26 Anti-inflammatory and immunosuppressant drugs
Overview
This chapter deals with the drugs used to treat inflammatory and immune disorders. While generally associated with diseases such as rheumatoid arthritis, it has become clear that inflammation forms a significant component of many, if not most, of the diseases encountered in the clinic and consequently anti-inflammatory drugs are extensively employed in virtually all branches of medicine.
The chief drugs used to treat inflammation may (somewhat arbitrarily) be divided into three major groups:
We first describe the therapeutic effects, mechanisms of action and unwanted effects common to all NSAIDs, and deal in a little more detail with aspirin, paracetamol and drugs that are selective for cyclo-oxygenase (COX)-2. The antirheumatoid drugs comprise a rather heterogeneous group, many of unknown mechanism of action. They include immunosuppressant drugs that are also used to prevent rejection of organ transplants. The glucocorticoids are covered in Chapter 32, but are briefly discussed in this chapter. We then consider the latest biologicals that are revolutionising treatment in many cases of severe disease. Finally, we consider drugs that do not fit easily into these categories: those used to treat gout and the histamine H1 receptor antagonists used to treat acute allergic conditions.
Cyclo-Oxygenase Inhibitors
This group includes the ‘traditional’ (in the historical sense) NSAIDs1 as well as the newer coxibs that are more selective for COX-2 (see below). These drugs, sometimes called the aspirin-like drugs, or antipyretic analgesics are among the most widely used of all agents. There are now more than 50 different NSAIDs on the global market; some current examples are listed in Table 26.1 and some structures given in Figure 26.1. These drugs provide symptomatic relief from pain and swelling in chronic joint disease such as occurs in osteo- and rheumatoid arthritis, as well as in more acute inflammatory conditions such as fractures, sprains, sports and other soft tissue injuries. They are also useful in the treatment of postoperative, dental and menstrual pain, and of headaches and migraine. Several NSAIDs are available over-the-counter and they are widely used for other types of minor aches and pains. There are many different NSAID formulations available, including tablets, injections and gels. Virtually all these drugs, particularly the ‘traditional’ NSAIDs, can have significant unwanted effects, especially in the elderly. Newer agents have fewer adverse actions.
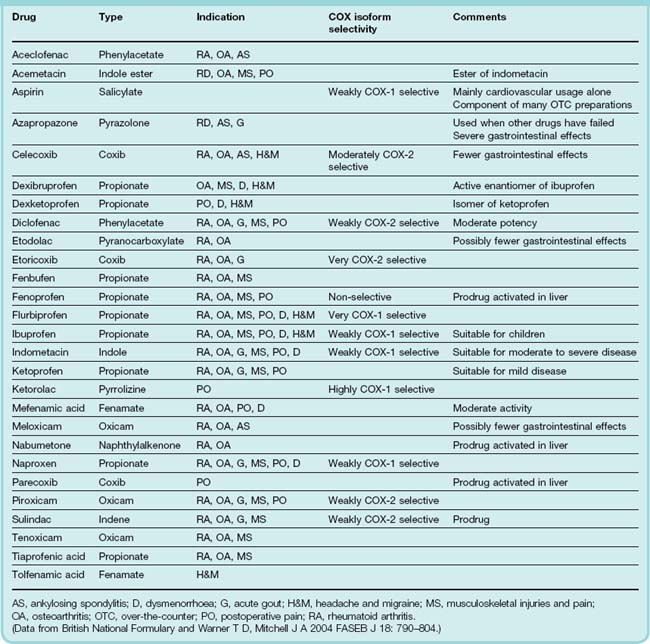
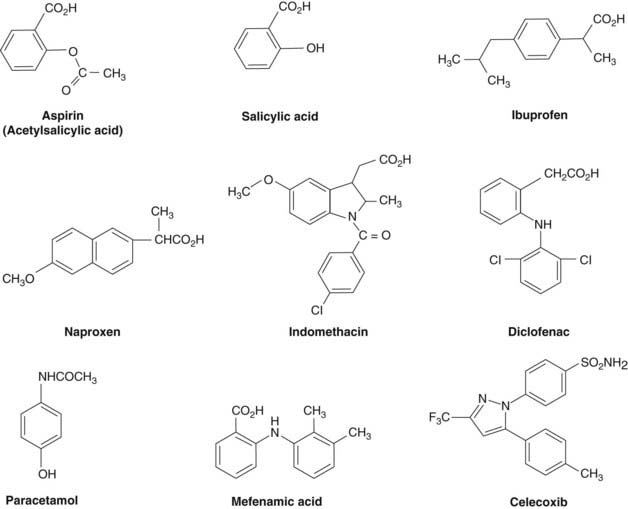
Fig. 26.1 Significant structural features of some non-steroidal anti-inflammatory drugs (NSAIDs) and coxibs.
Aspirin contains an acetyl group that is responsible for the inactivation of the COX enzyme. Salicylic acid is the end product when aspirin is de-acetylated. Oddly it has anti-inflammatory activity in its own right. Paracetamol is a commonly used analgesic agent also of simple structure. Most ‘classic’ NSAIDs are carboxylic acids. Coxibs (celecoxib shown here as an example), however, often contain sulfonamide or sulfone groups. These are important in the selectivity of the molecule as they impede access to the hydrophobic channel in the COX-1 enzyme (see Fig. 26.2).
While there are differences between individual NSAIDs, their primary pharmacology is related to their shared ability to inhibit the fatty acid COX enzyme, thereby inhibiting the production of prostaglandins and thromboxanes (see Ch. 17). There are two common isoforms of this enzyme, COX-1 and COX-2. There may also be other COX enzymes that can generate prostaglandins but these have not been completely characterised. While COX-1 and COX-2 are closely related (> 60% sequence identity) and catalyse the same reaction, it is clear that there are important differences between the expression and role of these two isoforms. COX-1 is a constitutive enzyme expressed in most tissues, including blood platelets. It has a ‘housekeeping’ role in the body, being involved in tissue homeostasis, and is responsible for the production of prostaglandins involved in, for example, gastric cytoprotection (see Ch. 29), platelet aggregation (Ch. 24), renal blood flow autoregulation (Ch. 28) and the initiation of parturition (Ch. 34).
In contrast, COX-2 is induced in inflammatory cells when they are injured, infected or activated by, for example, the inflammatory cytokines—interleukin (IL)-1 and tumour necrosis factor (TNF)-α (see Ch. 17). Thus the COX-2 isoform is mainly responsible for the production of the prostanoid mediators of inflammation (Vane & Botting, 2001), although there are some significant exceptions. For example, there is a considerable pool of ‘constitutive’ COX-2 present in the central nervous system (CNS) and some other tissues, although its function at these sites is not yet completely clear.
Most ‘traditional’ NSAIDs inhibit both COX-1 and COX-2, although they vary in the degree to which they inhibit each isoform. It is believed that the anti-inflammatory action (and probably most analgesic and antipyretic actions) of the NSAIDs are related to inhibition of COX-2, while their unwanted effects—particularly those affecting the gastrointestinal tract—are largely a result of their inhibition of COX-1. Compounds with a selective inhibitory action on COX-2 are now in clinical use, but while these drugs show fewer gastrointestinal side effects, they are by no means as well tolerated as was once hoped. This is partly because many patients taking these drugs have already been exposed to less selective inhibitors and have already suffered some gastrointestinal damage. As COX-2 seems to be important in healing and resolution, one can see how problems might still occur. There is also a concern about the cardiovascular effects of all NSAIDs when these are taken over a long time (see below). Some notes on the relative selectivity of some currently available NSAIDs and coxibs are given in Table 26.1.
 While the pharmacological actions of NSAIDs are broadly similar (although there are marked differences in toxicity and degree of patient tolerance), there are exceptions. Aspirin has other qualitatively different pharmacological actions (see below), and paracetamol is an interesting exception to the general NSAID ‘stereotype’. While it is an excellent analgesic and antipyretic, its anti-inflammatory activity is slight and seems to be restricted to a few special cases (e.g. inflammation following dental extraction; see Skjelbred et al., 1984). Paracetamol has been shown to inhibit prostaglandin biosynthesis in some experimental settings (e.g. during fever) but not in others. The main pharmacological actions and the common side effects of the NSAIDs are outlined below, followed by a more detailed coverage of aspirin and paracetamol and an outline of the pharmacology of the selective COX-2 inhibitors.
While the pharmacological actions of NSAIDs are broadly similar (although there are marked differences in toxicity and degree of patient tolerance), there are exceptions. Aspirin has other qualitatively different pharmacological actions (see below), and paracetamol is an interesting exception to the general NSAID ‘stereotype’. While it is an excellent analgesic and antipyretic, its anti-inflammatory activity is slight and seems to be restricted to a few special cases (e.g. inflammation following dental extraction; see Skjelbred et al., 1984). Paracetamol has been shown to inhibit prostaglandin biosynthesis in some experimental settings (e.g. during fever) but not in others. The main pharmacological actions and the common side effects of the NSAIDs are outlined below, followed by a more detailed coverage of aspirin and paracetamol and an outline of the pharmacology of the selective COX-2 inhibitors.
Cyclo-oxygenase inhibitors 
These drugs have three major therapeutic actions, stemming from the suppression of prostanoid synthesis in inflammatory cells through inhibition of the cyclo-oxygenase (COX)-2 isoform of the arachidonic acid COX. They are as follows:
Some important NSAIDs are aspirin, ibuprofen, naproxen, indometacin, piroxicam and paracetamol. Newer agents with more selective inhibition of COX-2 (and thus fewer adverse effects on the gastrointestinal tract) include celecoxib and etoricoxib.
Mechanism of Action
Vane and his colleagues established in 1971 that the main actions of NSAIDs were bought about through inhibition of arachidonic acid oxidation by the fatty acid COXs (see Fig. 26.2).
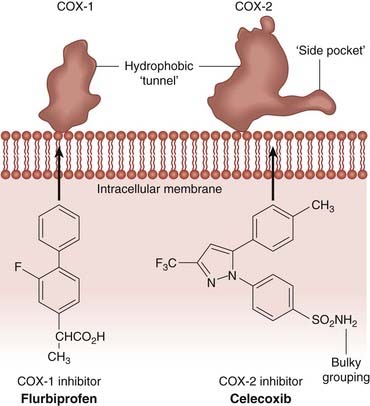
Fig. 26.2 Schematic diagram comparing the binding sites of cyclo-oxygenase (COX)-1 and COX-2.
The illustration shows the differences in NSAID binding sites in the two isoforms. Note that the COX-2 binding site is characterised by a ‘side pocket’ that can accommodate the bulky groups, such as the sulfonamide moiety of celecoxib, which would impede its access to the COX-1 site. Other NSAIDs, such as flurbiprofen (shown here), can enter the active site of either enzyme.
(After Luong et al. 1996 Nat Struct Biol 3: 927–933.)
These are bifunctional enzymes, having two distinct catalytic activities. The first, dioxygenase step incorporates two molecules of oxygen into the arachidonic (or other fatty acid substrate) chain at C11 and C15, giving rise to the highly unstable endoperoxide intermediate PGG2 with a hydroperoxy group at C15. A second, peroxidase function of the enzyme converts this to PGH2 with a hydroxy group at C15 (see Ch. 17), which can then be transformed in a cell-specific manner by separate isomerase, reductase or synthase enzymes into other prostanoids. Both COX-1 and COX-2 are haem-containing enzymes that exist as homodimers attached to intracellular membranes. Structurally, the isoforms are similar; both contain a hydrophobic channel into which the arachidonic or other substrate fatty acids dock so that the oxygenation reaction can proceed.
Most NSAIDs inhibit only the initial dioxygenation reaction. They are generally ‘competitive reversible’ inhibitors, but there are differences in their time courses. Generally, these drugs inhibit COX-1 rapidly, but the inhibition of COX-2 is more time-dependent and the inhibition is often irreversible. To block the enzymes, NSAIDs enter the hydrophobic channel, forming hydrogen bonds with an arginine residue at position 120, thus preventing substrate fatty acids from entering into the catalytic domain. However, a single amino acid change (isoleucine to valine at position 523) in the structure of the entrance of this channel in COX-2 results in a bulky side pocket that is not found in COX-1. This is important in understanding why some drugs, especially those with large sulphur-containing side groups, are more selective for the COX-2 isoform (Fig. 26.2). Aspirin is, however, an anomaly. It enters the active site and acetylates a serine at position 530, irreversibly inactivating COX. This is the basis for aspirin’s long-lasting effects on platelets (see below).
Other actions besides inhibition of COX may contribute to the anti-inflammatory effects of some NSAIDs. Reactive oxygen radicals produced by neutrophils and macrophages are implicated in tissue damage in some conditions, and some NSAIDs (e.g. sulindac) have oxygen radical-scavenging effects as well as COX inhibitory activity, so may decrease tissue damage. Aspirin also inhibits expression of the transcription factor NFκB (see Ch. 3), which has a key role in the transcription of the genes for inflammatory mediators.
Pharmacological Actions
All the NSAIDs have actions very similar to those of aspirin, the archetypal NSAID, which was introduced into clinical medicine in the 1890s. Their main pharmacological profile is listed in the clinical box.
Therapeutic Actions
Anti-Inflammatory Effects
As described in Chapter 17, many mediators coordinate inflammatory and allergic reactions. The NSAIDs reduce mainly those components of the inflammatory and immune response in which prostaglandins, mainly derived from COX-2, play a significant part. These include:
While the NSAIDs suppress the signs and symptoms of inflammation, they have little or no action on underlying chronic disease itself. As a class, they are generally without direct effect on other aspects of inflammation, such as cytokine/chemokine release, leukocyte migration, lysosomal enzyme release and toxic oxygen radical production, which contribute to tissue damage in chronic inflammatory conditions such as rheumatoid arthritis, vasculitis and nephritis.
Antipyretic Effect
A centre in the hypothalamus that controls the balance between heat loss and heat production regulates normal body temperature. Fever occurs when there is a disturbance of this hypothalamic ‘thermostat’, which leads to the set point of body temperature being raised. NSAIDs ‘reset’ this thermostat. Once there has been a return to the normal set point, the temperature-regulating mechanisms (dilatation of superficial blood vessels, sweating, etc.) then operate to reduce temperature. Normal body temperature in humans is not affected by NSAIDs.2
The NSAIDs exert their antipyretic action largely through inhibition of prostaglandin production in the hypothalamus. During an inflammatory reaction, bacterial endotoxins cause the release from macrophages of IL-1 (Ch. 17), which stimulates the generation, in the hypothalamus, of E-type prostaglandins that elevate the temperature set point. COX-2 may have a role here, because IL-1 induces it in vascular endothelium in the hypothalamus. There is some evidence that prostaglandins are not the only mediators of fever, hence NSAIDs may have an additional antipyretic effect by mechanisms as yet unknown.
Analgesic Effect
The NSAIDs are effective against mild or moderate pain, especially that arising from inflammation or tissue damage. Two sites of action have been identified.
First, peripherally, they decrease production of prostaglandins that sensitise nociceptors to inflammatory mediators such as bradykinin (see Chs 17 and 41) and they are therefore effective in arthritis, bursitis, pain of muscular and vascular origin, toothache, dysmenorrhoea, the pain of postpartum states and the pain of cancer metastases in bone. All conditions are associated with increased local prostaglandin synthesis probably as a result of COX-2 induction. Alone, or in combination with opioids, they decrease postoperative pain and in some cases can reduce the requirement for opioids by as much as one-third. Their ability to relieve headache may be related to the reduction in vasodilator prostaglandins acting on the cerebral vasculature.
In addition to these peripheral effects, there is a second, less well characterised central action, possibly in the spinal cord. Inflammatory lesions increase COX-2 and prostaglandin release within the cord, causing facilitation of transmission from afferent pain fibres to relay neurons in the dorsal horn.
Unwanted Effects
Overall, the burden of unwanted side effects is high, probably reflecting the fact that NSAIDs are used extensively in the more vulnerable elderly population, and often for extended periods of time. When used for joint diseases (which usually necessitates fairly large doses and long-continued use), there is a high incidence of side effects—particularly in the gastrointestinal tract but also in the liver, kidney, spleen, blood and bone marrow.
Because prostaglandins are involved in gastric cytoprotection, platelet aggregation, renal vascular autoregulation and induction of labour, among other effects, all NSAIDs share a broadly similar profile of mechanism-dependent side effects although there may be other additional unwanted effects peculiar to individual members of the group. COX-2-selective drugs have less, but not negligible, gastrointestinal toxicity (see below).
Gastrointestinal disturbances
Adverse gastrointestinal events are the commonest unwanted effects of the NSAIDs, and are believed to result mainly from inhibition of gastric COX-1, which is responsible for the synthesis of the prostaglandins that normally inhibit acid secretion and protect the mucosa (see Fig. 29.2).
These commonly include gastric discomfort, dyspepsia, diarrhoea (but sometimes constipation), nausea and vomiting, and in some cases gastric bleeding and ulceration. It has been estimated that 34–46% of users of NSAIDs will sustain some gastrointestinal damage that, while it may be asymptomatic, carries a risk of serious haemorrhage and/or perforation (Fries, 1983). Severe gastrointestinal effects (perforations, ulcers or bleeding) are said to result in the hospitalisation of over 100 000 people per year in the USA. Some 15% of these patients die from this iatrogenic disease (Fries, 1998). Damage is seen whether the drugs are given orally or systemically. However, in some cases (aspirin being a good example), local damage to the gastric mucosa caused directly by the drug itself may compound the damage. Figure 26.3 gives the relative risks of gastrointestinal damage with some common NSAIDs. Oral administration of prostaglandin analogues such as misoprostol (see Ch. 29) can diminish the gastric damage produced by these agents.
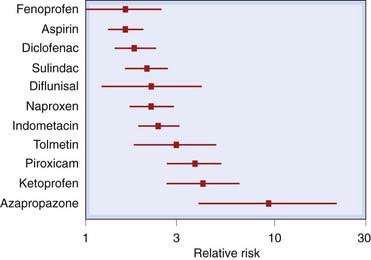
Fig. 26.3 The risk of gastrointestinal complications with various non-steroidal anti-inflammatory drugs.
The risk is shown relative to ibuprofen (relative risk = 1). Ibuprofen, given in a dose of 1200 mg daily, itself carries a risk double that of placebo. The lines represent 95% confidence intervals.
(From a figure by Hawkey, 2001; data derived from a meta-analysis of 12 comparative studies in Henry et al., 1996.)
Based on extensive experimental evidence, it had been predicted that COX-2-selective agents would provide good anti-inflammatory and analgesic actions with less gastric damage, and some older drugs (e.g. meloxicam) that were believed to be better tolerated in the clinic turned out to have some COX-2 selectivity. Two large prospective studies compared the gastrointestinal side effects of celecoxib and rofecoxib with those of standard comparator NSAIDs in patients with arthritis and showed some benefit, although the results were not as clear-cut as had been hoped.
Other ideas have been proposed to explain the gastric side effects of NSAIDs. The administration of COX-1 inhibitors themselves causes COX-2 induction and, on the basis of experimental evidence, Wallace (2000) has argued that selective inhibitors of either isozyme will cause less gastric damage than non-selective drugs.
Skin reactions
Rashes are common idiosyncratic unwanted effects of NSAIDs, particularly with mefenamic acid (10–15% frequency) and sulindac (5–10% frequency). They vary from mild erythematous, urticarial and photosensitivity reactions to more serious and potentially fatal diseases including Stevens–Johnson syndrome (a blistering rash that extends into the gut), and toxic epidermal necrolysis,3 characterised by widespread epithelial necrosis (fortunately rare). The mechanism is unclear.
Adverse renal effects
Therapeutic doses of NSAIDs in healthy individuals pose little threat to kidney function, but in susceptible patients they cause acute renal insufficiency, which is reversible on discontinuing the drug (see Ch. 57, Table 57.1). This occurs through the inhibition of the biosynthesis of those prostanoids (PGE2 and PGI2; prostacyclin) involved in the maintenance of renal blood flow, specifically in the PGE2-mediated compensatory vasodilatation that occurs in response to the action of noradrenaline (norepinephrine) or angiotensin II (see Ch. 28). Neonates and the elderly are especially at risk, as are patients with heart, liver or kidney disease, or a reduced circulating blood volume.
Chronic NSAID consumption, especially NSAID ‘abuse’,4 can cause analgesic nephropathy characterised by chronic nephritis and renal papillary necrosis (Ch. 28). Phenacetin, now withdrawn, was the main culprit; paracetamol, one of its major metabolites, is much less toxic. Regular use of prescribed doses of NSAIDs is less hazardous for the kidney than heavy and prolonged use of over-the-counter analgesics in a social context (e.g. Swiss workers manufacturing watches would hand round analgesics in the same way as sharing sweets or cigarettes!).
Cardiovascular side effects
While it had been recognised for some time that NSAIDs could oppose the effects of some antihypertensive drugs, there is currently fresh concern about the potential of these drugs, when given alone, to raise blood pressure, and therefore predispose to adverse cardiovascular events such as stroke and myocardial infarction.
This first arose during trials of the COX-2 inhibitor rofecoxib. Uncertainty about the cardiovascular risk posed by this drug during clinical trials led to the addition of a ‘warning label’ in 2002, but the results from a later long-term trial designed to assess the anticancer activity of rofecoxib showed that the risk of cardiovascular events increased significantly after 18 months of drug treatment. As a result of this, the drug was withdrawn in 2004.
It now seems that adverse cardiovascular pharmacology, especially following prolonged use or in patients with pre-existing cardiovascular risk, may be an effect common to all NSAIDs, although some (e.g. naproxen) appear to be better tolerated in this respect than others (see Ray et al., 2009). At the time of writing it seems that the most likely explanation for this effect is that the hypertension is secondary to inhibition of COX-2 in the renin-secreting macula densa region of the kidney (Ch. 28). The hypertensive effect is dose- and time-dependent.
General unwanted effects of cyclo-oxygenase inhibitors
Unwanted effects, many stemming from inhibition of the constitutive housekeeping enzyme cyclo-oxygenase (COX)-1 isoform of COX, are common, particularly in the elderly, and include the following:
Other unwanted effects
Approximately 5% of patients exposed to NSAIDs may experience aspirin-sensitive asthma. The exact mechanism is unknown, but inhibition of COX is implicated (see Ch. 27) and the presence of a sensitising, pre-existing viral infection may be the culprit. Aspirin is the worst offender, but there is cross-reaction with all other NSAIDs, except possibly COX-2 inhibitors (see Ch. 27). Other, much less common, unwanted effects of NSAIDs include CNS effects, bone marrow disturbances and liver disorders, the last being more likely if there is already renal impairment.5 Paracetamol overdose causes liver failure (see below). All NSAIDs (except COX-2 inhibitors) prevent platelet aggregation and therefore may prolong bleeding. Again, aspirin is the main problem in this regard (see below).
Some Important Nsaids and Coxibs
Table 26.1 lists commonly used NSAIDs, and the clinical uses of the NSAIDs are summarised in the clinical box.
Clinical uses of NSAIDs 
NSAIDs are widely used but cause serious adverse effects (especially gastrointestinal, renal, pulmonary and cardiovascular effects related to their main pharmacological actions, as well as idiosyncratic effects). Elderly patients and those with pre-existing disorders are at particular risk. The main uses are:
Aspirin
Aspirin (acetylsalicylic acid) was among the earliest drugs synthesised, and is still one of the most commonly consumed drugs worldwide. It is a common ingredient in many over-the-counter proprietary medicines. The drug itself is relatively insoluble, but its sodium and calcium salts are readily soluble.
While aspirin was previously thought of as an old anti-inflammatory workhorse, it is seldom used for this purpose now, having been supplanted by other, better tolerated NSAIDs. Today, in addition to its widespread use as an over-the-counter remedy, it is used clinically mainly as a cardiovascular drug because of its ability to provide a prolonged inhibition of platelet COX-1 and hence reduce aggregation.
While inhibition of platelet function is a feature of most NSAIDs, the effect of aspirin is longer lasting. This is because it irreversibly acetylates COX enzymes, and while these proteins can be replaced in most cells, the platelet is not able to accomplish de novo protein synthesis. This means that a small dose of the drug can permanently inactivate platelets for their lifetime (approximately 10 days). Since a proportion of platelets is replaced each day from the bone marrow, this inhibition gradually abates but a small daily dose (e.g. 75 mg) is all that is required to suppress platelet function to levels which benefit patients at risk for myocardial infarction and other cardiovascular problems (Ch. 24). The view that even patients not at risk would benefit from taking the drug prophylactically (primary prevention) was challenged by a recent meta-analysis (Baigent et al., 2009) suggesting that in the normal population, the risk from gastrointestinal bleeding outweighs the protective action. Whether or not this is the case, the use of aspirin to prevent recurrence (secondary prevention) seems unassailable.
Aspirin has also been canvassed for other conditions. These include:
Pharmacokinetic aspects
Aspirin, being a weak acid, is protonated in the acid environment of the stomach, thus facilitating its passage across the mucosa. Most absorption, however, occurs in the ileum, because of the extensive surface area of the microvilli.
Aspirin is rapidly (probably within 30 min) hydrolysed by esterases in plasma and tissues, particularly the liver, yielding salicylate. This compound itself has anti-inflammatory actions (indeed, it was the original anti-inflammatory from which aspirin was derived); the mechanism is not clearly understood, although it probably involves the COX system. Oral salicylate is no longer used for treating inflammation, although it is a component of some topical preparations. Approximately 25% of the salicylate is oxidised; some is conjugated to give the glucuronide or sulfate before excretion, and about 25% is excreted unchanged, the rate of excretion being higher in alkaline urine (see Ch. 8).
The plasma half-life of aspirin will depend on the dose, but the duration of action is not directly related to the plasma half-life because of the irreversible nature of the action of the acetylation reaction by which it inhibits COX activity.
Aspirin
Aspirin (acetylsalicylic acid) is the oldest non-steroidal anti-inflammatory drug. It acts by irreversibly inactivating both cyclo-oxygenase (COX)-1 and COX-2.
Unwanted effects
Salicylates (of which the main examples are aspirin, diflunisal and sulfasazine) may produce both local and systemic toxic effects. Aspirin shares many of the general unwanted effects of NSAIDs outlined above. In addition, there are certain specific unwanted effects that occur with aspirin and other salicylates.
Acute salicylate poisoning (a medical emergency, which occurs mainly in children and suicide attempts) causes major disturbance of acid–base and electrolyte balance. These drugs can uncouple oxidative phosphorylation (mainly in skeletal muscle), leading to increased oxygen consumption and thus increased production of carbon dioxide. This stimulates respiration, which is also stimulated by a direct action of the drugs on the respiratory centre. The resulting hyperventilation causes a respiratory alkalosis that is normally compensated by renal mechanisms involving increased bicarbonate excretion. Larger doses can cause a depression of the respiratory centre, which leads eventually to retention of carbon dioxide and thus an increase in plasma carbon dioxide. Because this is superimposed on a reduction in plasma bicarbonate, an uncompensated respiratory acidosis will occur. This may be complicated by a metabolic acidosis, which results from the accumulation of metabolites of pyruvic, lactic and acetoacetic acids (an indirect consequence of interference with carbohydrate metabolism). Hyperpyrexia secondary to the increased metabolic rate is also likely to be present, and dehydration may follow repeated vomiting. In the CNS, initial stimulation with excitement is followed eventually by coma and respiratory depression. Bleeding can also occur, mainly as a result of depressed platelet aggregation.
Drug interactions
Aspirin may cause a potentially hazardous increase in the effect of warfarin, partly by displacing it from plasma proteins (Ch. 56) and partly because its effect on platelets interferes with haemostatic mechanisms (see Ch. 24). Aspirin also interferes with the effect of some antihypertensives and with uricosuric agents such as probenecid and sulfinpyrazone. Because low doses of aspirin may, on their own, reduce urate excretion (Ch. 28), it should not be used in gout.
Paracetamol
Paracetamol (called acetaminophen in the USA) is one of the most commonly used non-narcotic analgesic–antipyretic agents and is a component of many over-the-counter proprietary preparations. In some ways, the drug constitutes an anomaly: while it has excellent analgesic and antipyretic activity, which can be traced to inhibition of CNS prostaglandin synthesis, it has weak anti-inflammatory activity (except in some specific instances) and does not share the gastric or platelet side effects of the other NSAIDs. For this reason, paracetamol is sometimes not classified as an NSAID at all. In man however, it is a selective though weak COX-2 inhibitor (Hinz et al 2008).
A potential solution to this puzzle was supplied by the observation that a further COX isoform, COX-3 (an alternate splice product of COX-1) existed predominantly in the CNS of some species, and that paracetamol, as well as some other drugs with similar properties (e.g. antipyrine and dipyrone), were selective inhibitors of this enzyme (Chandrasekharan et al., 2002). This elegant idea is still under investigation. Alternative explanations for the ability of paracetamol selectively to inhibit COX in the CNS alone have been provided by Ouellet & Percival (2001) and Boutaud et al. (2002).
Paracetamol
Paracetamol is a commonly used drug available over-the-counter. It has potent analgesic and antipyretic actions but rather weaker anti-inflammatory effects than other NSAIDs. It may act through inhibition of a central nervous system-specific cyclo-oxygenase (COX) isoform, although this is not yet conclusive.
Pharmacokinetic aspects
Paracetamol is given orally and is well absorbed, with peak plasma concentrations reached in 30–60 min. The plasma half-life of therapeutic doses is 2–4 h, but with toxic doses it may be extended to 4–8 h. Paracetamol is inactivated in the liver, being conjugated to give the glucuronide or sulfate.
Unwanted effects
With therapeutic doses, side effects are few and uncommon, although allergic skin reactions sometimes occur. It is possible that regular intake of large doses over a long period may cause kidney damage.
Toxic doses (10–15 g) cause potentially fatal hepatotoxicity. This occurs when the liver enzymes catalysing the normal conjugation reactions are saturated, causing the drug to be metabolised instead by mixed function oxidases. The resulting toxic metabolite, N-acetyl-p-benzoquinone imine, is inactivated by conjugation with glutathione, but when glutathione is depleted the toxic intermediate accumulates and causes necrosis in the liver and also in the kidney tubules.
The initial symptoms of acute paracetamol poisoning are nausea and vomiting, the hepatotoxicity being a delayed manifestation that occurs 24–48 h later. Further details of the toxic effects of paracetamol are given in Chapter 57. If the patient is seen sufficiently soon after ingestion, the liver damage can be prevented by giving agents that increase glutathione formation in the liver (acetylcysteine intravenously, or methionine orally). If more than 12 h have passed since the ingestion of a large dose, the antidotes, which themselves can cause adverse effects (nausea, allergic reactions), are less likely to be useful. Regrettably, ingestion of large amounts of paracetamol is a common method of suicide.
Coxibs
Three coxibs are currently available for clinical use in the UK; others may be available elsewhere. Several have been withdrawn, and the overall licensing situation is somewhat volatile. Current advice restricts the use of coxibs to patients for whom treatment with conventional NSAIDs would pose a high probability of serious gastrointestinal side effects, and they are prescribed only after an assessment of cardiovascular risk. Gastrointestinal disturbances may still occur with these agents, perhaps because COX-2 has been implicated in the healing of pre-existing ulcers, so inhibition could delay recovery from earlier lesions.
Celecoxib and etoricoxib
Celecoxib and etoricoxib are licensed in the UK for symptomatic relief in the treatment of osteoarthritis and rheumatoid arthritis and some other conditions. Both are administered orally and have similar pharmacokinetic profiles, being well absorbed with peak plasma concentrations being achieved within 1–3 h. They are extensively (> 99%) metabolised in the liver, and plasma protein binding is high (> 90%).
Common unwanted effects may include headache, dizziness, skin rashes and peripheral oedema caused by fluid retention. Consideration should be given to the possibility of adverse cardiovascular events prior to administration. Because of the potential role of COX-2 in the healing of ulcers, patients with pre-existing disease should avoid the drugs, if possible.
Parecoxib
Parecoxib is a prodrug of valdecoxib. The latter drug has now been withdrawn, but parecoxib is licensed for the short-term treatment of postoperative pain. It is given by intravenous or intramuscular injection, and is rapidly and virtually completely (> 95%) converted into the active valdecoxib by enzymatic hydrolysis in the liver. Maximum blood levels are achieved within approximately 30–60 min, depending on the route of administration. Plasma protein binding is high. The active metabolite, valdecoxib, is converted in the liver to various inactive metabolites, and has a plasma half-life of about 8 h.
Skin reactions, some of them serious, have been reported with the active metabolite valdecoxib, and patients should be monitored carefully. The drug should also be given with caution to patients with impaired renal function, and renal failure has been reported in connection with this drug. Postoperative anaemia may also occur.
Antirheumatoid Drugs
Arthritic disease is one of the commonest chronic inflammatory conditions in developed countries, and rheumatoid arthritis is a common cause of disability. One in three patients with rheumatoid arthritis is likely to become severely disabled. The joint changes, which are probably driven by an autoimmune reaction, involve inflammation, proliferation of the synovium and erosion of cartilage and bone. The primary inflammatory cytokines, IL-1 and TNF-α, have a major role in pathogenesis (Ch. 17). The pathogenesis of rheumatoid arthritis, and the action of therapeutic drugs, are summarised in Figure 26.4.
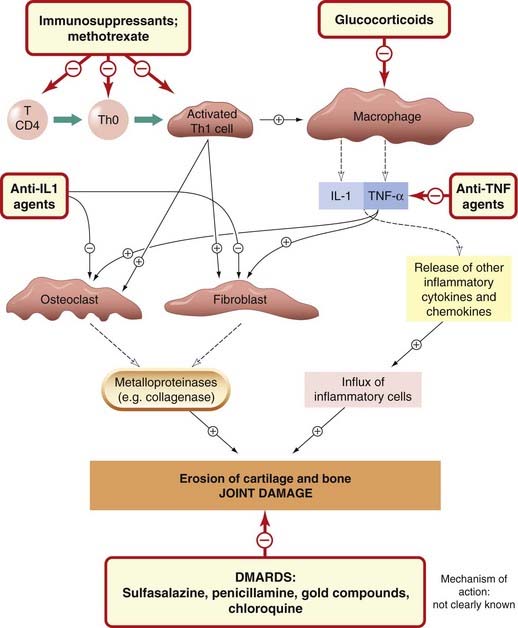
Fig. 26.4 A schematic diagram of the cells and mediators involved in the pathogenesis of rheumatoid joint damage, indicating the sites of action of antirheumatoid drugs.
DMARD, disease-modifying antirheumatic drug. For details of the anti-TNF, IL-1 and IL-2 receptor agents, see Table 26.3
The drugs most frequently used in initial therapy are the ‘disease-modifying antirheumatic drugs’ (DMARDs) and the NSAIDs. Unlike the NSAIDs, which reduce the symptoms but not the progress of the disease, the former group may halt or reverse the underlying disease itself. Although such claims may be optimistic, these drugs are nevertheless useful in the treatment of discrete groups of patients, and Rau (2005) has argued for their continuing use even when the newer anticytokine agents are available. Some immunosuppressants (e.g. azathioprine, ciclosporin) are also used, as are the glucocorticoids (covered in Chs 3 and 32).
Disease-Modifying Antirheumatic Drugs
The term ‘DMARD’ is a latex concept that can be stretched to cover a heterologous group of agents with unrelated chemical structures and different mechanisms of action. Included in this category are methotrexate, sulfasalazine, gold compounds, penicillamine and chloroquine and other antimalarials (see Table 26.2) and various immunosuppressant drugs.
Table 26.2 Comparison of some common ‘disease-modifying’ and immunosuppressive drugs used in the treatment of the arthritides
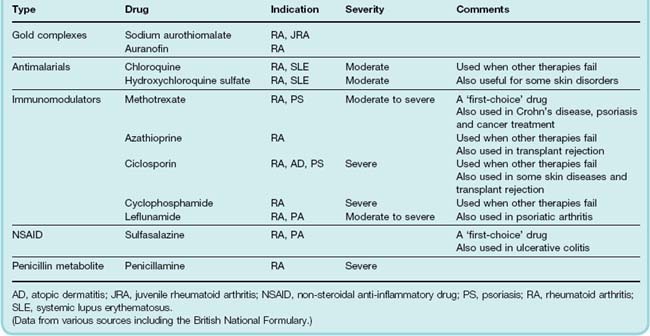
The antirheumatoid action of most of these agents was usually discovered through a mixture of serendipity and clinical intuition. When the drugs were introduced, nothing was known about their mechanism of action and decades of in vitro experiments have generally resulted in further bewilderment rather than understanding. DMARDs generally improve symptoms and can reduce disease activity in rheumatoid arthritis, as measured by reduction in the number of swollen and tender joints, pain score, disability score, X-ray appearance and serum concentration of acute-phase proteins and of rheumatoid factor (an immunoglobulin [Ig] M antibody against host IgG).
The DMARDs were often referred to as second-line drugs, with the implication that they are only resorted to when other therapies (e.g. NSAIDs) failed. Today, however, DMARD therapy may be initiated as soon as a definite diagnosis has been reached. Their clinical effects are usually slow (months) in onset, and it is usual to provide NSAID ‘cover’ during this induction phase. If therapy is successful (and the success rate is not invariably high), concomitant NSAID (or glucocorticoid) therapy can generally be dramatically reduced. Some DMARDs have a place in the treatment of other chronic inflammatory diseases, whereas others (e.g. penicillamine) are not thought to have a general anti-inflammatory action. Putative mechanisms of action of DMARDs have been reviewed by Bondeson (1997) and Cutolo (2002).
Methotrexate
Methotrexate is a folic acid antagonist with cytotoxic and immunosuppressant activity (see below and Chs 49 and 55) and potent antirheumatoid action. It is a common first-choice drug. It has a more rapid onset of action than other DMARDs, but treatment must be closely monitored because of potential blood dyscrasias (some fatal) and liver cirrhosis. It is, however, superior to most other DMARDs in terms of efficacy and unwanted effects, and is often given in conjunction with the anticytokine drugs.
Sulfasalazine
Sulfasalazine, a common first-choice DMARD in the UK, produces remission in active rheumatoid arthritis and is also used for chronic inflammatory bowel disease (see Ch. 29). It may act by scavenging the toxic oxygen metabolites produced by neutrophils. The drug is a complex of a sulfonamide (sulfapyridine) and salicylate. It is split into its component parts by bacteria in the colon, the 5-aminosalicylic acid being the putative radical scavenger. It is poorly absorbed after oral administration. The common side effects include gastrointestinal disturbances, malaise and headache. Skin reactions and leucopenia can occur but are reversible on stopping the drug. The absorption of folic acid is sometimes impaired; this can be countered by giving folic acid supplements. A reversible decrease in sperm count has also been reported. As with other sulfonamides, bone marrow depression and anaphylactic-type reactions may occur in a few patients. Hematological monitoring may be necessary.
Penicillamine
Penicillamine is dimethylcysteine; it is produced by hydrolysis of penicillin and appears in the urine after treatment with that drug. The D-isomer is used in the therapy of rheumatoid disease. About 75% of patients with rheumatoid arthritis respond to penicillamine. In responders, therapeutic effects are seen within weeks but do not reach a plateau for several months. Penicillamine is thought to modify rheumatoid disease partly by decreasing the immune response, IL-1 generation and/or partly by an effect on collagen synthesis, preventing the maturation of newly synthesised collagen. However, the precise mechanism of action is still a matter of conjecture. The drug has a highly reactive thiol group and also has metal-chelating properties, which are put to good use in the treatment of Wilson’s disease (pathological copper deposition causing neurodegeneration) or heavy metal poisoning.
Penicillamine is given orally, and only half the dose administered is absorbed. It reaches peak plasma concentrations in 1–2 h and is excreted in the urine. Dosage is started low and increased only gradually to minimise unwanted effects.
Unwanted effects occur in about 40% of patients treated and may necessitate cessation of therapy. Rashes and stomatitis are the most common unwanted effects but may resolve if the dosage is lowered. Anorexia, fever, nausea and vomiting, and disturbances of taste (the last related to the chelation of zinc) are seen, but often disappear with continued treatment. Proteinuria occurs in 20% of patients and should be monitored. Hematological monitoring is also required when treatment is initiated. Thrombocytopenia may require lowering the dose. Leucopenia or aplastic anaemia are absolute contraindications, as are the various autoimmune conditions (e.g. thyroiditis, myasthenia gravis) that sometimes supervene. Because penicillamine is a metal chelator, it should not be given with gold compounds.
Gold Compounds
Gold is administered in the form of organic complexes; sodium aurothiomalate and auranofin are the two most common preparations. The effect of gold compounds develops slowly over 3–4 months. Pain and joint swelling subside, and the progression of bone and joint damage diminishes. The mechanism of action is not clear, but auranofin, although not aurothiomalate, inhibits the induction of IL-1 and TNF-α.
Sodium aurothiomalate is given by deep intramuscular injection; auranofin is given orally. The compounds gradually become concentrated in the tissues, not only in synovial cells in joints but also in liver cells, kidney tubules, the adrenal cortex and macrophages throughout the body. The gold complexes remain in the tissues for some time after treatment is stopped. Excretion is mostly renal, but some is eliminated in the gastrointestinal tract. The half-life is 7 days initially but increases with treatment, so the drug is usually given first at weekly, then at monthly intervals.
Unwanted effects with aurothiomalate are seen in about one-third of patients treated, and serious toxic effects in about 1 patient in 10. Unwanted effects with auranofin are less frequent and less severe. Important unwanted effects include skin rashes (which can be severe), mouth ulcers, non-specific flu-like symptoms, proteinuria, thrombocytopenia and blood dyscrasias. Encephalopathy, peripheral neuropathy and hepatitis can occur. If therapy is stopped when the early symptoms appear, the incidence of serious toxic effects is relatively low.
Antimalarial Drugs
Hydroxychloroquine and chloroquine are 4-amino-quinoline drugs used mainly in the prevention and treatment of malaria (Ch. 53), but they are also used as DMARDs. Chloroquine is usually reserved for cases where other treatments have failed. They are also used to treat another autoimmune disease, lupus erythematosus, but are contraindicated in patients with psoriatic arthropathy because they make the skin lesions worse. The related compound, mepacrine, is also sometimes used for discoid lupus. The antirheumatic effects do not appear until a month or more after the drug is started, and only about half the patients treated respond. The pharmacokinetic aspects and unwanted effects of chloroquine are dealt with in Ch. 53; screening for ocular toxicity is particularly important.
Immunosuppressant Drugs
Immunosuppressants are used in the therapy of autoimmune disease and also to prevent and/or treat transplant rejection. Because they impair immune responses, they carry the hazard of a decreased response to infections and may facilitate the emergence of malignant cell lines. However, the relationship between these adverse effects and potency in preventing graft rejection varies with different drugs. The clinical use of immunosuppresants is summarised in the clinical box.
Clinical uses of immunosuppressant drugs
Immunosuppressant drugs are used by specialists, often in combination with glucorticoid and/or cytotoxic drugs:
Immunosuppressants
Most of these drugs act during the induction phase of the immunological response (see Ch. 6), reducing lymphocyte proliferation, although others also inhibit aspects of the effector phase. They can be roughly characterised as:
Ciclosporin
Ciclosporin is a naturally occurring compound first found in fungus. It is a cyclic peptide of 11 amino acid residues (including some not found in animals) with potent immunosuppressive activity but no effect on the acute inflammatory reaction per se. Its unusual activity, which (unlike most earlier immunosuppressants) does not involve cytotoxicity, was discovered in 1972 and was crucial for the development of transplant surgery (for a detailed review, see Borel et al., 1996). The drug has numerous actions on several cell types; in general, the actions of relevance to immunosuppression are:
The main action is a relatively selective inhibitory effect on IL-2 gene transcription, although a similar effect on interferon (IFN)-γ and IL-3 has also been reported. Normally, interaction of antigen with a T-helper (Th) cell receptor results in increased intracellular Ca2+ (Chs 2 and 6), which in turn stimulates a phosphatase, calcineurin. This activates various transcription factors that initiate IL-2 transcription. Ciclosporin binds to cyclophilin, a cytosolic protein member of the immunophilins (a group of proteins that act as intracellular receptors for such drugs). The drug–immunophilin complex binds to and inhibits calcineurin (a protein phosphatase that acts in opposition to the many protein kinases involved in signal transduction, see Ch. 3), thereby preventing activation of Th cells and production of IL-2 (Ch. 6).
Ciclosporin itself is poorly absorbed by mouth but can be given orally in a more readily absorbed formulation, or given by intravenous infusion. After oral administration, peak plasma concentrations are usually attained in about 3–4 h. The plasma half-life is approximately 24 h. Metabolism occurs in the liver, and most of the metabolites are excreted in the bile. Ciclosporin accumulates in most tissues at concentrations three to four times that seen in the plasma. Some of the drug remains in lymphomyeloid tissue and remains in fat depots for some time after administration has stopped.
The commonest and most serious unwanted effect of ciclosporin is nephrotoxicity, which is thought to be unconnected with calcineurin inhibition. It may be a limiting factor in the use of the drug in some patients (see also Ch. 57). Hepatotoxicity and hypertension can also occur. Less important unwanted effects include anorexia, lethargy, hirsutism, tremor, paraesthesia (tingling sensation), gum hypertrophy (especially when co-prescribed with calcium antagonists for hypertension; Ch. 22) and gastrointestinal disturbances. Ciclosporin has no depressant effects on the bone marrow.
Tacrolimus
Tacrolimus is a macrolide antibiotic of fungal origin with a very similar mechanism of action to ciclosporin, but higher potency. The main difference is that the internal receptor for this drug is not cyclophilin but a different immunophilin termed FKBP (FK-binding protein, so-called because tacrolimus was initially termed FK506). The tacrolimus–FKBP complex inhibits calcineurin with the effects described above. It is mainly used in organ transplantation and severe atopic eczema. Pimecrolimus (used topically for atopic eczema) acts in a similar way. Sirolimus (used to prevent organ rejection after transplantation, and also in coating on stents to prevent restenosis; Ch. 22) also combines with an immunophilin, but activates a protein kinase to produce its immunosuppressant effect.
Tacrolimus can be given orally, by intravenous injection or as an ointment for topical use in inflammatory disease of the skin. It is 99% metabolised by the liver and has a half-life of approximately 7 h.
The unwanted effects of tacrolimus are similar to those of ciclosporin but are more severe. The incidence of nephrotoxicity and neurotoxicity is higher, but that of hirsutism is lower. Gastrointestinal disturbances and metabolic disturbances (hyperglycaemia) can occur. Thrombocytopenia and hyperlipidaemia have been reported but decrease when the dosage is reduced.
Azathioprine
Azathioprine interferes with purine synthesis and is cytotoxic. It is widely used for immunosuppression, particularly for control of autoimmune diseases such as rheumatoid arthritis and to prevent tissue rejection in transplant surgery. This drug is metabolised to give mercaptopurine, a purine analogue that inhibits DNA synthesis (see Ch. 55). Both cell-mediated and antibody-mediated immune reactions are depressed by this drug, because it inhibits clonal proliferation during the induction phase of the immune response (see Ch. 6) through a cytotoxic action on dividing cells. As is the case with mercaptopurine itself, the main unwanted effect is depression of the bone marrow. Other toxic effects are nausea and vomiting, skin eruptions and a mild hepatotoxicity.
Mycophenolate Mofetil
Mycophenolate mofetil is a semisynthetic derivative of a fungal antibiotic used for preventing organ rejection. In the body, it is converted to mycophenolic acid, which restrains proliferation of both T and B lymphocytes and reduces the production of cytotoxic T cells by inhibiting inosine monophosphate dehydrogenase, an enzyme crucial for de novo purine biosynthesis in both T and B cells (other cells can generate purines through another pathway), so the drug has a fairly selective action. It is mainly used to curtail transplant rejection.
Mycophenolate mofetil is given orally and is well absorbed. Magnesium and aluminium hydroxides impair absorption, and colestyramine reduces plasma concentrations. The metabolite mycophenolic acid undergoes enterohepatic cycling and is eliminated by the kidney as the inactive glucuronide. Unwanted gastrointestinal effects are common.
Leflunomide
Leflunomide has a relatively specific inhibitory effect on activated T cells. It is transformed to a metabolite that inhibits de novo synthesis of pyrimidines by inhibiting dihydro-orotate dehydrogenase. It is orally active and well absorbed from the gastrointestinal tract. It has a long plasma half-life, and the active metabolite undergoes enterohepatic circulation. Unwanted effects include diarrhoea, alopecia, raised liver enzymes and indeed a risk of hepatic failure. The long half-life increases the risk of cumulative toxicity.
Glucocorticoids
Immunosuppression by glucocorticoids involves both their effects on the immune response and their anti-inflammatory actions. These are described in Chapter 32, and the sites of action of the agents on cell-mediated immune reactions are indicated in Figure 26.4. Glucocorticoids are immunosuppressant chiefly because, like ciclosporin, they restrain the clonal proliferation of Th cells, through decreasing transcription of the gene for IL-2. However, they also decrease the transcription of many other cytokine genes (including those for TNF-α, IFN-γ, IL-1 and many other interleukins) in both the induction and effector phases of the immune response. The synthesis and release of anti-inflammatory proteins (e.g. annexin 1, protease inhibitors, etc.) is also increased. These effects on transcription are mediated through inhibition of the action of transcription factors, such as activator protein-1 and NFκB.
Anticytokine Drugs and Other Biopharmaceuticals
The drugs in this section probably represent the greatest technological and conceptual breakthrough in the treatment of severe chronic inflammation for decades (see Maini, 2005). By their use, treatment can, for the first time, be targeted at specific aspects of the disease processes in rheumatoid arthritis and other inflammatory diseases. The drugs are biopharmaceuticals, that is to say, they are engineered recombinant antibodies and other proteins (see Ch. 59). As such, they are difficult and expensive to produce, and this limits their use. In the UK, their use (in the National Health Service) is generally restricted to patients who do not respond adequately to other DMARD therapy and they are usually provided under specialist supervision only. Some of these drugs are administered in combination with methotrexate.
The drugs currently available, and some of their characteristics and indications, are shown in Table 26.3. Adalimumab, etanercept and infliximab target TNF-α; anakinra targets IL-1. Rituximab, abatacept, natalizumab and efalizumab target receptors on leukocytes, disrupting immune signalling or cell trafficking or other functions. While they are not used for treating arthritis, basiliximab and daclizumab are included in the table as they act to prevent the rejection of transplanted organs in a similar way—by blocking the IL-2 receptor and suppressing T cell proliferation.
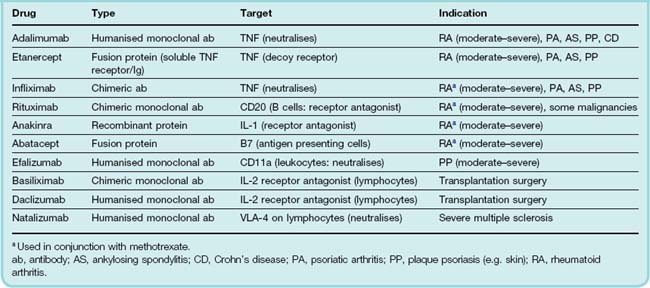
There is debate over the precise target of the anti-TNF agents. Some target both soluble and membrane-bound forms of TNF whereas others are more selective. Antibodies that target membrane-bound TNF (infliximab and adalimumab) may kill the host cell by complement-induced lysis. This produces a different quality of effect than simple immunoneutralisation of the soluble mediator (by, for example, etanercept). This fact is probably the reason why some of these drugs exhibit a slightly different pharmacological profile despite apparently acting through the same mechanism (see Arora et al., 2009, for further details).
As proteins, none of these drugs can be given orally. Administration is usually by subcutaneous injection or intravenous infusion and their pharmacokinetic profiles vary enormously. Dosing regimes differ but anakinra is usually given daily, efalizumab and etanercept once or twice per week, adalimumab, infliximab and rituximab every 2 weeks, and abatacept and natalizumab every month. Sometimes a loading dose of these drugs is given as a preliminary to regular administration. Some patients to not respond for reasons that are not entirely clear and therapy is generally discontinued if no therapeutic benefit is evident within a defined time span (usually 2–4 weeks).
Cytokines are crucial to the regulation of host defence systems (see Ch. 17), and leukocytes are key players in its functioning and execution. One might predict, therefore, that anticytokine or antileukocyte therapy—like any treatment that interferes with immune function—may precipitate latent disease (e.g. tuberculosis and hepatitis B) or encourage opportunistic infections. Reports suggest that this may be a problem with adalimumab, etanercept, infliximab, natalizumab and rituximab. The area has been reviewed by Bongartz et al. (2006). Another unexpected, but fortunately rare, effect seen with these drugs is the onset of psoriasis-like syndrome (Fiorino et al., 2009). Hypersensitivity, injection site reactions or mild gastrointestinal symptoms may be seen with any of these drugs.
Drugs Used in Gout
Gout is a metabolic disease in which plasma urate concentration is raised. Sometimes this is linked to overindulgence in alcoholic beverages, especially beer, or purine-rich foods such as offal. Increased cell turnover in haematological malignancies, particularly after treatment with cytotoxic drugs (see Ch. 55), or impaired excretion of uric acid are other causes. It is characterised by very painful intermittent attacks of acute arthritis produced by the deposition of crystals of sodium urate (a product of purine metabolism) in the synovial tissue of joints and elsewhere. An inflammatory response is evoked, involving activation of the kinin, complement and plasmin systems (see Ch. 17 and Fig. 6.1), generation of lipoxygenase products such as leukotriene B4 (Fig. 17.1), and local accumulation of neutrophil granulocytes. These engulf the crystals by phagocytosis, releasing tissue-damaging toxic oxygen metabolites and subsequently causing lysis of the cells with release of proteolytic enzymes. Urate crystals also induce the production of IL-1 and possibly other cytokines.
Drugs used to treat gout act in the following ways:
Their clinical uses are summarised in the clinical box, below.
Drugs used in gout and hyperuricaemia
Allopurinol
Allopurinol is an analogue of hypoxanthine that reduces the synthesis of uric acid by competitive inhibition of xanthine oxidase (Fig. 26.5). It is first converted to alloxanthine by xanthine oxidase, and this metabolite, which remains in the tissue for a considerable time, is an effective non-competitive inhibitor of the enzyme. Some inhibition of de novo purine synthesis also occurs.
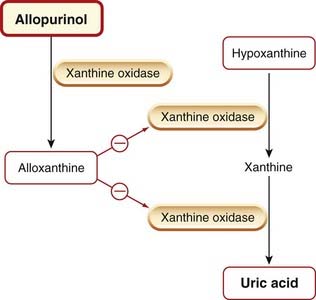
Allopurinol reduces the concentration of the relatively insoluble urates and uric acid in tissues, plasma and urine, while increasing the concentration of their more soluble precursors, the xanthines and hypoxanthines. The deposition of urate crystals in tissues (tophi) is reversed, and the formation of renal stones is inhibited. Allopurinol is the drug of choice in the long-term treatment of gout, but it is ineffective in the treatment of an acute attack and may even exacerbate the inflammation.
Allopurinol is given orally and is well absorbed. Its half-life is 2–3 h: its active metabolite alloxanthine (Fig. 26.5) has a half-life of 18–30 h. Renal excretion is a balance between glomerular filtration and probenecid-sensitive tubular reabsorption.
Unwanted effects are few. Gastrointestinal disturbances, allergic reactions (mainly rashes) and some blood problems can occur but usually disappear if the drug is stopped. Potentially fatal skin diseases such as Stevens–Johnson syndrome are rare—but devastating. Re-challenge under these circumstances is never justified. Acute attacks of gout occur commonly during the early stages of therapy (possibly as a result of physicochemical changes in the surfaces of urate crystals as these start to re-dissolve), so treatment with allopurinol is never initiated during an acute attack and is usually combined with an NSAID initially.
Allopurinol increases the effect of mercaptopurine, an antimetabolite used in cancer chemotherapy (Ch. 55), and also that of azathioprine (an immunosuppressant used to prevent transplant rejection; see below), which is metabolised to mercaptopurine. Allopurinol also enhances the effect of another anticancer drug, cyclophosphamide (Ch. 55). The effect of warfarin is increased because its metabolism is inhibited.
Uricosuric Agents
Uricosuric drugs increase uric acid excretion by a direct action on the renal tubule (see Ch. 28). Common drugs used are probenecid and sulfinpyrazone. Benzbromarone is also available on a named patient basis for treatment of patients with renal impairment. They remain useful as prophylaxis for patients with severe recurrent gout who have severe adverse reactions to allopurinol. Sulfinpyrazone also has NSAID activity. Treatment with uricosuric drugs is initiated with an NSAID, as for allopurinol. Aspirin and salicylates antagonise the action of uricosuric drugs and should not be used concurrently.
Although not strictly speaking in this group, rasburicase, a preparation containing the enzyme uric acid oxidase, is sometimes used for aggressive treatment. It oxidises uric acid in the blood to allantoin, which is more soluble and thus more readily excreted.
Colchicine
Colchicine is an alkaloid extracted from the autumn crocus. It has a specific effect in gouty arthritis and can be used both to prevent and to relieve acute attacks. It prevents migration of neutrophils into the joint by binding to tubulin, resulting in the depolymerisation of the microtubules and reduced cell motility. Colchicine-treated neutrophils develop a ‘drunken walk’. Colchicine may also prevent the production of a putative inflammatory glycoprotein by neutrophils that have phagocytosed urate crystals, and other mechanisms may also be important in bringing about its effects.
Colchicine is given orally, and is excreted partly in the gastrointestinal tract and partly in the urine.
The acute unwanted effects of colchicine are largely gastrointestinal and include nausea, vomiting and abdominal pain. Severe diarrhoea6 may be a problem, and with large doses may be associated with gastrointestinal haemorrhage and kidney damage. Prolonged treatment can, rarely, cause blood dyscrasias, rashes or peripheral neuropathy.
Antagonists of Histamine
There are three groups: H1, H2 and H3 receptor antagonists. The first group was introduced by Bovet and his colleagues in the 1930s, at a time when histamine receptors had not been classified (indeed, this was possible only because these agents were available). For historical reasons, then, the generic term antihistamine conventionally refers only to the H1 receptor antagonists that are used for treating various inflammatory and allergic conditions, and it is these drugs that are discussed in this section. The main clinical effect of H2 receptor antagonists is inhibition of gastric secretion (see Ch. 29). Several H3 receptor agonists and antagonists are now available, and the potential for their clinical use (mainly in CNS conditions) is being explored.
H1 Receptor Antagonists (Antihistamines)
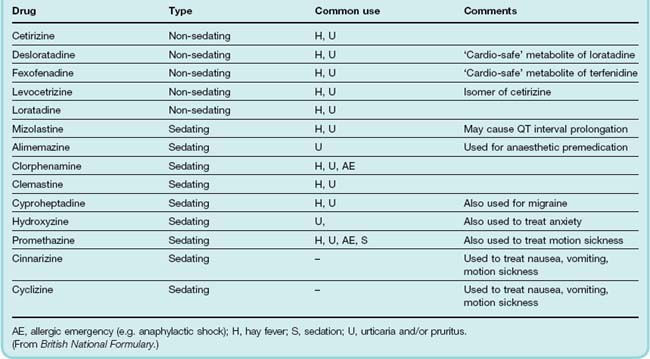
Details of some typical systemic H1 receptor antagonists are shown in Table 26.4. In addition to these there are several others that are primarily used topically (e.g. in nasal sprays or eye drops) in the treatment of hay fever and other allergic symptoms. These include antazoline, azelastine, epinastine, ketotifen, olapatadine and emadastine. In addition to H1 antagonist activities, some of these drugs (e.g. ketotifen) may also have ‘mast cell stabilising’ and other anti-inflammatory properties unrelated to histamine antagonism (see Assanasen & Naclerio, 2002).
Pharmacological actions
Conventionally, the antihistamines are divided into ‘first-generation’ drugs, that cross the blood–brain barrier and have sedating actions, and ‘second-generation’ drugs, which do not. Some second-generation agents (e.g. terfenadine) exhibited some cardiac toxicity (torsade de pointes, see Ch. 21). While the risk was extremely low, it was increased when the drug was taken with grapefruit juice or with agents that inhibit cytochrome P450 in the liver (see Chs 9 and 56). These drugs were withdrawn and replaced by ‘third-generation’ ‘cardio-safe’ drugs (often active metabolites of the original drugs, e.g. fexofenadine).
Pharmacologically, many of the actions of the H1 receptor antagonists follow from the actions of histamine outlined in Ch. 17. In vitro, for example, they decrease histamine-mediated contraction of the smooth muscle of the bronchi, the intestine and the uterus. They inhibit histamine-induced increases in vascular permeability and bronchospasm in the guinea pig in vivo, but are unfortunately of little value in allergic bronchospasm in humans. The clinical uses of H1 receptor antagonists are summarised in the clinical box, opposite.
Clinical uses of histamine H1 receptor antagonists
The CNS ‘side effects’ of some older H1 receptor antagonists are sometimes more clinically useful than the peripheral H1 antagonist effects. Some are fairly strong sedatives and may be used for this action (e.g. clorphenamine; see Table 26.4). Several are antiemetic and are used to prevent motion sickness (e.g. promethazine; see Ch. 29).
Several H1 receptor antagonists show weak blockade of α1-adrenoceptors (an example is the phenothiazine promethazine). Cyproheptadine is a 5-hydroxytryptamine antagonist as well as an H1 receptor antagonist.
Pharmacokinetic aspects
Most H1 receptor antagonists are well absorbed when given orally, and remain effective for 3–6 h, although there are exceptions. Most appear to be widely distributed throughout the body, but some do not penetrate the blood–brain barrier, for example the non-sedative drugs mentioned above (see Table 26.4). They are mainly metabolised in the liver and excreted in the urine.
When antihistamines are used to treat allergies, the sedative CNS effects are generally unwanted, but there are other occasions (e.g. in small children approaching bedtime) when such effects are more desirable. Even under these circumstances, other CNS effects, such as dizziness and fatigue, are unwelcome.
Many antihistamines have peripheral antimuscarinic side effects. The commonest of these is dryness of the mouth, but blurred vision, constipation and retention of urine can also occur. Unwanted effects that are not mechanism based are also seen; gastrointestinal disturbances are fairly common, while allergic dermatitis can follow topical application.
Possible Future Developments
Undoubtedly the most exciting area of current development is in ‘biologicals’ (see Ch. 59). The success of the anti-TNF agents has been very gratifying and the skilful use of recombinant and protein engineering to produce antibodies that neutralise inflammogens or block key leukocyte receptors or adhesion molecules is likely to continue. Particularly encouraging has been the news that early clinical results using rituximab and methotrexate in combination may actually abort the development of rheumatoid arthritis if given early enough. Several other biologics are in advanced state of clinical testing including toclizumab (anti-IL6), certilizumab-pegol (anti-TNF), golimumab (anti-TNF), ofatumumab (anti-CD 20) and ocrelizumab (anti-CD 20). The main problem with this sector is not the efficacy of the drugs but their cost and lack of oral availability, which places a severe strain on budgets and prevents them from being used as a first-line therapy. Hopefully, ways will be found to reduce the cost of production and development in this important technology.
Clearly a low-cost alternative to a neutralising anti-TNF antibody would be a welcome development. TNF converting enzyme (TACE) cleaves membrane-bound TNF thus releasing the soluble active form and so might be an attractive target. A number of putative small-molecule inhibitors of this enzyme are in phase II clinical trials and the results are awaited (see Moss et al., 2008, for a review).
A major blow to the NSAID area (and indeed to the pharmaceutical industry in general) has been the recent controversy surrounding the increased incidence of coronary thrombosis in patients taking COX-2 inhibitors and the withdrawal of some prominent members of this class for this and other reasons. The emerging evidence that ‘traditional’ NSAIDs may also have similar cardiovascular side effects has cast a pall over our existing therapies.7 At the time of writing, it is too early to say exactly how this awkward situation will be resolved (see Ray et al., 2009).
One of the few innovations in the beleaguered NSAID area has been the design and synthesis of nitric oxide (NO)-NSAIDs—conventional NSAIDs that have NO-donating groups attached to them by ester linkages. The ability of these drugs to release NO following hydrolysis in plasma and tissue fluid is aimed at reducing the risk of ulcerogenic events and increasing the anti-inflammatory activity, presumably due to the beneficial effects of low concentrations of NO (see Ch. 20). Some of these drugs (e.g. naproxcinod, a derivative of naproxen) are currently in clinical trial (see Stefano & Distrutti, 2007). Yedgar et al. (2007) discuss some alternative approaches to manipulating the production or action of eicosanoid mediators of inflammation.
References and Further Reading
Baigent C.L., Blackwell L., Collins R., et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849-1860. (An important study of the use of aspirin in the prevention of cardiovascular disease. Likely to be widely influential)
Bazan N.G. COX-2 as a multifunctional neuronal modulator. Nat. Med.. 2001;7:414-415. (Succinct treatment of possible role of COX-2 in the CNS; useful diagrams)
Boers M. NSAIDs and selective COX-2 inhibitors: competition between gastroprotection and cardioprotection. Lancet. 2001;357:1222-1223. (Editorial analysing crisply the results of two major randomised double-blind studies of gastrointestinal toxicity of selective COX-2 inhibitors as compared with non-selective NSAIDs)
Boutaud O., Aronoff D.M., Richardson J.H., et al. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H2 synthases. Proc. Natl. Acad. Sci. USA. 2002;99:7130-7135. (Proposes a solution to the paracetamol mystery: read together with Ouellet et al., 2001, below)
Chandrasekharan N.V., Dai H., Roos K.L., et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc. Natl. Acad. Sci. USA. 2002;99:13926-13931. (A new COX isozyme is described: COX-3. In humans, the COX-3 mRNA is expressed most abundantly in cerebral cortex and heart. It is selectively inhibited by analgesic/antipyretic drugs such as paracetamol and is inhibited by some other NSAIDs)
FitzGerald G.A., Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N. Engl. J. Med.. 2001;345:433-442. (Excellent coverage of the selective COX-2 inhibitors)
Flower R.J. The development of COX-2 inhibitors. Nat. Rev. Drug Discov.. 2003;2:179-191. (Reviews the work that led up to the development of the COX-2 inhibitors; several useful diagrams)
Fries J.F. Measuring the quality of life in relation to arthritis therapy. Postgrad. Med.. 1983;May:49-56.
Fries J.F. Quality-of-life considerations with respect to arthritis and nonsteroidal anti-inflammatory drugs. Am. J. Med.. 1998;104:14S-20S. discussion 21S–22S
Harris R.E., Beebe-Donk J., Doss H., Burr Doss D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: a critical review of non-selective COX-2 blockade. Oncol. Rep.. 2005;13:559-583. (This is an interesting paper that deals with the use of NSAIDs in the treatment of cancer; this is fast emerging as an alternative arena for NSAID therapy)
Hawkey C.J. Gastrointestinal toxicity of non-steroid anti-inflammatory drugs. In: Vane J.R., Botting R.M., editors. Therapeutic roles of selective COX-2 inhibitors. London: William Harvey Press; 2001:355-394. (Clear, detailed account of the adverse effects of NSAIDs)
Henry D., Lim L.L., Garcia Rodriguez L.A., et al. Variability in risk of gastrointestinal complications with individual non-steroidal anti-inflammatory drugs: results of a collaborative meta-analysis. BMJ. 1996;312:1563-1566. (Substantial analysis of the gastrointestinal effects of non-selective NSAIDs)
Hinz B., Cheremina O., Brune K. Acetaminophen (paracetamol) is a selective cyclooxgenase-2 inhibitor in man. FASEB J.. 2008;22:383-390.
Luong C., Miller A., Barnett J., et al. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol.. 1996;3:927-933. (An important research paper detailing the crystal structure of COX-2 and the relevance of this to NSAID and coxib action. Essential reading if you are seriously interested in this topic)
Ouellet M., Percival M.D. Mechanism of acetaminophen inhibition of cyclooxygenase isoforms. Arch. Biochem. Biophys.. 2001;387:273-280. (Proposes a solution to the paracetamol mystery: read together with Boutaud et al., 2002, above)
Ray W.A., Varas-Lorenzo C., Chung C.P., et al. Cardiovascular risks of non-steroidal anti-inflammatory drugs in patients after hospitalization for serious coronary heart disease. Circ. Cardiovasc. Qual. Outcomes. 2009;2:155-163. (This paper, together with an editorial on pages 146–147 of the same issue, present and comment on the findings from observational studies on the cardiovascular risk of a range of coxibs and NSAIDs. Likley to be an influential study)
Skjelbred P., Løkken P., Skoglund L.A. Post-operative administration of acetaminophen to reduce swelling and other inflammatory events. Curr. Ther. Res.. 1984;35:377-385. (A study showing that paracetamol can have anti-inflammatory properties under some circumstances)
Vane J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol.. 1971;231:232-239. (The definitive, seminal article that proposed cyclo-oxygenase inhibition as a mechanism of action for the aspirin-like drugs)
Vane J.R., Botting R.M., editors, Therapeutic roles of selective COX-2 inhibitors. 2001 William Harvey Press London. 584 (Outstanding multiauthor book covering all aspects of the mechanisms of action, actions, adverse effects and clinical role of COX-2 inhibitors in a range of tissues; excellent coverage)
Wallace J.L. How do NSAIDs cause ulcer disease? Baillière’s Best Pract. Res. Clin. Gastroenterol.. 2000;14:147-159. (Proposes an interesting idea concerning the role of the two COX isoforms in gastric homeostasis)
Warner T.D., Mitchell J.A. Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J.. 2004;18:790-804. (Excellent review of COX-1/-2 inhibitors and the relative merits of coxibs and the physiological role of COX-2)
Whittle B.J.R. Basis of gastrointestinal toxicity of non-steroid anti-inflammatory drugs. In: Vane J.R., Botting R.M., editors. Therapeutic roles of selective COX-2 inhibitors. London: William Harvey Press; 2001:329-354. (Excellent coverage; very good diagrams)
Wolfe M.M., Lichtenstein D.R., Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N. Engl. J. Med.. 1999;340:1888-1899. (Reviews epidemiology of the gastrointestinal complications of NSAIDs, covering the risk factors, the pathogenesis of gastrointestinal tract damage, and treatment; brief discussion of COX-2-selective drugs and NO-NSAIDs)
Yedgar S., Krimsky M., Cohen Y., Flower R.J. Treatment of inflammatory diseases by selective eicosanoid inhibition: a double-edged sword? Trends Pharmacol. Sci.. 2007;28:459-464. (A very accessible article that deals with the drawbacks of current NSAID therapy and reviews some potential solutions to the problems)
Alldred A., Emery P. Leflunomide: a novel DMARD for the treatment of rheumatoid arthritis. Expert Opin. Pharmacother.. 2001;2:125-137. (Useful review and update of this relatively new DMARD)
Bondeson J. The mechanisms of action of disease-modifying antirheumatic drugs: a review with emphasis on macrophage signal transduction and the induction of proinflammatory cytokines. Gen. Pharmacol.. 1997;29:127-150. (Good detailed review examining possible modes of action of these drugs)
Borel J.F., Baumann G., Chapman I., et al. In vivo pharmacological effects of ciclosporin and some analogues. Adv. Pharmacol.. 1996;35:115-246. (Borel was instrumental in the development of ciclosporin)
Cutolo M. Effects of DMARDs on IL-1Ra levels in rheumatoid arthritis: is there any evidence? Clin. Exp. Rheumatol.. 2002;20(5 Suppl 27):S26-S31. (Reviews the actions of DMARDs on the generation and release of the endogenous IL-1 antagonist. An interesting slant on the mechanism of action of these drugs)
Gummert J.F., Ikonen T., Morris R.E. Newer immunosuppressive drugs: a review. J. Am. Soc. Nephrol.. 1999;10:1366-1380. (Comprehensive review covering leflunomide, mycophenolate mofetil, sirolimus, tacrolimus and IL-2 receptor antibodies)
Hochberg M.C. Early aggressive DMARD therapy: the key to slowing disease progression in rheumatoid arthritis. Scand. J. Rheumatol. Suppl.. 1999;112:3-7. (Good general review of DMARD therapy and the use of these drugs in treating rheumatoid disease. Written before the recent explosion in anticytokine and other biologicals)
Morris R.E. Mechanisms of action of new immunosuppressive drugs. Ther. Drug Monit.. 1995;17:564-569. (Succinct, edifying review)
Rau R. Have traditional DMARDs had their day? Effectiveness of parenteral gold compared to biologic agents. Clin. Rheumatol.. 2005;24:189-202. (Argues for a continuing place of DMARDs in the clinic despite the introduction of the new biologicals)
Smolen J.S., Kalden J.R., Scott D.L., et al. Efficacy and safety of leflunomide compared with placebo and sulphasalazine in active rheumatoid arthritis: a double-blind, randomised, multicentre trial. Lancet. 1999;353:259-260. (Gives details of the results of a clinical trial showing the efficacy of leflunomide)
Snyder S.H., Sabatini D.M. Immunophilins and the nervous system. Nat. Med.. 1995;1:32-37. (Good coverage of mechanism of action of ciclosporin and related drugs)
Anticytokine drugs and other biologicals
Arora T., Padaki R., Liu L., et al. Differences in binding and effector functions between classes of TNF antagonists. Cytokine. 2009;45:124-131. (A research paper detailing the significance of the membrane-bound versus soluble TNF neutralising actions of the drugs)
Bongartz T., Sutton A.J., Sweeting M.J., et al. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295:2275-2285. (The title is self-explanatory)
Breedeveld F.C. Therapeutic monoclonal antibodies. Lancet. 2000;355:735-740. (Good review on the clinical potential of monoclonal antibodies)
Carterton N.L. Cytokines in rheumatoid arthritis: trials and tribulations. Mol. Med. Today. 2000;6:315-323. (Good review of agents modulating the action of TNF-α and IL-1; simple, clear diagram of cellular action of these cytokines, and summaries of the clinical trials of the agents in tabular form)
Choy E.H.S., Panayi G.S. Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med.. 2001;344:907-916. (Clear description of the pathogenesis of rheumatoid arthritis, emphasising the cells and mediators involved in joint damage; excellent diagrams of the interaction of inflammatory cells and of the mechanism of action of anticytokine agents)
Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol.. 2002;2:364-371. (Excellent review covering the role of cytokines in rheumatoid arthritis and the effects of anti-TNF therapy)
Fiorino G., Allez M., Malesci A., Danese E. Review article: anti TNF-alpha induced psoriasis in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther.. 2009;29:921-927. (Deals with this rare and unexpected side effect of anti-TNF therapy)
Maini R.N. The 2005 International Symposium on Advances in Targeted Therapies: what have we learned in the 2000s and where are we going? Ann. Rheum. Dis.. 2005;64(suppl 4):106-108. (An updated review dealing with the role of cytokines in the pathogenesis of rheumatoid arthritis and the results of clinical trials with anti-TNF and anti-IL-1 therapy)
O’Dell J.R. Anticytokine therapy—a new era in the treatment of rheumatoid arthritis. N. Engl. J. Med.. 1999;340:310-312. (Editorial with excellent coverage of the role of TNF-α in rheumatoid arthritis; summarises the differences between infliximab and etanercept)
Assanasen P., Naclerio R.M. Antiallergic anti-inflammatory effects of H1-antihistamines in humans. Clin. Allergy Immunol.. 2002;17:101-139. (An interesting paper that reviews several alternative mechanisms whereby antihistamines may regulate inflammation)
Leurs R., Blandina P., Tedford C., Timmerm N.H. Therapeutic potential of histamine H3 receptor agonists and antagonists Trends Pharmacol. Sci. 19:1998:177-183 (Describes the available H3 receptor agonists and antagonists, and their effects in a variety of pharmacological models, with discussion of possible therapeutic applications)
Simons F.E.R., Simons K.J. Drug therapy: the pharmacology and use of H1-receptor-antagonist drugs. N. Engl. J. Med.. 1994;23:1663-1670. (A bit dated now but contains effective coverage of the topic from the clinical viewpoint)
Moss M.L., Sklair-Tavron L., Nudelman R. Drug insight: tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat. Clin. Pract. Rheumatol.. 2008;4:300-309. (Accessible review dealing with this potentially important new concept. Some good diagrams)
Stefano F., Distrutti E. Cyclo-oxygenase (COX) inhibiting nitric oxide donating (CINODs) drugs: a review of their current status. Curr. Top. Med. Chem.. 2007;7:277-282. (The title is self-explanantory)
1Here, we use the term NSAID to include the coxibs but this is not a convention always followed in the literature.
2With possible exception of paracetamol which has been used clinically to lower body temperature during surgery.
3A horrible condition where skin peels away in sheets as if scalded.
4So called because the availability of NSAIDs in proprietary medicines over-the-counter, often in combination with other substances, such as caffeine, has tempted some people to consume them, often in prodigious quantities, for every conceivable malady.
5An odd side effect of the NSAID diclofenac came to light when a team of scientists investigated the curious decline in the population of several species of vulture in the Indian subcontinent. Dead cattle form an important part of the diet of these birds, and some animals had been treated with diclofenac for veterinary reasons. Apparently, residual amounts of the drug in the carcasses proved uniquely toxic to this species.
6Because the therapeutic margin is so small, it used to be said by rheumatologists that ‘patients must run before they can walk’.
7This does not, of course, apply to low-dose aspirin.