Coronary artery disease
Myocardial ischaemia occurs when there is an imbalance between the supply of oxygen (and other essential myocardial nutrients) and the myocardial demand for these substances.
Coronary blood flow to a region of the myocardium may be reduced by a mechanical obstruction that is due to:
There can be a decrease in the flow of oxygenated blood to the myocardium that is due to:
An increased demand for oxygen may occur owing to an increase in cardiac output (e.g. thyrotoxicosis) or myocardial hypertrophy (e.g. from aortic stenosis or hypertension).
Myocardial ischaemia most commonly occurs as a result of obstructive coronary artery disease (CAD) in the form of coronary atherosclerosis. In addition to this fixed obstruction, variations in the tone of smooth muscle in the wall of a coronary artery may add another element of dynamic or variable obstruction.
CAD is the largest single cause of death in the UK and many parts of the world. However, over the last decade, the mortality rate in the UK has fallen considerably. In 2009, 1 in 5 male and 1 in 8 female deaths were from coronary artery disease; approximately 82 000 deaths in the UK (www.bhf.org.uk/heart-health/statistics/mortality.aspx). It has been estimated that by 2010, 60% of the world’s heart disease occured in India. Sudden cardiac death is a prominent feature of CAD. One in every six coronary attacks present with sudden death as the first, last and only symptom.
Process of coronary atherosclerosis
Coronary atherosclerosis is a complex inflammatory process characterized by the accumulation of lipid, macrophages and smooth muscle cells in intimal plaques in the large and medium-sized epicardial coronary arteries. The vascular endothelium plays a critical role in maintaining vascular integrity and homeostasis. Mechanical shear stresses (e.g. from morbid hypertension), biochemical abnormalities (e.g. elevated LDL, diabetes mellitus), immunological factors (e.g. free radicals from smoking), inflammation (e.g. infection such as Chlamydophila pneumoniae) and genetic alteration may contribute to the initial endothelial ‘injury’ or dysfunction, which is believed to trigger atherogenesis.
The development of atherosclerosis follows the endothelial dysfunction, with increased permeability to and accumulation of oxidized lipoproteins, which are taken up by macrophages at focal sites within the endothelium to produce lipid-laden foam cells. Macroscopically, these lesions are seen as flat yellow dots or lines on the endothelium of the artery and are known as ‘fatty streaks’. The ‘fatty streak’ progresses with the appearance of extracellular lipid within the endothelium (‘transitional plaque’). Release of cytokines such as platelet-derived growth factor and transforming growth factor-β (TGF-β) by monocytes, macrophages or the damaged endothelium promotes further accumulation of macrophages as well as smooth muscle cell migration and proliferation. The proliferation of smooth muscle with the formation of a layer of cells covering the extracellular lipid separates it from the adaptive smooth muscle thickening in the endothelium. Collagen is produced in larger and larger quantities by the smooth muscle and the whole sequence of events cumulates as an ‘advanced or raised fibrolipid plaque’. The ‘advanced plaque’ may grow slowly and encroach on the lumen or become unstable, undergo thrombosis and produce an obstruction (‘complicated plaque’).
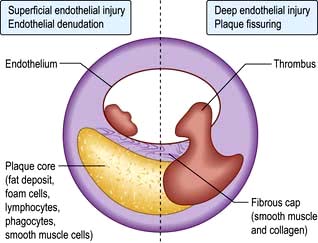
Two different mechanisms are responsible for thrombosis on the plaques (Fig. 14.58):
 The first process is superficial endothelial injury, which involves denudation of the endothelial covering over the plaque. Subendocardial connective tissue matrix is then exposed and platelet adhesion occurs because of reaction with collagen. The thrombus is adherent to the surface of the plaque.
The first process is superficial endothelial injury, which involves denudation of the endothelial covering over the plaque. Subendocardial connective tissue matrix is then exposed and platelet adhesion occurs because of reaction with collagen. The thrombus is adherent to the surface of the plaque.
The second process is deep endothelial fissuring, which involves an advanced plaque with a lipid core. The plaque cap tears (ulcerates, fissures or ruptures), allowing blood from the lumen to enter the inside of the plaque itself. The core with lamellar lipid surfaces, tissue factor (which triggers platelet adhesion and activation) produced by macrophages and exposed collagen, is highly thrombogenic. Thrombus forms within the plaque, expanding its volume and distorting its shape. Thrombosis may then extend into the lumen.
A 50% reduction in luminal diameter (producing a reduction in luminal cross-sectional area of approximately 70%) causes a haemodynamically significant stenosis. At this point the smaller distal intramyocardial arteries and arterioles are maximally dilated (coronary flow reserve is near zero), and any increase in myocardial oxygen demand provokes ischaemia.
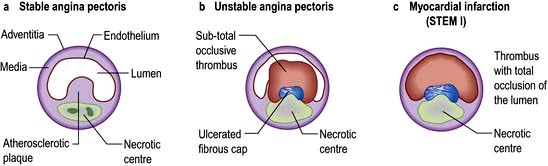
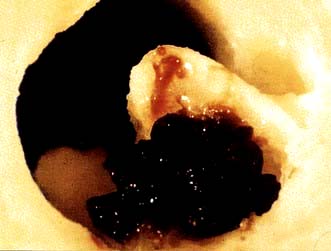
CAD gives rise to a wide variety of clinical presentations, ranging from relatively stable angina through to the acute coronary syndromes of unstable angina and myocardial infarction (Fig. 14.59). Figure 14.60 shows an actual plaque rupture.
Risk factors for coronary artery disease – primary and secondary prevention
CAD is an atherosclerotic disease that is multifactorial in origin, giving rise to the risk factor concept. Certain living habits promote atherogenic traits in genetically susceptible persons. A number of ‘risk’ factors are known to predispose to the condition (Table 14.26). Some of these, such as age, gender, race and family history, cannot be changed, whereas other major risk factors, such as serum cholesterol, smoking habits, diabetes and hypertension, can be modified.
Table 14.26 Risk factors for coronary disease
|
|
Atherosclerotic disease manifest in one vascular bed is often advanced in other territories. Patients with intermittent claudication have a two- to four-fold increased risk of CAD, stroke, or heart failure. Following initial myocardial infarction (MI), there is a 3–6-fold increase in the risk of heart failure and stroke. After stroke, the risk of heart failure and MI is increased two-fold.
The disease can be asymptomatic in its most severe form, with one in three myocardial infarctions going unrecognized. Some 30–40% of individuals who present with an acute coronary syndrome have had no prior warning symptom to suggest the presence of underlying disease.
Primary prevention can be defined as the prevention of the atherosclerotic disease process and secondary prevention as the treatment of the atherosclerotic disease process (i.e. treatment of the disease or its complications). The objective of prevention is to reduce the incidence of first or recurrent clinical events due to CAD, ischaemic stroke and peripheral artery disease.
Traditional risk factors
Age
CAD rates increase with age. Atherosclerosis is rare in childhood, except in familial hyperlipidaemia, but is often detectable in young men between 20 and 30 years of age. It is almost universal in the elderly in the West. Atheromatous lesions in the elderly are often complicated by calcification.
Gender
Men have a higher incidence of coronary artery disease than premenopausal women. However, after the menopause, the incidence of atheroma in women approaches that in men. The reasons for this gender difference are not clearly understood, but probably relate to the loss of the protective effect of oestrogen.
Family history
CAD is often found in several members of the same family. Because the disease is so prevalent and because other risk factors are familial, it is uncertain whether family history, per se, is an independent risk factor. A positive family history is generally accepted to refer to those in whom a first-degree relative has developed ischaemic heart disease before the age of 50 years.
Smoking
In men, the risk of developing CAD is directly related to the number of cigarettes smoked (see p. 807). It is estimated that about 20% of deaths from CAD in men and 17% of deaths from CAD in women are due to smoking. Evidence suggests that each person stopping smoking will reduce his/her own risk by 25%. The risk from smoking declines to almost normal after 10 years of abstention.
Diet and obesity
Diets high in fats are associated with ischaemic heart disease, as are those with low intakes of antioxidants (i.e. fruit and vegetables). Supplementation with antioxidants has been shown to be unhelpful in RCTs (p. 211).
It is estimated that up to 30% of deaths from CAD are due to unhealthy diets (see p. 200). The dietary changes which would help to reduce rates of CAD include a reduction in fat, particularly saturated fat intake, a reduction in salt intake and an increase in carbohydrate intake. The consumption of fruit and vegetables should be increased by 50%, to about 400 g/day, which is equivalent to at least five daily portions (see Box 5.2).
There is overwhelming evidence from clinical trials that modification of the diet has a significant impact on the risk of CVD in both the primary and secondary prevention settings.
Weight. Patients who are overweight and those who are obese have an increased risk of CAD. It is estimated that about 5% of deaths from CAD in men and that 6% of such deaths in women are due to obesity (a body mass index (BMI) of >30 kg/m2).
The adverse effect of excess weight is more pronounced when the fat is concentrated mainly in the abdomen. This is known as central obesity (visceral fat) and can be identified by a high waist/hip ratio.
Exercise. Reduction in weight by diet and exercise not only lowers the incidence of CVD but also diabetes/insulin resistance. It is estimated that about 36% of deaths from CAD in men and 38% of deaths from CAD in women are due to lack of physical activity. To produce the maximum benefit the activity needs to be regular and aerobic. Aerobic activity involves using the large muscle groups in the arms, legs and back steadily and rhythmically so that breathing and heart rate are significantly increased.
It is recommended that adults should participate in a minimum of 30 minutes of at least moderate intensity activity (such as brisk walking, cycling or climbing the stairs) on ≥5 days of the week.
Hypertension
Both systolic and diastolic hypertension are associated with an increased risk of CAD. Both drug treatment and lifestyle changes – particularly weight loss, an increase in physical activity and a reduction in salt and alcohol intake – can effectively lower blood pressure.
It is estimated that 14% of deaths from CAD in men and 12% of deaths from CAD in women are due to a raised blood pressure (defined as a systolic blood pressure of ≥140 mmHg, or a diastolic blood pressure of ≥90 mmHg) and that 6% of deaths from CAD in the UK could be avoided if the numbers of people who have high blood pressure were to be reduced by 50%.
Hyperlipidaemia
High serum cholesterol, especially when associated with a low value of high-density lipoproteins (HDL), is strongly associated with coronary atheroma. There is increasing evidence that high serum triglyceride (TG) is also independently linked with coronary atheroma (see p. 1034).
Familial hypercholesterolaemia combined with hypertriglyceridaemia and remnant hyperlipidaemia are also associated with increased risk of coronary atherosclerosis.
Measurement of the fasting lipid profile (total cholesterol, low- and high-density lipoproteins and triglycerides) should be performed on all people with an increased risk of cardiac disease.
The risk of CAD is directly related to serum cholesterol levels. Serum cholesterol levels can be reduced by drugs, physical activity and by dietary changes, in particular a reduction in the consumption of saturated fat. It is estimated that 45% of deaths from CAD in men and 47% of deaths from CAD in women are due to a raised serum cholesterol level (in this case >5.2 mmol/L) and that 10% of deaths from CAD in the UK could be avoided if everyone in the population had a serum cholesterol level of <6.5 mmol/L.
Different guidelines give slightly different advice for managing high levels of serum cholesterol (hyperlipidaemia). The National Service Framework for coronary heart disease in England includes guidelines on the prevention of CAD in clinical practice and suggests a cholesterol target of <5.0 mmol/L for both primary and secondary prevention.
High-density lipoprotein cholesterol (HDL-cholesterol) is the fraction of cholesterol that removes cholesterol (via the liver) from the blood. Low levels of HDL-cholesterol are associated with an increased risk of CAD and a worse prognosis after a heart attack. Guidelines on HDL-cholesterol generally recommend treatment for those with concentrations <1.0 mmol/L. HDL increases with exercise, alcohol in moderation, not smoking and when TG is lowered.
A 1% reduction in cholesterol levels reduces risks of CAD by 2–3%. Hyperlipidaemia can be treated as follows:
Statins: 24–30% reduction in mortality in primary and secondary prevention will be achieved if a statin (pravastatin or simvastatin) is given. Up to 50% reduction is achieved if the dose of statin (e.g. atorvastatin) is titrated to achieve a target LDL of <2.6 mmol/L.
Fibrates result in a significant reduction in CAD events in diabetics and patients with high TG and low HDL.
Diet: the so-called Mediterranean diet (p. 198) has resulted in a 75% reduction in CAD events in post-myocardial infarction patients.
Angiographic studies have shown that lowering the serum cholesterol can slow the progression of coronary atherosclerosis, and can cause regression of disease. Large clinical trials have shown that lipid lowering, usually with a statin, can decrease total mortality and new coronary events, and reduce the need for revascularization. Management of hypercholesterolaemia is described in detail on page 1037.
Diabetes mellitus
Diabetes, an abnormal glucose tolerance or raised fasting glucose, is strongly associated with vascular disease.
Diabetes substantially increases the risk of CAD. Men with type 2 diabetes have a two- to four-fold greater annual risk of CAD, with an even higher (3–5-fold) risk in women with type 2 diabetes.
Diabetes not only increases the risk of CAD but also magnifies the effect of other risk factors for CAD such as raised cholesterol levels, raised blood pressure, smoking and obesity.
Other risk factors
Although there is general agreement on established cardiovascular risk factors, epidemiological research continues to identify or evaluate additional risk factors that contribute to the occurrence of atherosclerotic CVD and warrant further clarification.
Sedentary lifestyle
Lack of exercise is an independent risk factor for CAD equal to hypertension, hyperlipidaemia and smoking. Regular exercise probably protects against its development (see above).
Psychosocial wellbeing
Four different types of psychosocial factors have been found to be most consistently associated with an increased risk of CAD: work stress, lack of social support, depression (including anxiety) and personality (particularly hostility).
Alcohol
Moderate alcohol consumption (one or two drinks per day) is associated with a reduced risk of CAD. At high levels of intake – particularly in ‘binges’ – the risk of CAD is increased. It is currently advised that ‘regular consumption of between three and four units a day by men’ and ‘between two and three units a day by women of all ages will not lead to any significant health risk’.
Genetic factors
A number of genetic factors have been linked with coronary artery disease. The angiotensin-converting enzyme (ACE) gene contains an insertion/deletion (I/D) polymorphism, the DD genotype of which has been associated with a predisposition to CAD and myocardial infarction.
Lipoprotein(a)
High plasma Lp(a) concentrations are associated with CAD and, although probably not an independent risk factor, elevated plasma Lp(a) increases the CAD risk associated with more traditional risk factors.
Coagulation factors
Serum fibrinogen is strongly, consistently and independently related to CAD risk. The pathophysiological mechanism by which fibrinogen levels mediate coronary disease risk is related to its effect on the coagulation cascade, platelet aggregation, endothelial function and smooth muscle cell proliferation and migration.
High levels of coagulation factor VII are also a risk factor. Polymorphisms of the factor VII gene may increase the risk of myocardial infarction.
Homocysteine, an amino acid regulated by vitamins B12, B6 and folate, is another factor that has been associated with CAD and atherosclerosis (see p. 212). Homocysteinaemia is a major risk factor in the pathogenesis of CAD and a strong predictor of mortality in this group. Plasma levels of homocysteine are influenced by a variety of genetic and non-genetic factors. The mechanism associating hyperhomocysteinaemia with atherosclerosis is its adverse effect on vascular endothelium. Folic acid in low doses may ameliorate this process.
Prevention policy
The priorities for CVD prevention in clinical practice are:
Patients with established CAD, PVD and cerebrovascular atherosclerotic disease
Asymptomatic individuals who are at high risk of developing atherosclerotic disease because of multiple risk factors resulting in a 10-year risk of >5% now (or if extrapolated to age 60) for developing a fatal event, i.e. those with markedly raised levels of single risk factors:
FURTHER READING
Blaha MJ, Budoff MJ, DeFilippis AP et al. Associations between C-reactive protein, coronary artery calcium, and cardiovascular events: implications for the JUPITER population from MESA, a population-based cohort study. Lancet 2011; 378:684–692.
Schmermund A, Voigtländer T. Predictive ability of coronary artery calcium and CRP. Lancet 2011; 378:641–643.
Risk estimation for CVD prevention in the asymptomatic population
Patients with established CVD have declared themselves to be at high total risk of further vascular events. Therefore they require the most intense lifestyle intervention, and where appropriate drug therapies.
However, in the majority of asymptomatic, apparently healthy people, preventative actions should be guided in accordance with the total CVD risk level. Indeed, risk factor management decisions should usually not be based on considerations of a single modestly raised factor.
To evaluate candidates for the major cardiovascular events cost-effectively, multivariate risk profiles have been formulated; these facilitate targeting those at high risk for preventative measures.
The Joint British Societies, the European Society of Cardiology and the American Heart Association have emphasized the importance of these risk profiles for motivating as well as reassuring patients and in assisting in selecting therapy. They concluded that these scores direct healthcare professionals to look at the whole patient and to recognize the cumulative nature of risk factors (Fig. 14.61). However, not all practitioners agree with this approach (see p. 1038).
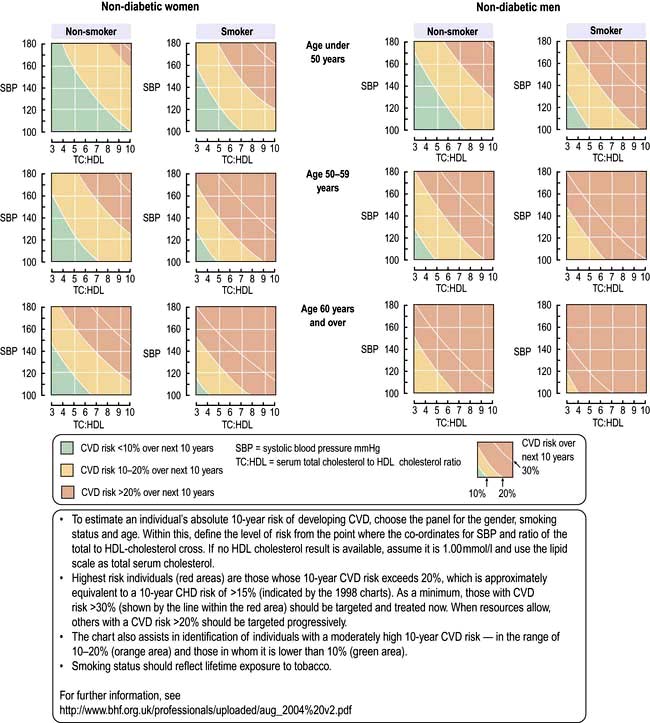
National Service Framework (NSF)
The UK’s NSF includes a nurse-led audited approach to reduce CAD by lowering saturated fat intake, increasing exercise and, most relevant, decreasing/stopping smoking. The hypertension treatment targets are 140/85 mmHg in patients at risk of or with established coronary artery disease and 130/80 mmHg in diabetics. The cholesterol target is either total cholesterol of <5.0 mmol/L (LDL-cholesterol <3 mmol/L) or a reduction of 30% (whichever is greater).
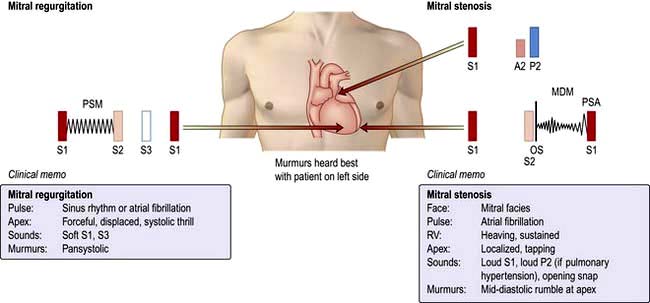
Angina
The diagnosis of angina (see also p. 675 and Table 14.27) is largely based on the clinical history. The chest pain is generally described as ‘heavy’, ‘tight’ or ‘gripping’. Typically, the pain is central/retrosternal and may radiate to the jaw and/or arms. Angina can range from a mild ache to a most severe pain that provokes sweating and fear. There may be associated breathlessness.
Table 14.27 Canadian cardiovascular society functional classification of angina
Class I |
No angina with ordinary activity. Angina with strenuous activity |
Class II |
Angina during ordinary activity, e.g. walking up hills, walking rapidly upstairs, with mild limitation of activities |
Class III |
Angina with low levels of activity, e.g. walking 50–100 yards on the flat, walking up one flight of stairs, with marked restriction of activities |
Class IV |
Angina at rest or with any level of exercise |
The Health Survey for England 2006 reported a prevalence of angina of 14.2% in men aged 65–74 years and 8.3% in women aged 65–74 years old. In the UK, an estimated 2 million people over the age of 35 years have had angina.
Classical or exertional angina pectoris is characterized by:
constricting discomfort in the front of the chest, arms, neck, jaw;
provoked by physical exertion, especially after meals and in cold, windy weather or by anger or excitement and
relieved (usually within minutes) with rest or glyceryl trinitrate. Occasionally, it disappears with continued exertion (‘walking through the pain’).
Typical angina has all three features, atypical angina two out of the three, and non-anginal chest pain one or less of these features.
Angina is stable when it is not a new symptom and when there is no change in the frequency or severity of attacks.
Unstable angina refers to angina of recent onset (<24 h) or a deterioration in previous stable angina with symptoms frequently occurring at rest, i.e. acute coronary syndrome (p. 730).
Refractory angina refers to patients with severe coronary disease in whom revascularization is not possible and angina is not controlled by medical therapy.
Variant (Prinzmetal’s) angina refers to an angina that occurs without provocation, usually at rest, as a result of coronary artery spasm. It occurs more frequently in women. Characteristically, there is ST segment elevation on the ECG during the pain. Specialist investigation using provocation tests (e.g. hyperventilation, cold-pressor testing or ergometrine challenge) may be required to establish the diagnosis.
Cardiac syndrome X refers to those patients with a good history of angina, a positive exercise test and angiographically normal coronary arteries. They form a heterogeneous group, and the syndrome is much more common in women than in men. Whilst they have a good prognosis, they are often highly symptomatic and can be difficult to treat. In women with this syndrome the myocardium shows an abnormal metabolic response to stress, consistent with the suggestion that the myocardial ischaemia results from abnormal dilator responses of the coronary microvasculature to stress. See Table 14.27 for the Canadian Cardiovascular Society Functional Classification of Angina.
Examination
There are usually no abnormal findings in angina, although occasionally a fourth heart sound may be heard. Signs to suggest anaemia, thyrotoxicosis or hyperlipidaemia (e.g. lipid arcus, xanthelasma, tendon xanthoma) should be sought. It is essential to exclude aortic stenosis (i.e. slow-rising carotid impulse and ejection systolic murmur radiating to the neck) as a possible cause for the angina. The blood pressure should be taken to identify co-existent hypertension.
Diagnosis and investigations for angina
Patients presenting with chest pain should have a 12-lead ECG performed to exclude an acute coronary syndrome. In many patients the ECG is normal between attacks although evidence of old myocardial infarction (e.g. pathological Q waves), left ventricular hypertrophy or left bundle branch block may be present. During an attack, transient ST depression, T wave inversion or other changes of the shape of the T wave may appear.
The diagnosis of stable angina can be made on clinical assessment alone OR by clinical assessment combined with anatomical (cardiac catheterization or CT coronary angiography) or functional imaging (SPECT, stress-echocardiography, stress-magnetic resonance imaging).
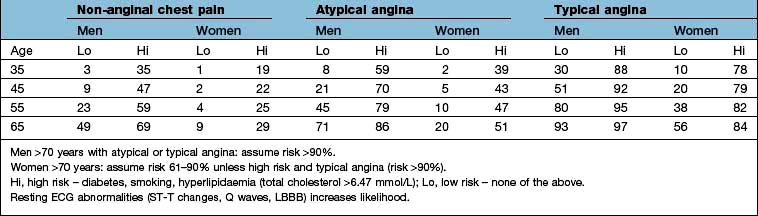
UK NICE guidance recommends assessing the likelihood of coronary artery disease in patients without known coronary artery disease who present with typical angina, atypical angina, or non-anginal chest pain (Table 14.28).
Patients with non-anginal chest pain (more likely if the pain is continuous, is unrelated to exertion, is exacerbated by respiration, is associated with dizziness, palpitations or difficulty in swallowing) should have alternate diagnoses considered and be investigated appropriately.
Patients with typical angina and a risk of disease of >90% do not need further diagnostic investigation and should be managed for stable angina.
Patients with typical or atypical angina and a risk of disease of 61–90% should have cardiac catheterization if appropriate.
Patients with typical or atypical angina and a risk of disease of 30–60% should be referred for functional testing (SPECT, stress-echocardiography, stress-magnetic resonance imaging).
Patients with typical or atypical angina and a risk of disease of 10–29% should be referred for CT coronary angiography. If CT calcium score is zero then angina is unlikely and other causes of chest pain should be sought (although young patients may have non-calcified plaque and CT angiography may be appropriate if symptomatic). If CT calcium score is 1–400, proceed to CT angiography but if the score is >400, invasive coronary angiography or functional imaging would be appropriate.
If stable angina cannot be diagnosed in patients with known coronary artery disease then functional assessment would be appropriate.
Table 14.28 Likelihood of coronary artery disease in relation to type of presentation, age and risk: data are percentage of people
NICE does not recommend exercise ECG as a diagnostic test in patients with chest pain symptoms although it has been present in the ESC guidelines from 2006. Using ST depression of <0.1 mV or 1 mm as a positive result, exercise ECG has a reported sensitivity of 67% and specificity of 72% for the detection of significant coronary disease in patients without prior myocardial infarction although interpretation of the findings is dependent on the prevalence of disease in the population and their presenting symptoms.
FURTHER READING
Gottlieb I, Miller JM, Arbab-Zadeh A et al. The absence of coronary calcification does not exclude obstructive coronary artery disease or the need for revascularization in patients referred for conventional coronary angiography. J Am Coll Cardiol 2010; 55:627–634.
Greenwood JD. Cardiovascular magnetic resonance and single photon computed tomography for the diagnosis of coronary artery disease (CE-MARC): a prospective Study. Lancet 2012; 379:453–460.
Skalidis El, Vardas PE. Guidelines on the management of stable angina pectoris. Eur Heart J 2006; 27(21):2606.
SIGNIFICANT WEBSITE
NICE clinical guideline 95, ‘Chest pain of recent onset’: http://www.nice.org.uk/nicemedia/live/12947/47938/47938.pdf
Management of stable angina
Patients should be informed as to the nature of their condition and reassured that the prognosis is good (annual mortality <2%). Underlying problems, such as anaemia or hyperthyroidism, should be treated. Management of co-existent conditions, such as diabetes and hypertension, should be optimized. Risk factors should be evaluated and steps made to correct them where possible; e.g. smoking must be stopped, hypercholesterolaemia should be identified and treated (see below), weight loss, where appropriate, and regular exercise should be encouraged. The stable angina algorithm in Figure 14.62 should be used to guide initial patient management, as well as Table 14.29.
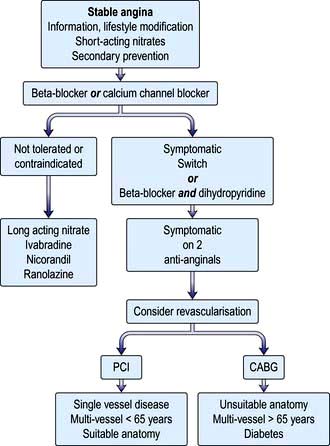
Figure 14.62 Algorithm for management of patients with stable angina. CABG, coronary artery bypass grafting; PCI, percutaneous coronary intervention.
(From NICE draft guideline: http://www.nice.org.uk/nicemedia/live/11878/52141/52141.pdf.)
Table 14.29 Pharmacological therapy in stable angina
| Drug | Dose | Indications/mechanism of action/cautions |
|---|---|---|
Vasodilator |
||
Glyceryl trinitrate |
0.3–1.0 mg sublingual |
Prophylaxis and treatment of angina – rapid onset |
Isosorbide mononitrate |
10–60 mg ×2 daily (slow release preparations are available) |
Prophylaxis of angina |
Beta-blocker |
||
Atenolol |
25–100 mg daily |
Inhibit beta-adrenoceptors, reduce heart rate and BP, reduce myocardial oxygen consumption |
Bisoprolol |
2.5–10 mg daily |
|
Metoprolol |
25–100 mg ×2 or 3 daily |
|
Calcium channel blockers |
||
Verapamil (phenylalkylamines) |
80–120 mg ×3 daily (or 240–480 mg daily slow release) |
Inhibit calcium channels in myocardium, cardiac conductive tissue and vascular smooth muscle |
Diltiazem (benzothiapines) |
60–120 mg ×3 daily (longer acting preparations are available) |
|
Amlodipine (dihydropyridines) |
5–10 mg daily |
|
Second-line anti-anginal drugs |
||
Ivabradine |
2.5–7.5 mg ×2 daily |
Inhibits the pacemaker If current in the SA node |
Nicorandil |
5–30 mg ×2 daily |
Activates ATP-sensitive potassium channels and has nitrate properties – peripheral and coronary vasodilatation |
Ranolazine |
375–750 mg ×2 daily |
May inhibit late sodium channels in cardiac cells |
Secondary prevention |
||
Aspirin |
75 mg daily |
Antiplatelet |
Angiotensin-converting enzyme inhibitor |
Variable |
Indicated if treating other condition, e.g. hypertension, heart failure, chronic kidney disease |
Statins |
Variable |
Use to reduce total cholesterol to below 4 mmol/L and LDL cholesterol to below 2 mmol/L |
Low-dose aspirin is indicated. Symptomatic treatment should be started with the vasodilator sublingual or buccal nitrate to relieve acute episodes. Prophylaxis should be started with either a beta-adrenoceptor antagonist (e.g. atenolol) OR calcium channel receptor antagonist (e.g. diltiazem). If patients remain symptomatic then a beta-blocker can be combined with a dihydropyridine calcium channel receptor antagonist (e.g. amlodipine). Patients intolerant of beta-blockers and/or calcium channel blockers are treated with long-acting nitrates (e.g. isosorbide mononitrate) or If current inhibitor ivabradine or potassium channel activator nicorandil or sodium channel inhibitor ranolazine.
The Courage Trial published in April 2007 randomized patients with stable but significant coronary artery disease and inducible ischaemia to percutaneous coronary intervention with stenting (n=1149) or optimal medical therapy (n=1138). The primary composite outcome (all-cause death and non-fatal myocardial infarction) occurred in 211 (18.3%) of the PCI patients and 202 (17.8%) of the medically treated patients. This supports an initial strategy of optimal medical therapy in patients with stable angina symptoms although revascularization should be used in patients who remain symptomatic despite two anti-anginals.
Revascularization
Percutaneous coronary intervention

Percutaneous coronary intervention (PCI) is the process of dilating a coronary artery stenosis using an inflatable balloon and metallic stent introduced into the arterial circulation via the femoral, radial or brachial artery (Fig. 14.63). A discrete, soft lesion in a straight vessel without involving a bifurcation has the best outcome. Unfavourable lesions are occluded vessels, stenoses that are calcified, tortuous, long, or involve a bifurcation. Complications of the procedure include bleeding, haematoma, dissection and pseudoaneurysm from the arterial puncture site although the use of the radial artery may reduce the risks. Serious complications include acute myocardial infarction (2%), stroke (0.4%) and death (1%). Thrombotic complications are reduced with the use of heparin or the direct thrombin inhibitor bivalirudin together with the antiplatelet agents, aspirin and the thienopyridine clopidogrel. In very high-risk acute coronary syndrome or diabetic patients the antiplatelet GPIIb/IIIa antagonists (tirofiban, eptifibatide and abciximab) are also used. Reductions in the need for repeat revascularization have been seen with the introduction of coated stents lined with substances that reduce coronary artery restenosis. The Cypher stent contains sirolimus, which is an immunosuppressant agent that reduces cellular proliferation; everolimus is a derivative of sirolimus which is used in the Xience V stent. The Taxus stent contains paclitaxel, which is a mitotic inhibitor drug that inhibits neointima formation. Some concerns have been raised about late-stent thrombosis (>6 months post-insertion) in patients with drug-eluting stents leading to acute myocardial infarction and frequently death. It has been suggested that inadequate endothelialization of the stent leads to exposure of thrombus stimulating surface when the patient discontinues clopidogrel therapy, leading to recommendations that patients take prolonged dual therapy (aspirin and clopidogrel) and avoid discontinuing therapy within 6–12 months of implantation.
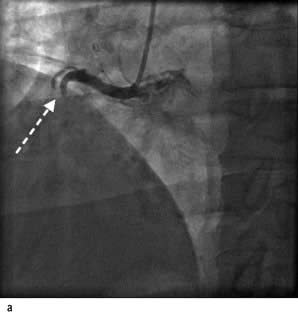
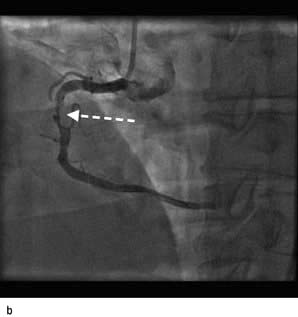
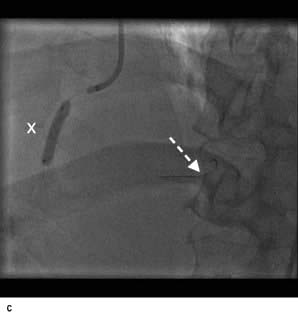
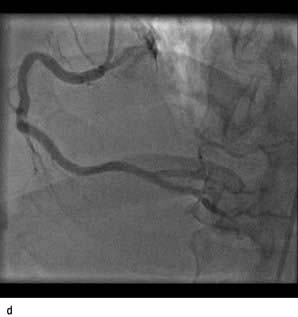
Figure 14.63 Percutaneous transluminal coronary angioplasty (PTCA). (a) Coronary angiography demonstrates an occluded right coronary artery (arrow). (b) A soft wire passed through the guide catheter reopens the artery but a severe stenosis remains (arrow). (c) A balloon (X) is inflated to dilate the stenosis. The soft guide-wire can be seen in the distal posterior descending artery (arrow). (d) The right coronary artery has now been successfully reopened with good antegrade flow.
FURTHER READING
Shahian DM et al. Percutaneous coronary interventions without on-site cardiac surgical backup. N Engl J Med 2012; 366:1814–1823.
Silber S, Windecker S, Vranckx P et al., on behalf of the RESOLUTE All Comers investigators. Unrestricted randomised use of two new generation drug-eluting coronary stents: 2-year patient-related versus stent-related outcomes from the RESOLUTE All Comers trial. Lancet 2011; 377:1241–1247.
Coronary artery bypass grafting
With coronary artery bypass grafting (CABG) autologous veins or arteries are anastomosed to the ascending aorta and to the native coronary arteries distal to the area of stenosis (Fig. 14.64). Improved graft survival can be obtained with in situ internal mammary and gastroepiploic arteries grafted onto the stenosed coronary artery. Three major randomized controlled trials compared CABG with medical therapy: the Coronary Artery Surgery Study (CASS); the Veterans Administration (VA) Cooperative Study and the European Coronary Surgery Study (ECSS). A meta-analysis has been performed that demonstrated that compared to medical therapy, CABG significantly improved angina symptoms, exercise capacity and reduced the need for antianginal therapy. Operative mortality is well below 1% in patients with normal left ventricular function. Perioperative strokes occur in up to 2% of cases, and more subtle neurological deficits are common. Off-pump coronary surgery is now performed; results show that it is as safe as on-pump surgery and causes less myocardial damage, but the graft patency rate is lower. Minimally invasive operative procedures for bypass grafting (‘MIDCAB’) are being developed, including laparoscopic approaches, and may be of use in certain subgroups of patients (e.g. previous CABG and those with co-existent medical conditions which would increase the operative risks of ‘full’ CABG).
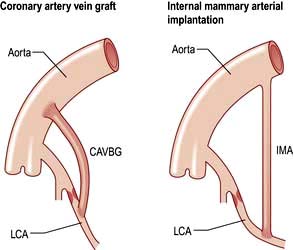
CABG versus PCI
Comparative trials between CABG and PCI have now been performed. All demonstrate a higher need for repeat revascularization with PCI than with CAGB. In the ERACI II study, PCI patients had fewer major adverse events initially and better 18-month survival than the CABG group. The SoS Trial reported a 2-year incidence of death or Q wave myocardial infarction of 9% in PCI versus 10% in CABG patients but fewer deaths in with CAGB (2% vs 5%). The SYNTAX study compared PCI with drug-eluting stents with CABG in patients with three-vessel disease and/or left main stem disease. The primary end-point (all cause death, stroke, myocardial infarction or repeat revascularization) occurred in more PCI patients. Although the draft guidance of stable angina from NICE recommends PCI for young patients with single or multi-vessel disease (and no diabetes), the European Society of Cardiology has recommended PCI ONLY in cases of single or double vessel disease NOT involving the proximal left anterior descending artery.
FURTHER READING
Weintraub WS et al. Comparative effectiveness of revascularization strategies. N Engl J Med 2012; 366:1467–1478.
Wijns W, Kolh P, Danchin N et al.; The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Guidelines on myocardial revascularization. Eur Heart J 2010; 31:2501–2555.
Acute coronary syndromes
Acute coronary syndromes (ACS) include:
The difference between UA and NSTEMI is that in the latter there is occluding thrombus, which leads to myocardial necrosis and a rise in serum troponins or CK-MB. Myocardial infarction (MI) occurs when cardiac myocytes die due to myocardial ischaemia, and can be diagnosed on the basis of appropriate clinical history, 12-lead ECG and elevated biochemical markers – troponin I and T, CK-MB. There are three types of MIs:
Type 1 – spontaneous MI with ischaemia due to a primary coronary event, e.g. plaque erosion/rupture, fissuring or dissection
Type 2 – MI secondary to ischaemia due to increased oxygen demand or decreased supply, such as coronary spasm, coronary embolism, anaemia, arrhythmias, hypertension, or hypotension
Type 3,4,5 – diagnosis of MI in sudden cardiac death, after percutaneous coronary intervention (PCI) and after coronary artery bypass graft (CABG), respectively.
Pathophysiology
The common mechanism to all ACS is rupture or erosion of the fibrous cap of a coronary artery plaque. This leads to platelet aggregation and adhesion, localized thrombosis, vasoconstriction and distal thrombus embolization. The presence of a rich lipid pool within the plaque and a thin fibrous cap are associated with an increased risk of rupture. Thrombus formation and the vasoconstriction produced by platelet release of serotonin and thromboxane A2, results in myocardial ischaemia due to reduction of coronary blood flow.
FURTHER READING
Bassand JP, Hamm CW, Ardissino D et al.; Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
Eagle KA, Lim MJ, Dabbous OH et al, for the GRACE investigators. A validated prediction model for all forms of acute coronary syndrome. Estimating the risk of 6-month postdischarge death in an international registry. JAMA 2004; 291:2727–2733.
Diagnosis
Clinical presentation
Patients with an ACS may complain of a new onset of chest pain, chest pain at rest, or a deterioration of pre-existing angina. However, some patients present with atypical features including indigestion, pleuritic chest pain or dyspnoea. Physical examination can detect alternative diagnoses such as aortic dissection, pulmonary embolism or peptic ulceration. In addition it can also detect adverse clinical signs such as hypotension, basal crackles, fourth heart sounds and cardiac murmurs.
Electrocardiogram
Although the 12-lead ECG may be normal in patients with an ACS, ST depression and T wave inversion are highly suggestive for an ACS, particularly if associated with anginal chest pain. The ECG should be repeated when the patient is in pain, and continuous ST-segment monitoring is recommended. With a STEMI, complete occlusion of a coronary vessel will result in persistent ST-elevation or left bundle branch block pattern, although transient ST elevation is seen with coronary vasospasm or Prinzmetal’s angina.
Biochemical markers
The cardiac troponin complex is made up of three distinct proteins (I, T and C) that are situated with tropomyosin on the thin actin filament that forms the skeleton of the cardiac myofilament. Troponin T attaches the complex to tropomyosin, troponin C binds calcium during excitation-contraction coupling, and troponin I inhibits the myosin binding site on the actin. The cardiac troponins are not detectable in normal people and so monoclonal antibody tests to cardiac-specific troponin I and cardiac-specific troponin T are highly sensitive markers of myocyte necrosis. If the initial troponin assay is negative, then it should be repeated 6–12 h after admission. The troponin assay has prognostic information, i.e. a high serum troponin level has an increased mortality risk in ACS (Box 14.1), and defines which patients may benefit from aggressive medical therapy and early coronary revascularization.
The measurement of the creatine-kinase-MB level was, until recently, the standard marker for myocyte death used in ACS. However, the presence of low levels of CK-MB in the serum of normal individuals and in patients with significant skeletal muscle damage, has limited its accuracy. It can be used to determine reinfarction as levels drop back to normal after 36–72 hours.
Myoglobin may be useful for a rapid diagnosis of an ACS as the levels become elevated very early in the time course of an MI, but because of the presence of myoglobin in skeletal muscle the test has poor specificity for ACS.
 Box 14.1
Box 14.1
Relationship between troponin I and risk of death in patients with acute coronary syndrome
| Serum troponin levels (ng/mL) | Mortality at 42 days (% of patients) |
|---|---|
0 to <0.4 |
1.0 |
0.4 to<1.0 |
1.7 |
1.0 to <2.0 |
3.4 |
2.0 to <5.0 |
3.7 |
5.0 to <9.0 |
6.0 |
>9.0 |
7.5 |
(Adapted from Antman et al. Cardiac-specific troponin I levels to predict the risk of mortality in patients with acute coronary syndromes. New England Journal of Medicine 1996; 335:1342–1349.)
NSTEMI and unstable angina
Risk stratification in NSTEMI and unstable angina
Initial risk in ACS is determined by complications of the acute thrombosis. This may produce recurrent myocardial ischaemia, marked ST depression, dynamic ST changes, a raised troponin level and be demonstrated with coronary angiography.
Long-term risks defined by clinical risk factors; age, prior myocardial infarction or bypass surgery, diabetes or heart failure. Biological markers, such as C-reactive protein, fibrinogen, brain natriuretic peptide, modified albumin and serum creatinine, can be used to further stratify patient risk. Left ventricular dysfunction and the presence of left main or triple vessel disease significantly increase the future cardiovascular risk. Both the Thrombolysis In Myocardial Infarction (TIMI) score and the Global Registry of Acute Coronary Events (GRACE) prediction score can be used in patients with ACS to define risk. TIMI is shown in Table 14.30. The GRACE score is based on age, heart rate, systolic blood pressure, serum creatinine and the Killip score.
Table 14.30 The TIMI risk score in acute coronary syndrome (NSTEMI/UA)
| Risk factor | Score |
|---|---|
Age >65 |
1 |
More than three coronary artery disease risk factors – hypertension, hyperlipidaemia, family history, diabetes, smoking |
1 |
Known coronary artery disease (coronary angiography stenosis >50%) |
1 |
Aspirin use in the last 7 days |
1 |
Severe angina (more than two episodes of rest pain in 24 h) |
1 |
ST deviation on ECG (horizontal ST depression or transient ST elevation >1 mm) |
1 |
Elevated cardiac markers (CK-MB or troponin) |
1 |
| Total score | Rate of death/MI in 14 days (%) | Rate of death/Ml/urgent revascularization (%) |
|---|---|---|
0–1 |
3 |
4.75 |
2 |
3 |
8.3 |
3 |
5 |
13.2 |
4 |
7 |
19.9 |
5 |
12 |
26.2 |
6–7 |
19 |
40.9 |
Investigation and treatment of NSTEMI and UA
All patients require immediate management of their chest pain as outlined on page 738 and in Table 14.31.
Table 14.31 Pharmacological therapy in acute coronary syndrome
| Drug | Notes | |
|---|---|---|
Myocardial oxygenation |
|
|
Oxygen |
35–50% |
Check ABG in severe COPD |
Antiplatelet |
|
|
Aspirin |
150–300 mg chewable or soluble aspirin, then 75–10 mg orally daily |
Caution if active peptic ulceration |
Clopidogrel |
300 mg orally loading dose, then 75 mg orally daily |
Caution: increased risk of bleeding, avoid if CABG planned |
Prasugrel |
60 mg oral loading dose, then 10 mg orally daily (5 mg daily if <60 kg or >75 years old) |
|
Ticagrelor |
Initially 180 mg, then 90 mg ×2 daily |
Risk of bleeding |
Antithrombin |
|
|
Heparin |
5000 units i.v. bolus, then 0.25 units/kg per hour |
Measure anticoagulant effect with APTT at 6 h |
Low-molecular-weight heparins, e.g. enoxaparin |
1 mg/kg s.c. ×2 daily |
|
Bivalirudin |
750 µg/kg i.v. bolus, then 1.75 mg/kg per hour for 4 h post PCI |
|
Fondaparinux |
2.5 mg SC daily, for up to 8 days |
|
Rivaroxaban |
Oral 2.5–10 mg daily |
Risk of bleeding |
Glycoprotein IIB/IIIA inhibitors |
|
|
Abciximab |
0.25 mg/kg i.v. bolus, then 0.125 µg/kg per min up to 10 µg/min i.v. ×12 h |
Indicated if coronary intervention likely within 24 h |
Eptifibatide |
180 µg/kg i.v. bolus, then 2 µg/kg per min ×72 h |
Indicated in high-risk patients managed without coronary intervention or during PCI |
Tirofiban |
0.4 µg/kg per min for 30 min, then 0.1 µg/kg per min ×48–108 h |
Indicated in high-risk patients managed without coronary intervention or during PCI |
Analgesia |
|
|
Diamorphine or morphine |
2.5–5.0 mg i.v. |
Prescribe with antiemetic, e.g. metoclopramide 10 mg i.v. |
Myocardial energy consumption |
|
|
Atenolol |
5 mg i.v. repeated after 15 min, then 25–50 mg orally daily |
Avoid in asthma, heart failure, hypotension, bradyarrhythmias |
Metoprolol |
5 mg i.v. repeated to a maximum of 15 mg, then 25–50 mg orally ×2 daily |
Avoid in asthma, heart failure, hypotension, bradyarrhythmias |
Coronary vasodilation |
|
|
Glyceryl trinitrate |
2–10 mg/h i.v./buccal/sublingual |
Maintain systolic BP >90 mmHg |
Plaque stabilization/ventricular remodeling |
|
|
HMG-CoA reductase inhibitors (statins) |
|
Combine with dietary advice and modification |
Simvastatin |
20–40 mg orally |
|
Pravastatin |
20–40 mg orally |
|
Atorvastatin |
80 mg orally |
|
ACE inhibitors |
|
Monitor renal function |
Ramipril |
2.5–10 mg orally |
|
Lisinopril |
5–10 mg orally |
High-risk patients for progression to myocardial infarction or death require urgent coronary angiography. These patients include those with persistent or recurrent angina with ST changes ≥2 mm or deep negative T wave changes, clinical signs of heart failure or haemodynamic instability, life-threatening arrhythmias (VF, VT).
Patients with immediate or high-risk TIMI or GRACE scores, elevated troponins, dynamic ST or T wave changes, diabetes mellitus, renal dysfunction, reduced left ventricular function, early post-infarction angina, previous myocardial infarction, PCI within 6 months, or previous CABG, should have early (<72 h) coronary angiography and interventions.
Low-risk patients can be managed with oral aspirin, clopidogrel, beta-blockers and nitrates. These include patients with no recurrence of chest pain during observation, no signs of heart failure, normal ECG or minor T wave changes on arrival and at 6–12 h, normal troponins on the initial assays and at 6–12 h post admission. An exercise test should be performed – a negative result has a good prognosis and an early positive test should direct the patient to an invasive strategy. If the patient is unable to exercise satisfactorily, or if the baseline ECG is abnormal (e.g. left ventricular hypertrophy or LBBB), then dobutamine stress echocardiography or myocardial perfusion scintigraphy are recommended. These tests are often used as the first line investigation.
Antiplatelet agents
The platelet is a key part of the thrombosis cascade involved in ACS. Rupture of the atheromatous plaque exposes the circulating platelets to ADP (adenosine diphosphate), thromboxane A2 (TxA2), epinephrine (adrenaline), thrombin and collagen tissue factor. This causes platelet activation, with thrombin as an especially potent stimulant of such activity. Platelet activation stimulates the expression of glycoprotein (GP) IIb/IIIa receptors on the platelet surface. These receptors bridge fibrinogen between adjacent platelets, causing platelet aggregates (Fig. 8.41).
Aspirin blocks the formation of thromboxane A2 and so prevents platelet aggregation. In ACS patients, 75–150 mg aspirin reduced the relative risk of death or myocardial infarction by about 35–50%. Ticagrelor, a nucleoside analogue, is used in combination with aspirin for the acute coronary syndrome.
Clopidrogrel is a thienopyridine that inhibits ADP-dependent activation of the GPIIb/IIIa complex that allows platelet aggregates to form. In the CURE study of 12 562 ACS patients, 9 months of 75 mg clopidogrel reduced the primary end-point of cardiovascular death, myocardial infarction, or stroke from 11.4% to 9.3% (p<0.0001), compared with placebo. However, clopidogrel is a pro-drug requiring conversion by hepatic cytochrome P450 enzymes to an active moiety that binds irreversibly to the P2Y12 receptor on platelet membranes and inhibits the ADP-dependent pathway of platelet activation. Proton-pump inhibitors, e.g. omeprazole, and genetic variation in the cytochrome P450 enzymes may theoretically reduce the effectiveness of clopidrogel. This has not been a problem in clinical practice. The TRITON-TIMI 38 study found that the thienopyridine prasugrel reduced ischemic events (12.1% with clopidogrel and 9.9% with prasugrel) but increased the risk of major bleeding (1.8% with clopidogrel and 2.4% with prasugrel).
Activated GP (glycoprotein) IIb/IIIa receptors on platelets bind to fibrinogen initiating platelet aggregation. Receptor antagonists have been developed that are powerful inhibitors of platelet aggregation. Abciximab is a monoclonal antibody that binds tightly and has a long half-life. Eptifibatide is a cyclic peptide that selectively inhibits GPIIb/IIIa receptors, but has a short half-life and wears off in 2–4 h. Tirofiban is a small non-peptide that rapidly blocks the GPIIb/IIIa receptors and is reversible in 4–6 h.
In the GUSTO-IV ACS study of 7800 patients, abciximab was administered but coronary intervention discouraged. At 30 days, 8.2% of abciximab patients and 8.0 % of placebo patients had reached the composite end-point of death or myocardial infarction. In the PRISM study of 3232 patients with angina, tirofiban reduced the 30-day death or myocardial infarction rate from 7.1% with placebo to 5.8%. Troponin-positive patients with diabetes scheduled to have coronary intervention benefit most from GPIIb/IIIa receptor antagonists.
Antithrombins
In ACS patients off aspirin, unfractionated heparin (UFH) produces a lower rate of refractory angina/myocardial infarction and death than placebo, and when used with aspirin reduces death and myocardial infarction from 10.3% to 7.9%. However, because of poor bioavailability and variable effects, frequent monitoring of APTT is necessary to ensure therapeutic levels. Low-molecular-weight heparins and in particular enoxaparin appear superior to UFH and can be given subcutaneously twice daily. Bivalirudin is a direct thrombin inhibitor that reversibly binds to thrombin and inhibits clot-bound thrombin. In the ACUITY trial, bivalirudin appeared as effective as heparin plus GPIIb/IIIa inhibitors in reducing ischaemic events in patients pretreated with a thienopyridine and undergoing diagnostic angiography or percutaneous intervention, but with less bleeding. Fondaparinux is a synthetic pentasaccharide that selectively binds to antithrombin, which inactivates factor Xa resulting in a strong inhibition of thrombin generation and clot formation. It does not inactivate thrombin and has no effect on platelets. The OASIS-5 trial evaluated the efficacy and safety of fondaparinux and enoxaparin in 20 078 high-risk patients with unstable angina or myocardial infarction without ST-segment elevation. Thrombotic endpoints were similar with both agents but fondaparinux was associated with less bleeding end-points at 2.2% compared with 4.1% with enoxaparin patients.
Rivaroxaban, a factor Xa inhibitor, is effective in reducing the risk of further cardiac events but with a risk of bleeding.
FURTHER READING
Mega JL et al. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 2012; 366:9–19.
Yusuf S, Mehta SR, Chrolavicius S et al.; Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
Anti-ischaemia agents
In patients with no contraindications (asthma, AV-block, acute pulmonary oedema), beta-blockers are administered intravenously or orally, to reduce myocardial ischaemia by blocking circulating catecholamines. This will reduce the heart rate and blood pressure, reducing myocardial oxygen consumption. The dose can be titrated to produce a resting heart rate of 50–60 b.p.m. In patients with ongoing angina, nitrates should be given either sublingually or intravenously. They effectively reduce preload and produce coronary vasodilation. However, tolerance can become a problem and patients should be weaned off intravenous administration if symptoms resolve.
Plaque stabilization/remodelling
HMG-CoA reductase inhibitor drugs (statins) and ACE inhibitors are routinely administered to patients with ACS. These agents may produce plaque stabilization, improve vascular and myocardial remodelling, and reduce future cardiovascular events. Starting the drugs whilst the patient is still in hospital increases the likelihood of patients receiving secondary drug therapy.
Coronary intervention
Coronary revascularization is recommended in high-risk patients with ACS. Coronary stenting may stabilize the disrupted coronary plaque; in the BENESTENT II trial it was demonstrated that stenting was superior to PTCA in reducing angiographic restenosis rates. Subgroup analysis of patients with unstable angina in the EPIC, EPILOG and CAPTURE trials confirmed the benefit of GPIIb/IIIa inhibitors at reducing the complication rate during PCI. The PCI-CURE study demonstrated that pretreatment with clopidogrel reduces the rate of cardiovascular death and MI. The current rate of CABG in ACS is low (5.4%). The mortality rates with CABG are greater in the high-risk group patients, particularly with a recent myocardial infarction. Single vessel lesions are usually treated with PCI, unless the anatomy is unfavourable. Conversely in patients with left main stem or triple vessel disease with impaired left ventricular function are best managed with surgery. Two studies have compared a conservative versus an invasive strategy in the modern era. In the FRISC-II study 2457 high-risk ACS patients were randomized to PTCA or CABG at 4 and 8 days, respectively, or a conservative approach with intervention only for severe angina. Revascularization within 10 days was performed in 71% of the invasive arm versus 9% of the conservative arm. After 1 year, there was a significant reduction in total mortality (2.2% vs 3.9%) in the invasive arm, as well a significant reduction in MI (8.6% vs 11.6%). In addition, the rate of angina or readmission was reduced by 50%. In the TACTICS study of 2220 high-risk ACS patients, similar findings were obtained with the rate of death or MI reduced from 9.5% to 7.3% by an invasive strategy. Patients with a troponin T >0.01 ng/mL obtained benefit, but not those who were troponin T negative.
ST elevation myocardial infarction (STEMI)
Myocardial infarction occurs when cardiac myocytes die due to prolonged myocardial ischaemia. The diagnosis can be made in patients with an appropriate clinical history together with findings from repeated 12-lead ECGs and elevated biochemical markers – troponin I and T, CK-MB.
Pathophysiology
Rupture or erosion of a vulnerable coronary artery plaque can produce prolonged occlusion of a coronary artery leading to myocardial necrosis within 15–30 minutes. The subendocardial myocardium is initially affected but with continued ischaemia the infarct zone extends through to the subepicardial myocardium, producing a transmural Q wave myocardial infarction. Early reperfusion may salvage regions of the myocardium, reducing future mortality and morbidity.
The 1-month mortality in patients with a myocardial infarction may be as high as 50% in the community, with 50% of deaths occurring in the first 2 hours of the event. In the pre-thrombolytic era the in-hospital mortality rate was nearly 20% but with modern therapy it may be as low as 6–7% at 1 month. Several risk factors can be identified that predict death rate at 30 days (TIMI STEMI score; Table 14.32).
Table 14.32 TIMI risk score in ST elevation myocardial infarction (STEMI)
Risk factor |
Score |
Age >65 |
2 |
Age >75 |
3 |
History of angina |
1 |
History of hypertension |
1 |
History of diabetes |
1 |
Systolic BP <100 |
3 |
Heart rate >100 |
2 |
Killip II–IV |
2 |
Weight >67 kg |
1 |
Anterior MI or LBBB |
1 |
Delay to treatment >4 h |
1 |
Total score |
Risk of death at 30 days |
0 |
0.8 |
1 |
1.6 |
2 |
2.2 |
3 |
4.4 |
4 |
7.3 |
5 |
12.4 |
6 |
16.1 |
7 |
23.4 |
8 |
26.8 |
9–16 |
35.9 |
Diagnosis
Symptoms and signs
Any patient presenting with severe chest pain lasting more than 20 minutes may be suffering from a myocardial infarction. The pain does not usually respond to sublingual GTN, and opiate analgesia is required. The pain may radiate to the left arm, neck or jaw. However, in some patients, particularly elderly or diabetic patients, the symptoms may be atypical and include dyspnoea, fatigue, pre-syncope or syncope. Autonomic symptoms are common and on examination the patient is pale and clammy, with marked sweating. In addition, the pulse is thready with significant hypotension, bradycardia or tachycardia.
Electrocardiography
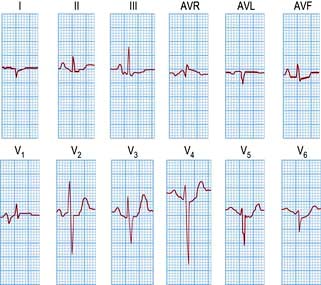
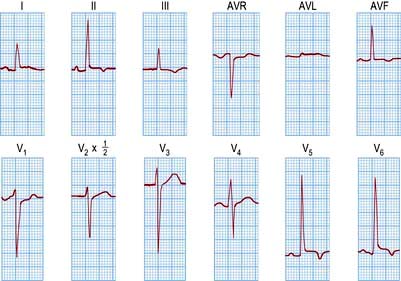
An ECG in patients with chest pain should be performed on admission to A&E. The baseline ECG is rarely normal, but if so should be repeated every 15 minutes, while the patient remains in pain. Continuous cardiac monitoring is required because of the high likelihood of significant cardiac arrhythmias. ECG changes (Table 14.33) are usually confined to the ECG leads that ‘face’ the infarction. The presence of new ST elevation (due to opening of the K+ channels) ≥0.2 mV at the J-point in leads V1–V3, and ≥0.1 mV in other leads, suggests anterior MI (Fig. 14.65). An inferior wall MI is diagnosed when ST elevation is seen in leads II, III and AVF (Fig. 14.66). Lateral MI produces changes in leads I, AVL and V5/V6. In patients with a posterior MI, there may be ST depression in leads V1–V3 with a dominant R wave, and ST elevation in lead V5/V6. New LBBB or presumed new LBBB is compatible with coronary artery occlusion requiring urgent reperfusion therapy. The evolution of the ECG during the course of STEMI is illustrated in Figure 14.67.
Table 14.33 Typical ECG changes in myocardial infarction (STEMI)
| Infarct site | Leads showing ST elevation |
|---|---|
Anterior: |
|
Small |
V3–V4 |
Extensive |
V2–V5 |
Anteroseptal |
V1–V3 |
Anterolateral |
V4–V6, I, AVL |
Lateral |
I, AVL |
Inferior |
II, III, AVF |
Posterior |
V1, V2 (reciprocal) |
Subendocardial |
Any lead |
Right ventricle |
VR4 |
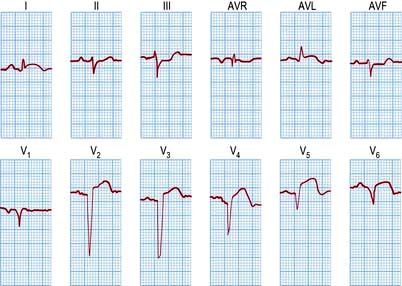
Figure 14.65 An acute anterolateral myocardial infarction shown by a 12-lead ECG. Note the ST segment elevation in leads I, AVL and V2–V6. The T wave is inverted in leads I, AVL and V3–V6. Pathological Q waves are seen in leads V2–V6.
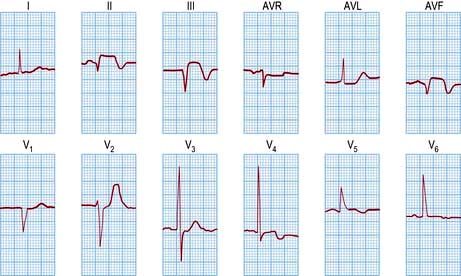
Figure 14.66 An acute inferior wall myocardial infarction shown by a 12-lead ECG. Note the raised ST segment and Q waves in the inferior leads (II, III and AVF). The additional T wave inversion in V4 and V5 probably represents anterior wall ischaemia.
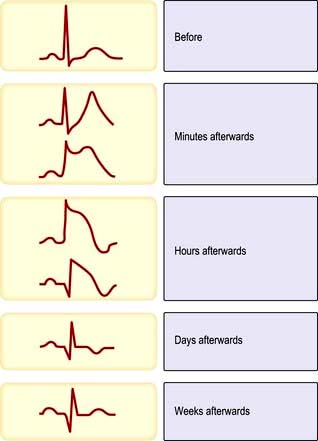
Figure 14.67 Electrocardiographic evolution of myocardial infarction (STEMI). After the first few minutes the T waves become tall, pointed and upright and there is ST segment elevation. After the first few hours the T waves invert, the R wave voltage is decreased and Q waves develop. After a few days the ST segment returns to normal. After weeks or months the T wave may return to upright but the Q wave remains.
Investigations
Blood samples should be taken for cardiac troponin I or T levels, although treatment should not be deferred until the results are available. Full blood count, serum electrolytes, glucose and lipid profile should be obtained. Transthoracic echocardiography (TTE) may be helpful to confirm a myocardial infarction, as wall-motion abnormalities are detectable early in STEMI. TTE may detect alternative diagnoses such as aortic dissection, pericarditis or pulmonary embolism.
Early medical management
Accident and emergency
Rapid triage for chest pain (Note: time is muscle):
Aspirin 150–300 mg chewed and clopidogrel 300 mg oral gel
Sublingual glyceryl trinitrate 0.3–1 mg. Repeat
Oxygen – nasal cannula 2–4 L/min (Fig. 16.21) if hypoxia is present
Brief history/risk factors. Examination
Intravenous access + blood for markers (plus FBC, biochemistry, lipids, glucose)
Intravenous opiate, e.g. diamorphine (or morphine) 2.5–5 mg + antiemetic, e.g. metoclopramide 10 mg
Beta-blocker (if no contraindication) for ongoing chest pain, hypertension, tachycardia
If primary PCI available (see p. 738), give GP IIb/IIIa inhibitor. Alternatively, give thrombolysis (see below).
Pre-hospital treatment, including thrombolysis, can be given by trained healthcare professionals under strict guidelines.
Percutaneous coronary intervention (PCI)
PCI performed within 90 minutes is the preferred reperfusion therapy in interventional cardiology centres that have the expertise available. In the PAMI (Primary Angioplasty in Myocardial Infarction) trial, patients with a myocardial infarction who presented within 12 hours of the onset of STEMI were randomized to primary PTCA or t-PA followed by conservative care. At 2-year follow-up the primary PTCA group had less recurrent ischaemia, lower re-intervention rates and reduced hospital readmission rates. Primary PTCA produced a combined end-point of death or re-infarction of 14.9% compared to 23% for t-PA. PCI with thrombus aspiration has recently been shown to result in better reperfusion and clinical outcomes.
The DANAMI 2 study investigated if rapid transfer of patients with STEMI for primary angioplasty in an interventional centre was superior to thrombolysis. Patients within 12 hours of a high-risk STEMI (>4 mm elevation) received front-loaded t-PA, primary angioplasty at a local centre, or primary angioplasty at an interventional centre after transfer. Primary PCI significantly reduced the death rate in both local and transferred patients as compared to thrombolytic therapy (8.0% vs 13.7%). The majority of the benefits with primary PCI are obtained by a reduction in recurrent myocardial infarction.
Coronary stenting in primary PCI reduces the need for repeat target vessel revascularization but did not appear to reduce mortality rates. However, one study using drug-eluting stents showed a decreased 2-year mortality rate.
The use of abciximab in STEMI patients undergoing primary angioplasty reduces immediate outcome (death, myocardial infarction, urgent revascularization), but this benefit is minimal by 6 months.
PCI following thrombolysis was initially discouraged but a trial has suggested that it is safe and improves the 1-year clinical outcome. Randomized trials have compared a strategy of thrombolysis (in hospitals without PCI capability) versus transfer to a PCI centre and have demonstrated a significant reduction in the combined end-point of death, reinfarction and stroke (although with a non-significant reduction in mortality) with transfer. The strategy of transfer for primary PCI is appropriate if the intervention can be performed within 90 minutes of presentation and this is now optimal therapy.
FURTHER READING
Montalescot G, Zeymer U, Silvain J et al.; for the ATOLL Investigators. Intravenous enoxaparin or unfractionated heparin in primary percutaneous coronary intervention for ST-elevation myocardial infarction: the international randomised open-label ATOLL trial. Lancet 2011; 378:693–703.
Prasad A, Herrmann J. Myocardial infarction due to percutaneous coronary intervention. N Engl J Med 2011; 364:453–464.
Schmermund A, Voigtländer T. Predictive ability of coronary artery calcium and CRP. Lancet 2011; 378:641–643.
Fibrinolysis
Fibrinolytic agents (p. 426) enhance the breakdown of occlusive thromboses by the activation of plasminogen to form plasmin. Fibrinolysis is still used if PCI is unavailable.
A meta-analysis of fibrinolytics (FTT), fibrinolysis within 6 h of STEMI or LBBB MI, prevented 30 deaths in every 1000 patients treated. Between 7 and 12 hours, 20 in every 1000 deaths were prevented. After 12 hours, the benefits are limited, and there is evidence to suggest less benefit for older patients, possibly because of the increased risk of strokes.
Prompt reperfusion therapy (door to needle time <30 min) will reduce the death rate following myocardial infarction. Double bolus r-PA (reteplase) and single bolus TNK-t-PA (tenecteplase) facilitate rapid administration of fibrinolytic therapy and can be used for pre-hospital thrombolysis. In patients who fail to reperfuse by 60–90 minutes, as demonstrated by 50% resolution of the ST segment elevation, rethrombolysis or referral for rescue coronary angioplasty is recommended.
Aspirin therapy should be prescribed with fibrinolysis, but there is little additional benefit in combining clopidogrel or abciximab therapy in patients with STEMI/new LBBB. Heparin is recommended with t-PA or tenecteplase, but not with streptokinase. Enoxaparin (low-molecular-weight heparin) appears to be superior to unfractionated i.v. heparin in patients receiving TNK-t-PA, with less reocclusion and better late patency (ASSENT 3).
The contraindications to thrombolysis are provided in Table 14.34.
Table 14.34 Contraindications to thrombolysis
|
|
Coronary artery bypass surgery
Cardiac surgery is usually reserved for the complications of myocardial infarction, such as ventricular septal defect or mitral regurgitation.
FURTHER READING
Mehilli M, Pache J, Abdel-Wahab M et al., for the Is Drug-Eluting-Stenting Associated with Improved Results in Coronary Artery Bypass Grafts? (ISAR-CABG) Investigators. Drug-eluting versus bare-metal stents in saphenous vein graft lesions (ISAR-CABG): a randomised controlled superiority trial. Lancet 2011; 378(9796):1071–1078.
Complications of myocardial infarction
Heart failure
Cardiac failure post STEMI is a poor prognostic feature that necessitates medical and invasive therapy to reduce the death rate (see Table 14.36). The Killip classification is used to assess patients with heart failure post-MI:
Killip I – no crackles and no third heart sound
Killip II – crackles in <50% of the lung fields or a third heart sound
Mild heart failure may respond to intravenous furosemide 40–80 mg i.v., with GTN administration if the blood pressure is satisfactory. Oxygen is required, with regular oxygen monitoring. ACE inhibitors can be given in <24–48 hours if the blood pressure is satisfactory. Patients with severe heart failure may require Swan–Ganz catheterization to determine the pulmonary wedge pressure. Intravenous inotropes such as dopamine or dobutamine are used in patients with severe heart failure. If the patient is in cardiogenic shock, then revascularization ± intra-aortic balloon pump insertion may be required.
Myocardial rupture and aneurysmal dilatation
Rupture of the free wall of the left ventricle is usually an early, catastrophic and fatal event. The patient will have a haemodynamic collapse, then an electromechanical cardiac arrest. A subacute rupture may allow for pericardiocentesis followed by the surgical repair of the rupture. Aneurysmal dilatation of the infarcted myocardium (Fig. 14.68) is a late complication that may require surgical repair.
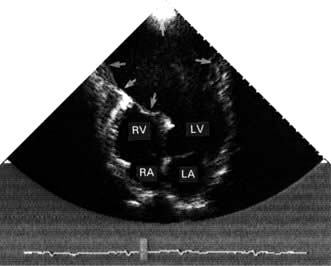
Figure 14.68 2-D Echocardiogram (apical four-chamber view) showing a very large apical left ventricular aneurysm (arrows). The relatively static blood in the aneurysm produces a swirling ‘smoke’ effect. This aneurysm was successfully resected surgically. LV, left ventricle; LA, left atrium; RV, right ventricle; RA, right atrium.
Ventricular septal defect (VSD)
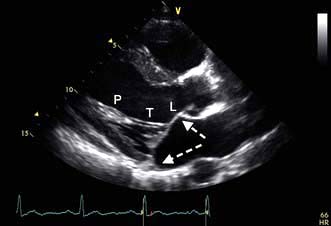
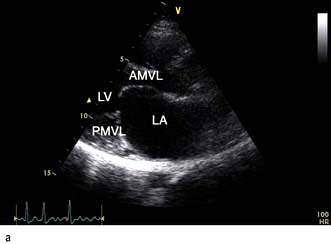
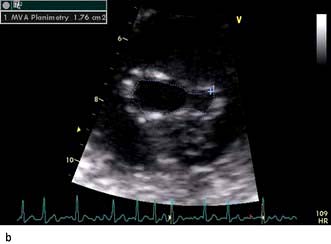
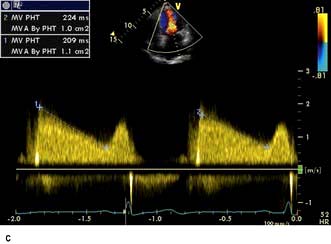
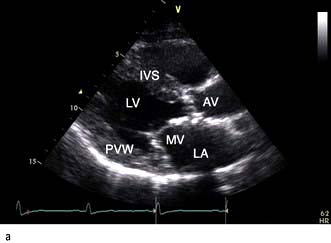
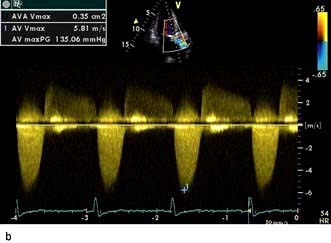
A VSD may occur in 1–2.0% of patients with STEMI, and may be associated with delayed or failed fibrinolysis. However, mortality is very high with a 12-month unoperative mortality of 92%. An intra-aortic balloon pump (IABP) and coronary angiography may allow for patient optimization prior to surgery. A post-infarct VSD is demonstrated in Figure 14.69.
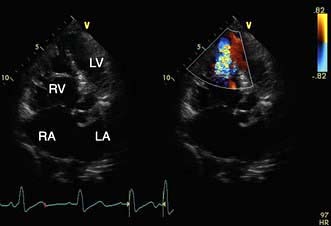
Mitral regurgitation
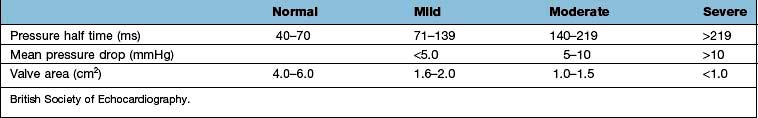
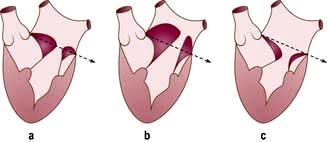
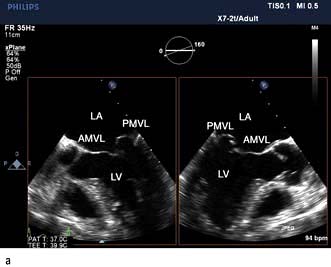
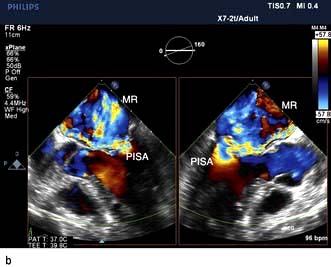
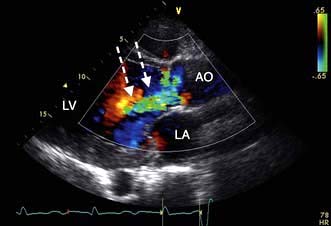
Severe mitral regurgitation can occur early in the course of STEMI. Three mechanisms may be responsible for the mitral regurgitation, and a transoesophageal echocardiogram (TOE) may be necessary to confirm the aetiology:
Severe left ventricular dysfunction and dilatation, causing annular dilatation of the valve and subsequent regurgitation
Myocardial infarction of the inferior wall, producing dysfunction of the papillary muscle that may respond to coronary intervention
Myocardial infarction of the papillary muscles, producing sudden severe pulmonary oedema and cardiogenic shock (IABP, coronary angiography and early surgery may improve patient survival).
Cardiac arrhythmias
Ventricular tachycardia and ventricular fibrillation are common in STEMI, particularly with reperfusion. Cardiac arrest requires defibrillation. Ventricular tachycardia should be treated with intravenous beta-blockers (metoprolol 5 mg, esmolol 50–200 µg/kg per min), lidocaine 50–100 mg, or amiodarone 900–1200 mg/24 hours. If the patient is hypotensive, synchronized cardioversion may be performed. Ensure that the serum potassium is above 4.5 mmol/L. Refractory ventricular tachycardia or fibrillation may respond to magnesium 8 mmol/L over 15 min i.v.
Atrial fibrillation occurs frequently and treatment with beta-blockers and digoxin may be required. Cardioversion is possible but relapse is frequent.
Bradyarrhythmias can be treated initially with i.v. atropine 0.5 mg repeated up to six times in 4 hours. Temporary transcutaneous or transvenous pacemaker insertion may be required in patients with symptomatic heart block.
Conduction disturbances
These are common following MI. AV block may occur during acute MI, especially of the inferior wall (the right coronary artery usually supplies the SA and AV nodes). Heart block, when associated with haemodynamic compromise, may need treatment with atropine or a temporary pacemaker. Such blocks may last for only a few minutes, but frequently continue for several days. Permanent pacing may need to be considered if heart block persists for over 2 weeks.
Post-ACS lifestyle modification
After recovery from an ACS patients should be encouraged to participate in a cardiac rehabilitation programme that provides education and information appropriate to the patients’ requirements. An exercise programme forms part of the rehabilitation.
Patients should be encouraged to eat a Mediterranean-style diet and to consume >7 g of omega-3 fatty acids/week from oily fish or >1 g daily of omega-3-acid ethyl esters.
Patients should maintain alcohol consumption within safe limits (≤21 units/week for men or <14 units for women) and avoid binge drinking.
Patients should be physically active for 20–30 min/day.
Overweight and obese patients should be offered advice and support to achieve and maintain a healthy weight.
Patients with hypertension should be treated to <140/90 or <130/80 if chronic kidney disease or diabetes.
Patients with diabetes should be treated to maintain HbA1c<7%.
Post-ACS drug therapy and assessment
Extensive clinical trial evidence has been gathered in post-myocardial infarction patients, demonstrating that a range of pharmaceuticals are advantageous in reducing mortality over the following years. Therefore, post-MI most patients should be taking most of the following medications:
A beta-blocker to maintain heart rate <60 b.p.m., e.g. metoprolol 50 mg twice daily
ACE inhibitors, e.g. ramipril 2.5 mg twice daily, titrated to maximum tolerated or target dose (if intolerant of ACE inhibitors use ARB, e.g. valsartan 20 mg twice daily titrated to maximum tolerated or target dose)
Statins, e.g. simvastatin 20–80 mg/day
Clopidogrel 75 mg/day for 9–12 months should be added in moderate–high-risk patients with non-ST elevation acute coronary syndrome (NST-ACS)
Aldosterone antagonist, e.g. eplerenone 25 mg/day, should be given in patients post-MI with clinical evidence of heart failure and reduced ejection fraction (renal function and potassium levels should be monitored).
FURTHER READING
Anderson JL, Adams CD, Antman EM et al. ACC/AHA 2007 Guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: Executive summary. Circulation 2007; 116:803–877.
Bassand JP, Hamm CW, Ardissino D et al.; for The Task Force for the Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of the European Society of Cardiology. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
Hansson SK. Inflammation, atherosclerosis and coronary artery disease. N Engl J Med 2005; 352:1685–1695.
Xavier D, Pais P, Devereauz PJ et al. Treatment and outcome of acute coronary syndromes in India (CREATE). Lancet 2008; 371:1435–1442.