60 Drug discovery and development
Overview
With the development of the pharmaceutical industry towards the end of the 19th century, drug discovery became a highly focused and managed process. Discovering new drugs moved from the domain of inventive doctors to that of scientists hired for the purpose. Today, the bulk of modern therapeutics, and of modern pharmacology, is based on drugs that came from the laboratories of pharmaceutical companies, without which neither the practice of therapeutics nor the science of pharmacology would be more than a pale fragment of what they have become.
In this chapter, we describe in outline the main stages of the process, namely (i) the discovery phase, i.e. the identification of a new chemical entity as a potential therapeutic agent; and (ii) the development phase, during which the compound is tested for safety and efficacy in one or more clinical indications, and suitable formulations and dosage forms devised. The aim is to achieve registration by one or more regulatory authorities, to allow the drug to be marketed legally as a medicine for human use.
Our account is necessarily brief and superficial, and more detail can be found elsewhere (Rang, 2006).
The Stages of a Project
Figure 60.1 shows in an idealised way the stages of a ‘typical’ project, aimed at producing a marketable drug that meets a particular medical need (e.g. to retard the progression of Parkinson’s disease or cardiac failure, or to prevent migraine attacks).
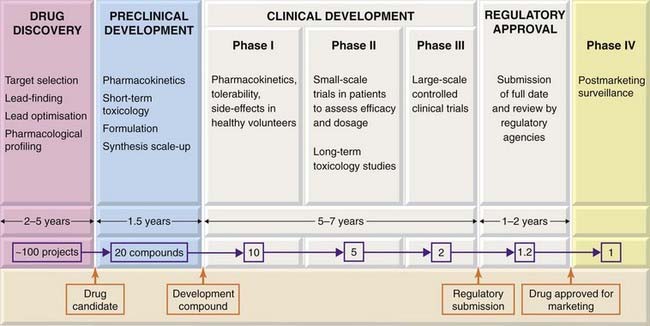
Fig. 60.1 The stages of development of a ‘typical’ new drug, i.e. a synthetic compound being developed for systemic use.
Only the main activities undertaken at each stage are shown, and the details vary greatly according to the kind of drug being developed.
Broadly, the process can be divided into three main components:
These phases do not necessarily follow in strict succession as indicated in Figure 60.1, but generally overlap.
The Drug Discovery Phase
Given the task of planning a project to discover a new drug to treat—say, Parkinson’s disease—where does one start? Assuming that we are looking for a novel drug rather than developing a slightly improved ‘me too’ version of a drug already in use,1 we first need to choose a new molecular target.
Target Selection
As discussed in Chapter 2, drug targets are, with few exceptions, functional proteins (e.g. receptors, enzymes, transport proteins). Although, in the past, drug discovery programmes were often based—successfully—on measuring a complex response in vivo, such as prevention of experimentally induced seizures, lowering of blood sugar or suppression of an inflammatory response, without the need for prior identification of a drug target, nowadays it is rare to start without a defined protein target, so the first step is target identification. This most often comes from biological intelligence. It was known, for example, that inhibiting angiotensin-converting enzyme lowers blood pressure by suppressing angiotensin formation, so it made sense to look for antagonists of the vascular angiotensin II receptor—hence the successful ‘sartan’ series of antihypertensive drugs (Ch. 22). Similarly, the knowledge that breast cancer is often oestrogen sensitive led to the development of aromatase inhibitors such as anastrazole, which prevents oestrogen synthesis. Current therapeutic drugs address about 120 distinct targets (see Hopkins & Groom, 2002; Rang, 2006), but there are still many proteins that are thought to play a role in disease for which we still have no cognate drug, and many of these represent potential starting points for drug discovery. Estimates range from a few hundred to several thousand potential drug targets that remain to be exploited therapeutically (see Betz, 2005). Selecting valid and ‘druggable’ targets from this plethora is a major challenge.
Conventional biological wisdom, drawing on a rich fund of knowledge of disease mechanisms and chemical signalling pathways, remains the basis on which novel targets are most often chosen. However, genomics is playing an increasing role by revealing new proteins involved in chemical signalling and new genes involved in disease. Space precludes discussion here of this burgeoning area; interested readers are referred to more detailed accounts (Lindsay, 2003; Kramer & Cohen, 2004; Betz, 2005; Rang, 2006).
Overall, it is evident that in the foreseeable future there is ample biological scope in terms of novel drug targets for therapeutic innovation. The limiting factor is not the biology and pharmacology, but other factors, such as the emergence of unexpected adverse effects during clinical testing, and the cost and complexity of drug discovery and development in relation to healthcare economics and increasing regulatory hurdles.
Lead Finding
When the biochemical target has been decided and the feasibility of the project has been assessed, the next step is to find lead compounds. The usual approach involves cloning of the target protein—normally the human form, because the sequence variation among species is often associated with pharmacological differences, and it is essential to optimise for activity in humans. An assay system must then be developed, allowing the functional activity of the target protein to be measured. This could be a cell-free enzyme assay, a membrane-based binding assay or a cellular response assay. It must be engineered to run automatically, if possible with an optical read-out (e.g. fluorescence or optical absorbance), and in a miniaturised multiwell plate format for reasons of speed and economy. Robotically controlled assay facilities capable of testing tens of thousands of compounds per day in several parallel assays are now commonplace in the pharmaceutical industry, and have become the standard starting point for most drug discovery projects. For details on high-throughput screening, see Sundberg (2000) and Hüser (2006).
To keep such hungry monsters running requires very large compound libraries. Large companies will typically maintain a growing collection of a million or more synthetic compounds, which will be routinely screened whenever a new assay is set up. Whereas, in the past, compounds were generally synthesised and purified one by one, often taking a week or more for each, the present tendency is to use combinatorial chemistry, which allows families of several hundreds or thousands of related compounds to be made simultaneously. By coupling such high-speed chemistry to high-throughput assay systems, the time taken over the initial lead-finding stage of projects has been reduced to a few months in most cases, having previously often taken several years. Despite the apparent mindlessness of the high-throughput random screening approach, it is often successful in identifying lead compounds that have the appropriate pharmacological activity and are amenable to further chemical modification. Building and maintaining huge compound libraries is, however, a costly business, and it has to be realised that even the largest practicable compound collection represents only a minute fraction of the number of ‘drug-like’ molecules that exists in theory—estimated at about 1060.
One problem with random screening is that many of the ‘hits’ detected in the initial screen turn out to be molecules that have features undesirable in a drug, such as too high a molecular weight, excessive polarity and possession of groups known to be associated with toxicity. Computational ‘prescreening’ of compound libraries is often used to eliminate such compounds.
The hits identified from the primary screen are used as the basis for preparing sets of homologues by combinatorial chemistry so as to establish the critical structural features necessary for binding selectively to the target. Several such iterative cycles of synthesis and screening are usually needed to identify one or more lead compounds for the next stage.
Natural products as lead compounds
Historically, natural products, derived mainly from fungal and plant sources, have proved to be a fruitful source of new therapeutic agents, particularly in the field of anti-infective, anticancer and immunosuppressant drugs. Familiar examples include penicillin, streptomycin and many other antibiotics; vinca alkaloids; paclitaxel; ciclosporin; and sirolimus (rapamycin). These substances presumably serve a specific protective function, having evolved so as to recognise with great precision vulnerable target molecules in an organism’s enemies or competitors. The surface of this resource has barely been scratched, and many companies are actively engaged in generating and testing natural product libraries for lead-finding purposes. Fungi and other microorganisms are particularly suitable for this, because they are ubiquitous, highly diverse, and easy to collect and grow in the laboratory. Compounds obtained from plants, animals or marine organisms are much more troublesome to produce commercially. The main disadvantage of natural products as lead compounds is that they are often complex molecules that are difficult to synthesise or modify by conventional synthetic chemistry, so that lead optimisation may be difficult and commercial production very expensive.
Lead Optimisation
Lead compounds found by random screening are the basis for the next stage, lead optimisation, where the aim (usually) is to increase the potency of the compound on its target and to optimise it with respect to other properties, such as selectivity and metabolic stability. In this phase, the tests applied include a broader range of assays on different test systems, including studies to measure the activity and time course of the compounds in vivo (where possible in animal models mimicking aspects of the clinical condition; see Ch. 7), and checking for unwanted effects in animals, evidence of genotoxicity and usually for oral absorption. The objective of the lead optimisation phase is to identify one or more drug candidates suitable for further development.
As shown in Figure 60.1, only about one project in five succeeds in generating a drug candidate, and it can take up to 5 years. The most common problem is that lead optimisation proves to be impossible; despite much ingenious and back-breaking chemistry, the lead compounds, like antisocial teenagers, refuse to give up their bad habits. In other cases, the compounds, although they produce the desired effects on the target molecule and have no other obvious defects, fail to produce the expected effects in animal models of the disease, implying that the target is probably not a good one. The virtuous minority proceed to the next phase, preclinical development.
Preclinical Development
The aim of preclinical development is to satisfy all the requirements that have to be met before a new compound is deemed ready to be tested for the first time in humans. The work falls into four main categories:
Much of the work of preclinical development, especially that relating to safety issues, is done under a formal operating code, known as Good Laboratory Practice (GLP), which covers such aspects as record-keeping procedures, data analysis, instrument calibration and staff training. The aim of GLP is to eliminate human error as far as possible, and to ensure the reliability of the data submitted to the regulatory authority, and laboratories are regularly monitored for compliance to GLP standards. The strict discipline involved in working to this code is generally ill-suited to the creative research needed in the earlier stages of drug discovery, so GLP standards are not usually adopted until projects get beyond the discovery phase.
Roughly half the compounds identified as drug candidates fail during the preclinical development phase; for the rest, a detailed dossier is prepared for submission to the regulatory authority such as the European Medicines Evaluation Agency or the US Food and Drugs Administration, whose permission is required to proceed with studies in humans. This is not lightly given, and the regulatory authority may refuse permission or require further work to be done before giving approval.
Non-clinical development work continues throughout the clinical trials period, when much more data, particularly in relation to long-term toxicity in animals, has to be generated. If a drug is intended for long-term use in the clinic, the toxicology studies may have to be extended for up to 2 years, and may include time-consuming studies for possible effects on fertility and fetal development. Failure of a compound at this stage is very costly, and considerable efforts are made to eliminate potentially toxic compounds much earlier in the drug discovery process by the use of in vitro, or even in silico, methods.
Clinical Development
Clinical development proceeds through four distinct phases (see Friedman et al., 1996, for details):
 The conduct of trials has to comply with an elaborate code known as Good Clinical Practice, covering every detail of the patient group, data collection methods, recording of information, statistical analysis and documentation.2
The conduct of trials has to comply with an elaborate code known as Good Clinical Practice, covering every detail of the patient group, data collection methods, recording of information, statistical analysis and documentation.2
Increasingly, phase III trials are being required to include a pharmacoeconomic analysis (see Ch. 1), such that not only clinical but also economic benefits of the new treatment are assessed.
At the end of phase III, the drug will be submitted to the relevant regulatory authority for licensing. The dossier required for this is a massive and detailed compilation of preclinical and clinical data. Evaluation by the regulatory authority normally takes a year or more, and further delays often arise when aspects of the submission have to be clarified or more data are required. Eventually, about two-thirds of submissions gain marketing approval. Overall, only 11.5% of compounds entering Phase I are eventually approved (see Munos, 2009). Increasing this proportion by better compound selection at the laboratory stage is one of the main challenges for the pharmaceutical industry.
Biopharmaceuticals
‘Biopharmaceuticals’, i.e. therapeutic agents produced by biotechnology rather than conventional synthetic chemistry, are discussed in Chapter 59. Such therapeutic agents comprise an increasing proportion—currently about 30%—of new products registered each year. The principles underlying the development and testing of biopharmaceuticals are basically the same as for synthetic drugs. In practice, biopharmaceuticals generally run into fewer toxicological problems than synthetic drugs,4 but more problems relating to production, quality control and drug delivery. Walsh (2003) covers this specialised field in more detail.
Commercial Aspects
Figure 60.1 shows the approximate time taken for such a project and the attrition rate (at each stage and overall) based on recent data from several large pharmaceutical companies. The key messages are (i) that it is a high-risk business, with only about one drug discovery project in 50 reaching its goal of putting a new drug on the market, (ii) that it takes a long time—about 12 years on average, and (iii) that it costs a lot of money to develop one drug (currently a mind-boggling $3.9 billion in 2008, see Munos, 2009).5 For any one project, the costs escalate rapidly as development proceeds, phase III trials and long-term toxicology studies being particularly expensive. The time factor is crucial, because the new drug has to be patented, usually at the end of the discovery phase, and the period of exclusivity (20 years in most countries) during which the company is free from competition in the market starts on that date. After 20 years, the patent expires, and other companies, which have not supported the development costs, are free to make and sell the drug much more cheaply, so the revenues for the original company decrease rapidly thereafter. Many profitable drugs will come to the end of their patents between 2010 and 2015, adding to the industry’s problems. Reducing the development time after patenting is a major concern for all companies, but so far it has remained stubbornly fixed at around 10 years, partly because the regulatory authorities are demanding more clinical data before they will grant a licence. In practice, only about one drug in three that goes on the market brings in enough revenue to cover its development costs. Success for the company relies on this one drug generating enough profit to pay for the rest.6
Future Prospects
Since about 1990, the drug discovery process has been in the throes of a substantial methodological revolution, following the rapid ascendancy of molecular biology, genomics and informatics, amid high expectations that this would bring remarkable dividends in terms of speed, cost and success rate. High-throughput screening has undoubtedly emerged as a powerful lead-finding technology, but overall the benefits are not yet clear: costs have risen steadily, the success rate has not improved (Fig. 60.2) and development times have not decreased.
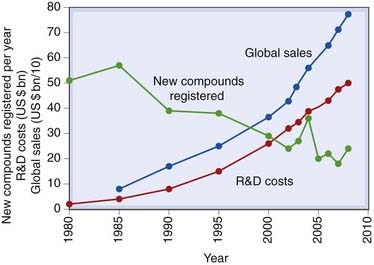
Fig. 60.2 Research and development (R&D) spend, sales and new drug registrations, 1980–2010.
Registrations refer to new chemical entities (including biopharmaceuticals, excluding new formulations and combinations of existing registered compounds).
(Data from various sources, including the Centre for Medicines Research, Pharmaceutical Research and Manufacturers Association of America.)
Figure 60.2 illustrates the steady decline in the number of new drugs launched in the major markets worldwide, despite escalating costs and improved technology. There has been much speculation as to the causes, the optimistic view (see below) being that fewer but better drugs are being introduced, and that the recent technological jump has yet to make its impact.
If the new drugs that are being developed improve the quality of medical care, there is room for optimism. In recent (‘prerevolutionary’) years, synthetic drugs aimed at new targets (e.g. selective serotonin reuptake inhibitors, statins and the kinase inhibitor imatinib) have made major contributions to patient care. Even if the new technologies do not improve productivity, we can reasonably expect that their ability to make new targets available to the drug discovery machine will have a real effect on patient care.
Trends to watch include the growing armoury of biopharmaceuticals, particularly monoclonal antibodies such as trastuzumab (an oestrogen receptor antibody used to treat breast cancer) and infliximab (a tumour necrosis factor antibody used to treat inflammatory disorders; see Ch. 26); these are successful recent examples, and more are in the pipeline. Another likely change will be the use of genotyping to ‘individualise’ drug treatments, so as to reduce the likelihood of administering drugs to ‘non-responders’ (see Ch. 11, which summarises the current status of ‘personalised medicine’). The implications for drug discovery will be profound, for the resulting therapeutic compartmentation of the patient population will mean that markets will decrease, bringing to an end the reliance on the ‘blockbusters’ referred to earlier. At the same time, clinical trials will become more complex (and expensive), as different genotypic groups will have to be included in the trial design. The hope is that therapeutic efficacy will be improved, not that it will be a route to developing drugs more cheaply and quickly. However, there is general agreement that the current modus operandi is commercially unsustainable (see Munos, 2009). Costs and regulatory requirements are continuing to rise, and the anticipated use of genomics to define subgroups of patients likely to respond to particular therapeutic agents (see Ch. 11) will mean fragmentation of the market, as we move away from the ‘one-drug-suits-all’ approach that has encouraged companies to focus their efforts on blockbuster drugs. More niche products targeted at smaller patient groups will be needed, though each costs as much to develop as a blockbuster and carries a similar risk of failure.
A Final Word
The pharmaceutical industry in recent years has attracted much adverse publicity, some of it well deserved, concerning drug pricing and profits, non-disclosure of adverse clinical trials data, reluctance to address major global heath problems such as tuberculosis and malaria, aggressive marketing practices and much else (see Angell, 2004). It needs to be remembered though that, despite its faults, the industry has been responsible for most of the therapeutic advances of the past half-century, without which medical care would effectively have stood still.
References and Further Reading
Angell M. The truth about the drug companies. New York: Random House; 2004. (A powerful broadside directed against the commercial practices of pharmaceutical companies)
Betz U.A.K. How many genomics targets can a portfolio afford? Drug Discov. Today. 2005;10:1057-1063. (Interesting analysis—despite its odd title—of approaches to target identification in drug discovery programmes)
Drews J. In quest of tomorrow’s medicines. New York: Springer; 1998. (Thoughtful and non-technical account of the history, principles and future directions of drug discovery)
Evans W.E., Relling M.V. Moving towards individualised medicine with pharmacogenomics. Nature. 2004;429:464-468. (Good review article discussing the likely influence of pharmacogenomics on therapeutics)
Friedman L.M., Furberg C.D., DeMets D.L. Fundamentals of clinical trials, third ed. St Louis: Mosby; 1996. (Standard textbook)
Hopkins A.L., Groom C.R. The druggable genome. Nat. Rev. Drug Discov.. 2002;1:727-730. (Interesting analysis of the potential number of drug targets represented in the human genome)
Hüser J., editor. High throughput screening in drug discovery. Vol. 35 of Methods and principles in drug discovery. Weinheim: Wiley-VCH, 2006. (Comprehensive textbook covering all aspects of this technology)
Kramer R., Cohen D. Functional genomics to new drug targets. Nat. Rev. Drug Discov.. 2004;3:965-972. (Describes the various approaches for finding new drug targets, starting from genomic data)
Lindsay M.A. Target discovery. Nat. Rev. Drug Discov.. 2003;2:831-836. (Well-balanced discussion of the application of genomics approaches to discovering new drug targets; more realistic in its stance than many)
Munos B. Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discov.. 2009;8:959-968. (Informative summary of the current status of the drug discovery industry, making clear that the modus operandi that has been successful in the past is no longer sustainable)
Rang H.P., editor. Drug discovery and development. Amsterdam: Elsevier, 2006. (Short textbook describing the principles and practice of drug discovery and development at the beginning of the 21st century)
Sundberg S.A. High-throughput and ultra-high-throughput screening: solution and cell-based approaches. Curr. Opin. Biotechnol.. 2000;11:47-53.
Walsh G. Biopharmaceuticals, second ed. Chichester: Wiley; 2003. (Comprehensive textbook covering all aspects of discovery, development and applications of biopharmaceuticals)
1Many commercially successful drugs have in the past emerged from exactly such ‘me too’ projects, examples being the dozen or so β-adrenoceptor-blocking drugs developed in the wake of propranolol, or the plethora of ‘triptans’ that followed the introduction of sumatriptan to treat migraine. Quite small improvements (e.g. in pharmacokinetics or side effects), coupled with aggressive marketing, have often proved enough, but the barriers to registration are getting higher, so the emphasis has shifted towards developing innovative (first in class) drugs aimed at novel molecular targets.
2Similar highly detailed codes must be followed in laboratory tests to determine safety (Good Laboratory Practice; see text) and drug manufacture (Good Manufacturing Practice).
3Recent high-profile cases include the withdrawal of rofecoxib (a cyclo-oxygenase-2 inhibitor; see Ch. 26) when it was found to increase the frequency of heart attacks, and of cerivastatin (Ch. 23), a cholesterol-lowering drug found to cause severe muscle damage in a few patients.
4The serious toxicity caused to human volunteers in the 2006 Phase I trials of the monoclonal antibody TGN 1412 (see Ch. 59) showed that this could not be relied on, and has led to substantial tightening of standards (and slowdown of the development of biopharmaceuticals).
5These cost estimates have been strongly challenged by commentators (see Angell, 2004) who argue that the pharmaceutical companies overestimate their costs several-fold in order to justify high drug prices.
6Actually, companies spend about twice as much on marketing and administration as on research and development.