Chapter 23 Changes in the carbon dioxide partial pressure
 Hypocapnia occurs when alveolar ventilation is excessive relative to carbon dioxide production and usually results from hyperventilation due to hypoxia, acidosis or lung disease. Hypercapnia most commonly occurs because of inadequate alveolar ventilation from a multitude of causes, or more rarely from increased carbon dioxide production.
Hypocapnia occurs when alveolar ventilation is excessive relative to carbon dioxide production and usually results from hyperventilation due to hypoxia, acidosis or lung disease. Hypercapnia most commonly occurs because of inadequate alveolar ventilation from a multitude of causes, or more rarely from increased carbon dioxide production.Routine monitoring of end-expiratory and arterial Pco2 means it should now be possible to avoid both hypo- and hypercapnia under almost all clinical circumstances. However, interest in hypercapnia has continued over recent years for two reasons. First, changes in the approach to artificial ventilation in severe lung injury have led to the use of ‘permissive hypercapnia’ (page 457). In order to minimise pulmonary damage, minute volume of ventilation is maintained deliberately low, and the arterial Pco2 is allowed to increase. Secondly, a massive expansion of laparoscopic surgical techniques, mostly using carbon dioxide for abdominal insufflation, has led to the anaesthetist having to control arterial Pco2 under conditions of significantly increased pulmonary carbon dioxide output (page 347).
Before describing the effects of carbon dioxide on various physiological systems, this chapter will briefly outline the causes of changes in arterial Pco2.
Causes of Hypocapnia1
Hypocapnia can result only from an alveolar ventilation that is excessive in relation to carbon dioxide production. Low values of arterial Pco2 are commonly found, resulting from artificial ventilation with an excessive minute volume or from voluntary hyperventilation due to psychological disturbance such as hysteria. A low arterial Pco2 may also result simply from hyperventilation during arterial puncture. Persistently low values may be due to an excessive respiratory drive resulting from one or more of the following causes.
Hypoxaemia is a common cause of hypocapnia, occurring in congenital heart disease with right-to-left shunting, residence at high altitude, pulmonary pathology or any other condition that reduces the arterial Po2 below about 8 kPa (60 mmHg). Hypocapnia secondary to hypoxaemia opposes the ventilatory response to the hypoxaemia (page 73).
Metabolic acidosis produces a compensatory hyper-ventilation (air hunger), which minimises the fall in pH that would otherwise occur. This is a pronounced feature of diabetic ketoacidosis; arterial Pco2 values below 3 kPa (22.5 mmHg) are not uncommon in severe metabolic acidosis. This is a vital compensatory mechanism. Failure to maintain the required hyperventilation, either from fatigue or inadequate artificial ventilation leads to a rapid life-threatening decrease in arterial pH.
Mechanical abnormalities of the lung may drive respiration through the vagus, resulting in moderate reduction of the Pco2. Thus conditions such as pulmonary fibrosis, pulmonary oedema and asthma are usually associated with a low to normal Pco2 until the patient passes into type 2 respiratory failure (page 393).
Neurological disorders may result in hyperventilation and hypocapnia. This is most commonly seen in those conditions that lead to the presence of blood in the cerebrospinal fluid, such as occurs following head injury or subarachnoid haemorrhage.
Causes of Hypercapnia
It is uncommon to encounter an arterial Pco2 above the normal range in a healthy subject. Any value of more than 6.1 kPa (46 mmHg) should be considered abnormal, but values up to 6.7 kPa (50 mmHg) may be attained by breath holding. It is difficult for the healthy subject to exceed this level by any respiratory manoeuvre other than by breathing mixtures of carbon dioxide in oxygen.
When a patient is hypercapnic, there are only four possible causes:
Though not strictly an increase in production, absorption of carbon dioxide from the peritoneum during laparoscopic surgery has the same respiratory effects and is described on page 347.
Effects of Carbon Dioxide on the Nervous System
A number of special difficulties hinder an understanding of the effects of changes in Pco2 on any physiological system. First, there is the problem of species difference, which is a formidable obstacle to the interpretation of animal studies in this field. The second difficulty arises from the fact that carbon dioxide can exert its effect either directly, or in consequence of (respiratory) acidosis. The third difficulty arises from the fact that carbon dioxide acts at many different sites in the body, sometimes producing opposite effects on a particular function, such as blood pressure (see below).
Carbon dioxide has at least five major effects on the brain:
 It is the main factor influencing the intracellular pH, which is known to have important effects on the metabolism, and therefore function, of the cell. It may be presumed to exert the inert gas narcotic effect in accord with its physical properties, which are similar to those of nitrous oxide. It influences the excitability of certain neurones, particularly relevant in the case of the reticuloactivating system.
It is the main factor influencing the intracellular pH, which is known to have important effects on the metabolism, and therefore function, of the cell. It may be presumed to exert the inert gas narcotic effect in accord with its physical properties, which are similar to those of nitrous oxide. It influences the excitability of certain neurones, particularly relevant in the case of the reticuloactivating system.The interplay of these effects is difficult to understand, although the gross changes produced are well established.
Effects On Consciousness
Carbon dioxide has long been known to cause unconsciousness in dogs entering the Grotto del Cane in Italy, where carbon dioxide issuing from a fumarole forms a layer near the ground. It has been widely used as an anaesthetic for short procedures in small laboratory animals. Inhalation of 30% carbon dioxide is sufficient for the production of anaesthesia in humans, but is complicated by the frequent occurrence of convulsions.2 In patients with ventilatory failure, carbon dioxide narcosis occurs when the Pco2 rises above 12–16 kPa (90–120 mmHg).3
Narcosis by carbon dioxide is probably not due primarily to its inert gas narcotic effects, because its oil solubility predicts a very much weaker narcotic than it seems to be. It is likely that the major effect on the central nervous system is by alteration of the intracellular pH, with consequent derangements of metabolic processes. In animals the narcotic effect correlates better with cerebrospinal fluid pH than with arterial Pco2.4
The effects of inhaling low concentrations of carbon dioxide for a prolonged period of time are described on page 305.
Cerebral Blood Flow5
Cerebral blood flow (CBF) increases with arterial Pco2 at a rate of about 7–15 ml.100g−1.min−1 for each kPa increase in Pco2 (1–2 ml.100g−1.min−1 per mmHg) within the approximate range 3–10 kPa (20–80 mmHg). The full response curve is S-shaped (Figure 23.1). The response at very low Pco2 is probably limited by the vasodilator effect of tissue hypoxia, and the response above 16 kPa (120 mmHg) seems to represent maximal vasodilatation. The changes shown in Figure 23.1 represent the brain as a whole and it is not possible to generalise about regional changes.
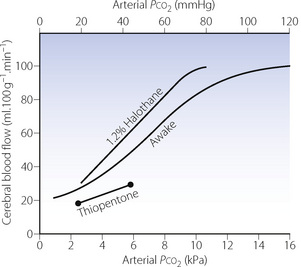
Mechanisms. In the intact animal, CBF is increased in response to Pco2 by a combination of vasodilatation of cerebral blood vessels and an increase in blood pressure (see below). Changes in Pco2 lead to a complex series of events that bring about vasodilation of cerebral blood vessels.5 In adults, the effect is initiated by changes in the extracellular pH in the region of the arterioles, which alters intracellular calcium levels both directly and indirectly via nitric oxide production and the formation of cyclic GMP. With prolonged hypocapnia, and to a lesser extent hypercapnia, changes in cerebral blood flow return towards baseline after a few hours,5,6 an effect thought to result from changes in cerebro-spinal fluid pH correcting the extracellular acidosis. Sensitivity of the cerebral circulation to carbon dioxide may be lost in a variety of pathological circumstances such as cerebral tumour, infarction or trauma. There is commonly a fixed vasodilatation in damaged areas of brain, which if widespread may cause dangerous increases in intracranial pressure.
Anaesthesia. Inhalational anaesthetics have a direct cerebral vasodilator effect and increase normocapnic CBF considerably.7,8 They also accentuate the response to both hypocapnia and hypercapnia; that is, they increase the slope of the relationship between Pco2 and CBF (Figure 23.1). In spite of the increased slope during hypocapnia, global CBF during anaesthesia with hyperventilation is normally still greater than when awake.5 Intravenous anaesthetics such as thiopentone and propofol9 reduce CBF at normal Pco2 in accordance with the reduced cerebral oxygen consumption. Vasoconstriction in response to hyperventilation continues to occur (Figure 23.1), but at deeper levels of anaesthesia the response is reduced compared with when awake.5
Intracranial pressure (ICP) tends to rise with increasing Pco2 as a result of cerebral vasodilatation. Hyperventilation was used for many years as a standard method of acutely reducing ICP after head injury, but the reduction in ICP may only be short-lived and the effects on CBF are variable. The possibility of increased CBF as a result of lowered ICP must be offset against reduced CBF from hypocapnic vasoconstriction. It is therefore preferable to monitor ICP, an invasive technique only available in specialised units dealing with head-injured patients. Recent recommendations on the management of head injury therefore advise that hyperventilation should only be used to reduce intracranial pressure when other therapeutic approaches have failed.10
Effects on the Autonomic and Endocrine Systems
Survival in severe hypercapnia is, to a large extent, dependent on the autonomic response. A great many of the effects of carbon dioxide on other systems are due wholly or in part to the autonomic response to carbon dioxide.
Animal studies11 have clearly shown an increase in plasma levels of both adrenaline and noradrenaline in response to an elevation of Pco2 during apnoeic mass-movement oxygenation (Figure 23.2). In moderate hypercapnia there is a proportionate rise of adrenaline and noradrenaline, but in gross hypercapnia (Pco2 more than 27 kPa or 200 mmHg) there is an abrupt rise of adrenaline. Similar, though variable, changes have been obtained over a lower range of Pco2 in human volunteers inhaling carbon dioxide mixtures.12,13 Animal studies indicate that the increase in catecholamine release is mediated by direct stimulation of ventrolateral medullary neurones, which respond to elevated Pco2 by increasing sympathetic nerve discharge.14
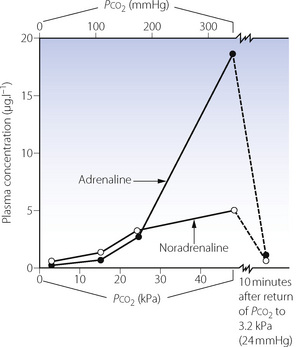
Fig. 23.2 This graph shows the changes in plasma catecholamine levels in the dog during the rise of Pco2 from 2.9 to 45 kPa (22 to 338 mmHg) in the course of 1 hour of apnoeic oxygenation. After 10 minutes of ventilation with oxygen, Pco2 returned to 3.2 kPa (24 mmHg), following which catecholamines were almost back to control values.
(Data from reference 11.)
The effect of an increased level of circulating catecholamines is, to a certain extent, offset by a decreased sensitivity of target organs when the pH is reduced. This is additional to the general depressant direct effect of carbon dioxide on target organs. Finally, acetylcholine hydrolysis is reduced at low pH and therefore certain parasympathetic effects may be enhanced during hypercapnia.
Effects on other Physiological Systems
Respiratory System
Chapter 5 describes the role of carbon dioxide in the control of breathing, and this is not discussed further here.
Pulmonary circulation. An elevated Pco2 causes vasoconstriction in the pulmonary circulation (page 110) but the effect is less marked than that of hypoxia.15 Nevertheless, in healthy subjects an end-expiratory Pco2 of 7 kPa (52 mmHg) increased pulmonary vascular resistance by 32%, which, along with elevated cardiac output, led to a 60% increase in mean pulmonary arterial pressure.16 Though regional variations in blood flow have not been demonstrated, this effect is believed to act in a similar fashion to hypoxic pulmonary vasoconstriction (HPV, page 108), tending to divert blood away from underventilated alveoli. Hypocapnia significantly attenuates HPV in animals, though this has not been described in humans. There is evidence from both animal17 and human18 studies that pH is the factor responsible for CO2-mediated changes in the pulmonary vasculature, rather than Pco2 per se.
Oxygenation of the blood. Quite apart from its effect on ventilation, carbon dioxide exerts three other important effects that influence the oxygenation of the blood. First, if the concentration of nitrogen (or other ‘inert’ gas) remains constant, the concentration of carbon dioxide in the alveolar gas can increase only at the expense of oxygen, which must be displaced. Secondly, an increase in Pco2 causes a displacement of the oxygen dissociation curve to the right (page 192). Finally, in animals, changes in Pco2 are known to affect the distribution of ventilation/perfusion ratios as measured by the multiple inert gas elimination technique (page 126). This results from changes in pH influencing pulmonary vessels as described in the previous paragraph, as well as causing changes in the size of small diameter bronchi.17
Cardiovascular System19
The effects of carbon dioxide on the circulation are complicated by the alternative modes of action on different components of the system. In general, both hypercapnia and acidosis have direct depressant effects on cardiac myocytes and vascular smooth muscle cells, effects that are normally opposed by the increase in catecholamines caused by elevated Pco2. Under different circumstances these opposing effects make the overall effect of carbon dioxide on the cardiovascular system unpredictable. Despite this problem, moderate degrees of hypercapnia have been proposed to have therapeutic potential in treating septic shock when its effects can mimic the commonly used inotrope dobutamine.20
Myocardial contractility and heart rate. Both contractility and heart rate are diminished by elevated Pco2 in the isolated preparation, probably as a result of change in pH. However, in the intact subject the direct depressant effect of carbon dioxide is overshadowed by the stimulant effect mediated through the sympathetic system. In artificially ventilated humans, increased Pco2 raises cardiac output and slightly reduces total peripheral resistance,13 and blood pressure therefore tends to be increased. Awake healthy subjects studied with non-invasive Doppler echocardiography show similar changes.16 With an end-expiratory Pco2 of 7 kPa (52 mmHg) cardiac output was increased by about 1 l.min−1 as a result of increases in both heart rate and stroke volume, and accompanied by a small rise in blood pressure. Measurements of left ventricular systolic and diastolic function were unchanged, confirming the dominance of catecholamine stimulation compared with direct depressant effects on the heart. The response of cardiac output to hypercapnia is diminished by most anaesthetics.13
Arrhythmias have been reported in awake humans during acute hypercapnia, but seldom seem to be of serious import. One study of normal subjects with modest degrees of hypercapnia did, however, demonstrate an increase in QT dispersion of the electrocardiogram during hypercapnia.16 This finding reflects regional repolarisation abnormalities of the ventricles and under other circumstances, such as ischaemic heart disease, indicates a propensity to develop life-threatening arrhythmias.
Blood pressure. As described above, an elevated Pco2 usually causes a small increase in blood pressure, an effect seen in both conscious and anaesthetised patients. However, the response is variable and certainly cannot be relied upon as an infallible diagnostic sign of hypercapnia. Hypotension accompanies an elevation of Pco2 if there is blockade of the sympathetic system by, for example, spinal anaesthesia. There is general agreement that hypotension follows a sudden fall of an elevated Pco2.
Effect on the Kidney
Renal blood flow and glomerular filtration rate are little influenced by minor changes of Pco2. However, at high levels of Pco2 there is constriction of the glomerular afferent arterioles, leading to anuria. Long-term hypercapnia results in increased resorption of bicarbonate by the kidneys, further raising the plasma bicarbonate level, and constituting a secondary or compensatory metabolic alkalosis. Long-term hypocapnia decreases renal bicarbonate resorption, resulting in a further fall of plasma bicarbonate and producing a secondary or compensatory metabolic acidosis. In each case the arterial pH returns towards the normal value but the bicarbonate ion concentration departs even further from normality.
Effect on Blood Electrolyte Levels
The acidosis that accompanies hypercapnia causes leakage of potassium ions from the cells into the plasma. Because it takes an appreciable time for the potassium ions to be transported back into the intracellular compartment, repeated bouts of hypercapnia at short intervals result in a stepwise rise in plasma potassium.
A reduction in the ionised fraction of the total calcium has, in the past, been thought to be the cause of the tetany that accompanies severe hypocapnia. However, the changes that occur are too small to account for tetany, which occurs in parathyroid disease only when there has been a fairly gross reduction of ionised calcium.21 Hyperexcitability affects all nerves, and spontaneous activity ultimately occurs. The muscle spasms probably result from activity in proprioceptive fibres causing reflex muscle contraction.
The effects of long-term small elevations in inspired carbon dioxide are described on page 305.
Hypercapnia in Clinical Practice
Clinical Signs
Hyperventilation is the cardinal sign of hypercapnia due to an increased concentration of carbon dioxide in the inspired gas, whether it be endogenous or exogenous. However, this sign will be absent in the paralysed patient and also in those in whom hypercapnia is the result of hypoventilation. Such patients, including those with COPD, constitute the great majority of those with hypercapnia.
Dyspnoea may or may not be present. In patients with central failure of respiratory drive, dyspnoea may be entirely absent. On the other hand, when hypoventilation results from mechanical failure in the respiratory system (airway obstruction, pneumothorax, pulmonary fibrosis, etc.), dyspnoea is usually obvious.
In patients with COPD hypercapnia is usually associated with a flushed skin and a full and bounding pulse with occasional extrasystoles. The blood pressure is often raised but this is not a reliable sign. Muscle twitchings and a characteristic flap of the hands may be observed when coma is imminent. Convulsions may occur. The patient will become comatose when the Pco2 is in the range 12–16 kPa (90–120 mmHg) (see above). Hypercapnia should always be considered in cases of unexplained coma.
Hypercapnia cannot be reliably diagnosed on clinical examination. This is particularly true when there is a neurological basis for hypoventilation. Now that it has become so simple to measure the arterial Pco2, an arterial sample should be taken in all cases of doubt.
Gross hypercapnia. Relatively, few cases of gross hypercapnia are documented, but there are sufficient to indicate that complete recovery from gross hypercapnia without hypoxia is possible and may even be the rule.22 One report from 199023 detailed five instances of hypercapnia without hypoxia in children with arterial Pco2 values in the range 21–36 kPa (155–269 mmHg). All were comatose or stuperose but recovered. A single case report of massive grain aspiration reported survival following a Pco2 of 66.8 kPa (501 mmHg).24 These cases indicate that, of the reported cases, full recovery seems to be the usual outcome. Hypoxia seems to be much more dangerous than hypercapnia.
References
1. Laffey JG, Kavanagh BP. Hypocapnia. N Engl J Med.. 2002;347:43-53.
2. Leake CD, Waters RM. The anesthetic properties of carbon dioxide. J Pharmacol Exp Ther. 928;33:280–281.
3. Refsum HE. Relationship between state of consciousness and arterial hypoxaemia and hypercapnia in patients with pulmonary insufficiency, breathing air. Clin Sci.. 1963;25:361-367.
4. Eisele JH, Eger EI, Muallem M. Narcotic properties of carbon dioxide in the dog. Anesthesiology. 1967;28:856-865.
*5. Brian JE. Carbon dioxide and the cerebral circulation. Anesthesiology. 1998;88:1365-1386.
6. Raichle ME, Posner JB, Plum F. Cerebral blood flow during and after hyperventilation. Arch Neurol.. 1970;23:394-403.
7. Ornstein E, Young WL, Fleischer LH, Ostapkovich N. Desflurane and isoflurane have similar effects on cerebral blood flow in patients with intracranial mass lesions. Anesthesiology. 1993;79:498-502.
8. Cho S, Kujigaki T, Uchiyama Y, Fukusai M, Shibata O, Somikawa K. Effects of sevoflurane with and without nitrous oxide on human cerebral circulation. Anesthesiology. 1996;85:755-760.
9. Eng C, Lam AM, Mayberg TS, Lee C, Mathisen T. The influence of propofol with and without nitrous oxide on cerebral blood flow velocity and CO2 reactivity in humans. Anesthesiology. 1992;77:872-879.
10. Yundt KD, Diringer MN. The use of hyperventilation and its impact on cerebral ischaemia in the treatment of traumatic brain injury. Crit Care Clin.. 1997;13:163-184.
11. Millar RA. Plasma adrenaline and noradrenaline during diffusion respiration. J Physiol.. 1960;150:79-90.
12. Sechzer PH, Egbert LD, Linde HW, Cooper DY, Dripps RD, Price HL. Effect of CO2 inhalation on arterial pressure ECG and plasma catecholamines and 17-OH corticosteroids in normal man. J Appl Physiol.. 1960;15:454-458.
13. Cullen DJ, Eger EI. Cardiovascular effects of carbon dioxide in man. Anesthesiology. 1974;41:345-349.
14. Toney GM. Sympathetic activation by the central chemoreceptor ‘reflex’: new evidence that RVLM vasomotor neurons are involved … but are they enough? J Physiol.. 2006;577:3.
15. Barer GR, Howard P, McCurrie JR. The effect of carbon dioxide and changes in blood pH on pulmonary vascular resistance in cats. Clin Sci.. 1967;32:361-376.
16. Kiely DG, Cargill RI, Lipworth BJ. Effects of hypercapnia on hemodynamic, inotropic, lusitropic, and electrophysiological indices in humans. Chest. 1996;109:1215-1221.
17. Domino KB, Swenson ER, Hlastala MP. Hypocapnia-induced ventilation/perfusion mismatch: a direct CO2 or pH-mediated effect. Am J Respir Crit Care Med.. 1995;152:1534-1539.
18. Loeppky JA, Scotto P, Riedel CE, Roach RC, Chick TW. Effects of acid-base status on acute hypoxic pulmonary vasoconstriction and gas exchange. J Appl Physiol.. 1992;72:1787-1797.
19. Fox P. Effects of carbon dioxide on the systemic circulation. In: Prys-Roberts C, editor. The Circulation in Anaesthesia. Oxford: Blackwell Scientific, 1980.
*20. Wang Z, Su F, Bruhn A, Yang X, Vincent J-L. Acute hypercapnia improves indices of tissue oxygenation more than dobutamine in septic shock. Am J Respir Crit Care Med.. 2008;177:178-183.
21. Tenney SM, Lamb TW. Physiological consequences of hypoventilation and hyperventilation. Handbk Physiol Sect 3. 1965;2:979.
22. Potkin RT, Swenson ER. Resuscitation from severe acute hypercapnia. Determinants of tolerance and survival. Chest. 1992;102:1742-1745.
23. Goldstein B, Shannon DC, Todres ID. Supercarbia in children: clinical course and outcome. Crit Care Med.. 1990;18:166-168.
24. Slinger P, Blundell PE, Metcalf IR. Management of massive grain aspiration. Anesthesiology. 1997;87:993-995.