Respiratory System, Mediastinum, and Pleurae
Diseases of the respiratory system (respiratory apparatus) are some of the leading causes of morbidity and mortality in animals and a major source of economic losses. Thus veterinarians are routinely called to diagnose, treat, and implement health management practices to reduce the impact of these diseases. In companion animals, diseases of the respiratory tract are also common and, although of little economic significance, are important to the health of the animals and thus to clinicians and owners.
Structure and Function
To facilitate the understanding of the structure and function, it is convenient to arbitrarily divide the respiratory system into conducting, transitional, and gas exchange systems (Fig. 9-1). The conducting system includes nostrils, nasal cavity, paranasal sinuses, nasopharynx, larynx, trachea, and extrapulmonary and intrapulmonary bronchi, all of which are largely lined by pseudostratified, ciliated columnar cells plus a variable proportion of secretory goblet (mucous) and serous cells (Figs. 9-2 and 9-3 and Web Fig. 9-1). The transitional system of the respiratory tract is composed of bronchioles, which serve as a transition zone between the conducting system (ciliated) and the gas exchange (alveolar) system (see Fig. 9-1). The disappearance of cilia in the transitional system is not abrupt; the ciliated cells in the proximal bronchiolar region become scarce and progressively attenuated, until the point where distal bronchioles no longer have ciliated cells. Normal bronchioles also lack goblet cells, but instead have other types of secretory cells, notably Clara and neuroendocrine cells. Clara cells, also referred to as secretory bronchiolar cells, contain numerous biosynthetic organelles that play an active role in detoxification of xenobiotics (foreign substances), similar to the role of hepatocytes (Fig. 9-4). Clara cells are also critical stem cells in the repair and remodeling of not only the bronchioles, but of most of the respiratory tract. In addition, Clara cells contribute to the innate immunity of the lung by secreting protective proteins (collectins) and pulmonary surfactant (Fig. 9-4, B). In carnivores and monkeys, and to a much lesser extent in horses and humans, the terminal portions of bronchioles are not only lined by cuboidal epithelium but also by segments of alveolar capillaries. These unique bronchioloalveolar structures are known as respiratory bronchioles (Fig. 9-5; also see Fig. 9-1). The gas exchange system of the respiratory tract in all mammals is formed by alveolar ducts and millions of alveoli (Fig. 9-6; also see Fig. 9-1). The surface of the alveoli is lined by two distinct types of epithelial cells known as type I pneumonocytes (membranous) and type II pneumonocytes (granular) (Fig. 9-7).
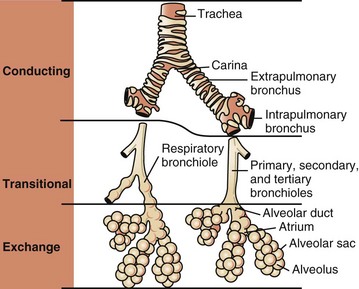
Fig. 9-1 Schematic diagram of airways from the trachea to the alveoli.
Conducting, transitional, and exchange components of the respiratory system. The transitional zone (bronchioles) is not as equally well developed in all species. (From Banks WJ: Applied veterinary histology, ed 3, St Louis, 1993, Mosby.)
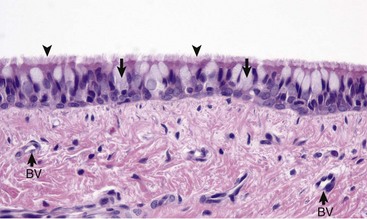
Fig. 9-2 Normal mucosa, trachea dog.
Mucosa consists of ciliated and nonciliated secretory cells. Goblet cells have a pale staining cytoplasm (arrows). The proportion of ciliated to nonciliated cells varies depending on the level of airways. Ciliated cells (arrowheads) are more abundant in proximal airways, whereas secretory cells are proportionally more numerous in distal portions of the conducting and transitional systems. The submucosa of the conducting system (nasal to bronchi) has abundant blood vessels (BV). (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
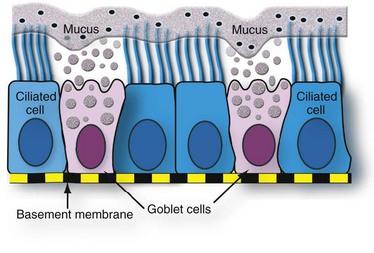
Fig. 9-3 Schematic representation of the mucociliary apparatus of the conducting system.
Both ciliated and goblet cells rest on the basement membrane. Mucus produced and released by goblet cells forms a carpet on which inhaled particles (dots) are trapped and subsequently expelled into the pharynx by the mucociliary apparatus. (Courtesy Dr. A. López, Atlantic Veterinary College.)
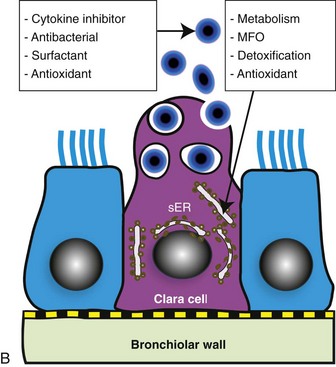
Fig. 9-4 Normal bronchiole, rat.
A, Bronchiole showing a thin wall composed of a basement membrane, smooth muscle, and connective tissue. On the luminal surface of the bronchiole note dome-shaped Clara cells (arrows) protruding into the lumen. H&E stain. B, Schematic representation of a Clara cell showing abundant smooth endoplasmic reticulum (SER) and cytoplasmic granules, which are extruded into the bronchiolar lumen. MFO, Mixed function oxidases. (Courtesy Dr. A. López, Atlantic Veterinary College.)
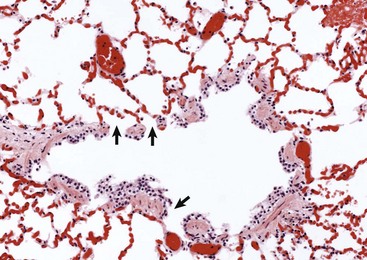
Fig. 9-5 Normal respiratory bronchiole, dog.
The wall of the bronchiole is covered by ciliated epithelium, which is supported by smooth muscle and connective tissue. Terminally, the wall becomes interrupted, forming lateral communications between the bronchiolar lumen and alveoli (arrows). (Courtesy Dr. A. López, Atlantic Veterinary College.)
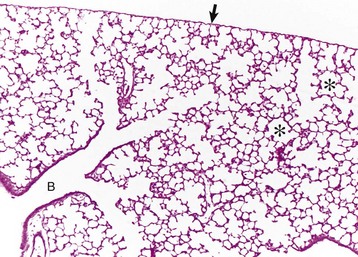
Fig. 9-6 Lung, rat.
Lungs were fixed by intratracheal perfusion of fixative to retain normal distention of airways. Note the dichotomous branching of the bronchioles and the thin visceral pleura (arrow) covering the surface of the lungs (B) that terminate as alveoli (asterisks). H&E stain. (Courtesy Dr. J. Martinez-Burnes, Atlantic Veterinary College.)
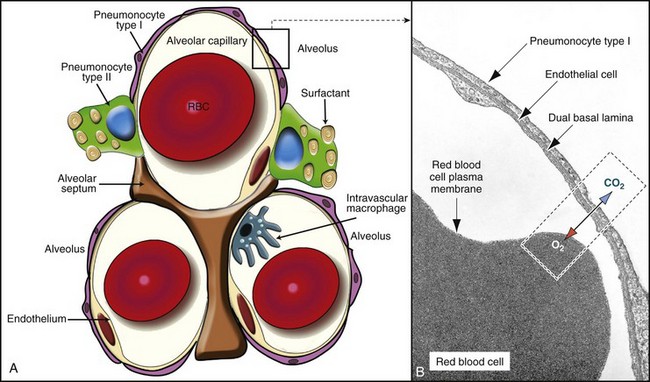
Fig. 9-7 The blood-air barrier.
A, In this schematic diagram, note the thin membrane (blood-air barrier) separating the blood compartment from the alveoli. Type I alveolar cells (membranous pneumonocytes) are remarkably thin and cover most of the alveolar wall. Note the endothelial cells lining the alveolar capillary. Alveolar interstitium supports the alveolar epithelium on one side and the endothelium on the other side of the blood-air barrier. Type II (granular) pneumonocytes appear as large cuboidal cells with lamellar bodies (surfactant) in the cytoplasm. A pulmonary intravascular macrophage, a component of the monocyte-macrophage system, is depicted on the wall of an alveolar capillary. A red blood cell (RBC) is present inside the lumen of the alveolar capillary. B, Alveolar wall. The blood-air barrier consists of cytoplasmic extensions of (1) type I alveolar cells (membranous pneumonocytes); (2) a dual basal lamina synthesized by type I alveolar cells; (3) cytoplasmic extensions of endothelial cells. TEM. Uranyl acetate and lead citrate stain. (A courtesy Dr. A. López, Atlantic Veterinary College. B from Kierszenbaum AL: Histology and cell biology, St Louis, 2002, Mosby.)
All three—the conducting, transitional, and exchange systems of the respiratory system—are vulnerable to injury because of constant exposure to a myriad of microbes, particles and fibers, and toxic gases and vapors present in the air. Vulnerability of the respiratory system to aerogenous (airborne) injury is primarily because of (1) the extensive area of the alveoli, which are the interface between the blood in alveolar capillaries and inspired air; (2) the large volume of air passing continuously into the lungs; and (3) the high concentration of noxious elements that can be present in the air (Table 9-1). For humans, it has been estimated that the surface of the pulmonary alveoli is approximately 200 m2, roughly the area of a tennis court. It has also been estimated that the volume of air reaching the human lung every day is around 9000 L. The surface of the equine lung is estimated to be around 2000 m2.
TABLE 9-1
Common Pathogens, Allergens, and Toxic Substances Present in Inhaled Air
| Category | Agents |
| Microbes | Viruses, Chlamydophila, bacteria, fungi, protozoa |
| Plant dust | Grain, flour, cotton, wood |
| Animal products | Dander, feathers, mites, insect chitin |
| Toxic gases | Ammonia (NH3), hydrogen sulfide (H2S), nitrogen dioxide (NO2), sulfur dioxide (SO2), chlorine |
| Chemicals | Organic and inorganic solvents, herbicides, asbestos, nickel, lead |
Lungs are also susceptible to blood-borne (hematogenous) microbes, toxins, and emboli. This fact is not surprising because the entire cardiac output of the right ventricle goes into the lungs, and approximately 9% of the total blood volume is within the pulmonary vasculature. The pulmonary capillary bed is the largest in the body, with a surface area of 70 m2 in the adult human; this area is equivalent to a length of 2400 km of capillaries, with 1 mL of blood occupying up to 16 km of capillary bed.
Normal Flora of the Respiratory System
The respiratory system has its own normal flora (microbiota), as does any other body system in contact with the external environment. If a sterile swab is passed deep into the nasal cavity of any healthy animal and cultured for microbes, yeasts, and fungi, many species of bacteria are recovered, such as Mannheimia (Pasteurella) haemolytica in cattle; Pasteurella multocida in cats, cattle, and pigs; and Bordetella bronchiseptica in dogs and pigs. The organisms that constitute the normal flora of the respiratory tract are restricted to the most proximal (rostral) region of the conducting system (nasal cavity, pharynx, and larynx). The thoracic portions of the trachea, bronchi, and lungs are considered to be essentially sterile. The types of bacteria present in the nasal flora vary considerably among animal species and in different geographic regions of the world. Some bacteria present in the nasal flora are pathogens that can cause important respiratory infections. For instance, Mannheimia (Pasteurella) haemolytica is part of the bovine nasal flora, yet this bacterium causes a devastating disease in cattle—pneumonic Mannheimiosis (shipping fever). Experimental studies have established that microorganisms from the nasal flora are continuously carried into the lungs via tracheal air. In spite of this constant bacterial bombardment from the nasal flora and from contaminated air, normal lungs remain sterile because of their remarkably effective defense mechanisms.
Portals of Entry into the Respiratory System
Microbes, toxins, and pneumotoxicants can gain access into the respiratory system by the following routes (also see Tables 9-1 and 9-2):
TABLE 9-2
Portals of Entry into the Respiratory System
| Route | Agents |
| Aerogenous (inhalation) | Virus, bacteria, Chlamydophila, fungi, toxic gases, and pneumotoxicants |
| Hematogenous (blood) | Virus, bacteria, fungi, parasites, toxins, and pneumotoxicants |
| Direct extension | Penetrating wounds, migrating awns, bites, and ruptured esophagus or perforated diaphragm (hardware) |
1. Aerogenous—Pathogens, such as bacteria, mycoplasmas, and viruses, along with toxic gases and foreign particles, including food, can gain access to the respiratory system via inspired air. This is the most common route in the transmission of most respiratory infections in domestic animals.
2. Hematogenous—Some viruses, bacteria, parasites, and toxins can enter the respiratory system via the circulating blood. This portal of entry is commonly seen in septicemias, bacteremias, and protozoa and viruses that target endothelial cells. Also, circulating leukocytes may release infectious organisms such as retroviruses and Listeria monocytogenes while traveling through the lungs.
3. Direct extension—In some instances, pathogenic organisms can also reach the pleura and lungs through penetrating injuries, such as gunshot wounds, migrating awns, or bites, or by direct extension from a ruptured esophagus or perforated diaphragm.
Defense Mechanisms of the Respiratory System
It is axiomatic that a particle, microbe, or toxic gas must first gain entry to a vulnerable region of the respiratory system before it can induce an adaptive immune response or have a pathologic effect. The characteristics of size, shape, dispersal, and deposition of particles present in inspired air are studied in aerobiology. It is important to recognize the difference between deposition, clearance, and retention of inhaled particles. Deposition is the process by which particles of various sizes and shapes are trapped within specific regions of the respiratory tract. Clearance is the process by which deposited particles are destroyed, neutralized, or removed from the mucosal surfaces. The difference between what is deposited and what is cleared from the respiratory tract is referred to as retention. The main mechanisms involved in clearance are sneezing, coughing, mucociliary transport, and phagocytosis (Table 9-3). Abnormal retention of particles resulting from increased deposition, decreased clearance, or a combination of both is the underlying pathogenetic mechanism in many pulmonary diseases (Fig. 9-8).
TABLE 9-3
Main Defense Mechanisms of the Respiratory System
| Regions of the Respiratory System | Defense Mechanisms |
| Conducting system (nose, trachea, and bronchi) | Mucociliary clearance, antibodies, lysozyme, mucus |
| Transitional system (bronchioles) | Clara cells, antioxidants, lysozyme, antibodies |
| Exchange system (alveoli) | Alveolar macrophages (inhaled pathogens), intravascular macrophages (circulating pathogens), opsonizing antibodies, surfactant, antioxidants |
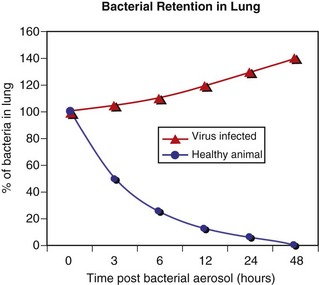
Fig. 9-8 Pulmonary clearance and retention of bacteria following inhalation of an experimental aerosol of bacteria.
When large numbers of bacteria are inhaled, the normal defense mechanisms promptly eliminate these microorganisms from the lungs (blue line). However, when the defense mechanisms are impaired by a viral infection, lung edema, stress, and so forth, the inhaled bacteria are not eliminated but colonize and multiply in the lung (red line). (Courtesy Dr. A. López, Atlantic Veterinary College.)
The anatomic configuration of the nasal cavity and bronchi plays a unique role in preventing or reducing the penetration of noxious material into the lungs, especially into the alveoli, which is the most vulnerable portion of the respiratory system. The narrow nasal meatuses and the coiled arrangement of the nasal conchae generate enormous turbulences of airflow and as a result, physical forces are created that forcefully impact particles larger than 10 µm onto the surface of the nasal mucosa (Fig. 9-9). Although particles smaller than 10 µm could escape trapping in the nasal cavity, these medium-sized particles meet a second barrier at the tracheal and bronchial bifurcations. Here, abrupt changes in the direction of air (inertia), which occurs at the branching of major airways, cause particles in the 2- to 10-µm size range to collide with the surface of bronchial mucosa (see Fig. 9-1). Because the velocity of inspired air at the level of the small bronchi and bronchioles has become rather slow, inertial and centrifugal forces no longer play a significant role in the trapping of inhaled particles. Here, in the transitional (bronchiolar) and exchange (alveolar) regions, particles 2 µm or smaller may come into contact with the mucosa by means of sedimentation because of gravitation or by diffusion as a result of Brownian movement. Infective aerosols containing bacteria and viruses are within the size ranges (0.01 to 2 µm) that typically gain access to the bronchioloalveolar region.
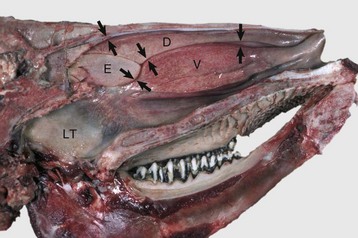
Fig. 9-9 Dorsal (D), ventral (V), and ethmoidal (E) conchae, midsagittal section of head, cow.
These meatuses (spaces between arrows) are narrow and the air turbulence produced in them by the coiled arrangement of the conchae causes suspended particles to impact on the mucus covering the surface of the nasal mucosa. These particles are then moved caudally by the mucociliary apparatus to the pharynx and finally swallowed. Note the abundant lymphoid tissue (LT) in the nasopharynx. (Courtesy Dr. R.G. Thomson, Ontario Veterinary College.)
In addition to size, other factors, such as shape, length, electrical charge, and humidity, play an important role in mucosal deposition, retention, and pathogenicity of inhaled particles. For example, particles longer than 200 µm may also reach the lower respiratory tract, provided their mean aerodynamic diameter is less than 1 µm. Asbestos is a good example of a large but slender fiber that can bypass the filtrating mechanisms by traveling parallel to the airstream. Once in the terminal bronchioles and alveoli, asbestos fibers cause asbestosis, a serious pulmonary disease in humans. In summary, the anatomic features of the nasal cavity and airways provide an effective barrier, preventing the penetration of most large particles into the lungs.
Once larger particles are trapped in the mucosa of conducting airways and small particles are deposited on the surface of the nasal, tracheal, or bronchioalveolar mucosa, it is crucial that these exogenous materials be removed to prevent or minimize injury to the respiratory system. For these purposes, the respiratory system is equipped with several defense mechanisms, all of which are provided by specialized cells operating in a remarkably well-coordinated manner.
Defense Mechanisms of the Conducting System (Nose, Trachea, and Bronchi)
Mucociliary clearance is the physical unidirectional movement and removal of deposited particles and gases dissolved in the mucus from the respiratory tract. Mucociliary clearance, also referred to as the waste disposal system, is provided by the mucociliary blanket (mucociliary escalator) and is the main defense mechanism of the conducting system (nasal cavity, trachea, and bronchi) (see Figs. 9-2 and 9-3). Mucus acts primarily as a barrier and a vehicle and is a complex mixture of water, glycoproteins, immunoglobulins, lipids, and electrolytes produced by goblet (mucous) cells, serous cells, submucosal glands, and fluid from transepithelial ion and water transport. Once serous fluid and mucus are secreted onto the surface of the respiratory mucosa, a thin, double-layer film of mucus is formed on top of the cells. The outer layer of this film is in a viscous gel phase, whereas the inner layer, which is in a fluid or sol phase, is directly in contact with cilia (see Fig. 9-3). A healthy human produces around 100 mL of mucus per day. Each ciliated cell in the conducting system has around 100 to 200 motile and chemosensory cilia (6 µm long), beating metachronously (forming a wave) at a ciliary beat frequency of approximately 1000 strokes per minute, and in a horse, for example, mucus moves longitudinally at a rate of up to 20 mm per minute. Rapid and powerful movement of cilia creates a series of waves that, in a continuous and synchronized manner, propel the mucus, exfoliated cells, and entrapped particles out of the respiratory tract to the pharynx. The mucus is finally swallowed, or when present in large amounts, it is coughed up out of the conducting system. If mucus flow were to move at the same rate in all levels of a conducting system, a “bottleneck” effect would be created in major airways as the minor but more numerous airways enter the bronchi. For this reason, the mucociliary transport in proximal (rostral) airways is physiologically faster than that of the distal (caudal) ones. Ciliary activity and mucus transport increase notably in response to stimuli such as in respiratory infections.
The mucociliary blanket of the nasal cavity, trachea, and bronchi also plays an important role in preventing injury from toxic gases. If a soluble gas contacts the mucociliary blanket, it mixes with the mucus, thus reducing the concentration of gas reaching deep into the alveoli. In other words, mucus acts as a “scavenger system,” whereby gases are solubilized and subsequently cleared from the respiratory tract via mucociliary transport. If ciliary transport is reduced (loss of cilia) or mucus production is excessive, coughing becomes an important mechanism for clearing the airways.
In addition to the mechanical barrier and physical transport provided by the mucociliary escalator, other cells closely associated with ciliated epithelium contribute to the defense mechanism of the conducting system. Among the most notable ones are the microfold (M) cells, which are modified epithelial cells covering the bronchial-associated lymphoid tissue (BALT), both of which are strategically situated at the corner of the bifurcation of bronchi and bronchioles, where inhaled particles often collide with the mucosa because of inertial forces. From here, inhaled particles and soluble antigens are phagocytosed and transported by macrophages, dendritic cells, and other professional antigen-presenting cells (APCs) into the BALT, thus providing a unique opportunity for B and T lymphocytes to enter into close contact with inhaled pathogenic substances. Pulmonary lymphocytes are not quiescent in the BALT but are in continual traffic to other organs and contribute to both cellular (cytotoxic, helper, suppressor T lymphocytes) and humoral immune responses. Immunoglobulin A (IgA), produced by mucosal plasma cells, and to a lesser extent, immunoglobulin G (IgG) and M (IgM) play important roles in the local immunity of the conducting system, especially with regard to preventing attachment of pathogens to the cilia. Chronic airway diseases, especially those caused by infection, such as those caused by mycoplasmas or retroviruses, are often accompanied by severe hyperplasia of the BALT.
The mucociliary clearance terminates at the pharynx, where mucus, propelled caudally from the nasal cavity and cranially from the tracheobronchial tree, is eventually swallowed and thus eliminated from the conducting system of the respiratory tract. Some respiratory pathogens, such as Rhodococcus equi, can infect the intestines after having been removed and swallowed from the respiratory tract into the alimentary system.
Defense Mechanisms of the Exchange System (Alveoli)
Alveoli lack ciliated and mucus-producing cells; thus the defense mechanism against inhaled particles in the alveolar region cannot be provided by mucociliary clearance. Instead, the main defense mechanism of alveoli (exchange system) is phagocytosis provided by the pulmonary alveolar macrophages (Fig. 9-10). These highly phagocytic cells, which are not to be confused with intravascular pulmonary macrophages, are derived largely from blood monocytes and to a much lesser extent, from a slowly dividing population of interstitial macrophages. After a temporary adaptive stage within alveolar interstitium, blood monocytes reduce their glycolytic metabolism and increase their oxidative metabolism to function in an aerobic rather than an anaerobic environment. Pulmonary alveolar macrophages contribute to the pulmonary innate and adaptive immune response rapidly attaching and phagocytosing bacteria and any other particle reaching the alveolar lumens. The number of free macrophages in the alveolar space is closely related to the number of inhaled particles reaching the lungs. This ability to increase, within hours, the number of available phagocytic cells is vital in protecting the distal lungs against foreign material, particularly when the inhaled particle load is high. Unlike that of tissue macrophages, the lifespan of alveolar macrophages in the alveoli is notably short, only a few days, and thus they are continuously being replaced by newly migrated blood monocytes.
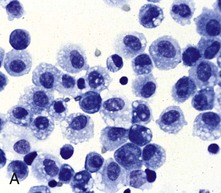
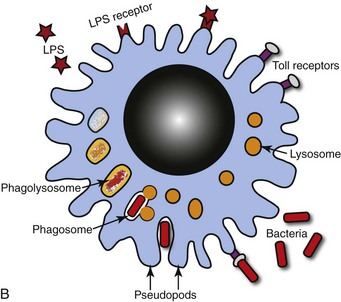
Fig. 9-10 Pulmonary alveolar macrophages.
A, Bronchoalveolar lavage, healthy pig. Alveolar macrophages characterized by abundant and vacuolated cytoplasm are the predominant cell in lavages from healthy lungs. Mayer’s hematoxylin counter stain. B, Schematic representation of a pulmonary alveolar macrophage. Note receptors in cell membrane, attachment of bacteria to cell receptor, bacteria being engulfed by cytoplasmic projections (pseudopods), formation of cytoplasmic phagosomes, and fusion of lysosomes with phagosome (phagolysosomes) which finally kill the ingested bacteria. (A courtesy Dr. L.A. Rijana-Ludbit, Tübingen; B courtesy of Dr. A. López, Atlantic Veterinary College.)
Alveolar phagocytosis plays a prominent role in the innate defense mechanism against inhaled bacteria without the need of an inflammatory reaction. Bacteria reaching the alveoli are rapidly phagocytosed, and bactericidal enzymes present in lysosomes are discharged into the phagosome containing the bacteria (see Fig. 9-10). Except for some facultative pathogens that are resistant to intracellular killing (e.g., Mycobacterium tuberculosis, Listeria monocytogenes, Brucella abortus, and some Salmonella spp.), most bacteria reaching the lungs are rapidly destroyed by activated alveolar macrophages. Similarly, inhaled particles, such as dust, pollen, spores, carbon, or erythrocytes from intraalveolar hemorrhage, are all phagocytosed and eventually removed from alveoli by pulmonary alveolar macrophages. Most alveolar macrophages leave the alveoli by migrating toward the bronchiolar (transitional) region until the mucociliary blanket is reached. Once there, pulmonary macrophages are removed in the same way as any other particle: along the mucociliary flow to the pharynx and swallowed. In the cat, as many as 1 million macrophages per hour move out from the alveoli into the conducting system and pharynx.
Destruction and removal of inhaled microbes and particles by alveolar macrophages is a well-orchestrated mechanism that engages many cells, receptors (i.e., Toll-like receptors [TLRs]) and pulmonary secretions in the lung. The cell-to-cell interactions are complex and involve pulmonary alveolar macrophages, pneumonocytes, endothelial cells, lymphocytes, plasma cells, natural killer (NK) cells, and dendritic cells. Antibodies are also important in the protection (acquired immune response) of the respiratory tract against inhaled pathogens. IgA is the most abundant antibody in the nasal and tracheal secretions and prevents the attachment and absorption of antigens (immune exclusion). IgG and to a lesser extent IgE and IgM promote the uptake and destruction of inhaled pathogens by phagocytic cells (immune elimination). IgG is the most abundant antibody in the alveolar surface and acts primarily as an opsonizing antibody for alveolar macrophages and neutrophils. In addition to antibodies, there are several secretory products locally released into the alveoli that constitute the alveolar lining material and contribute to the pulmonary defense mechanisms. The most important of these antimicrobial products are transferrin, anionic peptides, and pulmonary surfactant (Table 9-4).
TABLE 9-4
Defense Mechanisms Provided by Some Cells and Secretory Products Present in the Respiratory System
| Cell/Secretory Product | Action |
| Alveolar macrophage | Phagocytosis, main line of defense against inhaled particles and microbial pathogens in the alveoli |
| Intravascular macrophage | Phagocytosis, removal of particles, endotoxin, and microbial pathogens in the circulation |
| Ciliated cells | Expel mucus and inhaled particles and microbial pathogens by ciliary action |
| Clara cells | Detoxification of xenobiotics (mixed function oxidases) and protective secretions against oxidative stress and inflammation; production of surfactant |
| Mucus | Physical barrier; traps inhaled particles and microbial pathogens and neutralizes soluble gases |
| Surfactant | Protects alveolar walls and enhances phagocytosis |
| Lysozyme | Antimicrobial enzyme |
| Transferrin and lactoferrin | Inhibition and suppression of bacterial growth |
| α1-Antitrypsin | Protects against the noxious effects of proteolytic enzymes released by phagocytic cells; also inhibits inflammation |
| Interferon | Antiviral agent and modulator of the immune and inflammatory responses |
| Interleukins | Chemotaxis, upregulation of adhesion molecules |
| Antibodies | Prevent microbe attachment to cell membranes, opsonization |
| Complement | Chemotaxis; enhances phagocytosis |
| Antioxidants* | Prevent injury caused by superoxide anion, hydrogen peroxide, and free radicals (ROS) generated during phagocytosis, inflammation, or by inhalation of oxidant gases (ozone, nitrogen dioxide [NO2], sulfur dioxide [SO2]) |
*Superoxide dismutase, catalase, glutathione peroxidase, oxidant free radical scavengers (tocopherol, ascorbic acid).
To facilitate phagocytosis and discriminate between “self” and “foreign” antigens, pulmonary alveolar macrophages are furnished with a wide variety of specific receptors on their cell surfaces. Among the most important ones are Fc receptors for antibodies; complement receptors (C3b, C3a, C5a); tumor necrosis factor (TNF) receptor; and CD40 receptors, which facilitate phagocytosis and destruction of opsonized particles. TLRs recognize microbial components, and FAS receptors are involved in apoptosis and in the phagocytosis of apoptotic cells in the lung. “Scavenger receptors,” which are responsible for the recognition and uptake of foreign particulates, such as dust and fibers, are also present on pulmonary alveolar macrophages.
Defense Mechanisms Against Blood-Borne Pathogens (Intravascular Space)
Lungs are also susceptible to hematogenously borne microbes, toxins, or emboli. The hepatic (Kupffer cells) and splenic macrophages are the primary phagocytic cells responsible for removing circulating bacteria and other particles from the blood of dogs, some rodents, and humans. In contrast, the cell responsible for the removal of circulating particles, bacteria, and endotoxin from the blood of ruminants, cats, pigs, and horses is mainly the pulmonary intravascular macrophage, a distinct population of phagocytes normally residing within the pulmonary capillaries (see Fig. 9-7). In pigs, 16% of the pulmonary capillary surface is lined by pulmonary intravascular macrophages. In ruminants, 95% of intravenously injected tracer particles or bacteria are rapidly phagocytosed by these intravascular macrophages. Recent studies showed that an abnormally reduced number of Kupffer cells in diseased liver results in a compensatory increase in pulmonary intravascular macrophages, even in animal species in which these phagocytic cells are normally absent from the lung. In some abnormal conditions, such as sepsis, excessive release of cytokines by pulmonary intravascular macrophages may result in acute lung injury.
Defense Mechanisms Against Oxidant-Induced Lung Injury
Existing in an oxygen-rich environment and being the site of numerous metabolic reactions, the lungs also require an efficient defense mechanism against oxidant-induced cellular damage (oxidative stress). This form of damage is caused by inhaled oxidant gases (e.g., nitrogen dioxide, ozone, sulfur dioxide, or tobacco smoke), by xenobiotic toxic metabolites produced locally, by reaching the lungs via the bloodstream (e.g., 3-methylindole and paraquat), or by free radicals (reactive oxygen species) released by phagocytic cells during inflammation. Free radicals and reactive oxygen species (ROS) not only induce extensive pulmonary injury but also impair the defense and repair mechanisms in the lung. Oxygen and free radical scavengers, such as catalase, superoxide dismutase, ubiquinone, and vitamins E and C, are largely responsible for protecting pulmonary cells against peroxidation. These scavengers are present in alveolar and bronchiolar epithelial cells and in the extracellular spaces of the pulmonary interstitium.
In summary, the defense mechanisms are so effective in trapping, destroying, and removing bacteria that, under normal conditions, animals can be exposed to aerosols containing massive numbers of bacteria without any ill effects. If defense mechanisms are impaired, inhaled bacteria colonize and multiply in bronchi, bronchioles, and alveoli, and produce infection, which can result in fatal pneumonia. Similarly, when blood-borne pathogens, inhaled toxicants, or free radicals overwhelm the protective defense mechanisms, cells of the respiratory system are likely to be injured, often causing serious respiratory diseases.
Impairment of Defense Mechanisms in the Respiratory System
For many years, factors such as stress, viral infections, and pulmonary edema have been implicated in predisposing humans and animals to secondary bacterial pneumonia. There are many pathways by which the defense mechanisms can be impaired; only those relevant to veterinary species are discussed.
Viral Infections
Viral agents are notorious in predisposing humans and animals to secondary bacterial pneumonias by what is known as viral-bacterial synergism. A good example of this synergistic effect of combined virus-bacterial infections is documented from epidemics of humans with influenza virus in which the mortality rate has been significantly increased from secondary bacterial pneumonia. The most common viruses incriminated in predisposing animals to secondary bacterial pneumonia include influenza virus in pigs and horses; bovine herpesvirus 1 (BoHV-1), parainfluenza-3 (PI-3), and bovine respiratory syncytial virus (BRSV) in cattle; canine distemper virus in dogs; and herpesvirus and calicivirus in cats. The mechanism of the synergistic effect of viral-bacterial infections was previously believed to be the destruction of the mucociliary blanket and a concurrent reduction of mucociliary clearance, but in experimental studies, viral infections did not significantly reduce the physical removal of particles or bacteria out of the lungs. Now, it is known that 5 to 7 days after a viral infection, the mucociliary clearance and phagocytic function of pulmonary alveolar macrophages are notably impaired (see Fig. 9-8). Other mechanisms by which viruses impair defense mechanisms are multiple and remain poorly understood (Box 9-1). Immunization against viral infections in many cases prevents or reduces the synergistic effect of viruses and thus the incidence of secondary bacterial pneumonia
Toxic Gases
Certain gases also impair respiratory defense mechanisms, rendering animals more susceptible to secondary bacterial infections. For instance, hydrogen sulfide and ammonia, frequently encountered on farms, especially in buildings with poor ventilation, can impair pulmonary defense mechanisms and increase susceptibility to bacterial pneumonia. The effects of environmental pollutants on the defense mechanisms of humans and animals living in crowded and polluted cities remain to be determined.
Immunodeficiency
Immunodeficiency disorders, whether acquired or congenital, are often associated with increased susceptibility to viral, bacterial, and protozoal pneumonias. For example, humans with acquired immunodeficiency syndrome (AIDS) are notably susceptible to pneumonia caused by proliferation of Pneumocystis carinii. This ubiquitous organism, which under normal circumstances is not considered pathogenic, is also found in the pneumonic lungs of immunosuppressed pigs, foals, dogs, and rodents. Pigs infected with the porcine reproductive and respiratory syndrome (PRRS) virus frequently develop Pneumocystis carinii infection. Arabian foals born with combined immunodeficiency disease easily succumb to infectious diseases, particularly adenoviral pneumonia. Combined infections with two respiratory viruses, such as canine distemper and canine adenovirus (CAV-2), are sporadically reported in immunosuppressed puppies. Also, large doses of chemotherapeutic agents, such as steroids and alkylating agents, cause immunosuppression in dogs, cats, and other animals, increasing susceptibility to secondary viral and bacterial infections.
Other Conditions that Predispose to Secondary Bacterial Pneumonia
Uremia, endotoxemia, dehydration, starvation, hypoxia, acidosis, pulmonary edema, anesthesia, ciliary dyskinesia, and stress are only some of the many conditions that have been implicated in impairing respiratory defense mechanisms and consequently predisposing animals to develop secondary bacterial pneumonia. The mechanisms by which each of these factors suppresses pulmonary defenses are diverse and sometimes not well understood. For example, hypoxia and pulmonary edema decrease phagocytic function of pulmonary alveolar macrophages and alter the production of surfactant by type II pneumonocytes. Dehydration is thought to increase the viscosity of mucus, reducing or stopping mucociliary movement. Anesthesia induces ciliostasis, with concurrent loss of mucociliary function. Ciliary dyskinesia, an inherited defect in cilia, causes abnormal mucus transport; starvation, hypothermia, and tress can reduce humoral and cellular immune responses.
Structure and Function

Web Fig. 9-1 Ultrastructural morphology of respiratory mucosa.
A, Normal bronchial mucosa, bronchus, rat. The mucous layer was removed before fixation to expose the external surface of the epithelium. Mucosa consists of ciliated cells and nonciliated secretory cells. Ciliated cells have numerous slender cilia (arrows). Nonciliated secretory cells have a dome-shaped surface with abundant microvilli (arrowheads). The proportion of ciliated to nonciliated cells varies depending on the level of airways. Ciliated cells are more abundant in proximal airways, whereas secretory cells are more numerous in distal portions of the conducting and transitional systems. Scanning electron micrograph. Carbon-sputter coating method. B, Normal ciliated epithelium, trachea, cow. This trachea was specially fixed to preserve the mucous layer, which consists of an internal, clear, hypophase-fluid layer (not visible here) surrounding microvilli and kinocilia and an external mucous epiphase at the level of the tips of the kinocilia (cut in both transverse and longitudinal section here). TEM. Uranyl acetate and lead citrate stain. (A courtesy Dr. A. López, Atlantic Veterinary College. B from Sims DE, Westfall JA, Kiorpes AL, Horne MM: Biotech Histochem 66:173-180, 1991.)
Examination of the Respiratory Tract
The respiratory tract should always be examined in a systematic fashion. To determine whether negative pressure is present in the thoracic cavity, the diaphragm is punctured through the abdominal cavity before the thoracic cavity has been opened. When the diaphragm is punctured in a fresh carcass, the loss of negative pressure in the thorax causes the diaphragmatic cupola to drop back caudally toward the abdominal cavity, and at the same time, there is an audible sound caused by the inrush of air into the thorax. Lack of this movement may be an indication of advanced pneumothorax, pleural effusion, or the presence of uncollapsed lungs caused by pulmonary edema, pneumonia, fibrosis, or emphysema. In carcasses that have been dead for a long time, pulmonary air and gas produced by saprophytic bacteria leak into the pleural cavity, reducing the negative thoracic pressure and collapsing the lung.
The rib cage must be removed by cutting along the costosternal joints and along the neck of the ribs (close to the costovertebral joints) in such a way that pleural adhesions and abnormal thoracic contents can be observed and grossly quantified (e.g., 200 mL of clear, yellow fluid). The tongue, pharynx, esophagus, larynx, trachea, and thoracic viscera (lungs, heart, and thymus) should be removed as a unit (often called the pluck) and placed on the necropsy table.
The pharynx and esophagus are opened starting at the pharynx by a single cut with scissors along the dorsal midline and inspected for ulcers, foreign bodies, and neoplasms. The larynx and trachea must be examined by opening both along the dorsal midline from cranial to caudal ends and then extending the incision into the large bronchi of the caudal lung lobes. Normal tracheobronchial mucosa has a smooth and glistening pearl-colored surface with empty lumina in airways. The presence of foamy fluid in airways indicates pulmonary edema. Feed particles may suggest aspiration; however, careful examination of the mucosa is required because aspiration of ingesta from stomach or rumen into the lungs commonly takes place agonally or can be displaced into these areas when the carcass is moved.
The lungs should be examined before incision. Normal lungs typically have a homogeneous pink color (Web Fig. 9-2). External changes include the presence of rib imprints on the pleural surface when lungs fail to collapse. In addition, the lungs should be inspected for changes in color and texture and distribution of lesions. Color changes can be various shades of red, indicating hypostatic congestion, hyperemia (acute pneumonia), and hemorrhage; dark blue collapsed lobules or areas are indicative of atelectasis; pale pink to white lungs indicate notable anemia, fibrosis, or emphysema; and uniformly or patchy yellow-brown lungs indicate chronic passive congestion and pulmonary fibrosis likely secondary to chronic heart failure. Lungs from exsanguinated animals are generally paler than the normal pink color because of reduced blood in the pulmonary tissue. A covering of yellowish material on the pleural surface indicates accumulation of fibrin. Because it is impossible to describe the texture of normal lungs, experience in palpation is required to appreciate the actual texture of a normal lung. Texture is determined by gently palpating the surface and parenchyma of the lungs. Normal texture can change to firm, hard, elastic (rubbery), or crepitus (with a crackling sound or feeling). For a detailed description of lung texture, see the section on Classification of Pneumonias. Palpation of the lungs, which should be gentle, also permits detection of nonvisible nodules or abscesses in the parenchyma. Knowing the distribution of a lesion in the lungs also facilitates diagnosis because particular etiologic agents cause lesions with specific distribution. Distribution of lesions is generally described as focal, multifocal, locally extensive, or diffuse. According to their topography, pulmonary lesions can also be classified as cranioventral, dorsocaudal, and so on.
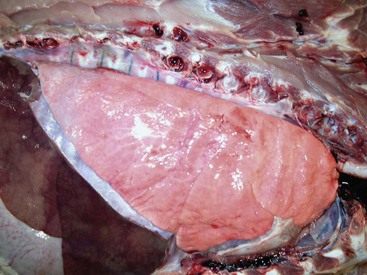
Web Fig. 9-2 Normal lung, pig.
The lung parenchyma appears homogeneously pink. The pale pink appearance of these normal lungs is due to exsanguination. The appearance of normal unexsanguinated lungs is bright pink to red. (Courtesy Dr. A. López, Atlantic Veterinary College.)
Reports must also contain an estimate of the extent of the pulmonary lesions, preferably expressed as a percentage of the volume of the lungs affected. For instance, a report may read “cranioventral consolidation involving 40% of the lungs.” If the lungs have focal lesions, a rough estimate of the number should also be included in the report. For instance, “numerous (approximately 25), small (1 to 2 cm in diameter), hard nodules were randomly distributed in all lung lobes.”
Two methods are used to examine the nasal structures. The first is making a midsagittal cut through the head and removing the nasal septum; the second is making several transverse sections of the nose at the level of the second premolar teeth. This latter method is preferred when examining pigs suspected of having atrophic rhinitis or animals suspected of having nasal neoplasms.
Histopathology and Biopsies
Microscopic examination of pulmonary tissue is routinely done in diagnostic laboratories. Samples of normal and abnormal lungs, along with other appropriate tissue, should always be submitted in 10% buffered-neutral formalin for histopathologic evaluation. A minimum of four lung samples (left cranial, left caudal, right cranial, and right caudal) should be taken for histopathologic examination in animals with a history of respiratory signs. To improve fixation, a paper towel can be placed over the samples of lung floating in fixative. When detailed evaluation of the alveolar walls is required, lungs can be fixed by a gentle intratracheal injection of fixative; however, this technique displaces transudates and exudates and can artificially cause distention of the perivascular and peribronchial spaces. Lung biopsy specimens are taken only sporadically because complications often outweigh the diagnostic value. However, the use of new techniques, such as endoscopic-directed biopsies, has notably reduced some of these complications. Biopsies of the lungs are recommended in cases of chronic persistent pulmonary disease unresponsive to treatment or intrathoracic masses of undetermined origin. Endoscopic-directed biopsies of the nasal and bronchial mucosa are routinely used in clinical practice and generally have a much better diagnostic value.
Bronchoalveolar Lavage and Tracheal Aspirates
Two valuable diagnostic tools in human medicine, bronchoalveolar lavage (BAL) and tracheal aspirates, have in recent years become more widely used in veterinary clinical diagnosis of respiratory ailments, particularly in horses, dogs, and cats. The basis of BAL is sampling to determine the cellular and biochemical composition of the lung in a respiratory patient live animal by infusing and retrieving sterile fluid via the trachea. BAL is done by inserting a tube directly through the larynx into a bronchus, or transtracheally by inserting a tube through a needle percutaneously into the cervical trachea. Microscopic examination of properly collected, stored, and processed samples may reveal many erythrocytes and siderophages in pulmonary hemorrhage or left-sided heart failure; inclusion bodies or syncytial cells in viral pneumonias; increased number of leukocytes in pulmonary inflammation; abundant mucus in asthma or equine heaves (chronic obstructive pulmonary disease [COPD]); presence of pulmonary pathogens, such as parasites, fungi, and bacteria; or tumor cells in cases of pulmonary neoplasia. In the healthy patient, 80% to 95% of the BAL cells are pulmonary alveolar macrophages (see Fig. 9-10).
Diseases of the Respiratory System
Pattern of Injury and Host Response
The conducting portion of the respiratory system is lined by pseudostratified columnar ciliated epithelium (most of the nasal cavity, paranasal sinuses, part of the larynx, and all of the trachea and bronchi), olfactory epithelium (part of the nasal cavity, particularly ethmoidal conchae), and squamous epithelium (nasal vestibulum and parts of the larynx). The pattern of injury, inflammation, and host response (wound healing) is characteristic for each of these three types of epithelium and independent of its anatomic location.
Pseudostratified ciliated epithelium, which lines most of the nasal cavity and nasopharynx, part of the larynx, and all of the trachea and bronchi, is exquisitely sensitive to injury. When these cells are irreversibly injured, whether caused by a viral infection, trauma, or inhalation of toxic gases, ciliated cells swell, typically lose their attachment to underlying basement membrane, and rapidly exfoliate (Fig. 9-11). A transient and mild exudate of fluid, plasma proteins, and neutrophils covers the ulcer. In the absence of complications or secondary bacterial infections, a specific type of progenitor cell known as nonciliated secretory cells, which is normally present in the mucosa, migrates to cover the denuded basement membrane and undergoes mitosis, eventually differentiating into new ciliated epithelial cells (see Fig. 9-11). Cellular migration, proliferation, and attachment are regulated by locally released growth factors and extracellular matrix (ECM) proteins such as collagen, integrins, and fibronectin. The capacity of ciliated epithelium to repair itself is remarkably effective. For example, epithelial healing in an uncomplicated ulcer of the tracheal mucosa can be completed in only 10 days. This sequence of cell degeneration, exfoliation, ulceration, mitosis, and repair is typically present in many viral infections in which viruses replicate in nasal, tracheal, and bronchial epithelium, causing extensive mucosal ulceration. Examples of transient infections of this type include human colds (rhinoviruses), infectious bovine rhinotracheitis (bovine herpesvirus 1), feline rhinotracheitis (feline herpesvirus 1), and canine infectious tracheobronchitis (CAV-2 and canine parainfluenza-2).
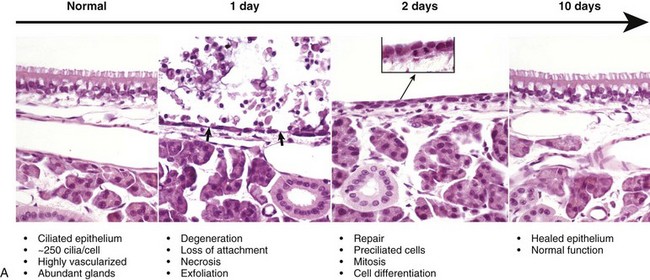

Fig. 9-11 Normal and injured nasal epithelium following exposure to air containing an irritant gas (hydrogen sulfide), nasal concha, rats.
A, Normal ciliated epithelium composed of tall columnar cells with numerous cilia. Day 1. Note detachment and exfoliation of ciliated cells, leaving a denuded basement membrane (arrows). This same type of lesion is seen in viral or mechanical injury to the mucosa of the conducting system. Two days after exposure, the basement membrane is lined by rapidly dividing preciliated cells, some of which exhibit mitotic activity (inset). Ten days after injury, the nasal epithelium is completely repaired. H&E stain. B, Schematic representation of the events of injury and repair in the respiratory mucosa of the conducting system. Blue cell = ciliated mucosal epithelial cell; pink cell = goblet cell; red cell = neutrophil. (A from López A, Prior M, Yong S et al: Am J Vet Res 49:1107-1111, 1988; B courtesy of Dr. A. López, Atlantic Veterinary College.)
If damage to the mucociliary blanket becomes chronic, goblet cell hyperplasia takes place, leading to excessive mucus production (hypersecretion) and reduced mucociliary clearance, and when there is loss of basement membrane, repair is by fibrosis and granulation tissue (scarring). In the most severe cases, prolonged injury causes squamous metaplasia, which together with scarring, causes airway obstruction and an impediment to mucociliary clearance. In laboratory rodents, hyperplastic and metaplastic changes, such as those seen in nasal polyps and squamous metaplasia, are considered a prelude to neoplasia.
The second type of epithelium lining the conducting system is the sensory olfactory epithelium, present in parts of the nasal mucosa, notably in the ethmoidal conchae. The patterns of degeneration, exfoliation, and inflammation in the olfactory epithelium are similar to those of the ciliated epithelium, except that olfactory epithelium has only limited capacity for regeneration. When olfactory epithelium has been irreversibly injured, olfactory cells swell, separate from adjacent sustentacular cells, and finally exfoliate into the nasal cavity. Once the underlying basement membrane of the olfactory epithelium is exposed, cytokines are released by leukocytes and endothelial cells, and inflammatory cells move into the affected area. When damage is extensive, ulcerated areas of olfactory mucosa are replaced by ciliated and goblet cells or squamous epithelium, or by fibrous tissue, all of which eventually cause reduction (hyposmia) or loss of olfactory function (anosmia). Repair of the olfactory epithelium is slower and less efficient than repair of the respiratory epithelium. Neurons in the olfactory mucosa have the unique ability to regenerate, a fact that is being explored as a potential source of new neurons in the treatment of spinal cord injury.
Squamous epithelium, located in the vestibular region of the nose (mucocutaneous junctions), is the third type of epithelium present in the nasal passages. Compared with ciliated and olfactory epithelia, nasal squamous epithelium is quite resistant to all forms of injury.
Anomalies of the Nasal Cavity
Localized congenital anomalies of the nasal cavity are rare in domestic animals and are often merely part of a more extensive craniofacial deformity (e.g., cyclops) or a component of generalized malformation (e.g., chondrodysplasia). Congenital anomalies involving the nasal cavity and sinuses, such as choanal atresia (lack of communication between the nasal cavity and pharynx), some types of chondrodysplasia, and osteopetrosis, are incompatible with life. Examples of nonfatal congenital anomalies include cystic nasal conchae, deviation of nasal septum, cleft upper lip (harelip, cheiloschisis), hypoplastic turbinates, and cleft palate (palatoschisis) (Fig. 7-1). Bronchoaspiration and aspiration pneumonia are common sequelae to cleft palate. Nasal and paranasal sinus cysts are slowly growing and expansive lesions that mimic neoplasia and cause severe cranial deformations in horses. These large cysts presumably originate congenitally from dentigerous tissue. As in other organs or systems, it is extremely difficult to determine the actual cause (genetic versus congenital) of anomalies based on pathologic evaluation.
Metabolic Disturbances of the Nasal Cavity
Metabolic disturbances affecting the nasal cavity and sinuses are also rare in domestic animals. Amyloidosis, the deposition of amyloid protein (fibrils with a β-pleated configuration) in various tissues, has been sporadically reported in the nasal cavity of horses and humans. Microscopic lesions are similar to those seen in other organs and consist of a deposition of hyaline amyloid material in nasal mucosa that is confirmed by a histochemical stain, such as Congo red. Unlike amyloidoses in other organs of domestic animals where amyloid is generally of the reactive type (amyloid AA), equine nasal amyloidosis appears to be of the immunocytic type (amyloid AL). Affected horses with large amyloid masses have difficulty breathing because of nasal obstruction, and may exhibit epistaxis and reduced athletic performance; on clinical examination, large, firm nodules resembling neoplasms (amyloidoma) can be observed in the alar folds, rostral nasal septum, and floor of nasal cavity.
Circulatory Disturbances of the Nasal Cavity
Congestion and Hyperemia: The nasal mucosa is well vascularized and is capable of rather dramatic variation in blood flow, whether passively as a result of interference with venous return (congestion) or actively because of vasodilation (hyperemia). Congestion of the mucosal vessels is a nonspecific lesion commonly found at necropsy and presumably associated with the circulatory failure preceding death (e.g., heart failure, bloat in ruminants in which the increased intraabdominal pressure causes increased intrathoracic pressure impeding the venous return from the head and neck). Hyperemia of the nasal mucosa is seen in early stages of inflammation, whether caused by irritations (e.g., ammonia, regurgitated feed), viral infections, secondary bacterial infections, allergy, or trauma.
Hemorrhage: Epistaxis is the clinical term used to denote blood flow from the nose (nosebleed) regardless of whether the blood originates from the nasal mucosa or from deep in the lungs such as in horses with “exercise-induced pulmonary hemorrhage.” Unlike blood in the digestive tract, where the approximate anatomic location of the bleeding can be estimated by the color the blood imparts to fecal material, blood in the respiratory tract is always red. This fact is due to the rapid transport of blood out of the respiratory tract by the mucociliary blanket and during breathing. Hemorrhages into the nasal cavity can be the result of local trauma, originate from erosions of submucosal vessels by inflammation (e.g., guttural pouch mycosis), or be caused by neoplasms. Hemoptysis refers to the presence of blood in sputum or saliva (coughing or spitting blood) and is most commonly the result of pneumonia, lung abscesses, ulcerative bronchitis, pulmonary thromboembolisms, and pulmonary neoplasia.
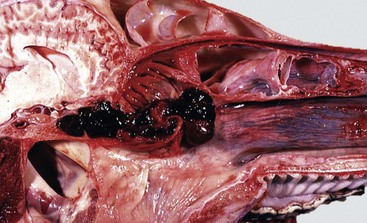
Ethmoidal (progressive) hematomas are important in older horses and are characterized clinically by chronic, progressive, often unilateral nasal bleeding. Grossly or endoscopically, an ethmoidal hematoma appears as a single, soft, tumorlike, pedunculated, expansive, dark red mass arising from the mucosa of the ethmoidal conchae (Fig. 9-12). Microscopic examination reveals a capsule lined by epithelium and hemorrhagic stromal tissue infiltrated with abundant macrophages, some of which are siderophages.
Inflammation of the Nasal Cavity
Inflammation of the nasal mucosa is called rhinitis, and inflammation of the sinuses is called sinusitis. These conditions usually occur together, although mild sinusitis can be undetected. Clinically, rhinosinusitis is characterized by nasal discharge.
The occurrence of infectious rhinitis presupposes an upset in the balance of the normal microbial flora of the nasal cavity. Innocuous bacteria present normally and protect the host through a process called competitive exclusion, whereby potential pathogens are kept at a harmless level. Disruption of this protective mechanism can be caused by respiratory viruses, pathogenic bacteria, fungi, irritant gases, environmental changes, immunosuppression, local trauma, stress, or prolonged antibacterial therapy.
Inflammatory processes in the nasal cavity are not life threatening and usually resolve completely. However, some adverse sequelae in cases of infectious rhinitis include bronchoaspiration of exudate leading to bronchopneumonia. Chronic rhinitis often leads to destruction of the nasal conchae (turbinates), deviation of the septum, and eventually, craniofacial deformation. Also, nasal inflammation may extend into the sinuses causing sinusitis; into facial bones causing osteomyelitis; through the cribriform plate causing meningitis; into the Eustachian tubes causing otitis media; and even into the inner ear causing otitis interna and vestibular syndrome (abnormal head tilt and abnormal gait), which in severe cases may lead to emaciation.
Based on the nature of exudate, rhinitis can be classified as serous, fibrinous, catarrhal, purulent, or granulomatous. These types of inflammatory reactions can progress from one to another in the course of the disease (i.e., serous to catarrhal to purulent), or in some instances exudates can be mixed, such as those seen in mucopurulent, fibrinohemorrhagic, or pyogranulomatous rhinitis. Microscopic examination of impression smears or nasal biopsy, and bacterial or fungal cultures are generally required in establishing the cause of inflammation. Common sequels to rhinitis are hemorrhage, ulcers, and in some cases polyps (hyperplasia) arising from inflamed nasal mucosa. Rhinitis also can be classified according to the age of the lesions as acute, subacute, or chronic; to the severity of the insult as mild, moderate, or severe; and to the etiologic agent as viral, allergic, bacterial, mycotic, parasitic, traumatic, or toxic.
Serous Rhinitis: Serous rhinitis is the mildest form of inflammation and is characterized by hyperemia and increased production of a clear fluid locally manufactured by serous glands present in the nasal submucosa. Serous rhinitis is of clinical interest only. It is caused by mild irritants or cold air, and it occurs during the early stages of viral infections, such as the common cold in humans, upper respiratory tract infections in animals, or in mild allergic reactions.
Catarrhal Rhinitis: Catarrhal rhinitis is a slightly more severe process and has, in addition to serous secretions, a substantial increase in mucus production by increased activity of goblet cells and mucous glands. A mucous exudate is a thick, translucent, or slightly turbid viscous fluid, sometimes containing a few exfoliated cells, leukocytes, and cellular debris. In chronic cases, catarrhal rhinitis is characterized microscopically by notable hyperplasia of goblet cells. As the inflammation becomes more severe, the mucus is infiltrated with neutrophils giving the exudate a cloudy mucopurulent appearance. This exudate is referred to as mucopurulent.
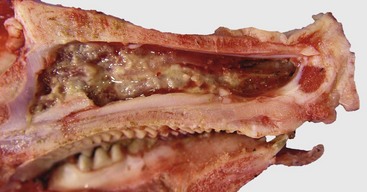
Purulent (Suppurative) Rhinitis: Purulent (suppurative) rhinitis is characterized by a neutrophilic exudate, which occurs when the nasal mucosa suffers a more severe injury that generally is accompanied by mucosal necrosis and secondary bacterial infection. Cytokines, leukotrienes, complement activation, and bacterial products cause exudation of leukocytes, especially neutrophils, which mix with nasal secretions, including mucus. Grossly, the exudate in suppurative rhinitis is thick and opaque, but it can vary from white to green to brown, depending on the types of bacteria and type of leukocytes (neutrophils or eosinophils) present in the exudate (Fig. 9-13 and Web Fig. 9-3). In severe cases, the nasal passages are completely blocked by the exudate. Microscopically, neutrophils can be seen in the submucosa and mucosa and form plaques of exudate on the mucosal surface. Neutrophils are commonly found marginated in vessels, in the lamina propria, and in between epithelial cells in their migration to the surface of the mucosa.
Fibrinous Rhinitis: Fibrinous rhinitis is a reaction that occurs when nasal injury causes a severe increase in vascular permeability, resulting in abundant exudation of plasma fibrinogen, which coagulates into fibrin. Grossly, fibrin appears like a yellow, tan, or gray rubbery mat on nasal mucosa. Fibrin accumulates on the surface and forms a distinct film of exudate sometimes referred to as pseudomembrane (Fig. 9-14). If this fibrinous exudate can be removed, leaving an intact underlying mucosa, it is termed a croupous or pseudodiphtheritic rhinitis. Conversely, if the pseudomembrane is difficult to remove and leaves an ulcerated mucosa, it is referred to as diphtheritic or fibrinonecrotic rhinitis. The term diphtheritic was derived from human diphtheria, which causes a severe and destructive inflammatory process of the nasal, tonsillar, pharyngeal, and laryngeal mucosa. Microscopically, the lesions include a perivascular edema with fibrin, a few neutrophils infiltrating the mucosa, and superficial plaques of exudate consisting of fibrin strands mixed with leukocytes and cellular debris covering a necrotic and ulcerated epithelium. Fungal infections, such as aspergillosis, can cause a severe fibrinonecrotizing rhinitis.
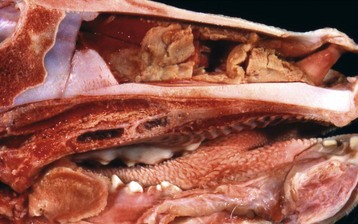
Fig. 9-14 Fibrinous rhinitis, midsagittal section of head, calf.
Infectious bovine rhinotracheitis (IBR; bovine herpesvirus 1). The nasal septum has been removed to expose nasal conchae. The nasal mucosa is covered by diphtheritic yellow membranes consisting of fibrinonecrotic exudate. Removal of these fibrinous membranes reveals focal ulcers in the underlying mucosa. (Courtesy Dr. Scott McBurney, Atlantic Veterinary College.)
Granulomatous Rhinitis: Granulomatous rhinitis is a reaction in the nasal mucosa and submucosa that is characterized by infiltration of numerous activated macrophages mixed with a few lymphocytes and plasma cells. In some cases, inflammation leads to the formation of polypoid nodules that in severe cases are large enough to cause obstruction of the nasal passages (Fig. 9-15). Granulomatous rhinitis is generally associated with chronic allergic inflammation or infection with specific organisms, such as those of the systemic mycoses (see the section on Lungs), tuberculosis, or rhinosporidiosis, and with foreign bodies. In some cases, the cause of granulomatous rhinitis cannot be determined.
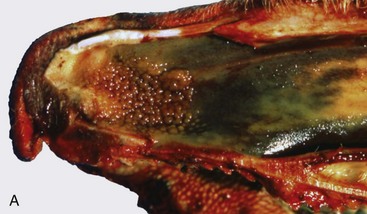
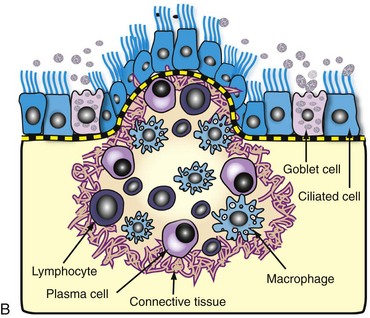
Fig. 9-15 Granulomatous rhinitis, midsagittal section of head, cow.
A, Note multiple and often confluent granulomas arising from the nasal mucosa. B, Schematic representation of a nasal granuloma showing the outer wall of the granuloma composed of connective tissue enclosing the center, which has been infiltrated with lymphocytes, plasma cells, and macrophages. (A courtesy Ontario Veterinary College. B courtesy of Dr. A. López, Atlantic Veterinary College.)
Sinusitis: Sinusitis occurs sporadically in domestic animals and is frequently combined with rhinitis (rhinosinusitis), or it occurs as a sequela to penetrating or septic wounds of the nasal, frontal, maxillary, or palatine bones; improper dehorning in young cattle, which exposes the frontal sinus; or maxillary tooth infection in horses and dogs (maxillary sinus). Based on the type of exudate, sinusitis is classified as serous, catarrhal, fibrinous (rare), purulent, or granulomatous. Paranasal sinuses have poor drainage; therefore exudate tends to accumulate, causing mucocele (accumulation of mucus) or empyema (accumulation of pus) (Fig. 9-16). Chronic sinusitis may extend into the adjacent bone (osteomyelitis) or through the ethmoidal conchae into the meninges and brain (meningitis and encephalitis).
Specific Diseases of the Nasal Cavity and Sinuses
Equine Viral Infections: Viruses, such as equine viral rhinopneumonitis virus, influenza virus, adenovirus, and rhinovirus, cause mild and generally transient respiratory infections in horses. The route of infection for these respiratory viruses is typically aerogenous. All these infections are indistinguishable clinically; signs consist mainly of malaise, fever, coughing, and nasal discharge varying from serous to purulent. Viral respiratory infections are common medical problems in adult horses.
Equine viral rhinopneumonitis: Equine viral rhinopneumonitis (EVR) is caused by two ubiquitous equine herpesviruses (EHV-1 and EHV-4) and may be manifested as a mild respiratory disease in weanling foals and young racehorses, as a neurologic disease (myeloencephalopathy), or as abortion in mares. The portal of entry for the respiratory form is typically aerogenous, and the disease is generally transient; thus the primary viral-induced lesions in the nasal mucosa and lungs are rarely seen at necropsy unless complicated by secondary bacterial rhinitis, pharyngitis, or bronchopneumonia. Studies with polymerase chain reaction (PCR) techniques have demonstrated that, like other herpesvirus, EHV-1 and EHV-4 persist latently in the trigeminal ganglia for long periods of time. Reactivation because of stress or immunosuppression and subsequent shedding of the virus are the typical source of infection for susceptible animals on the farm.
Equine influenza: Equine influenza is a common, highly contagious, and self-limiting upper respiratory infection of horses caused by aerogenous exposure to type A strains of influenza virus (H7N7 [A/equi-1] and H3N8 [A/equi-2]). Equine influenza has high morbidity (outbreaks) but low mortality, and it is clinically characterized by fever, conjunctivitis, and serous nasal discharge. It occurs mainly in 2- to 3-year-old horses at the racetrack. As with human influenza, equine influenza is usually a mild disease, but occasionally it can cause severe bronchointerstitial pneumonia with pulmonary edema. In some horses, impaired defense mechanisms caused by the viral infection are complicated by a secondary bacterial bronchopneumonia caused by opportunistic organisms (Streptococcus zooepidemicus, Staphylococcus aureus, or Bacteroides sp.) found in the normal flora of the upper respiratory tract. Uncomplicated cases of equine influenza are rarely seen in the postmortem room.
Other equine respiratory viruses: Equine rhinovirus, adenovirus, and parainfluenza virus produce mild and transient upper respiratory infections (nasopharynx and trachea) in horses, unless complicated by secondary pathogens. In addition to reduced athletic performance, infected horses may have a temporary suppression of cell-mediated immunity leading to opportunistic infections such as Pneumocystis carinii pneumonia. Fatal adenoviral infections with severe pneumonia or enteritis occur commonly in immunocompromised horses, particularly in Arabian foals with inherited combined immunodeficiency disease.
Equine Bacterial Infections: Strangles, Glanders, and Melioidosis: Strangles, glanders, and melioidosis of horses are all systemic bacterial diseases that cause purulent rhinitis and suppuration in various organs. These diseases are grouped as upper respiratory diseases because nasal discharge is often the most notable clinical sign.
Strangles: Strangles is an infectious and highly contagious disease of Equidae that is caused by Streptococcus equi ssp. equi (Streptococcus equi). It is characterized by suppurative rhinitis and lymphadenitis (mandibular and retropharyngeal lymph nodes) with occasional hematogenous dissemination to internal organs. Unlike Streptococcus equi ssp. zooepidemicus (Streptococcus zooepidemicus) and Streptococcus dysgalactiae ssp. equisimilis (Streptococcus equisimilis), Streptococcus equi is not part of the normal nasal flora. Infection occurs when susceptible horses come into contact with feed, exudate, or air droplets containing the bacterium. After penetrating through the nasopharyngeal mucosa, Streptococcus equi drains to the regional lymph nodes—mandibular and retropharyngeal lymph nodes—via lymphatic vessels. The gross lesions in horses with strangles (mucopurulent rhinitis) correlate with clinical findings and consist of copious amounts of mucopurulent exudate in the nasal passages with notable hyperemia of nasal mucosa. Affected lymph nodes are enlarged and may contain abscesses filled with thick purulent exudate (purulent lymphadenitis). The term bastard strangles is used in cases in which hematogenous dissemination of Streptococcus equi results in metastatic abscesses in such organs as the lungs, liver, spleen, kidneys, or brain or in the joints. This form of strangles is often fatal.
Common sequelae to strangles include bronchopneumonia caused by aspiration of nasopharyngeal exudate; laryngeal hemiplegia (“roaring”), resulting from compression of the recurrent laryngeal nerves by enlarged retropharyngeal lymph nodes; facial paralysis and Horner syndrome caused by compression of sympathetic nerves that run dorsal to the medial retropharyngeal lymph node; and purpura hemorrhagica as a result of vasculitis caused by deposition of Streptococcus equi antigen-antibody complexes in arterioles, venules, and capillaries of the skin and mucosal membranes. In severe cases, nasal infection extends directly into the paranasal sinuses or to the guttural pouches via the Eustachian tubes, causing inflammation and accumulation of pus (guttural pouch empyema). Rupture of abscesses in the mandibular and retropharyngeal lymph nodes leads to suppurative inflammation of adjacent subcutaneous tissue (cellulitis), and in severe cases the exudate escapes through cutaneous fistulas.
Strangles can affect horses of all ages, but it is most commonly seen in foals and young horses. It is clinically characterized by cough, nasal discharge, conjunctivitis, and painful swelling of regional lymph nodes. Some horses become carriers and a source of infection to other horses.
Glanders: Glanders is an infectious Office International des Epizooties (OIE)-notifiable disease of Equidae caused by Burkholderia mallei (Pseudomonas mallei) that can be transmitted to carnivores by consumption of infected horse meat. Humans are also susceptible, and the untreated infection is often fatal. This bacterium has been listed as a potential agent for biologic warfare and bioterrorism. In the past, Burkholderia mallei was found throughout the world, but today, glanders has been eradicated from most countries, except for some areas in North Africa, Asia, and Eastern Europe. There also have been some sporadic outbreaks reported in Brazil. The pathogenesis of glanders is not fully understood. Results from experimental infections suggest that infection occurs via the ingestion of contaminated feed and water and, very rarely, via inhalation of infectious droplets. The portals of entry are presumed to be the oropharynx or intestine, in which bacteria penetrate the mucosa and spread via lymph vessels to regional lymph nodes, then to the bloodstream, and thus hematogenously to the internal organs, particularly the lungs.
Lesions in the nasal cavity start as pyogranulomatous nodules in the submucosa; these lesions subsequently ulcerate, releasing copious amounts of Burkholderia mallei–containing exudate into the nasal cavity (see Fig. 4-24). Finally, ulcerative lesions in conchal mucosa heal and are replaced by typical stellate (star-shaped), fibrous scars. In some cases, the lungs also contain numerous gray, hard, small (2 to 10 mm), miliary nodules (resembling millet seeds), randomly distributed in one or more pulmonary lobes because of the hematogenous route. Microscopically, these nodules are typical chronic granulomas composed of a necrotic center, with or without calcification, surrounded by a layer of macrophages enclosed by a thick band of connective tissue infiltrated with macrophages, some giant cells, lymphocytes, and plasma cells. Cutaneous lesions, often referred to as equine farcy, are the result of severe suppurative lymphangitis characterized by nodular thickening of extended segments of lymph vessels in the subcutaneous tissue of the legs and ventral abdomen (see Fig. 4-24). Eventually, affected lymph vessels rupture and release large amounts of purulent exudate through sinuses to the surface of the skin.
Melioidosis (pseudoglanders): Melioidosis (pseudoglanders) is an important, life-threatening disease of humans, horses, cattle, sheep, goats, pigs, dogs, cats, and rodents caused by Burkholderia pseudomallei (Pseudomonas pseudomallei). This disease in horses is clinically and pathologically similar to glanders, hence the name pseudoglanders. In humans, this infection can cause severe sepsis and septic shock and has also been considered to have potential for biologic welfare. Melioidosis is currently present in Southeast Asia and to a much lesser extent in Northern Australia and some European countries where the causative organism is frequently found in rodents, feces, soil, and water. Ingestion of contaminated feed and water appears to be the main route of infection; direct transmission between infected animals and insect bites has also been postulated as a possible mechanism of infection. After gaining entrance to the animal, Burkholderia pseudomallei is disseminated by the bloodstream and causes suppuration and abscesses in most internal organs, such as nasal mucosa, joints, brain and spinal cord, lungs, liver, kidneys, spleen, and lymph nodes. The exudate is creamy or caseous and yellow to green. The pulmonary lesions in melioidosis are those of an embolic bacterial infection with the formation of pulmonary abscesses, which can become confluent. Focal adhesive pleuritis develops where abscesses rupture through the pleura and heal.
Other Causes of Equine Rhinitis: The protistan parasite, Rhinosporidium seeberi, causes nasal infection in humans, horses, mules, cattle, dogs, and cats. Gross lesions vary from barely visible granulomas to large expansive polypoid nodules that may be mistaken as tumors. These granulomatous nodules are detected by direct observation when present in the nasal mucosa close to the nares or by rhinoscopy when located in the deep nasal cavity. The offending organism, Rhinosporidium seeberi, is readily visible in histologic preparations and in impression smears, appearing as a large (400 µm), oval sporangium containing thousands of endospores (Web Fig. 9-4). Rhinosporidium seeberi was once considered a mycotic agent, but recent phylogenetic investigations suggest that it is an aquatic protistan parasite of the class Mesomycetozoea.
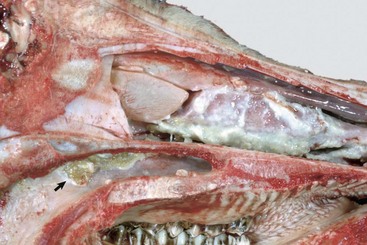
Web Fig. 9-3 Suppurative rhinitis, midsagittal section of head, calf.
The nasal septum has been removed to expose nasal conchae. The nasal mucosa is covered by yellow-white purulent exudate. There is also a large, round ulcer in mucosa of the nasopharynx (arrow). (Courtesy Western College of Veterinary Medicine.)
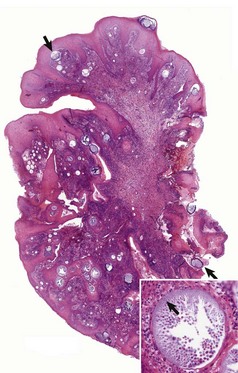
Web Fig. 9-4 Exophytic granulomatous mass surgically removed from the nasal mucosa (Rhinosporidium seeberi), mule.
Large pedunculated mass of granulomatous tissue containing numerous sporangia (arrows). Inset, Sporangium. Note a large encapsulated cyst filled with a myriad of Rhinosporidium seeberi endospores. H&E stain. (From Berrocal A, López A: Can Vet J 48:305-306, 2007.)
Infectious Bovine Rhinotracheitis: Infectious bovine rhinotracheitis (IBR), or “rednose,” occurs worldwide and is a disease of great importance to the cattle industry because of the synergism of the IBR virus with Mannheimia haemolytica in producing pneumonia. The causative agent, bovine herpesvirus 1 (BoHV-1), has probably existed as a mild venereal disease in cattle in Europe since at least the mid-1800s, but the respiratory form was not reported until intensive management feedlot systems were first introduced in North America around the 1950s. Typically, the disease is manifested as a transient, acute, febrile illness, which only in very severe cases results in inspiratory dyspnea caused by obstruction of the airways by exudate. Other forms of BoHV-1 infection include ulcerative rumenitis; enteritis; multifocal hepatitis in neonatal calves; nonsuppurative meningoencephalitis; infertility; and in experimental infections, mastitis, mammillitis, and ovarian necrosis. Except for the encephalitic form, the type of disease caused by BoHV-1 depends more on the site of entry than the viral strain. Like other herpesviruses, BoHV-1 also can remain latent in nerve ganglia, with recrudescence after stress or immunosuppression. This virus also causes bovine abortion, systemic infections of calves, and genital infections such as infectious pustular vulvovaginitis (IPV) and infectious balanoposthitis (IBP).
The respiratory form of IBR is characterized by severe hyperemia and focal necrosis of nasal, pharyngeal, laryngeal, tracheal, and sometimes bronchial mucosa (Figs. 9-17 and 9-18 and Web Fig. 9-5). As in other respiratory viral infections, IBR lesions are microscopically characterized by necrosis and exfoliation of ciliated cells followed by repair. Secondary bacterial infections of these areas of necrosis result in the formation of a thick layer of fibrinonecrotic material (diphtheritic) in the nasal, tracheal, and bronchial mucosa (see Figs. 9-17 and 9-18). Intranuclear inclusion bodies, commonly seen in herpesvirus infections, are rarely seen in field cases because inclusion bodies occur only during the early stages of the disease.
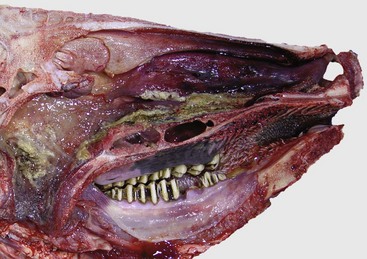
Fig. 9-17 Fibrinous rhinitis and pharyngitis, midsagittal section of head, steer.
The nasal and pharyngeal mucosae are covered by diphtheritic yellow membranes consisting of fibrinonecrotic exudate. The dorsal concha is markedly hyperemic. (Courtesy Dr. A. López Atlantic Veterinary College.)
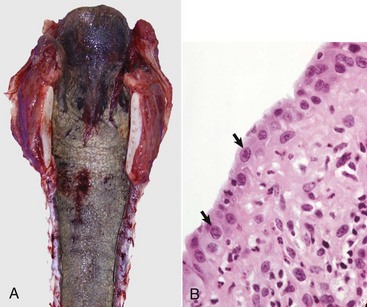
Fig. 9-18 Subacute fibrinonecrotic laryngitis and tracheitis, infectious bovine rhinotracheitis (IBR; bovine herpesvirus 1), longitudinal (dorsal) section of larynx and trachea, calf.
A, Thick plaques of fibrinonecrotic exudate cover the laryngeal and tracheal mucosae. B, Note the intranuclear inclusions (arrows), characteristic of herpesvirus infection, in a tracheal mucosal epithelial cell. Chronic inflammation is also present in the subjacent connective tissue. (A courtesy Dr. A. López, Atlantic Veterinary College. B courtesy College of Veterinary Medicine, University of Illinois.)
The most important sequela to IBR is pneumonia, which is caused either by direct aspiration of exudate from airways or as a result of an impairment in pulmonary defense mechanisms, thus predisposing the animal to secondary bacterial infection, most frequently Mannheimia haemolytica (see pneumonic Mannheimiosis discussion). Postmortem diagnosis of IBR is confirmed by isolation of the virus or its identification by immunohistochemistry or PCR in affected tissues.
Other Bovine Rhinitis: Nasal granulomas occur in cattle presumably as a result of repeated exposure to an as yet unidentified inhaled antigens. Nasal granulomas (atopic rhinitis) are reported mainly in cattle in Australia, South Africa, and the United Kingdom, where affected cattle develop multiple, small, pink or red, polypoid nodules, starting in the nasal vestibule that in time extend into the caudal aspect of the nasal septum. These nodules are composed of fibrovascular tissue mixed with lymphocytes (granulation tissue) superficially lined by hyperplastic epithelium with abundant mast cells and eosinophils in the lamina propria (nasal eosinophilia). The microscopic features suggest that hypersensitivity type I (immediate), type III (immune complex), and type IV (delayed) may be involved in nasal granulomas of cattle. Bovine (idiopathic) nasal granuloma must be differentiated from nasal mycetomas, nasal rhinosporidiosis, and nasal schistosomiasis, which also cause the formation of nodules in the nasal mucosa of cattle. An eosinophilic material consistent with the Splendore-Hoeppli phenomenon is occasionally observed in bovine mycotic granulomas. This phenomenon seen in some mycotic or bacterial infections is microscopically characterized by a deeply eosinophilic homogeneous material surrounded by bacteria or mycelia. It is thought to result from a localized antigen-antibody response in tissue.
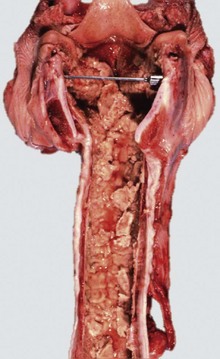
Web Fig. 9-5 Subacute fibrinous laryngitis and tracheitis, longitudinal (dorsal) section of larynx and trachea, calf.
Infectious bovine rhinotracheitis (IBR; bovine herpesvirus 1). Thick plaques of fibrinonecrotic exudate cover the laryngeal and tracheal mucosae. (Courtesy Dr. J.M. King, College of Veterinary Medicine, Cornell University.)
Ovine and Caprine Nasal Disease: Oestrus ovis (Diptera: Oestridae; nasal bot) is a brownish fly about the size of a honeybee that deposits its first-stage larvae in the nostrils of sheep in most parts of the world. Microscopic larvae mature into large bots (maggots), which spend most of their larval stages in nasal passages and sinuses, causing irritation, inflammation, and obstruction of airways. Matured larvae drop to the ground and pupate into flies. This type of parasitism in which living tissues are invaded by larvae of flies is known as myiasis (Fig. 9-19). Although Oestrus ovis is a nasal myiasis primarily of sheep, it sporadically affects goats, dogs, and sometimes humans (shepherds). The presence of the larvae in nasal passages and sinuses causes chronic irritation and erosive mucopurulent rhinitis and sinusitis; bots of Oestrus ovis can be found easily if the head is cut to expose the nasal passages and paranasal sinuses. Rarely, larvae of Oestrus ovis penetrate the cranial vault through the ethmoidal plate, causing direct or secondary bacterial meningitis.
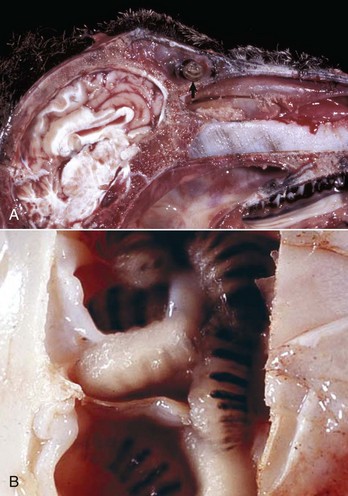
Fig. 9-19 Oestrus ovis, sheep.
A, Frontal sinus. Note the parasitic (fly) larvae in the frontal sinus (arrow). B, Nasal cavity. Higher magnification view of larvae of Oestrus ovis in a nasal cavity. (A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B courtesy Dr. M. Sierra and Dr. J. King, College of Veterinary Medicine, Cornell University.)
Infectious rhinitis is only sporadically reported in goats, and most of these cases are caused by Pasteurella multocida or Mannheimia haemolytica. The lesions range from a mild serous to catarrhal or mucopurulent inflammation.
Inclusion Body Rhinitis: Inclusion body rhinitis is a disease of young pigs with high morbidity and low mortality caused by a porcine cytomegalovirus (herpesvirus) and characterized by a mild rhinitis. This virus commonly infects the nasal epithelium of piglets younger than 5 weeks and causes a transient viremia. Because this disease is seldom fatal, lesions are seen only incidentally or in euthanized animals. In uncomplicated cases, the gross lesion is hyperemia of the nasal mucosa, but with secondary bacterial infections, mucopurulent exudate can be abundant. Microscopic lesions are typical and consist of a necrotizing, nonsuppurative rhinitis with giant, basophilic, intranuclear inclusion bodies in the nasal epithelium and glands (Fig. 9-20). Immunosuppressed piglets can develop a systemic cytomegalovirus infection characterized by necrosis of the liver, lungs, adrenal glands, and brain with intralesional inclusion bodies. Inclusion body rhinitis is clinically characterized by a mild and transient rhinitis, causing sneezing, nasal discharge, and excessive lacrimation.
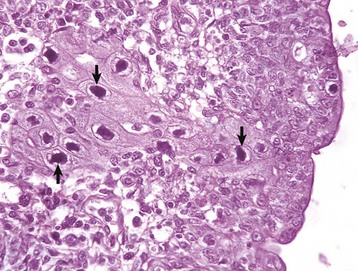
Atrophic Rhinitis: A common worldwide disease of pigs, atrophic rhinitis (progressive atrophic rhinitis) is characterized by inflammation and atrophy of nasal conchae (turbinates). In severe cases, atrophy of the conchae may cause a striking facial deformity in growing pigs because of deviation of the nasal septum and nasal bones. The etiopathogenesis of atrophic rhinitis is complex and has been a matter of controversy for many years. Pathogens historically associated with atrophic rhinitis include Bordetella bronchiseptica, Pasteurella multocida, Haemophilus parasuis, and viral infections such as porcine cytomegalovirus (inclusion body rhinitis). In addition, predisposing factors have included genetic makeup, environment, and nutritional deficiencies. The cause of atrophic rhinitis is currently believed to be a combined infection by specific strains of Bordetella bronchiseptica and toxigenic strains of Pasteurella multocida (types D and A). The only lesion associated with infection with Bordetella bronchiseptica alone is a mild-to-moderate turbinate atrophy (nonprogressive atrophic rhinitis), but this bacterium actively promotes the colonization of the nasal cavity by Pasteurella multocida. The toxigenic strains of Pasteurella multocida produce potent cytotoxins that inhibit osteoblastic activity and promote osteoclastic reabsorption in nasal bones, particularly in the ventral nasal conchae, where abnormal bone remodeling results in progressive atrophy of conchae.
The degree of conchal atrophy in pigs with atrophic rhinitis varies considerably, and in most pigs, the severity of the lesions does not correspond to the severity of the clinical signs. The best diagnostic method of evaluating this disease at necropsy is to make a transverse section of the snout between the first and second premolar teeth. In normal pigs, conchae are symmetric and fill most of the cavity, leaving only narrow air spaces (meatuses) between coiled conchae. The normal nasal septum is straight and divides the cavity into two mirror-imaged cavities. In contrast, the septum in pigs with atrophic rhinitis is generally deviated and the conchae appear smaller and asymmetric (Fig. 9-21). Conchal atrophy causes dorsal and ventral meatuses to appear rather enlarged, and in the most advanced cases, the entire nasal conchae may be missing, leaving a large, empty space.
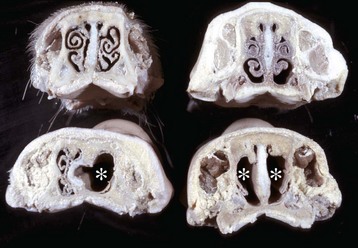
Fig. 9-21 Atrophic rhinitis, transverse sections of nasal passages and sinuses, caudal surfaces, level of the first or second premolar teeth, pigs.
Top left, Normal nasal cavity showing complete conchae (turbinates) that fill most of the nasal cavity and form narrow air passages (meatuses). Top right, Mild, symmetric atrophy of nasal conchae. Bottom left, Severe, unilateral atrophy of the right ventral nasal concha (asterisk) with deviation of the nasal septum to the left and widening of the ventral meatus. Bottom right, Severe, bilateral atrophy with complete loss of nasal conchae and extensive widening of the meatuses (asterisks). (Courtesy Dr. A. López, Atlantic Veterinary College.)
It may seem logical to assume that after loss of conchae in an obligate nasal breather, such as the pig, the filtration defense mechanism of the nasal cavity would be impaired, thus enhancing the chances of aerogenous infections in the lung. However, the relationship between atrophic rhinitis, pneumonia, and growth rates in pigs is still controversial.
Osteoclastic hyperplasia and osteopenia of the conchae are the key microscopic lesions in atrophic rhinitis. Depending on the stage of the disease, mucopurulent exudate may be found on the surface of the conchae. Hyperplastic or metaplastic changes can occur in the nasal epithelium and glands, and infiltrates of lymphoplasmacytic cells can be present in the lamina propria. In summary, atrophic rhinitis is an important disease in pigs worldwide; morphologic diagnosis is simple, but additional understanding of the pathogenesis will be necessary before effective preventive measures can be established.
Atrophic rhinitis is clinically characterized by sneezing, coughing, and nasal discharge as a result of inflammation and atrophy of nasal conchae (turbinates). Obstruction of the nasolacrimal duct is common and results in accumulation of dust and dried lacrimal secretions on the skin inferior to the medial canthus of the eye.
Canine Nasal Diseases: Dogs have no specific viral infections affecting exclusively the nasal cavity or sinuses. Acute rhinitis occurs as part of general respiratory disease caused by several distinct viruses, such as canine distemper virus, CAV-1 and -2, canine parainfluenza virus, reovirus, and canine herpesvirus. The viral lesions in the respiratory tract are generally transient, but the effect of the virus on other tissues and cells can be fatal, as in distemper encephalitis in dogs. As in other species, secondary bacterial rhinitis, sinusitis, and pneumonia are possible sequelae of respiratory viral infections; Bordetella bronchiseptica, Escherichia coli, and Pasteurella multocida are the most common isolates in dogs with bacterial rhinitis. A nonspecific (idiopathic) chronic lymphoplasmatic rhinitis is occasionally seen in dogs. Immotile cilia syndrome (ciliary dyskinesia), a congenital disease, reduces mucociliary clearance and is an important factor in recurrent canine rhinosinusitis, bronchitis, bronchiectasis, and pneumonia.
Other Causes of Canine Rhinitis: Aspergillus spp. and Penicillium spp. cause mycotic rhinitis and sinusitis in dogs (canine nasal aspergillosis) (Web Fig. 9-6). Nasal biopsies reveal extensive necrosis of the nasal epithelium and thick plaques of fibrinopurulent exudate mixed with many fungal hyphae. Cryptococcus neoformans and Rhinosporidium seeberi infections of the nasal cavity occur sporadically in dogs (Web Fig. 9-7). Lesions are characterized by mucosal granulomas containing periodic acid–Schiff (PAS)-positive organisms, and the infection is clinically characterized by mucopurulent nasal discharge.
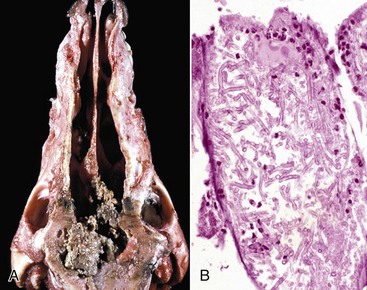
Web Fig. 9-6 Fibrinonecrotic sinusitis, aspergillosis, dorsal section of nasal cavities, dog.
A, The nasal conchae have been destroyed by chronic granulomatous inflammation. Mycotic exudate remaining in the caudal aspect of the nasal cavity is yellow-green and granular. B, Hyphae of Aspergillus spp. were isolated from the granulomatous inflammatory exudate. Note the neutrophils at the periphery of the fungal matt. H&E stain. (A courtesy College of Veterinary Medicine, University of Illinois. B courtesy Dr. M.A. Wallig, College of Veterinary Medicine, University of Illinois.)
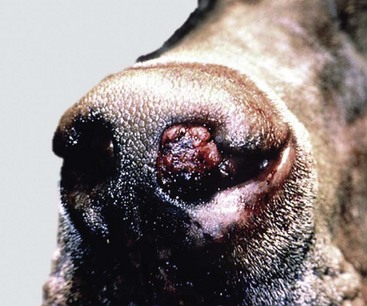
Web Fig. 9-7 Granulomatous rhinitis (Rhinosporidium seeberi), nasal cavity and nostril, dog.
A polypoid granulomatous mass fills the rostral part of the left nasal cavity. (Courtesy Dr. C. Bridges, College of Veterinary Medicine, Texas A&M University; and Dr. J.M. King, College of Veterinary Medicine, Cornell University.)
Linguatula serrata: Linguatula serrata is a rare but highly specialized pentastomid parasite that shares some morphologic features with arthropods and annelids and causes infection when dogs consume uncooked ruminant meat containing infective larvae. It occurs primarily in carnivores, although sheep and goats may become aberrant hosts. Humans can also acquire the infection by ingesting raw ovine or caprine meat. The adult parasite is found throughout the nasal passages and sometimes can reach the sinuses and middle ear by moving through the exudate in the eustachian tubes. In common with other nasal parasites, Linguatula serrata acts as an irritant, causing sneezing, catarrhal inflammation, and epistaxis. The eggs of this parasite leave the host in the exudate, which is coughed up or swallowed and eliminated in the feces.
The nasal cavity and paranasal sinuses of dogs can occasionally be infested with other parasites, including mites (Pneumonyssus caninum).
Allergic rhinitis: Allergic rhinitis (hay fever; nasolacrimal urticaria), which is so common in humans sensitized and reexposed to inhaled pollens or allergens, has been reported only sporadically in dogs and cats. Hay fever in humans and animals is a type I hypersensitivity reaction in which an IgE-mediated degranulation of mast cells results in an acute rhinitis and conjunctivitis. Microscopically the nasal mucosa is edematous and infiltrated with numerous eosinophils, neutrophils, and some macrophages. Clinically, allergic rhinitis is characterized by profuse serous nasal discharge and lacrimation.
Feline Viral Rhinotracheitis: Feline viral rhinotracheitis (FVR) is a common, worldwide respiratory disease of cats caused by a feline herpesvirus (FHV-1). The disease causes an impairment of pulmonary defense mechanisms, predisposing cats to secondary bacterial pneumonia or to a co-infection with feline calicivirus. The virus also can remain latent in ganglia. The vast majority of cats that recover from FVR become carriers and shed FHV-1, either spontaneously or after stress. Susceptible animals, particularly kittens with low maternal immunity, become infected after exposure to a diseased or a carrier cat. Replication of FHV-1 in the nasal, conjunctival, pharyngeal, and to a lesser extent, tracheal epithelium causes degeneration and exfoliation of cells.
Lesions caused by FHV-1 are fully reversible, but secondary infections with bacteria, such as Pasteurella multocida, Bordetella bronchiseptica, Streptococcus spp., and Mycoplasma felis, can cause a chronic, severe suppurative rhinitis, and conjunctivitis. Intranuclear inclusion bodies are rarely seen in cats with FVR because inclusions are only present during the early stages of infection and have already disappeared by the time the cat is presented for diagnosis.
Respiratory sequelae to FVR can include chronic bacterial rhinitis and sinusitis with persistent purulent discharges; lysis of nasal bones, which can lead to conchal atrophy; permanent damage to the olfactory epithelium; and secondary bacterial pneumonia. In addition to rhinitis and interstitial pneumonia, FVR also causes ulcerative keratitis, hepatic necrosis, emaciation, abortion, and stillbirths. Clinical signs of FVR infection are characterized by lethargy, oculonasal discharges, severe rhinitis, and conjunctivitis.
Feline Calicivirus: Feline calicivirus rhinitis (FCR) is caused by different strains of feline calicivirus (FCV). It is an important infection of the respiratory tract of cats, and depending on the virulence of the strain, lesions vary from a mild oculonasal discharge to severe rhinitis, mucopurulent conjunctivitis, and ulcerative gingivitis and stomatitis. The lesions, in addition to rhinitis and conjunctivitis, include acute, diffuse interstitial pneumonia with necrotizing bronchiolitis (see the section on Pneumonias of Cats) and prominent ulcers of the tongue and hard palate. Primary viral lesions are generally transient, but secondary bacterial infections (Bordetella bronchiseptica, Pasteurella multocida, or Escherichia coli) are a common complication. Some kittens develop lameness after infection or vaccination with calicivirus because of an acute and self-limiting arthritis (“limping kitten syndrome”). Carrier state and virus shedding from oronasal secretions and feces are natural sequelae after recovery from the acute phase of the disease. Clinical and pathologic features of FCV disease are strikingly similar, but not identical to those of FVR; these two viral infections account for 80% of all cases of feline respiratory diseases. A febrile systemic hemorrhagic syndrome with high mortality (up to 50%) has been recently reported in cats infected with virulent strains of FCV.
Feline Chlamydiosis: Feline chlamydiosis is a persistent respiratory infection of cats caused by Chlamydophila felis. Infection results in a conjunctivitis (similar to the conjunctivitis seen in human trachoma caused by Chlamydia trachomatis) and serous or mucopurulent rhinitis. In the past, Chlamydophila felis was incriminated as the agent responsible for “feline pneumonitis,” but its role in causing bronchointerstitial pneumonia in cats has been seriously challenged in recent years (see the section on Pneumonias of Cats).
Other Causes of Feline Rhinitis and Sinusitis:
Mycoplasma felis, feline leukemia virus, and feline immunodeficiency virus: Mycoplasma felis can also cause mucopurulent conjunctivitis and a mild upper respiratory infection, with clinical signs and lesions overlapping those seen with chlamydiosis, FVR, and FCR infections. Respiratory infections and bronchopneumonia in cats may also be associated with the immunosuppressive effects of feline retroviruses such as feline leukemia virus (FeLV) and feline immunodeficiency virus (FIV).
Mycotic rhinitis: The most common mycotic infection in the feline nasal cavity is caused by Cryptococcus neoformans, but not all animals exposed to this fungus necessarily develop cryptococcosis unless they are immunosuppressed. The lesions vary from discrete nasal granulomas to large confluent masses of mucopurulent exudate filling the entire nasal cavity and paranasal sinuses. Microscopic examination of the exudate reveals the typical thick-walled PAS-positive organisms (see the section on Pneumonias of Dogs). Nasal aspergillosis and allergic rhinosinusitis are sporadically reported in cats (see the section on Other Causes of Canine Rhinitis).
Neoplasia of the Nasal Cavity and Paranasal Sinuses
Neoplasms of the nasal cavity and paranasal sinuses may arise from any of the tissues forming these structures, including bone (osteoma or osteosarcoma), cartilage (chondroma or chondrosarcoma), connective tissue (fibroma or fibrosarcoma, myxoma or myxosarcoma), and blood vessels (hemangioma or hemangiosarcoma), and from all the different types of cells of glands and lining epithelium (adenoma, carcinoma, or adenocarcinoma). Nasal tumors originating from stromal tissues, such as bone, cartilage, and connective tissue, are morphologically indistinguishable from those seen in other sites. In general, nasal neoplasms are rare in domestic animals, except for endemic ethmoidal neoplasms (retroviral) in sheep and cattle, which can occur in several animals in a flock or herd (see the next section).
In companion animals, nasal neoplasms are most common in dogs, particularly in breeds such as the collie, Airedale terrier, basset hound, and German shepherd. The cat and the horse are less frequently affected. The main sites in order of frequency are the nasal passages and sinuses for dogs, the tip of the nose and nasal passages for cats, and the maxillary sinus and nasal passages for horses.
The majority of neoplasms in the nasal cavity are malignant. Benign nasal neoplasms (papilloma and adenoma) are rare and generally are either solitary or multiple, well-delineated nodules. In contrast, nasal carcinomas and nasal sarcomas are generally larger but vary in size and are often pale and multilobulated masses composed of fleshy to friable tissue (Fig. 9-22). Malignant neoplasms are locally invasive and tend to infiltrate sinuses, meninges, frontal brain, olfactory nerves, and vessels resulting in epistaxis. Carcinomas vary from anaplastic (poorly differentiated) to well differentiated, in which cell and tissue morphology retains some glandular (adenocarcinoma) or squamous cell patterns. Because nasal tumors in dogs and cats are usually large and invasive at the time of diagnosis, prognosis is usually poor and survival times are short. Sarcomas originating in the nasal cavity and paranasal sinuses are less common than carcinomas. Mesenchymal tumors can arise from bone (osteoma or osteosarcoma), cartilage (chondroma or chondrosarcoma), blood vessels (hemangioma or hemangiosarcoma), and connective tissue (fibroma or fibrosarcoma). Overall, benign epithelial and mesenchymal tumors are less common than their malignant counterparts. Secondary tumors in the nasal cavity are rare, with lymphoma being the most common metastatic tumor in the nasal cavity of domestic animals.
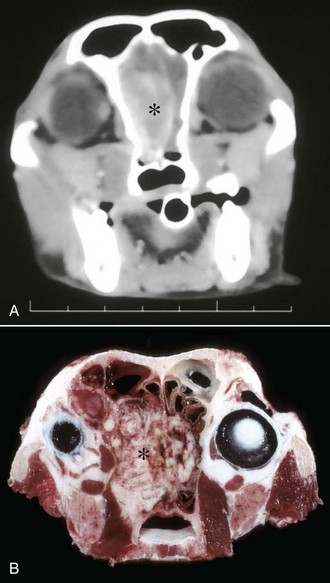
Fig. 9-22 Nasal carcinoma, transverse section of nasal passages and sinuses, 10-year-old dog.
A, Computerized tomography shows a large neoplastic mass (asterisk) infiltrating the nasal cavity and displacing the nasal septum laterally. Scale bar units in centimeters. B, Transverse section of head showing tumor diffusely infiltrating the nasal conchae and obliterating the meatuses (asterisk). (Courtesy Atlantic Veterinary College.)
Nasal neoplasms become secondarily infected by bacteria, and clinical signs often overlap with those of infectious rhinitis and include catarrhal or mucopurulent nasal discharge, periodic hemorrhage, increased lacrimation as a result of obstruction of nasolacrimal ducts, and sneezing. In some instances, it is not possible to clinically or grossly differentiate neoplasms from hyperplastic nodules or granulomatous rhinitis. Some neoplasms may infiltrate adjacent bone structures and produce notable facial deformities, loss of teeth, exophthalmus, and nervous signs. Large neoplasms also project into the meatuses, narrow the lumen, and interfere with airflow, causing stertorous breathing (Fig. 9-23). Biopsies, as well as brush and imprint cytology, have proved effective in the antemortem diagnosis of nasal neoplasms, particularly in those of epithelial lineage.
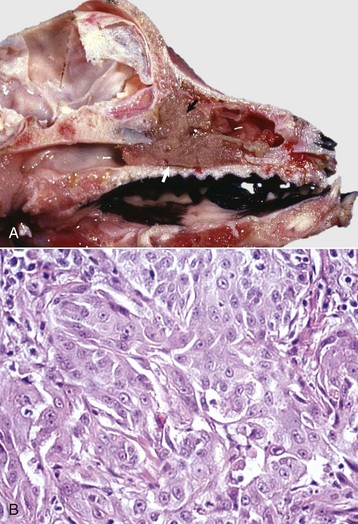
Fig. 9-23 Nasal adenocarcinoma, midsagittal section, head, adult dog.
A, A large neoplastic mass (arrows) has arisen from the ethmoidal concha and has infiltrated along the nasal passages. B, Multiple clusters of neoplastic epithelial cells with abundant eosinophilic cytoplasm and prominent nucleoli. H&E stain. (A courtesy Dr. J. M. King, College of Veterinary Medicine, Cornell University. B courtesy Dr. A. López, Atlantic Veterinary College.)
Endemic Ethmoidal Tumors: A unique group of nasal carcinomas (enzootic nasal tumors, enzootic intranasal tumors, and enzootic nasal carcinoma) of sheep, goats, and cattle arise from the surface epithelium and glands of the ethmoidal conchae. These types of carcinomas are caused by an oncogenic retrovirus, and the neoplasm has been successfully transmitted to susceptible animals by inoculation of virus or infected tissues. Endemic ethmoidal tumors are typically invasive but do not metastasize (Fig. 9-24). In some regions of the world, ethmoid tumors have been reported in horses and pigs, particularly in those farms where the endemic nasal tumors of ruminants are known to occur.
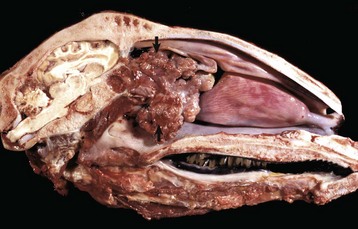
Fig. 9-24 Nasal adenocarcinoma (arrows), midsagittal section of the head, sheep.
The tumor has occluded the right nasal passage and choanae. The location (ethmoturbinates) and type of tumor (carcinoma) are typical of retrovirus-induced “enzootic nasal carcinoma.” (Courtesy Dr. L.E. Craig, College of Veterinary Medicine, University of Tennessee.)
Nasal Polyps and Nasal Cysts Resembling Neoplasms: Nonneoplastic exophytic masses that resemble neoplasms are commonly found in horses, cats, and to a lesser extent other species. In horses, polyps tend to form in the ethmoidal region, whereas in cats, polyps are most frequently found in the nasopharynx and Eustachian tubes. The pathogenesis of these benign growths is uncertain, although in many cases they follow chronic rhinitis or sinusitis. Grossly, polyps appear as firm, pedunculated nodules of various sizes protruding from the nasal mucosa into the nasal passages; the surface may be smooth, ulcerated, secondarily infected, and hemorrhagic. Microscopically, polyps are characterized by a core of well-vascularized stromal tissue that contains inflammatory cells and are covered by pseudostratified or squamous epithelium.
Nasal and paranasal sinus cysts are common in horses and are medically important because they clinically mimic neoplasms or infections. Although not considered a neoplastic growth, cysts are expansive and cause deformation or destruction of the surrounding bone. These cysts are typically composed of an epithelial cell capsule filled with yellow or hemorrhagic fluid and do not recur after surgical removal. Ethmoidal hematomas also resemble nasal tumors in horses.
Pharynx, Guttural Pouches, Larynx, and Trachea
Patterns of Injury and Host Response
The laryngeal mucosa is formed by pseudostratified columnar ciliated epithelium with goblet cells (mucociliary blanket) and squamous epithelium, whereas the trachea is exclusively lined by columnar ciliated epithelium. The pattern of injury and host response in the larynx and trachea is similar to those in the nasal mucosa (see the section on Defense Mechanisms of the Conducting System). The pharyngeal mucosa, composed of squamous epithelium, has similar patterns of necrosis and inflammation as the oral mucosa (see Chapter 7).
Anomalies
Congenital anomalies of this region are rare in all species. Depending on their location and severity, they may be inconsistent with postnatal life, pose little or no problem, interfere with quality of life, or manifest themselves in later life. If clinical signs of respiratory distress, such as stridor, coughing, dyspnea, or gagging, do occur, they are usually exacerbated by excitement, heat, stress, or exercise.
Brachycephalic Airway Syndrome: Brachycephalic airway syndrome is a clinical term that refers to increased air-flow resistance caused by stenotic nostrils and nasal meatuses and an excessively long soft palate. These abnormalities are present in brachycephalic canine breeds such as bulldogs, boxers, Boston terriers, pugs, Pekingese, and others. The defects are a result of a mismatch of the ratio of soft tissue to cranial bone and the obstruction of airflow by excessive length of the palatine soft tissue. Secondary changes, such as nasal and laryngeal edema caused by forceful inspiration, eventually lead to severe upper airway obstruction, respiratory distress, and exercise intolerance.
Hypoplastic Epiglottis, Epiglottic Entrapment, and Dorsal Displacement of the Soft Palate: Anomalies, such as hypoplastic epiglottis, epiglottic entrapment, and dorsal displacement of the soft palate, are important causes of respiratory problems and reduced athletic performance in horses. An undersized epiglottis is prone to being entrapped below the arytenoepiglottic fold, causing an equine syndrome known as epiglottic entrapment. This syndrome also occurs in horses with lateral deviation and deformity of epiglottis, epiglottic cysts, or necrosis of the tip of the epiglottis. Hypoplastic epiglottis also occurs in pigs. Dorsal displacement of the soft palate, particularly during exercise, narrows the lumen of the nasopharynx and creates abnormal air turbulence in the conducting system of horses. Epiglottic entrapment is clinically characterized by airway obstruction, exercise intolerance, respiratory noise, and cough.
Subepiglottic and Pharyngeal Cysts: Anomalous lesions, such as subepiglottic and pharyngeal cysts, are occasionally seen in horses, particularly in Standardbred and Thoroughbred race horses. These cysts vary in size (1 to 9 cm) and occur most commonly in the subepiglottic area and to a lesser extent in the dorsal pharynx, larynx, and soft palate. Cysts are lined by squamous or pseudostratified epithelium and contain thick mucus. Large cysts cause airway obstruction, reduced exercise tolerance, or dysphagia and predispose to bronchoaspiration of food.
Tracheal Agenesis and Tracheal Hypoplasia: Tracheal hypoplasia occurs most often in English bulldogs and Boston terriers; the tracheal lumen is decreased in diameter throughout its length.
Tracheal Collapse and Tracheal Stenosis: Tracheal collapse with reduction in tracheal patency occurs in toy, miniature, and brachycephalic breeds of dogs, in which the condition is also called tracheobronchial collapse or central airway collapse. The defect also occurs in horses, cattle, and goats. By radiographic, endoscopic, or gross examination, there is dorsoventral flattening of the trachea with concomitant widening of the dorsal tracheal membrane, which may then prolapse ventrally into the lumen (Fig. 9-25). Most commonly, the defect extends the entire length of the trachea and only rarely affects the cervical portion alone. Affected segments with a reduced lumen contain froth and even are covered by a diphtheritic membrane. In horses, the so-called scabbard trachea is characterized by lateral flattening, so that the tracheal lumen is reduced to a narrow vertical slit.
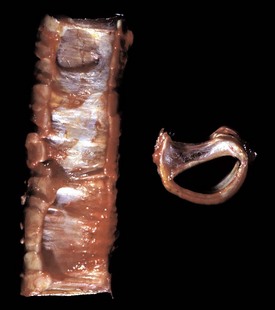
Fig. 9-25 Tracheal collapse, trachea, pony.
Left specimen, The dorsal surface of the trachea is flattened dorsoventrally, the dorsal ends of the C-shaped tracheal rings are widely separated, and the dorsal ligament between the two ends is lengthened and thinned. Right specimen (transverse section), The ends of the tracheal rings are widely separated, and the dorsal wall of the trachea is formed by the lengthened and thinned dorsal ligament. (Courtesy Dr. C.S. Patton, College of Veterinary Medicine, University of Tennessee.)
Segmental tracheal collapse causing stenosis has been associated with congenital and acquired abnormalities. In severe cases, abnormal cartilaginous glycoproteins and loss of elasticity of tracheal rings causes the trachea to collapse. In some other cases, it is an acquired tracheal lesion that follows trauma, compression caused by extraluminal masses, peritracheal inflammation, and flawed tracheotomy or transtracheal aspirate techniques.
Other tracheal anomalies include tracheoesophageal fistula, which is most commonly found in humans and sporadically in dogs and cattle. Congenital fistulas can occur at any site of the cervical or thoracic segments of the trachea. Acquired tracheoesophageal fistula can be a complication of improper intubation, tracheotomy, or esophageal foreign body.
Degenerative Diseases
Laryngeal Hemiplegia: Laryngeal hemiplegia (paralysis), sometimes called roaring in horses, is a common but obscure disease characterized by atrophy of the dorsal and lateral cricoarytenoid muscles (abductor and adductor of the arytenoid cartilage), particularly on the left side. Muscular atrophy is most commonly caused by a primary denervation (recurrent laryngeal neuropathy) of unknown cause (idiopathic axonopathy) and to a much lesser extent, secondary nerve damage (see the section on Denervation Atrophy in Chapters 14 and 15). Idiopathic laryngeal hemiplegia is an incurable axonal disease (axonopathy) of the cranial laryngeal nerve that affects mostly larger horses. Secondary laryngeal hemiplegia is rare and occurs after nerve damage caused by other pathologic processes such as compression or inflammation of the left recurrent laryngeal nerve. The medial retropharyngeal lymph nodes are located immediately ventral to the floor of the guttural pouches. As a result of this close anatomic relationship, swelling or inflammation of the guttural pouches and retropharyngeal lymph nodes often results in secondary damage to the laryngeal nerve. Common causes of secondary nerve damage (Wallerian degeneration) include guttural pouch mycosis, retropharyngeal abscesses, inflammation because of iatrogenic injection into the nerves, neck injury, and metastatic neoplasms involving the retropharyngeal lymph nodes (e.g., lymphosarcoma).
Grossly, the affected laryngeal muscle in a horse with laryngeal hemiplegia is pale and smaller than normal (muscle atrophy) (Fig. 9-26). Microscopically, muscle fibers have lesions of denervation atrophy (see Chapters 14 and 15). Atrophy of laryngeal muscles also occurs in dogs as an inherited condition (Siberian husky and Bouvier des Flandres), as a degenerative neuropathy in older dogs, secondary to laryngeal trauma in all species (e.g., choke chain damage), or to hepatic encephalopathy in horses.
Fig. 9-26 Laryngeal hemiplegia, larynx, dorsal surface, 2-year-old horse.
The left cricoarytenoideus dorsalis muscle is pale and atrophic (arrows), whereas the right cricoarytenoideus dorsalis muscle is normal. (Courtesy Dr. A. López, Atlantic Veterinary College.)
The abnormal inspiratory sounds (roaring) during exercise in horses with laryngeal hemiplegia are caused by paralysis of the left dorsal and lateral cricoarytenoid muscles, which cause incomplete dilation of the larynx, obstructing of airflow, and vibration of vocal cords.
Circulatory Disturbances
Laryngeal and Tracheal Hemorrhages: Hemorrhages in these sites occur as mucosal petechiae and are most commonly seen in coagulopathies; inflammation; septicemia and sepsis, particularly in pigs with classical swine fever (hog cholera); African swine fever or salmonellosis; and horses with equine infectious anemia. Severe dyspnea and asphyxia before death can cause congestion, ecchymosis, and petechiae in the laryngeal and tracheal mucosa; this lesion must be differentiated from postmortem imbibition of hemoglobin in autolyzed carcasses (see Chapter 1).
Laryngeal Edema: Laryngeal edema is a common feature of acute inflammation, but it is particularly important because swelling of the epiglottis and vocal cords can obstruct the laryngeal orifice, resulting in asphyxiation. Laryngeal edema occurs in pigs with edema disease; in horses with purpura hemorrhagica; in cattle with acute interstitial pneumonia; in cats with systemic anaphylaxis; and in all species as a result of trauma, improper endotracheal tubing, inhalation of irritant gases (e.g., smoke), local inflammation, and allergic reactions. Grossly the mucosa of the epiglottis and vocal cords is thickened and swollen, often protrudes dorsally onto the epiglottic orifice, and has a gelatinous appearance (Fig. 9-27).
Fig. 9-27 Laryngeal edema, larynx, mature cow.
Note the edematous thickening of the laryngeal mucosa of the vocal cords (arrows), which can cause respiratory distress due to the narrowing of the laryngeal lumen (rima glottidis). (Courtesy Dr. J. Andrews, College of Veterinary Medicine, University of Illinois.)
Tracheal Edema and Hemorrhage: Tracheal edema and hemorrhage syndrome of feeder cattle, also known as the honker syndrome or tracheal stenosis of feedlot cattle, is a poorly documented acute disease of unknown cause, most often seen during the summer months. Severe edema and a few hemorrhages are present in the mucosa and submucosa of the dorsal surface of the trachea, extending caudally from the midcervical area as far as the tracheal bifurcation. On a cut section, the tracheal mucosa is diffusely thickened and gelatinous. Clinical signs include inspiratory dyspnea that can progress to oral breathing, recumbency, and death by asphyxiation in less than 24 hours.
Inflammation
Pharyngitis, Laryngitis, and Tracheitis: Inflammation of the pharynx, larynx, and trachea are important because of their potential to obstruct airflow and to lead to aspiration pneumonia. The pharynx is commonly affected by infectious diseases of the upper respiratory and upper digestive tracts, and the trachea can be involved by extension from both the lungs and larynx.
Intraluminal foreign bodies in the pharynx, such as medicament boluses, apples, or potatoes, can move down and obstruct the larynx and trachea. Also, pharyngeal obstruction can be caused by masses in surrounding tissue such as neoplasms of the thyroid gland, thymus, and parathyroid glands.
Pharyngeal Perforation: A number of nonspecific insults can cause lesions and clinical signs. Trauma may take the form of penetrating wounds in any species: perforation of the caudodorsal wall of the pharynx from the improper use of drenching or balling guns in sheep, cattle, and pigs; choking injury because of the use of collars in dogs and cats; and the shearing forces of bite wounds. The results of the trauma may be minimal (local edema and inflammation) or as serious as complete luminal obstruction by exudate. Foreign bodies may be lodged anywhere in the pharyngeal region; the location and size determine the occurrence of dysphagia, regurgitation, dyspnea, or asphyxiation. Pigs have a unique structure known as the pharyngeal diverticulum (4 cm long in adult pigs), which is located in the pharyngeal wall rostral and dorsal to the esophageal entrance. It is important because barley awns may lodge in the diverticulum, causing an inflammatory swelling that affects swallowing. The diverticular wall may be perforated by awns or drenching syringes, which results in an exudate that can extend down the tissue planes between muscles of the neck and even into the mediastinum. The pharynx of the dog may also be damaged by trauma from chicken bones, sticks, and needles, resulting in the formation of a pharyngeal abscess.
Equine Pharyngeal Lymphoid Hyperplasia: Equine pharyngeal lymphoid hyperplasia, or pharyngitis with lymphoid follicular hyperplasia, is a common cause of partial upper airway obstruction in horses, particularly in 2- and 3-year-old racehorses. Lymphoid hyperplasia is also seen in healthy horses as part of a response to mild chronic pharyngitis, which in many instances tends to regress with age in older animals. The cause is undetermined, but chronic bacterial infection combined with environmental factors may cause excessive antigenic stimulation and lymphoid hyperplasia. The gross lesions, visible endoscopically or at necropsy, consist of variably sized (1 to 5 mm) white foci located on the dorsolateral walls of the pharynx and extend into the openings of the guttural pouches and onto the soft palate. In severe cases, lesions may appear as pharyngeal polyps. Microscopically, the lesions consist of large aggregates of lymphocytes and plasma cells in the pharyngeal mucosa. Clinical signs consist of stertorous inspiration, expiration, or both.
Inflammation of Guttural Pouches: The guttural pouches of horses are large diverticula (300 to 500 mL) of the ventral portion of the auditory (eustachian) tubes. These diverticula are therefore exposed to the same pathogens as the pharynx and have drainage problems similar to the pathogens of sinuses. Although it is probable that various pathogens, including viruses, can infect them, the most common pathogens are fungi, which cause guttural pouch mycosis and guttural pouch empyema in the horse. Because of the close anatomic proximity of guttural pouches to the internal carotid arteries, cranial nerves (VII, IX, X, XI, and XII), and atlantooccipital joint, disease of these diverticula may involve these structures and cause a variety of clinical signs in horses.
Guttural pouch mycosis occurs primarily in stabled horses and is caused by Aspergillus fumigatus and other Aspergillus sp. Infection is usually unilateral and presumably starts with the inhalation of spores from moldy hay. Grossly, the mucosal surfaces of the dorsal and lateral walls of the guttural pouch mucosa are first covered by focal, rounded, raised plaques of diphtheritic (fibrinonecrotic) exudate, which with time can become confluent and grow into a large fibrinonecrotic mass (Fig. 9-28). Microscopically, the lesions are severe necrotic inflammation of the mucosa and submucosa with widespread vasculitis and intralesional fungal hyphae. Necrosis of the wall of the guttural pouches can extend into the wall of the adjacent internal carotid artery causing hemorrhage into the lumen of the guttural pouch and recurrent epistaxis. Invasion of the internal carotid artery causes arteritis, which can also lead to formation of an aneurysm and fatal bleeding into the guttural pouches. In other cases, the fungi may be angioinvasive, leading to the release of mycotic emboli into the internal carotid artery, generally resulting in multiple cerebral infarcts. Dysphagia, another clinical sign seen in guttural pouch mycosis, is associated with damage to the pharyngeal branches of the vagus and glossopharyngeal nerves, which lie on the ventral aspect of the pouches. Horner’s syndrome results from damage to the cranial cervical ganglion and sympathetic fibers located in the caudodorsal aspect of the pouches. Finally, equine laryngeal paralysis (hemiplegia) can result from damage to the laryngeal nerves as previously described in the section on Laryngeal Hemiplegia.
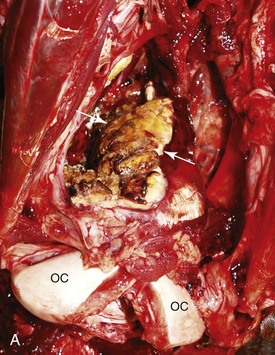
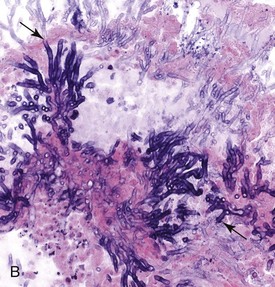
Fig. 9-28 Guttural pouch mycosis, ventral view of the head, horse.
A, Note the large mass filling the right guttural pouch (arrows). It is firmly attached to the wall and composed of fibrinonecrotic exudate and surrounded by clotted blood. OC, Occipital condyles. B, Fungal hyphae (arrows) are admixed with necrotic exudate. H&E stain. (Courtesy of Dr. A. López, Atlantic Veterinary College)
Empyema of guttural pouches is a sequela to suppurative inflammation of the nasal cavities, most commonly from Streptococcus equi infection (strangles). In severe cases, the entire guttural pouch can be filled with purulent exudate (Fig. 9-29). The sequelae are similar to those of guttural pouch mycosis except that there is no erosion of the internal carotid artery. It is clinically characterized by nasal discharge, enlarged retropharyngeal lymph nodes, painful swelling of the parotid region, dysphagia, and respiratory distress.
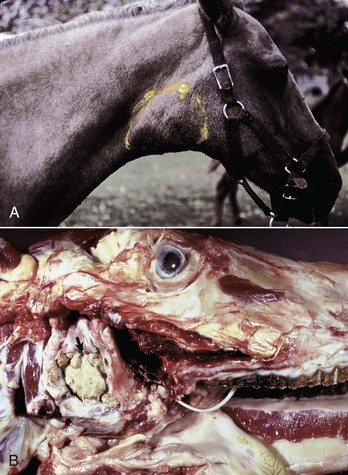
Fig. 9-29 Guttural pouch empyema, guttural pouch, horse.
A, Note the swollen right neck (outlined in yellow) in this horse with guttural pouch empyema. B, The guttural pouch is filled with masses of inspissated purulent exudate (arrow). (A courtesy College of Veterinary Medicine, University of Illinois. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Guttural pouch tympany develops sporadically in young horses when excessive air accumulates in the pouch from the one-way valve effect caused by inflammation or malformation of the Eustachian tube. It is generally unilateral and characterized by nonpainful swelling of the parotid region.
Necrotic Laryngitis: Necrotic laryngitis (calf diphtheria, laryngeal necrobacillosis) is a common disease of feedlot cattle and cattle affected with other diseases, with nutritional deficiencies, or housed under unsanitary conditions. It also occurs sporadically in sheep and pigs. Necrotic laryngitis, caused by Fusobacterium necrophorum, is part of the syndrome termed laryngeal necrobacillosis, which can include lesions of the tongue, cheeks, palate, and pharynx. An opportunistic pathogen, Fusobacterium necrophorum produces several exotoxins and endotoxins after gaining entry either through lesions of viral infections, such as IBR and vesicular stomatitis in cattle, or after traumatic injury produced by feed or careless use of specula or balling guns.
The gross lesions, regardless of location in the mouth or larynx (most common in the mucosa overlying the laryngeal cartilages), consist of well-demarcated, dry, yellow-gray, thick-crusted, and fibrinonecrotic exudate (Fig. 9-30) that in the early stages is bounded by a zone of active hyperemia. Deep ulceration develops, and if the lesion does not result in death, healing is by granulation tissue formation. Microscopically, the necrotic foci are first surrounded by hyperemic borders, then by a band of leukocytes, and finally the ulcers heal by granulation tissue and collagen (fibrosis). The lesions can extend deep into the submucosal tissue. Numerous bacteria are evident at the advancing edge.
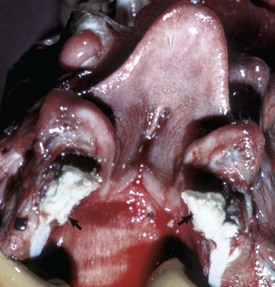
Fig. 9-30 Necrotic laryngitis, calf diphtheria (Fusobacterium necrophorum), larynx, calf.
Plaques of fibrinopurulent exudate are present on the mucosa of the arytenoid cartilages (arrows). Pieces of the exudate can be aspirated into the lungs and cause bronchopneumonia. (Courtesy Ontario Veterinary College.)
There are numerous and important sequelae to calf diphtheria; the most serious is death from severe toxemia or overwhelming fusobacteremia. Sometimes, the exudate may be copious enough to cause laryngeal obstruction and asphyxiation or be aspirated and cause bronchopneumonia. The clinical signs of necrotic laryngitis are fever, anorexia, depression, halitosis, moist painful cough, dysphagia, and inspiratory dyspnea and ventilatory failure because of fatigue of the respiratory muscles (diaphragm and intercostal).
Laryngeal Contact Ulcers: Ulcerative lesions in the larynx are commonly found in feedlot cattle. Grossly, the laryngeal mucosa reveals circular ulcers (up to 1 cm in diameter), which may be unilateral or bilateral and sometimes deep enough to expose the underlying arytenoid cartilages. The cause has not been established, but causal agents, such as viral, bacterial, and traumatic, have been proposed, along with increased frequency and rate of closure of the larynx (excessive swallowing and vocalization) when cattle are exposed to market and feedlot stresses such as dust, pathogens, and interruption of feeding. Contact ulcers predispose a calf to diphtheria (Fusobacterium necrophorum) and laryngeal papillomas. Ulceration of the mucosa and necrosis of the laryngeal cartilages have also been described in calves, sheep, and horses under the term laryngeal chondritis. Laryngeal abscesses involving the mucosa and underlying cartilage occur as a herd or flock problem in calves and sheep, presumably caused by a secondary infection with Arcanobacterium pyogenes (Actinomyces pyogenes; Corynebacterium pyogenes).
Tracheitis: The types of injury and host inflammatory responses in the trachea are essentially the same as those described for the nasal mucosa. Although tracheal mucosa is prone to aerogenous injury and necrosis, it has a remarkable capacity for repair. The most common causes of tracheitis are viral infections, such as those causing IBR (see Fig. 9-18), EVR, canine distemper, and feline rhinotracheitis. Viral lesions are generally mild and transient but often become complicated with secondary bacterial infections. At the early stages, the mucosa is notably hyperemic and can show white foci of necrosis. In the most severe cases, the affected mucosa detaches from the underlying basement membrane, causing extensive tracheal ulceration.
Chemical tracheitis is also commonly seen after aspiration (Fig. 9-31). Also, inhalation of fumes during barn fires can cause extensive injury and necrosis of the tracheal mucosa. In forensic cases, the presence of carbon pigment in the mucosal surface of trachea, bronchi, and bronchioles indicates that the burned animal was alive during the fire.
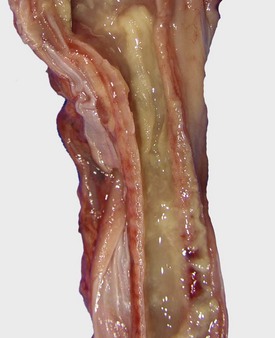
Fig. 9-31 Fibrinopurulent tracheitis, cat.
Note uniform plaque of fibrinopurulent exudate covering the entire tracheal mucosa. This cat also had suppurative bronchopneumonia and Pasteurella multocida was isolated from trachea and lung. (Courtesy Dr. L. Miller and Dr. A. López, Atlantic Veterinary College.)
According to the exudate, tracheitis in all animal species is classified as fibrinous, catarrhal, purulent, or granulomatous. Chronic polypoid tracheitis occurs in dogs and cats, probably secondary to chronic infection.
Canine Infectious Tracheobronchitis: Canine infectious tracheobronchitis (kennel cough) is a highly contagious infection that is clinically characterized by an acute onset of coughing notably exacerbated by exercise. The term is nonspecific, much like the “common cold” in humans or bovine respiratory disease (BRD) in cattle. The infection occurs commonly as a result of mixing dogs from different origins such as occurs at commercial kennels, animal shelters, and veterinary clinics. Between bouts of coughing, most animals appear normal, although some have rhinitis, pharyngitis, tonsillitis, or conjunctivitis; some with secondary pneumonia become quite ill.
The cause of canine infectious tracheobronchitis is complex, and many pathogens and environmental factors have been incriminated. Bordetella bronchiseptica, CAV-2, and canine parainfluenza virus-2 (CPIV-2) are most commonly implicated. The severity of the disease is increased when more than one agent is involved or if there are extreme environmental conditions (e.g., poor ventilation). For example, dogs asymptomatically infected with Bordetella bronchiseptica are more severely affected by superinfection with CAV-2 than those not carrying the bacterium. Other agents are sometimes isolated but of lesser significance and include CAV-1 (infectious canine hepatitis virus), reovirus type 1, canine herpesvirus, canine respiratory coronavirus (CRCoV), and Mycoplasma species.
Depending on the agents involved, gross and microscopic lesions are completely absent or they vary from catarrhal to mucopurulent tracheobronchitis, with enlargement of the tonsils and retropharyngeal and tracheobronchial lymph nodes. In dogs with Bordetella bronchiseptica infection, the lesions are suppurative or mucopurulent rhinitis and tracheobronchitis, and suppurative bronchiolitis. In contrast, when lesions are purely viral, microscopic changes are focal necrosis of the tracheobronchial epithelium. Sequelae can include spread either proximally or distally in the respiratory tract, the latter sometimes inducing chronic bronchitis and bronchopneumonia.
Parasitic Diseases of the Larynx and Trachea
Parasitic infections of the larynx and trachea can cause obstruction with dramatic consequences, but burdens sufficient to cause such effects are not commonly seen in veterinary practice.
Besnoitiosis (Besnoitia Bennetti; Besnoitia Besnoiti): Besnoitiosis (Besnoitia bennetti; Besnoitia besnoiti) is caused by an apicomplexan coccidian parasite, whose life cycle is still unknown. This parasite can cause pedunculated lesions on the skin, sclera, mucosa of the nasal cavity, and larynx of horses and donkeys, cattle, and wild animals. Besnoitiosis has been reported from Africa, Central and South America, and Britain. Grossly, pale, round, exophytic nodules up to 2 cm in diameter can be observed protruding from mucosal surfaces. These nodules microscopically consist of fingerlike projections covered by hyperplastic and sometimes ulcerated epithelium containing numerous thick-walled parasitic cysts with little inflammatory response.
Mammomonogamus (Syngamus) Laryngeus: Mammomonogamus (Syngamus) laryngeus) is a nematode that is seen attached to the laryngeal mucosa of cattle in tropical Asia and South America, and cats (gapeworm) in the Caribbean and southern United States (US). Occasionally, humans with a persistent cough or asthma-like symptoms have the parasite in the larynx or bronchi.
Oslerus (Filaroides) Osleri: Oslerus (Filaroides) osleri is a nematode parasite of dogs and other Canidae that causes characteristic protruding nodules into the lumen at the tracheal bifurcation. They are readily seen on endoscopic examination or at necropsy. In severe cases, these nodules can extend 5 cm cranially or caudally from the tracheal bifurcation and even into primary and secondary bronchi. The disease occurs worldwide, and Oslerus osleri is considered the most common respiratory nematode of dogs.
The gross lesions are variably sized, up to 1 cm, submucosal nodules that extend up to 1 cm into the tracheal lumen (Web Fig. 9-8). Microscopically, a mild mononuclear cell reaction is present when parasites are alive, but with the death of the parasite, an intense foreign body reaction develops with neutrophils and giant cells. Clinically, it can be asymptomatic, although it most often causes a chronic cough that can be exacerbated by exercise or excitement. Severe infestations can result in dyspnea, exercise intolerance, cyanosis, emaciation, and even death in young dogs.
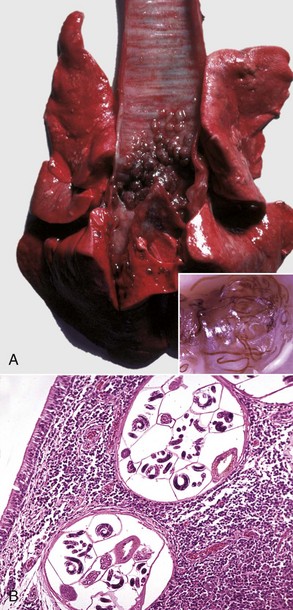
Web Fig. 9-8 Parasitic tracheobronchitis (Oslerus osleri), trachea and main bronchi, dog.
A, Note the numerous large parasitic nodules on the mucosal surface of the distal trachea and main bronchi. These nodules cause clinical signs only in severe infections. Inset, Filarial forms of Oslerus osleri can be seen in the nodule. B, Filarial forms of Oslerus osleri can be seen in the lamina propria of the tracheal mucosa. Numerous chronic inflammatory cells are also present. H&E stain. (A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. Inset and B courtesy College of Veterinary Medicine, University of Illinois.)
Neoplasms of Guttural Pouches, Larynx, and Trachea
Neoplasms of the Guttural Pouches: Neoplasms of the guttural pouches occur rarely in horses and are usually squamous cell carcinomas.
Neoplasms of the Larynx and Trachea: Laryngeal neoplasms are rare in dogs and extremely so in other species, although they have been reported in cats and horses.
The most common laryngeal neoplasms in dogs are papillomas and squamous cell carcinomas. Other less common tumors are laryngeal rhabdomyoma, previously referred to as laryngeal oncocytoma, and chondromas and osteochondromas. Lymphoma involving the laryngeal tissue is sporadically seen in cats.
When large enough to be obstructive, neoplasms may cause a change or loss of voice, cough, or respiratory distress with cyanosis, collapse, and syncope. Other signs include dysphagia, anorexia, and exercise intolerance. The neoplasm is sometimes visible from the oral cavity and causes swelling of the neck. The prognosis is poor, as most lesions recur after excision.
Tracheal neoplasms are even more uncommon than those of the larynx. The tracheal cartilage or mucosa can be the site of an osteochondroma, leiomyoma, osteosarcoma, mast cell tumor, and carcinoma. Lymphosarcomas in cats can extend from the mediastinum to involve the trachea.
Web Fig. 9-9 Acute to subacute embolic pneumonia, lung, dog.
The lung has numerous circular areas of hemorrhage distributed randomly throughout all lung lobes (embolic pattern [see Fig. 9-55]). These foci arise from injury to the microvasculature in alveolar septa and the visceral pleura secondary to lodgment of bacterial or fungal emboli (septic emboli) from valvular or mural endocarditis in the right heart or from other bacterial or fungal diseases where the bacterium or fungus gains access to the circulatory system as occurs in many bacterial and fungal enteritides or pneumonias caused by Salmonella spp., E coli, or Aspergillus spp. (Courtesy College of Veterinary Medicine, University of Illinois.)
Web Fig. 9-10 Fibrinous bronchopneumonia (pleuropneumonia), pneumonic mannheimiosis (Mannheimia haemolytica), right lung, steer.
Note the cranioventral pneumonia involving approximately 85% of the lung parenchyma. The lung is firm and swollen, and the visceral pleura is covered with a thick layer of fibrin. (Courtesy College of Veterinary Medicine, University of Illinois.)
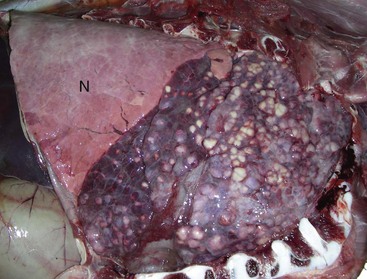
Web Fig. 9-11 Caseated granulomas, chronic-active bronchopneumonia, cow.
Note the cranioventral pneumonia and the sharp line of demarcation between normal (N) and pneumonic lung. The affected lung is firm and plum colored due to inflammation and contains numerous randomly distributed caseated granulomas. This cow had a dual bacterial infection with Mycoplasma sp. and Mannheimia haemolytica. (Courtesy Drs. V.E. Valli and S.J. Akare, College of Veterinary Medicine, University of Illinois.)
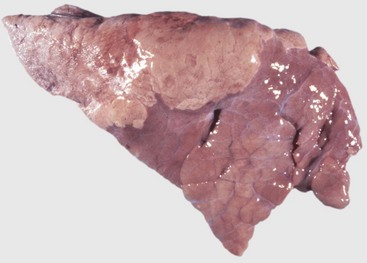
Web Fig. 9-12 Chronic suppurative bronchopneumonia (porcine enzootic pneumonia), lung, pig.
Cranioventral consolidation of 40% to 50% of pulmonary parenchyma. Consolidated lung (C) is firm, and the outlines of the lobules are accentuated by edema of the interlobular septa. N, Normal lung. (Courtesy Ontario Veterinary College.)
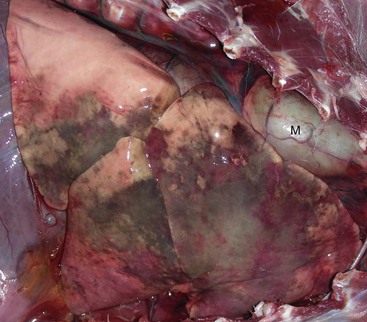
Web Fig. 9-13 Acute to subacute aspiration pneumonia, acute necrotizing bronchopneumonia, right lung, dog.
The cranioventral portions of the lung are firm, hemorrhagic, and necrotic. The necrotic areas are gray-yellow-brown and are caused by gastric content, especially hydrochloric acid, discoloring, coagulating, and digesting proteins in the tissues of the bronchi, bronchioles, and alveoli. The red regions around the necrotic foci are areas of active hyperemia in acute inflammation. This dog had myasthenia gravis and aspirated stomach content secondary to an acquired megaesophagus (M). (Courtesy Drs. C.A. Lichtensteiger and P.J. Roady, College of Veterinary Medicine, University of Illinois.)
Lungs
Each lung is subdivided into various numbers of pulmonary lobes. In the past, these were defined by anatomic fissures. However, in current anatomy, lobes are defined by the ramification of the bronchial tree. Following this criterion, the left lung of all domestic species is composed of cranial and caudal lobes, whereas the right lung, depending on species, is composed of cranial, middle (absent in horse), caudal, and accessory lobes. Each pulmonary lobe is further subdivided by connective tissue into pulmonary lobules, which in some species (cattle and pigs) are rather prominent and in others are much less conspicuous. From a practical point of view, identification of the lungs among different species could be achieved by carefully observing the degree of lobation (external fissures) and the degree of lobulation (connective tissue between lobules). Cattle and pigs have well-lobated and well-lobulated lungs; sheep and goats have well-lobated but poorly lobulated lungs; horses have both poorly lobated and poorly lobulated lungs and resemble human lungs; finally, dogs and cats have well-lobated but not well-lobulated lungs. The degree of lobulation determines the degree of air movement between the lobules. In pigs and cattle, movement of air between lobules is practically absent because of the thick connective tissue of the interlobular septa separating individual lobules. This movement of air between lobules and between adjacent alveoli (pores of Kohn) constitutes what is referred to as collateral ventilation. This collateral ventilation is poor in cattle and pigs and good in dogs. The functional implications of collateral ventilation are discussed in the section on Pulmonary Emphysema.
The lungs have an interconnecting network of stromal tissue supporting the blood and lymphatic vessels, nerves, bronchioles, and alveoli. For purposes of simplicity, the pulmonary interstitium can be anatomically divided into three contiguous compartments: (1) bronchovascular interstitium, where main bronchi and pulmonary vessels are situated; (2) interlobular interstitium separating pulmonary lobules and supporting small blood and lymph vessels; and (3) alveolar interstitium supporting the alveolar walls that contain pulmonary capillaries and alveolar epithelial cells (no lymphatic vessels here) (see discussion on the blood-air barrier in the section on Alveoli). Pulmonary changes, such as edema and emphysema, may affect one or more of these interstitial compartments.
Congenital Anomalies
Congenital anomalies of the lungs are rare in all species but are most commonly reported in cattle and sheep. Compatibility with life largely depends on the type of structures involved and the proportion of functional tissue present at birth. Accessory lungs are one of the most common anomalies and consist of distinctively lobulated masses of incompletely differentiated pulmonary tissue present in the thorax, abdominal cavity, or subcutaneous tissue virtually anywhere in the trunk. Large accessory lungs can cause dystocia. Ciliary dyskinesia (immotile cilia syndrome, Kartagener’s syndrome) is characterized by defective ciliary movement, which results in reduced mucociliary clearance because of a defect in the microtubules of all ciliated cells and most importantly, in the ciliated respiratory epithelium and spermatozoa. Primary ciliary dyskinesia often associated with situs inversus has been reported in dogs, which as a result usually have chronic recurrent rhinosinusitis, pneumonia, and infertility. Pulmonary agenesis, pulmonary hypoplasia, abnormal lobulation, congenital emphysema, lung hamartoma, and congenital bronchiectasis are occasionally seen in domestic animals. Congenital melanosis is a common incidental finding in pigs and ruminants and is usually seen at slaughter (Fig. 9-32). It is characterized by black spots, often a few centimeters in diameter, in various organs, mainly the lungs, meninges, intima of the aorta, and caruncles of the uterus. Melanosis has no clinical significance, and the texture of pigmented lungs remains unchanged.
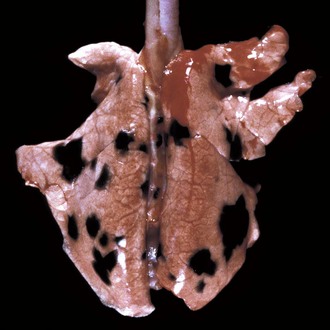
Fig. 9-32 Pulmonary melanosis, lungs, pig.
Note the areas of black (melanin pigment) discoloration of the pleural surface. This pigmentation extends into the lungs and is an incidental finding that has no clinical or pathologic significance. It is most common in “black-face” breeds of animals, especially sheep. (Courtesy College of Veterinary Medicine, University of Illinois.)
Metabolic Disturbances
Pulmonary Calcification (“Calcinosis”): Calcification of the lungs occurs in some hypercalcemic states, generally secondary to hypervitaminosis D or from ingestion of toxic (hypercalcemic) plants, such as Solanum malacoxylon (Manchester wasting disease) that contain vitamin D analogs. It is also a common sequela to uremia and hyperadrenocorticism in dogs and to pulmonary necrosis (dystrophic calcification) in most species. Calcified lungs may fail to collapse when the thoracic cavity is opened and have a characteristic “gritty” texture (Fig. 9-33). Microscopically, lesions vary from calcification of the alveolar basement membranes (see Fig. 9-33) to heterotopic ossification of the lungs. In most cases, pulmonary calcification in itself has little clinical significance, although its cause (e.g., uremia or vitamin D toxicosis) may be very important.
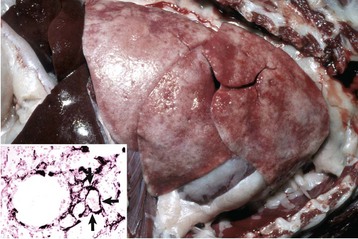
Fig. 9-33 Uremic pneumopathy from chronic renal failure, lung, 4-year-old dog.
The lungs have failed to collapse when the thorax was opened because of extensive mineralization of alveolar walls. Inset, Calcification of alveolar septa. Note the linear deposits of mineral in the alveolar septa (arrows). von Kossa stain with nuclear fast red counterstain. (Figure and Inset courtesy Dr. A. López, Atlantic Veterinary College.)
Abnormalities of Inflation
To achieve gaseous exchange, a balanced ratio of the volumes of air to capillary blood must be present in the lungs (ventilation/perfusion ratio), and the air and capillary blood must be in close proximity across the alveolar wall. A ventilation-perfusion mismatch occurs if pulmonary tissue is either collapsed (atelectasis) or overinflated (hyperinflation and emphysema).
Atelectasis (Congenital and Acquired): The term atelectasis means incomplete distention of alveoli and is used to describe lungs that have failed to expand with air at the time of birth (congenital atelectasis) or lungs that have collapsed after inflation has taken place (acquired atelectasis or alveolar collapse) (Figs. 9-34 and 9-35).
Fig. 9-34 Pulmonary atelectasis.
A, Multifocal neonatal atelectasis of the lung from 1-day-old calf. Note the prominent mosaic pattern of normally inflated (light) and atelectatic, uninflated (dark) lobules. Neonatal atelectasis is caused by aspiration of amniotic fluid, meconium, and squamous epithelial cells, causing obstruction of small bronchi and bronchioles at the time of birth. All pulmonary lobes are involved. Although focal lobular atelectasis is commonly seen in neonates, this lesion suggests that the fetus was acidotic and aspirated amniotic fluid. B, Atelectasis of a superficial pulmonary lobule in a cow. Note the absence of air in alveoli of this lobule that has resulted in its collapse and thus its darker beige color as shown in A. H&E stain. (A from López A, Bildfell R: Vet Pathol 29:104-111, 1992. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Fig. 9-35 Schematic representation of the types of atelectasis.
A, Normal alveolar distention. B, Obstructive atelectasis; obstruction of airways (i.e., exudate or parasite) affecting airflow and causing alveolar collapse. C, Compressive atelectasis; mass (i.e., abscess or tumor) compressing the lung parenchyma and causing alveolar collapse. (Redrawn from Dr. A. López, Atlantic Veterinary College.)
During fetal life, lungs are not fully distended, contain no air, and are partially filled with a locally produced fluid known as fetal lung fluid. Not surprisingly, lungs of aborted and stillborn fetuses sink when placed in water, whereas those from animals that have breathed float. At the time of birth, fetal lung fluid is rapidly reabsorbed and replaced by inspired air, leading to the normal distention of alveoli. Congenital atelectasis occurs in newborns who fail to inflate their lungs after taking their first few breaths of air; it is caused by obstruction of airways, often as a result of aspiration of amniotic fluid and meconium (described in the section on Meconium Aspiration Syndrome) (see Fig. 9-34). Congenital atelectasis also develops when alveoli cannot remain distended after initial aeration because of an alteration in quality and quantity of pulmonary surfactant produced by type II pneumonocytes and Clara cells. This form infant of congenital atelectasis is referred to in human neonatology as infant respiratory distress syndrome (IRDS) or as hyaline membrane disease because of the clinical and microscopic features of the disease. It commonly occurs in babies who are premature or born to diabetic or alcoholic mothers and is occasionally found in animals, particularly in foals and piglets. The pathetic, gasping attempts of affected foals and pigs to breathe have prompted the use of the name “barkers”; foals that survive may have brain damage from cerebral hypoxia (see Chapter 14) and are referred to as “wanderers,” owing to their aimless behavior and lack of a normal sense of fear.
Acquired atelectasis is much more common and occurs in two main forms: compressive and obstructive (see Fig. 9-35). Compressive atelectasis has two main causes: space-occupying masses in the pleural cavity, such as abscesses and tumors, or transferred pressures, such as that caused by bloat, hydrothorax, hemothorax, chylothorax, and empyema (Fig. 9-36). Another form of compressive atelectasis occurs when the negative pressure in the thoracic cavity is lost because of pneumothorax. This form generally has massive atelectasis and thus is also referred to as lung collapse.

Fig. 9-36 Compressive atelectasis and hydrothorax, lungs, dog.
Atelectatic lung appears as dark depressed pulmonary tissues (arrows). Also note a large volume of transudate in the ventral pleural cavity (*). (Courtesy Atlantic Veterinary College.)
Obstructive (absorption) atelectasis occurs when there is a reduction in the diameter of the airways caused by mucosal edema and inflammation, or when the lumen of the airway is blocked by mucus plugs, exudate, aspirated foreign material, or lungworms (see Fig. 9-35). When the obstruction is complete, trapped air in the lung eventually becomes reabsorbed. Unlike the compression type, obstructive atelectasis often has a lobular pattern as a result of blockage of the airway supplying that lobule. This lobular appearance of atelectasis is more common in species with poor collateral ventilation, such as cattle and sheep. The extent and location of obstructive atelectasis depends largely on the size of the affected airway (large versus small) and on the degree of obstruction (partial versus complete).
Atelectasis also occurs when large animals are kept recumbent for prolonged periods, such as during anesthesia (hypostatic atelectasis). The factors contributing to hypostatic atelectasis are a combination of blood-air imbalance, shallow breathing, airway obstruction because of mucus and fluid that has not been drained from bronchioles and alveoli, and from inadequate local production of surfactant. Atelectasis can also be a sequel to paralysis of respiratory muscles and prolonged use of mechanical ventilation or general anesthesia in intensive care.
In general, the lungs with atelectasis appear depressed below the surface of the normally inflated lung. The color is generally dark blue and the texture is flabby or firm; they are firm if there is concurrent edema or other processes, such as can occur in ARDS or “shock” lungs (see the section on Pulmonary Edema). Distribution and extent vary with the process, being patchy (multifocal) in congenital atelectasis, lobular in the obstructive type, and of various degrees in between in the compressive type. Microscopically, the alveoli are collapsed or slitlike and the alveolar walls appear parallel and close together, giving prominence to the interstitial tissue even without any superimposed inflammation.
Pulmonary Emphysema: Pulmonary emphysema, often simply referred to as emphysema, is an extremely important primary disease in humans, whereas in animals, it is always a secondary condition resulting from a variety of pulmonary lesions. In human medicine, emphysema is strictly defined as an abnormal permanent enlargement of airspaces distal to the terminal bronchiole, accompanied by destruction of alveolar walls (alveolar emphysema). This definition separates it from simple air space enlargement or hyperinflation, in which there is no destruction of alveolar walls and which can occur congenitally (Down syndrome) or be acquired with age (aging lung, sometimes misnamed “senile emphysema”). The pathogenesis of emphysema in humans is still controversial, but current thinking overwhelmingly suggests that destruction of alveolar walls is largely the result of an imbalance between proteases released by phagocytes and antiproteases produced in the lung as a defense mechanism (the protease-antiprotease theory). The destructive process is markedly accelerated by any factor, such as cigarette smoking, pollution, or defects in the synthesis of antiproteases in humans, that increases the recruitment of macrophages and leukocytes in the lungs. This theory originated when it was found that humans with homozygous α1-antitrypsin deficiency were remarkably susceptible to emphysema and that proteases (elastase) inoculated intratracheally into the lungs of laboratory animals produced lesions similar to those found in the disease. More than 90% of the problem relates to cigarette smoking, and airway obstruction is no longer considered to play a major role in the pathogenesis of emphysema in humans.
Primary emphysema does not occur in animals, and thus no animal disease should be called simply emphysema. In animals, this lesion is always secondary to obstruction of outflow of air or is agonal at slaughter. Secondary pulmonary emphysema occurs frequently in animals with bronchopneumonia, in which exudate plugging bronchi and bronchioles causes an airflow imbalance where the volume of air entering exceeds the volume leaving the lung. This airflow imbalance is often promoted by the so-called one-way valve effect caused by the exudate, which allows air into the lung during inspiration but prevents movement of air out of the lung during expiration.
Depending on the localization in the lung, emphysema can be classified as alveolar or interstitial. Alveolar emphysema characterized by distention and rupture of the alveolar walls, forming variably sized air bubbles in pulmonary parenchyma, occurs in all species. Interstitial emphysema occurs mainly in cattle, presumably because of their wide interlobular septa, and lack of collateral ventilation in these species does not permit air to move freely into adjacent pulmonary lobules. As a result, accumulated air penetrates the alveolar and bronchiolar walls and forces its way into the interlobular connective tissue, causing notable distention of the interlobular septa. It is also suspected that forced respiratory movements predispose to interstitial emphysema when air at high pressure breaks into the loose connective tissue of the interlobular septa (Fig. 9-37). Sometimes these bubbles of trapped air in alveolar or interstitial emphysema become confluent, forming large (several centimeters in diameter) pockets of air that are referred to as bullae (singular: bulla); the lesion is then called bullous emphysema. This lesion is not a specific type of emphysema and does not indicate a different disease process, but rather is a larger accumulation of air at one focus. In the most severe cases, air moves from the interlobular septa into the connective tissue surrounding the main stem bronchi and major vessels (bronchovascular bundles) and from here it leaks into the mediastinum, causing pneumomediastinum first, and eventually exits via the thoracic inlet into the cervical and thoracic subcutaneous tissue causing subcutaneous emphysema.
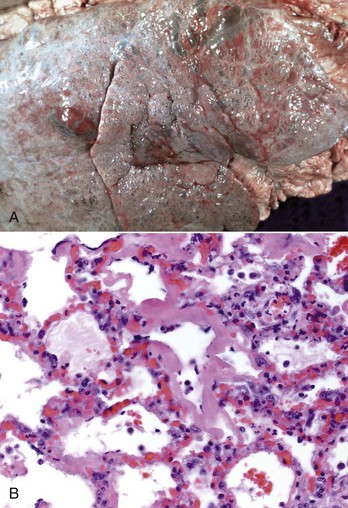
Fig. 9-37 Bovine pulmonary edema and emphysema (fog fever), lung, cow.
A, Emphysema, edema, and interstitial pneumonia involving all pulmonary lobes. Note the variably sized air bubbles in the interlobular septa and pulmonary parenchyma. The texture of these lungs would be notably crepitus as a result of the accumulation of air in pulmonary parenchyma. B, Note the thick eosinophilic hyaline membranes lining the alveoli. The alveoli are dilated and also contain some edema fluid, occasional pulmonary macrophages, and necrotic alveolar cells. H&E stain. (A courtesy Western College of Veterinary Medicine. B courtesy Dr. A. López, Atlantic Veterinary College.)
It should be noted that mild and even moderate alveolar emphysema is difficult to judge at necropsy and by light microscopy unless special techniques are used to prevent collapse of the lung when the thorax is opened. These techniques include plugging of the trachea or intratracheal perfusion of fixative (10% neutral-buffered formalin) before the thorax is opened to prevent collapse of the lungs. Important diseases that cause secondary pulmonary emphysema in animals include small airway obstruction (such as heaves) in horses and pulmonary edema and emphysema (fog fever) in cattle (see Fig. 9-37) and exudates in bronchopneumonia.
Circulatory Disturbances of the Lungs
Lungs are extremely well-vascularized organs with a dual circulation provided by pulmonary and bronchial arteries. Disturbances in pulmonary circulation have a notable effect on gaseous exchange, which may result in life-threatening hypoxemia and acidosis. In addition, circulatory disturbances in the lungs can have an impact on other organs, such as the heart and liver. For example, impeded blood flow in the lungs because of chronic pulmonary disease results in cor pulmonale, which is caused by unremitting pulmonary hypertension followed by cardiac dilation, right heart failure, chronic passive congestion of the liver (nutmeg liver), and generalized edema (anasarca).
Hyperemia and Congestion: Hyperemia is an active process that is part of acute inflammation, whereas congestion is the passive process resulting from decreased outflow of venous blood, as occurs in acute congestive heart failure (Fig. 9-38). In the early acute stages of pneumonia, the lungs appear notably red, and microscopically, blood vessels and capillaries are engorged with blood from hyperemia. Pulmonary congestion is most frequently caused by heart failure, which results in stagnation of blood in pulmonary vessels, leading to edema and egression of erythrocytes into the alveolar spaces. As with any other foreign particle, erythrocytes in alveolar spaces are rapidly phagocytosed (erythrophagocytosis) by pulmonary alveolar macrophages. When extravasation of erythrocytes is severe, large numbers of macrophages with brown cytoplasm may accumulate in the bronchoalveolar spaces. The brown cytoplasm is the result of accumulation of considerable amounts of hemosiderin; these macrophages filled with iron pigment (siderophages) are generally referred to as heart failure cells (Fig. 9-39). The lungs of animals with chronic heart failure usually have a patchy red appearance with foci of brown discoloration because of accumulated hemosiderin. In severe and persistent cases of heart failure, the lungs fail to collapse because of edema and pulmonary fibrosis. Terminal pulmonary congestion (acute) is frequently seen in animals euthanized with barbiturates and should not be mistaken for an antemortem lesion.
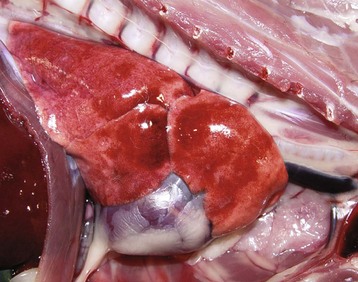
Fig. 9-38 Acute pulmonary congestion, lungs, dog.
The lung parenchyma is red because of congestion of pulmonary vasculature and alveolar capillaries. (Courtesy Dr. A. López, Atlantic Veterinary College.)
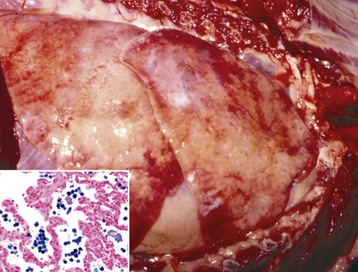
Fig. 9-39 Chronic pulmonary congestion and edema because of chronic heart failure (dilative cardiomyopathy), lungs, 5-year-old dog.
The lungs have failed to collapse (fibrosis) and have a mottled and brownish appearance (hemosiderosis). Inset, Microscopic view of alveoli. Large numbers of macrophages containing hemosiderin (heart failure cells) are present in alveoli. During heart failure, red blood cells gain access to alveoli where they are rapidly phagocytosed by pulmonary macrophages and the iron of the hemoglobin molecule is converted to hemosiderin. Hemosiderin gives a positive reaction for iron with the Prussian blue reaction. Prussian blue (iron) reaction with nuclear fast red counterstain. (Courtesy Dr. A. López, Atlantic Veterinary College.)
Hypostatic congestion is another form of pulmonary congestion that results from the effects of gravity and poor circulation on a highly vascularized tissue, such as the lung. This type of gravitational congestion is characterized by the increase of blood in the lower side of the lung, particularly the lower lung, of animals in lateral recumbency, particularly horses and cattle. The affected portions of the lung appear dark red and can have a firmer texture. In animals and particularly humans that have been prostrated for extended periods of time, hypostatic congestion may be followed by hypostatic edema, and hypostatic pneumonia as edema interferes locally with the bacterial defense mechanisms.
Pulmonary Hemorrhage: Pulmonary hemorrhages can occur as a result of trauma, coagulopathies, pulmonary thromboembolism from jugular thrombosis or from embolisms of exudate from a hepatic abscess that has eroded the wall and ruptured into the caudal vena cava (cattle), disseminated intravascular coagulation (DIC), vasculitis, or sepsis. A gross finding, often confused with intravital pulmonary hemorrhage is the result of severing both the trachea and carotid arteries simultaneously at slaughter. Blood is aspirated into the transected trachea and thence into the lungs and there forms a random pattern of scattered irregular red foci (1 to 10 mm), in one to more lobes. These red foci are readily visible on both the pleural and cut surfaces of the lung and free blood is visible in the lumens of bronchi and bronchioles.
Rupture of a major pulmonary vessel with resulting massive hemorrhage occurs occasionally in cattle when a growing abscess in a lung invades and disrupts the wall of a major pulmonary artery or vein (Fig. 9-40). In most cases, animals die rapidly, often with spectacular hemoptysis, and on postmortem examination, bronchi are filled with blood (see Fig. 9-40).
Fig. 9-40 Fatal pulmonary hemorrhage.
A, Schematic of an abscess (green) eroding the wall of a major pulmonary artery (red) and causing bleeding into the airways (blue). B, Cut surface of lung, cow. Major bronchi and the trachea are filled with clotted blood. This cow died unexpectedly, with severe respiratory distress and blood coming from the nose and mouth. A large abscess in the lung had eroded through the wall of a major pulmonary vessel. (A courtesy Dr. A. López, Atlantic Veterinary College. B courtesy Dr. R. Curtis, Atlantic Veterinary College.)
Exercise-induced pulmonary hemorrhage (EIPH) is a specific form of pulmonary hemorrhage in racehorses after exercise and clinically is characterized by epistaxis. Because only a small percentage of horses with bronchoscopic evidence of hemorrhage have clinical epistaxis, it is likely that EIPH goes undetected in many cases. The pathogenesis is still controversial, but current literature suggests laryngeal paralysis, bronchiolitis, extremely high pulmonary vascular and alveolar pressures during exercise, alveolar hypoxia, and preexisting pulmonary injury as possible causes. EIPH is seldom fatal; postmortem lesions in the lungs of horses that have been affected with several episodes of hemorrhage are characterized by large areas of dark brown discoloration, largely in the caudal lung lobes. Microscopically, lesions are alveolar hemorrhages, abundant alveolar macrophages containing hemosiderin (siderophages), and mild fibrosis of the alveolar walls.
Pulmonary Edema: In normal lungs, fluid from the vascular space slowly but continuously passes into the interstitial tissue where it is rapidly drained by the pulmonary and pleural lymphatic vessels. Recent investigations demonstrated that alveolar fluid clearance across the alveolar epithelium is also a major mechanism of fluid removal from the lung. Edema develops when the rate of fluid transudation from pulmonary vessels into interstitium or alveoli exceeds that of lymphatic and alveolar removal (Fig. 9-41). Pulmonary edema can be physiologically classified as cardiogenic (hydrostatic; hemodynamic) and noncardiogenic (permeability) types.
Fig. 9-41 Schematic representation of the pathogenesis of pulmonary edema. (Courtesy Dr. A. López, Atlantic Veterinary College)
Hydrostatic (cardiogenic) pulmonary edema develops when there is an elevated rate of fluid transudation because of increased hydrostatic pressure in the vascular compartment or decreased osmotic pressure in the blood. Once the lymph drainage has been overwhelmed, fluid accumulates in the perivascular spaces, causing distention of the bronchovascular bundles and alveolar interstitium, and eventually leaks into the alveolar spaces. Causes of hemodynamic pulmonary edema include congestive heart failure (increased hydrostatic pressure); iatrogenic fluid overload; disorders in which blood osmotic pressure is reduced, such as in the hypoalbuminemia seen in some hepatic diseases; nephritic syndrome; and protein-losing enteropathy. Hemodynamic pulmonary edema also occurs when lymph drainage is impaired, generally secondary to neoplastic invasion of lymph vessels.
Permeability edema (inflammatory) occurs when there is excessive opening of endothelial gaps or damage to the cells that constitute the blood-air barrier (endothelial cells or type I pneumonocytes). This type of edema is an integral and early part of the inflammatory response, primarily because of the effect of inflammatory mediators, such as leukotrienes, platelet-activating factor (PAF), cytokines, and vasoactive amines released by neutrophils, macrophages, mast cells, lymphocytes, endothelial cells, and type II pneumonocytes. These inflammatory mediators increase the permeability of the blood-air barrier. In other cases, permeability edema results from direct damage to the endothelium or type I pneumonocytes, allowing plasma fluids to move freely from the vascular space into the alveolar lumen (Fig. 9-42). Because type I pneumonocytes are highly vulnerable to some pneumotropic viruses (influenza, BRSV), toxicants (nitrogen dioxide [NO2], sulfur dioxide [SO2], hydrogen sulfide [H2S], 3-methylindole), and particularly to free radicals, it is not surprising that permeability edema commonly accompanies many viral or toxic pulmonary diseases. A permeability edema also occurs when endothelial cells in the lung are injured by bacterial toxins, sepsis, ARDS, DIC, anaphylactic shock, milk allergy, paraquat toxicity, and adverse drug reactions.
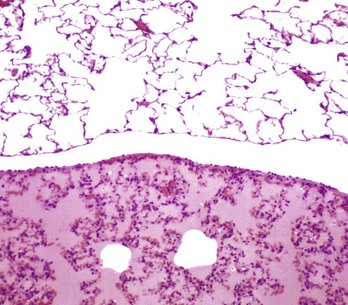
Fig. 9-42 Pulmonary edema, lung, rat.
Normal lung with alveoli filled with air (top) and lung with severe pulmonary edema characterized by transudation of protein-rich fluid (deeply eosinophilic) filling the alveoli and congested alveolar septa (bottom). H&E stain. (Courtesy Dr. A. López, Atlantic Veterinary College.)
The concentration of protein in edematous fluid is greater in permeability edema (exudate) than in hemodynamic edema (transudate); this difference has been used clinically in human medicine to differentiate one type of pulmonary edema from another. Microscopically, because of the higher concentration of protein, edema fluid in lungs with inflammation or damage to the blood-air barrier tends to stain more intensely eosinophilic than that of the hydrostatic edema from heart failure.
Grossly the edematous lungs—independent of the cause—are wet and heavy; the color varies, depending on the degree of congestion or hemorrhage; and fluid may be present in the pleural cavity. If edema is severe, bronchi and trachea contain considerable amounts of foamy fluid, which originates from the mixing of edema fluid and air (Fig. 9-43). On cut surfaces, the lung parenchyma oozes fluid like a wet sponge. In cattle and pigs that have distinct lobules, the lobular pattern becomes rather accentuated because of edematous distentions of lymphatic vessels in the interlobular septa and the edematous interlobular septum itself (Fig. 9-44). Severe pulmonary edema may be impossible to differentiate from peracute pneumonia; this fact is not surprising because pulmonary edema occurs in the very early stages of inflammation. Careful observation of the lungs at the time of necropsy is critical because diagnosis of pulmonary edema cannot be reliably performed microscopically. This is due in part to the loss of the edema fluid from the lungs during fixation with 10% neutral-buffered formalin and in part to the fact that the fluid itself stains very poorly or not at all with eosin because of its low protein content (hemodynamic edema). A protein-rich (permeability) edema is easier to visualize microscopically because it is deeply eosinophilic in hematoxylin and eosin (H&E)-stained sections (see Fig. 9-42), particularly if a fixative, such as Zenker’s solution, which precipitates protein, is used.
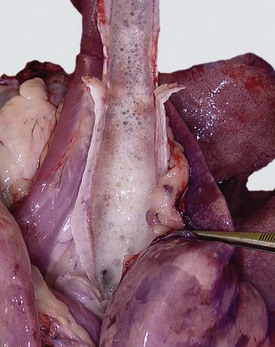
Fig. 9-43 Pulmonary edema, lungs and trachea, sheep.
Note large amounts of foamy fluid in the trachea and uncollapsed lungs with wet appearance. (Courtesy Dr. C. Legge and Dr. A. López, Atlantic Veterinary College.)
Fig. 9-44 Pulmonary edema, lungs, pig.
A, The lungs are distended by edema fluid, which has resulted in rounded edges and edematous distention of the interlobular septa. B, The cut surface is wet and the interlobular septa are markedly distended with edema fluid. Lung lobules are also congested. (A and B courtesy College of Veterinary Medicine, University of Illinois.)
Acute Respiratory Distress Syndrome: Acute (adult) respiratory distress syndrome (ARDS; shock lung) is an important condition in humans and animals characterized by pulmonary hypertension, intravascular aggregation of neutrophils in the lungs, diffuse alveolar damage, permeability edema, and formation of hyaline membranes, which are a mixture of plasma proteins, fibrin, surfactant, and cellular debris from necrotic pneumonocytes. The pathogenesis is complex and multifactorial but in general terms can be defined as diffuse alveolar damage that results from lesions in distant organs, from generalized systemic diseases, or from direct injury to the lung. Sepsis, major trauma, aspiration of gastric contents, extensive burns, and pancreatitis are some of the disease entities known to trigger ARDS. All these conditions provoke “hyperreactive macrophages” to directly or indirectly generate overwhelming amounts of cytokines (mainly TNF-α, interleukin [IL]-1, IL-6, and IL-8). Some of these cytokines prime neutrophils previously recruited in the lung capillaries and alveoli to release enzymes and free radicals, thus causing diffuse endothelial and alveolar damage that culminates in a fulminating pulmonary edema (Fig. 9-45). ARDS occurs in domestic animals and explains why pulmonary edema is one of the most common lesions found in many animals dying of sepsis, toxemia, aspiration of gastric contents, and pancreatitis, for example. A familial form of ARDS has been reported in Dalmatians. The pulmonary lesions in this syndrome are further discussed in the sections on Interstitial Pneumonia and Aspiration Pneumonia in Dogs.
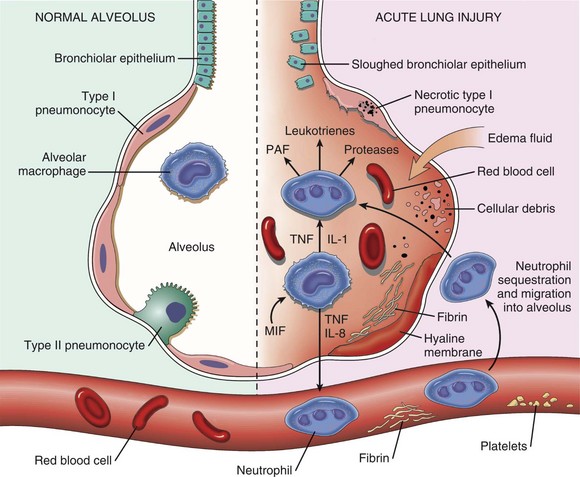
Fig. 9-45 Schematic representation of the cellular events leading to acute respiratory distress syndrome.
The normal alveolus (left side) compared to the injured alveolus in the early phase of acute lung injury and acute respiratory distress syndrome. Proinflammatory cytokines, such as interleukin-8 (IL-8) and interleukin-1 (IL-1), and tumor necrosis factor (TNF) (released by macrophages), cause neutrophils to adhere to pulmonary capillaries and extravasate into the alveolar space, where they go activate. Activated neutrophils release a variety of factors such as leukotrienes, oxidants, proteases, and platelet-activating factor (PAF), which contribute to local tissue damage, accumulation of edema fluid in the airspaces, surfactant activation, and hyaline membrane formation. Macrophage migration inhibition factor (MIF) released into the local milieu sustain the ongoing pro-inflammatory response. Subsequently, the release of macrophage-derived fibrogenic cytokines such as transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF) stimulate fibroblastic growth and collagen deposition associated with the healing phase of injury. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Neurogenic pulmonary edema is another distinctive but poorly understood form of life-threatening lung edema in humans that follows increased intracranial pressure (i.e., head injury, brain edema, brain tumors, or cerebral hemorrhage). This type of pulmonary edema can be experimentally reproduced in laboratory animals by injecting fibrin into the fourth ventricle. It involves both hemodynamic and permeability pathways presumably from massive sympathetic stimulation and overwhelming release of catecholamines. Neurogenic pulmonary edema has sporadically been reported in animals with brain injury or severe seizures or after severe stress and excitement.
Pulmonary Embolisms: With its vast capillary bed and position in the circulation, the lung acts as a safety net to catch emboli before they reach the brain and other tissues. However, this positioning is often to its own detriment. The most common pulmonary emboli in domestic animals are thromboemboli, septic (bacterial) emboli, fat emboli, and tumor cell emboli.
Pulmonary thromboembolism generally originates from a thrombus present elsewhere in the venous circulation (Fig. 9-46). Fragments released inevitably reach the lungs and become trapped in the pulmonary vasculature (Fig. 9-47). Small sterile thromboemboli are generally of little clinical or pathologic significance because they can be rapidly degraded and disposed of by the fibrinolytic system. Parasites, such as Dirofilaria immitis and Angiostrongylus vasorum; endocrinopathies, such as hyperadrenocorticism and hypothyroidism; glomerulopathies; and hypercoagulable states can be responsible for pulmonary arterial thrombosis and pulmonary thromboembolism in dogs. Pieces of thrombi breaking free from a jugular, femoral, or uterine vein can cause pulmonary thromboembolism. Pulmonary thromboembolisms occur in heavy horses after prolonged anesthesia (deep vein thrombosis), recumbent cows (“downer cow syndrome”), or in any animal undergoing long-term intravenous catheterization in which thrombi build up in the catheter and then break off (see Fig. 9-47).
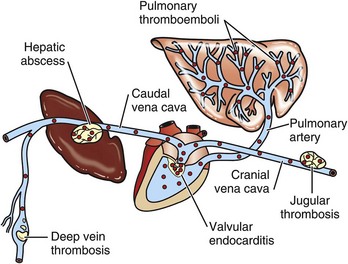
Fig. 9-46 Sources of pulmonary emboli.
Schematic diagram of pulmonary emboli (red dots) arising from: (1) rupture of a hepatic abscess into the caudal vena cava; (2) vegetative valvular endocarditis (tricuspid valve); (3) jugular thrombosis; and (4) deep vein thrombosis. Pulmonary infarcts are rare and often of little clinical significance because of the lung’s dual arterial circulation (i.e., pulmonary and bronchial arteries). (Redrawn with permission from Dr. A. López, Atlantic Veterinary College.)
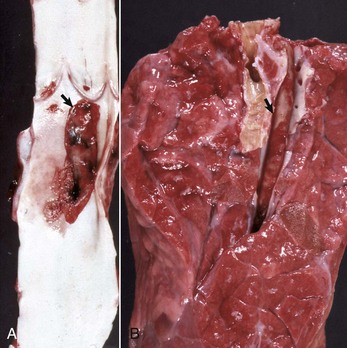
Fig. 9-47 Jugular thrombophlebitis and pulmonary thromboembolism, jugular vein and lung cut surface, cow.
A, The jugular vein has a large thrombus (arrow) attached to the wall at the site of prolonged catheterization. B, The pulmonary artery contains a large thrombus (arrow), presumably a thromboembolus that has broken off the jugular mural thrombus. Note that the pulmonary thromboembolus is not attached to the wall of the pulmonary artery. (Courtesy Dr. A. López, Atlantic Veterinary College.)
Septic emboli, pieces of thrombi contaminated with bacteria or fungi and broken free from infected mural or valvular thrombi in the heart and vessels, eventually become entrapped in the pulmonary circulation. These emboli originate most commonly from bacterial endocarditis (right side) and jugular thrombophlebitis in all species, hepatic abscesses that have eroded and discharge their contents into the caudal vena cava in cattle, and septic arthritis and omphalitis in farm animals (see Figs. 9-46 and 9-47). When present in large numbers, septic emboli may cause unexpected death because of massive pulmonary edema; survivors generally develop pulmonary arteritis and thrombosis and embolic (suppurative) pneumonia, which may lead to pulmonary abscesses.
Fat emboli can form after bone fractures or surgical interventions of bone. These are not as significant a problem in domestic animals as they are in humans. Brain emboli (i.e., pieces of brain tissue) in the pulmonary vasculature reported in severe cases of head injury in humans have recently been recognized in the bovine lung after strong pneumatic stunning at slaughter (captive bolt). Although obviously not important as an antemortem pulmonary lesion, brain emboli are intriguing as a potential risk for public health control of bovine spongiform encephalopathy (BSE). Hepatic emboli formed by circulating pieces of fragmented liver occasionally become trapped in the pulmonary vasculature after severe abdominal trauma and hepatic rupture. Tumor emboli (e.g., osteosarcoma and hemangiosarcoma in dogs and uterine carcinoma in cattle) can be numerous and striking and the ultimate cause of death in malignant neoplasia. In experimental studies, cytokines released during pulmonary inflammation are chemotactic for tumor cells and promote pulmonary metastasis.
Pulmonary (Lung) Infarcts: Because of a dual arterial supply to the lung, pulmonary infarction is rare and generally asymptomatic. However, pulmonary infarcts can be readily caused when pulmonary thrombosis and embolism are superimposed on an already compromised pulmonary circulation such as occurs in congestive heart failure. It also occurs in dogs with torsion of a lung lobe (Fig. 9-48). The gross features of infarcts vary considerably, depending on the stage, and they can be red to black, swollen, firm, and cone or wedge shaped, particularly at the lung margins. In the early acute stage, microscopic lesions are severely hemorrhagic, and this is followed by necrosis. In 1 to 2 days, a border of inflammatory cells develops, and a few days later, a large number of siderophages are present in the necrotic lung. If sterile, pulmonary infarcts heal as fibrotic scars; if septic, an abscess may form surrounded by a thick fibrous capsule.
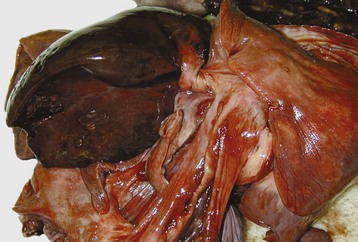
Fig. 9-48 Lobe torsion, middle lobe, lung, dog.
The right middle lung lobe is markedly congested and hemorrhagic from complete torsion. Although the right middle lobe is most frequently affected, other lobes can also rotate and undergo torsion. (Courtesy Dr. R. Fredrickson, College of Veterinary Medicine, University of Illinois.)
Patterns of Injury and Host Response in the Lungs
Bronchi: The patterns of necrosis, inflammation, and repair in intrapulmonary bronchi are similar to those previously described for the nasal and tracheal epithelium. In brief, injury to ciliated bronchial epithelium may result in degeneration, detachment, and exfoliation of necrotic cells. Under normal circumstances, cellular exfoliation is promptly followed by inflammation, mitosis, cell proliferation, cell differentiation, and finally by repair (Fig. 9-49). Depending on the type of exudate, bronchitis can be fibrinous, catarrhal, purulent, fibrinonecrotic (diphtheritic), and sometimes granulomatous. When epithelial injury becomes chronic, production of mucus is increased via goblet cell hyperplasia (chronic catarrhal inflammation). This form of chronic bronchitis is well illustrated in habitual smokers who continually need to cough out excessive mucus secretions (sputum). Unfortunately, in some cases, excessive mucus cannot be effectively cleared from airways, which leads to chronic obstructive bronchitis and emphysema (see Fig. 9-49). Chronic bronchial irritation causes squamous metaplasia of highly functional but vulnerable ciliated epithelium to nonfunctional, but more resistant, squamous epithelium. Squamous metaplasia has a calamitous effect on pulmonary clearance because it causes a structural loss and functional breakdown of portions of the mucociliary escalator. Hyperplasia of bronchial glands occurs frequently in chronic bronchitis, which translates to an increase of the Reid Index (bronchial-gland to bronchial-wall ratio). This index is less than 40% in the healthy human lung and in the lungs of most domestic species, except for cats, which generally have an index higher than 40%. The term airway remodeling encompasses all the structural changes that accompany chronic bronchitis such as hypertrophy and hyperplasia of smooth muscle, submucosal glands, and goblet cells; fibrosis; and increased bronchial vascularity.
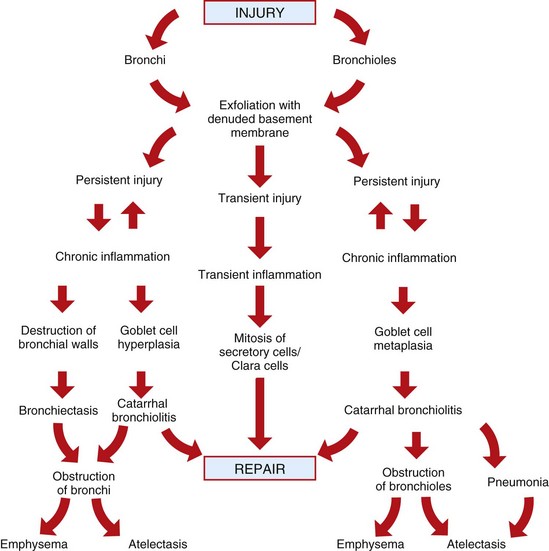
Fig. 9-49 Schematic diagram of the patterns of host response and possible sequelae to bronchial and bronchiolar injury. (Courtesy Dr. A. López, Atlantic Veterinary College.)
Bronchiectasis is one of the most devastating sequelae to chronic remodeling of the bronchi. It consists of a pathologic and permanent dilation of a bronchus with rupture of the bronchial wall as a result of obstruction or chronic inflammation. Destruction of walls occurs in part when proteolytic enzymes and oxygen radicals released from phagocytic cells during chronic inflammation degrade and weaken the smooth muscle and cartilage that help to maintain normal bronchial diameter (Fig. 9-50). Bronchiectasis may be saccular when destruction affects only a small localized portion of the bronchial wall or cylindric when destruction involves a large segment of a bronchus. Grossly, bronchiectasis is manifested by prominent lumps in the lungs (bosselated appearance or having rounded eminences) resulting from distention of bronchi with exudate, which results in a concurrent obstructive atelectasis of surrounding parenchyma (Fig. 9-51). The cut surfaces of dilated bronchi are filled with purulent exudates; for this reason, bronchiectasis is often mistaken for pulmonary abscesses. Careful inspection, usually requiring microscopic examination, confirms that exudate is contained and surrounded by remnants of a bronchial wall lined by squamous epithelium and not by a pyogenic membrane (connective tissue) as it is in the case of a pulmonary abscess. The squamous metaplasia further interferes with the normal function of the mucociliary escalator.
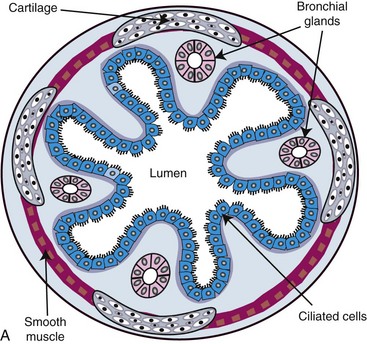
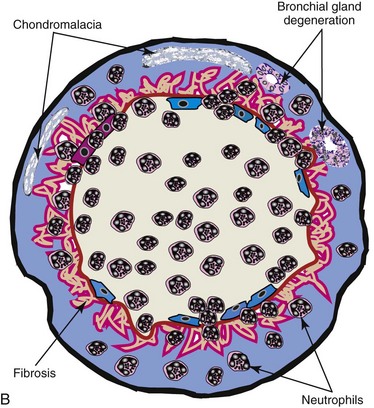
Fig. 9-50 Schematic representation of bronchiectasis.
A, Normal bronchus showing mucosa, submucosa, bronchial glands, and cartilage. B, Bronchiectasis. The affected bronchus is dilated and has lost its normal projections of the mucosa into the lumen. Note the inflammation, loss of mucosa, destruction of bronchial wall, and fibrosis with atrophy of cartilage and bronchial glands. (Courtesy Dr. A. López, Atlantic Veterinary College.)
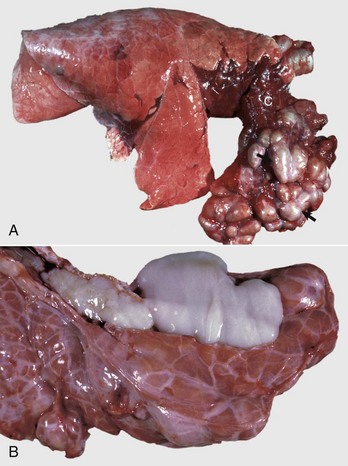
Fig. 9-51 Severe bronchiectasis with chronic bronchopneumonia, right lung, calf.
A, Note the segmentally distended (bosselated) bronchi (arrows) supplying the ventral portion of the cranial lung lobe. The lumens of affected bronchi are filled with purulent exudate. The surrounding lung parenchyma supplied by these bronchi is atelectatic (C). Bronchiectatic bronchi resemble pulmonary abscesses, but unlike abscesses, which contain a pyogenic exudate, the “exudate” in bronchiectasis is composed of remnants of the bronchial wall mixed with mucus. B, These distended bronchi are filled with purulent exudate. (A courtesy Ontario Veterinary College. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Bronchioles: The epithelial lining of the bronchiolar region (transitional zone) is exquisitely susceptible to injury, particularly to that caused by some respiratory viruses (PI-3, adenovirus, BRSV, or canine distemper), oxidant gases (NO2, SO2, or ozone [O3]), and toxic substances (3-methylindole, paraquat). The precise explanation as to why bronchiolar epithelium is so prone to injury is still not clear, but it is presumably due in part to (1) its high vulnerability to oxidants and free radicals; (2) the presence of Clara cells rich in mixed-function oxidases, which locally generate toxic metabolites; and (3) the tendency for pulmonary alveolar macrophages and leukocytes to accumulate in this region of the lungs. Depending on the types of injury and inflammatory response, bronchiolitis is classified as necrotizing, suppurative, catarrhal (mucous metaplasia), or granulomatous.
Repair in Acute and Mild Bronchiolar Injury: Once injury to the cells becomes irreversible, bronchiolar ciliated cells degenerate and exfoliate into the bronchiolar lumen, leaving a denuded basement membrane. Repair in the bronchiolar region is similar to but less effective than that in the tracheal or nasal mucosa. Under normal circumstances, recruited phagocytic cells remove exudate and cell debris from the lumina of affected bronchioles, thus preparing the basement membrane to be repopulated with new, undifferentiated cells originating from a rapidly dividing pool of Clara cells. After several days, these proliferating cells fully differentiate into normal bronchiolar ciliated cells (see Fig. 9-49).
Repair in Acute and Severe Bronchiolar Injury: In severe acute injury, such as that caused by aspiration pneumonia, exudate cannot be removed from the basement membrane of bronchioles. The exudate becomes infiltrated by fibroblasts, which form small masses of fibrovascular tissue that develop into well-organized, microscopic polyps inside the bronchiolar lumen. Their external surface eventually becomes covered by ciliated cells. This lesion is referred to as bronchiolitis obliterans, and the polyps may become so large as to cause airflow impairment (Fig. 9-52).
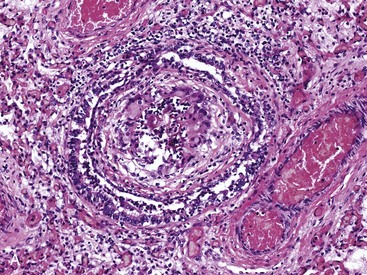
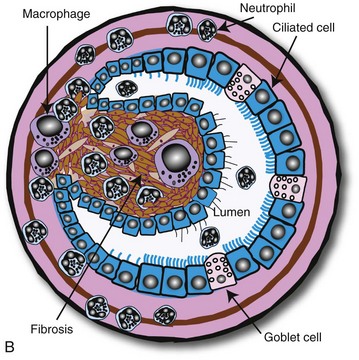
Fig. 9-52 Bronchiolitis obliterans.
A, Chronic inflammation in the bronchiolar wall resulting in the formation of a nodular mass of granulation tissue firmly attached to the airway wall and protruding into the bronchiolar lumen. H&E stain. B, Diagram illustrating organized exudate formed by connective tissue, macrophages, lymphocytes, and neutrophils, which is attached to the bronchiolar wall and covered by respiratory ciliated cells. (A courtesy Dr. A. López, Atlantic Veterinary College and Dr. Dominique Fournier, Ministère de l’Agriculture, des Pêcheries et de l’Alimentation, Quebec. B courtesy Dr. A. López, Atlantic Veterinary College.)
Repair in Chronic Bronchiolar Injury: In mild but persistent bronchiolar injury, goblet cells normally absent from bronchioles proliferate from basal cells, resulting in goblet cell metaplasia and causing a profound alteration in the physicochemical properties of bronchiolar secretions (Fig. 9-53). The normally serous bronchiolar fluid released by Clara cells becomes a tenacious material when mucus produced by goblet cells is added. As a result of increased viscoelasticity of the mucus, bronchiolar secretions cannot be removed effectively by ciliary action, leading to plugging and obstruction of distal airways. Under such conditions, often grouped as chronic obstructive pulmonary disease, coughing is required to clear mucus from obstructed bronchioles. Pulmonary emphysema and atelectasis are further sequelae to bronchiolar metaplasia and mucous hypersecretion blocking or partially blocking the lumens of these bronchioles. These two inflation abnormalities are characteristically present in COPD, which is called “heaves” in horses. Peribronchiolar proliferation of lymphocytes (BALT hyperplasia) is also a common microscopic lesion seen in chronic bronchiolitis.
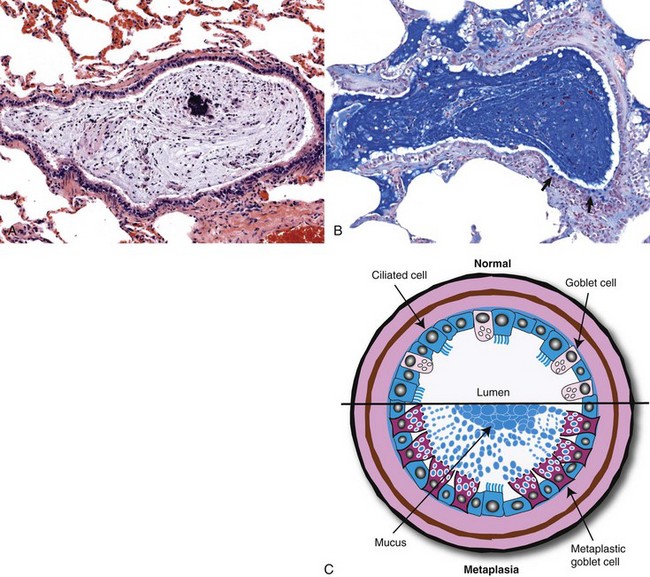
Fig. 9-53 Chronic obstructive pulmonary disease (heaves), recurrent airway obstruction (RAO), bronchiole, lung, horse.
A, This 15-year-old horse had a history of recurrent and progressive dyspnea unresponsive to treatment. Note how bronchiole is plugged with mucus admixed with cell debris and a few neutrophils. H&E stain. B, The bronchiole is filled with mucus and several goblet cells (arrows) are present in the mucosa. Healthy bronchioles do not have goblet cells or mucus. Alcian blue stain. C, Schematic diagram of a normal bronchiole (top half of the diagram) (arrows show normal goblet cells) and goblet cell metaplasia (bottom half of the diagram) (red cells = metaplastic goblet cells) in chronic obstructive pulmonary disease. Note how the mucus has obstructed the bronchiole. (A and B courtesy Dr. A. López and Dr. C. Legge, Atlantic Veterinary College; C courtesy Dr. A. López, Atlantic Veterinary College.)
Recurrent Airway Obstruction in Horses: Recurrent airway obstruction (RAO) of horses, such as COPD, heaves, chronic bronchiolitis-emphysema complex, chronic small airway disease, alveolar emphysema, and “broken wind,” is a common clinically asthma-like syndrome of horses and ponies, characterized by recurrent respiratory distress, chronic cough, poor athletic performance, airway neutrophilia, bronchoconstriction, mucus hypersecretion, and airway obstruction. The pathogenesis is still obscure, but genetic predisposition, TH2 (allergic) immune response, and the exceptional sensitivity of airways to environmental allergens (hyperreactive airway disease) have been postulated as the basic underlying mechanisms. What makes small airways hyperreactive to allergens is still a matter of controversy. Epidemiologic and experimental studies suggest that it could be the result of preceding bronchiolar damage caused by viral infections; ingestion of pneumotoxicants (3-methylindole); or prolonged exposure to organic dust, endotoxin, and environmental allergens (molds). It has been postulated that sustained inhalation of dust particles, whether antigenic or not, up-regulates the production of cytokines (TNF-α, IL-8, and monokine-inducible protein [MIP-2]) and neuropeptides (neurokinin A [NKA], neurokinin B [NKB], and substance P), attracting neutrophils into the bronchioloalveolar region and promoting leukocyte-induced bronchiolar injury. Summer pasture-associated obstructive pulmonary disease (SPAOPD) is a seasonal airway disease also reported in horses with similar clinical and pathologic findings. More recently, the term inflammatory airway disease has been introduced in equine medicine to describe RAO-like syndrome in young horses 2 to 4 years old.
The lungs of horses with heaves are grossly unremarkable, except for extreme cases in which alveolar emphysema may be present. Microscopically, the lesions include goblet cell metaplasia in bronchioles; plugging of bronchioles with mucus mixed with a few eosinophils (see Fig. 9-53); peribronchiolar infiltration with lymphocytes, plasma cells, and variable numbers of eosinophils; and hypertrophy of smooth muscle in bronchi and bronchioles. In severe cases, accumulation of mucus leads to the complete obstruction of bronchioles and alveoli, and resultant alveolar emphysema characterized by enlarged “alveoli” from the destruction of alveolar walls.
Airway Hyperresponsiveness: Airway hyperresponsiveness, or hyperreactive airway disease, is another sequela of bronchiolar injury. It develops in humans and animals (experimentally) after a transient and often innocuous viral infection of the lower respiratory tract or from exposure to certain allergens. Experimental work has shown that airway hyperreactivity in postviral bronchiolitis is associated with increased expression of TLRs and unusual susceptibility to inhaled endotoxin. Hyperreactive animals typically have an increased number of mast cells, eosinophils, and T lymphocytes in the airway mucosa. Clinically, airway hyperresponsiveness is characterized by an exaggerated bronchoconstriction after exposure to mild stimuli, such as cold air, or after animals are exposed to aerosols of histamine or methacholine.
Feline Asthma Syndrome: Feline asthma syndrome, also known as feline allergic bronchitis, is a clinical syndrome in cats of any age characterized by recurrent episodes of bronchoconstriction, cough, or dyspnea. The pathogenesis is not well understood but is presumed to originate, as in human asthma, as a type I hypersensitivity (IgE–mast cell reaction) to inhaled allergens. Dust, cigarette smoke, plant and household materials, and parasitic proteins have been incriminated as possible allergens. This self-limited allergic disease responds well to steroid therapy; thus it is rarely implicated as a primary cause of death except when suppressed defense mechanisms allow a secondary bacterial pneumonia. Bronchial biopsies from affected cats at the early stages reveal mild-to-moderate inflammation characterized by mucosal edema and infiltration of leukocytes, particularly eosinophils. Increased numbers of circulating eosinophil leukocytes (blood eosinophilia) are present in some but not all cats with feline asthma. In the most advanced cases, chronic bronchoconstriction and excess mucus production may result in smooth muscle hyperplasia and obstruction of the bronchi and bronchioles and infiltration of the airway mucosa by eosinophils. A syndrome known as canine asthma has been reported in dogs but is not as well characterized as the feline counterpart.
Alveoli: Because of their extremely delicate structure, alveoli are quite vulnerable to injury once the local defense mechanisms have been overwhelmed. The alveolar wall is a thin membrane formed by a core of interstitium supporting an extensive network of alveolar capillaries. Fibroblasts (septal cells), myofibroblasts, collagen, elastic fibers, and few interstitial macrophages and mast cells constitute the alveolar interstitium. The wall of the alveolar capillaries facing the airspace is remarkably thin and has three layers composed of vascular endothelium, basal lamina, and alveolar epithelium. These three layers of the alveolar capillaries constitute what is customarily referred to as the blood-air barrier (see Fig. 9-7). The epithelial side of the alveolus is primarily lined by rather thin type I pneumonocytes, which are arranged as a very delicate continuous membrane extending along the alveolar surface (see Fig. 9-7). Type I pneumonocytes are particularly susceptible to noxious agents that reach the alveolar region either aerogenously or hematogenously. Injury to type I pneumonocytes rapidly causes swelling and vacuolation of these cells. When cellular damage has become irreversible, type I cells detach, resulting in denudation of the basement membrane, increased alveolar permeability, and alveolar edema. Alveolar repair is possible as long as the basement membrane remains intact and lesions are not complicated by further injury or infection. Within 3 days, cuboidal type II (granular) pneumonocytes, which are the precursor cells and more resistant to injury, undergo mitosis and provide a large pool of new undifferentiated cells (Fig. 9-54). These new cells repave the denuded alveolar basement membrane and finally differentiate in type I pneumonocytes. When alveolar injury is diffuse, proliferation of type II pneumonocytes becomes so spectacular that the microscopic appearance of the alveolus resembles that of a gland or fetal lung; the lesion has been termed epithelialization or fetalization. Although it is part of the normal alveolar repair, hyperplasia of type II pneumonocytes can interfere in gas exchange and cause hypoxemia. In uncomplicated cases, type II pneumonocytes eventually differentiate into type I pneumonocytes, thus completing the last stage of alveolar repair. In some forms of chronic interstitial lung injury the surface of the alveolar basement membrane could become populated with migrating bronchiolar cells, a process known as alveolar bronchiolization or lambertosis. In severe cases, lambertosis, a metaplastic change, can be mistaken microscopically with alveolar adenomas.
Fig. 9-54 Hyperplasia of type II pneumonocytes.
A, Acute alveolar injury, crude oil aspiration, cow. Note proliferation of cuboidal epithelial cells (type II pneumonocytes) (arrows) along the luminal surface of the alveolar wall. During alveolar repair, type II pneumocytes are the precursor cell for necrotic and lost type I pneumonocytes. B, Chronic alveolar injury, interstitial pneumonia, horse. Note entire alveolar membrane lined with cuboidal type II pneumonocytes (arrowheads). The alveolar interstitium is expanded with inflammatory cells and the alveolar lumens contain cell debris mixed with leukocytes. (A courtesy Dr. A. López, Atlantic Veterinary College. B courtesy Drs. G. Hines, Provincial Veterinary Laboratory, New Brunswick and A. López, Atlantic Veterinary College.)
Type I pneumonocytes are one of the three structural components of the blood-air barrier, so when these epithelial cells are damaged, there is an increase in alveolar capillary permeability and transient leakage of plasma fluid, proteins, and fibrin into the alveolar lumen. Under normal circumstances, these fluids are rapidly cleared from the alveolus by alveolar and lymphatic absorption, and necrotic pneumonocytes (type I) and fibrin strands are phagocytosed and removed by pulmonary alveolar macrophages. When there is persistent and severe injury, fibroblasts and myofibroblasts may proliferate in the alveolar walls (alveolar interstitium), causing alveolar fibrosis, whereas in other forms of severe injury, fibroblasts and myofibroblasts actively migrate from the interstitium into the alveolar spaces, causing intraalveolar fibrosis. These two types of alveolar fibrosis are most commonly seen in toxic and allergic pulmonary diseases, and this alveolar fibrosis has a devastating effect on lung function.
Endothelial cells are also major players in the normal and abnormal physiology of the alveolus. These cells trap and share circulating antigens with intravascular and interstitial macrophages. The junction between alveolar endothelial cells is not as tight as that of the type I pneumonocytes, allowing some movement of fluid and small size molecular weight proteins into the alveolar interstitium. Endothelial cells maintain an intimate cell contact with erythrocytes and leukocytes passing through the lung, since the lumen of alveolar capillaries is slightly smaller (5.0 µm) than the diameter of red and white blood cells. Erythrocytes are easily deformable, so their transit time through the alveolar capillaries is shorter than leukocytes, which are less deformable cells. This longer transit time of leukocytes and their close cellular contact with alveolar endothelial cells has major impact in lung inflammation and ARDS.
On a minute-to-minute basis, the pulmonary defense mechanisms deal effectively with noxious stimuli and mild tissue injury without the need for an inflammatory response. However, if normal defense mechanisms are ineffective or insufficient (overwhelmed), the inflammatory process is rapidly turned on as a second line of defense.
General Aspects of Lung Inflammation
In the past three decades, an information explosion has increased the overall understanding of pulmonary inflammation, with so many proinflammatory and antiinflammatory mediators described to date that it would be impossible to review them all here (see Chapters 3 and 5).
Pulmonary inflammation is a highly regulated process that involves a complex interaction between cells imported from the blood (platelets, neutrophils, eosinophils, mast cells, and lymphocytes) and pulmonary cells (type I and II pneumonocytes; endothelial and Clara cells; alveolar and intravascular macrophages; and stromal interstitial cells, such as mast cells, interstitial macrophages, fibroblasts, and myofibroblasts). Blood-borne leukocytes, platelets, and plasma proteins are brought into the areas of inflammation by an elaborate network of chemical signals emitted by pulmonary cells and resident leukocytes. Long-distance communication between pulmonary cells and blood cells is largely done by soluble cytokines; once in the lung, imported leukocytes communicate with pulmonary and vascular cells through adhesion and other inflammatory molecules. The best known inflammatory mediators are the complement system (C3a, C3b, and C5a), coagulation factors (factors V, VII), arachidonic acid metabolites (leukotrienes and prostaglandins), cytokines (interleukins, monokines, and chemokines), adhesion molecules (ICAM, VCAM), neuropeptides (substance P, tachykinins, and neurokinins), enzymes and enzyme inhibitors (elastase, antitrypsin), oxygen metabolites (O2•, OH•, H2O2), antioxidants (glutathione), and nitric oxide (Web Table 9-1). Acting in concert, these and many other molecules send positive or negative signals to initiate, maintain, and hopefully resolve the inflammatory process without causing injury to the lung.
WEB TABLE 9-1
Main Chemical Mediators Involved in Pulmonary Inflammation
| Name | Source | Function in the Lung |
| Histamine | Mast cells, basophils, monocytes | Increase vascular permeability, pain |
| Prostaglandins and leukotrienes | Cell membranes | Increase permeability, platelet aggregation, vasoconstriction or vasodilation, lung edema, pain |
| L-selectin | Neutrophils, monocytes | Leukocyte attachment and migration, homing to areas of pulmonary inflammation |
| P-selectin, E-selectin; ICAM-1, ELAM-1 | Venules and capillary endothelium | Leukocyte attachment and migration to areas of pulmonary inflammation |
| IL-1 | Alveolar macrophages | ELAMs, leukocyte chemotaxis |
| IL-6 | Macrophages | Lymphocyte and fibroblast activation in the lung; downregulates TNF and reduces inflammation |
| IL-8 | Macrophages, fibroblasts | Leukocyte and lymphocyte chemotaxis |
| IL-9 | Macrophages, alveolar cells | Decreases cytokine production in pulmonary alveolar macrophages |
| TNF-α | Alveolar macrophages | ELAMs, endothelial adhesion, vascular permeability, lung edema; fever and acute-phase proteins, apoptosis |
| Eotaxin and IL-5 | Lymphocytes | Eosinophil chemotaxis, airway eosinophilia, asthma, pulmonary allergies |
| Epithelial secretory proteins | Type I and II pneumonocytes, Clara cells | Modulation of lung inflammation; regulation of fibroblasts-fibrosis and NO |
| Surfactant A, B (collectins) | Type II pneumonocytes and Clara cells | Chemotaxis, phagocytosis, immunomodulation, regulation of nitric oxide |
| NO | Pneumonocytes, macrophages, endothelium | Decreases cytokine production in pulmonary alveolar macrophages, modulation of apoptosis |
| TGF-α and TGF-β | Pneumonocytes, macrophages | Lung remodeling, deposition of connective tissue, fibrosis |
| Neuropeptides (Tachykinins: substance P, neurokinins | Macrophages | Lung permeability, lung edema, bronchoconstriction, bronchial secretions, inflammation |
ELAM, Endothelial adhesion molecule; ICAM, intercellular adhesion molecule; IL, interleukin; NO, nitric oxide; TGF, transforming growth factor; TNF, tumor necrosis factor.
Pulmonary macrophages (alveolar, intravascular, and interstitial), which have an immense biologic armamentarium, are the single most important effector cell and source of cytokines for all stages of pulmonary inflammation. These all-purpose phagocytic cells modulate the recruitment and trafficking of blood-borne leukocytes in the lung through the secretion of chemokines (see Web Table 9-1).
Before reviewing how inflammatory cells are recruited in the lungs, three significant features in pulmonary injury must be remembered: (1) leukocytes can exit the vascular system through the alveolar capillaries, unlike in other tissues, where postcapillary venules are the sites of leukocytic diapedesis (extravasation); (2) the intact lung contains within alveolar capillaries a large pool of resident leukocytes (marginal pool); and (3) additional neutrophils are sequestered within alveolar capillaries within minutes of local or systemic inflammatory response. These three pulmonary idiosyncrasies, along with the enormous length of the capillary network in the lung, explain why recruitment and migration of leukocytes into alveolar spaces develops so rapidly. Experimental studies with aerosols of endotoxin or Gram-negative bacteria have shown that within minutes of exposure, there is a significant increase in capillary leukocytes, and by 4 hours the alveolar lumen is filled with neutrophils. Not surprisingly, the BAL fluid collected from patients with acute pneumonia contains large amounts of inflammatory mediators such as TNF-α, IL-1, and IL-8. Also, the capillary endothelium of patients with acute pneumonia has increased “expression” of adhesion molecules, which facilitate the migration of leukocytes from capillaries into the alveolar interstitium and from there into the alveolar lumen. In allergic pulmonary diseases, eotaxin and IL-5 are primarily responsible for recruitment and trafficking of eosinophils in the lung (see Web Table 9-1).
Movement of plasma proteins into the pulmonary interstitium and alveolar lumen is a common but poorly understood phenomenon in pulmonary inflammation. Leakage of fibrinogen and plasma proteins into the alveolar space occurs when there is structural damage to the blood-air barrier. This leakage is also promoted by some types of cytokines that enhance procoagulant activity, whereas others reduce fibrinolytic activity. Excessive exudation of fibrin into the alveoli is particularly common in ruminants and pigs. The fibrinolytic system plays a major role in the resolution of pulmonary inflammatory diseases. In some cases, excessive plasma proteins leaked into alveoli mix with necrotic type I pneumonocytes and pulmonary surfactant, forming microscopic eosinophilic bands (membranes) along the lining of alveolar septa. These membranes, known as hyaline membranes, are found in specific types of pulmonary diseases, particularly in ARDS, and in cattle with acute interstitial pneumonias such as bovine pulmonary edema and emphysema and extrinsic allergic alveolitis (see Figs. 9-37 and 9-45).
In the last few years, nitric oxide has been identified as a major regulatory molecule of inflammation in a variety of tissues, including the lung. Produced locally by macrophages, pulmonary endothelium, and pneumonocytes, nitric oxide regulates the vascular and bronchial tone, modulates the production of cytokines, controls the recruitment and trafficking of neutrophils in the lung, and switches on/off genes involved in inflammation and immunity. Experimental work has also shown that pulmonary surfactant upregulates the production of nitric oxide in the lung, supporting the current view that pneumonocytes are also pivotal in amplifying and downregulating the inflammatory and immune responses in the lung (see Web Table 9-1).
As the inflammatory process becomes chronic, the types of cells making up cellular infiltrates in the lung change from mainly neutrophils to largely mononuclear cells. This shift in cellular composition is accompanied by an increase in specific cytokines, such as IL-4, interferon-γ (IFN-γ), and interferon-inducible protein (IP-10), which are chemotactic for lymphocytes and macrophages. Under appropriate conditions, these cytokines activate T lymphocytes, regulate granulomatous inflammation, and induce the formation of multinucleated giant cells such as in mycobacterial infections.
Inflammatory mediators locally released from inflamed lungs also have a biologic effect in other tissue. For example, pulmonary hypertension and right-sided heart failure (cor pulmonale) often follows chronic alveolar inflammation, not only as a result of increased pulmonary blood pressure but also from the effect of inflammatory mediators on the contractibility of smooth muscle of the pulmonary and systemic vasculature. Cytokines, particularly TNF-α, which are released during inflammation are associated, both as cause and effect, with the systemic inflammatory response syndrome (SIRS), sepsis, severe sepsis with multiple organ dysfunction, and septic shock (cardiopulmonary collapse).
As it occurs in any other sentinel system where many biologic promoters and inhibitors are involved (coagulation, the complement and immune systems), the inflammatory cascade could go into an “out-of-control” state, causing severe damage to the lungs. Acute lung injury (ALI), extrinsic allergic alveolitis, ARDS, pulmonary fibrosis, and asthma are archetypical diseases that ensue from an uncontrolled production and release of cytokines.
As long as acute alveolar injury is transient and there is no interference with the normal host response, the entire process of injury, degeneration, necrosis, inflammation, and repair can occur in less than 1 week. On the other hand, when acute alveolar injury becomes persistent or when the capacity of the host for repair is impaired, lesions can progress to an irreversible stage in which restoration of alveolar structure is no longer possible. In diseases, such as extrinsic allergic alveolitis, the constant release of proteolytic enzymes and free radicals by phagocytic cells perpetuates alveolar damage in a vicious circle. In other cases, such as in paraquat toxicity, the magnitude of alveolar injury can be so severe that type II pneumonocytes, basement membranes, and alveolar interstitium are so disrupted that the capacity for alveolar repair is lost. Fibronectins and transforming growth factors (TGFs) released from macrophages and other mononuclear cells at the site of chronic inflammation regulate the recruitment, attachment, and proliferation of fibroblasts. In turn, these cells synthesize and release considerable amounts of ECM (collagen, elastic fibers, or proteoglycans), eventually leading to fibrosis and total obliteration of normal alveolar architecture. In summary, in diseases in which there is chronic and irreversible alveolar damage, lesions invariably progress to a stage of terminal alveolar and interstitial fibrosis.
Alveolar Filling Disorders: Alveolar filling disorders are a heterogeneous group of lung diseases characterized by accumulation of various chemical compounds in the alveolar lumens. The most common are alveolar proteinosis in which the alveoli are filled with finely granular eosinophilic material; alveolar microlithiasis in which the alveoli contain numerous concentric calcified “microliths” or “calcospherites.” A similar but distinct concretion is known as corpora amylacea, which is an accumulation of cholesterol (cholesterol pneumonitis) or lipids (endogenous lipid pneumonia; see the section on Other Pneumonias of Cats). There is often little host response, and in many cases, it is only an incidental finding. Some of the alveolar filling disorders originate from inherited metabolic defects in which alveolar cells (epithelial or macrophages) cannot properly metabolize or remove lipids or proteins while others result from an excessive synthesis of these substances in the lung.