59 Biopharmaceuticals and gene therapy
Overview
In this chapter, we review the impact of two therapeutic concepts based on our growing understanding and skill in manipulating genes. Biopharmaceuticals is an umbrella term applied to the use of nucleic acids or ‘engineered’ proteins and antibodies in medicine, while gene therapy refers specifically to attempts to use those nucleic acids to reprogram cells to prevent, alleviate or cure disease. Of the two, the former has already proved itself in the clinic, whereas the latter has not yet led to licensed products,1 although there are many ongoing trials, and some convincing successes. It is clear that once the last remaining technical hurdles have been surmounted, it will hold great promise. In addition to introducing the central concepts in this chapter, we consider the considerable problems associated with developing these therapies, discuss safety issues and review the progress made to date.
Introduction
The ‘molecular biology revolution’, which had its roots in the discovery of the structure of DNA in the 1950s, and the advances in cell biology that followed in its train, has enabled us to manipulate the genetic material from cells in ways that are useful in practical therapeutics. The seductive notion that a gene of interest can be expressed in vitro to generate useful proteins that could not be prepared synthetically or, more daringly, that a gene could be directly introduced in vivo and persuaded to synthesise some crucial cellular component, has driven this field at breakneck speed.
Biopharmaceuticals (considered for the purposes of this chapter to comprise genetically engineered proteins and monoclonal antibodies) are already a well-recognised part of therapy, and we have already encountered them elsewhere in this book (see, for example, the anti-tumour necrosis factor [TNF] antibodies in Ch. 26). We still face many problems, not the least of which is the cost of manufacture, but the technology is established and maturing fast. Reviewing the area in 2004, Walsh noted that some 140 biopharmaceuticals had been licensed around the world by the previous year, and that 250 million patients were receiving these products at a cost of some US$30 billion.2
While the same basic concepts and technologies underpin both these approaches, gene therapy is the more considerable challenge. However, the idea commands such appeal that vast resources (both public and private) have been committed to its development. There are several reasons why it is so attractive. First, the approach offers the potential for radical cure of single-gene diseases such as cystic fibrosis and the haemoglobinopathies, which are collectively responsible for much misery throughout the world. Second, many other more common conditions, including malignant, neurodegenerative and infectious diseases, have a large genetic component. Conventional treatment of such disorders is, as readers of this book will have appreciated by now, woefully inadequate, so the promise of a completely new approach has enormous attraction. Finally, an ability to control gene expression could even revolutionise the management of diseases in which there is no genetic component at all.
The gurus are emphatic that ‘the conceptual part of the gene therapy revolution has indeed occurred …’—so where are the therapies? The devil, of course, is in the detail: in this case, the details of:
•
pharmacokinetics: delivery of the gene to appropriate target cells (especially in the CNS)
•
pharmacodynamics: the controlled expression of the gene in question
•
clinical efficacy and
long-term practicability.
But perhaps the most fundamental hurdle is the delivery problem; here, modern virology has helped with techniques borrowed from viruses that can be used to introduce functional nucleic acids into mammalian cells. The principle is so simple that any broadsheet reader can apprehend it, and the potential rewards (humanitarian, scientific and commercial) so great that it has led inevitably to great expectations and, perhaps equally inevitably, to frustration at the lack of practical progress.
There is a broad consensus that the Weismann barrier3 should not be breached and so gene therapy trials have focused on somatic cells. A moratorium has been agreed on therapies intended to alter the DNA of germ cells, which could influence future generations.
Biopharmaceuticals
We consider first the use of proteins as therapeutic agents. Of course, this in itself is not a novel idea; insulin, extracted from animal pancreas tissue (Ch. 30), and human growth hormone, extracted from human cadaver pituitary glands (Ch. 32), were among the first therapeutic proteins to be used, and for many years provided the only option for treating hormone deficiency disorders. However, there were problems. First, there were difficulties in extraction and disappointingly low yields. Second, in the case of insulin, administration of animal hormones to humans could evoke an immune response. Third, there was always a danger of the transmission of infectious agents across species, or between people. This was highlighted in the 1970s, when cases of Creutzfeldt–Jakob disease (see Ch. 39) occurred in patients treated with human growth hormone obtained from cadavers. This serious problem was later traced to contamination of the donor pituitary glands with infectious prions (Ch. 39). The advent of ‘genetic engineering’ techniques offered a new way to deal with these perennial problems.
Biopharmaceuticals and gene therapy Definition and potential uses 
•
Biopharmaceuticals include proteins, antibodies (and oligonucleotides) used as drugs:
–
first-generation biopharmaceuticals are mainly copies of endogenous proteins or antibodies, produced by recombinant DNA technology
–
second-generation biopharmaceuticals have been ‘engineered’ to improve the performance of the protein or antibody.
•
Applications:
–
therapeutic monoclonal antibodies
•
Gene therapy is the genetic modification of cells to prevent, alleviate or cure disease.
•
Potential applications:
–
radical cure of monogenic diseases (e.g. cystic fibrosis, haemoglobinopathies)
–
amelioration of diseases with or without a genetic component, including many malignant, neurodegenerative and infectious diseases.
Proteins and Polypeptides
The biopharmaceuticals in use today are generally classified as ‘first-’ or ‘second-’ generation agents. First-generation biopharmaceuticals are usually straightforward copies of human hormones or other proteins prepared by transfecting the human gene into a suitable expression system (a cell line that produces the protein in good yield), harvesting and purifying the recombinant protein produced and using this as the drug. The first agent to be produced in this way was human recombinant insulin in 1982.
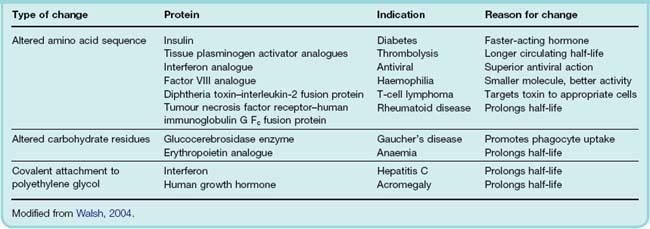
Second-generation biopharmaceuticals are those that have been engineered; that is to say, either the gene has been deliberately altered prior to transfection such that the structure of the expressed protein is changed, or some alteration is made to the purified end product. The reasons for making these changes are generally to improve some aspect of the protein’s activity profile. Human recombinant insulins designed to act faster or last longer were among the first in this class to be marketed; Table 59.1 contains other examples. Third-generation agents would be those in which proteins are designed from scratch to do a particular biological function. This technology is still some way off.
Problems in Manufacture
There are several problems associated with the manufacture of any type of recombinant protein, and one of the most pressing is the choice of expression system. Many recombinant proteins are expressed in bacterial systems (Escherichia coli, for example), which are useful because cultures grow quickly and they are generally easy to manipulate. Disadvantages include the fact that they may contain bacterial endotoxins, which must be scrupulously removed before administration to patients, and that bacterial cells do not accomplish the same type of post-translational processing (e.g. glycosylation) as mammalian cells. This could pose problems if the protein’s action is crucially dependent on this modification. To circumvent these problems, mammalian (e.g. Chinese hamster ovary, CHO) cells are also used as expression systems, although here the problem is often one of yield. Such cells require more careful culture, grow more slowly and produce less product, all of which contribute to the cost of the final medicine.
There are, however, a number of emergent technologies that could revolutionise the production process. The use of plants to produce recombinant proteins has attracted considerable interest (see Daniell et al., 2001, and Fischer et al., 2004). Several species have shown promise, including the tobacco plant. Human genes of interest can readily be transfected into the plant by using tobacco mosaic virus as a vector; the crop grows rapidly (yields a high biomass) and offers a number of other advantages. But attention has also focused on edible plants such as lettuce and bananas. The advantage here is that some orally active proteins, such as vaccines, expressed in the plant could be consumed directly without the need for prior purification. Several such proteins have already been produced in plants, and some are in clinical trial.
Another technology that could dramatically increase the yield of human recombinant proteins is the use of transgenic cattle. A dairy cow can produce some 10 000 litres of milk per year, and recombinant proteins introduced into the genome, and under the control of promoters that regulate production of other milk proteins, can generate yields as high as 1 g/l (see Brink et al., 2000).
Engineered Proteins
There are several ways in which proteins can be altered prior to expression. Alteration of the nucleotide sequence of the gene coding for the protein in question can be used to change single amino acids or, indeed, whole regions of the polypeptide chain. There are good reasons why it is an advantage to ‘engineer’ proteins prior to expression:
1
Modification of pharmacokinetic properties.
2
Generation of novel
fusion or other proteins.
3
Reducing immunogenicity, e.g. by
humanising.
It is frequently advantageous to modify the pharmacokinetic properties of recombinant proteins. Changes in the structure of human insulin, for example, provided diabetics with a form of the hormone that did not self-associate during storage and was thus faster acting and easier to manage. The half-life of proteins in the blood can often be extended by PEGylation (see Ch. 10), the addition of polyethylene glycol to the molecule. This post-translational engineering approach has been applied to some human hormones, such as recombinant growth hormone, interferons and others. Prolonging half-life is not merely a convenience to patients; it also reduces the overall cost of the treatment, and economic factors are important in the adoption of this type of therapy.
Fusion proteins comprise two or more proteins engineered to be expressed as one single polypeptide chain, sometimes joined by a short linker. An example is etanercept, an anti-inflammatory drug used in the treatment of rheumatoid arthritis and other conditions (see Ch. 26). This consists of the ligand-binding domain taken from the tumour necrosis factor receptor, joined to the Fc domain of a human immunoglobulin G antibody. The latter moiety increases its persistence in the blood. Reduction of immunogenicity through bioengineering is discussed below.
Monoclonal Antibodies
Although antibodies are used to confer passive immunity, there are a number of disadvantages inherent in their production and use that limit their utility. Conventionally, antisera are produced from the blood of immunised humans (e.g. to collect antitetanus serum) or from animals immunised with the antigen in question (e.g. with inactivated bacterial toxins). These are used to prepare antiserum containing high levels of specific antibodies, which can then be used clinically to neutralise pathogens or other dangerous substances in the blood of the patient.
Such preparations contain polyclonal antibodies—that is, a mixture of antibodies from all the plasma cell clones that reacted to that particular antigen. The actual composition and efficacy of these varies over time, and obviously there is a limit to how much plasma can be collected on any one occasion. The Nobel Prize-winning discovery by Milstein and Köhler, in 1975, of a method of producing from immunised mice an immortalised hybridoma, a fusion of one particular lymphocytic clone with an immortalised tumour cell, provided us for the first time with a method of producing monoclonal antibodies, comprising a single species of defined antibody at high abundance in vitro. Because these hybridomas were immortal, the cell line could be retained indefinitely and expanded to any density while preserving the integrity of its product.
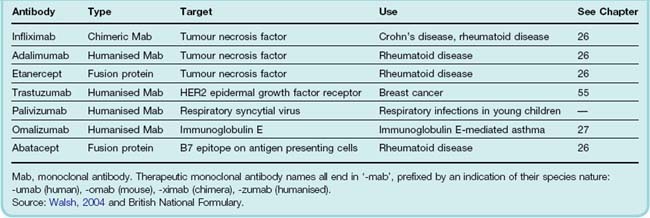
Monoclonal antibodies can be classified into first- or second-generation reagents along similar lines to other proteins discussed above. First-generation monoclonals were essentially murine monoclonals (or fragments thereof), but these suffered from several drawbacks. As mouse proteins, they provoked an immune response in 50–75% of all recipients. Other limiting factors were the short half-life in the circulation and the inability of the mouse antibodies to activate human complement.
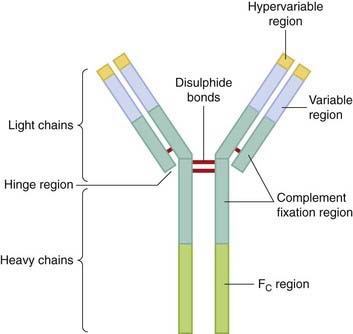
Most of these problems have been surmounted by using either chimeric or humanised monoclonals. The two terms refer to the degree to which the monoclonals have been engineered. Figure 59.1 shows how this is done; the antibody molecule consists of a constant domain (Fc) and the antibody-binding domain (Fab), with hypervariable regions that recognise and bind to the antigen in question. The genes for chimeric monoclonals are engineered to contain the cDNA of the murine Fab domain coupled with the human Fc domain sequences. This greatly (around five-fold) extends the plasma half-life and improves the functionality of the antibody in human medicine. A further development (and now the preferred approach) is to replace the entire Fc and Fab region with the human equivalent with the exception of the hypervariable regions, giving a molecule that, while essentially human in nature, contains the murine antibody-binding sites. The anticancer monoclonal herceptin (trastuzumab; see Ch. 55) is an example of such a therapeutically useful antibody, and some others are given in Table 59.2.
Safety Issues
We are, by now, accustomed to the concept of using proteins therapeutically, and many of the risks associated with (for example) anti-TNF therapy are well understood (see Ch. 26). For the most part, therapeutic proteins do not cause the range of toxic effects encountered with small molecules discussed in Chapter 57, but there are still very real dangers.
In 2006, for example, a UK clinical trial of a new monoclonal antibody (TGN 1412) designed to activate T cells (see Ch. 6) and thus treat B-cell lymphocytic leukaemia went badly wrong. All 6 subjects became severely ill following a ‘cytokine storm’ and suffered lasting damage. The incident provoked wide media publicity4 and, while the subsequent investigation blamed an ‘unpredictable’ biological reaction, it caused many to think hard about how such trials should be conducted in the future (see Muller & Brennan, 2009). Highly specific reagents such as monoclonals pose a particular problem as they may not cross-react with the corresponding antigens of other species, thus evading detection in the usual preclinical animal safety screens.
Gene Therapy
Gene Delivery
The transfer of recombinant nucleic acid into target cells—a special instance of the ‘drug distribution’ problem—is critical to the success of gene therapy. Nucleic acid must pass from the extracellular space across the plasma and nuclear membranes, and it must then be incorporated into the chromosomes. Because DNA is negatively charged and single genes have molecular weights around 104 times greater than conventional drugs, the problem is of a different order from the equivalent stage of routine drug development.
There are several important considerations in choosing a delivery system; these include:
•
the
capacity of the system (e.g. how much DNA it can carry)
•
the
transfection efficiency (its ability to enter and become utilised by cells)
•
the
lifetime of the transfected material (determined by the lifetime of the targeted cells)
•
the
safety issue, especially important in the case of viral delivery systems.
Various approaches have been developed (see Table 59.3) in an attempt to produce the optimal system.
Table 59.3 Characteristics of some delivery systems for gene therapy
| Vector |
Advantages |
Disadvantages |
| Liposomes |
Virus-free, cheap to produce |
Low efficiency, sometimes cytotoxic |
| DNA cassettes |
Virus-free |
Low efficiency, expression temporary |
| Herpes simplex virus type I |
Highly infective, persistent expression |
No integration with host DNA, cytotoxic, difficult to handle |
| Adenovirus |
Highly infective in epithelia |
Immunogenic and transient, requires readministration |
| Adeno-associated virus |
Stable |
Low capacity |
| Retrovirus |
Efficient, permanent |
Low capacity, unstable, must integrate into host DNA, requires dividing cells |
After Wolf & Jenkins, 2002.
There are two main strategies for delivering genes into patients: the in vivo and ex vivo approach. Using the in vivo strategy, the vector containing the therapeutic gene is injected into the patient, either intravenously (in which case some form of organ or tissue targeting is required) or directly into the target tissue (e.g. a malignant tumour). The ex vivo strategy is to remove cells from the patient (e.g. stem cells from bone marrow or circulating blood, or myoblasts from a biopsy of striated muscle), treat them with the vector and inject the genetically altered cells back into the patient.
An ideal vector should be safe, highly efficient (i.e. insert the therapeutic gene into a high proportion of target cells) and selective in that it would lead to expression of the therapeutic protein in the target cells but not to the expression of viral proteins. Provided that the cell into which it is inserted is itself long-lived, the vector should ideally cause persistent expression, avoiding the need for repeated treatment. The latter consideration can be a problem in some tissues. In the autosomal recessive disorder cystic fibrosis, for example, the airway epithelium malfunctions because it lacks a membrane Cl− transporter known as the cystic fibrosis transport regulator (CFTR). Epithelial cells in the airways are continuously dying off and being replaced, so even if the CFTR gene were stably transfected into the epithelium, there would still be a periodic need for further treatment unless the gene could be inserted into the progenitor (stem) cells. Similar problems are anticipated in other cells that turn over continuously, such as gastrointestinal epithelium and skin.
Viral Vectors
Many contemporary gene delivery strategies aim to capitalise on the capacity of viruses to subvert the transcriptional machinery of the cells they invade and their ability (in some cases) to fuse with the host genome. While producing a tantalising glimpse of the possible, there remain substantial practical problems with this approach, partly because as viruses have evolved the means to invade human cells, so humans have evolved immune responses and other protective mechanisms to thwart them. Although irritating in some respects, this is not all bad news from the point of view of safety.
As many of these viruses are pathogenic, they are usually modified such that they are ‘replication defective’ to avoid toxicity.
Retroviruses
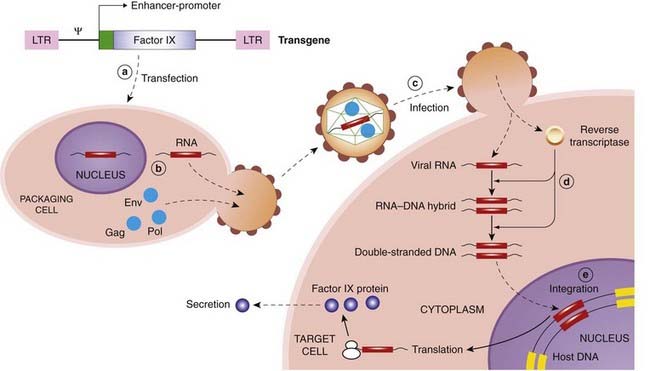
If introduced into stem cells, retroviral vectors have the attraction that their effects are persistent because they are incorporated into, and replicate with, host DNA, and so the ‘therapeutic’ gene is passed down to each daughter cell during division. Against this, the retroviral integrase randomly inserts the construct into chromosomes, so it may cause damage (see below). Also, since many retroviruses show little specificity, they could infect germ or non-target cells and produce undesired effects if administered in vivo. For this reason, retroviruses have been used mainly for ex vivo gene therapy. The life cycle of naturally occurring retroviruses may be exploited to create useful vectors for gene therapy (see Fig. 59.2).
Many viruses are equipped to infect specific cell types, though not necessarily the target cell of interest. It is possible to alter the retroviral envelope to alter specificity, such that the vector could be administered systemically but would target only the desired cell population. An example of this approach with a lentivirus (a type of retrovirus) is the substitution of the envelope protein of a non-pathogenic vector (e.g. mouse leukaemia virus) with the envelope protein of human vesicular stomatitis virus, in order specifically to target human epithelial cells.
Most retrovirus vectors are unable to penetrate the nuclear envelope, and because the nuclear membrane dissolves during cell division, they only infect dividing cells and not non-dividing cells such as adult neurons.
Adenovirus
Adenovirus vectors are popular because of the high transgene expression that can be achieved. They transfer genes to the nucleus of the host cell, but (unlike retroviruses) these are not inserted into the host genome and so do not produce effects that outlast the lifetime of the transfected cell. This property also obviates the risk of disturbing the function of other cellular genes and the theoretical risks of carcinogenicity and germ cell transfection, although at the cost of producing only a temporary effect. Because of these favourable properties, adenovirus vectors have been used for in vivo gene therapy. The vectors are genetically modified by making deletions in the viral genome, rendering it unable to replicate or cause widespread infection in the host while at the same time creating space in the viral genome for the therapeutic transgene to be inserted.
One of the first adenoviral vectors to be used lacked part of a growth-controlling region called E1. This defective virus was grown in a cell line that substitutes for the missing E1 function. Recombinant virus was produced by infecting target cells with a plasmid containing the cloned DNA of therapeutic interest plus an expression cassette and portions of adenoviral DNA. Recombination between this and the ‘backbone’ of the E1-deficient adenoviral genome resulted in a virus encoding the desired transgene. This approach led to seemingly spectacular results, demonstrating gene transfer to cell lines and animal models of disease, but it has been disappointing (e.g. in cystic fibrosis) in humans. The main problem is that low doses (administered by aerosol to patients with this disease) produce only a very low-efficiency transfer, whereas higher doses cause inflammation, a host immune response and short-lived gene expression. Furthermore, treatment cannot be repeated because of neutralising antibodies. This has led to recent attempts to manipulate adenoviral vectors to mutate or remove the genes that are most strongly immunogenic.
Other viral vectors
Other potential viral vectors under investigation include adeno-associated virus, herpes virus and disabled versions of human immunodeficiency virus (HIV). Adeno-associated virus associates with host DNA but is not activated unless the cell is infected with an adenovirus. It is less immunogenic than other vectors but is hard to mass produce and cannot be used to carry large transgenes. Herpes virus does not associate with host DNA but is very long lived in nervous tissue and could have a specific application in treating neurological disease. HIV, unlike most other retroviruses (see above), can infect non-dividing cells such as neurons. It is possible to remove the genes from HIV that control replication and substitute other genes. Alternatively, it may prove possible to transfer to other non-pathogenic retroviruses those genes that permit HIV to penetrate the nuclear envelope.
Non-Viral Vectors
Liposomes
Non-viral vectors include a variant of liposomes (Ch. 8). Plasmids (diameter up to approximately 2 µm) are too big to package in regular liposomes (diameter 0.025–0.1 µm), but larger particles can be made from positively charged lipids (‘lipoplexes’), which interact with both negatively charged cell membranes and DNA, improving delivery into the cell nucleus and incorporation into the host chromosome. Such particles have been used to deliver the genes for HLA-B7, interleukin-2 and CFTR. They are much less efficient than viruses, and attempts are currently under way to improve this by incorporating various viral signal proteins (membrane fusion proteins, for example) in their outer coat. Direct injection of these complexes into solid tumours (e.g. melanoma, breast, kidney and colon cancers) can, however, achieve high local concentrations within the tumour.
Microspheres
Biodegradable microspheres made from polyanhydride co-polymers of fumaric and sebacic acids (see Ch. 8) can be loaded with plasmid DNA. A plasmid with bacterial β-galactosidase activity formulated in this way and given by mouth to rats has resulted in systemic absorption and expression of the bacterial enzyme in the rat liver, raising the possibility of oral gene therapy.
Plasmid DNA
Surprisingly, plasmid DNA itself (‘naked DNA’) enters the nucleus of some cells and is expressed, albeit much less efficiently than when it is packaged in a vector. Such DNA carries no risk of viral replication and is not usually immunogenic (although autoantibodies to DNA do occur in systemic lupus erythematosus), but it cannot be targeted to a cell of interest. There is considerable interest in the possibility of using naked DNA for vaccines, because even very small amounts of foreign protein can stimulate an immune response. Such a vaccine for influenza is in clinical development, and more ambitious long-term targets include malaria, tuberculosis, Chlamydia, Helicobacter and hepatitis.
Controlling Gene Expression
To realise the full potential of gene therapy, it is not enough to transfer the gene selectively to the desired target cells and maintain acceptable expression of its product—difficult though these goals are. It is also essential that the activity of the gene is controlled. Historically, it was the realisation of the magnitude of this task that diverted attention from the haemoglobinopathies (which were the first projected targets of gene therapy). Correction of these disorders demands an appropriate balance of normal α- and β-globin chain synthesis to be effective, and for this, and many other potential applications, precisely controlled gene expression will be essential.
It has not yet proved possible to control transgenes in human recipients, but there are techniques that may eventually enable us to achieve this goal. One hinges on the use of an inducible expression system. This is a fairly standard technique whereby the inserted gene also includes a doxycycline-inducible promoter such that expression of the gene can be switched on or off by treatment with, or withdrawal of, doxycycline.
The control of transfected genes is important in gene targeting as well. By splicing the gene of interest with a tissue-specific promoter, it should be possible to restrict expression of the gene to the target tissue. Such an approach has been used in the design of gene therapy constructs for use in ovarian cancer, the cells of which express several proteins at high abundance, including the proteinase inhibitor SLP1. In combination with the SLP1 promoter, plasmids carrying various genes were successfully and selectively expressed in ovarian cancer cell lines (Wolf & Jenkins, 2002).
Gene delivery and expression
•
Gene delivery is one of the main hurdles to practical gene therapy.
•
Recombinant genes are transferred using a vector, often a suitably modified virus.
•
There are two main strategies for delivering genes into patients:
–
in vivo injection of the vector directly into the patient (e.g. into a malignant tumour)
–
ex vivo treatment of cells from the patient (e.g. stem cells from marrow or circulating blood), which are then returned to the patient.
•
An ideal vector would be safe, efficient, selective and produce long-lasting expression of the therapeutic gene.
•
Viral vectors include retroviruses, adenoviruses, adeno-associated virus, herpesvirus and disabled human immunodeficiency virus (HIV):
–
retroviruses infect many different types of dividing cells and become incorporated randomly into host DNA
–
adenoviruses are genetically modified to prevent replication and accommodate the therapeutic transgene. They transfer genes to the nucleus but not to the genome of the host cell. Problems include a strong host immune response, inflammation and short-lived expression. Treatment cannot be repeated because of neutralising antibodies
–
adeno-associated virus associates with host DNA and is non-immunogenic but is hard to mass-produce and has a small capacity
–
herpesvirus does not associate with host DNA but persists in nervous tissue and may be useful in treating neurological disease
–
disabled versions of HIV differ from most other retroviruses in that they infect non-dividing cells, including neurons.
•
Non-viral vectors include:
–
a variant of liposomes, made using positively charged lipids and called ‘lipoplexes’
–
biodegradable microspheres, which may offer orally active gene therapy
–
plasmid DNA (‘naked DNA’), which can be used as a vaccine.
•
A
tetracycline-inducible expression system or similar technique can control the activity of the therapeutic gene.
Safety Issues
Gene therapy raises a number of specific concerns that generally relate to the use of viral vectors. These are usually selected because they are non-pathogenic, or modified to render them innocuous, but there is a concern that such agents might still acquire virulence during use. Retroviruses, which insert randomly into host DNA, could damage the genome and interfere with the protective mechanisms that normally regulate the cell cycle (see Ch. 5), and if they happen to disrupt essential cellular functions, this could increase the risk of malignancy. This risk is more than a theoretical possibility; several children treated for severe combined immunodeficiency (SCID; see below) with a retrovirus vector developed a leukaemia-like illness (Woods et al., 2006). The retroviral vector was shown to have inserted itself into a gene called LMO-2. Mutations of LMO-2 are associated with childhood cancers.
Another problem is that immunogenic viral proteins may be expressed that elicit an inflammatory response, and this could be harmful in some situations (e.g. in the airways of patients with cystic fibrosis). Initial clinical experience was reassuring, but the tragic death of Jesse Gelsinger, an 18-year-old volunteer in a gene therapy trial for the non-fatal disease ornithine decarboxylase deficiency (which can be controlled by diet and drugs), led to the appreciation that safety concerns related to immune-mediated responses to vectors are very real (see Marshall, 1999).
But despite safety concerns, there have been some encouraging successes. We will finish by glancing at some of the areas where gene therapy has already proved its worth—as well as several areas of particular promise for the future.
Safety
•
There are those safety concerns that are
specific to any particular therapy (e.g. polycythaemia from overexpression of erythropoietin) and additional
general concerns relating, for example, to the nature of vectors.
•
Viral vectors:
–
might acquire virulence during use
–
contain viral proteins, which may be immunogenic
–
can elicit an inflammatory response
–
could damage the host genome and interfere with the cell cycle, provoking malignancy.
•
The limited clinical experience to date has not so far provided evidence of insurmountable problems.
Therapeutic Applications
Single-Gene Defects
Single-gene (monogenic) disorders were the obvious starting point for gene therapy trials. The haemoglobinopathies were the first projected targets, but early attempts (in the 1980s) were put ‘on hold’ because of the problem, mentioned above, posed by the need to control precisely the expression of the genes encoding the different polypeptide chains of the haemoglobin molecule. Patients with thalassaemia (the commonest monogenic disease) exhibit enormous phenotypic diversity and hence variable clinical symptoms because, even in monogenic disorders, other genes as well as environmental factors are also important.
Attention then shifted to a rare genetic disorder called adenine deaminase deficiency, which results in SCID. This led to the first therapeutic gene transfer protocol to be approved by the US National Institutes of Health, and subsequently a French team has treated 11 children with another form of SCID. The results provided the first proof that gene therapy can cure a life-threatening disease but also, less happily, evidence that retroviral vectors can cause malignancy.
Another early target was cystic fibrosis. Progress here has been slow: Atkinson (2008) has reviewed this area and explains the many problems associated with this approach.
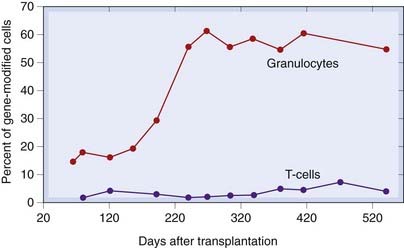
More recently, however, there have been several successes. For example, X-linked chronic granulomatous disease (see Ch. 17) has been successfully treated using a retroviral technique to deliver a functional version of the mutated NADPH oxidase protein (Ott et al., 2006 and Fig. 59.3) and a form of inherited blindness, Leber’s congenital amaurosis, associated with a mutation in a gene that produces retinal pigment, has been rectified using an adeno-associated virus vector bearing a cDNA coding for the intact gene (Maguire et al., 2008).
Gene Therapy for Cancer
Many current clinical gene therapy trials relate to its use in cancer. The first gene transfer experiment to be approved by the National Institutes of Health was a non-therapeutic protocol in the late 1980s designed to introduce a marker gene (conferring resistance to an analogue of neomycin) into a class of lymphocytes that infiltrate various tumours. Gene transfer was performed ex vivo and the cells reinjected into the patient in order to track their subsequent redistribution. This strategy was useful in tracking other cells and hence identifying the cause of relapse following bone marrow transplantation for various leukaemias. Several therapeutic approaches are under investigation. Promising approaches include:
•
restoring ‘protective’ proteins such as the tumour suppressor gene (see
Ch. 5)
•
inactivating oncogene expression (e.g. by using a retroviral vector bearing an antisense transcript RNA to the
k-ras oncogene; see below)
•
delivering a gene to malignant cells that renders them sensitive to drugs (e.g. thymidylate kinase, which activates
ganciclovir)—the so-called ‘suicide gene’ approach
•
delivery of proteins to healthy host cells in order to protect them (e.g. addition of the multidrug resistance channel to bone marrow cells ex vivo, thereby rendering them resistant to drugs used in chemotherapy)
•
tagging cancer cells with genes expressing proteins that render malignant cells more visible to the immune system (e.g. for antigens such as HLA-B7 or cytokines such as granulocyte macrophage colony-stimulating factor and interleukin-2).
Ovarian cancer is considered to be a good target for gene therapy because the vector can be directly introduced into the peritoneal cavity, where it is retained in a ‘closed’ environment. Several clinical trials are in progress or have been completed (see Wolf & Jenkins, 2002) with a variety of genes including p53 and the multidrug resistance gene, and utilising retroviral, adenoviral and liposome vectors. For a recent review of gene therapy in breast cancer, see Takahashi et al. (2006).
Gene therapy for cancer
•
Promising approaches include:
–
restoring protective proteins such as p53
–
delivering a gene to malignant cells that renders them sensitive to drugs
–
delivering a gene to healthy host cells to protect them from chemotherapy
–
tagging cancer cells with genes that make them immunogenic.
Gene Therapy and Infectious Disease
In addition to DNA vaccines mentioned above, there is considerable interest in the potential of gene therapy for HIV infection. Some 10% of all clinical gene therapy research is focused on this area and, by rendering stem cells (which differentiate into immune cells) resistant to HIV before they mature, aims to prevent HIV replication as well as its spread to uninfected cells. Various strategies are under investigation, including the use of genes that code for variants of HIV-directed proteins that serve as blocking agents (so-called ‘dominant-negative’ mutations, e.g. rev, which began clinical testing in 1995), RNA decoys and soluble forms of CD4 (the cellular receptor used by HIV to enter lymphocytes; Ch. 51) that will bind, and it is hoped inactivate, HIV extracellularly.
Gene Therapy and Cardiovascular Disease
Vascular gene transfer is attractive not least because cardiologists and vascular surgeons routinely perform invasive studies that offer the opportunity to administer gene therapy vectors ex vivo (e.g. to a blood vessel that has been removed to use as an autograft) or locally in vivo (e.g. by injection through a catheter directly into a diseased coronary or femoral artery). Vascular gene transfer offers potential new treatments for several cardiovascular diseases (see Ylä-Herttuala & Martin, 2000). The nature of many vascular disorders, such as restenosis following angioplasty (stretching up a narrowed artery using a balloon that can be inflated via a catheter), is such that transient gene expression might be all that is needed therapeutically. Extension of vein graft patency by gene therapy approaches has been reviewed by Chandiwal & Balasubramanian (2005). There is no shortage of attractive candidates for therapeutic overexpression in blood vessels, including nitric oxide synthase, prostacyclin synthase, thymidylate kinase, homeobox proteins and many others. Some of these have been studied in animal models of restenosis, finding that overexpression of vascular endothelial growth factor and fibroblast growth factor increases blood flow and collateral vessel growth in ischaemic leg muscle and myocardium. This is a promising area; for further details of angiogenic gene therapy, see Hammond & McKirnan (2001) and of peripheral vascular disease Ghosh et al. (2008).
Many trials are ongoing and these can be viewed online at Gene Therapy Review (http://www.genetherapyreview.com) and other sites (see Further Reading). Other uses of gene therapy include conditions as diverse as uterine leiomyoma (fibroids; Al-Hendy & Salama, 2006) and periodontal disease (Karthikeyan & Pradeep, 2006).
Other Gene-Based Approaches
So far, we have largely been considering the addition of entire genes, but there are other, related nucleic acid-based therapeutic strategies. One such attempt is to correct a gene that has been adversely altered by mutation. This has the enormous theoretical advantage that the corrected gene would remain under physiological control, avoiding many of the problems discussed above. This approach is in its infancy and is beyond the scope of this book.
Other therapeutic approaches that are, in effect, gene therapies are conventionally excluded from this category. These include organ transplantation to correct a gene deficiency (e.g. liver transplantation to correct low-density-lipoprotein receptor deficiency in homozygous familial hypercholesterolaemia; Ch. 23).
Another approach is the use of antisense oligonucleotides. These are short (15–25mer) oligonucleotides that are complementary to part of a gene or gene product that it is desired to inhibit. These snippets of genetic material can be designed to influence the expression of a gene either by forming a triplex (three-stranded helix) with a regulatory component of chromosomal DNA, or by complexing a region of mRNA. Oligonucleotides can cross plasma and nuclear membranes by endocytosis as well as by direct diffusion, despite their molecular size and charge. However, there are abundant enzymes that cleave foreign DNA in plasma and in cell cytoplasm, so methylphosphorate analogues have been synthesised in which a methyl group substitutes for an oxygen atom in the nucleotide backbone. Another approach is the use of phosphothiorate analogues in which a negatively charged sulfur atom substitutes for oxygen (so-called ‘S oligomers’). This increases water solubility as well as conferring resistance to enzymic degradation. The oligomer needs to be at least 15 bases long to confer specificity and tight binding.
Following parenteral administration, such oligomers distribute widely (although not to the central nervous system) and work in part by interfering with the transcription of mRNA and in part by stimulating its breakdown by ribonuclease H, which cleaves the bound mRNA. This approach is being used in clinical studies in patients with viral disease (including HIV infection) and malignancy (including the use of Bcl-2 antisense therapy administered subcutaneously in patients with non-Hodgkin’s lymphoma). A related approach (see Castanatto & Rossi, 2009), which provides more efficient gene silencing than antisense oligonucleotides, is the use of short interfering RNA (siRNA),5 whereby short lengths of double-stranded RNA recruit an enzyme complex, known as RISC, which selectively degrades the corresponding mRNA produced by the cell, thereby blocking expression. Clinical trials of siRNA therapeutics are in progress.
Other gene-based approaches
•
Correction of a mutated gene. This is in its infancy.
•
Antisense oligonucleotides are short (15–25) oligonucleotides that are complementary to part of the target gene and influence expression by forming a triplex (three-stranded helix) with a regulatory component of chromosomal DNA or by complexing a region of mRNA. siRNA, which acts by a different mechanism, can be used in the same way.
•
Oligonucleotides can cross plasma and nuclear membranes but there are abundant enzymes that cleave foreign DNA, so water-soluble methylphosphorate or phosphothiorate analogues, which are resistant to enzymic degradation, are used. This approach is being used in clinical trials in HIV infection and malignancy.
References and Further Reading
General reviews on biopharmaceuticals, gene therapy and utilities
Scientific American published an issue devoted to gene therapy in June 1997, which is an excellent introduction, including articles by T Friedmann (on ‘overcoming the obstacles to gene therapy’), P L Felgner (on non-viral strategies for gene therapy), R M Blaese (on gene therapy for cancer) and D Y Ho and R M Sapolsky (on gene therapy for the nervous system).
Brink M.F., Bishop M.D., Pieper F.R. Developing efficient strategies for the generation of transgenic cattle which produce biopharmaceuticals in milk. Theriogenology. 2000;53:139-148. (A bit specialised, as it focuses mainly on the husbandry of transgenic cattle, but interesting nonetheless)
Castanatto D., Rossi J.J. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426-433. (Useful review of the mechanism, current status and potential applications of RNAi as a means of controlling gene expression)
Daniell H., Streatfield S.J., Wycoff K. Medical molecular farming: production of antibodies, biopharmaceuticals and edible vaccines in plants. Trends Plant Sci.. 2001;6:219-226. (Interesting paper with some good examples)
Fischer N., Leger O. Bispecific antibodies: molecules that enable novel therapeutic strategies. Pathobiology. 2007;74:3-14. (Interesting new twist to the therapeutic monoclonal story)
Fischer R., Stoger E., Schillberg S., et al. Plant-based production of biopharmaceuticals. Curr. Opin. Plant Biol.. 2004;7:152-158. (Interesting general review on the use of plants for the production of biopharmaceuticals)
Guttmacher A.E., Collins F.S. Genomic medicine: a primer. N. Engl. J. Med.. 2002;347:1512-1520. (First in a series on genomic medicine)
Verma I.M., Somia N. Gene therapy—promises, problems and prospects. Nature. 1997;389:239-242. (The authors, from the Salk Institute, describe the principle of getting corrective genetic material into cells to alleviate disease, the practical obstacles to this and the hopes that better delivery systems will overcome them)
Walsh G. Second-generation biopharmaceuticals. Eur. J. Pharm. Biopharm.. 2004;58:185-196. (Excellent overview of therapeutic proteins and antibodies; some good tables and figures)
Weatherall D.J. Single gene disorders or complex traits: lessons from the thalassaemias and other monogenic diseases. BMJ. 2000;321:1117-1120. (Argues that relating genotype to phenotype is the challenge for genetic medicine over the next century)
Problems
Check E. A tragic setback. Nature. 2002;420:116-118. (News feature describing efforts to explain the mechanism underlying a leukaemia-like illness in a child previously cured of SCID by gene therapy)
Marshall E. Gene therapy death prompts review of adenovirus vector. Science. 1999;286:2244-2245. (Deals with the tragic ‘Gelsinger affair’)
Muller P.Y., Brennan F.R. Safety assessment and dose selection for first-in-human clinical trials with immunomodulatory monoclonal antibodies. Clin. Pharmacol. Ther.. 2009;85:247-258. (A sober and, at times, very technical assessment of the safety precedures required for the ‘first-in-man’ testing of therapeutic monoclonals. Written in the wake of the TGN 1412 affair)
Stobbart L., Murtagh M.J., Rapley T., et al. ‘We saw human guinea pigs explode’. BMJ. 2007;334:566-567. (Analysis of the press coverage of the above clinical trial)
Woods N.B., Bottero V., Schmidt M., von Kalle C., Verma I.M. Gene therapy: therapeutic gene causing lymphoma. Nature. 2006;440:1123.
Therapeutic uses
Al-Hendy A., Salama S. Gene therapy and uterine leiomyoma: a review. Hum. Reprod. Update. 2006;12:385-400.
Athanasopoulos T., Fabb S., Dickson G. Gene therapy vectors based on adeno-associated virus: characteristics and applications to acquired and inherited diseases (review). Int. J. Mol. Med.. 2000;6:363-375. (Good review)
Atkinson T.J. Cystic fibrosis, vector-mediated gene therapy, and relevance of toll-like receptors: a review of problems, progress, and possibilities. Curr. Gene Ther.. 2008;8:201-207.
Bauerschmitz G.J., Barker S.D., Hemminki A. Adenoviral gene therapy for cancer: from vectors to targeted and replication competent agents (review). Int. J. Oncol.. 2002;21:1161-1174. (Superb review: very comprehensive)
Chandiwal A., Balasubramanian V., Baldwin Z.K., Conte M.S., Schwartz L.B. Gene therapy for the extension of vein graft patency: a review. Vasc. Endovascular Surg.. 2005;39:1-14.
Ghosh R., Walsh S.R., Tang T.Y., Noorani A., Hayes P.D. Gene therapy as a novel therapeutic option in the treatment of peripheral vascular disease: systematic review and meta-analysis. Int. J. Clin. Pract.. 2008;62:1383-1390.
Hammond H.K., McKirnan M.D. Angiogenic gene therapy for heart disease: a review of animal studies and clinical trials. Cardiovasc. Res.. 2001;49:561-567. (Comprehensive review spanning animal and human trials of gene therapy for myocardial ischaemia)
Karthikeyan B.V., Pradeep A.R. Gene therapy in periodontics: a review and future implications. J. Contemp. Dent. Pract.. 2006;7:83-91.
Li F., Hayes J.K., Wong K.C. Gene therapy: a novel method for the treatment of myocardial ischemia and reperfusion injury—mini-review. Acta Anaesthesiol. Sin.. 2000;38:207-215. (The title is self-explanatory)
Maguire A.M., Simonelli F., Pierce E.A., et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med.. 2008;358:2240-2248. (Clinical trial of gene therapy to correct a cause of congenital blindness)
Nathwani A.C., Davidoff A.M., Linch D.C. A review of gene therapy for haematological disorders. Br. J. Haematol.. 2005;128:3-17. (The title is self-explanantory; easy to read and comprehensive in scope)
Ott M.G., Schmidt M., Schwarzwaelder K., et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med.. 2006;12:401-409. (Clinical trial of gene therapy to correct hereditary neutrophil dysfunction)
Roth J.A., Grammer S.F. Gene replacement therapy for non-small cell lung cancer: a review. Hematol. Oncol. Clin. North Am.. 2004;18:215-229. (Useful and readable paper on ‘replacement therapy’ in cancer therapy)
Takahashi S., Ito Y., Hatake K., Sugimoto Y. Gene therapy for breast cancer. Review of clinical gene therapy trials for breast cancer and MDR1 gene therapy trial in Cancer Institute Hospital. Breast Cancer. 2006;13:8-15.
Wolf J.K., Jenkins A.D. Gene therapy for ovarian cancer (review). Int. J. Oncol.. 2002;21:461-468. (Excellent review and broad introduction to gene therapy in general)
Ylä-Herttuala S., Martin J.F. Cardiovascular gene therapy. Lancet. 2000;355:213-222. (Reviews rationale, vectors, delivery, therapeutic targets, human trials, ethics and future directions)
Useful Web resources
http://www.genetherapynet.com (Gene Therapy Net—a fantastic resource for both patients and professionals. It is a veritable clearing house for information and up-to-date news on all aspects of gene therapy. It even advertises for volunteers and has a ‘jobs’ section, in case you are tempted! Has links to other related sites)