22 The vascular system
Overview
This chapter is concerned with the pharmacology of blood vessels. The walls of arteries, arterioles, venules and veins contain smooth muscle whose contractile state is controlled by circulating hormones and by mediators released locally from sympathetic nerve terminals (Ch. 12) and endothelial cells. These work mainly by regulating Ca2+ in vascular smooth muscle cells, as described in Chapter 4. In the present chapter, we first consider the control of vascular smooth muscle by the endothelium and by the renin–angiotensin system, followed by the actions of vasoconstrictor and vasodilator drugs. Finally, we briefly consider clinical uses of vasoactive drugs in some important diseases, namely hypertension (pulmonary as well as systemic), heart failure, shock, peripheral vascular disease and Raynaud’s disease. The use of vasoactive drugs to treat angina is covered in Chapter 21.
Introduction
In this chapter we briefly describe the structure and function of the vascular system. Actions of drugs on the vascular system can be broken down into effects on:
Drug effects considered in this chapter are caused by actions on vascular smooth muscle cells. Like other muscles, vascular smooth muscle contracts when cytoplasmic Ca2+ ([Ca2+]i) rises, but the coupling between [Ca2+]i and contraction is less tight than in striated or cardiac muscle (Ch. 4). Vasoconstrictors and vasodilators act by increasing or reducing [Ca2+]i, and/or by altering the sensitivity of the contractile machinery to [Ca2+]i. Figure 4.10 summarises cellular mechanisms that are involved in the control of smooth muscle contraction and relaxation. The control of vascular smooth muscle tone by various mediators is described in other chapters (noradrenaline in Ch. 14, 5-HT in Ch. 15, prostanoids in Ch. 17, nitric oxide [NO] in Ch. 20, cardiac natriuretic peptides in Ch. 21, antidiuretic hormone in Ch. 32). Here we focus on endothelium-derived mediators and on the renin–angiotensin–aldosterone system, before describing the actions of vasoactive drugs and their uses in some important clinical disorders (hypertension, heart failure, shock, peripheral vascular disease and Raynaud’s disease).
Vascular Structure and Function
Blood is ejected with each heartbeat from the left ventricle into the aorta, whence it flows rapidly to the organs via large conduit arteries. Successive branching leads via muscular arteries to arterioles (endothelium surrounded by a layer of smooth muscle only one cell thick) and capillaries (naked tubes of endothelium), where gas and nutrient exchanges occur. Capillaries coalesce to form postcapillary venules, venules and progressively larger veins leading, via the vena cava, to the right heart. Deoxygenated blood ejected from the right ventricle travels through the pulmonary artery, pulmonary capillaries and pulmonary veins back to the left atrium.1 Small muscular arteries and arterioles are the main resistance vessels, while veins are capacity vessels that contain a large fraction of the total blood volume. In terms of cardiac function, therefore, arteries and arterioles regulate the afterload, while veins and pulmonary vessels regulate the preload of the ventricles.
Viscoelastic properties of large arteries determine arterial compliance (i.e. the degree to which the volume of the arterial system increases as the pressure increases). This is an important factor in a circulatory system that is driven by an intermittent pump such as the heart. Blood ejected from the left ventricle is accommodated by distension of the aorta, which absorbs the pulsations and delivers a relatively steady flow to the tissues. The greater the compliance of the aorta, the more effectively are fluctuations damped out,2 and the smaller the oscillations of arterial pressure with each heartbeat (i.e. the difference between the systolic and diastolic pressure, known as the ‘pulse pressure’). Reflection of the pressure wave from branch points in the vascular tree also sustains arterial pressure during diastole. In young people, this helps to preserve a steady perfusion of vital organs, such as the kidney, during diastole.
However, excessive reflection can pathologically augment aortic systolic pressure. This results from stiffening of the aorta due to loss of elastin during ageing, especially in people with hypertension. Elastin is replaced by inelastic collagen. Cardiac work (see Ch. 21) can be reduced by increasing arterial compliance or by reducing arterial wave reflection, even if the cardiac output and mean arterial pressure are unchanged. Over around 55 years of age, pulse pressure and aortic stiffness are important risk factors for cardiac disease.
Control of Vascular Smooth Muscle Tone
Two important physiological systems regulate vascular tone, namely the vascular endothelium and the renin–angiotensin system.
The Vascular Endothelium
A new chapter in our understanding of vascular control opened with the discovery that vascular endothelium acts not only as a passive barrier between plasma and extracellular fluid, but also as a source of numerous potent mediators. These actively control the contraction of the underlying smooth muscle as well as influencing platelet and mononuclear cell function: the roles of the endothelium in haemostasis and thrombosis are discussed in Chapter 24. Several distinct classes of mediator are involved (Fig. 22.1).
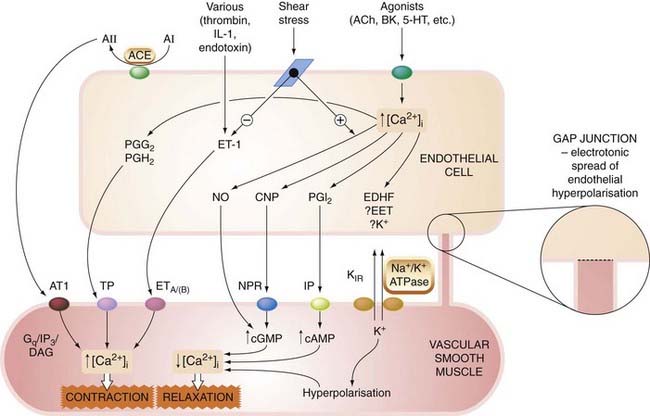
Fig. 22.1 Endothelium-derived mediators.
The schematic shows some of the more important endothelium-derived contracting and relaxing mediators; many (if not all) of the vasoconstrictors also cause smooth muscle mitogenesis, while vasodilators commonly inhibit mitogenesis. 5-HT, 5-hydroxytryptamine; A, angiotensin; ACE, angiotensin-converting enzyme; ACh, acetylcholine; AT1, angiotensin AT1 receptor; BK, bradykinin; CNP, C-natriuretic peptide; DAG, diacylglycerol; EDHF, endothelium-derived hyperpolarising factor; EET, epoxyeicosatetraenoic acid; ET-1, endothelin-1; ETA/(B), endothelium A (and B) receptors; Gq, G-protein; IL-1, interleukin-1; IP, I prostanoid receptor; IP3, inosinol 1,4,5-trisphosphate; KIR, inward rectifying potassium channel; Na+/K+ ATPase, electrogenic pump; NPR, natriuretic peptide receptor; PG, prostaglandin; TP, T prostanoid receptor.
Vascular smooth muscle 
In addition to secreting vasoactive mediators, endothelial cells express several enzymes and transport mechanisms that act on circulating hormones and are important targets of drug action. ACE is a particularly important example (see below).
Many endothelium-derived mediators are mutually antagonistic, conjuring an image of opposing rugby football players swaying back and forth in a scrum; in moments of exasperation, one sometimes wonders whether all this makes sense or whether the designer simply could not make up her mind! An important distinction is made between mechanisms that are tonically active in resistance vessels under basal conditions, as is the case with the noradrenergic nervous system (Ch. 14), NO (Ch. 20) and endothelin (see below), and those that operate mainly in response to injury, inflammation, etc., as with PGI2. Some of the latter group may be functionally redundant, perhaps representing vestiges of mechanisms that were important to our evolutionary forebears, or they may simply be taking a breather on the touchline and are ready to rejoin the fray if called on by the occurrence of some vascular insult. Evidence for such a ‘back-up’ role comes, for example, from mice that lack the IP receptor for PGI2, and that have a normal blood pressure and do not develop spontaneous thrombosis, but are more susceptible to vasoconstrictor and thrombotic stimuli than their wild-type litter mates (Murata et al., 1997).
The Endothelium in Angiogenesis
As touched on in Chapter 8, the barrier function of vascular endothelium differs markedly in different organs, and its development during angiogenesis is controlled by several growth factors, including vascular endothelial growth factor (VEGF) and various tissue-specific factors such as endocrine gland VEGF. These are involved in repair processes and in pathogenesis (e.g. tumour growth and in neovascularisation in the eye—an important cause of blindness in patients with diabetes mellitus). These factors and their receptors are potentially fruitful targets for drug development and new therapies (including gene therapies; Ch. 59).
Endothelin
Discovery, biosynthesis and secretion
Hickey et al. described a vasoconstrictor factor produced by cultured endothelial cells in 1985. This was identified as endothelin, a 21-residue peptide, by Yanagisawa et al. (1988), who achieved the isolation, analysis and cloning of the gene for this peptide, which at that time was the most potent vasoconstrictor known,3 in an impressively short space of time.
 Three genes encode different sequences (ET-1, ET-2 and ET-3), each with a distinctive ‘shepherd’s crook’ structure produced by two internal disulfide bonds. These isoforms are differently expressed in organs such as brain and adrenal glands (Table 22.1), suggesting that endothelins have functions beyond the cardiovascular system, and this is supported by observations of mice in which the gene coding for ET-1 is disrupted (see below). ET-1 is the only endothelin present in endothelial cells, and is also expressed in many other tissues. Its synthesis and actions are summarised schematically in Figure 22.2. ET-2 is much less widely distributed: it is present in kidney and intestine. ET-3 is present in brain, lung, intestine and adrenal gland. ET-1 is synthesised from a 212-residue precursor molecule (prepro-ET), which is processed to ‘big ET-1’ and finally cleaved by an endothelin-converting enzyme to yield ET-1. Cleavage occurs not at the usual Lys–Arg or Arg–Arg position, but at a Trp–Val pair, implying a very atypical endopeptidase. The converting enzyme is a metalloprotease and is inhibited by phosphoramidon (a pharmacological tool but not used therapeutically). Big ET-1 is converted to ET-1 intracellularly and also on the surface of endothelial and smooth muscle cells.
Three genes encode different sequences (ET-1, ET-2 and ET-3), each with a distinctive ‘shepherd’s crook’ structure produced by two internal disulfide bonds. These isoforms are differently expressed in organs such as brain and adrenal glands (Table 22.1), suggesting that endothelins have functions beyond the cardiovascular system, and this is supported by observations of mice in which the gene coding for ET-1 is disrupted (see below). ET-1 is the only endothelin present in endothelial cells, and is also expressed in many other tissues. Its synthesis and actions are summarised schematically in Figure 22.2. ET-2 is much less widely distributed: it is present in kidney and intestine. ET-3 is present in brain, lung, intestine and adrenal gland. ET-1 is synthesised from a 212-residue precursor molecule (prepro-ET), which is processed to ‘big ET-1’ and finally cleaved by an endothelin-converting enzyme to yield ET-1. Cleavage occurs not at the usual Lys–Arg or Arg–Arg position, but at a Trp–Val pair, implying a very atypical endopeptidase. The converting enzyme is a metalloprotease and is inhibited by phosphoramidon (a pharmacological tool but not used therapeutically). Big ET-1 is converted to ET-1 intracellularly and also on the surface of endothelial and smooth muscle cells.

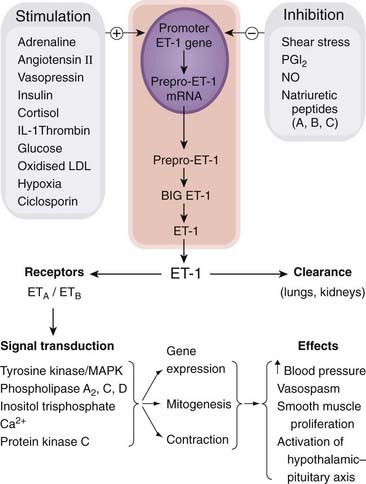
Fig. 22.2 Endothelin-1 (ET-1) synthesis and actions.
The schematic shows some of the more important actions only. IL-1, interleukin-1; LDL, low-density lipoprotein; MAPK, mitogen-activated protein kinase; NO, nitric oxide; PGI2, prostaglandin I2.
Stimuli of endothelin synthesis include many vasoconstrictor mediators released by trauma or inflammation, including activated platelets, endotoxin, thrombin, various cytokines and growth factors, angiotensin II, antidiuretic hormone (ADH), adrenaline, insulin, hypoxia and low shear stress. Inhibitors of ET synthesis include NO, natriuretic peptides, PGE2, PGI2, heparin and high shear stress.
Release mechanisms of ET-1 are poorly understood. There is evidence that preformed ET-1 can be stored in endothelial cells, although probably not in granules. ET-1 concentration in plasma is low (< 5 pmol/l) compared with concentrations that activate endothelin receptors, but concentrations in the extracellular space between endothelium and vascular smooth muscle are presumably much higher, and endothelin receptor antagonists (see below) cause vasodilatation when infused directly into the brachial artery, consistent with tonic ET-1-mediated vasoconstrictor activity in resistance vasculature. ET-1 has an elimination half life of < 5 min, despite a much longer duration of action, and clearance occurs mainly in the lung and kidneys.
Endothelin receptors and responses
There are two types of endothelin receptor, designated ETA and ETB (Table 22.2), both of which are G-protein coupled (Ch. 3). The predominant overall response is vasoconstriction.
Table 22.2 Endothelin receptors
| Receptor | Affinity | Pharmacological response |
|---|---|---|
| ETA | ET-1 = ET-2 > ET-3 | Vasoconstriction, bronchoconstriction, stimulation of aldosterone secretion |
| ETB | ET-1 = ET-2 = ET-3 | Vasodilatation, inhibition of ex vivo platelet aggregation |
(From Masaki T 1993 Endocr Rev 14: 256–268.)
Endothelin-1 preferentially activates ETA receptors. Messenger RNA for the ETA receptor is expressed in many human tissues, including vascular smooth muscle, heart, lung and kidney. It is not expressed in endothelium. ETA-mediated responses include vasoconstriction, bronchoconstriction and aldosterone secretion. ETA receptors are coupled to phospholipase C, which stimulates Na+/H+ exchange, protein kinase C and mitogenesis, as well as causing vasoconstriction through inositol trisphosphate-mediated Ca2+ release (Ch. 3). There are several partially selective ETA-receptor antagonists, including BQ-123 (a cyclic pentapeptide) and several orally active non-peptide drugs (e.g. bosentan, a mixed ETA/ETB antagonist used in treating pulmonary arterial hypertension—see below). ETB receptors are activated to a similar extent by each of the three endothelin isoforms, but sarafotoxin S6c (a 21-residue peptide that shares the shepherd’s crook structure of the endothelins and was isolated from the venom of the burrowing asp) is a selective agonist and has proved useful as a pharmacological tool for studying the ETB receptor. Messenger RNA for the ETB receptor is mainly expressed in brain (especially cerebral cortex and cerebellum), with moderate expression in aorta, heart, lung, kidney and adrenals. In contrast to the ETA receptor, it is highly expressed in endothelium, where it may initiate vasodilatation by stimulating NO and PGI2 production, but it is also present in vascular smooth muscle, where it initiates vasoconstriction like the ETA receptor. ETB receptors play a part in clearing ET-1 from the circulation, and ET antagonists with appreciable affinity for ETB receptors consequently increase plasma concentrations of ET-1, complicating interpretation of experiments with these drugs.
Functions of endothelin
Endothelin-1 is a paracrine mediator rather than a circulating hormone, although it stimulates secretion of several hormones (see below). Administration of an ETA-receptor antagonist or of phosphoramidon into the brachial artery increases forearm blood flow, and ETA receptor antagonists lower arterial blood pressure, suggesting that ET-1 contributes to vasoconstrictor tone and the control of peripheral vascular resistance. Endothelins have several other possible functions, including roles in:
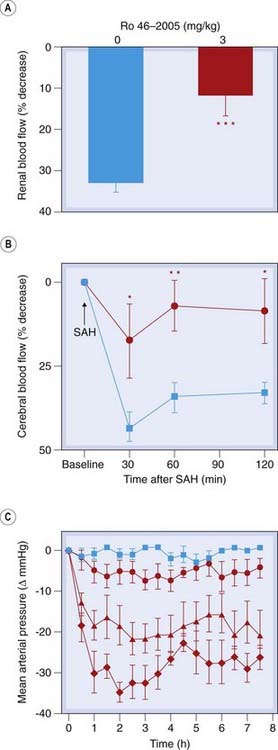
Fig. 22.3 In vivo effects of a potent non-peptide endothelin-1 ETA- and ETB-receptor antagonist, Ro 46-2005, in three animal models.
[A] Prevention by Ro 46-2005 of postischaemic renal vasoconstriction in rats. [B] Prevention by Ro 46-2005 of the decrease in cerebral blood flow after subarachnoid haemorrhage (SAH) in rats treated with placebo (blue) or with Ro 46-2005 (red). [C] Effect of orally administered Ro 46-2005 on mean arterial pressure in sodium-depleted squirrel monkeys treated with placebo (blue) or increasing doses of antagonist (red: • <  < ♦.
< ♦.
(From Clozel M et al. 1993 Nature 365: 759–761.)
The role of the endothelium in controlling vascular smooth muscle
The Renin–Angiotensin System
The renin–angiotensin system synergises with the sympathetic nervous system, for example by increasing the release of noradrenaline from sympathetic nerve terminals. It stimulates aldosterone secretion and plays a central role in the control of Na+ excretion and fluid volume, as well as of vascular tone.
The control of renin secretion (Fig. 22.4) is only partly understood. It is a proteolytic enzyme that is secreted by the juxtaglomerular apparatus (see Fig. 28.2) in response to various physiological stimuli including reduced renal perfusion pressure, or reduced Na+ concentration in distal tubular fluid which is sensed by the macula densa (a specialised part of the distal tubule apposed to the juxtaglomerular apparatus). Renal sympathetic nerve activity, β-adrenoceptor agonists and PGI2 all stimulate renin secretion directly, whereas angiotensin II causes feedback inhibition. Atrial natriuretic peptide (Ch. 21) also inhibits renin secretion. Renin is cleared rapidly from plasma. It acts on angiotensinogen (a plasma globulin made in the liver), splitting off a decapeptide, angiotensin I.
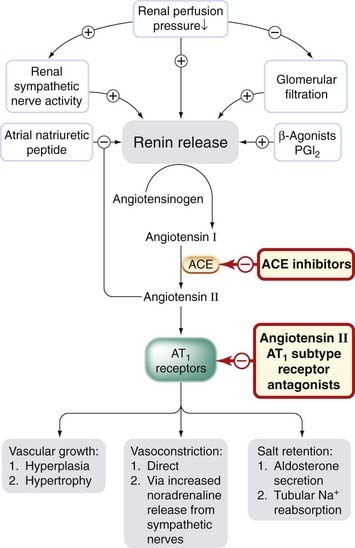
Fig. 22.4 Control of renin release and formation, and action of angiotensin II.
Sites of action of drugs that inhibit the cascade are shown. ACE, angiotensin-converting enzyme; AT1, angiotensin II receptor subtype 1.
Angiotensin I has no appreciable activity per se, but is converted by angiotensin-converting enzyme (ACE) to an octapeptide, angiotensin II, which is a potent vasoconstrictor. Angiotensin II is a substrate for enzymes (aminopeptidase A and N) that remove single amino acid residues, giving rise, respectively, to angiotensin III and angiotensin IV (Fig. 22.5). These had been regarded as of little importance, but it is now known that angiotensin III stimulates aldosterone secretion and is involved in thirst. Angiotensin IV also has distinct actions, probably via its own receptor, including release of plasminogen activator inhibitor-1 from the endothelium (Ch. 21). Receptors for angiotensin IV have a distinctive distribution, including the hypothalamus.
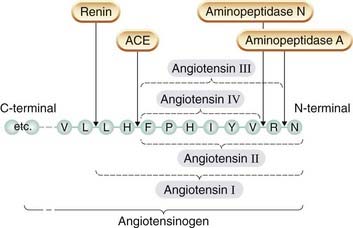
Fig. 22.5 Formation of angiotensins I–IV from the N-terminal of the precursor protein angiotensinogen.
Angiotensin-converting enzyme is a membrane-bound enzyme on the surface of endothelial cells, and is particularly abundant in the lung, which has a vast surface area of vascular endothelium.4 The common isoform of ACE is also present in other vascular tissues, including heart, brain, striated muscle and kidney, and is not restricted to endothelial cells.5 Consequently, local formation of angiotensin II can occur in different vascular beds, and it provides local control independent of blood-borne angiotensin II. ACE inactivates bradykinin (see Ch. 19) and several other peptides. This may contribute to the pharmacological actions of ACE inhibitors, as discussed below. The main actions of angiotensin II are mediated via AT1 and/or AT2 receptors, which belong to the family of G-protein-coupled receptors. Effects mediated by AT1 receptors include:
AT2 receptors are expressed during fetal life and in distinct brain regions in adults. They may be involved in growth, development and exploratory behaviour. Cardiovascular effects of AT2 receptors (inhibition of cell growth and lowering of blood pressure) are relatively subtle and oppose those of AT1 receptors.
The renin–angiotensin–aldosterone pathway contributes to the pathogenesis of heart failure, and several leading classes of therapeutic drug act on it at different points (see below).
Vasoactive Drugs
Drugs can affect vascular smooth muscle by acting either directly on smooth muscle cells, or indirectly, for example on endothelial cells, on sympathetic nerve terminals or on the central nervous system (CNS) (Table 22.3). Mechanisms of directly acting vasoconstrictors and vasodilators are summarised in Figure 4.10. Many indirectly acting drugs are discussed in other chapters (see Table 22.3). We concentrate here on agents that are not covered elsewhere.
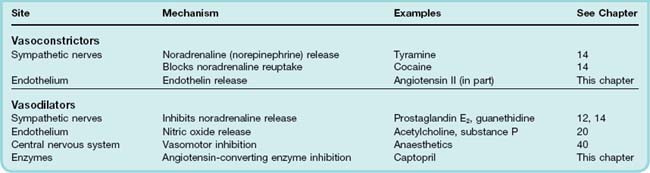
Vasoconstrictor Drugs
The α1-adrenoceptor agonists and drugs that release noradrenaline from sympathetic nerve terminals or inhibit its reuptake (sympathomimetic amines) cause vasoconstriction and are discussed in Chapter 14. Some eicosanoids (e.g. thromboxane A2; see Chs 17 and 24) and several peptides, notably endothelin, angiotensin and ADH, are also predominantly vasoconstrictor. Sumatriptan and ergot alkaloids acting on certain 5-hydroxytryptamine receptors (5-HT2 and 5-HT1D) also cause vasoconstriction (Ch. 15).
Angiotensin II
The physiological role of the renin–angiotensin system is described above. Angiotensin II is roughly 40 times as potent as noradrenaline in raising blood pressure. Like α1-adrenoceptor agonists, it constricts mainly cutaneous, splanchnic and renal vasculature, with less effect on blood flow to brain and skeletal muscle. It has no routine clinical uses, its therapeutic importance lying in the fact that other drugs (e.g. captopril and losartan, see below) affect the cardiovascular system by reducing its production or action.
Antidiuretic Hormone
Antidiuretic hormone (ADH, also known as vasopressin) is a posterior pituitary peptide hormone (Ch. 32). It is important for its antidiuretic action on the kidney (Ch. 28) but is also a powerful vasoconstrictor in skin and some other vascular beds. Its effects are initiated by two distinct receptors (V1 and V2). Water retention is mediated through V2 receptors, occurs at low plasma concentrations of ADH and involves activation of adenylyl cyclase in renal collecting ducts. Vasoconstriction is mediated through V1 receptors, requires higher concentrations of ADH and involves activation of phospholipase C (see Ch. 3). ADH causes generalised vasoconstriction, including the coeliac, mesenteric and coronary vessels. It also affects other (e.g. gastrointestinal and uterine) smooth muscle and causes abdominal cramps for this reason. It is sometimes used to treat patients with bleeding oesophageal varices and portal hypertension before more definitive treatment, although many gastroenterologists prefer to use octreotide (unlicensed indication; see Ch. 32) for this. It may also have a place in treating hypotensive shock (see below).
Endothelin
Endothelins are discussed above in the context of their physiological roles; as explained above, they have vasodilator and vasoconstrictor actions, but vasoconstriction predominates. Intravenous administration causes transient vasodilatation followed by profound and long-lived vasoconstriction. The endothelins are even more potent vasoconstrictors than angiotensin II. As yet, they have no clinical uses, and ET antagonists are licensed only for the rare disease primary pulmonary hypertension (see below).
Vasoconstrictor substances
Vasodilator Drugs
Vasodilator drugs play a major role in the treatment of common conditions including hypertension, cardiac failure and angina pectoris, as well as several less common but severe diseases including pulmonary hypertension and Raynaud’s disease.
Directly Acting Vasodilators
Targets on which drugs act to relax vascular smooth muscle include plasma membrane voltage-dependent calcium channels, sarcoplasmic reticulum channels (Ca2+ release or reuptake) and enzymes that determine Ca2+ sensitivity of the contractile proteins (see Fig. 4.10).7
Calcium antagonists
L-type calcium antagonists are discussed in Chapter 21. As well as their actions on the heart they cause generalised arterial vasodilatation, although individual agents exhibit distinct patterns of regional potency. Dihydropyridines (e.g. nifedipine) act preferentially on vascular smooth muscle, whereas verapamil acts directly on the heart (negative chronotropic and inotropic effects) in addition to causing vasodilatation; diltiazem is intermediate in specificity. Consequently, rapid-acting dihydropyridines usually produce reflex tachycardia, whereas verapamil and diltiazem do not.
Drugs that activate potassium channels
Some drugs (e.g. minoxidil, diazoxide) relax smooth muscle by opening KATP channels (see Fig. 22.6). This hyperpolarises the cells and switches off voltage-dependent calcium channels.8 Potassium channel activators work by antagonising the action of intracellular ATP on these channels.
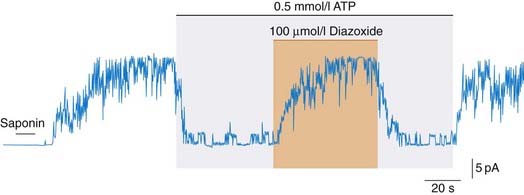
Fig. 22.6 ATP-sensitive potassium channels.
Patch clamp (see Ch. 3) record from insulin-secreting pancreatic B cell: saponin permeabilised the cell, with loss of intracellular ATP, causing the channels to open (upward deflection) until they were inhibited by ATP. Addition of diazoxide, a vasodilator drug (which also inhibits insulin secretion; see text) reopens the channels. In smooth muscle, this causes hyperpolarisation and relaxation.
(Redrawn from Dunne et al. 1990 Br J Pharmacol 99: 169.)
Minoxidil is a very potent and long-acting vasodilator, used as a drug of last resort in treating severe hypertension unresponsive to other drugs. It causes hirsutism (its active metabolite is actually used as a rub-on cream to treat baldness). It also causes marked salt and water retention, and is usually prescribed with a loop diuretic. It causes reflex tachycardia, and a β-adrenoceptor antagonist is used to prevent this. Nicorandil (Ch. 21) combines KATP channel activation with NO donor activity, and is used in refractory angina. Levosimendan combines KATP channel activation with sensitisation of the cardiac contractile mechanism to Ca2+ by binding troponin C (Ch. 21), and is used in decompensated heart failure (see below).
Drugs that act via cyclic nucleotides
Cyclase activation
Many drugs relax vascular smooth muscle by increasing the cellular concentration of either cGMP or cAMP. For example, NO, nitrates and the natriuretic peptides act through cGMP (see Chs 20 and 21); BAY41-2272, a pyrazolopyridine, activates soluble guanylyl cyclase via an NO-independent site (see Ch. 20). The β2 agonists, adenosine and PGI2 increase cytoplasmic cAMP (see Ch. 14). Dopamine has mixed vasodilator and vasoconstrictor actions. It selectively dilates renal vessels, where it increases cAMP by activating adenylyl cyclase. It is the precursor of noradrenaline (Ch. 14), and is also a transmitter in its own right in the brain (Ch. 38) and probably also in the periphery (Ch. 12). Dopamine, when administered as an intravenous infusion, produces a mixture of cardiovascular effects resulting from agonist actions on α- and β-adrenoceptors, as well as on dopamine receptors. Blood pressure increases slightly, but the main effects are vasodilatation in the renal circulation and increased cardiac output. Dopamine was widely used in intensive care units in patients in whom renal failure associated with decreased renal perfusion appeared imminent; despite its beneficial effect on renal haemodynamics, it does not, however, improve survival in these circumstances and this use is obsolete (Liang et al., 2008). Nesiritide, a recombinant form of human B-type natriuretic peptide (BNP) (see Ch. 21), has been approved in the USA for the treatment of acutely decompensated heart failure, but a pooled analysis of randomised controlled trials has suggested that it too may increase mortality, and indications for its use remain controversial (Potter et al., 2009).
Nitroprusside (nitroferricyanide) is a powerful vasodilator with little effect outside the vascular system. It reacts with tissue sulfhydryl groups under physiological conditions to yield NO. Unlike the organic nitrates, which preferentially dilate capacitance vessels and muscular arteries, it acts equally on arterial and venous smooth muscle. Its clinical usefulness is limited because it must be given intravenously. In solution, particularly when exposed to light, nitroprusside hydrolyses with formation of cyanide. The intravenous solution must therefore be made up freshly from dry powder and protected from light. Nitroprusside is rapidly converted to thiocyanate in the body, its plasma half-life being only a few minutes, so it must be given as a continuous infusion with careful monitoring to avoid hypotension. Prolonged use causes thiocyanate accumulation and toxicity (weakness, nausea and inhibition of thyroid function); consequently, nitroprusside is useful only for short-term treatment (usually up to 72 h maximum). It is used in intensive care units for hypertensive emergencies, to produce controlled hypotension during surgery, and to reduce cardiac work during the reversible cardiac dysfunction that occurs after cardiopulmonary bypass surgery.
Phosphodiesterase inhibition
Phosphodiesterases (PDEs; see Ch. 3) include at least 14 distinct isoenzymes. Methylxanthines (e.g. theophylline) and papaverine are non-selective PDE inhibitors (and have other actions, too). Methylxanthines exert their main effects on bronchial smooth muscle and on the CNS, and are discussed in Chapters 27 and 47. In addition to inhibiting PDE, some methylxanthines are also purine receptor antagonists (Ch. 16). Papaverine is produced by opium poppies (see Ch. 41) and is chemically related to morphine. However, pharmacologically it is quite unlike morphine, its main action being to relax smooth muscle. Its mechanism is poorly understood but seems to involve a combination of PDE inhibition and block of calcium channels. Selective PDE type III inhibitors (e.g. milrinone) increase cAMP in cardiac muscle. They have a positive inotropic effect but, despite short-term haemodynamic improvement, increase mortality in heart failure, possibly by causing dysrhythmias. Cilostazol, a related drug, improves symptoms in patients with peripheral vascular disease (see below). Dipyridamole, as well as enhancing the actions of adenosine (see Ch. 16), also causes vasodilatation by inhbiting phosphodiesterase. It is used to prevent stroke, but can provoke angina. Selective PDE type V inhibitors (e.g. sildenafil) inhibit the breakdown of cGMP. Penile erection is caused by increased activity in nitrergic nerves in the pelvis. These release NO (Ch. 20), which activates guanylyl cyclase in smooth muscle in the corpora cavernosa. Taken by mouth about an hour before sexual stimulation, sildenafil increases penile erection by potentiating this pathway. It has revolutionised treatment of erectile dysfunction (see Ch. 34) and has therapeutic potential in other situations, including pulmonary hypertension (see below) via potentiation of NO-mediated effects.
Vasodilators with Unknown Mechanism of Action
Hydralazine
Hydralazine acts mainly on arteries and arterioles, causing a fall in blood pressure accompanied by reflex tachycardia and an increased cardiac output. It interferes with the action of inositol trisphosphate on Ca2+ release from the sarcoplasmic reticulum. Its original clinical use was in hypertension. It is still used for short-term treatment of severe hypertension in pregnancy but can cause an immune disorder resembling systemic lupus erythematosus,9 so alternative agents are now usually preferred for long-term treatment of hypertension. It has a place in treating heart failure in patients of African origin in combination with a long-acting organic nitrate (see below).
Indirectly Acting Vasodilator Drugs
The two main groups of indirectly acting vasodilator drugs are inhibitors of:
The central control of sympathetically mediated vasoconstriction is believed to involve not only α2 adrenoceptors but also another class of receptor, termed the imidazoline I1 receptor, present in the brain stem in the rostral ventrolateral medulla. Drugs can inhibit the sympathetic pathway at any point from the CNS to the peripheral sympathetic nerve terminal (see Ch. 14). In addition, many vasodilators (e.g. acetylcholine, bradykinin, substance P) exert some or all of their effects by stimulating biosynthesis of vasodilator prostaglandins or of NO (or of both) by vascular endothelium (see above and Ch. 20), thereby causing functional antagonism of the constrictor tone caused by sympathetic nerves and angiotensin II.
The renin–angiotensin–aldosterone system (RAAS—see Table 22.4 for a summary of selective antagonists) can be inhibited at several points:
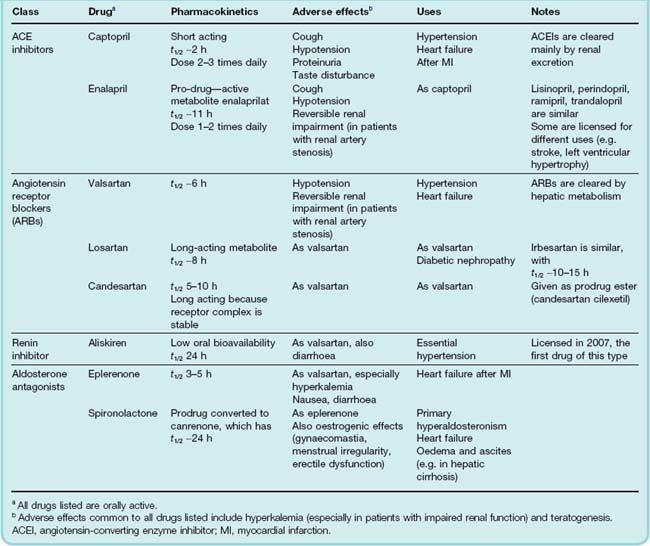
Renin inhibitors
Orally active renin inhibitors reduce plasma renin activity. One such drug, aliskiren, is licensed for essential hypertension.
Angiotensin-converting enzyme inhibitors
The first ACEI to be marketed was captopril (Fig. 22.7), an early example of successful drug design based on a chemical knowledge of the target molecule. Various small peptides had been found to be weak inhibitors of the enzyme.10 Captopril was designed to combine the steric properties of such peptide antagonists in a non-peptide molecule that was active when given by mouth. Captopril has a short plasma half-life (about 2 h) and must be given 2 or 3 times daily. Later ACE inhibitors (Table 22.4), which are widely used in the clinic, have a longer duration of action.
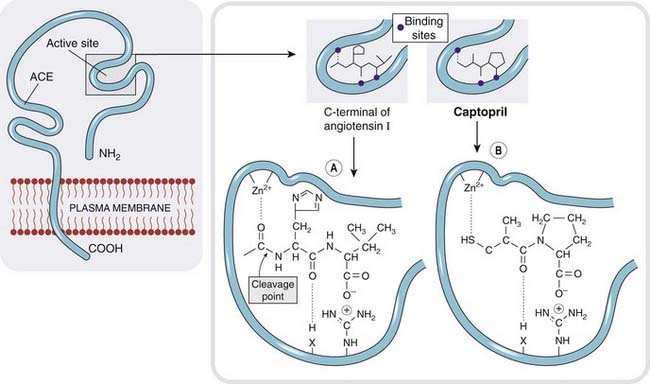
Fig. 22.7 The active site of angiotensin-converting enzyme.
[A] Binding of angiotensin I. [B] Binding of the inhibitor captopril, which is an analogue of the terminal dipeptide of angiotensin I.
Pharmacological effects
ACE inhibitors cause only a small fall in arterial pressure in healthy human subjects who are consuming the amount of salt contained in a usual Western diet, but a much larger fall in hypertensive patients, particularly those in whom renin secretion is enhanced (e.g. in patients receiving diuretics). ACEIs affect capacitance and resistance vessels, and reduce cardiac load as well as arterial pressure. They do not affect cardiac contractility, so cardiac output normally increases. They act preferentially on angiotensin-sensitive vascular beds, which include those of the kidney, heart and brain. This selectivity may be important in sustaining adequate perfusion of these vital organs in the face of reduced perfusion pressure. Critical renal artery stenosis11 represents an exception to this, where ACE inhibition results in a fall in glomerular filtration rate (see below).
Clinical uses of ACE inhibitors are summarised in the clinical box.
Unwanted effects
Adverse effects (Table 22.4) directly related to ACE inhibition are common to all drugs of this class. These include hypotension, especially after the first dose and especially in patients with heart failure who have been treated with loop diuretics, in whom the renin–angiotensin system is highly activated. A dry cough, possibly the result of accumulation of bradykinin (Ch. 17), is the commonest persistent adverse effect. Kinin accumulation may also underly angioedema (painful swelling in tissues which can be life-threatening if it involves the airway). Patients with severe bilateral renal artery stenosis predictably develop renal failure if treated with ACEIs, because glomerular filtration is normally maintained, in the face of low afferent arteriolar pressure, by angiotensin II, which selectively constricts efferent arterioles; hyperkalaemia may be severe owing to reduced aldosterone secretion. Such renal failure is reversible provided that it is recognised promptly and treatment with ACEI discontinued.
Angiotensin II receptor antagonists
Losartan, candesartan, valsartan and irbesartan (sartans) are non-peptide, orally active AT1 receptor antagonists (ARBs). ARBs differ pharmacologically from ACEIs (Fig. 22.8) but behave similarly to ACEIs in clinical practice, apart from not causing cough—consistent with the ‘bradykinin accumulation’ explanation of this side effect, mentioned above. ACE is not the only enzyme capable of forming angiotensin II, chymase (which is not inhibited by ACE inhibitors) providing one alternative route. It is not known if alternative pathways of angiotensin II formation are important in vivo, but if so, then ARBs could be more effective than ACE inhibitors in such situations. It is not known whether any of the beneficial effects of ACE inhibitors are bradykinin/NO mediated, so it is unwise to assume that ARBs will necessarily share all the therapeutic properties of ACE inhibitors. However, there is considerable overlap in the clinical indications for ARBs and ACEIs (Table 22.4).
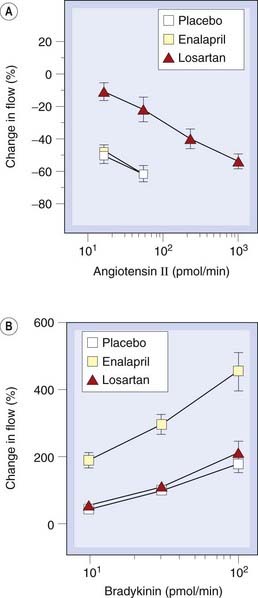
Fig. 22.8 Comparison of effects of angiotensin-converting enzyme inhibition and angiotensin receptor blockade in the human forearm vasculature.
[A] Effect of brachial artery infusion of angiotensin II on forearm blood flow after oral administration of placebo, enalapril (10 mg) or losartan (100 mg). [B] Effect of brachial artery infusion of bradykinin, as in [A].
(From Cockcroft J R et al. 1993 J Cardiovasc Pharmacol 22: 579–584.)
Types of vasodilator drug
Directly acting vasodilators
Indirectly acting vasodilators
Clinical Uses of Vasoactive Drugs
It is beyond the scope of this book to provide a detailed account of the clinical uses of vasoactive drugs, but it is nonetheless useful to consider briefly the treatment of certain important disorders, namely:
Systemic Hypertension
Systemic hypertension is a common disorder that, if not effectively treated, increases the risk of coronary thrombosis, strokes and renal failure. Until about 1950, there was no effective treatment, and the development of antihypertensive drugs has been a major therapeutic success story. Systemic blood pressure is an excellent ‘surrogate marker’ for increased cardiovascular risk in that there is good evidence from randomised controlled trials that common antihypertensive drugs (diuretics, ACEIs, calcium antagonists) combined with lifestyle changes not only lowers blood pressure but also reduces the extra risks of heart attacks and strokes associated with high blood pressure.
Correctable causes of hypertension include phaeochromocytoma,12 steroid-secreting tumours of the adrenal cortex or narrowing (coarctation) of the aorta, but most cases involve no obvious cause and are grouped as essential hypertension (so-called because it was originally, albeit incorrectly, thought that the raised blood pressure was ‘essential’ to maintain adequate tissue perfusion). Increased cardiac output may be an early feature, but by the time essential hypertension is established (commonly in middle life) there is usually increased peripheral resistance and the cardiac output is normal. Blood pressure control is intimately related to the kidneys, as demonstrated in humans requiring renal transplantation: hypertension ‘goes with’ the kidney from a hypertensive donor, and donating a kidney from a normotensive to a hypertensive corrects hypertension in the recipient (see also Ch. 28). Persistently raised arterial pressure leads to hypertrophy of the left ventricle and remodelling of resistance arteries, with narrowing of the lumen, and predisposes to atherosclerosis.
Figure 22.9 summarises physiological mechanisms that control arterial blood pressure and shows sites at which antihypertensive drugs act, notably the sympathetic nervous system, the renin–angiotensin–aldosterone system and endothelium-derived mediators. Remodelling of resistance arteries in response to raised pressure reduces the ratio of lumen diameter to wall thickness and increases the peripheral vascular resistance. The role of cellular growth factors (including angiotensin II) and inhibitors of growth (e.g. NO) in the evolution of these structural changes is of great interest to vascular biologists, and is potentially important for ACEIs and ARBs.
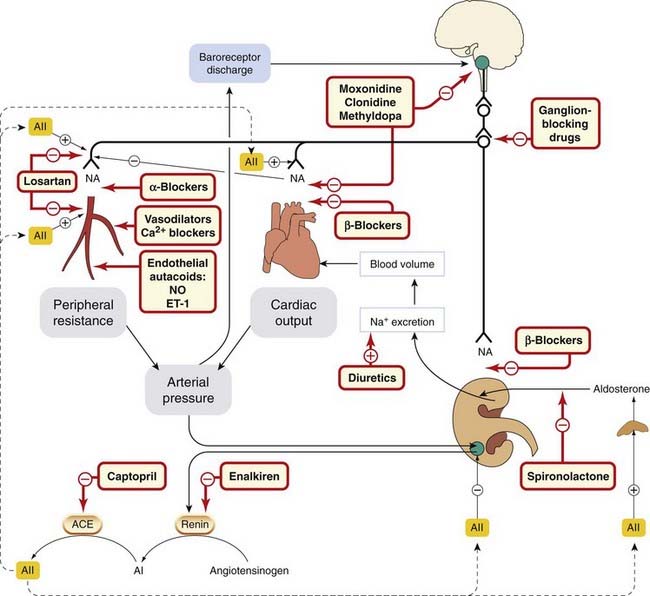
Fig. 22.9 Diagram showing the main mechanisms involved in arterial blood pressure regulation
(black lines), and the sites of action of antihypertensive drugs (hatched boxes + orange lines). ACE, angiotensin-converting enzyme; AI, angiotensin I; AII, angiotensin II; ET-1, endothelin-1; NA, noradrenaline; NO, nitric oxide.
Reducing arterial blood pressure greatly improves the prognosis of patients with hypertension. Controlling hypertension (which is asymptomatic) without producing unacceptable side effects is therefore an important clinical need, which is, in general, well catered for by modern drugs. Treatment involves non-pharmacological measures (e.g. increased exercise, reduced dietary salt and saturated fat with increased fruit and fibre, and weight and alcohol reduction) followed by the staged introduction of drugs, starting with those of proven benefit and least likely to produce side effects. Some of the drugs that were used to lower blood pressure in the early days of antihypertensive therapy, including ganglion blockers, adrenergic neuron blockers and reserpine (see Ch. 14), produced a fearsome array of adverse effects and are now obsolete. The preferred regimens have changed progressively as better-tolerated drugs have become available. One rational strategy with some evidence to support it, and recommended by the current British Hypertension Society guidelines, is to start treatment with either an ACEI or an AT1 antagonist in patients who are likely to have normal or raised plasma renin (i.e. younger white people), and with either a thiazide diuretic or a calcium antagonist in older people and people of African origin (who are more likely to have low plasma renin). If the target blood pressure is not achieved but the drug is well tolerated, then a drug of the other group is added. It is best not to increase the dose of any one drug excessively, as this often causes adverse effects and engages homeostatic control mechanisms (e.g. renin release by a diuretic) that limit efficacy.
β-Adrenoceptor antagonists are less well tolerated than ACEIs or ARBs, and the evidence supporting their routine use is less strong than for other classes of antihypertensive drugs. They are useful for hypertensive patients with some additional indication for β blockade, such as angina or heart failure.
Addition of a third or fourth drug (e.g. to ARB/diuretic or ARB/calcium antagonist combination) is often needed, and a long-acting α1-adrenoceptor antagonist (Ch. 14) such as doxazosin is one option in this setting. The α1 antagonists additionally improve symptoms of benign prostatic hypertrophy,13 common in older men , albeit at the expense of some postural hypotension, which is the main unwanted effect of these agents. Doxazosin is used once daily and has a mild but theoretically desirable effect on plasma lipids (reducing the ratio of low- to high-density lipoproteins; see Ch. 23). Spironolactone (a competitive antagonist of aldosterone; Ch. 32) has staged something of a comeback in treating severe hypertension. Careful monitoring of plasma K+ concentration is required, because spironolactone inhibits urinary K+ excretion as well as causing oestrogen-related adverse effects, but it is usually well tolerated in low doses. Methyldopa is now used mainly for hypertension during pregnancy because of the lack of documented adverse effects on the baby (in contrast to ACEIs, ARBs and standard β-adrenoceptor antagonists, which are contraindicated during pregnancy). Clonidine (a centrally acting α2 agonist) is now seldom used. Moxonidine, a centrally acting agonist at imidazoline I1 receptors that causes less drowsiness than α2 agonists, is licensed for mild or moderate hypertension, but there is little evidence from clinical end-point trials to support its use. Minoxidil, combined with a diuretic and β-adrenoceptor antagonist, is sometimes effective where other drugs have failed in severe hypertension resistant to other drugs. Fenoldopam, a selective dopamine D1 receptor agonist, is approved in the USA for the short-term management in hospital of severe hypertension. Its effect is similar in magnitude to that of intravenous nitroprusside, but it lacks thiocyanate-associated toxicity and is slower in onset and offset.
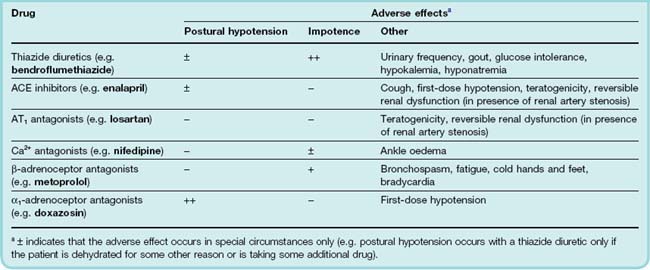
Commonly used antihypertensive drugs and their common adverse effects are summarised in Table 22.5.
Heart Failure
Heart failure is a clinical syndrome characterised by symptoms of breathlessness and/or fatigue, usually with signs of fluid overload (edema, crackles heard when listening to the chest). The underlying physiological abnormality (see also Ch. 21) is a cardiac output that is inadequate to meet the metabolic demands of the body, initially during exercise but, as the syndrome progresses, also at rest. It may be caused by disease of the myocardium itself (most commonly secondary to coronary artery disease), or by circulatory factors such as volume overload (e.g. leaky valves, or arteriovenous shunts caused by congenital defects) or pressure overload (e.g. stenosed—i.e. narrowed—valves, arterial or pulmonary hypertension). Some of these underlying causes are surgically correctable, and in some either the underlying disease (e.g. hyperthyroidism; Ch. 33), or an aggravating factor such as anaemia (Ch. 25) or atrial fibrillation (Ch. 21), is treatable with drugs. Here, we focus on drugs used to treat heart failure irrespective of the underlying cause.
When cardiac output is insufficient to meet metabolic demand, an increase in fluid volume occurs, partly because increased venous pressure causes increased formation of tissue fluid, and partly because reduced renal blood flow activates the renin–angiotensin–aldosterone system, causing Na+ and water retention. Irrespective of the cause, the outlook for adults with cardiac failure is grim: 50% of those with the most severe grade are dead in 6 months, and of those with ‘mild/moderate’ disease, 50% are dead in 5 years. Non-drug measures, including dietary salt restriction and exercise training in mildly affected patients,14 are important, but drugs are needed to improve symptoms of oedema, fatigue and breathlessness, and to improve prognosis.
A highly simplified diagram of the sequence of events is shown in Figure 22.10. A common theme is that several of the feedbacks that are activated are ‘counter-regulatory’—i.e. they make the situation worse not better. This occurs because the body fails to distinguish the haemodynamic state of heart failure from haemorrhage, in which release of vasoconstrictors such as angiotensin II and ADH would be appropriate.15 ACEIs and ARBs, β-adrenoceptor and aldosterone antagonists interrupt these counter-regulatory neurohormonal mechanisms and have each been shown to prolong life in heart failure, although prognosis remains poor despite optimal management.
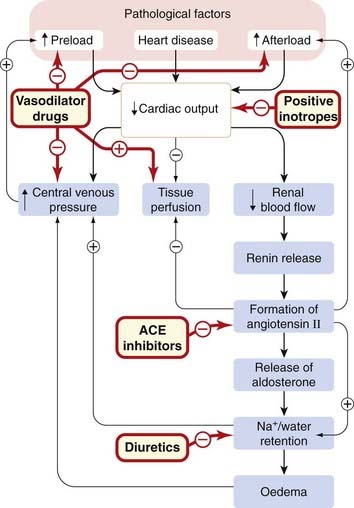
Fig. 22.10 Simplified scheme showing the pathogenesis of heart failure, and the sites of action of some of the drugs used to treat it.
The symptoms of heart failure are produced by reduced tissue perfusion, oedema and increased central venous pressure. ACE, angiotensin-converting enzyme.
Drugs used to treat heart failure act in various complementary ways to do the following.
Increase natriuresis
Diuretics, especially loop diuretics (Ch. 28), are important in increasing salt and water excretion, especially if there is pulmonary oedema. In chronic heart failure, drugs that have been shown to improve prognosis were all studied in patients treated with diuretics.
Inhibit the renin–angiotensin–aldosterone system
The renin–angiotensin–aldosterone system is inappropriately activated in patients with cardiac failure, especially when they are treated with diuretics. The β-adrenoceptor antagonists inhibit renin secretion and are used in clinically stable patients with chronic heart failure (see below). ACEIs and ARBs block the formation of angiotensin II and inhibit its action, respectively, thereby reducing vascular resistance, improving tissue perfusion and reducing cardiac afterload. They also cause natriuresis by inhibiting secretion of aldosterone and by reducing the direct stimulatory effect of angiotensin II on reabsorption of Na+ and HCO3− in the early part of the proximal convoluted tubule. Most important of all, they prolong life. The question of whether ACEIs and ARBs can usefully be combined is being evaluated. Angiotensin II is not the only stimulus to aldosterone secretion, and during chronic treatment with ACEIs, circulating aldosterone concentrations return towards pretreatment values (a phenomenon known as ‘aldosterone escape’). This provided a rationale for studying the effect of combining spironolactone (an aldosterone antagonist; see Ch. 32) with ACEI treatment, and this further reduces mortality. Eplerenone is an aldosterone antagonist with less oestrogen-like adverse effects than sprironolactone; it too has been shown to improve survival in patients with heart failure when added to conventional therapy. Patients with impaired renal function were excluded from these trials, and careful monitoring of plasma K+ concentration is important when they are treated with an ACEI or an ARB in combination with an aldosterone antagonist.
Block β adrenoceptors
Heart failure is accompanied by potentially harmful activation of the sympathetic nervous system as well as of the renin–angiotensin system, providing a rationale for using β-adrenoceptor antagonists. Most clinicians were very wary of this approach because of the negative inotropic action of these drugs, but when started in low doses that are increased slowly, metoprolol, carvedilol and bisoprolol each improves survival when added to optimal treatment in clinically stable patients with chronic heart failure.
Antagonise ADH
ADH (see above) is released in heart failure and may contribute to undesirable vasoconstriction (via V1A receptors) and hyponatraemia (via V2 receptors).16 Two non-peptide vasopressin receptor antagonists (‘vaptans’) have been licensed by the Food and Drug Administration and many more are in development (Finley et al., 2008). Conivaptan is a non-selective V1A /V2 antagonist licensed for treatment of the syndrome of inappropriate ADH secretion (SIADH) and intravenously for short-term treatment of hypervolaemic (or euvolaemic) heart failure. Tolvaptan is a selective V2 receptor antagonist approved for oral treatment of clinically significant hypervolaemic (or euvolaemic) hyponatraemia. Neither has been shown to improve long-term survival in heart failure, and their possible place in therapy is currently the subject of intense investigation (Jessup et al., 2009).
Relax vascular smooth muscle
Glyceryl trinitrate (Ch. 21) is infused intravenously to treat acute cardiac failure. Its venodilator effect reduces venous pressure, and its effects on arterial compliance and wave reflection reduce cardiac work. The combination of hydralazine (to reduce afterload) with a long-acting organic nitrate (to reduce preload) in patients with chronic heart failure improved survival in a randomised controlled trial, but the results suggested that the benefit was restricted to African-American patients. This ethnic group is genetically very heterogeneous, and it is unknown what other groups will benefit from such treatment.
Increase the force of cardiac contraction
Cardiac glycosides (Ch. 21) are used either in patients with heart failure who also have chronic rapid atrial fibrillation, or in patients who remain symptomatic despite treatment with a diuretic and ACEI. Digoxin does not reduce mortality in heart failure patients in sinus rhythm who are otherwise optimally treated, but does improve symptoms and reduce the need for hospital admission. In contrast, PDE inhibitors (see Ch. 21) increase cardiac output, but increase mortality in heart failure, probably through cardiac dysrhythmias. Dobutamine (a β1-selective adrenoceptor agonist; see Ch. 21) is used intravenously when a rapid response is needed in the short term, for example following heart surgery.
Drugs used in chronic heart failure 
Shock and Hypotensive States
Shock is a medical emergency characterised by inadequate perfusion of vital organs, usually because of a very low arterial blood pressure. This leads to anaerobic metabolism and hence to increased lactate production. Mortality is very high, even with optimal treatment in an intensive care unit. Shock can be caused by various insults, including haemorrhage, burns, bacterial infections, anaphylaxis (Ch. 17) and myocardial infarction (Fig. 22.11). The common factor is reduced effective circulating blood volume (hypovolaemia) caused either directly by bleeding or by movement of fluid from the plasma to the gut lumen or extracellular fluid. The physiological (homeostatic) response to this is complex: vasodilatation in a vital organ (e.g. brain, heart or kidney) favours perfusion of that organ, but at the expense of a further reduction in blood pressure, which leads to reduced perfusion of other organs. Survival depends on a balance between vasoconstriction in non-essential vascular beds and vasodilatation in vital ones. The dividing line between the normal physiological response to blood loss and clinical shock is that in shock tissue hypoxia produces secondary effects that magnify rather than correct the primary disturbance. Therefore patients with established shock have profound and inappropriate vasodilatation in non-essential organs, and this is difficult to correct with vasoconstrictor drugs. The release of mediators (e.g. histamine, 5-hydroxytryptamine, bradykinin, prostaglandins, cytokines including interleukins and tumour necrosis factor, NO and undoubtedly many more as-yet-unidentified substances) that cause capillary dilatation and leakiness is the opposite of what is required to improve function in this setting. Mediators promoting vasodilatation in shock converge on two main mechanisms:
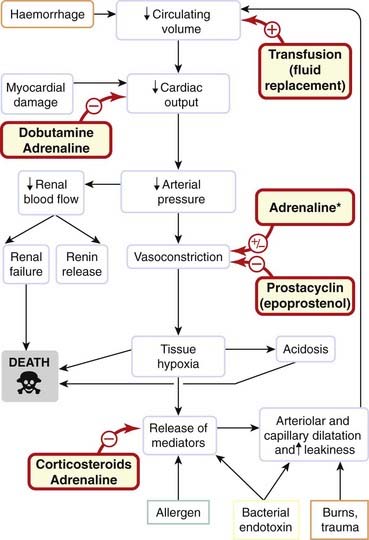
Fig. 22.11 Simplified scheme showing the pathogenesis of hypovolaemic shock.
*Adrenaline causes vasodilatation in some vascular beds, vasoconstriction in others.
A third key mechanism seems to be a relative deficiency of ADH, which is secreted acutely in response to haemorrhage but subsequently declines, probably because of depletion from the neurohypophysis (see Ch. 32)—contrast this with the situation in chronic heart failure discussed above where excess (rather than deficient) ADH may contribute to problems.
Patients with shock are not a homogeneous population, making it hard to perform valid clinical trials, and in contrast to hypertension and heart failure there is very little evidence to support treatment strategies based on hard clinical end points (such as improved survival). Hypoperfusion leads to multiple organ failure (including renal failure), and intensive therapy specialists spend much effort supporting the circulations of such patients with cocktails of vasoactive drugs designed to optimise flow to vital organs. Trials of antagonists designed to block or neutralise endotoxin, interleukins, tumour necrosis factor and the inducible form of NO synthase have so far been disappointing. Volume replacement is of benefit if there is hypovolaemia; antibiotics are essential if there is persistent bacterial infection; adrenaline can be life-saving in anaphylaxis; a preparation of recombinant activated protein C, drotrecogin alpha (activated) (see Ch. 24) improves mortality in severe septic shock with multiple organ failure and is licensed for this indication; vasopressin may be effective in increasing blood pressure even when there is resistance to adrenaline; corticosteroids suppress the formation of NO and of prostaglandins but are not of proven benefit once shock is established; epoprostenol (PGI2) may be useful in patients with inappropriate platelet activation (e.g. meningococcal sepsis); positive inotropic agents, including adrenaline and dobutamine, may help in individual patients, as may levosimendan (Mebazaa et al., 2007).
Peripheral Vascular Disease
When atheroma involves peripheral arteries, the first symptom is usually pain in the calves on walking (claudication), followed by pain at rest, and in severe cases gangrene of the feet or legs. Treatment is often surgical. Other vascular beds (e.g. coronary, cerebral and renal) are often also affected by atheromatous disease in patients with peripheral vascular disease. Drug treatment includes antiplatelet drugs (e.g. aspirin, clopidogrel; see Ch. 24), a statin (e.g. simvastatin; see Ch. 23) and an ACEI (e.g. ramipril; see above). These reduce the excess risk of ischaemic coronary and cerebral events. Additionally, several placebo-controlled studies have demonstrated that cilostazol, a type III PDE inhibitor (see above), improves pain-free and maximum walking distance in such patients, but its effect on mortality is unknown.
Raynaud’s Disease
Inappropriate vasoconstriction of small arteries and arterioles gives rise to Raynaud’s phenomenon (blanching of the fingers during vasoconstriction, followed by blueness owing to deoxygenation of the static blood and redness from reactive hyperaemia following return of blood flow). This can be mild, but if severe causes ulceration and gangrene of the fingers. It can occur in isolation (Raynaud’s disease) or in association with a number of other diseases, including several so-called connective tissue diseases (e.g. systemic sclerosis, systemic lupus erythematosus). Treatment of Raynaud’s phenomenon hinges on stopping smoking (crucially) and on avoiding the cold; β-adrenoceptor antagonists are contraindicated. Vasodilators (e.g. nifedipine; see Ch. 21) are of some benefit in severe cases, and evidence from several small studies suggests that other vasodilators (e.g. PGI2, CGRP) can have surprisingly prolonged effects, but are difficult to administer.
Pulmonary Hypertension
After birth, pulmonary vascular resistance is much lower than systemic vascular resistance, and systolic pulmonary artery pressure in adults is normally approximately 20 mmHg.17
Pulmonary artery pressure is much less easy to measure than is systemic pressure, often requiring cardiac catheterisation, so only severe and symptomatic pulmonary hypertension usually gets diagnosed. Pulmonary hypertension usually causes some regurgitation of blood from the right ventricle to the right atrium. This tricuspid regurgitation can be used to estimate the pulmonary artery pressure indirectly by ultrasonography. Pulmonary hypertension may be idiopathic (i.e. of unknown cause, analogous to essential hypertension in the systemic circulation), or associated with some other disease. Increased pulmonary pressure can result from an increased cardiac output (such as occurs, for example, in patients with hepatic cirrhosis—where vasodilatation may accompany intermittent subclinical exposure to bacterial endotoxin—or in patients with congenital connections between the systemic and pulmonary circulations). Vasoconstriction and/or structural narrowing of the pulmonary resistance arteries increase pulmonary arterial pressure, even if cardiac output is normal. In some situations, both increased cardiac output and increased pulmonary vascular resistance are present.
In contrast to systemic hypertension, pulmonary hypertension associated with other diseases is much more common than idiopathic pulmonary hypertension, which is a rare, severe and progressive disease. Endothelial dysfunction (see above, and also Chs 23 and 24) is implicated in its aetiology. Drugs (e.g. anorexic drugs including dexfenfluramine, now withdrawn) and toxins (e.g. monocrotaline) can cause pulmonary hypertension. Occlusion of the pulmonary arteries, for example with recurrent pulmonary emboli (Ch. 24), is a further cause, and anticoagulation (see Ch. 24) is an important part of treatment. Aggregates of deformed red cells in patients with sickle cell anaemia (Ch. 25) can also occlude small pulmonary arteries.
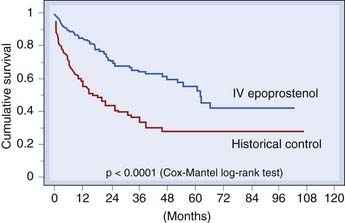
Increased pulmonary vascular resistance may, alternatively, result from vasoconstriction and/or structural changes in the walls of pulmonary resistance arteries. Many of the diseases (e.g. systemic sclerosis) associated with Raynaud’s phenomenon mentioned in the section above are also associated with pulmonary hypertension. Vasoconstriction may precede cellular proliferation and medial hypertrophy which causes wall thickening in the pulmonary vasculature. Treatment with vasodilators (e.g. nifedipine) is used. Vasodilators with an antiproliferative action (e.g. epoprostenol, drugs that potentiate NO, or antagonise endothelin) are more promising.
Drugs used in treating pulmonary arterial hypertension and clinical disorders for which vasoactive drugs are important are shown in the clinical boxes.
Drugs used in pulmonary hypertension
Drugs are used where indicated to treat any underlying cause; in addition, consider the following:
Clinical disorders for which vasoactive drugs are important
References and Further Reading
Vascular structure and function, control of vascular smooth muscle tone
Guimarães S., Moura D. Vascular adrenoceptors: an update. Pharmacol. Rev.. 2001;53:319-356. (Functional perspective)
Ward J.P.T., Knock G.A., Snetkov V.A., Aaronson P.I. Protein kinases in vascular smooth muscle tone—role in the pulmonary vasculature and hypoxic pulmonary vasoconstriction Pharmacol. Ther. 104:2004:207-231 (Concludes that the strongest evidence for direct involvement of protein kinases in the mechanisms of hypoxic pulmonary vasoconstriction concerns a central role for Rho kinase in Ca2+ sensitisation)
Vascular endothelium (see Ch. 20 for further reading on nitric oxide)
Bunting S., Gryglewski R., Moncada S., Vane J.R. Arterial walls generate from prostaglandin endoperoxides a substance (prostaglandin X) which relaxes strips of mesenteric and celiac arteries and inhibits platelet aggregation. Prostaglandins. 1976;12:897-913. (Classic)
Murata T., Ushikubi F., Matsuoka T., et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678-682. (I prostanoid receptor-deficient mice are viable, reproductive and normotensive; however, their susceptibility to thrombosis is increased … the results establish that prostacyclin is an endogenous antithrombotic agent)
Endothelium-derived hyperpolarising factor
Félétou M., Vanhoutte P.M. EDHF: an update Clin. Sci. 117:2009:139-155 (Reviews briefly the many endothelial mediators that can cause hyperpolarisation, but focuses on the pathway that works by increasing endothelial [Ca2+]i thereby activating KCa channels with consequent hyperpolarisation of underlying vascular smooth muscle both by electrotonic spread through gap junctions and release of K+ with activation of KIR and electrogenic sodium-potassium pump)
Carmeliet P., Jain R.K. Angiogenesis in cancer and other diseases. Nature. 2000;407:249-257. (New approaches to treatment of cancer and other diseases, via a growing number of pro- and antiangiogenic molecules; see also (in same issue) Yancopoulos, G.D., et al., 2000 Vascular specific growth factors and blood vessel formation, pp. 242–248)
Ge Y., Bagnall A., Stricklett P.K., et al. Collecting duct-specific knockout of the endothelin B receptor causes sodium retention and hypertension. Am. J. Physiol.. 2006;291:1274-1280.
Hickey K.A., Rubanyi G., Paul R.J., Highsmith R.F. Characterization of a coronary vasoconstrictor produced by cultured endothelial cells. Am. J. Physiol.. 1985;248(part 1):C550-C556. (Key discovery)
Kirchengast M., Luz M. Endothelin receptor antagonists—clinical realities and future directions. J. Cardiovasc. Pharmacol.. 2005;5:182-191. (Critically reviews clinical data on endothelin receptor antagonism in cardiovascular indications against the background of preclinical research)
Yanagisawa M., Kurihara H., Kimura S., et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411-415. (Tour de force)
Burnier M., Brunner H.R. Angiotensin II receptor antagonists. Lancet. 2000;355:637-645. (Reviews this class of drugs)
Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N. Engl. J. Med.. 2000;342:145-153. (Ramipril significantly lowers rates of death, myocardial infarction and stroke in a wide range of high-risk patients)
Watanabe T., Barker T.A., Berk B.C. Angiotensin II and the endothelium—diverse signals and effects. Hypertension. 2005;45:163-169. (Reviews the renin–angiotensin system in the endothelium based on the diverse signals and effects mediated by multiple angiotensin I- and angiotensin II-derived peptides, multiple angiotensin-metabolising enzymes, multiple receptors and vascular bed-specific intracellular signals)
Holmes C.L., Russell J.A. Vasopressin. Semin. Respir. Crit. Care Med.. 2004;25:705-711. (‘A deficiency of vasopressin exists in some shock states and replacement of physiological levels of vasopressin can restore vascular tone. Vasopressin is therefore emerging as a rational therapy for vasodilatory shock.’ Reviews rationale, evidence and uncertainties for using vasopressin in shock)
Vasodilator drugs (see Ch. 21 for further reading on calcium antagonists)
Chan C.K.S., Burke S.L., Zhu H., et al. Imidazoline receptors associated with noradrenergic terminals in the rostral ventrolateral medulla mediate the hypotensive responses of moxonidine but not clonidine. Neuroscience. 2005;132:991-1007. (The hypotensive and bradycardic actions of moxonidine but not clonidine are mediated through imidazoline receptors and depend on noradrenergic CNS pathways; noradrenergic innervation may be associated with imidazoline receptor protein)
Murphy M.B., Murray C., Shorten G.D. Fenoldopam—a selective peripheral dopamine receptor agonist for treatment of severe hypertension. N. Engl. J. Med.. 2001;345:1548-1557. (Similar effectiveness as that of nitroprusside but without thiocyanate toxicity or instability in light; however, it is slower in onset and offset than nitroprusside)
Finley J.J., Konstam M.A., Udelson J.E. Arginine vasopressin antagonists for the treatment of heart failure and hyponatraemia. Circulation. 2008;118:410-421.
Gheorghiade M., Gattis W.A., O’Connor C.M., et al. Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure—a randomized controlled trial. JAMA. 2004;291:1963-1971. (Tolvaptan plus standard therapy has promise for heart failure)
Jessup M., Brozena S. Heart failure. N. Engl. J. Med.. 2003;348:2007-2018.
Jessup M., Abraham W.T., Casey D.E., et al. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119:1977-2016.
McMurray J.J.V. Val-HeFT: do angiotensin-receptor blockers benefit heart failure patients already receiving ACE inhibitor therapy? Nat. Clin. Pract. Cardiovasc. Med.. 2005;2:128-129.
Taylor A.L., Ziesche S., Yancy C., et al. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N. Engl. J. Med.. 2004;351:2049-2057. (Addition of a fixed dose of isosorbide dinitrate plus hydralazine to standard therapy for heart failure including neurohormonal blockers increased survival among black patients with advanced heart failure)
Landry D.W., Oliver J.A. Mechanisms of disease: the pathogenesis of vasodilatory shock. N. Engl. J. Med.. 2001;345:588-595. (Reviews mechanisms promoting inappropriate vasodilation in shock, including activation of ATP-sensitive potassium channels, increased synthesis of NO and depletion of ADH)
Australian and New Zealand Intensive Care Society Clinical Trials Group. Low-dose dopamine in patients with early renal dysfunction: a placebo-controlled randomized trial. Lancet. 2000;356:2139-2143. (No clinically significant protection; see also accompanying editorial, Renal-dose dopamine: will the message now get through?)
Bernard G.R., Vincent J.L., Laterre P.F., et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N. Engl. J. Med.. 2001;344:699-709. (Continuous intravenous infusion of activated protein C, a vitamin K-dependent anticoagulant that promotes fibrinolysis, inhibits thrombosis and is anti-inflammatory, significantly reduced risk of death at 28 days, from 30.8 to 24.7%)
Liang K.V., Williams A.W., Greene E.L., Redfield M.M. Acute decompensated heart failure and the cardiorenal syndrome. Crit. Care Med.. 2008;36(Suppl. 1):S75-S88. (Review from Mayo Clinic)
Mebazaa A., Nieminen M.S., Packer M., et alfor the SURVIVE investigators. Levosimendan vs dobutamine for patients with acute decompensated heart failure. JAMA. 2007;297:1883-1891. (Randomized, double-blind trial comparing the efficacy and safety of intravenous levosimendan versus dobutamine in 1327 patients hospitalised with acute decompensated heart failure who required inotropic support. Compared with patients in the dobutamine group, levosimendan did not significantly reduce all-cause mortality at 180 days or affect any secondary clinical outcomes)
Potter L.R., Yoder A.R., Flora D.R., et al. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp. Pharmacol.. 2009;191:341-366. (Reviews the history, structure, function and clinical applications of natriuretic peptides and their receptors)
Peripheral vascular disease, Raynaud’s disease and pulmonary hypertension
Hiatt W.R. Medical treatment of peripheral arterial disease and claudication. N. Engl. J. Med.. 2001;344:1608-1621. (Discusses risk factor modification, summarises evidence for efficacy of cilostazol, and describes rationale for several investigational drugs, for example propionyl levocarnitine)
Badesch D.B., Abman S.H., Ahearn G.S., et al. Medical therapy for pulmonary arterial hypertension—ACCP evidence-based clinical practice guidelines. Chest. 2004;126(Suppl.):35S-62S. (Evidence-based treatment recommendations for physicians involved in the care of these complex patients)
Beppu H., Ichinose F., Kawai N., et al. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am. J. Physiol. Lung Cell Mol. Physiol.. 2004;287:L1241-L1247. (‘Heterozygous mutations of the bone morphogenetic protein type II receptor, BMPR-II, gene have been identified in patients with primary pulmonary hypertension … in mice, mutation of one copy of the BMPR-II gene causes pulmonary hypertension but impairs the ability of the pulmonary vasculature to remodel in response to prolonged hypoxic breathing.’)
Higenbottam T., Laude L., Emery C., Essener M. Pulmonary hypertension as a result of drug therapy. Clin. Chest Med.. 2004;25:123-131. (Reviews anorectic drug-induced pulmonary arterial hypertension and considers mechanisms)
Humbert M., Sitbon O., Simonneau G. Drug therapy: treatment of pulmonary arterial hypertension. N. Engl. J. Med.. 2004;351:1425-1436.
Lee A.J., Chiao T.B., Tsang M.P. Sildenafil for pulmonary hypertension. Ann. Pharmacother. 2005;39:869-884. (Sildenafil is a promising and well-tolerated treatment for pulmonary hypertension; well-designed trials are needed)
McLaughlin V.V., Sitbon O., Badesch D.B., et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur. Respir. J.. 2005;25:244-249. (Bosentan improved survival in patients with advanced primary pulmonary hypertension)
Napoli C., Loscalzo J. Nitric oxide and other novel therapies for pulmonary hypertension. J. Cardiovasc. Pharmacol. Ther.. 2004;9:1-8. (Focus on endothelial NO, NO replacement and related therapies)
Papp Z., Csapo K., Pollesello P., et al Pharmacological mechanisms contributing to the clinical efficacy of levosimendan Cardiovasc. Drug Rev. 23:2005:71-98 (Levosimendan is a Ca2+ sensitiser, binding to troponin C in the myocardium and, additionally, opening ATP-sensitive potassium channels in vascular smooth muscle. Clinical studies have suggested long-term benefits on mortality following short-term administration in patients with decompensated heart failure)
Rich S., McLaughlin V.V. Chapter 67. In: Zipes D.P., Libby P., Bonow R.O., Braunwald E., editors. Braunwald’s heart disease. seventh ed. Philadelphia: Elsevier; 2005:1807-1842.
Task-force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. Eur. Heart J.. 2004;25:2243-2278.
West J., Fagan K., Steudel W., et al Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle Circ. Res. 94:2004:1109-1114 (Bone morphogenetic peptides, BMPs, a family of cytokines critical to normal development, are implicated in the pathogenesis of familial pulmonary arterial hypertension; deletion of BMPRII results in early fetal death. To study BMP signalling in postnatal vascular disease, these authors constructed a smooth muscle-specific transgenic mouse expressing a dominant-negative BMPRII under control of a tetracycline gene switch. When the mutation was activated after birth, mice developed increased pulmonary artery pressure, indicating that loss of BMPRII signalling is sufficient to produce the pulmonary hypertensive phenotype)
1William Harvey (physician to King Charles I) inferred the circulation of the blood on the basis of superbly elegant quantitative experiments long before the invention of the microscope enabled visual confirmation of the tiny vessels he had predicted. This intellectual triumph did his medical standing no good at all, and Aubrey wrote that ‘he fell mightily in his practice, and was regarded by the vulgar as crack-brained’. Plus ça change …
2This cushioning action is called the ‘windkessel’ effect. The same principle was used to deliver a steady rather than intermittent flow from old-fashioned fire pumps.
3Subsequently an 11-amino acid peptide (urotensin) was isolated from the brains of bony fish and found to be 50–100 times more potent a vasoconstrictor than endothelin in some blood vessels. It and its receptor are expressed in human tissue but its function, if any, in man remains enigmatic.
4Approximately that of a football field.
5A different isoform of ACE is also present in testis, and male mice lacking this ACE have markedly reduced fertility.
6These effects are initiated by the G-protein-coupled AT1 receptor acting via the same intracellular tyrosine phosphorylation pathways as are used by cytokines, for example the Jak/Stat pathway (Ch. 3).
7A pyridine drug, Y27632, causes vasorelaxation by inhibiting a Rho-associated protein kinase, thereby selectively inhibiting smooth muscle contraction by inhibiting Ca2+ sensitisation.
8KATP channels in pancreatic islet B cells form an important link between the metabolic state of the cell and membrane function, and sulfonylurea drugs cause insulin secretion by mimicking the action of ATP on these channels (see Ch. 30). Conversely, potassium channel activators such as diazoxide (Fig. 22.6) increase blood glucose by inhibiting insulin secretion from the pancreas.
9An autoimmune disease affecting one or more tissues, including joints, skin and pleural membranes. It is characterised by antibodies directed against DNA.
10The lead compound was a nonapeptide derived from the venom of Bothrops jacaraca—a South American snake. It was originally characterised as a bradykinin-potentiating peptide (ACE inactivates bradykinin).
11Severe narrowing of the renal artery caused, for example, by atheroma (Ch. 23).
12Catecholamine-secreting tumours of chromaffin tissue, usually the adrenal medulla (Ch. 13).
13Difficulty starting the stream, poor stream, terminal dribbling and needing to get up often in the night to pass urine—all depressingly common in ageing men.
14Bed rest used to be recommended but results in deconditioning, and regular exercise has been shown to be beneficial in patients who can tolerate it.
15Natural selection presumably favoured mechanisms that would benefit young hunter gatherers at risk of haemorrhage; middle-aged or elderly people at high risk of heart failure are past their reproductive prime.
16Inappropriate secretion of ADH causes hyponatraemia because the kidney retains water while continuing to excrete sodium ions, whereas drinking, which is largely determined by habit in addition to thirst, continues. This leads to reduction of the plasma sodium concentration as a result of dilution.
17In fetal life, pulmonary vascular resistance is high; failure to adapt appropriately at birth is associated with prematurity, lack of pulmonary surfactant and hypoxaemia. The resulting pulmonary hypertension is treated by paediatric intensive care specialists with measures including replacement of surfactant and ventilatory support, sometimes including inhaled NO—see Ch. 20.