2 How drugs act
General principles
Overview
The emergence of pharmacology as a science came when the emphasis shifted from describing what drugs do to explaining how they work. In this chapter, we set out some general principles underlying the interaction of drugs with living systems (Ch. 3 goes into the molecular aspects in more detail). The interaction between drugs and cells is described, followed by a more detailed examination of different types of drug–receptor interaction. We are still far from the holy grail of being able to predict the pharmacological effects of a novel chemical substance, or to design ab initio a chemical to produce a specified therapeutic effect; nevertheless, we can identify some important general principles, which is our purpose in this chapter.
Introduction
To begin with, we should gratefully acknowledge Paul Ehrlich for insisting that drug action must be explicable in terms of conventional chemical interactions between drugs and tissues, and for dispelling the idea that the remarkable potency and specificity of action of some drugs put them somehow out of reach of chemistry and physics and required the intervention of magical ‘vital forces’. Although many drugs produce effects in extraordinarily low doses and concentrations, low concentrations still involve very large numbers of molecules. One drop of a solution of a drug at only 10−10 mol/l still contains about 3 × 109 drug molecules, so there is no mystery in the fact that it may produce an obvious pharmacological response. Some bacterial toxins (e.g. diphtheria toxin) act with such precision that a single molecule taken up by a target cell is sufficient to kill it.
One of the basic tenets of pharmacology is that drug molecules must exert some chemical influence on one or more constituents of cells in order to produce a pharmacological response. In other words, drug molecules must get so close to these constituent cellular molecules that the two interact chemically in such a way that the function of the latter is altered. Of course, the molecules in the organism vastly outnumber the drug molecules, and if the drug molecules were merely distributed at random, the chance of interaction with any particular class of cellular molecule would be negligible. Pharmacological effects, therefore, require, in general, the non-uniform distribution of the drug molecule within the body or tissue, which is the same as saying that drug molecules must be ‘bound’ to particular constituents of cells and tissues in order to produce an effect. Ehrlich summed it up thus: ‘Corpora non agunt nisi fixata’ (in this context, ‘A drug will not work unless it is bound’).1
These critical binding sites are often referred to as ‘drug targets’ (an obvious allusion to Ehrlich’s famous phrase ‘magic bullets’, describing the potential of antimicrobial drugs). The mechanisms by which the association of a drug molecule with its target leads to a physiological response constitute the major thrust of pharmacological research. Most drug targets are protein molecules. Even general anaesthetics (see Ch. 40), which were long thought to produce their effects by an interaction with membrane lipid, now appear to interact mainly with membrane proteins (see Franks, 2008). All rules need exceptions, and many antimicrobial and antitumour drugs (Chs 50 and 55), as well as mutagenic and carcinogenic agents (Ch. 57), interact directly with DNA rather than protein; bisphosphonates, used to treat osteoporosis (Ch. 35), bind to calcium salts in the bone matrix, rendering it toxic to osteoclasts, much like rat poison.
Protein Targets for Drug Binding
Four main kinds of regulatory protein are commonly involved as primary drug targets, namely:
There are some exceptions, particularly among the new generation of biopharmaceutical drugs (see Ch. 59). Furthermore, many drugs bind (in addition to their primary targets) to plasma proteins (see Ch. 8) and other tissue proteins, without producing any obvious physiological effect. Nevertheless, the generalisation that most drugs act on one or other of the four types of protein listed above serves as a good starting point.
Further discussion of the mechanisms by which such binding leads to cellular responses is given in Chapters 3–4.
Targets for drug action 
Drug Receptors
What Do We Mean by Receptors?
 As emphasised in Chapter 1, the concept of receptors is central to pharmacology, and the term is most often used to describe the target molecules through which soluble physiological mediators—hormones, neurotransmitters, inflammatory mediators, etc.—produce their effects. Examples such as acetylcholine receptors, cytokine receptors, steroid receptors, and growth hormone receptors abound in this book, and generally the term receptor indicates a recognition molecule for a chemical mediator.
As emphasised in Chapter 1, the concept of receptors is central to pharmacology, and the term is most often used to describe the target molecules through which soluble physiological mediators—hormones, neurotransmitters, inflammatory mediators, etc.—produce their effects. Examples such as acetylcholine receptors, cytokine receptors, steroid receptors, and growth hormone receptors abound in this book, and generally the term receptor indicates a recognition molecule for a chemical mediator.
‘Receptor’ is sometimes used to denote any target molecule with which a drug molecule (i.e. a foreign compound rather than an endogenous mediator) has to combine in order to elicit its specific effect. For example, the voltage-sensitive sodium channel is sometimes referred to as the ‘receptor’ for local anaesthetics (see Ch. 42), or the enzyme dihydrofolate reductase as the ‘receptor’ for methotrexate (Ch. 49). The term drug target, of which receptors are one type, is preferable in this context.
In the more general context of cell biology, the term receptor is used to describe various cell surface molecules (such as T-cell receptors, integrins, Toll receptors, etc; see Ch. 6) involved in the cell-to-cell interactions that are important in immunology, cell growth, migration and differentiation, some of which are also emerging as drug targets. These receptors differ from conventional pharmacological receptors in that they respond to proteins attached to cell surfaces or extracellular structures, rather than to soluble mediators.
Various carrier proteins are often referred to as receptors, such as the low-density lipoprotein receptor that plays a key role in lipid metabolism (Ch. 23) and the transferrin receptor involved in iron absorption (Ch. 25). These entities have little in common with pharmacological receptors. Though quite distinct from pharmacological receptors, these proteins play an important role in the action of drugs such as statins (Ch. 23).
Receptors in Physiological Systems
Receptors form a key part of the system of chemical communication that all multicellular organisms use to coordinate the activities of their cells and organs. Without them, we would resemble a bucketful of amoebae.
Some fundamental properties of receptors are illustrated by the action of adrenaline (epinephrine) on the heart. Adrenaline first binds to a receptor protein (the β-adrenoceptor, see Ch. 14) that serves as a recognition site for adrenaline and other catecholamines. When it binds to the receptor, a train of reactions is initiated (see Ch. 3) leading to an increase in force and rate of the heartbeat. In the absence of adrenaline, the receptor is functionally silent. This is true of most receptors for endogenous mediators (hormones, neurotransmitters, cytokines, etc.), although there are examples (see Ch. 3) of receptors that are ‘constitutively active’—that is, they exert a controlling influence even when no chemical mediator is present.
There is an important distinction between agonists, which ‘activate’ the receptors, and antagonists, which combine at the same site without causing activation, and block the effect of agonists on that receptor. The distinction between agonists and antagonists only exists for receptors with this type of physiological regulatory role; we cannot usefully speak of ‘agonists’ for the more general class of drug targets described above.
The characteristics and accepted nomenclature of pharmacological receptors are described by Neubig et al. (2003). The origins of the receptor concept and its pharmacological significance are discussed by Rang (2006).
Drug Specificity
For a drug to be useful as either a therapeutic or a scientific tool, it must act selectively on particular cells and tissues. In other words, it must show a high degree of binding site specificity. Conversely, proteins that function as drug targets generally show a high degree of ligand specificity; they bind only molecules of a certain precise type.
These principles of binding site and ligand specificity can be clearly recognised in the actions of a mediator such as angiotensin (Ch. 22). This peptide acts strongly on vascular smooth muscle, and on the kidney tubule, but has very little effect on other kinds of smooth muscle or on the intestinal epithelium. Other mediators affect a quite different spectrum of cells and tissues, the pattern in each case reflecting the specific pattern of expression of the protein receptors for the various mediators. A small chemical change, such as conversion of one of the amino acids in angiotensin from L to D form, or removal of one amino acid from the chain, can inactivate the molecule altogether, because the receptor fails to bind the altered form. The complementary specificity of ligands and binding sites, which gives rise to the very exact molecular recognition properties of proteins, is central to explaining many of the phenomena of pharmacology. It is no exaggeration to say that the ability of proteins to interact in a highly selective way with other molecules—including other proteins—is the basis of living machines. Its relevance to the understanding of drug action will be a recurring theme in this book.
Finally, it must be emphasised that no drug acts with complete specificity. Thus tricyclic antidepressant drugs (Ch. 46) act by blocking monoamine transporters but are notorious for producing side effects (e.g. dry mouth) related to their ability to block various receptors. In general, the lower the potency of a drug and the higher the dose needed, the more likely it is that sites of action other than the primary one will assume significance. In clinical terms, this is often associated with the appearance of unwanted side effects, of which no drug is free.
Since the 1970s, pharmacological research has succeeded in identifying the protein targets of many different types of drug. Drugs such as opioid analgesics (Ch. 41), cannabinoids (Ch. 18) and benzodiazepine tranquillisers (Ch.43), whose actions had been described in exhaustive detail for many years, are now known to target well-defined receptors, which have been fully characterised by gene-cloning techniques (see Ch. 3).
Receptor Classification
Where the action of a drug can be associated with a particular receptor, this provides a valuable means for classification and refinement in drug design. For example, pharmacological analysis of the actions of histamine (see Ch. 17) showed that some of its effects (the H1 effects, such as smooth muscle contraction) were strongly antagonised by the competitive histamine antagonists then known. Black and his colleagues suggested in 1970 that the remaining actions of histamine, which included its stimulant effect on gastric secretion, might represent a second class of histamine receptor (H2). Testing a number of histamine analogues, they found that some were selective in producing H2 effects, with little H1 activity. By analysing which parts of the histamine molecule conferred this type of specificity, they were able to develop selective H2 antagonists, which proved to be potent in blocking gastric acid secretion, a development of major therapeutic significance (Ch. 29). Two further types of histamine receptor (H3 and H4) were recognised later.
Receptor classification based on pharmacological responses continues to be a valuable and widely used approach. Newer experimental approaches have produced other criteria on which to base receptor classification. The direct measurement of ligand binding to receptors (see below) has allowed many new receptor subtypes to be defined that could not easily be distinguished by studies of drug effects. Molecular cloning (see Ch. 3) provided a completely new basis for classification at a much finer level of detail than can be reached through pharmacological analysis. Finally, analysis of the biochemical pathways that are linked to receptor activation (see Ch. 3) provides yet another basis for classification.
The result of this data explosion was that receptor classification suddenly became much more detailed, with a proliferation of receptor subtypes for all the main types of ligand. As alternative molecular and biochemical classifications began to spring up that were incompatible with the accepted pharmacologically defined receptor classes, the International Union of Pharmacological Sciences (IUPHAR) convened expert working groups to produce agreed receptor classifications for the major types, taking into account the pharmacological, molecular and biochemical information available. These wise people have a hard task; their conclusions will be neither perfect nor final but are essential to ensure a consistent terminology. To the student, this may seem an arcane exercise in taxonomy, generating much detail but little illumination. There is a danger that the tedious lists of drug names, actions and side effects that used to burden the subject will be replaced by exhaustive tables of receptors, ligands and transduction pathways. In this book, we have tried to avoid detail for its own sake and include only such information on receptor classification as seems interesting in its own right or is helpful in explaining the actions of important drugs. A comprehensive IUPHAR database of known receptor classes is available (see http://www.iuphar-db.org), as well as a regularly updated summary (Alexander et al., 2009).
Drug–Receptor Interactions
Occupation of a receptor by a drug molecule may or may not result in activation of the receptor. By activation, we mean that the receptor is affected by the bound molecule in such a way as to elicit a tissue response. The molecular mechanisms associated with receptor activation are discussed in Chapter 3. Binding and activation represent two distinct steps in the generation of the receptor-mediated response by an agonist (Fig. 2.1). If a drug binds to the receptor without causing activation and thereby prevents the agonist from binding, it is termed a receptor antagonist. The tendency of a drug to bind to the receptors is governed by its affinity, whereas the tendency for it, once bound, to activate the receptor is denoted by its efficacy. These terms are defined more precisely below (p. 13). Drugs of high potency generally have a high affinity for the receptors and thus occupy a significant proportion of the receptors even at low concentrations. Agonists also possess significant efficacy, whereas antagonists, in the simplest case, have zero efficacy. Drugs with intermediate levels of efficacy, such that even when 100% of the receptors are occupied the tissue response is submaximal, are known as partial agonists, to distinguish them from full agonists, the efficacy of which is sufficient that they can elicit a maximal tissue response. These concepts, though clearly an oversimplified description of events at the molecular level (see Ch. 3), provide a useful basis for characterising drug effects.
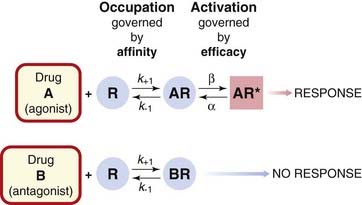
Fig. 2.1 The distinction between drug binding and receptor activation.
Ligand A is an agonist, because when it is bound, the receptor (R) tends to become activated, whereas ligand B is an antagonist, because binding does not lead to activation. The rate constants k+1, k−1, α and β for the binding and activation steps vary between drugs. For an antagonist, which does not activate the receptor, β = 0.
We now discuss certain aspects in more detail, namely drug binding, agonist concentration–effect curves, competitive antagonism, partial agonists and the nature of efficacy. Understanding these concepts at a qualitative level is sufficient for many purposes, but for more detailed analysis a quantitative formulation is needed (see p. 16).
The Binding of Drugs to Receptors
The binding of drugs to receptors can often be measured directly by the use of drug molecules (agonists or antagonists) labelled with one or more radioactive atoms (usually 3H, 14C or 125I). The usual procedure is to incubate samples of the tissue (or membrane fragments) with various concentrations of radioactive drug until equilibrium is reached. The bound radioactivity is measured after removal of the supernatant.
In such experiments, there is invariably a certain amount of ‘non-specific binding’ (i.e. drug taken up by structures other than receptors), which obscures the specific component and needs to be kept to a minimum. The amount of non-specific binding is estimated by measuring the radioactivity taken up in the presence of a saturating concentration of a (non-radioactive) ligand that inhibits completely the binding of the radioactive drug to the receptors, leaving behind the non-specific component. This is then subtracted from the total binding to give an estimate of specific binding (Fig. 2.2). The binding curve (Fig. 2.2B) defines the relationship between concentration and the amount of drug bound (B), and in most cases it fits well to the relationship predicted theoretically (see Fig. 2.11, below), allowing the affinity of the drug for the receptors to be estimated, as well as the binding capacity (Bmax), representing the density of receptors in the tissue. When combined with functional studies, binding measurements have proved very valuable. It has, for example, been confirmed that the spare receptor hypothesis (p. 13) for muscarinic receptors in smooth muscle is correct; agonists are found to bind, in general, with rather low affinity, and a maximal biological effect occurs at low receptor occupancy. It has also been shown, in skeletal muscle and other tissues, that denervation leads to an increase in the number of receptors in the target cell, a finding that accounts, at least in part, for the phenomenon of denervation supersensitivity. More generally, it appears that receptors tend to increase in number, usually over the course of a few days, if the relevant hormone or transmitter is absent or scarce, and to decrease in number if it is in excess, a process of adaptation to drugs or hormones resulting from continued administration (see p. 15).
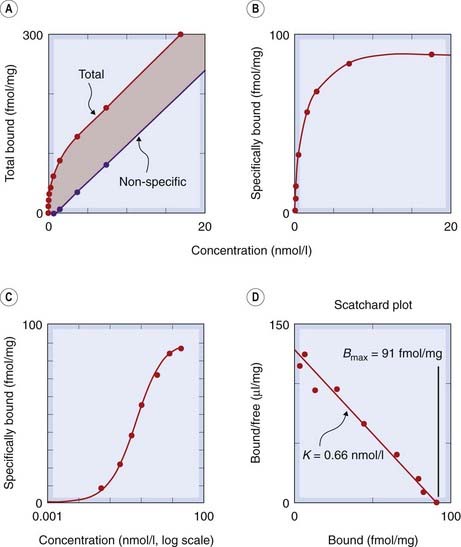
Fig. 2.2 Measurement of receptor binding (β adrenoceptors in cardiac cell membranes).
The ligand was [3H]-cyanopindolol, a derivative of pindolol (see Ch. 14). [A] Measurements of total and non-specific binding at equilibrium. Non-specific binding is measured in the presence of a saturating concentration of a non-radioactive β-adrenoceptor agonist, which prevents the radioactive ligand from binding to β adrenoceptors. The difference between the two lines represents specific binding. [B] Specific binding plotted against concentration. The curve is a rectangular hyperbola (equation 2.5). [C] Specific binding plotted against concentration (log scale). The sigmoid curve is a logistic curve representing the logarithmic scaling of the rectangular hyperbola plotted in panel B. [D] Scatchard plot (equation 2.7). This gives a straight line from which the binding parameters K and Bmax can be calculated.
Non-invasive imaging techniques, such as positron emission tomography (PET), can also be used to investigate the distribution of receptors in structures such as the living human brain. This technique has been used, for example, to measure the degree of dopamine receptor blockade produced by antipsychotic drugs in the brains of schizophrenic patients (see Ch. 45).
Binding curves with agonists often reveal an apparent heterogeneity among receptors. For example, agonist binding to muscarinic receptors (Ch. 13) and also to β-adrenoceptors (Ch. 14) suggests at least two populations of binding sites with different affinities. This may be because the receptors can exist either unattached or coupled within the membrane to another macromolecule, the G-protein (see Ch. 3), which constitutes part of the transduction system through which the receptor exerts its regulatory effect. Antagonist binding does not show this complexity, probably because antagonists, by their nature, do not lead to the secondary event of G-protein coupling. Because agonist binding results in activation, agonist affinity has proved to be a surprisingly elusive concept, about which afficionados love to argue.
The Relation between Drug Concentration and Effect
Although binding can be measured directly, it is usually a biological response, such as a rise in blood pressure, contraction or relaxation of a strip of smooth muscle in an organ bath, the activation of an enzyme, or a behavioural response, that we are interested in, and this is often plotted as a concentration–effect curve (in vitro) or dose–response curve (in vivo), as in Figure 2.3. Such curves allow us to estimate the maximal response that the drug can produce (Emax), and the concentration or dose needed to produce a 50% maximal response (EC50 or ED50), parameters that are useful for comparing the potencies of different drugs that produce qualitatively similar effects (see Ch. 7). Although they look similar to the binding curve in Figure 2.2C, concentration–effect curves cannot be used to measure the affinity of agonist drugs for their receptors, because the physiological response produced is not, as a rule, directly proportional to receptor occupancy. For an integrated physiological response, such as a rise in arterial blood pressure produced by adrenaline (epinephrine), many factors interact. Adrenaline (see Ch. 14) increases cardiac output and constricts some blood vessels while dilating others, and the change in arterial pressure itself evokes a superimposed reflex response. The final effect is clearly not a direct measure of receptor occupancy in this instance, and the same is true of most drug-induced effects.
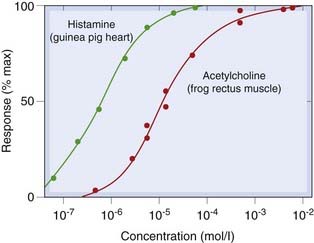
Fig. 2.3 Experimentally observed concentration–effect curves.
Although the lines, drawn according to the binding equation 2.5, fit the points well, such curves do not give correct estimates of the affinity of drugs for receptors. This is because the relationship between receptor occupancy and response is usually non-linear.
In interpreting concentration–effect curves, it must be remembered that the concentration of the drug at the receptors may differ from the known concentration in the bathing solution. Agonists may be subject to rapid enzymic degradation or uptake by cells as they diffuse from the surface towards their site of action, and a steady state can be reached in which the agonist concentration at the receptors is very much less than the concentration in the bath. In the case of acetylcholine, for example, which is hydrolysed by cholinesterase present in most tissues (see Ch. 13), the concentration reaching the receptors can be less than 1% of that in the bath, and an even bigger difference has been found with noradrenaline (norepinephrine), which is avidly taken up by sympathetic nerve terminals in many tissues (Ch. 14). Thus, even if the concentration–effect curve, as in Figure 2.3, looks just like a facsimile of the binding curve (Fig. 2.2C), it cannot be used directly to determine the affinity of the agonist for the receptors.
Competitive Antagonism
Though one drug can inhibit the response to another in several ways (see below), competition at the receptor level is particularly important, both in the laboratory and in the clinic, because of the high potency and specificity that can be achieved.
In the presence of a competitive antagonist, the agonist occupancy at a given agonist concentration is reduced, because the receptor can accommodate only one molecule at a time. However, because the two are in competition, raising the agonist concentration can restore the agonist occupancy (and hence the tissue response). The antagonism is therefore said to be surmountable, in contrast to other types of antagonism (see below) where increasing the agonist concentration fails to overcome the blocking effect. A simple theoretical analysis (see p. 17) predicts that in the presence of a fixed concentration of the antagonist, the log concentration–effect curve for the agonist will be shifted to the right, without any change in slope or maximum—the hallmark of competitive antagonism (Fig. 2.4A). The shift is expressed as a dose ratio, r, (the ratio by which the agonist concentration has to be increased in the presence of the antagonist in order to restore a given level of response). Theory predicts that the dose ratio increases linearly with the concentration of the antagonist (see p. 17). These predictions are often borne out in practice (see Fig. 2.5), and examples of competitive antagonism are very common in pharmacology. The surmountability of the block by the antagonist may be important in practice, because it allows the functional effect of the agonist to be restored by an increase in concentration. With other types of antagonism (see below), the block is usually insurmountable.
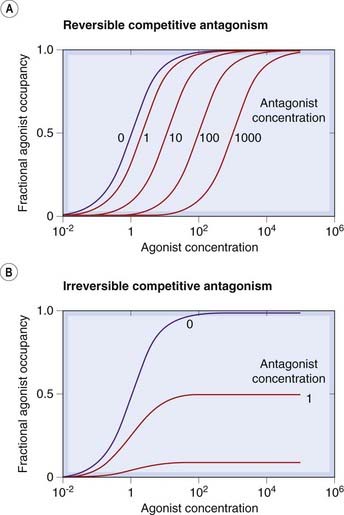
Fig. 2.4 Hypothetical agonist concentration–occupancy curves in the presence of reversible [A] and irreversible [B] competitive antagonists.
The concentrations are normalised with respect to the equilibrium constants, K (i.e. 1.0 corresponds to a concentration equal to K and results in 50% occupancy). Note that increasing the agonist concentration overcomes the effect of a reversible antagonist (i.e. the block is surmountable), so that the maximal response is unchanged, whereas the effect of an irreversible antagonist is unsurmountable and full agonist occupancy cannot be achieved.
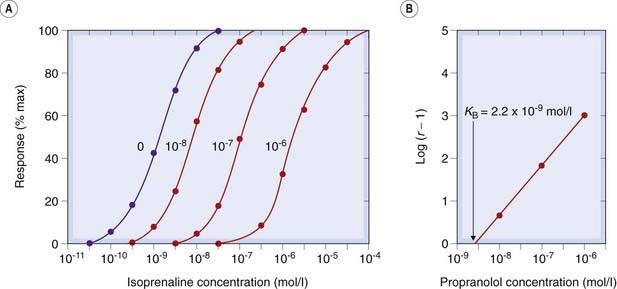
Fig. 2.5 Competitive antagonism of isoprenaline by propranolol measured on isolated guinea pig atria.
[A] Concentration–effect curves at various propranolol concentrations (indicated on the curves). Note the progressive shift to the right without a change of slope or maximum. [B] Schild plot (equation 2.10). The equilibrium constant (K) for propranolol is given by the abscissal intercept, 2.2 × 10−9 mol/l.
(Results from Potter L T 1967 Uptake of propranolol by isolated guinea-pig atria. J Pharmacol Exp Ther 55: 91–100.)
The salient features of competitive antagonism are:
Competitive antagonism is the most direct mechanism by which one drug can reduce the effect of another (or of an endogenous mediator), and several examples are listed in Table 3.1.
The characteristics of reversible competitive antagonism described above reflect the fact that the rate of dissociation of the antagonist molecules is sufficiently high that a new equilibrium is rapidly established on addition of the agonist. In effect, the agonist is able to displace the antagonist molecules from the receptors, although it cannot, of course, evict a bound antagonist molecule. Displacement occurs because, by occupying a proportion of the vacant receptors, the agonist reduces the rate of association of the antagonist molecules; consequently, the rate of dissociation temporarily exceeds that of association, and the overall antagonist occupancy falls.
Irreversible, or non-equilibrium, competitive antagonism occurs when the antagonist dissociates very slowly, or not at all, from the receptors, with the result that no change in the antagonist occupancy takes place when the agonist is applied.2
The predicted effects of reversible and irreversible antagonists are compared in Figure 2.4.
Competitive antagonism
In some cases (Fig. 2.6A), the theoretical effect is accurately reproduced, but the distinction between reversible and irreversible competitive antagonism (or even non-competitive antagonism; see below) is not always so clear. This is because of the phenomenon of spare receptors (see p. 13); if the agonist occupancy required to produce a maximal biological response is very small (say 1% of the total receptor pool), then it is possible to block irreversibly nearly 99% of the receptors without reducing the maximal response. The effect of a lesser degree of antagonist occupancy will be to produce a parallel shift of the log concentration–effect curve that is indistinguishable from reversible competitive antagonism (Fig. 2.6B).
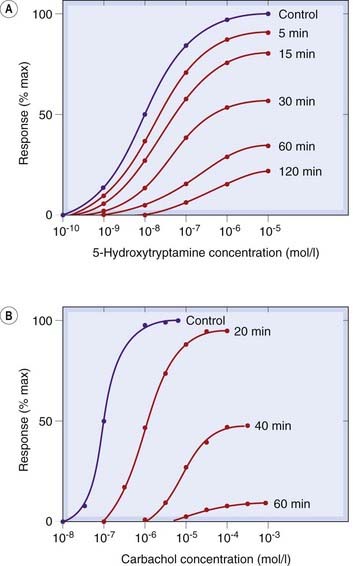
Fig. 2.6 Effects of irreversible competitive antagonists on agonist concentration–effect curves.
[A] Rat stomach smooth muscle responding to 5-hydroxytryptamine at various times after addition of methysergide (10−9 mol/l). [B] Rabbit stomach responding to carbachol at various times after addition of dibenamine (10−5 mol/l).
([A] After Frankhuijsen A L, Bonta I L 1974 Eur J Pharmacol 26: 220; [B] After Furchgott R F 1965 Adv Drug Res 3: 21.)
Irreversible competitive antagonism occurs with drugs that possess reactive groups that form covalent bonds with the receptor. These are mainly used as experimental tools for investigating receptor function, and few are used clinically. Irreversible enzyme inhibitors that act similarly are clinically used, however, and include drugs such as aspirin (Ch. 26), omeprazole (Ch. 29) and monoamine oxidase inhibitors (Ch. 46).
Allosteric Effects
In addition to the agonist binding site, to which competitive antagonists bind, receptor proteins possess many other (allosteric) binding sites (see Ch. 3) through which drugs can influence receptor function in various ways, increasing or decreasing the affinity of agonists for the agonist binding site, or by modifying efficacy. Depending on the direction of the effect, the ligands may be allosteric antagonists or allosteric facilitators of the agonist effect, and the effect may be to alter the slope and maximum of the agonist log concentration–effect curve. This type of allosteric modulation of receptor function has attracted much attention recently (see review by May et al., 2007), and may prove to be more widespread than previously envisaged. Well-known examples of allosteric facilitation include the action of glycine (allosteric ligand) on glutamate receptors and of benzodiazepines on GABAA receptors (Ch. 37).
Partial Agonists and the Concept of Efficacy
So far, we have considered drugs either as agonists, which in some way activate the receptor when they occupy it, or as antagonists, which cause no activation. However, the ability of a drug molecule to activate the receptor is actually a graded, rather than an all-or-nothing, property. If a series of chemically related agonist drugs acting on the same receptors is tested on a given biological system, it is often found that the largest response that can be produced by the drug in high concentration differs from one drug to another. Some compounds (known as full agonists) can produce a maximal response (the largest response that the tissue is capable of giving), whereas others (partial agonists) can produce only a submaximal response. Figure 2.7A shows concentration effect curves for several α-adrenoceptor agonists (see Ch. 14) which cause contraction of isolated strips of rabbit aorta. The full agonist phenylephrine produced the maximal effect of which the tissue was capable; the other compounds could only produce submaximal responses and are partial agonists. The difference between full and partial agonists lies in the relationship between receptor occupancy and response. In the experiment shown in Figure 2.7 it was possible to estimate the affinity of the various drugs for the receptor, and hence (based on the theoretical model described later; p. 17) to calculate the fraction of receptors occupied (known as occupancy) as a function of drug concentration. Plots of response as a function of occupancy for the different compounds are shown in Figure 2.7B, showing that for partial agonists the response at a given level of occupancy is less than for full agonists. The weakest partial agonist, tolazoline, produces a barely detectable response even at 100% occupancy, and is usually classified as a competitive antagonist (see p. 10 and Ch. 14).
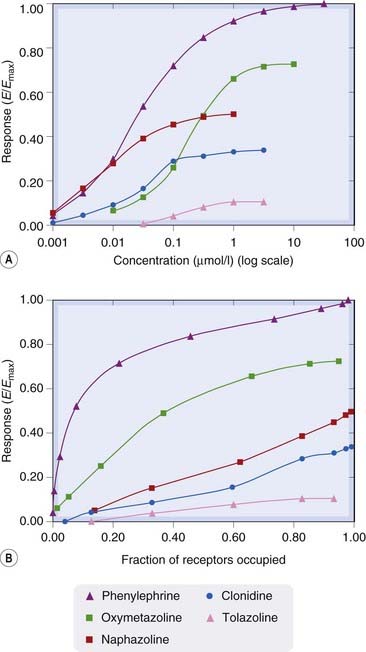
[A] Log concentration–effect curves for a series of α-adrenoceptor agonists causing contraction of an isolated strip of rabbit aorta. Phenylephrine is a full agonist. The others are partial agonists with different efficacies. [B] The relationship between response and receptor occupancy for the series. Note that the full agonist, phenylephrine, produces a near-maximal response when only about half the receptors are occupied, whereas partial agonists produce submaximal responses even when occupying all of the receptors. The efficacy of tolazoline is so low that it is classified as an α-adrenoceptor antagonist (see Ch. 14). In these experiments, receptor occupancy was not measured directly, but was calculated from pharmacological estimates of the equilibrium constants of the drugs.
(Data from Ruffolo et al. 1979 J Pharmacol Exp Ther 209: 429–436.)
These differences can be expressed quantitatively in terms of efficacy (e), a parameter originally defined by Stephenson (1956) that describes the ‘strength’ of the agonist–receptor complex in evoking a response of the tissue. In the simple scheme shown in Figure 2.1, efficacy describes the tendency of the drug–receptor complex to adopt the active (AR*), rather than the resting (AR) state. A drug with zero efficacy (e = 0) has no tendency to cause receptor activation, and causes no tissue response. A drug with efficacy3 is a full agonist, while partial agonists lie in between.
Subsequently, it was appreciated that characteristics of the tissue (e.g. the number of receptors that it possesses and the nature of the coupling between the receptor and the response; see Ch. 3), as well as of the drug itself, were important, and the concept of intrinsic efficacy was developed (see Jenkinson, 1996; Kenakin, 1997), which can account for a number of anomalous findings. For example, depending on tissue characteristics, a given drug may appear as a full agonist in one tissue but a partial agonist in another, and drugs may differ in their relative agonist potencies in different tissues, though the receptor is the same.
It would be nice to be able to explain what efficacy means in physical terms, and to understand why one drug may be an agonist while another, chemically very similar, is an antagonist. We are beginning to understand the molecular events underlying receptor activation (described in Ch. 3) but can still give no clear answer to the question of why some ligands are agonists and some are antagonists, although the simple theoretical two-state model described below provides a useful starting point.
Despite its uncertain mechanistic basis, efficacy is a concept of great practical importance. Adrenaline (epinephrine) and propranolol (see Ch. 14) have comparable affinities for the β-adrenoceptor but differ in efficacy. Woe-betide the doctor—and the student, for that matter—who confuses them. Efficacy matters!
Constitutive Receptor Activation and Inverse Agonists
Although we are accustomed to thinking that receptors are activated only when an agonist molecule is bound, there are examples (see De Ligt et al., 2000) where an appreciable level of activation may exist even when no ligand is present. These include receptors for benzodiazepines (see Ch. 43), cannabinoids (Ch. 18), serotonin (Ch. 15) and several other mediators. Furthermore, receptor mutations occur—either spontaneously, in some disease states (see Bond & Ijzerman, 2006) or experimentally created (see Ch. 4)—that result in appreciable activation in the absence of any ligand (constitutive activation). Resting activity may be too low to have any effect under normal conditions but become evident if receptors are overexpressed, a phenomenon clearly demonstrated for β-adrenoceptors (see Bond et al., 1995), a result that may prove to have major pathophysiological implications. Thus if, say, 1% of receptors are active in the absence of any agonist, in a normal cell expressing perhaps 10 000 receptors, only 100 will be active. Increasing the expression level 10-fold will result in 1000 active receptors, producing a significant effect. Under these conditions, it may be possible for a ligand to reduce the level of constitutive activation; such drugs are known as inverse agonists (Fig. 2.8; see De Ligt et al., 2000) to distinguish them from neutral antagonists, which do not by themselves affect the level of activation. Inverse agonists can be regarded as drugs with negative efficacy, to distinguish them from agonists (positive efficacy) and neutral antagonists (zero efficacy). New examples of constitutively active receptors and inverse agonists are emerging with increasing frequency (mainly among G-protein-coupled receptors; Seifert & Wenzel-Seifert, 2002). In theory, an inverse agonist, by silencing constitutively active receptors, should be more effective than a neutral antagonist in disease states associated with receptor mutations or with receptor-directed autoantibodies that result in enhanced constitutive activation. These include certain types of hyperthyroidism, precocious puberty and parathyroid diseases (see Bond & Ijzerman, 2006). This remains to be verified, but it turns out that most of the receptor antagonists in clinical use are actually inverse agonists when tested in systems showing constitutive receptor activation. However, most receptors—like cats—show a preference for the inactive state, and for these there is no practical difference between a competitive antagonist and an inverse agonist. It remains to be seen whether the inverse agonist principle will prove to be generally important in therapeutics, but interest is running high. So far, nearly all the examples come from the family of G-protein-coupled receptors (see Ch. 3 and the review by Costa & Cotecchia, 2005), and it is not clear whether similar phenomena occur with other receptor families.
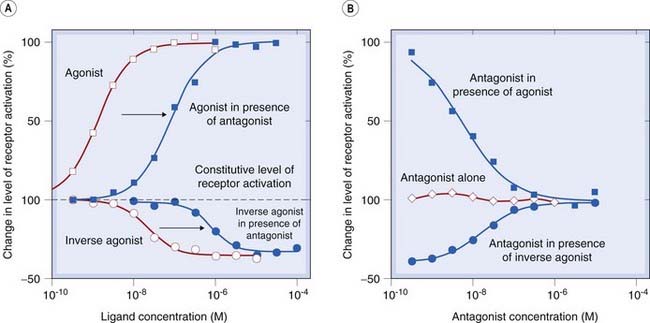
Fig. 2.8 Inverse agonism. The interaction of a competitive antagonist with normal and inverse agonists in a system that shows receptor activation in the absence of any added ligands (constitutive activation).
[A] The degree of receptor activation (vertical scale) increases in the presence of an agonist (open squares) and decreases in the presence of an inverse agonist (open circles). Addition of a competitive antagonist shifts both curves to the right (closed symbols). [B] The antagonist on its own does not alter the level of constitutive activity (open symbols), because it has equal affinity for the active and inactive states of the receptor. In the presence of an agonist (closed squares) or an inverse agonist (closed circles), the antagonist restores the system towards the constitutive level of activity. These data (reproduced with permission from Newman-Tancredi A et al. 1997 Br J Pharmacol 120: 737–739) were obtained with cloned human 5-hydroxytryptamine (5-HT) receptors expressed in a cell line. (Agonist, 5-carboxamidotryptamine; inverse agonist, spiperone; antagonist, WAY 100635; ligand concentration [M = mol/l]; see Ch. 15 for information on 5-HT receptor pharmacology.)
The following section describes a simple model that explains full, partial and inverse agonism in terms of the relative affinity of different ligands for the resting and activated states of the receptor.
The two-state receptor model
As illustrated in Figure 2.1, agonists and antagonists both bind to receptors, but only agonists activate them. How can we express this difference, and account for constitutive activity, in theoretical terms? The two-state model (Fig. 2.9) provides a simple but useful approach.
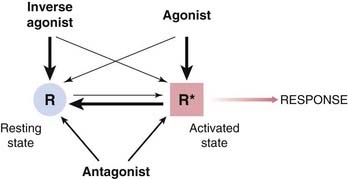
The receptor is shown in two conformational states, ‘resting’ (R) and ‘activated’ (R*), which exist in equilibrium. Normally, when no ligand is present, the equilibrium lies far to the left, and few receptors are found in the R* state. For constitutively active receptors, an appreciable proportion of receptors adopt the R* conformation in the absence of any ligand. Agonists have higher affinity for R* than for R, so shift the equilibrium towards R*. The greater the relative affinity for R* with respect to R, the greater the efficacy of the agonist. An inverse agonist has higher affinity for R than for R* and so shifts the equilibrium to the left. A ‘neutral’ antagonist has equal affinity for R and R* so does not by itself affect the conformational equilibrium but reduces by competition the binding of other ligands.
As shown in Figure 2.1, we envisage that the occupied receptor can switch from its ‘resting’ (R) state to an activated (R*) state, R* being favoured by binding of an agonist but not an antagonist molecule.
As described above, receptors may show constitutive activation (i.e. the R* conformation can exist without any ligand being bound), so the added drug encounters an equilibrium mixture of R and R* (Fig. 2.9). If it has a higher affinity for R* than for R, the drug will cause a shift of the equilibrium towards R* (i.e. it will promote activation and be classed as an agonist). If its preference for R* is very large, nearly all the occupied receptors will adopt the R* conformation and the drug will be a full agonist (positive efficacy); if it shows only a modest degree of selectivity for R* (say 5–10-fold), a smaller proportion of occupied receptors will adopt the R* conformation and it will be a partial agonist; if it shows no preference, the prevailing R:R* equilibrium will not be disturbed and the drug will be a neutral antagonist (zero efficacy), whereas if it shows selectivity for R it will shift the equilibrium towards R and be an inverse agonist (negative efficacy). We can therefore think of efficacy as a property determined by the relative affinity of a ligand for R and R*, a formulation known as the two-state model, which is useful in that it puts a physical interpretation on the otherwise mysterious meaning of efficacy, as well as accounting for the existence of inverse agonists.
A major problem with the two-state model is that, as we now know, receptors are not actually restricted to two distinct states but have much greater conformational flexibility, so that there is more than one inactive and active conformation. The different conformations that they can adopt may be preferentially stabilised by different ligands, and may produce different functional effects by activating different signal transduction pathways (see Ch. 3). Redefining efficacy for such a multistate model is difficult, however, and requires a more complicated state transition model than that described here.
Agonists, antagonists and efficacy
Spare Receptors
Stephenson (1956), studying the actions of acetylcholine analogues in isolated tissues, found that many full agonists were capable of eliciting maximal responses at very low occupancies, often less than 1%. This means that the mechanism linking the response to receptor occupancy has a substantial reserve capacity. Such systems may be said to possess spare receptors, or a receptor reserve. This is common with drugs that elicit smooth muscle contraction but less so for other types of receptor-mediated response, such as secretion, smooth muscle relaxation or cardiac stimulation, where the effect is more nearly proportional to receptor occupancy. The existence of spare receptors does not imply any functional subdivision of the receptor pool, but merely that the pool is larger than the number needed to evoke a full response. This surplus of receptors over the number actually needed might seem a wasteful biological arrangement. It means, however, that a given number of agonist–receptor complexes, corresponding to a given level of biological response, can be reached with a lower concentration of hormone or neurotransmitter than would be the case if fewer receptors were provided. Economy of hormone or transmitter secretion is thus achieved at the expense of providing more receptors.
Drug Antagonism and Synergism
Frequently, the effect of one drug is reduced or enhanced in the presence of another. Competitive antagonism, described earlier, is a common and important mechanism, which will be encountered frequently in this book. However, a variety of other mechanisms can account for inhibitory or facilitatory interactions between drugs. The following list includes the most important ones:
Chemical Antagonism
Chemical antagonism refers to the uncommon situation where the two substances combine in solution; as a result, the effect of the active drug is lost. Examples include the use of chelating agents (e.g. dimercaprol) that bind to heavy metals and thus reduce their toxicity, and the use of the neutralising antibody infliximab which has an anti-inflammatory action due to its ability to sequester the inflammatory cytokine, tumour necrosis factor (TNF; see Ch. 17).
Pharmacokinetic Antagonism
Pharmacokinetic antagonism describes the situation in which the ‘antagonist’ effectively reduces the concentration of the active drug at its site of action. This can happen in various ways. The rate of metabolic degradation of the active drug may be increased (e.g. the reduction of the anticoagulant effect of warfarin when an agent that accelerates its hepatic metabolism, such as phenobarbital, is given; see Chs 9 and 56). Alternatively, the rate of absorption of the active drug from the gastrointestinal tract may be reduced, or the rate of renal excretion may be increased. Interactions of this sort, discussed in more detail in Chapter 56, are common and can be important in clinical practice.
Block of Receptor–Effector Linkage
Non-competitive antagonism describes the situation where the antagonist blocks at some point, downstream from the receptor, the chain of events that leads to the production of a response by the agonist. For example, drugs such as verapamil and nifedipine prevent the influx of Ca2+ through the cell membrane (see Ch. 22) and thus block non-specifically the contraction of smooth muscle produced by other drugs. As a rule, the effect will be to reduce the slope and maximum of the agonist log concentration–response curve although it is quite possible for some degree of rightward shift to occur as well.
Physiological Antagonism
Physiological antagonism is a term used loosely to describe the interaction of two drugs whose opposing actions in the body tend to cancel each other. For example, histamine acts on receptors of the parietal cells of the gastric mucosa to stimulate acid secretion, while omeprazole blocks this effect by inhibiting the proton pump; the two drugs can be said to act as physiological antagonists.
Desensitisation and Tachyphylaxis
Often, the effect of a drug gradually diminishes when it is given continuously or repeatedly. Desensitisation and tachyphylaxis are synonymous terms used to describe this phenomenon, which often develops in the course of a few minutes. The term tolerance is conventionally used to describe a more gradual decrease in responsiveness to a drug, taking days or weeks to develop, but the distinction is not a sharp one. The term refractoriness is also sometimes used, mainly in relation to a loss of therapeutic efficacy. Drug resistance is a term used to describe the loss of effectiveness of antimicrobial or antitumour drugs (see Chs 49 and 55). Many different mechanisms can give rise to this type of phenomenon. They include:
Change in Receptors
Among receptors directly coupled to ion channels (see Ch. 3), desensitisation is often rapid and pronounced. At the neuromuscular junction (Fig. 2.10A), the desensitised state is caused by a conformational change in the receptor, resulting in tight binding of the agonist molecule without the opening of the ionic channel. Phosphorylation of intracellular regions of the receptor protein is a second, slower mechanism by which ion channels become desensitised.
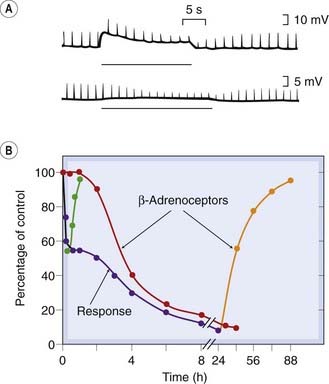
Fig. 2.10 Two kinds of receptor desensitisation.
[A] Acetylcholine (ACh) at the frog motor endplate. Brief depolarisations (upward deflections) are produced by short pulses of ACh delivered from a micropipette. A long pulse (horizontal line) causes the response to decline with a time course of about 20 s, owing to desensitisation, and it recovers with a similar time course. [B] β-Adrenoceptors of rat glioma cells in tissue culture. Isoproterenol (1 µmol/l) was added at time zero, and the adenylate cyclase response and β-adrenoceptor density measured at intervals. During the early uncoupling phase, the response (blue line) declines with no change in receptor density (red line). Later, the response declines further concomitantly with disappearance of receptors from the membrane by internalisation. The green and orange lines show the recovery of the response and receptor density after the isoproterenol is washed out during the early or late phase.
(From: [A] Katz B, Thesleff S 1957 J Physiol 138: 63; [B] Perkins J P 1981 Trends Pharmacol Sci 2: 326.)
Most G-protein-coupled receptors (see Ch. 3) also show desensitisation (see Fig. 2.10B). Phosphorylation of the receptor interferes with its ability to activate second messenger cascades, although it can still bind the agonist molecule. The molecular mechanisms of this ‘uncoupling’ are described by Lefkowitz et al. (1998) and considered further in Chapter 3. This type of desensitisation usually takes a few minutes to develop, and recovers at a similar rate when the agonist is removed.
It will be realised that the two-state model in its simple form, discussed earlier, needs to be further elaborated to incorporate additional ‘desensitised’ states of the receptor.
Translocation of Receptors
Prolonged exposure to agonists often results in a gradual decrease in the number of receptors expressed on the cell surface, as a result of internalisation of the receptors. This is shown for β-adrenoceptors in Figure 2.10B and is a slower process than the uncoupling described above. In studies on cell cultures, the number of β-adrenoceptors can fall to about 10% of normal in 8 h in the presence of a low concentration of isoprenaline, and recovery takes several days. Similar changes have been described for other types of receptor, including those for various peptides. The internalised receptors are taken into the cell by endocytosis of patches of the membrane, a process that also depends on receptor phosphorylation. This type of adaptation is common for hormone receptors and has obvious relevance to the effects produced when drugs are given for extended periods. It is generally an unwanted complication when drugs are used clinically, but it can be exploited. For example, gonadotrophin-releasing hormone (see Ch. 34) is used to treat endometriosis or prostatic cancer; given continuously, this hormone paradoxically inhibits gonadotrophin release (in contrast to the normal stimulatory effect of the physiological secretion, which is pulsatile).
Exhaustion of Mediators
In some cases, desensitisation is associated with depletion of an essential intermediate substance. Drugs such as amphetamine, which acts by releasing amines from nerve terminals (see Chs 14 and 47), show marked tachyphylaxis because the amine stores become depleted.
Altered Drug Metabolism
Tolerance to some drugs, for example barbiturates (Ch. 43) and ethanol (Ch. 48), occurs partly because repeated administration of the same dose produces a progressively lower plasma concentration, because of increased metabolic degradation. The degree of tolerance that results is generally modest, and in both of these examples other mechanisms contribute to the substantial tolerance that actually occurs. On the other hand, the pronounced tolerance to nitrovasodilators (see Chs 20 and 22) results mainly from decreased metabolism, which reduces the release of the active mediator, nitric oxide.
Physiological Adaptation
Diminution of a drug’s effect may occur because it is nullified by a homeostatic response. For example, the blood pressure-lowering effect of thiazide diuretics is limited because of a gradual activation of the renin–angiotensin system (see Ch. 22). Such homeostatic mechanisms are very common, and if they occur slowly the result will be a gradually developing tolerance. It is a common experience that many side effects of drugs, such as nausea or sleepiness, tend to subside even though drug administration is continued. We may assume that some kind of physiological adaptation is occurring, presumably associated with altered gene expression resulting in changes in the levels of various regulatory molecules, but little is known about the mechanisms involved.
Quantitative Aspects of Drug–Receptor Interactions
Here we present some aspects of so-called receptor theory, which is based on applying the Law of Mass Action to the drug–receptor interaction and which has served well as a framework for interpreting a large body of quantitative experimental data.
The binding reaction
The first step in drug action on specific receptors is the formation of a reversible drug–receptor complex, the reactions being governed by the Law of Mass Action. Suppose that a piece of tissue, such as heart muscle or smooth muscle, contains a total number of receptors, Ntot, for an agonist such as adrenaline. When the tissue is exposed to adrenaline at concentration xA and allowed to come to equilibrium, a certain number, NA, of the receptors will become occupied, and the number of vacant receptors will be reduced to Ntot − NA. Normally, the number of adrenaline molecules applied to the tissue in solution greatly exceeds Ntot, so that the binding reaction does not appreciably reduce xA. The magnitude of the response produced by the adrenaline will be related (even if we do not know exactly how) to the number of receptors occupied, so it is useful to consider what quantitative relationship is predicted between NA and xA. The reaction can be represented by:
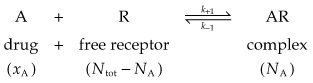
The Law of Mass Action (which states that the rate of a chemical reaction is proportional to the product of the concentrations of reactants) can be applied to this reaction.
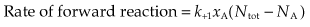
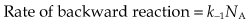
At equilibrium, the two rates are equal:
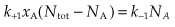
The proportion of receptors occupied, or occupancy (pA), is NA/Ntot, which is independent of Ntot.
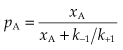
Defining the equilibrium constant for the binding reaction, KA = k−1/k+1, equation 2.4 can be written:
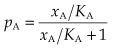
This important result is known as the Hill–Langmuir equation.4
The equilibrium constant,5 KA, is a characteristic of the drug and of the receptor; it has the dimensions of concentration and is numerically equal to the concentration of drug required to occupy 50% of the sites at equilibrium. (Verify from equation 2.5 that when xA = KA, pA = 0.5.) The higher the affinity of the drug for the receptors, the lower will be the value of KA. Equation 2.5 describes the relationship between occupancy and drug concentration, and it generates a characteristic curve known as a rectangular hyperbola, as shown in Figure 2.11A. It is common in pharmacological work to use a logarithmic scale of concentration; this converts the hyperbola to a symmetrical sigmoid curve (Fig. 2.11B).
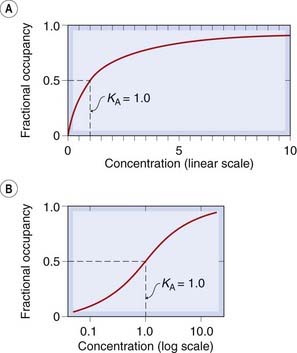
Fig. 2.11 Theoretical relationship between occupancy and ligand concentration.
The relationship is plotted according to equation 2.5. [A] Plotted with a linear concentration scale, this curve is a rectangular hyperbola. [B] Plotted with a log concentration scale, it is a symmetrical sigmoid curve.
The same approach is used to analyse data from experiments in which drug binding is measured directly (see p. 9, Fig. 2.2). In this case, the relationship between the amount bound (B) and ligand concentration (xA) should be:
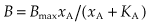
where Bmax is the total number of binding sites in the preparation (often expressed as pmol/mg of protein). To display the results in linear form, equation 2.6 may be rearranged to:
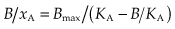
A plot of B/xA against B (known as a Scatchard plot; Fig. 2.2C) gives a straight line from which both Bmax and KA can be estimated. Statistically, this procedure is not without problems, and it is now usual to estimate these parameters from the untransformed binding values by an iterative non-linear curve-fitting procedure.
To this point, our analysis has considered the binding of one ligand to a homogeneous population of receptors. To get closer to real-life pharmacology, we must consider (a) what happens when more than one ligand is present, and (b) how the tissue response is related to receptor occupancy.
Binding when more than one drug is present
Suppose that two drugs, A and B, which bind to the same receptor with equilibrium constants KA and KB, respectively, are present at concentrations xA and xB. If the two drugs compete (i.e. the receptor can accommodate only one at a time), then, by application of the same reasoning as for the one-drug situation described above, the occupancy by drug A is given by:
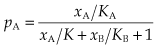
Comparing this result with equation 2.5 shows that adding drug B, as expected, reduces the occupancy by drug A. Figure 2.4A shows the predicted binding curves for A in the presence of increasing concentrations of B, demonstrating the shift without any change of slope or maximum that characterises the pharmacological effect of a competitive antagonist (see Fig. 2.5). The extent of the rightward shift, on a logarithmic scale, represents the ratio (rA, given by xA′/xA where xA′ is the increased concentration of A) by which the concentration of A must be increased to overcome the competition by B. Rearranging 2.8 shows that
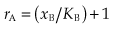
Thus rA depends only on the concentration and equilibrium constant of the competing drug B, not on the concentration or equilibrium constant of A.
If A is an agonist, and B is a competitive antagonist, and we assume that the response of the tissue will be an unknown function of pA, then the value of rA determined from the shift of the agonist concentration–effect curve at different antagonist concentrations can be used to estimate the equilibrium constant KB for the antagonist. Such pharmacological estimates of rA are commonly termed agonist dose ratios (more properly concentration ratios, although most pharmacologists use the older term). This simple and very useful equation (2.9) is known as the Schild equation, after the pharmacologist who first used it to analyse drug antagonism.
Equation 2.9 can be expressed logarithmically in the form:
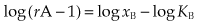
Thus a plot of log (rA − 1) against log xB, usually called a Schild plot (as in Fig. 2.5), should give a straight line with unit slope and an abscissal intercept equal to log KB. Following the pH and pK notation, antagonist potency can be expressed as a pA2 value; under conditions of competitive antagonism, pA2 = −log KB. Numerically, pA2 is defined as the negative logarithm of the molar concentration of antagonist required to produce an agonist dose ratio equal to 2. As with pH notation, its principal advantage is that it produces simple numbers, a pA2 of 6.5 being equivalent to a KB of 3.2 × 10−7 mol/l.
For competitive antagonism, r shows the following characteristics:
These predictions have been verified for many examples of competitive antagonism (Fig. 2.5).
In this section, we have avoided going into great detail and have oversimplified the theory considerably. As we learn more about the actual molecular details of how receptors work to produce their biological effects (see Ch. 3), the shortcomings of this theoretical treatment become more obvious. The two-state model can be incorporated without difficulty, but complications arise when we include the involvement of G-proteins (see Ch. 3) in the reaction scheme, and when we allow for the fact that receptor ‘activation’ is not a simple on–off switch, as the two-state model assumes, but may take different forms. It is as though the same receptor can turn on a tap or a light bulb, depending on which agonist does the talking. Despite strenuous efforts by theoreticians to allow for such possibilities, the molecules always seem to remain one step ahead. Nevertheless, this type of basic theory applied to the two-state model remains a useful basis for developing quantitative models of drug action. The book by Kenakin (1997) is recommended as an introduction, and his later review (Kenakin, 2002) presents a more elaborate theoretical approach.
Binding of drugs to receptors
The Nature of Drug Effects
In discussing how drugs act in this chapter, we have focused mainly on the consequences of receptor activation. Details of the receptors and their linkage to effects at the cellular level are described in Chapter 3. We now have a fairly good understanding at this level. It is important, however, particularly when considering drugs in a therapeutic context, that their direct effects on cellular function generally lead to secondary, delayed effects, which are often highly relevant in a clinical situation in relation to both therapeutic efficacy and harmful effects (see Fig. 2.12). For example, activation of a β-adrenoceptor in the heart (see Chs 3 and 21) causes rapid changes in the functioning of the heart muscle, but also slower (minutes to hours) changes in the functional state of the receptors (e.g. desensitisation), and even slower (hours to days) changes in gene expression that produce long-term changes (e.g. hypertrophy) in cardiac structure and function. Similarly, antidepressant drugs, which have immediate effects on transmitter metabolism in the brain (see Ch. 46) take weeks to produce therapeutic benefit. Opioids (see Ch. 41) produce an immediate analgesic effect but, after a time, tolerance and dependence ensue, and in some cases long-term addiction. In these and many other examples, the nature of the intervening mechanism is unclear, although as a general rule any long-term phenotypic change necessarily involves alterations of gene expression. Drugs are often used to treat chronic conditions, and understanding long-term as well as acute drug effects is becoming increasingly important. Pharmacologists have traditionally tended to focus on short-term physiological responses, which are much easier to study, rather than on delayed effects. The focus is now clearly shifting.
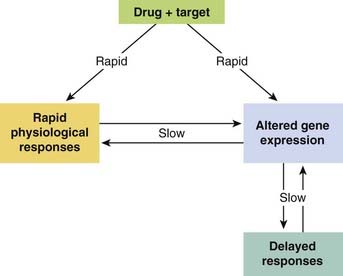
Fig. 2.12 Early and late responses to drugs.
Many drugs act directly on their targets (left-hand arrow) to produce a rapid physiological response. If this is maintained, it is likely to cause changes in gene expression that give rise to delayed effects. Some drugs (right-hand arrow) have their primary action on gene expression, producing delayed physiological responses. Drugs can also work by both pathways. Note the bidirectional interaction between gene expression and response.
Drug effects
References and Further Reading
Alexander S.P.H., Mathie A., Peters J.A. Guide to receptors and channels (GRAC), fourth ed. Br. J. Pharmacol.. 2009;158(Suppl. 1):S1-S254.
Changeux J.-P., Giraudat J., Dennis M. The nicotinic acetylcholine receptor: molecular architecture of a ligand-regulated ion channel. Trends Pharmacol. Sci.. 1987;8:459-465. (One of the first descriptions of receptor action at the molecular level)
Colquhoun D. The quantitative analysis of drug–receptor interactions: a short history. Trends Pharmacol. Sci.. 2006;27:149-157. (An illuminating account for those interested in the origins of one of the central ideas in pharmacology)
Franks N.P. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci.. 2008;9:370-386.
Jenkinson D.H. Classical approaches to the study of drug–receptor interactions. In: Foreman J.C., Johansen T., editors. Textbook of Receptor Pharmacology. Boca Raton: CRC Press, 1996. (Good account of pharmacological analysis of receptor-mediated effects)
Kenakin T. Pharmacologic analysis of drug–receptor interactions, third ed. New York: Lippincott-Raven; 1997. (Useful and detailed textbook covering most of the material in this chapter in greater depth)
Neubig R., Spedding M., Kenakin T., Christopoulos A. International Union of Pharmacology Committee on receptor nomenclature and drug classification: XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol. Rev.. 2003;55:597-606. (Summary of IUPHAR-approved terms and symbols relating to pharmacological receptors—useful for reference purposes)
Rang H.P. The receptor concept: Pharmacology’s Big Idea. Br. J. Pharmacol.. 2006;147(Supp. 1):9-16. (Short review of the origin and status of the receptor concept)
Stephenson R.P. A modification of receptor theory. Br. J. Pharmacol.. 1956;11:379-393. (Classic analysis of receptor action introducing the concept of efficacy)
Teitler M., Herrick-Davis K., Purohit A. Constitutive activity of G-protein coupled receptors: emphasis on serotonin receptors. Curr. Top Med. Chem.. 2002;2:529-538.
Receptor mechanisms: agonists and efficacy
Bond R.A., Ijzerman A.P. Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol. Sci.. 2006;27:92-96. (Discussion of pathophysiological consequences of constitutive receptor activation and therapeutic potential of inverse agonists—mainly hypothetical so far, but with important implications)
Bond R.A., Leff P., Johnson T.D., et al. Physiological effects of inverse agonists in transgenic mice with myocardial overexpression of the β2-adrenoceptor. Nature. 1995;374:270-276. (A study with important clinical implications, showing that overexpression of β-adrenoceptors results in constitutive receptor activation)
Costa T., Cotecchia S. Historical review: negative efficacy and the constitutive activity of G-protein-coupled receptors. Trends Pharmacol. Sci.. 2005;26:618-624. (A clear and thoughtful review of ideas relating to constitutive receptor activation and inverse agonists)
De Ligt R.A.F., Kourounakis A.P., Ijzerman A.P. Inverse agonism at G protein-coupled receptors: (patho)physiological relevance and implications for drug discovery. Br. J. Pharmacol.. 2000;130:1-12. (Useful review article giving many examples of constitutively active receptors and inverse agonists, and discussing the relevance of these concepts for disease mechanisms and drug discovery)
Kenakin T. Drug efficacy at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol.. 2002;42:349-379. (A theoretical treatment that attempts to take into account recent knowledge of receptor function at the molecular level)
May L.T., Leach K., Sexton P.M., Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol.. 2007;47:1-51. (Comprehensive review describing the characteristics, mechanisms and pharmacological implications of allosteric interactions at GPCRs)
Milligan G., Bond R.A., Lee M. Inverse agonism: pharmacological curiosity or potential therapeutic strategy? Trends Pharmacol. Sci.. 1995;16:10-13. (Excellent review of the significance of constitutive receptor activation and the effects of inverse agonists)
Seifert R., Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common properties of wild-type receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol.. 2002;366:381-416. (Detailed review article emphasising that constitutively active receptors occur commonly and are associated with several important disease states)
Lefkowitz R.J., Pitcher J., Krueger K., Daaka Y. Mechanisms of β-adrenergic receptor desensitization and resensitization. Adv. Pharmacol.. 1998;42:416-420.
Swope S.L., Moss S.I., Raymond I.A., Huganir R.L. Regulation of ligand-gated ion channels by protein phosphorylation. Adv. Second Messenger Phosphoprotein Res.. 1999;33:49-78. (Comprehensive review article describing the role of phosphorylation in desensitisation)
1There are, if one looks hard enough, exceptions to Ehrlich’s dictum—drugs that act without being bound to any tissue constituent (e.g. osmotic diuretics, osmotic purgatives, antacids and heavy metal chelating agents). Nonetheless, the principle remains true for the great majority.
2This type of antagonism is sometimes called non-competitive, but that term is ambiguous and best avoided in this context.
3In Stephenson’s formulation, efficacy is the reciprocal of the occupancy needed to produce a 50% maximal response, thus e = 25 implies that a 50% maximal response occurs at 4% occupancy. There is no theoretical upper limit to efficacy.
4A V Hill first published it in 1909, when he was still a medical student. Langmuir, a physical chemist working on gas adsorption, derived it independently in 1916. Both subsequently won Nobel prizes. Until recently, it was known to pharmacologists as the Langmuir equation, even though Hill deserves the credit.
5The equilibrium constant is sometimes called the dissociation constant. Some authors prefer to use the reciprocal of KA, referred to as an affinity constant, in these expressions, which can cause confusion to the unwary.