Congenital heart disease
A congenital cardiac malformation occurs in about 1% of live births. There is an overall male predominance, although some individual lesions (e.g. atrial septal defect and persistent ductus arteriosus) occur more commonly in females. As a result of improved medical and surgical management, more children with congenital cardiac disease are surviving into adolescence and adulthood. Thus, there is a need for an increased awareness among general physicians and cardiologists of the problems posed by these individuals.
Fetal circulation (Fig. 14.84)
In the developing fetus, oxygenated blood and nutrients are supplied to the fetus via the placenta and the umbilical vein. Half of that blood is directed to the fetal ductus venosus and carried to the inferior vena cava (IVC), the other half enters the liver.
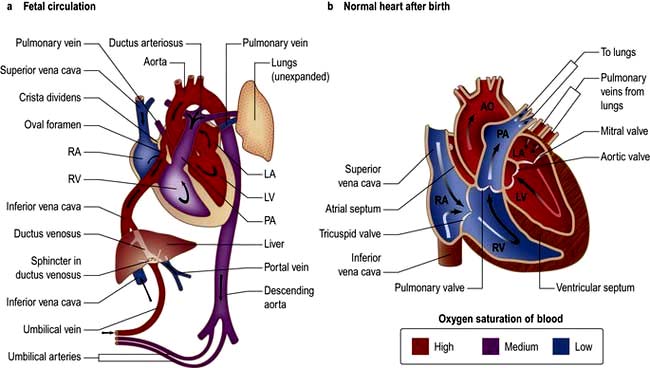
Figure 14.84 Anatomy showing circulation. AO, aorta; LA, left atrium; LV, left ventricle; PA, pulmonary artery; RA, right atrium; RV, right ventricle.
Blood moves from the IVC to the right atrium of the heart. In the fetus, there is an opening between the right and left atrium (the foramen ovale), and most of the blood (which is a mixture of oxygenated and de-oxygenated blood) flows from the right into the left atrium, bypassing pulmonary circulation. This blood goes into the left ventricle and is pumped through the aorta into the fetal body. Some of the blood flows from the aorta through the internal iliac arteries to the umbilical arteries and re-enters the placenta, where carbon dioxide and other waste products from the fetus are taken up and enter the woman’s circulation.
Some of the blood from the right atrium does not enter the left atrium, but enters the right ventricle and is pumped into the pulmonary artery. In the fetus, there is a connection between the pulmonary artery and the aorta, the ductus arteriosus, which directs most of this blood away from the lungs. With the first breath after delivery the vascular resistance in the pulmonary arteries falls and more blood moves from the right atrium to right ventricle and pulmonary arteries and oxygenated blood travels back to the left atrium through the pulmonary veins. The decrease in right atrial pressure and relative increase in left atrial pressure results in closure of the foramen ovale.
The ductus arteriosus usually closes off within one or two days of birth completely separating the left and right system. The umbilical vein and the ductus venosus closes off within 2–5 days after birth, leaving behind the ligamentum teres and the ligamentum venosus of the liver, respectively.
Aetiology
The aetiology of congenital cardiac disease is often unknown, but recognized associations include:
 Maternal prenatal rubella infection (persistent ductus arteriosus, and pulmonary valvular and arterial stenosis)
Maternal prenatal rubella infection (persistent ductus arteriosus, and pulmonary valvular and arterial stenosis)
Maternal alcohol misuse (septal defects)
Maternal drug treatment and radiation
Genetic abnormalities (e.g. The familial form of atrial septal defect and congenital heart block)
Chromosomal abnormalities (e.g. septal defects and mitral and tricuspid valve defects are associated with Down’s syndrome (trisomy 21) or coarctation of the aorta in Turner’s syndrome (45, XO).
Classification
See Table 14.41.
Table 14.41 Classification of congenital heart disease
| Acyanotic | Cyanotic |
|---|---|
With shunts |
With shunts |
Atrial septal defect |
Fallot’s tetralogy |
Ventricular septal defect |
Transposition of the great vessels |
Patent ductus arteriosus |
|
Partial anomalous venous drainage |
Severe Ebstein’s anomaly |
Without shunts |
Without shunts |
Coarctation of the aorta |
Severe pulmonary stenosis |
Congenital aortic stenosis |
Tricuspid atresia |
|
Pulmonary atresia |
|
Hypoplastic left heart |
Symptoms and signs
Congenital heart disease should be recognized as early as possible, as the response is usually better the earlier the treatment is initiated. Some symptoms, signs and clinical problems are common in congenital heart disease:
Central cyanosis occurs because of right-to-left shunting of blood or because of complete mixing of systemic and pulmonary blood flow. In the latter case, e.g. Fallot’s tetralogy, the abnormality is described as cyanotic congenital heart disease.
Pulmonary hypertension results from large left-to-right shunts. The persistently raised pulmonary flow leads to the development of increased pulmonary artery vascular resistance and consequent pulmonary hypertension. This is known as the Eisenmenger’s reaction (or the Eisenmenger’s complex when due specifically to a ventricular septal defect). The development of pulmonary hypertension significantly worsens the prognosis.
Clubbing of the fingers occurs in congenital cardiac conditions associated with prolonged cyanosis.
Paradoxical embolism of thrombus from the systemic veins to the systemic arterial system may occur when a communication exists between the right and left heart. There is therefore an increased risk of cerebrovascular emboli and also abscesses (as with endocarditis).
Polycythaemia can develop secondary to chronic hypoxaemia, leading to a hyperviscosity syndrome and an increased thrombotic risk, e.g. strokes.
Growth retardation is common in children with cyanotic heart disease.
Syncope is common when severe right or left ventricular outflow tract obstruction is present. Exertional syncope, associated with deepening central cyanosis, may occur in Fallot’s tetralogy. Exercise increases resistance to pulmonary blood flow but reduces systemic vascular resistance. Thus, the right-to-left shunt increases and cerebral oxygenation falls.
Squatting is the posture adopted by children with Fallot’s tetralogy. It results in obstruction of venous return of desaturated blood and an increase in the peripheral systemic vascular resistance. This leads to a reduced right-to-left shunt and improved cerebral oxygenation.
Presentation
Adolescents and adults with congenital heart disease present with specific common problems related to the long-standing structural nature of these conditions and any surgical treatment:
Endocarditis (particularly in association with otherwise innocuous lesions such as small VSDs or bicuspid aortic valve that can give up to 10% lifetime risk)
Progression of valvular lesions (calcification and stenosis of congenitally deformed valves, e.g. bicuspid aortic valve)
Atrial and ventricular arrhythmias (often quite resistant to treatment)
Right heart failure (especially when surgical palliation results in the right ventricle providing the systemic supply)
End-stage heart failure (rarely managed by heart or heart-lung transplantation).
Genetic counselling
These conditions necessitate active follow-up of adult patients. Pregnancy is normally safe except if pulmonary hypertension or vascular disease is present, when the prognosis for both mother and fetus is poor.
Table 14.42 lists the most common congenital lesions and their occurrence in first-degree relatives.
Table 14.42 Common congenital lesions
| Percentage of congenital lesions | Occurrence in first-degree relatives (%) | |
|---|---|---|
Ventricular septal defect |
39 |
4 |
Atrial septal defect |
10 |
2 |
Persistent ductus |
10 |
4 |
Arteriosus |
|
|
Pulmonary stenosis |
7 |
|
Coarctation of the aorta |
7 |
2 |
Aortic stenosis |
6 |
4 |
Fallot’s tetralogy |
6 |
4 |
Others |
15 |
|
Genetic factors should be considered in all patients presenting with congenital heart disease. For example, parents with a child suffering from Fallot’s tetralogy stand a 4% chance of conceiving another child with the disease, and so fetal ultrasound screening of the mother during pregnancy is essential. Parents with congenital heart disease are also more likely to have affected offspring. Fathers have a 2% risk, while mothers have a higher risk (around 5%). Individual families can exhibit even higher risks of recurrence.
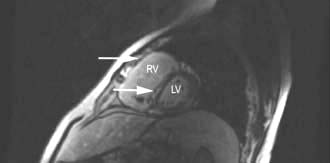
Ventricular septal defect (VSD) (Fig. 14.85)
VSD is the most common congenital cardiac malformation (1 : 500 live births). The haemodynamic consequences of this are dependent on the shunt size. Left ventricular pressure is higher than right and blood therefore moves from left to right and pulmonary blood flow increases. In large defects the large volumes of blood flow through the pulmonary vasculature leads to pulmonary hypertension and eventual Eisenmenger’s complex when right ventricular pressure becomes higher than left and as a result blood starts to shunt from right to left leading to cyanosis.
Small restrictive VSDs (‘Maladie de Roger’) are often found incidentally as patients are asymptomatic. They are associated with a loud pan-systolic murmur. The majority close spontaneously by the age of 10 years.
Large (non-restrictive) VSDs result in significant LA and LV dilatation (due to LV volume overload). Large defects usually present with heart failure symptoms in childhood and eventually lead to pulmonary hypertension and Eisenmenger’s complex. As pressures equalize the murmur becomes softer.
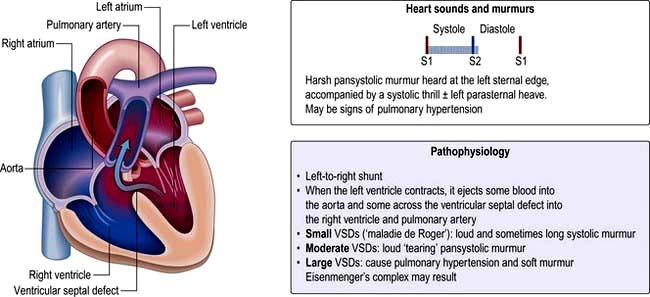
Investigations and intervention
A small VSD produces no abnormal X-ray or ECG findings. In larger defects, the chest X-ray may demonstrate prominent pulmonary arteries owing to increased pulmonary blood flow but with pulmonary hypertension there may be ‘pruned’ pulmonary arteries. Cardiomegaly occurs when a moderate or a large VSD is present and the ECG may show both left and right ventricular hypertrophy. Echocardiography can assess the size and location of the VSD and its haemodynamic consequences. Interventional options are either surgical patch repair or device closure if it is an isolated muscular VSD. Indications for intervention include left atrial and ventricular enlargement with or without early LV dysfunction; reversible pulmonary hypertension (mild) where there is a residual left to right shunt and no significant desaturation with exercise; infective endocarditis. Patients with a restrictive VSD and patients after successful closure have an excellent long term outcome. Prophylaxis of endocarditis is discussed on page 87.
Atrial septal defect (ASD)
This condition is often first diagnosed in adulthood and represents one-third of congenital heart disease. It is two to three times more common in women than in men. There are three main types of ASD (Fig. 14.86):
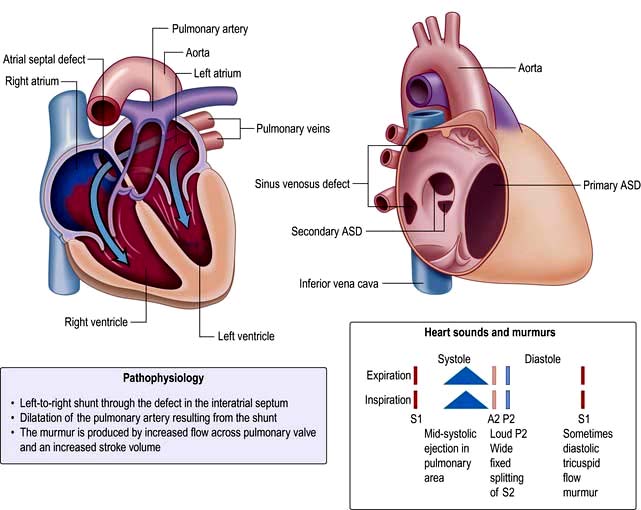
Sinus venosus defects – located in the superior part of the septum near the SVC (superior sinus venosus defect) or the inferior part of the septum near the IVC (inferior sinus venosus defect) entry point.
Ostium secundum defects (75%) – located in the mid-septum (fossa ovalis). This should not be confused with the patent foramen ovale (PFO), which is a normal variant and not a true septal defect. PFO is usually asymptomatic but is associated with paradoxical emboli and an increased incidence of embolic stroke.
Ostium primum (atrioventricular septal) defects (15%) – located in the lower part of atrial septum.
Adult patients with an unrepaired ASD with significant left to right shunt develop right heart overload and dilatation. These patients develop symptoms of dyspnoea and exercise intolerance and may develop atrial arrhythmias from right atrial dilatation. There is also increased pulmonary vascular flow, but significant pulmonary hypertension develops in <5% of patients and it is thought that these patients have additional factors including genetic predisposition to pulmonary hypertension. The physical signs of ASD reflect the volume overloading of the right ventricle (Fig. 14.86). A right ventricular heave can usually be felt.
Investigations and intervention
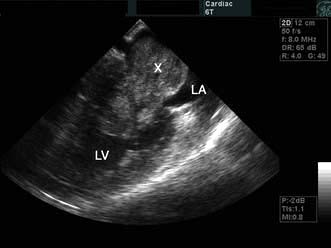
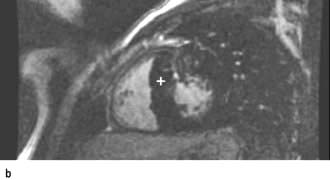
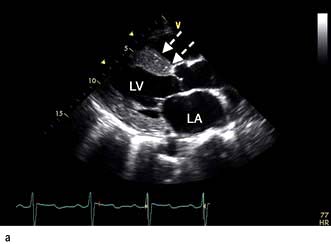
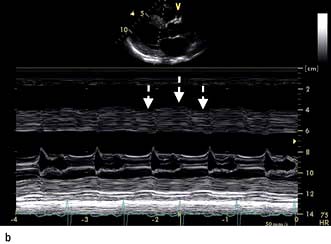
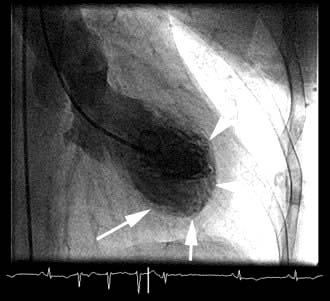
The chest X-ray may demonstrate prominent pulmonary arteries with pulmonary plethora. Right bundle branch block and right axis deviation may be present on the ECG (because of dilatation of the right ventricle). Ostium primum defects may have left axis deviation on the ECG. Echocardiography may demonstrate hypertrophy and dilatation of the right heart and pulmonary arteries. Subcostal views with 2D and colour Doppler demonstrates the ASD (Fig. 14.87) and allow calculation of the left-right shunt (QP : QS ratio). CMR and CT are helpful to assess for anomalous pulmonary venous drainage which may accompany an ASD. Indications for intervention include: an ASD with significant left to right shunting resulting in right atrial/ventricular enlargement which should be closed irrespective of symptoms; thromboembolic events including certain patients with a patent foramen ovale. The options for intervention include device closure using a transcatheter clamshell device (Fig. 14.88) for most secundum ASDs (if suitable size) or surgical closure for all other ASD types.
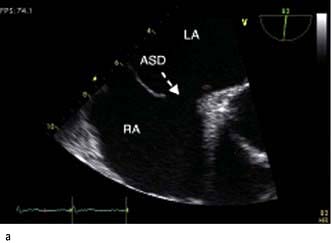
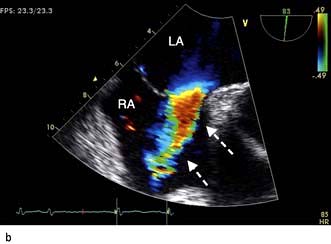
Figure 14.87 Ostium secundum atrial septal defect (arrows) in a young girl. (a) Shown by a 2-D echocardiogram subcostal four-chamber view. (b) Colour Doppler can demonstrate the left-to-right shunt. LA, left atrium; RA, right atrium.
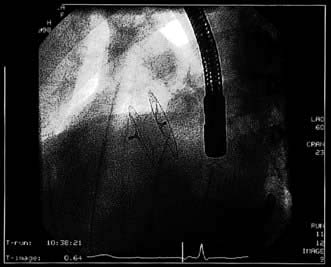
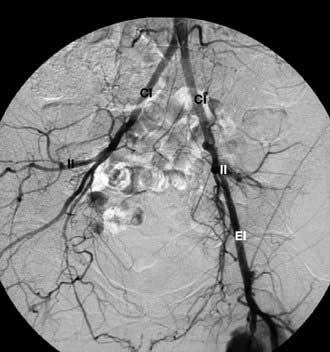
Figure 14.88 Angiographic appearance of a fully deployed ASD closure device. The device bridges the ASD and wedges against the surfaces of the right and left atrial septa, occluding flow. The metal object in frame is the distal end of a transoesophageal echocardiography probe.
(Courtesy of Dr D Ward, St George’s, University of London.)
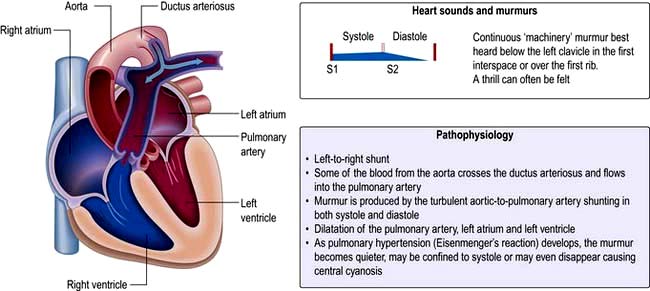
Patent ductus arteriosus (PDA)
This is a persistent communication between the proximal left pulmonary artery and the descending aorta resulting in a continuous left to right shunt (Fig. 14.89). Normally the ductus arteriosus closes within a few hours of birth in response to decreased pulmonary resistance however in some cases (particularly premature babies and in cases with maternal rubella) the ductus persists. Indometacin (a prostaglandin inhibitor) is given to stimulate duct closure. If the shunt is moderate to large it will result in left heart volume overload overload and in some cases pulmonary hypertension and Eisenmenger’s syndrome. The characteristic clinical signs are a bounding pulse and continuous ‘machinery murmur’ however as pulmonary hypertension develops in a large PDA the murmur becomes softer.
Investigations and intervention
With a large shunt, the aorta and pulmonary arterial system may be prominent on chest X-ray. The ECG may demonstrate left atrial abnormality and left ventricular hypertrophy. The development of Eisenmenger reaction will produce right ventricular hypertrophy. Echocardiography may show a dilated left atrium and left ventricle with right heart changes occurring late. Colour Doppler imaging of the proximal pulmonary arteries may demonstrate the shunt. Indications for intervention (usually with percutaneous devices) include left ventricular dilatation; mild–moderate pulmonary arterial hypertension (not Eisenmenger). Small defects may predispose to endarteritis and should be considered for device closure unless clinically silent.
Coarctation of the aorta
A coarctation of the aorta is a narrowing of the aorta at or just distal to the insertion of the ductus arteriosus (distal to the origin of the left subclavian artery (Fig. 14.90). Rarely it can occur proximal to the left subclavian. It occurs twice as commonly in men as in women. It is also associated with Turner’s syndrome (p. 978). In 80% of cases, the aortic valve is bicuspid (and potentially stenotic or endocarditic). Other associations include patent ductus arteriosus, ventricular septal defect, mitral stenosis or regurgitation and circle of Willis aneurysms. Severe narrowing of the aorta encourages the formation of a collateral arterial circulation involving the periscapular and intercostal arteries. Decreased renal perfusion can lead to the development of systemic hypertension that persists even after surgical correction.
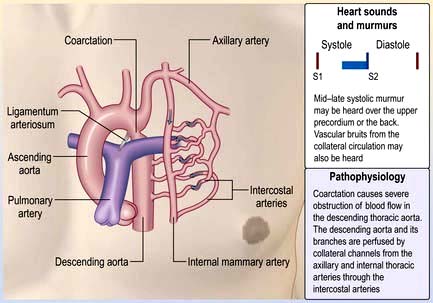
Coarctation of the aorta is often asymptomatic for many years. Headaches and nosebleeds (due to hypertension), and claudication and cold legs (due to poor blood flow in the lower limbs) may be present. Physical examination reveals hypertension in the upper limbs, and weak, delayed (radiofemoral delay) pulses in the legs. If coarctation is present in the aorta, proximal to the left subclavian artery, there will be asynchronous radial pulses in right and left arms. Poor peripheral pulses are seen in severe cases. For heart sounds and murmurs in coarctation of aorta, see Figure 14.90.
Investigations and intervention
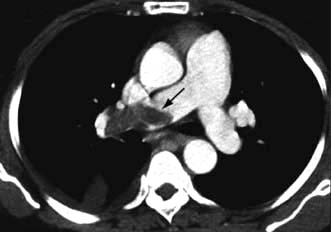
Chest X-ray may reveal a dilated aorta indented at the site of the coarctation. This is manifested by an aorta (seen in the upper right mediastinum) shaped like a ‘figure 3’. In adults, tortuous and dilated collateral intercostal arteries may erode the undersurfaces of the ribs (‘rib notching’). ECG demonstrates left ventricular hypertrophy. Echocardiography sometimes shows the coarctation and other associated anomalies. CT and CMR (Fig. 14.91) scanning can accurately demonstrate the coarctation and quantify flow.
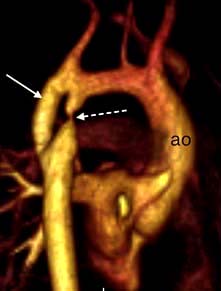
Intervention is required if there is a peak–peak gradient across the coarctation of >20 mmHg and/or proximal hypertension. In neonates coarctation is treated with surgical repair. In older children and adults, balloon dilatation and stenting is an option although many centres still prefer surgery. Balloon dilatation is preferred for recoarctation.
Cyanotic congenital heart disease
Fallot’s tetralogy
Tetralogy of Fallot (Fig. 14.92) consists of:
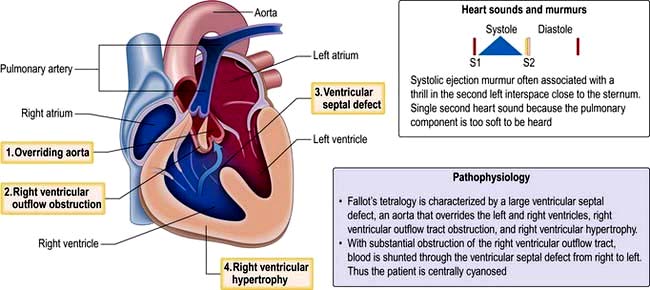
Symptoms depend on the degree of pulmonary stenosis. Often this is progressive in the first year of life and cyanosis develops due to increased right-sided pressures, resulting in a right to left shunt. Fallot’s spells are episodes of severe cyanosis noted in children due to spasm of the subpulmonary muscle – these can be relieved by increasing systemic resistance by postural manoeuvres, e.g. squatting. In babies with severe pulmonary stenosis systemic-to-pulmonary artery shunts (i.e. Blalock–Taussig – subclavian to pulmonary artery shunt) may be used initially to increase pulmonary blood flow in severe cases of pulmonary stenosis. The majority of adults with tetralogy of Fallot will have undergone complete repair which involves relief of the right ventricular outflow tract obstruction and closure of the VSD. The overall survival of those who have had operative repair is excellent. DiGeorge’s syndrome is found in 15% of those with tetralogy of Fallot.
Transposition of the great arteries
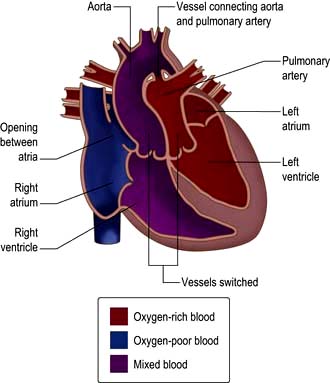
Complete transposition of the great arteries (TGA)
In complete transposition of the great arteries (TGA), the right atrium connects to the morphological right ventricle, which gives rise to the aorta and the left atrium connects to the morphological left ventricle, which gives rise to the pulmonary artery (Fig. 14.93). This is incompatible with life as blood circulates in two parallel circuits, i.e. deoxygenated blood from the systemic veins passes into the right heart and then via the aorta back to the systemic circulation. Oxygenated blood from the pulmonary veins passes through the left heart and back to the lungs. Babies with transposition are usually born cyanosed – if there is a significant ASD, VSD or PDA allowing a shunt (i.e. mixing of oxygenated and deoxygenated blood) the diagnosis might be delayed. In those without a shunt an atrial septostomy is performed. A Rashkind’s balloon is used to dilate the foramen ovale and is used to maintain saturations at 50–80% until a definitive procedure can be performed. The arterial switch procedure is now performed in the first 2 weeks of life – the aorta is reconnected to the left ventricle and the pulmonary artery is connected to the right ventricle. The coronary arteries are re-implanted.
Currently, the majority of adult patients with transposition of the great arteries will have had an ‘atrial switch’ operation. The right ventricle remains the systemic ventricle in this situation. Although most of these patients do well for many years, life expectancy is clearly limited by eventual failure of the systemic right ventricle.
Congenitally corrected transposition of the great arteries (ccTGA)
In congenitally corrected transposition of the great arteries (ccTGA), systemic venous return to the right atrium enters a morphological left ventricle, which pumps into the pulmonary artery. Pulmonary venous blood returns to the left atrium and then via the morphological right ventricle to the aorta. The circulation is physiologically corrected but the systemic circulation is supported by a morphologic right ventricle. ccTGA is often associated with cardiac lesions; systemic (tricuspid) atrio-ventricular valve abnormalities with valve insufficiency; VSD; subpulmonary stenosis; complete heart block; Wolff–Parkinson–White syndrome; dextrocardia. Many patients with ccTGA live a normal life, although other patients require pacemaker insertion (the AV node is abnormal leading to heart block), surgery for a regurgitant tricuspid valve, or develop heart failure from the systemic (right ventricle).
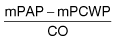
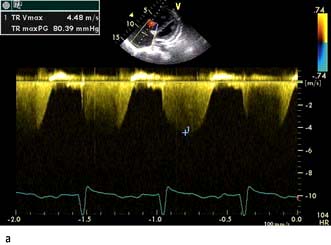
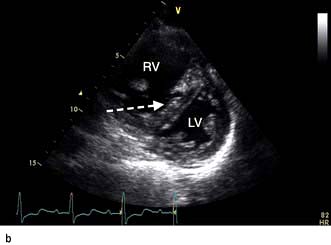
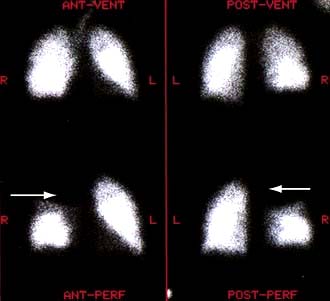
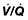 scan) is a good and widely available diagnostic investigation. Pulmonary 99mTc scintigraphy demonstrates underperfused areas (
scan) is a good and widely available diagnostic investigation. Pulmonary 99mTc scintigraphy demonstrates underperfused areas (