Chapter 31 Acute lung injury
 Acute lung injury is lung inflammation that develops in response to a variety of both pulmonary and generalised acute diseases. The clinical features of acute lung injury vary from mild, self-limiting dyspnoea to rapidly progressive and fatal respiratory failure.
Acute lung injury is lung inflammation that develops in response to a variety of both pulmonary and generalised acute diseases. The clinical features of acute lung injury vary from mild, self-limiting dyspnoea to rapidly progressive and fatal respiratory failure.Acute lung injury (ALI) describes a characteristic form of parenchymal lung disease, and represents a wide range of severity from short-lived dyspnoea to a rapidly terminal failure of the respiratory system, when the term acute respiratory distress (ARDS) is normally used. The syndrome was first described in 1967 when Ashbaugh et al1 reported a condition in adults that seemed similar to the respiratory distress syndrome seen in infants. One of the subjects reported in this original paper was aged only 11 years, and in recognition of the fact that respiratory distress syndrome is known to occur in children the term adult respiratory distress syndrome should be avoided. There are a great many other synonyms for ALI, including acute respiratory failure, shock lung, respirator lung, pump lung and Da Nang lung.
Clinical Aspects of Acute Lung Injury2,3,4
Definition
There is no single diagnostic test, and confusion has arisen in the past from differing diagnostic criteria. This has complicated comparisons of incidence, mortality, aetiology and efficacy of therapy in different centres. To address this problem, European–American consensus conferences produced the following widely accepted definitions.5
Acute lung injury diagnosis requires the presence of four criteria:
Acute respiratory distress syndrome is defined in almost identical terms except that the impairment of gas exchange is worse with a Pao2 to FiO2 ratio of ≤26.7 (kPa) or ≤200 (mmHg).
These definitions are now widely accepted and have been extremely helpful in researching ALI, particularly epidemiological studies. However, there are several provisos to their use in the clinical situation. For example, it is possible for patients with diseases that elevate left atrial pressure to also have ALI, but they would fall outside the strict definition. Also, many earlier definitions suggest that one or more of the known predisposing conditions should have been present and that the clinical course has followed the recognised pattern (see below). Finally, it is noted that the histology is usually diagnostic but it is seldom indicated or advisable to take a lung biopsy. There is no reliable laboratory test to confirm the diagnosis (see below).
In part, the diagnosis of ALI depends on exclusion of other conditions. Sometimes it is not easy to separate it from other diseases such as pulmonary embolus, pulmonary oedema, fibrosing alveolitis, or diffuse pneumonia, which may present many similar features.
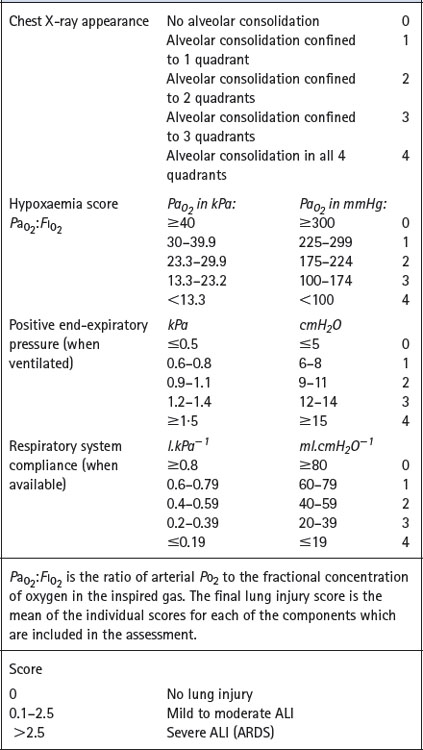
Scoring systems. Various attempts have been made to derive a single numerical value to assess the severity of ALI. Murray et al6 proposed an expanded three-part definition comprising distinction between acute and chronic phases, identification of aetiological and associated conditions and a numerical lung injury score, details of which are shown in Table 31.1.
Table 31.1 Lung injury score6
Predisposing Conditions and Risk Factors for ALI
Although the clinical and histopathological pictures of ALI are remarkably consistent, they have been described as the sequel to a large range of predisposing conditions (Table 31.2). There are, however, very important differences in the progression of ALI and its response to treatment, depending on the underlying cause and associated pathology. Nevertheless, recognition of the predisposing conditions is crucially important for predicting which patients are at risk and the establishment of early diagnosis.
Table 31.2 Some predisposing conditions for ALI
| DIRECT LUNG INJURY | INDIRECT LUNG INJURY |
|---|---|
| Common: Pneumonia Aspiration of gastric contents |
Common: Sepsis Severe non-thoracic trauma Multiple transfusions of blood-products |
| Less common: Lung contusion Near drowning Inhalation of toxic gases or vapours Fat or amniotic fluid embolus Reperfusion oedema, e.g. following lung transplantation |
Less common: Acute pancreatitis Cardiopulmonary bypass Severe burns Drug overdose Disseminated intravascular coagulation |
The conditions listed in Table 31.2 are not equally likely to proceed to ALI. Studies have consistently identified sepsis syndrome (see below) as the condition most likely to result in development of ALI with about 40% of patients being affected.7 Patients who have aspirated gastric contents, received multiple emergency transfusions or incurred pulmonary contusions have a 17–24% chance of developing ALI. Overall, 25% of patients with a single risk factor develop ARDS but this rises to 42% with two factors and 85% with three. Age and sex do not affect the likelihood of developing ALI.
Sepsis syndrome is defined as a systemic response to proven or presumed infection, with hyper- (or hypo-) thermia, tachycardia, tachypnoea, and one or more organs exhibiting signs of hypoperfusion or dysfunction. There is usually altered cerebral function, arterial hypoxaemia, lactacidosis and oliguria. Many cases of ALI represent the pulmonary manifestation of the multi-organ dysfunction syndrome (MODS) that is a feature of this condition, and ALI is frequently associated with circulatory failure (septic shock).
Pulmonary and extrapulmonary ALI. Gattinoni et al8 proposed that patients with ALI should be considered as two separate groups. Pulmonary ALI results from clinical conditions that cause direct lung injury, whilst extrapulmonary ALI follows indirect lung injury (Table 31.2). These two sub-groups of ALI have been shown to differ with respect to pathological mechanisms, appearances on chest radiographs and CT scans, abnormalities of respiratory mechanics and response to ventilatory strategies.9
Incidence and Mortality10
In the past, the lack of accepted definitions of lung injury led to widely varying estimates of the incidences of ALI and ARDS. Despite more consistent diagnostic criteria in recent years, the estimated incidence of ALI remains variable at 18–79 cases per year per 100 000 population.11 The reasons for this variation in estimates of the incidence of ALI is unknown.2 Around 70% of cases of ALI are severe enough to be classified as ARDS.
There is, however, considerable agreement that mortality from ALI is high: two decades ago in excess of 50% of patients died whatever the criteria of diagnosis. Outcome has improved in recent years with current estimates of around a 40% mortality rate, but still with wide variability,3,12 and only slow progress.13 There appears to be no difference in the mortality of ALI arising from pulmonary or extrapulmonary causes.14
Clinical Course
Four phases may be recognised in the development of severe ALI. In the first the patient is dyspnoeic and tachypnoeic but there are no other abnormalities. The chest radiograph is normal at this stage, which lasts for about 24 hours. In the second phase there is hypoxaemia but the arterial Pco2 remains normal or subnormal. There are minor abnormalities of the chest radiograph. This phase may last for 24–48 hours. Diagnosis is easily missed in these prodromal stages and is very dependent on the history of one or more predisposing conditions.
It is only in phase three that the diagnostic criteria of ALI become established. There is significant arterial hypoxaemia due to an increased alveolar/arterial Po2 gradient, and the arterial Pco2 may be slightly elevated. The lungs become stiff and the chest radiograph shows the characteristic bilateral diffuse infiltrates. Ventilatory support is usually instituted at this stage, but many patients escape this intervention and are managed on respiratory wards rather than in intensive care.15
The fourth phase is often terminal and comprises massive bilateral consolidation with unremitting hypoxaemia even when ventilated with very high inspired oxygen concentrations. Dead space is substantially increased and the arterial Pco2 is only with difficulty kept in the normal range.
Not every patient passes through all these phases and the condition may resolve at any stage. It is difficult to predict whether the condition will progress and there is currently no useful laboratory test. Serial observations of the chest radiograph, the alveolar/arterial Po2 gradient and the function of other compromised organs are the best guides to progress. The more systems in failure, the worse is the outlook.
Pathophysiology
Alveolar/capillary permeability is increased substantially throughout the course of ALI.16 This may be demonstrated by the enhanced transit of various tracer molecules across the alveolar/capillary membrane (page 425).17
Maldistribution of ventilation and perfusion.18 Computerised tomography (CT) of patients with ARDS shows that opacities representing collapsed areas are distributed throughout the lungs in a heterogeneous manner but predominantly in the dependent regions.19 Following a change in posture, the opacities move to the newly dependent zones within a few minutes.20 The most conspicuous functional disability is the shunt (Figure 31.1), which is usually so large (often more than 40%) that increasing the inspired oxygen concentration cannot produce a normal arterial Po2 (see the isoshunt chart, Figure 8.11). CT scans of patients with ALI also demonstrate substantial areas of lung overdistension.21 These areas contribute to the increased dead space, which may exceed 70% of tidal volume and requires a large increase in minute volume to attempt to preserve a normal arterial Pco2. Both shunt and dead space correlate strongly with the non-inflated lung tissue seen with CT (Figure 31.1).
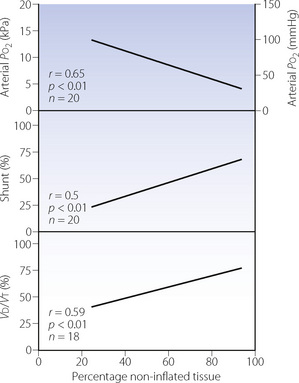
Fig. 31.1 Relationship of arterial Po2, shunt and physiological dead space (Vd/Vt) to the percentage of non-inflated lung tissue seen by computed tomography in patients with acute respiratory distress syndrome, artificially ventilated with positive end-expiratory pressure of 0.5 kPa (5 cmH2O).
(After reference 19.)
Lung mechanics. In established ARDS, lung compliance is greatly reduced and the static compliance of the respiratory system (lungs + chest wall) is of the order of 300 ml.kPa−1 (30 ml.cmH2O−1).22 Patients with pulmonary and extrapulmonary forms of ARDS (see above) have different abnormalities of respiratory system mechanics.8 Respiratory system compliance is reduced to a similar extent in both groups, but the abnormality is mostly with lung compliance when lung disease is the cause, and chest wall compliance with extrapulmonary causation.
Functional residual capacity is reduced by collapse and increased elastic recoil.
Mean total resistance to airflow was found to be 1.5–2 kPa.l−1.s (15–20 cmH2O.l−1.s),22 or about three times that of anaesthetised patients with normal lungs, measured by the same technique. Using the model shown in Figure 4.4, some two-thirds of the total resistance in patients with ARDS could be assigned to visco-elastic resistance of tissue, although the airway resistance was still about twice normal.
Oxygen consumption by the lung. Pulmonary oxygen consumption (page 212) can be very high in patients with severe ALI. It is possible that some of this represents reactive oxygen species formation (see Chapter 26), but the increase in pulmonary oxygen consumption does not seem to correlate with various markers of pulmonary inflammation at the time the measurement is made.23
Mechanisms of Acute Lung Injury
Histopathology
Although of diverse aetiology, the histological appearances of ARDS are remarkably consistent and this lends support for ARDS being considered a discrete clinical entity. Histological changes at autopsy may be divided into two stages, as follows.24
Acute stage. The acute stage is characterised by damaged integrity of the blood–gas barrier. The changes are primarily in the inter-alveolar septa and cannot be satisfactorily seen with light microscopy. Electron microscopy shows extensive damage to the type I alveolar epithelial cells (page 22), which may be totally destroyed. Meanwhile the basement membrane is usually preserved and the endothelial cells still tend to form a continuous layer with apparently intact cell junctions. Endothelial permeability is nevertheless increased and interstitial oedema is found, predominantly on the ‘service’ side of the capillary as seen in other forms of pulmonary oedema (page 421). Protein-containing fluid leaks into the alveoli, which also contain red blood cells, leukocytes and strands of fibrin. Intravascular coagulation is common,25 and, in patients with septicaemia, capillaries may be completely plugged with leukocytes, and the underlying endothelium damaged.
Chronic or fibroproliferative stage. Attempted repair and proliferation predominate in the chronic stage of ARDS. Within a few days of the onset of the condition, there is a thickening of endothelium, epithelium and the interstitial space. The type I epithelial cells are destroyed and replaced by type II cells, which proliferate but do not differentiate into type I cells as usual. They remain cuboidal and about ten times the thickness of the type I cells they have replaced. This appears to be a non-specific response to damaged type I cells, and is similar to that which results from exposure to high concentrations of oxygen (page 387).
The interstitial space is greatly expanded by oedema fluid, fibres and a variety of proliferating cells. In the same way as for other causes of pulmonary fibrosis, extracellular matrix remodelling begins (page 437). Fibrosis commences after the first week and ultimately fibrocytes predominate: extensive fibrosis is seen in resolving cases. These fibroproliferative changes may occur earlier in pulmonary, compared with extrapulmonary, causes of ARDS.26
Cellular Mechanisms27
The diversity of predisposing conditions suggests that there may be several possible mechanisms, at least in the early stages of development of ALI, but the end-result is remarkably similar. In all cases, lung injury seems to begin with damage to the alveolar/capillary membrane. This is followed by progressive inflammation leading to alveolar epithelial cell injury, alveolar transudation, pulmonary vasoconstriction and capillary obstruction.
Cells that are capable of damaging the alveolar capillary membrane include neutrophils, basophils, macrophages and platelets. Damage may be inflicted by a large number of substances, including bacterial endotoxin, reactive oxygen species, proteases, thrombin, fibrin, fibrin degradation products, arachidonic acid metabolites and innumerable pro-inflammatory cytokines. It seems improbable that any one mechanism is responsible for all cases of ALI. It is more likely that different mechanisms operate in different predisposing conditions and in different animal models of ALI.
Neutrophils have a key role in human ALI.28 Although ALI can still be induced in neutrophil depleted animals, patients with ARDS have large numbers of neutrophils and associated cytokines in bronchoalveolar lavage (BAL) fluid samples. Neutrophil activation may occur in response to a large number of substances, some of which are illustrated in Figure 31.2. Which of these are important in ALI is unknown, but likely to depend on the predisposing condition; for example, complement component C5a is known to be involved in sepsis-related ALI. Margination of neutrophils from the pulmonary capillary into the lung parenchyma is the first stage of neutrophil activation and has been described on page 435. During margination and once in the interstitium, the neutrophil is ‘primed’– that is, stimulated to produce preformed mediators ready for release, and to establish the bactericidal contents of their lysosomes. Finally, stimulation results from a whole host of cytokines, some derived from other inflammatory cells (macrophages, lymphocytes or endothelial cells) and some from other neutrophils, so amplifying the process. Stimulation causes release of a whole host of inflammatory mediators (Figure 31.2), and is also associated with inappropriate release of lysosomal contents. Instead of being released into phagocytic vesicles containing bacteria, they come into direct contact with the endothelium, which is thereby damaged.
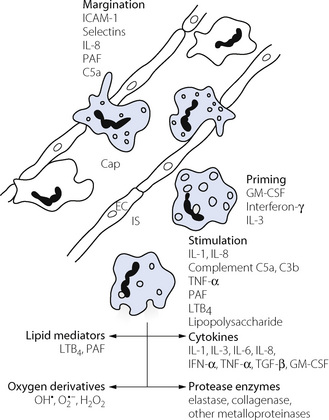
Fig. 31.2 Neutrophil activation and the main cytokines and mediators involved. This takes place in three stages. Margination, when neutrophils adhere to the capillary (Cap) wall and migrate between endothelial cells (EC) into the interstitial space (IS); priming, when the cells generate preformed mediators and lysozomal contents; and stimulation, when neutrophils release the various mediators shown. The scheme shown is based on studies of both systemic and pulmonary inflammation. Neutrophil margination may occur by different mechanisms in pulmonary capillaries (see page 435). For explanation of abbreviations see text.
Four groups of substances released from neutrophils (Figure 31.2) are considered to contribute to lung damage, as follows.
Macrophages and basophils. Macrophages are already present in the normal alveolus (page 23) but their numbers increase greatly in ALI. They produce a wide range of bactericidal agents and cytokines similar to those of the neutrophil. Lung macrophages produce IL-10, which suppresses gene expression of many cytokines, and so acts as one of the very few anti-inflammatory cytokines so far identified in ALI.29
Platelets are present in the pulmonary capillaries in large number in ARDS. Aggregation in the capillary is associated with increased capillary hydrostatic pressure, possibly due to release of arachidonic acid metabolites.
Besides giving rise to pulmonary oedema, many of the mediators released by these inflammatory cells have other effects that contribute to the pulmonary changes seen in ALI. For example, arachidonic acid metabolites cause pulmonary venoconstriction, which will raise pulmonary capillary pressure and compound the effect of increased permeability. Accumulation of platelets and neutrophils along with intravascular coagulation will occlude pulmonary vessels, producing pulmonary hypertension and unperfused lung units. It has also been noted that many proteins, including albumin, but particularly fibrin monomer, can antagonise the action of surfactant so fundamentally altering lung mechanics.30
The potential contribution to ALI of lung damage secondary to artificial ventilation is described on page 481.
Principles of Therapy31,32
Aggressive treatment of the underlying cause in conjunction with supportive therapy remains the mainstay of current management. Optimal management of the cardiovascular system and fluid balance10 is a vital component of ALI treatment as any increase in pulmonary capillary pressure (e.g. from fluid overload) may lead to catastrophic pulmonary oedema. Respiratory support requires artificial ventilation in all but the most minor degrees of ALI.
Artificial Ventilation in ALI33,34,35
General principles of artificial ventilation and the resulting physiological effects are described in detail in the next chapter. In this section, only the problems associated with ventilation of patients with ALI are described.
The lungs of patients with severe ALI may be conveniently divided into three hypothetical sections.36 First, there will be some ‘normal’ areas, usually in the non-dependent region. Second, there will be areas of lung, usually in dependent regions, with such severe collapse and alveolar flooding that ventilation of these areas will be impossible. Finally, there will be an intermediate area with poorly ventilated or collapsed alveoli that are capable of being ‘recruited’ by appropriate artificial ventilation, with a resultant improvement in gas exchange. Though the relative amounts of each section will vary greatly according to the severity of the ALI, there will always be some lung in the final area, and so capable of recruitment.
Tidal volume. The recognition that positive pressure ventilation can lead to lung damage (page 481) has led to a change in ventilatory technique used in patients with ALI. Overdistension of alveoli by application of large tidal volumes is a significant factor in lung damage. In particular, because of the extensive areas of pulmonary collapse a typical patient with ALI may only have approximately one-third of the lung being ventilated. Thus use of a normal tidal volume (10–12 ml.kg−1) will, for the few alveoli being ventilated, equate to a tidal volume of three times usual for normal healthy lungs, which equates to over 2 litres in a 70 kg subject.
Pressure-controlled ventilation (page 465) is now the preferred technique in most centres to avoid the problems outlined in the previous paragraph. However, with pressure-controlled ventilation in lungs with low compliance, such as ALI, the delivery of an adequate minute volume may be difficult. Two techniques are advocated to deal with this problem. First, inverse I/E ratios may be used, in which expiratory time is shorter than inspiratory time, allowing the delivery of a larger tidal volume. Second, the hypercapnia that results from the inadequate minute volume may be partially ignored. Known as permissive hypercapnia,37 arterial Pco2 is allowed to increase until such time as the respiratory acidosis is deemed detrimental, though the impact of this strategy remains controversial.38
Positive end expiratory pressure (PEEP). In patients with ALI PEEP reduces the amount of non-inflated lung tissue seen at CT scan,19 particularly in dependent lung regions.39 Shunt fraction and therefore the arterial Po2 (Figure 31.3) also improve. Reduced pulmonary compliance means that cardiac output is better maintained than might be expected (page 48), with a reduction of about 20% with PEEP of 1.5 kPa (15 cm H2O) (Figure 31.3).
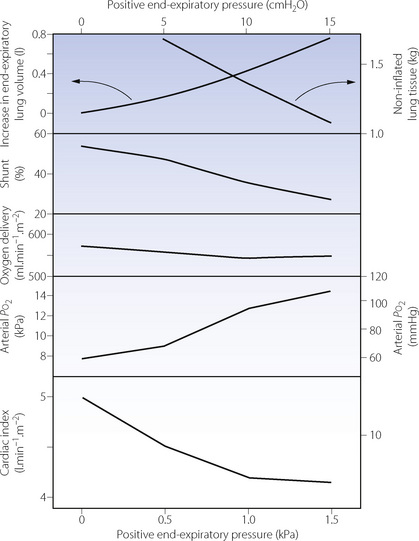
Fig. 31.3 Effect of positive end-expiratory pressure on various factors influencing oxygen delivery in patients with acute respiratory distress syndrome. Although arterial Po2 is increased, cardiac output is decreased and there is no significant change in oxygen transport.
(Data on non-inflated lung tissue are from reference 19; remaining data from reference 39.)
The ideal PEEP value to use has been controversial for decades. Differing end-points (shown here in parentheses) have given rise to numerous terms such as ‘optimal’ PEEP (lowest physiological shunt fraction), ‘best’ PEEP (optimal static lung compliance), ‘preferred’ PEEP (best oxygen delivery), and ‘least’ PEEP (acceptable values for arterial Po2, inspired oxygen and cardiac output). High levels of PEEP should result in increased alveolar recruitment and improved oxygenation, but normal alveoli can only enlarge in response to PEEP to a certain extent, above which dramatic increases in alveolar pressure, and possible damage, occur (pages 482 et seq). Identifying this point has vexed intensivists for some time. It has been suggested that PEEP be increased until the lower inflection of the patient’s respiratory system static compliance curve is reached (point A in Figure 31.4), which is normally between 10–15 cmH2O.40 The pressure seen at the lower point of inflection is believed to represent the pressure at which most recruitable alveoli have been opened, while the upper inflection point (B in Figure 31.4) designates the point above which overdistension of alveoli may occur.33 A pressure-volume curve in ARDS patients has also been compared to chest CT scans, and may prove useful in ascertaining which patients have significant potentially recruitable regions of lung.41
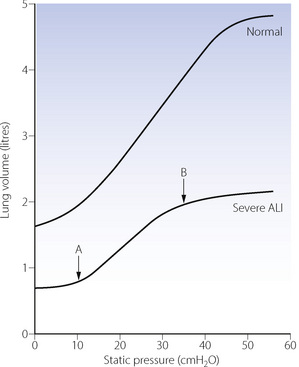
Fig. 31.4 Static pressure versus lung volume curves for patients receiving positive pressure ventilation. Note the severely reduced lung volume and compliance in ALI. Point A indicates the lower inflection of the curve, above which compliance is considerably improved. Application of positive end-expiratory pressure of approximately 12 cmH2O in this patient will therefore improve tidal volume relative to the ventilatory pressure required. Point B indicates the upper inflection point, above which alveolar overdistension may occur, therefore, in this patient, airway pressure should ideally be maintained below 35 cmH2O.
In clinical practice using higher levels of PEEP (13.2 cmH2O) versus lower levels (8.3 cmH2O) has been shown to improve oxygenation, but this did not result in any difference in survival.42 Possible reasons for this lack of outcome benefit include adverse effects of the higher PEEP on the lungs or cardiovascular system, or that some patients with ALI may have very little ‘recruitable’ volume of lung.43
Protective ventilation strategy.44 In ALI, the ventilatory strategy used must balance the conflicting requirements of maintaining adequate gas exchange in severely diseased lungs while simultaneously avoiding damaging the lungs by the use of large tidal volumes, high airway pressures or harmful levels of inspired oxygen. Protective ventilation describes a widely advocated ventilatory strategy that may achieve the best compromise, and involves using small tidal volumes to prevent alveolar overdistension and moderate levels of PEEP to maintain alveolar recruitment. Initial tidal volumes used for ventilation should be between 6–8 ml.kg−1 using pressure controlled ventilation. PEEP is set using a pressure–volume curve or increased until arterial Po2 is adequate or cardiovascular depression occurs. If plateau airway pressure exceeds 35 cmH2O, or the inspired oxygen level required to obtain acceptable arterial Po2 exceeds 0.65, then an alternative ventilatory strategy should be considered.33 Protective ventilation is the only intervention in the management of ALI that has consistently been found to improve mortality.35,44,45
Alternative ventilatory strategies.4,33 Many other techniques have been described as part of respiratory support for patients with ARDS who continue to have unacceptably poor gas exchange despite the use of protective ventilation. None of these have been shown to improve clinical outcome. Possible interventions include:
 recruitment manoeuvres, similar to those described for atelectasis during anaesthesia (page 336), may be used. Transient, self-limiting, bradycardia and/or desaturation is common, but the manoeuvres do improve oxygenation,46 though this has not been shown to improve outcomes from severe ALI. prone position. In the prone position both ventilation and perfusion become more uniform, and the areas of atelectasis in dependent lung regions change position, re-forming in the anterior (now dependent) regions.20 About two-thirds of patients demonstrate an improvement in oxygenation on turning prone, though this is normally not sustained and repeated turning of the patient may be required
recruitment manoeuvres, similar to those described for atelectasis during anaesthesia (page 336), may be used. Transient, self-limiting, bradycardia and/or desaturation is common, but the manoeuvres do improve oxygenation,46 though this has not been shown to improve outcomes from severe ALI. prone position. In the prone position both ventilation and perfusion become more uniform, and the areas of atelectasis in dependent lung regions change position, re-forming in the anterior (now dependent) regions.20 About two-thirds of patients demonstrate an improvement in oxygenation on turning prone, though this is normally not sustained and repeated turning of the patient may be requiredOther Therapeutic Options
Specific therapy for ALI is the goal of much research, which is directed particularly towards the control of sepsis and the development of antagonists to the various mediators considered above.31 In most cases it has proved difficult to demonstrate their efficacy in the clinical setting. Detailed description of these, and several other pharmacological approaches to the treatment of ALI, is beyond the scope of this book, but a summary is shown in Table 31.3.
Table 31.3 Summary of pharmacological interventions suggested for the treatment of ALI or ARDS
| THERAPY | EXAMPLES | PROPOSED MECHANISM |
|---|---|---|
| Pulmonary vasodilators | Prostacyclin | Non-specific pulmonary vasodilator |
| Nitric oxide | Regional pulmonary vasodilator (see text) | |
| Almitrine | Enhancement of hypoxic pulmonary vasoconstriction | |
| Surfactant | Artificial surfactants | Replace depleted alveolar surfactant, may also have anti-inflammatory properties |
| Anti-inflammatory | Steroids | General anti-inflammatory |
| Ketoconazole | Inhibits thromboxane synthesis | |
| Ibuprofen/Indomethacin | Inhibits prostaglandin production | |
| Prostaglandin E1 | Inhibits platelet aggregation, vasodilator | |
| Pentoxifylline | Reduces neutrophil chemotaxis and activation | |
| Endotoxin/TNF/IL-1 antagonists | Inhibition of specific aspects of inflammatory response | |
| Anti-oxidants | N-acetylcysteine | Increased glutathione activity (page 385) |
| Recombinant human manganese SOD | Replaces epithelial extracellular SOD (page 385) | |
| Anticoagulants | Heparin | Reduce fibrin deposition in alveoli |
N.B. All the therapies listed have been shown to have beneficial effects in in-vitro or animal studies of ALI. There is insufficient evidence of improved outcome for any of the therapies listed to be recommended for routine use in human ALI.
SOD, superoxide dismutase (page 385); TNF, tumour necrosis factor; IL-1, interleukin 1.
References
1. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. 1967;2:319-323.
2. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med.. 2000;342:1334-1349.
*3. Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 2007;369:1553-1565.
4. Leaver K, Evans TW. Acute respiratory distress syndrome. BMJ. 2007;335:389-395.
5. Bernard GR, Artigas A, Brigham KL, et al. The American–European consensus conference on ARDS: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med.. 1994;149:818-824.
6. Murray JF, Matthay MA, Luce JM, Flick MR. An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis.. 1988;138:720-723.
7. Hudson LD, Millberg JA, Anardi D, Maunder RJ. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med.. 1995;151:293-301.
8. Gattinoni L, Pelosi P, Suter PM, Pedoto A, Vercesi P, Lissoni A. Acute respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different syndromes? Am J Respir Crit Care Med.. 1998;158:3-11.
*9. Pelosi P, D’Onofrio D, Chiumello D, et al. Pulmonary and extrapulmonary acute respiratory distress syndrome are different. Eur Respir J. 2003;22(suppl 42):48S-56S.
10. Rivers EP. Fluid-management strategies in acute lung injury – liberal, conservative, or both? N Engl J Med.. 2006;354:2598-2600.
11. Milberg JA, Davis DR, Steinberg KP, Hudson LD. Improved survival of patients with acute respiratory distress syndrome (ARDS): 1983–1993. JAMA. 1995;273:306-309.
12. Rubenfeld GD, Herridge MS. Epidemiology and outcomes of acute lung injury. Chest. 2007;131:554-562.
13. Brochard L, Rouby J-J. Changing mortality in acute respiratory distress syndrome? Yes, we can!. Am J Respir Crit Care Med.. 2009;179:177-178.
14. Agarwal R, Srinivas R, Nath A, Jindal S. Is the mortality higher in the pulmonary vs the extrapulmonary ARDS? A meta-analysis. Chest. 2008;133:1463-1473.
15. Quartin AA, Campos MA, Maldonado DA, Ashkin D, Cely CM, Schein RMH. Acute lung injury outside of the ICU. Incidence in respiratory isolation on a general ward. Chest. 2009;135:261-268.
16. Sinclair DG, Braude S, Haslam PL, Evans TW. Pulmonary endothelial permeability in patients with severe lung injury: Clinical correlates and natural history. Chest. 1994;106:535-539.
17. Rocker GM. Bedside measurement of pulmonary capillary permeability in patients with acute lung injury. What have we learned? Intensive Care Med.. 1996;22:619-621.
18. Sinclair SE, Albert RK. Altering ventilation-perfusion relationships in ventilated patients with acute lung injury. Intensive Care Med.. 1997;23:942-950.
19. Gattinoni L, Pesenti A, Bombino M, et al. Relationships between lung computed tomographic density, gas exchange, and PEEP in acute respiratory failure. Anesthesiology. 1988;69:824-832.
20. Gattinoni L, Pelosi P, Vitale G, Pesenti A, D’Andrea L, Mascheroni D. Body position changes redistribute lung computed-tomographic density in patients with acute respiratory failure. Anesthesiology. 1991;74:15-23.
21. Vieira SRR, Puybasset L, Richecoeur J, et al. A lung computed tomographic assessment of positive end-expiratory pressure-induced lung overdistension. Am J Respir Crit Care Med.. 1998;158:1571-1577.
22. Tantucci C, Corbeil C, Chasse M, et al. Flow and volume dependence of respiratory system flow resistance in patients with adult respiratory distress syndrome. Am Rev Respir Dis.. 1992;145:355-360.
23. Jolliet P, Thorens JB, Nicod L, Pichard C, Kyle U, Chevrolet JC. Relationship between pulmonary oxygen consumption, lung inflammation, and calculated venous admixture in patients with acute lung injury. Intensive Care Med.. 1996;22:277-285.
24. Bachofen M, Weibel ER. Structural alterations and lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med.. 1982;3:35-43.
25. Schultz MJ, Dixon B, Levi M. The pulmonary protein C system: preventive or therapeutic target in acute lung injury? Thorax. 2009;64:95-97.
26. Negri EM, Hoelz C, Barbas CSV, Montes GS, Saldiva PHN, Capelozzi VL. Acute remodeling of parenchyma in pulmonary and extrapulmonary ARDS. An autopsy study of collagen and elastic system fibres. Pathol Res Pract.. 2002;198:355-361.
*27. Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol.. 2004;202:145-156.
*28. Abraham E. Neutrophils and acute lung injury. Crit Care Med.. 2003;31(supp):S195-S199.
29. Ward PA. Role of complement, chemokines, and regulatory cytokines in acute lung injury. Ann N Y Acad Sci.. 1996;796:104-112.
30. Seeger W, Stöhr G, Wolf HRD, Neuhof H. Alteration of surfactant function due to protein leakage. J Appl Physiol.. 1985;58:326-338.
31. Artigas A, Bernard GR, Carlet J, et al. The American–European consensus conference on ARDS, Part 2: Ventilatory, pharmacologic, supportive therapy, study design strategies and issues related to recovery and remodelling. Intensive Care Med.. 1998;24:378-398.
32. Brower RG, Ware LB, Berthiaume Y, Matthay MA. Treatment of ARDS. Chest. 2001;120:1347-1367.
33. Malarkkan N, Snook NJ, Lumb AB. New aspects of ventilation in acute lung injury. Anaesthesia. 2003;58:647-667.
*34. Rouby J-J, Constantin J-M, Giradi CrdeA, Zhang M, Lu Q. Mechanical ventilation in patients with acute respiratory distress syndrome. Anesthesiology. 2004;101:228-234.
35. Girard TD, Bernard GR. Mechanical ventilation in ARDS. A state-of-the-art review. Chest. 2007;131:921-929.
36. Gattinoni L, Pesenti A. ARDS: the non-homogenous lung: facts and hypotheses. Intensive Crit Care Digest.. 1987;6:1-4.
37. Laffey JG, O’Croinin D, McLoughlin P, Kavanagh BP. Permissive hypercapnia – role in protective lung ventilatory strategies. Intensive Care Med.. 2004;30:347-356.
38. Kavanagh BP. Therapeutic hypercapnia: careful science, better trials. Am J Respir Crit Care Med.. 2005;171:96-97.
39. Gattinoni L, D’Andrea L, Pelosi P, Vitale G, Pesenti A, Fumagalli R. Regional effects and mechanism of positive end-expiratory pressure in early adult respiratory distress syndrome. JAMA. 1993;269:2122-2127.
40. Rupie E, Dambrosio M, Servillo G, et al. Titration of tidal volume and induced hypercapnia in acute respiratory distress syndrome. Am J Respir Crit Care Med.. 1995;152:121-128.
41. Rouby J-J, Lu Q, Vieira S. Pressure/volume curves and lung computed tomography in acute respiratory distress syndrome. Eur Respir J. 2003;22(suppl 42):27S-36S.
42. Brower RG, Lanken PN, MacIntyre N, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med.. 2004;351:327-336.
43. Slutsky AS, Hudson LD. PEEP or no PEEP – lung recruitment may be the solution. N Engl J Med.. 2006;354:1839-1841.
44. Matthay MA, Calfee CS. Therapeutic value of a lung protective ventilation strategy in acute lung injury. Chest. 2005;128:3089-3091.
45. Petrucci N, Iacovelli W. Ventilation with lower tidal volumes versus traditional tidal volumes in adults for acute lung injury and acute respiratory distress syndrome. Cochrane Database Syst Rev. 2003;3. CD003844.
46. Fan E, Wilcox E, Brower RG, et al. Recruitment maneuvers for acute lung injury: a systematic review. Am J Respir Crit Care Med.. 2008;178:1156-1163.
47. Adhikari N, Granton JT. Inhaled nitric oxide for acute lung injury. No place for NO? JAMA. 2004;291:1629-1631.
48. Chan KPW, Stewart TE, Mehta S. High-frequency oscillatory ventilation for adult patients with ARDS. Chest. 2007;131:1907-1916.
49. Kacmarek RM, Wiedemann HP, Lavin PT, Wedel MK, Tütüncü AS, Slutsky AS. Partial liquid ventilation in adult patients with acute respiratory distress syndrome. Am J Respir Crit Care Med.. 2006;173:882-889.