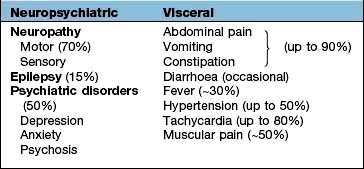
Complications of diabetes
People with diabetes still have a considerably reduced life expectancy. The major cause of death in treated patients is due to cardiovascular problems (60–70%) followed by renal failure (10%) and infections (6%). There is no doubt that the duration and degree of hyperglycaemia play a major role in the production of complications. Better diabetic control can reduce the rate of progression of both nephropathy and retinopathy and the DCCT showed a 60% reduction in developing complications over 9 years when the HbA1c was kept at around 7% in type 1 diabetes.
Pathophysiology
The mechanisms leading to damage are ill-defined. The following are consequences of hyperglycaemia and may play a role:
 Non-enzymatic glycosylation of a wide variety of proteins, e.g. haemoglobin, collagen, LDL and tubulin in peripheral nerves. This leads to an accumulation of advanced glycosylated end-products causing injury and inflammation via stimulation of pro-inflammatory factors, e.g. complement, cytokines.
Non-enzymatic glycosylation of a wide variety of proteins, e.g. haemoglobin, collagen, LDL and tubulin in peripheral nerves. This leads to an accumulation of advanced glycosylated end-products causing injury and inflammation via stimulation of pro-inflammatory factors, e.g. complement, cytokines.
Polyol pathway. The metabolism of glucose by increased intracellular aldose reductase leads to accumulation of sorbitol and fructose. This causes changes in vascular permeability, cell proliferation and capillary structure via stimulation of protein kinase C and TGF-β.
Abnormal microvascular blood flow impairs supply of nutrients and oxygen. Microvascular occlusion is due to vasoconstrictors, e.g. endothelins and thrombogenesis, and leads to endothelial damage.
Other factors include the formation of reactive oxygen species and growth factors stimulation (TGF-β) and vascular endothelial growth factor (VEGF). These growth factors are released by ischaemic tissues and cause endothelial cells to proliferate.
Haemodynamic changes, e.g. in kidney (see p. 1025).
It has been proposed that all of the above mechanisms stem from a single hyperglycaemia-induced process of overproduction of superoxide by the mitochondrial electron chain. This paradigm offers an integrated explanation of how complications of diabetes develop.
Macrovascular complications (Table 20.10)
Diabetes is a risk factor for the development of atherosclerosis. This risk is related to that of the background population. For example, people with diabetes in Japan are less likely than European patients to develop atherosclerosis, but more likely to develop it than non-diabetic Japanese.
Myocardial infarction is 3–5 times as likely, and women with diabetes lose their premenopausal protection from coronary artery disease.
Table 20.10 Diabetic risk factors for macrovascular complications
Several large trials have shown that intensive glucose-lowering treatment of diabetes has a relatively minor effect upon cardiovascular risk. Since the effect of other cardiovascular risk factors is enhanced in diabetes, it is vital to tackle all cardiovascular risk factors together in diabetes, and not just to focus on glucose levels.
Hypertension. The UKPDS demonstrated that aggressive treatment of hypertension produces a marked reduction in adverse cardiovascular outcomes, both microvascular and macrovascular. To achieve the target for blood pressure (Table 20.8), the UKPDS found that one-third of patients needed three or more antihypertensive drugs in combination, and two-thirds of treated patients needed two or more.
Smoking: the avoidable risk factor (see p. 806). Never give up efforts to help diabetic patients stop smoking.
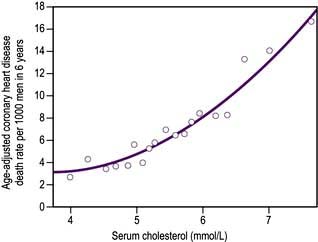
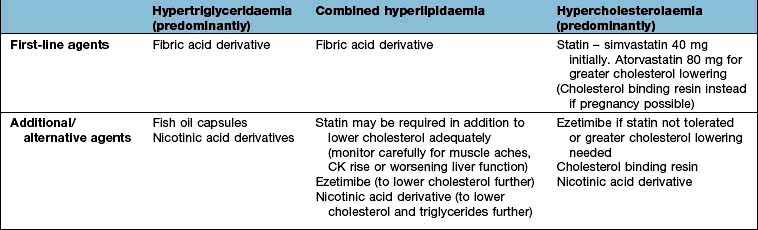
Lipid abnormalities. Clinical trials suggest that there is no ‘safe’ cut-off for serum cholesterol. The lowest achievable level seems best to aim for, and in practice this means that almost all people with type 2 diabetes will be treated with a statin.
Low-dose aspirin can reduce macrovascular risk, but is associated with a morbidity and mortality from bleeding. The benefits of aspirin outweigh the bleeding risk when the risk of a cardiovascular end-point is >30% in the next 10 years. This risk is reached in patients aged under 45 with three strong additional cardiovascular risk factors, aged 45–54 with three additional risk factors, aged 54–65 with two additional risk factors or aged over 65 with just one additional risk factor.
ACE inhibitors/angiotensin II receptor antagonists. Treating people with diabetes and at least one other major cardiovascular risk factor with an ACE inhibitor produces a 25–35% lowering of the risk of heart attack, stroke, overt nephropathy or cardiovascular death. Angiotensin II receptor antagonists are sometimes preferred initially and are also used for those intolerant to ACE inhibitors.
Microvascular complications
In contrast to macrovascular disease, which is prevalent in the West as a whole, microvascular disease is specific to diabetes. Small blood vessels throughout the body are affected but the disease process is of particular danger in three sites:
Diabetic retinopathy, nephropathy and neuropathy tend to manifest 10–20 years after diagnosis in young patients, but may present earlier in older patients, probably because these have had unrecognized diabetes for months or even years prior to diagnosis. Genetic factors appear to contribute to the susceptibility to microvascular disease. Diabetic siblings of diabetic patients with renal and eye disease have a three- to five-fold increased risk of the same complication in both type 1 and type 2 patients. There are racial differences in the overall prevalence of nephropathy. In the USA, prevalence is: Pima American Indians > Hispanic/Mexican > US black > US white patients.
Diabetic eye disease
At least 90% of young patients with type 1 diabetes will develop retinal changes, but these only progress to sight-threatening retinopathy in a minority. Some 30–50% will require laser photocoagulation to prevent or limit progression to proliferative retinopathy, and good control of blood pressure is essential. Diabetes is still the commonest cause of blindness in under 65 year olds. It affects the eye in a variety of ways:
Diabetic retinopathy is damage to the retina and iris caused by diabetes, which can lead to blindness.
Cataract is denaturation of the protein and other components of the lens of the eye which render it opaque.
External ocular palsies (p. 1081). The sixth and the third nerve are the most commonly affected. Third nerve palsy is not associated with pain. These nerve palsies usually recover spontaneously within a period of 3–6 months.
Natural history
Cataract develops earlier in people with diabetes than in the general population. Sustained very poor diabetes control with a degree of ketosis can cause an acute cataract (snowflake cataract), which comes on rapidly. Fluctuations in blood glucose concentration can cause refractive variability, as a result of osmotic changes within the lens (the absorption of water into the lens causes temporary hypermetropica). This presents as fluctuating difficulty in reading. It resolves with better metabolic control of the diabetes.
Diabetic retinopathy (Fig. 20.13) is the most commonly diagnosed diabetes-related complication. Its prevalence increases with the duration of diabetes (Fig. 20.14). Some 20% of people with type 1 diabetes will have retinal changes after 10 years, rising to >95% after 20 years (see Table 20.11); 20–30% of people with type 2 diabetes have retinopathy at diagnosis. The metabolic consequences of poorly-controlled diabetes cause intramural pericyte death, and thickening of the basement membrane in the small blood vessels of the retina. This leads initially to incompetence and increased permeability of the vascular walls, and later to occlusion of the vessels (capillary closure). This process has somewhat different consequences in the peripheral retina and in the macular area.
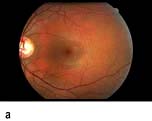
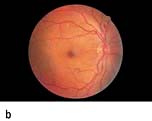
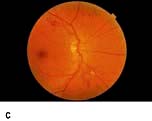
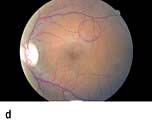
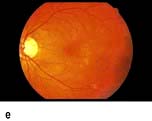
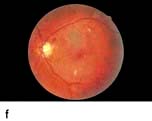
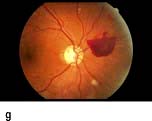
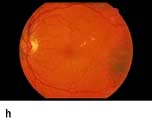
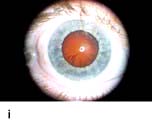
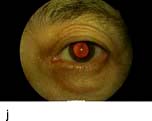
Figure 20.13 Features of diabetic eye disease. (a) The normal macula (centre) and optic disc (to left). (b) Microaneurysms (small circles) and blot haemorrhage (larger circle) – early background retinopathy. (c) Hard exudates (circled) and single cotton wool spot (arrowed) in addition to multiple blot haemorrhages in background retinopathy. (d) Intra-retinal microvascular abnormalities (IRMA) – pre-proliferative retinopathy (circled). (e) Venous loop (circled) also indicates pre-proliferative change. (f) Fronds of new vessels on the disc and elsewhere (proliferative). (g) Pre-retinal haemorrhage in proliferative disease. (h) Hard exudates within a disc-width of the macula (maculopathy). (i) Cortical and (j) central cataracts can be seen against the red reflex with the ophthalmoscope.
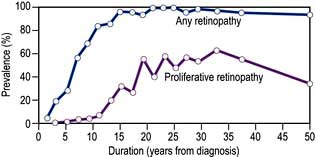
Figure 20.14 Prevalence of retinopathy in relation to duration of the disease in patients with type 1 diabetes mellitus diagnosed under the age of 33 years. Almost all eventually develop background change and 60% progress to proliferative retinopathy.
(Data from Archives of Ophthalmology 1984; 102:520.)
Table 20.11 Grading of pathological changes in the retina in diabetic retinopathy: the action needed
| Retinopathy grade | Retinal abnormality (cause) | Action needed |
|---|---|---|
Peripheral retina |
|
|
Background (R1) |
Annual screening only |
|
Pre-proliferative (R2) |
Non-urgent referral to an ophthalmologist |
|
Proliferative (R3) |
Urgent referral to an ophthalmologist |
|
Advanced retinopathy |
Urgent referral to an ophthalmologist – but much vision already lost |
|
Central retina |
|
|
Maculopathy (M1) |
Referral to an ophthalmologist soon |
Note: Hard exudates have a bright yellowish-white colour and are often irregular in outline with a sharply defined margin. Cotton-wool spots are greyish-white, have indistinct margins and a dull matt surface, unlike the glossy appearance of hard exudates. R, retinopathy; M, maculopathy.
Damage to the wall of small vessels causes microaneurysms (small red dots) within the retina. When vessel walls are breached superficial (blot) haemorrhages occur in the ganglion cell and outer plexiform layers. Damaged blood vessels leak fluid into the retina. The fluid is cleared into the retinal veins leaving behind protein and lipid deposits causing hard exudates. These are eventually cleared by macrophages.
Micro-infarcts within the retina due to occluded vessels cause cotton wool spots. The spot itself is due to the accumulation of axoplasmic debris. This debris is removed by macrophages. As this occurs, there may be white dots at the site of the previous cotton wool spot (cytoid bodies). Damage to the walls of veins causes their calibre to vary (venous beading), and elongation to occur causing venous loops. Blockage of blood vessels leads to areas of capillary non-perfusion. Ischaemia in these areas causes the release of vascular growth factors such as VEGF (vascular endothelial growth factor). These factors cause new blood vessels to grow in the retina (neovascularization). Some of these new blood vessels are inside the retina and are helpful. These new intraretinal vessels, and other vessels whose walls are damaged and dilated, give the appearance of intraretinal microvascular abnormalities (IRMAs).
Other new vessels emerge through the retina and lie on its surface, usually at the margin of an area of capillary closure. The normal shearing stresses that occur within the eye can cause these poorly supported new vessels to bleed. Small haemorrhages give rise to pre-retinal haemorrhages (boat-shaped haemorrhages). With further bleeding vitreous haemorrhage occurs with consequent sudden loss of vision. Later collagen tissue grows along the margins of the new vessels and giving rise to fibrotic bands. These bands may contract and pull on the retina causing further haemorrhage and retinal detachment. Sometimes vessels may be induced to grow on the pupil margin (rubeosis) and in the angle of the anterior chamber of the eye giving rise to rapid increase in intraocular pressure (rubeotic glaucoma). These features of retinopathy in the peripheral retina are grouped, according to the risk of visual loss, into three stages (Table 20.11).
Fluid from leaking vessels is cleared poorly in the macular area due to its anatomy differing from the rest of the retina. Above a certain rate of formation, clearance fails and macular oedema occurs. This distorts and thickens the retina at the macula. If sustained, this distortion causes loss of central vision. Macular oedema is not visible with the ophthalmoscope or with retinal photography. For this reason surrogate markers for the presence of macular oedema are used (Table 20.11). Capillary occlusion in the macular area will also cause loss of central vision.
Examination
Bedside examination of the eye. Visual acuity should be checked using both a pinhole and the patient’s distance spectacles. The ocular movements are assessed to detect any ocular motor palsies. The iris is examined for rubeosis and then the pupils dilated with 1% tropicamide. About 20 minutes later the eye is examined for the presence of a cataract by looking at the lens with a +10.00 lens in the ophthalmoscope and viewing the lens against the red reflex. The retina is then examined systematically looking at the disc, then all four quadrants, and finally the macula. The macula is examined last because this induces the greatest discomfort, and pupillary constriction.
Eye screening
Screening for sight-threatening eye disease with universal access is seen as offering the best hope of displacing diabetes as the commonest cause of blindness in those under 65 years of age. The National Screening Committee in the UK has helped establish digital photography-based screening across the country, based on a national set of standards. All people with diabetes, over the age of 12 are offered annual measurement of their acuity, and photographs of their retina. Box 20.5 shows standardized criteria for screening schemes; these are regularly inspected.
 Box 20.5
Box 20.5
Criteria for a successful local screening scheme for sight-threatening diabetic retinopathy
Clearly defined geographical area for the screening programme
Adequate number of people with diabetes for viability (>12 000)
An identified screening programme manager
An identified clinical screening lead
An identified hospital eye service for diagnosis and laser treatment
Computer software capable of supporting call/recall of patients and image grading
Centralized appointment administration
Single collated list of all people with diabetes in the area over the age of 12
Equipment to obtain adequate disc and macula centred images of each eye
Process to manage people with poor quality images
Clear route of referral for treatment, and for feedback from treatment centre to screening unit
Management of diabetic eye disease (Table 20.11)
Extraction and intraocular lens implantation is indicated if the cataract is causing visual disability to the patient or is giving rise to inability to view the retina adequately. Cataract extraction is straightforward if there is no retinopathy present. Pre-existing retinopathy may worsen after cataract extraction.
The DCCT and UKPDS show that the risk of developing diabetic eye disease, and the risk of established retinopathy developing further, can be reduced by tight metabolic control of both diabetes and blood pressure. Development or progression of retinopathy may be accelerated by rapid improvement in glycaemic control, pregnancy and in those with nephropathy, and these groups need frequent monitoring. Fluorescein angiography (a fluorescent dye is injected into an arm vein and photographed in transit through the retinal vessels) is used to define the extent of the potentially sight-threatening diabetic retinopathy. Ocular coherence tomography (OCT) is used to image the content of the layers of the retina at the macula, and in particular to measure retinal thickness. It can detect macular oedema and other macular abnormalities.
Treatment of proliferative retinopathy
New vessels are an indication for laser photocoagulation therapy. New vessels on the disc carry the worst prognosis and warrant urgent laser therapy. The laser should be directed at the new vessels and, in addition, to the associated areas of capillary non-perfusion (ischaemia). If the proliferative retinopathy has progressed to new vessels developing on the optic disc then a technique known as panretinal photocoagulation (PRP) is carried out. This involves multiple laser burns to the peripheral retina, especially in the areas of capillary non-perfusion. Rubeosis is also treated with panretinal photocoagulation. If some bleeding has occurred but there is a good view then laser treatment should be applied. Vitreoretinal surgery is used if bleeding is recurrent and preventing laser therapy. It is also used to try to salvage some vision after vitreous haemorrhage and to treat fibrotic traction retinal detachment in advanced retinopathy.
Extrafoveal exudates can be watched. However, if they are beginning to encroach on the fovea then the centre of any rings of exudates, should be treated by laser photocoagulation. If oedema has spread into the centre of the macula, then a technique known as grid photocoagulation is used, where a number of laser burns are scattered around the macula. This limits deterioration in vision, and on occasion results in some visual improvement. Ischaemic maculopathy is not treatable, and leads to central visual loss.
The future
Anti-VEGF drugs, such as bevacizumab and ranibizumab (see p. 1064) are being used to control diabetic retinopathy and diabetic maculopathy, particularly that which involves the centre of the macula and is causing sight loss. Recent studies have shown benefit over laser for this type of maculopathy.
FURTHER READING
Googe J, Brucker AJ, Bressler NM et al.; of the Diabetic Retinopathy Clinical Research Network. Randomized trial evaluating short-term effects of intravitreal ranibizumab or triamcinolone acetonide on macular edema after focal/grid laser for diabetic macular edema in eyes also receiving panretinal photocoagulation. Retina 2011; 31:1009–1027.
Michaelides M, Kaines A, Hamilton RD et al. A prospective randomized trial of intravitreal bevacizumab or laser therapy in the management of diabetic macular edema (BOLT study) 12-month data: report 2. Ophthalmology 2010; 117:1078–1086.
The diabetic kidney
The kidney may be damaged by diabetes in three main ways:
Diabetic nephropathy
Epidemiology
Clinical nephropathy secondary to glomerular disease usually manifests 15–25 years after diagnosis of diabetes and affects 25–35% of patients diagnosed under the age of 30 years. It is the leading cause of premature death in young diabetic patients. Older patients also develop nephropathy, but the proportion affected is smaller. The incidence of end-stage kidney disease has fallen in recent decades, probably due to better control of blood glucose and blood pressure, but this benefit has been cancelled out by the rising incidence of both types of diabetes.
Pathophysiology
The earliest functional abnormality in the diabetic kidney is renal hypertrophy associated with a raised glomerular filtration rate. This appears soon after diagnosis and is related to poor glycaemic control. As the kidney becomes damaged by diabetes, the afferent arteriole (leading to the glomerulus) becomes vasodilated to a greater extent than the efferent glomerular arteriole. This increases the intraglomerular filtration pressure, further damaging the glomerular capillaries. This increased intraglomerular pressure also leads to increased local shearing forces which are thought to contribute to mesangial cell hypertrophy and increased secretion of extracellular mesangial matrix material. This process eventually leads to glomerular sclerosis. The initial structural lesion in the glomerulus is thickening of the basement membrane. Associated changes result in disruption of the protein cross-linkages which normally make the membrane an effective filter. In consequence, there is a progressive leak of large molecules (particularly protein) into the urine.
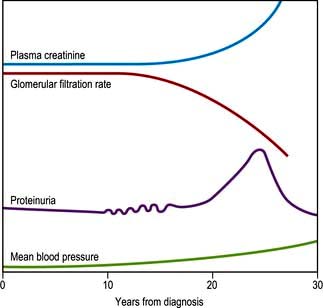
The earliest evidence of this is ‘microalbuminuria’ – amounts of urinary albumin so small as to be undetectable by standard dipsticks (see p. 309). Microalbuminuria may be tested for by radioimmunoassay or by using special dipsticks. It is a predictive marker of progression to nephropathy in type 1 diabetes, and of increased cardiovascular risk in type 2 diabetes. Microalbuminuria may, after some years, progress to intermittent albuminuria followed by persistent proteinuria. Light-microscopic changes of glomerulosclerosis become manifest; both diffuse and nodular glomerulosclerosis can occur. The latter is sometimes known as the Kimmelstiel–Wilson lesion. At the later stage of glomerulosclerosis, the glomerulus is replaced by hyaline material.
At the stage of persistent proteinuria, the plasma creatinine is normal but the average patient is only some 5–10 years from end-stage kidney disease. The proteinuria may become so heavy as to induce a transient nephrotic syndrome, with peripheral oedema and hypoalbuminaemia.
Patients with nephropathy typically show a normochromic normocytic anaemia and a raised erythrocyte sedimentation rate (ESR). Hypertension is a common development and may itself damage the kidney still further. A rise in plasma creatinine is a late feature that progresses inevitably to renal failure, although the rate of progression may vary widely between individuals.
The natural history of this process is shown in Figure 20.15.
Ischaemic lesions
Arteriolar lesions, with hypertrophy and hyalinization of the vessels, can occur in patients with diabetes. The appearances are similar to those of hypertensive disease and lead to ischaemic damage to the kidneys.
Infective lesions
Urinary tract infections are relatively more common in women with diabetes, but not in men. Ascending infection may occur because of bladder stasis resulting from autonomic neuropathy, and infections more easily become established in damaged renal tissue. Autopsy material frequently reveals interstitial changes suggestive of infection, but ischaemia may produce similar changes and the true frequency of pyelonephritis in diabetes is uncertain. Untreated infections in diabetics can result in renal papillary necrosis, in which renal papillae are shed in the urine, but this complication is now very rare.
Diagnosis
The urine of all diabetic patients should be checked regularly (at least annually) for the presence of protein. Many centres also screen younger patients for microalbuminuria since there is evidence that meticulous glycaemic control and early antihypertensive treatment, particularly with ACE inhibitors and angiotensin 2 blockers, may delay the onset of frank proteinuria. The albumin creatinine ratio (ACR) (tested on a mid-stream first morning urine sample) is <2.5 in healthy men, <3.5 mg/mmol in healthy women. Once proteinuria is present, other possible causes should be considered (see below), but once these are excluded, a presumptive diagnosis of diabetic nephropathy can be made. For practical purposes this implies inevitable progression to end-stage kidney disease, although the time course can be markedly slowed by early aggressive antihypertensive therapy. Clinical suspicion of a non-diabetic cause of nephropathy may be provoked by an atypical history, the absence of diabetic retinopathy (usually but not invariably present with diabetic nephropathy) and the presence of red-cell casts in the urine. Renal biopsy should be considered in such cases, but is rarely necessary or helpful. A 24-hour urine collection is often performed to quantify protein loss. Regular measurement is made of the plasma creatinine level with estimated glomerular filtration rate (eGFR).
Management
The management of diabetic nephropathy is similar to that of other causes of chronic kidney disease, with the following provisos:
Aggressive treatment of blood pressure with a target below 130/80 mmHg has been shown to slow the rate of deterioration of renal failure considerably. Angiotensin-converting enzyme inhibitors or an angiotensin receptor II antagonist are the drugs of choice (see Chapter 8). These drugs should also be used in normotensive patients with persistent microalbuminuria. Reduction in albuminuria occurs with this treatment.
Oral hypoglycaemic agents partially excreted via the kidney (e.g. glibenclamide and metformin) should be avoided.
Insulin sensitivity increases and drastic reductions in insulin dosage may be needed.
Associated diabetic retinopathy tends to progress rapidly, and frequent ophthalmic supervision is essential.
Management of end-stage disease is made more difficult by the fact that patients often have other complications of diabetes such as blindness, autonomic neuropathy or peripheral vascular disease. Vascular shunts tend to calcify rapidly and hence chronic ambulatory peritoneal dialysis may be preferable to haemodialysis. The failure rate of renal transplants is somewhat higher than in non-diabetic patients. A segmental pancreatic or islet graft is sometimes performed under cover of the immunosuppression needed for the renal graft, and this has been shown to improve survival as well as offering freedom from insulin injections.
Diabetic neuropathy
Diabetes can damage peripheral nervous tissue in a number of ways. The vascular hypothesis postulates occlusion of the vasa nervorum as the prime cause. This seems likely in isolated mononeuropathies, but the diffuse symmetrical nature of the common forms of neuropathy implies a metabolic cause. Since hyperglycaemia leads to increased formation of sorbitol and fructose in Schwann cells, accumulation of these sugars may disrupt function and structure.
The earliest functional change in diabetic nerves is delayed nerve conduction velocity; the earliest histological change is segmental demyelination, caused by damage to Schwann cells. In the early stages axons are preserved, implying prospects of recovery, but at a later stage irreversible axonal degeneration develops.
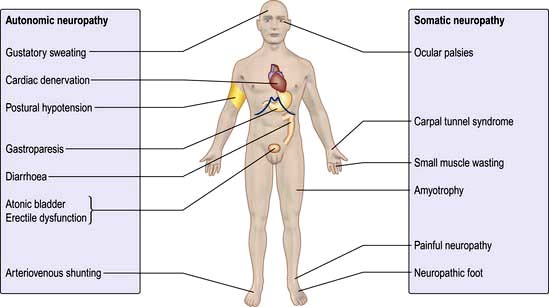
The following varieties of neuropathy occur (Fig. 20.16):
Symmetrical mainly sensory polyneuropathy (distal)
Mononeuropathy and mononeuritis multiplex
Diabetic amyotrophy (asymmetrical motor diabetic neuropathy)
Symmetrical mainly sensory polyneuropathy
This is often unrecognized by the patient in its early stages. Early clinical signs are loss of vibration sense, pain sensation (deep before superficial) and temperature sensation in the feet. At later stages patients may complain of a feeling of ‘walking on cotton wool’ and can lose their balance when washing the face or walking in the dark owing to impaired proprioception. Involvement of the hands is much less common and should prompt a search for non-diabetic causes. Complications include unrecognized trauma, beginning as blistering due to an ill-fitting shoe or a hot-water bottle, and leading to ulceration.
Sequelae of neuropathy. Involvement of motor nerves to the small muscles of the feet gives rise to interosseous wasting. Unbalanced traction by the long flexor muscles leads to a characteristic shape of the foot, with a high arch and clawing of the toes, which in turn leads to abnormal distribution of pressure on walking, resulting in callus formation under the first metatarsal head or on the tips of the toes and perforating neuropathic ulceration. Neuropathic arthropathy (Charcot’s joints) may sometimes develop in the ankle. The hands show small-muscle wasting as well as sensory changes, but these signs and symptoms must be differentiated from those of the carpal tunnel syndrome, which occurs with increased frequency in diabetes and may be amenable to surgery.
Acute painful neuropathy
A diffuse, painful neuropathy is less common. The patient describes burning or crawling pains in the feet, shins and anterior thighs. These symptoms are typically worse at night, and pressure from bedclothes may be intolerable. It may present at diagnosis or develop after sudden improvement in glycaemic control (e.g. when insulin is started). It usually remits spontaneously after 3–12 months if good control is maintained. A more chronic form, developing later in the course of the disease, is sometimes resistant to almost all forms of therapy. Neurological assessment is difficult because of the hyperaesthesia experienced by the patient, but muscle wasting is not a feature and objective signs can be minimal.
Management is firstly to explore for non-diabetic causes (see p. 1146). Explanation and reassurance about the high likelihood of remission within months may be all that is needed. Duloxetine (NICE recommend as first-line therapy), tricyclics, gabapentin or pregabalin, mexiletine, valproate and carbamazepine all reduce the perception of neuritic pain somewhat, but usually not as much as patients hope for. Transepidermal nerve stimulation (TENS) benefits some patients. Topical capsaicin-containing creams help occasionally. A few report that acupuncture has helped.
Mononeuritis and mononeuritis multiplex (multiple mononeuropathy)
Any nerve in the body can be involved in diabetic mononeuritis; the onset is typically abrupt and sometimes painful. Radiculopathy (i.e. involvement of a spinal root) may also occur.
Isolated palsies of nerves to the external eye muscles, especially the third and sixth nerves, are more common in diabetes. A characteristic feature of diabetic third nerve lesions is that pupillary reflexes are retained owing to sparing of pupillomotor fibres. Full spontaneous recovery is the rule for most episodes of mononeuritis over 3–6 months. Lesions are more likely to occur at common sites for external pressure palsies or nerve entrapment (e.g. the median nerve in the carpal tunnel, see p. 1074).
Diabetic amyotrophy
This condition is usually seen in older men with diabetes. Presentation is with painful wasting, usually asymmetrical, of the quadriceps muscles or occasionally in the shoulders. The wasting may be very marked and knee reflexes are diminished or absent. The affected area is often extremely tender. Extensor plantar responses sometimes develop and CSF protein content is elevated. Diabetic amyotrophy is usually associated with periods of poor glycaemic control and may be present at diagnosis. It often resolves in time with careful metabolic control of the diabetes.
Autonomic neuropathy
Asymptomatic autonomic disturbances can be demonstrated on testing in many patients (Box 20.6), but symptomatic autonomic neuropathy is rare. It affects both the sympathetic and parasympathetic nervous systems and can cause disabling postural hypotension.
Box 20.6
Bedside testing of autonomic function
| Normal | Abnormal | |
|---|---|---|
10 |
≥30 |
|
Heart rate responses to: |
|
|
≥15 |
≤10 |
|
≥1.21 |
≤1.20 |
|
≥1.04 |
≤1.00 |
R–R, time between R and next R on ECG; HR, heart rate.
Vagal neuropathy results in tachycardia at rest and loss of sinus arrhythmia. At a later stage, the heart may become denervated (resembling a transplanted heart). Cardiovascular reflexes such as the Valsalva manoeuvre are impaired. Postural hypotension occurs owing to loss of sympathetic tone to peripheral arterioles. A warm foot with a bounding pulse is sometimes seen in a polyneuropathy as a result of peripheral vasodilatation.
Vagal damage can lead to gastroparesis, often asymptomatic, but sometimes leading to intractable vomiting. Implantable devices which stimulate gastric emptying, and injections of botulinum toxin into the pylorus (to partly paralyse the sphincter), have each shown benefit in cases of this previously intractable problem. Autonomic diarrhoea characteristically occurs at night accompanied by urgency and incontinence. Diarrhoea and steatorrhoea may occur owing to small bowel bacterial overgrowth; treatment is with antibiotics such as tetracycline.
Loss of tone, incomplete emptying and stasis (predisposing to infection) can occur, and may ultimately result in an atonic, painless, distended bladder. Treatment is with intermittent self-catheterization, permanent catheterization if that fails and prophylactic antibiotic therapy for those prone to recurrent infection.
This is common. The first manifestation is incomplete erection which may in time progress to total failure; retrograde ejaculation also occurs in patients with autonomic neuropathy. Erectile dysfunction in diabetes has many causes including anxiety, depression, alcohol excess, drugs (e.g. thiazides and beta-blockers), primary or secondary gonadal failure, hypothyroidism and inadequate vascular supply owing to atheroma in pudendal arteries. The history and examination should focus on these possible causes. Blood is taken for LH, FSH, testosterone, prolactin and thyroid function. Treatment should ideally include sympathetic counselling of both partners.
Phosphodiesterase type-5 inhibitors (sildenafil, tadalafil, vardenafil, avanafil), which enhance the effects of nitric oxide on smooth muscle and increase penile blood flow, are used in those who do not take nitrates for angina. Some 60% of patients can be expected to benefit from this therapy.
Alternatives for those who fail to improve with a phosphodiesterase inhibitor, who dislike the side-effects (headache and a green tinge to vision the next day) or those in whom it is contraindicated, are:
Apomorphine 2 or 3 mg sublingually 20 min before sexual activity
Alprostadil (prostaglandin E1 preparation) given as a small pellet inserted with a device into the urethra (125 µg initially with a maximum of 500 µg). If the partner is pregnant, barrier contraception must be used to keep prostaglandin away from the fetus
Intracavernosal injection or insertion of a pellet of alprostadil urethra into the urethra (2.5 µg initially with a maximum of 40 µg). Side-effects include priapism which needs urgent treatment should erection last more than 3 hours
The diabetic foot
A total of 10–15% of diabetic patients develop foot ulcers at some stage in their lives. Diabetic foot problems are responsible for nearly 50% of all diabetes-related hospital admissions. Many diabetic limb amputations could be delayed or prevented by more effective patient education and medical supervision. Ischaemia, infection and neuropathy combine to produce tissue necrosis. Although all these factors may co-exist, the ischaemic and the neuropathic foot (Table 20.12) can be distinguished. In rural India, foot ulcers are commonly due to neuropathic and infective causes rather than vascular causes.
Table 20.12 Distinguishing features between ischaemia and neuropathy in the diabetic foot
| Ischaemia | Neuropathy | |
|---|---|---|
Symptoms |
Claudication |
Usually painless |
Rest pain |
Sometimes painful neuropathy |
|
Inspection |
Dependent rubor |
High arch |
Trophic changes |
Clawing of toes |
|
|
No trophic changes |
|
Palpation |
Cold |
Warm |
Pulseless |
Bounding pulses |
|
Ulceration |
Painful |
Painless |
Heels and toes |
Plantar |
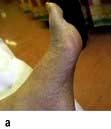
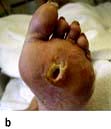

Diabetic foot. (a) High arch and clawing of toes. (b) Typical neuropathic plantar ulceration. (c) Vascular pattern of ulceration.
Management
Many diabetic foot problems are avoidable, so patients need to learn the principles of foot care (Table 20.13). Older patients should visit a chiropodist/podiatrist regularly and should not cut their own toenails. Once tissue damage has occurred in the form of ulceration or gangrene, the aim is preservation of viable tissue. The four main threats to the skin and subcutaneous tissues are:
Infection. This can take hold rapidly in a diabetic foot. Early antibiotic treatment is essential, with antibiotic therapy adjusted in the light of culture results. The organisms grown from the skin surface may not be the organism causing deeper infection. Collections of pus are drained and excision of infected bone is needed if osteomyelitis develops and does not respond to appropriate antibiotic therapy. Regular X-rays of the foot are needed to check on progress.
Ischaemia. The blood flow to the feet is assessed clinically and with Doppler ultrasound. Femoral angiography is used to localize areas of occlusion amenable to bypass surgery or angioplasty. Relatively few patients fall into this category.
Abnormal pressure. An ulcerated site must be kept non-weight-bearing. Resting the affected leg may need to be supplemented with special deep shoes and insoles to move pressure away from critical sites, or by removable or non-removable casts of the leg. After healing, special shoes and insoles are likely to continue to be needed to protect the feet and prevent abnormal pressure repeating damage to a healed area. In neuropathic feet particularly, sharp surgical debridement by a chiropodist is necessary to prevent callus distorting the local wound architecture and causing damage through abnormal pressure on normal skin nearby.
Wound environment. Dressings are used to absorb or remove exudate, maintain moisture, and protect the wound from contaminating agents, and should be easily removable. Expensive new dressings containing growth factors and other biologically active agents may have a role to play in future, but their place is still being assessed.
Table 20.13 Principles of diabetic foot care
Good liaison between physician, chiropodist/podiatrist and surgeon is essential if periods in hospital are to be used efficiently. When irreversible arterial insufficiency is present, it is often quicker and kinder to opt for an early major amputation rather than subject the patient to a debilitating sequence of conservative procedures.
Infections
There is no evidence that diabetic patients with good glycaemic control are more prone to infection than normal subjects. However, poorly-controlled diabetes entails increased susceptibility to the following infections:
One reason why poor control leads to infection is that chemotaxis and phagocytosis by polymorphonuclear leucocytes are impaired because at high blood glucose concentrations neutrophil superoxide generation is impaired.
Conversely, infections may lead to loss of glycaemic control, and are a common cause of ketoacidosis. Insulin-treated patients need to increase their dose by up to 25% in the face of infection, and non-insulin-treated patients may need insulin cover while the infection lasts. Patients should be told never to omit their insulin dose, even if they are nauseated and unable to eat; instead they should test their blood glucose frequently and seek urgent medical advice. Diabetic patients should receive pneumococcal vaccine and yearly influenza vaccine.
Diabetes and cancer
Certain types of cancer are more common in type 2 diabetes. The risk of carcinoma of the uterus and of the pancreas is approximately doubled, and there is a 20–50% increase in the risk of colorectal and breast cancer. These associations appear to be mediated by obesity, which confers similar levels of risk in the absence of hyperglycaemia, although there is also an element of reverse causation with carcinoma of the pancreas, which can precipitate or cause diabetes. Metformin-treated patients have been reported to have a lower cancer risk than those on other therapies, and this agent is under investigation for possible anti-tumour properties.
Skin and joints
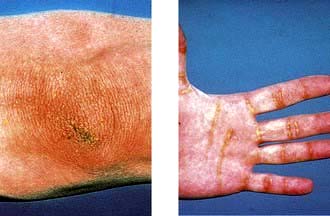
Joint contractures in the hands are a common consequence of childhood diabetes. The sign may be demonstrated by asking the patient to join the hands as if in prayer; the metacarpophalangeal and interphalangeal joints cannot be opposed. Thickened, waxy skin can be noted on the backs of the fingers. These features may be due to glycosylation of collagen and are not progressive (see also p. 1220). The condition is sometimes referred to as diabetic cheiroarthropathy.
Osteopenia in the extremities is also described in type 1 diabetes but rarely leads to clinical consequences.
FURTHER READING
The ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
Camilleri M. Diabetic gastroparesis. N Engl J Med 2007; 356:820–829.
The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Prolonged effect of intensive therapy on the risk of retinopathy complications in patients with type 1 diabetes mellitus. Arch Ophthalmol 2008; 126:1707–1715.
Duckworth W, Abraira C, Moritz T et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–39.
McVary KT. Erectile dsyfunction. N Engl J Med 2007; 357:2473–2481.