23 Atherosclerosis and lipoprotein metabolism
Overview
Atheromatous disease is ubiquitous and underlies the commonest causes of death (myocardial infarction caused by thrombosis—Ch. 24—on ruptured atheromatous plaque in a coronary artery) and disability (stroke, heart failure) in industrial societies. Hypertension is one of the most important risk factors for atheroma, and is discussed in Chapter 22. Here, we consider other risk factors, especially dyslipidaemia,1 which, like hypertension, is amenable to drug therapy. We describe briefly the processes of atherogenesis and of lipid transport as a basis for understanding the actions of lipid-lowering drugs. Important agents (statins, fibrates, cholesterol absorption inhibitors, nicotinic acid derivatives, fish oil derivatives) are described, with emphasis on the statins, which reduce the incidence of arterial disease and prolong life.
Introduction
In this chapter we summarise the pathological process of atherogenesis and approaches to the prevention of atherosclerotic disease. Lipoprotein transport forms the basis for understanding drugs used to treat dyslipidaemia. We emphasise the statins, which have been a major success story, not only lowering plasma cholesterol but also reducing cardiovascular events by approximately 25–50% and prolonging life. However, some patients cannot tolerate them, and they are not effective in all patients. Evidence that other drugs that influence dyslipidaemia improve clinical outcomes is less secure than for the statins, and two recent setbacks described below call into question the reliability of changes in circulating lipid concentrations in response to drugs as surrogates predicting clinical improvement. In the absence of hard evidence of clinical improvement, other classes of lipid-lowering drugs remain second line to statins, so there is rather a lot of ‘small print’ in this section.
Atherogenesis
Atheroma is a focal disease of the intima of large and medium-sized arteries. Lesions evolve over decades, during most of which time they are clinically silent, the occurrence of symptoms signalling advanced disease. Presymptomatic lesions are often difficult to detect non-invasively, although ultrasound is useful in accessible arteries (e.g. the carotids), and associated changes such as reduced aortic compliance and arterial calcification can be detected by measuring, respectively, aortic pulse wave velocity and coronary artery calcification. Until recently, there have been no good subprimate models, but transgenic mice (see Ch. 7) deficient in apolipoproteins or receptors that play key roles in lipoprotein metabolism have transformed this scene. Nevertheless, most of our current understanding of atherogenesis comes from human epidemiology and pathology, and from clinical investigations.
Epidemiological studies have identified numerous risk factors for atheromatous disease. Some of these cannot be altered (e.g. a family history of ischaemic heart disease), but others are modifiable (see Table 23.1) and are potential targets for therapeutic drugs. Clinical trials have shown that improving risk factors can reduce the consequences of atheromatous disease. Many risk factors (e.g. type 2 diabetes, dyslipidaemia, cigarette smoking) cause endothelial dysfunction (see Ch. 22), evidenced by reduced vasodilator responses to acetylcholine or to increased blood flow (so-called ‘flow-mediated dilatation’, responses that are inhibited by drugs that block nitric oxide [NO] synthesis; Ch. 20). Healthy endothelium produces NO and other mediators that protect against atheroma, so it is likely that metabolic cardiovascular risk factors act by causing endothelial dysfunction.
Table 23.1 Modifiable risk factors for atheromatous disease
| Raised low-density lipoprotein cholesterol |
| Reduced high-density lipoprotein cholesterol |
| Hypertension (Ch. 22) |
| Diabetes mellitus (Ch. 30) |
| Cigarette smoking (Ch. 48) |
| Obesity (Ch. 31) |
| Physical inactivity |
| Raised C-reactive proteina |
| Raised coagulation factors (e.g. factor VII, fibrinogen) |
| Raised homocysteine |
| Raised lipoprotein(a)b |
a Strongly associated with atheromatous disease but unknown if this is causal.
b Potentially modifiable but strongly genetically determined: nicotinic acid does lower lipoprotein(a).
Atherogenesis involves the following processes:
To understand how drugs prevent atheromatous disease, it is necessary briefly to review lipoprotein transport.
Lipoprotein Transport
Lipids and cholesterol are transported in the bloodstream as complexes of lipid and protein known as lipoproteins. These consist of a central core of hydrophobic lipid (including triglycerides and cholesteryl esters) encased in a hydrophilic coat of polar phospholipid, free cholesterol and apoprotein. There are four main classes of lipoprotein, differing in the relative proportion of the core lipids and in the type of apoprotein (various kinds of apoA and apoB, see below). Apoproteins bind to receptors specific for each that mediate uptake of lipoprotein particles into liver, blood or other tissues. Lipoproteins differ in size and density, and this latter property, measured originally by ultracentrifugation but now commonly estimated by simpler methods, is the basis for their classification into:
Each class of lipoprotein has a specific role in lipid transport, and there are different pathways for exogenous and for endogenous lipids, as well as a pathway for reverse cholesterol transport (Fig. 23.1). In the exogenous pathway, cholesterol and triglycerides absorbed from the ileum are transported as chylomicrons in lymph and then blood, to capillaries in muscle and adipose tissue. Here, triglycerides are hydrolysed by lipoprotein lipase, and the tissues take up the resulting free fatty acids and glycerol. The chylomicron remnants, still containing their full complement of cholesteryl esters, pass to the liver, bind to receptors on hepatocytes and undergo endocytosis. Cholesterol liberated in hepatocytes is stored, oxidised to bile acids, secreted unaltered in bile, or can enter the endogenous pathway.
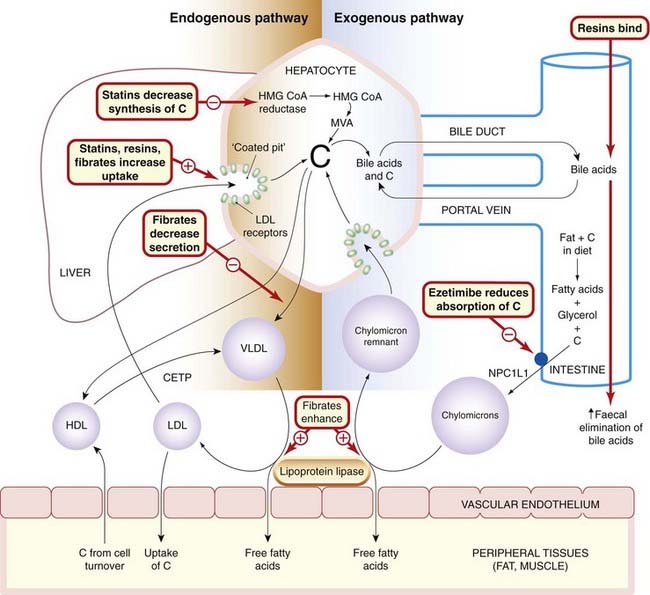
Fig. 23.1 Schematic diagram of cholesterol transport in the tissues, with sites of action of the main drugs affecting lipoprotein metabolism.
C, cholesterol; CETP, cholesteryl ester transport protein; HDL, high-density lipoprotein; HMG-CoA reductase, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; LDL, low-density lipoprotein; MVA, mevalonate; NPC1L1, a cholesterol transporter in the brush border of enterocytes; VLDL, very-low-density lipoprotein.
In the endogenous pathway, cholesterol and newly synthesised triglycerides are transported from the liver as VLDL to muscle and adipose tissue, where triglyceride is hydrolysed to fatty acids and glycerol; these enter the tissues as described above. During this process, the lipoprotein particles become smaller but retain a full complement of cholesteryl esters and become LDL particles. LDL provides the source of cholesterol for incorporation into cell membranes and for synthesis of steroids (see Chs 32 and 34) but is also key in atherogenesis. Cells take up LDL by endocytosis via LDL receptors that recognise apoB-100. Cholesterol can return to plasma from the tissues in HDL particles (reverse cholesterol transport). Cholesterol is esterified with long-chain fatty acids in HDL particles, and the resulting cholesteryl esters are transferred to VLDL or LDL particles by a transfer protein present in the plasma and known as cholesteryl ester transfer protein (CETP). Lipoprotein(a), or Lp(a), is a species of LDL that is associated with atherosclerosis and is localised in atherosclerotic lesions. Lp(a) contains a unique apoprotein, apo(a), with structural similarities to plasminogen (Ch. 24). Lp(a) competes with and inhibits the binding of plasminogen to its receptors on the endothelial cell. Plasminogen is normally the substrate for plasminogen activator, which is secreted by and bound to endothelial cells, generating the fibrinolytic enzyme plasmin (see Fig. 24.10). The effect of the binding of Lp(a) is that less plasmin is generated, fibrinolysis is inhibited and thrombosis promoted.
 There is current interest in four lipid transfer proteins that have been implicated in atherogenesis (reviewed by Stein & Stein, 2005). ACAT (acyl coenzyme A: cholesterol acyltransferase), which is expressed in two forms, catalyses the intracellular synthesis of cholesteryl ester in macrophages, adrenal cortex, gut and liver. LCAT (lecithin cholesterol acyltransferase) catalyses cholesteryl ester synthesis in HDL particles. CETP and PLTP (phospholipid transfer protein) are involved in transfer of cholesterol between different classes of lipoprotein particle in plasma. Tamoxifen, used in the treatment and prevention of breast cancer (Chs 34 and 55), is a potent ACAT inhibitor (de Medina et al., 2004).
There is current interest in four lipid transfer proteins that have been implicated in atherogenesis (reviewed by Stein & Stein, 2005). ACAT (acyl coenzyme A: cholesterol acyltransferase), which is expressed in two forms, catalyses the intracellular synthesis of cholesteryl ester in macrophages, adrenal cortex, gut and liver. LCAT (lecithin cholesterol acyltransferase) catalyses cholesteryl ester synthesis in HDL particles. CETP and PLTP (phospholipid transfer protein) are involved in transfer of cholesterol between different classes of lipoprotein particle in plasma. Tamoxifen, used in the treatment and prevention of breast cancer (Chs 34 and 55), is a potent ACAT inhibitor (de Medina et al., 2004).
Dyslipidaemia
Dyslipidaemia may be primary or secondary. The primary forms are due to a combination of diet and genetics (often but not always polygenic). They are classified into six phenotypes (the Frederickson classification; Table 23.2). An especially great risk of ischaemic heart disease occurs in a subset of primary type IIa hyperlipoproteinaemia caused by single-gene defects of LDL receptors; this is known as familial hypercholesterolaemia (FH), and the plasma cholesterol concentration in affected adults is typically > 8 mmol/l in heterozygotes and 12–25 mmol/l in homozygotes. Study of FH enabled Brown & Goldstein (1986) to define the LDL receptor pathway of cholesterol homeostasis (for which they shared a Nobel Prize). Drugs used to treat primary dyslipidaemia are described below.

Secondary forms of dyslipidaemia are a consequence of other conditions, such as diabetes mellitus, alcoholism, nephrotic syndrome, chronic renal failure, hypothyroidism, liver disease and administration of drugs, for example isotretinoin (an isomer of vitamin A given by mouth as well as topically in the treatment of severe acne), tamoxifen (Mikhailidis et al., 1997), ciclosporine (Ch. 26) and protease inhibitors used to treat infection with human immunodeficiency virus (Ch. 51). Secondary forms are treated where possible by correcting the underlying cause.
Lipoprotein metabolism and dyslipidaemia 
Lipids, including cholesterol and triglycerides, are transported in the plasma as lipoproteins, of which there are four classes:
Prevention of Atheromatous Disease
Drug treatment is often justified, to supplement healthy habits. Treatment of hypertension (Ch. 22) and, to a lesser extent, diabetes mellitus (Ch. 30) reduces the incidence of symptomatic atheromatous disease, and antithrombotic drugs (Ch. 24) reduce arterial thrombosis. Reducing LDL is also effective and is the main subject of this present chapter, but several other steps in atherogenesis are also potential targets for pharmacological attack.
Angiotensin-converting enzyme inhibitors (Ch. 22) improve endothelial function and prolong life in patients with atheromatous disease. Other drugs that also increase NO biosynthesis or availability are under investigation.
Measures to increase HDL: moderate alcohol consumption increases HDL, and epidemiological evidence favours moderate alcohol consumption in older people.2 Regular exercise also increases circulating HDL; drug treatment to increase HDL is of uncertain benefit. Fibrates and nicotinic acid derivatives—see below—modestly increase HDL, and reduce LDL and triglycerides. In subjects with low HDL, inhibition of cholesteryl ester transfer protein (CETP) with torcetrapib markedly increased HDL, but also increased blood pressure and was associated with a 60% increase in all-cause mortality (leading to abrupt discontinuation of its development). It is unclear if this is a class effect, but anacetrapib markedly increases HDL without increasing blood pressure; its effect on mortality is not yet known. ApoA-I Milano is a variant of apolipoprotein A-I identified in individuals in rural Italy with very low levels of HDL but almost no cardiovascular disease. Infusion of recombinant ApoA-I Milano–phospholipid complexes causes rapid regression of atherosclerosis in animal models, and administered intravenously caused regression of atherosclerosis in patients with acute coronary syndrome. It is expensive to produce and must be administered intravenously, but the strategy continues to be a focus of intense interest (see review by Duffy & Rader, 2009).
Antioxidants (e.g. vitamin C and vitamin E) are of interest, both because of evidence that they improve endothelial function in patients with increased oxidant stress, and because of epidemiological evidence that a diet rich in antioxidants is associated with reduced risk of coronary artery disease. Results from clinical trials have been negative, however, and several antioxidants reduce HDL. Oestrogen, used to prevent symptoms of the menopause (Ch. 34) and to prevent postmenopausal osteoporosis, has antioxidant properties and exerts other vascular effects that could be beneficial. Epidemiological evidence suggested that women who use such hormone replacement might be at reduced risk of atheromatous disease, but controlled trials showed significant adverse effects on cardiovascular mortality (Ch. 34 and see commentary by Dubey et al., 2004).
Anti-inflammatory approaches: drug treatment to lower C-reactive protein has been mooted, but it is possible that elevated C-reactive protein is a marker of vascular inflammation rather than playing an active part in disease progression. Other anti-inflammatory measures are being investigated; for example, acyl coenzyme A, cholesterol acyltransferase (ACAT) inhibitors.
Other novel therapies in development include drugs that inhibit squalene synthesis, microsomal transport protein (MTP) inhibitors and drugs that alter apoB. Among drugs that alter apoB, mipomersen is notable as an antisense oligonucleotide complementary to the coding region for apoB-100 of mRNA. It is an interfering RNA (iRNA; see Ch. 59) modified to render it resistant to nuclease enzymes. Injected once weekly, it has a marked effect in lowering LDL in patients with homoxyzgous FH, who are highly resistant to drug treatment (Kastelein et al., 2006).
Atheromatous disease
Lipid-Lowering Drugs
Several drugs decrease plasma LDL. Drug therapy is used in addition to dietary measures and correction of other modifiable cardiovascular risk factors.
The main agents used clinically are:
Fish oil lowers plasma triglyceride concentration but can increase plasma cholesterol.
Statins: Hmg-COa Reductase Inhibitors
The rate-limiting enzyme in cholesterol synthesis is HMG-CoA reductase, which catalyses the conversion of HMG-CoA to mevalonic acid (see Fig. 23.1). Simvastatin, lovastatin and pravastatin are specific, reversible, competitive HMG-CoA reductase inhibitors with Ki values of approximately 1 nmol/l. Atorvastatin and rosuvastatin are long-lasting inhibitors. Decreased hepatic cholesterol synthesis upregulates LDL receptor synthesis, increasing LDL clearance from plasma into liver cells. The main biochemical effect of statins is therefore to reduce plasma LDL. There is also some reduction in plasma triglyceride and increase in HDL. Several large randomised placebo-controlled trials of the effects of HMG-CoA reductase inhibitors on morbidity and mortality have been positive.
The Scandinavian Simvastatin Survival Study (4S) recruited patients with ischaemic heart disease and plasma cholesterol of 5.5–8.0 mmol/l: simvastatin lowered serum LDL by 35% and death by 30% (Fig. 23.2). This was accounted for by a 42% reduction in death from coronary disease over the median follow-up period of 5.4 years. Other large trials have confirmed reduced mortality both in patients with established ischaemic heart disease (e.g. the Cholesterol and Recurrent Events [CARE] trial) and in healthy people at risk of coronary disease, with a wide range of plasma cholesterol values and other risk factors, and treated with different statins (e.g. the West of Scotland Coronary Prevention Study [WOSCOPS], the Heart Protection Study and the Anglo-Scandinavian Cardiac Outcomes Trial [ASCOT]). Intensive lowering of LDL with atorvastatin 80 mg had a greater effect on event rate than did a 10 mg dose, but with a greater incidence of abnormally raised plasma transaminase activity (LaRosa et al., 2005). In secondary prevention trials of statins, cardiovascular event rate is approximately linearly related to the achieved plasma LDL over a concentration range from approximately 1.8 to 4.9 mmol/l, and the event rate falls on the same line in placebo- and statin-treated patients.
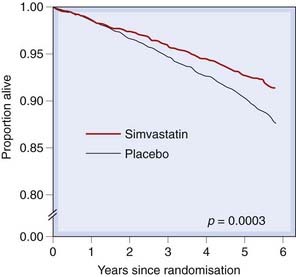
Fig. 23.2 Survival in patients with coronary heart disease and serum cholesterol 5.5–8.0 mmol/l treated either with placebo or with simvastatin.
The relative risk of death in the simvastatin group was 0.70 (95% confidence intervals 0.58–0.85).
(Based on 4S study 1994 Lancet 344: 1383–1389.)
Other actions of statins
Products of the mevalonate pathway react with protein (‘lipidation’, which is the addition to a protein of hydrophobic groups such as prenyl or farnesyl moieties). Several important membrane-bound enzymes (e.g. endothelial NO synthase; see Ch. 20) are modified in this way. The fatty groups serve as anchors, localising the enzyme in organelles such as caveoli and Golgi apparatus. Consequently, there is currently great interest in actions of statins that are unrelated, or indirectly related, to their effect on plasma LDL (sometimes referred to as pleiotropic effects). Some of these actions are undesirable (e.g. HMG-CoA reductase guides migrating primordial germ cells, and statin use is contraindicated during pregnancy), but some offer therapeutic promise, for example in Alzheimer’s disease where a role for statins is controversial (see review by Querfurth & LaFerla, 2010) and prevention of prostate cancer (Shannon et al., 2005). Such actions include:
The extent to which these effects contribute to the antiatheromatous actions of statins is unknown.
Pharmacokinetics
Short-acting statins are given by mouth at night to reduce peak cholesterol synthesis in the early morning. They are well absorbed and extracted by the liver, their site of action, and are subject to extensive presystemic metabolism via cytochrome P450 and glucuronidation pathways. Simvastatin is an inactive lactone prodrug; it is metabolised in the liver to its active form, the corresponding β-hydroxy fatty acid.
Adverse effects
Statins are well tolerated; mild unwanted effects include muscle pain (myalgia), gastrointestinal disturbance, raised concentrations of liver enzymes in plasma, insomnia and rash. More serious adverse effects are rare but include severe myositis (rhabdomyolysis) and angio-oedema. Myositis is a class effect of statins, occurs also with other lipid-lowering drugs (especially fibrates) and is dose related.3 It is commoner in patients with low lean body mass or uncorrected hypothyroidism.
Clinical uses of HMG-CoA reductase inhibitors (statins, e.g. simvastatin, atorvastatin)
Fibrates
Several fibric acid derivatives (fibrates) are available, including bezafibrate, ciprofibrate, gemfibrozil, fenofibrate and clofibrate. These markedly reduce circulating VLDL, and hence triglyceride, with a modest (approximately 10%) reduction in LDL and an approximately 10% increase in HDL. Their mechanism of action is complex (see Fig. 23.1). They are agonists at PPARα nuclear receptors4 (Ch. 3); in humans, the main effects are to increase transcription of the genes for lipoprotein lipase, apoA1 and apoA5. They increase hepatic LDL uptake. In addition to effects on lipoproteins, fibrates reduce plasma C-reactive protein and fibrinogen, improve glucose tolerance and inhibit vascular smooth muscle inflammation by inhibiting the expression of the transcription factor nuclear factor κB. As with the pleiotropic effects of statins (see above), there is great interest in these actions, although again it is unknown if they are clinically important.
In one study, gemfibrozil reduced coronary heart disease by approximately one-third compared with placebo in middle-aged men with primary hyperlipoproteinaemia, but fibrates have not been shown to improve survival. An HDL intervention trial performed by the US Veterans Affairs Department in some 2500 men with coronary heart disease and low HDL together with low LDL showed that gemfibrozil increased HDL and reduced coronary disease and stroke. Event rates were linked to changes in HDL but not to triglycerides or to LDL, suggesting that increasing HDL with a fibrate reduces vascular risk.
Adverse effects
Myositis is unusual but can be severe (rhabdomyolysis), with myoglobinuria and acute renal failure. It occurs particularly in patients with renal impairment, because of reduced protein binding and impaired drug elimination. Fibrates should be avoided in such patients and also in alcoholics, who are predisposed to hypertriglyceridaemia but are at risk of rhabdomyolysis.5 Myositis can also be caused (rarely) by statins (see above), and the combined use of fibrates with this class of drugs is therefore generally inadvisable (although it is sometimes undertaken by specialists). Gastrointestinal symptoms, pruritus and rash are more common than with statins. Clofibrate predisposes to gallstones, and its use is therefore limited to patients who have had a cholecystectomy (i.e. removal of the gall bladder).
Clinical uses of fibrates (e.g. gemfibrozil, fenofibrate)
Drugs That Inhibit Cholesterol Absorption
Historically, bile acid-binding resins (e.g. colestyramine, colestipol) were the only agents available to reduce cholesterol absorption and were among the few means to lower plasma cholesterol. Taken by mouth, they sequester bile acids in the intestine and prevent their reabsorption and enterohepatic recirculation (Fig. 23.1). The concentration of HDL is unchanged, and they cause an unwanted increase in triglycerides.
The American Lipid Research Clinics’ trial of middle-aged men with primary hypercholesterolaemia showed that addition of a resin to dietary treatment caused a mean 13% fall in plasma cholesterol and a 20–25% fall in coronary heart disease over 7 years, but no studies have shown improved survival. Decreased absorption of exogenous cholesterol and increased metabolism of endogenous cholesterol into bile acids in the liver lead to increased expression of LDL receptors on hepatocytes, and hence to increased clearance of LDL from the blood and a reduced concentration of LDL in plasma. Resins are bulky, unpalatable and often cause diarrhoea. They interfere with the absorption of fat-soluble vitamins, and of thiazide diuretics (Chs 22 and 28), digoxin (Ch. 21) and warfarin (Ch. 24), which should therefore be taken at least 1 h before or 4–6 h after the resin. With the introduction of statins, their use in treating dyslipidaemia was relegated largely to additional treatment in patients with severe disease (e.g. FH) and (a separate use) treating bile salt-associated symptoms of pruritus (itch) and diarrhoea—see clinical box. Colesevelam (introduced recently) is less bulky (daily dose up to 4 g compared with a dose up to 36 g for colestyramine) but more expensive. Subsequently, plant sterols and stanols have been marketed; these are isolated from wood pulp and used to make margarines or yoghurts. They reduce plasma cholesterol to a small extent and are tastier than resins.6 Their mechanism is unclear; sitostanol in the gut lumen competes with cholesterol for uptake and sitosterol interferes with cholesterol transfer within the enterocyte.
Ezetimibe
Ezetimibe is one of a group of azetidinone cholesterol absorption inhibitors, and is used as an adjunct to diet and statins in hypercholesterolaemia. It inhibits absorption of cholesterol (and of plant stanols) from the duodenum by blocking a transport protein (NPC1L1) in the brush border of enterocytes, without affecting the absorption of fat-soluble vitamins, triglycerides or bile acids. Because of its high potency compared with resins (a daily dose of 10 mg compared with a dose of resin of up to 36 g of colestyramine), it should represent a very real advance as a substitute for resins as supplementary treatment to statins in patients with severe dyslipidaemia. However, disappointingly, in a study of 720 patients with heterozygous FH comparing simvastatin alone with the combination of simvastatin with ezetimibe, whereas ezetimibe did indeed have the desired effect on LDL (approximately an extra 20% reduction), it did not retard thickening in the inner layers of the carotid artery over 2 years of follow-up (Kastelein et al., 2008). Such thickening is closely linked to atherosclerosis. A larger trial evaluating its effect on cardiovascular outcome is ongoing and eagerly (anxiously?) awaited. The mechanism of ezetimibe is distinct from that of phytosterol and phytostanol esters, which interfere with the micellar presentation of sterols to the cell surface.
Ezetimibe is administered by mouth and is absorbed into intestinal epithelial cells, where it localises to the brush border, which is its presumed site of action. It is also extensively (> 80%) metabolised to an active metabolite. Enterohepatic recycling results in slow elimination. The terminal half-life is approximately 22 h. It enters milk (at least in animal studies) and is contraindicated for women who are breastfeeding. It is generally well tolerated but can cause diarrhoea, abdominal pain or headache; rash and angio-oedema have been reported.
Nicotinic Acid
Nicotinic acid is a vitamin, and as such is essential for many important metabolic processes. Quite separately from this, it has been used in gram quantities as a lipid-lowering agent. It is converted to nicotinamide, which inhibits hepatic VLDL secretion (see Fig. 23.1), with consequent reductions in circulating triglyceride and LDL including Lp(a), and an increase in HDL. The mechanism is poorly understood but is believed to be initiated by an effect on lipolysis via a G-protein-coupled orphan receptor called HM74A and present in adipocyte membranes (see review by Karpe & Frayn, 2004). It also influences hepatic diacylglycerol transferase. Long-term administration to survivors of myocardial infarction reduced mortality in the Coronary Drug Project trial, but unwanted effects limit its clinical use. A modified-release preparation is better tolerated, and is a real, if modest, advance.
Adverse effects include flushing, palpitations and gastrointestinal disturbance. Flushing is associated with production of PGD2 (Ch. 17) and is reduced by taking with aspirin or with laropiprant (a PGD2 antagonist)—Figure 23.3. High doses can disturb liver function, impair glucose tolerance, and precipitate gout by increasing circulating urate concentration.
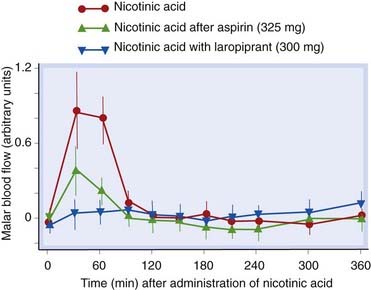
Fig 23.3 Vasodilatation caused by nicotinic acid (1.5 g, extended-release preparation) is attenuated by aspirin or by laropiprant, an antagonist of prostaglandin D2 (PGD2).
Blood flow in the cheeks of human subjects was measured by laser Doppler perfusion imaging after either placebo or nicotinic acid. Aspirin (325 mg 30 min before nicotinic acid) or laropiprant (300 mg with nicotinic acid) reduced the increase in malar blood flow caused by nicotinic acid.
(Redrawn from Lai E et al. 2007 Suppression of niacin-induced vasodilation with an antagonist to prostaglandin D2 receptor subtype 1. Clin Pharmacol Therap 81: 849–857.)
Fish Oil Derivatives
Omega-3 marine triglycerides reduce plasma triglyceride concentrations but increase cholesterol. Plasma triglyceride concentrations are less strongly associated with coronary artery disease than is cholesterol, but there is epidemiological evidence that eating fish regularly does reduce ischaemic heart disease, and dietary supplementation with ω-3 polyunsaturated fatty acids (PUFAs) improves survival in patients who have recently had a myocardial infarction (GISSI-Prevenzione Investigators, 1999). The mechanism may be the potent antiarrhythmic effects of PUFA (reviewed by Leaf et al., 2003). The mechanism of action of fish oil on plasma triglyceride concentrations is unknown. Fish oil is rich in PUFA, including eicosapentaenoic and docosahexaenoic acid, and it has other potentially important effects including inhibition of platelet function, prolongation of bleeding time, anti-inflammatory effects and reduction of plasma fibrinogen. Eicosapentaenoic acid substitutes for arachidonic acid in cell membranes and gives rise to 3-series prostaglandins and thromboxanes (that is, prostanoids with three double bonds in their side chains rather than the usual two), and 5-series leukotrienes. This probably accounts for their effects on haemostasis, because thromboxane A3 is much less active as a platelet-aggregating agent than is thromboxane A2, whereas PGI3 is similar in potency to PGI2 as an inhibitor of platelet function. The alteration in leukotriene biosynthesis probably underlies the anti-inflammatory effects of fish oil. Fish oil is contraindicated in patients with type IIa hyperlipoproteinaemia because of the increase in LDL that it causes. A preparation of omega 3-acid ethyl esters is licensed in the UK for prevention of recurrent events after myocardial infarction in addition to treatment of hypertriglyceridaemia; it causes less increase in LDL and fewer problems with fishy odour, weight gain and dyspepsia than the older fish oil preparations.
Drugs in dyslipidaemia
The main drugs used in patients with dyslipidaemias are:
References and Further Reading
Atherosclerosis and dyslipidaemia
Brown M.S., Goldstein J.L. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34-47. (Classic from these Nobel Prize winners; see also Goldstein, J.L., Brown, M.S., 1990. Regulation of the mevalonate pathway. Nature 343, 425–430)
Durrington P.N. Hyperlipidaemia: diagnosis and management, third ed. London: Hodder Arnold; 2005. (Extremely readable, authoritative book)
Ross R. Atherosclerosis—an inflammatory disease. N. Engl. J. Med.. 1999;340:115-126.
Stein O., Stein Y. Lipid transfer proteins (LTP) and atherosclerosis. Atherosclerosis. 2005;178:217-230. (Reviews four lipid transfer proteins—ACAT, CETP, LCAT and PLTP—and the therapeutic potential of modulating them)
LaRosa J.C., Grundy S.M., Waters D.D., et alFor the Treating to New Targets (TNT) Investigators. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N. Engl. J. Med.. 2005;352:1425-1435. (Intensive lipid-lowering therapy atorvastatin 80 mg daily in coronary heart disease patients provided significant clinical benefit beyond that afforded by 10 mg; this occurred with a greater incidence of elevated aminotransferase levels)
Liao J.K., Laufs U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol.. 2005;45:89-118. (‘Many pleiotropic effects are mediated by inhibition of isoprenoids, which serve as lipid attachments for intracellular signalling molecules. In particular, inhibition of small GTP-binding proteins, Rho, Ras, and Rac, whose proper membrane localisation and function are dependent on isoprenylation, may play an important role in mediating the pleiotropic effects of statins.’)
Merx M.W., Liehn E.A., Graf J., et al. Statin treatment after onset of sepsis in a murine model improves survival. Circulation. 2005;112:117-124. (Statins offer the potential of effective sepsis treatment)
Querfurth H.W., LaFerla F.M. Mechanisms of disease: Alzheimer’s disease. N. Engl. J. Med.. 2010;362:329-344.
Hague W., Emberson J., Ridker P.M., For the Air Force/Texas Coronary Atherosclerosis Prevention Study Investigators. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute events. N. Engl. J. Med.. 2001;344:1959-1965. (Statins may be effective in preventing coronary events in people with unremarkable serum lipid concentrations but with elevated C-reactive protein, a marker of inflammation and risk factor for coronary disease; see also accompanying editorial, Munford, R.S., Statins and the acute phase response, pp. 2016–2018)
Sabbagh M.N., Connor D.J., Undas A., et al. Simvastatin depresses blood clotting by activation of prothrombin, factor V, and factor XIII and by enhancing factor Va inactivation. Circulation. 2001;103:2248-2253. (Simvastatin effects on blood clotting independent of cholesterol reduction)
Shannon J., Tewoderos S., Garzotto M., et al. Statins and prostate cancer risk: a case-control study. Am. J. Epidemiol.. 2005;162:318-325. (Statin use was associated with a reduction in prostate cancer risk—odds ratio, 0.38; 95% confidence interval, 0.21, 0.69—especially of more aggressive prostate cancer)
Van Doren M., Broihier H.T., Moore L.A., et al. HMG-CoA reductase guides migrating primordial germ cells. Nature. 1998;396:466-469. (Regulated expression of HMG-CoA reductase provides spatial guide to migrating primordial germ cells)
Vasa M., Fichtlscherer S., Adler K., et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103:2885-2890. (May participate in repair after ischaemic injury)
Canner P.L., Furberg C.D., Terrin M.L., et al. Benefits of niacin by glycemic status in patients with healed myocardial infarction (from the Coronary Drug Project). Am. J. Cardiol.. 2005;95:254-257. (The Coronary Drug Project, conducted during 1966 to 1974, was a randomised, double-blind, placebo-controlled trial in 8341 men with previous myocardial infarction; nicotinic acid significantly reduced total mortality during 6.2 years’ treatment plus an additional 9 years of post-trial follow-up)
Karpe F., Frayn K.N. The nicotinic acid receptor—a new mechanism for an old drug. Lancet. 2004;363:1892-1894. (Brief review of recent evidence that nicotinic acid acts via a G-protein-coupled orphan receptor)
Taylor A.J., Sullenberger L.E., Lee H.J., et al. Arterial biology for the investigation of the treatment effects of reducing cholesterol (ARBITER) 2—A double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110:3512-3517. (A double-blind, randomised, placebo-controlled study of once-daily extended-release nicotinic acid 1 g added to background statin therapy in 167 patients with coronary heart disease and low HDL. The primary end point was the change in common carotid intima media thickness after 1 year. Nicotinic acid slowed the progression of atherosclerosis)
Gervois P., Torra I.P., Fruchart J.C., et al. Regulation of lipid and lipoprotein metabolism by PPAR activators. Clin. Chem. Lab. Med.. 2000;38:3-11. (Review)
Bloomfield Rubins H., Davenport J., Babikian V., et al. Reduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol. The Veterans Affairs HDL Intervention Trial (VA-HIT). Circulation. 2001;103:2828-2833. (Evidence that increasing HDL reduces stroke)
GISSI-Prevenzione Investigators (Gruppo Italiano per lo Studio della Sopravivenza nell’Infarto Miocardico). Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Lancet. 1999;354:447-455. (11 324 patients surviving myocardial infarction were randomly assigned supplements of n-3 PUFA, 1 g daily, vitamin E, both or neither for 3.5 years. The primary end point was death, non-fatal myocardial infarction and stroke combined. Dietary supplementation with n-3 PUFA led to a clinically important and statistically significant benefit. Vitamin E had no benefit)
Leaf A., Kang J.X., Xiao Y.F., et al. Clinical prevention of sudden cardiac death by n-3 polyunsaturated fatty acids and mechanism of prevention of arrhythmias by n-3 fish oils. Circulation. 2003;107:2646-2652. (Reviews antiarrhythmic action of PUFA, including electrophysiological effects on voltage-dependent sodium and L-type calcium channels)
Kastelein J.J., Akdim F., Stroes E.S., et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N. Engl. J. Med.. 2008;358:1431-1443. (In patients with FH, combined therapy with ezetimibe and simvastatin did not result in a significant difference in changes in intima media thickness, as compared with simvastatin alone, despite decreases in LDL cholesterol and C-reactive protein)
Kosoglou T., Statkevich P., Johnson-Levonas A.O., et al. Ezetimibe—a review of its metabolism, pharmacokinetics and drug interactions. Clin. Pharmacokinetics. 2005;44:467-494.
de Medina P., Payrá B.L., Bernad J., et al. Tamoxifen is a potent inhibitor of cholesterol esterification and prevents the formation of foam cells. J. Pharmacol. Exp. Ther.. 2004;308:1542-1548. (Molecular modelling revealed similarity between tamoxifen and ACAT inhibitor)
Duffy D., Rader D.J. Update on strategies to increase HDL quantity and function. Nature. Rev. Cardiol.. 2009;6:455-463.
Kharbanda R.K., Wallace S., Walton B., et al. Systemic acyl-CoA: cholesterol acyltransferase inhibition reduces inflammation and improves vascular function in hypercholesterolemia. Circulation. 2005;111:804-807. (Systemic ACAT inhibition reduced tumour necrosis factor-α levels in hypercholesterolaemic subjects and improved resistance vessel endothelial function, with small effects on circulating cholesterol)
Wierzbicki A.S. Lipid lowering therapies in development. Expert Opin. Investig. Drugs. 2004;13:1405-1408.
1The term dyslipidaemia is preferred to hyperlipidaemia because a low plasma concentration of high-density lipoprotein cholesterol is believed to be harmful and is a therapeutic target.
2’Sinful, ginful, rum-soaked men, survive for three score years and ten’—or longer, we rather hope…
3Cerivastatin, a potent statin introduced at relatively high dose, was withdrawn because of severe myositis occurring particularly in patients treated with gemfibrozil—discussed later in the chapter.
4Standing for peroxisome proliferator-activated receptors—don’t ask! (Peroxisomes are organelles that are not present in human cells, so something of a misnomer!) Thiazolidinedione drugs used in treating diabetes act on related PPARγ receptors; see Ch. 30.
5For several reasons, including a tendency to lie immobile for prolonged periods followed by generalised convulsions—‘rum fits’—and delirium tremens.
6This is not, however, saying much.