontinence and total urinary incontinence episodes, although the difference was not statistically significant. The large standard errors and the low placebo results are atypical for OAB trials. Aprepitant was generally well tolerated, and the incidence of side effects, including dry mouth, was low. The results of this initial proof of concept study suggest that NK-1 receptor antagonism holds promise as a potential treatment approach for OAB.
Drug Treatment of Overactivity in Augmented or Intestinal Neobladders
With regard to the subject of overactivity in bowel augmented or intestinal neobladders, Andersson and associates (1992) reviewed this subject and its pharmacologic treatment. They noted a few instances of positive results with drugs given systematically, but locally applied agents were believed to offer more promise. Pure antimuscarinic agents had produced few good results, either locally or systemically. Oxybutynin had shown some good results with local therapy but poor results with systemic therapy. The α- and β-AR agonists had shown little or no effects. Other possibilities mentioned included opioid agonists (diphenoxylate and loperamide), Ca2+ antagonists, K+ channel openers, and nitric oxide donors. Apostolidis and colleagues (2007) reported a case of successful treatment with BoNT-A after failed augmentation ileocystoplasty.
Increasing Outlet Resistance
Many factors seem to be involved in the pathogenesis of SUI: urethral support, vesical neck function, and function of the nerves and musculature of the bladder, urethra, and pelvic floor (DeLancey, 1997; Mostwin et al, 2005). Anatomic factors cannot be treated pharmacologically. However, women with SUI have lower resting urethral pressures than age-matched continent women (Henriksson et al, 1979; Hilton and Stanton, 1983), and because it seems likely that there is a reduced urethral closure pressure in most women with SUI it seems logical to increase urethral pressure to improve the condition.
Factors that may contribute to urethral closure include tone of urethral smooth and striated muscle and the passive properties of the urethral lamina propria, in particular its vasculature. The relative contribution to intraurethral pressure of these factors is still subject to debate. However, there is ample pharmacologic evidence that a substantial part of urethral tone is mediated through stimulation of α-ARs in the urethral smooth muscle by released norepinephrine (Andersson, 1993; Andersson and Wein, 2004). A contributing factor to SUI, mainly in elderly women with lack of estrogen, may be lack of mucosal function. The pharmacologic treatment of SUI aims at increasing intraurethral closure forces by increasing the tone in the urethral smooth and striated muscles. Several drugs may contribute to such an increase (Andersson, 1988; Zinner et al, 2004b), but relative lack of efficacy and/or side effects have limited their clinical use (Table 68–4).
Table 68–4 Drugs Used in the Treatment of Stress Incontinence (ICI, 2008; Andersson et al, 2009)
| DRUG | LEVEL OF EVIDENCE | GRADE OF RECOMMENDATION |
|---|---|---|
| Duloxetine | 1 | B |
| Imipramine | 3 | D |
| Clenbuterol | 3 | C |
| Methoxamine | 2 | D |
| Midodrine | 2 | C |
| Ephedrine | 3 | D |
| Norephedrine (phenylpropanolamine) | 3 | D |
| Estrogen | 2 | D |
α-Adrenergic Receptor Agonists
Several drugs with agonistic effects on α-ARs have been used in the treatment of SUI (Table 68–5). However, ephedrine and norephedrine (phenylpropanolamine [PPA]) seem to have been the most widely used (Andersson et al, 2002). The original Agency for Healthcare Policy and Research Guidelines (1992) reported eight RCTs with PPA 50 mg twice daily for SUI in women. Percent cures (all figures refer to percent effect on drug minus percent effect on placebo) were listed as 0% to 14%, percent reduction in continence as 19% to 60%, and percent side effects and percent dropouts as 5% to 33% and 0% to 4.3%, respectively. The most recent Cochrane review on the subject (Alhasso et al, 2005) assessed RCTs or quasi-RCTs in adults with SUI that included an adrenergic agonist drug in at least one arm of the trial. There were no controlled studies reported on the use of such drugs in men. Twenty-two eligible trials were identified, 11 of which were crossover trials, which included 1099 women, 673 of whom received an adrenergic drug (PPA in 11, midrodrine in 2, norepinephrine in 3, clenbuterol in 3, terbutaline in 1, eskornade in 1, and RO 115-1240 in 1). The authors concluded “there was weak evidence to suggest that use of an adrenergic agonist was better than placebo treatment.” The limited evidence suggested that such drugs were better than placebo in reducing the number of pad changes and incontinence episodes and in improving subjective symptoms. There was not enough evidence to evaluate the merits of an adrenergic agonist compared with estrogen, whether used alone or in combination. Regarding adverse events, the review reported similar numbers with adrenergic, placebo, or alternative drug treatment. Over 25% of subjects reported such effects, but when these consisted of effects due to adrenergic stimulation they caused discontinuation in only 4% of the total.
Table 68–5 Drugs Used in the Treatment of Overflow Incontinence (ICI 2004)
| DRUG | LEVEL OF EVIDENCE | GRADE OF RECOMMENDATION |
|---|---|---|
| α-Adrenoceptor Antagonists | ||
| Alfuzosin | 4 | C |
| Doxazosin | 4 | C |
| Prazosin | 4 | C |
| Terazosin | 4 | C |
| Tamsulosin | 4 | C |
| Phenoxybenzamine | 4 | NR |
| Muscarinic Receptor Agonists | ||
| Bethanechol | 4 | D |
| Carbachol | 4 | D |
| Cholinesterase Inhibitors | ||
| Distigmine | 4 | D |
| Other Drugs | ||
| Baclofen | 4 | C |
| Benzodiazepines | 4 | C |
| Dantrolene | 4 | C |
NR, not recommended.
The date of the most recent amendment to this review is listed as May 15, 2007; no new trials were identified at that time. The update of 2005 (Alhasso et al, 2005) included seven new RCTs over the 2003 report, cited in the last consultation. The final statement on “what’s new” reads, “The conclusions still provide limited support for the use of adrenergics but side effects may cause dropout, and some side effects may be dangerous. Further trials are needed.”
Ephedrine and PPA lack selectivity for urethral α-ARs and can increase blood pressure and cause sleep disturbances, headache, tremor, and palpitations (Andersson et al, 2002). Kernan and coworkers (2000) reported the risk of hemorrhagic stroke to be 16 times higher in women younger than 50 years of age who had been taking PPA as an appetite suppressant (statistically significant) and 3 times higher in women who had been taking the drug for less than 24 hours as a cold remedy (not statistically significant). There was no increased risk in men. PPA has been removed from the market in the United States.
Numerous case reports of adverse reactions due to ephedra alkaloids exist, and some (Bent et al, 2003) had suggested that sale of these compounds as a dietary supplement be restricted or banned. In December 2003 the U.S. FDA decreed such a ban, a move that has survived legal appeal.
Midodrine and methoxamine stimulate α1-ARs with some degree of selectivity. According to the RCTs available, the effectiveness of these drugs is moderate and the clinical usefulness seems to be limited by adverse effects (Weil et al, 1998; Radley et al, 2001; Alhasso et al, 2003).
Attempts continue to develop agonists with relative selectivity for the human urethra. Musselman and associates (2004) reported on a phase 2 randomized crossover study with Ro 115-1240, a peripheral active selective α1A/1L adrenergic receptor partial agonist (Blue et al, 2004) in 37 women with mild to moderate SUI. A moderate, positive effect was demonstrated, but side effects have apparently curtailed further development of the drug.
β-Adrenergic Receptor Antagonists
The theoretical basis for the use of β-AR antagonists in the treatment of SUI is that blockade of urethral β-ARs may enhance the effects of norepinephrine on urethral α-ARs. Propranolol has been reported to have beneficial effects in the treatment of SUI (Gleason et al, 1974; Kaisary, 1984) but there are no RCTs supporting such an action. In Gleason and colleagues’ (1974) study the beneficial effects become manifest only after 4 to 10 weeks of treatment—a phenomenon difficult to explain. Donker and Van der Sluis (1976) reported that β blockade did not change the urethral pressure profile in normal women. Although suggested as an alternative to α-AR agonists in patients with SUI and hypertension, these agents may have major potential cardiac and pulmonary side effects of their own, related to their therapeutic β-AR blockade.
β-Adrenergic Receptor Agonists
β-AR stimulation is generally conceded to decrease urethral pressure (Andersson, 1993), but β2-AR agonists have been reported to increase the contractility of some fast-contracting striated muscle fibers and suppress that of slow-contracting fibers of others (Fellenius et al, 1980). Some β-AR agonists also stimulate skeletal muscle hypertrophy—in fast-twitch more so than slow-twitch fibers (Kim et al, 1992). Clenbuterol has been reported to potentiate the field stimulation–induced contraction in rabbit isolated periurethral muscle preparations, an action that is suppressed by propranolol and greater than that produced by isoproterenol (Kishimoto et al, 1991). These authors were the first to report an increase in urethral pressure with clinical use of clenbuterol and to speculate on its potential for the treatment of SUI. Yamanishi and associates (1994) reported an inotropic effect of clenbuterol and terbutaline on the fatigued striated urethral sphincter of dogs that was abolished by β-AR blockade.
Yasuda and coworkers (1993) described the results of a double-blind placebo-controlled trial with clenbuterol agent in 165 women with SUI. Positive statistical significance was achieved for subjective evaluation of incontinence frequency, pad usage per day, and overall global assessment. Pad weight decreased from 11.7 ± 17.9 g to 6.0 ± 12.3 g for drug and from 18.3 ± 29.0 g to 12.6 ± 24.7 g for placebo, raising questions about the comparability of the two groups. The “significant” increase in maximum urethral closure pressure was from 46.0 ± 18.2 cm H2O to 49.3 ± 19.1 cm H2O, versus a change of −1.5 cm H2O in the placebo group. Fifty-six of 77 patients in the clenbuterol group reported some degree of improvement versus 48 of 88 in the placebo group. The positive effects were suggested to be a result of an action on urethral striated muscle and/or the pelvic floor muscles. Ishiko and associates (2000) investigated the effects of clenbuterol on 61 female patients with SUI in a 12-week randomized study, comparing drug therapy to pelvic floor exercises. The frequency and volume of SUI and the patient’s own impression were used as the basis for the assessment of efficacy. The improvement of incontinence was 76.9%, 52.6%, and 89.5% in the respective groups. In an open study, Noguchi and colleagues (1997) reported positive results with clenbuterol (20 mg twice daily for 1 month) in 9 of 14 patients with mild to moderate SUI after radical prostatectomy. No subsequent published reports have appeared. Further well-designed RCTs documenting the effects of clenbuterol are needed to adequately assess its potential as a treatment for SUI.
Serotonin-Norepinephrine Uptake Inhibitors
Imipramine
Imipramine, among several other pharmacologic effects, inhibits the reuptake of norepinephrine and serotonin in adrenergic nerve endings. In the urethra this can be expected to enhance the contractile effects of norepinephrine on urethral smooth muscle. Gilja and colleagues (1984) reported in an open study on 30 women with SUI that imipramine 75 mg daily produced subjective continence in 21 patients and increased mean maximum urethral closure pressure from 34 to 48 mm Hg. A 35% cure rate was reported by pad test and, in an additional 25%, a 50% or more improvement.
Lin and coworkers (1999) assessed the efficacy of imipramine (25 mg imipramine three times a day for 3 months) as a treatment of genuine SUI in 40 women. A 20-minute pad test, uroflowmetry, filling and voiding cystometry, and stress urethral pressure profile were performed before and after treatment. The efficacy of “successful treatment” was 60% (95% CI 11.8 to 75.2). There are no RCTs on the effects of imipramine on SUI. No subsequent published reports have appeared.
Duloxetine
As mentioned previously, duloxetine is a serotonin-norepinephrine reuptake inhibitor that has been shown to significantly increase sphincteric muscle activity during the filling/storage phase of micturition in the cat acetic acid model of irritated bladder function (Thor et al, 1995; Katofiasc et al, 2002). The sphincteric effects were reversed by α1-adrenergic (prazosin) and 5-HT2 serotonergic (LY 53857) antagonism, whereas the bladder effects were mediated by temporal prolongation of the actions of serotonin and norepinephrine in the synaptic cleft (Fraser et al, 2003). Duloxetine is lipophilic, well absorbed, and extensively metabolized (CYP2D6). Its plasma half-life is approximately 12 hours (Sharma et al, 2000).
There are many studies, including RCTs, documenting the effects of duloxetine in SUI (Norton et al, 2002; Dmochowski et al, 2003a; Cardozo et al, 2004a; Millard et al, 2004; Van Kerrebroeck et al, 2004; Ghoneim et al, 2005; Kinchen et al, 2005; Hurley et al, 2006; Castro-Diaz et al, 2007; Bump, 2008). A Cochrane review of the effects of duloxetine for SUI in women is available, the last substantive amendment listed as May 25, 2005 (Mariappan et al, 2005). Fifteen reports were deemed eligible for analysis, nine primary studies, and six additional reports related to one or two of the primary references. An additional analysis “performed under the auspices of the Cochrane Incontinence Group” was performed on just the nine primary trials comparing duloxetine and placebo and published separately (Mariappan et al, 2007). The results can be summarized as follows. Subjective “cure” in the duloxetine 80 mg daily (40 mg twice daily) group was higher than in the placebo group (10.8% vs. 7.7%, overall RR = 1.42; 95% CI, 1.02 to 1.98; P = .04). The estimated absolute size of effect was about 3 more patients cured of every 100 treated. Objective cure data, available from only one trial, showed no clear drug/placebo difference. Duloxetine showed greater improvement in Incontinence Quality of Life score (weighted mean difference for 80 mg: 4.5; 95% CI 2.83 to 6.18, P < .00001). Patient global impression of improvement (PGI-I) data also favored the drug (RR for better health status 1.24, 95% CI 1.14 to 1.36; P < .00001). Adverse effects in six trials were analyzed. These were reported by 71% of drug subjects and 59% of those allocated to placebo. Nausea was the most common adverse event, and the incidence ranged from 23% to 25% and was the main reason for discontinuation. Other side effects reported were vomiting, constipation, dry mouth, fatigue, dizziness, and insomnia for an overall RR of 1.30 (95% CI 1.23 to 1.37). Across these six trials 17% in the drug group and 4% in the placebo study arms withdrew. In the 2007 article the authors conclude by saying that further research is needed as to whether management policies incorporating duloxetine are clinically effective and cost effective compared with other current minimally invasive and more invasive approaches in patients with varying severity of SUI and that “longer term experience is now a priority to determine whether there is sustained efficacy during and after duloxetine use and to rule out complications.” Hashim and Abrams (2006) suggested that to reduce the risk of nausea that therapy start with a dose of 20 mg twice daily for 2 weeks and then to increase to the recommended 40 mg twice-daily dosage.
Duloxetine is licensed at 40 mg twice daily for the treatment of SUI in the European Union (European Medicines Agency, 2005) for women with moderate to severe SUI (defined as 14 or more episodes per week). It was withdrawn from the U.S. FDA consideration process for the treatment of SUI but is approved for the treatment of major depressive disorder (20 to 30 mg twice daily initially, 60 mg once-daily maintenance), diabetic peripheral neuropathic pain (60 mg once daily), and generalized anxiety disorder (60 mg once daily). The product information contains a “black box” warning of “increased risk of suicidal thinking and behavior in children, adolescents, and young adults taking antidepressants for major depressive disorder and other psychiatric disorders,” noting also that “depression and certain other psychiatric disorders are themselves associated with increases in the risk of suicide” (Prescribing Information, revised December 2007, Eli Lilly and Company, Indianapolis, IN). Other warnings and precautions in the U.S. product information for psychiatric indications, not SUI, include hepatotoxicity (not to be used in patients with substantial alcohol use or chronic liver disease), orthostatic hypotension, serotonin syndrome (general statement regarding selective serotonin reuptake inhibitors, abrupt discontinuation (may result in dizziness, paresthesias, irritability, and headache), inhibitors of CYP1H2 or thioridazine (do not administer concomitantly), potent inhibitors of CYP2D6 (may increase concentration), and others.
Male Stress Urinary Incontinence
Although a problem of significant magnitude, especially after total prostatectomy for cancer, the pharmacologic treatment of male SUI is an area that has received relatively little attention. Tsakiris and coworkers (2008) searched for articles on this subject published between 1966 and June 2007 and did a generalized database search in addition. Nine trials were identified using α-AR agonists, β2 antagonists, or serotonin-norepinephrine reuptake inhibitors. Only one of these included a comparison arm (Filocamo et al, 2007), 40 mg twice daily duloxetine plus pelvic floor exercises versus pelvic floor exercises with placebo. The results suggested a positive effect of drug but were a bit confusing. Of those patients completing the 4-month trial (92/112), 78% of the drug-treated patients versus 52% of those in the placebo group were dry. However, 1 month after the end of the study the corresponding figures were 46% versus 73%, a shift still observed 2 months later. The authors of the review article suggest further larger and well-designed studies on duloxetine for this potential usage.
Estrogens for Stress Incontinence
Estrogen-sensitive tissues of the bladder, urethra, and pelvic floor are all potentially important in maintaining the continence mechanism in women. For women to remain continent during increases in intra-abdominal pressure the urethral pressure must exceed the intravesical pressure at all times except during micturition. There are four estrogen-sensitive functional layers that could play a part in the maintenance of a positive urethral pressure and closure mechanism: (1) the epithelium, including the mucosal seal mechanism; (2) the vasculature; (3) the connective tissue component; and (4) the muscular components, including the smooth muscle of the bladder neck and urethra and the striated muscle, which is both intrinsic to the urethra and extrinsic (a part of the pelvic floor striated musculature). If estrogen did have a positive effect on SUI it could be by means of an effect on any or all of the structures (Andersson et al, 2009).
The role of estrogen in the treatment of SUI has been controversial, even though there are a number of reported studies (see Hextall, 2000). Some have given promising results, but this may be because they were observational, not randomized, blinded, or controlled. The situation is further complicated by the fact that a number of different types of estrogen have been used with varying doses, routes of administration, and durations of treatment. Fantl and coworkers (1996) treated 83 hypoestrogenic women with urodynamic evidence of genuine SUI and/or detrusor instability with conjugated equine estrogens 0.625 mg and medroxyprogesterone 10 mg cyclically for 3 months. Controls received placebo tablets. At the end of the study period the clinical and quality of life variables had not changed significantly in either group. Jackson and associates (1999) treated 67 postmenopausal women with genuine SUI or mixed incontinence with estradiol valerate 2 mg or placebo daily for 6 months. There was no significant change in objective outcome measures.
There have been two meta-analyses performed that have helped to clarify the situation further. In the first, a report by the Hormones and Urogenital Therapy (HUT) committee, the use of estrogens to treat all causes of incontinence in postmenopausal women was examined (Fantl et al, 1994). Of 166 articles identified, which were published in English between 1969 and 1992, only 6 were controlled trials and 17 uncontrolled series. The results showed that there was a significant subjective improvement for all patients and those with genuine SUI. However, assessment of the objective parameters revealed that there was no change in the volume of urine lost. Maximum urethral closure pressure did increase significantly, but this result was influenced by only one study showing a large effect. In the second meta-analysis, Sultana and Walters (1990) reviewed 8 controlled and 14 uncontrolled prospective trials and included all types of estrogen treatment. They also found that estrogen therapy was not an efficacious treatment of SUI but may be useful for the often associated symptoms of urgency and frequency. Estrogen when given alone therefore does not appear to be an effective treatment for SUI. However, several studies have shown that it may have a role in combination with other therapies (for combination with α-AR agonists, see earlier). In a randomized trial, Ishiko and coworkers (2001) compared the effects of the combination of pelvic floor exercise and estriol (1 mg/day) in 66 patients with postmenopausal SUI. Efficacy was evaluated every 3 months based on stress scores obtained from a questionnaire. They found a significant decrease in stress score in mild and moderate SUI patients in both groups 3 months after the start of therapy and concluded that combination therapy with estriol plus pelvic floor exercise was effective and capable of serving as first-line treatment for mild SUI.
These conclusions still seem valid. Thus reviews of recent literature agree on the fact that “estrogen therapy has little effect in the management of urodynamic SUI” (Al-Badr et al, 2003; Robinson and Cardozo, 2003a, 2003b).
Circumventing the Problem
Vasopressin Analogues: Desmopressin
Desmopressin (1-desamino-8-D-arginine vasopressin; DDAVP) is a synthetic vasopressin analogue with a pronounced antidiuretic effect but practically lacking vasopressor actions (Andersson et al, 1988; Vande Walle et al, 2006). It is now widely used as a treatment for primary nocturnal enuresis (Nevéus et al, 1999; Glazener and Evans, 2002; Andersson et al, 2009). Studies have shown that one of the factors that can contribute to nocturnal enuresis in children, and probably in adults, is lack of a normal nocturnal increase in plasma vasopressin, which results in high nocturnal urine production (Rittig et al, 1989; Matthiesen et al, 1996; Nørgaard et al, 1997; Hjalmas, 1999). By decreasing the nocturnal production of urine, beneficial effects may be obtained in enuresis and nocturia. However, the drug may also have stimulatory effects on the CNS, as found in rats (DiMichele et al, 1996). Several, controlled, double-blind investigations have shown intranasal administration of desmopressin to be effective in the treatment of nocturnal enuresis in children (Nevéus et al, 1999; Glazener and Evans, 2002; Andersson et al, 2009). The dose used in most studies has been 20 µg intranasally at bedtime. However, the drug is orally active, even if the bioavailability is low (less than 1% compared with 2% to 10% after intranasal administration), and its efficacy in primary nocturnal enuresis in children and adolescents has been documented in randomized, double-blind, placebo-controlled studies (Janknegt et al, 1997; Skoog et al, 1997; Andersson et al, 2009).
Positive effects of desmopressin on nocturia in adults have been documented (Månsson et al, 1980; Hilton and Stanton, 1982; Asplund and Aberg, 1993; Asplund et al, 1999; Chancellor et al, 1999; Andersson et al, 2009). Nocturnal frequency and enuresis due to idiopathic DO responded favorably to intranasal desmopressin therapy even when previous treatment with “antispasmodics” had been unsuccessful (Hilton and Stanton, 1982). Also in patients with multiple sclerosis, desmopressin was shown in controlled studies to reduce nocturia and micturition frequency (Hilton et al, 1983; Kinn and Larsson 1990; Eckford et al, 1994; Fredrikson et al, 1996; Ferreira and Letwin, 1998; Andersson et al, 2009). In multiple sclerosis, effects on daytime frequency also have been documented (Hoverd and Fowler, 1998). Furthermore, desmopressin was shown to be successful in treating nocturnal enuresis in patients with spina bifida with diurnal incontinence (Horowitz et al, 1997).
Oral desmopressin also has proved to be effective in the treatment of nocturia. In a randomized double-blind study, Mattiasson and colleagues (2002) investigated the efficacy and safety of oral desmopressin in the treatment of nocturia in men. A 3-week dose-titration phase established the optimum desmopressin dose (0.1, 0.2, or 0.4 mg); and, after a 1-week “washout” period, patients who responded in the dose-titration period were randomized to receive the optimal dose of desmopressin or placebo in a double-blind design for 3 weeks. In all, 151 patients entered the double-blind period (86 treated with desmopressin, 65 with placebo). In the desmopressin group, 28 (34%) patients and in the placebo group, 2 (3%) patients had significantly fewer than half the number of nocturnal voids relative to baseline; the mean number of nocturnal voids decreased from 3.0 to 1.7 and from 3.2 to 2.7, respectively, reflecting a mean decrease of 43% and 12%. The mean duration of the first sleep period increased by 59% (from 2.7 to 4.5 hours) in the desmopressin group, compared with an increase of 21% (from 2.5 to 2.9 hours) in the placebo group. The mean nocturnal diuresis decreased by 36% (from 1.5 to 0.9 mL/min) in the desmopressin group and by 6% (from 1.7 to 1.5 mL/min) in the placebo group. The mean ratio of night per 24-hour urine volume decreased by 23% and 1%, and the mean ratio of night/day urine volume decreased by 27% and increased by 3% for the desmopressin and placebo groups, respectively. In the double-blind treatment period, similar numbers of patients had adverse events: 15 (17%) patients in the desmopressin and 16 (25%) patients in the placebo group. Most adverse events were mild. Serum sodium levels were less than 130 mmol/L in 10 (4%) patients, and this occurred during dose titration. The authors concluded that orally administered desmopressin is an effective and well-tolerated treatment for nocturia in men.
Lose and associates (2003) found similar results in women. In the double-blind phase of their study, 144 patients were randomly assigned to groups (desmopressin, n = 72; placebo, n = 72). For desmopressin, 33 (46%) patients had a 50% or greater reduction in nocturnal voids against baseline levels compared with 5 (7%) patients receiving placebo. The mean number of nocturnal voids, duration of sleep until the first nocturnal void, nocturnal diuresis, and ratios of nocturnal per 24 hours and nocturnal per daytime urine volumes changed significantly in favor of desmopressin versus placebo. In the dose-titration phase, headache (22%), nausea (8%), and hyponatremia (6%) were reported.
Robinson and colleagues (2004) introduced antidiuresis as a new concept in managing female daytime urinary incontinence. In a multicenter, multinational, randomized, double-blind, placebo-controlled, crossover exploratory study of women (aged 18 to 80 years) complaining of severe daytime urinary incontinence, 60 received study medication (safety population) and 57 completed the study. The primary efficacy end point was the number of periods with no leakage for 4 hours after dosing. There was a higher mean incidence of periods with no leakage in the first 4 hours on desmopressin, at 62 (35)%, than on placebo, at 48 (40)%, and during the first 8 hours, at 55 (37)% versus 40 (41)%. There was also a higher frequency of dry days on desmopressin than on placebo; 36% of patients had no leakage on virtually all treatment days (6 or 7) for 4 hours after dosing. The time from dosing to first incontinence episode was longer on desmopressin, at 6.3 (2.5) hours versus 5.2 (3.3) hours whereas the volume leaked per incontinence episode was lower on desmopressin than placebo. The total volume voided was consistently lower on desmopressin, at 1180 (58) mL versus 1375 (57) mL, over the 24-hour period after administration. There were no serious or severe adverse events reported, and it was concluded that desmopressin is an effective and safe treatment in women with daytime urinary incontinence.
Even if side effects are uncommon during desmopressin treatment there is a risk of water retention and hyponatremia (Robson et al, 1996; Schwab and Ruder, 1997). In elderly patients it was recommended that the serum sodium concentration should be measured before and after a few days of treatment (Rembratt et al, 2003). Currently there are no predictive factors about who may be at increased risk of developing hyponatremia (Andersson et al, 2009). However, to reduce the risk of hyponatremia it is recommended that patients older than age 79 years or with a 24-hour urine volume more than 28 mL/kg should not be given desmopressin (Rembratt et al, 2006). In those older than age 65 years, serum sodium levels should be checked at baseline and at 3 days and 7 days after beginning treatment or changing dose. It is probably good medical practice that these serum sodium measurements are also applied to those younger than 65 years of age, as well as checking sodium levels at 3 weeks after treatment or change of dose because there is the potential for levels of sodium to mean change within a 3-week period (Callreus et al, 2005). One study showed that hyponatremia can develop after 6 months of administration, although this is not clinically significant (Bae et al, 2007), and therefore serum sodium levels may be checked at 6 months after administration and then every 6 months thereafter. Long-term use of desmopressin does not seem to affect baseline antidiuretic hormone secretion (Bae et al, 2007). The fluid intake will need to be limited to a minimum from 1 hour before the dose until 8 hours afterward, and there needs to be periodic blood pressure and weight measurements to monitor for fluid overload.
Desmopressin is a well-documented therapeutic alternative in pediatric nocturnal enuresis and also is effective in adults with nocturia of polyuric origin. Whether it will be an alternative for managing daytime OAB symptomatology, including urgency, frequency, and urinary incontinence, requires further documentation.
Diuretics
Another circumventive pharmacologic approach is to give a rapid-acting loop diuretic 4 to 6 hours before bedtime. This, of course, assumes the nocturia is not caused by obstructive uropathy. A randomized, double-blind crossover study on this approach using bumetanide in a group of 14 general practice patients was reported by Pedersen and Johansen (1988). Control nocturia episodes per week averaged 17.5; with placebo this decreased to 12 (!) and with drug to 8. Bumetanide was preferred to placebo by 11 of 14 patients.