11 Pharmacogenetics, pharmacogenomics and ‘personalised medicine’
Overview
The concept of individualising drug therapy in light of genomic information (‘personalised medicine’) is introduced. We explain relevant elementary genetic concepts and describe briefly several single-gene pharmacogenetic disorders (plasma cholinesterase deficiency, acute intermittent porphyria, drug acetylation deficiency and aminoglycoside ototoxicity). These prove the concept that inherited factors influence individual drug response. We then cover pharmacogenomic tests that are currently clinically available, including tests for variations in human leukocyte antigen (HLA) genes (adverse drug reaction susceptibility to abacavir, anticonvulsants and clozapine); in genes influencing drug metabolism—thiopurine-S-methyltransferase (TPMT), dihydropyrimidine dehydrogenase (DPYD) and CYP isoenzymes (CYP2D6 and CYP2C9); and for drug targets such as the epidermal growth factor receptor HER2, tyrosine kinase inhibitors and the main target for warfarin, vitamin K epoxide reductase (VKOR). Future prospects and challenges to the wider utilisation of pharmacogenomic tests are explained.
Introduction
Responses to some therapeutic agents, including most vaccines, the oral contraceptive (Ch. 34) and low-dose aspirin to prevent arterial thrombosis (Ch. 24), are sufficiently predictable to enable adoption of a standard dose regimen. Other useful drugs, such as lithium (Ch. 46) or antihypertensive drugs (Ch. 22), are individualised and their doses adjusted on the basis of monitoring the drug concentration in the plasma or a response such as change in blood pressure, together with any adverse effects.
Interindividual variation in response to drugs is a serious problem; if not taken into account, it can result in lack of efficacy or unexpected side effects. Such variation can be pharmacokinetic (too much or not enough of the drug at its site of action), pharmacodynamic (greater or less effect from a given concentration at the site of action because of differences between individuals at the level of the primary drug target or downstream events) or idiosyncratic (a qualitatively abnormal reaction that occurs in only a few exposed individuals). Variation is partly caused by environmental factors, and these are discussed in Chapter 56. However, studies comparing identical with non-identical twins have shown that much of the variation of drug response is genetically determined; for example, elimination half-lives of antipyrine, a probe of hepatic drug oxidation, and of warfarin, an oral anticoagulant (Ch. 24), differ much less between identical than between fraternal twins.
Genes influence pharmacokinetics by altering the expression of proteins involved in absorption, distribution, metabolism or excretion (ADME); pharmacodynamic variation reflects differences in drug targets, G-proteins or other downstream pathways; and susceptibility to idiosyncratic reactions results from differences in enzymes or immune mechanisms. It is hoped that as our understanding of the human genome improves, together with the introduction of simpler methods to identify genetic differences between individuals, it will become possible to use genetic information specific to an individual patient to preselect a drug that will be effective and not cause toxicity, rather than relying on trial and error supported by physiological clues as at present—an aspiration referred to as ‘personalised medicine’. Thus far this approach, which was initially over-hyped, has yielded relatively little in the way of clinical benefit, and it is unlikely to provide the rapid revolution that was trumpeted when the human genome was first sequenced. Progress has been slow, but the US Food and Drug Administration (FDA) has approved addition of pharmacogenomics labelling information to the package inserts of over 50 drugs, and although the use of pharmacogenomic tests is patchy and not yet supported by evidence of improved outcomes from clinical trials, the approach seems very likely to make important contributions in the medium term. This is our justification for including this separate chapter on the topic.
We begin by revisiting some elementary genetics as a basis for understanding pharmacogenetic disorders, followed by examples of inherited diseases where drug response is abnormal (pharmacogenetic disorders) and conclude with a brief account of drugs with available genomic tests and how these are beginning to be applied to individualise drug therapy in clinical practice (pharmacogenomics).
Relevant Elementary Genetics
Genes are the fundamental units of heredity; they consist of ordered sequences of nucleotides (adenine, guanine, thymidine and cytosine—A, G, T, C) located in particular positions in a particular DNA strand. Genes are conventionally abbreviated as for the protein they code for, but are written in italics—for example ‘CYP2D6’ represents a protein while ‘CYP2D6’ is the gene that encodes it. Most cellular DNA is located in the chromosomes in cell nuclei, but a small amount is present in mitochondria and is inherited from the mother (since the ovum contributes mitochondria to the gamete). DNA is transcribed to complementary messenger RNA (mRNA) which is translated in rough endoplasmic reticulum into a sequence of amino acids. The resulting peptide undergoes folding and sometimes post-translational modification to form the final protein product. The DNA sequence of a gene that codes protein is known as the exon. Introns are DNA sequences that interrupt the exon; an intron is transcribed into mRNA but this sequence is excised from the message and not translated into protein. The rate of transcription is controlled by promoter regions in the DNA to which RNA polymerase binds to initiate transcription.
Mutations are heritable changes in the base sequence of DNA. This may, or may not,1 result in a change in the amino acid sequence of the protein for which the gene codes. Most changes in protein structure are deleterious, and so the altered gene dies out in succeeding generations as a result of natural selection. A few changes may confer an advantage, however, at least under some environmental circumstances. A pharmocogenetically relevant example is the X-linked gene for glucose 6-phosphate dehydrogenase (G6PD); deficiency of this enzyme confers partial resistance to malaria (a considerable selective advantage in parts of the world where this disease is common) at the expense of susceptibility to haemolysis as an idiosyncratic reaction in response to oxidative stress in the form of exposure to various dietary constituents, including several drugs (e.g. the antimalarial drug primaquine; see Ch. 53). This ambiguity gives rise to the abnormal gene being preserved in future generations, at a frequency that depends on the balance of selective pressures in the environment. Thus the distribution of G6PD deficiency is similar to the geographical distribution of malaria. The situation where several functionally distinct forms of a gene are common in a population is called a ‘balanced’ polymorphism (balanced because disadvantage, for example in a homozygote, is balanced by an advantage, for example in a heterozygote).
Polymorphisms are different alternative sequences at a locus within the DNA strand (alleles) that persist in a population through several generations. They arise initially because of a mutation, and are stable if they are non-functional, or die out during subsequent generations if (as is usually the case) they are disadvantageous. However, if the prevailing selective pressures in the environment are favourable, leading to a selective advantage, a polymorphism may increase in frequency over successive generations. Now that genes can be sequenced readily, it has become apparent that single nucleotide polymorphisms (SNPs, DNA sequence variations that occur when a single nucleotide in the genome sequence is altered) are very common (see Web links in the reference list for a useful ‘fact sheet’ about SNPs). They may entail substitution of one nucleotide for another (usually substitution of C for T), or deletion or insertion of a nucleotide. Insertions and deletions result in a ‘frame shift’ in translation—for example, after an insertion the first element of the next triplet in the code becomes the second and all subsequent bases are shifted one ‘to the right’. The result can be loss of protein synthesis, abnormal protein synthesis or an abnormal rate of protein synthesis.
SNPs occur every 100–300 bases along the 3 billion base human genome. Approximately two-thirds of SNPs involve C for T substitution. SNPs can occur in coding (gene) and non-coding regions of the genome. A single SNP can be an important determinant of disease—for example, a common genetic variant due to an SNP in one of the coagulation factors, known as factor V Leiden, is the commonest form of inherited thrombophilia (Ch. 24). This confers an increased risk of venous thrombosis in response to environmental factors such as prolonged immobility, but might perhaps have been an advantage to ancestors more at risk of haemorrhage than of thrombosis. Alternatively, predisposition to disease may depend on a combination of several SNPs in or near a gene. Such combinations are known as haplotypes and are inherited from each parent.
See Web links in the reference list for a useful source of basic information, including definitions, from the Human Genome Project.
Single-Gene Pharmacokinetic Disorders
Where a mutation disrupts gene function profoundly this may result in a ‘single-gene disorder’ which is inherited in Mendelian fashion. This was recognised for albinism (albinos lack an enzyme that is needed to synthesise the brown pigment melanin) and other ‘inborn errors of metabolism’ in the early part of the 20th century by Archibald Garrod, a British physician who initiated the study of biochemical genetics. Investigation of this large group of individually rare diseases has contributed disproportionately to our understanding of molecular pathology—familial hypercholesterolaemia and the mechanism of action of statins (Ch. 23) is one example.
Plasma Cholinesterase Deficiency
In the 1950s Walter Kalow discovered that suxamethonium sensitivity is due to genetic variation in the rate of drug metabolism as a result of a Mendelian autosomal recessive trait. This short-acting neuromuscular-blocking drug is widely used in anaesthesia and is normally rapidly hydrolysed by plasma cholinesterase (Ch. 13). About 1 in 3000 individuals fail to inactivate suxamethonium rapidly and experience prolonged neuromuscular block if treated with it; this is because a recessive gene gives rise to an abnormal type of plasma cholinesterase. The abnormal enzyme has a modified pattern of substrate and inhibitor specificity. It is detected by a blood test that measures the effect of the inhibitor dibucaine, which inhibits the abnormal enzyme less than the normal enzyme. Heterozygotes hydrolyse suxamethonium at a more or less normal rate, but their plasma cholinesterase has reduced sensitivity to dibucaine, intermediate between normal subjects and homozygotes. Only homozygotes express the disease: they appear completely healthy unless exposed to suxamethonium (or, presumably, closely related chemicals) but experience prolonged paralysis if exposed to a dose that would cause neuromuscular block for only a few minutes in a healthy person.2 For a biological perspective on heritable variation in response to xenobiotics, especially in insects and bacteria, see Kalow 1997. There are other reasons why responses to suxamethonium may be abnormal in an individual patient, notably malignant hyperpyrexia (Ch. 13), a genetically determined idiosyncratic adverse drug reaction involving the ryanodine receptor (Ch. 4). It is important to test family members who may be affected, but the disorder is so rare that it is currently impractical to screen for it routinely before therapeutic use of suxamethonium.
Acute Intermittent Porphyria
The hepatic porphyrias are prototypic pharmacogenetic disorders in which patients may be symptomatic even if they are not exposed to a drug, but where a wide spectrum of drugs can provoke very severe worsening of the course of the disease. Although uncommon, they are clinically important. They are inherited disorders involving the biochemical pathway of porphyrin haem biosynthesis. Acute intermittent prophyria is the commonest and most severe form. It is autosomal dominant (in contrast to plasma cholinesterase deficiency) and is due to one of many different mutations in the gene coding porphobilinogen deaminase (PBGD), a key enzyme in haem biosynthesis in red cell precursors, hepatocytes and other cells. All of these mutations reduce activity of this enzyme, and clinical features are caused by the resulting build-up of haem precursors including porphyrins. There is a strong interplay with the environment through exposure to drugs, hormones and other chemicals. The use of sedative, anticonvulsant or other drugs in patients with undiagnosed porphyria can be lethal, though with appropriate supportive management most patients recover completely.3 Many drugs, especially but not exclusively those that induce CYP enzymes (e.g. barbiturates, griseofulvin, carbamazepine, estrogens—see Ch. 9), can precipitate acute attacks in susceptible individuals. Porphyrins are synthesised from δ-amino laevulinic acid (ALA) which is formed by ALA synthase in the liver. This enzyme is induced, like various other hepatic enzymes, by drugs such as barbiturates, resulting in increased ALA production and, hence, increased porphyrin accumulation. As mentioned above the genetic trait is inherited as an autosomal dominant, but frank disease is approximately five times more common in women than in men, because hormonal fluctuations precipitate acute attacks.
Drug Acetylation Deficiency
Both examples considered so far are uncommon diseases. However, in the 1960s Price-Evans demonstrated that the rate of drug acetylation varied in different populations as a result of balanced polymorphism. Figure 11.1 contrasts the approximately Gaussian distribution of plasma concentrations achieved 3 h after administration of a dose of salicylate with the bimodal distribution of plasma concentrations after a dose of isoniazid. The isoniazid concentration was < 20 µmol/l in about half the population, and in this group the mode was approximately 9 µmol/l. In the other half of the population (plasma concentration > 20 µmol/l), the mode was approximately 30 µmol/l. Elimination of isoniazid depends mainly on acetylation, catalysed by an acetyltransferase enzyme (Ch. 9). White populations contain roughly equal numbers of ‘fast acetylators’ and ‘slow acetylators’. The characteristic of fast or slow acetylation is controlled by a single recessive gene associated with low hepatic acetyltransferase activity. Other ethnic groups have different proportions of fast and slow acetylators. Isoniazid causes two distinct forms of toxicity. One is peripheral neuropathy, which is produced by isoniazid itself and is commoner in slow acetylators. The other is hepatotoxicity, caused by conversion of the acetylated metabolite to acetylhydrazine and is commoner in fast acetylators, at least in some populations. This genetic variation thus produces a qualitative change in the pattern of toxicity caused by the drug in different populations.
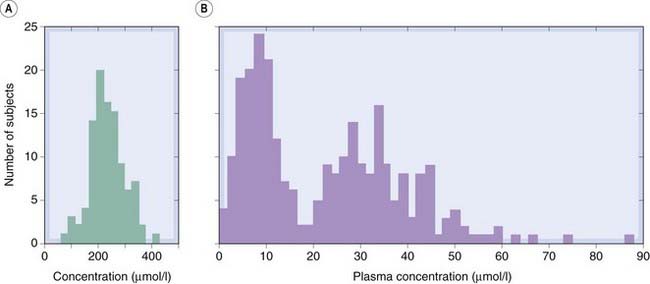
Fig. 11.1 Distribution of individual plasma concentrations for two drugs in humans.
[A] Plasma salicylate concentration 3 h after oral dosage with sodium salicylate. [B] Plasma isoniazid concentration 6 h after oral dosage. Note the normally distributed values for salicylate, compared with the bimodal distribution of isoniazid.
(From: (A) From Evans & Clarke 1961 Br Med Bull 17: 234–280; (B) From Price-Evans D A 1963 Am J Med 3: 639.)
Acetyltransferase is also important in the metabolism of other drugs, including hydralazine (Ch. 22), procainamide (Ch. 21), dapsone and various other sulfonamides (Ch. 50) and acetylator status influences drug-induced lupus, an autoimmune disorder affecting many organs including skin, joints and kidneys which is an idiopathic adverse reaction caused by several of these agents. However, neither phenotyping (by measuring kinetics of drug transformation) nor genotyping for acetyltransferase has found a way into routine clinical practice, probably because these drugs are relatively little used and there are several alternative treatments available that are usually preferred.
Aminoglycoside Ototoxicity
In the examples above, variations in chromosomal genes, sex-linked or inherited in autosomal dominant or autosomal recessive fashion, cause variations in drug response. Increased susceptibility to hearing loss caused by aminoglycoside antibiotics (see Ch. 50) is, in some families, inherited quite differently, namely exclusively through the mother to all her children. This is the pattern expected of a mitochondrial gene, and indeed the most common predisposing mutation is m.1555A>G, a mitochondrial DNA mutation. This mutation accounts for 30–60% of aminoglycoside ototoxicity in China, where use of aminoglycosides is common because they are cheap. Aminoglycosides work by binding to bacterial ribosomes (Ch. 50), which share properties with human mitochondrial ribosomes; aminoglycosides cause ototoxity in all individuals exposed to too high a dose. The m.1555A>G mutation makes mitochondrial ribosomes even more similar to their bacterial counterpart, increasing the affinity of the drug which remains bound to ribosomes in the hair cells in the ear for several months following a single dose in susceptible individuals. Screening for this variant may be appropriate in children who are likely to require treatment with aminoglycosides (Bitner-Glindzicz & Rahman, 2007).
Therapeutic Drugs and Clinically Available Pharmacogenomic Tests
Clinical tests to predict drug responsiveness were anticipated to be one of the first applications of sequencing the human genome, but their development has been slowed by various scientific, commercial, political and educational barriers (Flockhart et al., 2009). Reimbursement for expensive drugs, whether provided by the state or by insurance schemes, depends increasingly on evidence of cost-effectiveness. New tests need to improve demonstrably on our current ability to prescribe optimally, and must lead to a clear-cut change in prescribing, such as using a different drug or a different dosing regimen. So far the evidence in support of any pharmacogenetic test is less convincing than the ideal of a randomised controlled trial of a pharmacogenomics-informed prescribing strategy versus current best practice, but several of the tests mentioned below are increasingly used in clinical practice. They include tests for (a) variants of different human leukocyte antigens (HLAs) that have been strongly linked to susceptibilities to several severe idiosyncratic reactions; (b) genes controlling aspects of drug metabolism; and (c) genes encoding drug targets. For one drug (warfarin), a test combines genetic information about metabolism with information about its target. The genetic susceptibility of collie dogs to neurotoxic effects of ivermectin mentioned in Chapter 8 (footnote, p. 99) is of importance in veterinary medicine. It results from a variant of P-glycoprotein that alters the properties of the blood–brain barrier of dogs with collie ancestry, and in future genes coding for proteins influencing drug distribution in man may also be fertile territory for new tests.
Pharmacogenetics 
Methodology. Mutations in the germline are passed to the next generation where they are present in all cells; in practice, tests for such germline mutations are usually made on venous blood samples which contain chromosomal and mitochondrial DNA in white blood cells. Somatic cell mutations underlie the pathogenesis of some tumours (Ch. 5), and the presence or absence of such somatic cell mutations guides drug selection. The genomic tests are performed on DNA from samples of the tumour obtained surgically. The tests themselves involve amplification of the relevant sequence(s) and molecular biological methods, often utilising chip technology, to identify the various polymorphisms.
Hla Gene Tests
Abacavir and HLAB*5701
 Abacavir (Ch. 51) is a reverse transcriptase inhibitor which is highly effective in treating HIV infection. Its use has been limited by severe rashes. Susceptibility to this adverse effect is closely linked to the human leukocyte antigen (HLA) variant HLAB*5701, and testing for this variant is used widely and supported by prospective trials; see Figure 11.2 (Lai-Goldman & Faruki, 2008).
Abacavir (Ch. 51) is a reverse transcriptase inhibitor which is highly effective in treating HIV infection. Its use has been limited by severe rashes. Susceptibility to this adverse effect is closely linked to the human leukocyte antigen (HLA) variant HLAB*5701, and testing for this variant is used widely and supported by prospective trials; see Figure 11.2 (Lai-Goldman & Faruki, 2008).
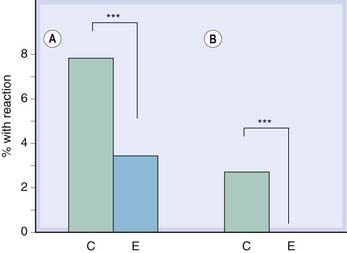
Fig. 11.2 Incidence of abacavir hypersensitivity is reduced by pharmacogenetic screening.
In the PREDICT-1 study (Mallal et al., 2008), patients were randomised to standard care (control group, C) or prospective pharmacogenetic screening (experimental group, E). All the control subjects were treated with abacavir, but only those experimental subjects who were HLA-B*5701 negative were treated with abacavir. There were two prespecified end points: clinically suspected hypersensitivity reactions [A] and clinically suspected reactions that were immunologically confirmed by a positive patch test [B]. Both end points favoured the experimental group (P < 0.0001).
Figure redrawn from Hughes AR et al 2008 Pharmacogenetics Journal 8: 365-374.
Anticonvulsants and HLAB*1502
Carbamazepine (Ch. 44) can also cause severe (life-threatening) rashes including Stevens Johnson syndrome (in which a multiform rash with blistering and other lesions extends into the gastrointestinal tract) and toxic epidermal necrolysis (in which the outer layer of the skin peels away from the dermis as though it has been scalded). These are associated with a particular HLA allele, HLAB*1502, which occurs almost only in people with Asian ancestry (Man et al., 2007); the FDA recommends that Chinese patients should be screened for this allele before starting treatment. People who develop such a reaction to carbamazepine may develop a similar problem if treated with phenytoin, and the same allele has been associated with hypersensitivity reactions to this drug too.
Clozapine and HLA-DQB1*0201
Clozapine is a uniquely effective antipsychotic drug with a different pattern of adverse effects from classical antipsychotic drugs (Ch. 45); its use is limited by agranulocytosis in approximately 1% of patients. This idiosyncratic adverse effect has been associated with HLA-DQB1*0201, but so far studies have been small and the specificity and sensitivity of the test are yet to be established.
Drug Metabolism-Related Gene Tests
Thiopurines and TPMT
Thiopurine drugs (tioguanine, mercaptopurine and its prodrug azathioprine; Ch. 55) have been used for the past 50 years to treat leukaemias, including acute lymphoblastic leukaemia (ALL, which accounts for approximately one-fifth of all childhood malignancies), and more recently for even more common diseases including inflammatory bowel disease, and to cause immunosuppression. All of these drugs cause bone marrow and liver toxicity, and are detoxified by thiopurine-S-methyltransferase (TPMT) which is present in blood cells, as well as by xanthine oxidase. There are large inherited variations in TPMT activity with a trimodal frequency distribution (Weinshilboum & Sladek, 1980); low TPMT activity in blood is associated with high concentrations of active 6-thioguanine nucleotides (TGN) in blood and with bone marrow toxicity whereas high TPMT activity is associated with lower concentrations of TGN and reduced efficacy (Lennard et al., 1989, 1990). Before treatment with these drugs, phenotyping (by a blood test for TPMT activity) or genotyping of TMPT alleles TPMT*3A, TPMT*3C, TPMT*2, is recommended. Even with such testing, careful monitoring of the white blood cell count is needed because of environmental factors (e.g. drug interaction with allopurinol via its effect on xanthine oxidase 0 (Ch. 56, Table 56.3).
5-Fluorouracil (5-FU) And DPYD
5-FU (Ch. 55, Fig. 55.6) is used extensively to treat solid tumours, but has variable efficacy and unpredictable mucocutaneous toxicity. It is detoxified by dihydropyrimidine dehydrogenase (DPYD), which has multiple clinically identifiable functional genetic variants. Currently available genetic information is neither highly sensitive nor specific, but the FDA recommends that the drug not be given to patients with DPYD deficiency.
Tamoxifen and CYP2D6
Tamoxifen (Chs 34 and 55) is metabolised to an oestrogen antagonist endoxifen by CYP2D6 which is subject to marked polymorphic variation (see Ch. 9); several small association studies have suggested a link between CYP2D6 genotype and efficacy. Genotyping tests for CYP2D6 are available, but genetic results from larger comparative trials of tamoxifen versus aromatase inhibitors are awaited. Treatment with other CYP2D6 substrates, for example tetrabenazine, used to treat Huntington’s disease (Ch. 39) may also be influenced by knowledge of CYP2D6 genotype: the FDA recommends that patients who are CYP2D6 poor metabolisers should not be prescribed more than 50 mg daily because of the risk of severe depression.
Irinotecan and UGT1A1*28
Irinotecan, a topoisomerase I inhibitor (Ch. 55) has marked activity against colorectal and lung cancers in a minority of patients, but toxicity (diarrhoea and bone marrow suppression) can be severe. It works through an active metabolite (SN-38) which is detoxified by glucuronidation by UDP-glucuronyltransferase (UGT; Ch. 9, Fig 9.3). Reduced activity of this enzyme is common and gives rise to the inherited benign condition of hyperbilirubinaemia known as Gilbert’s syndrome in which unconjugated bilirubin accumulates in plasma. UGT1A1 genetic testing is clinically available and predicts irinotecan pharmacokinetics and clinical outcomes. The best way to use information from the test is still uncertain, however.
Drug Target-Related Gene Tests
Trastuzumab and HER2
Trastuzumab (‘Herceptin’; Ch. 55) is a monoclonal antibody that antagonises epidermal growth factor (EGF) by binding to one of its receptors (human epidermal growth factor receptor 2—HER2) which can occur in tumour tissue as a result of somatic mutation. It is used in patients with breast cancer whose tumour tissue overexpresses this receptor. Other patients have not been found to benefit from it.
Dasatinib, Imatinib and BCR-ABL1
Dasatinib is a dual BCR/ABL and Src tyrosine kinase inhibitor used in haematological malignancies characterised by the presence of a Philadelphia chromosome, namely chronic myeloid leukaemia (CML) and some adults with acute lymphoblastic leukaemia (ALL). The Philadelphia chromosome results from a translocation defect when parts of two chromosomes (9 and 22) swap places; part of a ‘breakpoint cluster region’ (BCR) in chromosome 22 links to the ‘Abelson-1’ (ABL) region of chromosome 9. A mutation (T315I) in BCR/ABL confers resistance to the inhibitory effect of dasatinib and patients with this variant do not benefit from this drug. Pharmacogenetic testing is also being evaluated for imatinib (Ch. 55), another tyrosine kinase inhibitor used in patients with CML and other myelodysplastic disorders associated with rearrangements in the gene for platelet-derived growth factor receptor or for BCR-ABL.
Combined (Metabolism and Target) Gene Tests
Warfarin and CYP2C9 + VKORC1 Genotyping
Warfarin is par excellence a drug where dosing must be individualised. This is done by measuring the international normalised ratio (INR), a measure of its effect on blood coagulability (Ch. 24), but thrombotic events despite treatment (lack of efficacy) and serious adverse effects (usually bleeding) remain all too common. Surely we can do better? Warfarin is the most widely used drug for which pharmacogenetic testing has been proposed, based on a study showing that polymorphisms in its key target, vitamin K epoxide reductase (VKOR; see Fig. 24.5) and in CYP2C9, involved in its metabolism, are associated with outcomes. Figure 11.3 shows the effects of VKOR haplotype and of CYP2C9 genotype on the mean dose of warfarin needed to achieve therapeutic INR. Dosing algorithms have been proposed based on the results of testing for polymorphisms of these genes (Schwarz et al., 2008), and may come into general use.
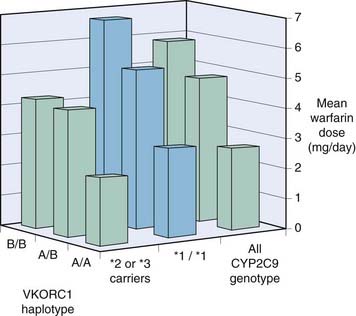
Fig. 11.3 Effect of VKOR haplotype and CYP2C9 genotype on warfarin dose.
186 patients on long-term warfarin treatment who had already been studied for CYP2C9 were studied retrospectively for genetic variants of VKOR (Rieder et al., 2005). VKOR haplotype as well as CYP2C9 genotype influenced the mean warfarin dose (which had been adjusted to achieve therapeutic INR). A, haplotypes 1 and 2; B, haplotypes 7, 8 and 9. A/A, A/B and B/B represent haplotype combinations. *1/*1 represents CYP2C9 wild type homozygotes; *2 and *3 represent CYP2C9 variants.
Figure redrawn from Beitelshees AL, McLeod HL 2006 Applying pharmacogenomics to enhance the use of biomarkers for drug effect and drug safety. TIPS 27: 498–502.
Twin studies as well as several well-documented single-gene disorders (including Mendelian chromosomal—autosomal recessive, dominant or sex-linked—and maternally inherited mitochondrial disorders) prove the concept that susceptibility to adverse drug effects, whether pharmacodynamic, pharmacokinetic or idiosyncratic, can be genetically determined. Pharmacogenomic testing offers the possibility of more precise ‘personalised’ therapeutics for several drugs and disorders. This is a field of intense research activity, rapid progress and high expectations, but proving that these tests add to present best practice and improve outcomes remains a challenge.