Chapter 29 Pulmonary vascular disease
 Pulmonary oedema occurs when increases in pulmonary capillary pressure or the permeability of the alveolar/capillary membrane cause fluid to accumulate in the interstitium and alveoli.
Pulmonary oedema occurs when increases in pulmonary capillary pressure or the permeability of the alveolar/capillary membrane cause fluid to accumulate in the interstitium and alveoli.Pulmonary Oedema
Pulmonary oedema is defined as an increase in pulmonary extravascular water, which occurs when transudation or exudation exceeds the capacity of the lymphatic drainage. In its more severe forms there is free fluid in the alveoli.
Anatomical Factors
The pulmonary capillary endothelial cells abut against one another at fairly loose junctions which are of the order of 5 nm wide.1 These junctions permit the passage of quite large molecules and the pulmonary lymph contains albumin at about half the concentration in plasma. Alveolar epithelial cells are connected by tight junctions at their alveolar surface with a gap of only about 1 nm. Under normal circumstances the tightness of these junctions prevents the escape of large molecules, such as albumin, from the interstitial fluid into the alveoli. However, the proteins that make up the tight junction are not simply passive structural units, and can, for example in response to nitric oxide, be modified and allow an increase in permeability across the tight junction.2,3
The lung has a well-developed lymphatic system draining the interstitial tissue through a network of channels around the bronchi and pulmonary vessels towards the hilum. Lymphatic vessels are seen in the juxtaseptal alveolar region (see below) and are commonly found in association with bronchioles. Down to airway generation 11 (see Table 2.1), the lymphatics lie in a potential space around the air passages and vessels, separating them from the lung parenchyma. In the hilum of the lung, the lymphatic drainage passes through several groups of tracheobronchial lymph glands, where they receive tributaries from the superficial subpleural plexus. Most of the lymph from the left lung usually enters the thoracic duct where it can be conveniently sampled in animals. The right side drains into the right lymphatic duct.
The normal lymphatic drainage from human lungs is astonishingly small – only about 10 ml per hour. However, lymphatic flow can increase up to ten times this value when transudation into the interstitial spaces is increased.4 This presumably occurs when pulmonary oedema is threatened but it cannot be conveniently measured in man.
Pulmonary Fluid Dynamics
For intravascular fluid to enter the alveoli it must traverse three barriers. First, it must move from the microcirculation into the interstitial space (across the endothelium), secondly through the interstitium and finally from the interstitial space into the alveoli (across the epithelium) (Figure 29.1).
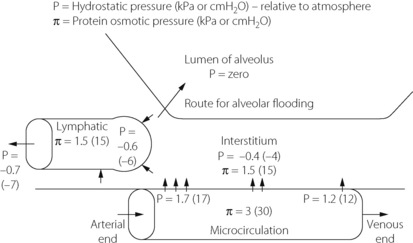
Fig. 29.1 Normal values for hydrostatic and plasma protein osmotic pressures in the pulmonary microcirculation and interstitium.
(Values taken from reference 5.)
Fluid exchange across the endothelium. This is promoted by the hydrostatic pressure difference between capillary and interstitium but counteracted by the osmotic pressure of the plasma proteins. The balance of pressures is normally sufficient to prevent any appreciable transudation but it may be upset in a wide variety of pathological circumstances.
It is customary to display the relationship between fluid flow and the balance of pressures in the form of the Starling equation. For the endothelial barrier this is as follows:
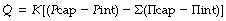
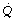 |
is the flow rate of transudated fluid which, in equilibrium, will be equal to the lymphatic drainage. |
| K | is the hydraulic conductance (i.e. flow rate of fluid per unit pressure gradient across the endothelium). |
| Pcap | is the hydrostatic pressure in the pulmonary capillary. |
| Pint | is the hydrostatic pressure in the interstitium. |
| Σ | is the reflection coefficient, in this case applying to albumin. It is an expression of the permeability of the endothelium to the solute (albumin). A value of unity indicates total reflection corresponding to zero concentration of the solute in the interstitial fluid. A value of zero indicates free passage of the solute across the membrane and, with equal concentrations on both sides of the membrane, the solute could exert no osmotic pressure across the membrane. This normally applies to the crystalloids in plasma. |
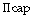 |
is the osmotic pressure the solute exerts within the pulmonary capillary. |
 |
is the osmotic pressure the solute exerts in the interstitium. |
Under normal circumstances in humans, the pulmonary lymph flow 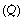 is about 10 ml per hour with a protein content about half that of plasma. The pulmonary microvascular pressure (Pcap) is in the range 0–2 kPa (0–15 mmHg), relative to atmosphere, depending on the vertical height in the lung field. Furthermore, there is a progressive decrease in capillary pressure from its arterial to its venous end, since approximately one-half the pulmonary vascular resistance is across the capillary bed (see Figures 7.2 and 29.1). In this context, it is meaningless to think of a single value for the mean pulmonary capillary pressure.
is about 10 ml per hour with a protein content about half that of plasma. The pulmonary microvascular pressure (Pcap) is in the range 0–2 kPa (0–15 mmHg), relative to atmosphere, depending on the vertical height in the lung field. Furthermore, there is a progressive decrease in capillary pressure from its arterial to its venous end, since approximately one-half the pulmonary vascular resistance is across the capillary bed (see Figures 7.2 and 29.1). In this context, it is meaningless to think of a single value for the mean pulmonary capillary pressure.
The hydrostatic pressure in the interstitial space (Pint) of the lung is not easy to measure. Animal studies using micropuncture techniques obtained subatmospheric pressures of −0.40 to −1.25 kPa (−4 to −12.5 cmH2O).6 In the excised lung there was no vertical gradient in interstitial pressures such as might have been expected from the effect of gravity, but this was observed when measurements were made with the chest and pleura intact.6
The reflection coefficient for albumin (Σ) in the healthy lung is about 0.5. The overall osmotic pressure gradient between blood and interstitial fluid is about 1.5 kPa (11.5 mmHg). Thus there is a fine balance between forces favouring and opposing transudation. There is a considerable safety marginin the upper part of the lung where the capillary hydrostatic pressure is lowest. However, in the dependent part of the lung, where the hydrostatic pressure is highest, the safety margin is slender.
Fluid dynamics within the interstitium. It is now accepted that the interstitium does not simply act as a passive conduit for fluid transfer to the lymphatics.7,8 Proteoglycan and hyaluron molecules are present in the pulmonary interstitium of animals, and function like a gel to absorb water to minimise increase in interstitial pressure and prevent hydration of other extracellular structures such as collagen.9 Regional differences in the properties of these molecules are believed to be responsible for the establishment of a pressure gradient between the septal interstitium and the juxtaseptal region where lymphatic channels originate. This gradient may promote, and allow some control of, fluid flow from the endothelium to the lymphatics in the normal lung.7
With increased fluid transfer across the endothelium, the interstitial space can accommodate large volumes of water with only small increases in pressure, the interstitial compliance being high. Some 500 ml can be accommodated in the interstitium and lymphatics of the human lungs with a rise of pressure of only about 0.2 kPa (2 cmH2O).5 Eventually, the capacity of the molecules to absorb water is exceeded, and the proteoglycan structure breaks down, possibly leading to disturbances of nearby collagen molecules and therefore basement membrane function, producing alveolar oedema.7 Alterations of interstitial proteoglycan structure during lung injury may contribute to the greater likelihood of pulmonary oedema under these circumstances (Chapter 31).
Fluid exchange across the alveolar epithelium. The permeability of this barrier to gases is considered in Chapter 9. It is freely permeable to gases, water and hydrophobic substances but virtually impermeable to albumin.
There is now considerable evidence of active fluid clearance from the alveoli in normal human lungs.3,10,11 For methodological reasons, most studies of this system have involved Type II alveolar epithelial cells, but the same processes are believed to occur in Type I cells and in clara cells in the distal airways. On the alveolar side of these cells, the cell membrane contains epithelial sodium channels (ENaC)12 and cystic fibrosis transmembrane conductance regulator (CFTR) channels (page 413), which actively pump sodium and chloride ions respectively into the cell.3 On the interstitial border of the cells, chloride moves passively out of the cell and the Na+/K+-ATPase channel actively removes sodium from the cell. Water from the alveolus follows these ion transfers down an osmotic gradient into the interstitium. Aquaporins are found in human alveolar epithelial cells, suggesting that water movement may be facilitated by these water channel proteins, but their role in normal adult lung remains unclear.3
A small amount of active clearance of fluid from the alveoli occurs under normal circumstances, but these systems become vital when pulmonary oedema threatens. Active removal of alveolar fluid by alveolar epithelial cells increases within one hour of the onset of oedema.13 Stimulation of β2 adrenoceptors by catecholamines increases the affinity of existing Na+/K+-ATPase channels for sodium and causes new channels to be incorporated into the cell membrane from intracellular endosomal stores. After a few hours, a variety of hormones13 (e.g. thyroxine, aldosterone, glucocorticoids) and cytokines3,11 (e.g. tumour necrosis factor) induce the transcription of new Na+/K+-ATPase channels and increase fluid clearance. These mechanisms are important both for minimising the severity of pulmonary oedema and clearing oedema fluid once the precipitating cause has resolved.
Stages of Pulmonary Oedema
There is presumably a prodromal stage in which pulmonary lymphatic drainage is increased, but there is no increase in extravascular water. This may progress to the following stages.
Stage I. Interstitial pulmonary oedema. In its mildest form, there is an increase in interstitial fluid but without passage of oedema fluid into the alveoli. With the light microscope this is first detected as cuffs of distended lymphatics, typically ‘8’-shaped around the adjacent branches of the bronchi and pulmonary artery (Figure 29.2). Electron microscopy shows fluid accumulation in the alveolar septa but this is characteristically confined to the ‘service’ side of the pulmonary capillary which contains the stroma, leaving the geometry of the ‘active’ side unchanged (see page 21 and Figure 2.7). Thus, gas exchange is better preserved than might be expected from the overall increase in lung water. Interstitial swelling is however not without risks, and swelling on the service side will eventually cause narrowing of the capillary lumen, though this does not occur until pulmonary oedema is advanced.
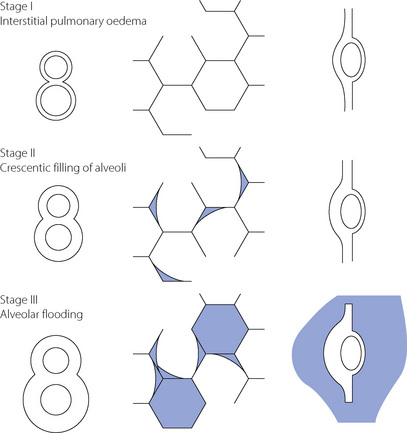
Fig. 29.2 Schematic diagram of the stages in the development of pulmonary oedema. On the left is shown the development of the cuff of distended lymphatics around the branches of the bronchi and pulmonary arteries. In the middle is the appearance of the alveoli by light microscopy (fixed in inflation). On the right is the appearance of the pulmonary capillaries by electron microscopy. The active side of the capillary is to the right. See text for details.
Physical signs are generally minimal in stage I, except perhaps for mild dyspnoea, particularly with exercise. The alveolar/arterial Po2 gradient is normal or only slightly increased.
Stage II. Crescentic filling of the alveoli. With further increase in extravascular lung water, interstitial oedema of the alveolar septa is increased and fluid begins to pass into some alveolar lumina. It first appears as crescents in the angles between adjacent septa, at least in lungs which have been fixed in inflation (Figure 29.2). The centre of the alveoli and most of the alveolar walls remain clear, and gas exchange is not grossly abnormal, but dyspnoea at rest is likely and the characteristic butterfly shadow may be visible on the chest radiograph.
Stage III. Alveolar flooding. In the third stage, there is quantal alveolar flooding. Some alveoli are totally flooded while others, frequently adjacent, have only the crescentic filling or else no fluid at all in their lumina. It seems that fluid accumulates up to a point at which a critical radius of curvature results in surface tension sharply increasing the transudation pressure gradient. This produces flooding on an all-or-none basis for each individual alveolus. Due to the effect of gravity on pulmonary vascular pressures (page 123), alveolar flooding tends to occur in the dependent parts of the lungs. Râles can be heard during inspiration and the lung fields show an overall opacity superimposed on the butterfly shadow.
Clearly there can be no effective gas exchange in the capillaries of an alveolar septum which is flooded on both sides, and blood flow through these alveoli constitutes venous admixture or shunt. This results in an increased alveolar/arterial Po2 gradient and hypoxaemia, which may be life threatening. Blood flow to the oedematous lung regions is slightly reduced by hypoxic pulmonary vasoconstriction (page 108), possibly in conjunction with interstitial swelling causing capillary narrowing (see above), but the shunt commonly remains substantial.
Hypercapnia is not generally a problem. In less severe pulmonary oedema, there is usually an increased respiratory drive, due partly to hypoxaemia and partly to stimulation of J receptors (page 68). As a result the Pco2 is usually normal or somewhat decreased.
Stage IV. Froth in the air passages. When alveolar flooding is extreme, the air passages become blocked with froth, which moves to and fro with breathing. This effectively stops all gas exchange and is rapidly fatal unless treated.
Aetiology of Pulmonary Oedema
On the basis of the Starling equation, it is possible to make a rational approach to the aetiology of pulmonary oedema. There are three groups of aetiological factors, classified according to their effect on factors in the Starling equation.
Increased capillary pressure (haemodynamic pulmonary oedema). This group comprises the commonest causes of pulmonary oedema. There is an elevation of the hydrostatic pressure gradient across the pulmonary capillary wall, until it exceeds the osmotic pressure of the plasma proteins. Interstitial fluid accumulates until it overwhelms the ability of the interstitium to absorb fluid and transport it to the lymphatics. Fluid then begins to enter the alveoli and will initially be actively removed by the alveolar epithelial cells until this system is also overwhelmed. The oedema fluid has a protein content which is less than that of normal pulmonary lymph or plasma.5 Apart from transudation in accord with the Starling equation, severe pulmonary capillary hypertension may result in loss of structural integrity (see below).
Causes of an increase in pulmonary capillary pressure are numerous:
 Absolute hypervolaemia may result from overtransfusion, from excessive and rapid administration of other blood volume expanders or from acute renal failure. Relative pulmonary hypervolaemia may result from redistribution of the circulating blood volume into the lungs. Examples of how this may occur include use of the Trendelenburg position or vasopressor drugs that act on the systemic circulation to a greater extent than the pulmonary circulation and so redirect blood into the pulmonary circulation. Raised pulmonary capillary pressure will inevitably result from an increase in pulmonary venous pressure.4,8 This may occur from any form of left heart failure, most commonly left ventricular failure or mitral valve lesions. Increased pulmonary blood flow may raise the pulmonary capillary pressure sufficiently to precipitate pulmonary oedema. This may result from a left-to-right cardiac shunt, anaemia or, rarely, as a result of exercise.
Absolute hypervolaemia may result from overtransfusion, from excessive and rapid administration of other blood volume expanders or from acute renal failure. Relative pulmonary hypervolaemia may result from redistribution of the circulating blood volume into the lungs. Examples of how this may occur include use of the Trendelenburg position or vasopressor drugs that act on the systemic circulation to a greater extent than the pulmonary circulation and so redirect blood into the pulmonary circulation. Raised pulmonary capillary pressure will inevitably result from an increase in pulmonary venous pressure.4,8 This may occur from any form of left heart failure, most commonly left ventricular failure or mitral valve lesions. Increased pulmonary blood flow may raise the pulmonary capillary pressure sufficiently to precipitate pulmonary oedema. This may result from a left-to-right cardiac shunt, anaemia or, rarely, as a result of exercise.Increased permeability of the alveolar/capillary membrane (permeability oedema). This group comprises the next commonest causes of pulmonary oedema. The mechanism is the loss of integrity of the alveolar/capillary membrane, allowing albumin and other macro-molecules to enter the alveoli. The osmotic pressure gradient which opposes transudation is then lost. The oedema fluid has a protein content that approaches that of plasma.
The alveolar/capillary membrane can be damaged either directly or indirectly by many agents, which are reviewed in Chapter 31. Apart from the possibility of the condition progressing to acute lung injury, permeability pulmonary oedema is always potentially very dangerous. The presence of protein in the alveoli tends to make the oedema refractory and the protein may become organised into a so-called hyaline membrane.
‘Stress failure’ of the pulmonary capillaries occurs when the pulmonary capillary pressure is increased in the range 3–5 kPa (30–50 cmH2O). Discontinuities appear in the capillary endothelium and type I alveolar epithelial cells, while the basement membrane often remains intact.4,14 This would seem to result in increased permeability and leakage of protein into the alveoli. The gaps tend to occur in the cell body, rather than at the junctions between the cells. High-altitude pulmonary oedema (page 286) is an example of this mechanism.
Decreased osmotic pressure of the plasma proteins. The Starling equation indicates that the osmotic pressure of the plasma proteins is a crucial factor opposing transudation. Although seldom the primary cause of pulmonary oedema, a reduced plasma albumin concentration is very common in the seriously ill patient and it must inevitably decrease the microvascular pressure threshold at which transudation commences.
Neurogenic pulmonary oedema may follow head injuries or other cerebral lesions. Evidence for the existence of pulmonary venous sphincters has provided a possible mechanism for neurogenic pulmonary oedema.15 Constriction of these sphincters, either due to circulating adrenaline or a neural response, could cause an abrupt increase in pulmonary capillary pressure. A study of neurogenic pulmonary oedema in humans supported this hypothesis by demonstrating that the oedema fluid often has a low protein content suggesting a haemodynamic mechanism (see above).15
Re-expansion pulmonary oedema is described on page 445 and pulmonary oedema following lung resection on page 495.
Principles of Therapy
Immediate treatment aims to restore the arterial Po2 to normal values. The inspired oxygen concentration should be increased, up to 100% if necessary. Sitting the patient up is a simple way to reduce central blood volume. Treatment of the underlying cause of pulmonary oedema follows directly from the Starling equation and an understanding of the aetiology.
Haemodynamic pulmonary oedema. Treatment aims to reduce left atrial pressure. Depending on the precise aetiology, treatment is directed towards improvement of left ventricular function and/or reduction of blood volume. The latter may be quickly and easily achieved by peripheral vasodilatation. Drugs that predominantly dilate the capacitance (venous) system, such as nitrates or angiotensin-converting enzyme inhibitors, will be most effective. This mechanism is probably also responsible for the beneficial effects of furosemide and diamorphine in the acute situation. Diuretics act more slowly but are useful for long-term treatment. Essentially the patient is titrated to the left along his Frank–Starling curve (Figure 29.3). In addition the curve is moved upwards and to the left, if this is possible, using positive inotropes as an adjunct to correction of left ventricular malfunction, for example from ischaemia. The further the curve can be moved, the greater will be that part of it lying in the safe quadrant between low cardiac output on one hand and pulmonary oedema on the other.
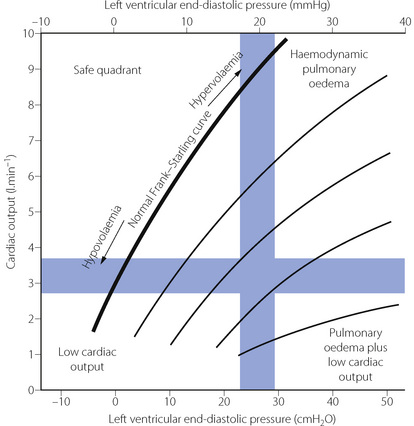
Fig. 29.3 Quadrant diagram relating cardiac output to left ventricular end-diastolic pressure. The thick curve is a typical normal Frank–Starling curve. To the right are shown curves representing progressive left ventricular failure. Top left is the safe quadrant, which contains a substantial part of the normal curve, but much less of the curves representing ventricular failure. Top right is the quadrant representing normal cardiac output but raised left atrial pressure, attained at the upper end of relatively normal Frank–Starling curves (e.g. hypervolaemia). There is a danger of haemodynamic pulmonary oedema. Bottom left is the quadrant representing normal or low left atrial pressure but low cardiac output, attained at the lower end of all curves (e.g. hypovolaemia). The patient is in shock. Bottom right is the quadrant representing both low cardiac output and raised left atrial pressure. There is simultaneous danger of pulmonary oedema and shock, and the worst Frank–Starling curves hardly leave this quadrant.
Permeability pulmonary oedema. Treatment should be directed towards restoration of the integrity of the alveolar/capillary membrane. Unfortunately, no particularly successful measures are available towards this end. It is, however, important to minimise left atrial pressure even though this is not the primary cause of the oedema. Attempts may be made to increase the plasma albumin concentration if it is reduced.
Artificial ventilation and positive end-expiratory pressure (PEEP). Severe pulmonary oedema causes degrees of hypoxia that may quickly be lethal. Tracheal intubation and positive pressure ventilation is therefore commonly required, and the results are often spectacular. Froth in the airways may be aspirated, and any areas of atelectasis occurring along with the oedema improved. Artificial ventilation is often combined with PEEP, resulting in further improvements in arterial Po2. It was originally thought that the positive pressures drove the fluid back into the circulation, but evidence that extravascular lung water is reduced by PEEP is contradictory, with few human studies. Animal studies of pulmonary oedema indicate that by increasing the lung volume, the capacity of the interstitium to hold liquid is increased.16 Similarly, with haemodynamic pulmonary oedema in dogs, PEEP does not alter the total amount of lung water but a greater proportion is in the extra-alveolar interstitial space,17 and lymphatic drainage is increased.18 With haemodynamic pulmonary oedema positive pressure ventilation has beneficial effects on the function of the failing heart (page 480) and it is probably this effect, rather than any effect on the lungs, that causes the clinical benefit in humans.
Clinical Measurement
Pulmonary vascular pressures. As an indication of impending or actual haemodynamic pulmonary oedema, the most physiological measurement is the pulmonary artery occlusion pressure (page 113), which equates to pulmonary venous pressure. In clinical practice the use of this technique is declining, with less invasive estimates of pulmonary vascular pressures being preferred such as echocardiography.
Permeability of the alveolar/capillary membrane. The only practical approach in humans is measurement of the rate of loss of a gamma-emitting tracer molecule from the lung into the circulation. The most sensitive tracer is 99mTcDTPA (metastable technetium-99-labelled diethylene triamine penta-acetate, molecular weight 492 daltons).19 The half-time of clearance from the lung fields is usually in the range 40–100 minutes in the healthy non-smoker. The half-time is reduced below 40 minutes following a variety of lung insults. However, it is within the range 10–40 minutes in apparently healthy smokers and this limits its scope for the early detection of a damaged alveolar/capillary membrane.
Lung water. Measurement of lung water in the intact subject has proved difficult. A great deal of effort has been devoted to the double indicator method. This uses the techniques for the measurement of pulmonary or central blood volume by dye dilution but with two indicators. One indicator is chosen to remain within the circulation while the other (usually ‘coolth’ or tritiated water) diffuses into the interstitial fluid. Extravascular lung water is then derived as the difference between the volumes as measured with the two indicators. These methods are limited because a high level of accuracy is required to demonstrate small changes in lung water, though clinically acceptable results may now be obtained reasonably conveniently.20
Pulmonary Embolism
The pulmonary circulation may be occluded by embolism, which may be gas, thrombus, fat, tumour or foreign body. The architecture of the microvasculature is well adapted to minimise the effects of embolism. Large numbers of pulmonary capillaries tend to arise at right angles from metarterioles and there are abundant anastomoses throughout the microcirculation. This tends to preserve circulation distal to the impaction of a small embolus. Nevertheless, a large pulmonary embolus is a serious and potentially lethal condition.
Thromboembolism21,22
The commonest pulmonary embolus consists of detached venous thromboses from veins in the thigh and the pelvic venous plexuses. Smaller thrombi are filtered in the lungs without causing symptoms but larger emboli may impact in major vessels, typically at a bifurcation forming a saddle embolus. There may be a catastrophic increase in pulmonary vascular resistance with acute right heart failure or cardiac arrest.
Diagnosis of pulmonary thrombo-embolus.22 Massive pulmonary thrombo-embolus causes rapid cardiac arrest and death, and over half the cases are undiagnosed except at autopsy.23 Similarly, small pulmonary emboli may be completely asymptomatic, but often precede more significant, or lethal, embolism later. For patients with intermediate-sized emboli, a combination of pleuritic chest pain, dyspnoea and tachypnoea is indicative of pulmonary embolus. Changes in the electrocardiogram following pulmonary embolus reflect disturbed right-sided cardiac function secondary to elevated pulmonary arterial pressure and are generally non-specific and only occur after a large embolism. A variety of imaging techniques are available, with computed tomography (CT) scanning now regarded as the investigation of choice for diagnosis of pulmonary thrombo-embolism (Figure 29.4). Pulmonary radioisotope perfusion or ventilation-perfusion scans may detect smaller emboli not seen with CT scanning, but the technique has low sensitivity,24 particularly in patients with pre-existing lung disease.
Fig. 29.4 Spiral computed tomographic scan of pulmonary thomboemboli. Intravenous contrast injected immediately before scanning makes the blood vessels appear white. Emboli then appear as darker areas within the blood vessel lumen. Saddle embolus (SE) situated mainly in the right pulmonary artery (RPA). AA, ascending aorta; DA, descending aorta; LPA, left pulmonary artery. (I am indebted to Celia Craven, Superintendent Radiographer, St James’s Hospital, Leeds for supplying the scans.)
Pathophysiology.21,25 Three mechanisms give rise to the physiological changes seen in pulmonary embolism. First is physical occlusion of the pulmonary vascular system. Second, platelet activation within the thrombus leads to release of 5-hydroxytryptamine (5-HT, serotonin) and thromboxane A2, causing a further increase in pulmonary vascular resistance. Finally, the right ventricle commonly is unable to overcome the raised pulmonary vascular resistance and cardiac output falls, eventually culminating in right heart failure.
The primary respiratory lesion is an increase in alveolar dead space with an increased arterial/end-tidal Pco2 gradient. Carbon dioxide elimination is therefore reduced and if ventilation remains unchanged arterial Pco2 slowly climbs, until elimination is restored in spite of the large dead space.26 However, in awake patients hypercapnia is unusual because hyperventilation is almost always present and arterial Pco2 is usually below the normal range.25 The cause of respiratory stimulation is unclear, but may involve stimulation of J-receptors as in air embolism (see below), or hypoxia if present.
Arterial Po2 is also decreased. This results from derangement of normal ventilation perfusion relationships. Initially, whilst cardiac output remains normal, partial obstruction of the pulmonary circulation results in excessive blood flow to those lung regions that are still perfused, giving a low ventilation/perfusion ratio in these areas. When cardiac output begins to decrease as a result of a failing right ventricle, pulmonary perfusion will fall below normal levels and low mixed venous oxygen content will exacerbate the abnormal ventilation/perfusion relationships (page 134). Elevated right atrial pressures, as a consequence of pulmonary hypertension, may cause right-to-left intracardiac shunting21 through an unsuspected patent foramen ovale (page 217).
Bronchospasm is a well recognised complication27 and has been attributed to the 5-HT released from platelets and also to local hypocapnia in the part of the lung without effective pulmonary circulation. Pulmonary compliance may be reduced with large pulmonary emboli, but the mechanism of this change is unknown. Pulmonary infarction, which might be expected to occur, is rarely a problem. The lung can obtain oxygen directly from air within the airways and alveoli, from backflow along pulmonary veins, and from the bronchial circulation. Only when these sources are also impaired does infarction occur, for example when localised pulmonary oedema or pulmonary haemorrhage into the airways occurs in conjunction with embolism.
Principles of therapy.22 Anticoagulation with intravenous heparin is the mainstay of treatment and prevents further clot from forming, either in lung or elsewhere, and allows endogenous fibrinolysis to proceed. If right ventricular dysfunction occurs, which is relatively easy to assess using echocardiography,28 or haemodynamic instability is present due to a low cardiac output, thrombolytic therapy may also be used. Surgical embolectomy is now reserved for patients with significant pulmonary embolism who are unable to receive, or unresponsive to, other forms of treatment.
Air Embolism
An embolus may arise from pneumothorax or pulmonary barotrauma but is most commonly iatrogenic. In neurosurgery, the usual cause of air embolism is the use of the sitting position for posterior fossa surgery. A subatmospheric venous pressure at the operative site allows air to enter dural veins, which are held open by their structure. In open cardiac surgery, it is almost impossible to remove all traces of air from the cardiac chambers before closing the heart. Some small degree of air embolism is almost inevitable in all types of intravenous therapy, but catastrophic air embolism can occur when compression bags are used to accelerate the flow rate of intravenous fluids or blood bags that accidentally already contain air.
Detection of air embolism. Early diagnosis of air embolism is essential in neurosurgery, and there are three principal methods in routine use. Bubbles in circulating blood give a very characteristic sound with a precordial Doppler probe. The method is, if anything, too sensitive, because a shower of very small bubbles produces a particularly large signal. The simplest method is based on the end-expired CO2 concentration, which is easily measured from capnography. Many factors influence the end-expiratory concentration (page 169) but a sudden decrease is likely to be either cardiac arrest or air embolism. Transoesophageal echocardiography is an efficient method of detecting air embolism29 and, furthermore, it is the only practicable method of detecting paradoxical air embolism (see below).
Pathophysiology of air embolus. Provided there is no major intracardiac right-to-left shunt, small quantities of air are filtered out by the lungs where they are gradually excreted and little harm results. Alveolar dead space is increased according to the proportion of the pulmonary circulation that is occluded. The resultant increase in arterial/end-expiratory Pco2 gradient is the basis of detection of air embolism by capnography as described above. Pulmonary arterial pressure is increased by a large embolus due to the right ventricle working against an increased pulmonary vascular resistance. Finally, in animal studies, airway resistance is increased following air embolism, an effect mediated by arachidonic acid metabolites, possibly in conjunction with platelet activation and stimulation of pulmonary irritant receptors.30
Massive air embolism (probably in excess of 100 ml) may cause cardiac arrest by accumulation in the right ventricle, where compression of the air bubble prevents ventricular ejection of blood. Treatment then requires aspiration of air through a cardiac catheter, which is difficult. In lesser degrees of embolisation during surgery, reduced cardiac output probably also contributes to the sudden reduction in end-expiratory Pco2.
Paradoxical air embolism. Rarely, there may be passage of air emboli from the right to left heart without there being an overt right-to-left shunt. This is important because air then enters the systemic arterial circulation where there may be embolism and infarction, particularly of the brain. It is possible to pass a probe through such a foramen ovale in over 25% of the adult population (page 217), but paradoxical embolism does not usually occur because pressure is slightly higher in the left atrium than the right. However, under many circumstances, such as following pulmonary embolism, right atrial pressure may be elevated to the point that a right-to-left shunt occurs.
Fat Embolism31
Fracture of long bones or major orthopaedic surgery may be associated with fat embolism.32 This term is not strictly correct, as the features of ‘fat embolism syndrome’ result from release of bone marrow micro-emboli. Some degree of fat embolism occurs in almost all patients having hip and knee replacement surgery, but clinical sequelae occur in less than 1% of these.
Microscopic intravascular bone marrow fragments promote intravascular coagulation and platelet adherence, particularly under the conditions of venous stasis present during surgery, and so develop into larger ‘mixed’ emboli. There is initially an increase in physiological dead space but this is soon accompanied by an increase in shunt. Release of inflammatory mediators in the lung causes bronchospasm, increases capillary permeability and leads to localised pulmonary oedema.32
Lipid seems to pass through the pulmonary circulation to invade the systemic circulation. Surface forces between blood and lipid are much less than between blood and air and so would not offer the same hindrance to passage through the lungs. In the systemic circulation, fat emboli cause characteristic petechiae in the anterior axillary folds and there is often evidence of cerebral involvement.31
Amniotic Fluid Embolism33,34
Amniotic fluid embolism occurs rarely during delivery, but is fatal in over half of cases. Death normally results from cardiovascular disturbances and haemorrhage secondary to coagulopathy. Pulmonary vascular resistance is increased, but animal studies indicate that pulmonary hypertension is only transient, returning to normal after just a few minutes. The reasons for this effect on the pulmonary circulation remain unclear. Amniotic fluid and fetal cells in the circulation may cause no cardiovascular changes, and either an immune-mediated response, or the release of vasoactive mediators such as endothelin or prostaglandins have been suggested as mechanisms causing the clinical syndrome.
Pulmonary Hypertension
There are many causes of pulmonary hypertension, which are classified as either primary or secondary (Table 29.1). The latter is much more common, and is therefore considered first.
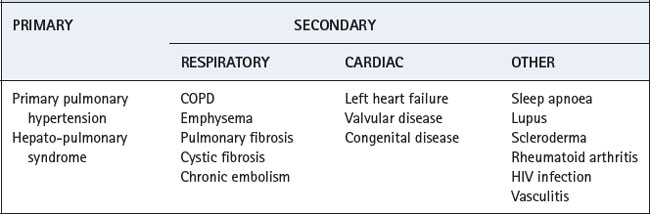
Secondary Pulmonary Hypertension
Respiratory disease. Pulmonary vascular resistance is increased by almost any pulmonary disease that results in chronic hypoxia (Table 29.1). Similar changes occur with intermittent hypoxia caused, for example, by sleep apnoea (Chapter 16). The change is initially temporary and reversible but progresses to become permanent. Nitric oxide (NO) production by pulmonary endothelium contributes to the normal low resistance of the pulmonary circulation (page 107). Hypoxia has been shown to reduce this basal NO secretion,35 and further work has identified reduced production of constitutive nitric oxide synthase as the mechanism.36
Cardiac disease. Valvular disease of the left heart leads to an elevation of pressure in the left atrium and pulmonary veins. Increases in pulmonary capillary pressure from this cause tend to be long-term, and lead to remodelling of the pulmonary circulation. Smooth muscle hypertrophy and fibrosis of the pulmonary vasculature cause pulmonary arterial hypertension and, eventually, right heart failure (cor pulmonale).4,8 A low cardiac output, either from the original valvular heart disease or the resulting right heart failure, results in reduction of mixed venous Po2, which then causes further increases in pulmonary vascular resistance.
Treatment should first be directed towards improving the underlying condition, particularly if this is causing chronic or intermittent hypoxia. Long-term administration of oxygen, during the day and during sleep, is beneficial and recommended for any patient with hypoxia and pulmonary hypertension.37 Vasodilator therapy is complicated by the lack of drugs with specific action on the pulmonary circulation, and is discussed below.
Primary Pulmonary Hypertension38,39
Pulmonary hypertension occurring in the absence of hypoxia is termed primary pulmonary hypertension (PPH) and has a prevalence of approximately 1300 per million. It is a progressive disease, which normally presents in early adulthood with worsening shortness of breath and eventually right heart failure. There is a familial contribution to PPH, and it may rarely be associated with advanced liver disease or the use of some older appetite suppressant drugs. Prognosis is poor, with most patients dying within three years of diagnosis.
Pathophysiology. The disease is characterised by proliferation of endothelial cells, hypertrophy of pulmonary arterial smooth muscle, and by thrombosis within pulmonary vessels.40 Abnormal endothelial function is believed to be where the primary defect occurs, and nitric oxide related functions are abnormal. The defect seems to arise in communication between endothelial and smooth muscle cells, though this has yet to be fully characterised.41
Treatment.37,38,42,43 In recent years a variety of drugs have been developed that lower pulmonary arterial pressures (page 111), all of which are now used for treating PPH. Prostacyclin and its analogues are the mainstays of PPH therapy, particularly now that orally active agents are becoming available.44 Endothelin receptor antagonists are also now in routine use, though the debate continues about whether selective or non-selective drugs are best.45 PPH remains a common indication for lung transplantation (Chapter 33).
Hepatopulmonary Syndrome (HPS)
This clinical syndrome describes the combination of liver disease, pulmonary vascular dilatation and impaired oxygenation.46 In this syndrome the pulmonary circulation becomes abnormally dilated. Pulmonary capillary diameter at rest is normally <7 μm, though with physiological increases in cardiac output, for example during exercise, this may increase up to 15 μm. In HPS pulmonary capillaries can be as large as 100 μm in diameter, and pulmonary arteriovenous or portopulmonary shunts may develop. Hypoxia is therefore the result of widespread areas with low ventilation/perfusion ratios, including shunts. With a high cardiac output state, which is common in liver failure, a diffusion barrier also develops when a large blood flow passes through dilated pulmonary capillaries leaving insufficient time for diffusion of oxygen into the blood (Chapter 9).
Pulmonary capillary dilatation in HPS results from excessive nitric oxide production, levels of which are increased in expired breath analysis (page 414). What stimulates the excess NO production is unknown, with contenders including production of endothelin-1 or tumour necrosis factor alpha (TNFα). Excessive production of carbon monoxide by the haemoxygenase system has also been implicated as carboxyhaemoglobin levels are high in patients with HPS.47
Treatment of HPS with NO antagonists has shown variable results, though using a TNFα antagonist may be useful for its prevention. Liver transplantation reverses the syndrome, though the time taken for the lung to recover is variable.
References
1. DeFouw DO. Ultrastructural features of alveolar epithelial transport. Am Rev Respir Dis.. 1983;127(suppl 5):S9-S13.
2. Bhattacharya J. The alveolar water gate. Am J Physiol Lung Cell Mol Physiol.. 2004;286:L257-L258.
*3. Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev.. 2002;82:569-600.
4. Gehlbach BK, Geppert E. The pulmonary manifestations of left heart failure. Chest. 2004;125:669-682.
5. Staub NA. Pathophysiology of pulmonary oedema. In: Staub NA, Taylor AE, editors. Edema. New York: Raven Press, 1984.
6. Miserocchi G, Negrini D, Gonano C. Direct measurement of interstitial pulmonary pressure in in situ lung with intact pleural space. J Appl Physiol.. 1990;69:2168-2174.
7. Bhattacharya J. The microphysiology of lung liquid clearance. Adv Exp Med Biol.. 1995;381:95-108.
8. Drake RE, Doursout MF. Pulmonary edema and elevated left atrial pressure: Four hours and beyond. News Physiol Sci.. 2002;17:223-226.
9. Negrini D, Passi A, De Luca G, Miserocchi G. Pulmonary interstitial pressure and proteoglycans during development of pulmonary oedema. Am J Physiol.. 1996;270:H2000-H2007.
10. Matthay MA, Clerici C, Saumon G. Active fluid clearance from the distal air spaces of the lung. J Appl Physiol.. 2002;93:1533-1541.
11. Sznajder JI, Factor P, Ingbar DH. Lung edema clearance: role of Na+-K+-ATPase. J Appl Physiol.. 2002;93:1860-1866.
12. Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol.. 2009;71:403-423.
13. Crandall ED, Matthay MA. Alveolar epithelial transport. Basic science to clinical medicine. Am J Respir Crit Care Med.. 2001;162:1021-1029.
14. Tsukimoto K, Mathieu-Costello O, Prediletto R, Elliott AR, West JB. Ultrastructural appearances of pulmonary capillaries at high transmural pressures. J Appl Physiol.. 1991;71:573-582.
15. Smith WS, Matthay MA. Evidence for a hydrostatic mechanism in human neurogenic pulmonary edema. Chest. 1997;111:1326-1333.
16. Gee MH, Williams DO. Effect of lung inflation on perivascular cuff fluid volume in isolated dog lung lobes. Microvasc Res.. 1979;17:192-196.
17. Paré PD, Warriner B, Baile EM, Hogg JC. Redistribution of pulmonary extravascular water with positive end-expiratory pressure in canine pulmonary edema. Am Rev Respir Dis.. 1983;127:590-593.
18. Mondéjar EF, Mata GV, Cärdenas A, Mansilla A, Cantalejo F, Rivera R. Ventilation with positive end-expiratory pressure reduces extravascular lung water and increases lymphatic flow in hydrostatic pulmonary oedema. Crit Care Med.. 1996;24:1562-1567.
19. Jones JG, Royston D, Minty BD. Changes in alveolar-capillary barrier function in animals and humans. Am Rev Respir Dis.. 1983;127(suppl 5):S51-S59.
20. Godje O, Peyerl M, Seebauer T, Dewald O, Reichart B. Reproducibility of double indicator dilution measurements of intrathoracic blood volume compartments, extravascular lung water, and liver function. Chest. 1998;113:1070-1077.
21. Goldhaber SZ, Elliott CG. Acute pulmonary embolism: Part I. Epidemiology, pathophysiology, and diagnosis. Circulation. 2003;108:2726-2729.
*22. Tapson VF. Acute pulmonary embolism. N Engl J Med.. 2008;358:1037-1052.
23. Ryu JH, Olson EJ, Pellikka PA. Clinical recognition of pulmonary embolism: problem of unrecognized and asymptomatic cases. Mayo Clin Proc.. 1998;73:873-879.
24. PIOPED investigators. Value of the ventilation/perfusion scan in acute pulmonary embolism: results of the Prospective Investigation of Pulmonary Embolism Diagnosis (PIOPED). JAMA. 1990;263:2753-2759.
25. Santolicandro A, Prediletto R, Fornai E, et al. Mechanisms of hypoxemia and hypocapnia in pulmonary embolism. Am J Respir Crit Care Med.. 1995;152:336-347.
26. Breen PH, Mazumdar B, Skinner SC. How does experimental pulmonary embolism decrease CO2 elimination? Respir Physiol.. 1996;105:217-224.
27. Windebank WJ, Boyd G, Moran F. Pulmonary thromboembolism presenting as asthma. BMJ. 1973;1:90-94.
28. Goldhaber SZ. Assessing the prognosis of acute pulmonary embolism. Tricks of the trade. Chest. 2008;133:334-336.
29. Furuya H, Okumura F. Detection of paradoxical air embolism by transesophageal echocardiography. Anesthesiology. 1984;60:374-377.
30. Chen HF, Lee BP, Kou YR. Mechanisms underlying stimulation of rapidly adapting receptors during pulmonary air embolism in dogs. Respir Physiol.. 1997;109:1-13.
31. Mellor A, Soni N. Fat embolism. Anaesthesia. 2001;56:145-154.
32. Hofmann S, Huemer G, Salzer M. Pathophysiology and management of the fat embolism syndrome. Anaesthesia. 1998;53(suppl 2):35-37.
33. Davies S. Amniotic fluid embolus: a review of the literature. Can J Anaesth.. 2001;48:88-98.
34. O’Shea A, Eappen S. Amniotic fluid embolism. Int Anesthesiol Clin.. 2007;45:17-28.
35. Adnot S, Raffestin B. Pulmonary hypertension: NO therapy? Thorax. 1996;51:762-764.
36. McQuillan L, Leung G, Marsden P, Kostyk S, Kourembanas S. Hypoxia inhibits expression of NOS via transcriptional and posttranscriptional mechanisms. Am J Physiol.. 1994;36:H1921-H1927.
37. Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV. Medical therapy for pulmonary arterial hypertension. Updated ACCP evidence-based clinical practice guidelines. Chest. 2007;131:1917-1928.
38. Humbert M. Update in pulmonary arterial hypertension 2007. Am J Respir Crit Care Med.. 2008;177:574-579.
39. Gaine SP, Rubin LJ. Primary pulmonary hypertension. Lancet. 1998;352:719-725.
40. Rich S. Clinical insights into the pathogenesis of primary pulmonary hypertension. Chest. 1998;114:237S-241S.
*41. Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159-165.
42. Peacock AJ. Treatment of pulmonary hypertension. BMJ. 2003;326:835-836.
43. National Pulmonary Hypertension Centres of the UK and Ireland. Consensus statement on the management of pulmonary hypertension in clinical practice in the UK and Ireland. Thorax. 2008;63:ii1-ii41.
44. Gomberg-Maitland M, Olschewski H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur Respir J. 2008;31:891-901.
45. Dupuis J, Hoeper MM. Endothelin receptor antagonists in pulmonary arterial hypertension. Eur Respir J. 2008;31:407-415.
46. Rodriguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome – a liver-induced lung vascular disorder. N Engl J Med.. 2008;358:2378-2387.
47. Arguedas MR, Drake BB, Kapoor A, Fallon MB. Carboxyhemoglobin levels in cirrhotic patients with and without hepatopulmonary syndrome. Gastroenterology. 2005;128:328-333.