7 Method and measurement in pharmacology
Overview
We emphasised in Chapters 2 and 3 that drugs, being molecules, produce their effects by interacting with other molecules. This interaction can lead to effects at all levels of biological organisation, from molecules to human populations (Fig. 7.1).1
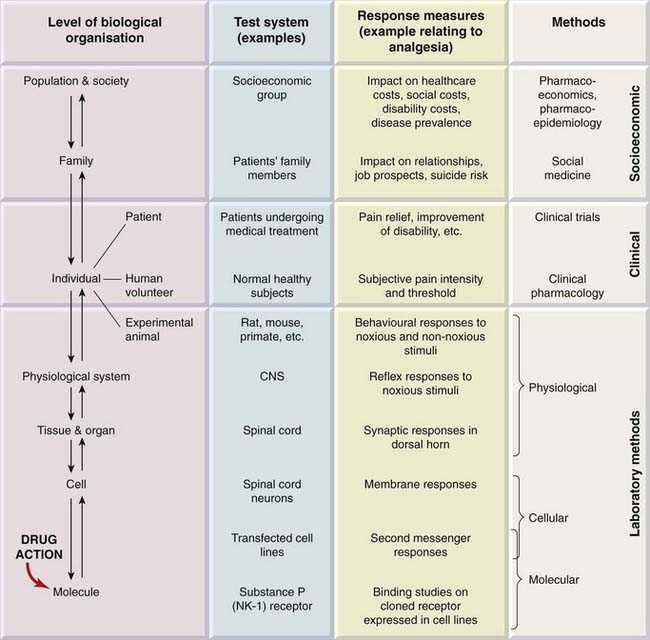
Gaddum, a pioneering pharmacologist, commented in 1942: ‘A branch of science comes of age when it becomes quantitative.’ In this chapter, we cover the principles of metrication at the various organisational levels, ranging from laboratory methods to clinical trials. Assessment of drug action at the population level is the concern of pharmacoepidemiology and pharmacoeconomics (see Ch. 1), disciplines that are beyond the scope of this book.
We consider first the general principles of bioassay, and its extension to studies in human beings; we describe the development of animal models to bridge the predictive gap between animal physiology and human disease; we next discuss aspects of clinical trials used to evaluate therapeutic efficacy in a clinical setting; finally, we consider the principles of balancing benefit and risk. Experimental design and statistical analysis are central to the interpretation of all types of pharmacological data. Kirkwood & Sterne (2003) provide an excellent introduction.
Bioassay
Bioassay, defined as the estimation of the concentration or potency of a substance by measurement of the biological response that it produces, has played a key role in the development of pharmacology. Quantitation of drug effects by bioassay is necessary to compare the properties of different substances, or the same substance under different circumstances. It is used:
 Bioassay plays a key role in the development of new drugs, discussed in Chapter 60.
Bioassay plays a key role in the development of new drugs, discussed in Chapter 60.
The use of bioassay to measure the concentration of drugs and other active substances in the blood or other body fluids—once an important technology—has now been largely replaced by analytical chemistry techniques.
New hormones and other chemical mediators are often discovered by the biological effects that they produce. The first clue may be the finding that a tissue extract or some other biological sample produces an effect on an assay system. For example, the ability of extracts of the posterior lobe of the pituitary to produce a rise in blood pressure and a contraction of the uterus was observed at the beginning of the 20th century. Quantitative assay procedures based on these actions enabled a standard preparation of the extract to be established by international agreement in 1935. By use of these assays, it was shown that two distinct peptides—vasopressin and oxytocin—were responsible, and they were eventually identified and synthesised in 1953. Biological assay had already revealed much about the synthesis, storage and release of the hormones, and was essential for their purification and identification. Nowadays, it does not take 50 years of laborious bioassays to identify new hormones before they are chemically characterised,2 but bioassay still plays a key role. The recent growth of biopharmaceuticals (see Ch. 59) as registered therapeutic agents has relied on bioassay techniques and the establishment of standard preparations. Biopharmaceuticals, whether derived from natural sources (e.g. monoclonal antibodies, vaccines) or by recombinant DNA technology (e.g. erythropoietin), tend to vary from batch to batch, and need to be standardised with respect to their biological activity. Varying glycosylation patterns, for example, which are not detected by immunoassay techniques, may affect biological activity.
Biological Test Systems
Nowadays, an important use of bioassay is to provide information that will predict the effect of the drug in the clinical situation (where the aim is to improve function in patients suffering from the effects of disease). The choice of laboratory test systems (in vitro and in vivo ‘models’) that provide this predictive link is an important aspect of quantitative pharmacology. As our understanding of drug action at the molecular level advances (Ch. 3), this knowledge, and the technologies underlying it, have greatly extended the range of models that are available for measuring drug effects. By the 1960s, pharmacologists had become adept at using isolated organs and laboratory animals (usually under anaesthesia) for quantitative experiments, and had developed the principles of bioassay to allow reliable measurements to be made with these sometimes difficult and unpredictable test systems.
Bioassays on different test systems may be run in parallel to reveal the profile of activity of an unknown mediator. This was used to great effect by Vane and his colleagues, who studied the generation and destruction of endogenous active substances such as prostanoids (see Ch. 17) by the technique of cascade superfusion (Fig. 7.2). In this technique, the sample is run sequentially over a series of test preparations chosen to differentiate between different active constituents of the sample. The pattern of responses produced identifies the active material, and the use of such assay systems for ‘on-line’ analysis of biological samples has been invaluable in studying the production and fate of short-lived mediators such as prostanoids and nitric oxide (Ch. 20).
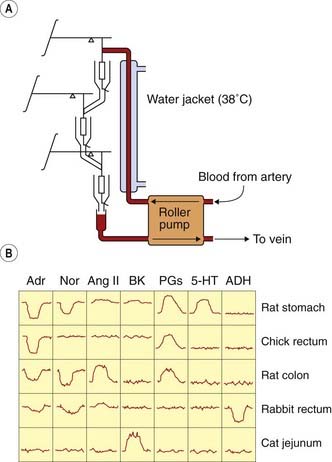
Fig. 7.2 Parallel assay by the cascade superfusion technique.
Blood is pumped continuously from the test animal over a succession of test organs, whose responses are measured by a simple transducer system. The response of these organs to a variety of test substances (at 0.1–5 ng/ml) is shown. Each active substance produces a distinct pattern of responses, enabling unknown materials present in the blood to be identified and assayed. 5-HT, 5-hydroxytryptamine; ADH, antidiuretic hormone; Adr, adrenaline (epinephrine); Ang II, angiotensin II; BK, bradykinin; Nor, noradrenaline (norepinephrine); PG, prostaglandin.
(From Vane J R 1969 Br J Pharmacol 35: 209–242.)
These ‘traditional’ assay systems address drug action at the physiological level—roughly, the mid-range of the organisational hierarchy shown in Fig. 7.1. Subsequent developments have extended the range of available models in both directions, towards the molecular and towards the clinical. The introduction of binding assays (Ch. 3) in the 1970s was a significant step towards analysis at the molecular level. Subsequently, use of cell lines engineered to express specific human receptor subtypes has become widespread as a screening tool for drug discovery (see Ch. 60). Indeed, the range of techniques for analysing drug effects at the molecular and cellular levels is now very impressive. Bridging the gap between these levels and effects at the physiological and the therapeutic levels has, however, proved much more difficult, because human illness cannot, in many cases, be accurately reproduced in experimental animals. The use of transgenic animals to model human disease represents a real advance, and is discussed in more detail below.
Bioassay 
General Principles of Bioassay
The Use of Standards
J H Burn wrote in 1950: ‘Pharmacologists today strain at the king’s arm, but they swallow the frog, rat and mouse, not to mention the guinea pig and the pigeon.’ He was referring to the fact that the ‘king’s arm’ had been long since abandoned as a standard measure of length, whereas drug activity continued to be defined in terms of dose needed to cause, say, vomiting of a pigeon or cardiac arrest in a mouse. A plethora of ‘pigeon units’, ‘mouse units’ and the like, which no two laboratories could agree on, contaminated the literature.3 Even if two laboratories cannot agree—because their pigeons differ—on the activity in pigeon units of the same sample of an active substance, they should nonetheless be able to agree that preparation X is, say, 3.5 times as active as standard preparation Y on the pigeon test. Biological assays are therefore designed to measure the relative potency of two preparations, usually a standard and an unknown. Maintaining stable preparations of various hormones, antisera and other biological materials, as reference standards, is the task of the UK National Board for Biological Standards Control.
The Design of Bioassays
Given the aim of comparing the activity of two preparations, a standard (S) and an unknown (U) on a particular preparation, a bioassay must provide an estimate of the dose or concentration of U that will produce the same biological effect as that of a known dose or concentration of S. As Figure 7.3 shows, provided that the log dose–effect curves for S and U are parallel, the ratio, M, of equiactive doses will not depend on the magnitude of response chosen. Thus M provides an estimate of the potency ratio of the two preparations. A comparison of the magnitude of the effects produced by equal doses of S and U does not provide an estimate of M (see Fig. 7.3).
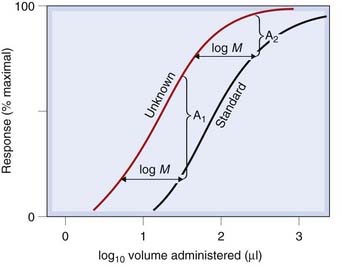
Fig. 7.3 Comparison of the potency of unknown and standard by bioassay.
Note that comparing the magnitude of responses produced by the same dose (i.e. volume) of standard and unknown gives no quantitative estimate of their relative potency. (The differences, A1 and A2, depend on the dose chosen.) Comparison of equieffective doses of standard and unknown gives a valid measure of their relative potencies. Because the lines are parallel, the magnitude of the effect chosen for the comparison is immaterial; i.e. log M is the same at all points on the curves.
The main problem with all types of bioassay is that of biological variation, and the design of bioassays is aimed at:
Commonly, comparisons are based on analysis of dose–response curves, from which the matching doses of S and U are calculated. The use of a logarithmic dose scale means that the curves for S and U will normally be parallel, and the potency ratio (M) is estimated from the horizontal distance between the two curves (Fig. 7.3). Assays of this type are known as parallel line assays, the minimal design being the 2 + 2 assay, in which two doses of standard (S1 and S2) and two of unknown (U1 and U2) are used. The doses are chosen to give responses lying on the linear part of the log dose–response curve, and are given repeatedly in randomised order, providing an inherent measure of the variability of the test system, which can be used, by means of straightforward statistical analysis, to estimate the confidence limits of the final result.
Problems arise if the two log dose–response curves are not parallel, for example if the assay is used to compare two drugs whose mechanism of action is not the same, or if one is a partial agonist (see Ch. 2). In this case it is not possible to define the relative potencies of S and U unambiguously in terms of a simple ratio and the experimenter must then face up to the fact that the comparison requires measurement of more than a single dimension of potency. An example of this kind of difficulty is met when diuretic drugs (Ch. 28) are compared. Some (‘low ceiling’) diuretics are capable of producing only a small diuretic effect, no matter how much is given; others (‘high ceiling’) can produce a very intense diuresis (described as ‘torrential’ by authors with vivid imaginations). A comparison of two such drugs requires not only a measure of the doses needed to produce an equal low-level diuretic effect, but also a measure of the relative heights of the ceilings.
A simple example of an experiment to compare two analgesic drugs, morphine and codeine (see Ch. 41) in humans, based on a modified 2 + 2 design is shown in Figure 7.4. Each of the four doses was given on different occasions to each of the four subjects, the order being randomised and both subject and observer being unaware of the dose given. Subjective pain relief was assessed by a trained observer, and the results showed morphine to be 13 times as potent as codeine. This, of course, does not prove its superiority, but merely shows that a smaller dose is needed to produce the same effect. Such a measurement is, however, an essential preliminary to assessing the relative therapeutic merits of the two drugs, for any comparison of other factors, such as side effects, duration of action, tolerance or dependence, needs to be done on the basis of doses that are equiactive as analgesics.
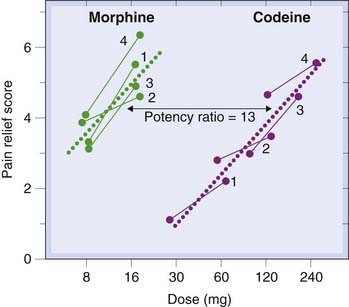
Fig. 7.4 Assay of morphine and codeine as analgesics in humans.
Each of four patients (numbered 1–4) was given, on successive occasions in random order, four different treatments (high and low morphine, and high and low codeine) by intramuscular injection, and the subjective pain relief score calculated for each. The calculated regression lines gave a potency ratio estimate of 13 for the two drugs.
(After Houde R W et al. 1965. In: Analgetics. Academic Press, New York.)
Animal Models of Disease
There are many examples where simple intuitive models predict with fair accuracy therapeutic efficacy in humans. Ferrets vomit when placed in swaying cages, and drugs that prevent this are also found to relieve motion sickness and other types of nausea in humans. Irritant chemicals injected into rats’ paws cause them to become swollen and tender, and this model predicts very well the efficacy of drugs used for symptomatic relief in inflammatory conditions such as rheumatoid arthritis in humans. As discussed elsewhere in this book, models for many important disorders, such as epilepsy, diabetes, hypertension and gastric ulceration, based on knowledge of the physiology of the condition, are available, and have been used successfully to produce new drugs, even though their success in predicting therapeutic efficacy is far from perfect.4
Ideally, an animal model should resemble the human disease in the following ways:
In practice, there are many difficulties, and the shortcomings of animal models are one of the main roadblocks on the route from basic medical science to improvements in therapy. The difficulties include the following.
Animal models
Genetic and Transgenic Animal Models
Nowadays, genetic approaches are increasingly used as an adjunct to conventional physiological and pharmacological approaches to disease modelling.
By selective breeding, it is possible to obtain pure animal strains with characteristics closely resembling certain human diseases. Genetic models of this kind include spontaneously hypertensive rats, genetically obese mice, epilepsy-prone dogs and mice, rats with deficient vasopressin secretion, and many other examples. In many cases, the genes responsible have not been identified.
The obese mouse, which arose from a spontaneous mutation in a mouse-breeding facility, is one of the most widely used models for the study of obesity and type 2 diabetes (see Ch. 30). The phenotype results from inactivation of the leptin gene, and shows good face validity (high food intake, gross obesity, impaired blood glucose regulation, vascular complications—features characteristic of human obesity) and good predictive validity (responding to pharmacological intervention similarly to humans), but poor construct validity, since obese humans are not leptin deficient.
Deliberate genetic manipulation of the germline to generate transgenic animals (see Rudolph & Moehler, 1999; Offermanns & Hein, 2004) is of growing importance as a means of replicating human disease states in experimental animals, and thereby providing animal models that are expected to be more predictive of therapeutic drug effects in humans. This versatile technology, first reported in 1980, can be used in many different ways, for example:
Currently, most transgenic technologies are applicable in mice but much more difficult in other mammals. Other vertebrates (e.g. zebrafish) and invertebrates (Drosophila, Caenorhabditis elegans) are increasingly used for drug screening purposes.
Examples of such models include transgenic mice that overexpress mutated forms of the amyloid precursor protein or presenilins, which are important in the pathogenesis of Alzheimer’s disease (see Ch. 39). When they are a few months old, these mice develop pathological lesions and cognitive changes resembling Alzheimer’s disease, and provide very useful models with which to test possible new therapeutic approaches to the disease. Another neurodegenerative condition, Parkinson’s disease (Ch. 39) has been modelled in transgenic mice that overexpress synuclein, a protein found in the brain inclusions that are characteristic of the disease. Transgenic mice with mutations in tumour suppressor genes and oncogenes (see Ch. 5) are widely used as models for human cancers. Mice in which the gene for a particular adenosine receptor subtype has been inactivated show distinct behavioural and cardiovascular abnormalities, such as increased aggression, reduced response to noxious stimuli and raised blood pressure. These findings serve to pinpoint the physiological role of this receptor, whose function was hitherto unknown, and to suggest new ways in which agonists or antagonists for these receptors might be developed for therapeutic use (e.g. to reduce aggressive behaviour or to treat hypertension). Transgenic mice can, however, be misleading in relation to human disease. For example, the gene defect responsible for causing cystic fibrosis (a disease affecting mainly the lungs in humans), when reproduced in mice, causes a disorder that mainly affects the intestine.
Pharmacological Studies in Humans
Studies involving human subjects range from experimental pharmacodynamic or pharmacokinetic investigations to formal clinical trials. Non-invasive recording methods, such as functional magnetic resonance imaging to measure regional blood flow in the brain (a surrogate for neuronal activity) and ultrasonography to measure cardiac performance, have greatly extended the range of what is possible. The scientific principles underlying experimental work in humans, designed, for example, to check whether mechanisms that operate in other species also apply to humans, or to take advantage of the much broader response capabilities of a person compared with a rat, are the same as for animals, but the ethical and safety issues are paramount, and ethical committees associated with all medical research centres tightly control the type of experiment that can be done, weighing up not only safety and ethical issues, but also the scientific importance of the proposed study. At the other end of the spectrum of experimentation on humans are formal clinical trials, often involving thousands of patients, aimed at answering specific questions regarding the efficacy and safety of new drugs.
Clinical Trials
Clinical trials are an important and highly specialised form of biological assay, designed specifically to measure therapeutic efficacy. The need to use patients undergoing treatment for experimental purposes raises serious ethical considerations, and imposes many restrictions. Here, we discuss some of the basic principles involved in clinical trials; the role of such trials in the course of drug development is described in Chapter 60.
A clinical trial is a method for comparing objectively, by a prospective study, the results of two or more therapeutic procedures. For new drugs, this is carried out during phase III of clinical development (Ch. 60). It is important to realise that, until about 50 years ago, methods of treatment were chosen on the basis of clinical impression and personal experience rather than objective testing.6 Although many drugs, with undoubted effectiveness, remain in use without ever having been subjected to a controlled clinical trial, any new drug is now required to have been tested in this way before being licensed for general clinical use.7
On the other hand, digitalis (see Ch. 21) was used for 200 years to treat cardiac failure before a controlled trial showed it to be of very limited value except in a particular type of patient.
A good account of the principles and organisation of clinical trials is given by Friedman et al. (1996). A clinical trial aims to compare the response of a test group of patients receiving a new treatment (A) with that of a control group receiving an existing ‘standard’ treatment (B). Treatment A might be a new drug or a new combination of existing drugs, or any other kind of therapeutic intervention, such as a surgical operation, a diet, physiotherapy and so on. The standard against which it is judged (treatment B) might be a currently used drug treatment or (if there is no currently available effective treatment) a placebo or no treatment at all.
The use of controls is crucial in clinical trials. Claims of therapeutic efficacy based on reports that, for example, 16 out of 20 patients receiving drug X got better within 2 weeks are of no value without a knowledge of how 20 patients receiving no treatment, or a different treatment, would have fared. Usually, the controls are provided by a separate group of patients from those receiving the test treatment, but sometimes a crossover design is possible in which the same patients are switched from test to control treatment or vice versa, and the results compared. Randomisation is essential to avoid bias in assigning individual patients to test or control groups. Hence, the randomised controlled clinical trial is now regarded as the essential tool for assessing clinical efficacy of new drugs.
Concern inevitably arises over the ethics of assigning patients at random to particular treatment groups (or to no treatment). However, the reason for setting up a trial is that doubt exists whether the test treatment offers greater benefit than the control treatment. All would agree on the principle of informed consent,8 whereby each patient must be told the nature and risks of the trial, and agree to participate on the basis that he or she will be randomly and unknowingly assigned to either the test or the control group.
Unlike the kind of bioassay discussed earlier, the clinical trial does not normally give any information about potency or the form of the dose–response curve, but merely compares the response produced by two stipulated therapeutic regimens. Survival curves provide one commonly used measure. Figure 7.5 shows rates of disease-free survival in two groups of breast cancer patients treated with conventional chemotherapy with and without the addition of paclitaxel (see Ch. 55). The divergence of the curves shows that paclitaxel significantly improved the clinical response. Additional questions may be posed, such as the prevalence and severity of side effects, or whether the treatment works better or worse in particular classes of patient, but only at the expense of added complexity and numbers of patients, and most trials are kept as simple as possible. The investigator must decide in advance what dose to use and how often to give it, and the trial will reveal only whether the chosen regimen performed better or worse than the control treatment. It will not say whether increasing or decreasing the dose would have improved the response; another trial would be needed to ascertain that. The basic question posed by a clinical trial is thus simpler than that addressed by most conventional bioassays. However, the organisation of clinical trials, with controls against bias, is immeasurably more complicated, time-consuming and expensive than that of any laboratory-based assay.
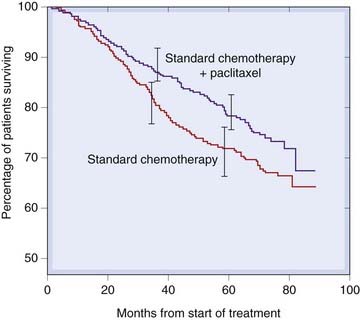
Fig. 7.5 Disease-free survival curves followed for 8 years in matched groups of breast cancer patients treated with a standard chemotherapy regime alone (629 patients), or with addition of paclitaxel (613 patients), showing a highly significant (P = 0.006) improvement with paclitaxel.
Error bars represent 95% confidence intervals.
(Redrawn from Martin et al. 2008 J Natl Cancer Inst 100:805–814.)
Avoidance of Bias
There are two main strategies that aim to minimise bias in clinical trials, namely:
If two treatments, A and B, are being compared on a series of selected patients, the simplest form of randomisation is to allocate each patient to A or B by reference to a series of random numbers. One difficulty with simple randomisation, particularly if the groups are small, is that the two groups may turn out to be ill-matched with respect to characteristics such as age, sex or disease severity. Stratified randomisation is often used to avoid the difficulty. Thus the subjects might be divided into age categories, random allocation to A or B being used within each category. It is possible to treat two or more characteristics of the trial population in this way, but the number of strata can quickly become large, and the process is self-defeating when the number of subjects in each becomes too small. As well as avoiding error resulting from imbalance of groups assigned to A and B, stratification can also allow more sophisticated conclusions to be reached. B might, for example, prove to be better than A in a particular group of patients even if it is not significantly better overall.
The double-blind technique, which means that neither subject nor investigator is aware at the time of the assessment which treatment is being used, is intended to minimise subjective bias. It has been repeatedly shown that, with the best will in the world, subjects and investigators both contribute to bias if they know which treatment is which, so the use of a double-blind technique is an important safeguard. It is not always possible, however. A dietary regimen or a surgical operation, for example, can seldom be disguised, and even with drugs, pharmacological effects may reveal to patients what they are taking and predispose them to report accordingly.9 In general, however, the use of a double-blind procedure, with precautions if necessary to disguise such clues as the taste or appearance of the two drugs, is an important principle.10
The Size of the Sample
Both ethical and financial considerations dictate that the trial should involve the minimum number of subjects, and much statistical thought has gone into the problem of deciding in advance how many subjects will be required to produce a useful result. The results of a trial cannot, by their nature, be absolutely conclusive. This is because it is based on a sample of patients, and there is always a chance that the sample was atypical of the population from which it came. Two types of erroneous conclusion are possible, referred to as type I and type II errors. A type I error occurs if a difference is found between A and B when none actually exists (false positive). A type II error occurs if no difference is found although A and B do actually differ (false negative). A major factor that determines the size of sample needed is the degree of certainty the investigator seeks in avoiding either type of error. The probability of incurring a type I error is expressed as the significance of the result. To say that A and B are different at the P < 0.05 level of significance means that the probability of obtaining a false positive result (i.e. incurring a type I error) is less than 1 in 20. For most purposes, this level of significance is considered acceptable as a basis for drawing conclusions.
The probability of avoiding a type II error (i.e. failing to detect a real difference between A and B) is termed the power of the trial. We tend to regard type II errors more leniently than type I errors, and trials are often designed with a power of 0.8–0.9. To increase the significance and the power of a trial requires more patients. The second factor that determines the sample size required is the magnitude of difference between A and B that is regarded as clinically significant. For example, to detect that a given treatment reduces the mortality in a certain condition by at least 10 percentage points, say from 50% (in the control group) to 40% (in the treated group), would require 850 subjects, assuming that we wanted to achieve a P < 0.05 level of significance and a power of 0.9. If we were content only to reveal a reduction by 20 percentage points (and very likely miss a reduction by 10 points), only 210 subjects would be needed. In this example, missing a real 10-point reduction in mortality could result in abandonment of a treatment that would save 100 lives for every 1000 patients treated—an extremely serious mistake from society’s point of view. This simple example emphasises the need to assess clinical benefit (which is often difficult to quantify) in parallel with statistical considerations (which are fairly straightforward) in planning trials.
A trial may give a significant result before the planned number of patients have been enrolled, so it is common for interim analyses to be carried out (by an independent team so that the trial team remains unaware of the results). If this analysis gives a conclusive result, or if it shows that continuation is unlikely to give a conclusive result, the trial can be terminated, thus reducing the number of subjects tested. In one such large-scale trial (Beta-blocker Heart Attack Trial Research Group, 1982) of the value of long-term treatment with the β-adrenoceptor-blocking drug propranolol (Ch. 14) following heart attacks, the interim results showed a significant reduction in mortality, which led to the early termination of the trial. In sequential trials, the results are computed case by case (each case being paired with a control) as the trial proceeds, and the trial stopped as soon as a result (at a predetermined level of significance) is achieved.
Various ‘hybrid’ trial designs, which have the advantage of sequential trials in minimising the number of patients needed but do not require strict pairing of subjects, have been devised (see Friedman et al., 1996).
Recently, the tendency has been to perform very large-scale trials, to allow several different treatment protocols, in various different patient groups to be compared. An example is the ALLHAT trial of various antihypertensive and lipid-lowering drugs to improve the outcome in cardiovascular disease (see Ch. 22). This ran from 1994 to 2002, cost US$130 million, and involved more than 42 000 patients in 623 treatment centres, with an army of coordinators and managers to keep it on track. One of its several far-reaching conclusions was that a cheap and familiar diuretic drug in use for more than 50 years was more effective than more recent and expensive antihypertensive drugs.11
Clinical Outcome Measures
The measurement of clinical outcome can be a complicated business, and is becoming increasingly so as society becomes more preoccupied with assessing the efficacy of therapeutic procedures in terms of improved quality of life, and societal and economic benefit, rather than in terms of objective clinical effects, such as lowering of blood pressure, improved airways conductance or increased life expectancy. Various scales for assessing ‘health-related quality of life’ have been devised and tested (see Walley & Haycocks, 1997), and the tendency is to combine these with measures of life expectancy to arrive at the measure ‘quality-adjusted life years’ (QALYs) as an overall measure of therapeutic efficacy, which attempts to combine both survival time and relief from suffering in assessing overall benefit.12 In planning clinical trials, it is necessary to decide the purpose of the trial in advance, and to define the outcome measures accordingly.
Frequentist and Bayesian Approaches
The conventional approach to analysis of scientific data (including clinical trials data) is known as ‘frequentist’ and is based on a null hypothesis, for example of the form: treatment A is no more effective than treatment B. Rejection of the hypothesis implies that A is more effective than B. Suppose that a trial shows, on average, that patients treated with A live longer than patients treated with B. Conventional frequentist statistics addresses the question: If A were actually no more effective than B, what is the probability (P) of obtaining the results that were actually obtained in the trial? In other words, given that treatment A is no better than B, how often, had we repeated the trial many times, would we have obtained results suggesting that A is better? If this probability is low (say, less than 0.05), we reject the null hypothesis and conclude that A is most likely better. If P is larger, the results could quite easily have been obtained without there being any true difference between A and B, and we cannot reject the null hypothesis.
If we have no prior reason for thinking that A will be better than B, the frequentist approach is perfectly appropriate, and it is the usual principle on which trials of unknown drugs are based. But often, in real life, there will be good reason, based on previous trials or clinical experience, to believe that A is actually better than B. Using a Bayesian approach allows this to be taken into account formally and explicitly by defining a prior probability for the effect of A. The data from the new trial, which can be smaller than a conventional trial, are then statistically superimposed on the prior probability curve to produce a posterior probability curve, in effect an update of the prior probability curve that takes account of the new data. The Bayesian approach is controversial, depending as it does on expressing the (often subjective) prior assumption in explicit mathematical terms, and the statistical analysis is complex. Nevertheless, it can be argued that to ignore altogether prior knowledge and experience when interpreting new data is unjustified, and even unethical, and the Bayesian approach is consequently gaining acceptance.
For an explanation of the principles underlying Bayesian approaches, which are being increasingly applied to clinical trials, see Spiegelhalter et al. (1999) and Lilford & Braunholtz (2000).
Placebos
A placebo is a dummy medicine containing no active ingredient (or alternatively, a dummy surgical procedure, diet or other kind of therapeutic intervention), which the patient believes is (or could be, in the context of a controlled trial) the real thing. The ‘placebo response’ is widely believed to be a powerful therapeutic effect,13 producing a significant beneficial effect in about one-third of patients. While many clinical trials include a placebo group that shows improvement, few have compared this group directly with untreated controls. A survey of these trial results (Hróbjartsson & Grøtsche, 2001) concluded (controversially) that the placebo effect was generally insignificant, except in the case of pain relief, where it was small but significant. They concluded that the popular belief in the strength of the placebo effect is misplaced, and probably reflects in part the tendency of many symptoms to improve spontaneously and in part the reporting bias of patients who want to please their doctors. The ethical case for using placebos as therapy, which has been the subject of much public discussion, may therefore be weaker than has been argued. The risks of placebo therapies should not be underestimated. The use of active medicines may be delayed. The necessary element of deception risks undermining the confidence of patients in the integrity of doctors. A state of ‘therapy dependence’ may be produced in people who are not ill, because there is no way of assessing whether a patient still ‘needs’ the placebo.
Meta-Analysis
It is possible, by the use of statistical techniques, to combine the data obtained in several individual trials (provided each has been conducted according to a randomised design) in order to gain greater power and significance. This procedure, known as meta-analysis or overview analysis, can be very useful in arriving at a conclusion on the basis of several published trials, of which some claimed superiority of the test treatment over the control while others did not. As an objective procedure, it is certainly preferable to the ‘take your pick’ approach to conclusion forming adopted by most human beings when confronted with contradictory data. It has several drawbacks, however (see Naylor, 1997), the main one being ‘publication bias’, because negative studies are generally considered less interesting, and are therefore less likely to be published, than positive studies. Double counting, caused by the same data being incorporated into more than one trial report, is another problem.
The organisation of large-scale clinical trials involving hundreds or thousands of patients at many different centres is a massive and expensive undertaking that makes up one of the major costs of developing a new drug, and can easily go wrong.
An early large trial (Anturane Reinfarction Trial Research Group, 1978) involved 1620 patients at 26 research centres in the USA and Canada, 98 collaborating researchers and a formidable list of organising committees, including two independent audit committees to check that the work was being carried out in conformity with the strict protocols established. The conclusion was that the drug under test (sulfinpyrazone) reduced by almost one-half the mortality from repeat heart attacks in the 8-month period after a first attack, and could save many lives. The US Food and Drug Administration, however, refused to grant a licence for the use of the drug, criticising the trial as unreliable and biased in several respects. Their independent analysis of the data showed the beneficial effect of the drug to be slight and insignificant. Further analysis and further trials, however, supported the original conclusion, but by then the efficacy of aspirin in this condition had been established, so the use of sulfinpyrazone never found favour. Much larger trials are now regularly conducted, exemplified by the ALLHAT trial mentioned above (p. 96).
Clinical trials
Balancing Benefit and Risk
Therapeutic Index
The concept of therapeutic index aims to provide a measure of the margin of safety of a drug, by drawing attention to the relationship between the effective and toxic doses:

where LD50 is the dose that is lethal in 50% of the population, and ED50 is the dose that is ‘effective’ in 50%. Obviously, it can only be measured in animals, and it is not a useful guide to the safety of a drug in clinical use for several reasons:
Other Measures of Benefit and Risk
Alternative ways of quantifying the benefits and risks of drugs in clinical use have received much attention. One useful approach is to estimate from clinical trial data the proportion of test and control patients who will experience (a) a defined level of clinical benefit (e.g. survival beyond 2 years, pain relief to a certain predetermined level, slowing of cognitive decline by a given amount) and (b) adverse effects of defined degree. These estimates of proportions of patients showing beneficial or harmful reactions can be expressed as number needed to treat (NNT; i.e. the number of patients who need to be treated in order for one to show the given effect, whether beneficial or adverse). For example, in a recent study of pain relief by antidepressant drugs compared with placebo, the findings were: for benefit (a defined level of pain relief), NNT = 3; for minor unwanted effects, NNT = 3; for major adverse effects, NNT = 22. Thus of 100 patients treated with the drug, on average 33 will experience pain relief, 33 will experience minor unwanted effects, and 4 or 5 will experience major adverse effects, information that is helpful in guiding therapeutic choices. One advantage of this type of analysis is that it can take into account the underlying disease severity in quantifying benefit. Thus if drug A halves the mortality of an often fatal disease (reducing it from 50% to 25%, say), the NNT to save one life is 4; if drug B halves the mortality of a rarely fatal disease (reducing it from 5% to 2.5%, say), the NNT to save one life is 40. Notwithstanding other considerations, drug A is judged to be more valuable than drug B, even though both reduce mortality by one-half. Furthermore, the clinician must realise that to save one life with drug B, 40 patients must be exposed to a risk of adverse effects, whereas only 4 are exposed for each life saved with drug A.
Determination of risk and benefit
References and Further Reading
Colquhoun D. Lectures on biostatistics. Oxford: Oxford University Press; 1971. (Standard textbook)
Kirkwood B.R., Sterne J.A.C. Medical statistics, second ed. Malden: Blackwell; 2003. (Clear introductory textbook covering statistical principles and methods)
Lilford R.J., Braunholtz D. Who’s afraid of Thomas Bayes? J. Epidemiol. Community Health. 2000;54:731-739. (Explains the principles of Bayesian analysis in a non-mathematical way)
Walley T., Haycocks A. Pharmacoeconomics: basic concepts and terminology. Br. J. Clin. Pharmacol.. 1997;43:343-348. (Useful introduction to analytical principles that are becoming increasingly important for therapeutic policy makers)
Yanagisawa M., Kurihara H., Kimura S., et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411-415. (The first paper describing endothelin—a remarkably full characterisation of an important new mediator)
Offermanns S., Hein L. Transgenic models in pharmacology 2004 Handb. Exp. Pharmacol. 159. (A comprehensive series of review articles describing transgenic mouse models used to study different pharmacological mechanisms and disease states)
Ristevski S. Making better transgenic models: conditional, temporal, and spatial approaches. Mol. Biotechnol.. 2005;29:153-164. (Description of methods for controlling transgene expression)
Rudolph U., Moehler H. Genetically modified animals in pharmacological research: future trends. Eur. J. Pharmacol.. 1999;375:327-337. (Good review of uses of transgenic animals in pharmacological research, including application to disease models)
Anturane Reinfarction Trial Research Group. Sulfinpyrazone in the prevention of cardiac death after myocardial infarction. N. Engl. J. Med.. 1978;298:289-295. (Example of an early large-scale clinical trial)
Beta-blocker Heart Attack Trial Research Group. A randomised trial of propranolol in patients with acute myocardial infarction. 1. Mortality results. JAMA. 1982;247:1707-1714. (A trial that was terminated early when clear evidence of benefit emerged)
Friedman L.M., Furberg C.D., DeMets D.L. Fundamentals of clinical trials, third ed. St Louis: Mosby; 1996. (Standard textbook)
Hróbjartsson A., Gøtzsche P.C. Is the placebo powerless? An analysis of clinical trials comparing placebo with no treatment. N. Engl. J. Med.. 2001;344:1594-1601. (An important meta-analysis of clinical trial data, which shows, contrary to common belief, that placebos in general have no significant effect on clinical outcome, except—to a small degree—in pain relief trials. Confirmed in an extended analysis: J. Int. Med. 2004, 256, 91–100)
Naylor C.D. Meta-analysis and the meta-epidemiology of clinical research. Br. Med. J.. 1997;315:617-619. (Thoughtful review on the strengths and weaknesses of meta-analysis)
Sackett D.L., Rosenburg W.M.C., Muir-Gray J.A., et al. Evidence-based medicine: what it is and what it isn’t. Br. Med. J.. 1996;312:71-72. (Balanced account of the value of evidence-based medicine—an important recent trend in medical thinking)
Spiegelhalter D.J., Myles J.P., Jones D.R., Abrams K.R. An introduction to Bayesian methods in health technology assessment. Br. Med. J.. 1999;319:508-512. (Short non-mathematical explanation of the Bayesian approach to data analysis)
1Consider the effect of cocaine on organised crime, of organophosphate ‘nerve gases’ on the stability of dictatorships or of anaesthetics on the feasibility of surgical procedures for examples of molecular interactions that affect the behaviour of populations and societies.
2In 1988, a Japanese group (Yanagisawa et al., 1988) described in a single remarkable paper the bioassay, purification, chemical analysis, synthesis and DNA cloning of a new vascular peptide, endothelin (see Ch. 19).
3More picturesque examples of absolute units of the kind that Burn would have frowned on are the PHI and the mHelen. PHI, cited by Colquhoun (1971), stands for ‘purity in heart index’ and measures the ability of a virgin pure-in-heart to transform, under appropriate conditions, a he-goat into a youth of surpassing beauty. The mHelen is a unit of beauty, 1 mHelen being sufficient to launch 1 ship.
4There have been many examples of drugs that were highly effective in experimental animals (e.g. in reducing brain damage following cerebral ischaemia) but ineffective in humans (stroke victims). Similarly, substance P antagonists (Ch. 19) are effective in animal tests for analgesia, but they proved inactive when tested in humans. How many errors in the opposite direction may have occurred we shall never know, because such drugs will not have been tested in humans.
5With conventional transgenic technology, the genetic abnormality is expressed throughout development, sometimes proving lethal or causing major developmental abnormalities. Conditional transgenesis is now possible (see Ristevski, 2005), allowing the transgene to remain silent until triggered by the administration of a chemical promoter (e.g. the tetracycline analogue, doxycycline, in the most widely used Cre-Lox conditional system). This avoids the complications of developmental effects and long-term adaptations, and may allow adult disease to be modelled more accurately.
6Not exclusively. James Lind conducted a controlled trial in 1753 on 12 mariners, which showed that oranges and lemons offered protection against scurvy. However, 40 years passed before the British Navy acted on his advice, and a further century before the US Navy did.
7It is fashionable in some quarters to argue that to require evidence of efficacy of therapeutic procedures in the form of a controlled trial runs counter to the doctrines of ‘holistic’ medicine. This is a fundamentally antiscientific view, for science advances only by generating predictions from hypotheses and by subjecting the predictions to experimental test. ‘Alternative’ medical procedures, such as homeopathy, aromatherapy, acupuncture or ‘detox’, have rarely been so tested, and where they have they generally lack efficacy. Standing up for the scientific approach is the evidence-based medicine movement (see Sackett et al., 1996), which sets out strict criteria for assessing therapeutic efficacy, based on randomised, controlled clinical trials, and urges scepticism about therapeutic doctrines whose efficacy has not been so demonstrated.
8Even this can be contentious, because patients who are unconscious, demented or mentally ill are unable to give such consent, yet no one would want to preclude trials that might offer improved therapies to these needy patients. Clinical trials in children are particularly problematic but are necessary if the treatment of childhood diseases is to be placed on the same evidence base as is judged appropriate for adults. There are many examples where experience has shown that children respond differently from adults, and there is now increasing pressure on pharmaceutical companies to perform trials in children, despite the difficulties of carrying out such studies. The same concerns apply to trials in elderly patients.
9The distinction between a true pharmacological response and a beneficial clinical effect produced by the knowledge (based on the pharmacological effects that the drug produces) that an active drug is being administered is not easy to draw, and we should not expect a mere clinical trial to resolve such a fine semantic issue.
10Maintaining the blind can be problematic. In an attempt to determine whether melatonin is effective in countering jet lag, a pharmacologist selected a group of fellow pharmacologists attending a congress in Australia, providing them with unlabelled capsules of melatonin or placebo, with a jet lag questionnaire to fill in when they arrived. Many of them (one of the authors included), with analytical resources easily to hand, opened the capsules and consigned them to the bin if they contained placebo. Pharmacologists are only human.
11Though without much impact so far on prescribing habits, owing to the marketing muscle of pharmaceutical companies.
12As may be imagined, trading off duration and quality of life raises issues about which many of us feel decidedly squeamish. Not so economists, however. They approach the problem by asking such questions as: ‘How many years of life would you be prepared to sacrifice in order to live the rest of your life free of the disability you are currently experiencing?’ Or, even more disturbingly: ‘If you could gamble on surviving free of disability for your normal lifespan, or (if you lose the gamble) dying immediately, what odds would you accept?’ Imagine being asked this by your doctor. ‘But I only wanted something for my sore throat,’ you protest weakly.
13Its opposite, the nocebo effect, describes the adverse effects reported with dummy medicines.
14Ironically, thalidomide—probably the most harmful drug ever marketed—was promoted specifically on the basis of its exceptionally high therapeutic index (i.e. it killed rats only when given in extremely large doses).