Cardiac arrhythmias
An abnormality of the cardiac rhythm is called a cardiac arrhythmia. Arrhythmias may cause sudden death, syncope, heart failure, chest pain, dizziness, palpitations or no symptoms at all. There are two main types of arrhythmia:
Tachycardias are more symptomatic when the arrhythmia is fast and sustained. Tachycardias are subdivided into supraventricular tachycardias, which arise from the atrium or the atrioventricular junction, and ventricular tachycardias, which arise from the ventricles.
Some arrhythmias occur in patients with apparently normal hearts, and in others arrhythmias originate from diseased tissue, such as scar, as a result of underlying structural heart disease. When myocardial function is poor, arrhythmias are more symptomatic and are potentially life-threatening.
Sinus node function
The normal cardiac pacemaker is the sinus node (p. 669) and, like most cardiac tissue, it depolarizes spontaneously.
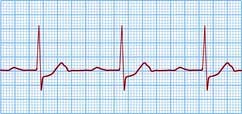
The rate of sinus node discharge is modulated by the autonomic nervous system. Normally the parasympathetic system predominates, resulting in slowing of the spontaneous discharge rate from approximately 100 to 70 b.p.m. A reduction of parasympathetic tone or an increase in sympathetic stimulation leads to tachycardia; conversely, increased parasympathetic tone and decreased sympathetic stimulation produces bradycardia. The sinus rate in women is slightly faster than in men. Normal sinus rhythm is characterized by P waves that are upright in leads I, and II of the ECG (Fig. 14.18), but inverted in the cavity leads AVR and V1 (Fig. 14.36a).
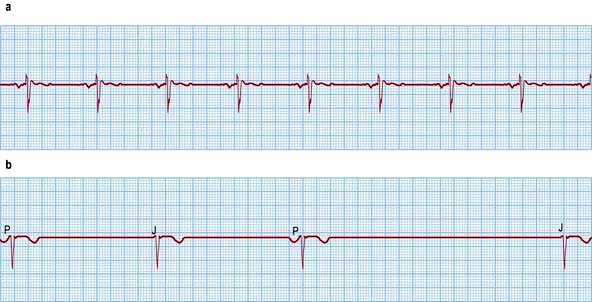
Figure 14.36 (a) An ECG showing normal sinus rhythm (PR interval <0.2 s). P wave preceding each QRS complex. (b) A patient with sick sinus syndrome. This shows sinus arrest (only occasional sinus P waves) and junctional escape beats (J). P waves are inverted in the cavity leads that are shown here.
Sinus arrhythmia
Fluctuations of autonomic tone result in phasic changes of the sinus discharge rate. During inspiration, parasympathetic tone falls and the heart rate quickens, and on expiration the heart rate falls. This variation is normal, particularly in children and young adults. Typically sinus arrhythmia results in predictable irregularities of the pulse.
Sinus bradycardia
A sinus rate of <60 b.p.m. during the day or <50 b.p.m. at night is known as sinus bradycardia.
It is usually asymptomatic unless the rate is very slow. It is normal in athletes owing to increased vagal tone. Other causes may be divided into systemic or cardiac and are discussed below in the section entitled ‘Bradycardias and heart block’ (see p. 698).
Sinus tachycardia
Sinus rate acceleration to more than 100 b.p.m. is known as sinus tachycardia. Again, causes may be divided into systemic or cardiac and are discussed below in the section entitled ‘Supraventricular tachycardias’ (p. 702).
Mechanisms of arrhythmia production
Abnormalities of automaticity, which could arise from a single cell, and abnormalities of conduction, which require abnormal interaction between cells, account for both bradycardia and tachycardia. Sinus bradycardia is a result of abnormally slow automaticity while bradycardia due to AV block is caused by abnormal conduction within the AV node or the intraventricular conduction system. The mechanisms generating tachycardia are shown in Figure 14.37.
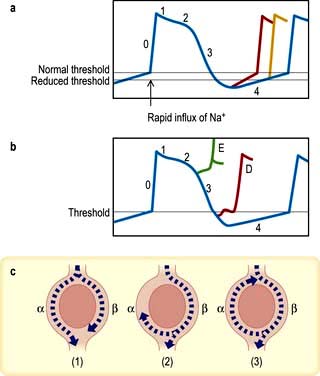
Figure 14.37 Mechanisms of arrhythmogenesis. (a,b) Action potentials (i.e. the potential difference between intracellular and extracellular fluid) of ventricular myocardium after stimulation. (a) Increased (accelerated) automaticity due to reduced threshold potential or an increased slope of phase 4 depolarization (see p. 680). (b) Triggered activity due to early (E) or delayed (D) ‘after depolarizations’ reaching threshold potential. (c) Mechanism of circus movement or re-entry. In panel (1) the impulse passes down both limbs of the potential tachycardia circuit. In panel (2) the impulse is blocked in one pathway (α) but proceeds slowly down pathway β, returning along pathway α until it collides with refractory tissue. In panel (3) the impulse travels so slowly along pathway β that it can return along pathway α and complete the re-entry circuit, producing a circus movement tachycardia.
Accelerated automaticity (Fig. 14.37a)
The normal mechanism of spontaneous cardiac rhythmicity is slow depolarization of the transmembrane voltage during diastole until the threshold potential is reached and the action potential of the pacemaker cells takes off. This mechanism may be accelerated by increasing the rate of diastolic depolarization or changing the threshold potential. For example, sympathetic stimulation releases epinephrine (adrenaline), which enhances automaticity. Such changes are thought to produce sinus tachycardia, escape rhythms and accelerated AV nodal (junctional) rhythms.
Triggered activity (Fig. 14.37b)
Myocardial damage can result in oscillations of the transmembrane potential at the end of the action potential. These oscillations, which are called ‘after depolarizations’, may reach threshold potential and produce an arrhythmia. If they occur before the transmembrane potential reaches its threshold (at the end of phase 3 of the action potential), they are called ‘early after depolarizations’ (E in Fig. 14.37b). When they develop after the transmembrane potential is completed, they are called ‘delayed after depolarizations’ (D in Fig. 14.37b).
The abnormal oscillations can be exaggerated by pacing, catecholamines, electrolyte disturbances, hypoxia, acidosis and some medications, which may then trigger arrhythmia. The atrial tachycardias produced by digoxin toxicity are due to triggered activity. The initiation of ventricular arrhythmia in the long QT syndrome (p. 708) may be caused by this mechanism.
Re-entry (or circus movements) (Fig. 14.37c)
The mechanism of re-entry occurs when a ‘ring’ of cardiac tissue surrounds an inexcitable core (e.g. in a region of scarred myocardium). Tachycardia is initiated if an ectopic beat finds one limb refractory (α), resulting in unidirectional block, and the other limb excitable. Provided conduction through the excitable limb (β) is slow enough, the other limb (α) will have recovered and will allow retrograde activation to complete the re-entry loop. If the time to conduct around the ring is longer than the recovery times (refractory periods) of the tissue within the ring, circus movement will be maintained, producing a run of tachycardia. The majority of regular paroxysmal tachycardias are produced by this mechanism.
Bradycardias and heart block
Bradycardias may be due to failure of impulse formation (sinus bradycardia) or failure of impulse conduction from the atria to the ventricles (atrioventricular block).
Bradycardia
Sinus bradycardia
Sinus bradycardia is due to extrinsic factors influencing a relatively normal sinus node or due to intrinsic sinus node disease. The mechanism can be acute and reversible or chronic and degenerative. Common causes of sinus bradycardia include:
Intrinsic causes
 Acute ischaemia and infarction of the sinus node (as a complication of acute myocardial infarction)
Acute ischaemia and infarction of the sinus node (as a complication of acute myocardial infarction)
Chronic degenerative changes such as fibrosis of the atrium and sinus node (sick sinus syndrome).
Sick sinus syndrome or sinoatrial disease is usually caused by idiopathic fibrosis of the sinus node. Other causes of fibrosis such as ischaemic heart disease, cardiomyopathy or myocarditis can also cause the syndrome. Patients develop episodes of sinus bradycardia or sinus arrest (Fig. 14.36) and commonly, owing to diffuse atrial disease, experience paroxysmal atrial tachyarrhythmias (tachy-brady syndrome).
Neurally mediated syndromes
Neurally mediated syndromes are due to a reflex (called Bezold–Jarisch) that may result in both bradycardia (sinus bradycardia, sinus arrest and AV block) and reflex peripheral vasodilatation. These syndromes usually present as syncope or pre-syncope (dizzy spells).
Carotid sinus syndrome occurs in the elderly and mainly results in bradycardia. Syncope occurs (see p. 676).
Neurocardiogenic (vasovagal) syncope (syndrome) usually presents in young adults but may present for the first time in elderly patients (see p. 676). It results from a variety of situations (physical and emotional) that affect the autonomic nervous system. The efferent output may be predominantly bradycardic, predominantly vasodilatory or mixed.
Postural orthostatic tachycardia syndrome (POTS) is a sudden and significant increase in heart rate associated with normal or mildly reduced blood pressure produced by standing. The underlying mechanism is a failure of the peripheral vasculature to appropriately constrict in response to orthostatic stress, which is compensated by an excessive increase in heart rate.
Many medications, such as antihypertensives, tricyclic antidepressants and neuroleptics, can be the cause of syncope, particularly in the elderly. Careful dose titration and avoidance of combining two agents with potential to cause syncope help to prevent iatrogenic syncope.
Treatment
The management of sinus bradycardia is first to identify and if possible remove any extrinsic causes. Temporary pacing may be employed in patients with reversible causes until a normal sinus rate is restored and in patients with chronic degenerative conditions until a permanent pacemaker is implanted.
Chronic symptomatic sick sinus syndrome requires permanent pacing (DDD), with additional antiarrhythmic drugs (or ablation therapy) to manage any tachycardia element. Thromboembolism is common in tachy-brady syndrome and patients should be anticoagulated unless there is a contraindication.
Patients with carotid sinus hypersensitivity (asystole >3 s), especially if symptoms are reproduced by carotid sinus massage, and in whom life-threatening causes of syncope have been excluded, benefit from pacemaker implantation.
Treatment options in vasovagal attacks include avoidance, if possible, of situations known to cause syncope in a particular patient, and sitting/lying down and applying counter pressure manoeuvres (pushing palms together or crossing legs) if an attack threatens. Increased salt intake, compression of the lower legs with hose and drugs such as beta-blockers, alpha-agonists, e.g. midodrine, or myocardial negative inotropes (such as disopyramide) may be helpful.
In selected patients with ‘malignant’ neurocardiogenic syncope (syncope associated with injuries and demonstrated asystole) permanent pacemaker therapy is helpful. These patients benefit from dual chamber pacemakers with a feature called ‘rate drop response’ which, once activated, paces the heart at a fast rate for a set period of time in order to prevent syncope.
Heart block
Heart block or conduction block may occur at any level in the conducting system. Block in either the AV node or the His bundle results in AV block, whereas block lower in the conduction system produces bundle branch block.
Atrioventricular block
First-degree AV block
This is simple prolongation of the PR interval to >0.22 s. Every atrial depolarization is followed by conduction to the ventricles but with delay (Fig. 14.38).
Second-degree AV block
This occurs when some P waves conduct and others do not. There are several forms (Fig. 14.39):
Mobitz I block (Wenckebach block phenomenon) is progressive PR interval prolongation until a P wave fails to conduct. The PR interval before the blocked P wave is much longer than the PR interval after the blocked P wave.
Mobitz II block occurs when a dropped QRS complex is not preceded by progressive PR interval prolongation. Usually the QRS complex is wide (>0.12 s).
2 : 1 or 3 : 1 (advanced) block occurs when every second or third P wave conducts to the ventricles. This form of second-degree block is neither Mobitz I nor II.
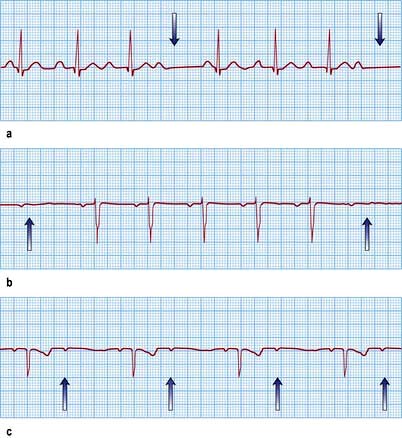
Figure 14.39 Three varieties of second-degree atrioventricular (AV) block. (a) Wenckebach (Mobitz type I) AV block. The PR interval gradually prolongs until the P wave does not conduct to the ventricles (arrows). (b) Mobitz type II AV block. The P waves that do not conduct to the ventricles (arrows) are not preceded by gradual PR interval prolongation. (c) Two P waves to each QRS complex. The PR interval prior to the dropped P wave is always the same. It is not possible to define this type of AV block as type I or type II Mobitz block and it is, therefore, a third variety of second-degree AV block (arrows show P waves).
Wenckebach AV block in general is due to block in the AV node, whereas Mobitz II block signifies block at an infra-nodal level such as the His bundle. The risk of progression to complete heart block is greater and the reliability of the resultant escape rhythm is less with Mobitz II block. Therefore pacing is usually indicated in Mobitz II block, whereas patients with Wenckebach AV block are usually monitored.
Acute myocardial infarction may produce second-degree heart block. In inferior myocardial infarction, close monitoring and transcutaneous temporary back-up pacing are all that is required. In anterior myocardial infarction, second-degree heart block is associated with a high risk of progression to complete heart block, and temporary pacing followed by permanent pacemaker implantation is usually indicated; 2 : 1 Heart block may either be due to block in the AV node or at an infra-nodal level. Management depends on the clinical setting in which it occurs.
Third-degree (complete) AV block
Complete heart block occurs when all atrial activity fails to conduct to the ventricles (Fig. 14.40). In patients with complete heart block the aetiology needs to be established (Table 14.7). In this situation life is maintained by a spontaneous escape rhythm.
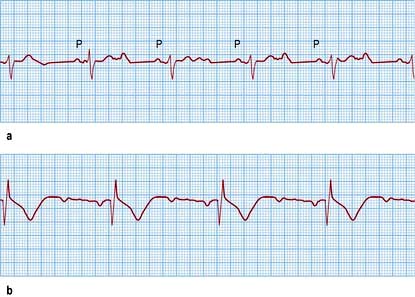
Figure 14.40 Two examples of complete heart block. (a) Congenital complete heart block. The QRS complex is narrow (0.08 s) and the QRS rate is relatively rapid (52 b.p.m.). (b) Acquired complete heart block. The QRS complex is broad (0.13 s) and the QRS rate is relatively slow (38 b.p.m.).
Table 14.7 Aetiology of complete heart block
|
|
|
SLE, systemic lupus erythematosus; CABG, coronary artery bypass graft surgery; VSD, ventricular septal defect; AV, atrioventricular.
Narrow complex escape rhythm (<0.12 s QRS complex) implies that it originates in the His bundle and therefore that the region of block lies more proximally in the AV node. The escape rhythm occurs with an adequate rate (50–60 b.p.m.) and is relatively reliable.
Treatment depends on the aetiology. Recent-onset narrow-complex AV block due to transient causes may respond to intravenous atropine, but temporary pacing facilities should be available for the management of these patients. Chronic narrow-complex AV block requires permanent pacing (dual chamber, p. 695) if it is symptomatic or associated with heart disease. Pacing is also advocated for isolated, congenital AV block, even if asymptomatic.
Broad complex escape rhythm (>0.12 s) implies that the escape rhythm originates below the His bundle and therefore that the region of block lies more distally in the His-Purkinje system.
The resulting rhythm is slow (15–40 b.p.m.) and relatively unreliable. Dizziness and blackouts (Stokes–Adams attacks) often occur. In the elderly, it is usually caused by degenerative fibrosis and calcification of the distal conduction system (Lev’s disease). In younger individuals, a proximal progressive cardiac conduction disease due to the inflammatory process is known as Lenegre’s disease. Sodium channel abnormalities have been identified in both syndromes. Broad-complex AV block may also be caused by ischaemic heart disease, myocarditis or cardiomyopathy. Permanent pacemaker implantation (p. 695) is indicated, as pacing considerably reduces the mortality. Because ventricular arrhythmias are not uncommon, an implantable cardioverter-defibrillator (ICD) may be indicated in those with severe left ventricular dysfunction (>0.30 s).
Bundle branch block
The His bundle gives rise to the right and left bundle branches. The left bundle subdivides into the anterior and posterior divisions of the left bundle. Various conduction disturbances can occur.
Bundle branch conduction delay
This produces slight widening of the QRS complex (up to 0.11 s). It is known as incomplete bundle branch block.
Complete block of a bundle branch
This is associated with a wider QRS complex (≥0.12 s). The shape of the QRS depends on whether the right or the left bundle is blocked.
Right bundle branch block (Fig. 14.41a) produces late activation of the right ventricle. This is seen as deep S waves in leads I and V6 and as a tall late R wave in lead V1 (late activation moving towards right- and away from left-sided leads).
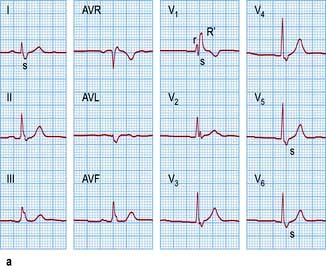
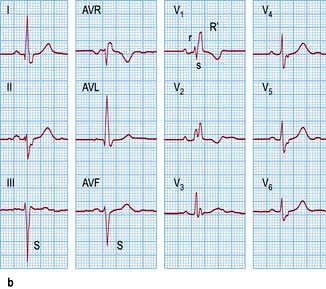
Figure 14.41 Right bundle branch block versus bifascicular block. (a) A 12-lead ECG showing right bundle branch block. Note an rsR′ pattern with the tall R′ in lead V1–V2 and the broad S waves in leads I and V5–V6. (b) Compare with an ECG showing bifascicular block. In addition to right bundle branch block, note left axis deviation and deep S waves in leads III and AVF typical for left anterior hemiblock.
Left bundle branch block (Fig. 14.42) produces the opposite – a deep S wave in lead V1 and a tall late R wave in leads I and V6. Because left bundle branch conduction is normally responsible for the initial ventricular activation, left bundle branch block also produces abnormal Q waves.
Hemiblock
Delay or block in the divisions of the left bundle branch produces a swing in the direction of depolarization (electrical axis) of the heart. When the anterior division is blocked (left anterior hemiblock), the left ventricle is activated from inferior to superior. This produces a superior and leftwards movement of the axis (left axis deviation). Delay or block in the postero-inferior division swings the QRS axis inferiorly to the right (right axis deviation).
Bifascicular block (Fig. 14.41b)
This is a combination of a block of any two of the following: the right bundle branch, the left antero-superior division and the left postero-inferior division. Block of the remaining fascicle will result in complete AV block.
Clinical features of heart blocks
Bundle branch blocks are usually asymptomatic. Right bundle branch block causes wide but physiological splitting of the second heart sound. Left bundle branch block may cause reverse splitting of the second sound. Patients with intraventricular conduction disturbances may complain of syncope. This is due to intermittent complete heart block or to ventricular tachyarrhythmias. ECG monitoring and electrophysiological studies are needed to determine the cause of syncope in these patients.
Causes
Right bundle branch block occurs as an isolated congenital anomaly or is associated with cardiac or pulmonary conditions (Table 14.8). Right bundle branch block alone does not alter the electrical axis of the heart. Axis deviations signify right ventricular hypertrophy (RV overload) or co-existent fascicular block. The combination of right bundle branch block with left axis deviation is associated with ostium primum atrial septal defects. Complete left bundle branch block is often associated with extensive left ventricular disease. The most common causes are listed in Table 14.8 and are similar to those of complete heart block.
Table 14.8 Causes of bundle branch block
| Right | Left |
|---|---|
It is a normal finding in 1% of young adults and 5% of elderly adults |
Left ventricular outflow obstruction |
Supraventricular tachycardias
Supraventricular tachycardias (SVTs) arise from the atrium or the atrioventricular junction. Conduction is via the His–Purkinje system; therefore the QRS shape during tachycardia is usually similar to that seen in the same patient during baseline rhythm. A classification of supraventricular tachycardia is listed in Table 14.9. Some of these are discussed in more detail below.
Table 14.9 Causes of supraventricular tachycardia (SVT)
| Tachycardia | ECG features | Comment |
|---|---|---|
Sinus tachycardia |
P wave morphology similar to sinus rhythm preceding QRS |
Need to determine underlying cause |
AV nodal re-entry tachycardia (AVNRT) |
No visible P wave, or inverted P wave immediately before or after QRS complex |
Commonest cause of palpitations in patients with normal hearts |
AV reciprocating tachycardia (AVRT) complexes |
P wave visible between QRS and T wave |
Due to an accessory pathway. If pathway conducts in both directions, ECG during sinus rhythm may be pre-excited |
Atrial fibrillation |
Irregularly irregular RR intervals and absence of organized atrial activity |
Commonest tachycardia in patients over 65 years |
Atrial flutter |
Visible flutter waves at 300/min (sawtooth appearance) usually with a 2 : 1 AV conduction |
Suspect in any patient with regular SVT at 150/min |
Atrial tachycardia |
Organized atrial activity with P wave morphology different from sinus rhythm preceding QRS |
Usually occurs in patients with structural heart disease |
Multifocal atrial tachycardia |
Multiple P wave morphologies (≥3) and irregular RR intervals |
Rare arrhythmia; most commonly associated with significant chronic lung disease |
Accelerated junctional tachycardia |
ECG similar to AVNRT |
Rare in adults |
Inappropriate sinus tachycardia
Inappropriate sinus tachycardia is a persistent increase in resting heart rate unrelated to or out of proportion with the level of physical or emotional stress. It is found predominantly in young women and is not uncommon in health professionals. Sinus tachycardia due to intrinsic sinus node abnormalities such as enhanced automaticity, or abnormal autonomic regulation of the heart with excess sympathetic and reduced parasympathetic input, is extremely rare.
In general, sinus tachycardia is a secondary phenomenon and the underlying causes need to be actively investigated. Depending on the clinical setting, acute causes include exercise, emotion, pain, fever, infection, acute heart failure, acute pulmonary embolism and hypovolaemia. Chronic causes include pregnancy, anaemia, hyperthyroidism and catecholamine excess. The underlying cause should be found and treated, rather than treating the compensatory physiological response. If necessary, beta-blockers may be used to slow the sinus rate, e.g. in hyperthyroidism (p. 937); ivabradine, an IF (pacemaker current) blocker, may be useful when beta-blockade cannot be tolerated.
Atrioventricular junctional tachycardias
AV nodal re-entry and AV re-entry tachycardias are usually referred to as paroxysmal SVTs and are often seen in young patients with no or little structural heart disease, although congenital heart abnormalities (e.g. Ebstein’s anomaly, atrial septal defect, Fallot’s tetralogy) can co-exist in a small proportion of patients with these arrhythmias. The first presentation is commonly between ages 12 and 30, and the prevalence is approximately 2.5/1000.
In these tachycardias the AV node is an essential component of the re-entry circuit.
Atrioventricular nodal re-entry tachycardia (AVNRT)
This tachycardia is twice as common in women. Clinically, the tachycardia often strikes suddenly without obvious provocation, but exertion, coffee, tea and alcohol may aggravate or induce the arrhythmia. An attack may stop spontaneously or may continue indefinitely until medical intervention.
In AVNRT, there are two functionally and anatomically different pathways predominantly within the AV node: one is characterized by a short effective refractory period and slow conduction, and the other has a longer effective refractory period and conducts faster. In sinus rhythm, the atrial impulse that depolarizes the ventricles usually conducts through the fast pathway. If the atrial impulse (e.g. an atrial premature beat) occurs early when the fast pathway is still refractory, the slow pathway takes over in propagating the atrial impulse to the ventricles. It then travels back through the fast pathway which has already recovered its excitability, thus initiating the most common ‘slow-fast’, or typical, AVNRT.
The rhythm is recognized on ECG by normal regular QRS complexes, usually at a rate of 140–240/minute (Fig. 14.43a). Sometimes the QRS complexes will show typical bundle branch block. P waves are either not visible or are seen immediately before or after the QRS complex because of simultaneous atrial and ventricular activation. Less commonly observed (5–10%) is tachycardia when the atrial impulse conducts anterogradely through the fast pathway and returns through the slow pathway, producing a long RP’ interval (‘fast-slow’, or long RP’ tachycardia).
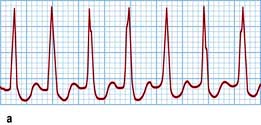
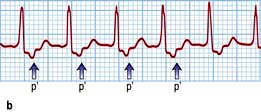
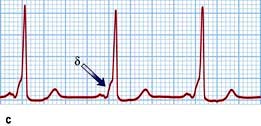
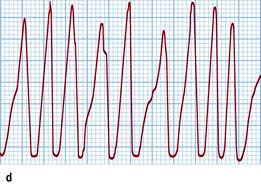
Figure 14.43 Atrioventricular junctional tachycardia. (a) Atrioventricular nodal re-entry tachycardia. The QRS complexes are narrow and the P waves cannot be seen. (b) Atrioventricular re-entry tachycardia (Wolff–Parkinson–White syndrome, WPW). The tachycardia P waves (arrows) are clearly seen after narrow QRS complexes. (c) An electrocardiogram taken in a patient with WPW syndrome during sinus rhythm. Note the short PR interval and the δ wave (arrow). (d) Atrial fibrillation in the WPW syndrome. Note tachycardia with broad QRS complexes with fast and irregular ventricular rate.
Atrioventricular reciprocating tachycardia (AVRT)
This large circuit comprises the AV node, the His bundle, the ventricle and an abnormal connection of myocardial fibres from the ventricle back to the atrium. It is called an accessory pathway or bypass tract and results from an incomplete separation of the atria and the ventricles during fetal development.
In contrast to AVNRT, this tachycardia is due to a macro-reentry circuit and each part of the circuit is activated sequentially. As a result atrial activation occurs after ventricular activation and the P wave is usually clearly seen between the QRS and T wave (Fig. 14.43b).
Accessory pathways are most commonly situated on the left but may occur anywhere around the AV groove. The most common accessory pathways, known as Kent bundles, are in the free wall or septum. In about 10% of cases multiple pathways occur. Mahaim fibres are atrio-fascicular or nodo-fascicular fibres entering the ventricular myocardium in the region of the right bundle branch. Accessory pathways that conduct from the ventricles to the atria only are not visible on the surface ECG during sinus rhythm and are therefore ‘concealed’. Accessory pathways that conduct bidirectionally usually are manifest on the surface ECG. If the accessory pathway conducts from the atrium to the ventricle during sinus rhythm, the electrical impulse can conduct quickly over this abnormal connection to depolarize part of the ventricles abnormally (pre-excitation). A pre-excited ECG is characterized by a short PR interval and a wide QRS complex that begins as a slurred part known as the δ wave (Fig. 14.43c). Patients with a history of palpitations and a pre-excited ECG have a syndrome known as Wolff–Parkinson–White (WPW) syndrome.
During AVRT the AV node and ventricles are activated normally (orthodromic AVRT), resulting usually in a narrow QRS complex. Less commonly, the tachycardia circuit can be reversed, with activation of the ventricles via the accessory pathway and atrial activation via retrograde conduction through the AV node (antidromic AVRT). This results in a broad complex tachycardia. These patients are also prone to atrial fibrillation.
During atrial fibrillation, the ventricles may be depolarized by impulses travelling over both the abnormal and the normal pathways. This results in pre-excited atrial fibrillation, a characteristic tachycardia that is characterized by irregularly irregular broad QRS complexes (Fig. 14.43d). If an accessory pathway has a short antegrade effective refractory period (<250 ms), it may conduct to the ventricles at an extremely high rate and may cause ventricular fibrillation. The incidence of sudden death is 0.15–0.39% per patient-year and it may be a first manifestation of the disease in younger individuals. Verapamil and digoxin may allow a higher rate of conduction over the abnormal pathway and precipitate ventricular fibrillation. Therefore, neither verapamil nor digoxin should be used to treat atrial fibrillation associated with the WPW syndrome.
Symptoms
The leading symptom of most SVTs, in particular AV node re-entry and AV re-entry tachycardias, is rapid regular palpitations, usually with abrupt onset and sudden termination, which can occur spontaneously or be precipitated by simple movements. A common feature is termination by Valsalva manoeuvres. In younger individuals with no structural heart disease, the rapid heart rate can be the main pathological finding.
Irregular palpitations may be due to atrial premature beats, atrial flutter with varying AV conduction block, atrial fibrillation or multifocal atrial tachycardia. In patients with depressed ventricular function, uncontrolled atrial fibrillation can reduce cardiac output and cause hypotension and congestive heart failure.
Other symptoms may include anxiety, dizziness, dyspnoea, neck pulsation, central chest pain and weakness. Polyuria may occur because of release of atrial natriuretic peptide in response to increased atrial pressures during the tachycardia, especially during AVNRT and atrial fibrillation. Prominent jugular venous pulsations due to atrial contractions against closed atrioventricular valves may be observed during AVNRT.
Syncope has been reported in 10–15% of patients, usually just after initiation of the arrhythmia or in association with a prolonged pause following its termination. It is more common if the patient is standing. However, in older patients with concomitant heart disease, such as aortic stenosis, hypertrophic cardiomyopathy and cerebrovascular disease, significant hypotension and syncope may result from moderately fast ventricular rates.
Acute management
In an emergency, distinguishing between AVNRT and AVRT may be difficult, but it is usually not critical as both tachycardias respond to the same treatment. Patients presenting with SVTs and haemodynamic instability (e.g. hypotension, pulmonary oedema) require emergency cardioversion. If the patient is haemodynamically stable, vagal manoeuvres, including right carotid massage (Practical Box 14.3), Valsalva manoeuvre (Practical Box 14.4) and facial immersion in cold water can be successfully employed.
 Practical Box 14.4
Practical Box 14.4
Valsalva manoeuvre
Valsalva manoeuvre is an abrupt voluntary increase in intrathoracic and intra-abdominal pressures by straining.
Provide continuous electrocardiographic monitoring.
Patient is in supine position.
Patient should not take deep inspiration before straining.
Ideally, the patient blows into the mouthpiece of a manometer against a pressure of 30–40 mmHg for 15 s.
Alternatively, the patient strains for 15 s while breath-holding.
Transient acceleration of tachycardia usually occurs during the strain phase as a result of sympathetic excess.
On release of strain, the rate of tachycardia slows because of the compensatory increase in vagal tone (baroreceptor reflex), and it may be terminated in about 50% of patients.
Termination of tachycardia may be followed by pauses and transient ventricular ectopics.
Of these techniques, the Valsalva manoeuvre is the best and is often easier for the patient to perform successfully. It should be undertaken when the patient is resting in the supine position (thus avoiding elevated background sympathetic tone). Several seconds after the release of strain, the resulting intense vagal effect may terminate AVNRT or AVRT or may produce sufficient AV block to reveal an underlying atrial tachyarrhythmia.
If physical manoeuvres have not been successful, intravenous adenosine (initially 6 mg by i.v. push, followed by 12 mg if needed) should be tried. This is a very short-acting (half-life <10 s) naturally occurring purine nucleoside that causes complete heart block for a fraction of a second following i.v. administration. It is highly effective at terminating AVNRT and AVRT or unmasking underlying atrial activity. It rarely affects ventricular tachycardia. The side-effects of adenosine are very brief but include:
It is contraindicated in patients with a history of asthma. In some patients, adenosine can induce atrial fibrillation.
An alternative treatment is verapamil 5–10 mg i.v. over 5–10 min, i.v. diltiazem, or beta-blockers (esmolol, propranolol, metoprolol). Verapamil (or diltiazem) must not be given after beta-blockers or if the tachycardia presents with broad (>0.12 s) QRS complexes.
Long-term management
Patients with suspected cardiac arrhythmias should always be referred to the cardiologist for electrophysiological evaluation and long-term management, as both pharmacological and non-pharmacological alternatives, including ablation of an accessory pathway, are readily available. Verapamil, diltiazem and beta-blockers have proven efficacy in 60–80% of patients. Sodium-channel blockers (flecainide and propafenone), potassium repolarization current blockers (sotalol, dofetilide, azimilide), and the multichannel blocker amiodarone may also prevent the occurrence of tachycardia.
Refinement of catheter ablation techniques has rendered many AV junctional tachycardias entirely curable. Modification of the slow pathway is successful in 96% of patients with AVNRT, although a 1% risk of AV block is present. In AVRT, the target for catheter ablation is the accessory pathway(s). The success rate for ablation of a single accessory pathway is approximately 95%, with a recurrence rate of 5%, requiring a repeat procedure.
Atrial tachyarrhythmias
Atrial tachyarrhythmias including atrial fibrillation, atrial flutter, atrial tachycardia and atrial ectopic beats all arise from the atrial myocardium (Fig. 14.44). They share common aetiologies, which are listed in Table 14.10.
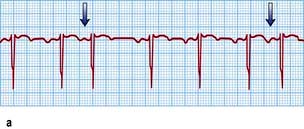
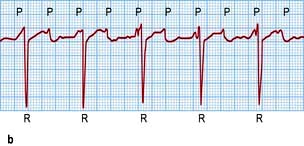
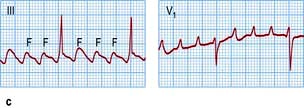
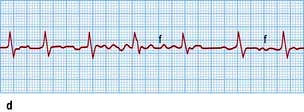
Figure 14.44 ECGs of a variety of atrial arrhythmias. (a) Atrial premature beats (arrows). The premature P wave is different from the sinus P wave and conducts to the ventricle with a slightly prolonged PR interval. (b) Atrial tachycardia with second-degree atrioventricular block. Note the fast atrial (P wave) rate of 150/min and the slower ventricular (R wave) rate of 75/min. (c) Atrial flutter. Some flutter waves are marked with an F. In this case the flutter frequency is 240/min. Every fourth flutter wave is transmitted to the ventricles and the ventricular rate is therefore 60/min. (d) Atrial fibrillation. Note absolute rhythm irregularity and baseline undulations (f waves).
Table 14.10 Causes of atrial tachyarrhythmias
Atrial fibrillation
This is a common arrhythmia, occurring in 1–2% of the general population and 5–10% of patients over 75 years of age. It also occurs, particularly in a paroxysmal form, in younger patients. Any condition resulting in raised atrial pressure, increased atrial muscle mass, atrial fibrosis, or inflammation and infiltration of the atrium, may cause atrial fibrillation. There are also many genetic and systemic causes of atrial fibrillation (Table 14.10).
Although rheumatic heart disease, alcohol intoxication and thyrotoxicosis are the ‘classic’ causes of atrial fibrillation, hypertension and heart failure are the most common causes. Hyperthyroidism may provoke atrial fibrillation, sometimes as virtually the only feature of the disease, and thyroid function tests are mandatory in any patient with unaccounted atrial fibrillation. Atrial fibrillation occurs in one-third of patients after cardiac surgery. It usually manifests during the first 4 days and is associated with increased morbidity and mortality, largely due to stroke and circulatory failure, a longer hospital stay, and later recurrences.
In some patients no cause can be found, and this group is labelled as ‘lone’ atrial fibrillation. The pathogenesis of ‘lone’, or ‘idiopathic’, atrial fibrillation is unknown but genetic predisposition or even specific genetically predetermined forms of the arrhythmia have been proposed. About 30–40% of those with AF, especially those who present at a young age, have at least one parent with AF and genes associated with the sodium channel, the potassium channel, gap junction proteins and right left isomerism) have been implicated. Gene defects linked to chromosomes 10, 6, 5 and 4 have been associated with familial AF.
Atrial fibrillation is maintained by continuous, rapid (300–600/min) activation of the atria by multiple meandering re-entry wavelets, often driven by rapidly depolarizing automatic foci, located predominantly within the pulmonary veins. The atria respond electrically at this rate but there is no coordinated mechanical action and only a proportion of the impulses are conducted to the ventricles. The ventricular response depends on the rate and regularity of atrial activity, particularly at the entry to the AV node, the refractory properties of the AV node itself, and the balance between sympathetic and parasympathetic tone.
Symptoms and signs
Symptoms attributable to atrial fibrillation are highly variable. In some patients (about 30%) it is an incidental finding, whilst others attend hospital as an emergency with rapid palpitations dyspnoea and/or chest pain following the onset of atrial fibrillation. Most patients with ongoing atrial fibrillation experience some deterioration of exercise capacity or wellbeing, but this may only be appreciated once sinus rhythm is restored. When caused by rheumatic mitral stenosis, the onset of atrial fibrillation results in considerable worsening of cardiac failure.
The patient has a very irregular pulse, as opposed to a basically regular pulse with an occasional irregularity (e.g. extrasystoles) or recurring irregular patterns (e.g. Wenckebach block). The irregular nature of the pulse in atrial fibrillation is maintained during exercise.
The ECG shows fine oscillations of the baseline (so-called fibrillation or f waves) and no clear P waves. The QRS rhythm is rapid and irregular. Untreated, the ventricular rate is usually 120–180/minute, but it slows with treatment.
The clinical classification of atrial fibrillation includes first detected, paroxysmal (stops spontaneously within 7 days), persistent (requires cardioversion to stop), and permanent (no spontaneous or induced cardioversion) forms of the arrhythmia and is helpful for the decision-making between rhythm restoration and rate control. Atrial fibrillation may be asymptomatic and the ‘first detected episode’ should not be regarded as necessarily the true onset.
Management
When atrial fibrillation is due to an acute precipitating event such as alcohol toxicity, chest infection or hyperthyroidism, the provoking cause should be treated. Strategies for the acute management of AF are:
Ventricular rate control: this is achieved by drugs which block the AV node (see below)
Cardioversion (± anticoagulation): this is achieved electrically by DC shock (p. 694) or medically either by intravenous infusion of an antiarrhythmic drug such as flecainide, propafenone, vernakalant or amiodarone or by taking an oral agent (flecainide or propafenone) previously tested in hospital and found to be safe in a particular patient (‘pill-in-pocket’ approach).
how well the arrhythmia is tolerated (is cardioversion urgent?)
whether anticoagulation is required before considering elective cardioversion
whether spontaneous cardioversion is likely (previous history? reversible cause?).
Conversion to sinus rhythm can be achieved by electrical DC cardioversion (p. 694) in about 80% of patients. Biphasic waveform defibrillation is more effective than conventional (monophasic) defibrillation, and biphasic defibrillators are now standard. To minimize the risk of thromboembolism associated with cardioversion patients are fully anticoagulated with warfarin (INR 2.0–3.0) or with dabigatran 150 mg twice daily for 3 weeks before cardioversion (unless atrial fibrillation is of <48 hours duration) and at least 4 weeks after the procedure. The patient is then assessed for the necessity for long-term anticoagulation. If cardioversion is urgent, transoesophageal echocardiography may be used to document the presence or absence of atrial thrombus as a guide to possibly avoiding pre-cardioversion anticoagulation.
Long-term management of atrial fibrillation
‘Rate control’ (AV nodal slowing agents plus oral anticoagulation)
‘Rhythm control’ (antiarrhythmic drugs plus DC cardioversion plus oral anticoagulation).
Major randomized studies in patients predominantly over the age of 65 years (AFFIRM) or in patients with heart failure (AF-CHF) have shown that there is no net mortality or symptom benefit to be gained from one strategy compared with the other. Which strategy to adopt needs to be assessed for each individual patient. Factors to consider include the likelihood of maintaining sinus rhythm and the safety/tolerability of antiarrhythmic drugs in a particular patient.
Rhythm control
This is advocated for younger, symptomatic and physically active patients. Recurrent paroxysms may be prevented by oral medication. In general, patients with no significant heart disease can be treated with any class Ia, 1c or III antiarrhythmic drug, although it is recommended that amiodarone (because of its substantial extracardiac adverse effect profile) should be reserved until other drugs have failed. For patients with heart failure or left ventricular hypertrophy only amiodarone is recommended. Patients with coronary artery disease may be treated with sotalol or amiodarone. Patients with paroxysmal atrial fibrillation or with early persistent atrial fibrillation (little left atrial dilatation) may be treated with left atrial ablation. The ectopic triggers for atrial fibrillation are generally found in the pulmonary veins which can be isolated from the atria using radiofrequency or cryothermal energy. Occasionally, more extensive ablation within the left atrium is needed. These techniques are more successful than antiarrhythmic drugs and may represent a ‘cure’ in some patients. However, the procedure is invasive and carries some hazard of serious complications such as stroke, and bleeding in about 2% of cases. In the long term, recurrence is not uncommon and an apparently successful ablation does not remove the obligation for appropriate anticoagulation. Ablation has not been shown to improve long-term cardiovascular outcome but it does successfully treat symptoms due to atrial fibrillation.
Rate control
As a primary strategy this is appropriate in patients who:
have the permanent form of the arrhythmia associated with symptoms which can be further improved by slowing heart rate or are older than 65 years with recurrent atrial tachyarrhythmias (‘accepted’ atrial fibrillation)
have persistent tachyarrhythmias and have failed cardioversion(s) and serial prophylactic antiarrhythmic drug therapy and in whom the risk/benefit ratio from using specific antiarrhythmic agents is shifted towards increased risk.
Rate control is usually achieved by a combination of digoxin, beta-blockers or nondihydropyridine calcium-channel blockers (verapamil or diltiazem). Digoxin monotherapy may be sufficient for elderly non-ambulant patients. In younger patients the effect of catecholamines easily overwhelms the vagotonic effect of digoxin and additional AV nodal slowing agents are needed. The ventricular rate response is generally considered controlled if the resting heart rate is <110 b.p.m. but a more strict control between 60 and 80 b.p.m. at rest and <110 b.p.m. during moderate exercise may be needed if symptoms persist.. To assess the adequacy of rate control, an ECG rhythm strip may be sufficient in an elderly patient but ambulatory 24-hour Holter monitoring and an exercise stress test (treadmill) are needed in younger individuals. Older patients with poor rate control despite optimal medical therapy should be considered for AV node ablation and pacemaker implantation (‘ablate and pace’ strategy). These patients usually experience a marked symptomatic improvement but because of the ongoing risk of thromboembolism require life-long anticoagulation.
Anticoagulation
This is indicated in patients with atrial fibrillation related to rheumatic mitral stenosis or in the presence of a mechanical prosthetic heart valve. A scoring system known as CHADS2 is used as the first step to determine the need for anticoagulation. CHADS2 is an acronym standing for Congestive heart failure, Hypertension, Age ≥75, Diabetes mellitus and previous Stroke or TIA. Each factor scores 1 except previous stroke or TIA which scores 2. A total score of 2 implies that oral anticoagulation is needed. When the score is <2 the CHADS2VASc scoring system should be applied. CHADS2VASc adds Vascular disease (aorta, coronary or peripheral arteries), Age 65–74, and female Sex category. Each factor scores 1 except previous Age >75 and stroke or TIA which score 2. A CHADS2VASc score of 2 requires oral anticoagulation and a score of 1 merits consideration for oral anticoagulation or aspirin. A score of 0 should not require any antithrombotic prophylaxis.
When oral anticoagulation is required either warfarin (dose adjusted to maintain an INR between 2.0 and 3.0) or dabigatran 150 mg two times daily (or 110 mg two times daily in elderly patients (>75 years), those with moderate renal impairment (creatinine clearance: 30 to 50 mL/min) or those taking P-glycoprotein inhibitors such as amiodarone and verapamil) can be considered. Other new oral anticoagulants, such as apixaban and rivaroxaban, are now also being used.
FURTHER READING
Camm AJ, Kirchhof P, Lip GY et al. European Heart Rhythm Association; European Association for Cardio-Thoracic Surgery. Guidelines for the management of atrial fibrillation: the Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Europace 2010; 12(10):1360–1420.
Dobrev D, Nattel S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet 2010; 375:1212–1223.
Mega JL. A new era for anticoagulation in atrial fibrillation (Editorial). N Engl J Med 2011; 365:1052–1054.
Patel C, Yan GX, Kowey PR. Dronedarone. Circulation 2009; 120(7):636–644.
Patel MR, Mahaffey KW, Garg J et al.; ROCKET AF Steering Committee for the ROCKET AF Investigators. Rivaroxaban versus Warfarin in Nonvalvular Atrial Fibrillation. N Engl J Med 2011; 365:883–891.
Atrial flutter
Atrial flutter is often associated with atrial fibrillation and often requires a similar initial therapeutic approach. Atrial flutter is usually an organized atrial rhythm with an atrial rate typically between 250 and 350 b.p.m. Typical, or isthmus-dependent, atrial flutter involves a macro re-entrant right atrial circuit around the tricuspid annulus. The wavefront circulates down the lateral wall of the right atrium, through the Eustachian ridge between the tricuspid annulus and the inferior vena cava, and up the interatrial septum, giving rise to the most frequent pattern, referred to as counter-clockwise flutter. Re-entry can also occur in the opposite direction (clockwise or reverse flutter).
The ECG shows regular sawtooth-like atrial flutter waves (F waves) between QRS complexes (Fig. 14.44c). In typical counter-clockwise atrial flutter, the F waves are negative in the inferior leads and positive in leads V1 and V2. In clockwise atrial flutter, the deflection of the F waves is the opposite. If F waves are not clearly visible, it is worth trying to reveal them by slowing AV conduction by carotid sinus massage or by the administration of AV nodal blocking drugs such as adenosine or verapamil.
Symptoms are largely related to the degree of AV block. Most often, every second flutter beat conducts, giving a ventricular rate of 150 b.p.m. Occasionally, every beat conducts, producing a heart rate of 300 b.p.m. More often, especially when patients are receiving treatment, AV conduction block reduces the heart rate to approximately 75 b.p.m.
Management
Treatment of a symptomatic acute paroxysm is electrical cardioversion. Patients who have been in atrial flutter more than 1–2 days should be treated in a similar manner to patients with atrial fibrillation and anticoagulated for 3 weeks prior to cardioversion.
Recurrent paroxysms may be prevented by class III antiarrhythmic agents. AV nodal blocking agents may be used to control the ventricular rate if the arrhythmia persists. However, the treatment of choice for patients with recurrent atrial flutter is catheter ablation (p. 712), which permanently interrupts re-entry by creating a line of conduction block within the isthmus between the inferior vena cava and the tricuspid valve ring. This technique offers patients whose only arrhythmia is typical atrial flutter an almost certain chance of a cure, although the later occurrence of atrial fibrillation is not uncommon.
Atrial tachycardia
This is an uncommon arrhythmia. Its prevalence is believed to be <1% in patients with arrhythmias. It is usually associated with structural heart disease but in many cases it is referred to as idiopathic. Macro re-entrant tachycardia often occurs after surgery for congenital heart disease. Atrial tachycardia with block is often a result of digitalis poisoning.
The mechanisms of atrial tachycardia are attributed to enhanced automaticity, triggered activity or intra-atrial reentry. Atrial re-entry tachycardia is usually relatively slow (125–150 b.p.m.) and can be initiated and terminated by atrial premature beats. The P′P′ intervals are regular. The PR interval depends on the rate of tachycardia and is longer than in sinus rhythm at the same rate.
Automatic tachycardia usually presents with higher rates (125–250 b.p.m.) and is often characterized by a progressive increase in the atrial rate with onset of the tachycardia (‘warm-up’) and progressive decrease prior to termination (‘cool-down’). Atrial tachycardia is typically caused by a focus which is frequently located along the crista terminalis in the right atrium, adjacent to a pulmonary vein in the left atrium, or around one of the atrial appendages. Automatic atrial tachycardia may also present as an incessant variety leading to tachycardia-induced cardiomyopathy. Short runs of atrial tachycardia may provoke more sustained episodes of atrial fibrillation.
Figure 14.44b demonstrates an atrial tachycardia at an atrial rate of 150/minute. The P waves are abnormally shaped and occur in front of the QRS complexes. Carotid sinus massage may increase AV block during tachycardia, thereby facilitating the diagnosis, but does not usually terminate the arrhythmia. Treatment options include cardioversion, antiarrhythmic drug therapy to maintain sinus rhythm, AV nodal slowing agents to control rate and, in selected cases, radiofrequency catheter ablation.
Atrial ectopic beats
These often cause no symptoms, although they may be sensed as an irregularity or heaviness of the heart beat. On the ECG they appear as early and abnormal P waves, and are usually, but not always, followed by normal QRS complexes (Fig. 14.44a). Treatment is not normally required unless the ectopic beats provoke more significant arrhythmias, when beta-blockers may be effective.
Ventricular tachyarrhythmias
Ventricular tachyarrhythmias can be discussed under the following headings:
Life-threatening ventricular tachyarrhythmias
Normal heart ventricular tachycardia
Life-threatening ventricular tachyarrhythmias
Sustained ventricular tachycardia and ventricular fibrillation with haemodynamic instability (e.g. syncope, hypotension) are life-threatening ventricular tachyarrhythmias.
Sustained ventricular tachycardia
Sustained ventricular tachycardia (>30 s) often results in pre-syncope (dizziness), syncope, hypotension and cardiac arrest, although it may be remarkably well tolerated in some patients. Examination reveals a pulse rate typically between 120 and 220 b.p.m. Usually, there are clinical signs of atrioventricular dissociation (i.e. intermittent cannon ‘a’ waves in the neck) and variable intensity of the first heart sound).
The ECG shows a rapid ventricular rhythm with broad (often ≥0.14 s), abnormal QRS complexes. AV dissociation may result in visible P waves which appear to march through the tachycardia, capture beats (intermittent narrow QRS complex owing to normal ventricular activation via the AV node and conducting system) and fusion beats (intermediate between ventricular tachycardia beat and capture beat).
Supraventricular tachycardia with bundle branch block may resemble ventricular tachycardia on the ECG. However, if a broad complex tachycardia is due to SVT with either right or left bundle branch block, then the QRS morphology should resemble a typical RBBB or LBBB pattern (Figs 14.41a, 14.42). Other ECG criteria to differentiate VT from SVT with aberrancy are indicated in Table 14.11. Some 80% of all broad complex tachycardias are due to ventricular tachycardia, and the proportion is even higher in patients with structural heart disease. Therefore, in all cases of doubt, ventricular tachycardia should be diagnosed.
Table 14.11 ECG distinction between supraventricular tachycardia (SVT) with bundle branch block and ventricular tachycardia (VT)
Treatment may be urgent, depending on the haemodynamic situation. If the patient is haemodynamically compromised (e.g. hypotensive or pulmonary oedema), emergency DC cardioversion may be required. On the other hand, if the blood pressure and cardiac output are well maintained, intravenous therapy with class I drugs or amiodarone is usually used. First-line drug treatment consists of lidocaine (50–100 mg i.v. over 5 minutes) followed by a lidocaine infusion (2–4 mg i.v. per minute). Amiodarone is given as a dilute intravenous infusion (5 mg/kg over 1 hour (loading dose)) followed by a maintenance infusion of 1200 mg over 24 hours. DC cardioversion is necessary if medical therapy is unsuccessful.
Ventricular fibrillation
This is very rapid and irregular ventricular activation with no mechanical effect. The patient is pulseless and becomes rapidly unconscious, and respiration ceases (cardiac arrest). The ECG shows shapeless, rapid oscillations and there is no hint of organized complexes (Fig. 14.45). It is usually provoked by a ventricular ectopic beat. Ventricular fibrillation rarely reverses spontaneously. The only effective treatment is electrical defibrillation. Basic and advanced cardiac life support is needed (p. 691).

Figure 14.45 Ventricular fibrillation. Four beats of sinus rhythm followed by a ventricular ectopic beat that initiates ventricular fibrillation. The ST segment during sinus rhythm is elevated owing to acute myocardial infarction in this case.
If the attack of ventricular fibrillation occurs during the first day or two of an acute myocardial infarction, it is probable that prophylactic therapy will be unnecessary. If the ventricular fibrillation was not related to an acute infarction, the long-term risk of recurrent cardiac arrest and sudden death is high.
Survivors of these ventricular tachyarrhythmias are, in the absence of an identifiable reversible cause (e.g. acute myocardial infarction, severe metabolic disturbance), at high risk of sudden death. Implantable cardioverter-defibrillators (ICDs) are first-line therapy in the management of these patients (p. 713).
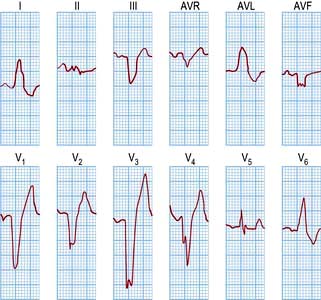
The Brugada syndrome
This inheritable condition accounts for part of a group of patients with idiopathic ventricular fibrillation who have no evidence of causative structural cardiac disease. It is more common in young male adults and in South-east Asia. The diagnosis is made by identifying the classic ECG changes that may be present spontaneously or be provoked by the administration of a class I antiarrhythmic (flecainide or ajmaline): right bundle branch block with coved ST elevation in leads V1–V3 (Fig. 14.46). Atrial fibrillation may occur.
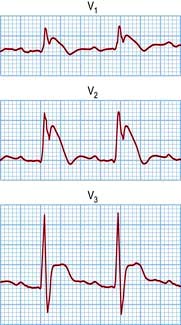
In 20% of cases it is a monogenic inheritable condition associated with loss of sodium channel function due to a mutation in the SCN5A gene. Recently other mutations in the SCN1B gene, glycerol-3-phosphate dehydrogenase-1-like gene (GPD1LL-type) and genes related to calcium channel subunits CACNA1C and CACNB2 have also been implicated in the genesis of this syndrome. It can present with sudden death during sleep, resuscitated cardiac arrest and syncope, or the patient may be asymptomatic and diagnosed incidentally or during familial assessment. There is a high risk of sudden death, particularly in the symptomatic patient or those with spontaneous ECG changes. The only successful treatment is an ICD. Beta-blockade is not helpful and may be harmful in this syndrome.
Long QT syndrome
This describes an ECG where the ventricular repolarization (QT interval) is greatly prolonged. The causes of long QT syndrome are listed in Table 14.12.
Table 14.12 Causes of long QT syndrome
| Congenital | Acquired |
|---|---|
Jervell–Lange–Nielsen (autosomal recessive) |
Electrolyte abnormalities |
Hypokalaemia |
|
Hypomagnesaemia |
|
Hypocalcaemia |
|
|
Drugs |
|
Quinidine, disopyramide |
|
Sotalol, amiodarone |
|
Tricyclic antidepressants, e.g. amitriptyline |
|
Phenothiazine drugs, e.g. chlorpromazine |
|
Antipsychotics, e.g. haloperidol, olanzapine |
|
Macrolides, e.g. erythromycin |
|
Quinolones, e.g. ciprofloxacin |
|
Methadone |
|
Poisons |
|
Organophosphate insecticides |
|
Miscellaneous |
|
Bradycardia |
|
Mitral valve prolapse |
|
Acute myocardial infarction |
|
Diabetes |
|
Prolonged fasting and liquid protein diets (long-term) |
|
Central nervous system diseases, e.g. dystrophia myotonica |
Congenital long QT syndrome
Two major syndromes have been described, which may (Jervell and Lange–Nielsen syndrome) or may not (Romano–Ward syndrome) be associated with congenital deafness.
The molecular biology of the congenital long QT syndromes has been shown to be heterogeneous. It is usually a monogenic disorder and has been associated with mutations in cardiac potassium and sodium channel genes (Table 14.13). The different genes involved appear to correlate with different phenotypes (Fig. 14.47b) that can exhibit such variable penetrance that carriers may have completely normal ECGs. There are three major subtypes: LQT1 in which the arrhythmia is usually provoked by exercise, particularly swimming; LQT2 with provocation associated with emotion and acoustic stimuli; and LQT3 where the arrhythmias occur during rest or when asleep. It is likely that identification of the mutation involved will not only improve diagnostic accuracy, particularly with cascade screening in affected families, but also guide future therapy for the congenital long QT syndrome.
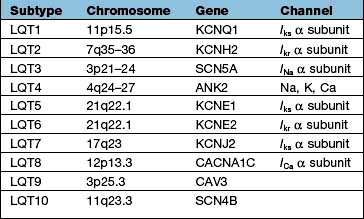
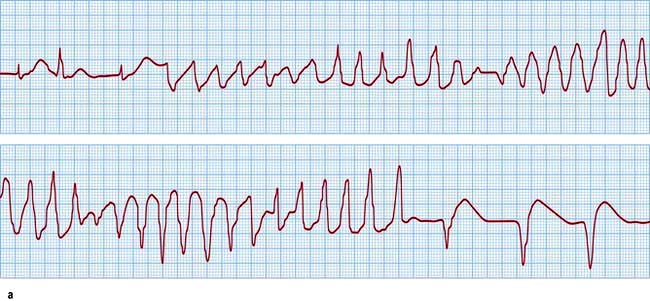
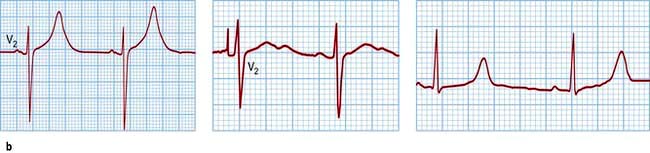
Figure 14.47 Prolonged QT. (a) An ECG demonstrating a supraventricular rhythm with a long QT (LQT) interval giving way to atypical ventricular tachycardia (torsades de pointes). The tachycardia is short-lived and is followed by a brief period of idioventricular rhythm. (b) Further three examples of a prolonged QT interval corresponding to three different types of LQT.
Acquired long QT syndrome
QT prolongation and torsades de pointes are usually provoked by bradycardia.
FURTHER READING
Ackerman MJ, Mohler PJ. Defining a new paradigm for human arrhythmia syndromes: phenotypic manifestations of gene mutations in ion channel- and transporter-associated proteins. Circul Res 2010; 107(4):457–465.
Heist EK, Ruskin JN. Drug-induced arrhythmia. Circulation 2010; 122(14):1426–1435.
Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet 2008; 372:750–763.
Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med 2008; 358:169–176.
Clinical features
Patients with a long QT develop syncope and palpitations as a result of polymorphic ventricular tachycardia (torsades de pointes). They usually terminate spontaneously but may degenerate to ventricular fibrillation, resulting in sudden death.
Torsades de pointes is characterized on the ECG by rapid, irregular, sharp complexes that continuously change from an upright to an inverted position (Fig. 14.47a).
Between spells of tachycardia or immediately preceding the onset of tachycardia the ECG shows a prolonged QT interval; the corrected QT (Table 14.4, p. 682) is usually >0.50 s.
Management
Any electrolyte disturbance is corrected
The heart rate is maintained with atrial or ventricular pacing
Magnesium sulphate 8 mmol (Mg2+) over 10–15 min for acquired long QT
Intravenous isoprenaline may be effective when QT prolongation is acquired (isoprenaline is contraindicated for congenital long QT syndrome).
Long-term, congenital long QT syndrome is generally treated by beta-blockade, pacemaker therapy and occasionally left cardiac sympathetic denervation. LQT1 patients seem to respond well to beta-blockade and LQT3 patients are better treated with sodium channel blockers. All LQT patients should avoid drugs known to prolong the QT interval. Patients who remain symptomatic despite conventional therapy and those with marked QT prolongation or a strong family history of sudden death usually need ICD therapy.
Short QT syndrome
Five types have been described caused by genetic abnormalities leading to faster repolarization. Ventricular arrhythmias and sudden death may occur and an ICD is the best treatment.
Normal heart ventricular tachycardia
Monomorphic ventricular tachycardia in patients with structurally normal hearts (idiopathic VT) is usually a benign condition with an excellent long-term prognosis. Occasionally, it is incessant (so called Galavardin’s tachycardia) and if untreated may lead to cardiomyopathy.
Normal heart VT either arises from a focus in the right ventricular outflow tract or in the left ventricular septum. Treatment of symptoms is usually with beta-blockers. There is a special form of verapamil-sensitive tachycardia that responds well to non-dihydropyridine calcium antagonists. In symptomatic patients, radiofrequency catheter ablation is highly effective, resulting in a cure in >90% cases. It is sometimes difficult to distinguish arrhythmogenic right ventricular hypertrophy (p. 770) from this seemingly benign disorder.
Non-sustained ventricular tachycardia
Non-sustained ventricular tachycardia (NSVT) is defined as ventricular tachycardia that is ≥5 consecutive beats but lasts <30 s (Fig. 14.48d). NSVT can be found in 6% of patients with normal hearts and usually does not require treatment. NSVT is documented in up to 60–80% of patients with heart disease. There is insufficient evidence on prognosis, but an ICD has been shown to improve survival of patients with particularly poor left ventricular function (ejection fraction ≤30%) by preventing arrhythmic death. Antiarrhythmic suppression of NSVT is not usually advocated but beta-blockers may improve quality-of-life in symptomatic individuals.
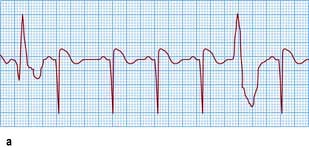
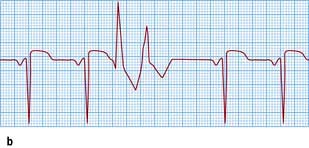
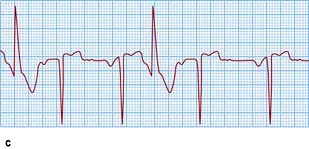
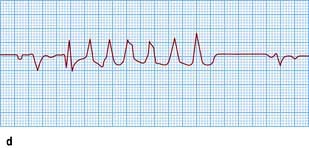
Figure 14.48 Varieties of ventricular ectopic activity. (a) Two ventricular ectopic beats of different morphology (multimorphological). (b) Two ventricular premature beats (VPBs) occurring one after the other (a pair or couplet of VPBs). (c) Frequently repetitive ventricular ectopic activity of a single morphology. (d) Brief run of ventricular tachycardia (non-sustained ventricular tachycardia) that follows previous ectopic activity.
Ventricular premature beats
These may be uncomfortable, especially when frequent. The patient complains of extra beats, missed beats or heavy beats because it may be the premature beat, the post-ectopic pause or the next sinus beat that is noticed by the patient. The pulse is irregular owing to the premature beats. Some early beats may not be felt at the wrist. When a premature beat occurs regularly after every normal beat, ‘pulsus bigeminus’ may occur. If premature ventricular beats are highly symptomatic, treatment with beta-blockade may be helpful. If the ectopics are very frequent left ventricular dysfunction may develop, and if the ectopics stem from a single focus, especially when in the right ventricle, catheter ablation can be very effective.
These premature beats (Fig. 14.48a–c) have a broad (>0.12 s) and bizarre QRS complex because they arise from an abnormal (ectopic) site in the ventricular myocardium. Following a premature beat there is usually a complete compensatory pause because the AV node or ventricle is refractory to the next sinus impulse. Early ‘R-on-T’ ventricular premature beats (occurring simultaneously with the upstroke or peak of the T wave of the previous beat) may induce ventricular fibrillation in patients with heart disease, particularly in patients following myocardial infarction.
Ventricular premature beats are usually treated only if symptomatic. Usually simple measures such as reassurance and beta-blocker therapy are all that is required.
Long-term management of cardiac tachyarrhythmias
Options for the long-term management of cardiac tachyarrhythmias include:
To determine the optimal strategy for a given patient the following questions must be addressed:
Is the principal aim of treatment symptom relief or prevention of sudden death?
Is maintaining sinus rhythm or controlling ventricular rates the treatment goal?
Commonly employed treatment strategies for the management of specific tachyarrhythmias are outlined in Table 14.14.
Table 14.14 Long-term management of tachyarrhythmias
| Tachycardia | Management aims | Management strategies |
|---|---|---|
AV node re-entry tachycardia (AVNRT) |
Relieve symptoms |
AV-node-blocking agents |
Class Ic or class III |
||
Catheter ablation |
||
AV reciprocating tachycardia (AVRT) |
Relieve symptoms |
AV-node-blocking agents |
Class Ic or class III |
||
Catheter ablation |
||
Wolff–Parkinson–White (WPW) syndrome |
Relieve symptoms |
Class Ic or class III |
Catheter ablation |
||
Atrial fibrillation |
Relieve symptoms |
Maintenance of sinus rhythm: |
class Ic or class III ± cardioversion |
||
catheter ablation of ectopic focus |
||
Rate control: |
||
AV-node-slowing agents |
||
AV node ablation plus pacemaker |
||
Anticoagulation |
||
Atrial flutter |
Relieve symptoms |
Class Ic or class III |
Catheter ablation |
||
AV-node-blocking agents |
||
Anticoagulation |
||
Atrial tachycardia |
Relieve symptoms |
Class Ic or class III |
Catheter ablation |
||
AV-node-blocking agents |
||
Anticoagulation |
||
Life-threatening ventricular tachyarrhythmias |
Prevent sudden death |
Implantable cardioverter-defibrillator (ICD) |
Beta-blockers |
||
Congenital long QT |
Prevent sudden death |
Beta-blockers ± pacemaker ICD |
Correct bradycardia |
||
Correct electrolytes |
||
Acquired long QT |
Prevent sudden death |
Avoidance of all QT-prolonging drugs |
Normal heart ventricular tachycardias |
Relieve symptoms |
Beta-blockers, calcium channel blockers |
Catheter ablation |
||
Non-sustained VT (NSVT) |
Relieve symptoms |
Beta-blockers |
Class Ic and class III |
||
ICD in clearly defined subgroups |
Antiarrhythmic drugs
Drugs that modify the rhythm and conduction of the heart are used to treat cardiac arrhythmias. Antiarrhythmic drugs may aggravate or produce arrhythmias (proarrhythmia) and they may also depress ventricular contractility and must therefore be used with caution. They are classified according to their effect on the action potential (Vaughan Williams’ classification; Table 14.15 and Fig. 14.49).
Table 14.15 Vaughan Williams’ classification of antiarrhythmic drugs
Class I |
Membrane-depressant drugs (sodium-channel blockers) |
Ia |
Disopyramide |
Ib |
Lidocaine, mexiletine |
Ic |
Flecainide, propafenone |
Class II |
β-Adrenoceptor blocking drugs, e.g. atenolol, propranolol, esmolol |
Class III |
Prolong action potential, e.g. amiodarone, dronedarone, sotalol |
Class IV |
Calcium-channel blockers, e.g. verapamil, diltiazem |
(Other |
Adenosine, digoxin) |
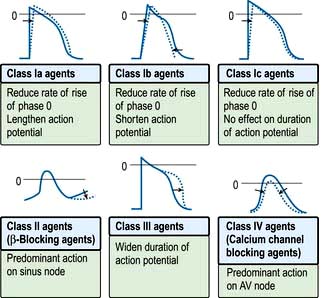
Figure 14.49 Vaughan Williams’ classification of antiarrhythmic drugs based on their effect on cardiac action potentials. 0 = 0 mV. The dotted curves indicate the effects of the drugs.
Class I drugs
These are membrane-depressant drugs that reduce the rate of entry of sodium into the cell (sodium-channel blockers). They may slow conduction, delay recovery or reduce the spontaneous discharge rate of myocardial cells. Class Ia drugs (e.g. disopyramide) lengthen the action potential, class Ib drugs (e.g. lidocaine) shorten the action potential, and class Ic (flecainide, propafenone) do not affect the duration of the action potential. Class I agents have been found to increase mortality compared to placebo in post-myocardial infarction patients with ventricular ectopy (Cardiac Arrhythmia Suppression Trial (CAST) trials – class Ic agents) and in patients treated for atrial fibrillation (class Ia agent, quinidine). In view of this, class Ic agents such as flecainide and all other class I drugs should be reserved for patients who do not have significant coronary artery disease, left ventricular dysfunction, or other forms of significant structural heart disease.
Class II drugs
These antisympathetic drugs prevent the effects of catecholamines on the action potential. Most are β-adrenoceptor antagonists. Cardioselective beta-blockers (β1) include metoprolol, bisoprolol, atenolol and acebutolol. Beta-blockers suppress AV node conduction, which may be effective in preventing attacks of junctional tachycardia, and may help to control the ventricular rate during paroxysms of other forms of SVT (e.g. atrial fibrillation). In general beta-blockers are anti-ischaemic and anti-adrenergic, and have proven beneficial effects in patients post-myocardial infarction (by preventing ventricular fibrillation) and in patients with congestive heart failure. It is therefore advisable to use beta-blocker therapy either alone or in combination with other antiarrhythmic drugs in patients with symptomatic tachyarrhythmias, particularly in patients with coronary artery disease.
Class III drugs
These prolong the action potential, usually by blocking the rapid component of the delayed rectifier potassium current (IKr) and do not affect sodium transport through the membrane. The drugs in this class are amiodarone and sotalol. Sotalol is also a beta-blocker.
Some drugs, such as ibutilide and dofetilide, are only available in some countries. Sotalol may result in acquired long QT syndrome and torsades de pointes. The risk of torsades is increased in the setting of hypokalaemia, and particular care should be taken in patients taking diuretic therapy. Amiodarone therapy in contrast to most other antiarrhythmic drugs carries a low risk of proarrhythmia in patients with significant structural heart disease. Because it has many toxic and potentially serious side-effects, patients need to be counselled prior to commencing amiodarone and monitored at regular follow-up intervals. Dofetilide has been used to treat atrial fibrillation and flutter in patients with recent myocardial infarction and poor LV function. Dronedarone is a multichannel blocking drug which suppresses the recurrence of atrial fibrillation and reduces hospital admissions in patients with cardiovascular risk (ATHENA). However, it has proven harmful in patients with left ventricular dysfunction and heart failure (ANDROMEDA and PALLAS) and must not be used when heart failure is present.
Vernakalant is a multichannel blocker which is approved for the rapid intravenous medical cardioversion of new onset atrial fibrillation.
Class IV drugs
The non-dihydropyridine calcium channel blockers that reduce the plateau phase of the action potential are particularly effective at slowing conduction in nodal tissue. Verapamil and diltiazem are two drugs in this group. These drugs can prevent attacks of junctional tachycardia (AVNRT and AVRT) and may help to control ventricular rates during paroxysms of other forms of SVT (e.g. atrial fibrillation).
Drug therapy is commonly used for symptomatic relief in patients who do not have life-threatening tachyarrhythmias. Antiarrhythmic drugs have not been shown to prolong life. Patient safety is the main factor determining the choice of antiarrhythmic therapy and proarrhythmic risks need to be carefully assessed prior to initiating therapy. As a generalization, class Ic agents are employed in patients with structurally normal hearts and class III agents are used in patients with structural heart disease, although exceptions occur.
In order to administer antiarrhythmic agents as safely as possible it is helpful to be familiar with the different proarrhythmia mechanisms and their main predisposing risk factors (Table 14.16).
Table 14.16 Proarrhythmia mechanisms and the predisposing risk factors
| Proarrhythmia mechanism | Drug | Predisposing risk factors |
|---|---|---|
Patients with structurally normal hearts and normal QT intervals, or with implantable defibrillators, are either at very low risk of proarrhythmia or are protected from any life-threatening consequences, and in these patients it is possible to persevere with drug therapy until an efficacious, well-tolerated agent is identified.
Catheter ablation
Radiofrequency catheter ablation is frequently employed in the management of symptomatic tachyarrhythmias. Ablations are performed by placing three or four electrode catheters into the heart chambers in order to record and pace from various sites. Pacing the atria or the ventricles is used to trigger the tachycardia and to study the tachycardia mechanism. Successful ablation depends on accurate identification of either the site of origin of a focal tachycardia or of a critical component of a macro-re-entry tachycardia. The following tachyarrhythmias can be readily ablated:
AV node re-entry tachycardia (AVNRT)
AV re-entry tachycardia (AVRT) with an accessory pathway, including WPW syndrome
Symptomatic patients with a pre-excited ECG because of accessory pathway conduction (WPW syndrome) are advised to undergo catheter ablation as first-line therapy, owing to the risk of sudden death associated with this condition. This is especially the case in patients with pre-excited atrial fibrillation. Patients with accessory pathways that only conduct retrogradely from the ventricles to the atrium are not at increased risk of sudden death but experience symptoms due to AVRT. These patients are commonly offered an ablation procedure if simple measures such as AV nodal slowing agents fail to suppress tachycardia. Asymptomatic patients with the WPW ECG pattern are now frequently offered an ablation procedure for prophylactic reasons. The main risk associated with accessory pathway ablation is thromboembolism in patients with left-sided accessory pathways. The success rate for catheter ablation of AVNRT and accessory pathways is >95%.
Patients with normal hearts and documented ventricular tachycardia should be referred for specialist evaluation. Unlike VT in patients with structural heart disease, normal heart VT is not associated with increased risk of sudden death and is easily cured by catheter ablation.
Catheter ablation is recommended in patients with atrial flutter that is not easily managed medically. Ablation of typical flutter is effective in 90–95% cases. In the direct comparison of catheter ablation and antiarrhythmic therapy, the rate of recurrence was significantly lower following ablation. Atrial tachycardia, especially in patients with structurally normal hearts, may also be cured by catheter ablation. In atrial fibrillation, adequate control of ventricular rates is sometimes not possible despite optimal medical therapy. These patients experience a marked symptomatic improvement following AV node ablation and pacemaker implantation. Unlike other forms of catheter ablation this does not cure the arrhythmia; atrial fibrillation continues and anticoagulation is still required following ablation.
In younger patients with structurally normal hearts, atrial ectopic beats, which commonly arise from a focus situated in the pulmonary veins, may trigger atrial fibrillation. Catheter ablation of this ectopic focus includes the application of radiofrequency energy in or around the pulmonary veins in order to abolish the connection between the sleeves of arrhythmogenic atrial myocardium surrounding or extending into the veins from the atrium (pulmonary vein isolation). The trigger is therefore eliminated and the arrhythmia does not recur. These techniques appear to be highly effective, especially in young patients with paroxysmal atrial fibrillation, normal atrial size and no underlying heart disease (70–80% long-term success), but are presently time-consuming procedures (4 h or more) and are associated with some serious complications such as stroke, pericardial haemorrhage, pulmonary vein stenosis and atrio-oesophageal fistula in a small minority of patients (in experienced centres <2% altogether).
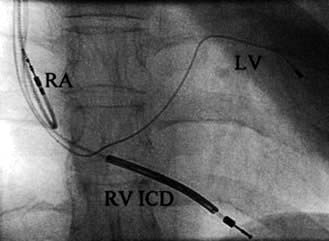
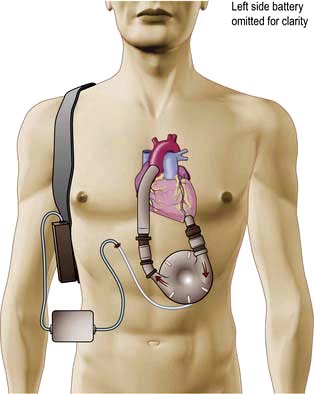
Implantable cardioverter-defibrillator (ICD)
Life-threatening ventricular arrhythmias (ventricular fibrillation or rapid ventricular tachycardia with hypotension) result in death in up to 40% within 1 year of diagnosis. Large multicentre prospective trials such as the Antiarrhythmics (amiodarone) Versus Implantable Defibrillator (AVID) trial have proven that implantable defibrillators improve overall survival in patients who have experienced an episode of life-threatening ventricular tachyarrhythmia.
The ICD recognizes ventricular tachycardia or fibrillation and automatically delivers pacing or a shock to the heart to cause cardioversion to sinus rhythm. Modern ICDs are only a little larger than a pacemaker and are implanted in a pectoral position (Fig. 14.50). The device may have leads to sense and pace both the right atrium and ventricle, and the lithium batteries employed are able to provide energy for over 100 shocks each of around 30 J. ICD discharges are painful if the patient is conscious. However, ventricular tachycardia may often be terminated by overdrive pacing the heart, which is painless. The ICD is superior to all other treatment options at preventing sudden cardiac death. The use of this device has cut the sudden death rate in patients with a history of serious ventricular arrhythmias to approximately 2% per year. However, the majority of these patients have significant structural heart disease and overall cardiac mortality due to progressive heart failure remains high. As a result the ICD is now first-line therapy in the secondary prevention of sudden death.
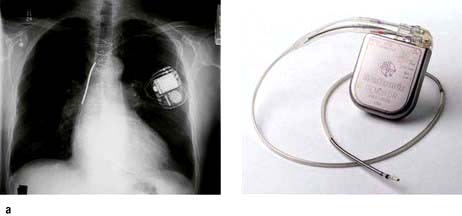
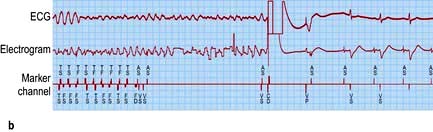
Figure 14.50 Implantable cardioverter-defibrillator. (a) X-ray of a ‘dual-chamber’ defibrillator in a left pectoral position with atrial and ventricular leads. (b) ECG showing the termination of ventricular fibrillation by the direct current shock at 20 J. Electrogram (EG) recorded internally from the ventricular lead of an ICD reveals chaotic ventricular activity consistent with ventricular fibrillation. This is confirmed by an electrocardiogram (lead V2). The marker channel (MC) demonstrates that the device detects ventricular fibrillation correctly (FS, fibrillation sensing, lower line) and delivers an appropriate shock (CD) that terminates the arrhythmia and restores normal sinus rhythm (VS, ventricular sensing, lower line). Incidentally, atrial fibrillation is also detected before shock delivery (upper line on the marker channel).
Implantable cardioverter-defibrillators are also employed in the primary prevention of sudden cardiac death. The chances of surviving an out-of-hospital cardiac arrest are as low as 10%. Therefore selected patients who have never experienced a spontaneous episode of life-threatening ventricular tachyarrhythmia but who are assessed to be at high risk of sudden death are advised to undergo ICD implantation. In two large primary prevention ICD trials, Multicenter Automated Defibrillator Implantation Trial (MADIT II) and Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT), therapy with an ICD reduced mortality by 23–31% on top of conventional treatment, which included revascularization, beta-blockers and angiotensin-converting enzyme inhibitors. ICD combined with cardiac resynchronization therapy may improve both symptoms and life expectancy of patients with any degree of heart failure (COMPANION, CARE-HF and MADIT-CRT).
The following groups of patients may merit prophylactic ICD placement:
Those with coronary artery disease; significant impairment of left ventricular function (LVEF ≤35–40%), spontaneous non-sustained ventricular tachycardia in whom sustained ventricular tachycardia was induced by pacing the heart during an electrophysiological study
Those with NYHA class III–IV heart failure (LVEF ≤35%) in combination with cardiac resynchronization therapy, and in class I–II NYHA heart failure (LVEF ≤35%) when LBBB is present
Those with very poor LV function post-MI (LVEF ≤30%)
Those with dilated and hypertrophic cardiomyopathy, long QT syndrome and Brugada syndrome or other channelopathies who have a strong family history of sudden cardiac death.
FURTHER READING
Vardas PE, Auricchio A, Blanc JJ et al. European Society of Cardiology; European Heart Rhythm Association. Guidelines for cardiac pacing and cardiac resynchronization therapy. The Task Force for Cardiac Pacing and Cardiac Resynchronization Therapy of the European Society of Cardiology. Developed in collaboration with the European Heart Rhythm Association. Europace 2007; 9:959–998.
Zipes DP, Camm AJ, Borggrefe M et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Europace 2006; 8:746–837.
SIGNIFICANT WEBSITE
National Institute of Health and Clinical Excellence. Implantable cardioverter defibrillators (ICDs) for the treatment of arrhythmias. Technology appraisal review TA11; 2006: www.nice.org.uk