Chapter 19 Endocrine disease
Introduction
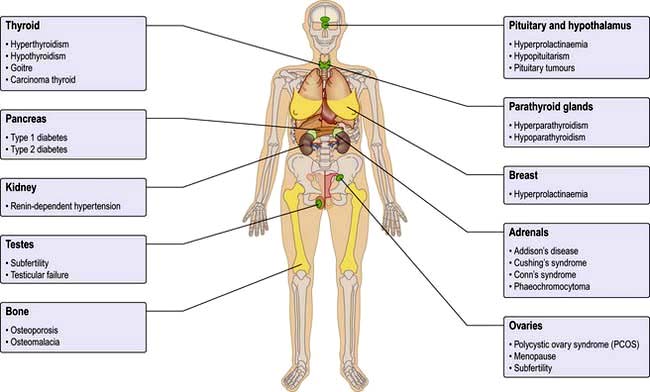
The endocrine system consists of glands that exert their actions at distant parts of the body via the production of biologically active hormones secreted into the bloodstream. Unlike the neurological system, which produces an immediate response, the endocrine system typically has a slower and longer lasting effect on the body. The main endocrine glands are the pituitary, thyroid, adrenals, gonads, parathyroids and pancreas and the common endocrine problems seen in clinical practice are shown in Figure 19.1. The pituitary gland, a pea-sized structure situated at the base of the brain, plays a key role in the control and feedback mechanisms of the endocrine system and has been termed the ‘conductor of the endocrine orchestra’.
Clinical presentation of endocrine disease
History
Hormones produce widespread effects in the body, and states of hormonal deficiency or excess typically present with symptoms that are generalized, diffuse and nonspecific. Symptoms of tiredness, weakness or lack of energy or drive and changes in appetite or thirst are common presentations. Other typical ‘hormonal’ symptoms include changes in body size and shape, problems with libido and potency, periods or sexual development, and changes in the skin (dry, greasy, acne, bruising, thinning or thickening) and hair (loss or excess). The differential diagnosis is often wide but endocrine disorders should be always considered when assessing a patient with any of these common complaints.
The past, family and social history is essential for making the diagnosis, planning appropriate management and interpreting results of borderline hormonal blood tests.
 The past history should include previous surgery or radiation involving endocrine glands, menstrual history, pregnancy and growth and development in childhood.
The past history should include previous surgery or radiation involving endocrine glands, menstrual history, pregnancy and growth and development in childhood.
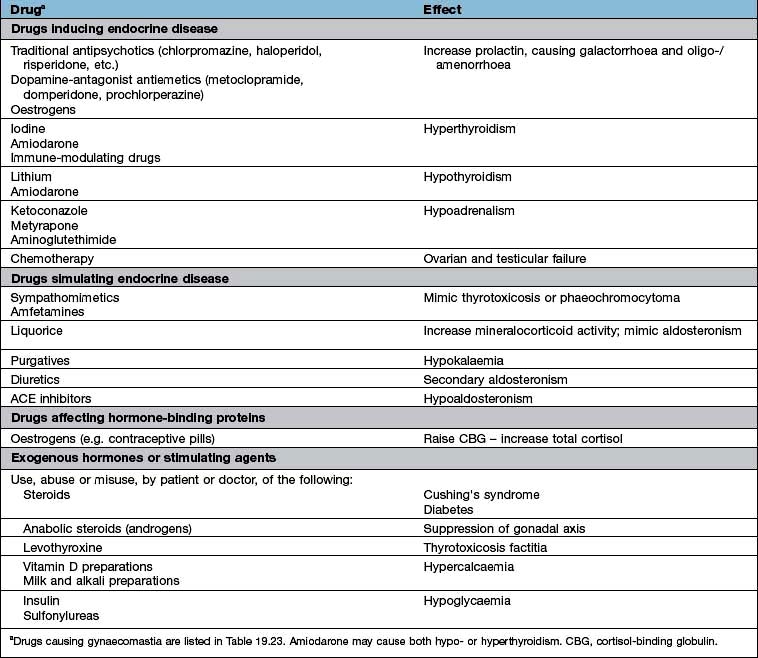
A full drug history will exclude common iatrogenic endocrine problems (Table 19.1).
A family history of autoimmune disease, endocrine disease including tumours, diabetes and cardiovascular disease is frequently relevant, and knowledge of family members’ height, weight, body habitus, hair growth and age of sexual development may aid interpretation of the patient’s own symptoms.
Table 19.1 Drugs and endocrine disease
Examination
A full general examination is essential to endocrine assessment because endocrine disorders affect all organ systems. Weight, height, body mass index (BMI), blood pressure and general habitus should all be documented, together with presence or absence of specific signs of deficiency or excess of individual hormone axes (signs of hyper- or hypo-thyroidism, acromegaly, Cushing’s).
In people with suspected pituitary disease, visual fields and adjacent cranial nerves should be assessed clinically. In thyroid disease, presence of goitre or thyroid eye disease should be documented. Skin changes may give clinical clues, including pigmentation (Addison’s and Nelson’s), vitiligo (autoimmune endocrinopathies), acanthosis nigricans (polycystic ovary syndrome (PCOS) and diabetes), skin thinning (Cushing’s, hypogonadism) or thickening (acromegaly, PCOS) and bruising and striae (Cushing’s). Hirsutism is a key sign in women and signs of hair loss from the head (following a change in thyroid function, androgen excess in women or normal virilization in men) or from the body, axillary and pubic areas (hypogonadism) may occur in both sexes.
In children, height and pubertal status are an essential part of the clinical examination.
Specific clinical signs associated with particular endocrine diseases are discussed in detail in the appropriate sections.
Common endocrine conditions
The most common endocrine disorders, excluding obesity (Ch. 5) and diabetes mellitus (Ch. 20), are:
Thyroid disorders: hypothyroidism, hyperthyroidism, goitre
Hirsutism, usually due to polycystic ovary syndrome
While most other endocrine conditions are uncommon, they often affect young people and are usually curable or completely controllable with appropriate therapy.
Aetiology of endocrine disease
Aetiological mechanisms common to many endocrine disorders include:
Autoimmune disease
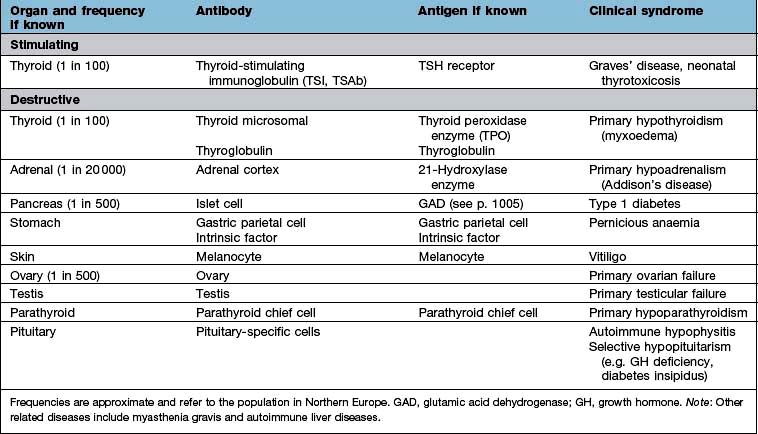
Organ-specific autoimmune diseases can affect every major endocrine organ (Table 19.2). They are characterized by the presence of specific antibodies in the serum, often present years before clinical symptoms are evident, are usually more common in women and have a strong genetic component, often with an identical-twin concordance rate of 50% and with HLA associations (see individual diseases). Several of the autoantigens have been identified.
Endocrine tumours
Most endocrine tumours are benign, although a cytological or histological diagnosis may be needed if there is clinical or radiological suspicion of malignancy. Clinical presentation depends on whether the tumour is functional or non-functional, the latter presenting only as a mass clinically or on imaging. Palpable thyroid nodules are common, and mass effects are a frequent presentation of pituitary adenomas, but the increased use of high-resolution ultrasound and detailed cross-sectional imaging has revealed a very high prevalence of asymptomatic, incidentally-discovered thyroid, adrenal and pituitary lesions, commonly termed ‘incidentalomas’ (see p. 989).
Functional tumours cause their effects via excess secretion of the relevant hormone. While often considered to be ‘autonomous’, i.e. independent of the physiological control mechanisms, many functional tumours do show evidence of feedback occurring at a higher ‘set-point’ than normal (e.g. ACTH secretion from a pituitary basophil adenoma). This is relevant in the dynamic assessment of endocrine diseases such as in the differential diagnosis of Cushing’s syndrome.
Endocrine adenomas typically present in a single gland, although rarer multiple endocrine neoplasia (MEN) syndromes exist due to very specific mutations of a single gene, such as the mutations of the RET proto-oncogene in MEN 2 or the MEN1 gene mutation in MEN 1 (see p. 997).
Enzyme defects
The biosynthesis of most hormones involves many stages. Deficient or abnormal enzymes can lead to absent or reduced production of the secreted hormone. In general, severe deficiencies present early in life with obvious signs; partial deficiencies usually present later with mild signs or are only evident under stress. An example of an enzyme deficiency is congenital adrenal hyperplasia (CAH), where the molecular basis has also been identified as mutations or deletions of the gene encoding the relevant enzymes (see p. 987).
Receptor abnormalities
There are rare conditions in which hormone secretion and control are normal but the receptors are defective; thus, if androgen receptors are defective, normal levels of androgen will not produce masculinization (e.g. testicular feminization). There are also a number of rare syndromes of diabetes and insulin resistance from receptor abnormalities (see p. 1006); other examples include nephrogenic diabetes insipidus, pseudohypoparathyroidism and thyroid hormone resistance which can cause an unusual pattern of thyroid blood results.
Hormonal activity
Synthesis, storage and release of hormones
Hormones may be of several chemical structures: polypeptide, glycoprotein, steroid or amine. Hormone release is the end-product of a long cascade of intracellular events. In the case of polypeptide hormones, neural or endocrine stimulation of the cell leads to increased transcription from DNA to a specific mRNA, which is in turn translated to the peptide product. This is often in the form of a precursor molecule that may itself be biologically inactive. This ‘prohormone’ is then further processed before being packaged into granules, in the Golgi apparatus. These granules are then transported to the plasma membrane before release, which is itself regulated by a complex combination of intracellular regulators. Hormone release may be in a brief spurt caused by the sudden stimulation of granules, often induced by an intracellular Ca2+-dependent process, or it is ‘constitutive’ (immediate and continuous secretion).
Plasma transport
Most classical hormones are secreted into the systemic circulation. In contrast, hypothalamic releasing hormones are released into the pituitary portal system so that much higher concentrations of the releasing hormones reach the pituitary than occur in the systemic circulation.
Many hormones are bound to proteins within the circulation. In most cases, only the free (unbound) hormone is available to the tissues and thus biologically active. This binding serves to buffer against very rapid changes in plasma levels of the hormone, and some binding protein interactions are also involved in the active regulation of hormone action. Many tests of endocrine function measure total rather than free hormone, which can give rise to difficulties in interpretation when binding proteins are altered in disease states or by drugs.
Binding proteins comprise both specific, high-affinity proteins of limited capacity, such as thyroxine-binding globulin (TBG), cortisol-binding globulin (CBG), sex-hormone-binding globulin (SHBG) and IGF-binding proteins (e.g. IGF-BP3) and other less specific, low-affinity ones, such as prealbumin and albumin.
Hormone action and receptors
Hormones act by binding to specific receptors in the target cell. Most hormone receptors are proteins with complex tertiary structures. The structure of the hormone-binding domain of the receptor complements the tertiary structure of the hormone, while changes in other parts of the receptor in response to hormone binding are responsible for the effects of the activated receptor within the cell. The structure of common hormones and their receptors is described under individual hormone axes.
Hormone receptors are broadly divided into:
Cell surface or membrane receptors: typically transmembrane receptors which contain hydrophobic sections spanning the lipid-rich plasma membrane and which trigger internal cellular messengers (see also p. 18)
Nuclear receptors which typically bind hormones and translocate them to the nucleus where they bind hormone response elements of nuclear DNA via characteristic amino-acid sequences (e.g. so-called ‘zinc fingers’, see p. 27).
Abnormal receptors are an occasional cause of endocrine disease (see p. 43).
Mechanisms of hormone-receptor action
Common structural mechanisms of hormone-receptor action are illustrated in Figure 2.9 (p. 25) and include:
G-protein coupled receptors (7-transmembrane or serpentine receptors). These bind hormones on their extracellular domain and activate the membrane G-protein complex with their intracellular domain. The activated complex may then:
stimulate cyclic AMP (cAMP) generation by adenylate cyclase – activating further intracellular kinases and leading to phosphorylation
activate phospholipase C (PLC) leading to generation of inositol 1,4,5-triphosphate (IP3) and release of intracellular calcium – in turn leading to calmodulin-dependent kinase activity and phosphorylation
lead to diacylglycerol (DAG) activation of C-kinase and subsequent protein phosphorylation.
Most peptide hormones act via G-protein coupled receptors.
Dimeric transmembrane receptors, from several receptor superfamilies, bind hormone in their extracellular components (sometimes causing the dimerization of the receptor monomer) and directly phosphorylate intracellular messengers via their intracellular components, leading to a variety of intracellular activation cascades. Growth hormone, prolactin and insulin-like growth factor-1 (IGF-1) act via this type of receptor.
Lipid-soluble molecules pass through the cell membrane and typically bind with their nuclear receptors in the cell cytoplasm before translocation of the activated hormone-receptor complex to the nucleus where it binds to nuclear DNA, often in combination with a multi-component complex of promoters, inhibitors and transcription factors. This interaction usually leads to increased transcription of the relevant gene product. Steroid and thyroid hormones act via this type of receptor.
Hormone release and binding to receptors. The activation of intracellular kinases, phosphorylation, release of intracellular calcium and other ‘second messenger’ pathways and the direct stimulation of DNA transcription results in some or all of the following:
Stimulation or release of pre-formed hormone from storage granules
Stimulation or synthesis of hormone and other cellular components
Opening or closing of ion or water channels in the cell membrane (e.g. calcium channels or aquaporin water channels)
Activation or deactivation of other DNA binding proteins leading to stimulation or inhibition of DNA transcription.
In each case, binding of the hormone to its receptor is the first step in a complex cascade of interrelated intracellular events which eventually lead to the overall effects of that hormone on cellular function.
The sensitivity and/or number of receptors for a hormone are often decreased after prolonged exposure to a high hormone concentration, the receptors thus becoming less sensitive (’downregulation’, e.g. angiotensin II receptors, β-adrenoceptors). The reverse is true when stimulation is absent or minimal, the receptors showing increased numbers or sensitivity (‘upregulation’).
Control and feedback
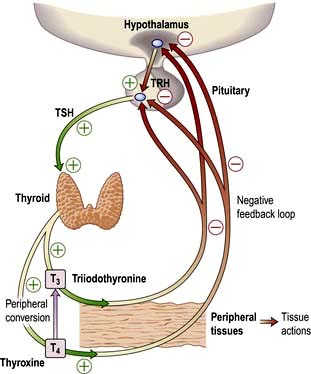
Most hormone systems are under tight regulatory control (typically by the hypothalamo-pituitary (HP) axis) by a system known as negative feedback. An example of the negative feedback system in the hypothalamo-pituitary-thyroid axis is demonstrated in Figure 19.2 and described here:
TRH (thyrotrophin-releasing hormone) is secreted in the hypothalamus and travels via the portal system to the pituitary where it stimulates the thyrotrophs to produce thyroid-stimulating hormone (TSH).
TSH is secreted into the systemic circulation where it stimulates increased thyroidal iodine uptake by the thyroid and the synthesis and release of thyroxine (T4) and triiodothyronine (T3).
Serum levels of T3 and T4 are increased by TSH; in addition, the conversion of T4 to T3 (the more active hormone) in peripheral tissues is stimulated by TSH.
T3 and T4 then enter cells where they bind to nuclear receptors and promote increased metabolic and cellular activity.
Levels of T3, from the blood and from local conversion of T4, are sensed by receptors in the pituitary and the hypothalamus. If they rise above normal, TRH and TSH production is suppressed, leading to reduced T3 and T4 secretion.
Peripheral T3 and T4 levels fall to normal.
If, however, T3 and T4 levels are low, for example after thyroidectomy, increased amounts of TRH and TSH are secreted, stimulating the remaining thyroid to produce more T3 and T4; blood levels of T3 and T4 may be restored to normal, at the expense of increased TSH drive, reflected by a high TSH level, ‘compensated euthyroidism’.
Conversely, in thyrotoxicosis when factors other than TSH itself are maintaining high T3 and T4 levels, the same mechanisms lead to suppression of TSH secretion.
Primary and secondary gland failure
It is useful in clinical endocrinology to distinguish between ‘primary’ disease of the end-organ gland (e.g. due to auto-immune destruction, atrophic change, infiltration or surgical removal of the gland), and ‘secondary’ disorders of the same axis caused by disease of the pituitary gland. An understanding of the negative feedback system is key to interpreting endocrine blood results and diagnosing the site of the disease process in clinical practice. In general terms:
‘Primary’ hormone deficiency due to a disease process in the endocrine end-organ (thyroid, adrenal or gonad) will lead to a loss of negative feedback and subsequent elevation in the corresponding anterior pituitary hormone. Conversely, an abnormal hormone excess due to a disease process in the primary endocrine gland, or excess amount of exogenous hormone, will lead to increased negative feedback and suppression of the corresponding pituitary hormones.
In ‘secondary gland failure’ there are low or ‘inappropriately normal’ levels of the pituitary trophic hormone in the face of a low end-organ hormone level. For example, if a patient has low circulating free T3 (fT3) and T4 levels in the context of a low TSH, pituitary disease should be suspected. Equally, the presence of a non-suppressed plasma ACTH in the context of Cushing’s syndrome implies that the pituitary rather than the adrenal itself is the cause.
Hormone resistance
In certain situations, receptor abnormalities can give rise to abnormal negative feedback due to hormone resistance, which can lead to an unusual pattern of blood results. For example, thyroid hormone resistance, due to mutations in the thyroid hormone receptor, is characterized by an elevation in thyroid hormones with a non-suppressed TSH. With this pattern of thyroid results, the clinician should also consider the rare diagnosis of a TSH secreting pituitary tumour.
Measurement of hormones
Hormones are measured in routine clinical practice by biochemical assays in the laboratory. It is possible to measure pituitary trophic hormones and the hormones produced by the end-organ glands, but hypothalamic hormones are not routinely measured in practice because of their low concentration and local action within the hypothalamo-pituitary axis. Circulating levels of most hormones are very low (10−9– 10−12 mol/L) and cannot be measured by simple chemical techniques. Hormones are therefore usually measured by immunoassays, which rely on highly specific polyclonal or monoclonal antibodies, which bind to the hormone being measured during the assay incubation. This hormone-antibody interaction is measured by use of labelled hormone after separation of bound and free fractions (Fig. 19.3).
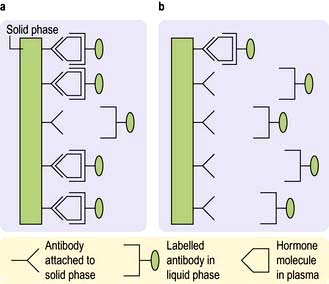
Figure 19.3 Principles of measurement of hormone levels in plasma by immunoassay (precise details vary with different assays and manufacturers). Immunoassays use two antibodies specific to the hormone being measured – one typically attached to a solid phase and one labelled antibody in the liquid phase. (a) High hormone levels in plasma: large amount of hormone binds to antibody on solid phase – large amount of labelled antibody linked to solid phase via molecules of the hormone. (b) Low hormone levels in plasma: less hormone, and therefore less labelled antibody, is linked to the solid phase. Label (radioactive, chemiluminescent, enzymatic or fluorescent) can be measured in either solid or liquid phase after separation of phases; levels of label will be proportional to the amount of hormone in the sample.
Immunoassays are sensitive but have limitations. In particular, the immunological activity of a hormone, as used in developing the antibody, may not necessarily correspond to biological activity and there may be false positive and negative results. The patient’s blood may also contain heterophile antibodies which interact with the animal antibodies used in the assay, and result in falsely low or high values. When there is a discrepancy between endocrine blood results and the clinical presentation, the clinician must question the validity of an endocrine result, and a close relationship with the relevant laboratory is essential. It may be necessary for the sample to be measured in a different laboratory using an alternative antibody, or to measure hormones in ways other than by immunoassay. Examples of alternative techniques to accurately quantify and characterize hormone levels include equilibrium dialysis, high-pressure liquid chromatography (HPLC) and, increasingly, mass spectroscopy.
Hormone binding proteins
Many hormones are transported in the bloodstream from the primary gland to their distant target organ attached to a specific binding protein (p. 940). It is more helpful to measure the free hormone rather than total bound hormone level, as this is the part that is biologically active. Some modern assays attempt to measure the free hormone level directly (e.g. free T4) and are therefore a more accurate reflection of biological activity, although there are often technical problems with this approach and many assays still measure total hormone level.
Cortisol, which is bound to cortisol binding globulin (CBG), and testosterone, which is bound to sex hormone binding globulin (SHBG), are still usually measured in their total form and can be affected by alterations in binding protein levels. In women who are pregnant or on the combined oral contraceptive pill, high oestrogen levels may lead to an elevation in CBG which can overestimate cortisol and give the false impression of hypercortisolaemia. In people with diabetes mellitus or other insulin-resistant states which may lower SHBG levels, low total testosterone levels may give the false impression of androgen deficiency. Conversely, hyperthyroidism or oestrogen excess can cause an elevation in SHBG, leading to apparently high total testosterone levels. As with all endocrine results, the data need to be interpreted in the clinical context.
Patterns of hormonal secretion
Hormone secretion can be continuous or intermittent, for example:
Continuous secretion is shown by the thyroid hormones, with a half-life of 7–10 days for T4 and 6–10 hours for T3, and with little variation in levels over the day, month and year
Pulsatile secretion is the normal pattern for the gonadotrophins, LH and FSH, with major pulses released every 1–2 hours depending on the phase of the menstrual cycle. Growth hormone is also secreted in a pulsatile fashion, with undetectable levels in between pulses. A single measurement is therefore not helpful to diagnose GH deficiency or excess.
Biological rhythms
Circadian means changes over the 24 hours of the day–night cycle and is best shown for the pituitary–adrenal axis. Figure 19.4 shows plasma cortisol levels measured over 24 hours – levels are highest in the early morning and lowest overnight. Additionally, cortisol release is pulsatile, following the pulsatility of pituitary ACTH. Thus ‘normal’ cortisol levels vary during the day and great variations can be seen in samples taken only 30 minutes apart.
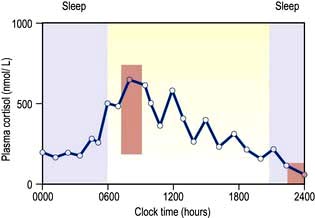
Figure 19.4 Plasma cortisol levels during a 24-hour period. Note both the pulsatility and the shifting baseline. Normal ranges for 09:00 hours (180–700 nmol/L) and 24:00 hours (<100 nmol/L; must be taken when asleep) are shown in the orange boxes. Purple shading shows sleep.
The menstrual cycle is an example of a longer and more complex (28-day) biological rhythm (see p. 972).
Other regulatory factors
Stress. Physiological ‘stress’ and acute illness produce rapid increases in ACTH and cortisol, growth hormone (GH), prolactin, adrenaline (epinephrine) and noradrenaline (norepinephrine). These can occur within seconds or minutes.
Sleep. Secretion of GH and prolactin is increased during sleep, especially the rapid eye movement (REM) phase.
Feeding and fasting. Many hormones regulate the body’s control of energy intake and expenditure and are therefore profoundly influenced by feeding and fasting. Secretion of insulin is increased and growth hormone decreased after ingestion of food, and secretion of a number of hormones is altered during prolonged food deprivation.
Testing endocrine function
Endocrine function is assessed by measurement of hormone levels in blood (or more precisely in plasma or serum) and sometimes in other body fluids on samples obtained basally and in response to stimulation and suppression tests.
Basal blood levels
Assays for all clinically relevant pituitary and end-organ hormones are available.
The time, day and condition of measurement make great differences to hormone levels, and the method and timing of samples therefore depends upon the characteristics of the endocrine system involved. There are also sex, developmental and age differences.
Basal levels are especially useful for systems with long half-lives (e.g. T4 and T3, IGF-1, androstenedione, SHBG). These vary little over the short term and random samples are therefore satisfactory.
Basal samples for many hormones need to be interpreted with respect to normal ranges for the time of day/month, diet or posture concerned. Hormones with a marked circadian rhythm (testosterone in men, cortisol, ACTH, 17αOH-progesterone) must be measured at appropriate time of day (typically at 08:00 to 10:00 but, e.g. at 24:00 to demonstrate normal low levels of cortisol at this time). LH/FSH, oestrogen and progesterone vary with time of menstrual cycle and renin/aldosterone vary with sodium intake, posture and age. For these hormones, all relevant details must be recorded or the results may prove uninterpretable.
Stress-related hormones
Measurement of stress-related hormones may be problematic either because the patient is stressed by hospital attendance or venepuncture, leading to falsely high levels (e.g. catecholamines, prolactin where sampling via an indwelling needle some time after initial venepuncture may be required) or because low levels in a non-stressed individual are unable to confirm an adequate reserve required for normal physiological stress (cortisol and GH).
Urine collections
Collections over 24 hours have the advantage of providing an ‘integrated mean’ of a day’s secretion but in practice are often incomplete or wrongly timed. They also vary with sex and body size or age. Written instructions should be provided for the patient to ensure accurate collection. Examples of hormones measured in this way are catecholamines and urinary free cortisol levels.
Saliva
Saliva is sometimes used for steroid estimations, especially in children or for samples taken at home. Midnight salivary cortisol levels are increasingly used for the diagnosis of Cushing’s syndrome due to the practical difficulties in obtaining a midnight blood sample.
Stimulation and suppression tests
These tests are used when basal levels give equivocal information. In general, stimulation tests are used to confirm suspected deficiency, and suppression tests to confirm suspected excess of hormone secretion. These tests are valuable in many instances.
For example, where the secretory capacity of a gland is damaged, maximal stimulation by the trophic hormone will give a diminished output. Thus, in the short ACTH stimulation test for adrenal reserve (Box 19.1, Fig. 19.5a), the healthy subject shows a normal response while the subject with primary hypoadrenalism (Addison’s disease) demonstrates an impaired cortisol response to tetracosactide (an ACTH analogue).
 Box 19.1
Box 19.1
Short ACTH (tetracosactide) stimulation test
a Precise cortisol normal ranges are variable between laboratories and assays – appropriate local reference ranges must be used.
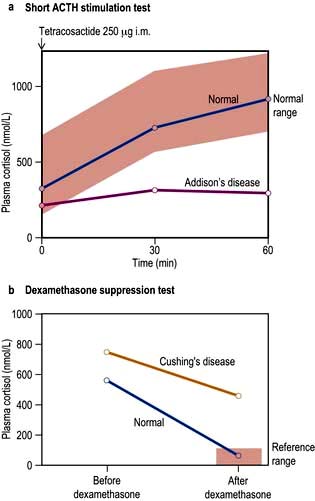
Figure 19.5 Short ACTH stimulation and dexamethasone tests. (a) This test shows a normal response in a healthy subject and a decreased response in a patient with Addison’s disease. (b) Dexamethasone suppression tests in a normal subject and in a patient with Cushing’s disease showing inadequate suppression.
A patient with a hormone-producing tumour usually fails to show normal negative feedback. A patient with Cushing’s disease (excess pituitary ACTH) will thus fail to suppress ACTH and cortisol production when given a dose of synthetic steroid, in contrast to normal subjects. Figure 19.5b shows the response of a normal subject given dexamethasone 1 mg at midnight; cortisol is suppressed the following morning. The subject with Cushing’s disease shows inadequate suppression.
The detailed protocol for each test must be followed exactly, since differences in technique will produce variations in results.
The pituitary gland and hypothalamus
Anatomy
Most peripheral hormone systems are controlled by the hypothalamus and pituitary. The hypothalamus is sited at the base of the brain around the third ventricle and above the pituitary stalk, which leads down to the pituitary itself, carrying the hypophyseal-pituitary portal blood supply.
The anatomical relations of the hypothalamus and pituitary (Fig. 19.6) include the optic chiasm just above the pituitary fossa; any expanding lesion from the pituitary or hypothalamus can thus produce visual field defects by pressure on the chiasm. Such upward expansion of the gland through the diaphragma sellae is termed ‘suprasellar extension’. Lateral extension of pituitary lesions may involve the vascular and nervous structures in the cavernous sinus and may rarely reach the temporal lobe of the brain. The pituitary is itself encased in a bony box, therefore any lateral, anterior or posterior expansion must cause bony erosion.
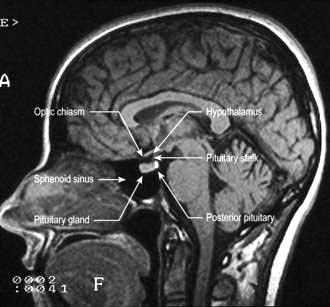
Figure 19.6 MR image of a sagittal section of the brain, showing the pituitary fossa and adjacent structures.
(By kind permission of Dr Martin Jeffree.)
Embryologically, the anterior pituitary is formed from an upgrowth of Rathke’s pouch (ectodermal) which meets an outpouching of the third ventricular floor which becomes the posterior pituitary. This unique combination of primitive gut and neural tissue provides an essential link between the rapidly responsive central nervous system and the longer-acting endocrine system. Several transcription factors – LHX3, HESX1, PROP1, POU1F1 – are responsible for the differentiation and development of the pituitary cells. Mutation of these produces pituitary disease.
Physiology
Hypothalamus
This contains many vital centres for such functions as appetite, thirst, thermal regulation and sleeping/waking. It acts as an integrator of many neural and endocrine inputs to control the release of pituitary hormone-releasing factors. It plays a role in the circadian rhythm, menstrual cyclicity, and responses to stress, exercise and mood.
Hypothalamic neurones secrete pituitary hormone-releasing and -inhibiting factors and hormones (Table 19.3) into the portal system which run down the stalk to the pituitary. As well as the classical hormones illustrated in Figure 19.7, the hypothalamus also contains large amounts of other neuropeptides and neurotransmitters such as neuropeptide Y, vasoactive intestinal peptide (VIP) and nitric oxide that can also alter pituitary hormone secretion.
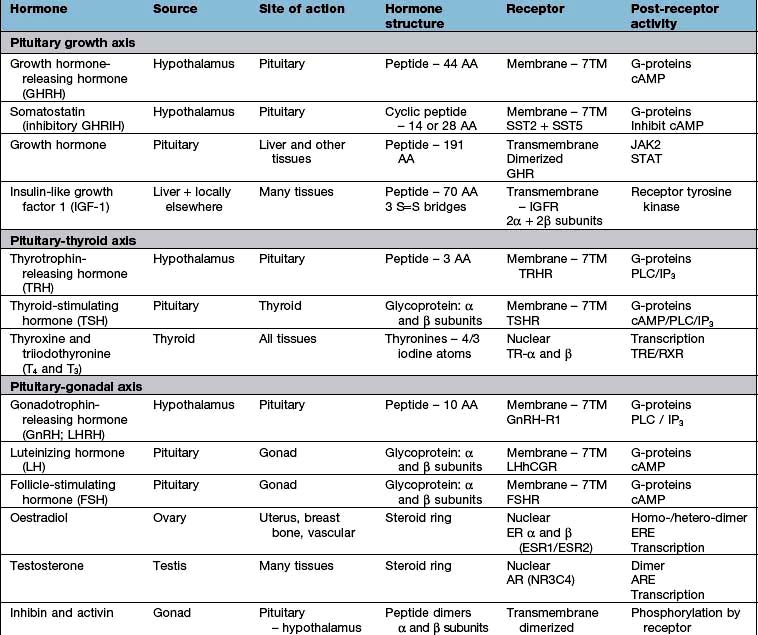
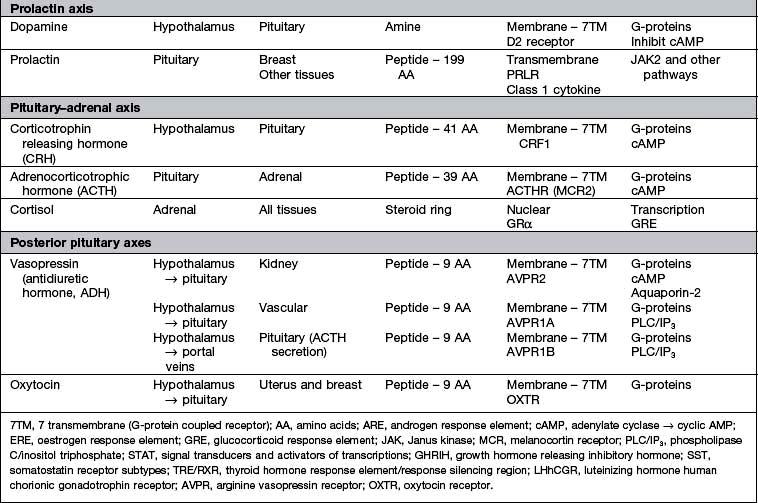
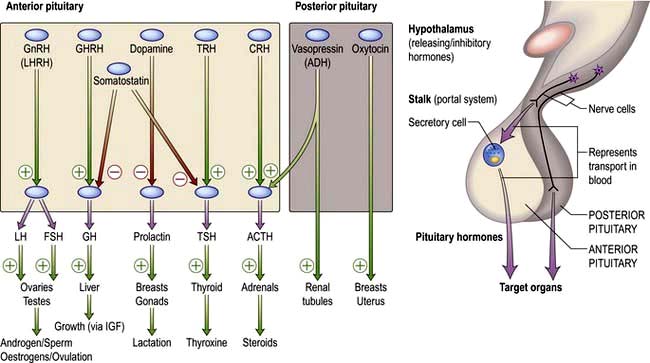
Figure 19.7 Hypothalamic releasing hormones and the pituitary trophic hormones. See the text for an explanation.
Synthetic hypothalamic hormones and their antagonists are available for the testing of many aspects of endocrine function and for treatment.
Anterior pituitary
The majority of anterior pituitary hormones are under predominantly positive control by the hypothalamic releasing hormones apart from prolactin, which is under tonic inhibition by dopamine. Pathological conditions interrupt the flow of hormones between the hypothalamus and pituitary gland and therefore cause deficiency of most hormones but oversecretion of prolactin. There are five major anterior pituitary axes: the gonadotrophin axis, the growth axis, prolactin, the thyroid axis and the adrenal axis.
Posterior pituitary
The posterior pituitary is neuro-anatomically connected to specific hypothalamic nuclei, and acts merely as a storage organ. Antidiuretic hormone (ADH, also called vasopressin) and oxytocin, both nonapeptides, are synthesized in the supraoptic and paraventricular nuclei in the anterior hypothalamus. They are then transported along the axon and stored in the posterior pituitary (Fig. 19.7). This means that damage to the stalk or pituitary alone does not prevent synthesis and release of ADH and oxytocin. ADH is discussed on page 991; oxytocin produces milk ejection and uterine myometrial contraction.
Presentations of pituitary and hypothalamic disease
Diseases of the pituitary can cause under- or overactivity of each of the hypothalamo-pituitary-end-organ axes which are under the control of this gland. The clinical features of the syndromes associated with such altered pituitary function, e.g. Cushing’s syndrome, can be the presenting symptom of pituitary disease or of end-organ disease and are discussed later. First, however, we look at clinical features of pituitary disease which are common to all hormonal axes.
Pituitary space-occupying lesions and tumours
Pituitary tumours (Table 19.4) are the most common cause of pituitary disease, and the great majority of these are benign pituitary adenomas, usually monoclonal in origin. Problems are caused by:
the result of inadequate production of hormone by the remaining normal pituitary, i.e. hypopituitarism.
Table 19.4 Characteristics of common pituitary and related tumours
| Tumour or condition | Usual size | Most common clinical presentation |
|---|---|---|
Prolactinoma |
Most <10 mm (microprolactinoma) |
Galactorrhoea, amenorrhoea, hypogonadism, erectile dysfunction |
|
Some >10 mm (macroprolactinoma) |
As above plus headaches, visual field defects and hypopituitarism |
Acromegaly |
Few mm to several cm |
Change in appearance, visual field defects and hypopituitarism |
Cushing’s disease |
Most small: few mm (some cases are hyperplasia) |
Central obesity, cushingoid appearance (local symptoms rare) |
Nelson’s syndrome |
Often large: >10 mm |
Post-adrenalectomy, pigmentation, sometimes local symptoms |
Non-functioning tumours |
Usually large: >10 mm |
Visual field defects; hypopituitarism (microadenomas may be incidental finding) |
Craniopharyngioma |
Often very large and cystic (skull X-ray abnormal in >50%; calcification common) |
Headaches, visual field defects, growth failure (50% occur below age 20; about 15% arise from within sella) |
Investigations (of a possible or proven mass)
Is there a tumour?
If there is, how big is it and what local anatomical effects is it exerting? Pituitary and hypothalamic space-occupying lesions, hormonally active or not, can cause symptoms by pressure on, or infiltration of:
the visual pathways, with field defects and visual loss (most common)
the cavernous sinus, with III, IV and VI cranial nerve lesions
bony structures and the meninges surrounding the fossa, causing headache
hypothalamic centres: altered appetite, obesity, thirst, somnolence/wakefulness or precocious puberty
the ventricles, causing interruption of cerebrospinal fluid (CSF) flow leading to hydrocephalus
Investigations
MRI of the pituitary. MRI is superior to CT scanning (Fig. 19.8) and will readily show any significant pituitary mass. Small lesions within the pituitary fossa on MRI consistent with small pituitary microadenomas are very common (10% of normal individuals in some studies). Such small lesions are sometimes detected during MRI scanning of the head for other reasons – so-called ‘pituitary incidentalomas’.
Visual fields. These should be plotted formally by automated computer perimetry or Goldmann perimetry, but clinical assessment by confrontation using a small red pin as target is also sensitive and valuable. Common defects are upper temporal quadrantanopia and bitemporal hemianopia (see p. 1073).
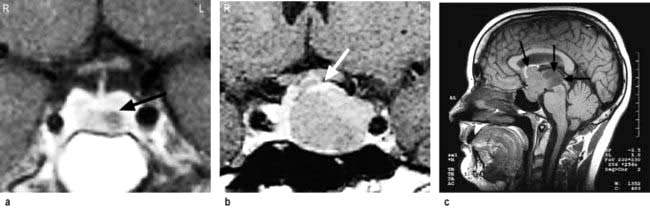
Figure 19.8 (a) Coronal MRI of pituitary, showing a left-sided lucent intrasellar microadenoma (arrowed). The pituitary stalk is deviated slightly to the right. (b) Coronal MRI of pituitary, showing macroadenoma with moderate suprasellar extension, and lateral extension compressing left cavernous sinus. The top of the adenoma is compressing the optic chiasm (arrowed). (c) Sagittal MRI of head, showing a pituitary macroadenoma with massive suprasellar extension (arrows).
Is there a hormonal excess?
There are three major conditions usually caused by secretion from pituitary adenomas which will show positive immunostaining for the relevant hormone:
Prolactin excess (prolactinoma or hyperprolactinaemia): histologically, prolactinomas are ‘chromophobe’ adenomas (a description of their appearance on classical histological staining)
GH excess (acromegaly or gigantism): somatotroph adenomas, usually ‘acidophil’, and sometimes due to specific G-protein mutations (see p. 945)
excess ACTH secretion (Cushing’s disease and Nelson’s syndrome): corticotroph adenomas, usually ‘basophil’.
Many tumours are able to synthesize several pituitary hormones, and occasionally more than one hormone is secreted in clinically significant excess (e.g. both GH and prolactin).
The clinical features of acromegaly, Cushing’s disease or hyperprolactinaemia are usually (but not always) obvious, and are discussed on pages 953, and 957. Hyperprolactinaemia may be clinically ‘silent’. Tumours producing LH, FSH or TSH are well described but very rare.
Some common pituitary tumours, usually ‘chromophobe’ adenomas, cause no clinically apparent hormone excess and are referred to as ‘non-functioning’ tumours. Laboratory studies such as immunocytochemistry or in situ hybridization show that these tumours may often produce small amounts of LH and FSH or the α-subunit of LH, FSH and TSH, and occasionally ACTH.
Is there a deficiency of any hormone?
Clinical examination may give clues; thus, short stature in a child with a pituitary tumour is likely to be due to GH deficiency. A slow, lethargic adult with pale skin is likely to be deficient in TSH and/or ACTH. Milder deficiencies may not be obvious, and require specific testing (see Table 19.7).
Treatment
Treatment depends on the type and size of tumour (Table 19.5). In general, therapy has three aims:
Table 19.5 Comparisons of primary treatments for pituitary tumours
| Treatment method | Advantages | Disadvantages |
|---|---|---|
Surgical |
|
|
Trans-sphenoidal adenomectomy or hypophysectomy |
Relatively minor procedure Potentially curative for microadenomas and smaller macroadenomas |
Some extrasellar extensions may not be accessible |
Transcranial (usually transfrontal) |
Good access to suprasellar region |
Major procedure; danger of frontal lobe damage |
Radiotherapy |
|
|
External (40–50 Gy) |
Non–invasive |
Slow action, often over many years |
Stereotactic |
Precise administration of high dose to lesion |
Long-term follow-up data limited |
Medical |
|
|
Dopamine agonist therapy (e.g. bromocriptine, cabergoline) |
Non-invasive; reversible |
Usually not curative; significant side-effects in minority |
Somatostatin analogue therapy (octreotide, lanreotide) |
Non-invasive; reversible |
Usually not curative; gallstones; expensive |
Growth hormone receptor antagonist (pegvisomant) |
Highly selective |
Usually not curative; very expensive |
Removal/control of tumour
Surgery via the trans-sphenoidal route is usually the treatment of choice. Very large tumours are occasionally removed via the open transcranial (usually transfrontal) route.
Radiotherapy – by conventional linear accelerator or newer stereotactic techniques – is usually employed when surgery is impracticable or incomplete, as it controls but rarely abolishes tumour mass. The conventional regimen involves a dose of 45 Gy, given as 20–25 fractions via three fields. Stereotactic techniques use either a linear accelerator or multiple cobalt sources (‘gamma-knife’).
Medical therapy with somatostatin analogues and/or dopamine agonists sometimes causes shrinkage of specific types of tumour (see p. 954) and if successful can be used as primary therapy.
Reduction of excess hormone secretion
Reduction is usually obtained by surgical removal but sometimes by medical treatment. Useful control can be achieved with dopamine agonists for prolactinomas or somatostatin analogues for acromegaly, but ACTH secretion usually cannot be controlled by medical means. Growth hormone antagonists are also available for acromegaly (p. 955).
Replacement of hormone deficiencies
Replacement of hormone deficiencies, i.e. hypopituitarism, is discussed below (see Table 19.8).
Table 19.8 Replacement therapy for hypopituitarism
| Axis | Usual replacement therapies |
|---|---|
Adrenal |
Hydrocortisone 15–40 mg daily (starting dose 10 mg on rising/5 mg lunchtime/5 mg evening) |
(Normally no need for mineralocorticoid replacement) |
|
Thyroid |
Levothyroxine 100–150 µg daily |
Gonadal |
|
Male |
Testosterone intramuscularly, orally, transdermally or implant |
Female |
Cyclical oestrogen/progestogen orally or as patch |
Fertility |
HCG plus FSH (purified or recombinant) or pulsatile GnRH to produce testicular development, spermatogenesis or ovulation |
Growth |
Recombinant human GH used routinely to achieve normal growth in children |
Also advocated for replacement therapy in adults where GH has effects on muscle mass and wellbeing |
|
Thirst |
Desmopressin 10–20 µg one to three times daily by nasal spray or orally 100–200 µg three times daily |
Carbamazepine, thiazides and chlorpropamide are very occasionally used in mild diabetes insipidus |
|
Breast (prolactin inhibition) |
Dopamine agonist (e.g. cabergoline, 500 µg weekly) |
Small tumours producing no significant symptoms, pressure or endocrine effects may be observed with appropriate clinical, visual field, imaging and endocrine assessments.
Differential diagnosis of pituitary or hypothalamic masses
Although pituitary adenomas are the most common mass lesion of the pituitary (90%), a variety of other conditions may also present as a pituitary or hypothalamic mass and form part of the differential diagnosis.
Other tumours
Craniopharyngioma (1–2%), a usually cystic hypothalamic tumour, often calcified, arising from Rathke’s pouch, often mimics an intrinsic pituitary lesion. It is the most common pituitary tumour in children but may present at any age.
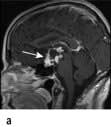
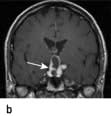
Craniopharyngioma: a partially cystic pituitary and suprasellar mass. (a) Sagittal, (b) coronal MRI.
Uncommon tumours include meningiomas, gliomas, chondromas, germinomas and pinealomas. Primary pituitary carcinomas are very rare, but occasionally prolactin and ACTH secreting tumours can present in an aggressive manner which may require chemotherapy in addition to conventional treatment. Secondary deposits occasionally present as apparent pituitary tumours, typically presenting with headache and diabetes insipidus.
Hypophysitis and other inflammatory masses
A variety of inflammatory masses occur in the pituitary or hypothalamus. These include rare pituitary-specific conditions (e.g. autoimmune [lymphocytic] hypophysitis, giant cell hypophysitis, postpartum hypophysitis) or pituitary manifestations of more generalized disease processes (sarcoidosis, Langerhans’ cell histiocytosis, Wegener’s granulomatosis). These lesions may be associated with diabetes insipidus and/or an unusual pattern of hypopituitarism.
Hypopituitarism
Pathophysiology
Deficiency of hypothalamic releasing hormones or of pituitary trophic hormones can be selective or multiple. Thus isolated deficiencies of GH, LH/FSH, ACTH, TSH and vasopressin (ADH) are all seen, some cases of which are genetic and congenital and others sporadic and autoimmune or idiopathic in nature.
Multiple deficiencies usually result from tumour growth or other destructive lesions. There is generally a progressive loss of anterior pituitary function. GH and gonadotrophins are usually first affected. Hyperprolactinaemia, rather than prolactin deficiency, occurs relatively early because of loss of tonic inhibitory control by dopamine. TSH and ACTH are usually last to be affected.
Panhypopituitarism refers to deficiency of all anterior pituitary hormones; it is most commonly caused by pituitary tumours, surgery or radiotherapy. Vasopressin (ADH) and oxytocin secretion will be significantly affected only if the hypothalamus is involved by a hypothalamic tumour or major suprasellar extension of a pituitary lesion, or if there is an infiltrative/inflammatory process. Posterior pituitary deficiency is rare in an uncomplicated pituitary adenoma.
Genetics of hypopituitarism
Specific genes are responsible for the development of the anterior pituitary involving interaction between signalling molecules and transcription factors. For example, mutations in PROP1 and POU1F1 (previously PIT-1) prevent the differentiation of anterior pituitary cells (precursors to somatotroph, lactotroph, thyrotroph and gonadotroph cells), leading to deficiencies of GH, prolactin, TSH and GnRH. In addition, novel mutations within GH and GHRH receptor genes have been identified which may explain the pathogenesis of isolated GH deficiency in children. Despite these advances, most cases of hypopituitarism do not have specific identifiable genetic causes.
Causes
Disorders causing hypopituitarism are listed in Table 19.6. Pituitary and hypothalamic tumours, and surgical or radiotherapy treatment, are the most common.
Table 19.6 Causes of hypopituitarism
Congenital |
Traumatic |
Isolated deficiency of pituitary hormones (e.g. Kallmann’s syndrome) |
Skull fracture through base |
Infiltrations |
|
Infective |
Sarcoidosis |
Basal meningitis (e.g. tuberculosis) |
|
Vascular |
|
Pituitary apoplexy |
|
Others |
|
Radiation damage |
|
Immunological |
‘Functional’ |
Autoimmune (lymphocytic) hypophysitis |
Anorexia nervosa |
Neoplastic |
|
Pituitary or hypothalamic tumours |
|
Craniopharyngioma |
|
Meningiomas |
|
Gliomas |
|
Pinealoma |
|
Secondary deposits, especially breast |
|
Lymphoma |
Clinical features
Symptoms and signs depend upon the extent of hypothalamic and/or pituitary deficiencies, and mild deficiencies may not lead to any complaint by the patient. In general, symptoms of deficiency of a pituitary-stimulating hormone are the same as primary deficiency of the peripheral endocrine gland (e.g. TSH deficiency and primary hypothyroidism cause similar symptoms due to lack of thyroid hormone secretion).
Secondary hypothyroidism and adrenal failure both lead to tiredness and general malaise.
Hypothyroidism causes weight gain, slowness of thought and action, dry skin and cold intolerance.
Hypoadrenalism causes mild hypotension, hyponatraemia and ultimately cardiovascular collapse during severe intercurrent stressful illness.
Gonadotrophin and thus gonadal deficiencies lead to loss of libido, loss of secondary sexual hair, amenorrhoea and erectile dysfunction.
Hyperprolactinaemia may cause galactorrhoea and hypogonadism.
GH deficiency causes growth failure in children and impaired wellbeing in some adults.
Weight may increase (due to hypothyroidism, see above) or decrease in severe combined deficiency (pituitary cachexia).
Longstanding panhypopituitarism gives the classic picture of pallor with hairlessness (‘alabaster skin’).
Kallmann’s syndrome. This syndrome is isolated gonadotrophin (GnRH) deficiency (p. 976).This syndrome arises due to mutations in the KAL1 gene which is located on the short (p) arm of the X chromosome. Kallmann’s is classically characterized by anosmia because the KAL1 gene provides instructions to make anosmin, which has a role in development of both the olfactory system as well as migration of GnRH secreting neurones.
Septo-optic dysplasia. This is a rare congenital syndrome (associated with mutations in the HESX1 gene) presenting in childhood with a clinical triad of midline forebrain abnormalities, optic nerve hypoplasia and hypopituitarism.
Sheehan’s syndrome is due to pituitary infarction following postpartum haemorrhage and is rare in developed countries.
Pituitary apoplexy. A pituitary tumour occasionally enlarges rapidly owing to infarction or haemorrhage. This may produce severe headache, double vision and sudden severe visual loss, sometimes followed by acute life-threatening hypopituitarism. Often pituitary apoplexy can be managed conservatively with replacement of hormones and close monitoring of vision, although if there is a rapid deterioration in visual acuity and fields, surgical decompression of the optic chiasm may be necessary.
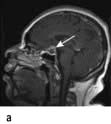
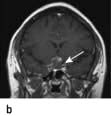
Pituitary apoplexy, showing a bright area of haemorrhage at the top of a pituitary adenoma. (a) Sagittal, (b) coronal.
The ‘empty sella’ syndrome. An ‘empty sella’ is sometimes reported on pituitary imaging. This is sometimes due to a defect in the diaphragma and extension of the subarachnoid space (cisternal herniation) or may follow spontaneous infarction or regression of a pituitary tumour. All or most of the sella turcica is devoid of apparent pituitary tissue, but, despite this, pituitary function is usually normal, the pituitary being eccentrically placed and flattened against the floor or roof of the fossa.
Investigations
Each axis of the hypothalamic-pituitary system requires separate investigation. However, the presence of normal gonadal function (ovulation/menstruation or normal libido/erections) suggests that multiple defects of anterior pituitary function are unlikely.
Tests range from the simple basal levels (e.g. free T4 for the thyroid axis), to stimulatory tests for the pituitary, and tests of feedback for the hypothalamus (Table 19.7). Assessment of the hypothalamic-pituitary-adrenal axis is complex: basal 09:00 hours cortisol levels above 400 nmol/L usually indicate an adequate reserve, while levels below 100 nmol/L predict an inadequate stress response. In many cases basal levels are equivocal and a dynamic test is essential: the insulin tolerance test (Box 19.2) is widely regarded as the ‘gold standard’ but the short ACTH stimulation test (Box 19.1), though an indirect measure, is used by many as a routine test of hypothalamic-pituitary-adrenal status. Occasionally, the difference between ACTH deficiency and normal HPA axis can be subtle, and the assessment of adrenal reserve is best left to an experienced endocrinologist.
Box 19.2
Insulin tolerance test
Procedure
Test explained to patient and consent obtained
Should only be performed in experienced, specialist units
Exclude cardiovascular disease (ECG), epilepsy or unexplained blackouts; exclude severe untreated hypopituitarism (basal cortisol must be >100 nmol/L; normal free T4)
Intravenous hydrocortisone and glucose available for emergency
Overnight fast, begin at 08:00–09:00 hours
Soluble insulin, 0.15 U/kg, i.v. at time 0
Glucose, cortisol and GH levels at 0, 30, 45, 60, 90, 120 min
Normal response
Cortisol rises above 550 nmol/La
GH rises above 7 ng/L (severe deficiency = <3 ng/L (<9 mU/L))
Glucose must be <2.2 mmol/L to achieve adequate stress response
a Precise cortisol normal ranges are variable between laboratories and assays – appropriate local reference ranges must be used.
Treatment
Steroid and thyroid hormones are essential for life. Both are given as oral replacement drugs, as in primary thyroid and adrenal deficiency, aiming to restore the patient to clinical and biochemical normality (Table 19.8) and levels are monitored by routine hormone assays. Note: Thyroid replacement should not commence until normal glucocorticoid function has been demonstrated or replacement steroid therapy initiated, as an adrenal ‘crisis’ may otherwise be precipitated.
Sex hormones are replaced with androgens and oestrogens, both for symptomatic control and to prevent long-term problems related to deficiency (e.g. osteoporosis).
When fertility is desired, gonadal function is stimulated directly by human chorionic gonadotrophin (HCG, mainly acting as LH), purified or biosynthetic gonadotrophins, or indirectly by pulsatile gonadotrophin-releasing hormone (GnRH – also known as luteinizing hormone-releasing hormone, LHRH); all are expensive and time-consuming and should be restricted to specialist units.
GH therapy is given in the growing child, under the care of a paediatric endocrinologist. In adult GH deficiency, GH therapy also produces improvements in body composition, work capacity and psychological wellbeing, together with reversal of lipid abnormalities associated with a high cardiovascular risk, and often results in significant symptomatic benefit in some cases. NICE recommends GH replacement for people with severe GH deficiency and significant quality of life impairment. It is expensive and in the UK costs £2500–6000 per annum.
Glucocorticoid deficiency may mask impaired urine concentrating ability, diabetes insipidus only becoming apparent after steroid replacement because steroids are required for excretion of free water.
FURTHER READING
Dattani MT. Growth hormone deficiency and combined pituitary deficiency: does the genotype matter? Clin Endocrinol 2005; 63:121–130.
National Institute for Health and Clinical Excellence. Human growth hormone (somatotropin) in adults with growth hormone deficiency; 2003: Technology appraisal 64; http://guidance.nice.org.uk/TA64/Guidance/pdf/English
Schneider HJ, Aimaretti G, Kreitschmann-Andermahr I et al. Hypopituitarism. Lancet 2007; 369:1151–1470.
Growth and abnormal stature
Physiology and control of growth hormone (GH) (Fig. 19.9)
GH is the pituitary factor responsible for stimulation of body growth in humans. Its secretion is stimulated by GHRH, released into the portal system from the hypothalamus; it is also under inhibitory control by somatostatin. A separate GH stimulating system involves a distinct receptor (GH secretogogue receptor), which interacts with ghrelin (see p. 259). It is not known how these two systems interact but because ghrelin is synthesized in the stomach, it suggests a nutritional role for GH.
GH acts by binding to a specific (single transmembrane) receptor located mainly in the liver (Table 19.3). This induces an intracellular phosphorylation cascade involving the JAK/STAT (Janus kinase/signal transducing activators of transcription) pathway (p. 32). STAT proteins are translocated from the cytoplasm into the cell nucleus and cause GH-specific effects by binding to nuclear DNA.
IGF-1 (insulin-like growth factor-1), a somatomedin stimulates growth and its hepatic secretion is stimulated by a tissue-specific effect of GH on the liver. There are multiple IGF-binding proteins (IGF-BP) in plasma – IGF-BP3 can be measured clinically to improve assessment of GH status, particularly in children.
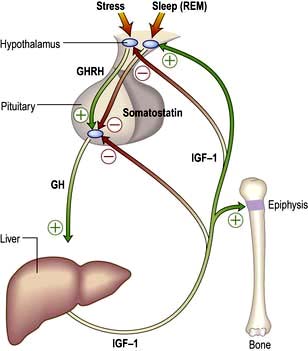
Figure 19.9 The control of growth hormone (GH) and insulin-like growth factor-1 (IGF-1). Pituitary GH is secreted under dual control of growth hormone-releasing hormone (GHRH) and somatostatin and stimulates release of IGF-1 in liver and elsewhere. IGF-1 has peripheral actions including bone growth and exerts negative feedback to hypothalamus and pituitary.
The metabolic actions of the system are:
Increasing collagen and protein synthesis
Promoting retention of calcium, phosphorus and nitrogen, necessary substrates for anabolism
Opposing the action of insulin (a ‘counter-regulatory’ hormone effect).
GH release is intermittent and mainly nocturnal, especially during REM sleep. The frequency and size of GH pulses increase during the growth spurt of adolescence and decline thereafter. Acute stress and exercise both stimulate GH release while, in the normal subject, hyperglycaemia suppresses it.
IGF-1 may, in addition, play a major role in maintaining neoplastic growth. A relationship has been shown between circulating IGF-1 concentrations and breast cancer in premenopausal women and prostate cancer in men.
Normal growth
There are factors other than GH involved in linear growth in the human.
Genetic factors. Children of two short parents will probably be short and vice-versa.
Nutritional factors. Adequate nutrients must be available. Impaired growth can result from inadequate dietary intake or small bowel disease (e.g. coeliac disease).
General health. Any serious systemic disease in childhood is likely to reduce growth (e.g. chronic kidney disease or chronic infection).
Intrauterine growth retardation. These infants often grow poorly in the long term, while infants with simple prematurity usually catch up. There is some evidence that low birthweight may predispose to hypertension, diabetes and other health problems in later adult life (p. 195).
Emotional deprivation and psychological factors. These can impair growth by complex, poorly understood mechanisms, probably involving temporarily decreased GH secretion.
In general, there are three overlapping phases of growth: infantile (0–2 years), which appears largely substrate (food) dependent; childhood (age 2 years to puberty), which is largely GH dependent; and the adolescent ‘growth spurt’, dependent on GH and sex hormones.
The relevant aspects of history and examination in the assessment of problems are shown in Box 19.3.
Assessment of growth
Charts showing normal centiles of height and weight are essential to monitor growth; they are available for normal British children (Fig. 19.10) and many other national and ethnic groups. Height must be measured, ideally at the same time of day on the same instrument by the same observer.
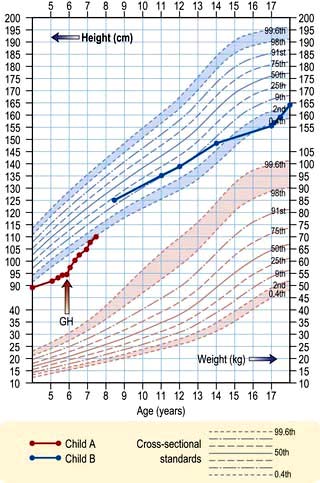
Figure 19.10 A height chart for boys. Child A illustrates the course of a child with hypopituitarism, initially treated with cortisol and thyroxine, but showing growth only after growth hormone treatment. Child B shows the course of a child with constitutional growth delay without treatment.
Height velocity is more helpful than current height. It requires at least two measurements some months apart and, ideally, multiple serial measurements. Height velocity is the rate of current growth (cm per year), while the current attained height is largely dependent upon previous growth. Standard deviation scores (SDS) based on the degree of deviation from age–sex norms are widely used. Computer programs also allow calculation of many of these indices.
The approximate future height of a child (‘mid-parental height’) can be simply predicted from the parental heights. For a boy, this is:
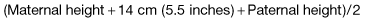
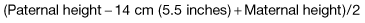
Thus, with a father of 180 cm and mother of 154 cm, the predicted heights are 174 cm for a son and 160 cm for a daughter.
Growth failure: short stature
When children or their parents complain of short stature, particular attention should focus on:
Intrauterine growth retardation, weight and gestation at birth
Possible systemic disorders – any system, but especially small bowel disease
Evidence of skeletal, chromosomal or other congenital abnormalities
Endocrine status – particularly thyroid
Dietary intake and use of drugs, especially steroids for asthma
School, general practitioner, clinic and home records of height and weight should be obtained if possible to allow growth-velocity calculation. If unavailable, such data must be obtained prospectively.
A child with normal growth velocity is unlikely to have significant endocrine disease and the commonest cause of short stature in this situation is pubertal or ‘constitutional’ delay. However, low growth velocity without apparent systemic cause requires further investigation. Sudden cessation of growth suggests major physical disease; if no gastrointestinal, respiratory, renal or skeletal abnormality is apparent, then a cerebral tumour or hypothyroidism is likeliest.
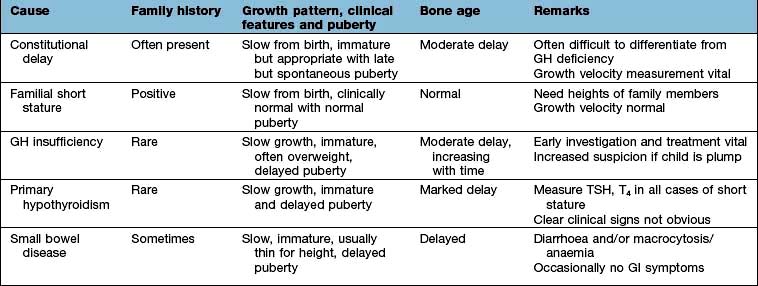
Consistently slow-growing children require full endocrine assessment. Features of the more common causes of growth failure are given in Table 19.9.
Around the time of puberty, where constitutional delay is clearly shown and symptoms require intervention, then very-low-dose sex steroids in 3- to 6-month courses will usually induce acceleration of growth.
Investigations
Systemic disease having been excluded, perform:
Thyroid function tests: serum TSH and free T4 to exclude hypothyroidism
GH status. Basal levels are of little value. Dynamic tests include the GH response to insulin (the ‘gold standard’; Box 19.2), glucagon, arginine, exercise and clonidine. Tests should only be performed in centres experienced in their use and interpretation. Normal responses depend on test and GH assay used
Blood levels of IGF-1 (insulin-like growth factor-1) and IGF-BP3 (binding protein 3) may provide evidence of GH undersecretion
Assessment of bone age. Non-dominant hand and wrist X-rays allow assessment of bone age by comparison with standard charts
Karyotyping in females. Turner’s syndrome (p. 978) is associated with short stature. It is thought that this is due to a defect in the short stature homeobox (SHOX) gene which has a role in non-GH mediated growth.
Treatment
Systemic illness should be treated and primary hypothyroidism treated with levothyroxine.
For GH insufficiency, recombinant GH (somatropin) is given as nightly injections in doses of 0.17–0.35 mg/kg per week, with dose adjustments made according to clinical response and IGF-1 levels. Treatment is expensive and should be supervised in expert centres.
GH treatment in so-called ‘short normal’ children has not been shown to produce any worthwhile increase in final height. In Turner’s syndrome (see p. 983) large doses of GH are effective in increasing final height, especially in combination with appropriate very-low-dose oestrogen replacement. Familial cases of resistance to GH owing to an abnormal GH receptor (Laron-type dwarfism) are well described. They are very rare but may respond to therapy with synthetic IGF-1 (mecasermin).
Tall stature
The most common causes are hereditary (two tall parents!), idiopathic (constitutional) or early development. It can occasionally be due to hyperthyroidism. Other causes include chromosomal abnormalities (e.g. Klinefelter’s syndrome, Marfan’s syndrome) or metabolic abnormalities. GH excess is a very rare cause and is usually clinically obvious.
Pituitary hypersecretion syndromes
Acromegaly and gigantism
Growth hormone stimulates skeletal and soft tissue growth. GH excess therefore produces gigantism in children (if acquired before epiphyseal fusion) and acromegaly in adults. Both are due to a GH secreting pituitary tumour (somatotroph adenoma) in almost all cases. Hyperplasia due to ectopic GHRH excess is very rare. Overall incidence is approximately 3–4/million per year and the prevalence is 50–80/million worldwide. Acromegaly usually occurs sporadically, although gene mutations can rarely give rise to familial acromegaly, typically the AIP gene in familial isolated pituitary adenoma.
Clinical features
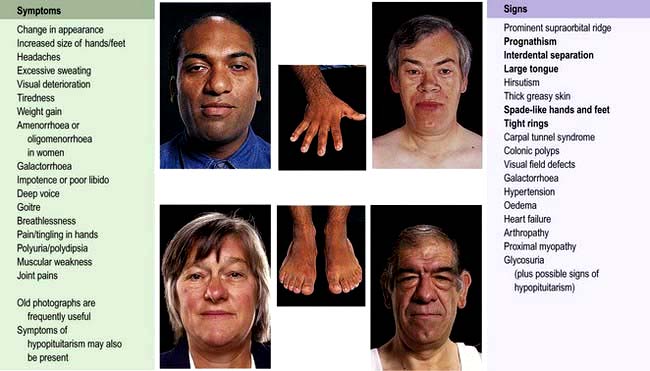
Symptoms and signs of acromegaly are shown in Figure 19.11. One-third of patients present with changes in appearance, one-quarter with visual field defects or headaches; in the remainder the diagnosis is made by an alert observer in another clinic, e.g. GP, diabetic, hypertension, dental, dermatology. Sleep apnoea is common and requires investigation and treatment if there are suggestive symptoms (see p. 818). Sweating, headaches and soft tissue swelling are particularly useful symptoms of persistent growth hormone secretion. Headache is very common in acromegaly and may be severe even with small tumours; it is often improved after surgical cure or with somatostatin analogues.
Investigations
GH levels may exclude acromegaly if undetectable but a detectable value is non-diagnostic taken alone. Normal adult levels are <0.5 µg/L for most of the day except during stress or a ‘GH pulse’.
A glucose tolerance test is diagnostic if there is no suppression of GH. Acromegalics fail to suppress GH below 0.3 µg/L and some show a paradoxical rise; about 25% of acromegalics have a positive diabetic glucose tolerance test.
IGF-1 levels are almost always raised in acromegaly – a single plasma level of IGF-1 reflects mean 24-hour GH levels and is useful in diagnosis. A normal IGF-1 together with random growth hormone <1 µg/L may be taken to exclude acromegaly if the diagnosis is clinically unlikely.
Visual field examination: defects are common, e.g. bitemporal hemianopia.
MRI scan of pituitary if above tests abnormal. This will almost always reveal the pituitary adenoma.
Pituitary function: partial or complete anterior hypopituitarism is common.
Prolactin: mild to moderate hyperprolactinaemia occurs in 30% of patients (see Fig. 19.13). In some, the adenoma secretes both GH and prolactin.
Management and treatment
Untreated acromegaly results in markedly reduced survival. Most deaths occur from heart failure, coronary artery disease and hypertension-related causes. In addition, there is an increase in deaths due to neoplasia, particularly large bowel tumours (some specialist centres advocate regular colonoscopy to detect and remove colonic polyps to reduce the risk of colonic cancer). Treatment is therefore indicated in all except the elderly or those with minimal abnormalities. The aim of therapy is to achieve a mean growth hormone level below 2.5 µg/L; this is not always ‘normal’ but has been shown to reduce mortality to normal levels and is therefore considered a ‘safe’ GH level. A normal IGF-1 is also a goal of therapy. Occasionally there can be discordance between GH and IGF-1 levels, which can create management dilemmas.
When present, hypopituitarism should be corrected (see p. 950) and concurrent diabetes and/or hypertension should be treated conventionally; both usually improve with treatment of the acromegaly.
The general advantages and disadvantages of surgery, radiotherapy and medical treatment are discussed on page 947. Progress can be assessed by monitoring GH and IGF-1 levels.
Surgery. Trans-sphenoidal surgery is the appropriate first-line therapy. It will result in clinical remission in a majority of cases (60–90%) with pituitary microadenoma, but in only 50% of those with macroadenoma. Very high preoperative GH and IGF-1 levels are also poor prognostic markers of surgical cure. Surgical success rates are variable and highly dependent upon experience, and a specialist pituitary surgeon is essential. Transfrontal surgery is rarely required except for massive macroadenomas. There is approximately a 10% recurrence rate.
Pituitary radiotherapy. External radiotherapy is normally used after pituitary surgery fails to normalize GH levels rather than as primary therapy. It is often combined with medium-term treatment with a somatostatin analogue, dopamine agonist or GH antagonist because of the slow biochemical response to radiotherapy, which may take 10 years or more and is often associated with hypopituitarism which makes it unattractive in patients of reproductive age. Stereotactic radiotherapy is used in some centres.
Medical therapy. There are three receptor targets for the treatment of acromegaly: pituitary somatostatin receptors and dopamine (D2) receptors and growth hormone receptors in the periphery.
Hyperprolactinaemia
The hypothalamic-pituitary control of prolactin secretion is illustrated in Figure 19.12. Prolactin is a large peptide secreted in the pituitary and acts via a transmembrane receptor stimulating JAK2 and other pathways (Table 19.3).
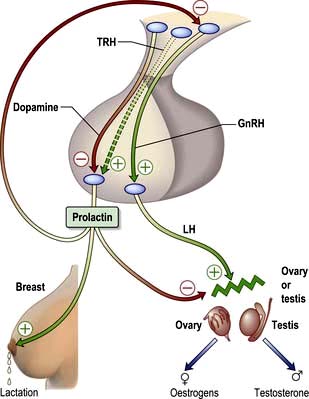
Figure 19.12 The control and actions of prolactin. Prolactin is mainly controlled by tonic inhibition by hypothalamic dopamine. Prolactin stimulates lactation but also inhibits both hypothalamic GnRH secretion and the gonadal actions of LH.
Prolactin is under tonic dopamine inhibition: factors known to increase prolactin secretion (e.g. TRH) are probably of less relevance. Prolactin stimulates milk secretion (but not breast tissue development) but also inhibits gonadal activity. It decreases GnRH pulsatility at the hypothalamic level and, to a lesser extent, blocks the action of LH on the ovary or testis, producing hypogonadism even when the pituitary gonadal axis itself is intact.
The role of prolactin outside pregnancy and lactation is not well defined, although there is some epidemiological evidence of a link between high prolactin levels and breast cancer which has led to an interest in the development of prolactin receptor antagonists.
Physiological hyperprolactinaemia occurs in pregnancy, lactation and severe stress, as well as during sleep and coitus. The range of serum prolactin seen in common causes of hyperprolactinaemia is illustrated in Figure 19.13. Mildly increased prolactin levels (400–600 mU/L) may be physiological and asymptomatic but higher levels require a diagnosis. Levels above 5000 mU/L always imply a prolactin-secreting pituitary tumour.
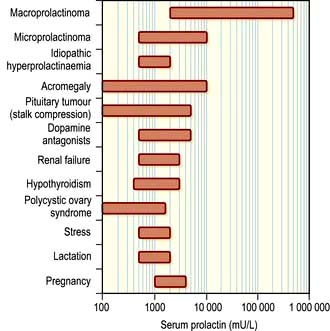
Causes
Hyperprolactinaemia has many causes. Common pathological causes include prolactinoma, co-secretion of prolactin in tumours causing acromegaly, stalk compression due to pituitary adenomas and other pituitary masses, polycystic ovary syndrome, primary hypothyroidism and ‘idiopathic’ hyperprolactinaemia. Rarer causes are oestrogen therapy (e.g. the ‘pill’), renal failure, liver failure, post-ictal state and chest wall injury. Dopamine antagonist drugs (metoclopramide, domperidone, most antipsychotics) are a common iatrogenic cause, as well as most other antiemetics (except cyclizine) and opiates.
Clinical features
Hyperprolactinaemia stimulates milk production in the breast and inhibits GnRH and gonadotrophin secretion per se. It usually presents with:
Galactorrhoea, spontaneous or expressible (60% of cases)
Oligomenorrhoea or amenorrhoea
Decreased libido in both sexes
Symptoms or signs of oestrogen or androgen deficiency – in the long term osteoporosis may result, especially in women
Delayed or arrested puberty in the peripubertal patient
Mild gynaecomastia is often seen in men due to the associated hypogonadism rather than a direct effect of prolactin.
Additionally, headaches and/or visual field defects occur if there is a pituitary tumour (more common in men). Note that many people with galactorrhoea do not have hyperprolactinaemia – ‘normoprolactinaemic galactorrhoea’ – and the causes are poorly understood.
Investigations
Hyperprolactinaemia should be confirmed by repeat measurement. If there are no clinical features of hyperprolactinaemia, the possibility of macroprolactinaemia should be considered. This is a higher molecular weight complex of prolactin bound to IgG, which is physiologically inactive but occurs in a small proportion of normal people and can therefore lead to unnecessary treatment. Macroprolactinaemia can be diagnosed in the laboratory by precipitation of IgG with polyethylene glycol, after which prolactin levels will be normal on testing; most laboratories will do this routinely.
Further tests are appropriate after physiological and drug causes have been excluded:
Visual fields should be checked.
Primary hypothyroidism must be excluded since this is a cause of hyperprolactinaemia.
Anterior pituitary function should be assessed if there is any clinical evidence of hypopituitarism or radiological evidence of a pituitary tumour (Table 19.7; Box 19.1, Box 19.2).
MRI of the pituitary is necessary if there are any clinical features suggestive of a pituitary tumour, and desirable in all cases when prolactin is significantly elevated (above 1000 mU/L).
In the presence of a pituitary mass on MRI, the level of prolactin helps determine whether the mass is a prolactinoma or a non-functioning pituitary tumour causing stalk-disconnection hyperprolactinaemia: levels of above 5000 mU/L in the presence of a macroadenoma, or above 2000 mU/L in the presence of a microadenoma (or with no radiological abnormality), strongly suggest a prolactinoma (see p. 946). Macroprolactinoma refers to tumours above 10 mm diameter, microprolactinoma to smaller ones.
Occasionally, very large prolactinomas can be associated with such high serum prolactin levels that some assays give an artefactual falsely low result (known as the ‘hook effect’). If suspected, this can be excluded by serial dilutions of the serum sample.
Treatment
Hyperprolactinaemia is usually treated to avoid the long-term effects of oestrogen deficiency (even if the patient would otherwise welcome the lack of periods!) or testosterone deficiency in the male. Exceptions include minor elevations (400–1000 mU/L) with preservation of normal regular menstruation (or normal male testosterone levels) and postmenopausal people with microprolactinomas who are not taking oestrogen replacement.
Medical treatment. Hyperprolactinaemia is controlled with a dopamine agonist.
Cabergoline (500 µg once or twice a week judged on clinical response and prolactin levels) is the best tolerated and longest-acting drug and is the first drug of choice.
Bromocriptine is the longest-established therapy and therefore preferred if pregnancy is planned: initial doses should be small (e.g. 1 mg), taken with food and gradually increased to 2.5 mg two or three times daily. Side-effects, which prevent effective therapy in a minority of cases, include nausea and vomiting, dizziness and syncope, constipation and cold peripheries.
Complications seen, when cabergoline is used in higher doses in Parkinson’s disease, include pulmonary, retroperitoneal and pericardial fibrotic reactions and cardiac valve lesions. Patients need monitoring, although such adverse effects appear to be very rare in patients on lower, ‘endocrine’ doses.
In most cases a dopamine agonist will be the first and only therapy and can be used in the long term. Prolactinomas usually shrink in size on a dopamine agonist, and in macroadenomas any pituitary mass effects commonly resolve (Fig. 19.14). Microprolactinomas may not recur after several years of dopamine agonist therapy in a minority of cases, but in the majority hyperprolactinaemia will recur if treatment is stopped.
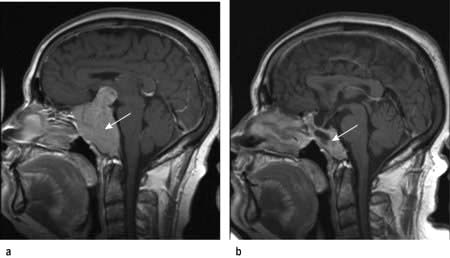
Figure 19.14 Macroprolactinoma before (a) and after (b) 2 years treatment with the dopamine agonist cabergoline showing marked adenoma shrinkage.
Trans-sphenoidal surgery may restore normoprolactinaemia in people with microadenoma, but is rarely completely successful with macroadenomas and risks damage to normal pituitary function. Therefore most patients and physicians elect to continue medical therapy rather than proceed to surgery. Prolactin should therefore always be measured before surgery on any mass in the pituitary region. Some surgeons believe that long-term bromocriptine increases the hardness of the adenoma and makes resection more difficult, but others dissent from this view.
Radiotherapy usually controls adenoma growth and is slowly effective in lowering prolactin but causes progressive hypopituitarism. It may be advocated after medical tumour shrinkage or after surgery in larger tumours, especially where families are complete or if the drug treatment is poorly tolerated, but most workers simply advocate continuation of dopamine agonist therapy in responsive cases.
In patients planning pregnancy, it is useful to know the size of the pituitary lesion before starting dopamine agonist therapy. Rarely, tumours enlarge during pregnancy to produce headaches and visual field defects. Dopamine agonists, which are traditionally stopped after conception, can be restarted if there are any signs of tumour growth during pregnancy.
Cushing’s syndrome
Cushing’s syndrome is the term used to describe the clinical state of increased free circulating glucocorticoid. It occurs most often following the therapeutic administration of synthetic steroids or ACTH (see below).
Pathophysiology and causes
Spontaneous Cushing’s syndrome is rare, with an incidence of <5/million per year.
Causes of Cushing’s syndrome are usually subdivided into two groups (Table 19.10):
1. Increased circulating ACTH from the pituitary (65% of cases), known as Cushing’s disease, or from an ‘ectopic’, non-pituitary, ACTH-producing tumour elsewhere in the body (10%) with consequent glucocorticoid excess (‘ACTH dependent’ Cushing’s)
2. A primary excess of endogenous cortisol secretion (25%) by an adrenal tumour or nodular hyperplasia, with subsequent (physiological) suppression of ACTH (‘non-ACTH-dependent’ Cushing’s). Rare cases are due to aberrant expression of receptors for other hormones (e.g. glucose-dependent insulinotrophic peptide (GIP), LH or catecholamines) in adrenal cortical cells.
Table 19.10 Causes of Cushing’s syndrome
Clinical features
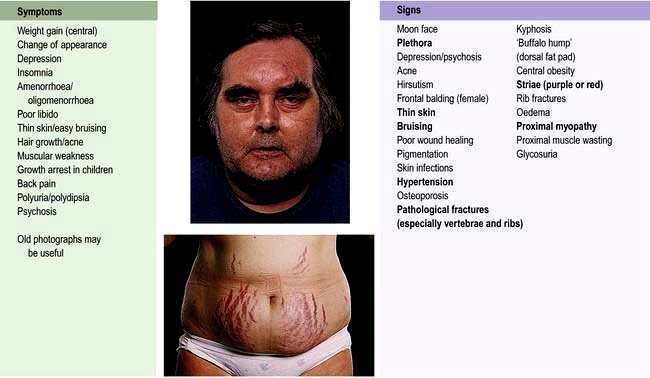
The clinical features of Cushing’s syndrome are those of glucocorticoid excess and are illustrated in Figure 19.15.
Pigmentation occurs only with ACTH-dependent causes.
A cushingoid appearance can be caused by excess alcohol consumption (pseudo-Cushing’s syndrome) – the pathophysiology is poorly understood.
Impaired glucose tolerance or frank diabetes is common, especially in the ectopic ACTH syndrome.
Hypokalaemia due to the mineralocorticoid activity of cortisol is common with ectopic ACTH secretion.
Diagnosis
There are two phases to the investigation.
1. Confirmation
Most obese, hirsute, hypertensive patients do not have Cushing’s syndrome, and some cases of genuine Cushing’s have relatively subtle clinical signs. Confirmation rests on demonstrating inappropriate cortisol secretion, not suppressed by exogenous glucocorticoids: difficulties occur with obesity and depression where cortisol dynamics are often abnormal. Random cortisol measurements are of no value. Occasional patients are seen with so-called ‘cyclical Cushing’s’ where the abnormalities come and go.
Investigations to confirm the diagnosis include:
48-hour low-dose dexamethasone test (see Table 19.11). Normal individuals suppress plasma cortisol to <50 nmol/L. People with Cushing’s syndrome fail to show complete suppression of plasma cortisol levels (although levels may fall substantially in a few cases). This test is highly sensitive (>97%). The overnight dexamethasone test is slightly simpler, but has a higher false-positive rate.
24-hour urinary free cortisol measurements. This is simple, but less reliable – repeatedly normal values render the diagnosis most unlikely, but some people with Cushing’s syndrome have normal values on some collections (approximately 10%).
Circadian rhythm. After 48 hours in hospital, cortisol samples are taken at 09:00 hours and 24:00 hours (without warning the patient). Normal subjects show a pronounced circadian variation (see Fig. 19.4, p. 942); those with Cushing’s syndrome have high midnight cortisol levels (>100 nmol/L), though the 09:00 hours value may be normal. Midnight salivary cortisol collected at home gives the same information more simply where the assay is available.
Other tests. There are frequent exceptions to the classic responses to diagnostic tests in Cushing’s syndrome. If any clinical suspicion of Cushing’s remains after preliminary tests then specialist investigations are still indicated. These may include insulin stress test, desmopressin stimulation test (p. 993) and CRH tests.
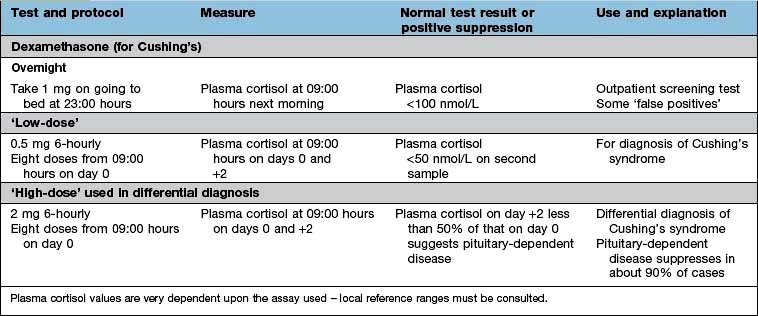
2. Differential diagnosis of the cause
This can be extremely difficult since all causes can result in clinically identical Cushing’s syndrome. The classical ectopic ACTH syndrome is distinguished by a short history, pigmentation and weight loss, unprovoked hypokalaemia, clinical or chemical diabetes and plasma ACTH levels above 200 ng/L, but many ectopic tumours are benign and mimic pituitary disease closely, both clinically and biochemically. Severe hirsutism/virilization suggests an adrenal tumour.
Biochemical and radiological procedures for diagnosis include:
Plasma ACTH levels. Low or undetectable ACTH levels (<10 ng/L) on two or more occasions are a reliable indicator of non-ACTH-dependent disease.
Adrenal CT or MRI scan. Adrenal adenomas and carcinomas causing Cushing’s syndrome are relatively large and always detectable by CT scan. Carcinomas are distinguished by large size, irregular outline and signs of infiltration or metastases. Bilateral adrenal hyperplasia may be seen in ACTH-dependent causes or in ACTH-independent nodular hyperplasia.
Pituitary MRI. A pituitary adenoma may be seen but the adenoma is often small and not visible in a significant proportion of cases.
Plasma potassium levels. Hypokalaemia is common with ectopic ACTH secretion. (All diuretics must be stopped.)
High-dose dexamethasone test (Table 19.11). Failure of significant plasma cortisol suppression suggests an ectopic source of ACTH or an adrenal tumour.
CRH test. An exaggerated ACTH and cortisol response to exogenous CRH suggests pituitary-dependent Cushing’s disease, as ectopic sources rarely respond.
Chest X-ray to look for a carcinoma of the bronchus or a bronchial carcinoid. Carcinoid lesions may be very small; if ectopic ACTH is suspected, whole-lung, mediastinal and abdominal CT scanning should be performed.
Further investigations may involve selective catheterization of the inferior petrosal sinus to measure ACTH for pituitary lesions, or blood samples taken throughout the body in a search for ectopic sources. Radiolabelled octreotide (111In octreotide) is occasionally helpful in locating ectopic ACTH sites.
Treatment
Successful treatment of Cushing’s syndrome with a normal biochemical profile should lead to reversal of the presenting clinical features. However, untreated Cushing’s syndrome has a very bad prognosis, with death from hypertension, myocardial infarction, infection and heart failure. Whatever the underlying cause, cortisol hypersecretion should be controlled prior to surgery or radiotherapy. Considerable morbidity and mortality is otherwise associated with operating on unprepared patients, especially when abdominal surgery is required.
The usual drug is metyrapone, an 11β-hydroxylase blocker, which is given in doses of 750 mg to 4 g daily in 3–4 divided doses. Ketoconazole (200 mg three times daily) is also used and is synergistic with metyrapone. Plasma cortisol should be monitored, aiming to reduce the mean level during the day to 150–300 nmol/L, equivalent to normal production rates. Aminoglutethimide and trilostane (which reversibly inhibits 3-hydroxysteroid dehydrogenase/5–5,4 isomers) are occasionally used.
Choice of further treatment depends upon the cause.
Cushing’s disease (pituitary-dependent hyperadrenalism)
Trans-sphenoidal removal of the tumour is the treatment of choice. Selective adenomectomy nearly always leaves the patient ACTH deficient immediately postoperatively, and this is a good prognostic sign. Overall, pituitary surgery results in remission in 75–80% of cases, but results vary considerably and an experienced surgeon is essential.
External pituitary irradiation alone is slow acting, only effective in 50–60% even after prolonged follow-up and mainly used after failed pituitary surgery. Children, however, respond much better to radiotherapy, 80% being cured. Stereotactic radiotherapy can be useful in selected cases.
Medical therapy to reduce ACTH (e.g. bromocriptine, cabergoline, cyproheptadine, somatostatin analogues such as pasireotide) is rarely effective. There is some evidence that aggressive corticotroph adenomas may respond to temozolomide chemotherapy.
Bilateral adrenalectomy is an effective last resort if other measures fail to control the disease (see Nelson’s syndrome). This can be performed laparoscopically.
Cushing’s syndrome due to other causes
Adrenal adenomas should be resected after achievement of clinical remission with metyrapone or ketoconazole. Contralateral adrenal suppression may last for a year or more.
Adrenal carcinomas are highly aggressive and the prognosis is poor. In general, if there are no widespread metastases, tumour bulk should be reduced surgically. The adrenolytic drug mitotane may inhibit growth of the tumour and prolong survival, though it can cause nausea and ataxia. Some would also give radiotherapy to the tumour bed after surgery.
Tumours secreting ACTH ectopically should be removed if possible. Otherwise chemotherapy/radiotherapy may be used, depending on the tumour. Control of the Cushing’s syndrome with metyrapone or ketoconazole is beneficial for symptoms, and bilateral adrenalectomy may be appropriate to give complete control of Cushing’s syndrome if prognosis from the tumour itself is reasonable.
If the source of ACTH is not clear, cortisol hypersecretion should be controlled with medical therapy until a diagnosis can be made.
Nelson’s syndrome
Nelson’s syndrome is increased pigmentation (because of high levels of ACTH) associated with an enlarging pituitary tumour, which occurs in about 20% of cases after bilateral adrenalectomy for Cushing’s disease. The syndrome is rare now that adrenalectomy is an uncommon primary treatment, and its incidence may be reduced by pituitary radiotherapy soon after adrenalectomy. The Nelson’s adenoma may be treated by pituitary surgery and/or radiotherapy (unless given previously).
Hypersecretion of other pituitary hormones
Pituitary tumours may rarely secrete TSH (and cause thyrotoxicosis). Such ‘TSHomas’ lead to the unusual biochemical pattern of elevated fT4 levels with normal or high circulating TSH levels. FSH and LH secreting pituitary tumours may cause elevated sex steroids but are extremely uncommon.
The thyroid axis
The metabolism of virtually all nucleated cells of many tissues is controlled by the thyroid hormones. Overactivity or underactivity of the gland are the most common of all endocrine problems.
Anatomy
The thyroid gland consists of two lateral lobes connected by an isthmus. It is closely attached to the thyroid cartilage and to the upper end of the trachea, and thus moves on swallowing. It is often palpable in normal women.
Embryologically it originates from the base of the tongue and descends to the middle of the neck. Remnants of thyroid tissue can sometimes be found at the base of the tongue (lingual thyroid) and along the line of descent. The gland has a rich blood supply from superior and inferior thyroid arteries.
The thyroid gland consists of follicles lined by cuboidal epithelioid cells. Inside is the colloid (the iodinated glycoprotein thyroglobulin) which is synthesized by the follicular cells. Each follicle is surrounded by basement membrane, between which are parafollicular cells containing calcitonin-secreting C cells.
Physiology
The thyroid synthesizes two hormones:
Inorganic iodide is trapped by the gland by an enzyme dependent system, oxidized and incorporated into the glycoprotein thyroglobulin to form mono- and diiodotyrosine and then T4 and T3 (Fig. 19.16).
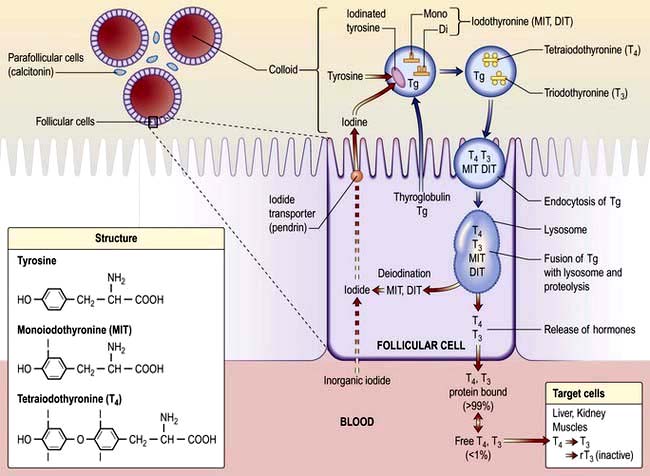
More T4 than T3 is produced, but T4 is converted in some peripheral tissues (liver, kidney and muscle) to the more active T3 by 5′-monodeiodination; an alternative 3′-monodeiodination yields the inactive reverse T3 (rT3). The latter step occurs particularly in severe non-thyroidal illness (see below).
In plasma, more than 99% of all T4 and T3 is bound to hormone-binding proteins (thyroxine-binding globulin, TBG; thyroid-binding prealbumin, TBPA; and albumin). Only free hormone is available for action in the target tissues, where T3 binds to specific nuclear receptors within target cells. Many drugs and other factors affect TBG; all may result in confusing total T4 levels in blood, and most laboratories therefore now measure free T4 levels.
Control of the hypothalamic-pituitary-thyroid axis. Thyrotrophin-releasing hormone (TRH), a peptide produced in the hypothalamus, stimulates the pituitary to secrete thyroid-stimulating hormone (TSH) (see Fig. 19.2). TSH in turn stimulates growth and activity of the thyroid follicular cells via the G-protein coupled TSH membrane receptor (see Table 19.3). The T3 and T4 subsequently secreted into the circulation by follicular cells exert negative feedback on the hypothalamus, as described on page 941.
Circulating T4 is peripherally deiodinated to T3 which binds to the thyroid hormone nuclear receptor (TR) on target organ cells to cause modified gene transcription. There are two TR receptors (TR-α and TR-β) and the tissue-specific effects of T3 are dependent upon the local expression of these TR receptors. TR-α knockout mice show poor growth, bradycardia and hypothermia, whilst TR-β knockout mice show thyroid hyperplasia and high T4 levels in the presence of inappropriately normal circulating TSH, suggesting a role for the latter receptors in thyroid hormone resistance (see p. 967).
Physiological effects of thyroid hormones. The physiological effects of thyroid hormones are summarized in Table 19.12.
Table 19.12 Physiological effects of thyroid hormone
| Target | Effect |
|---|---|
Cardiovascular system |
Increases heart rate and cardiac output |
Bone |
Increases bone turnover and resorption |
Respiratory system |
Maintains normal hypoxic and hypercapnic drive in respiratory centre |
Gastrointestinal system |
Increases gut motility |
Blood |
Increases red blood cell 2,3-BPGa facilitating oxygen release to tissues |
Neuromuscular function |
Increases speed of muscle contraction and relaxation and muscle protein turnover |
Carbohydrate metabolism |
Increases hepatic gluconeogenesis/glycolysis and intestinal glucose absorption |
Lipid metabolism |
Increases lipolysis and cholesterol synthesis and degradation |
Sympathetic nervous system |
Increases catecholamine sensitivity and β-adrenergic receptor numbers in heart, skeletal muscle, adipose cells and lymphocytes |
a 2,3-BPG, 2,3-bisphosphoglyceric acid.
Dietary iodine requirement. Globally, dietary iodine deficiency is a major cause of thyroid disease, as iodine is an essential requirement for thyroid hormone synthesis. The recommended daily intake of iodine should be at least 140 µg, and dietary supplementation of salt and bread has reduced the number of areas where ‘endemic goitre’ still occurs (see below).
SIGNIFICANT WEBSITE
International Council for the Control of Iodine Deficiency Disorders: http://www.iccidd.org
Thyroid function tests
Immunoassays for free T4, free T3 and TSH are widely available. There are only minor circadian rhythms, and measurements may be made at any time. Particular uses of the tests are summarized in Table 19.13, with typical findings in common disorders.
Table 19.13 Characteristics of thyroid function tests in common thyroid disorders (the clinically most informative tests in each situation are shown in bold)
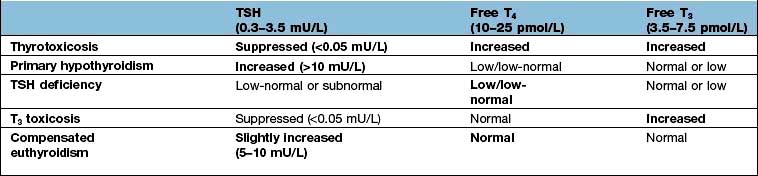
TSH measurement
In most circumstances, TSH levels can discriminate between hyperthyroidism, hypothyroidism and euthyroidism (normal thyroid gland function). Exceptions are hypopituitarism, and the ‘sick euthyroid’ syndrome where low levels (which normally imply hyperthyroidism) occur in the presence of low or normal T4 and T3 levels. As a single test of thyroid function TSH is the most sensitive in most circumstances, but accurate diagnosis requires at least two tests, e.g. TSH plus free T4 or free T3 where hyperthyroidism is suspected, TSH plus serum free T4 where hypothyroidism is likely.
TRH test
This has been rendered almost obsolete by modern sensitive TSH assays except for investigation of hypothalamic-pituitary dysfunction. TRH (protirelin) is occasionally used to differentiate between thyroid hormone resistance and TSHoma in the context of raised fT4 and TSH levels. Typically, after TRH administration there is a rise in TSH in thyroid hormone resistance, whilst in TSHoma there is a flat response due to continued autonomous TSH secretion which does not respond to TRH.
Problems in interpretation of thyroid function tests
There are three major areas of difficulty.
1.Serious acute or chronic illness
Thyroid function is affected in several ways:
Reduced concentration and affinity of binding proteins
Systemically ill patients can therefore have an apparently low total and free T4 and T3 with a normal or low basal TSH (the ‘sick euthyroid’ syndrome). Levels are usually only mildly below normal and are thought to be mediated by interleukins IL-1 and IL-6; the tests should be repeated after resolution of the underlying illness.
2.Pregnancy and oral contraceptives
These lead to greatly increased TBG levels and thus to high or high-normal total T4. Free T4 is usually normal. Normal ranges for free T4 and TSH alter with the normal physiological changes during pregnancy and TSH is often slightly suppressed in the first trimester, but this rarely causes clinical problems.
3.Drugs
Amiodarone decreases T4 to T3 conversion and free T4 levels may therefore be above normal in a euthyroid patient; conversely amiodarone may induce both hyper- and hypothyroidism – the TSH level is usually reliable.
Many drugs affect thyroid function tests by interfering with protein binding but this now rarely causes a problem with free T4 assays.
Antithyroid antibodies
Serum antibodies to the thyroid are common and may be either destructive or stimulating; both occasionally co-exist in the same patient.
Destructive antibodies are directed against the microsomes or against thyroglobulin; the antigen for thyroid microsomal antibodies is the thyroid peroxidase (TPO) enzyme. TPO antibodies are found in up to 20% of the normal population, especially older women, but only 10–20% of these develop overt hypothyroidism.
TSH receptor IgG antibodies (TRAb) typically stimulate, but occasionally block, the receptor; they can be measured in two ways:
Hypothyroidism
Pathophysiology
Underactivity of the thyroid is usually primary, from disease of the thyroid, but may be secondary to hypothalamic-pituitary disease (reduced TSH drive) (Table 19.14). Primary hypothyroidism is one of the most common endocrine conditions with an overall UK prevalence of over 2% in women, but under 0.1% in men; lifetime prevalence for an individual is higher – perhaps as high as 9% for women and 1% for men with mean age at diagnosis around 60 years. The worldwide prevalence of subclinical hypothyroidism varies from 1% to 10%.
Table 19.14 Causes of hypothyroidism
Primary disease of thyroid |
InfectivePost-subacute thyroiditis |
Congenital |
|
Agenesis |
Post-surgery |
Post-irradiation |
|
Defects of hormone synthesis |
|
Iodine deficiency |
|
Infiltration |
|
Tumour |
|
Secondary (to hypothalamic-pituitary disease) |
|
Autoimmune |
|
Hypopituitarism |
|
Isolated TSH deficiency |
|
Peripheral resistance to thyroid hormone |
Causes of primary hypothyroidism (Table 19.14)
Autoimmune
Atrophic (autoimmune) hypothyroidism. This is the most common cause of hypothyroidism and is associated with antithyroid autoantibodies leading to lymphoid infiltration of the gland and eventual atrophy and fibrosis. It is six times more common in females and the incidence increases with age. The condition is associated with other autoimmune disease such as pernicious anaemia, vitiligo and other endocrine deficiencies (p. 939). Occasionally intermittent hypothyroidism occurs with subsequent recovery; antibodies which block the TSH receptor may sometimes be involved in the aetiology.
Hashimoto’s thyroiditis. This form of autoimmune thyroiditis, again more common in women and most common in late middle age, produces atrophic changes with regeneration, leading to goitre formation. The gland is usually firm and rubbery but may range from soft to hard. TPO antibodies are present, often in very high titres (>1000 IU/L). Patients may be hypothyroid or euthyroid, though they may go through an initial toxic phase, ‘Hashi-toxicity’. Levothyroxine therapy may shrink the goitre even when the patient is not hypothyroid.
Postpartum thyroiditis. This is usually a transient phenomenon observed following pregnancy. It may cause hyperthyroidism, hypothyroidism or the two sequentially. It is believed to result from the modifications to the immune system necessary in pregnancy, and histologically is a lymphocytic thyroiditis. The process is normally self-limiting, but when conventional antibodies are found there is a high chance of this proceeding to permanent hypothyroidism. Postpartum thyroiditis may be misdiagnosed as postnatal depression, emphasizing the need for thyroid function tests in this situation.
Defects of hormone synthesis
Iodine deficiency. Dietary iodine deficiency still exists (p. 213) in some areas as ‘endemic goitre’ where goitre, occasionally massive, is common. The patients may be euthyroid or hypothyroid depending on the severity of iodine deficiency. The mechanism is thought to be borderline hypothyroidism leading to TSH stimulation and thyroid enlargement in the face of continuing iodine deficiency. Iodine deficiency is still a problem in the Netherlands, Western Pacific, India, South East Asia, Russia and parts of Africa. Efforts to prevent deficiency by providing iodine in salt continue worldwide but often with incomplete success. Even in the late 20th century of the 500 million with iodine deficiency in India, about 2 million had cretinism (see below).
Dyshormonogenesis. This rare condition is due to genetic defects in the synthesis of thyroid hormones; patients develop hypothyroidism with a goitre. One particular familial form is associated with sensorineural deafness due to a deletion mutation in chromosome 7, causing a defect of the transporter pendrin (Pendred’s syndrome) (see Fig. 19.16).
Clinical features (Fig. 19.17)
Hypothyroidism produces many symptoms. The alternative term ‘myxoedema’ refers to the accumulation of mucopolysaccharide in subcutaneous tissues. The classic picture of the slow, dry-haired, thick-skinned, deep-voiced patient with weight gain, cold intolerance, bradycardia and constipation makes the diagnosis easy. Milder symptoms are, however, more common and hard to distinguish from other causes of nonspecific tiredness. Many cases are detected on biochemical screening.
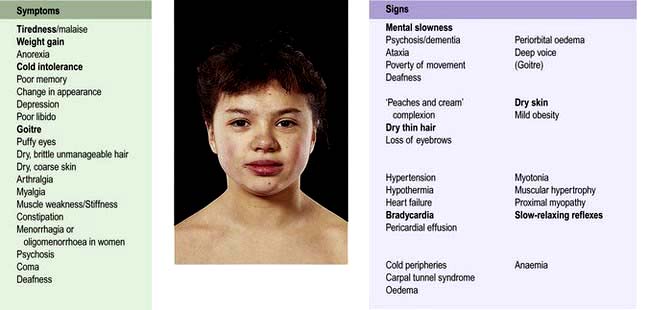
Figure 19.17 Hypothyroidism: symptoms and signs. Bold type indicates symptoms or signs of greater discriminant value. A history from a relative is often revealing. Symptoms of other autoimmune disease may be present.
Special difficulties in diagnosis may arise in certain circumstances:
Children with hypothyroidism may not show classic features but often have a slow growth velocity, poor school performance and sometimes arrest of pubertal development.
Young women with hypothyroidism may not show obvious signs. Hypothyroidism should be excluded in all people with oligomenorrhoea/amenorrhoea, menorrhagia, infertility or hyperprolactinaemia.
The elderly show many clinical features that are difficult to differentiate from normal ageing.
Investigation of primary hypothyroidism
Serum TSH is the investigation of choice; a high TSH level confirms primary hypothyroidism. A low free T4 level confirms the hypothyroid state (and is also essential to exclude TSH deficiency if clinical hypothyroidism is strongly suspected and TSH is normal or low).
Thyroid and other organ-specific antibodies may be present. Other abnormalities include the following:
Anaemia, which is usually normochromic and normocytic in type but may be macrocytic (sometimes this is due to associated pernicious anaemia) or microcytic (in women, due to menorrhagia)
Increased serum aspartate transferase levels, from muscle and/or liver
Increased serum creatine kinase levels, with associated myopathy
Hypercholesterolaemia and hypertriglyceridaemia
Hyponatraemia due to an increase in ADH and impaired free water clearance.
Treatment
Replacement therapy with levothyroxine (thyroxine, i.e. T4) is given for life. The starting dose will depend upon the severity of the deficiency and on the age and fitness of the patient, especially their cardiac performance: 100 µg daily for the young and fit, 50 µg (increasing to 100 µg after 2–4 weeks) for the small, old or frail. People with ischaemic heart disease require even lower initial doses, especially if the hypothyroidism is severe and longstanding. Most physicians would then begin with 25 µg daily and perform serial ECGs, increasing the dose at 3- to 4-week intervals if angina does not occur or worsen and the ECG does not deteriorate. Occasional patients develop ‘thyrotoxic’ (hyperthyroid) symptoms despite normal fT4 levels if dose if increased too rapidly.
Monitoring. The aim is to restore T4 and TSH to well within the normal range. Adequacy of replacement is assessed clinically and by thyroid function tests after at least 6 weeks on a steady dose. If serum TSH remains high, the dose of T4 should be increased in increments of 25–50 µg with the tests repeated at 6–8 weeks intervals until TSH becomes normal. Complete suppression of TSH should be avoided because of the risk of atrial fibrillation and osteoporosis. The usual maintenance dose is 100–150 µg given as a single daily dose. An annual thyroid function test is recommended – this is usually performed in the primary care setting, often assisted and prompted by district ‘thyroid registers’.
Clinical improvement on T4 may not begin for 2 weeks or more, and full resolution of symptoms may take 6 months. The necessity of lifelong therapy must be emphasized and the possibility of other autoimmune endocrine disease developing, especially Addison’s disease or pernicious anaemia, should be considered. During pregnancy, an increase in T4 dosage of about 25–50 µg is often needed to maintain normal TSH levels, and the necessity of optimal replacement during pregnancy is emphasized by the finding of reductions in cognitive function in children of mothers with elevated TSH during pregnancy.
A few people with primary hypothyroidism complain of incomplete symptomatic response to T4 replacement. Combination T4 and T3 replacement has been advocated in this context, but randomized clinical trials show no consistent benefit in quality of life symptoms.
Borderline hypothyroidism or ‘compensated euthyroidism’
Patients are frequently seen with low-normal serum T4 levels and slightly raised TSH levels. Sometimes this follows surgery or radioactive iodine therapy when it can reasonably be seen as ‘compensatory’. Treatment with levothyroxine is normally recommended where the TSH is consistently above 10 mU/L, or when possible symptoms, high-titre thyroid antibodies, or lipid abnormalities are present. Where the TSH is only marginally raised, the tests should be repeated 3–6 months later. Conversion to overt hypothyroidism is more common in men or when TPO antibodies are present. In practice, vague symptoms in people with marginally elevated TSH (below 10 mU/L) rarely respond to treatment, but a ‘therapeutic trial’ of replacement may be needed to confirm that symptoms are unrelated to the thyroid. It is also considered best to normalize TSH during (and ideally before) pregnancy to avoid fetal adverse effects.
Myxoedema coma
Severe hypothyroidism, especially in the elderly, may present with confusion or even coma. Myxoedema coma is very rare: hypothermia is often present and the patient may have severe cardiac failure, pericardial effusions, hypoventilation, hypoglycaemia and hyponatraemia. The mortality was previously at least 50% and patients require full intensive care. Optimal treatment is controversial and data lacking; most physicians would advise T3 orally or intravenously in doses of 2.5–5 µg every 8 hours, then increasing as above. Large intravenous doses should not be used. Additional measures, though unproven, should include:
Oxygen (by ventilation if necessary)
Monitoring of cardiac output and pressures
Myxoedema madness. Depression is common in hypothyroidism. Rarely, with severe hypothyroidism in the elderly, the patient may become frankly demented or psychotic, sometimes with striking delusions. This may occur shortly after starting T4 replacement.
Screening for hypothyroidism
The incidence of congenital hypothyroidism is approximately 1 in 3500 births. Untreated, severe hypothyroidism produces permanent neurological and intellectual damage (‘cretinism’). Routine screening of the newborn using a blood spot, as in the Guthrie test, to detect a high TSH level as an indicator of primary hypothyroidism is efficient and cost-effective; cretinism is prevented if T4 is started within the first few months of life.
Screening of elderly patients for thyroid dysfunction has a low pick-up rate and is controversial and not currently recommended. However, patients who have undergone thyroid surgery or received radioiodine should have regular thyroid function tests, as should those receiving lithium or amiodarone therapy.
Hyperthyroidism
Hyperthyroidism (thyroid overactivity, thyrotoxicosis) is common, affecting perhaps 2–5% of all females at some time and with a sex ratio of 5 : 1, most often between the ages of 20 and 40 years. Nearly all cases (>99%) are caused by intrinsic thyroid disease; a pituitary cause is extremely rare (Table 19.15).
Table 19.15 Causes of hyperthyroidism
|
|
Graves’ disease
This is the most common cause of hyperthyroidism and is due to an autoimmune process. Serum IgG antibodies bind to TSH receptors in the thyroid, stimulating thyroid hormone production, i.e. they behave like TSH. These TSH receptor antibodies (TSHR-Ab) are specific for Graves’ disease. Persistent high levels predict a relapse when drug treatment is stopped. There is an association with HLA-B8, DR3 and DR2 and a 40% concordance rate amongst monozygotic twins with a 5% concordance rate in dizygotic twins. There is a weak association with cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), HLA-DRB*08 and DRB3*0202 on chromosome 6.
Yersinia enterocolitica, Escherichia coli and other Gram-negative organisms contain TSH binding sites. This raises the possibility that the initiating event in the pathogenesis may be an infection with possible ‘molecular mimicry’ in a genetically susceptible individual, but the precise initiating mechanisms remain unproven in most cases.
Thyroid eye disease accompanies the hyperthyroidism in many cases (see below) but other components of Graves’ disease, e.g. Graves’ dermopathy, are very rare. Rarely lymphadenopathy and splenomegaly may occur. Graves’ disease is also associated with other autoimmune disorders such as pernicious anaemia, vitiligo and myasthenia gravis.
The natural history is one of fluctuation, many patients showing a pattern of alternating relapse and remission; perhaps only 40% of subjects have a single episode. Many patients eventually become hypothyroid.
FURTHER READING
Bartalena L, Baldeschi L, Dickinson A et al. Consensus statement of the European Group on Graves’ orbitopathy (EUGOGO) on the management of GO. Eur J Endocrinol 2008; 158:273–285.
Brent GA. Graves’ disease. N Engl J Med 2008; 358:2594–2605.
Brent GA. Thyroid function and screening in pregnancy. N Engl J Med 2012; 366:562–568.
Other causes of hyperthyroidism/thyrotoxicosis
Solitary toxic adenoma/nodule
This is the cause of about 5% of cases of hyperthyroidism. It does not usually remit after a course of antithyroid drugs.
Toxic multinodular goitre
This commonly occurs in older women. Again, antithyroid drugs are rarely successful in inducing a remission, although they can control the hyperthyroidism.
de Quervain’s thyroiditis
This is transient hyperthyroidism from an acute inflammatory process, probably viral in origin. Apart from the toxicosis, there is usually fever, malaise and pain in the neck with tachycardia and local thyroid tenderness. Thyroid function tests show initial hyperthyroidism, the erythrocyte sedimentation rate (ESR) and plasma viscosity are raised, and thyroid uptake scans show suppression of uptake in the acute phase. Hypothyroidism, usually transient, may then follow after a few weeks. Treatment of the acute phase is with aspirin, using short-term prednisolone in severely symptomatic cases.
Amiodarone-induced thyrotoxicosis
Amiodarone, a class III antiarrhythmic drug (see p. 699), causes two types of hyperthyroidism.
Type I amiodarone-induced thyrotoxicosis (AIT) is associated with pre-existing Graves’ disease or multinodular goitre. In this situation hyperthyroidism is probably triggered by the high iodine content of amiodarone.
Type II AIT is not associated with previous thyroid disease and is thought to be due to a direct effect of the drug on thyroid follicular cells leading to a destructive thyroiditis with release of T4 and T3. Type II AIT may be associated with a hypothyroid phase several months after presentation. Because amiodarone inhibits the deiodination of T4 to T3, biochemical presentation of both types of AIT may be associated with higher T4:T3 ratios than usual.
Clinical features of hyperthyroidism
The symptoms and signs of hyperthyroidism affect many systems (Fig. 19.18).

Figure 19.18 Hyperthyroidism: symptoms and signs. Bold type indicates symptoms or signs of greater discriminant value.
Symptomatology and signs vary with age and with the underlying aetiology.
The eye signs, of lid lag and ‘stare’ may occur with hyperthyroidism of any cause but other features of thyroid eye disease (see below) occur only in Graves’ disease.
Graves’ dermopathy is rare: pretibial myxoedema is an infiltration of the skin on the shin. Thyroid acropachy is very rare and consists of clubbing, swollen fingers and periosteal new bone formation.
In the elderly, a frequent presentation is with atrial fibrillation, other tachycardias and/or heart failure, often with few other signs. Thyroid function tests are mandatory in any patient with atrial fibrillation.
Children frequently present with excessive height or excessive growth rate, or with behavioural problems such as hyperactivity. They may also show weight gain rather than loss.
So-called ‘apathetic thyrotoxicosis’ in some elderly patients presents with a clinical picture more like hypothyroidism. There may be very few signs and a high degree of clinical suspicion is essential.
Differential diagnosis
Hyperthyroidism is often clinically obvious but treatment should never be instituted without biochemical confirmation.
Differentiation of the mild case from anxiety states may be difficult; useful positive clinical markers are eye signs, a diffuse goitre, proximal myopathy and wasting. Weight loss, despite a normal or increased appetite, is a very useful clinical symptom of hyperthyroidism. The hyperdynamic circulation with warm peripheries seen with hyperthyroidism can be contrasted with the clammy hands of anxiety.
Investigations
Serum TSH is suppressed in hyperthyroidism (<0.05 mU/L), except for the very rare instances of TSH hypersecretion.
A raised free T4 or T3 confirms the diagnosis; T4 is almost always raised but T3 is more sensitive as there are occasional cases of isolated ‘T3 toxicosis’.
Thyroid peroxidase (TPO) and thyroglobulin antibodies are present in most cases of Graves’ disease.
TSHR-Ab are not measured routinely, but are commonly present: thyroid-stimulating immunoglobulin (TSI) 80% positive, TSH-binding inhibitory immunoglobulin (TBII) 60–90% in Graves’ disease (see p. 961).
Treatment
Three possibilities are available: antithyroid drugs, radioiodine and surgery. Practices and beliefs differ widely within and between countries.
Antithyroid drugs
\xEF"\xBF\xEF"\xBFCarbimazole\xEF"\xBF\xEF"\xBF is most often used in the UK, and propylthiouracil (PTU) is also used. Thiamazole (methimazole), the active metabolite of carbimazole, is used in the USA. These drugs inhibit the formation of thyroid hormones and also have other minor actions; carbimazole/thiamazole is also an immunosuppressive agent. Initial doses and side-effects are detailed in Table 19.16.
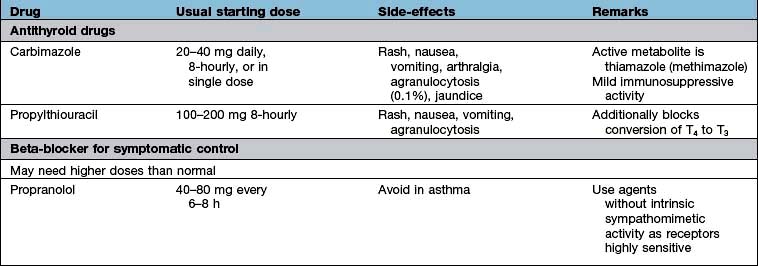
Although thyroid hormone synthesis is reduced very quickly, the long half-life of T4 (7 days) means that clinical benefit is not apparent for 10–20 days. As many of the manifestations of hyperthyroidism are mediated via the sympathetic system, beta-blockers are used to provide rapid partial symptomatic control; they also decrease peripheral conversion of T4 to T3. Drugs preferred are those without intrinsic sympathomimetic activity, e.g. propranolol (Table 19.16). They should not be used alone for hyperthyroidism except when the condition is self-limiting, as in subacute thyroiditis.
Subsequent management is either by gradual dose titration or a ‘block and replace’ regimen. Neither regimen has been shown to be unequivocally superior. TSH often remains suppressed for many months after clinical improvement and normalization of T4 and T3.
Dosage regimen
1. Start carbimazole 20–40 mg daily.
2. Review after 4–6 weeks and reduce the dose of carbimazole, depending on clinical state and fT4/fT3 levels. TSH levels may remain suppressed for several months and are unhelpful at this stage.
3. When clinically and biochemically euthyroid, stop beta-blockers.
4. Review thyroid function regularly during the planned course of treatment (typically 18 months – but some use courses between 6 and 24 months).
5. Reduce carbimazole if fT4 falls below or TSH rises above normal – and when approaching the end of the planned course.
6. Increase carbimazole if fT4 or fT3 are above normal (and consider if TSH remains suppressed after several months with a normal fT4).
7. Stop treatment at end of course if the patient is euthyroid on 5 mg daily carbimazole.
PTU is used in similar fashion (Table 19.16).
‘Block and replace’ regimen. With this policy, full doses of antithyroid drugs, usually carbimazole 40 mg daily, are given to suppress the thyroid completely while replacing thyroid activity with 100 µg of levothyroxine daily once euthyroidism has been achieved. This is continued usually for 18 months, the claimed advantages being the avoidance of over- or undertreatment and the better use of the immunosuppressive action of carbimazole. This regimen is contraindicated in pregnancy as T4 crosses the placenta less well than carbimazole.
Relapse
About 50% of patients will relapse after a course of carbimazole or PTU, mostly within the following 2 years but occasionally much later. Long-term antithyroid therapy is then used or surgery or radiotherapy is considered (see below). Most patients (90%) with hyperthyroidism have a diffuse goitre but those with large single or multinodular goitres are unlikely to remit after a course of antithyroid drugs and will usually require definitive treatment. Severe biochemical hyperthyroidism is also less likely to remain in remission.
Toxicity
The major side-effect of drug therapy is agranulocytosis that occurs in approximately 1 in 1000 patients, usually within 3 months of treatment. All patients must be warned to seek immediate medical attention for a white blood cell count if they develop unexplained fever or sore throat – written information is essential. Rashes are more frequent and usually require a change of drug. If toxicity occurs on carbimazole, PTU may be used and vice-versa; side-effects are only occasionally repeated on the other drug.
Radioactive iodine
Radioactive iodine (RAI) is given to patients of all ages, although it is contraindicated in pregnancy and while breastfeeding. RAI is the most common treatment modality in the USA whereas antithyroid drugs tend to be favoured in Europe.
131Iodine is given in an empirical dose (usually 200–550 MBq) because of variable uptake and radiosensitivity of the gland. It accumulates in the thyroid and destroys the gland by local radiation although it takes several months to be fully effective.
Patients must be rendered euthyroid before treatment. They should stop antithyroid drugs at least 4 days before radioiodine, and not recommence until 3 days after radioiodine. Patients on PTU should stop antithyroid medication earlier than those on carbimazole before RAI because it has a radioprotective action. Many patients do not need to restart antithyroid medication after treatment.
Early discomfort in the neck and immediate worsening of hyperthyroidism are sometimes seen; if worsening occurs, the patient should receive propranolol (Table 19.16); if necessary carbimazole can be restarted. Euthyroidism normally returns in 2–3 months. People with dysthyroid eye disease are more likely to show worsening of eye problems after radioiodine than after antithyroid drugs; this represents a partial contraindication to RAI, although worsening can usually be prevented by steroid administration.
Long-term surveillance. Hypothyroidism affects the majority of subjects over the following 20 years. Some 75% of patients are rendered euthyroid in the short term but a small proportion remain hyperthyroid and may require a second dose of radioiodine. Long-term surveillance of thyroid function is necessary with frequent tests in the first year after therapy, and at least annually thereafter.
Risk of carcinogenesis has been long debated, but the overwhelming evidence suggests that overall cancer incidence and mortality are not increased after radioactive iodine (and indeed are significantly reduced in some studies).
Surgery
Thyroidectomy should be performed only in patients who have previously been rendered euthyroid. Conventional practice is to stop the antithyroid drug 10–14 days before operation and to give potassium iodide (60 mg three times daily), which reduces the vascularity of the gland.
The operation should be performed only by experienced surgeons to reduce the chance of complications:
Early postoperative bleeding causing tracheal compression and asphyxia is a rare emergency requiring immediate removal of all clips/sutures to allow escape of the blood/haematoma.
Laryngeal nerve palsy occurs in 1%. Vocal cord movement should be checked preoperatively. Mild hoarseness is more common and thyroidectomy is best avoided in professional singers!
Transient hypocalcaemia occurs in up to 10% but with permanent hypoparathyroidism in fewer than 1%.
Ongoing thyroid function depends on the operation performed. With a single toxic nodule excision of the lesion is curative. With Graves’ disease or multinodular goitre the traditional ‘subtotal’ thyroidectomy, aiming for euthyroidism on no treatment, results in recurrent hyperthyroidism in 1–3% within 1 year, then 1% per year and hypothyroidism in about 10% of patients within 1 year, and then increasing with time. ‘Near-total’ thyroidectomy is therefore now preferred with inevitable hypothyroidism but a much reduced risk of recurrence.
Indications for either surgery or radioiodine are given in Box 19.4.
Box 19.4
Choice of surgery or radioiodine therapy for hyperthyroidism
FURTHER READING
Royal College of Physicians. Report of a Working Party: Radioiodine in the management of benign thyroid disease – Clinical guidelines. London: Royal College of Physicians; 2007, http://www.bnms.org.uk/rcp-guidelines/
Special situations in hyperthyroidism
Thyroid crisis or ‘thyroid storm’
This rare condition, with a mortality of 10%, is a rapid deterioration of hyperthyroidism with hyperpyrexia, severe tachycardia, extreme restlessness, cardiac failure and liver dysfunction. It is usually precipitated by stress, infection or surgery in an unprepared patient, or radioiodine therapy. With careful management it should no longer occur and most cases referred as ‘crisis’ are simply severe but uncomplicated thyrotoxicosis.
Treatment is urgent. Propranolol in full doses is started immediately together with potassium iodide, antithyroid drugs, corticosteroids (which suppress many of the manifestations of hyperthyroidism) and full supportive measures. Control of cardiac failure and tachycardia is also necessary.
Occasionally, hyperthyroidism can lead to a thyrotoxic cardiomyopathy which causes ischaemic changes on a 12-lead ECG which reverse after reversion to euthyroidism.
Hyperthyroidism in pregnancy and neonatal life
Since hyperthyroidism typically affects young women, pregnancies, both planned and unplanned, inevitably occur during antithyroid treatment. PTU is usually the preferred antithyroid drug during pregnancy or in any woman planning pregnancy, due to rare reports of congenital abnormalities with carbimazole which have not been described with PTU.
The high level of HCG found in normal pregnancy is a weak stimulator of the TSH receptor, commonly causing suppressed TSH with slightly elevated fT4/fT3 in the first trimester which may be associated with hyperemesis gravidarum. True maternal hyperthyroidism occurring de novo during pregnancy is however uncommon and usually mild. Diagnosis can be difficult because of the overlap with symptoms of normal pregnancy and misleading thyroid function tests, although TSH is largely reliable. The pathogenesis is almost always Graves’ disease.
Thyroid-stimulating immunoglobulin (TSI) crosses the placenta to stimulate the fetal thyroid. Carbimazole and PTU (see below) also cross the placenta, but T4 does so poorly, so a ‘block-and-replace’ regimen is contraindicated. The smallest dose necessary of PTU (see above) is used and the fetus must be monitored. If necessary (high doses needed, poor patient compliance or drug side-effects), surgery can be performed, preferably in the second trimester. Radioactive iodine is absolutely contraindicated.
The fetus and maternal Graves’ disease
Any mother with a history of Graves’ disease may have circulating TSI. Even if she is euthyroid after surgery or RAI, the immunoglobulin may still be present to stimulate the fetal thyroid, and the fetus can thus become hyperthyroid.
Any such patient should therefore be monitored during pregnancy. Fetal heart rate provides a direct biological assay of fetal thyroid status, and monitoring should be performed at least monthly. Rates above 160/minute are strongly suggestive of fetal hyperthyroidism, and maternal treatment with PTU and/or propranolol is used. Direct measurement of TSHR-Ab may be helpful to predict neonatal thyrotoxicosis in this situation. To prevent a euthyroid mother becoming hypothyroid, T4 may be given as this does not easily cross the placenta. Sympathomimetics, used to prevent premature labour, are contraindicated as they may provoke fatal tachycardia in the fetus. The paediatrician should be informed and the infant checked immediately after birth – overtreatment with PTU or carbimazole can cause fetal goitre. Breast-feeding while on usual doses of carbimazole or PTU appears to be safe.
Hyperthyroidism may also develop in the neonatal period as TSI has a half-life of approximately 3 weeks. Manifestations in the newborn include irritability, failure to thrive and persisting weight loss, diarrhoea and eye signs. Thyroid function tests are difficult to interpret as neonatal normal ranges vary with age.
Untreated neonatal hyperthyroidism is probably associated with hyperactivity in later childhood.
Thyroid hormone resistance
Thyroid hormone resistance is an inherited condition caused by an abnormality of the thyroid hormone receptor. Mutations to the receptor (TR β) result in the need for higher levels of thyroid hormones to achieve the same intracellular effect. As a result, the normal feedback control mechanisms (see Fig. 19.2, p. 941) result in high blood levels of T4 with a normal TSH in order to maintain a euthyroid state. This has two consequences:
1. Thyroid function tests appear abnormal even when the patient is euthyroid and requires no treatment. Specialist review is required to differentiate from hyperthyroidism due to inappropriate TSH secretion.
2. Different tissues contain different thyroid hormone receptors and, in some families, receptors in certain tissues may have normal activity. In this case the level of thyroid hormones to maintain euthyroidism at pituitary and hypothalamic levels (which controls secretion of TSH) may be higher than that required in other tissues such as heart and bone, so that these tissues may exhibit ‘thyrotoxic’ effects in spite of a normal serum TSH. This ‘partial thyroid hormone resistance’ can be very difficult to manage effectively.
Long-term consequences of hyperthyroidism
Long-term follow-up studies of hyperthyroidism show a slight increase in overall mortality, which affects all age groups, is not fully explained and tends to occur in the first year after diagnosis. Thereafter, the only long-term risk of adequately treated hyperthyroidism appears to be an increased risk of osteoporosis. People with persistently suppressed TSH levels have an increased likelihood of developing atrial fibrillation which may predispose to thromboembolic disease.
Thyroid eye disease
This is also known as dysthyroid eye disease or ophthalmic Graves’ disease.
Pathophysiology
The ophthalmopathy of Graves’ disease is due to a specific immune response that causes retro-orbital inflammation (Fig. 19.19). Swelling and oedema of the extraocular muscles lead to limitation of movement and to proptosis which is usually bilateral but can sometimes be unilateral. Ultimately increased pressure on the optic nerve may cause optic atrophy. Histology of the extraocular muscles shows focal oedema and glycosaminoglycan deposition followed by fibrosis. The precise autoantigen which leads to the immune response remains to be identified, but it appears to be an antigen in retro-orbital tissue with similar immunoreactivity to the TSH receptor.
Figure 19.19 Model of the Initiation of Thyrotrophin-Receptor Autoimmunity in Graves’ Ophthalmopathy and Its Consequences.Thyrotrophin receptor is internalized and degraded by antigen-presenting cells that present thyrotrophin-receptor peptides, in association with major histocompatibility complex (MHC) class II antigens, to helper T cells. These cells become activated, interact with autoreactive B cells through CD154-CD40 bridges, and secrete interleukin-2 and interferon-γ. These cytokines induce the differentiation of B cells into plasma cells that secrete anti-thyrotrophin-receptor antibodies. Anti-thyrotrophin-receptor antibodies recognize the thyrotrophin receptor on orbital fibroblasts and, in conjunction with the secreted type 1 helper T cytokines interferon-γ and tumour necrosis factor (TNF), initiate the tissue changes characteristic of Graves’ ophthalmopathy. These antibodies stimulate the thyrotrophin receptor on thyroid follicular epithelial cells, leading to hyperplasia and increased production of the thyroid hormones triiodothyronine (T3) and thyroxine (T4). IL, interleukin; IFN, interferon.
(Adapted from Bahn RS 2010 Graves’ ophthalmology. N Engl J Med 362:726–738.)
Eye disease is a manifestation of Graves’ disease and can occur in patients who may be hyperthyroid, euthyroid or hypothyroid. Thyroid dysfunction and ophthalmopathy usually occur within two years of each other although sometimes a gap of many years is seen. TSH receptor antibodies are almost invariably found in the serum but their role in the pathogenesis is becoming clearer (Fig. 19.19). Ophthalmopathy is more common and more severe in smokers.
Clinical features
The clinical appearances are characteristic (Fig. 19.18) but thyroid eye disease demonstrates a wide range of severity. A high proportion of people with Graves’ disease notice some soreness, painful watering or prominence of the eyes, and the ‘stare’ of lid retraction is relatively common. More severe proptosis occurs in a minority of cases, and limitation and discomfort of eye movement and visual impairment due to optic nerve compression are relatively uncommon. Proptosis and lid retraction may limit the ability to close the eyes completely so that corneal damage may occur. There is periorbital oedema and conjunctival oedema and inflammation.
Eye manifestations do not parallel the degree of biochemical thyrotoxicosis, or the need for antithyroid therapy, but exacerbation of eye disease is more common after radioiodine treatment (15% versus 3% on antithyroid drugs). Sight is threatened in only 5–10% of cases, but the discomfort and cosmetic problems cause great patient anxiety.
Investigations
Few investigations are necessary if the appearances are characteristic and bilateral. TSH, fT3 and fT4 are measured.
There are a variety of grading systems but none is universally accepted. It is essential to clearly document eye movements and the degree of oedema and inflammation. The exophthalmos should be measured to allow progress to be monitored. MRI or CT of the orbits will exclude retro-orbital space-occupying lesions, show enlarged muscles and oedema, and may show a taut optic nerve due to raised intra-orbital pressure.
Treatment
If the patient is thyrotoxic this should be treated, but this will not directly result in an improvement of the ophthalmopathy, and hypothyroidism must be avoided as it may exacerbate the eye problem. Smoking should be stopped. Treatment of the eyes may be either local or systemic, and always requires close liaison between specialist endocrinologist and ophthalmologist:
Methylcellulose or hypromellose eyedrops are given to aid lubrication and improve comfort.
Some patients gain relief by sleeping upright.
The eyelids can be taped to ensure closure at night.
Systemic steroids (prednisolone 30–120 mg daily) usually reduce inflammation if more severe symptoms are present. Pulse intravenous methylprednisolone may be used initially and is more rapidly effective in severe cases.
Surgical decompression of the orbit(s) may be required, particularly if pressure of orbital contents on the optic nerve threatens vision, and at a later, stable stage for cosmetic reasons.
Lid surgery will protect the cornea if lids cannot be closed, and can be useful later for cosmetic reasons.
Corrective eye muscle surgery may improve diplopia due to muscle changes, but should be deferred until the situation has been stable for 6 months and should follow any orbital decompression.
Irradiation of the orbits (20 Gy in divided doses) is used in some centres. This improves inflammation and ocular motility but has little effect on proptosis and its precise role is debated.
Immunomodulatory agents may produce a response in some patients when conventional treatments fail, although clinical trial evidence is inconsistent.
Goitre (thyroid enlargement)
Goitre is more common in women than in men and may be either physiological or pathological.
Clinical features
Goitres are present on examination in up to 9% of the population. Most commonly a goitre is noticed as a cosmetic defect by the patient or by friends or relatives. The majority are painless, but pain or discomfort can occur in acute varieties. Large goitres can produce dysphagia and difficulty in breathing, implying oesophageal or tracheal compression.
A small goitre may be more easily visible (on swallowing) than palpable. Clinical examination should record the size, shape, consistency and mobility of the gland as well as whether its lower margin can be demarcated (thus implying the absence of retrosternal extension). A bruit may be present. Associated lymph nodes should be sought and the tracheal position determined if possible. Examination should never omit an assessment of the patient’s clinical thyroid status.
Specific enquiry should be made about any medication, especially iodine-containing preparations, and possible exposure to radiation.
Particular points of note are:
Puberty and pregnancy may produce a diffuse increase in size of the thyroid.
Pain in a goitre may be caused by thyroiditis, bleeding into a cyst or (rarely) a thyroid tumour.
Excessive doses of carbimazole or PTU will induce goitre.
Iodine deficiency and dyshormonogenesis (see above) can also cause goitre.
Assessment
There are two major aspects of any goitre: its pathological nature and the patient’s thyroid status.
The nature can often be judged clinically. Goitres (Table 19.17) are usually separable into diffuse and nodular types, the causes of which differ.
Table 19.17 Goitre: causes and types
Diffuse |
Nodular |
Simple |
Multinodular goitre |
Physiological (puberty, pregnancy) |
Solitary nodular |
Tumours |
|
Adenomas |
|
Miscellaneous |
|
Sarcoidosis |
Diffuse goitre
Simple goitre
In this instance, no clear cause is found for enlargement of the thyroid, which is usually smooth and soft. It may be associated with thyroid growth-stimulating antibodies.
Autoimmune thyroid disease
Hashimoto’s thyroiditis and thyrotoxicosis are both associated with firm diffuse goitre of variable size. A bruit is often present in thyrotoxicosis.
Thyroiditis
Acute tenderness in a diffuse swelling, sometimes with severe pain, is suggestive of an acute viral thyroiditis (de Quervain’s). It may produce transient clinical hyperthyroidism with an increase in serum T4 (see p. 964).
Nodular goitres
Multinodular goitre
Most common is the multinodular goitre, especially in older patients. The patient is usually euthyroid but may be hyperthyroid or borderline with suppressed TSH levels but normal free T4 and T3. Multinodular goitre is the most common cause of tracheal and/or oesophageal compression and can cause laryngeal nerve palsy. It may also extend retrosternally.
Tc scan showing multinodular goitre, with predominant involvement of the left lobe of the thyroid (anterior view with arrowed anatomical marker at the sternal notch).
The classical ‘multinodular goitre’ is usually readily apparent clinically, but it should be noted that modern, high-resolution ultrasound frequently reports multiple small nodules in glands which are clinically diffusely enlarged and associated with autoimmune thyroid disease. These nodules are also found in up to 40% of the normal population.
Solitary nodular goitre
Such a goitre presents a difficult problem of diagnosis. Malignancy should be of concern in any solitary nodule – however, the majority of such nodules are cystic or benign and, indeed, may simply be the largest nodule of a multinodular goitre. The diagnostic challenge is to identify the small minority of malignant nodules, which require surgery, from the majority of benign nodules, which do not. Sometimes a history of rapid enlargement, associated lymph nodes or pain may suggest an aggressive malignancy, but most thyroid cancers are painless and slow-growing so that investigations are paramount. Risk factors for malignancy include previous irradiation, longstanding iodine deficiency and occasional familial cases.
Solitary toxic nodules are quite uncommon and may be associated with T3 toxicosis.
Fibrotic goitre (Riedel’s thyroiditis)
Fibrotic goitre is a rare condition, usually producing a ‘woody’ gland. It is associated with other midline fibrosis and is often difficult to distinguish from carcinoma, being irregular and hard. Clinical clues include systemic symptoms of inflammation and elevation in inflammatory markers; it has been shown to be an IgG4-related disease.
Investigations
Clinical findings will dictate appropriate initial tests:
Thyroid function tests: TSH plus free T4 or T3 (see Table 19.7).
Thyroid antibodies: to exclude autoimmune aetiology.
Ultrasound. Ultrasound with high resolution is a sensitive method for delineating nodules and can demonstrate whether they are cystic or solid. In addition, a multinodular goitre may be demonstrated when only a single nodule is palpable. Unfortunately, even cystic lesions can be malignant and thyroid tumours may arise within a multinodular goitre; therefore fine-needle aspiration (see below) is often required and performed under ultrasound control at the same time as the scan.
Chest and thoracic inlet X-rays or CT scan to detect tracheal compression and large retrosternal extensions in people with very large goitre or clinical symptoms.
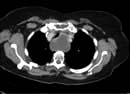
Fine-needle aspiration (FNA). In people with a solitary nodule or a dominant nodule in a multinodular goitre, there is a 5% chance of malignancy; in view of this, FNA should be performed in the outpatient clinic or during ultrasound. Cytology in expert hands can usually differentiate the suspicious or definitely malignant nodule.
FNA reduces the necessity for surgery, but there is a 5% false-negative rate, which must be borne in mind (and the patient appropriately counselled). Continued observation is required when an isolated thyroid nodule is assumed to be benign without excision.
Thyroid scan (99mTc, 125I or 131I) can be useful to distinguish between functioning (hot) or non-functioning (cold) nodules. A hot nodule is only rarely malignant; however, a cold nodule is malignant in only 10% of cases and FNA has largely replaced isotope scans in the diagnosis of thyroid nodules.
Treatment
Euthyroid goitre
Many goitres are small, cause no symptoms and can be observed (including self-monitoring by the patient in the long term). In particular, during puberty and pregnancy a goitre associated with euthyroidism rarely requires intervention and the patient can be reassured that spontaneous resolution is likely. Indications for surgical intervention are:
The possibility of malignancy. A positive or suspicious FNA makes surgery mandatory and surgery may be necessary if doubt persists even in the presence of a negative FNA (especially if the patient is concerned by the false-negative rate).
Pressure symptoms on the trachea or, more rarely, oesophagus. The possibility of retrosternal extension should be excluded.
Cosmetic reasons. A large goitre is often a considerable anxiety to the patient even though functionally and anatomically benign.
Radioactive iodine has also been advocated for the treatment of euthyroid goitre, particularly when surgery is an unattractive option.
Thyroid carcinoma
Types of thyroid carcinoma, their characteristics and treatment are listed in Table 19.18. While not common, these tumours are responsible for 400 deaths annually in the UK and an annual incidence of 30 000 cases in the USA. Over 75% occur in women. In 90% of cases they present as thyroid nodules (see above), but occasionally with cervical lymphadenopathy (about 5%), or with lung, cerebral, hepatic or bone metastases.
Carcinomas derived from thyroid epithelium may be papillary or follicular (differentiated) or anaplastic (undifferentiated). Medullary carcinomas (about 5% of all thyroid cancers) arise from the calcitonin-producing C cells. The pathogenesis of thyroid epithelial carcinomas is not understood except for occasional familial papillary carcinoma and those cases related to previous head and neck irradiation or ingestion of radioactive iodine (e.g. post-Chernobyl). These tumours are minimally active hormonally and are extremely rarely associated with hyperthyroidism; over 90%, however, secrete thyroglobulin, which can therefore act as a tumour marker after thyroid ablation.
Papillary and follicular carcinomas
The primary treatment is surgical, normally total or near-total thyroidectomy for local disease. Regional or more extensive neck dissection is needed where there is local nodal spread or involvement of local structures.
Most tumours will take up iodine, and UK and other guidelines currently recommend radioactive iodine (RAI) ablation of residual thyroid tissue postoperatively for most people with differentiated thyroid cancer. After ablation of normal thyroid in this way, RAI may be used to localize residual disease (scanning using low doses) or to treat it (using high doses: 5.5–7.5 GBq). When recurrence does occur, local invasion and lymph node involvement is most common, and lungs and bone are the most common sites of distant metastases.
To minimize risk of recurrence patients are treated with suppressive doses of levothyroxine (sufficient to suppress TSH levels below the normal range). Patient progress is monitored both clinically and biochemically using serum thyroglobulin levels as a tumour marker. The measurement of thyroglobulin is most sensitive when TSH is high but this requires the withdrawal of levothyroxine therapy. Recombinant TSH (thyrotropin alfa, rhTSH) 900 µg (2 doses over 48 hours) is used to stimulate thyroglobulin without stopping thyroxine therapy. Detectable thyroglobulin suggests recurrence, in which case whole body 131I scanning is required. Unfortunately the presence of anti-thyroglobulin antibodies can make the assay unreliable.
The prognosis is extremely good when differentiated thyroid cancer is excised while confined to the thyroid gland, and the specific therapies available lead to a relatively good prognosis even in the presence of metastases at diagnosis. Accepted markers of high risk include greater age (>40 years), larger primary tumour size (>4 cm) and macroscopic invasion of capsule and surrounding tissues.
Anaplastic carcinomas and lymphoma
These do not respond to radioactive iodine, and external radiotherapy produces only a brief respite.
Medullary carcinoma
Medullary carcinoma (MTC) is a neuroendocrine tumour of the calcitonin-producing C cells of the thyroid. This condition is often associated with multiple endocrine neoplasia type 2 (MEN 2, see p. 998) – approximately 25% of patients diagnosed with MTC have a mutation of the RET proto-oncogene, although the other manifestations of MEN 2 may be absent, hence the importance of genetic counselling and family screening. People with MEN2 mutations are advised to have a prophylactic thyroidectomy as early as 5 years of age to prevent the development of MTC.
Total thyroidectomy and wide lymph node clearance is usually indicated in MTC. Local invasion or metastasis is frequent, and the tumour responds poorly to treatment, although progression is often slow.
FURTHER READING
British Thyroid Association and Royal College of Physicians Guidelines 2007. The management of thyroid cancer 2007. London: BTA
Wartofsky, L. Highlights of the American Thyroid Association Guidelines for patients with thyroid nodules or differentiated thyroid carcinoma: the 2009 revision. Thyroid 2009; 19:1139–43.
Reproduction and sex
Terminology in reproductive medicine is shown in Box 19.5.
Box 19.5
Definitions in reproductive medicine
Erectile dysfunction |
Inability of the male to achieve or sustain an erection adequate for satisfactory intercourse |
Azoospermia |
Absence of sperm in the ejaculate |
Oligospermia |
Reduced numbers of sperm in the ejaculate |
Libido |
Sexual interest or desire; often difficult to assess and is greatly affected by stress, tiredness and psychological factors |
Menarche |
Age at first period |
Primary amenorrhoea |
Failure to begin spontaneous menstruation by age 16 |
Secondary amenorrhoea |
Absence of menstruation for 3 months in a woman who has previously had cycles |
Oligomenorrhoea |
Irregular long cycles; often used for any length of cycle above 32 days |
Dyspareunia |
Pain or discomfort in the female during intercourse |
Menstruation |
Onset of spontaneous (usually regular) uterine bleeding in the female |
Virilization |
Occurrence of male secondary sexual characteristics in the female |
Embryology
Up to 8 weeks of gestation the sexes share a common development, with a primitive genital tract including the Wolffian and Müllerian ducts. There are additionally a primitive perineum and primitive gonads.
In the presence of a Y chromosome, the potential testis develops while the ovary regresses.
In the absence of a Y chromosome, the potential ovary develops and related ducts form a uterus and the upper vagina.
Production of Müllerian inhibitory factor from the early ‘testis’ produces atrophy of the Müllerian duct, while, under the influence of testosterone and dihydrotestosterone, the Wolffian duct differentiates into an epididymis, vas deferens, seminal vesicles and prostate. Androgens induce transformation of the perineum to include a penis, penile urethra and scrotum containing the testes, which descend in response to androgenic stimulation. At birth, testicular volume is 0.5–1 mL.
Physiology
The male
An outline of the hypothalamic-pituitary-gonadal axis is shown in Figure 19.20.
1. Pulses of gonadotrophin-releasing hormone (GnRH) are released from the hypothalamus and stimulate LH and FSH release from the pituitary. LH and FSH are composed of two glycoprotein chains (α and β subunits). The α subunits are identical and are shared with TSH, whilst the β subunit confers specific biological activity.
2. LH stimulates testosterone production from Leydig cells of the testis.
3. Testosterone acts via nuclear androgen receptors which interact with coregulatory proteins to produce the appropriate tissue responses: male secondary sexual characteristics, anabolism and the maintenance of libido. It also acts locally within the testis to aid spermatogenesis. Testosterone circulates largely bound to sex hormone-binding globulin (SHBG) (see p. 942). Testosterone feeds back on the hypothalamus/pituitary to inhibit GnRH secretion.
4. FSH stimulates the Sertoli cells in the seminiferous tubules to produce mature sperm and the inhibins A and B.
5. Inhibin feeds back to the pituitary to decrease FSH secretion. Activin, a related peptide, counteracts inhibin.
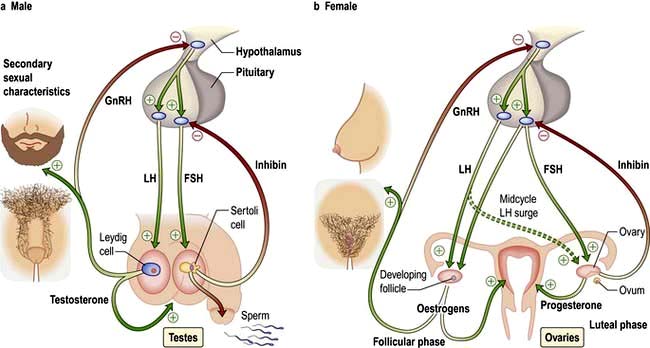
Figure 19.20 Male and female hypothalamic-pituitary-gonadal axes. LH and FSH are secreted in pulses in response to hypothalamic GnRH and have differential effects on the gonads (see text). Testosterone and oestrogens have multiple peripheral effects and exert negative feedback on LH secretion. Inhibin exerts negative feedback on FSH secretion.
The secondary sexual characteristics of the male. for which testosterone is necessary are the growth of pubic, axillary and facial hair, enlargement of the external genitalia, deepening of the voice, sebum secretion, muscle growth and frontal balding.
The female
Female physiology is more complex (Figs 19.20, 19.21).
1. In the adult female, higher brain centres impose a menstrual cycle of 28 days upon the activity of hypothalamic GnRH.
2. Pulses of GnRH, at about 2-hour intervals, stimulate release of pituitary LH and FSH.
3. LH stimulates ovarian androgen production by the ovarian theca cells.
4. FSH stimulates follicular development and aromatase activity (an enzyme required to convert ovarian androgens to oestrogens) in the ovarian granulosa cells. FSH also stimulates release of inhibin from ovarian stromal cells, which inhibits FSH release. Activin counteracts inhibin (Fig. 19.20).
5. Although many follicles are ‘recruited’ for development in early folliculogenesis, by day 8–10 a ‘leading’ (or ‘dominant’) follicle is selected for development into a mature Graafian follicle.
6. Oestrogens have a double feedback action on the pituitary (Fig. 19.20). Initially they inhibit gonadotrophin secretion (negative feedback), but later high-level exposure results in increased GnRH secretion and increased LH sensitivity to GnRH (positive feedback), which leads to the mid-cycle LH surge inducing ovulation from the leading follicle (Fig. 19.21).
7. The follicle then differentiates into a corpus luteum, which secretes both progesterone and oestradiol during the second half of the cycle (luteal phase).
8. Oestrogen initially and then progesterone cause uterine endometrial proliferation in preparation for possible implantation; if implantation does not occur, the corpus luteum regresses and progesterone secretion and inhibin levels fall so that the endometrium is shed (menstruation) allowing increased GnRH and FSH secretion.
9. If implantation and pregnancy follow, human chorionic gonadotrophin (HCG) production from the trophoblast maintains corpus luteum function until 10–12 weeks of gestation, by which time the placenta will be making sufficient oestrogen and progesterone to support itself.
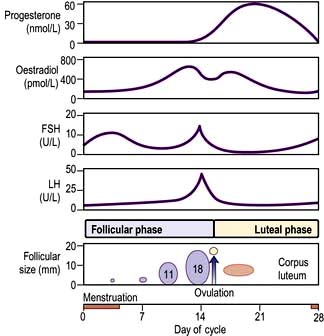
Secondary sexual characteristics of the female. These are induced by oestrogens, especially development of the breast and nipples, vaginal and vulval growth and pubic hair development. Oestrogens also induce growth and maturation of the uterus and fallopian tubes. They circulate largely bound to SHBG.
Puberty
The mechanisms initiating puberty are poorly understood but are thought to result from withdrawal of central inhibition of GnRH release. Environmental and physical factors are involved in the timing of puberty (including body fat changes, physical exercise) as well as genetic factors (e.g. a G protein-coupled receptor gene, GPR54) required for pubertal maturation. Kisspeptin is the endogenous ligand for kisspeptin receptor KISSIR formerly known as GPR54 and this peptide is believed to play a crucial role in the regulation of GnRH production and the timing of puberty.
LH and FSH are both low in the prepubertal child. In early puberty, FSH begins to rise first, initially in nocturnal pulses; this is followed by a rise in LH with a subsequent increase in testosterone/oestrogen levels. The milestones of puberty in the two sexes are shown in Figure 19.22.
Figure 19.22 The age of development of features of puberty. Stages and testicular size show mean ages, and all vary considerably between individuals. The same is true of height spurt, shown here in relation to other data. Numbers 2–5 indicate stages of development.
In boys, pubertal changes begin at between 10 and 14 years and are complete at between 15 and 17 years. The genitalia develop, testes enlarge and the area of pubic hair increases. Peak height velocity is reached between ages 12 and 17 years during stage 4 of testicular development. Full spermatogenesis occurs comparatively late.
In girls, events start a year earlier. Breast bud enlargement begins at age 9–13 years and continues to 12–18 years. Pubic hair growth commences at ages 9–14 years and is completed at 12–16 years. Menarche occurs relatively late (age 11–15 years) but peak height velocity is reached earlier (at age 10–13 years), and growth is completed much earlier than in boys.
Precocious puberty
Development of secondary sexual characteristics, or menarche in girls, at or before the age of 9 years is premature. All cases require assessment by a paediatric endocrinologist.
Idiopathic (true) precocity is most common in girls and very rare in boys. This is a diagnosis of exclusion. With no apparent cause for premature breast or pubic hair development, and an early growth spurt, it may be normal and may run in families. Treatment with long-acting GnRH analogues (given by nasal spray, by subcutaneous injection or by implant) causes suppression of gonadotrophin release via downregulation of the receptor – and therefore reduced sex hormone production – and is moderately effective; cyproterone acetate, an antiandrogen with progestational activity, is also used.
Other forms of precocity include:
Cerebral precocity. Many causes of hypothalamic disease, especially tumours, present in this way. In boys this must be rigorously excluded. MRI scan is almost always indicated to exclude this diagnosis.
McCune–Albright syndrome. This usually occurs in girls, with precocity, polyostotic fibrous dysplasia and skin pigmentation (café-au-lait). See also page 998.
Premature thelarche. This is early breast development alone, usually transient, at age 2–4 years. It may regress or persist until puberty. There is no evidence of follicular development.
Premature adrenarche. This is early development of pubic hair without significant other changes, usually after the age of 5 years and more commonly in girls. It is also more common in obese children due to reduced SHBG levels leading to higher free circulating androgens. In boys with precocious adrenarche, the rare possibility of an androgen-secreting testicular tumour should be looked for if serum androgens are high and LH is suppressed.
Delayed puberty
Over 95% of children show signs of pubertal development by the age of 14 years. In its absence, investigation should begin by the age of 15 years. Causes of hypogonadism (see below) are clearly relevant but most cases represent constitutional delay.
In constitutional delay, pubertal development, bone age and stature are in parallel. A family history may confirm that other family members experienced the same delayed development, which is common in boys but very rare in girls.
In boys, a testicular volume >5 mL indicates the onset of puberty. A rising serum testosterone is an earlier clue.
In girls, the breast bud is the first sign. Ultrasound allows accurate assessment of ovarian and uterine development.
Elevated LH/FSH levels will identify a primary gonadal defect, and in other cases, GnRH (LHRH) tests can indicate the stage of early puberty.
If any progression into puberty is evident clinically, investigations are not required. When delay is great and problems are serious (e.g. severe teasing at school), low-dose short-term sex hormone therapy is used to induce puberty.
The menopause
The menopause, or cessation of periods, naturally occurs at about the age of 45–55 years. During the late 40s, FSH initially, and then LH concentrations begin to rise, probably as follicle supply diminishes. Oestrogen levels fall and the cycle becomes disrupted. Most women notice irregular scanty periods coming on over a variable period, though in some sudden amenorrhoea or menorrhagia occurs. Eventually the menopausal pattern of low oestradiol levels with grossly elevated LH and FSH levels (usually >50 and >25 U/L, respectively) is established. Premature menopause may also occur surgically, with radiotherapy to the ovaries and with ovarian disease.
Clinical features and treatment
Features of oestrogen deficiency are hot flushes (which occur in most women and can be disabling), vaginal dryness and atrophy of the breasts. There may also be symptoms of loss of libido, loss of self-esteem, nonspecific aches and pains, sleep disturbance, irritability, depression, loss of concentration and weight gain.
Women show a rapid loss of bone density in the 10 years following the menopause (osteoporosis, see p. 552) and the premenopausal protection from ischaemic heart disease disappears.
Hormone replacement therapy (HRT). Symptomatic patients should usually be treated but the previous widespread use of HRT has been thrown into doubt by a number of large prospective studies which have reported in recent years. Although scientific debate continues, the overall benefits and risks are summarized as in Box 19.6.
Box 19.6
Risks and benefits of hormone replacement therapya
Potential benefits
Symptomatic improvement in most menopausal symptoms for the majority of women. Oestrogen-deficient symptoms respond well to oestrogen replacement, the vaguer general symptoms may or may not improve. Vaginal symptoms also respond to local oestrogen preparations.
Protection against fractures of wrist, spine and hip, secondary to osteoporosis (~24–33%) (see p. 552), owing to protection of predominantly trabecular bone (p. 549). However, this is not a recommended indication for therapy.
Significant reduction in the risk of large bowel cancer (~33%).
Possible increase in general wellbeing, although hopes for a reduction in the incidence of Alzheimer’s disease have not been confirmed.
Potential risks
Significant increase in the risk of breast cancer (+26%), but no change in breast cancer mortality. This is primarily a risk of combined oestrogen-progesterone HRT. Some studies suggest that breast cancers diagnosed on HRT are easier to treat effectively.
Significant increase in the risk of endometrial cancer when unopposed oestrogens are given to women with a uterus.
Significant increase in the risk of ischaemic heart disease (+29%) and stroke (+41%).
Inconvenience of withdrawal bleeds, unless a hysterectomy has been performed or regimens used which include continuous oestrogen and progesterone.
a Percentages are from the Women’s Health Initiative (WHI) study of 16 600 women.
Absolute risks and benefits for individual women clearly depend on their background risk of that disease, and there is as yet no evidence on the relative risks of different hormone preparations or routes of administration (oral, transdermal or implant). Overall, the WHI study estimated that, over 5 years of treatment, an extra one woman in every 100 would develop an illness that would not have occurred had she not been taking HRT. However, the decision about whether or not a woman takes HRT is now very much an individual decision based on:
the severity of the woman’s menopausal symptoms
her personal risk of conditions which may be prevented or made more likely by HRT
HRT is not recommended purely for prevention of postmenopausal osteoporosis in the absence of menopausal symptoms.
Symptomatic treatment is the main indication with the lowest effective dose given for short-term rather than long-term treatment.
Selective oestrogen receptor modulators, SERMs (e.g. raloxifene), offer a potentially attractive combination of positive oestrogen effects on bone and cardiovascular system with no effects on oestrogen receptors of uterus and breast and possible reduction in breast cancer incidence; long-term outcome studies, however, are still awaited.
FURTHER READING
Beral V; Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 2003; 362:419–427.
Chlebowski RT, Schwartz AG, Wakelee H et al. Oestrogen plus progestin and lung cancer in postmenopausal women (Women’s Health Initiative trial): a post-hoc analysis of a randomised controlled trial. Lancet 2009; 374:1243–1251.
Palmert MR, Dunkel L. Delayed puberty. N Engl J Med 2012; 336:443–453.
Premature menopause and ovarian failure
The most common cause of premature menopause in women (before age 40) is ovarian failure. This may be autoimmune, and is rarely caused by identifiable genetic causes such as the fragile X pre-mutation but is most commonly of unknown aetiology, although often familial. Repeat LH/FSH levels are necessary before giving a diagnosis of premature menopause because of the psychological impact of this diagnosis and the possibility that a single elevation of LH/FSH might simply be the mid-cycle ovulatory surge. Bilateral oophorectomy and some chemotherapy regimens cause the same oestrogen deficiency state.
Treatment. HRT should almost always be given, as the risk of osteoporosis and other conditions related to oestrogen deficiency almost always outweighs the risks of HRT at this younger age. HRT may still also be actively recommended when normal menopause occurs relatively early (e.g. before the age of 50).
The ageing male
In the male, there is no sudden ‘change of life’. However, there is a progressive loss in sexual function with reduction in morning erections and frequency of intercourse.
The age of onset varies widely. Typically, overall testicular volume diminishes and sex hormone-binding globulin (SHBG) and gonadotrophin levels gradually rise, but other men present with low or borderline testosterone without elevation of LH/FSH. Low testosterone certainly increases the risk of osteoporosis and in some studies is associated with increased cardiovascular risk, but it remains unclear to what extent general symptoms of lack of energy, drive, muscle strength and general wellbeing may relate to these hormonal changes. Loss of libido and erectile dysfunction are common symptoms even when hormones are normal, and long-term outcome studies of testosterone replacement are still awaited. Therefore, the decision to offer testosterone replacement to an ageing male is currently based on full clinical and biochemical assessment and full discussion of potential risks (including prostate disease) as well as benefits. If testosterone is unequivocally low (<7 pmol/L) most authorities would recommend replacement. However, few would treat if testosterone is >12 pmol/L with normal LH/FSH. Clinically, a large proportion of cases are in the borderline range (7–12 pmol/L), which can lead to difficulties in reaching a firm diagnosis.
Clinical features of disorders of sex and reproduction
A detailed history and examination of all systems is required (Box 19.7). A man having regular satisfactory intercourse or a woman with regular ovulatory periods is most unlikely to have significant endocrine disease, assuming the history is accurate.
Tests of gonadal function
Much can be deduced from basal measurements of the gonadotrophins, oestrogens/testosterone and prolactin:
Low testosterone or oestradiol with high gonadotrophins indicates primary gonadal disease.
Low levels of testosterone/oestradiol with low or normal LH/FSH imply hypothalamic-pituitary disease.
Demonstration of ovulation (by measurement of luteal phase serum progesterone and/or by serial ovarian ultrasound in the follicular phase) or a healthy sperm count (20–200 million/mL, >60% grade I motility and <20% abnormal forms), provide absolute confirmation of normal female or male reproductive endocrinology, but these tests are not always essential.
Pregnancy provides complete demonstration of normal male and female function.
Hyperprolactinaemia can be confirmed or excluded by direct measurement. Levels may increase with stress; if this is suspected, a cannula should be inserted and samples taken through it 30 min later.
More detailed tests are indicated in Table 19.19.
Table 19.19 Tests of gonadal function
| Test | Uses/comments |
|---|---|
Male |
|
Basal testosterone |
Normal levels exclude hypogonadism |
Sperm count |
Normal count excludes deficiencyMotility and abnormal sperm forms should be noted |
Female |
|
Basal oestradiol |
Normal levels exclude hypogonadism |
Luteal phase progesterone (days 18–24 of cycle) |
If >30 nmol/L, suggests ovulation |
Ultrasound of ovaries |
To confirm ovulation |
Both sexes |
|
Basal LH/FSH |
Demonstrates state of feedback system for hormone production (LH) and germ cell production |
(FSH) |
|
HCG test (testosterone or oestradiol measured) |
Response shows potential of ovary or testis; failure demonstrates primary gonadal problem |
Clomifene test (LH and FSH measured) |
Tests hypothalamic negative feedback system; clomifene is oestrogen antagonist and causes LH/FSH to rise |
LHRH test (rarely used) |
Shows adequacy (or otherwise) of LH and FSH stores in pituitary |