Chapter 8 Haematological disease
Introduction and general aspects
Plasma is the liquid component of blood, which contains soluble fibrinogen. Serum is what remains after the formation of the fibrin clot.
The formation of blood cells (haemopoiesis)
The haemopoietic system includes the bone marrow, liver, spleen, lymph nodes and thymus. There is huge turnover of cells with the red cells surviving 120 days, platelets around 7 days but granulocytes only 7 hours. The production of as many as 1013 new myeloid cells (all blood cells except for lymphocytes) per day in the normal healthy state requires tight regulation according to the needs of the body.
Blood islands are formed in the yolk sac in the 3rd week of gestation and produce primitive blood cells, which migrate to the liver and spleen. These organs are the chief sites of haemopoiesis from 6 weeks to 7 months, when the bone marrow becomes the main source of blood cells. However, in childhood and adult life, the bone marrow is the only source of blood cells in a normal person.
At birth, haemopoiesis is present in the marrow of nearly every bone. As the child grows, the active red marrow is gradually replaced by fat (yellow marrow) so that haemopoiesis in the adult becomes confined to the central skeleton and the proximal ends of the long bones. Only if the demand for blood cells increases and persists do the areas of red marrow extend. Pathological processes interfering with normal haemopoiesis may result in resumption of haemopoietic activity in the liver and spleen, which is referred to as extramedullary haemopoiesis.
All blood cells are derived from pluripotent stem cells. These stem cells are supported by stromal cells (see below), which also influence haemopoiesis. The stem cell has two properties – the first is self-renewal, i.e. the production of more stem cells, and the second is its proliferation and differentiation into progenitor cells, committed to one specific cell line.
There are two major ancestral cell lines derived from the pluripotential stem cell: lymphocytic and myeloid (non-lymphocytic) cells (Fig. 8.1). The former gives rise to T and B cells. The myeloid stem cell gives rise to the progenitor CFU-GEMM (colony-forming unit, granulocyte–erythrocyte–monocyte–megakaryocyte). The CFU-GEMM can go on to form CFU-GM, CFU-Eo, and CFU-Meg, each of which can produce a particular cell type (i.e. neutrophils, eosinophils and platelets) under appropriate growth conditions. The progenitor cells such as CFU-GEMM cannot be recognized in bone marrow biopsies but are recognized by their ability to form colonies when haemopoietic cells are immobilized in a soft gel matrix. Haemopoiesis is under the control of growth factors and inhibitors, and the microenvironment of the bone marrow also plays a role in its regulation.
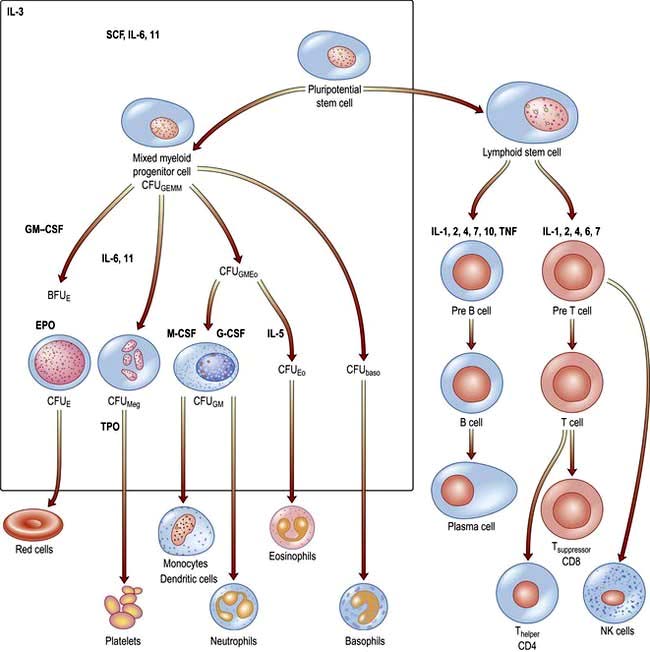
Figure 8.1 Role of growth factors in normal haemopoiesis. Some of the multiple growth factors acting on stem cells and early progenitor cells are shown. baso, basophil; BFU, burst-forming unit; CFU, colony-forming unit; CSF, colony-stimulating factor; E, erythroid; Eo, eosinophil; EPO, erythropoietin; G, granulocyte; GEMM, mixed granulocyte, erythroid, monocyte, megakaryocyte; GM, granulocyte, monocyte; IL, interleukin; M, monocyte; Meg, megakaryocyte; SCF, stem cell (Steel) factor or C-kit ligand; TNF, tumour necrosis factor; TPO, thrombopoietin.
Haemopoietic growth factors
Haemopoietic growth factors are glycoproteins, which regulate the differentiation and proliferation of haemopoietic progenitor cells and the function of mature blood cells. They act on the cytokine-receptor superfamily expressed on haemopoietic cells at various stages of development to maintain the haemopoietic progenitor cells and to stimulate increased production of one or more cell lines in response to stresses such as blood loss and infection (Fig. 8.1).
These haemopoietic growth factors including erythropoietin, interleukin 3 (IL-3), IL-6, -7, -11, -12, β-catenin, stem cell factor (SCF, Steel factor or C-kit ligand) and Fms-tyrosine kinase 3 (Flt3) act via their specific receptor on cell surfaces to stimulate the cytoplasmic janus kinase (JAK) (see p. 25). This major signal transducer activates tyrosine kinase causing gene activation in the cell nucleus. Colony-stimulating factors (CSFs, the prefix indicating the cell type, see Fig. 8.1), as well as interleukins and erythropoietin (EPO) regulate the lineage committed progenitor cells.
Thrombopoietin (TPO, which, like erythropoietin, is produced in the kidneys and the liver) controls platelet production, along with IL-6 and IL-11. In addition to these factors stimulating haemopoiesis, other factors inhibit the process and include tumour necrosis factor (TNF) and transforming growth factor-β (TGF-β). Many of the growth factors are produced by activated T cells, monocytes and bone marrow stromal cells such as fibroblasts, endothelial cells and macrophages; these cells are also involved in inflammatory responses. Bone marrow stem cells can differentiate into other organ cell types, e.g. heart, liver, nerves, bone and this is called stem cell plasticity.
Uses in treatment
Many growth factors have been produced by recombinant DNA techniques and are being used clinically. Examples include granulocyte-colony-stimulating factor (G-CSF), which is used to accelerate haemopoietic recovery after chemotherapy and haemopoietic cell transplantation, and erythropoietin, which is used to treat anaemia in patients with chronic kidney disease. Thrombopoietin receptor agonists are being used to treat patients with immune thrombocytopenic purpura.
Stem cell diseases
The clonal proliferation of bone marrow stem cells leads to diseases including leukaemia (see p. 451), polycythaemia vera (see p. 402), myelofibrosis (see p. 404), paroxysmal nocturnal haemoglobinuria (see p. 401). Failure of stem cell growth leads to aplastic anaemia (see p. 385).
Peripheral blood
Automated cell counters are used to measure the haemoglobin concentration (Hb) and the number and size of red cells, white cells and platelets (Table 8.1). Other indices can be derived from these values. A carefully evaluated blood film is still an essential adjunct to the above, as definitive abnormalities of cells can be seen.
 The mean corpuscular volume (MCV) of red cells is a useful index and is used to classify anaemia (see p. 376).
The mean corpuscular volume (MCV) of red cells is a useful index and is used to classify anaemia (see p. 376).
The red cell distribution width (RDW) is calculated by dividing the standard deviation of the red cell width by the mean cell width × 100. An elevated RDW suggests variation in red cell size, i.e. anisocytosis, and this is seen in iron deficiency. In β-thalassaemia trait, the RDW is usually normal.
The white cell count (WCC), (or WBC, white blood count) gives the total number of circulating leucocytes, and many automated cell counters produce differential counts as well.
Reticulocytes are young red cells and usually comprise <2% of the red cells. The reticulocyte count gives a guide to the erythroid activity in the bone marrow. An increased count is seen with increased marrow maturity, e.g. following haemorrhage or haemolysis, and during the response to treatment with a specific haematinic. A low count in the presence of anaemia indicates an inappropriate response by the bone marrow and may be seen in bone marrow failure (from whatever cause) or where there is a deficiency of a haematinic.
Erythrocyte sedimentation rate (ESR) is the rate of fall of red cells in a column of blood and is a measure of the acute-phase response. The pathological process may be immunological, infective, ischaemic, malignant or traumatic. A raised ESR reflects an increase in the plasma concentration of large proteins, such as fibrinogen and immunoglobulins. These proteins cause rouleaux formation, with red cells clumping together and therefore falling more rapidly. The ESR increases with age, and is higher in females than in males.
Plasma viscosity is a measurement used instead of the ESR in some laboratories. It is also dependent on the concentration of large molecules such as fibrinogen and immunoglobulins. It is not affected by the level of Hb.
C-reactive protein (CRP) is a pentraxin, one of the proteins produced in the acute-phase response. It is synthesized exclusively in the liver and rises within 6 hours of an acute event. The CRP level rises with fever (possibly triggered by IL-1, IL-6 and TNF-α and other cytokines), in inflammatory conditions and after trauma. It follows the clinical state of the patient much more rapidly than the ESR and is unaffected by the level of Hb, but it is less helpful than the ESR or plasma viscosity in monitoring chronic inflammatory diseases. The measurement of CRP is easy and quick to perform using an immunoassay that can be automated. High-sensitivity assays have shown that increased levels may predict future cardiovascular disease (see p. 728).
Table 8.1 Normal values for peripheral blood
| Male | Female | |
|---|---|---|
Hb (g/L) |
135–175 |
115–160 |
PCV (haematocrit; L/L) |
0.4–0.54 |
0.37–0.47 |
RCC (1012/L) |
4.5–6.0 |
3.9–5.0 |
MCV (fL) |
80–96 |
|
MCH (pg) |
27–32 |
|
MCHC (g/L) |
320–360 |
|
RDW (%) |
11–15 |
|
WBC (109/L) |
4.0–11.0 |
|
Platelets (109/L) |
150–400 |
|
ESR (mm/h) |
<20 |
|
Reticulocytes |
0.5–2.5% (50–100 × 109/L) |
|
ESR, erythrocyte sedimentation rate; Hb, haemoglobin; MCH, mean corpuscular haemoglobin; MCHC, mean corpuscular haemoglobin concentration; MCV, mean corpuscular volume of red cells; PCV, packed cell volume; RCC, red cell count; RDW, red blood cell distribution width; WBC, white blood count.
The red cell
Erythropoiesis
Red cell precursors pass through several stages in the bone marrow. The earliest morphologically recognizable cells are pronormoblasts. Smaller normoblasts result from cell divisions, and precursors at each stage progressively contain less RNA and more Hb in the cytoplasm. The nucleus becomes more condensed and is eventually lost from the late normoblast in the bone marrow when the cell becomes a reticulocyte.
Reticulocytes contain residual ribosomal RNA and are still able to synthesize Hb. They remain in the marrow for about 1–2 days and are released into the circulation, where they lose their RNA and become mature red cells (erythrocytes) after another 1–2 days. Mature red cells are non-nucleated biconcave discs.
Nucleated red cells (normoblasts) are not normally present in peripheral blood, but are present if there is extramedullary haemopoiesis and in some marrow disorders (see leucoeryothroblastic anaemia, p. 413).
About 10% of erythroblasts die in the bone marrow even during normal erythropoiesis. Such ineffective erythropoiesis is substantially increased in some anaemias such as thalassaemia major and megaloblastic anaemia.
Erythropoietin is a hormone which controls erythropoiesis. The gene for erythropoietin is on chromosome 7 and codes for a heavily glycosylated polypeptide of 165 amino acids. Erythropoietin has a molecular weight of 30 400 and is produced in the peritubular cells in the kidneys (90%) and in the liver (10%). Its production is regulated mainly by tissue oxygen tension. Production is increased if there is hypoxia from whatever cause, e.g. anaemia or cardiac or pulmonary disease. The erythropoietin gene is one of a number of genes that is regulated by the hypoxic sensor pathway. The 3′-flanking region of the erythropoietin gene has a hypoxic response element, which is necessary for the induction of transcription of the gene in hypoxic cells. Hypoxia-inducible factor 1 (HIF-1) is a transcription factor, which binds to the hypoxia response element and acts as a master regulator of several genes that are responsive to hypoxia. Erythropoietin stimulates an increase in the proportion of bone marrow precursor cells committed to erythropoiesis, and CFU-E are stimulated to proliferate and differentiate. Increased ‘inappropriate’ production of erythropoietin occurs in certain tumours such as renal cell carcinoma and other causes (see Table 8.15).
Haemoglobin synthesis
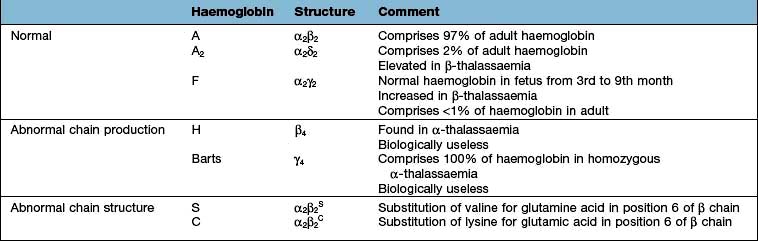
Haemoglobin performs the main functions of red cells – carrying O2 to the tissues and returning CO2 from the tissues to the lungs. Each normal adult Hb molecule (HbA) has a molecular weight of 68 000 and consists of two α and two β globin polypeptide chains (α2β2). HbA comprises about 97% of the Hb in adults. Two other haemoglobin types, HbA2 (α2δ2) and HbF (α2γ2), are found in adults in small amounts (1.5–3.2% and <1%, respectively) (see p. 390).
Haemoglobin synthesis occurs in the mitochondria of the developing red cell (Fig. 8.2). The major rate-limiting step is the conversion of glycine and succinic acid to δ-aminolaevulinic acid (ALA) by ALA synthase. Vitamin B6 is a coenzyme for this reaction, which is inhibited by haem and stimulated by erythropoietin. Two molecules of δ-ALA condense to form a pyrrole ring (porphobilinogen). These rings are then grouped in fours to produce protoporphyrins and with the addition of iron haem is formed. Haem is then inserted into the globin chains to form a haemoglobin molecule. The structure of Hb is shown in Figure 8.3.
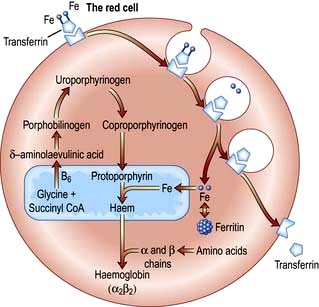
Figure 8.2 Haemoglobin synthesis. Transferrin attaches to a surface receptor on developing red cells. Iron is released and transported to the mitochondria, where it combines with protoporphyrin to form haem. Protoporphyrin itself is manufactured from glycine and succinyl-CoA. Haem combines with α and β chains (formed on ribosomes) to make haemoglobin.
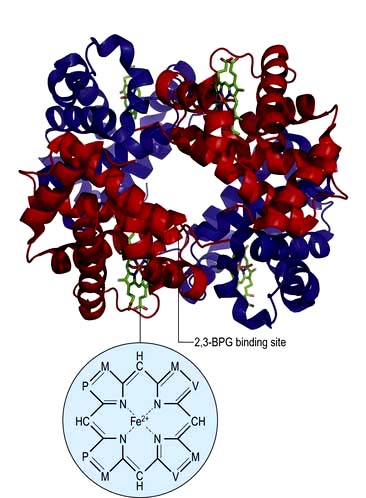
Figure 8.3 Model of the haemoglobin molecule showing α (red) and β (blue) chains. 2,3-BPG (bisphosphoglycerate) binds in the centre of the molecule and stabilizes the deoxygenated form by cross-linking the β chains (also see Fig. 8.4). M, methyl; P, propionic acid; V, vinyl.
Haemoglobin function
The biconcave shape of red cells provides a large surface area for the uptake and release of oxygen and carbon dioxide. Haemoglobin becomes saturated with oxygen in the pulmonary capillaries where the partial pressure of oxygen is high and Hb has a high affinity for oxygen. Oxygen is released in the tissues where the partial pressure of oxygen is low and Hb has a low affinity for oxygen.
In adult haemoglobin (Hb), a haem group is bound to each of the four globin chains; the haem group has a porphyrin ring with a ferrous atom which can reversibly bind one oxygen molecule. The haemoglobin molecule exists in two conformations, R and T. The T (taut) conformation of deoxyhaemoglobin is characterized by the globin units being held tightly together by electrostatic bonds (Fig. 8.4). These bonds are broken when oxygen binds to haemoglobin, resulting in the R (relaxed) conformation in which the remaining oxygen binding sites are more exposed and have a much higher affinity for oxygen than in the T conformation. The binding of one oxygen molecule to deoxyhaemoglobin increases the oxygen affinity of the remaining binding sites – this property is known as ‘cooperativity’ and is the reason for the sigmoid shape of the oxygen dissociation curve. Haemoglobin is, therefore, an example of an allosteric protein. The binding of oxygen can be influenced by secondary effectors – hydrogen ions, carbon dioxide and red-cell 2,3-bisphosphoglycerate (2,3-BPG). Hydrogen ions and carbon dioxide added to blood cause a reduction in the oxygen-binding affinity of haemoglobin (the Bohr effect). Oxygenation of haemoglobin reduces its affinity for carbon dioxide (the Haldane effect). These effects help the exchange of carbon dioxide and oxygen in the tissues.
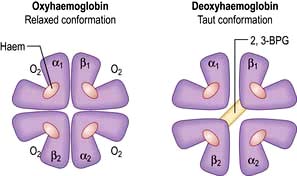
Figure 8.4 Oxygenated and deoxygenated haemoglobin molecule. The haemoglobin molecule is predominantly stabilized by α-β chain bonds rather than α-α and β-β chain bonds. The structure of the molecule changes during O2 uptake and release. When O2 is released, the β chains rotate on the α1β2 and α2β1 contacts, allowing the entry of 2,3-BPG which causes a lower affinity of haemoglobin for O2 and improved delivery of O2 to the tissues.
Red cell metabolism produces 2,3-BPG from glycolysis. 2,3-BPG accumulates because it is sequestered by binding to deoxyhaemoglobin. The binding of 2,3-BPG stabilizes the T conformation and reduces its affinity for oxygen. The P50 is the partial pressure of oxygen at which the haemoglobin is 50% saturated with oxygen. P50 increases with 2,3-BPG concentrations, which increase when oxygen availability is reduced in conditions such as hypoxia or anaemia. P50 also rises with increasing body temperature, which may be beneficial during prolonged exercise. Haemoglobin regulates oxygen transport as shown in the oxyhaemoglobin dissociation curve. When the primary limitation to oxygen transport is in the periphery, e.g. heavy exercise, anaemia, the P50 is increased to enhance oxygen unloading. When the primary limitation is in the lungs, e.g. lung disease, high altitude exposure, the P50 is reduced to enhance oxygen loading.
A summary of normal red cell production and destruction is given in Figure 8.5.
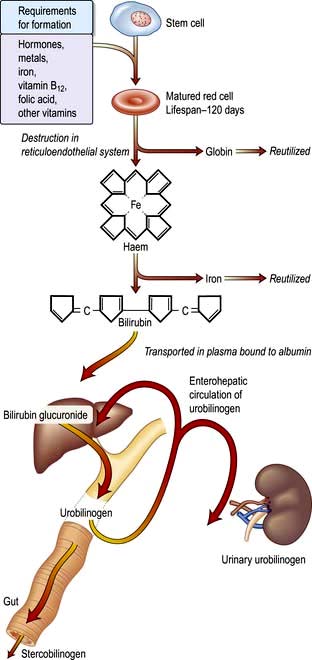
Anaemia
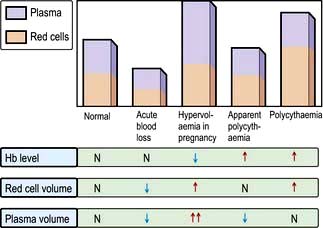
Anaemia is present when there is a decrease in Hb in the blood below the reference level for the age and sex of the individual (Table 8.1). Alterations in the Hb may occur as a result of changes in the plasma volume, as shown in Figure 8.6. A reduction in the plasma volume will lead to a spuriously high Hb – this is seen with dehydration and in the clinical condition of apparent polycythaemia (see p. 404). A raised plasma volume produces a spurious anaemia, even when combined with a small increase in red cell volume as occurs in pregnancy.
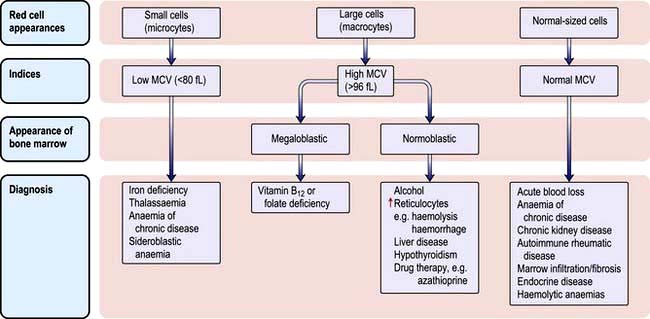
The various types of anaemia, classified by MCV, are shown in Figure 8.7. There are three major types:
Clinical features
Patients with anaemia may be asymptomatic. A slowly falling level of Hb allows for haemodynamic compensation and enhancement of the oxygen-carrying capacity of the blood. A rise in 2,3-BPG causes a shift of the oxygen dissociation curve to the right, so that oxygen is more readily given up to the tissues. Where blood loss is rapid, more severe symptoms will occur, particularly in elderly people.
Symptoms (all nonspecific)
Anaemia exacerbates cardiorespiratory problems especially in the elderly. For example, angina or intermittent claudication may be precipitated by anaemia. A good way to assess the effects of anaemia is to ask about breathlessness in relation to different levels of exercise (e.g. walking on the flat or climbing one flight of stairs).
Signs
Specific signs seen in the different types of anaemia will be discussed in the appropriate sections. Examples include:
Koilonychia – spoon-shaped nails seen in longstanding iron deficiency anaemia
Jaundice – found in haemolytic anaemia
It must be emphasized that anaemia is not a final diagnosis, and a cause should be sought.
Investigations
Peripheral blood
A low Hb should always be evaluated with:
The white blood cell (WBC) count
The reticulocyte count (as this indicates marrow activity)
The blood film, as abnormal red cell morphology (see Fig. 8.9) may indicate the diagnosis. Where two populations of red cells are seen, the blood film is said to be dimorphic. This may, for example, be seen in patients with ‘double deficiencies’ (e.g. combined iron and folate deficiency in coeliac disease, or following treatment of anaemic patients with the appropriate haematinic).
Bone marrow
Techniques for obtaining bone marrow are shown in Practical Box 8.1.
 Practical Box 8.1
Practical Box 8.1
Techniques for obtaining bone marrow
The technique should be explained to the patient and consent obtained.
Examination of the bone marrow is performed to further investigate abnormalities found in the peripheral blood (Practical Box 8.1). Aspiration provides a film which can be examined by microscopy for the morphology of the developing haemopoietic cells. The trephine provides a core of bone which is processed as a histological specimen and allows an overall view of the bone marrow architecture, cellularity and presence/absence of abnormal infiltrates.
Type of erythropoiesis (e.g. normoblastic or megaloblastic)
Cellularity of the various cell lines
Infiltration of the marrow, i.e. presence of non-haematopoietic cells such as cancer cells
Special tests may be performed for further diagnosis: cytogenetic, immunological, cytochemical markers, biochemical analyses and microbiological culture.
Microcytic anaemia
Iron deficiency is the most common cause of anaemia in the world, affecting 30% of the world’s population. This is because of the body’s limited ability to absorb iron and the frequent loss of iron owing to haemorrhage. Although iron is abundant, most is in the insoluble ferric (Fe3+) form, which has poor bioavailability. Ferrous (Fe2+) is more readily absorbed.
The other causes of a microcytic hypochromic anaemia are anaemia of chronic disease, sideroblastic anaemia and thalassaemia. In thalassaemia (see p. 390), there is a defect in globin synthesis, in contrast to the other three causes of microcytic anaemia where the defect is in the synthesis of haem.
Iron
Dietary intake
The average daily diet in the UK contains 15–20 mg of iron, although normally only 10% of this is absorbed. Absorption may be increased to 20–30% in iron deficiency and pregnancy.
Non-haem iron is mainly derived from cereals, which are commonly fortified with iron; it forms the main part of dietary iron. Haem iron is derived from haemoglobin and myoglobin in red or organ meats. Haem iron is better absorbed than non-haem iron, whose availability is more affected by other dietary constituents.
Absorption
Factors influencing iron and haem iron absorption (Fig. 8.8) are shown in Table 8.2.
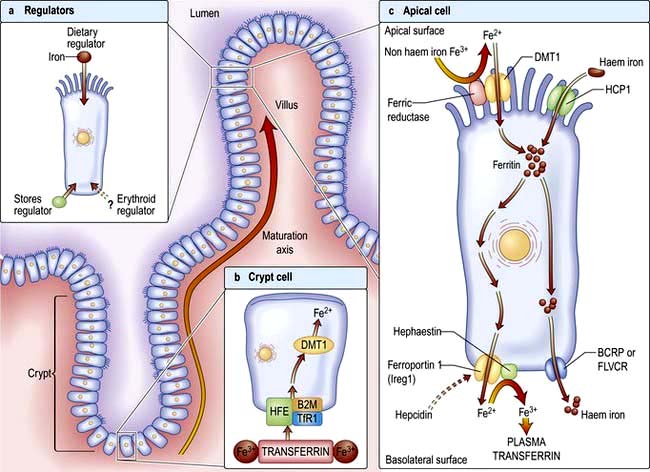
Figure 8.8 (a) Regulation of the absorption of intestinal iron. The iron-absorbing cells of the duodenal epithelium originate in the intestinal crypts and migrate toward the tip of the villus as they differentiate (maturation axis). Absorption of intestinal iron is regulated by at least three independent mechanisms, although the protein hepcidin is key. First, iron absorption is influenced by recent dietary iron intake (dietary regulator). After a large dietary bolus, absorptive cells are resistant to iron uptake for several days. Second, iron absorption can be modulated considerably in response to body iron stores (stores regulator). Third, a signal communicates the state of bone marrow erythropoiesis to the intestine (erythroid regulator). (b) Duodenal crypt cells sense body iron status through the binding of transferrin to the HFE/B2M/TfR1 gene complex. Cytosolic enzymes change the oxidative state of iron from ferric (Fe3+) to ferrous (Fe2+). A decrease in crypt cell iron concentration upregulates the divalent metal transporter (DMT1). This increases as crypt cells migrate up the villus and become mature absorptive cells. (c) Apical cell. Dietary iron is reduced from the ferric to the ferrous state by the brush border ferrireductase. DMT1 facilitates iron absorption from the intestinal lumen. The export proteins, e.g. ferroportin 1 and hephaestin, transfer iron from the enterocyte into the circulation depending on the hepcidin level. A second pathway absorbs intact haem iron into the circulation via BRCP and FLVCR. BCRP, breast cancer resistant protein; B2M, β2-microglobulin; FLVCR, feline leukaemia virus subgroup C; HCP1, haem carrier protein-1; HFE, hereditary haemochromatosis gene; TfR1, transferrin receptor.
Table 8.2 Factors influencing iron absorption
Dietary haem iron is more rapidly absorbed than non-haem iron derived from vegetables and grain. Most haem is absorbed in the proximal intestine, with absorptive capacity decreasing distally. The intestinal haem transporter HCP1 (haem carrier protein 1) has been identified and found to be highly expressed in the duodenum. It is upregulated by hypoxia and iron deficiency. Some haem iron may be reabsorbed intact into circulation via the cell by two exporter proteins – BCRP (breast cancer resistant protein) and FLVCR (feline leukaemia virus subgroup C) (Fig. 8.8).
Non-haem iron absorption occurs primarily in the duodenum. Non-haem iron is dissolved in the low pH of the stomach and reduced from the ferric to the ferrous form by a brush border ferrireductase. Cells in duodenal crypts are able to sense the body’s iron requirements and retain this information as they mature into cells capable of absorbing iron at the tips of the villi. A protein, divalent metal transporter 1 (DMT1) or natural resistance-associated macrophage protein (NRAMP2), transports iron (and other metals) across the apical (luminal) surface of the mucosal cells in the small intestine.
Once inside the mucosal cell, iron may be transferred across the cell to reach the plasma, or be stored as ferritin; the body’s iron status at the time the absorptive cell developed from the crypt cell is probably the crucial deciding factor. Iron stored as ferritin will be lost into the gut lumen when the mucosal cells are shed; this regulates iron balance. The mechanism of transport of iron across the basolateral surface of mucosal cells involves a transporter protein, ferroportin 1 (FPN 1) through its iron-responsive element (IRE). This transporter protein requires an accessory, multicopper protein, hephaestin (Fig. 8.8).
The body iron content is closely regulated by the control of iron absorption but there is no physiological mechanism for eliminating excess iron from the body. The key molecule regulating iron absorption is hepcidin, a 25 amino acid peptide synthesized in the liver. Hepcidin acts by regulating the activity of the iron exporting protein ferroportin by binding to ferroportin causing its internalization and degradation, thereby decreasing iron efflux from iron exporting tissues into plasma. Therefore, high levels of hepcidin (occurring in inflammation states) via inflammatory cytokines, e.g. IL-6 will destroy ferroportin and limit iron absorption, and low levels of hepcidin (e.g. in anaemia, low iron stores, hypoxia) will encourage iron absorption. For example, in patients with haemochromatosis, mutations in the genes HFE, HJV and TfR2 will interrupt hepcidin synthesis. Therefore, in the intestinal cells, a deficiency of hepcidin would lead to less ferroportin being bound and thus more iron will be released into the plasma.
A longstanding mystery is why anaemias characterized by ineffective erythropoiesis such as thalassaemia are associated with excessive and inappropriate iron absorption. Preliminary evidence again suggests that the increased iron absorption in β-thalassaemia is mediated by downregulation of hepcidin and upregulation of ferroportin.
Transport in the blood
The normal serum iron level is about 13–32 µmol/L; there is a diurnal rhythm with higher levels in the morning. Iron is transported in the plasma bound to transferrin, a β-globulin that is synthesized in the liver. Each transferrin molecule binds two atoms of ferric iron and is normally one-third saturated. Most of the iron bound to transferrin comes from macrophages in the reticuloendothelial system and not from iron absorbed by the intestine. Transferrin-bound iron becomes attached by specific receptors to erythroblasts and reticulocytes in the marrow and the iron is removed (Fig. 8.2).
In an average adult male, 20 mg of iron, chiefly obtained from red cell breakdown in the macrophages of the reticuloendothelial system, is incorporated into Hb every day.
Iron stores
About two-thirds of the total body iron is in the circulation as haemoglobin (2.5–3 g in a normal adult man). Iron is stored in reticuloendothelial cells, hepatocytes and skeletal muscle cells (500–1500 mg). About two-thirds of this is stored as ferritin and one-third as haemosiderin in normal individuals. Small amounts of iron are also found in plasma (about 4 mg bound to transferrin), with some in myoglobin and enzymes.
Ferritin is a water-soluble complex of iron and protein. It is more easily mobilized than haemosiderin for Hb formation. It is present in small amounts in plasma.
Haemosiderin is an insoluble iron–protein complex found in macrophages in the bone marrow, liver and spleen. Unlike ferritin, it is visible by light microscopy in tissue sections and bone marrow films after staining by Perls’ reaction.
Requirements
Each day 0.5–1.0 mg of iron is lost in the faeces, urine and sweat. Menstruating women lose 30–40 mL of blood/month, an average of about 0.5–0.7 mg of iron/day. Blood loss through menstruation in excess of 100 mL will usually result in iron deficiency as increased iron absorption from the gut cannot compensate for such losses of iron. The demand for iron also increases during growth (about 0.6 mg/day) and pregnancy (1–2 mg/day). In the normal adult the iron content of the body remains relatively fixed. Increases in the body iron content (haemochromatosis) are classified into:
Iron deficiency
Iron deficiency anaemia develops when there is inadequate iron for haemoglobin synthesis. The causes are:
Most iron deficiency is due to blood loss, usually from the uterus or gastrointestinal tract. Premenopausal women are in a state of precarious iron balance owing to menstruation. A common cause of iron deficiency worldwide is blood loss from the gastrointestinal tract resulting from parasites such as hookworm infestation. The poor quality of the diet, predominantly containing vegetables, also contributes to the high prevalence of iron deficiency in developing countries. Even in developed countries, iron deficiency is not uncommon in infancy where iron intake is insufficient for the demands of growth. It is more prevalent in infants born prematurely or where the introduction of mixed feeding is delayed.
Clinical features
The symptoms of anaemia are described on page 375. The well known clinical features of iron deficiency listed below are generally only seen in cases of very longstanding iron deficiency:
Spoon-shaped nails (koilonychia)
Atrophy of the papillae of the tongue
A syndrome of dysphagia and glossitis (Plummer–Vinson or Paterson–Brown–Kelly syndrome; see p. 243).
The diagnosis of iron deficiency anaemia relies on a clinical history which should include questions about dietary intake, self-medication with non-steroidal anti-inflammatory drugs (which may give rise to gastrointestinal bleeding), and the presence of blood in the faeces (which may be a sign of haemorrhoids or carcinoma of the lower bowel). In women, a careful enquiry about the duration of periods, the occurrence of clots and the number of sanitary towels or tampons (normal 3–5/day) used should be made.
Investigations
Blood count and film. A characteristic blood film is shown in Figure 8.9. The red cells are microcytic (MCV <80 fL) and hypochromic (MCH (mean corpuscular haemoglobin) <27 pg). There is poikilocytosis (variation in shape) and anisocytosis (variation in size). Target cells are seen.
Serum iron and iron-binding capacity. The serum iron falls and the total iron-binding capacity (TIBC) rises in iron deficiency compared with normal. Iron deficiency is regularly present when the transferrin saturation (i.e. serum iron divided by TIBC) falls below 19% (Table 8.3).
Serum ferritin. The level of serum ferritin reflects the amount of stored iron. The normal values for serum ferritin are 30–300 µg/L (11.6–144 nmol/L) in males and 15–200 µg/L (5.8–96 nmol/L) in females. In simple iron deficiency, a low serum ferritin confirms the diagnosis. However, ferritin is an acute-phase reactant, and levels increase in the presence of inflammatory or malignant diseases. Very high levels of ferritin may be observed in hepatitis and in a rare disease, haemophagocytic lymphohistiocytosis (p. 80).
Serum soluble transferrin receptors. The number of transferrin receptors increases in iron deficiency. The results of this immunoassay compare well with results from bone marrow aspiration at estimating iron stores. This assay can help to distinguish between iron deficiency and anaemia of chronic disease (Table 8.3), and may avoid the need for bone marrow examination even in complex cases. It may sometimes be helpful in the investigation of complicated causes of anaemia.
Other investigations. These will be indicated by the clinical history and examination. Investigations of the gastrointestinal tract are often required to determine the cause of the iron deficiency (see p. 257).
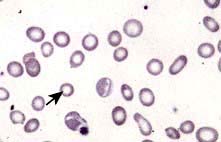
Differential diagnosis
The presence of anaemia with microcytosis and hypochromia does not necessarily indicate iron deficiency. The most common other causes are thalassaemia, sideroblastic anaemia and anaemia of chronic disease, and in these disorders the iron stores are normal or increased. The differential diagnosis of microcytic anaemia is shown in Table 8.3.
Treatment
The correct management of iron deficiency is to find and treat the underlying cause, and to give iron to correct the anaemia and replace iron stores. Patients with iron deficiency taking iron will increase their Hb level by approximately 10 g/L/week unless of course other factors such as bleeding are present.
Oral iron is all that is required in most cases. The best preparation is ferrous sulphate (200 mg three times daily, a total of 180 mg ferrous iron), which is absorbed best when the patient is fasting. If the patient has side-effects such as nausea, diarrhoea or constipation, taking the tablets with food or reducing the dose using a preparation with less iron such as ferrous gluconate (300 mg twice daily, only 70 mg ferrous iron) is all that is usually required to reduce the symptoms.
In developing countries, distribution of iron tablets and fortification of food are the main approaches for the alleviation of iron deficiency. However, iron supplementation programmes have been ineffective, probably mainly because of poor compliance.
Oral iron should be given for long enough to correct the Hb level and to replenish the iron stores; this can take 6 months. The commonest causes of failure to respond to oral iron are:
These possibilities should be considered before parenteral iron is used. However, parenteral iron is required by occasional patients, e.g. intolerant to oral preparation, severe malabsorption, chronic disease (e.g. inflammatory bowel disease). Iron stores are replaced much faster with parenteral iron than with oral iron, but the haematological response is no quicker. Parenteral iron can be given by slow intravenous infusion of low-molecular-weight iron dextran (test dose required), iron sucrose, ferric carboxymaltose, iron isomaltoside 1000; oral iron should be discontinued.
Anaemia of chronic disease
One of the most common types of anaemia, particularly in hospital patients, is the anaemia of chronic disease, occurring in patients with chronic infections such as tuberculosis or chronic inflammatory disease such as Crohn’s disease, rheumatoid arthritis, systemic lupus erythematosus (SLE), polymyalgia rheumatica and malignant disease. There is decreased release of iron from the bone marrow to developing erythroblasts, an inadequate erythropoietin response to the anaemia, and decreased red cell survival.
The exact mechanisms responsible for these effects are not clear but it seems likely that high levels of hepcidin expression play a key role (see iron absorption).
The serum iron and the TIBC are low, and the serum ferritin is normal or raised because of the inflammatory process. The serum soluble transferrin receptor level is normal (Table 8.3). Stainable iron is present in the bone marrow, but iron is not seen in the developing erythroblasts. Patients do not respond to iron therapy, and treatment is, in general, that of the underlying disorder. Recombinant erythropoietin therapy is used in the anaemia of renal disease (see p. 623), and occasionally in inflammatory disease (rheumatoid arthritis, inflammatory bowel disease).
Sideroblastic anaemia
Sideroblastic anaemias are inherited or acquired disorders characterized by a refractory anaemia, a variable number of hypochromic cells in the peripheral blood, and excess iron and ring sideroblasts in the bone marrow. The presence of ring sideroblasts is the diagnostic feature of sideroblastic anaemia. There is accumulation of iron in the mitochondria of erythroblasts owing to disordered haem synthesis forming a ring of iron granules around the nucleus that can be seen with Perls’ reaction. The blood film is often dimorphic; ineffective haem synthesis is responsible for the microcytic hypochromic cells. Sideroblastic anaemias can be inherited as an X-linked disease transmitted by females. Acquired causes include myelodysplasia, myeloproliferative disorders, myeloid leukaemia, drugs (e.g. isoniazid), alcohol misuse and lead toxicity. It can also occur in other disorders such as rheumatoid arthritis, carcinomas, megaloblastic and haemolytic anaemias. A structural defect in δ-aminolaevulinic acid (ALA) synthase, the pyridoxine-dependent enzyme responsible for the first step in haem synthesis (Fig. 8.2), has been identified in one form of inherited sideroblastic anaemia. Primary acquired sideroblastic anaemia is one of the myelodysplastic syndromes (see p. 405) and this is the cause of the vast majority of cases of sideroblastic anaemia in adults. Lead toxicity is described in Chapter 17.
Normocytic anaemia
Normocytic, normochromic anaemia is seen in anaemia of chronic disease, in some endocrine disorders (e.g. hypopituitarism, hypothyroidism and hypoadrenalism) and in some haematological disorders (e.g. aplastic anaemia and some haemolytic anaemias) (Fig. 8.7). In addition, this type of anaemia is seen acutely following blood loss.
Macrocytic anaemias
These can be divided into megaloblastic and non-megaloblastic types, depending on bone marrow findings.
Megaloblastic anaemia
Megaloblastic anaemia is characterized by the presence in the bone marrow of erythroblasts with delayed nuclear maturation because of defective DNA synthesis (megaloblasts). Megaloblasts are large and have large immature nuclei. The nuclear chromatin is more finely dispersed than normal and has an open stippled appearance (Fig. 8.10). In addition, giant metamyelocytes are frequently seen in megaloblastic anaemia. These cells are about twice the size of normal cells and often have twisted nuclei. Megaloblastic changes occur in:
Vitamin B12 deficiency or abnormal vitamin B12 metabolism
Folic acid deficiency or abnormal folate metabolism
Other defects of DNA synthesis, such as congenital enzyme deficiencies in DNA synthesis (e.g. orotic aciduria), or resulting from therapy with drugs interfering with DNA synthesis (e.g. hydroxycarbamide (hydroxyurea), azathioprine, zidovudine – AZT)
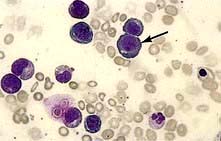
Haematological findings
Anaemia may be present. The MCV is characteristically >96 fL unless there is a co-existing cause of microcytosis when there may be a dimorphic picture with a normal/low average MCV.
The peripheral blood film shows oval macrocytes with hypersegmented polymorphs with six or more lobes in the nucleus (Fig. 8.11).
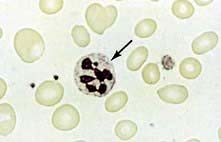
Biochemical basis of megaloblastic anaemia
The key biochemical problem common to both vitamin B12 and folate deficiency is a block in DNA synthesis owing to an inability to methylate deoxyuridine monophosphate to deoxythymidine monophosphate, which is then used to build DNA (Fig. 8.12). The methyl group is supplied by the folate coenzyme, methylene tetrahydrofolate.
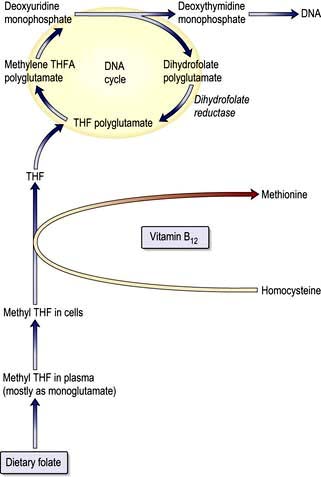
Figure 8.12 Biochemical basis of megaloblastic anaemia. The metabolic relationship between vitamin B12 and folate and their role in DNA synthesis. THF, tetrahydrofolate.
Deficiency of folate reduces the supply of this coenzyme; deficiency of vitamin B12 also reduces its supply by slowing the demethylation of methyltetrahydrofolate (methyl THF) and preventing cells receiving tetrahydrofolate for synthesis of methylene tetrahydrofolate polyglutamate.
Other congenital and acquired forms of megaloblastic anaemia are due to interference with purine or pyrimidine synthesis causing an inhibition in DNA synthesis.
Vitamin B12
Vitamin B12 is synthesized by certain microorganisms, and humans are ultimately dependent on animal sources. It is found in meat, fish, eggs and milk, but not in plants. Vitamin B12 is not usually destroyed by cooking. The average daily diet contains 5–30 µg of vitamin B12, of which 2–3 µg is absorbed. The average adult stores some 2–3 mg, mainly in the liver, and it may take 2 years or more after absorptive failure before B12 deficiency develops, as the daily losses are small (1–2 µg).
Structure and function
Cobalamins consist of a planar group with a central cobalt atom (corrin ring) and a nucleotide set at right-angles (Fig. 8.13). Vitamin B12 was first crystallized as cyanocobalamin, but the main natural cobalamins have deoxyadenosyl-, methyl- and hydroxocobalamin groups attached to the cobalt atom.
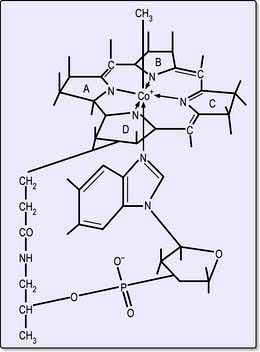
The main function of B12 is the methylation of homocysteine to methionine with the demethylation of methyl THF polyglutamate to THF. THF is a substrate for folate polyglutamate synthesis.
Deoxyadenosylcobalamin is a coenzyme for the conversion of methylmalonyl CoA to succinyl CoA. Measurement of methylmalonic acid in urine was used as a test for vitamin B12 deficiency but it is no longer carried out routinely.
Absorption and transport
Vitamin B12 is liberated from protein complexes in food by gastric enzymes and then binds to a vitamin B12-binding protein (‘R’ binder), which is related to plasma transcobalamin I (TCI) and is derived from saliva. Vitamin B12 is released from the ‘R’ binder by pancreatic enzymes and then becomes bound to intrinsic factor.
Intrinsic factor is a glycoprotein with a molecular weight of 45 000. It is secreted by gastric parietal cells along with H+ ions. It combines with vitamin B12 and carries it to specific receptors on the surface of the mucosa of the ileum. Vitamin B12 enters the ileal cells and intrinsic factor remains in the lumen and is excreted. Vitamin B12 is transported from the enterocytes to the bone marrow and other tissues by the glycoprotein transcobalamin II (TCII). Although TCII is the essential carrier protein for vitamin B12, the amount of B12 on TCII is low. However, it has a rapid clearance and is able to deliver cobalamin to all cells of the body. Vitamin B12 in plasma is mainly bound to TCI (70–90%), but the functional role of this protein is unknown. About 1% of an oral dose of B12 is absorbed ‘passively’ without the need for intrinsic factor.
Vitamin B12 deficiency
There are a number of causes of B12 deficiency and abnormal B12 metabolism (Table 8.4). The most common cause of vitamin B12 deficiency in adults is pernicious anaemia. Malabsorption of vitamin B12 because of pancreatitis, coeliac disease or treatment with metformin is mild and does not usually result in significant vitamin B12 deficiency.
Table 8.4 Vitamin B12 deficiency and abnormal B12 utilization: further causes (see text)
| Low dietary intake | Abnormal utilization |
|---|---|
Vegans |
Congenital transcobalamin II deficiency |
|
Nitrous oxide (inactivates B12) |
Impaired absorption |
|
Stomach |
|
Pernicious anaemia |
|
Gastrectomy |
|
Congenital deficiency of intrinsic factor |
|
Small bowel |
|
Ileal disease or resection |
|
Bacterial overgrowth |
|
Tropical sprue |
|
Fish tapeworm (Diphyllobothrium latum) |
|
Pernicious anaemia
Pernicious anaemia (PA) is an autoimmune disorder in which there is atrophic gastritis with loss of parietal cells in the gastric mucosa with consequent failure of intrinsic factor production and vitamin B12 malabsorption.
Pathogenesis of pernicious anaemia
This disease is common in the elderly, with 1 in 8000 of the population aged over 60 years being affected in the UK. It can be seen in all races, but occurs more frequently in fair-haired and blue-eyed individuals, and those who have the blood group A. It is more common in females than males.
There is an association with other autoimmune diseases, particularly thyroid disease, Addison’s disease and vitiligo. Approximately one-half of all patients with PA have thyroid antibodies. There is a higher incidence of gastric carcinoma with PA (1–3%) than in the general population.
Parietal cell antibodies are present in the serum in 90% of patients with PA – and also in many older patients with gastric atrophy. Conversely, intrinsic factor antibodies, although found in only 50% of patients with PA, are specific for this diagnosis. Two types of intrinsic factor antibodies are found: a blocking antibody, which inhibits binding of intrinsic factor to B12, and a precipitating antibody, which inhibits the binding of the B12-intrinsic factor complex to its receptor site in the ileum.
B12 deficiency may rarely occur in children from a congenital deficiency or abnormality of intrinsic factor, or as a result of early onset of the adult autoimmune type.
Pathology
Autoimmune gastritis (see p. 247) affecting the fundus is present with plasma cell and lymphoid infiltration. The parietal and chief cells are replaced by mucin-secreting cells. There is achlorhydria and absent secretion of intrinsic factor. The histological abnormality can be improved by corticosteroid therapy, which supports an autoimmune basis for the disease.
Clinical features
The onset of PA is insidious, with progressively increasing symptoms of anaemia. Patients are sometimes said to have a lemon-yellow colour owing to a combination of pallor and mild jaundice caused by excess breakdown of haemoglobin. A red sore tongue (glossitis) and angular stomatitis are sometimes present.
The neurological changes, if left untreated for a long time, can be irreversible. These neurological abnormalities occur only with very low levels of serum B12 (<60 ng/L or 50 pmol/L) and occasionally occur in patients who are not clinically anaemic. The classical neurological features are those of a polyneuropathy progressively involving the peripheral nerves and the posterior and eventually the lateral columns of the spinal cord (subacute combined degeneration, see p. 1147). Patients present with symmetrical paraesthesiae in the fingers and toes, early loss of vibration sense and proprioception, and progressive weakness and ataxia. Paraplegia may result. Dementia, psychiatric problems, hallucinations, delusions, and optic atrophy may occur from vitamin B12 deficiency.
Investigations
Haematological findings show the features of a megaloblastic anaemia as described on page 381.
Bone marrow shows the typical features of megaloblastic erythropoiesis (Fig. 8.10), although it is frequently not performed in cases of straightforward macrocytic anaemia and a low serum vitamin B12.
Serum bilirubin may be raised as a result of ineffective erythropoiesis. Normally a minor fraction of serum bilirubin results from premature breakdown of newly formed red cells in the bone marrow. In many megaloblastic anaemias, where the destruction of developing red cells is much increased, the serum bilirubin can be increased.
LDH can also be reised due to haemolysis.
Serum methylmalonic acid (MMA) and homocysteine (HC) are raised in B12 deficiency. They are only useful in cases where the B12 and folate levels are not conclusive with only HC raised in folate deficiency.
Serum vitamin B12 is usually well below 160 ng/L, which is the lower end of the normal range. Serum vitamin B12 can be assayed using radioisotope dilution or immunological assays.
Serum folate level is normal or high, and the red cell folate is normal or reduced owing to inhibition of normal folate synthesis.
Absorption tests
Vitamin B12 absorption tests are performed only occasionally when the underlying cause of the B12 deficiency is not obvious. They cannot be performed in the UK as radioactive B12 is not available. However, the principle of the absorption test demonstrates how B12 is absorbed.
Schilling test. Radioactive B12 is given orally followed by an i.m. injection of non-radioactive B12 to saturate B12 binding proteins and to flush out 58Co-B12. The urine is collected for 24 h and >10% of the oral dose would be excreted in a normal person. If this is abnormal, the test is repeated with the addition of oral intrinsic factor capsules. If the excretion is now normal, the diagnosis is pernicious anaemia or gastrectomy. If the excretion is still abnormal, the lesion must be in the terminal ileum or there may be bacterial overgrowth. The latter could be confirmed by repeating the test after a course of antibiotics.
Differential diagnosis
Vitamin B12 deficiency must be differentiated from other causes of megaloblastic anaemia, principally folate deficiency, but usually this is quite clear from the blood level of these two vitamins.
Pernicious anaemia should be distinguished from other causes of vitamin B12 deficiency (Table 8.4). Any disease involving the terminal ileum or bacterial overgrowth in the small bowel can produce vitamin B12 deficiency (see p. 268). Gastrectomy can lead, in the long term, to vitamin B12 deficiency.
Folic acid
Folic acid monoglutamate is not itself present in nature but occurs as polyglutamates. Folates are present in food as polyglutamates in the reduced dihydrofolate or tetrahydrofolate (THF) forms (Fig. 8.14), with methyl (CH3), formyl (CHO) or methylene (CH2) groups attached to the pteridine part of the molecule. Polyglutamates are broken down to monoglutamates in the upper gastrointestinal tract, and during the absorptive process these are converted to methyl THF monoglutamate, which is the main form in the serum. The methylation of homocysteine to methionine requires both methylcobalamin and methyl THF as coenzymes. This reaction is the first step in which methyl THF entering cells from the plasma is converted into folate polyglutamates. Intracellular polyglutamates are the active forms of folate and act as coenzymes in the transfer of single carbon units in amino acid metabolism and DNA synthesis (Fig. 8.12).
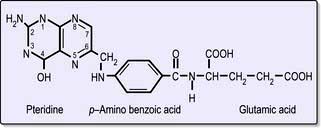
Figure 8.14 Folic acid structure. This is formed from three building blocks as shown. Tetrahydrofolate has additional hydrogen atoms at positions 5, 6, 7 and 8.
Dietary intake
Folate is found in green vegetables, such as spinach and broccoli, and offal, such as liver and kidney. Cooking causes a loss of 60–90% of the folate. The minimal daily requirement is about 100 µg.
Folate deficiency
The causes of folate deficiency are shown in Table 8.5. The main cause is poor intake, which may occur alone or in combination with excessive utilization or malabsorption. The body’s reserves of folate, unlike vitamin B12, are low (about 10 mg). On a deficient diet, folate deficiency develops over the course of about 4 months, but folate deficiency may develop rapidly in patients who have both a poor intake and excess utilization of folate (e.g. patients in intensive care units).
Table 8.5 Causes of folate deficiency
| Nutritional (major cause) | Excess utilization |
|---|---|
Poor intake |
Physiological |
Old age |
Pregnancy |
Poor social conditions |
Lactation |
Starvation |
Prematurity |
Alcohol excess (also causes impaired utilization) |
Pathological |
Haematological disease with excess red cell production, e.g. haemolysis |
|
Poor intake due to anorexia |
|
Gastrointestinal disease, e.g. partial gastrectomy, coeliac disease, Crohn’s disease |
Malignant disease with increased cell turnover |
Inflammatory disease |
|
Cancer |
Metabolic disease, e.g. homocystinuria |
Antifolate drugs |
|
Anticonvulsants: |
Haemodialysis or peritoneal dialysis |
Phenytoin |
|
Primidone |
Malabsorption |
Methotrexate |
Occurs in small bowel disease, but the effect is minor compared with that of anorexia |
Pyrimethamine |
|
Trimethoprim |
|
|
There is no simple relationship between maternal folate status and fetal abnormalities but folic acid supplements at the time of conception and in the first 12 weeks of pregnancy reduce the incidence of neural tube defects. In the USA and Canada, mandatory fortification of grain products, e.g. bread, flour and rice, has substantially improved folate status and has been associated with a significant fall in neural tube defects.
Clinical features
Patients with folate deficiency may be asymptomatic or present with symptoms of anaemia or of the underlying cause. Glossitis can occur. Unlike with B12 deficiency, neuropathy does not occur.
Investigations
The haematological findings are those of a megaloblastic anaemia as discussed on page 382.
Treatment and prevention of megaloblastic anaemia
Treatment depends on the type of deficiency. Blood transfusion is not indicated in chronic anaemia; indeed, it is dangerous to transfuse elderly patients, as heart failure may be precipitated. Folic acid may produce a haematological response in vitamin B12 deficiency but may aggravate the neuropathy. Large doses of folic acid alone should not be used to treat megaloblastic anaemia unless the serum vitamin B12 level is known to be normal. In severely ill patients, it may be necessary to treat with both folic acid and vitamin B12 while awaiting serum levels.
Treatment of vitamin B12 deficiency
Hydroxocobalamin 1000 µg can be given intramuscularly to a total of 5–6 mg over the course of 3 weeks; 1000 µg is then necessary every 3 months for the rest of the patient’s life. Alternatively, oral B12 2 mg/day is used, as 1–2% of an oral dose is absorbed by diffusion and therefore does not require intrinsic factor.
Compliance with an oral daily regimen may be a problem, particularly in elderly patients. The use of sublingual nuggets of B12 (2 × 1000 µg daily) has been suggested to be an effective and more convenient option.
Clinical improvement may occur within 48 hours and a reticulocytosis can be seen some 2–3 days after starting therapy, peaking at 5–7 days. Improvement of the polyneuropathy may occur over 6–12 months, but longstanding spinal cord damage is irreversible. Hypokalaemia can occur and, if severe, supplements should be given. Iron deficiency often develops in the first few weeks of therapy. Hyperuricaemia also occurs but clinical gout is uncommon. In patients who have had a total gastrectomy or an ileal resection, vitamin B12 should be monitored; if low levels occur, prophylactic vitamin B12 should be given. Vegans may require B12 supplements.
Treatment of folate deficiency
Folate deficiency can be corrected by giving 5 mg of folic acid daily; the same haematological response occurs as seen after treatment of vitamin B12 deficiency. Treatment should be given for about 4 months to replace body stores. Any underlying cause, e.g. coeliac disease, should be treated.
Prophylactic folic acid (400 µg daily) is recommended for all women planning a pregnancy to reduce neural tube defects. Many authorities also recommend prophylactic administration of folate throughout pregnancy.
Women who have had a child with a neural tube defect should take 5 mg folic acid daily before and during a subsequent pregnancy.
Prophylactic folic acid is also given in chronic haematological disorders where there is rapid cell turnover.
Macrocytosis without megaloblastic changes
A raised MCV with macrocytosis on the peripheral blood film can occur with a normoblastic rather than a megaloblastic bone marrow.
A common physiological cause of macrocytosis is pregnancy. Macrocytosis may also occur in the newborn. Common pathological causes are:
Some haematological disorders (e.g. aplastic anaemia, sideroblastic anaemia, pure red cell aplasia)
Drugs (e.g. hydroxycarbamide, azathioprine)
Cold agglutinins due to autoagglutination of red cells (see p. 398) (the MCV decreases to normal with warming of the sample to 37°C).
In all these conditions, normal levels of vitamin B12 and folate will be found. The exact mechanisms in each case are uncertain, but in some there is increased lipid deposition in the red cell membrane.
An increased number of reticulocytes also leads to a raised MCV because they are large cells.
A high alcohol consumption is a frequent cause of a raised MCV, and in such patients the MCV can be used as a surrogate marker for monitoring excessive alcohol consumption. A full blown megaloblastic anaemia can also occur in people who use alcohol to excess; this is due to a toxic effect of alcohol on erythropoiesis and/or to dietary folate deficiency.
Anaemia due to marrow failure (aplastic anaemia)
Aplastic anaemia is defined as pancytopenia with hypocellularity (aplasia) of the bone marrow; there are no leukaemic, cancerous or other abnormal cells in the peripheral blood or bone marrow. It is usually an acquired condition but may rarely be inherited.
Aplastic anaemia is due to a reduction in the number of pluripotential stem cells (Fig. 8.1) together with a fault in those remaining or an immune reaction against them so that they are unable to repopulate the bone marrow. Failure of only one cell line may also occur, resulting in isolated deficiencies such as the absence of red cell precursors in pure red cell aplasia. Evolution to myelodysplasia, paroxysmal nocturnal haemoglobinuria (PNH) or acute myeloblastic leukaemia occurs in some cases, probably owing to the emergence of an abnormal clone of haemopoietic cells.
Causes
A list of causes of aplasia is given in Table 8.6. Immune mechanisms are probably responsible for most cases of idiopathic acquired aplastic anaemia and play a part in at least the persistence of many secondary cases. Activated cytotoxic T cells in blood and bone marrow are responsible for the bone marrow failure.
Table 8.6 Causes of aplastic anaemia
Many drugs may cause marrow aplasia, including cytotoxic drugs such as busulfan and doxorubicin, which are expected to cause transient aplasia as a consequence of their therapeutic use. However, some individuals develop aplasia due to sensitivity to non-cytotoxic drugs such as chloramphenicol, gold, carbimazole, chlorpromazine, phenytoin, ribavirin, tolbutamide, non-steroidal anti-inflammatory agents, and many others which have been reported to cause occasional cases of aplasia.
Inherited aplastic anaemias are rare. Multiple gene mutations have been identified. Several are tumour suppressor genes and have been seen in one-third of aplastic anaemias. Fanconi’s anaemia is inherited as an autosomal recessive and is associated with skeletal, skin, eye, renal and central nervous system abnormalities. It usually presents between the ages of 5 and 10 years.
Clinical features
The clinical manifestations of marrow failure from any cause are anaemia, bleeding and infection. Bleeding is often the predominant initial presentation of aplastic anaemia with bruising with minimal trauma or blood blisters in the mouth. Physical findings include ecchymoses, bleeding gums and epistaxis. Mouth infections are common. Lymphadenopathy and hepatosplenomegaly are rare in aplastic anaemia.
Investigations
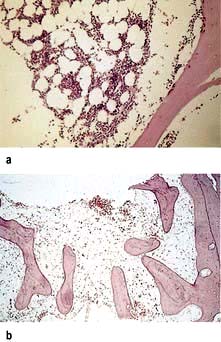
The virtual absence of reticulocytes
A hypocellular or aplastic bone marrow with increased fat spaces (Fig. 8.15).
Differential diagnosis
This is from other causes of pancytopenia (Table 8.7). A bone marrow trephine is essential for assessment of the bone marrow cellularity.
Table 8.7 Causes of pancytopenia
|
Aplastic anaemia (see Table 8.6) Bone marrow infiltration or replacement: |
Treatment and prognosis
The treatment of aplastic anaemia depends on providing supportive care while awaiting bone marrow recovery and specific treatment to accelerate marrow recovery.
The main danger is infection and stringent measures should be undertaken to avoid this (see also p. 448). Any suspicion of infection in a severely neutropenic patient should lead to immediate institution of broad-spectrum parenteral antibiotics. Supportive care including transfusions of red cells and platelets should be given as necessary. The cause of the aplastic anaemia must be eliminated if possible.
The course of aplastic anaemia can be variable, ranging from a rapid spontaneous remission to a persistent increasingly severe pancytopenia, which may lead to death through haemorrhage or infection. The most reliable determinants for the prognosis are the number of neutrophils, reticulocytes, platelets, and the cellularity of the bone marrow.
A bad prognosis (i.e. severe aplastic anaemia) is associated with the presence of two of the following three features:
Bone marrow transplantation is the treatment of choice for patients under the age of 40 with an HLA-identical sibling donor, where it gives a 75–90% chance of long-term survival.
Immunosuppressive therapy is recommended for:
Patients with severe disease over the age of 40
Younger patients with severe disease without an HLA-identical sibling donor
Patients who do not have severe disease but who are transfusion-dependent.
The standard immunosuppressive treatment is antithymocyte globulin (ATG) and ciclosporin, which results in response rates of 60–80% with 5-year survival rates of 75–85%.
Bone marrow transplantation using matched unrelated donors is an option for patients under the age of 50 who have no matched sibling donor, and who have failed to respond to immunosuppression with ATG and ciclosporin, and the results are improving (5-year survival of 65–73%). The main problems are graft rejection, graft-versus-host disease and viral infections.
Levels of haemopoietic growth factors (Fig. 8.1) are normal or increased in most patients with aplastic anaemia, and are ineffective as primary treatment.
Steroids should not be used to treat severe aplastic anaemia except for serum sickness due to ATG. They are also used to treat children with congenital pure red cell aplasia (Diamond–Blackfan syndrome).
Adult pure red cell aplasia is associated with a thymoma in 5–15% of cases and thymectomy occasionally induces a remission. It may also be associated with autoimmune disease or be idiopathic. Steroids, cyclophosphamide, azathioprine and ciclosporin are effective treatment in some cases.
Haemolytic anaemias: an introduction
Haemolytic anaemias are caused by increased destruction of red cells. The red cell normally survives about 120 days, but in haemolytic anaemias the red cell survival times are considerably shortened.
Breakdown of normal red cells occurs in the macrophages of the bone marrow, liver and spleen (Fig. 8.5).
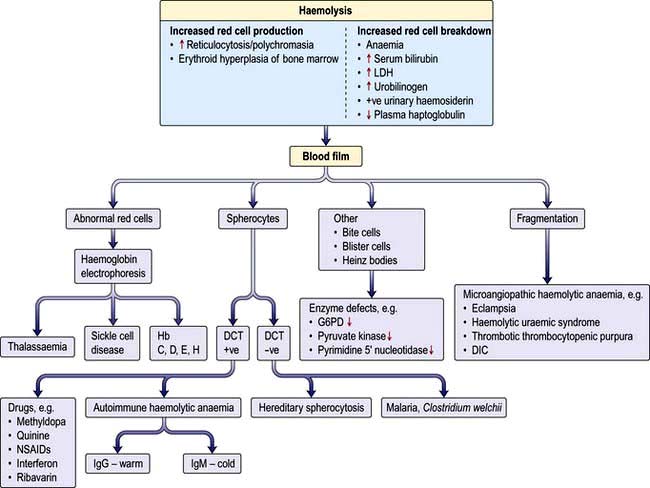
Consequences of haemolysis (Fig. 8.16)
Shortening of red cell survival does not always cause anaemia as there is a compensatory increase in red cell production by the bone marrow. If the red cell loss can be contained within the marrow’s capacity for increased output, then a haemolytic state can exist without anaemia (compensated haemolytic disease). The bone marrow can increase its output by six to eight times by increasing the proportion of cells committed to erythropoiesis (erythroid hyperplasia) and by expanding the volume of active marrow. In addition, immature red cells (reticulocytes) are released prematurely. These cells are larger than mature cells and stain with a light blue tinge on a peripheral blood film (the description of this appearance on the blood film is polychromasia) due to the presence of residual ribosomal RNA. Reticulocytes may be counted accurately as a percentage of all red cells on a blood film using a supravital stain for residual RNA (e.g. new methylene blue).
Sites of haemolysis
Extravascular haemolysis
In most haemolytic conditions red cell destruction is extravascular. The red cells are removed from the circulation by macrophages in the reticuloendothelial system, particularly the spleen.
Intravascular haemolysis
When red cells are rapidly destroyed within the circulation, haemoglobin is liberated (Fig. 8.17). This is initially bound to plasma haptoglobins but these soon become saturated.
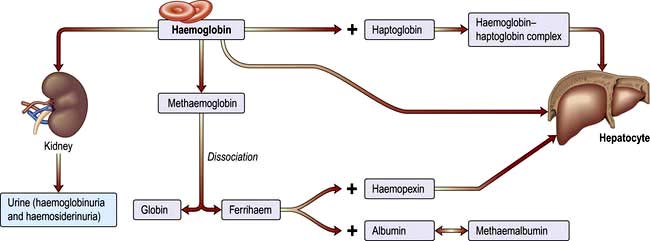
Excess free plasma haemoglobin is filtered by the renal glomerulus and enters the urine, although small amounts are reabsorbed by the renal tubules. In the renal tubular cell, haemoglobin is broken down and becomes deposited in the cells as haemosiderin. This can be detected in the spun sediment of urine using Perls’ reaction. Some of the free plasma haemoglobin is oxidized to methaemoglobin, which dissociates into ferrihaem and globin. Plasma haemopexin binds ferrihaem; but if its binding capacity is exceeded, ferrihaem becomes attached to albumin, forming methaemalbumin. On spectrophotometry of the plasma, methaemalbumin forms a characteristic band; this is the basis of Schumm’s test.
The liver plays a major role in removing haemoglobin bound to haptoglobin and haemopexin and any remaining free haemoglobin.
Evidence for haemolysis
Increased red cell breakdown is accompanied by increased red cell production. This is shown in Figure 8.16.
Demonstration of shortened red cell lifespan
Red cell survival can be estimated from 51Cr-labelled red cells given intravenously but is rarely performed.
Intravascular haemolysis
This is suggested by raised levels of plasma haemoglobin, haemosiderinuria, very low or absent haptoglobins, and the presence of methaemalbumin (positive Schumm’s test).
Various laboratory studies will be necessary to determine the exact type of haemolytic anaemia present. The causes of haemolytic anaemias are shown in Table 8.8.
Table 8.8 Causes of haemolytic anaemia
Inherited |
Acquired |
Miscellaneous |
Inherited haemolytic anaemia
Red cell membrane defects
The normal red cell membrane consists of a lipid bilayer crossed by integral proteins with an underlying lattice of proteins (or cytoskeleton), including spectrin, actin, ankyrin and protein 4.1, attached to the integral proteins (Fig. 8.18).
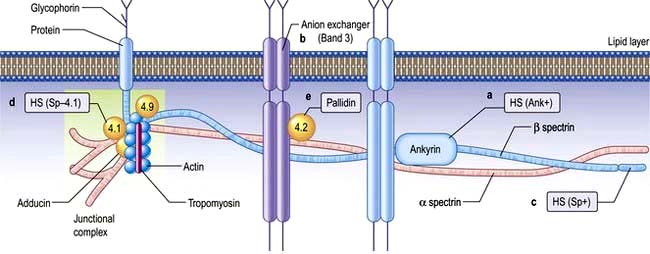
Figure 8.18 Hereditary spherocytosis (HS) and hereditary elliptocytosis (HE): red cell membrane showing the sites of the principal defects. Vertical interactions producing HS: (a) ankyrin mutation, HS (Ank+) producing deficiency (45% of cases); (b) HS band 3 deficiency (20%); (c) β spectrin deficiency, HS (Sp+) (<20%); (d) abnormal spectrin/protein 4.1 binding, HS (Sp–4.1); (e) protein 4.2 (pallidin) deficiency (Japanese). These produce various autosomal dominant and recessive forms of the disease. Horizontal interactions producing HE: α spectrin (80%), protein 4.1 (15%), β spectrin (5%).
Hereditary spherocytosis (HS)
HS is the most common inherited haemolytic anaemia in northern Europeans, affecting 1 in 5000. It is inherited in an autosomal dominant manner, but in 25% of patients, neither parent is affected and it is presumed that HS has occurred by spontaneous mutation or is truly recessive. HS is due to defects in the red cell membrane, resulting in the cells losing part of the cell membrane as they pass through the spleen, possibly because the lipid bilayer is inadequately supported by the membrane skeleton. The best-characterized defect is a deficiency in the structural protein spectrin, but quantitative defects in other membrane proteins have been identified (Fig. 8.18), with ankyrin defects being the most common. The abnormal red cell membrane in HS is associated functionally with an increased permeability to sodium, and this requires an increased rate of active transport of sodium out of the cells which is dependent on ATP produced by glycolysis. The surface-to-volume ratio decreases, and the cells become spherocytic. Spherocytes are more rigid and less deformable than normal red cells. They are unable to pass through the splenic microcirculation so they have a shortened lifespan.
Clinical features
The condition may present with jaundice at birth. However, the onset of jaundice can be delayed for many years and some patients may go through life with no symptoms and are detected only during family studies. The patient may eventually develop anaemia, splenomegaly and ulcers on the leg. As in many haemolytic anaemias, the course of the disease may be interrupted by aplastic, haemolytic and megaloblastic crises. Aplastic anaemia usually occurs after infections, particularly with erythro (parvo) virus, whereas megaloblastic anaemia is the result of folate depletion owing to the hyperactivity of the bone marrow. Chronic haemolysis leads to the formation of pigment gallstones (see p. 351).
Investigations
Anaemia. This is usually mild, but occasionally can be severe.
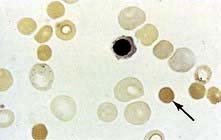
Blood film. This shows spherocytes (Fig. 8.19) and reticulocytes.
Haemolysis is evident (e.g. the serum bilirubin and urinary urobilinogen will be raised).
Osmotic fragility. When red cells are placed in solutions of increasing hypotonicity, they take in water, swell, and eventually lyse. Spherocytes tolerate hypotonic solutions less well than do normal biconcave red cells. Osmotic fragility tests are infrequently carried out in routine practice, but may be useful to confirm a suspicion of spherocytosis on a blood film.
Direct antiglobulin (Coombs’) test is negative in hereditary spherocytosis, virtually ruling out autoimmune haemolytic anaemia where spherocytes are also commonly present.
Treatment
Splenectomy is indicated in hereditary spherocytosis to relieve symptoms due to anaemia or splenomegaly, reverse growth failure and prevent recurrent gallstones. It is best to postpone splenectomy until after childhood, as sudden overwhelming fatal infections, usually due to encapsulated organisms such as pneumococci, may occur (see p. 406). Splenectomy should be preceded by appropriate immunization and followed by lifelong penicillin prophylaxis (see Box 8.3). In addition to the well known risk of bacterial infection, there is also some evidence that there is a significant risk of adverse arterial and venous thromboembolic events after splenectomy performed for hereditary spherocytosis.
Following splenectomy, the spherocytes persist but the Hb usually returns to normal as the red cells are no longer destroyed.
Hereditary elliptocytosis
This disorder of the red cell membrane is inherited in an autosomal dominant manner and has a prevalence of 1 in 2500 in Caucasians. The red cells are elliptical due to deficiencies of protein 4.1 or the spectrin/actin/4.1 complex which leads to weakness of the horizontal protein interaction and to the membrane defect (Fig. 8.18). Clinically it is a similar condition to HS but milder. Only a minority of patients have anaemia and only occasional patients require splenectomy.
Rarely, hereditary spherocytosis or elliptocytosis may be inherited in a homozygous fashion giving rise to a severe haemolytic anaemia sometimes necessitating splenectomy in early childhood.
Hereditary stomatocytosis
Stomatocytes are red cells in which the pale central area appears slit-like. Their presence in large numbers may occur in a hereditary haemolytic anaemia associated with a membrane defect, but excess alcohol intake is also a common cause. Although these hereditary conditions are very rare, a correct diagnosis is required since splenectomy is contraindicated as it may result in fatal thromboembolic events.
Haemoglobin abnormalities
In early embryonic life, haemoglobins Gower 1, Gower 2 and Portland predominate. Later, fetal haemoglobin (HbF), which has two α and two γ chains, is produced (Fig. 8.20). There is increasing synthesis of β chains from 13 weeks’ gestation and at term there is 80% HbF and 20% HbA. The haemoglobin switch from HbF to HbA occurs after birth when the genes for γ chain production are further suppressed and there is rapid increase in the synthesis of β chains. BCL IIA, a zinc finger protein, is one of a number of proteins that suppress γ gene expression. There is little HbF produced (normally <1%) from 6 months after birth. The δ chain is synthesized just before birth and HbA2 (α2δ2) remains at a level of about 2% throughout adult life (Table 8.9).
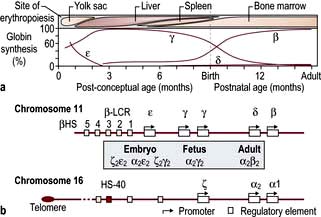
Figure 8.20 Normal development switches in globin expression.
(a) Sites of haemopoiesis and globin changes at various gestational ages. (b) Structure of α-like and β-like globin gene clusters. LCR, locus control region; HS, major upstream regulatory element.
(From Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet 2012; 379:373–383, with permission.)
Globin chains are synthesized in the same way as any protein (see p. 42). A normal individual has four α-globin chain genes (Fig. 8.20) with two α-globin genes on each haploid genome (genes derived from one parent). These are situated close together on chromosome 16. The genes controlling the production of ε, γ, δ and β chains are close together on chromosome 11. The globin genes are arranged on chromosomes 16 and 11 in the order in which they are expressed and combine to give different haemoglobins. Normal haemoglobin synthesis is discussed on page 374.
The thalassaemias
The thalassaemias affect people throughout the world (Fig. 8.21). Normally, there is balanced (1:1) production of α and β chains. The defective synthesis of globin chains in thalassaemia leads to ‘imbalanced’ globin chain production, leading to precipitation of globin chains within the red cell precursors and resulting in ineffective erythropoiesis. Precipitation of globin chains in mature red cells leads to haemolysis.
β-Thalassaemia
In homozygous β-thalassaemia, either no normal β chains are produced (β0) or β-chain production is very reduced (β+). There is an excess of α chains, which precipitate in erythroblasts and red cells causing ineffective erythropoiesis and haemolysis. The excess α chains combine with whatever β, δ and γ chains are produced, resulting in increased quantities of HbA2 and HbF and, at best, small amounts of HbA. In heterozygous β-thalassaemia there is usually symptomless microcytosis with or without mild anaemia. Table 8.10 shows the findings in the homozygote and heterozygote for the common types of β-thalassaemia.
Table 8.10 β-Thalassaemia: common findings
| Type of thalassaemia | Findings in homozygote | Findings in heterozygote |
|---|---|---|
β+ |
Thalassaemia major HbA + F + A2 |
Thalassaemia minor HbA2 raised |
β0 |
Thalassaemia major HbF + A2 |
Thalassaemia minor HbA2 raised |
δβ |
Thalassaemia intermedia |
Thalassaemia minor HbF 5–15% |
HbF only |
HbA2 normal |
|
δβ+ (Lepore) |
Thalassaemia major or intermedia |
Thalassaemia minor |
HbF and Lepore |
Hb Lepore 5–15% |
|
HbA2 normal |
Hb Lepore is a cross-fusion product of δ and β globin genes.
Adapted with permission from Weatherall DJ. Disorders of the synthesis of function of haemoglobin. In: Weatherall DJ, Warrell DA, Cox TM, Firth JD (eds) Oxford Textbook of Medicine, 5th edn. Oxford: Oxford University Press; 2010.
Molecular genetics
The molecular errors accounting for over 200 genetic defects leading to β-thalassaemia have been characterized. Unlike in α-thalassaemia, the defects are mainly point mutations rather than gene deletions. The mutations result in defects in transcription, RNA splicing and modification, translation via frame shifts and nonsense codons producing highly unstable β-globin, which cannot be utilized.
Clinical syndromes
Clinically, β-thalassaemia can be divided into the following:
Thalassaemia minor (or trait), the symptomless heterozygous carrier state
Thalassaemia intermedia, a moderate anaemia, not requiring regular transfusions (with a number of different genotypes)
Thalassaemia major (generally homozygous β-thalassaemia), severe anaemia requiring regular transfusions.
Thalassaemia minor (trait)
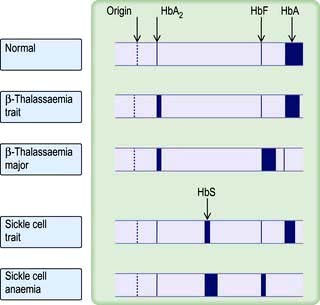
This common carrier state (heterozygous β-thalassaemia) is asymptomatic. Anaemia is mild or absent. The red cells are hypochromic and microcytic with a low MCV and MCH, and it may be confused with iron deficiency. However, the two are easily distinguished, as in thalassaemia trait the serum ferritin and the iron stores are normal (Table 8.2). The RDW is usually normal (see p. 373). Hb electrophoresis usually shows a raised HbA2 and often a raised HbF (Fig. 8.22). Iron should not be given to these patients unless they also have proven coincidental iron deficiency.
Thalassaemia intermedia
Thalassaemia intermedia includes patients who are symptomatic with moderate anaemia (Hb 70–100 g/L), who do not require regular transfusions.
Thalassaemia intermedia may be due to a combination of homozygous mild β+- and α-thalassaemia, where there is reduced α-chain precipitation and less ineffective erythropoiesis and haemolysis. The inheritance of hereditary persistence of HbF with homozygous β-thalassaemia also results in a milder clinical picture than unmodified β-thalassaemia major because the excess α chains are partially removed by the increased production of γ chains.
Patients may have splenomegaly and bone deformities. Recurrent leg ulcers, gallstones and infections are also seen. It should be noted that these patients may be iron overloaded despite a lack of regular blood transfusions. This is caused by excessive iron absorption which results from the underlying dyserythropoiesis (see iron absorption, p. 377).
Thalassaemia major (Cooley’s anaemia)
Most children affected by homozygous β-thalassaemia present during the first year of life with:
Failure to thrive and recurrent bacterial infections
Severe anaemia from 3 to 6 months when the switch from γ- to β-chain production should normally occur
Extramedullary haemopoiesis that soon leads to hepatosplenomegaly and bone expansion, giving rise to the classical thalassaemic facies (Fig. 8.23a).
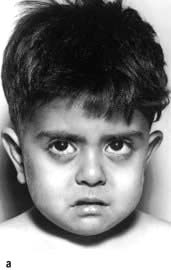
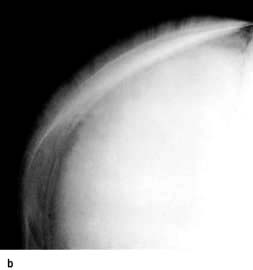
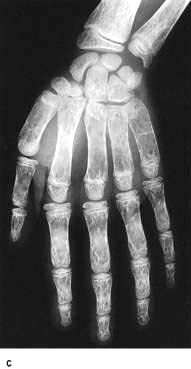
Figure 8.23 Thalassaemia. (a) A child with thalassaemia, showing the typical facial features. (b) Skull X-ray of a child with β-thalassaemia, showing the ‘hair on end’ appearance. (c) X-ray of hand, showing expansion of the marrow and a thinned cortex.
Skull X-rays in these children show the characteristic ‘hair on end’ appearance of bony trabeculation as a result of expansion of the bone marrow into cortical bone (Fig. 8.23b). The expansion of the bone marrow is also shown in an X-ray of the hand (Fig. 8.23c).
The classic features of untreated thalassaemia major are generally only observed in patients from countries without good blood transfusion support.
Management
The aims of treatment are to suppress ineffective erythropoiesis, prevent bony deformities and allow normal activity and development.
Long-term folic acid supplements are required.
Regular transfusions should be given to keep the Hb above 100 g/L. Blood transfusions may be required every 4–6 weeks.
If transfusion requirements increase, splenectomy may help, although this is usually delayed until after the age of 6 years because of the risk of infection. Prophylaxis against infection is required for patients undergoing splenectomy (see p. 406).
Iron overload caused by repeated transfusions (transfusion haemosiderosis) may lead to damage to the endocrine glands, liver, pancreas and the myocardium by the time patients reach adolescence. Magnetic resonance imaging (myocardial T2- relaxation time) is useful for monitoring iron overload in thalassaemia; both the heart and the liver can be monitored. The standard iron-chelating agent remains desferrioxamine, although it has to be administered parenterally. Desferrioxamine is given as an overnight subcutaneous infusion on 5–7 nights each week. Ascorbic acid 200 mg daily is given, as it increases the urinary excretion of iron in response to desferrioxamine. Often young children have a very high standard of chelation as it is organized by their parents. However, when the children become adults and take on this role themselves they often rebel and chelation with desferrioxamine may become problematic. Deferiprone, an oral iron chelator, has been available for some years, and results on a new once-daily oral iron chelator, deferasirox, indicate that it is safe, similar in effectiveness to desferrioxamine and is being increasingly used.
Intensive treatment with desferrioxamine has been reported to reverse damage to the heart in patients with severe iron overload, but excessive doses of desferrioxamine may cause cataracts, retinal damage and nerve deafness. Infection with Yersinia enterocolitica occurs in iron-loaded patients treated with desferrioxamine. Iron overload should be periodically assessed by measuring the serum ferritin and by assessment of hepatic iron stores by MRI.
Bone marrow transplantation has been used in young patients with HLA-matched siblings. It has been successful in patients in good clinical condition with a 3-year mortality of <5%, but there is a high mortality (>50%) in patients in poor condition with iron overload and liver dysfunction.
Prenatal diagnosis and gene therapy are discussed on page 43.
Patients’ partners should be tested. If both partners have β-thalassaemia trait, there is a one in four chance of such pregnancy resulting in a child having β-thalassaemia major. Therefore, couples in this situation must be offered prenatal diagnosis (see p. 43).
α-Thalassaemia
Molecular genetics
In contrast to β-thalassaemia, α-thalassaemia is often caused by gene deletions, although mutations of the α-globin genes may also occur. The gene for α-globin chains is duplicated on both chromosomes 16, i.e. a normal person has a total of four α-globin genes. Deletion of one α-chain gene (α+) or both α-chain genes (α0) on each chromosome 16 may occur (Table 8.11). The former is the most common of these abnormalities.
Four-gene deletion (deletion of both genes on both chromosomes); there is no α-chain synthesis and only Hb Barts (γ4) is present. Hb Barts cannot carry oxygen and is incompatible with life (Table 8.9 and Table 8.11). Infants are either stillborn at 28–40 weeks or die very shortly after birth. They are pale, oedematous and have enormous livers and spleens – a condition called hydrops fetalis.
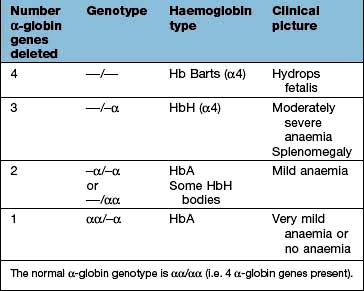
Three-gene deletion; HbH disease, which is common in parts of Asia, has four β chains with low levels of HbA and Hb Barts. HbA2 is normal or reduced. HbH does not transport oxygen and precipitates in erythroblasts and erythrocytes. There is moderate anaemia (Hb 70–100 g/L) and splenomegaly (thalassaemia intermedia). The patients are not usually transfusion-dependent.
Two-gene deletion (α-thalassaemia trait); there is microcytosis with or without mild anaemia. HbH bodies may be seen on staining a blood film with brilliant cresyl blue.
Globin chain synthesis studies for the detection of a reduced ratio of α to β chains may be necessary for the definitive diagnosis of α-thalassaemia trait.
Less commonly, α-thalassaemia may result from genetic defects other than deletions, for example mutations in the stop codon producing an α chain with many extra amino acids (Hb Constant Spring). It has a more severe clinical course than HbH with severe anaemia often precipitated by infection.
Sickle syndromes
Sickle cell haemoglobin (HbS) results from a single-base mutation of adenine to thymine, which produces a substitution of valine for glutamic acid at the sixth codon of the β-globin chain (α2β26glu→val). In the homozygous state (sickle cell anaemia), both genes are abnormal (HbSS), whereas in the heterozygous state (sickle cell trait, HbAS) only one chromosome carries the gene. As the synthesis of HbF is normal, the disease usually does not manifest itself until the HbF decreases to adult levels at about 6 months of age.
The sickle gene is commonest in Africans (up to 25% gene frequency in some populations) but is also found in India, the Middle East and Southern Europe.
Pathogenesis
Deoxygenated HbS molecules are insoluble and polymerize. The flexibility of the cells is decreased and they become rigid and take up their characteristic sickle appearance (Fig. 8.24). This process is initially reversible but, with repeated sickling, the cells eventually lose their membrane flexibility and become irreversibly sickled. This is due to dehydration, partly caused by potassium leaving the red cells via calcium activated potassium channels called the Gados channel. These irreversibly sickled cells are dehydrated and dense and will not return to normal when oxygenated. Sickling can produce:
impaired passage of cells through the microcirculation, leading to obstruction of small vessels and tissue infarction.
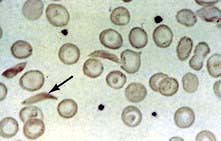
Sickling is precipitated by infection, dehydration, cold, acidosis or hypoxia. In many cases, the cause is unknown, but adhesion proteins on activated endothelial cells (VCAM-1) may play a causal role, particularly in vaso-occlusion when rigid cells are trapped, facilitating polymerization. HbS releases its oxygen to the tissues more easily than does normal Hb, and patients therefore feel well despite being anaemic (except of course during crises or complications).
Depending on the type of haemoglobin chain combinations, three clinical syndromes occur:
Homozygous HbSS have the most severe disease
Combined heterozygosity (HbSC) for HbS and C (see below) who suffer intermediate symptoms
Heterozygous HbAS (sickle cell trait) have no symptoms (see p. 395).
Sickle cell anaemia
Clinical features
Vaso-occlusive crises
An early presentation may be acute pain in the hands and feet (dactylitis) owing to vaso-occlusion of the small vessels. Severe pain in other bones, e.g. femur, humerus, vertebrae, ribs, pelvis, occurs in older children/adults. These attacks vary greatly in frequency from patient to patient and sometimes in the same patient from year to year; however, as a generalization, a patient with moderately severe sickle cell anaemia may have around three hospital admissions a year from painful vaso-occlusive crises. Fever often accompanies the pain.
Pulmonary hypertension
Pulmonary hypertension is a known consequence of sickle cell anaemia, occurring in 30–40% of patients, and is associated with an increased mortality. The ‘hyperhaemolytic paradigm’ (HHP) proposes that haemolysis in sickle cell disease leads to increased cell-free plasma Hb, which consumes NO, leading to a state of NO deficiency, endothelial dysfunction and a high prevalence of pulmonary hypertension. The basis of the HHP has recently been questioned.
Acute chest syndrome
This occurs in up to 30%, and pulmonary hypertension and chronic lung disease are the commonest causes of death in adults with sickle cell disease. The acute chest syndrome is caused by infection, fat embolism from necrotic bone marrow or pulmonary infarction due to sequestration of sickle cells. It comprises shortness of breath, chest pain, hypoxia and new chest X-ray changes due to consolidation. The presentation may be gradual or very rapid, leading to death in a few hours. Management is with pain relief, high-flow supplemental oxygen and antibiotics (cefotaxime and clarithromycin), which should be started immediately. Exchange transfusion will reduce the amount of HbS to <20% if there is no improvement. Ventilation (CPAP) may be necessary. Infections can be due to Chlamydia and mycoplasma, as well as Streptococcus pneumoniae.
Anaemia
Chronic haemolysis produces a stable haemoglobin level, usually in the 60–80 g/L range, but an acute fall in the haemoglobin level can occur owing to:
Splenic sequestration
Vaso-occlusion produces an acute painful enlargement of the spleen. There is splenic pooling of red cells and hypovolaemia, leading in some to circulatory collapse and death. The condition occurs in childhood before multiple infarctions have occurred. The latter eventually leads to a fibrotic nonfunctioning spleen. Liver sequestration can also occur.
Long-term problems
Growth and development. Young children are short but regain their height by adulthood. However, they remain below the normal weight. There is often delayed sexual maturation, which may require hormone therapy.
Bones are a common site for vaso-occlusive episodes, leading to chronic infarcts. Avascular necrosis of hips, shoulders, compression of vertebrae and shortening of bones in the hands and feet occur. These episodes are the common cause for the painful crisis. Osteomyelitis is commoner in sickle cell disease and is caused by Staphylococcus aureus, Staph. pneumoniae and salmonella (see p. 534). Occasionally, hip joint replacement may be required.
Infections are common in tissues susceptible to vasoocclusion, e.g. bones, lungs, kidneys.
Leg ulcers occur spontaneously (vaso-occlusive episodes) or following trauma and are usually over the medial or lateral malleoli. They often become infected and are quite resistant to treatment, sometimes blood transfusion may facilitate ulcer healing.
Cardiac problems occur, with cardiomegaly, arrhythmias and iron overload cardiomyopathy. Myocardial infarctions occur due to thrombotic episodes which are not secondary to atheroma.
Neurological complications occur in 25% of patients, with transient ischaemic attacks, fits, cerebral infarction, cerebral haemorrhage and coma. Strokes occur in about 11% of patients under 20 years of age. The most common finding is obstruction of a distal intracranial internal carotid artery or a proximal middle cerebral artery. About 10% of children without neurological signs or symptoms have abnormal blood-flow velocity indicative of clinically significant arterial stenosis; such patients have very high risk of stroke. It has now been demonstrated that if children with stenotic cranial artery lesions, as demonstrated on transcranial Doppler ultrasonography, are maintained on a regular programme of transfusion that is designed to suppress erythropoiesis so that no more than 30% of the circulating red cells are their own, about 90% of strokes in such children could be prevented.
Cholelithiasis. Pigment stones occur as a result of chronic haemolysis.
Liver. Chronic hepatomegaly and liver dysfunction are caused by trapping of sickle cells.
Renal. Chronic tubulointerstitial nephritis occurs (see p. 596).
Priapism. An unwanted painful erection occurs from vaso-occlusion and can be recurrent. This may result in impotence. Treatment is with an α-adrenergic blocking drug, analgesia and hydration.
Eye. Background retinopathy, proliferative retinopathy, vitreous haemorrhages and retinal detachments all occur. Regular yearly eye checks are required.
Pregnancy. Impaired placental blood flow causes spontaneous abortion, intrauterine growth retardation, preeclampsia and fetal death. Painful episodes, infections and severe anaemia occur in the mother.
Investigations
Blood count. The level of Hb is in the range 60–80 g/L with a high reticulocyte count (10–20%).
Blood films can show features of hyposplenism (see Fig. 8.29) and sickling (Fig. 8.24).
Sickle solubility test. A mixture of HbS in a reducing solution such as sodium dithionite gives a turbid appearance because of precipitation of HbS, whereas normal Hb gives a clear solution. A number of commercial kits such as Sickledex are available for rapid screening for the presence of HbS, e.g. before surgery in appropriate ethnic groups and in the A&E department.
Hb electrophoresis (Fig. 8.22) is always needed to confirm the diagnosis. There is no HbA, 80–95% HbSS and 2–20% HbF.
The parents of the affected child will show features of sickle cell trait.
Management
Precipitating factors (see above) should be avoided or treated quickly. The complications requiring inpatient management are shown in Table 8.12.
Table 8.12 Complications requiring inpatient management
Acute painful attacks require supportive therapy with intravenous fluids, and adequate analgesia. Oxygen and antibiotics are only given if specifically indicated. Crises can be extremely painful and require strong, usually narcotic, analgesia. Morphine is the drug of choice. Milder pain can sometimes be relieved by codeine, paracetamol and NSAIDs (Box 8.1).
 Box 8.1
Box 8.1
Management of acute painful crisis in opioid naive adults with sickle cell disease
a Caution advised with NSAIDs in renal impairment. (Adapted from Rees DC, Olujohungbe AD, Parker NE et al. Guidelines for the management of the acute painful crisis in sickle cell disease. British Journal of Haematology 2003; 120(5):744–752.)
Prophylaxis is with penicillin 500 mg daily and vaccination with polyvalent pneumococcal and Haemophilus influenzae type b vaccine (see p. 406). Folic acid is given to all patients with haemolysis.
Anaemia
Blood transfusions should only be given for clear indications. Patients with steady state anaemia, those having minor surgery or having painful episodes without complications should not be transfused. Transfusions should be given for heart failure, TIAs, strokes, acute chest syndrome, acute splenic sequestration and aplastic crises. Before elective operations and during pregnancy, repeated transfusions may be used to reduce the proportion of circulating HbS to <20% to prevent sickling. Exchange transfusions may be necessary in patients with severe or recurrent crises, or before emergency surgery. Transfusion and splenectomy may be life-saving for young children with splenic sequestration. A full blood crossmatching compatibility screen should always be performed.
Hydroxycarbamide (hydroxyurea) is the first drug which has been widely used as therapy for sickle cell anaemia. It acts, at least in part, by increasing HbF concentrations. Hydroxycarbamide has been shown in trials to reduce the episodes of pain, the acute chest syndrome, and the need for blood transfusions.
Inhaled nitric oxide is a new approach to the treatment of painful crises in sickle cell anaemia based on the hyperhaemolytic paradigm discussed briefly above. However, it is yet to become an established therapy based on randomized controlled trials.
Stem cell transplantation has been used to treat sickle cell anaemia although in fewer numbers than for thalassaemia. Children and adolescents younger than 16 years of age who have severe complications (strokes, recurrent chest syndrome or refractory pain) and have an HLA-matched donor are the best candidates for transplantation.
Sickle cell trait
These individuals have no symptoms unless extreme circumstances cause anoxia, such as flying in non-pressurized aircraft. Sickle cell trait gives some protection against Plasmodium falciparum malaria (see p. 144), and consequently the sickle gene has been seen as an example of a balanced polymorphism (where the advantage of the malaria protection in the heterozygote is balanced by the mortality of the homozygous condition). Typically there is 60% HbA and 40% HbS. It should be emphasized that unlike thalassaemia trait, the blood count and film of a person with sickle cell trait are normal. The diagnosis is made by a positive sickle test or by Hb electrophoresis (Fig. 8.22).
Other structural globin chain defects
There are very many Hb variants and most are not associated with any clinical manifestations. However, some Hb variants may interact with HbS, e.g. compound heterozygosity for HbC and HbS gives rise to HbSC disease. The clinical course of HbSC disease is generally somewhat milder than that of HbSS disease, but there is an increased likelihood of thrombosis, which may lead to thrombosis in pregnancy and to retinopathy.
Combined defects of globin chain production and structure
Abnormalities of Hb structure (e.g. HbS, C) can occur in combination with thalassaemia. The combination of β-thalassaemia trait and sickle cell trait (sickle cell β-thalassaemia) resembles sickle cell anaemia (HbSS) clinically.
HbE (α2β226glu → lys) is the most common Hb variant in South-east Asia, and the second most prevalent haemoglobin variant worldwide. HbE heterozygotes are asymptomatic; the haemoglobin level is normal, but red cells are microcytic. Homozygous HbE causes a mild microcytic anaemia, but the combination of heterozygosity for HbE and β-thalassaemia produces a variable anaemia, which can be as severe as β-thalassaemia major.
Prenatal screening and diagnosis of severe haemoglobin abnormalities
Of the offspring of parents who both have either β-thalassaemia or sickle cell trait, 25% will have β-thalassaemia major or sickle cell anaemia, respectively. Recognition of these heterozygous states in parents and family counselling provide a basis for antenatal screening and diagnosis (p. 44).
Prognosis
Pregnant women with either sickle cell trait or thalassaemia trait must be identified at antenatal booking either by selective screening of high-risk groups on the basis of ethnic origin or by universal screening of all pregnant women. β-Thalassaemia trait can always be detected by a low MCV and MCH and confirmed by haemoglobin electrophoresis. However, sickle cell trait is undetectable from a blood count and the laboratory need a specific request to screen for sickle cell trait. Clearly, universal antenatal screening as practised in the UK avoids such problems.
If a pregnant woman is found to have a haemoglobin defect, her partner should be tested. Antenatal diagnosis is offered if both are affected as there is a risk of a severe fetal Hb defect, particularly β-thalassaemia major. Fetal DNA analysis can be carried out using amniotic fluid, chorionic villus or fetal blood samples. Abortion is offered if the fetus is found to be severely affected. Chorionic villus biopsy has the advantage that it can be carried out in the first trimester, thus avoiding the need for second trimester abortions.
Metabolic disorders of the red cell
Red cell metabolism
The mature red cell has no nucleus, mitochondria or ribosomes and is therefore unable to synthesize proteins. Red cells have only limited enzyme systems but they maintain the viability and function of the cells. In particular, energy is required in the form of ATP for the maintenance of the flexibility of the membrane and the biconcave shape of the cells to allow passage through small vessels, and for regulation of the sodium and potassium pumps to ensure osmotic equilibrium. In addition, it is essential that Hb be maintained in the reduced state.
The enzyme systems responsible for producing energy and reducing power are (Fig. 8.25):
The glycolytic (Embden–Meyerhof) pathway, in which glucose is metabolized to pyruvate and lactic acid with production of ATP
The hexose monophosphate (pentose phosphate) pathway, which provides reducing power for the red cell in the form of NADPH.
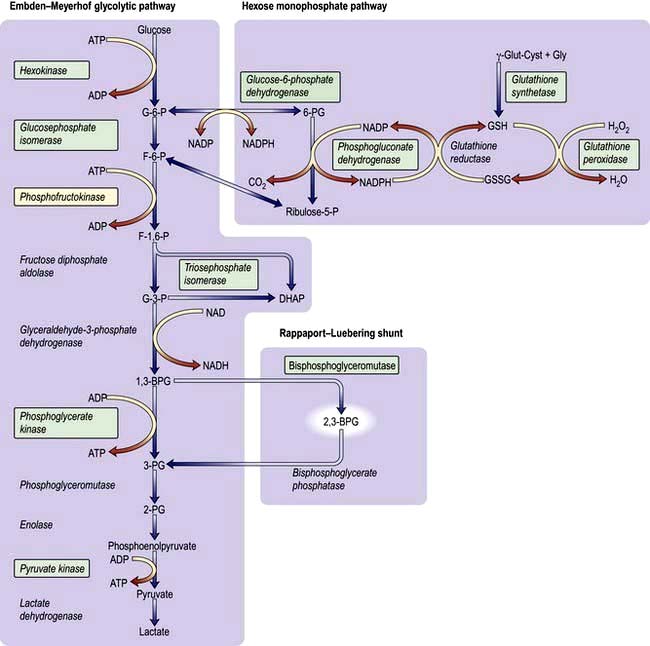
Figure 8.25 Glucose metabolism pathways in red cells. The enzymes in green boxes indicate documented hereditary deficiency diseases. BPG, bisphosphoglycerate; DHAP, dihydroxyacetone phosphate; F, fructose; G, glucose; GSSG, oxidized glutathione; GSH, reduced glutathione; P, phosphate; PG, phosphoglycerate.
About 90% of glucose is metabolized by the former and 10% by the latter. The hexose monophosphate shunt maintains glutathione (GSH) in a reduced state. Glutathione is necessary to combat oxidative stress to the red cell, and failure of this mechanism may result in:
Rigidity due to cross-linking of spectrin, which decreases membrane flexibility (see Fig. 8.18) and causes ‘leakiness’ of the red cell membrane
Oxidation of the Hb molecule, producing methaemoglobin and precipitation of globin chains as Heinz bodies localized on the inside of the membrane; these bodies are removed from circulating red cells by the spleen.
2,3-BPG is formed from a side-arm of the glycolytic pathway (Fig. 8.25). It binds to the central part of the Hb tetramer, fixing it in the low-affinity state (Fig. 8.4). A decreased affinity with a shift in the oxygen dissociation curve to the right enables more oxygen to be delivered to the tissues.
In addition to the G6PD, pyruvate kinase and pyrimidine 5′ nucleotidase deficiencies described below, there are a number of rare enzyme deficiencies that need specialist investigation.
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
The enzyme G6PD holds a vital position in the hexose monophosphate shunt (Fig. 8.25), oxidizing glucose-6-phosphate to 6-phosphoglycerate with the reduction of NADP to NADPH. The reaction is necessary in red cells where it is the only source of NADPH, which is used via glutathione to protect the red cell from oxidative damage. G6PD deficiency is a common condition that presents with a haemolytic anaemia and affects millions of people throughout the world, particularly in Africa, around the Mediterranean, the Middle East (around 20%) and South-east Asia (up to 40% in some regions).
The gene for G6PD is localized to chromosome Xq28 near the factor VIII gene. The deficiency is more common in males than in females. However, female heterozygotes can also have clinical problems due to lyonization, whereby because of random X-chromosome inactivation female heterozygotes have two populations of red cells – a normal one and a G6PD-deficient one.
There are over 400 structural types of G6PD, and mutations are mostly single amino acid substitutions (missense point mutations). WHO has classified variants by the degree of enzyme deficiency and severity of haemolysis. The most common types with normal activity are called type B+, which is present in almost all Caucasians and about 70% of black Africans, and type A+, which is present in about 20% of black Africans. There are many variants with reduced activity but only two are common. In the African, or A− type, the degree of deficiency is mild and more marked in older cells. Haemolysis is self-limiting as the young red cells newly produced by the bone marrow have nearly normal enzyme activity. However, in the Mediterranean type, both young and old red cells have very low enzyme activity. After an oxidant shock the Hb level may fall precipitously; death may follow unless the condition is recognized and the patient is transfused urgently.
Clinical syndromes
Acute drug-induced haemolysis (Table 8.13) – usually dose-related
Favism (ingestion of fava beans)
Infections and acute illnesses will also precipitate haemolysis in patients with G6PD deficiency.
Table 8.13 Drugs causing haemolysis in glucose-6-phosphate deficiency
|
|
Mothballs containing naphthalene can also cause haemolysis.
The clinical features are due to rapid intravascular haemolysis with symptoms of anaemia, jaundice and haemoglobinuria.
FURTHER READING
Abboud MR. Hematopoietic stem-cell transplantation for adults with sickle cell disease. N Engl J Med 2009; 361:2380–2381.
Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008; 371:64–72.
Gladwin MT, Vichinsey E. Pulmonary complications of sickle cell disease. N Engl J Med 2008; 357:2254–2265.
Shander A, Sazama K. Clinical consequences of iron overload from chronic red blood cell transfusions, its diagnosis, and its management by chelation therapy. Transfusion 2010; 50:1144–1155.
Taher AT, Musallam KM, Cappellini MD et al. Optimal management of β thalassaemia intermedia. Br J Haematol 2011; 152:512–523.
Investigations
Blood count is normal between attacks.
During an attack the blood film may show irregularly contracted cells, bite cells (cells with an indentation of the membrane), blister cells (cells in which the Hb appears to have become partially detached from the cell membrane; Fig. 8.26), Heinz bodies (best seen on films stained with methyl violet) and reticulocytosis.
Haemolysis is evident (see p. 387).
G6PD deficiency can be detected using several screening tests, such as demonstration of the decreased ability of G6PD-deficient cells to reduce dyes. The level of the enzyme may also be directly assayed. There are two diagnostic problems. Immediately after an attack the screening tests may be normal (because the oldest red cells with least 6GPD activity are destroyed selectively). The diagnosis of heterozygous females may be difficult because the enzyme level may range from very low to normal depending on lyonization. However, the risk of clinically significant haemolysis is minimal in patients with borderline G6PD activity.
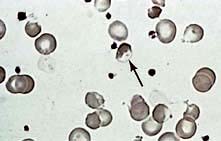
Pyruvate kinase deficiency
This is the most common defect of red cell metabolism after G6PD deficiency; it affects thousands rather than millions of people. The site of the defect is shown in Figure 8.25. There is reduced production of ATP, causing rigid red cells. Homozygotes have haemolytic anaemia and splenomegaly. It is inherited as an autosomal recessive.
Investigations
Anaemia of variable severity is present (Hb 50–100 g/L). The oxygen dissociation curve is shifted to the right as a result of the rise in intracellular 2,3-BPG, and this reduces the severity of symptoms due to anaemia.
Blood film shows distorted (‘prickle’) cells and a reticulocytosis.
Pyruvate kinase activity is low (affected homozygotes have levels of 5–20%).
Pyrimidine 5′ nucleotidase deficiency
This autosomal disorder produces a haemolytic anaemia with basophilic stippling of the red cells. The enzyme degrades pyrimidine nucleotides to cytidine and uridine (pentose phosphate shunt), which in turn leads to the degradation of RNA in the reticulocytes. Lack of the enzyme results in accumulation of partially degraded RNA, which shows as basophilic stippling in mature red cells. The enzyme is also inhibited by lead (see p. 922) and thus basophilic stippling is seen in lead poisoning. The hereditary form can be diagnosed by measuring the enzyme in erythrocytes. A screening test using the ultraviolet absorption spectrum of red cells is available.
Acquired haemolytic anaemia
These anaemias may be divided into those due to immune, non-immune, or other causes (Table 8.8).
Miscellaneous causes
Various toxic substances can disrupt the red cell membrane and cause haemolysis (e.g. arsenic, and products of Clostridium welchii).
Malaria frequently causes anaemia owing to a combination of a reduction in red cell survival and reduced production of red cells.
Hypersplenism (see p. 406) results in a reduced red cell survival, which may also contribute to the anaemia seen in malaria.
Extensive burns result in denaturation of red cell membrane proteins and reduced red cell survival.
Some drugs (e.g. dapsone, sulfasalazine) cause oxidative haemolysis with Heinz bodies.
Some ingested chemicals (e.g. weedkillers such as sodium chlorate) can cause severe oxidative haemolysis leading to acute kidney injury.
Autoimmune haemolytic anaemias
Autoimmune haemolytic anaemias (AIHA) are acquired disorders resulting from increased red cell destruction due to red cell autoantibodies. These anaemias are characterized by the presence of a positive direct antiglobulin (Coombs’) test, which detects the autoantibody on the surface of the patient’s red cells (Fig. 8.27).
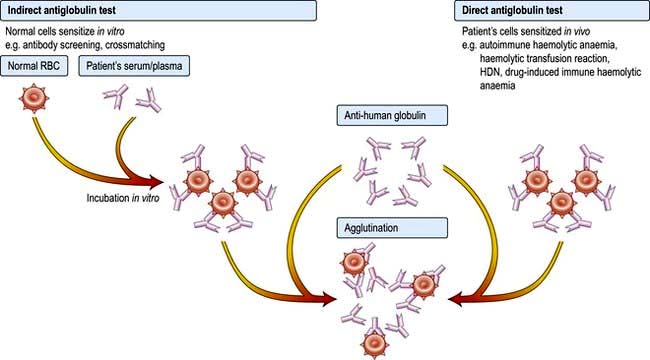
Figure 8.27 Antiglobulin (Coombs’) tests. The anti-human globulin forms bridges between the sensitized cells causing visible agglutination. The direct test detects patients’ cells sensitized in vivo. The indirect test detects normal cells sensitized in vitro. HDN, haemolytic disease of newborn.
AIHA is divided into ‘warm’ and ‘cold’ types, depending on whether the antibody attaches better to the red cells at body temperature (37°C) or at lower temperatures. The major features and the causes of these two forms of AIHA are shown in Table 8.14. In warm AIHA, IgG antibodies predominate and the direct antiglobulin test is positive with IgG alone, IgG and complement or complement only. In cold AIHA, the antibodies are usually IgM. They easily elute off red cells, leaving complement, which is detected as C3d.
Table 8.14 Causes and major features of autoimmune haemolytic anaemias
| Warm | Cold | |
|---|---|---|
Temperature at which antibody attaches best to red cells |
37°C |
Lower than 37°C |
Type of antibody |
IgG |
IgM |
Direct Coombs’ test |
Strongly positive |
Positive |
Causes of primary conditions |
Idiopathic |
Idiopathic |
Causes of secondary condition |
Autoimmune rheumatic disorders, e.g. SLE |
Infections, e.g. infectious mononucleosis, Mycoplasma pneumoniae, other viral infections (rare) |
Chronic lymphocytic leukaemia |
||
Lymphomas |
||
Hodgkin’s lymphoma |
||
Carcinomas |
||
Drugs, many including methyldopa, penicillins, cephalosporins, NSAIDs, quinine, interferon |
Immune destruction of red cells
IgM or IgG red cell antibodies which fully activate the complement cascade cause lysis of red cells in the circulation (intravascular haemolysis).
IgG antibodies frequently do not activate complement and the coated red cells undergo extravascular haemolysis (Fig. 8.28). They are either completely phagocytosed in the spleen through an interaction with Fc receptors on macrophages, or they lose part of the cell membrane through partial phagocytosis and circulate as spherocytes until they become sequestered in the spleen. Some IgG antibodies partially activate complement, leading to deposition of C3b on the red cell surface, and this may enhance phagocytosis as macrophages also have receptors for C3b.
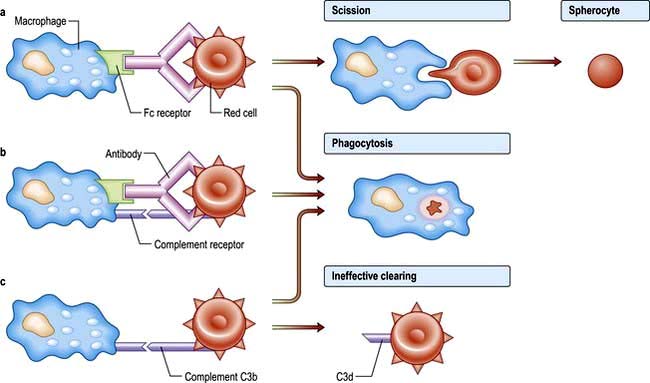
Figure 8.28 Extravascular haemolysis is due to interaction of antibody-coated cells with cells in the reticuloendothelial system, predominantly in the spleen. (a) Spherocytosis results from partial phagocytosis. (b) Complete phagocytosis may occur and this is enhanced if there is complement as well as antibody on the cell surface. (c) Cells coated with complement only are ineffectively removed and circulate with C3d or C3b on their surface.
Non-complement-binding IgM antibodies are rare and have little or no effect on red cell survival. IgM antibodies which partially rather than fully activate complement cause adherence of red cells to C3b receptors on macrophages, particularly in the liver, although this is an ineffective mechanism of haemolysis. Most of the red cells are released from the macrophages when C3b is cleaved to C3d and then circulate with C3d on their surface.
‘Warm’ autoimmune haemolytic anaemias
Clinical features
These anaemias may occur at all ages and in both sexes, although they are most frequent in middle-aged females. They can present as a short episode of anaemia and jaundice but they often remit and relapse and may progress to an intermittent chronic pattern. The spleen is often palpable. Infections or folate deficiency may provoke a profound fall in the haemoglobin level. Autoimmune haemolytic anaemias are primary or secondary. A history of blood transfusions and infections, exposure to drugs or vaccination and the general clinical condition make the secondary form likely. If there is a suspicion of a drug related anaemia then stopping the drug is the obvious measure. The clinical examination will reveal any lymphadenopathy or splenomegaly. The commonest underlying cause is a lymphoproliferative disorder (Table 8.14).
Investigations
Haemolytic anaemia is evident (see p. 387).
Spherocytosis is present as a result of red cell damage.
Direct antiglobulin test is positive, with either IgG alone (67%), IgG and complement (20%) or complement alone (13%) being found on the surface of the red cells.
Autoantibodies may have specificity for the Rh blood group system (e.g. for the e-antigen).
Autoimmune thrombocytopenia and/or neutropenia may also be present (Evans’ syndrome).
Abdominal CT scan for the detection of splenomegaly or abdominal lymphoma.
Treatment and prognosis
Corticosteroids (e.g. prednisolone in doses of 1 mg/kg daily) are effective in inducing a remission in about 80% of patients. Steroids reduce both production of the red cell autoantibody and destruction of antibody-coated cells. Splenectomy is the most effective second-line therapy. Other immunosuppressive drugs, such as azathioprine and rituximab, may be effective in patients who fail to respond to steroids and splenectomy. Blood transfusion may be necessary if there is severe anaemia although compatibility testing is complicated by the presence of red cell autoantibodies.
‘Cold’ autoimmune haemolytic anaemias
Normally, low titres of IgM cold agglutinins reacting at 4°C are present in plasma and are harmless. At low temperatures, these antibodies can attach to red cells and cause their agglutination in the cold peripheries of the body. In addition, activation of complement may cause intravascular haemolysis when the cells return to the higher temperatures in the core of the body.
After certain infections (such as Mycoplasma, cytomegalovirus, Epstein–Barr virus (EBV)), there is increased synthesis of polyclonal cold agglutinins producing a mild to moderate transient haemolysis.
Chronic cold haemagglutinin disease (CHAD)
This usually occurs in the elderly with a gradual onset of haemolytic anaemia owing to the production of monoclonal IgM cold agglutinins. After exposure to cold, the patient develops an acrocyanosis similar to Raynaud’s as a result of red cell autoagglutination.
Investigations
Red cells agglutinate in the cold or at room temperature. Agglutination is sometimes seen in the sample tube after cooling but is more easily seen on the peripheral blood film made at room temperature. The agglutination is reversible after warming the sample. The agglutination may cause a spurious increase in the MCV (see p. 376).
Cold agglutinin test (the titre is markedly elevated in CHAD to >1:512 )
Direct antiglobulin test is positive with complement (C3d) alone.
Monoclonal IgM antibodies with specificity for the Ii blood group system, usually for the I antigen but occasionally for the i antigen.
Treatment
The underlying cause should be treated, if possible. Patients should avoid exposure to cold. Steroids, alkylating agents and splenectomy are usually ineffective. Treatment with anti-CD20 (rituximab) has been successful in some cases. Blood transfusion may be necessary, and if so, the patient should be in a warm environment; compatibility testing may be difficult due to the cold agglutinin.
Paroxysmal cold haemoglobinuria (PCH)
This is a rare condition associated with common childhood infections, such as measles, mumps and chickenpox. Intravascular haemolysis is associated with polyclonal IgG complement-fixing antibodies. These antibodies are biphasic, reacting with red cells in the cold in the peripheral circulation, with lysis occurring due to complement activation when the cells return to the central circulation. The antibodies have specificity for the P red cell antigen. The lytic reaction is demonstrated in vitro by incubating the patient’s red cells and serum at 4°C and then warming the mixture to 37°C (Donath–Landsteiner test). Haemolysis is self-limiting but red cell transfusions may be necessary.
Drug-induced immune haemolytic anaemia
Drug-induced haemolytic anaemias are rare, although over 100 drugs have been reported to cause immune haemolytic anaemia. The interaction between a drug and red cell membrane may produce three types of antibodies:
Antibodies to the drug only, e.g. quinidine, rifampicin. Immune complexes attach to red cells, and may cause acute and severe intravascular haemolysis, sometimes associated with kidney injury. The haemolysis usually resolves quickly once the drug is withdrawn.
Antibodies to the cell membrane only, e.g. methyldopa, fludarabine. There is extravascular haemolysis and the clinical course tends to be more protracted.
Antibodies to part-drug, part-cell membrane, e.g. penicillin. This develops only in patients receiving large doses of penicillin. The haemolysis typically develops over 7–10 days, and recovery is gradual after drug withdrawal.
Confirmation of the diagnosis of drug-induced immune haemolytic anaemia requires:
A temporal association between administration of a drug and haemolytic anaemia
Recovery after withdrawal of the drug
The direct antiglobulin test should be positive
Drug-dependent red cell antibodies are detectable in the first and third mechanisms described above. In the second, the antibodies are not drug-dependent and are indistinguishable from autoantibodies.
Alloimmune haemolytic anaemia
Antibodies produced in one individual react with the red cells of another. This situation occurs in haemolytic disease of the newborn, haemolytic transfusion reactions (see p. 400) and after allogeneic bone marrow, renal, liver, cardiac or intestinal transplantation when donor lymphocytes transferred in the allograft (‘passenger lymphocytes’) may produce red cell antibodies against the recipient and cause haemolytic anaemia.
Haemolytic disease of the newborn (HDN)
HDN is due to fetomaternal incompatibility for red cell antigens. Maternal alloantibodies against fetal red cell antigens pass from the maternal circulation via the placenta into the fetus, where they destroy the fetal red cells. Only IgG antibodies are capable of transplacental passage from mother to fetus.
The most common type of HDN is that due to ABO incompatibility, where the mother is usually group O and the fetus group A.
HDN due to ABO incompatibility is usually mild and exchange transfusion is rarely needed. HDN due to RhD incompatibility has become much less common in developed countries following the introduction of anti-D prophylaxis (see below). HDN may be caused by antibodies against antigens in many blood group systems (e.g. other Rh antigens such as c and E, and Kell, Duffy and Kidd; see p. 407).
Sensitization occurs as a result of passage of fetal red cells into the maternal circulation (which most readily occurs at the time of delivery), so that first pregnancies are rarely affected. However, sensitization may occur at other times, for example after a miscarriage, ectopic pregnancy or blood transfusion, or due to episodes during pregnancy which cause transplacental bleeding such as amniocentesis, chorionic villus sampling and threatened miscarriage.
Clinical features
These vary from a mild haemolytic anaemia of the newborn to intrauterine death from 18 weeks’ gestation with the characteristic appearance of hydrops fetalis (hepatosplenomegaly, oedema and cardiac failure).
Kernicterus occurs owing to severe jaundice in the neonatal period, where the unconjugated (lipid-soluble) bilirubin exceeds 250 µmol/L and bile pigment deposition occurs in the basal ganglia. This can result in permanent brain damage, choreoathetosis and spasticity. In mild cases, it may present as deafness.
Investigations
Routine antenatal serology
All mothers should have their ABO and RhD groups determined and tested for atypical antibodies after attending the antenatal booking clinic. These tests should be repeated at 28 weeks’ gestation.
If an antibody is detected, its blood group specificity should be determined and the mother should be retested at least monthly. A rising antibody titre of IgG antibodies or a history of HDN in a previous pregnancy is an indication for referral to a fetal medicine unit.
Antenatal assessment and treatment
If a clinically significant antibody capable of causing HDN, e.g. ant-D, anti-c or anti-K, is detected, the father’s phenotype provides useful information to predict the likelihood of the fetus carrying the relevant red cell antigen. If the father is heterozygous, the genotype of the fetus can be determined from fetal DNA obtained by amniocentesis, chorionic villus sampling or fetal blood sampling. Soluble fetal DNA in maternal plasma can also be used for RhD typing, avoiding an invasive procedure.
The severity of anaemia is assessed by Doppler flow velocity of the fetal middle cerebral artery; measurement of bile pigments in the amniotic fluid is no longer routinely used. If the infant appears to have severe anaemia by non-invasive monitoring, ultrasound-guided fetal blood sampling is used to confirm this directly, and if necessary, an intravascular fetal transfusion of red cells is given.
Postnatal management
In mild cases, phototherapy may be used to convert bilirubin to water-soluble biliverdin. Biliverdin can be excreted by the kidneys and this therefore reduces the chance of kernicterus. In more severely affected cases, exchange transfusion may be necessary to replace the infant’s red cells and to remove bilirubin. Indications for exchange transfusion include:
a cord Hb of <120 g/L (normal cord Hb is 136–196 g/L)
a cord bilirubin of >60 µmol/L
Further exchange transfusions may be necessary to remove the unconjugated bilirubin.
The blood used for exchange transfusions should be ABO-compatible with the mother and infant, lack the antigen against which the maternal antibody is directed, be fresh (no more than 5 days from the day of collection) and be CMV-seronegative to prevent transmission of cytomegalovirus.
Prevention of RhD immunization in the mother
Anti-D should be given after delivery when all of the following are present:
There is no maternal anti-D detectable in the mother’s serum; i.e. the mother is not already immunized.
The dose is 500 IU of IgG anti-D intramuscularly within 72 hours of delivery. The Kleihauer test is used to assess the number of fetal cells in the maternal circulation. A blood film prepared from maternal blood is treated with acid, which elutes HbA. HbF is resistant to this treatment and can be seen when the film is stained with eosin. If large numbers of fetal red cells are present in the maternal circulation, a higher or additional dose of anti-D will be necessary.
It may be necessary to give prophylaxis to RhD-negative women at other times when sensitization may occur, e.g. after an ectopic pregnancy, threatened miscarriage or termination of pregnancy. The dose of anti-D is 250 IU before 20 weeks’ gestation and 500 IU after 20 weeks. A Kleihauer test should be carried out after 20 weeks to determine if more anti-D is required.
Of previously non-immunized RhD-negative women carrying RhD-positive fetuses, 1–2% became immunized by the time of delivery. Antenatal prophylaxis with anti-D has been shown to reduce the incidence of immunization during pregnancy, and its routine use has been implemented in the UK. It can be given as two doses of anti-D immunoglobulin of 500 IU or 1500 IU (one at 28 weeks’ and one at 34 weeks’ gestation) or as a single dose of 1500 IU either at 28 weeks’ or between 28 and 30 weeks’ gestation. Monoclonal anti-D could in principle replace polyclonal anti-D, which is collected from RhD-negative women immunized in pregnancy and deliberately immunized RhD-negative males, but it is likely to be some years before trials have been completed to confirm its safety and effectiveness.
Non-immune haemolytic anaemia
Paroxysmal nocturnal haemoglobinuria (PNH)
Paroxysmal nocturnal haemogloblinuria is a rare form of haemolytic anaemia which results from the clonal expression of haematopoietic stems cells that have mutations in the X-linked gene PIG-A. These mutations result in an impaired synthesis of glycosylphosphatidylinositol (GPI), which anchors many proteins to the cell surface such as decay accelerating factor (DAF; CD55) and membrane inhibitor of reactive lysis (MIRL; CD59) to cell membranes. CD55 and CD59 and other proteins are involved in complement degradation (at the C3 and C5 levels), and in their absence the haemolytic action of complement continues.
Clinical features
The major clinical signs are intravascular haemolysis, venous thrombosis and haemoglobinuria. Haemolysis may be precipitated by infection, iron therapy or surgery. Characteristically, only the urine voided at night and in the morning on waking is dark in colour, although the reason for this phenomenon is not clear. In severe cases all urine samples are dark. Urinary iron loss may be sufficient to cause iron deficiency. Some patients present insidiously with signs of anaemia and recurrent abdominal pains.
Venous thrombotic episodes may occur at atypical sites and severe thromboses may occur, for example in hepatic (Budd–Chiari syndrome), mesenteric or cerebral veins. The cause of the increased predisposition to thrombosis is not known, but may be due to complement-mediated activation of platelets deficient in CD55 and CD59. Another suggestion is that intravascular haemolysis, which releases haemoglobin in the plasma, lowers plasma nitric oxide, which causes the symptoms and venous thrombosis.
Investigations
Intravascular haemolysis is evident (see p. 387).
Flow cytometric analysis of red cells with anti-CD55 and anti-CD59.
Bone marrow is sometimes hypoplastic (or even aplastic) despite haemolysis.
Treatment and prognosis
PNH is a chronic disorder requiring supportive measures such as blood transfusions, which are necessary for patients with severe anaemia. However, treatment with eculizumab has revolutionized therapy. It is a recombinant humanized monoclonal antibody that prevents the cleavage of C5 (and therefore the formation of the membrane attack complex). It reduces intravascular haemolysis, haemoglobinuria, the need for transfusion and gives an improved quality of life. Vaccinate against Neisseria meningitidis 2 weeks before treatment.
Long-term anticoagulation may be necessary acutely for patients with recurrent thrombotic episodes. Its long-term value is unclear with the use of eculizumab. In patients with bone marrow failure, treatment options include immunosuppression with antilymphocyte globulin, ciclosporin or bone marrow transplantation. Bone marrow transplantation has been successfully carried out using either HLA-matched sibling donors in patients under the age of 50 or matched unrelated donors in patients under the age of 25.
The course of PNH is variable. PNH may transform into aplastic anaemia or acute leukaemia, but it may remain stable for many years and the PNH clone may even disappear, which must be taken into account if considering potentially dangerous treatments such as bone marrow transplantation. The median survival is 10–15 years.
Mechanical haemolytic anaemia
Red cells may be injured by physical trauma in the circulation. Direct injury may cause immediate cell lysis or be followed by resealing of the cell membrane with the formation of distorted red cells or ‘fragments’. These cells may circulate for a short period before being destroyed prematurely in the reticuloendothelial system.
The causes of mechanical haemolytic anaemia include:
Damaged artificial heart valves
March haemoglobinuria, where there is damage to red cells in the feet associated with prolonged marching or running
Microangiopathic haemolytic anaemia (MAHA), where fragmentation of red cells occurs in an abnormal microcirculation caused by malignant hypertension, eclampsia, haemolytic uraemic syndrome, thrombotic thrombocytopenic purpura, vasculitis or disseminated intravascular coagulation.