Chapter 27 Ventilatory failure
 Ventilatory failure occurs when alveolar ventilation becomes too low to maintain normal arterial blood gas partial pressures.
Ventilatory failure occurs when alveolar ventilation becomes too low to maintain normal arterial blood gas partial pressures.Definitions
Respiratory failure is defined as a failure of maintenance of normal arterial blood gas partial pressures. Hypoxia as a result of cardiac and other extra-pulmonary forms of shunting are excluded from this definition. Respiratory failure may be subdivided according to whether the arterial Pco2 is normal or low (type 1) or elevated (type 2). Mean of the normal arterial Pco2 is 5.1 kPa (38.3 mmHg) with 95% limits (2 s.d.) of±1.0 kPa (7.5 mmHg). The normal arterial Po2 is more difficult to define because it decreases with age (page 194), and is strongly influenced by the concentration of oxygen in the inspired gas. Mechanisms that contribute to respiratory failure include ventilatory failure (reduced alveolar ventilation) and venous admixture as a result of either pure intrapulmonary shunt or ventilation perfusion mismatch (Chapter 8).
Ventilatory failure is defined as a pathological reduction of the alveolar ventilation below the level required for the maintenance of normal alveolar gas partial pressures. Because arterial Po2 (unlike arterial Pco2) is so strongly influenced by shunting, the adequacy of ventilation is conveniently defined by the arterial Pco2, although it is also reflected in end-expiratory Pco2 and Po2. This chapter is concerned mainly with pure ventilatory failure; other causes of respiratory failure are described in the next four chapters.
Pattern of Changes in Arterial Blood Gas Tensions
Figure 27.1 shows, on a Po2/Pco2 diagram, the typical patterns of deterioration of arterial blood gases in respiratory failure. The shaded area indicates the normal range of values with increasing age corresponding to a leftward shift. Pure ventilatory failure in a young person with otherwise normal lungs would result in changes along the broken line. Chronic obstructive pulmonary disease (COPD), the most common cause of predominantly ventilatory failure, occurs in older people, and the observed pattern of change is shown within the upper arrow in Figure 27.1. The limit of survival, while breathing air, is reached at a Po2 of about 2.7 kPa (20 mmHg) and Pco2 of 11 kPa (83 mmHg). The limiting factor is not Pco2 but Po2. This prevents the rise of Pco2 to higher levels except when the patient’s inspired oxygen concentration is increased. It may also be raised above 11 kPa by the inhalation of carbon dioxide. In either event, a Pco2 in excess of 11 kPa may be considered an iatrogenic disorder. Figure 27.1 also shows the pattern of blood gas changes caused by shunting or pulmonary venous admixture (Chapter 8).
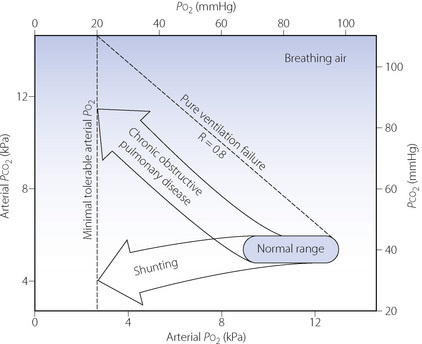
Fig. 27.1 Pattern of deterioration of arterial blood gases in chronic obstructive pulmonary disease and pulmonary shunting. The shaded area indicates the normal range of arterial blood gas partial pressures from 20 to 80 years of age. The oblique broken line shows the theoretical changes in alveolar Po2 and Pco2 resulting from pure ventilatory failure. In chronic obstructive pulmonary disease, the arterial Po2 is always less than the value that would be expected in pure ventilatory failure at the same Pco2 value. Discussion of shunting is to be found in Chapter 8 and further discussion of chronic obstructive pulmonary disease in Chapter 28.
In general, the arterial Po2 indicates the severity of respiratory failure (assuming that the patient is breathing air), while the Pco2 indicates the differential diagnosis between ventilatory failure and shunting as shown in Figure 27.1. In respiratory disease it is, of course, common for ventilatory failure and shunting to coexist in the same patient, and the relative contribution of each mechanism will determine whether type 1 or 2 respiratory failure develops.
Time Course of Changes in Blood Gas Tensions in Acute Ventilatory Failure
Although the upper arrow in Figure 27.1 shows the effect of established ventilatory failure on arterial blood gas tensions, short-term deviations from this pattern occur in acute ventilatory failure. This is because the time courses of changes of Po2 and Pco2 in response to acute changes in ventilation are quite different.
Body stores of oxygen are small, amounting to about 1550 ml while breathing air. Therefore, following a step change in the level of alveolar ventilation, the alveolar and arterial Po2 rapidly reach the new value and the half-time for the change is only 30 seconds (see page 205 and Figure 11.19). In contrast, the body stores of carbon dioxide are very large: of the order of 120 litres. Therefore, following a step change in the level of alveolar ventilation, the alveolar and arterial Pco2 only slowly attain the value determined by the new alveolar ventilation. Furthermore, the time course is slower following a reduction of ventilation than an increase (Figure 10.11) and the half-time of rise of Pco2 following a step reduction of ventilation is of the order of 16 minutes.
The practical point is that, during the early phase of acute hypoventilation, there may be a low Po2 while the Pco2 is increasing but is still within the normal range. Thus the pulse oximeter may, under certain circumstances such as when breathing air, give an earlier warning of hypoventilation than the capnograph. This breaks the rule that the Pco2 is the essential index of alveolar ventilation, and it may be erroneously believed that the diagnosis is shunting rather than hypoventilation.
Causes of Ventilatory Failure1,2
The causes of ventilatory failure may be conveniently considered under the headings of the anatomical sites where they arise. These sites are indicated in Figure 27.2. Lesions or malfunctions at sites A to E result in a reduction of input to the respiratory muscles. Dyspnoea may not be apparent and the diagnosis of ventilatory failure may be overlooked on superficial inspection of the patient. Lesions or malfunctions at sites G to J result in evident dyspnoea and no one is likely to miss the diagnosis of hypoventilation. The various sites will now be considered individually.
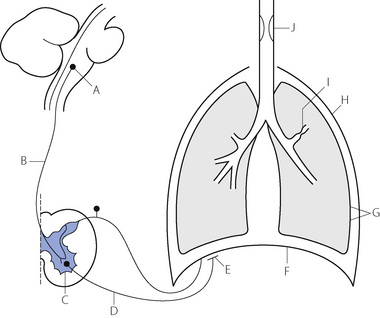
Fig. 27.2 Summary of sites at which lesions, drug action or malfunction may result in ventilatory failure. (A) Respiratory centre. (B) Upper motor neuron. (C) Anterior horn cell. (D) Lower motor neuron. (E) Neuromuscular junction. (F) Respiratory muscles. (G) Altered elasticity of lungs or chest wall (H) Loss of structural integrity of chest wall and pleural cavity. (I) Increased resistance of small airways. (J) Upper airway obstruction.
A wide variety of drugs may cause central apnoea or respiratory depression (page 77) and these include opioids, barbiturates and most anaesthetic agents, whether intravenous or inhalational. The respiratory neurones may also be affected by a variety of neurological conditions such as raised intracranial pressure, stroke, trauma or neoplasm.
Cardiac failure may result in respiratory muscle weakness due to reduced blood supply,6 often coupled with low compliance lungs due to pulmonary oedema (Chapter 29).
Assessment of respiratory muscle strength is described on page 97.
Closed pneumothorax causes interference with ventilation in proportion to the quantity of air in the chest, and is described on page 445.
Increased Dead Space
Very rarely, a large increase in the respiratory dead space may cause ventilatory failure. Minute volume may be increased but the alveolar ventilation is reduced and the patient presents with a high Pco2 accompanied by a high minute volume. An increase in the arterial/end-expiratory Pco2 gradient (more than 2 kPa or 15 mmHg) indicates an increase in the alveolar dead space. This condition may be caused by ventilation of large unperfused areas of lung (e.g. an air cyst communicating with the bronchus), pulmonary emboli or pulmonary hypotension. External or apparatus dead space also tends to reduce alveolar ventilation and may be added either intentionally or accidentally.
Relationship between Ventilatory Capacity and Ventilatory Failure
Tests for the measurement of ventilatory capacity are described on pages 96 et seq. However, a severe reduction in ventilatory capacity does not necessarily mean that a patient will be in ventilatory failure. Figure 27.3 shows the lack of correlation between FEV1 and arterial Pco2 in the grossly abnormal range of FEV1 0.3–1 litre from a series of patients with COPD.7
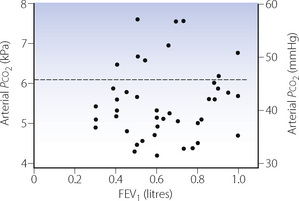
Fig. 27.3 Lack of correlation between arterial Pco2 and forced expiratory volume in one second (FEV1) in 44 patients with chronic obstructive pulmonary disease. The broken line indicates the upper limit of normal for Pco2.
(Data from reference 7.)
It should again be stressed that the usual tests of ventilatory capacity depend on the expiratory muscles while the work of breathing is normally achieved by the action of inspiratory muscles.
Metabolic Demand and Ventilatory Failure
In renal failure, protein intake is a major factor in the onset of uraemia. Similarly, in ventilatory failure, the onset of hypoxia and hypercapnia is directly related to the metabolic demand. Just as patients with renal failure benefit from a low protein diet, so patients with a severe reduction of ventilatory capacity protect themselves by limiting the exercise they take.
As COPD progresses, the ventilatory capacity decreases and the minute volume of breathing required for a particular level of activity increases. The increased ventilatory requirement is because both the dead space and the oxygen cost of breathing increase. The patient is thus trapped in a pincer movement of decreasing ventilatory capacity and increasing ventilatory requirement. As the jaws of the pincer close, there is first a limitation on heavy exercise, then on moderate exercise and so on until the patient is dyspnoeic at rest. At any time his work capacity is limited by the fraction of his ventilatory capacity that he is able to maintain for a given level of oxygen uptake.
The complex interaction between these factors is demonstrated in Figure 27.4, where the upper part shows the normal state. Assuming that an untrained subject can comfortably maintain a minute volume equal to about 30% of his maximal breathing capacity (MBC) without dyspnoea, he has a reserve of ventilatory capacity that is adequate for rest and a power output of 100 watts. However, a power output of 200 watts requires a minute volume that exceeds a third of his MBC and he becomes aware of his breathing at this level of exercise.
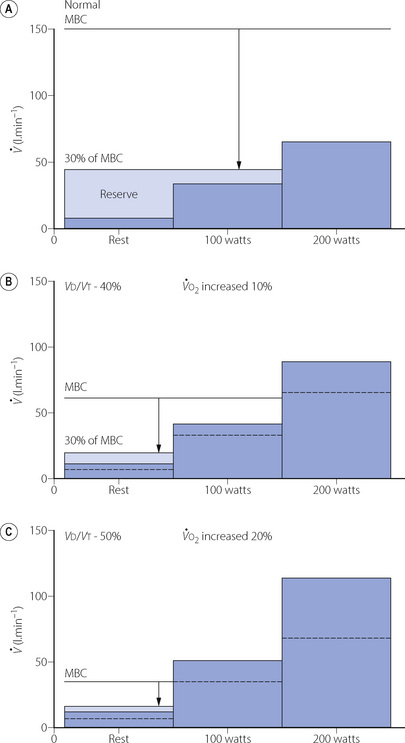
Fig. 27.4 Relationship between maximal breathing capacity (MBC) and ventilatory requirements at rest and work at 100 and 200 watts. The tips of the arrows indicate 30% of MBC, which can usually be maintained without dyspnoea. Ventilatory reserve is between this level and the various ventilatory requirements. (A) Normal. (B) Moderate loss of ventilatory capacity with some increase in oxygen cost of breathing. (C) Severe loss of ventilatory capacity with considerable increase in the oxygen cost of breathing.
Figure 27.4b shows moderately severe COPD with the following changes:
Factors 2 and 3 together result in an increased minute volume for each level of activity.
Again, on the assumption that dyspnoea will not be apparent until the minute volume is 30% of MBC, the reserve of ventilation is now sufficient for rest, but 100 watts of power output will result in dyspnoea.
Finally, in Figure 27.4c, the changes have progressed to the point where resting minute volume exceeds 30% of MBC and the patient is dyspnoeic at rest.
Breathlessness8
Breathlessness or dyspnoea has been defined as ‘undue awareness of breathing or awareness of difficulty in breathing’.9 This definition applies to both the awareness of breathing during severe exercise in the healthy subject and the dyspnoea of a patient with respiratory failure or heart failure. In the first case the sensation is normal and to be expected. However, in the latter, it is pathological and should be considered as a symptom.
The Origin of the Sensation
Hypoxia and hypercapnia may force the patient to breathe more deeply but they are not per se responsible for the sensation of dyspnoea, which arises from the ventilatory response rather than the stimulus itself. Patients with respiratory paralysis caused by poliomyelitis did not usually complain of dyspnoea in spite of abnormal blood gas tensions. Campbell & Guz9 advanced their reasons for believing that dyspnoea is not akin to pain, though recent studies are now supporting this analogy. Like pain, dyspnoea includes a distressing perception usually referred to as ‘air-hunger’, which seems to result more from hypercapnia than from the work of breathing.10 Some patients have dyspnoea at relatively low levels of work of breathing, whereas others show no dyspnoea at high levels of work. Functional imaging of the brain also shows that the cortical areas activated by dyspnoea are close to regions activated by pain.11 Finally, as for pain, there is undoubtedly a psychological component to the sensation of breathlessness.12 Dyspnoea arising from respiratory disease, particularly acutely, is often associated with anxiety and panic, which exacerbate the symptom. Conversely, many patients with primary psychological complaints such as panic disorder present with dyspnoea in the absence of any respiratory disease.
In 1963 it was suggested that a major factor in the origin of dyspnoea was an ‘inappropriateness’ between the tension generated in the respiratory muscles and the resultant shortening of the muscle fibres.13 This sensory input from muscle spindles would indicate to the brain that breathing was in some way hindered. The theory has since been widened to include other sensory receptors in the respiratory system, again suggesting that dyspnoea results from a mismatch between motor output and sensory input in the respiratory centre.2 These theories seem to fit observations made during breath holding (pages 76 et seq), which provide some insight into the origin of the sensation of breathlessness. Blood gas tensions are by no means the only factor limiting breath-holding time, although Po2 is more important than Pco2. The sensation that terminates breath holding can be relieved by ventilation without change of blood gas tensions, by bilateral vagal block and by curarisation. Diaphragmatic afferents appear to be more important than those from the intercostals.
It cannot be said that the problem of breathlessness is completely understood at the present time.2 The origin seems to be multifactorial and the mechanisms of its generation are clearly complex.
Treatment of breathlessness. Optimal treatment of the underlying disease process that is causing the dyspnoea is clearly the first approach to managing the symptom. However, in the later stages of many respiratory diseases, and in almost all patients with malignancy, breathlessness becomes an intractable and distressing problem. Palliation of breathlessness is now recognised as a valuable form of therapy, and offers further insight into the multifactorial nature of the symptom.8,14 Simple measures such as a fan blowing on the face are effective, and breathing oxygen relieves dyspnoea in many patients, even some who are not hypoxic.14 Opioids are effective, whether exogenous or endogenous as a result of exercising.15 Whether this opioid effect is mediated by reducing the respiratory drive or by simply altering the patient’s perception of their breathlessness is unknown.16
Principles of Therapy for Ventilatory Failure
Many patients lead normal lives with arterial Pco2 levels as high as 8 kPa (60 mmHg). Higher levels are associated with increasing disability, largely due to the accompanying hypoxaemia when the patient is breathing air (see Figure 27.1). Treatment may be divided into symptomatic relief of hypoxaemia and attempts to improve the alveolar ventilation.
Treatment of Hypoxaemia due to Hypoventilation by Administration of Oxygen
Hypoxia must be treated as the first priority, and administration of oxygen is the fastest and most effective method.
The relationship between alveolar Po2 and alveolar ventilation is explained on pages 180 et seq. and illustrated in Figure 11.2. If other factors remain constant, an increase in inspired gas Po2 will result in an equal increase in alveolar gas Po2. Therefore only small increases in inspired oxygen concentration are required for the relief of hypoxia due to hypoventilation. Figure 27.5 shows the rectangular hyperbola relating Pco2 and alveolar ventilation (as in Figure 10.9), but superimposed are the concentrations of inspired oxygen required to restore a normal alveolar Po2 for different degrees of alveolar hypoventilation. It will be seen that 30% is sufficient for the degree of alveolar hypoventilation that will result in an alveolar Pco2 of 13 kPa (almost 100 mmHg). Clearly this is an unacceptable Pco2, and therefore 30% can be regarded as the upper limit of inspired oxygen concentration to be used in the palliative relief of hypoxia due to ventilatory failure, without attempting to improve the alveolar ventilation.
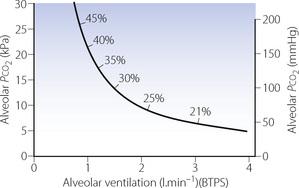
Fig. 27.5 Alveolar Pco2 as a function of alveolar ventilation at rest. The percentages indicate the inspired oxygen concentration that is then required to restore normal alveolar Po2.
The use of very high concentrations of inspired oxygen will prevent hypoxia even in gross alveolar hypoventilation, which carries the risk of dangerous hypercapnia. Although this is itself a strong contraindication to the use of high concentrations of oxygen under these circumstances, the effect may be even worse in patients who have lost their ventilatory sensitivity to carbon dioxide and rely upon their hypoxic drive to maintain ventilation (page 412). Recognition of this potential problem unfortunately resulted in a tendency to withhold oxygen for fear of causing hypercapnia. The rule is that hypoxia must be treated first, because hypoxia kills quickly while hypercapnia kills slowly. However, it must always be remembered that administration of oxygen to a patient with ventilatory failure will do nothing to improve the Pco2 and may make it worse. The arterial Pco2 must be measured if there is any doubt.
Improvement of Alveolar Ventilation
The only way to reduce the arterial Pco2 is to improve the alveolar ventilation. The first line of therapy is to improve ventilatory capacity by treatment of the underlying cause while simultaneously providing carefully controlled oxygen therapy and avoiding the use of drugs that depress breathing.
The second line is chemical stimulation of breathing. Doxapram stimulates breathing via an action on the peripheral chemoreceptors (page 78) and is effective in treating exacerbations of COPD, but only for the first few hours after admission to hospital.17
The third line of treatment is by tracheal intubation or tracheostomy, which may improve alveolar ventilation by reducing dead space and facilitating the control of secretions.
The fourth line of therapy is the institution of artificial ventilation (considered in detail in Chapter 32). It is difficult to give firm guidelines for the institution of artificial ventilation and the arterial Pco2 should not be considered in isolation. Nevertheless, a Pco2 in excess of 10 kPa (75 mmHg) that cannot be reduced by other means in a patient who is deemed recoverable is generally considered as a firm indication. However, artificial ventilation may be required at much lower levels of Pco2 if there is actual or impending respiratory fatigue as a result of increased work of breathing. This may be difficult to diagnose or predict. Although it is now recognised that intense activity by the respiratory muscles results in fatigue, as in the case of other skeletal muscles under similar conditions, it is also thought that ventilatory failure from this cause occurs only very late in the course of most respiratory diseases.
References
1. Roussos C, Koutsoukou A. Respiratory failure. Eur Respir J. 2003;22(Supp 47):3S-14S.
*2. Laghi F, Tobin MJ. Disorders of the respiratory muscles. Am J Respir Crit Care Med.. 2003;168:10-48.
3. Sharsar T, Chevret S, Bourdain F, Raphael JC. Early predictors of mechanical ventilation in Guillain–Barré syndrome. Crit Care Med.. 2003;31:278-283.
4. Mier A, Brophy C, Green M. Respiratory muscle function in myasthenia gravis. Am Rev Respir Dis.. 1988;138:867-874.
5. Roussos C, Zakynthinos S. Fatigue of the respiratory muscles. Intensive Care Med.. 1996;22:134-155.
6. Hammond MD, Bauer KA, Sharp JT, Rocha RD. Respiratory muscle strength in congestive heart failure. Chest. 1990;98:1091-1094.
7. Nunn JF, Milledge JS, Chen D, Doré C. Respiratory criteria of fitness for surgery and anaesthesia. Anaesthesia. 1988;43:543-551.
*8. Meek PM, Schwartzstein RM, Adams L, et al. Dyspnea. mechanisms, assessment, and management: A consensus statement. Am J Respir Crit Care Med.. 1999;159:321-340.
9. Campbell EJM, Guz A. Breathlessness. In: Hornbein TF, editor. Regulation of breathing, Part II. New York: Marcel Dekker, 1981.
10. Banzett RB, Pedersen SH, Schwartzstein RM, Lansing RW. The affective dimension of laboratory dyspnea: air hunger is more unpleasant than work/effort. Am J Respir Crit Care Med.. 2008;177:1384-1390.
11. von Leupoldt A, Dahme B. Cortical substrates for the perception of dyspnea. Chest. 2005;128:345-354.
12. Smoller JW, Pollack MH, Otto MW, Rosenbaum JF, Kradin RL. Panic anxiety, dyspnea, and respiratory disease – Theoretical and clinical considerations. Am J Respir Crit Care Med.. 1996;154:6-17.
13. Campbell EJM, Howell JBL. The sensation of breathlessness. Br Med Bull.. 1963;19:36-40.
14. Booth S, Wade R. Oxygen or air for palliation of breathlessness in advanced cancer. J R Soc Med.. 2002;96:215-218.
15. Mahler DA, Murray JA, Waterman LA, et al. Endogenous opioids modify dyspnoea during treadmill exercise in patients with COPD. Eur Respir J. 2009;33:771-777.
16. Jennings A-L, Davies AN, Higgins JPT, Gibbs JSR, Broadley KE. A systematic review of the use of opioids in the management of dyspnoea. Thorax. 2002;57:939-944.
17. Greenstone M, Lasserson TJ. Doxapram for ventilatory failure due to exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2003;1:CD000223.