Chapter 13 Water, electrolytes and acid–base balance
Distribution and composition of body water
In normal, healthy people, the total body water constitutes 50–60% of lean bodyweight in men and 45–50% in women. In a healthy 70 kg male, total body water is approximately 42 L. This is contained in three major compartments:
 Intracellular fluid (28 L, about 35% of lean bodyweight)
Intracellular fluid (28 L, about 35% of lean bodyweight)
Extracellular – the interstitial fluid that bathes the cells (9.4 L, about 12%)
In addition, small amounts of water are contained in bone, dense connective tissue, and epithelial secretions, such as the digestive secretions and cerebrospinal fluid.
The intracellular and interstitial fluids are separated by the cell membrane; the interstitial fluid and plasma are separated by the capillary wall (Fig. 13.1). In the absence of solute, water molecules move randomly and in equal numbers in either direction across a semi-permeable membrane. However, if solutes are added to one side of the membrane, the intermolecular cohesive forces reduce the activity of the water molecules. As a result, water tends to stay in the solute-containing compartment because there is less free diffusion across the membrane. This ability to hold water in the compartment can be measured as the osmotic pressure.
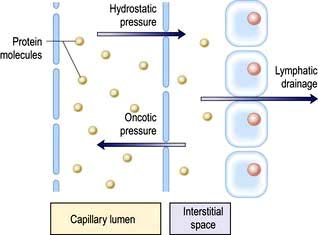
Figure 13.1 Distribution of water between the vascular and extravascular (interstitial) spaces. This is determined by the equilibrium between hydrostatic pressure, which tends to force fluid out of the capillaries, and oncotic pressure, which acts to retain fluid within the vessel. The net flow of fluid outwards is balanced by ‘suction’ of fluid into the lymphatics, which returns it to the bloodstream. Similar principles govern the volume of the peritoneal and pleural spaces.
Osmotic pressure
Osmotic pressure is the primary determinant of the distribution of water among the three major compartments. The concentrations of the major solutes in the compartments differ, each having one solute that is primarily limited to that compartment and therefore determines its osmotic pressure:
The intracellular fluid contains mainly potassium (K+) (most of the cell Mg2+ is bound and osmotically inactive)
In the extracellular compartment, Na+ salts predominate in the interstitial fluid, and proteins in the plasma.
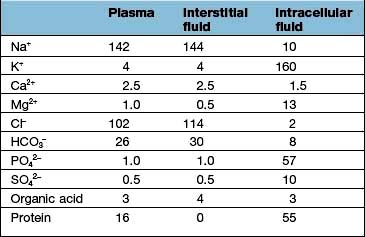
Regulation of the plasma volume is somewhat more complicated because of the tendency of the plasma proteins to hold water in the vascular space by an oncotic effect which is, in part, counterbalanced by the hydrostatic pressure in the capillaries that is generated by cardiac contraction (Fig. 13.1). The composition of intracellular and extracellular fluids is shown in Table 13.1.
A characteristic of an osmotically active solute is that it cannot freely leave its compartment. The capillary wall, for example, is relatively impermeable to plasma proteins, and the cell membrane is ‘impermeable’ to Na+ and K+ because the Na+/K+-ATPase pump largely restricts Na+ to the extracellular fluid and K+ to the intracellular fluid. By contrast, Na+ freely crosses the capillary wall and achieves similar concentrations in the interstitium and plasma; as a result, it does not contribute to fluid distribution between these compartments. Similarly, urea crosses both the capillary wall and the cell membrane and is osmotically inactive. Thus, the retention of urea in renal failure does not alter the distribution of the total body water.
A conclusion from these observations is that body Na+ stores are the primary determinant of the extracellular fluid volume. Thus, the extracellular volume – and therefore tissue perfusion – are maintained by appropriate alterations in Na+ excretion. For example, if Na+ intake is increased, the extra Na+ will initially be added to the extracellular fluid. The associated increase in extracellular osmolality will cause water to move out of the cells, leading to extracellular volume expansion. Balance is restored by excretion of the excess Na+ in the urine.
Distribution of different types of replacement fluids
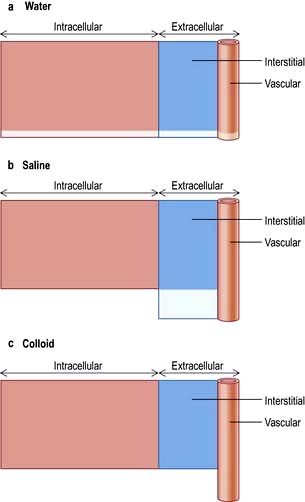
Figure 13.2 shows the relative effects on the compartments of the addition of identical volumes of water, saline and colloid solutions. Thus, 1 L of water given intravenously as 5% glucose is distributed equally into all compartments, whereas the same amount of 0.9% saline remains in the extracellular compartment. The latter is thus the correct treatment for extracellular water depletion – sodium keeping the water in this compartment. The addition of 1 L of colloid with its high oncotic pressure stays in the vascular compartment and is a treatment for hypovolaemia.
Regulation of extracellular volume (Fig. 13.3)
The extracellular volume is determined by the sodium concentration. The regulation of extracellular volume is dependent upon a tight control of sodium balance, which is exerted by normal kidneys. Renal Na+ excretion varies directly with the effective circulating volume. In a 70 kg man:
Plasma fluid constitutes one-third of extracellular volume (4.6 L), and of this,
85% (3.9 L) lies in the venous side and only 15% (0.7 L) resides in the arterial circulation.
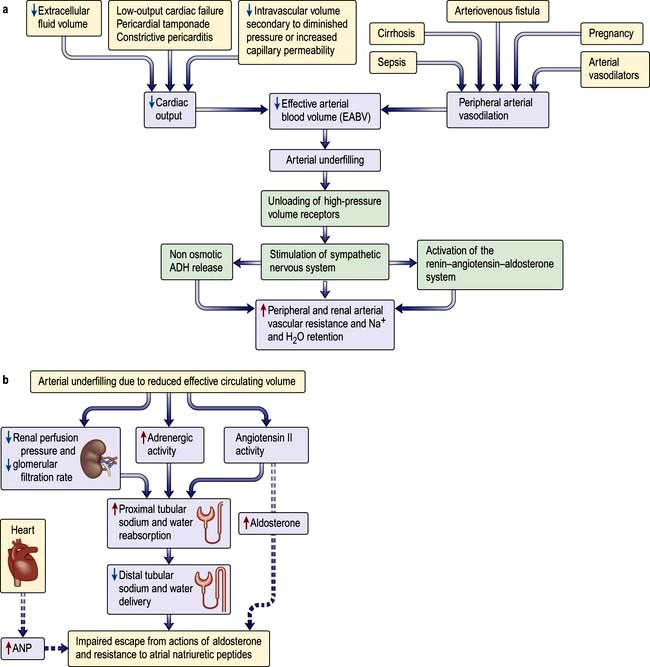
Figure 13.3 Regulation of extracellular volume. (a) Sequence of events in which a decrease in cardiac output or peripheral arterial dilatation initiates renal sodium and water retention. (b) Mechanism of impaired escape from the actions of aldosterone and resistance to atrial natriuretic peptides (ANP).
(Modified from Schrier RW. Renal and Electrolyte Disorders, 7th edn. Philadelphia: Lippincott Williams and Wilkins; 2010, with permission.)
The unifying hypothesis of extracellular volume regulation in health and disease proposed by Schrier states that the fullness of the arterial vascular compartment – or the so-called effective arterial blood volume (EABV) – is the primary determinant of renal sodium and water excretion. Thus effective arterial blood volume constitutes effective circulatory volume for the purposes of body fluid homeostasis.
The fullness of the arterial compartment depends upon a normal ratio between cardiac output and peripheral arterial resistance. Thus, diminished EABV is initiated by a fall in cardiac output or a fall in peripheral arterial resistance (an increase in the holding capacity of the arterial vascular tree). When the EABV is expanded, the urinary Na+ excretion is increased and can exceed 100 mmol/L. By contrast, the urine can be rendered virtually free of Na+ in the presence of EABV depletion and normal renal function.
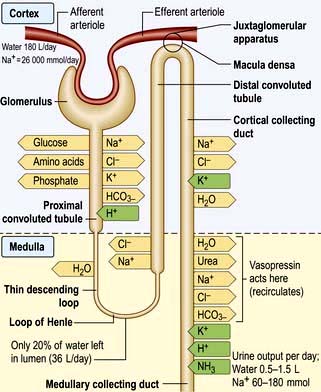
These changes in Na+ excretion can result from alterations both in the filtered load, determined primarily by the glomerular filtration rate (GFR), and in tubular reabsorption, which is affected by multiple factors. In general, it is changes in tubular reabsorption that constitute the main adaptive response to fluctuations in the effective circulating volume. How this occurs can be appreciated from Table 13.2 and Figure 13.4 and Figure 12.2 (see p. 563), which depicts the sites and determinants of segmental Na+ reabsorption. Although the loop of Henle and distal tubules make a major overall contribution to net Na+ handling, transport in these segments primarily varies with the amount of Na+ delivered; that is, reabsorption is flow-dependent. In comparison, the neurohumoral regulation of Na+ reabsorption according to body needs occurs primarily in the proximal tubules and collecting ducts.

Neurohumoral regulation of extracellular volume
This is mediated by volume receptors that sense changes in the EABV rather than alterations in the sodium concentration. These receptors are distributed in both the renal and cardiovascular tissues.
Intrarenal receptors. Receptors in the walls of the afferent glomerular arterioles respond, via the juxtaglomerular apparatus, to changes in renal perfusion, and control the activity of the renin-angiotensin-aldosterone system (see p. 1006). In addition, sodium concentration in the distal tubule and sympathetic nerve activity alter renin release from the juxtaglomerular cells. Prostaglandins I2 and E2 are also generated within the kidney in response to angiotensin II, acting to maintain glomerular filtration rate and sodium and water excretion, modulating the sodium-retaining effect of this hormone.
Extrarenal receptors. These are located in the vascular tree in the left atrium and major thoracic veins, and in the carotid sinus body and aortic arch. These volume receptors respond to a slight reduction in effective circulating volume and result in increased sympathetic nerve activity and a rise in catecholamines. In addition, volume receptors in the cardiac atria control the release of a powerful natriuretic hormone – atrial natriuretic peptide (ANP) – from granules located in the atrial walls (see p. 943).
High-pressure arterial receptors (carotid, aortic arch, juxtaglomerular apparatus) predominate over low-pressure volume receptors in volume control in mammals. The low-pressure volume receptors are distributed in thoracic tissues (cardiac atria, right ventricle, thoracic veins, pulmonary vessels) and their role in the volume regulatory system is marginal.
Aldosterone and possibly ANP are responsible for day-to-day variations in Na+ excretion, by their respective ability to augment and diminish Na+ reabsorption in the collecting ducts.
A salt load, for example, leads to an increase in the effective circulatory and extracellular volume, raising both renal perfusion pressure, and atrial and arterial filling pressure. The increase in the renal perfusion pressure reduces the secretion of renin, and subsequently that of angiotensin II and aldosterone (see Fig. 12.5), whereas the rise in atrial and arterial filling pressure increases the release of ANP. These factors combine to reduce Na+ reabsorption in the collecting duct, thereby promoting excretion of excess Na+.
By contrast, in patients on a low Na+ intake or in those who become volume-depleted as a result of vomiting and diarrhoea, the ensuing decrease in effective volume enhances the activity of the renin-angiotensin-aldosterone system and reduces the secretion of ANP. The net effect is enhanced Na+ reabsorption in the collecting ducts, leading to a fall in Na+ excretion. This increases the extracellular volume towards normal.
With more marked hypovolaemia, a decrease in GFR leads to an increase in proximal and thin ascending limb Na+ reabsorption which contributes to Na+ retention. This is brought about by enhanced sympathetic activity acting directly on the kidneys and indirectly by stimulating the secretion of renin/angiotensin II (see Fig. 13.3b) and non-osmotic release of antidiuretic hormone (ADH), also called vasopressin. The pressure natriuresis phenomenon may be the final defence against changes in the effective circulating volume. Marked persistent hypovolaemia leads to systemic hypotension and increased salt and water absorption in the proximal tubules and ascending limb of Henle. This process is partly mediated by changes in renal interstitial hydrostatic pressure and local prostaglandin and nitric oxide production.
Volume regulation in oedematous conditions
Sodium and water are retained despite increased extracellular volume in oedematous conditions such as cardiac failure, hepatic cirrhosis and hypoalbuminaemia. Here the principal mediator of salt and water retention is the concept of arterial underfilling due either to reduced cardiac output or diminished peripheral arterial resistance. Arterial underfilling in these settings leads to reduction of pressure or stretch (i.e. ‘unloading’ of arterial volume receptors), which results in activation of the sympathetic nervous system, activation of the renin-angiotensin-aldosterone system and non-osmotic release of ADH. These neurohumoral mediators promote salt and water retention in the face of increased extracellular volume. The common nature of the degree of arterial fullness and neurohumoral pathway in the regulation of extracellular volume in health and disease states forms the basis of Schrier’s unifying hypothesis of volume homeostasis (Fig. 13.3a).
Mechanism of impaired escape from actions of aldosterone and resistance to ANP
Not only is the activity of the renin-angiotensin-aldosterone system increased in oedematous conditions such as cardiac failure, hepatic cirrhosis and hypoalbuminaemia, but also the action of aldosterone is more persistent than in normal subjects and patients with Conn’s syndrome, who have increased aldosterone secretion (see p. 989).
In normal subjects, high doses of mineralocorticoids initially increase renal sodium retention so that the extracellular volume is increased by 1.5–2 L. However, renal sodium retention then ceases, sodium balance is re-established, and there is no detectable oedema. This escape from mineralocorticoid-mediated sodium retention explains why oedema is not a characteristic feature of primary hyperaldosteronism (Conn’s syndrome). The escape is dependent on an increase in delivery of sodium to the site of action of aldosterone in the collecting ducts. The increased distal sodium delivery is achieved by high extracellular volume-mediated arterial overfilling. This suppresses sympathetic activity and angiotensin II generation, and increases cardiac release of ANP with resultant increase in renal perfusion pressure and GFR. The net result of these events is reduced sodium absorption in the proximal tubules and increased distal sodium delivery which overwhelms the sodium-retaining actions of aldosterone.
In patients with the above oedematous conditions, e.g. heart failure, escape from the sodium-retaining actions of aldosterone does not occur and therefore they continue to retain sodium in response to aldosterone. Accordingly they have substantial natriuresis when given spironolactone, which blocks mineralocorticoid receptors. Alpha-adrenergic stimulation and elevated angiotensin II increase sodium transport in the proximal tubule, and reduced renal perfusion and GFR further increases sodium absorption from the proximal tubules by presenting less sodium and water in the tubular fluid. Sodium delivery to the distal portion of the nephron, and thus the collecting duct, is reduced. Similarly, increased cardiac ANP release in these conditions requires optimum sodium concentration at the site of its action in the collecting duct for its desired natriuretic effects. Decreased sodium delivery to the collecting duct is therefore the most likely explanation for the persistent aldosterone-mediated sodium retention, absence of escape phenomenon and resistance to natriuretic peptides in these patients (Fig. 13.3b).
Regulation of water excretion
Body water homeostasis is affected by thirst and the urine concentrating and diluting functions of the kidney. These in turn are controlled by intracellular osmoreceptors, principally in the hypothalamus, to some extent by volume receptors in capacitance vessels close to the heart, and via the renin-angiotensin system. Of these, the major and best-understood control is via osmoreceptors. Changes in the plasma Na+ concentration and osmolality are sensed by osmoreceptors that influence both thirst and the release of ADH (vasopressin) from the supraoptic and paraventricular nuclei of the anterior hypothalamus.
ADH plays a central role in urinary concentration by increasing the water permeability of the normally impermeable cortical and medullary collecting ducts. There are three major G-protein coupled receptors for vasopressin (ADH):
V1A found in vascular smooth muscle cells
V1B in anterior pituitary and throughout the brain
V2 receptors in the principal cells of the kidney distal convoluting tubule and collecting ducts (see below).
Activation of the V1A receptors induces vasoconstriction while V1B receptors appear to mediate the effect of ADH on the pituitary, facilitating the release of ACTH. The V2 receptors mediate the antidiuretic response as well as other functions.
The ability of ADH to increase the urine osmolality is related indirectly to transport in the ascending limb of the loop of Henle, which reabsorbs NaCl without water. This process, which is the primary step in the countercurrent mechanism, has two effects: it makes the tubular fluid dilute and the medullary interstitium concentrated. In the absence of ADH, little water is reabsorbed in the collecting ducts, and a dilute urine is excreted. By contrast, the presence of ADH promotes water reabsorption in the collecting ducts down the favourable osmotic gradient between the tubular fluid and the more concentrated interstitium. As a result, there is an increase in urine osmolality and a decrease in urine volume.
The cortical collecting duct has two cell types (see also p. 597) with very different functions:
Principal cells (about 65%) have sodium and potassium channels in the apical membrane and, as in all sodium-reabsorbing cells, Na+/K+-ATPase pumps in the basolateral membrane.
Intercalated cells, in comparison, do not transport NaCl (since they have a lower level of Na+/K+-ATPase activity) but play a role in hydrogen and bicarbonate handling and in potassium reabsorption in states of potassium depletion.
The ADH-induced increase in collecting duct water permeability occurs primarily in the principal cells. ADH acts on V2 (vasopressin) receptors located on the basolateral surface of principal cells, resulting in the activation of adenyl cyclase. This leads to protein kinase activation and to preformed cytoplasmic vesicles that contain unique water channels (called aquaporins) moving to and then being inserted into the luminal membrane. Four renal aquaporins have been well characterized and are localized in different areas of the cells of the collecting duct. The water channels span the luminal membrane and permit water movement into the cells down a favourable osmotic gradient (Fig. 13.5). This water is then rapidly returned to the systemic circulation across the basolateral membrane. When the ADH effect has worn off, the water channels aggregate within clathrin-coated pits, from which they are removed from the luminal membrane by endocytosis and returned to the cytoplasm. A defect in any step in this pathway, such as in attachment of ADH to its receptor or the function of the water channel, can cause resistance to the action of ADH and an increase in urine output. This disorder is called nephrogenic diabetes insipidus.
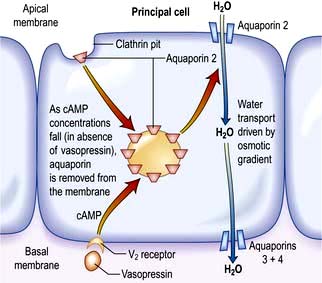
Figure 13.5 Aquaporin-mediated water transport in the renal collecting duct. Stimulation of the vasopressin 2 receptor causes cAMP-mediated insertion of the aquaporin into the apical membrane, allowing water transport down the osmotic gradient.
(Adapted from Connolly DL, Shanahan CM, Weissberg PL. Water channels in health and disease. Lancet 1996; 347:211, with permission from Elsevier.)
Plasma osmolality
In addition to influencing the rate of water excretion, ADH plays a central role in osmoregulation because its releaseis directly affected by the plasma osmolality. At a plasma osmolality of <275 mosmol/kg, which usually represents a plasma Na+ concentration of <135–137 mmol/L, there is essentially no circulating ADH. As the plasma osmolality rises above this threshold, however, the secretion of ADH increases progressively.
Two simple examples will illustrate the basic mechanisms of osmoregulation, which is so efficient that the plasma Na+ concentration is normally maintained within 1–2% of its baseline value.
1. Ingestion of a water load leads to an initial reduction in the plasma osmolality, thereby diminishing the release of ADH. The ensuing reduction in water reabsorption in the collecting ducts allows the excess water to be excreted in a dilute urine.
2. Water loss resulting from sweating is followed by, in sequence, a rise in both plasma osmolality and ADH secretion, enhanced water reabsorption, and the appropriate excretion of a small volume of concentrated urine. This renal effect of ADH minimizes further water loss but does not replace the existing water deficit. Thus, optimal osmoregulation requires an increase in water intake, which is mediated by a concurrent stimulation of thirst. The importance of thirst can also be illustrated by studies in patients with central diabetes insipidus, who are deficient in ADH. These patients often complain of marked polyuria, which is caused by the decline in water reabsorption in the collecting ducts. However, they do not typically become hypernatraemic, because urinary water loss is offset by the thirst mechanism.
Osmoregulation versus volume regulation
A common misconception is that regulation of the plasma Na+ concentration is closely correlated with the regulation of Na+ excretion. However, it is related to volume regulation, which has different sensors and effectors (volume receptors) from those involved in water balance and osmoregulation (osmoreceptors).
The roles of these two pathways should be considered separately when evaluating patients.
A water load is rapidly excreted (in 4–6 h) by inhibition of ADH release so that there is little or no water reabsorption in the collecting ducts. This process is normally so efficient that volume regulation is not affected and there is no change in ANP release or in the activity of the renin-angiotensin-aldosterone system. Thus, a dilute urine is excreted, and there is little alteration in the excretion of Na+.
0.9% saline administration, by contrast, causes an increase in volume but no change in plasma osmolality. In this setting, ANP secretion is increased, aldosterone secretion is reduced and ADH secretion does not change. The net effect is the appropriate excretion of the excess Na+ in a relatively iso-osmotic urine.
In some cases, both volume and osmolality are altered and both pathways are activated. For example, if a person with normal renal function eats salted potato chips and peanuts without drinking any water, the excess Na+ will increase the plasma osmolality, leading to osmotic water movement out of the cells and increased extracellular volume. The rise in osmolality will stimulate both ADH release and thirst (the main reason why many restaurants and bars supply free salted foods), whereas the hypervolaemia will enhance the secretion of ANP and suppress that of aldosterone. The net effect is increased excretion of Na+ without water.
This principle of separate volume and osmoregulatory pathways is also evident in the syndrome of inappropriate ADH secretion (SIADH). Patients with SIADH (see p. 993) have impaired water excretion and hyponatraemia (dilutional) caused by the persistent presence of ADH. However, the release of ANP and aldosterone is not impaired and, thus, Na+ handling remains intact. These findings have implications for the correction of the hyponatraemia in this setting which initially requires restriction of water intake.
ADH is also secreted by non-osmotic stimuli such as stress (e.g. surgery, trauma), markedly reduced effective circulatory volume (e.g. cardiac failure, hepatic cirrhosis), psychiatric disturbance and nausea, irrespective of plasma osmolality. This is mediated by the effects of sympathetic overactivity on supraoptic and paraventricular nuclei. In addition to water retention, ADH release in these conditions promotes vasoconstriction owing to the activation of V1A (vasopressin) receptors distributed in the vascular smooth muscle cells.
Regulation of cell volume
Most cells respond to swelling or shrinkage by activating specific metabolic or membrane-transport processes that return cell volume to its normal resting state. Within minutes after exposure to hypotonic solutions and resulting cell swelling, a common feature of many cells is the increase in plasma membrane potassium and chloride conductance. Although extrusion of intracellular potassium certainly contributes to a regulatory volume decrease, the role of chloride efflux itself is modest, given the relatively low intracellular chloride concentration. Other intracellular osmolytes, such as taurine and other amino acids, are transported out of the cell to achieve a regulatory volume decrease. By contrast, these regulatory mechanisms are operative in reverse to protect cell volume under hypertonic conditions, as is the case in the renal medulla. The tubular cells at the tip of renal papillae, which are constantly exposed to a hypertonic extracellular milieu, maintain their cell volume on a long-term basis by actively taking up smaller molecules, such as betaine, taurine and myoinositol, and by synthesizing more sorbitol and glycerophosphocholine.
Increased extracellular volume
Increased extracellular volume occurs in numerous disease states. The physical signs depend on the distribution of excess volume and on whether the increase is local or systemic. According to Starling principles, distribution depends on:
Venous tone, which determines the capacitance of the blood compartment and thus hydrostatic pressure
Depending on these factors, fluid accumulation may result in expansion of interstitial volume, blood volume or both.
Clinical features
Peripheral oedema is caused by expansion of the extracellular volume by at least 2 L (15%). The ankles are normally the first part of the body to be affected, although they may be spared in patients with lipodermatosclerosis (where the skin is tethered and cannot expand to accommodate the oedema). Oedema may be noted in the face, particularly in the morning. In a patient in bed, oedema may accumulate in the sacral area. Expansion of the interstitial volume also causes pulmonary oedema, pleural effusion, pericardial effusion and ascites. Expansion of the blood volume (overload) causes a raised jugular venous pressure, cardiomegaly, added heart sounds, basal crackles as well as a raised arterial blood pressure in certain circumstances.
Causes
Extracellular volume expansion is due to sodium chloride retention. Increased oral salt intake does not normally cause volume expansion because of rapid homeostatic mechanisms which increase salt excretion. However, a rapid intravenous infusion of a large volume of saline will cause volume expansion. Most causes of extracellular volume expansion are associated with renal sodium chloride retention.
Heart failure
Reduction in cardiac output and the consequent fall in effective circulatory volume and arterial filling lead to activation of the renin-angiotensin-aldosterone system, non-osmotic release of ADH, and increased activity of the renal sympathetic nerves via volume receptors and baroreceptors (Fig. 13.3a). Sympathetic overdrive also indirectly augments ADH and renin-angiotensin-aldosterone response in these conditions. The cumulative effect of these mediators results in increased peripheral and renal arteriolar resistance and water and sodium retention. These factors result in extracellular volume expansion and increased venous pressure, causing oedema formation.
Hepatic cirrhosis
The mechanism is complex, but involves peripheral vasodilatation (possibly owing to increased nitric oxide generation) resulting in reduced effective arterial blood volume (EABV) and arterial filling. This leads to an activation of a chain of events common to cardiac failure and other conditions with marked peripheral vasodilatation (Fig. 13.3). The cumulative effect results in increased peripheral and renal resistance, water and sodium retention, and oedema formation.
Nephrotic syndrome
Interstitial oedema is a common clinical finding with hypoalbuminaemia, particularly in the nephrotic syndrome. Expansion of the interstitial compartment is secondary to the accumulation of sodium in the extracellular compartment. This is due to an imbalance between oral (or parenteral) sodium intake and urinary sodium output, as well as alterations of fluid transfer across capillary walls. The intrarenal site of sodium retention is the cortical collecting duct (CCD) where Na+/K+-ATPase expression and activity are increased threefold along the basolateral surface (Fig. 13.4). In addition, amiloride-sensitive epithelial sodium channel activity is also increased in the CCD. The renal sodium retention should normally be counterbalanced by increased secretion of sodium in the inner medullary collecting duct, brought about by the release of ANP. This regulatory pathway is altered in patients with nephrotic syndrome by enhanced kidney specific catabolism of cyclic GMP (the second messenger for ANP) following phosphodiesterase activation.
Oedema generation was classically attributed to the decrease in the plasma oncotic pressure and the subsequent increase in the transcapillary oncotic gradient. However, the oncotic pressure and transcapillary oncotic gradient remain unchanged and the transcapillary hydrostatic pressure gradient is not altered. Conversely, capillary hydraulic conductivity (a measure of permeability) is increased. This is determined by intercellular macromolecular complexes between the endothelial cells consisting of tight junctions (made of occludins, claudins and ZO proteins) and adherens junctions (made of cadherin, catenins and actin cytoskeleton). Elevated TNF-α levels in nephrotic syndrome activate protein kinase C, which changes phosphorylation of occludin and capillary permeability. In addition, increased circulating ANP can increase capillary hydraulic conductivity by altering the permeability of intercellular junctional complexes. Furthermore, reduction in effective circulatory volume and the consequent fall in cardiac output and arterial filling can lead to a chain of events as in cardiac failure and cirrhosis (see above and Fig. 13.3). These factors result in extracellular volume expansion and oedema formation.
Sodium retention
A decreased GFR decreases the renal capacity to excrete sodium. This may be acute, as in the acute nephritic syndrome (see p. 582), or may occur as part of the presentation of chronic kidney disease. In end-stage renal failure, extracellular volume is controlled by the balance between salt intake and its removal by dialysis.
Numerous drugs cause renal sodium retention, particularly in patients whose renal function is already impaired:
Oestrogens cause mild sodium retention, due to a weak aldosterone-like effect. This is the cause of weight gain in the premenstrual phase.
Mineralocorticoids and liquorice (the latter potentiates the sodium-retaining action of cortisol) have aldosterone-like actions.
NSAIDs cause sodium retention in the presence of activation of the renin-angiotensin-aldosterone system by heart failure, cirrhosis and in renal artery stenosis.
Thiazolidinediones (TZD) (see p. 1011) are widely used to treat type 2 diabetes. Their mechanism of action is attributed to binding and activation of the PPAR-γ system. PPARs are nuclear transcription factors essential to the control of energy metabolism that are modulated via binding with tissue-specific fatty acid metabolites. Of the three PPAR isoforms, γ has been extensively studied and is expressed at high levels in adipose and liver tissues, macrophages, pancreatic-β cells and principal cells of the collecting duct. These drugs have been asociated with salt and water retention and are contraindicated in patients with heart failure. Recent evidence suggests that TZD-induced oedema (like insulin) is also due to upregulation of epithelial Na transporter channel (ENaC) but by different pathways. Diuretics of choice for TZD-induced oedema are amiloride and triamterene.
Substantial amounts of sodium and water may accumulate in the body without clinically obvious oedema or evidence of raised venous pressure. In particular, several litres may accumulate in the pleural space or as ascites; these spaces are then referred to as ‘third spaces’. Bone may also act as a ‘sink’ for sodium and water.
Other causes of oedema
Initiation of insulin treatment for type 1 diabetes and refeeding after malnutrition are both associated with the development of transient oedema. The mechanism is complex but involves upregulation of ENaC in the principal cell of the collecting duct. This transporter is amiloride sensitive which makes amiloride or triamterene the diuretic of choice in insulin-induced oedema.
Oedema may result from increased capillary pressure owing to relaxation of precapillary arterioles. The best example is the peripheral oedema caused by dihydropyridine calcium-channel blockers such as nifedipine which affects up to 10% of the patients. Oedema is usually resolved by stopping the offending drug.
Oedema is also caused by increased interstitial oncotic pressure as a result of increased capillary permeability to proteins. This can occur as part of a rare complement-deficiency syndrome; with therapeutic use of interleukin 2 in cancer chemotherapy; or in ovarian hyperstimulation syndrome (see p. 981).
Idiopathic oedema of women
This, by definition, occurs in women without heart failure, hypoalbuminaemia, renal or endocrine disease. Oedema is intermittent and often worse in the premenstrual phase. The condition remits after the menopause. Patients complain of swelling of the face, hands, breasts and thighs, and a feeling of being bloated. Sodium retention during the day and increased sodium excretion during recumbency are characteristic; an abnormal fall in plasma volume on standing caused by increased capillary permeability to proteins may be the cause of this. The oedema may respond to diuretics, but returns when they are stopped. A similar syndrome of diuretic-dependent sodium retention can be caused by abuse of diuretics, for instance as part of an attempt to lose weight; but not all women with idiopathic oedema admit to having taken diuretics, and the syndrome was described before diuretics were introduced for clinical use, so the cause remains unclear.
Local increase in oedema
This does not reflect disturbances of extracellular volume control per se, but can cause clinical confusion. Examples are ankle oedema due to venous damage following thrombosis or surgery, ankle or leg oedema due to immobility, oedema of the arm due to subclavian thrombosis, and facial oedema due to superior vena caval obstruction.
Treatment
The underlying cause should be treated where possible. Heart failure, for example, should be treated, and offending drugs such as NSAIDs withdrawn.
Sodium restriction has only a limited role, but is useful in patients who are resistant to diuretics. Sodium intake can easily be reduced to approximately 100 mmol (2 g) daily; reductions below this are often difficult to achieve without affecting the palatability of food.
Manoeuvres that increase venous return (e.g. strict bed rest or water immersion) stimulate salt and water excretion by effects on cardiac output and ANP release, but they are seldom of practical value.
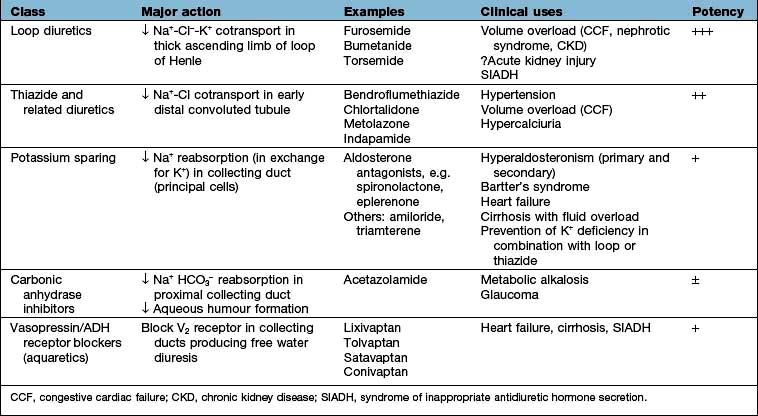
The mainstay of treatment is the use of diuretic agents, which increase sodium, chloride and water excretion in the kidney (Table 13.3). These agents act by interfering with membrane ion pumps which are present on numerous cell types; they mostly achieve specificity for the kidney by being secreted into the proximal tubule, resulting in much higher concentrations in the tubular fluid than in other parts of the body.
Clinical use of diuretics
Loop diuretics
These potent diuretics are useful in the treatment of any cause of systemic extracellular volume overload. They stimulate excretion of both sodium chloride and water by blocking the sodium-potassium-2-chloride (NKCC2) channel in the thick ascending limb of Henle (Fig. 13.6) and are useful in stimulating water excretion in states of relative water overload. They also act by causing increased venous capacitance, resulting in rapid clinical improvement in patients with left ventricular failure, preceding the diuresis. Unwanted effects include:
Allergic tubulointerstitial nephritis and other allergic reactions
Myalgia – especially with high-dose bumetanide
Ototoxicity (due to an action on sodium pump activity in the inner ear) – particularly with furosemide
Interference with excretion of lithium, resulting in toxicity.
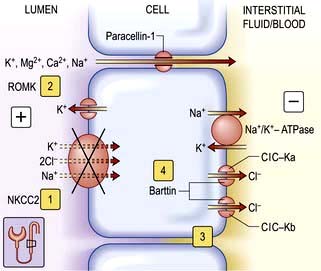
Figure 13.6 Transport mechanisms in the thick ascending limb of the loop of Henle. Sodium chloride is reabsorbed in the thick ascending limb by the bumetanide-sensitive sodium-potassium-2-chloride cotransporter (NKCC2). The electroneutral transporter is driven by the low intracellular sodium and chloride concentrations generated by the Na+/K+-ATPase and the kidney-specific basolateral chloride channel (ClC-Kb). The availability of luminal potassium is rate-limiting for NKCC2, and recycling of potassium through the ATP-regulated potassium channel (ROMK – renal outer medulla K+ channel) ensures the efficient functioning of the NKCC2 and generates a lumen-positive transepithelial potential. Genetic studies have identified putative loss of function mutations in the genes encoding NKCC2 1, ROMK 2, ClC-Kb 3, and barttin 4 in subgroups of patients with Bartter’s syndrome. In contrast to the normal condition, loss of function of NKCC2 impairs reabsorption of sodium and potassium. Inactivation of the basolateral ClC-Kb and barttin reduces transcellular reabsorption of chloride. Loss of function of any of these will reduce the transepithelial potential and thus decrease the driving force for the paracellular reabsorption of cations (K+, Mg2+, Ca2+ and Na+). Paracellin-1 is necessary for the paracellular transport of Ca2+ and Mg2+. In most patients with Bartter’s syndrome, urinary calcium excretion is increased. Hypercalcaemia or increased activation of calcium-sensing receptor inactivates ROMK and causes Bartter’s syndrome. Ka and Kb, kidney-specific basolateral chloride channel. ROMK, renal outer medullary potassium channel.
There is little to choose between the drugs in this class. Bumetanide has a better oral bioavailability than furosemide, particularly in patients with severe peripheral oedema, and has more beneficial effects than furosemide on venous capacitance in left ventricular failure.
Thiazide diuretics (see p. 719)
These are less potent than loop diuretics. They act by blocking a sodium chloride channel in the distal convoluted tubule (Fig. 13.7). They cause relatively more urate retention, glucose intolerance and hypokalaemia than loop diuretics. They interfere with water excretion and may cause hyponatraemia, particularly if combined with amiloride or triamterene. This effect is clinically useful in diabetes insipidus. Thiazides reduce peripheral vascular resistance by mechanisms that are not completely understood but do not appear to depend on their diuretic action, and are widely used in the treatment of essential hypertension. They are also used extensively in mild to moderate cardiac failure. Thiazides reduce calcium excretion. This effect is useful in patients with idiopathic hypercalciuria, but may cause hypercalcaemia. Numerous agents are available, with varying half-lives but little else to choose between them. Metolazone is not dependent for its action on glomerular filtration, and therefore retains its potency in renal impairment.
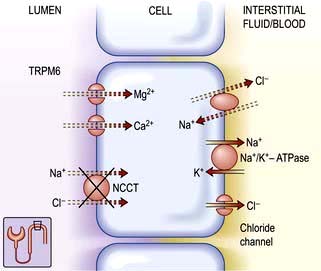
Figure 13.7 Transport mechanisms in the distal convoluted tubule. Under normal conditions, sodium chloride is reabsorbed by the apical thiazide-sensitive sodium-chloride cotransporter (NCCT) in the distal convoluted tubule. The electroneutral transporter is driven by the low intracellular sodium and chloride concentrations generated by the Na+/K+-ATPase and an, as yet, undefined basolateral chloride channel. In this nephron segment, there is an apical calcium channel and a basolateral sodium-coupled exchanger. Physiological evidence indicates that the mechanisms for the transport of magnesium are similar to those for calcium. In Gitelman’s syndrome, putative loss of function mutations in the sodium-chloride cotransporter (NCCT (X)) lead to decreased reabsorption of sodium chloride and increased reabsorption of calcium. Functional overactivity of NCCT leads to Gordon’s syndrome by a new mechanism (see text). TRPM6, a member of the transient receptor potential family.
Potassium-sparing diuretics (see Fig. 13.8)
Aldosterone antagonists, which compete with aldosterone in the collecting ducts and reduce sodium absorption, e.g. spironolactone and eplerenone (which has a shorter half-life). Spironolactone is used in patients with heart failure because it significantly reduces the mortality in these patients by antagonizing the fibrotic effect of aldosterone on the heart. Eplerenone is devoid of antiandrogenic or antiprogesterone properties.
Amiloride and triamterene inhibit sodium uptake by blocking epithelial sodium channels in the collecting duct and reduce renal potassium excretion by reducing lumen-negative transepithelial voltage. They are mainly used as potassium-sparing agents with thiazide or loop diuretics.
Carbonic anhydrase inhibitors
These are relatively weak diuretics and are seldom used except in the treatment of glaucoma. They cause metabolic acidosis and hypokalaemia.
Aquaretics (vasopressin or antidiuretic hormone antagonists)
Vasopressin V2 receptor antagonists are very useful agents in the treatment of conditions associated with elevated levels of vasopressin, such as heart failure, cirrhosis and SIADH (see p. 993). Non-peptide vasopressin V2 receptor antagonists are efficacious in producing free water diuresis in humans. Studies in patients with heart failure and cirrhosis suggest that such agents will allow normalization of serum osmolality with less water restriction (see p. 650).
Resistance to diuretics
Resistance may occur as a result of:
Reduced GFR, which may be due to decreased circulating volume despite oedema (e.g. nephrotic syndrome, cirrhosis with ascites) or intrinsic renal disease
Activation of sodium-retaining mechanisms, particularly aldosterone.
Management. Intravenous administration of diuretics may establish a diuresis. High doses of loop diuretics are required to achieve adequate concentrations in the tubule if GFR is depressed. However, the daily dose of furosemide must be limited to a maximum of 2 g for an adult, because of ototoxicity. Intravenous albumin solutions restore plasma oncotic pressure temporarily in the nephrotic syndrome and allow mobilization of oedema but do not increase the natriuretic effect of loop diuretics.
Combinations of various classes of diuretics are extremely helpful in patients with resistant oedema. A loop diuretic plus a thiazide inhibit two major sites of sodium reabsorption; this effect may be further potentiated by addition of a potassium-sparing agent. Metolazone in combination with a loop diuretic is particularly useful in refractory congestive cardiac failure, because its action is less dependent on glomerular filtration. However, this potent combination can cause severe electrolyte imbalance. Both aminophylline and dopamine increase renal blood flow and may be useful in refractory cardiogenic sodium retention. In addition, theophyllines, by inhibiting phosphodiesterase activity in the inner medullary collecting duct, prolong the action of cyclic GMP (a second messenger of ANP).
Decreased extracellular volume
Deficiency of sodium and water causes shrinkage both of the interstitial space and of the blood volume and may have profound effects on organ function.
Clinical features
Symptoms. Thirst, muscle cramps, nausea and vomiting, and postural dizziness occur. Severe depletion of circulating volume causes hypotension and impairs cerebral perfusion, causing confusion and eventual coma.
Signs can be divided into those due to loss of interstitial fluid and those due to loss of circulating volume.
Loss of interstitial fluid leads to loss of skin elasticity (’turgor’) – the rapidity with which the skin recoils to normal after being pinched. Skin turgor decreases with age, particularly at the peripheries. The turgor over the anterior triangle of the neck or on the forehead is a very useful sign in all ages.
Loss of circulating volume leads to decreased pressure in the venous and (if severe) arterial compartments. Loss of up to 1 L of extracellular fluid in an adult may be compensated for by venoconstriction and may cause no physical signs.
Loss of more than this causes the following:
Postural hypotension
Normally the blood pressure rises if a subject stands up, as a result of increased venous return due to venoconstriction (this maintains cerebral perfusion). Loss of extracellular fluid (underfill) prevents this and causes a fall in blood pressure. This is one of the earliest and most reliable signs of volume depletion, as long as the other causes of postural hypotension are excluded (Table 13.4).
Table 13.4 Postural hypotension: some causes of a fall in blood pressure from lying to standing
Low jugular venous pressure
In hypovolaemic patients, the jugular venous pulsation can be seen only with the patient lying completely flat, or even head down, because the right atrial pressure is lower than 5 cmH2O.
Causes
Salt and water may be lost from the kidneys, from the gastrointestinal tract, or from the skin. Examples are given in Table 13.5.
Table 13.5 Causes of extracellular volume depletion
In addition, there are a number of situations where signs of volume depletion occur despite a normal or increased body content of sodium and water.
Septicaemia causes vasodilatation of both arterioles and veins, resulting in greatly increased capacitance of the vascular space. In addition, increased capillary permeability to plasma proteins leads to loss of fluid from the vascular space to the interstitium.
Diuretic treatment of heart failure or nephrotic syndrome may lead to rapid reduction in plasma volume. Mobilization of oedema may take much longer.
There may be inappropriate diuretic treatment of oedema (e.g. when the cause is local rather than systemic).
Investigations
Blood tests are in general not helpful in the assessment of extracellular volume. Plasma urea may be raised owing to increased urea reabsorption and, later, to prerenal failure (when the creatinine rises as well), but this is very nonspecific. Urinary sodium is low if the kidneys are functioning normally, but is misleading if the cause of the volume depletion involves the kidneys (e.g. diuretics, intrinsic renal disease). Urine osmolality is high in volume depletion (owing to increased water reabsorption), but may also often mislead.
Assessment of volume status is shown in Box 13.1.
 Box 13.1
Box 13.1
Assessment of volume status
Best achieved by simple clinical observations which you should do yourself. Check:
Central venous pressure both basal and after intravenous fluid challenge (p. 873)
Treatment
The overriding principle is to replace what is missing.
Haemorrhage
The rational treatment of acute haemorrhage is the infusion of a combination of red cells and a plasma substitute or (if unavailable) whole blood. (Chronic anaemia causes salt and water retention rather than volume depletion by a mechanism common to conditions with peripheral vasodilatation.)
Loss of plasma
Loss of plasma, as occurs in burns or severe peritonitis, should be treated with human plasma or a plasma substitute (see p. 390).
Loss of water and electrolytes
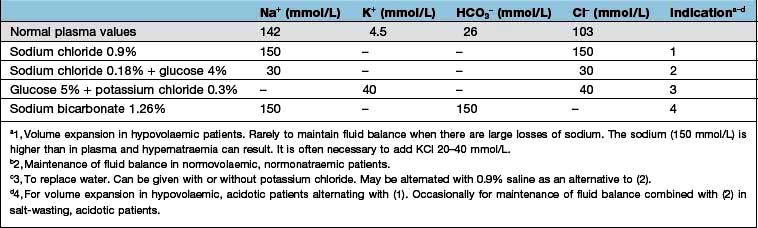
Loss of water and electrolytes, as occurs with vomiting, diarrhoea, or excessive renal losses, should be treated by replacement of the loss. If possible, this should be with oral water and sodium salts. These are available as slow sodium (600 mg, approximately 10 mmol each of Na+ and Cl− per tablet), the usual dose of which is 6–12 tablets/day with 2–3 L of water. It is used in mild or chronic salt and water depletion, such as that associated with renal salt wasting.
Sodium bicarbonate (500 mg, 6 mmol each of Na+ and HCO3− per tablet) is used in doses of 6–12 tablets/day with 2–3 L of water. This is used in milder chronic sodium depletion with acidosis (e.g. chronic kidney disease, post-obstructive renal failure, renal tubular acidosis). Sodium bicarbonate is less effective than sodium chloride in causing positive sodium balance. Oral rehydration solutions are described in Box 4.10.
Intravenous fluids are sometimes required (Table 13.6). Rapid infusion (e.g. 1000 mL per hour or even faster) is necessary if there is hypotension and evidence of impaired organ perfusion (e.g. oliguria, confusion); in these situations, plasma expanders (colloids) are often used in the first instance to restore an adequate circulating volume (see p. 887). Repeated clinical assessments are vital in this situation, usually complemented by frequent measurements of central venous pressure (see p. 872, for the management of shock). Severe hypovolaemia induces venoconstriction, which maintains venous return; over-rapid correction does not give time for this to reverse, resulting in signs of circulatory overload (e.g. pulmonary oedema) even if a total body extracellular fluid (ECF) deficit remains. In less severe ECF depletion (such as in a patient with postural hypotension complicating acute tubular necrosis), the fluid should be replaced at a rate of 1000 mL every 4–6 h, again with repeated clinical assessment. If all that is required is avoidance of fluid depletion during surgery, 1–2 L can be given over 24 h, remembering that surgery is a stimulus to sodium and water retention and that over-replacement may be as dangerous as under-replacement. Regular monitoring by fluid balance charts, bodyweight and plasma biochemistry is mandatory.
Loss of water alone
This causes extracellular volume depletion only in severe cases, because the loss is spread evenly among all the compartments of body water. In the rare situations where there is a true deficiency of water alone, as in diabetes insipidus or in a patient who is unable to drink (e.g. after surgery), the correct treatment is to give water.
If intravenous treatment is required, water is given as 5% glucose with K+, because pure water would lead to osmotic lysis of blood cells.
FURTHER READING
Ahmed MS, Wong CF, Pai P. Cardiorenal syndrome – a new classification and current evidence on its management. Clin Nephrol 2010; 74(4):245–257.
Bie P. Blood volume, blood pressure and total body sodium: internal signaling and output control. Acta Physiol (Oxford) 2009; 195(1):187–196.
Bie P, Damkjaer M. Renin secretion and total body sodium: pathways of integrative control. Clin Exp Pharmacol Physiol 2010; 37(2):e34–42.
Schrier RW. Molecular mechanisms of clinical concentrating and diluting disorders. Prog Brain Res 2008; 170:539–550.
Wakil A, Atkin SL. Serum sodium disorders: safe management. Clin Med 2010; 10:79–82.
Disorders of sodium concentration
These are best thought of as disorders of body water content. As discussed above, sodium content is regulated by volume receptors; water content is adjusted to maintain, in health, a normal osmolality and (in the absence of abnormal osmotically active solutes) a normal sodium concentration. Disturbances of sodium concentration are caused by disturbances of water balance.
Hyponatraemia
Hyponatraemia (Na <135 mmol/L) is a common biochemical abnormality. The causes depend on the associated changes in extracellular volume:
Hyponatraemia with hypovolaemia (Table 13.7)
Hyponatraemia with euvolaemia (Table 13.8)
Hyponatraemia with hypervolaemia (Table 13.9).
Table 13.7 Causes of hyponatraemia with decreased extracellular volume (hypovolaemia)
| Extrarenal (urinary sodium <20 mmol/L) | Kidney (urinary sodium >20 mmol/L) |
|---|---|
Table 13.8 Causes of hyponatraemia with normal extracellular volume (euvolaemia)
|
Syndrome of inappropriate antidiuretic hormone (see Table 19.36) |
Table 13.9 Causes of hyponatraemia with increased extracellular volume (hypervolaemia)
Heart failure |
Oliguric kidney injury |
Liver failure |
Hypoalbuminaemia |
Rarely, hyponatraemia may be a ‘pseudo-hyponatraemia’. This occurs in hyperlipidaemia (either high cholesterol or high triglyceride) or hyperproteinaemia where there is a spuriously low measured sodium concentration, the sodium being confined to the aqueous phase but having its concentration expressed in terms of the total volume of plasma. In this situation, plasma osmolality is normal and therefore treatment of ‘hyponatraemia’ is unnecessary. Note: Artefactual ‘hyponatraemia’, caused by taking blood from the limb into which fluid of low sodium concentration is being infused, should be excluded.
Hyponatraemia with hypovolaemia
This is due to salt loss in excess of water loss; the causes are listed in Table 13.7. In this situation, ADH secretion is initially suppressed (via the hypothalamic osmoreceptors); but as fluid volume is lost, volume receptors override the osmoreceptors and stimulate both thirst and the release of ADH. This is an attempt by the body to defend circulating volume at the expense of osmolality.
With extrarenal losses and normal kidneys, the urinary excretion of sodium falls in response to the volume depletion, as does water excretion, leading to concentrated urine containing <10 mmol/L of sodium. However, in salt-wasting kidney disease, renal compensation cannot occur and the only physiological protection is increased water intake in response to thirst.
Clinical features
With sodium depletion the clinical picture is usually dominated by features of volume depletion (see p. 638). The diagnosis is usually obvious where there is a history of gut losses, diabetes mellitus or diuretic abuse. Examination of the patient is often more helpful than the biochemical investigations, which include plasma and urine electrolytes and osmolality.
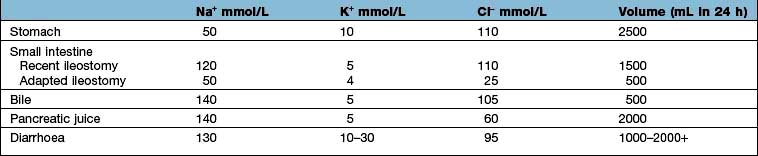
Table 13.10 shows the potential daily losses of water and electrolytes from the gut. Losses due to renal or adrenocortical disease may be less easily identified but a urinary sodium concentration of >20 mmol/L, in the presence of clinically evident volume depletion, suggests a renal loss.
Treatment
This is directed at the primary cause whenever possible.
Give oral electrolyte-glucose mixtures (see p. 122)
Hyponatraemia with euvolaemia (see Table 13.8)
This results from an intake of water in excess of the kidney’s ability to excrete it (dilutional hyponatraemia) with no change in body sodium content but the plasma osmolality is low.
With normal kidney function, dilution hyponatraemia is uncommon even if a patient drinks approximately 1 L per hour.
The most common iatrogenic cause is overgenerous infusion of 5% glucose into postoperative patients; in this situation it is exacerbated by an increased ADH secretion in response to stress.
Postoperative hyponatraemia is a common clinical problem (almost 1% of patients) with symptomatic hyponatraemia occurring in 20% of these patients.
Marathon runners drinking excess water and ‘sports drinks’ can become hyponatraemic.
Premenopausal females are at most risk for developing hyponatraemic encephalopathy postoperatively, with postoperative ADH values in young females being 40 times higher than in young males.
To prevent hyponatraemia, avoid using hypotonic fluids postoperatively and administer 0.9% saline unless otherwise clinically contraindicated. The serum sodium should be measured daily in any patient receiving continuous parenteral fluid.
Some degree of hyponatraemia is usual in acute oliguric kidney injury, while in chronic kidney disease (CKD) it is most often due to ill-given advice to ‘push’ fluids.
Clinical features
Dilutional hyponatraemia symptoms are common when hyponatraemia develops acutely (<48 h, often postoperatively). Symptoms rarely occur until the serum sodium is less than 120 mmol/L and are more usually associated with values around 110 mmol/L or lower, particularly when chronic. They are principally neurological and are due to the movement of water into brain cells in response to the fall in extracellular osmolality.
Hyponatraemic encephalopathy symptoms and signs include headache, confusion and restlessness leading to drowsiness, myoclonic jerks, generalized convulsions and eventually coma. MRI scan of the brain reveals cerebral oedema but, in the context of electrolyte abnormalities and neurological symptoms, it can help to make a confirmatory diagnosis.
Risk factors for developing hyponatraemic encephalopathy. The brain’s adaptation to hyponatraemia initially involves extrusion of blood and CSF, as well as sodium, potassium and organic osmolytes, in order to decrease brain osmolality. Various factors can interfere with successful adaptation. These factors rather than the absolute change in serum sodium predict whether a patient will suffer hyponatraemic encephalopathy.
Children under 16 years are at increased risk due to their relatively larger brain-to-intracranial volume ratio compared with adults.
Premenopausal women are more likely to develop encephalopathy than postmenopausal females and males because of inhibitory effects of sex hormones and the effects of vasopressin on cerebral circulation resulting in vasoconstriction and hypoperfusion of brain.
Hypoxaemia is a major risk factor for hyponatraemic encephalopathy. Patients with hyponatraemia, who develop hypoxia due to either non-cardiac pulmonary oedema or hypercapnic respiratory failure, have a high risk of mortality. Hypoxia is the strongest predictor of mortality in patients with symptomatic hyponatraemia.
Investigations
The cause of hyponatraemia with apparently normal extracellular volume requires investigation:
Plasma and urine electrolytes and osmolalities. The plasma concentrations of sodium, chloride and urea are low, giving a low osmolality. The urine sodium concentration is usually high and the urine osmolality is typically higher than the plasma osmolality. However, maximal dilution (<50 mosmol/kg) is not always present.
Further investigations to exclude Addison’s disease, hypothyroidism, ‘syndrome of inappropriate ADH secretion’ (SIADH) and drug-induced water retention, e.g. chlorpropamide.
Remember, potassium and magnesium depletion potentiate ADH release and are causes of diuretic-associated hyponatraemia.
The syndrome of inappropriate ADH secretion is often over-diagnosed. Some causes are associated with a lower set-point for ADH release, rather than completely autonomous ADH release – an example is chronic alcohol use.
Treatment
The underlying cause should be corrected where possible.
Most cases are simply managed by restriction of water intake (to 1000 or even 500 mL/day) with review of diuretic therapy. Magnesium and potassium deficiency must be corrected. In mild sodium deficiency, 0.9% saline given slowly (1 L over 12 hours) is sufficient.
Acute onset with symptoms. The most common cause of acute hyponatraemia in adults is postoperative iatrogenic hyponatraemia. Excessive water intake associated with psychosis, marathon running and use of Ecstasy (a recreational drug) are other causes. All are acute medical emergencies and should be treated aggressively and immediately. In patients in whom there are severe neurological signs, such as fits or coma or cerebral oedema, hypertonic saline (3%, 513 mmol/L) should be used. It must be given very slowly (not more than 70 mmol/h), the aim being to increase the serum sodium by 4–6 mmol/L in the first 4 hours, but the absolute change should not exceed 15–20 mmol/L over 48 hours. In general, the plasma sodium should not be corrected to >125–130 mmol/L. 1 mL/kg of 3% sodium chloride will raise the plasma sodium by 1 mmol/L, assuming that total body water comprises 50% of total bodyweight.
Symptomatic hyponatraemia in patients with intracranial pathology should be managed aggressively and immediately with 3% saline like acute hyponatraemia.
Chronic/asymptomatic. If hyponatraemia has developed slowly, as it does in the majority of patients, the brain will have adapted by decreasing intracellular osmolality and the hyponatraemia can be corrected slowly (without use of hypertonic saline).
However, clinically it can be difficult to know how long the hyponatraemia has been present and 3% of hypertonic saline is still required.
Osmotic demyelination syndrome (ODS)
Avoiding ODS
A rapid rise in extracellular osmolality, particularly if there is an ‘overshoot’ to high serum sodium and osmolality, will result in the osmotic demyelination, syndrome (ODS), formally known as central pontine demyelination, which is a devastating neurologic complication. Plasma sodium concentration in patients with hyponatraemia should not rise by more than 8 mmol/L per day. The rate of rise of plasma sodium should be even lower in patients at higher risk for ODS, e.g. patients with alcohol excess, cirrhosis, malnutrition, or hypokalaemia. Other factors predisposing to demyelination are pre-existing hypoxaemia and CNS radiation (see above). ODS is diagnosed by the appearance of characteristic hypointense lesions on T1-weighted images and hyperintense on T2-weighted images on MRI; these take up to 2 weeks or longer to appear.
The pathophysiology of ODS is not fully understood. The most plausible explanation is that the brain loses organic osmolytes very quickly in order to adapt to hyponatraemia so that osmolarity is similar between the intracellular and extracellular compartments. However, neurones reclaim organic osmolytes slowly in the phase of rapid correction of hyponatraemia, resulting in an hypo-osmolar intracellular compartment and lead to shrinkage of cerebral vascular endothelial cells. Consequently the blood–brain barrier is functionally impaired, allowing lymphocytes, complement, and cytokines to enter the brain, damage oligodendrocytes, activate microglial cells and cause demyelination.
The most crucial issue in the treatment of hyponatraemia is to prevent rapid correction. A rapid rise in plasma sodium is almost always due to a water diuresis, which happens when vasopressin (ADH) action stops suddenly, for example with volume repletion in patients with intravascular volume depletion, cortisol replacement in patients with Addison disease, resolution of non-osmotic stimuli for vasopressin release such as nausea or pain. However, sometimes chronic hyponatraemia can develop in the absence of vasopressin excess. Even in these cases, water diuresis due to increased distal delivery of filtrate is the main cause of rapid rise in plasma sodium.
In the absence of vasopressin, it is generally assumed that the total urine volume is equal to the volume of filtrate delivered to the distal nephron, which is the GFR minus the volume reabsorbed in the proximal convoluted tubule (PCT). Approximately 80% of the GFR is reabsorbed in PCT under normal circumstance (increases even more in the presence of intravascular volume depletion). However, in real life water excretion will be less than the volume of distal delivery of filtrate, even in the absence of vasopressin, because a significant degree of water is reabsorbed in the inner medullary collecting duct through its residual water permeability, prompted by a very high osmotic force in the interstitium (see Fig. 12.2).
Even a modest water diuresis in the elderly with reduced muscle mass is large enough to cause a rapid rise in plasma sodium. Moreover, there is a higher risk for ODS if hypokalaemia is present. In such cases if plasma sodium rises too quickly due to anticipated water diuresis, administration of desmopressin to stop the water diuresis is beneficial. If plasma sodium rises regardless then lowering plasma sodium to the maximum limit of correction (<8 mmol/L per day) with the administration of 5% glucose solution is the best strategy.
Reversible hyponatraemia culminating in hypernatraemia
In many patients, the cause of water retention is reversible (e.g. hypovolaemia, thiazide diuretics). On correction of the cause, vasopressin levels fall and plasma sodium rises by up to 2 mmol/L per hour as a result of excretion of dilute urine. This excessive water diuresis should be anticipated and prevented by use of desmopressin.
Patients who are chronically hyponatraemic with concomitant hypokalaemia are especially susceptible to overcorrection. Plasma sodium is a function of the ratio of exchangeable body sodium plus potassium to total body water, so potassium administration increases sodium concentration. For example, a mildly symptomatic hyponatraemic patient with a plasma sodium of <120 mmol/L and potassium of <2 mmol/L can potentially develop ODS as a result of overcorrection of hyponatraemia simply as a direct result of replacing the large potassium deficit.
Antidiuretic hormone antagonists (vasopressin antagonists)
Vasopressin V2 receptor antagonists (see p. 645), which produce a free water diuresis, are being used in clinical trials for the treatment of hyponatraemic encephalopathy. Three oral agents, lixivaptan, tolvaptan and satavaptan, are selective for the V2 (antidiuretic) receptor, while conivaptan blocks both the V1A and V2 receptors.
These agents produce a selective water diuresis without affecting sodium and potassium excretion; they raise the plasma sodium concentration in patients with hyponatraemia caused by the SIADH, heart failure and cirrhosis.
The efficacy of oral tolvaptan in ambulatory patients has been demonstrated in patients with hyponatraemia (mean plasma sodium 129 mmol/L) caused by the SIADH, heart failure, or cirrhosis who had a sustained rise in plasma sodium to 136 mmol/L for 4 weeks. Tolvaptan is now approved for use in patients with euvolaemic hyponatraemia and those with SIADH. In addition, intravenous conivaptan is available and is also approved for the treatment of euvolaemic hyponatraemia (i.e. SIADH) in some countries. The approved dosing for conivaptan is a 20 mg bolus followed by continuous infusion of 20 mg over 1–4 days. The continuous infusion increases the risk of phlebitis, which requires the use of large veins and changing the infusion site every 24 hours.
FURTHER READING
Nemerovski C, Hutchinson DJ. Treatment of hypervolemic or euvolemic hyponatremia associated with heart failure, cirrhosis, or the syndrome of inappropriate antidiuretic hormone with tolvaptan: a clinical review. Clin Ther 2010; 32(6):1015–1032.
Rozen-Zvi B, Yahav D, Gheorghiade M et al. Vasopressin receptor antagonists for the treatment of hyponatremia: systematic review and meta-analysis. Am J Kidney Dis 2010; 56(2):325–337.
Hyponatraemia with hypervolaemia
The common causes of hyponatraemia due to water excess are shown in Table 13.9. In all these conditions, there is usually an element of reduced glomerular filtration rate with avid reabsorption of sodium and chloride in the proximal tubule. This leads to reduced delivery of chloride to the ‘diluting’ ascending limb of Henle’s loop and a reduced ability to generate ‘free water’, with a consequent inability to excrete dilute urine. This is commonly compounded by the administration of diuretics that block chloride reabsorption and interfere with the dilution of filtrate either in Henle’s loop (loop diuretics) or distally (thiazides).
Syndrome of inappropriate ADH secretion
This is described in Chapter 19 (p. 746). There is inappropriate secretion of ADH, causing water retention and hyponatraemia.
Hypernatraemia
This is much rarer than hyponatraemia and nearly always indicates a water deficit. Causes are listed in Table 13.11).
Table 13.11 Causes of hypernatraemia
|
|
|
Hypernatraemia is always associated with increased plasma osmolality, which is a potent stimulus to thirst. None of the above cause hypernatraemia unless thirst sensation is abnormal or access to water limited. For instance, a patient with diabetes insipidus will maintain a normal serum sodium concentration by maintaining a high water intake until an intercurrent illness prevents this. Thirst is frequently deficient in elderly people, making them more prone to water depletion. Hypernatraemia may occur in the presence of normal, reduced or expanded extracellular volume, and does not necessarily imply that total body sodium is increased.
Clinical features
Symptoms of hypernatraemia are nonspecific. Nausea, vomiting, fever and confusion may occur. A history of longstanding polyuria, polydipsia and thirst suggests diabetes insipidus. Assessment of extracellular volume status guides resuscitation. Mental state should be assessed. Convulsions occur in severe hypernatraemia.
Investigations
Simultaneous urine and plasma osmolality and sodium should be measured. Plasma osmolality is high in hypernatraemia. Passage of urine with an osmolality lower than that of plasma in this situation is clearly abnormal and indicates diabetes insipidus. In pituitary diabetes insipidus, urine osmolality will increase after administration of desmopressin; the drug (a vasopressin analogue) has no effect in nephrogenic diabetes insipidus. If urine osmolality is high this suggests either an osmotic diuresis due to an unmeasured solute (e.g. in parenteral feeding) or excessive extrarenal loss of water (e.g. heat stroke).
Treatment
Treatment is that of the underlying cause, e.g.
In ADH deficiency, replace ADH in the form of desmopressin, a stable non-pressor analogue of ADH
Remember to withdraw nephrotoxic drugs where possible and replace water either orally or, if necessary, intravenously.
In severe (>170 mmol/L) hypernatraemia, 0.9% saline (150 mmol/L) should be used initially. Avoid too rapid a drop in serum sodium concentration; the aim is correction over 48 h, as over-rapid correction may lead to cerebral oedema.
In less severe (e.g. >150 mmol/L) hypernatraemia, the treatment is 5% glucose or 0.45% saline; the latter is obviously preferable in hyperosmolar diabetic coma. Very large volumes – 5 L/day or more – may need to be given in diabetes insipidus.
If there is clinical evidence of volume depletion (see p. 646), this implies that there is a sodium deficit as well as a water deficit. Treatment of this is discussed on page 647.
Disorders of potassium concentration
Regulation of serum potassium concentration
The usual dietary intake varies between 80 and 150 mmol daily, depending upon fruit and vegetable intake. Most of the body’s potassium (3500 mmol in an adult man) is intracellular. Serum potassium levels are controlled by:
Uptake of potassium into cells is governed by the activity of the Na+/K+-ATPase in the cell membrane and by H+ concentration.
acidosis – K+ exchanged for H+ across cell membrane
cell damage or cell death – resulting in massive K+ release.
Kidney plays the pivotal role in the maintenance of potassium balance by varying its secretion with changes in dietary intake. Over 90% of the filtered potassium is reabsorbed in the proximal tubule and the loop of Henle and only <10% of the filtered load is delivered to the early distal tubule. Potassium absorption on proximal tubule is entirely passive and follows that of sodium and water, while its reabsorption in the thick ascending limb of the loop of Henle is mediated by the sodium-potassium-2-chloride cotransporter. However, potassium is secreted by the principal cells in the cortical and outer medullary collecting tubule. Secretion in these segments is very tightly regulated in health and can be varied according to individuals needs and is responsible for most of urinary potassium excretion.
Renal excretion of potassium is increased by aldosterone, which stimulates K+ and H+ secretion in exchange for Na+ in the principal cells of the collecting duct (Fig. 13.8). Because H+ and K+ are interchangeable in the exchange mechanism, acidosis decreases and alkalosis increases the secretion of K+. Aldosterone secretion is stimulated by hyperkalaemia and increased angiotensin II levels, as well as by some drugs, and this acts to protect the body against hyperkalaemia and against extracellular volume depletion. The body adapts to dietary deficiency of potassium by reducing aldosterone secretion. However, because aldosterone is also influenced by volume status, conservation of potassium is relatively inefficient, and significant potassium depletion may therefore result from prolonged dietary deficiency.
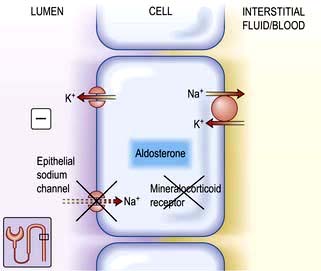
Figure 13.8 Aldosterone-regulated transport in the cortical collecting ducts. Under normal conditions, the epithelial sodium channel is the rate-limiting barrier for the normal entry of sodium from the lumen into the cell. The resulting lumen-negative transepithelial voltage (indicated by the minus sign) drives potassium secretion from the principal cells and proton secretion from the α-intercalated cells (see Fig. 13.11). In Liddle’s syndrome, a mutation in the gene encoding the epithelial sodium channel results in persistent unregulated reabsorption of sodium and increased secretion of potassium (not shown). In pseudohypoaldosteronism type I autosomal recessive, loss of function mutations (X) in this gene inactivate the channel. In the autosomal dominant variety, the mutation is in the gene encoding the mineralocorticoid regulation of the activity of the epithelial sodium channel. Either mechanism reduces the activity of the epithelial sodium channel, thus causing salt wasting and decreasing the secretion of potassium and protons.
A number of drugs affect K+ homeostasis by affecting aldosterone release (e.g. heparin, NSAIDs) or by directly affecting renal potassium handling (e.g. diuretics).
Recent evidence has shown that other endogenous proteins and metabolites also affect potassium homeostasis. Klotho, an anti-ageing protein expressed in the distal tubule (and other organs), increases potassium excretion. CD63, a tetra-spanning protein, inhibits its excretion. Moreover, protein kinase A and C mediated phosphorylation inhibits conductance K channels in the principal cells of the collecting duct but the cytochrome p450-epoxygenase-mediated metabolite of arachidonic acid (11–12-epoxyeicosatrienoic acid) activates these channels and plays a role in overall potassium homeostasis.
Normally, only about 10% of daily potassium intake is excreted in the gastrointestinal tract. Vomit contains around 5–10 mmol/L of K+, but prolonged vomiting causes hypokalaemia by inducing sodium depletion, stimulating aldosterone, which increases renal potassium excretion. Potassium is secreted by the colon, and diarrhoea contains 10–30 mmol/L of K+; profuse diarrhoea can therefore induce marked hypokalaemia. Colorectal villous adenomas may rarely produce profuse diarrhoea and K+ loss.
Hypokalaemia
Common causes
Causes
The most common causes of chronic hypokalaemia are diuretic treatment (particularly thiazides) and hyperaldosteronism. Acute hypokalaemia is often caused by intravenous fluids without potassium and redistribution into cells. The common causes are shown in Table 13.12.
Table 13.12 Causes of hypokalaemia
|
|
Rare causes
These rare causes are discussed in detail because they show the mechanisms of how diuretics can affect the kidney.
Bartter’s syndrome (clinically similar to loop diuretics)
This consists of metabolic alkalosis, hypokalaemia, hypercalciuria, occasionally hypomagnesaemia (see p. 657), normal blood pressure, and an elevated plasma renin and aldosterone. The primary defect in this disorder is an impairment in sodium and chloride reabsorption in the thick ascending limb of the loop of Henle (Fig. 13.6). Mutation in the genes encoding either the sodium-potassium-2-chloride cotransporter (NKCC2), the ATP-regulated renal outer medullary potassium channel (ROMK) or kidney-specific basolateral chloride channels (ClC-Kb) – Bartter types I, II and III, respectively – causes loss of function of these channels, with consequent impairment of sodium and chloride reabsorption. There is also an increased intrarenal production of prostaglandin E2 which is secondary to sodium and volume depletion, hypokalaemia and the consequent neurohumoral response rather than a primary defect. PGE2 causes vasodilatation and may explain why the blood pressure remains normal.
Barttin, a β-subunit for ClC-Ka and ClC-Kb chloride channels, is encoded by the BSND (Bartter’s syndrome with sensorineural deafness) gene. Loss of function mutations cause type IV Bartter’s syndrome associated with sensorineural deafness and renal failure. Barttin co-localizes with a subunit of the chloride channel in basolateral membranes of the renal tubule and inner ear epithelium. It appears to mediate chloride exit in the thick ascending limb (TAL) of the loop of Henle and chloride recycling in potassium-secreting strial marginal cells in the inner ear. A very rare variant of type IV is a disorder with an impairment of both chloride channels (ClC-Ka and ClC-Kb) producing the same phenotypic defects.
A gain of function mutation of the calcium sensing receptor (CaSR) which leads to autosomal dominant hypocalcaemia has also been recognized in Bartter’s syndrome. In the kidney, the CaSR is expressed mainly in the basolateral membrane of cortical TAL. Activation of CaSR by high calcium or magnesium or by gain of function mutation triggers intracellular signalling, including release of arachidonic acid and inhibition of adenylate cyclase. Both actions result in inhibition of ROMK activity, which in turn leads to reduction in the lumen-positive electrical potential and transcellular absorption of calcium. This effect of CaSR explains why patients with mutations in this receptor may present with both hypocalcaemia, hypercalciuria and renal wasting of NaCl, resulting in a Bartter-like syndrome.
In summary, these defects in sodium chloride transport are thought to initiate the following sequence, which is almost identical to that seen with chronic ingestion of a loop diuretic. The initial salt loss leads to mild volume depletion, resulting in activation of the renin-angiotensin-aldosterone system. The combination of hyperaldosteronism and increased distal flow (owing to the reabsorptive defect) enhances potassium and hydrogen secretion at the secretory sites in the collecting tubules, leading to hypokalaemia and metabolic alkalosis.
Diagnostic pointers include high urinary potassium and chloride despite low serum values as well as increased plasma renin (NB: in primary aldosteronism, renin levels are low). Hyperplasia of the juxtaglomerular apparatus is seen on renal biopsy (careful exclusion of diuretic abuse is necessary). Hypercalciuria is a common feature but magnesium wasting, though rare, also occurs.
Treatment is with combinations of potassium supplements, amiloride and indomethacin.
Gitelman’s syndrome (similar to thiazide diuretics)
Gitelman’s syndrome is a phenotype variant of Bartter’s syndrome characterized by hypokalaemia, metabolic alkalosis, hypocalciuria, hypomagnesaemia, normal blood pressure, and elevated plasma renin and aldosterone. There are striking similarities between the Gitelman’s syndrome and the biochemical abnormalities induced by chronic thiazide diuretic administration. Thiazides act in the distal convoluted tubule to inhibit the function of the apical sodium-chloride cotransporter (NCCT) (Fig. 13.7). Analysis of the gene encoding the NCCT has identified loss of function mutations in Gitelman’s syndrome.
Like Bartter’s syndrome, defective NCCT function leads to increased solute delivery to the collecting duct, with resultant solute wasting, volume contraction and an aldosterone-mediated increase in potassium and hydrogen secretion. Unlike Bartter’s syndrome, the degree of volume depletion and hypokalaemia is not sufficient to stimulate prostaglandin E2 production. Impaired function of NCCT is predicted to cause hypocalciuria, as does thiazide administration. Impaired sodium reabsorption across the apical membrane, coupled with continued intracellular chloride efflux across the basolateral membrane, causes the cell to become hyperpolarized. This in turn stimulates calcium reabsorption via apical, voltage-activated calcium channels. Decreased intracellular sodium also facilitates calcium efflux via the basolateral sodium-calcium exchanger. The mechanism for urinary magnesium losses is described on page 656.
Treatment consists of potassium and magnesium supplementation (MgCl2) and a potassium-sparing diuretic. Volume resuscitation is usually not necessary, because patients are not dehydrated. Elevated prostaglandin E2 does not occur (see above) and, therefore, NSAIDs are not indicated in this disorder.
Liddle’s syndrome
This is characterized by potassium wasting, hypokalaemia and alkalosis, but is associated with low renin and aldosterone production, and high blood pressure. There is a mutation in the gene encoding for the amiloride-sensitive epithelial sodium channel in the distal tubule/collecting duct. This leads to constitutive activation of the epithelial sodium channel, resulting in excessive sodium reabsorption with coupled potassium and hydrogen secretion. Unregulated sodium reabsorption across the collecting tubule results in volume expansion, inhibition of renin and aldosterone secretion and development of low renin hypertension (Fig. 13.8).
Therapy consists of sodium restriction along with amiloride or triamterene administration. Both are potassium-sparing diuretics which directly close the sodium channels. The mineralocorticoid antagonist spironolactone is ineffective, since the increase in sodium-channel activity is not mediated by aldosterone.
Clinical features
Hypokalaemia is usually asymptomatic, but severe hypokalaemia (<2.5 mmol) causes muscle weakness. Potassium depletion may also cause symptomatic hyponatraemia (see p. 648).
Hypokalaemia is associated with an increased frequency of atrial and ventricular ectopic beats. This association may not always be causal, because adrenergic activation (for instance after myocardial infarction) causes both hypokalaemia and increased cardiac irritability. Hypokalaemia in patients without cardiac disease is unlikely to lead to serious arrhythmias.
Hypokalaemia seriously increases the risk of digoxin toxicity by increasing binding of digoxin to cardiac cells, potentiating its action, and decreasing its clearance.
Chronic hypokalaemia is associated with interstitial renal disease, but the pathogenesis is not completely understood.
Treatment
The underlying cause should be identified and treated where possible. Table 13.13 shows some examples.
Table 13.13 Treatment of hypokalaemia
| Cause | Treatment |
|---|---|
Dietary deficiency |
Increase intake of fresh fruit/vegetables or oral potassium supplements (20–40 mmol daily). (Potassium supplements can cause gastrointestinal irritation) |
Hyperaldosteronism, e.g. cirrhosis, thiazide therapy |
Spironolactone/eplerenone. Co-prescription of a potassium-sparing diuretic with a similar onset and duration of action |
Intravenous fluid replacement |
Add 20 mmol of K+/L of fluid with monitoring |
Acute hypokalaemia may correct spontaneously. In most cases, withdrawal of oral diuretics or purgatives, accompanied by the oral administration of potassium supplements in the form of slow-release potassium or effervescent potassium, is all that is required. Intravenous potassium replacement is required only in conditions such as cardiac arrhythmias, muscle weakness or severe diabetic ketoacidosis. When using intravenous therapy in the presence of poor renal function, replacement rates <2 mmol per hour should be used only, with hourly monitoring of serum potassium and ECG changes. Ampoules of potassium should be thoroughly mixed in 0.9% saline; do not use a glucose solution as this would make hypokalaemia worse.
The treatment of adrenal disorders is described on page 958.
Failure to correct hypokalaemia may be due to concurrent hypomagnesaemia. Serum magnesium should be measured and any deficiency corrected.
Hyperkalaemia
Causes
Acute self-limiting hyperkalaemia occurs normally after vigorous exercise and is of no pathological significance. Hyperkalaemia in all other situations is due either to increased release from cells or to failure of excretion (Table 13.14). The most common causes are renal impairment and drug interference with potassium excretion. The combination of ACE inhibitors with potassium-sparing diuretics or NSAIDs is particularly dangerous.
Table 13.14 Causes of hyperkalaemia
|
|
Rare causes
Hyporeninaemic hypoaldosteronism
This is also known as type 4 renal tubular acidosis (see p. 664). Hyperkalaemia occurs because of acidosis and hypoaldosteronism.
Pseudohypoaldosteronism type 1 (autosomal recessive and dominant types)
This is a disease of infancy, apparently due to resistance to the action of aldosterone. It is characterized by hyperkalaemia and evidence of sodium wasting (hyponatraemia, extracellular volume depletion). Autosomal recessive forms result from loss of function because of mutations in the gene for epithelial sodium channel activity (opposite to Liddle’s syndrome). This disorder involves multiple organ systems and is especially marked in the neonatal period. With aggressive salt replacement and control of hyperkalaemia, these children can survive and the disorder appears to become less severe with age. The autosomal dominant type is due to mutations affecting the mineralocorticoid receptor (Fig. 13.8). These patients present with salt wasting and hyperkalaemia but do not have other organ-system involvement.
Hyperkalaemic periodic paralysis (see p. 1153)
This is precipitated by exercise, and is caused by an autosomal dominant mutation of the skeletal muscle sodium channel gene.
Gordon’s syndrome (familial hyperkalaemic hypertension, pseudohypoaldosteronism type 2)
This appears to be a mirror image of Gitelman’s syndrome (see p. 653), in which primary renal retention of sodium causes hypertension, volume expansion, low renin/aldosterone, hyperkalaemia and metabolic acidosis. There is also an increased sensitivity of sodium reabsorption to thiazide diuretics, suggesting that the thiazide-sensitive sodium-chloride cotransporter (NCCT) is involved. Genetic analyses, however, have excluded abnormalities in NCCT. The involvement of two loci on chromosomes 1 and 12 and further genetic heterogeneity has also been found. These genes do not correspond to ionic transporters but to unexpected proteins, WNK (With No lysine Kinase) 1 and WNK 4, which are two closely related members of a novel serine–threonine kinase family. WNK 4 normally inhibits NCCT by preventing its membrane translocation from the cytoplasm. Loss of function mutation in WNK 4 results in escape of NCCT from normal inhibition and its overactivity as seen from the patient’s phenotype. WNK 1 is an inhibitor of WNK 4 and in some patients with Gordon’s syndrome, gain of function mutation in WNK 1 results in functional deficiency of WNK 4 and overactivity of NCCT.
Clinical features
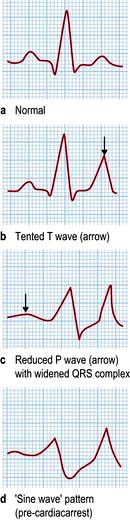
Serum potassium of >7.0 mmol/L is a medical emergency and is associated with ECG changes. Severe hyperkalaemia may be asymptomatic and may predispose to sudden death from asystolic cardiac arrest. Muscle weakness is often the only symptom, unless (as is commonly the case) the hyperkalaemia is associated with metabolic acidosis, causing Kussmaul respiration. Hyperkalaemia causes depolarization of cell membranes, leading to decreased cardiac excitability, hypotension, bradycardia and eventual asystole.
Treatment
Treatment for severe hyperkalaemia requires both urgent measures to save lives and maintenance therapy to keep potassium down, as summarized in Emergency Box 13.1. The cause of the hyperkalaemia should be found and treated.
 Emergency Box 13.1
Emergency Box 13.1
Correction of severe hyperkalaemia
High potassium levels are cardiotoxic as they inactivate sodium channels. Divalent cations, e.g. calcium, restore the voltage dependability of the channels. Calcium ions protect the cell membranes from the effects of hyperkalaemia but do not alter the potassium concentration.
Supraphysiological insulin (20 units) drives potassium into the cell and lowers plasma potassium by 1 mmol in 60 min, but must be accompanied by glucose to avoid hypoglycaemia. Regular measurements of blood glucose for at least 6 h after use of insulin should be performed and extra glucose must be available for immediate use. The use of glucose alone in non-diabetic patients, to stimulate endogenous insulin release, does not produce the high levels of insulin required and therefore is not recommended.
Intravenous or nebulized salbutamol (10–20 mg) has not yet found widespread acceptance and may cause disturbing muscle tremors at the doses required.
Correction of acidosis with hypertonic (8.4%) sodium bicarbonate causes volume expansion and should not be used; 1.26% is used with severe acidosis (pH <6.9). Gastric aspiration will remove potassium and leads to alkalosis.
Ion-exchange resins (polystyrene sulphonate resins) are used as maintenance therapy to keep potassium down after emergency treatment. They make use of the ion fluxes which occur in the gut to remove potassium from the body, and are the only way short of dialysis of removing potassium from the body. They may cause fluid overload (resonium contains Na+) or hypercalcaemia (calcium resonium). Resins do not appear to significantly enhance the excretion of potassium beyond the effect of diarrhoea induced by osmotic or secretory cathartics.
In general, all of these measures are simply ways of buying time either to correct the underlying disorder or to arrange removal of potassium by dialysis, which is the definitive treatment for hyperkalaemia in renal failure.
FURTHER READING
Bubien JK. Epithelial Na+ channel (ENaC), hormones, and hypertension. J Biol Chem 2010; 285(31):23527–23531.
Flatman PW. Co-transporters, WNKs and hypertension: an update. Curr Opin Nephrol Hypertens 2008; 17(2):186–192.
Furgeson SB, Linas S. Mechanisms of type I and type II pseudohypoaldosteronism. J Am Soc Nephrol 2010; 21(11):1842–1845.
Wang WH, Yue P, Sun P et al. Regulation and function of potassium channels in aldosterone-sensitive distal nephron. Curr Opin Nephrol Hypertens 2010; 19(5):463–470.
Disorders of magnesium concentration
Magnesium (Mg2+) plays a pivotal role in many biological processes such as enzymatic reactions, gene transcription, bone remodelling, and neuromuscular stability. Approximately 99% of the Mg2+ in the body is in the intracellular compartment, mainly in bone (∼85%) and muscle and soft tissues (∼14%). The other 1% is in the extracellular fluid.
Plasma magnesium levels are normally maintained within the range 0.7–1.1 mmol/L (1.4–2.2 mEq/L). The average daily magnesium intake is 15 mmol, which is absorbed mainly in the small intestine and to a lesser extent in the colon. In the healthy adult, there is no net gain or loss of magnesium from bone, so that balance is achieved by the urinary excretion of the net magnesium absorbed. The kidney reabsorbs between approximately 95% and 98% of the filtered Mg2+ and plays a major role in maintaining plasma Mg2+ concentrations within the normal range.
Renal handling of magnesium
Cortical thick ascending limb of Henle (cTAL)
Approximately 30% of Mg2+ is bound to plasma proteins but the remaining fraction is freely filterable. The major site of magnesium transport is the cortical thick ascending limb (cTAL) of the loop of Henle, where 65–70% of the filtered load is reabsorbed with only 10–20% being reabsorbed in the proximal tubule (Fig. 13.6). This transport is passive, paracellular and carried out by tight junction proteins (paracellin-1 and claudins). This process is driven by the lumen-positive electrochemical gradient, characteristic of this segment. This voltage gradient is created by the apical disproportionate net transport of two Cl− to one Na+ (by the bumetanide-sensitive sodium-potassium-2-chloride transporter) and the secretion of K+ (via the ROMK) (see Fig. 13.6). Loss of function mutations in these key reabsorptive processes lead to hypomagnesaemia as part of distinctive clinical syndromes described below.
Bartter’s syndrome (p. 652). Hypomagnesaemia is not present in all patients with Bartter’s syndrome because expected dissipation of the luminal positive voltage gradient is prevented by lack of dilution of tubular fluid which maintains transepithelial voltage in the normal range. Compensatory increased absorption of Mg2+ in the distal convoluted tubule (DCT) can also partly prevent hypomagnesaemia in this condition.
Familial hypomagnesaemia, hypercalciuria and nephrocalcinosis (FHHNC) is characterized by excessive renal magnesium and calcium wasting. Individuals develop bilateral nephrocalcinosis and progressive chronic kidney disease. Patients also have elevated PTH levels, which precedes any reduction in GFR. A substantial proportion of patients show incomplete distal renal tubular acidosis, hypocitraturia and hyperuricaemia. Extrarenal involvement such as myopia, nystagmus, chorioretinitis has been reported. The main defect in magnesium and calcium reabsorption lies in cTAL. Ten different mutations have been identified in a novel gene which encodes for paracellin-1 and claudins 16/19 complex, members of the claudin family of tight junction proteins (see p. 23).
Distal convoluted tubule (DCT)
The reabsorption rate in the DCT (10%) is much lower than in the cTAL, but it defines the final urinary excretion, as there is no significant reabsorption in the collecting duct: 3–5% of filtered magnesium is finally excreted in the urine. Magnesium reabsorption in the DCT is transcellular and active (Fig. 13.7). The DCT has a slight lumen-negative voltage of approximately −5 mV. The luminal Mg2+ concentration in the DCT ranges between 0.2 and 0.7 mmol/L, whereas the intracellular concentration of Mg2+ is estimated to be maintained around 0.2–1.0 mmol/L. Therefore the voltage difference across the apical membrane plays a key role in Mg2+ transport within the DCT. Magnesiotropic proteins, including the transient receptor potential channel melastatin member 6 (TRPM6), the pro-epidermal growth factor (EGF), the potassium channels Kir4.1 as well as the hepatocyte nuclear factor 1B (HNF1B) are situated in DCT.
TRPM6 is a member of the transient receptor potential channel family. It is an Mg2+-permeable channel that is also expressed in the luminal membrane of the intestinal epithelium. Inactivating mutations of TRPM6, a rare autosomal recessive disease, thus cause a combination of impaired gut absorption of Mg2+ and renal wasting known as hypomagnesaemia with secondary hypocalcaemia (HSH). It is characterized by disturbed neuromuscular excitability, muscle spasms, tetany and generalized convulsions. Severe hypomagnesaemia is observed (0.1–0.4 mmol/L) due to impaired intestinal Mg2+ absorption and renal reabsorption.
The epidermal growth factor (EGF) resides on the basolateral surface of the DCT cells. It markedly stimulates the activity of TRPM6. Loss of function mutation results in an autosomal recessive form of isolated renal hypomagnesaemia (IRH). IRH presents with hypomagnesaemia (0.53–0.66 mmol/L) and an inappropriately high fractional excretion of Mg2+ with epileptic seizures and moderate mental retardation. In contrast to HSH, Ca2+ handling is not affected in these patients. Cancer therapies which inhibit EGF also cause hypomagnesaemia by the above mechanism.
Thiazide-sensitive Na+–Cl− cotransporter in the DCT plays a role in sodium and chloride absorption and maintenance of lumen-negative voltage. Loss of function mutation in this cotransporter results in Gitelman’s syndrome (p. 645; see Fig. 13.7). Hypomagnesaemia is likely due to a reduced abundance of TRPM6. The observed hypocalciuria is caused by an increased proximal tubular reabsorption, a process that occurs in response to the mild volume depletion.
The γ-subunit of the Na+–K+-ATPase on the basolateral aspect of DCT plays a pivotal role in the sodium and chloride absorption and maintenance of lumen-negative voltage (a key requirement for magnesium absorption) in this segment of the nephron. A loss of function mutation in the FXYD2 gene (transcription factor for gamma chain of Na+–K+-ATPase) causes isolated dominant hypomagnesaemia. The affected individuals present with renal Mg2+ wasting, accompanied by hypocalciuria.
The HNF1b gene encodes a transcription factor linked to the regulation of the FXYD2 gene. Defects in HNF1b gene have been implicated in genetic defects of beta-cell function (p. 1007). Interestingly, almost half of the carriers of a mutation in the HNF1b gene display hypomagnesaemia (<0.65 mmol/L) due to renal wasting of Mg2+. As in patients with FXYD2 mutations, hypocalciuria is present.
ATP-sensitive inward rectifier potassium channel 10 (Kir4.1) is present on the basolateral surface of DCT. It allows K+ ions to recycle across the basolateral membrane, thereby maintaining an adequate supply of K+ to sustain the high Na+–K+-ATPase activity observed in this segment. Loss of its function has been linked to a new hypomagnesaemic syndrome, EAST syndrome. The impaired electrogenic Na+–K+-ATPase transport causes depolarization of the apical membrane and reduces inward transport of Mg2+ via TRPM6. Patients have epilepsy, ataxia, sensorineural deafness (Kir 4.1 is present in the inner ear), and tubulopathy (of a Gitelman-like phenotype).
Hypomagnesaemia
In addition to the familial causes above, hypomagnesaemia most often develops as a result of deficient intake, defective gut absorption, or excessive gut or urinary loss (Table 13.15). It can also occur with acute pancreatitis, possibly owing to the formation of magnesium soaps in the areas of fat necrosis. The serum magnesium is usually <0.7 mmol/L (1.4 mEq/L). The phenotypes can in many cases be mimicked by drug treatment such as aminoglycosides and cisplatinum compounds. Due to the severe effects of hypomagnesaemia, routine measurements of serum Mg2+ should be conducted in the critically ill as well as in patients who are exposed to drugs and other conditions associated with Mg2+ deficiency.
Table 13.15 Causes of hypomagnesaemia
|
|
SIADH, syndrome of inappropriate antidiuretic hormone secretion.
Clinical features
Symptoms and signs (indicating a deficit of 0.5–1 mmol/kg) include irritability, tremor, ataxia, carpopedal spasm, hyperreflexia, confusional and hallucinatory states and epileptiform convulsions. An ECG may show a prolonged QT interval, broad flattened T waves, and occasional shortening of the ST segment.
Treatment
This involves the withdrawal of precipitating agents such as diuretics or purgatives. If symptomatic (or with hypocalcaemia), give a parenteral infusion of 50 mmol of magnesium chloride in 1 L of 5% glucose or other isotonic fluid over 12–24 h. This should be repeated daily and continued for 2 days after normal plasma levels have been achieved.
Relationship between hypomagnesaemia and plasma calcium
Calcium deficiency usually but not always develops with hypomagnesaemia. Hypomagnesaemia can be further subdivided into three main groups:
Hypercalciuria with hypomagnesaemia: this originating from defects in Mg2+ absorption in cTAL such as several forms of Bartter’s syndrome, loop diuretics and familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC).
Normocalciuria: this includes autosomal dominant hypomagnesaemia due to mutations in Kv1.1, isolated renal hypomagnesaemia (IRH) due to mutations in the EGF gene.
Hypocalciuria with hypomagnesaemia: is a hallmark feature of thiazide diuretic use, Gitelman’s syndrome (GS), and has also been reported in the EAST syndrome, due to mutations in NCC and Kir4.1, respectively. Genetic defects of beta-cell function and isolated dominant hypomagnesaemia (IDH) caused by mutations in HNF1b and FXYD2 also lead to hypomagnesaemia with accompanying hypocalciuria.
Relationship between hypomagnesaemia and plasma potassium
Magnesium depletion can lead to refractory hypokalaemia. The intracellular magnesium blocks secretory K+ currents through ROMK channels; therefore, magnesium depletion promotes K+ loss. Furthermore, this magnesium-mediated inhibition of K+ secretion increases as extracellular K+ decreases, which appropriately reduces K+ loss in the presence of K+ deficiency. Close monitoring, with potassium supplements if necessary, is required in patients presenting with primary symptomatic low plasma magnesium levels.
Hypermagnesaemia
This primarily occurs in patients with acute or chronic kidney disease given magnesium-containing laxatives or antacids. It can also be induced by magnesium-containing enemas. Mild hypermagnesaemia may occur in patients with adrenal insufficiency. Causes are given in Table 13.16.
Table 13.16 Causes of hypermagnesaemia
Clinical features
Symptoms and signs relate to neurological and cardiovascular depression, and include weakness with hyporeflexia proceeding to narcosis, respiratory paralysis and cardiac conduction defects. Symptoms usually develop when the plasma magnesium level exceeds 2 mmol/L (4 mEq/L).
Treatment
Treatment requires withdrawal of any magnesium therapy. An intravenous injection of 10 mL of calcium gluconate 10% (2.25 mmol calcium) is given to antagonize the effects of hypermagnesaemia, along with glucose and insulin (as for hyperkalaemia) to lower the plasma magnesium level. Dialysis may be required in patients with severe kidney disease.
FURTHER READING
Glaudemans B, van der Wiist J, Scola RH et al. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest 2009; 119:936–942.
San-Cristobal P, Dimke H, Hoenderop JG et al. Novel molecular pathways in renal Mg2+ transport: a guided tour along the nephron. Curr Opin Nephrol Hypertens 2010; 19(5):456–462.
Yang L, Frindt G, Palmer LG. Magnesium modulates ROMK channel–mediated potassium secretion. J Am Soc Nephrol 2010; 21:2109–2116.
Disorders of phosphate concentration
Phosphate forms an essential part of most biochemical systems, from nucleic acids downwards. The regulation of plasma phosphate level is both direct and closely linked to calcium.
About 85% of all body phosphorus is within bone, plasma phosphate normally ranging from 0.80 to 1.15 mmol/L (2.5–3.6 mg/dL) and accounts for only 1% of the total body phosphate. However, plasma phosphate levels correlate in most circumstances with total body sodium. Phosphate reabsorption from the glomerular filtrate occurs entirely and actively in the renal proximal tubule and is hormonally regulated. It is decreased by parathyroid hormone (PTH), mediated by a cyclic AMP-dependent mechanism; thus hyperparathyroidism is associated with low plasma levels of phosphate. Other factors that are known to control phosphate reabsorption in the proximal tubule are 1,25-dihydroxyvitamin D3, sodium delivery to the proximal tubule, serum concentrations of calcium, bicarbonate, carbon dioxide tension, glucose, alanine, serotonin, dopamine and sympathetic activity.
Osteoblast-secreted phosphaturic factors (phosphatonins) such as fibroblast growth factor 23 (FGF23), matrix extracellular phosphoglycoprotein (MEPG) and frizzled related protein 4 (FRP-4) play a role in phosphate homeostasis. FGF23 is the most extensively investigated phosphatonin which binds to its receptors FGFR1 in the kidney and causes phosphaturia and also regulates vitamin D by inactivation of 1α-hydroxylase (CYP27B1) and upregulation of 24 hydroxylase (CYP24A1) enzymes with the net result of low 1,25 vitamin D synthesis. Moreover, FGF23 requires Klotho (p. 652) to act as a coreceptor with FGFR1 for its activity. Loss of function mutation in either FGF23 or Klotho results in a similar phenotype of shortened lifespan, premature ageing (p. 37) including hyperphosphataemia and as expected from mode of action increased 1–25 vitamin D levels. Klotho can also inhibit phosphate absorption directly in the absence of FGF23 or PTH.
Phosphate absorption is an active process carried out by a family of sodium-phosphate cotransporters (NPT) in the gut and kidneys. NPT2a and NPT2c are expressed in the brush border of the renal proximal tubule whilst NPT2b is expressed in lungs and intestine. NPT2a plays a central role in the renal reabsorption of phosphate but essentially requires a companion protein called sodium hydrogen exchanger regulatory factor 1 (NHERF1) for membrane sorting. Intestinal absorption is carried out by NPT2b but its mutation does not cause any phosphate abnormalities because of the compensation taking place by the renal expression of NPT2a and possibly NPT2c. Under normal circumstances plasma phosphate levels are kept constant, e.g. after a phosphate-rich meal, the intestinal bone axis releases FGF23 from bone which inhibits NPT2a and causes phosphate excretion. Moreover, phosphate in the plasma either directly or indirectly by lowering ionized calcium causes the release of PTH which also inhibits NPT2a and NHERF1 resulting in phosphaturia (Fig. 13.9). These two principal mechanisms keep plasma phosphate levels within normal limits on a daily basis.
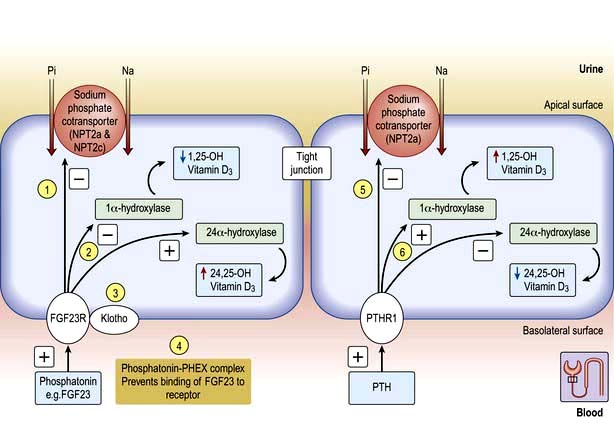
Figure 13.9 PHEX and PTH regulation of phosphate transport and vitamin D metabolism in the proximal tubule. (1) The hormone phosphatonin, e.g. FGF23, is an inhibitor of sodium-phosphate cotransporter (NPT2a, c) causing phosphaturia and also inhibits (2) 1 alpha hydroxylase and lowers 1,25 vitamin D synthesis when engaged to FGFR1-klotho complex (3). When bound to PHEX (phosphate-regulating gene with homologies to endopeptidase on the X chromosome) it is bioinactive and cannot bind to FGF23R1-klotho complex thus preventing phosphatonin activity (4). Over-production of phosphatonins occurs in tumour-induced osteomalacia, gain of function mutation as in autosomal dominant hypophosphataemic rickets and loss of function mutation in PHEX as X-linked hypophosphataemia. This causes renal phosphate wasting, as well as increased 1,25-dihydroxyvitamin D degradation. Parathormone (PTH) also inhibits NPT2a (5) and companion protein sodium hydrogen exchanger regulatory factor 1 (not shown) and causes phosphaturia but has opposite effects on vitamin D metabolism (6) compared with phosphatonin (FGF23).
Hypophosphataemia
Significant hypophosphataemia (<0.4 mmol/L or <1.25 mg/dL) occurs in a number of clinical situations, owing to redistribution into cells, to renal losses, or to decreased intake (Table 13.17).
Table 13.17 Causes of hypophosphataemia
|
|
Muscle weakness, e.g. diaphragmatic weakness, decreased cardiac contractility, skeletal muscle rhabdomyolysis
A left shift in the oxyhaemoglobin dissociation curve (reduced 2,3-bisphosphoglycerate, 2,3-BPG) and rarely haemolysis
Mild hypophosphataemia often resolves without specific treatment. However, diaphragmatic weakness may be severe in acute hypophosphataemia, and may impede weaning a patient from a ventilator. Interestingly, chronic hypophosphataemia (in X-linked hypophosphataemia) is associated with normal muscle power.
Causes
Primary hyperparathyroidism is a common cause of hypophosphataemia. Very rarely, gain of function mutations of PTH1 receptor cause hypophosphataemia and Jansen’s metaphyseal chondrodysplasia due to constitutive activation of PTH signaling even in the presence of low or absent circulating PTH levels.
Hypophosphataemia can be part of osteomalacia and rickets due to vitamin D deficiency either dietary (globally the commonest cause) or genetic and is usually accompanied by hypocalcaemia (calcipenic) and secondary hyperparathyroidism.
Vitamin D-dependent rickets type I
Also known as pseudovitamin D-deficient rickets, this is caused by 1α-hydroxylase deficiency due to inactivating mutations in its gene. It can be corrected with high daily doses of vitamin D. This condition manifests clinically in the first year of life with severe hypocalcaemia often complicated by tetany, moderate hypophosphataemia and enamel hypoplasia. The characteristic biochemical findings are normal serum levels of 25-hydroxyvitamin D and low values of 1,25–dihydroxyvitamin D and usually relatively high PTH levels. The treatment of choice is replacement therapy with calcitriol.
Vitamin D-dependent rickets type II
This is a form of vitamin D resistance and is now known as hereditary vitamin D-resistant rickets. It is an autosomal recessive disorder and is usually caused by loss of function mutations in the gene encoding the vitamin D receptor. The clinical manifestations vary widely, depending upon the type of mutation within the vitamin D receptor and the amount of residual vitamin D receptor activity. Affected children usually develop rickets within the first 2 years of life with alopecia in two-thirds of cases which is due to lack of vitamin D receptor action within keratinocytes. The treatment involves a therapeutic trial of calcitriol and calcium supplementation. Long-term infusion of calcium into a central vein is a possible alternative for severely resistant patients. Oral calcium therapy may be sufficient once radiographic healing has been observed.
Decreased renal reabsorption of phosphate
Excessive phosphatonins (FGF23)
This condition also occurs in patients with tumour-induced osteomalacia (TIO), X-linked dominant hypophosphataemic rickets (XLR), and autosomal-dominant hypophosphataemic rickets (ADHR). These syndromes have similar biochemical and osseous phenotypes. Patients have osteomalacia or rickets, reduced tubular phosphate reabsorption, hypophosphataemia, normal or low serum calcium, normal PTH and PTH-related protein concentrations, and normal or low 1,25-dihydroxyvitamin D3. Urinary cyclic AMP levels are generally in the normal range.
In TIO, there is excessive production of phosphaturic agents (which are normally produced by osteoblasts and act as hormones by acting on kidneys and regulating phosphate absorption and vitamin D activation), e.g. fibroblast growth factor 23 (FGF23), matrix extracellular phosphoglycoprotein (MEPG) and frizzled related protein 4 (FRP-4). These cannot be degraded by normal concentrations of PHEX (phosphate regulating gene with homologies to endopeptidases on the X chromosome). This results in net excess of inhibitors of the sodium-phosphate cotransporter in the proximal tubule, and phosphaturia.
In ADHR, FGF-23 is mutated so that it is resistant to PHEX proteolysis. In XLR, mutations in PHEX prevent binding to FGF23 and FRP-4, resulting in a net relative excess of phosphatonins.
Normal adaptive increases in 1,25-dihydroxyvitamin D3 synthesis in response to low phosphate levels do not occur in TIO, ADHR and XLR, aggravating phosphaturia (Fig. 13.9).
Autosomal recessive hypophosphataemia with high FGF23 levels has been described in patients with mutations in the gene encoding dentin matrix protein 1 (DMP1), which is a transcription factor produced by dental cells, osteoblasts and osteocytes. It is also secreted and can modulate the formation of mineralized matrix.
Reduced NPT2a/NHERF1 activity
Mutations in NPT2a (sodium phosphate cotransporter 2a) and NHERF1 (sodium hydrogen regulator factor 1) have been identified in patients with low phosphate levels and reduced ratio of the maximum tubular reabsorption of phosphate (TmP) normalised for GFR (TmP/GFR ratio). Heterozygous loss of function mutation in NPT2a and NHERF1 in patients is characterized by hypophosphataemia, low TmP/GFR and normal PTH. Carriers of these mutations do not have hypercalcaemia, which means these mutation do not affect the action of PTH on bones.
Dent’s disease
Dent’s disease is the generally accepted name for a group of hereditary tubular disorders including X-linked recessive nephrolithiasis with renal failure, X-linked recessive hypophosphataemic rickets, and idiopathic low-molecular-weight proteinuria. It is characterized by low-molecular-weight proteinuria, hypercalciuria, hyperphosphaturia, nephrocalcinosis, kidney stones and eventual renal failure, with some patients developing rickets or osteomalacia.
Dent’s disease is caused by loss of function mutation of a proximal tubular endosomal chloride channel, ClC5. This chloride channel, along with the proton pump, is essential for acidification of proximal tubular endosomes. The process is linked with normal endocytosis, degradation and recycling of absorbed proteins, vitamins and hormones. Defective endosomal acidification (owing to the mutated ClC5 gene) results in impaired endosomal degradation and recycling of endocytosed hormones such as PTH with clinical phenotype of hyperparathyroidism. Moreover, the receptors (megalin and cubilin) for reabsorption of low-molecular-weight proteins and albumin in the proximal tubules are decreased in Dent’s disease. This explains low-molecular-weight proteinuria and excessive urinary leaks of cytokines, hormones and chemokines. This urinary profile is associated with progressive renal fibrosis and more rapid decline in renal function.
Diagnostic approach
Patients with hypophosphataemia should have their TmP/GFR measured. A value of <0.7 mmol/L indicates renal phosphate wasting. If PTH levels are high then it is very likely to be hyperparathyroidism either primary or secondary to vitamin D deficiency (acquired or genetic) or Dent’s disease. If PTH levels are low then the only possibility is gain of function mutation in PTH1R. If PTH levels are normal then assess FGF23 levels. High FGF23 levels will indicate possible mutated genes in FGF23/Klotho, PHEX and DMP1. Normal FGF23 and PTH levels point to mutations in NPT2a, NPT2c and NHERF1.
Treatment
Treatment for hypophosphataemia includes combined therapy with phosphate supplementation and calcitriol (1,25-dihydroxyvitamin D) administration.
Treatment of acute hypophosphataemia, if warranted, is with intravenous phosphate at a maximum rate of 9 mmol every 12 hours, with repeated measurements of calcium and phosphate, as over-rapid administration of phosphate may lead to severe hypocalcaemia, particularly in the presence of alkalosis. Chronic hypophosphataemia can be corrected, if warranted, with oral effervescent sodium phosphate.
Hyperphosphataemia
Hyperphosphataemia is common in patients with CKD (see p. 618 and Table 13.18). Hyperphosphataemia is usually asymptomatic but may result in precipitation of calcium phosphate, particularly in the presence of a normal or raised calcium or of alkalosis. Uraemic itching may be caused by a raised calcium phosphate product. Prolonged hyperphosphataemia causes hyperparathyroidism, and periarticular and vascular calcification.
Table 13.18 Causes of hyperphosphataemia
Familial tumoral calcinosis is characterized by calcifications of muscles, skin, eyelids and vessels as well hyperostosis. Absence of glycosylation of FGF23 makes it unstable and more sensitive to proteolysis. This results in its deficiency and hyperphosphataemia due to increased renal phosphate reabsorption through increased NPT2a activity.
Usually, no treatment is required for acute hyperphosphataemia, as the causes are self-limiting. Treatment of chronic hyperphosphataemia is with gut phosphate binders and dialysis (see p. 622).
FURTHER READING
Markadieu N, Bindels RJ, Hoenderop JG. The renal connecting tubule: resolved and unresolved issues in Ca(2+) transport. Intl J Biochem Cell Biol 2011; 43(1):1–4.
Nakatani T, Sarraj B, Ohnishi M et al. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)-mediated regulation of systemic phosphate homeostasis. FASEB J 2009; 23:433–441.
Prié D, Friedlander G. Genetic disorders of renal phosphate transport. N Engl J Med 2010; 362:2399–2409.
Tiosano D, Hochberg Z. Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab 2009; 27:392–401.
Acid–base disorders
The concentration of hydrogen ions in both extracellular and intracellular compartments is extremely tightly controlled, and very small changes lead to major cell dysfunction. The blood pH is tightly regulated and is normally maintained at between 7.38 and 7.42. Any deviation from this range indicates a change in the hydrogen ion concentration [H+] because blood pH is the negative logarithm of [H+] (Table 13.19). The [H+] at a physiological blood pH of 7.40 is 40 nmol/L. An increase in the [H+] – a fall in pH – is termed acidaemia. A decrease in [H+] – a rise in the blood pH – is termed alkalaemia. The disorders that cause these changes in the blood pH are acidosis and alkalosis, respectively.
Table 13.19 Relationship between [H+] and pH
| pH | [H+] (nmol/L) |
|---|---|
6.9 |
126 |
7.0 |
100 |
7.1 |
79 |
7.2 |
63 |
7.3 |
50 |
7.4 |
40 |
7.5 |
32 |
7.6 |
25 |
Normal acid–base physiology
The normal adult diet contains 70–100 mmol of acid. Throughout the body, there are buffers that minimize any changes in blood pH that these ingested hydrogen ions might cause. Such buffers include intracellular proteins (e.g. haemoglobin) and tissue components (e.g. the calcium carbonate and calcium phosphate in bone) as well as the bicarbonate-carbonic acid buffer pair generated by the hydration of carbon dioxide. This buffer pair is clinically most relevant, in part because its contribution can be measured and because alterations in this buffer pair reveal changes in all other buffer systems. Bicarbonate ions [HCO3−] and carbonic acid (H2CO3) exist in equilibrium; and in the presence of carbonic anhydrase, carbonic acid dissociates to carbon dioxide and water, as expressed in the following equation:
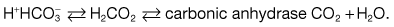
The addition of hydrogen ions drives the reaction to the right, decreasing the plasma bicarbonate concentration [HCO3−] and increasing the arterial carbon dioxide pressure (PaCO2). As shown in the following Henderson–Hasselbalch equation, a fall in the plasma [HCO3−] increases [H+] and thus lowers blood pH:
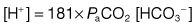
where [H+] is expressed in nmol/L, PaCO2 in kilopascals, [HCO3−] in mmol/L and 181 is the dissociation coefficient of carbonic acid. Alternatively the equation can be expressed as:
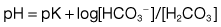
where pK = 6.1. Thus, the bicarbonate used in the buffering process must be regenerated to maintain normal acid–base balance.
Although the acidaemia stimulates an increase in ventilation, which blunts this change in pH, increased ventilation does not regenerate the bicarbonate used in the buffering process. Consequently, the kidney must excrete hydrogen ions to return the plasma [HCO3−] to normal. Maintenance of a normal plasma [HCO3−] under physiological conditions depends not only on daily regeneration of bicarbonate but also on reabsorption of all bicarbonate filtered across the glomerular capillaries.
Renal reabsorption of bicarbonate
The plasma [HCO3−] is normally maintained at approximately 25 mmol/L. In individuals with a normal glomerular filtration rate (120 mL/min), about 4500 mmol of bicarbonate is filtered each day. If this filtered bicarbonate were not reabsorbed, the plasma [HCO3−] would fall, along with blood pH. Thus, maintenance of normal plasma [HCO3−] requires that essentially all of the bicarbonate in the glomerular filtrate be reabsorbed (Fig. 13.10).
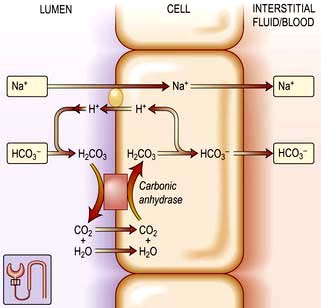
Figure 13.10 Resorption of sodium bicarbonate in the renal (mainly proximal) tubule. Bicarbonate is reclaimed by the secretion of H+ in exchange for Na+ into the tubule. This results in the formation of H2CO3, which is then broken down to CO2. This is reabsorbed and converted back to H2CO3, which now dissociates into H+ and HCO3−. The net result is reabsorption of Na+ and HCO3−. This process is dependent on carbonic anhydrase within the cells and on the luminal surface of the tubular cell.
The proximal convoluted tubule reclaims 85–90% of filtered bicarbonate; by contrast, the distal nephron reclaims very little. This difference is caused by the greater quantity of luminal (brush border) carbonic anhydrase in the proximal tubule than in the distal nephron. As a result of these quantitative differences, bicarbonate that escapes reabsorption in the proximal tubule is excreted in the urine.
Proximal tubular bicarbonate reabsorption is catalysed by the Na+/K+-ATPase pump located in the basolateral cell membrane. By exchanging peritubular potassium ions for intracellular sodium ions, the pump keeps the intracellular sodium concentration low, allowing sodium ions to enter the cell by moving down the sodium concentration gradient from the tubule lumen to the cell interior. Hydrogen ions are transported in the opposite direction (at the Na+-H+ antiporter), thereby maintaining electroneutrality. Before bicarbonate enters the proximal tubule, it combines with secreted hydrogen ions, forming carbonic acid. In the presence of luminal carbonic anhydrase (CA-IV) carbonic acid rapidly dissociates into carbon dioxide and water, which can then rapidly enter the proximal tubular cell. In the cell, carbon dioxide is hydrated by cytosolic carbonic anhydrase (CA-II), ultimately forming bicarbonate, which is then transported down an electrical gradient from the cell interior, across the membrane into the peritubular fluid, and into the blood. In this process, each hydrogen ion secreted into the proximal tubule lumen is reabsorbed and can be resecreted; there is no net loss of hydrogen ions or net gain of bicarbonate ions.
Renal excretion of [H+] (Fig. 13.11)
More acid is secreted into the proximal tubule (up to 4500 nmol of hydrogen ions each day) than into any other nephron segment. However, the hydrogen ions secreted into the proximal tubule are almost completely reabsorbed with bicarbonate; consequently, proximal tubular hydrogen ion secretion does not contribute significantly to hydrogen ion elimination from the body. The excretion of the daily acid load requires hydrogen ion secretion in more distal nephron segments.
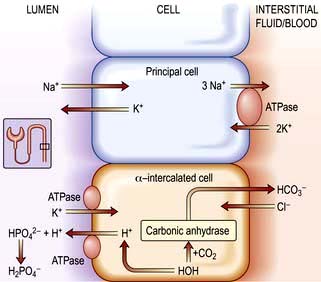
Figure 13.11 Renal excretion of H+. Secretion of H+ from the cortical collecting ducts is indirectly linked to Na+ reabsorption. Intracellular potassium is exchanged for sodium in the principal cell. Aldosterone stimulates H+ secretion by entering the principal cell, where it opens Na+ channels in the luminal membrane and increases Na+/K+-ATPase activity. The movement of cationic Na+ into the principal cells then creates a negative charge within the tubule lumen. K+ moves from the electrochemical gradient and into the lumen. Aldosterone apparently also stimulates the H+-ATPase directly in the intercalated cell, further enhancing H+ secretion. When the urinary pH falls to 4.0–4.5, further H+ secretion by the α-intercalated cells ceases. The filtration of titratable acids (e.g. phosphoric acid, H2PO4) raises the intraluminal pH and permits this process to continue. Secreted H+ binds to the conjugate anion of a titratable acid (HPO42− in this case) and is excreted in the urine. The H+ to be secreted arises from the reassociation of H2O and CO2 in the presence of carbonic anhydrase; thus, a bicarbonate molecule is regenerated each time an H+ is eliminated in the urine.
Most dietary hydrogen ions come from sulphur-containing amino acids that are metabolized to sulphuric acid (H2SO4), which then reacts with sodium bicarbonate as follows:
Excess sulphate is excreted in the urine, whereas excess hydrogen ions are buffered by bicarbonate and lower the plasma [HCO3−]. This fall in plasma [HCO3−] leads to a slight decrease in the blood pH, although a smaller decrease in the blood pH than would have occurred if buffer were unavailable. The subsequent excretion of hydrogen ions takes place primarily in the collecting duct and results in the regeneration of 1 mmol of bicarbonate for every mmol of hydrogen ions excreted in the urine.
The collecting duct has three types of cells:
The principal cell with an aldosterone-sensitive Na+ absorption site. These cells reabsorb Na+ and H2O and secrete K+ under the influence of aldosterone.
The α-intercalated cell, which possesses the proton pump for the active secretion of hydrogen ions in exchange for reabsorption of K+ ions. Aldosterone increases H+ ion secretion.
The β-intercalated cells are mirror images of α-intercalated cells where the H+-ATPase pump is located in the basolateral rather than the apical membrane whereby H+ ions are secreted into the peritubular capillary. The HCO3− ions, on the other hand, are secreted into the tubular lumen by an anion exchanger in the apical membrane. The identity of this transporter is uncertain, however, as it does not appear to represent the same Cl−-HCO3− exchanger that is present in the basolateral membrane of the H+-secreting intercalated cells.
Secretion of hydrogen ions from the cortical collecting duct is indirectly linked to sodium reabsorption. Aldosterone has several facilitating effects on hydrogen ion secretion. Aldosterone opens sodium channels in the luminal membrane of the principal cell and increases Na+/K+-ATPase activity. The subsequent movement of cationic sodium into the principal cell creates a negative charge within the tubule lumen. Potassium ions from the principal cells and hydrogen ions from the α-intercalated cells move out from the cells down the electrochemical gradient and into the lumen. Aldosterone also stimulates directly the H+-ATPase in the α-intercalated cell, further enhancing hydrogen ion secretion.
When hydrogen ions are secreted into the lumen of the collecting tubule, a tiny, but physiologically critical, fraction of these excess hydrogen ions remains in solution. Here, they increase the urinary [H+] and lower urinary pH below 4.0. Nevertheless, below this urine pH, inhibition of proton-secreting pumps such as H+-ATPase severely restricts kidney secretion of more hydrogen ions. Consequently, secretion of hydrogen ions depends on the presence of buffers in the urine that maintain the urine pH at a level higher than 4.0.
In the presence of alkali excess, the homeostatic needs are reversed. Although the kidney can excrete excess alkaline load by reducing reabsorption of filtered bicarbonate in the proximal and distal tubule, the collecting ducts also contribute by secreting bicarbonate brought about by switching to β-intercalated cells. This switch enables kidneys to secrete bicarbonate and conserve H+ ions.
Buffer systems in acid excretion
Two buffer systems are involved in acid excretion: the titratable acids such as phosphate, and the ammonia system. Each system is responsible for excreting about half of the daily acid load of 50–100 mmol under physiological conditions (Fig. 13.11).
Titratable acid
A titratable acid is a filtered buffer substance having a conjugate anion that can be titrated within the pH range occurring physiologically in the urine. Phosphoric acid (pKa 6.8) is the usual titratable urinary buffer. Hydrogen ions bind to the conjugate anions of the titratable acids and are excreted in the urine. For each hydrogen ion excreted in this form, a bicarbonate ion is regenerated within the cell and returned to the blood (Fig. 13.11).
Ammonium (NH4+)
In the setting of metabolic acidosis, titratable acids cannot increase significantly because the availability of titratable acid is fixed by the plasma concentration of the buffer and by the GFR. The ammonia buffer system, by contrast, can increase several hundred-fold when necessary. Consequently, impaired renal excretion of hydrogen ions is always associated with a defect in ammonium excretion (Fig. 13.12).
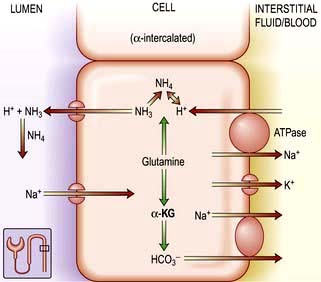
Figure 13.12 The ammonia buffering system in the kidney. All ammonia used to buffer H+ in the collecting duct is synthesized in the proximal convoluted tubule, and glutamine is the main source of this ammonia. As glutamine is metabolized, α-ketoglutarate (α-KG) is formed, which ultimately breaks down to bicarbonate that is then secreted into the peritubular fluid at an Na+-HCO3− cotransporter.
All ammonia used to buffer urinary hydrogen ions in the collecting tubule is synthesized in the proximal convoluted tubule. Glutamine is the primary source of ammonia. It undergoes deamination catalysed by glutaminase, resulting in α-ketoglutaric acid (Fig. 13.12) and ammonia. Once formed, ammonia can diffuse into the proximal tubule lumen and become acidified, forming ammonium. Once in the proximal tubule lumen, ammonium flows along the tubule to the thick ascending limb of Henle’s loop. Here, it is transported out of the tubule into the medullary interstitium. Ammonium then dissociates to ammonia, leading to a high interstitial ammonia concentration. The notion that ammonia diffuses down its concentration gradient into the lumen of the collecting tubule has recently been challenged by the discovery of rhesus (Rh) associated glycoproteins acting as ammonia transport proteins also called RhCG/Rhcg which are expressed in the basolateral and apical surfaces of DCT, inner medullary collecting duct and type A intercalated cells. These proteins play a fundamental role in renal ammonia excretion under both basal and acidosis states. Once secreted, NH3 reacts with the hydrogen ions secreted by the collecting tubular cells to form ammonium. Because ammonium (NH4) is not lipid-soluble, it is trapped in the lumen and excreted in the urine as ammonium chloride. Two conditions predominantly promote ammonia synthesis by the proximal tubular cell: systemic acidosis and hypokalaemia.
Glutamine metabolism and ureagenesis in the liver were thought to play a role in acid–base homeostasis. The liver was believed to contribute to regulation of acid–base balance by controlling the rate of ureagenesis and therefore bicarbonate consumption in response to changes in plasma acidity. Studies in human volunteers have concluded that ureagenesis is a maladaptive process for acid–base regulation and that ureagenesis has no discernible homeostatic effect on acid–base equilibrium in humans.
Causes of acid–base disturbance
Acid–base disturbance may be caused by:
Abnormal CO2 removal in the lungs (’respiratory’ acidosis and alkalosis)
Abnormalities in the regulation of bicarbonate and other buffers in the blood (‘metabolic’ acidosis and alkalosis).
Both may, and usually do, co-exist. For instance, metabolic acidosis causes hyperventilation (via medullary chemoreceptors, see p. 794), leading to increased removal of CO2 in the lungs and partial compensation for the acidosis. Conversely, respiratory acidosis is accompanied by renal bicarbonate retention, which could be mistaken for primary metabolic alkalosis. The situation is even more complex if a patient has both respiratory disease and a metabolic disturbance.
Diagnosis
Clinical history and examination usually point to the correct diagnosis. Table 13.20 shows the typical blood changes, but in complicated patients the acid–base nomogram (Fig. 13.13) is invaluable. The [H+] and PaCO2 are measured in arterial blood (for precautions see p. 659) as well as the bicarbonate. If the values from a patient lie in one of the bands in the diagram, it is likely that only one abnormality is present. If the [H+] is high (pH low) but the PaCO2 is normal, the intercept lies between two bands: the patient has respiratory dysfunction, leading to failure of CO2 elimination, but this is partly compensated for by metabolic acidosis, stimulating respiration and CO2 removal (this is the most common ‘combined’ abnormality in practice).
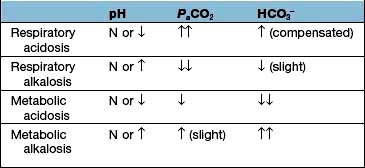
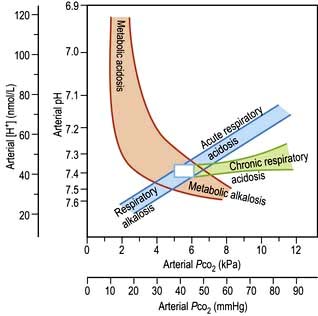
Figure 13.13 The Flenley acid–base nomogram. This was derived from a large number of observations in patients with ‘pure’ respiratory or metabolic disturbances. The bands show the 95% confidence limits representing the individual varieties of acid–base disturbance. The central white box shows the approximate limits of arterial pH and PCO2 in normal individuals.
Respiratory acidosis and alkalosis
Respiratory acidosis
This is caused by retention of CO2. The PaCO2 and [H+] rise. Renal retention of bicarbonate may partly compensate, returning the [H+] towards normal (see p. 876).
Respiratory alkalosis
Increased removal of CO2 is caused by hyperventilation, so there is a fall in PaCO2 and [H+] (see p. 876).
Metabolic acidosis
This is due to the accumulation of any acid other than carbonic acid, and there is a primary decrease in the plasma [HCO3−]. Several disorders can lead to metabolic acidosis: acid administration, acid generation (e.g. lactic acidosis during shock or cardiac arrest), impaired acid excretion by the kidneys, or bicarbonate losses from the gastrointestinal tract or kidneys. Calculation of the plasma anion gap is extremely useful in narrowing this differential diagnosis.
The anion gap
The first step is to identify whether the acidosis is due to retention of H+Cl− or to another acid. This is achieved by calculation of the anion gap.
The normal cations present in plasma are Na+, K+, Ca2+, Mg2+.
The normal anions present in plasma are Cl−, HCO3−, negative charges present on albumin, phosphate, sulphate, lactate, and other organic acids.
The sums of the positive and negative charges are equal.
Measurement of plasma [Na+], [K+], [Cl−] and [HCO3−] is usually easily available.
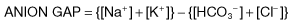
Because there are more unmeasured anions than cations, the normal anion gap is 10–18 mmol/L, although calculations with more sensitive methods place this at 6–12 mmol/L. Albumin normally makes up the largest portion of these unmeasured anions. As a result, a fall in the plasma albumin concentration from the normal value of about 40 g/L to 20 g/L may reduce the anion gap by as much as 6 mmol/L, because each 1 g/L of albumin has a negative charge of 0.2–0.28 mmol/L.
Metabolic acidosis with a normal anion gap
If the anion gap is normal in the presence of acidosis, this suggests that H+Cl− is being retained or that Na+HCO3− is being lost. Causes of a normal-anion-gap acidosis are given in Table 13.21. In these conditions, plasma bicarbonate decreases and is replaced by chloride to maintain electroneutrality. Consequently, these disorders are sometimes referred to collectively as hyperchloraemic acidoses.
Table 13.21 Causes of metabolic acidosis with a normal anion gap
Renal tubular acidosis (RTA)
This term refers to systemic acidosis caused by impairment of the ability of the renal tubules to maintain acid–base balance. This group of disorders is uncommon and only rarely a cause of significant clinical disease.
Type 4 renal tubular acidosis
Also called ‘hyporeninaemic hypoaldosteronism’, this is probably the most common of these disorders. The cardinal features are hyperkalaemia and acidosis occurring in a patient with mild chronic kidney disease, usually caused by tubulo-interstitial disease (e.g. reflux nephropathy) or diabetes. Gordon’s syndrome (see p. 655) shares biochemical abnormalities but differs in having normal GFR and hypertension. Plasma renin and aldosterone are found to be low, even after measures which would normally stimulate their secretion. The features for the diagnosis are shown in Table 13.22. An identical syndrome is caused by chronic ingestion of NSAIDs, which impair renin and aldosterone secretion. In the presence of acidosis, urine pH may be low. Treatment is with fludrocortisone, sodium bicarbonate, diuretics, or ion exchange resins to remove potassium, or a combination of these. Dietary potassium restriction alone is ineffective.
Table 13.22 Features of hyporeninaemic hypoaldosteronism (type 4 renal tubular acidosis)
|
Low plasma bicarbonate and hyperchloraemia Normal ACTH stimulation test (p. 944) Low basal 24-hour urinary aldosterone Subnormal response of plasma renin and plasma aldosterone to stimulation |
Type 3 renal tubular acidosis (RTA)
This condition is vanishingly rare, and represents a combination of type 1 and type 2. Inherited type 3 RTA is caused by mutations resulting in carbonic anhydrase type II deficiency, which is characterized by osteopetrosis, RTA of mixed type, cerebral calcification, and mental retardation.
Type 2 (‘proximal’) renal tubular acidosis
This is very rare in adult practice. It is caused by failure of sodium bicarbonate reabsorption in the proximal tubule. The cardinal features are acidosis, hypokalaemia, an inability to lower the urine pH below 5.5 despite systemic acidosis, and the appearance of bicarbonate in the urine despite a subnormal plasma bicarbonate. This disorder normally occurs as part of a generalized tubular defect, together with other features such as glycosuria and amino-aciduria. Inherited forms of isolated type 2 RTA are described as both autosomal dominant and recessive patterns of inheritance, where putative mutations are in the Na+-H+ antiporter in the apical membrane and Na+-HCO3− cotransporter in the basolateral membrane of proximal tubular cells respectively (see Fig. 13.10). Treatment is with sodium bicarbonate: massive doses may be required to overcome the renal ‘leak’.
Type 1 (’distal’) renal tubular acidosis
This is due to a failure of H+ excretion in the distal tubule (Table 13.23). It consists of:
Table 13.23 Causes of distal renal tubular acidosis (type 1 RTA)
|
|
a May also cause proximal renal tubular acidosis.
These features may be present only in the face of increased acid production; hence the need for an acid load test in diagnosis (Practical Box 13.1). Other features include:
Low urinary citrate (owing to increased citrate absorption in the proximal tubule where it can be converted to bicarbonate)
 Practical Box 13.1
Practical Box 13.1
Diagnosis of renal tubular acidosis
Plasma HCO3− <21 mmol/L, urine pH >5.3 = renal tubular acidosis.
To differentiate between proximal (very rare) and distal (rare) requires bicarbonate infusion test Plasma HCO3−>21 mmol/L but suspicion of partial renal tubular acidosis (e.g. nephrocalcinosis-associated diseases): acid load test required as follows:
Give 100 mg/kg ammonium chloride by mouth
Check urine pH hourly and plasma HCO3− at 3 h
Plasma HCO3− should drop below 21 mmol/L unless the patient vomits (in which case the test should be repeated with an antiemetic)
If urine pH remains >5.3 despite a plasma HCO3− of 21 mmol/L, the diagnosis is confirmed
These abnormalities result in osteomalacia, renal stone formation and recurrent urinary infections. Osteomalacia is caused by buffering of H+ by Ca2+ in bone, resulting in depletion of calcium from bone. Renal stone formation is caused by hypercalciuria, hypocitraturia (citrate inhibits calcium phosphate precipitation), and alkaline urine (which favours precipitation of calcium phosphate). Recurrent urinary infections are caused by renal stones.
Both autosomal dominant and recessive inheritance patterns have been reported in primary distal RTA. In the autosomal recessive distal RTA, a substantial proportion of patients have sensorineural deafness, and this is associated with a loss of function mutation in the H+-ATPase at the apical surface of intercalated cells.
Treatment is with sodium bicarbonate, potassium supplements and citrate. Thiazide diuretics are useful by causing volume contraction and increased proximal sodium bicarbonate reabsorption.
Urinary anion gap
Another useful tool in the evaluation of metabolic acidosis with a normal anion gap is the urinary anion gap:
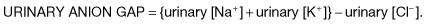
This calculation can be used to distinguish the normal anion-gap acidosis caused by diarrhoea (or other gastrointestinal alkali loss) from that caused by distal renal tubular acidosis. In both disorders, the plasma [K+] is characteristically low. In patients with renal tubular acidosis, urinary pH is always greater than 5.3.
Although excretion of urinary hydrogen ions in the patient with diarrhoea should acidify the urine, hypokalaemia leads to enhanced ammonia synthesis by the proximal tubular cells. Despite acidaemia, the excess urinary buffer increases the urine pH to a value above 5.3 in some patients with diarrhoea.
Whenever urinary acid is excreted as ammonium chloride, the increase in urinary chloride excretion decreases the urinary anion gap. Thus, the urinary anion gap should be negative in the patient with diarrhoea, regardless of the urine pH. On the other hand, although hypokalaemia may result in enhanced proximal tubular ammonia synthesis in distal renal tubular acidosis, the inability to secrete hydrogen ions into the collecting duct in this condition limits ammonium chloride formation and excretion; thus, the urinary anion gap is positive in distal renal tubular acidosis.
Metabolic acidosis with a high anion gap
If the anion gap is increased, there is an unmeasured anion present in increased quantities. This is either one of the acids normally present in small, but unmeasured quantities, such as lactate, or an exogenous acid. Causes of a high-anion-gap acidosis are given in Table 13.24.
Table 13.24 Causes of metabolic acidosis with an increased anion gap
|
Kidney failure (serum sulphate and phosphate) Accumulation of organic acids Type B – decreased hepatic lactate metabolism deficiency (decreased pyruvate dehydrogenase activity)
|
Uraemic acidosis
Kidney disease causes acidosis in several ways. Reduction in the number of functioning nephrons decreases the capacity to excrete ammonia and H+ in the urine. In addition, tubular disease may cause bicarbonate wasting. Acidosis is a particular feature of those types of CKD in which the tubules are particularly affected, such as reflux nephropathy and chronic obstructive uropathy.
Chronic acidosis is most often caused by chronic kidney disease, where there is a failure to excrete fixed acid. Up to 40 mmol of hydrogen ions may accumulate daily. These are buffered by bone, in exchange for calcium. Chronic acidosis is therefore a major risk factor for renal osteodystrophy and hypercalciuria.
Chronic acidosis has also been shown to be a risk factor for muscle wasting in renal failure, and may also contribute to the inexorable progression of some types of renal disease.
Uraemic acidosis should be corrected because of these effects on growth, muscle turnover and bones. Oral sodium bicarbonate 2–3 mmol/kg daily is usually enough to maintain serum bicarbonate above 20 mmol/L, but may contribute to sodium overload. Calcium carbonate improves acidosis and also acts as a phosphate binder and calcium supplement, and is commonly used. Acidosis in end-stage kidney failure is usually fully corrected by adequate dialysis.
Lactic acidosis
Increased lactic acid production occurs when cellular respiration is abnormal, because of either a lack of oxygen in the tissues (‘type A’) or a metabolic abnormality, such as drug-induced (‘type B’) (Table 13.24). The most common cause in clinical practice is type A lactic acidosis, occurring in septic or cardiogenic shock. Significant acidosis can occur despite a normal blood pressure and PaCO2, owing to splanchnic and peripheral vasoconstriction. Acidosis worsens cardiac function and vasoconstriction further, contributing to a downward spiral and fulminant production of lactic acid.
Diabetic ketoacidosis (see p. 1017)
There is a high-anion-gap acidosis due to the accumulation of acetoacetic and hydroxybutyric acids, owing to increased production and some reduced peripheral utilization.
Mixed metabolic acidosis
Both types of acidosis may co-exist. For instance, cholera would be expected to cause a normal-anion-gap acidosis owing to massive gastrointestinal losses of bicarbonate, but the anion gap is often increased owing to renal failure and lactic acidosis as a result of hypovolaemia.
Clinical features
Clinically, the most obvious effect is stimulation of respiration, leading to the clinical sign of ‘air hunger’, or Kussmaul respiration. Interestingly, patients with profound hyperventilation may not complain of breathlessness, although in others it may be a presenting complaint.
Acidosis increases delivery of oxygen to the tissues by shifting the oxyhaemoglobin dissociation curve to the right, but it also leads to inhibition of 2,3-BPG production, which returns the curve towards normal (see p. 870). Cardiovascular dysfunction is common in acidotic patients, although it is often difficult to dissociate the numerous possible causes of this. Acidosis is negatively inotropic. Severe acidosis also causes venoconstriction, resulting in redistribution of blood from the peripheries to the central circulation, and increased systemic venous pressure, which may worsen pulmonary oedema caused by myocardial depression. Arteriolar vasodilatation also occurs, further contributing to hypotension.
Cerebral dysfunction is variable. Severe acidosis is often associated with confusion and fits, but numerous other possible causes are usually present.
As mentioned earlier, acidosis stimulates potassium loss from cells, which may lead to potassium deficiency if renal function is normal, or to hyperkalaemia if renal potassium excretion is impaired.
General treatment of acidosis
Treatment should be aimed at correcting the primary cause. In lactic acidosis caused by poor tissue perfusion (’type A’), treatment should be aimed at maximizing oxygen delivery to the tissues by protecting the airway, improving breathing and circulation. This usually requires inotropic agents, mechanical ventilation and invasive monitoring. In ‘type B’ lactic acidosis, treatment is that of the underlying disorder; e.g.:
The question of whether severe acidosis should be treated with bicarbonate is extremely controversial:
Rapid correction of acidosis may result in tetany and fits owing to a rapid decrease in ionized calcium.
Administration of sodium bicarbonate (8.4%) provides 1 mmol/mL of sodium, which may lead to extracellular volume expansion, exacerbating pulmonary oedema.
Bicarbonate therapy increases CO2 production and will therefore correct acidosis only if ventilation can be increased to remove the added CO2 load.
The increased amounts of CO2 generated may diffuse more readily into cells than bicarbonate, worsening intracellular acidosis.
Administration of sodium bicarbonate (50 mmol, as 50 mL of 8.4% sodium bicarbonate intravenously) is still occasionally given during cardiac arrest and is often necessary before arrhythmias can be corrected. Correction of hyperkalaemia associated with acidosis is also of undoubted benefit. In other situations there is no clinical evidence to show that correction of acidosis improves outcome, but it is standard practice to administer sodium bicarbonate when [H+] is above 126 nmol/L (pH <6.9), using intravenous 1.26% (150 mmol/L) bicarbonate infused over 2–3 h with electrolyte and pH monitoring. Intravenous sodium lactate should never be given.
Metabolic alkalosis
Metabolic alkalosis is common, comprising half of all the acid–base disorders in hospitalized patients. This observation should not be surprising since vomiting, the use of diuretics, and nasogastric suction are common among hospitalized patients. The mortality associated with metabolic alkalosis is substantial; the mortality rate is 45% in patients with an arterial pH of 7.55 and 80% when the pH is >7.65. Although this relationship is not necessarily causal, severe alkalosis should be viewed with concern.
Classification and definitions
Metabolic alkalosis has been classified on the basis of underlying pathophysiology (Table 13.25).
Table 13.25 Causes of metabolic alkalosis
The most common group is due to chloride depletion which can be corrected without potassium repletion. The other major grouping is that due to potassium depletion, usually with mineralocorticoid excess. Metabolic alkalosis due to both potassium and chloride depletion also occurs.
Chloride may be lost from the gut, kidney or skin. The loss of gastric fluid rich in acid results in alkalosis because bicarbonate generated during the production of gastric acid returns to the circulation. In Zollinger–Ellison syndrome (see p. 370) or gastric outflow obstruction these losses can be massive. Although sodium and potassium loss in the gastric juice is variable, the obligate urinary loss of these cations is intensified by bicarbonaturia, which occurs during disequilibrium.
Chloruretic agents all directly produce loss of chloride, sodium and fluid in the urine. These losses in turn promote metabolic alkalosis by several mechanisms:
Diuretic-induced increases in sodium delivery to the distal nephron enhance potassium and hydrogen ion secretion
Extracellular volume contraction stimulates renin and aldosterone secretion, which blunts sodium losses but accelerates potassium and hydrogen ion secretion
Potassium depletion augments bicarbonate reabsorption in the proximal tubule and
Stimulates ammonia production which in turn will increase urinary net acid excretion.
Urinary losses of chloride exceed those for sodium and are associated with alkalosis even when potassium depletion is prevented. The cessation of events that generate alkalosis is not necessarily accompanied by resolution of the alkalosis. A widely accepted hypothesis for the maintenance of alkalosis is chloride depletion rather than volume depletion. Although normal functioning of the proximal tubule is essential for bicarbonate absorption, the collecting duct appears to be the major nephron site for altered electrolyte and proton transport in both maintenance and recovery from metabolic alkalosis. During maintenance, the α-intercalated cells in the cortical collecting duct do not secrete bicarbonate because insufficient chloride is available for bicarbonate exchange. When chloride is administered and luminal or cellular chloride concentration increases, bicarbonate is promptly excreted and alkalosis is corrected.
Metabolic alkalosis in hypokalaemia is generated primarily by an increased intracellular shift of hydrogen ion causing intracellular acidosis. Potassium depletion is also associated with enhanced ammonia production with increased obligate net acid excretion. However, the role of intracellular acidosis is supported by the correction of the alkalosis by infusion of potassium without any suppression of renal net excretion. The correction is assumed to occur by the movement of potassium into and hydrogen ion out of the cell, which titrates extracellular fluid bicarbonate.
Milk–alkali syndrome in which both bicarbonate and calcium are ingested produces alkalosis by vomiting, calcium-induced bicarbonate absorption and reduced GFR. Cationic antibiotics in high doses can cause alkalosis by obligatory bicarbonate loss in the urine.
Clinical features
The symptoms of metabolic alkalosis per se are difficult to separate from those of chloride, volume or potassium depletion. Tetany (see p. 997), apathy, confusion, drowsiness, cardiac arrhythmias and neuromuscular irritability are common when alkalosis is severe. The oxyhaemoglobin dissociation curve is shifted to the left. Respiration may be depressed.
Treatment
Chloride-responsive metabolic alkalosis
Although replacement of the chloride deficit is essential in chloride depletion states, selection of the accompanying cation – sodium, potassium or proton – is dependent on the assessment of extracellular fluid volume status (see p. 646), the presence or absence of associated potassium depletion, and the degree and reversibility of any depression of GFR. If kidney function is normal, bicarbonate and base equivalents will be excreted with sodium or potassium, and metabolic alkalosis will be rapidly corrected as chloride is made available.
If chloride and extracellular depletion co-exist then isotonic saline solution is appropriate therapy.
In the clinical settings of fluid overload, saline is contraindicated. In such situations, intravenous use of hydrochloride acid or ammonium chloride can be given. If GFR is adequate, acetazolamide, which causes bicarbonate diuresis by inhibiting carbonic anhydrase, can also be used. When the kidney is incapable of responding to chloride repletion, dialysis is necessary.
Chloride-resistant metabolic alkalosis
Metabolic alkalosis due to potassium depletion is managed by the correction of the underlying cause (see hypokalaemia). Mild to moderate alkalosis requires oral potassium chloride administration. However, the presence of cardiac arrhythmia or generalized weakness requires intravenous potassium chloride.
Greenlee MM, Lynch IJ, Gumz ML, et al. The renal H,K-ATPases. Curr Opin Nephrol Hypertens. 2010;19(5):478–482.
Kaplan LJ, Kellum JA. Fluids, pH, ions and electrolytes. Curr Opin Crit Care. 2010;16(4):323–331.
Karet FE. Disorders of water and acid-base homeostasis. Nephron Physiol. 2011;118(1):28–34.
Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol. 2007;2(1):162–174.