Chapter 18 Bronchiectasis, primary ciliary dyskinesia and cystic fibrosis
BRONCHIECTASIS
For many years, non-cystic fibrosis (CF) bronchiectasis has been a poorly evaluated condition for which management has been somewhat empirical and the patients have been included in a general respiratory clinic. The British Thoracic Society Bronchiectasis Guidelines (2007) have been developed with three aims:
With over a thousand references, these guidelines are an excellent resource for managing non-CF bronchiectasis. The recently published guidelines by the American College of Physicians have emphasized the paucity of evidence-based clinical information for the management of non-CF bronchiectasis (Rosen 2006).
‘Bronchiectasis’ is the term used for permanent dilatation of one or more bronchi, whereby the elastic and muscular tissue of the bronchial walls is destroyed by acute or chronic infection (Cole 1995). This damage leads to impaired drainage of bronchial secretions. These secretions often become chronically infected, producing a persistent host inflammatory response. The combination of infection and a chronic inflammatory host response results in a progressive destructive lung disease. Depending upon the aetiology, bronchiectasis can affect specific lobes or both lungs.
The incidence and prevalence of bronchiectasis are unknown. Chest radiography — although usually the first investigation — is an insensitive method for evaluating bronchiectasis. Computed tomography (CT) is the gold standard in the detection of bronchiectasis (Smith & Flower 1996), but population screening using CT is not justified. With the decline in childhood tuberculosis, measles and whooping cough there is an impression that bronchiectasis is less prevalent. However, treatment of bronchiectasis has improved and it is important to try and establish the exact cause. Diagnosing the cause may define a specific approach to treatment and provide a prognosis as in the case of cystic fibrosis. A survey of the causative factors of bronchiectasis identified that 29% of cases were post infectious, 8% due to an immune defect and 7% due to allergic bronchopulmonary aspergillosis (ABPA), but for 53% of patients no cause was found (Pasteur et al 2000). A list of the common causes of bronchiectasis is set out in Table 18.1.
Table 18.1 Common causes of bronchiectasis
| Post-infective | |
| Mucociliary clearance defects | |
| Immune defects | |
| Allergic bronchopulmonary aspergillosis |
|
| Localized bronchial obstruction | |
| Gastric aspiration |
Delivery of care for complex diseases requires experience, expertise and teamwork. This maxim applies to lung cancer, transplantation and interstitial lung disease and it may improve the outcome for patients with bronchiectasis if they are cared for by a specialist multidisciplinary team, in a similar way to patients with cystic fibrosis.
Clinical features
The range of disease expression may vary from patients who are totally asymptomatic to those who have severe disease with a cough productive of large amounts of purulent sputum, which is sometimes bloodstained. The latter require treatment of high intensity with frequent hospital attendance. Severe exacerbations may be accompanied by chest pain, breathlessness and fevers. Patients with inherited diseases such as cystic fibrosis and primary ciliary dyskinesia will often have accompanying sinus disease with nasal blockage, a purulent discharge and facial pain.
Clinical signs are non-specific. On auscultation there may be localized or widespread inspiratory and expiratory crackles with occasional wheezing. Clubbing is infrequent except with severe disease and cystic fibrosis.
Diagnosis and investigations
 The chest radiograph may be normal or there may be signs of thickened bronchial walls (tramlining), crowding of vessels with loss of volume and cyst-like shadows with fluid levels. The chest radiograph on its own is an insensitive test, detecting less than 50% of patients with bronchiectasis (Currie et al 1987). High-resolution CT is the imaging method of choice as a diagnostic tool in bronchiectasis. It has a high specificity, greater than 90% (Smith & Flower 1996). Sputum specimens for examination and culture to identify the micro-organisms and their sensitivity to antibiotics. The most common bacteria found in bronchiectatic sputum are Haemophilus influenzae (70%), Streptococcus pneumoniae and Pseudomonas (Ps) aeruginosa. The latter is found in patients with diffuse bronchiectasis and associated with accelerated lung disease (Evans et al 1996). Patients infected with Ps. aeruginosa require a higher intensity of treatment. The diagnosis of allergic bronchopulmonary aspergillosis (ABPA) is difficult. The routine investigations should include: skin prick tests, eosinophil count, Aspergillus precipitins, total IgE levels with specific IgG and IgE levels to Aspergillus. Plain radiography may show fleeting shadows responsive to steroids. CT scanning may show the typical proximal bronchiectasis. Gene mutation analysis. This should be performed on all cases of idiopathic bronchiectasis to exclude some of the more benign mutations of cystic fibrosis (Pasteur et al 2000). Electron microscopy of the cilia (if the diagnosis of primary ciliary dyskinesia (PCD) is being considered) and evaluation of the patient in a specialist centre (Ferkol et al 2006).
The chest radiograph may be normal or there may be signs of thickened bronchial walls (tramlining), crowding of vessels with loss of volume and cyst-like shadows with fluid levels. The chest radiograph on its own is an insensitive test, detecting less than 50% of patients with bronchiectasis (Currie et al 1987). High-resolution CT is the imaging method of choice as a diagnostic tool in bronchiectasis. It has a high specificity, greater than 90% (Smith & Flower 1996). Sputum specimens for examination and culture to identify the micro-organisms and their sensitivity to antibiotics. The most common bacteria found in bronchiectatic sputum are Haemophilus influenzae (70%), Streptococcus pneumoniae and Pseudomonas (Ps) aeruginosa. The latter is found in patients with diffuse bronchiectasis and associated with accelerated lung disease (Evans et al 1996). Patients infected with Ps. aeruginosa require a higher intensity of treatment. The diagnosis of allergic bronchopulmonary aspergillosis (ABPA) is difficult. The routine investigations should include: skin prick tests, eosinophil count, Aspergillus precipitins, total IgE levels with specific IgG and IgE levels to Aspergillus. Plain radiography may show fleeting shadows responsive to steroids. CT scanning may show the typical proximal bronchiectasis. Gene mutation analysis. This should be performed on all cases of idiopathic bronchiectasis to exclude some of the more benign mutations of cystic fibrosis (Pasteur et al 2000). Electron microscopy of the cilia (if the diagnosis of primary ciliary dyskinesia (PCD) is being considered) and evaluation of the patient in a specialist centre (Ferkol et al 2006).Medical management
Progression of bronchiectasis is related to poor clearance of infected secretions. Physiotherapy (see below) is probably the most important component of long-term treatment. Antibiotics are fundamental to treating infective exacerbations and controlling the severity of bronchiectasis. The choice of antibiotic will be determined by the frequency and sensitivity of micro-organisms grown in sputum culture. The route and frequency of delivery will be decided by the severity of the disease. Antibiotics can be given orally, nebulized and intravenously. The best results will depend on the skill and experience of the team looking after the patient.
Indications for antibiotics
Oral antibiotics can be given as prophylaxis, for occasional infective exacerbations or continuously for repeated severe infections. Viral infections can produce a bacterial infective exacerbation, which will often require a course of prophylactic oral antibiotics. Patients with severe disease and persistent purulent sputum, who repeatedly relapse following a short course of antibiotics, can be maintained on long-term oral antibiotics. Those commonly used are the penicillins and more recently the macrolides.
Nebulized antibiotics may be used for patients with severe bronchiectasis whose disease is progressive and difficult to control (Currie 1997). Nebulized antibiotics can delay persistent infection with Ps. aeruginosa in patients with cystic fibrosis if instituted at the time of first colonization (Valerius et al 1991). Although there are no randomized clinical trials, this practice may be used in bronchiectasis as acquisition of Ps. aeruginosa is associated with greater morbidity (Evans et al 1996). Randomized controlled trials, using nebulized antibiotics for bronchiectatic patients chronically infected with Ps. aeruginosa, have shown a reduction in sputum density of Ps. aeruginosa (Barker et al 2000) and a lessening of disease severity (Oriols et al 1999). Tobramycin solution for inhalation (TOBI) has been used in a pilot study in patients with severe bronchiectasis (Scheinberg & Shore 2005). Although there was an improvement in respiratory symptoms, there was a significant intolerance of the medication, with 10 withdrawals from the study (n = 41). Intravenous antibiotics are used for severe disease, patients who fail to respond to oral antibiotics and those chronically infected with Ps. aeruginosa.
Other treatment measures
Influenza vaccination should be given annually to all patients with bronchiectasis unless there is a medical contraindication.
Topical medication may be indicated for chronic mucopurulent rhinosinusitis and the recommended technique for inhaled topical deposition of drugs is the head-down and forward position to encourage entry of the drops to the ethmoid and maxillary sinuses (Wilson et al 1987).
Where there is an immunoglobulin deficiency, replacement therapy should be given in an attempt to prevent further lung damage.
Surgical resection should be considered only if the bronchiectasis is localized, but there are no randomized controlled trials to compare surgical versus conservative treatment in the decision-making process (Corless & Warburton 2000). In very severe widespread bronchiectasis with respiratory failure, lung transplantation may be considered.
The inhalation of recombinant human deoxyribonuclease (rhDNase) does not appear to improve ciliary transportability, spirometry, dyspnoea or quality of life in patients with non-CF bronchiectasis (Wills et al 1996).
Inhaled steroids have been evaluated, with a trend to improving some respiratory parameters, but larger studies are needed (Kolbe & Wells 2000). A subset of bronchiectatic patients respond to bronchodilators and all patients should be tested for a response (Hassan et al 1999).
Physiotherapy management
Physiotherapy may help in the treatment of patients’ problems of excess bronchial secretions, breathlessness, reduced exercise tolerance and chest wall pain of musculoskeletal origin.
Excess bronchial secretions
It is important that the patient understands the pathology of the condition and the reasons for treatment. Clinically, effective physiotherapy should reduce the episodes of superimposed infection and may help to minimize further lung damage.
An airway clearance technique (ACT), for example the active cycle of breathing techniques (ACBT) or autogenic drainage (AD) (Chapter 5), should be introduced and self-treatment encouraged (Rosen 2006). Each patient should be assessed to determine the positions that may increase the efficiency of secretion clearance and a CT scan, if available, may facilitate this. The sitting position may be adequate for patients with minimal secretions. The horizontal position may be a more acceptable and comfortable alternative to the head-down tipped position and has been shown to be equally effective in patients who expectorate >20 g of sputum per day (Cecins et al 1999).
In patients who present with gastro-oesophageal reflux (GOR) there may be concern that the head-down tipped position will exacerbate the problem. The head-down tipped position is now rarely indicated. Chen et al (1998) reported no difference in the duration or frequency of symptoms in the various drainage positions. Other techniques that may be used to facilitate airway clearance have proven efficacy when used in gravity-assisted positions, e.g. Flutter and the active cycle of breathing techniques (Eaton et al 2007, Thompson et al 2002) and Acapella (Patterson et al 2004, 2005, 2007). These are discussed in Chapter 5. It has been suggested that a test of incremental respiratory endurance (TIRE) may be a useful method of airway clearance. In a single treatment session comparing the TIRE with ACBT, significantly more sputum was expectorated with ACBT (Patterson et al 2004). In contrast, Chatham et al (2004) demonstrated that TIRE was more effective than the ACBT. Further research is needed in this field.
Regular daily treatment is often necessary, but the frequency will vary among individuals. For many patients, treatment once a day is sufficient but the frequency should be increased during episodes of superimposed infection. Some patients find their chest is ‘dry’ at the beginning of the day. The timing of treatment should take into consideration both the time of day that the chest is most productive and the patient’s lifestyle. Adherence may be increased by agreeing a suitable home programme with the patient. Elderly or frail patients may require assistance from a relative or carer who should be carefully instructed by a physiotherapist.
It is important that physiotherapy techniques and positions for treatment are reassessed at regular intervals (Currie et al 1986). Most patients should be reassessed within 3 months of initial instruction and then at least annually. A diagnosis of bronchiectasis may be confirmed in the absence of a daily productive cough. For these patients it would seem advisable to teach an ACT to be used during acute exacerbations of pulmonary infection (British Thoracic Society Guidelines 2007). In some patients with bronchiectasis it may be the increase in ventilation to the bronchiectatic area (use of the dependent position) rather than any ‘drainage’ from the area in an uppermost position that will increase airway clearance. Individual assessment is imperative for effective management and the end-point of a treatment session must be recognized by self-assessment.
Hypertonic saline
Hypertonic saline has been shown to increase the ciliary transportability of bronchiectatic sputum, probably through its action of altering sputum rheology (Wills & Greenstone 2006). Kellet et al (2005) compared ACBT with three other treatment arms: ACBT preceded by nebulized terbutaline alone or in combination with either isotonic or 7% saline, in stable bronchiectatic patients producing less than 10 mg sputum per day. Hypertonic saline was shown to improve sputum weight, ease of expectoration, and lung function and reduce viscosity compared with the other treatment arms. Longer-term studies are required to determine the place of hypertonic saline on infection rates, quality of life and lung function.
Acute exacerbation of infection
Patients may be admitted to hospital with an acute exacerbation of pulmonary infection. The patient will probably be expectorating an increased amount of more purulent sputum, may be febrile, dehydrated and breathless. Haemoptysis is not uncommon and pleuritic chest pain may be present. The most severely affected may present with respiratory failure.
Mechanical adjuncts will be required in addition to an airway clearance technique to assist the clearance of excess bronchial secretions. A nebulized bronchodilator and/or humidification (Conway et al 1992) before treatment may help in the mobilization of tenacious secretions. Intermittent positive pressure breathing (IPPB) may help both in the clearance of secretions and in reducing the work of breathing. Patients who, many years ago, received the more radical treatment of resection of more than one lobe will probably have very poor respiratory reserve by the time they reach middle age. A superimposed infection in these patients may precipitate respiratory failure. Modified positioning, for example side lying or high side lying, combined with IPPB may be an effective form of treatment in minimizing the effort of clearing secretions. Non-invasive ventilation (Chapter 11) may be indicated in acute respiratory failure, although the outcome may be less successful in the presence of excess bronchial secretions.
Following resection of lung tissue, the anatomy of the bronchial tree may alter and the traditional positions for drainage of segments of the remaining lobes may be unsuitable or inappropriate. The physiotherapist should try various positions until the optimal ones are found.
The presence of blood streaking in the sputum is not a contraindication to physiotherapy and treatment should be continued. If there is frank haemoptysis, physiotherapy should be temporarily discontinued but resumed as soon as the sputum is only mildly blood stained to avoid retention of old blood and mucus. Before discharge from hospital, it is important that the patient is able to take the responsibility for their treatment and is confident with the positions and techniques required to continue regularly at home. If a bronchodilator has been prescribed, this should be taken before treatment and a few patients with bronchiectasis may also be prescribed nebulized antibiotic drugs, which should be inhaled after airway clearance. A breath-enhanced nebulizer or adaptive aerosol delivery (AAD) device is recommended for delivery of antibiotics (Chapter 5). If a patient is on the waiting list for lung transplantation, a preoperative rehabilitation programme should be established and postoperative treatment would be as outlined in Chapter 15.
Breathlessness
Some patients with bronchiectasis also demonstrate a degree of bronchospasm and may benefit from the inhalation of a bronchodilator before physiotherapy to clear secretions. Instruction in the use of an appropriate device for drug delivery is important.
A minority of patients with bronchiectasis complain of breathlessness. For these patients rest positions to relieve breathlessness and breathing control while walking and stair climbing should be included in the treatment programme (Chapter 5).
Reduced exercise tolerance
Exercise should be encouraged to improve general physical fitness. It will also assist the mobilization of bronchial secretions. Patients with severe bronchiectasis may benefit from a group pulmonary rehabilitation programme (Chapter 13). There is little research evaluating the place of exercise in bronchiectasis although there is a suggestion that inspiratory muscle training (IMT) may improve endurance exercise and quality of life (Bradley et al 2002). Newall et al (2005) evaluated an 8-week high-intensity pulmonary rehabilitation programme in combination with IMT or IMT sham or a control group. Both exercise groups showed significant improvements in exercise performance and inspiratory muscle strength. This was maintained at 3 months only in the IMT group. In the short term there would appear to be no advantage of including IMT in an exercise programme although it may prove beneficial in the longer term.
Evaluation of physiotherapy
Effective treatment can be recognized by a decrease in the quantity and purulence of sputum, absence of fever, improvements in spirometry, a reduction in breathlessness, an increase in exercise tolerance, increased energy levels and a reduction or absence of chest wall pain. Improvements in oxygen saturation and blood gas tensions may also be identified.
PRIMARY CILIARY DYSKINESIA
Primary ciliary dyskinesia (PCD) is an autosomal recessive disorder with an incidence of between 1 in 15 000 and 1 in 30 000 (Cole 1995) and an expected prevalence of 3000 cases in the United Kingdom. Many cases are underdiagnosed. However, there is an increasing understanding of the complex genetics of PCD (Bush & Ferkol 2006) and therefore an improvement in the diagnosis and understanding of the phenotypic features (Noone et al 2004).
PCD is characterized by abnormal structure of the cilia; normal structure of the cilia but with abnormal function; absence of the cilia. These abnormalities result in recurrent infections in the nose, ears, sinuses and lungs. Fertility may be affected, both in the female because the fallopian tubes are lined with cilia and in the male due to reduced sperm motility. In 50% of cases, PCD is associated with dextrocardia or situs inversus. Kartagener described a syndrome of bronchiectasis, sinusitis and situs inversus in 1933. Later it was recognized that there was also a ciliary abnormality and this could occur without situs inversus. Cilia defects were described first in spermatozoa and later in nasal and bronchial cilia and the term ‘immotile cilia syndrome’ was applied to this group of conditions. With the discovery of a range of cilial defects, with variation in beat frequency and ultrastructure and the recognition that not all abnormal cilia are immotile, the term primary ciliary dyskinesia was adopted (Greenstone et al 1988).
The age of presentation can vary from newborn to 51 years (Turner et al 1981). Chronic sputum production and nasal symptoms are the main presenting symptoms. Other presentations include pneumonia and rhinitis in the newborn, ‘asthma’ with a productive cough, chronic and severe secretory otitis media (with associated hearing problems), severe oesophageal reflux in the older child and problems of infertility and ectopic pregnancy in the adult. Specific investigations which may clarify the diagnosis of PCD include a nasal mucociliary clearance test such as the saccharin test (Stanley et al 1984), photometric determination of ciliary beat frequency (Rutland & Cole 1980) and electron micrographic analysis. Genetic testing should be undertaken to exclude the diagnosis of cystic fibrosis. Exhaled and nasal nitric oxide is very low in PCD (Karadag et al 1997) but increased in bronchiectasis and asthma. Although the measurement is not recommended as a diagnostic test, if levels are low in a patient with bronchiectasis then the diagnosis of PCD should be excluded.
Medical management
Early diagnosis is essential and medical treatment centres around the prevention of lung damage and bronchiectasis with aggressive use of antibiotics and daily chest physiotherapy. Intravenous treatment may be necessary for unresponsive infections and long-term nebulized antibiotics should be considered for patients colonized with Ps. aeruginosa. In childhood, careful regular assessment and monitoring of hearing should indicate the requirement for hearing aids or grommet insertion due to the build-up of fluid in the middle ear. Hearing aids are considered preferable because grommets may cause additional discharge. Hearing loss is temporary and resolves spontaneously later in childhood.
Recent studies have focused on the influence of drugs on cough clearance (Houtmeyers et al 1999, Noone et al 1999). In PCD airway clearance is dependent on cough, but an increased amount of secretion is necessary to ensure effective clearance with coughing. Aerolized uridine-5′-triphosphate has been shown to improve whole lung clearance during cough after a single dose when compared to 0.12% saline (Noone et al 1999). Further trials of this drug are required to determine the clinical significance of long-term administration. Two case reports have suggested benefit from inhalation of rhDNase in the acute situation (Desai et al 1995, ten Berge et al 1999). However, its use has not been validated in PCD in a controlled trial. Inhaled b2-agonists are frequently prescribed in PCD for their effect on bronchodilation, mucociliary transport and thinning of secretions (Rubin 1988). Regular use in asthma may be associated with increased bronchial responsiveness and decreased airway calibre. Koh et al (2000) have shown that no such adverse effects or decrease in lung function were seen in PCD over a 6-week period. Severe gastro-oesophageal reflux, which can compromise airway clearance, is a problem for some patients and requires appropriate management with a proton pump inhibitor.
Referral for assisted conception may be necessary for both males and females who are infertile or subfertile. Psychosocial support will include help with benefits, liaison with schools about infections and possible deafness and counselling may be appropriate to cope with the problem of infertility. Care centres around daily airway clearance and the control of infection by the general practitioner. Periodic review in a specialist centre by a multidisciplinary team, with expertise in respiratory disorders, is recommended.
Physiotherapy management
Daily physiotherapy is usually necessary in the child with PCD. It is important that parents are taught to recognize signs of infection early: for example, the child may be lethargic, ‘off colour’ and feel abnormally hot (pyrexia). In a study to examine cough frequency in children who were clinically stable, parental scoring equated to ambulatory monitoring. The cough frequency was shown to correlate with inflammatory markers but not with FEV1 (Zihlif et al 2005). Physiotherapy should be increased during infective episodes and parents must understand that effective treatment is not achieved by antibiotics alone.
Due to the cilial defect, secretions are most likely to collect in the dependent areas: the lower lobes and often the middle lobe and lingula. The middle lobe, which may be situated on the left side owing to situs inversus, is more commonly affected than the lingula. The goal of treatment should be to assist clearance of secretions from the dependent parts of the lungs using an effective airway clearance technique. Both children and adults should be encouraged to blow their noses regularly.
Huffing games and airway clearance devices can usually be introduced at an early age and by 8 or 9 years the child can begin to do most of the treatment themselves, gradually becoming independent. It has been suggested that the PEP mask may be a useful technique, based on the theoretical benefits of peripheral mobilization of secretions, and can be used at any age including babies (Gremmo & Guenza 1999). Some patients may require nebulized antibiotics and inhaled b2-agonists for their beneficial effect on mucociliary clearance. Beta-2 agonists should be inhaled before and antibiotics after airway clearance. Exercise, which increases bronchodilation to a greater extent than b2-agonists (Phillips et al 1996), should be encouraged from the time of diagnosis and its importance emphasized to parents and patients (Fig. 18.1). Even with grommets in place children can enjoy swimming (Pringle 1992).

Very occasionally, nasopharyngeal suction may be indicated in the infant when it is impossible to clear nasal and bronchial secretions by any other means.
Regular assessment of techniques, remotivation of the patient and support for the parents are important aspects of physiotherapy. It is probable that chronic lung damage will be minimized if physiotherapy is continued on a regular basis.
Evaluation of physiotherapy
In the young patient with PCD, effective treatment in the stable condition may be recognized by the presence of only minimal coughing on exertion. During an infective episode signs and symptoms of effective treatment include a reduction in shortness of breath, coughing, wheeze and fever if either or both have been present.
In the older patient, a constant volume of sputum is usually expectorated while stable. During an infective episode, the increased volume of expectorated sputum should lessen with effective treatment.
CYSTIC FIBROSIS
Cystic fibrosis (CF) is the most frequent cause of suppurative lung disease in Caucasian children and young adults and is characterized by chronic pulmonary disease, pancreatic insufficiency and increased concentrations of electrolytes in the sweat (HØiby & Koch 1990).
Cystic fibrosis is an autosomal recessive condition most commonly found in Caucasian populations with a carrier rate of 1 in 25 and the disease occurring in approximately 1 in 2500 live births (Dodge et al 1993). Carriers of the genetic defect show no signs of cystic fibrosis, but if both parents carry the abnormal gene each child born has a 1 in 4 chance of inheriting the condition. When the condition was first described by Anderson (1938), life expectancy was less than 2 years. Increased recognition of the disease, especially in its milder forms, and improved treatment has resulted in a median age of survival of approximately 31 years (Shale 1997a). Current survival figures from the Cystic Fibrosis Foundation (2006) report a median survival in the USA of over 35 years of age. Over the last 20 years in the Manchester Centre (United Kingdom), the number of patients living into the fourth and fifth decade of life has increased from 5% to 35%. Cohort survival graphs indicate an improvement in survival, with time, in the UK in all age groups (Dodge et al 1993). If the trend for improved survival continues, many of the patients born in 2000 now have a predicted median survival exceeding 50 years (Dodge et al 2007).
Before identification of the gene in 1989, a diagnosis of cystic fibrosis was made using the sweat test, which measures the amount of sodium in the sweat (Di Sant-Agnese & Davis 1979). The basic defect for CF lies on chromosome 7 and was identified in 1989 (Rommens et al 1989). The faulty gene in CF codes for the transmembrane conductance regulator (CFTR). The abnormality in this protein leads to changes in ion transport (McBride 1990), which produce changes in the nature of the mucus and serous secretions produced by the exocrine glands, cells of the respiratory system and digestive tract.
Ion transport in human airways is dominated by the absorption of sodium ions from the mucosal surface (Alton et al 1992), and this is associated with the movement of water into the epithelial cells. The balance between the movement of sodium and chloride probably determines the volume and composition of the airway surface liquid and may affect mucociliary clearance (Alton et al 1992).
It has proved extremely difficult to provide an accepted unifying hypothesis as to how defective CFTR function translates into the lethal pathophysiology of the lung. In particular, how it results in the aggressive suppurative lung disease so characteristic of CF and different from the indolent non-CF bronchiectasis. Two conflicting theories currently prevail. One is the airway surface liquid (ASL) tonicity hypothesis (Zabner et al 1998), which relates the pathophysiology to altered tonicity (high salt content) of the ASL layer. However, the weight of evidence now favours the ASL volume hypothesis, which suggests that a depletion of the volume of the ASL is a significant factor in the pathogenesis of CF pulmonary disease (Coakley & Boucher 2007).
The lungs are structurally normal at birth (Reid & De Haller 1967), but studies have demonstrated evidence of inflammation and infection in infants and children with CF (Birrer et al 1994, Khan et al 1995) and in asymptomatic adults with normal lung function (Konstan et al 1994). Infection stimulates further mucus secretion and a generalized obstructive, suppurative cycle becomes established. Repeated infections result in a neutrophil bronchiolitis. The neutrophils are ineffective at eliminating the micro-organisms which chronically infect the small airways. They break down, releasing numerous peptides, and in particular neutrophil elastase, which destroy lung tissue. The consequences are a destructive progressive suppurative bronchiectasis. The cycle of infection and inflammation impairs ciliary function and reduces mucus clearance.
As the suppurative bronchiectasis progresses, chronic hypoxia may lead to pulmonary hypertension. The majority of patients die from respiratory failure when they no longer respond to medical treatment or they do not receive a transplant.
Diagnosis and presentation
Newborn screening for CF is now possible and, where available, it most often uses measurement of immunoreactive trypsin (IRT) (which is abnormally high in CF) followed by DNA testing for a limited number of CFTR mutations. National newborn screening programmes for CF currently exist in New Zealand, France and Denmark, and will be available nationally in the United Kingdom by 2008. Regional or local programmes exist in parts of the United States of America, Australia and other areas of Europe. The results of a large randomized controlled trial of newborn screening for CF reported improved height, weight and head circumference in a screened group compared with a non-screened group (Farrell et al 2001) as well as higher cognitive function (Koscik et al 2004).
In the absence of newborn screening, the majority of patients continue to be diagnosed early in life with symptoms related to either the respiratory or gastrointestinal systems. Gastrointestinal abnormalities are often the earliest and most common presenting feature. The finding of echogenic bowel, during routine antenatal ultrasound, is associated with CF (although only in a minority of cases). Karyotyping and CF screening should be considered in this situation. In the neonate failure to pass meconium (meconium ileus) is the most common presenting feature, occurring in about 10–15% of cases (Park & Grand 1981). Signs of intestinal obstruction usually occur within 48 hours of birth. The infant fails to pass meconium after birth because the bowel is obstructed by sticky inspissated intestinal contents. In milder cases there may only be a delay in the passage of meconium. Blood should be taken for genotyping in infants with meconium ileus, as this condition can also occur in infants who do not have CF.
Another presenting sign in infants and young children is a voracious appetite and failure to thrive due to pancreatic insufficiency and malabsorption. Abnormalities in ion transport in the pancreas lead to inflammation and later to fibrosis of the acinar portion of the gland and to hyposecretion of the major digestive enzymes secreted by the pancreas. The presenting symptom is steatorrhoea with the passage of characteristically fatty and offensive stools. The majority of patients (85%) with CF are pancreatic insufficient (Davidson 2000). The remaining 15% usually have better nutrition and pulmonary function and a better survival prognosis. Occasionally older children present, having been managed for other respiratory conditions, for example asthma. In adulthood a late diagnosis may be made when the patient presents with infertility.
Classically the diagnosis is based on clinical findings, a high concentration of sweat chloride (>60 mmol/l) and/or identification of two disease-associated CFTR mutations. Early diagnosis is important to facilitate early access to specialist services in order to initiate appropriate treatment and to access prognostic and genetic services.
Signs and symptoms
Respiratory
The respiratory signs and symptoms of cystic fibrosis vary. The majority of older children and adults have a cough productive of sputum with varying degrees of purulence. The respiratory pathogens most commonly isolated in sputum are Pseudomonas aeruginosa (61%), Staphylococcus aureus (28.3%), Haemophilus influenzae (8.9%) and Burkholderia cepacia (3.2%) (FitzSimmons 1993). Infection with B. cepacia is often associated with accelerated pulmonary disease and a worse prognosis (Muhdi et al 1996).
Chest pain is common as the disease progresses and may be musculoskeletal or pleuritic. Breathlessness may be associated with infective exacerbations and increasing disease severity. Pneumothorax should be considered if there is an acute onset of breathlessness and pain. As breathlessness increases, appetite may fall and weight loss is common. Haemoptysis is common in adults but is usually mild, although episodes of frank haemoptysis may occur. Most patients develop finger clubbing which is associated with more severe disease.
Auscultation is often unrewarding when compared with the severity of radiological disease. Coarse inspira tory crackles are often heard. A pleural rub may be heard in association with infective exacerbations. The chest radiograph is often normal at birth but early changes include bronchial wall thickening, initially in the upper zones. As the disease progresses, hyperinflation may be noticeable with ill-defined nodular shadows, numerous ring and parallel line shadows indicating bronchial wall thickening and bronchiectasis (Chapter 2). High-resolution computed tomography (HRCT) imaging provides detailed evaluation of the lungs in CF (Chapter 2). Studies using HRCT in infants with CF have demonstrated early structural changes, even in those who have minimal symptoms (Long et al 2004).
Pulmonary function tests initially show signs of airways obstruction, but with advanced disease a restrictive pattern may be superimposed on the obstructive defect and a diffusion abnormality will also become apparent. Pulmonary function tests in infants with CF have shown early changes with diminished airway function soon after diagnosis, even in infants with no clinical history of clinical infection. These changes seem to persist during early childhood (Ranganathan et al 2001, 2004). More recently the use of multiple breath washout techniques to measure lung clearance index (LCI) (Chapter 3) have been shown to be effective and sensitive in terms of detecting early lung disease in cystic fibrosis (Aurora et al 2005a, Lum et al 2007). Pulmonary function measurements (FEV1, PaO2, PaCO2) have been shown to be predictors of mortality (Kerem et al 1992). As the disease progresses ventilation/perfusion imbalance occurs, leading to hypoxaemia and pulmonary hypertension. Carbon dioxide retention occurs in patients with severe disease.
Asthma is as common in patients with CF as it is among the general population. Many patients with CF have a positive skin test to Aspergillus fumigatus. This is often seen in the sputum of patients and can be isolated in 40–60% of patients (Chen et al 2001). Colonization of the lower airway with Aspergillus fumigatus and the complication of allergic bronchopulmonary aspergillosis (ABPA) is well recognized in patients with cystic fibrosis (Skov et al 2005) and occurs in up to 10% of patients (Mastella et al 2000). ABPA is recognized by recurrent wheezing, deteriorating chest symptoms, fleeting fluffy shadows on the chest radiograph and elevated IgE levels which are specifically raised to Aspergillus.
Some patients develop nasal polyps. These may grow rapidly, are frequently recurrent and may require surgical removal. They may be related to chronic sinus infection.
Gastrointestinal
Distal intestinal obstruction syndrome (DIOS) is obstruction of the small bowel occurring in children and adults and is similar to that seen in neonates presenting with meconium ileus. This may be related to poor adherence with pancreatic supplements. It presents as small bowel obstruction with abdominal distension and discomfort, vomiting and reduced or absent bowel signs. Diagnosis is confirmed by the classic radiographic appearances of small bowel obstruction.
Cystic fibrosis-related diabetes (CFRD) has become a significant complication as a consequence of improved survival and is a result of progressive fibrosis damaging the endocrine cells that produce insulin (Bridges & Spowart 2006). The onset of diabetes, if not detected and treated promptly, can result in a decline in the patient’s clinical condition (Lanng et al 1992). The basic defect also affects the hepatobiliary system, which can result in a biliary cirrhosis. Patients with severe disease can develop portal hypertension. The main complication is bleeding from gastric or oesophageal varices. Liver transplantation may be required.
Other
Puberty may be delayed for both male and female patients. Most women with cystic fibrosis have normal or near-normal fertility. Improving survival has resulted in an increasing number of the female population having children. Outcome of pregnancy is improved if pulmonary function is greater than 60% predicted (Edenborough et al 1995). Pregnancy has been reported to have a slight adverse effect on the health of women with CF (Gillet et al 2002). Women with CF require a greater intensity of treatment during pregnancy (McMullen et al 2006).
Most males are infertile because of developmental defects of the vas deferens, which is either absent or blocked, but they can produce sperm. Improved technology, whereby sperm can be aspirated from either the testis or epididymal sac in conjunction with intracytoplasmic sperm injection (ICSI), has resulted in CF biological fathers (Phillipson et al 2000).
Approximately one-third of adult patients with CF develop rheumatic symptoms (Bourke et al 1987). The two most common forms are an episodic and recurrent arthralgia /arthritis and hypertrophic pulmonary osteoarthropathy. They are characterized by joint pain, tenderness, swelling and limitation of movement, usually symmetrical and affecting particularly the knees, ankles and wrists (Johnson & Knox 1994). More important has been the recent recognition of the high prevalence of low bone mineral density in children and adults (Bachrach et al 1994, Bhudmkanok et al 1996, Haworth et al 1999), which leads to a high incidence of fractures. Rib fractures can result in considerable pain, sputum retention and morbidity.
Medical management
Paediatric and adult patients with CF should receive care from a specialist CF centre. Pulmonary function and nutrition, the two main prognostic indicators for survival, are better when care is delivered from paediatric and adult CF centres (Mahadeva et al l998). Models for shared care, between the CF centre and the district hospital, at the paediatric level have worked extremely well for many years but this process is not commonly practised at the adult level. Most specialist units have a system of annual review when comprehensive assessment and testing is undertaken.
Cystic fibrosis is an extremely complex disease. Care is best delivered by a multidisciplinary team comprising doctors, physiotherapists, dietitians, nurses, social workers, psychologists and other disciplines who will complement each other in their individual areas of expertise. The patient should also be closely involved in choice of care and self-care at home.
Morbidity and mortality are primarily related to chronic progressive respiratory infection. Therefore the mainstay of treatment is oral, nebulized and intravenous antibiotics.
Long-term oral anti-staphylococcal antibiotics are given in the early years to treat Staphylococcus aureus, which is often the main micro-organism causing chronic infection. A considerable advance over the last few years has been the introduction of the macrolides as a long-term treatment for cystic fibrosis. Although an antibiotic, it is probable that the modulatory anti-inflammatory properties of the drug are responsible for the improvement in clinical status and preserved pulmonary function demonstrated in well-conducted clinical trials (Equi et al 2002, Saiman et al 2003, Wolter et al 2002).
Subsequently patients become chronically infected with Ps. aeruginosa, which increases treatment requirements and morbidity. The practice of starting nebulized and oral antibiotics at time of first culture of Ps. aeruginosa has been shown to be effective in eradicating and delaying persistent infection (Valerius et al 1991).
Nebulized antibiotics (Webb & Dodd 1997) have been shown to be effective in the treatment of chronic Ps. aeruginosa infection (Mukhopadhyay et al 1996, Touw et al 1995). Antibiotics are usually inhaled twice daily and should follow airway clearance. For many years, colistin has been the standard drug used for nebulization but there have been no large randomized controlled trials to unequivocally demonstrate benefit. High-dose preservative-free tobramycin (TOBI) has been used for inhalation with demonstrated benefit in CF patients (Ramsey et al 1999). However, the high cost and occasional intolerance may preclude its use in all CF patients infected with Ps. aeruginosa.
Intravenous antibiotics are frequently used for acute infective exacerbations but opinions differ as to the regular or symptomatic use of intravenous antibiotics (Elborn et al 2000). Treatment usually needs to continue for at least 14 days and can be evaluated by monitoring respiratory function, sputum quantity, bodyweight, blood gases and blood inflammatory markers such as C-reactive protein (Hodson 1996).
Patients needing frequent or prolonged antipseudomonal treatment, and who have poor venous access, may require implantable intravenous access devices. These devices can maintain continuity of antibiotic infusions and quality of life for the patient undertaking treatment at home (Shale 1997b, Stead et al 1987).
Segregation of patients colonized with Burkholderia cepacia, from other patients with cystic fibrosis, limits the spread of the organism by social contact (Govan et al 1993, LiPuma et al 1990, Muhdi et al 1996). It is now recognized that organisms classified as B. cepacia comprise a number of distinct genomic species each known as a genomovar of the B. cepacia complex (BCC). Currently there are 10 different described genomovars in the BCC. Disease progression and survival may be influenced by the genomovar status of the CF patient (Jones et al 2004). In some units adults are segregated in outpatient clinics according to their genomovar status on the basis that some genomars are transmissible and can superinfect patients with non-transmissible genomovars (Ledson et al 1998).
Many CF units adopt a general segregation policy for outpatient clinics. This is based either according to microbiological status or on a total segregation approach (where all patients are segregated regardless of microbiological culture). Health professionals must pay particular attention to hygiene and thorough hand washing between examining patients (Cystic Fibrosis Trust 2004a).
There has been concern regarding the emergence of transmissible strains of Ps. aeruginosa in large CF centres despite the use of the correct infection control measures (Jones et al 2001, McCallum et al 2001). As a consequence, many CF clinics practice inpatient and outpatient segregation by microbiological status (Cystic Fibrosis Trust 2004b). At the Manchester Centre (United Kingdom), a 4-year prospective surveillance demonstrated ongoing transmissible Ps. aeruginosa cross-infection between inpatients despite established conventional infection control measures. As a consequence, communal areas such as the day room and kitchen were closed and the patients are now required to stay in their rooms and not mix irrespective of their individual microbiological status (Jones et al 2005).
Contamination of nebulizers is common and patients must be given instruction in the cleaning and care of nebulizer equipment. To minimize contamination, cleaning and drying of this equipment after use are essential (Hutchinson et al 1996).
Some patients benefit from the inhalation of bronchodilator drugs. Steroids may be indicated if asthma or ABPA complicates cystic fibrosis. The use and value of inhaled steroids, to treat the inflammatory component of airflow obstruction in the long-term management of cystic fibrosis, are still under review with prospective controlled trials.
As a consequence of lung infection, there are large quantities of DNA from the breakdown of inflammatory cells, e.g. neutrophils. The inhalation of rhDNase acts on the DNA in the purulent lung secretions (Range & Knox 1995). It has been shown to improve lung function (Shah et al 1996), reduce viscoelasticity of the mucus (Shah et al 1996) and decrease exacerbations of bronchopulmonary infection (Fuchs et al 1994). Occasionally alteration in voice and episodes of pharyngitis may be experienced, but these are usually minor and transient (Hodson & Shah 1995). Alternate day therapy may be as effective as daily treatment in some patients (Suri 2005).
Hypertonic saline, inhaled before physiotherapy, may also assist in clearance of secretions (Eng et al 1996, Robinson et al 1996). There is a potential logic in the use of hypertonic saline for inhalation, whereby it will restore to normal the disrupted airway surface liquid of the CF airways. Two randomized trials have shown, in a small number of CF patients, preservation of lung function and in one of the trials a reduction in infective exacerbations (Donaldson et al 2006, Elkins et al 2006a). Hypertonic saline is inexpensive, safe and there is a reasonably high level of evidence to support its use. However, it does have an unpleasant taste and adds another treatment burden to the already overloaded self-care plan of all CF patients.
There is little evidence to support the use of other mucolytic agents such as acetylcysteine (Parvolex®). Some mucolytic agents may induce bronchoconstriction and a bronchial challenge should be undertaken at the time of the first inhalation.
In CF a high energy intake is needed as a result of malabsorption and the increased metabolic requirements during infection. The dietary energy intake should exceed the normal daily recommendation to sustain and maintain adequate weight, muscle bulk and function (Poole 1995). Supplements of fat-soluble vitamins and vitamin K are usually necessary in addition to pancreatic enzymes, which should be taken with all meals and snacks (Wolfe & Collins 2007). When nasal obstruction by polyps is incomplete, a corticosteroid nasal spray may be tried. Complete obstruction is unusual and polypectomy may be indicated.
Haemoptysis will usually stop spontaneously, but if bleeding is severe and prolonged, bronchial artery embolization by an experienced operator in a specialist centre can be a life-saving procedure (Ashleigh & Webb 2007) The current use of short courses of oral or intravenous tranexamic acid for moderate haemoptysis is effective.
Pneumothorax can occur spontaneously in the older patient. Small pneumothoraces may resolve without treatment, but most pneumothoraces require the insertion of an intercostal drain. Surgical intervention is required for large non-resolving leaks. Video-assisted thorascopic surgery (VATS) may be used to avoid a thoracotomy.
Heart-lung and double lung transplantation (Chapter 15) have been successfully carried out in patients with end-stage lung disease but there is a critical shortage of donor organs. Non-invasive ventilation may be life-saving and indicated for patients developing severe respiratory failure to bridge the waiting time to transplantation (Hodson et al 1991, Madden et al 2002).
If medical treatment has failed and the patient is distressed, palliative care must be expertly employed to allow the patient to die comfortably and with dignity. It is important not to withhold such care even if the patient is listed for transplantation.
Home treatment
In many countries the emphasis on treatment is moving from hospital to home. The benefits for patients of treatment at home include less disruption to school, work and family life while avoiding the isolation from friends and family that hospitalization incurs. Increasing numbers of CF patients are receiving their intravenous antibiotics at home usually because quality of life is better, but clinical outcome is better for the patient treated in hospital and patients treated at home require close supervision (Nazer et al 2006, Thornton et al 2004).
For the newly diagnosed or newly referred patient, home visits by members of the specialist team (usually the clinical nurse specialist) provide an opportunity for advice, education and support for the patient and family, as necessary. Domiciliary physiotherapy services are sometimes available and can provide the opportunity for discussion and demonstration of physiotherapy techniques in the home, an opportunity for a more effective assessment of the necessity and appropriateness of equipment and the possibility of specialist physiotherapy during terminal care. There is also evidence of improved adherence with treatment and a reduction in the stress of coping with the disease (Rogers & Goodchild 1996).
Many patients awaiting heart-lung transplantation can be cared for at home, with a clinical nurse specialist visiting to provide assessment and to identify the need for changes in treatment to maintain optimal health status. It may be appropriate for a patient to receive either a course of intravenous antibiotics at home or to continue a course started in hospital.
The future
Cystic fibrosis is a complex disease. An enormous amount of effort is being expended to improve standards of care (de Boeck 2000), provide guidelines for antibiotic treatment (Cystic Fibrosis Trust 2002a, Doring et al 2000) and evidence, based upon controlled trials, for different aspects of treatment (Cheng et al 2000). More patients (but not enough) are being transplanted and survival figures are improving with greater experience (Vizza et al 2000). The physicians and scientists are continuously evaluating current care (Davis et al 1996) and searching for new therapies to improve quality of life and long-term survival (Rubin 1998).
Gene therapy aims to correct the basic defect by inserting the appropriate DNA or RNA to compensate for the defective gene. The gene is transferred via a ‘carrier’ or vector. To date both viral and non-viral vectors have been extensively investigated but difficulties have been experienced with both. Viral vectors such as the adenovirus can stimulate an inflammatory response and non-viral vectors such as liposomes are not as efficient (Du Bois 1995). Theoretically the transfer of sufficient normal copies of the CFTR gene, to sufficient numbers of affected cells, should result in the production of enough normal protein to reduce the clinical manifestations of cystic fibrosis (Stern & Geddes 1994). Despite several in-vitro and in-vivo studies it has been difficult to convert this theory into practice. Significant advances have, however, been made and there is considerable research in progress which may lead to effective gene therapy in the future (Boyd 2006).
Stem cell therapy aims to permanently correct the genetic defect by developing a cell line that continually produces cells to re-establish a normal epithelium. To date research in stem cell therapy for CF is in its infancy and little is known about the stem cell biology of the lung. Stem cell research is attracting interest and is a hope for future treatment (Boyd 2006).
Physiotherapy management
Advances in the medical management of cystic fibrosis have increased the expectation of survival into the fifth decade of life (Dodge et al 2007). As the science of the basic defect is translated into a greater understanding of the pathophysiology of the disease and novel complications of an ageing population emerge, the physiotherapist’s role is continually challenged. The management encompasses the treatment from birth through childhood and adolescence into adulthood and parenting. It is adapted through changing lifestyles, disease severity and the changes of the acute exacerbation and stable state of the disease. Physiotherapy requires detailed accurate assessment and treatment, tailored to the individual as lifestyle and disease severity change. In parallel with the advances in the medical management, the role of the physiotherapist has expanded from the clearance of bronchial secretions to include the assessment of exercise capacity and the prescription of safe and effective exercise programmes, assessment and education of inhalation therapy and in the later stages of the disease the use of oxygen therapy and non-invasive ventilation (NIV).
More recently the problems of musculoskeletal pain, low bone mineral density and urinary incontinence have emerged. The physiotherapist’s treatment is confounded by the many complications of this multisystem disease, e.g. diabetes, distal intestinal obstruction syndrome and arthropathy. Improved survival is also attributed to the enormous burden of self-care imposed on patients. To enhance adherence to this treatment regimen it is crucial that the physiotherapist works with the patient and their family/carers to encourage an effective but realistic treatment plan, balanced with their wishes to lead a normal life.
Infants and small children
Historically a diagnosis of CF was confirmed in babies at some time during the first year of life, often following a symptomatic presentation of respiratory infection and failure to thrive. Traditionally ‘chest physiotherapy’ was instigated twice daily as soon as the diagnosis had been confirmed and treatment comprised the use of gravity-assisted positions and chest clapping. In the absence of specific radiological signs, most physiotherapists adopted a general drainage regimen (alternate side lying and prone in a head-down tipped position and supine flat) with the addition of the sitting position for the apical segments of the upper lobes in infants, as they spend much of their time lying down. Once the child began to sit and stand, the apical segments were omitted from treatment.
In the past few years, the approach to airway clearance in babies with CF has changed considerably. Many other airway clearance techniques have been developed (Chapter 5) and some of these, such as positive expiratory pressure (PEP) applied via a facemask, assisted autogenic drainage (AD) and physical activity are now used in the infant/paediatric population. Many centres throughout the world have modified the traditional approach by omitting the use of gravity-assisted positioning in the drainage regimen. This has been to a large extent due to growing clinical concerns regarding gastro-oesophageal reflux. Gravitational effects in the tipped position theoretically lead to a lowering of intra-abdominal pressure and an increase in intrathoracic pressure. This, together with the increase in diaphragmatic activity, may enhance the competence of the oesophageal sphincter (Sindel et al 1989). Despite this theoretical assumption, infants with CF are known to have a higher incidence of gastro-oesophageal reflux (GOR) and studies have suggested that this is exacerbated by use of the head-down tipped position during airway clearance (Button et al 1997). Other groups have not reproduced these findings, albeit in slightly differing cohorts (Phillips 1996, Taylor & Threlfall 1997). Longer-term follow-up data from the subjects included in the study by Button et al (1997) suggest that GOR may result in both short- and long-term sequelae in terms of respiratory status (Button et al 2003, 2004). These results have led many centres to discontinue using the head-down tipped position. This remains a slightly contentious issue, and there are centres that continue to use gravity-assisted positioning judiciously. When GOR is suspected, it should be investigated rigorously and if confirmed the airway clearance regimen may need to be modified and anti-reflux medication should be started.
Whichever airway clearance treatment is chosen, it is usually advised that it be undertaken before feeds and for approximately 10–15 minutes. If an infant or child has specific radiological signs, or in the presence of a lower respiratory tract infection, treatment may need to be intensified in terms of frequency and duration.
Early diagnosis of CF, particularly since the introduction of neonatal screening in many parts of the world, along with early and aggressive multidisciplinary care, has led to a novel cohort of infants who are apparently free of any respiratory symptoms and are nutritionally healthy. The appropriateness of implementing a routine daily airway clearance regimen in this cohort of infants has raised much debate. Unfortunately there are no studies that have evaluated whether the routine instigation of airway clearance regardless of clinical status is beneficial. The debate continues widely on the international stage and opinion remains divided (Prasad & Main 2006).
Arguments for routine treatment are threefold. First, many feel it is essential to establish a daily routine in order to ensure adherence to therapy in the long term. Secondly, the anatomical and physiological differences in the infant respiratory system (Chapter 10) may make them more vulnerable to chest complications. Finally, there is conclusive evidence that pathophysiological changes in the lungs occur very early, before the onset of clinical signs, with evidence of early inflammation and infection (Armstrong et al 1995), altered respiratory function (Ranganathan et al 2001) and structural changes radiologically (Martinez et al 2005).
However, while the evidence that pathophysiological changes occur early in the disease process is compelling, the early picture is usually not one associated with copious secretions. The value of a daily airway clearance regimen is therefore unclear (Bush & Gotz 2006). In addition there is no doubt that a significant burden of care is imposed on families by routine treatment regimens.
There is little robust evidence with respect to best physiotherapy practice in this group of infants (Button 1999, Constantini et al 2001). The few existing studies that attempt to evaluate the efficacy of airway clearance or to compare the various airway clearance modalities have been undertaken in older populations with established disease (Desmond et al 1983), as have the majority of studies comparing the efficacy of the various techniques (Elkins et al 2006b, Main et al 2005). It is unlikely to be appropriate to extrapolate findings from these studies to a healthy infant who shows no overt signs of respiratory involvement. In the United Kingdom the proposal of a national neonatal screening programme provided a unique opportunity to undertake a randomized controlled trial of the efficacy of routine daily chest physiotherapy in screened infants with CF. Unfortunately this trial did not come to fruition but in attempting to address this important issue, the Association of Chartered Physiotherapists in Cystic Fibrosis (United Kingdom) undertook a consensus exercise based on the Delphi process (Jones & Hunter 1995) in order to provide expert opinion and guidance for the future care of these infants (Prasad et al 2008). The results of the Delphi process have resulted in guidelines which state that physiotherapists are not required to initiate routine airway clearance if, following careful assessment, the child is well and felt not to have symptoms which would respond to respiratory physiotherapy. It is important to stress that all parents and carers should be taught an appropriate airway clearance regimen, and this is practised and revised to maintain competency. Airway clearance is instigated whenever there are respiratory symptoms and an assessment tool is being developed to assist parents with assessment (Fergusson, personal communication 2007). The Delphi process has also recognized that the more recently developed airway clearance modalities may be more appropriate for these infants, rather than traditional postural drainage or modified postural drainage and percussion, and that physical activity should be greatly emphasized from the outset. The suggestion is not that treatment should be withdrawn, but that a different emphasis be placed on the management to include physical activity and a more flexible and holistic approach supported by an easily accessible specialist physiotherapy service.
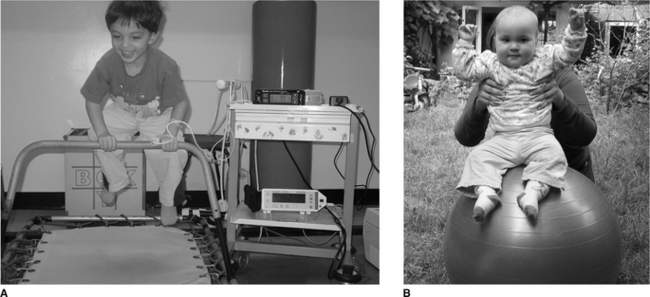
Even at a young age treatment should be fun. The young child can be bounced up and down on the parent’s knees, exercises such as ‘wheelbarrows’, jumping on a mini-trampoline (Fig. 18.2A) or the use of a gym ball (Fig 18.2B). Laughing often also stimulates coughing. From the age of 2 years the child can be encouraged to actively participate in breathing techniques in the form of play and other airway clearance modalities (Chapter 5) can be introduced as the child grows. PEP can be made more enjoyable if administrated in the form of bubble PEP (Chapter 5). From as early an age as possible children should be encouraged to play a more active role in their treatment. With increased cooperation, the child can be introduced to various airway clearance techniques and become independent with treatment (Box 18.1).
Box 18.1 Airway clearance techniques
Active cycle of breathing techniques (ACBT)
Modified autogenic drainage (M AD)
High-frequency chest wall oscillation (HFCWO)
Intrapulmonary percussive ventilation (IPV)
Oscillating positive expiratory pressure:
Infants and small children swallow their bronchial secretions, but as soon as possible expectoration should be encouraged. Nasopharyngeal suction should only be used in babies if it is essential to obtain a sputum specimen or if the infant is distressed by secretions. Learning to blow the nose is also important, to keep the upper airways clear.
Airway clearance
The removal of bronchial secretions remains the mainstay of physiotherapy management as bronchial infection and respiratory failure continue to be the major causes of morbidity and mortality.
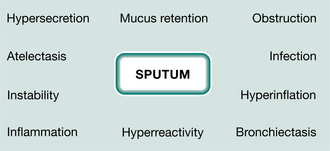
Infected bronchial secretions are responsible for many complications in the airways and lung tissue (Fig. 18.3). Obstruction occurs initially in the small airways, with repeated infections and hypersecretion resulting in damage to the airway wall, central airway instability and hyperreactivity. Infected secretions in cystic fibrosis are dehydrated, hyperadhesive and hyperviscoelastic. Studies of airway clearance techniques have attempted to identify characteristics to address some of the problems of the airway and secretions. The presenting pathological problem should be considered when choosing an airway clearance technique (Lapin 2000).
The physiological principles of airway clearance are discussed in Chapter 5. In order to achieve effective treatment, secretions should be mobilized and removed without causing an increase in airway obstruction or fatigue. The evidence-based airway clearance techniques (see Box 18.1) all aim to enhance airflow, increase lung volume and may alter the rheological properties of mucus. In people with CF who have bron chial wall instability, expiratory airflow is likely to be reduced when intrathoracic pressures are high (Zach et al 1985). Huffing generates lower intrathoracic pressure than coughing (Langlands 1967) and it has been suggested that it is as effective as coughing for mucociliary clearance (Hasani et al 1994). By altering the intrathoracic pressure and the lung volume at which the expiratory manoeuvre is performed, the physiotherapist can tailor the point of compression to the area of obstruction without causing airway collapse. Autogenic drainage prevents airway collapse by maximizing airflow at different lung volumes and avoiding high-pressure peaks (Schöni 1989).
The changing clinical picture of people with CF, with improved clinical status and a generally more active lifestyle, together with an increased understanding of the pathophysiological concepts of airway clearance techniques, have obviated the emphasis on gravity-assisted positioning for airway clearance. Research has shown that ventilation to the dependent area of the lung may be more effective than gravity-assisted ‘drainage’ from the uppermost lung (Lannefors & Wollmer 1992). Elkins et al (2005a) have shown that huffing and coughing are compromised in the ‘head-down tilt’ and side-lying positions.
Currently there is a lack of robust evidence to support the long-term benefit of airway clearance. Trials are confounded by small numbers, inconsistency of techniques and no control population (Prasad & Main 1998). To date the majority of trials have been short term and undertaken mainly in adults. A meta-analysis of chest physiotherapy suggested that airway clearance produced significantly greater sputum expectoration than no treatment and the addition of exercise further improved lung function (Thomas et al 1995). However, a systematic review was not able to demonstrate a benefit for treatment compared with no treatment, although short-term studies indicate that there may be deterioration in lung function during periods without treatment in those with established disease (van der Schans et al 2000).
Systematic reviews have not identified any single airway clearance technique as being superior (Elkins et al 2006b, Main et al 2005) and longer-term RCTs support these findings (Accurso et al 2004, Pryor et al 2006a). However, patient preference is likely to be an important factor in terms of adherence. Constant review of techniques is essential to determine the most effective regimen to meet changing lifestyles and disease progression during the clinically stable and acute state.
Exercise offers an important contribution to sputum expectoration (Baldwin et al 1994, Sahl et al 1989), but in the majority of patients it should be complementary and not exclusive (Bilton et al 1992). Patients perceive exercise differently to other forms of treatment (Abbott et al 1996) and some prefer this method of airway clearance. It is important for the physiotherapist to be sensitive to the patient’s beliefs (Carr et al 1996), but encourage formal airway clearance during an acute exacerbation when the patient is unable to exercise at his normal level.
The frequency and duration of treatment will vary. When secretions are minimal, treatment once a day may be sufficient but additionally some form of exercise should be encouraged. Many patients will require treatment two or three times a day, but the programme should be realistic and allow for other normal activities. Some techniques may be more time consuming to perform, difficult to learn and may be position dependent. Others involve equipment that requires meticulous cleaning. The choice of technique should be individualized to suit the patient’s age, lifestyle, preference and disease severity.
Maintenance / increase in exercise tolerance
The value of exercise in the management of CF is now well established. Short-term studies of exercise training programmes in cystic fibrosis have been shown to have considerable therapeutic benefit and the majority of patients wish to include exercise in their routine self-care (Webb & Dodd 2000). Studies have shown improved exercise tolerance (Andreasson et al 1987, Edlund et al 1986, Freeman et al 1993), ventilatory muscle endurance (Keens et al 1977), cardiorespiratory fitness (Orenstein et al 1981), muscle bulk and body image (Strauss et al 1987), decreased breathlessness (O’Neill et al 1987) and improved quality of life (de Jong et al 1997). Two randomized controlled trials of home exercise programmes have demonstrated the long-term value of exercise (Moorcroft et al 2004, Schneiderman-Walker et al 2000). Early studies demonstrated that patients with mild to moderate disease (FEV1 = 55% predicted) could exercise to the same level as their peers, but those with more severe disease (FEV1 < 55% predicted) would require individualized recommendations and supervised exercise programmes (Cropp et al 1982). Everyone can exercise and no patient should be excluded because of disease severity (Webb & Dodd 2000). The benefits of exercise should be introduced and emphasized from a very early age.
Assessment of exercise capacity
The patient’s baseline exercise capacity should be assessed, when the patient is clinically stable, to determine their level of fitness and limitations. The results will give guidance for effective and safe exercise recommendations. The test will provide a baseline measure for further testing to monitor improvement or change in any values and evaluate an intervention. Assessment should consider the choice of protocol, the type of test and the measurements required. The choice of protocol (Table 18.2) depends on the information required, the facilities available and the patient’s clinical condition. It may be desirable to determine a functional level of exercise in preference to peak performance (Jones 1988).
Table 18.2 Types of exercise test
| Endurance exercise | Progressive maximal Sub maximal |
| Peak power output | Wingate protocol |
| Strength |
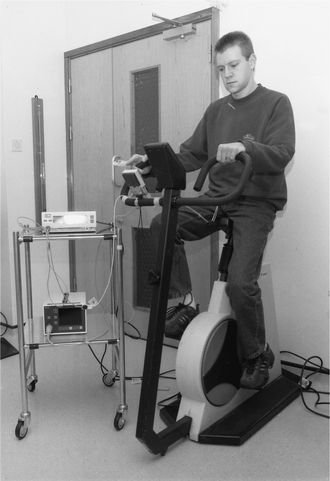
Space may be limited and a cycle ergometer may therefore be more appropriate than the modified shuttle test (Bradley et al 1999). The patient may be too breathless to perform a maximal test and some tests may be too difficult for children to perform. The step test may be a validated alternative to measure functional exercise capacity (Balfour-Lynn et al 1998). The measurements and equipment required are outlined in Table 18.3. The standard measures of work capacity (distance walked, wattage), pulse, oxygen saturation and a subjective measure of breathlessness and muscle fatigue are sufficient for routine assessment (Fig. 18.4). More sophisticated measures give additional information but are not necessary for routine use. Safety precautions during testing should include personnel trained in resuscitation; oxygen and appropriate drugs for resuscitation should be immediately available in the exercise area. From the results of the test, the physiotherapist can recommend a level of exercise to provide an appropriate training effect that is safe. It is also useful to establish the pattern of habitual activity. Some patients are unwilling to participate in formal exercise programmes, but are happy to increase everyday activity.
Table 18.3 Measurements and equipment required for assessing exercise capacity
| Measurements | Equipment |
|---|---|
| Peak work capacity | |
| Peak heart rate | |
| Oxygen saturation | • Pulse oximeter |
| Spirometry (pre- and post-exercise) | • Spirometer |
| Perceived breathlessness | |
| Perceived muscular fatigue | • Borg or VAS scores |
| Respiratory rate | • Count or ‘on-line’ |
| • On-line system | |
| Blood lactate | • Lactate analyser or blood to laboratory |
| PaO2 and PaCO2 | • Arterial line |
VAS, visual analogue scale;  E, minute ventilation; VT, tidal volume; TiTOT, ratio of time spent in inspiration to total respiratory time; RR, respiratory rate; CO2, carbon dioxide; PaO2, partial pressure of oxygen in arterial blood; PaCO2, partial pressure of carbon dioxide in arterial blood
E, minute ventilation; VT, tidal volume; TiTOT, ratio of time spent in inspiration to total respiratory time; RR, respiratory rate; CO2, carbon dioxide; PaO2, partial pressure of oxygen in arterial blood; PaCO2, partial pressure of carbon dioxide in arterial blood
Exercise programmes
Exercise is limited by the symptoms of breathlessness or muscle fatigue. The aim of an exercise programme is to improve exercise performance, make a given level of exercise more comfortable and increase the activities of daily living. It is important to establish the goals of an exercise programme for the individual patient, which may be different for carer and patient (Table 18.4). Exercise programmes must be tailored to the individual, based on disease severity, level of fitness and patient preference.
Table 18.4 The differing aims of exercise programmes for the patient and carer
| Aims of carer | Aims of patient |
|---|---|
Types of exercise
An exercise progamme should combine endurance and strength-training exercises for upper and lower body. Attention has also been paid to the value of anaerobic exercise and strength training, especially in children whose natural activity patterns are characterized by short bursts of vigorous physical activity (Klijn et al 2004, Orenstein et al 2004).
Endurance exercise aims to improve the capacity to endure more exercise without discomfort, e.g. swimming, running, cycling, skipping, aerobic classes, step aerobics, trampolines (Edlund et al 1986, Orenstein et al 1981, Sahl et al 1989) (Fig. 18.5). Strength training aims to increase muscle mass and strength, e.g. weights and sprint training (Strauss et al 1987) (Fig. 18.6). Interval training may be useful for those patients unable to sustain long periods of exercise. Short bursts of exercise at higher rates will enhance a training response. It may be of benefit for those patients with prolonged periods of desaturation. The evidence for inspiratory muscle training (IMT) has previously been conflicting (Asher et al 1982, Sawyer & Clayton 1993) and to date has shown no advantage over general upper body muscle training (Keens et al 1977). Two randomized controlled trials of IMT in adults and children have shown a positive outcome on exercise performance and quality of life (Albini et al 2004, Enright et al 2004) There are no studies to date evaluating the benefits of a lifestyle change. Parents of children and adolescents with CF have been reported to be less positive about the benefits of exercise than the parents of healthy children (Boas et al 1999). It is essential to establish from the time of diagnosis the importance of the contribution that exercise makes to a healthy lifestyle and to encourage participation of the whole family. There is a decline in physical activity in late adolescence (Britto et al 2000). Careful consideration and encouragement should be given to this age group at this time. Contact by the physiotherapist with local gym and sports facilities may offer reduced fees for exercise sessions.
Intensity, duration and frequency
Endurance exercise.
An effective exercise programme should make reasonable demands on the patient’s physical capacity and be progressive. The intensity can be derived from the results of the exercise test. Various recommendations have been suggested:
50% of peak work capacity is below the anaerobic threshold for most patients and represents a functional level of activity (Marcotte et al 1986) 50–60% peak oxygen consumption (V. Opeak) is necessary to improve physical fitness (Astrand & Rodahl 1977) and the patient’s capability is related to the percentage of their individual V.O2peak 70–85% of the measured maximum heart rate is sufficient to achieve a training effect (Orenstein et al 1981) ‘breathlessness without distress’ for those patients with severe disease who are limited by ventilatory mechanics (Godfrey & Mearns 1971).Exercise should begin at the chosen intensity for a period of time sufficient to cause breathlessness or muscle fatigue without undue stress and progress to 20–30 minutes 3–4 days a week. Progression for the patient who is limited by breathlessness will be at a slower rate than the unfit patient who is limited by muscular fatigue.
Strength training.
This should begin with a weight that can be lifted comfortably 10–15 times and progressed by increasing the repetitions to 20–30 and then by increasing the weight. The intensity should be sufficient to leave the patient ‘pleasantly tired without soreness’. The duration and frequency can progress to 15–30 minutes on alternate days. It is important to ensure correct positioning, correct technique and coordination of breathing for each exercise. A warm-up, stretching exercises and cool-down should be incorporated into each session to avoid injury. A more aerobic programme would include low weights and high repetitions whereas high weights and low repetitions constitute an anaerobic programme.
Strength training in children should be approached with care. Growing bones are sensitive to repetitive loading and the epiphysial plate is susceptible to injury before full growth is complete. Overstrenuous resistance training is as dangerous for children as it is for adults and joints should not be subjected to repetitive stress. Heavy weights and high resistance eccentric exercises should be avoided and machines are generally safer than free weights. To minimize the risk, a resistance should be selected which can be lifted 10–15 times without fatigue (or 8–12 repetitions in children). As with adults, exercises should be carefully planned and well performed in a correct position.
Precautions
There are no absolute contraindications but exercise should cease temporarily for the following medical problems: abdominal obstruction, an acute bronchopulmonary exacerbation associated with fever, transient arthralgia and arthritis, pneumothorax, persistent haemoptysis and surgery including a Caesarean section. Exercise-induced bronchoconstriction is rare in CF, but can usually be controlled with pre-exercise bronchodilators. Patients undertaking exercise in hot climates should be well hydrated and advised to supplement their salt intake (Bar-Or et al 1992). Exercise for the diabetic patient should be encouraged (see section on CF related diabetes). Certain sporting activities carry a medical risk (Webb & Dodd 1999). Contact sports, bungee jumping and parachute jumping are not advised for those patients with diagnosed low bone mineral density, portal hypertension and significant enlargement of the spleen and liver. Scuba diving could be hazardous for patients with air trapping and sinus disease. In the hypoxic patient, exercise at altitude poses a potential risk (Speechly-Dick et al 1992). Careful advice should be given to patients contemplating skiing and any strenuous/high-intensity aerobic and anaerobic exercise at altitude.
Exercising the patient with advancing disease
There is no evidence that carefully tailored and supervised exercise is harmful in these patients and they should not be excluded from a training programme. For patients with severe disease and those awaiting transplantation, deconditioning occurs rapidly. Maintaining mobility is crucial and strength and endurance exercises should be encouraged (Webb et al 1996). A maximal exercise test will define the limits of breathlessness and muscle fatigue. Exercise programmes should then be planned with these in mind. Positions for breathing control (Chapter 5) should be introduced to alleviate exertion breathlessness and increase mobility.
Studies have reported the benefits of oxygen supplementation for patients with severe disease (Heijerman et al 1992, Marcus et al 1992). Oxygen may be required to ease the symptom of breathlessness and increase exercise performance. Many patients are reluctant to use oxygen for activity, especially outdoors. Considerable benefits are achieved using oxygen before and after exercise for recovery. Patients can use small oxygen cylinders discreetly to enable them to travel and socialize in the community. During exercise and periods of re-covery, inspiratory flow rates increase, so the oxygen prescription and delivery device should be adjusted to relieve exercise-induced breathlessness. Patients mouth breathe during exercise and periods of breathlessness and a fixed concentration mask at low concentration and high flows may provide greater relief than nasal cannulae (Dodd et al 1998). The oxygen flow should be titrated to a level of comfort which provides an arterial oxygen saturation >90% (Heijerman et al 1992). For patients who are dependent on non-invasive ventilation, exercise should also be performed using the equipment (Webb et al 1996). This can take the form of weight training in sitting and step-ups in standing or walking on a treadmill (Fig. 18.7). Careful assessment with attention to respiratory rate, perceived breathlessness and exertion can provide a comfortable level of exercise without undue breathlessness, with or without oxygen to maintain mobility and quality of life.

Figure 18.7 Exercising with non-invasive ventilation.
(Reproduced with permission from Webb et al 1996.)
Inhalation therapy
Patients are prescribed an ever-increasing number of medications delivered by inhalation. Delivery is potentially maximized by matching the patient’s inspiratory flow rate and drug characteristics to the delivery device. Inhalers are more convenient and less time consuming than nebulizers, but some drugs can be delivered only by the nebulized route.
Bronchodilators
Bronchodilators are prescribed to relieve airway obstruction. Careful evaluation by reversibility testing and subjective benefit will determine the response and the merits of the inhaled or nebulized route for the individual patient. Beta-adrenergic drugs increase cilial action, improve mucociliary clearance (Wood et al 1975) and may be of benefit before chest clearance (Kuhn & Nahata 1985). During an acute exacerbation, when secretions are excessive and obstruction is increased, the nebulized route may be more effective (Conway & Watson 1997). Inhalers are available in a range of doses. It is preferable and less time consuming for the patient to be prescribed a device that will deliver a higher dose of medication than to increase the number of puffs of a lower dose.
The value of long-acting b2-agonists has been described (Bargon et al 1997) but the place of inhaled corticosteroids is questionable (Balfour-Lynn et al 1997, 2000, Bisgaard et al 1997). Withdrawing steroids in selective patients would however appear to be safe and not affect respiratory exacerbation rates (Balfour-Lynn et al 2006). CF patients often show a dip in PEFR and report symptoms of tightness in the early evening. One can speculate that this is because drugs are metabolized faster in patients with CF so that the benefits of long-acting drugs are shortened. It is important to select a device that is both acceptable to the individual patient and matches their inspiratory flow rate. Generally CF patients have high inspiratory flow rates. There are various teaching devices to assess patient technique and suitability for the required flow rate of the device, but careful consideration must be given to the risk of cross-infection in this patient group. Some commercially prescribed devices require specific inspiratory flow rates for effective deposition. The Accuhaler®(GlaxoSmith-Kline) is a dry powder inhaler, which is less dependent on inspiratory flow (Hill & Slater 1998). It is less confusing for the patient if the same device is prescribed for all medications. It is sometimes possible to combine two types of drug in one preparation, e.g. Seretide® (GlaxoSmithKline) combining a corticosteroid and long-acting bronchodilator. The physiotherapist should always be mindful of the treatment burden on CF patients.
Mucolytic agents
Inhalation of hypertonic saline by an ultrasonic nebulizer has been shown to improve lung function and perceived effectiveness of chest physiotherapy over a 2-week period (Eng et al 1996) and to improve mucociliary clearance after one inhalation (Robinson et al 1996). In both of these studies the positive effects were significantly increased when compared with 0.9% saline. Hypertonic saline may induce bronchoconstriction (Rodwell & Anderson 1996). A test dose should be given, with recordings of PEFR or FEV1 before and 5 minutes after inhalation, to identify any increase in airflow obstruction. In a large controlled long-term study of twice daily 7% saline, respiratory exacerbations were reduced and the treatment was considered to be safe if preceded by a bronchodilator (Elkins et al 2006a).
The optimal drug effect of rhDNase is different for each individual and can vary from 30 minutes to several hours. It is recommended initially that inhalation should be performed before early evening to avoid coughing during sleeping hours and 30 minutes to 2 hours before chest clearance (Conway & Watson 1997). Some patients who have difficulty clearing sputum in the morning gain considerable benefit from inhalation of rhDNase at night, without coughing during sleeping hours. Treatment regimens should therefore be individualized and frequently assessed as the disease changes. When the optimum time has been determined, inhalation in relation to chest clearance can be recommended. The effect on lung function has been shown to be equally as effective with alternate day dosing as daily (Suri et al 2002). This may decrease the treatment burden and cost, but the effect on sputum viscosity should be carefully monitored. rhDNase should not be mixed with other drugs as it requires isotonic conditions and a neutral pH for maximal activity. If nebulized antibiotics or inhaled steroids are part of the treatment regimen, the pH of these solutions is acidic and may denature the protein (Ramsey & Dorkin 1994). At least 30 minutes should separate antibiotic and rhDNase inhalation (Conway & Watson 1997). A sidestream nebulizer (Respironics) has been recommended for inhalation (Shah et al 1997). The nebulizer chamber used for rhDNase should not be used for any other drug. rhDNase should be stored at 0–4°C and should be removed from the refrigerator and brought to room temperature (approximately 15 minutes) before inhalation to avoid bronchoconstriction. Other mucolytic agents, for example acetylcysteine (Parvolex®), are also said to reduce mucus viscosity, but have been less frequently used since the introduction of rhDNase and hypertonic saline.
Antibiotics
Aerosolized antibiotics should be inhaled following airway clearance. Studies have shown that bronchoconstriction may be induced by some preparations of inhaled antibiotics. (Chua et al 1990, Cunningham et al 2001, Maddison et al 1994, Nikolaizik et al 1996). A test dose should be performed when the patient is clinically stable, to detect any increase in airflow obstruction. Spirometry should be performed before, immediately and up to 30 minutes following inhalation (Webb & Dodd 1997). Bronchoconstriction may be prevented by altering the tonicity of the solution to iso- or hypotonic (Dodd et al 1997). The long-term effects of repeated bronchoconstriction are unknown and it is now recommended that the inhalation of antibiotics should be preceded by an inhaled bronchodilator (Chua et al 1990, Webb & Dodd 1997).
The details of inhaling several drugs can be complex. It is crucial that the patient is given verbal and written instructions of how to use (including the cleaning and maintenance of any equipment), when to use and the order of use in relation to airway clearance and other drugs. This complexity may be simplified by the admixture of inhaled solutions /suspensions (Wolfgang et al 2006). While the physico-chemical compatibility is known, the aerodynamic characteristics of a mixture require further study.
Intelligent nebulizers
The development of a new generation of nebulizer delivery devices utilizing vibrating mesh technology e.g. eFlow® rapid (PARI), I-nebTM AAD® System (Respironics UK), has revolutionized the efficiency and efficacy of drug delivery, deposition and patient acceptability of this burdensome treatment. Treatment times are shorter with audible feedback on completion of inhalation. The devices are small, silent and battery driven. Incorporated within this technology in some devices (e.g. I-nebTM AAD®) is an adaptive aerosol delivery (AAD) and ‘controlled inhalations’. These devices adapt to the individual’s breathing pattern and deliver a preset dose during a proportion of the inhalation phase only. A precise dose is always delivered irrespective of the breathing pattern and environmental contamination is minimized. A controlled inhalation by the device restricts the patient to a slow deep inhalation to maximize peripheral airway deposition (Mullinger et al 2004).
Acute bronchopulmonary infection
Increased cough and sputum production with a fall in spirometry, decreased exercise tolerance, weight loss, lack of energy and increased breathlessness are common signs and symptoms of an acute exacerbation. Fever and chest pain may also be present. It is likely that during an acute infection, the duration and frequency of airway clearance will need to be increased and, if necessary, assistance given with manual techniques. Periods of breathing control may need to be lengthened to avoid fatigue and the treatment should be discontinued before the patient feels exhausted. Positions for treatment may need to be modified to reduce breathlessness. During periods of rest the positions of high side lying and forward lean sitting may facilitate contraction of the diaphragm, by altering the length-tension status (Sharp et al 1980), and ease the work of breathing. There are such wide variations in pathology, signs and symptoms that for the inexperienced physiotherapist it is very difficult to know when to discontinue a treatment session. As a guide, a treatment session may range from about 15 to 45 minutes. This may include the use of devices: for example, non-invasive ventilation and intermittent positive pressure breathing.
Airway obstruction should be minimized before instigating airway clearance. Regular nebulized bronchodilators may be recommended (Conway & Watson 1997) and benefits have been reported following the use of intravenous terbutaline (Finnegan et al 1992) or aminophylline (Hodson 2000).
Mucolytic agents such as hypertonic saline and rhDNase have been shown to be effective mucolytic agents (Fuchs et al 1994) as discussed above. Although rhDNase is recognized to be the treatment of choice (Suri et al 2002), there is individual variability and both drugs are worthy of consideration. There is little evidence that inhaling bland aerosols, e.g. normal saline as a mucolytic is effective in improving the rheological properties of mucus, increasing ciliary clearance or increasing mucus transport in CF (Wanner & Rao 1980).
Intermittent positive pressure breathing (IPPB) or non-invasive ventilation (NIV) may be indicated for the tiring breathless patient who is having difficulty clearing secretions because of compromised ventilatory mechanics. Short-term trials of NIV in adults and children have shown improved oxygen saturation and muscle pressure during airway clearance, compared with the active cycle of breathing techniques (ACBT) and the forced expiration technique (FET) alone. There was no difference in lung function or sputum expectoration, but NIV was preferred by the patients (Fauroux et al 1999, Holland et al 2003). It is important to note, however, that in both studies supplemental oxygen was not used during airway clearance. Careful consideration must be given in patients who have had a previous pneumothorax.
Supplemental oxygen, during airway clearance, is advised if the PaO2 is below 8.0 kPa (60 mmHg) or SpO2 is below 92%. Theoretically, improving hypoxia should decrease respiratory rate and improve tidal volume, thus promoting a more effective treatment. Airway clearance techniques may be better tolerated and breathlessness reduced with the use of supplemental oxygen. The high total gas flows of low-concentration Venturi masks may be more acceptable, and provide greater relief than nasal cannulae for recovery from coughing (Dodd et al 1998).
Oxygen therapy for respiratory failure
CF patients have a supra-normal drive to breathe, with an increased ventilatory response to increasing hypoxia resulting in a lower than usual PaCO2. Inspiratory time is increased, which further increases the work of breathing. Patients are therefore unlikely to chronically retain carbon dioxide, but at a PaO2 below 50 mmHg, ventilation decreases and respiratory failure easily ensues (Bureau et al 1981) (presumably due to muscle fatigue in the presence of an increasing respiratory drive). Patients with CF have high inspiratory flow rates and the normal recommended flow rates of fixed-concentration masks may be insufficient to meet that demand. It is important to increase the normal recommended flow rate to meet the peak inspiratory flow rate, i.e. a 24% mask may require settings of 2–4 litres or more and a 28% mask 4–6 litres or more. Failure to increase the flow rate will result in a decreased oxygen concentration and increased work of breathing. For the severely breathless patient requiring >28% concentration, oxygen delivery by a high-flow generator at low concentrations of oxygen (e.g. 32%) may be necessary (Dodd et al 1998).
CF patients with severe disease may hypoventilate at night, leading to a significant rise in PaCO2 (Spier et al 1984). The ventilatory response to hypercapnia is blunted in CF and although the long-term consequences of increasing hypercapnia are unclear, it is possible that ventilatory drive and CO2 sensitivity may be blunted (Gozal 1997). A fixed-concentration mask should be considered for the safe delivery of oxygen at night-time, but NIV may be required for those patients with increasing levels of PaCO2 in response to oxygen. CF patients may not be hypercapnic during the day, but may retain carbon dioxide at night and may therefore respond differently to supplemental oxygen administered at night than during the day. An accurate prescription will be necessary for night-time, exercise and rest with details of delivery device, concentration and flow rate.
Non-invasive ventilation
Non-invasive ventilation (NIV) has reported benefits for patients who are listed for transplantation (Hill et al 1998, Hodson et al 1991) and for the stable hypercapnic patient at home (Piper et al 1992). The physiotherapist may be involved with introducing the patient to the ventilator and adjusting the settings to meet the patient’s comfort and give adequate oxygenation. For some patients NIV may be continued during airway clearance with appropriate adjustment to the settings, but for others airway clearance alone or in conjunction with IPPB is just as effective. There is evidence, from short-term studies, for the use of NIV in airway clearance but the long-term effect is unknown (Bradley et al 2006). The development of a pneumothorax in patients who are dependent on NIV causes a medical management dilemma. If a patent intercostal tube is in situ, NIV can be delivered safely with a pressure preset ventilator. Each patient will require supervised and individualized management with the risks of mechanical ventilation being balanced against the potential benefits (Haworth et al 2000).
Humidification of oxygen
It is recommended that oxygen should be humidified in CF, due to the basic defect in the airway. It is suggested that water vapour humidity is preferable to nebulized humidity for maintenance of PaO2 (Kuo et al 1991). Oxygen delivered by nasal cannulae can be humidified with a bubble through water vapour humidifier to a maximum flow of 6 litres. A heated humidifier (e.g. Aerodyne, Kendall, Basingstoke, Hampshire; Fisher & Paykel, Maidenhead, Berkshire) is necessary to humidify the large volumes of gas delivered by 24% and 28% concentration masks and a high-flow generator because the time interval for moisture transfer decreases as flow rate increases above 20 l/min.
The large volumes of air delivered by NIV may cause drying of secretions and require humidification by a heated water vapour source. This may increase the problem of breakdown of the skin on the bridge of the nose, and in those patients humidification may have to be reserved for an acute exacerbation. In the terminal stage of the disease, when secretions are very tenacious, it may be necessary to humidify the oxygen delivered to the nasal mask with a bubble-through humidifier.
Domiciliary oxygen
Transferring oxygen into the community can be a challenge (Dodd et al 1998). Oxygen concentrators are usually prescribed for night-time use. The driving pressure is much lower than that of a cylinder and although adequate for use with nasal cannulae and NIV, there may be difficulties with the use of Venturi masks. Cylinders and 24% or 28% Venturi masks are recommended for relief of breathlessness. Liquid oxygen is the ideal system for CF patients and allows mobility outside the home. Careful assessment is required for overnight, ambulatory and short-burst oxygen to determine the required flow rate, concentration and hours of use. In the United Kingdom, explicit details are given to the contractors for domiciliary oxygen to enable them to work with the patient to decide which technology would best suit their therapeutic need (Department of Health 2004).
Complications of cystic fibrosis
Advanced cystic fibrosis
The late stages of the disease are characterized by repeated exacerbations, often with continuous intravenous antibiotics and increased hospitalization, respiratory failure and reduced mobility. It is essential to maximize the reversal of airway obstruction with airway clearance techniques, nebulized and intravenous bronchodilators and steroids. Sputum viscosity is reduced with rhDNase and intravenous fluids, and expectoration facilitated with hypertonic saline.
Shortness of breath may be reduced with the use of humidified oxygen, careful positioning and relaxation techniques. Type I respiratory failure requires meticulous evaluation at rest and during mobility for the requirements of oxygen. Hypercapnic respiratory failure may be reversed with NIV. Mobility is crucial for patients who are listed for transplantation and to maintain quality of life. Some patients have difficulty coming to terms with their loss of independence and input from a clinical psychologist experienced in the treatment of cystic fibrosis may prove beneficial.
Despite optimal medical and physiotherapeutic care, the terminal stage of the disease will ensue. It is inappropriate to withdraw support in the terminal stages even though physiotherapy may no longer be effective. Assistance can be given with positioning breathless patients in high side lying or sitting leaning forward, to make them as comfortable as possible. Occasionally IPPB can be used to assist the clearance of secretions from the upper airways, but care must be taken to use it only as a part of physiotherapy treatment and not as a form of pseudoventilation. Nasotracheal suction is not indicated as it would serve no useful purpose at this stage. Morphine or one of its derivatives will help to relieve anxiety and breathlessness.
Allergic bronchopulmonary aspergillosis
Allergic bronchopulmonary aspergillosis (ABPA) leads to airway narrowing, gas trapping and small airways disease. Tenacious mucus plugs, often brown in colour, and wheezing are common presenting symptoms of ABPA. Airway obstruction is increased and there is often a marked reduction in lung function (Kraemer et al 2006). Medical management includes oral steroids and antifungal agents (e.g. itraconazole or voriconazole). Mucus plugging can lead to lobar or segmental collapse. Bronchodilators may be helpful and airway clearance techniques should be adapted to minimize airway obstruction. IPPB may be indicated. Exercise tolerance may be reduced (Simmonds et al 1990) and programmes may need to be modified.
Arthropathy and joint pain
CF-related arthropathy can present as hypertrophic pulmonary osteoarthropathy (HPOA) or periodic arthritis (Koch & Lanng 2000). HPOA is related to pulmonary disease and presents most frequently in adults. Episodes of transient arthritis appear to be unrelated to pulmonary disease. HPOA is characterized by pain, swelling and warmth of the involved area and occasionally small joint effusions. Arthritis can be associated with fever and vasculitis. Active physiotherapy is not generally indicated. Joints should be rested and gentle mobilization encouraged, within the limits of the symptoms. The medical management for a pulmonary exacerbation may lead to a resolution of symptoms (Rush et al 1986) and CF arthritis usually resolves spontaneously (Turner et al 1997).
Cystic fibrosis-related diabetes
The specific changes in glucose metabolism in CF are well described (Bridges & Spowart 2006, Koch & Lanng 2000). The incidence increases with age and it is suggested that survival is reduced in this group. Hyperglycaemia causes polyuria, which leads to dehydration and a possible increase in sputum viscosity. Expectoration is difficult and lung function declines. The physiotherapist should be alert to the results of blood glucose monitoring (BM) in the analysis of chest symptoms and formulation of a treatment plan. It is essential that the medical management includes control of blood sugar and rehydration with intravenous fluids. When diabetes is difficult to control, attention should be directed to the application of airway clearance techniques which reduce viscosity and the consideration of rhDNase.
Insulin requirements will change with exercise. Exercise can sometimes improve overall blood glucose control by reducing insulin requirements. When diabetes is well controlled, exercise may induce hypoglycaemia either during exercise or up to 24–36 hours after exercise (Cystic Fibrosis Trust 2004c). The balance of insulin requirement and carbohydrate intake should be discussed with the CF dietitian and physician and individualized for both normal activity and exercise programmes. It is important to maintain exercise during admission to hospital because reduced activity will result in an increase in BM values. If the diabetes is labile during an acute exacerbation, a pre-exercise BM is advisable in addition to close liaison with the dietitian.
Distal intestinal obstruction syndrome
This complication describes intestinal obstruction occurring after the neonatal period (Davidson 2000). It is characterized by abdominal pain, distension, vomiting, palpable fecal masses and partial or complete intestinal obstruction. Urgent medical management includes intravenous rehydration, Gastrografin® and possibly a balanced electrolyte solution. The symptoms of immobility, pain, decreased functional residual capacity and dehydration compromise the clearance of secretions. It is important for airway clearance to continue, but techniques should be modified until the problem resolves. Mobilization is encouraged and may help bowel movement. For patients with a persistent problem, a lower body exercise programme may be useful.
Gastro-oesophageal reflux
This is well recognized in CF with a higher incidence in infants. The cause and effect of physiotherapy positioning for this group is unclear and has been discussed earlier. Treatment is directed at medical management with the use of prokinetic agents and/or gastric suppressants, thickening of feeds and appropriate positioning. The adult is often unaware that gastro-oesophageal reflux (GOR) is a complication of CF and the symptoms may be under-reported. The physiotherapist may be alerted to the possibility of GOR if the patient reports symptoms during airway clearance (for example upper gastrointestinal discomfort). In adults with more severe disease GOR, both silent and symptomatic, can be a significant problem (Button et al 2005a). Oxygenation and reflux scores have been shown to deteriorate in the head-down tilt position. This may be of significance when undertaking airway clearance treatments, particularly in the acutely unwell individual with CF (Elkins et al 2005a).
Haemoptysis
Blood streaking of sputum frequently occurs, especially at the time of an acute pulmonary exacerbation. It is appropriate to continue with the normal airway clearance regimen and to reassure the patient. Frank haemoptysis can be considered moderate at <250 ml or severe at >250 ml (Hodson 1994) and it is important to establish the volume expectorated over 24 hours to determine the treatment plan (Table 18.5). It is important to continue with chest clearance to remove the blood and infected secretions (Hodson 1994). Management is aimed at clearing secretions without increasing the bleeding. Noting the activity and position at the time of haemoptysis will provide a clue to the cause and influence management. The weakened artery may rupture due to increasing heart rate or increasing the flow of blood when the area of lung supplied by the artery is dependent (the bronchial arteries lie posteriorly). If the patient can establish the location of the bleed, it is advisable to avoid chest clearance with the affected lobe dependent (Bilton et al 1990).
Table 18.5 The treatment of haemoptysis
| Mild Streaking |
• Normal airway clearance regimen |
| Moderate <250 ml |
|
| Severe >250 ml |
|
| Embolization | • Chest clearance can resume after the procedure in consultation with the radiologist |
When haemoptysis is severe, the location of the bleeding should be dependent to avoid asphyxiation (Jones & Davis 1990). When the bleeding has subsided, chest clearance can be resumed. Bronchoscopy will locate the bleeding for those patients with persistent haemoptysis and the management usually requires embolization (King et al 1989, Vidal et al 2006). Chest clearance can resume following the procedure in consultation with the radiologist. Implantable venous access devices, which require flushing with an anticoagulant to maintain patency, may increase the risk of haemoptysis during intravenous therapy. If this occurs, the use of normal saline for flushing may lead to cessation of bleeding.
Liver disease
Liver disease usually presents as biliary cirrhosis, but fatty infiltration is a recognized feature. Hepatosplenomegaly and portal hypertension ensue with the development of oesophageal varices and potentially life-threatening haematemesis (Westaby 2000). Liver transplantation is an increasing feature of adult CF care. An enlarged liver and spleen will compromise respiratory mechanics and cause breathlessness. Careful consideration should be given to positioning during airway clearance. Airway clearance should be discontinued during acute haemorrhage. Careful exercise advice is recommended (see exercise section).
Low bone mineral density
Low bone mineral density (BMD) is a recognized complication of CF in both children and adults (Bachrach et al 1994, Bhudmkanok et al 1996). The correlates of low BMD include poor absorption of vitamins, minerals and protein from the gut, oral steroids, inactivity, increased cytokines from infection and hypogonadism (Haworth et al 1999). The problem is one of reduced bone accretion in childhood and early and increased rate of bone resorption in adult life. Bone mineral density appears to be normal until the age of about 10 years and seems to decrease during the adolescent years (Cystic Fibrosis Trust 2007). Lack of physical activity is a known correlate, but there is no direct evidence that lack of physical activity leads to low BMD and it is currently unknown if weight-bearing activities can increase peak bone mass, preserve BMD or increase BMD in those with low BMD. However, it would seem prudent to encourage weight-bearing exercises based on studies in the healthy population (Cystic Fibrosis Trust 2007) There is an increased risk of rib fractures (Bachrach et al 1994) and there is some concern that manual techniques during airway clearance could cause a rib fracture. While there are no reports to date of manual techniques causing rib fractures in CF, there are other methods of airway clearance with proven efficacy which should be considered as alternatives (Cystic Fibrosis Trust 2002b). Coughing has been reported to cause a rib fracture in CF (Elkin et al 2001). A regular review of airway clearance techniques is essential in this vulnerable group of patients.
Rib and vertebral fractures are extremely painful and inhibit airway clearance and mobility. A chest X-ray should always be performed to exclude a pneumothorax. Mucus plugging and an infective exacerbation may quickly ensue. Effective analgesia is a priority before airway clearance and mobilization. Effective coughing should be facilitated by support to the chest wall and the use of antibiotics and rhDNase should reduce sputum volume and ease the mobilization of secretions.
Musculoskeletal dysfunction
Patients with CF develop alterations in chest wall mechanics and spinal deformities due to progressive lung disease, malnutrition and poor bone mineralization (Tattersall et al 2000). This leads to abnormal posture, pain and poor quality of life (Dodd & Prasad 2005). Postural screening and specific testing will direct the design of targeted interventions to improve musculoskeletal and neuromuscular impairments (Massery 2005). Musculoskeletal abnormalities are secondary to respiratory impairment. An understanding of the relationship between the respiratory and postural muscles of the trunk is critical to planning an effective strategy to address the musculoskeletal problems in CF. Current clinical evidence would suggest that an optimum time to screen and intervene, to minimize or prevent musculoskeletal problems, would be during the prepubescent years. Postural adjustments may correct kyphosis and lordosis, but the value of postural therapy needs further evaluation in CF. The flexible ruler technique has been validated in adults with CF to assess spinal deformity and may provide a simple non-invasive tool for routine use in the clinical setting (Boyle et al 2003). Other outcome measures to be considered would include optimal postural development, improved breathing mechanics, prevention of joint conditions, development of fewer pulmonary complications and functional and quality of life issues (Massery 2005). Manual therapy techniques may increase thoracic mobility and may improve lung function (Vibekk 1991). Musculoskeletal abnormalities are a consequence of improved survival and the physiotherapist should be proactive in addressing postural concerns and symptoms and treat appropriately. Flexibility and postural exercises should be introduced in childhood as part of an exercise regimen to prevent the problems that are reported with increasing age and disease severity.
Pneumothorax
In people with CF a pneumothorax usually occurs with advanced disease and chronic Pseudomonas infection (Penketh et al 1982). It is associated with increased mortality and may be an independent indicator of prognosis. A pneumothorax may occur due to rupture of a subpleural bleb through the visceral pleura or, rarely, as a result of misplacement of a central line. The physiotherapist may be alerted to the problem by a sudden increase in breathlessness and shoulder tip pain.
The aim of physiotherapy management is to clear secretions, aid re-expansion of the collapsed lung and prevent infection in the re-expanding lung (Table 18.6). It is usual for prophylactic intravenous antibiotics to be given at this time because airway clearance and mobility are compromised. When the pneumothorax is small (<20% of lung volume) treatment is conservative and airway clearance should continue with the aim of clearing secretions without increasing the size of the pneumothorax. Techniques that increase intrathoracic pressure and paroxysmal coughing should be avoided and positive pressure devices should be used with caution. Mobility is encouraged but exercise programmes should temporarily cease. An increase in symptoms may be an indication that the pneumothorax has increased in size. This should be immediately reported and an urgent chest X-ray requested.
Table 18.6 The treatment of a pneumothorax
| Small (up to 20% of lung volume) |
Chest clearance techniques are aimed at clearing secretions without increasing the size of the pneumothorax. ↑ Intrathoracic pressure should be minimized. |
| Large (with intercostal drain) |
Treatment is aimed at clearing secretions of all lung areas with adequate pain relief. |
| Removal of intercostal drain | |
| Unresolved | • VATS or thoracotomy |
VATS, video-assisted thoracoscopic surgery
Larger pneumothoraces will require an intercostal drain and, if persistent, video-assisted thoracoscopic surgery (VATS) is the preferred treatment. In the light of lung transplantation, pleurodesis should be avoided. While the drain is in situ, airway clearance techniques are reintroduced as indicated at assessment and with appropriate analgesia. Mobilization and gentle exercise are encouraged. When the intercostal tube is removed, care should be taken with airway clearance, in the short term, to prevent any reoccurrence of the resolving pneumothorax. Normal activities are advised, but strenuous exercise and lifting are not recommended. Patients should be cautioned about driving and air travel is contraindicated for 6 weeks (British Thoracic Society 2004). Following surgery, airway clearance should be restarted as soon as possible with adequate analgesia.
Pregnancy
Improved survival has resulted in more females reaching reproductive age. Pregnancy is well tolerated by patients with an FEV1 >60% predicted, but associated with increased maternal and fetal complications in those with an FEV1 <60% predicted (Edenborough et al 1995). As pregnancy can stress the pulmonary, nutritional and cardiovascular reserves of the patient with CF, knowledge of the normal changes (Elkus & Popovich 1992, Weinberger et al 1980) and their implications in CF (Kotloff et al 1992) is useful (Table 18.7). Airway clearance techniques should continue throughout pregnancy and be modified as pregnancy progresses with consideration of the degree of breathlessness and discomfort. Breathlessness commonly occurs in the first and second trimesters and may be normal. Careful monitoring of the vital capacity is essential to identify the onset of an acute exacerbation. Elevation of the diaphragm reduces functional residual capacity and causes early airway closure. Attention should be paid to clearance of the lung bases to prevent the possibility of trapped secretions.
Table 18.7 The normal changes of pregnancy and the implications for patients with cystic fibrosis
| Normal changes | Implications in cystic fibrosis |
|---|---|
| Thoracic cage |
• Normal excursion of the diaphragm |
| Cardiovascular | |
| • In the presence of cor pulmonale and pulmonary hypertension the risk of cardiovascular collapse is greater, due to the inability to cope with the increased circulating blood volume, particularly during and immediately following delivery | |
| Hormones (peak in the 3rd trimester) | |
| • ↑ Oestrogen results in hypersecretion and mucosal oedema of airways | • Potential ↑ airway obstruction |
| • ↑ Progesterone = ↑ respiratory drive, ↑ minute ventilation, resulting in chronic hyperventilation by ↑ TV | • Breathlessness most commonly occurs in the 1st and 2nd trimesters and may be normal |
| • ↑ PaO2 ↓ PaCO2 | • Careful monitoring of vital capacity determines the cause, i.e. normal mechanism of pregnancy or ↑ disease severity |
| • ↓ Bronchomotor tone | • Instability of airways may be increased |
| • Threefold ↑ in cortisol level and prostaglandins | • This may improve airway obstruction |
| Lung volumes | |
| • ↓ ERV ↓ RV ↓ FRC (due to diaphragmatic displacement) | • ↓ FRC results in early airway closure at lung bases and possible retention of secretions |
| • Normal VC ↓ TLC | • Hypoxaemia may result from ventilation–perfusion mismatch |
AP, anteroposterior; TV, tidal volume; PaO2, partial pressure of oxygen in arterial blood; PaCO2, partial pressure of carbon dioxide in arterial blood; ERV, expiratory reserve volume; RV, residual volume; FRC, functional residual capacity; VC, vital capacity; TLC, total lung capacity
Gentle exercise during pregnancy should be encouraged and established aerobic programmes may be continued. Non-weight-bearing exercises, e.g. swimming and cycling, have a lower energy cost than weight-bearing exercises (Kotloff et al 1992). Weight training should not be introduced during pregnancy, but an established programme should be modified and discussed with the medical team. The importance of pelvic floor exercises should be stressed at the beginning of pregnancy and reinforced at each clinic visit. Leakage of urine is a problem for many women (Orr et al 2001) and pregnancy will further stress the pelvic floor. Abdominal supports are available for patients who suffer lower back pain or sacroiliac strain, in combination with postural advice and gentle exercise. Gastro-oesophageal reflux may present for the first time. Medication should relieve symptoms and prevent potential aspiration.
Following the birth, airway clearance begins immediately and physiotherapy may be required while the patient is in the delivery room. It is important to liaise with the midwife and medical team to ensure that pain relief is adequate. Sputum production may increase as the diaphragm descends and ventilation to the lower lobes improves. Gravity-assisted positioning, if indicated, can resume as soon as the mother feels comfortable. Pelvic floor exercises should continue and foot and leg exercises are introduced while the mother is immobile. Mobility begins as soon as the mother feels comfortable. The return to physical activity depends on prenatal fitness, maternal health during pregnancy and progress during the postnatal period. It is usual to begin with gentle forms of exercise such as walking and to build up to low-impact aerobics. Returning to an established programme will depend on the discomfort of the women’s abdomen and perineum; every mother will require individualized advice. In the postnatal period pelvic floor exercises should continue for at least 12 weeks, but many mothers give up after the postnatal check up at 6 weeks.
Surgery
Routine surgical procedures, for example insertion of totally implantable venous access ports and gastrostomy, have become increasingly common. In addition the improved longevity of people with CF has led to an increased frequency of surgical interventions either directly or indirectly associated with the disease. Careful pre- and postoperative assessment of this patient group is essential to minimize postoperative pulmonary complications. A preoperative admission for intravenous antibiotics and intensive physiotherapy is often undertaken before elective surgery. Physiotherapists in some centres use these elective surgical procedures as an opportunity to perform perioperative airway clearance during intubation and anaesthesia. Effective airway clearance during anaesthesia may in theory compensate for any postoperative respiratory deterioration related to the anaesthetic and surgery or compromised airway clearance techniques because of postoperative discomfort. In addition, bronchial lavage and suction during anaesthesia can provide valuable sputum samples for microbiological culture.
Relatively little is known about the immediate or postoperative effects of physiotherapy treatments under anaesthesia. A small paediatric study reported a significant reduction in lung function parameters (compliance and peak inspiratory pressure) associated with physiotherapy during anaesthesia compared with a group receiving airway care as deemed appropriate by the attending anaesthetist (Tannenbaum et al 2007). Both groups showed a non-significant decline in FEV1 the day after surgery, compared with preoperative values, although there were no longer-term effects on FEV1. Perioperative chest physiotherapy should not be performed routinely in patients undergoing elective surgical procedures; however, it should be considered if the benefits of removing secretions are deemed to outweigh the short-term risk. In these circumstances it may be prudent for the anaesthetist to consider modifying ventilatory support to counteract any short-term negative effects of treatment (Tannenbaum et al 2007).
Transplantation
The physiotherapist should be involved both before and after heart-lung or lung transplantation as outlined in Chapter 15.
Urinary incontinence
Leakage of urine is recognized internationally as a problem for women and girls with CF (Cornaccia et al 2001, Orr et al 2001, Prasad et al 2006), with higher prevalence rates (30–68%) than the normal population (12.8% in women aged 16–20 years and 35% in women over 35 years of age) (Chiarelli et al 1999, Thomas et al 1980). The studies report no relationship between prevalence and age, with onset occurring as early as 10 years (Dodd & Langman 2005). Leakage in men is described in CF, with higher than normal prevalence rates of 16% (Gumery et al 2005) compared with 1.7% in the general population aged 15–64 years (Maral et al 2001). Coughing is the major cause of leakage for the majority of women with increased frequency and severity during acute pulmonary exacerbations.
Studies have addressed the assessment and treatment of the pelvic floor muscles (Button et al 2005b, 2005c, McVean et al 2003). Button and colleagues measured pelvic floor muscle activity using electromyographic (EMG) activity, and ultrasound imaging during coughing and huffing. The authors concluded that the problem of leakage was likely to be related to reduced endurance during prolonged coughing rather than the strength or the timing of the contraction. Following a 3-month intervention of pelvic floor muscle training (exercise, electrical stimulation, biofeedback and bladder training), EMG and ultrasound imaging measures all improved and patients reported significantly fewer incontinent episodes. The improvement was sustained over the following 3 months. Over the same time period, McVean et al (2003) reported an improvement in pelvic floor muscle endurance (Oxford grade), with no change in strength, in a self-selected group of patients.
Despite the physical and social implications of urinary incontinence (UI), women report the major impact of the problem to be their ability to perform airway clearance and spirometry. The physiotherapist should pay particular attention to teaching controlled and effective coughing during ACTs (Langman et al 2006). Cornaccia et al reported no association between airway clearance and leakage, but all subjects were taught to practise ‘the knack’ (Miller et al 1998) before performing a cough. Continence during a cough depends on the complex interaction of the muscles of the abdomino-pelvic capsule, which work to their greatest advantage with the lumbar spine in a neutral lordotic position (Sapsford et al 2001). Careful positioning advice, to optimize cough and continence, will avoid the flexed spinal posture encouraged by the intense prolonged coughing in women with CF (Figs 18.8 and 18.9).

Figure 18.9 Corrected positioning during A ACT and B coughing. Support is given to the pelvic floor.
Studies have revealed that women and girls are rarely forthcoming about the problem, are tolerant of their symptoms and face assessment and treatment with apprehension and embarrassment. A sensitive and open approach with early recognition of symptoms needs to be developed to evaluate this distressing problem.
As a result of our emerging knowledge it would seem prudent to adopt both preventative and active strategies to the management of UI (Button et al 2005b, Dodd & Langman 2005). This requires the collaborative expertise of specialist respiratory, incontinence and musculoskeletal physiotherapists.
Infection control
The physiotherapist should have knowledge of the respiratory pathogen harboured by the patient, the risk of transmission and the potential for cross-infection. Infection control policies, specific to cystic fibrosis, should be in place and attention paid to the risk of cross-infection between patients and from physiotherapy equipment. All physiotherapy treatment sessions should be with individual patients (Cystic Fibrosis Trust 2004a, 2004b).
Evaluation of physiotherapy
Sputum weight
In clinical practice sputum weight or volume is a useful outcome measure of airway clearance in an individual in a stable state. It provides a baseline measure of the normal sputum production for the individual. Any increase in sputum, either during treatment or out of treatment, is a possible indication of an acute exacerbation of pulmonary infection. Total daily sputum weight during an acute exacerbation is less informative, but, together with lung function, will determine if the problem is one of increased infection or sputum retention (Table 18.8). During an admission for an acute exacerbation, the goal is to maximize sputum expectoration during periods of airway clearance and minimize expectoration at other times. Measures of treatment and out-of-treatment sputum weight will provide this information and indicate, to the less adherent patient, the value of airway clearance techniques. Radiolabelled aerosols have been used to measure rate of mucus clearance in efficacy studies of airway clearance techniques (Elkins et al 2005b, Lannefors & Wollmer 1992), but remain a research tool rather than a practical clinical measurement.
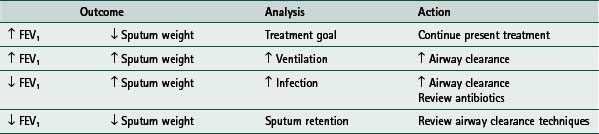
Lung function
FEV1 is considered a gold standard measure of lung function. However, as an outcome measure in airway clearance studies it is not ideal as it may not be sufficiently sensitive to detect changes caused by sputum clearance. In addition, there are a number of other confounding factors that may influence this outcome in patients with chronic respiratory disease such as CF (Gibson 2005, Prasad & Main 1998, van der Schans et al 1999). However, measurements of FEV1 in an individual, as a clinical outcome tool, are useful, in terms of assessing the response to an intervention (for example initiation of rhDNase therapy), and longitudinal measurements are helpful in monitoring disease progression (rate of decline). In children particularly, measurement of FEV1 depends on effort and cooperation and is not always a reliable or sensitive measure of early lung disease (Aurora et al 2004). It cannot therefore be considered a robust tool for paediatric physiotherapy intervention studies. Lung clearance index (LCI) calculated from multiple breath inert gas washout has recently been proposed as a more sensitive alternative measure of peripheral lung disease (Aurora et al 2005b). Measurement of lung function is now possible in infants and preschool children (Ranganathan 2006), although it is not widely available.
Other spirometric measurements are also used. The expiratory flow volume curve identifies the location of any flow limitation throughout expiration from total lung capacity (TLC) to residual volume (RV). This will vary from the early detection of small airway limitation to large airway collapse and gives the physiotherapist an invaluable insight into the problems of airway clearance. The manoeuvre is performed with an open glottis and therefore mimics a huff from TLC to RV. In the long term, measurements of small airway function (FEF25 and FEF75) are not comparable due to changes in TLC and RV and should therefore be interpreted with caution.
Arterial / capillary blood gases
Serial measurements of PaO2 and PaCO2 monitor decreases in oxygen tension and ensuing hypercapnia. PaCO2 levels inform the physiotherapist of ventilatory drive and the work of breathing. Pulse oximetry gives a quick measure of oxygen saturation, but should not be used to prescribe oxygen as the corresponding PaCO2 is unknown.
Subjective measures
Visual analogue scales (VAS) and Borg scores (Chapters 3 and 14) provide important subjective measures of breathlessness, chest tightness, muscle fatigue and pain. The 15-count breathlessness score and ease of breathing scores have been validated in children (Orenstein 2002, Prasad et al 2000).
Other useful measures include health status/quality of life questionnaires (Congleton et al 1997, Gee et al 2000, Orenstein et al 1989, Shepherd et al 1992) and activities of daily living. More recently emphasis has been placed on the importance of patient-reported outcomes that are disease-specific and one such measure has been validated in the CF population (Quittner et al 2000).
Adherence
The daily timetable of self-care can be an enormous burden for the patient and carer who are trying to balance treatment with family, work and social commitments. This can be particularly difficult for the adolescent and young adult, and studies show a decline in adherence with increasing age (Gudas et al 1991). Adherence has been shown to be treatment-specific in people with CF (Abbott et al 1994) and adherence with one treatment does not guarantee adherence with all self-care. Treatments that are complex, requiring time and effort with no immediate benefit, e.g. airway clearance and nebulized antibiotics, have lower adherence rates than other treatments that are quick and give immediate relief of symptoms (Dodd & Webb 2000). Over the years, studies have reported only 50% of patients to have ‘good’ adherence with airway clearance (Abbott et al 1994, Conway et al 1996, Passero et al 1981). Large sputum production, feeling better following treatment and help with treatment were significant factors influencing adherence with chest clearance (Abbott et al 1994).
Exercise is perceived differently; adherence is higher and should be encouraged for all its therapeutic benefits. Self-management and self-efficacy should be encouraged to improve adherence, but support and praise are essential to avoid the feeling of loneliness and isolation (McIlwaine & Davidson 1996).
Knowledge is considered an important precursor to adherence and it is important to appraise knowledge of treatments and correct any misunderstandings (Conway et al 1996). Assessing patients’ understanding of specific treatments and providing individualized specific teaching has been shown to improve knowledge and reported adherence in the short term (Unsworth et al 1998).
The reasons for non-adherence are complex and the World Health Organization has identified interacting factors that may affect adherence (WHO 2003). Psychological influences, patients’ perceptions and health beliefs (Abbott et al 1996) and coping styles (Abbott et al 2001) influence adherence (Prasad & Cerny 2002). It is important to recognize that non-adherence is normal and to encourage openness with self-reporting by adopting a non-judgmental approach. The multidisciplinary team should examine all levels of care from their centre policies, social and economic factors, treatment complexities and individual patient-related factors to seek opportunities to encourage adherence (Downs 2006). Treatment goals and plans tailored to daily lifestyle should be agreed and compromise accepted by the medical team.
Continuity of care
Continuous assessment and reassessment of patients with cystic fibrosis is essential for effective management. Patients’ needs will change as they progress from infancy through school, higher education, work and parenting. The emphasis is on leading as normal a life as possible while making time to include the many aspects of treatment. Visits to schools to support individual patients and to increase the awareness and knowledge of the teachers are beneficial (Dyer & Morais 1996). Children should be encouraged to attend normal schools.
Although the majority of parents and patients can take the responsibility for physiotherapy treatments, it is essential that there is a regular review of the techniques by a physiotherapist. This also provides an opportunity to update the techniques and to discuss any problems. It is important for the physiotherapist to work with carers and patients to encourage autonomy with treatments from an early age and to ensure a successful transfer to adult care. There are times when the parents or patient are unable to cope effectively on their own and the need for assistance during these periods should be recognized and help should be arranged.
If the patient is treated at both a local hospital and a specialist cystic fibrosis unit, communication among physiotherapists is essential to avoid confusion about treatment.
The physiotherapist is part of a multidisciplinary team and must be aware of the roles of the other members. Good communication within the team is essential. Members of the team may experience considerable stress from long-term involvement with patients with a chronic progressive illness. Coping with this stress can be helped by the team members recognizing each other’s needs and providing the necessary support.
In caring for the patient with cystic fibrosis, the physical care is important but the psychological effects on both the family and the patient must also be considered. Many countries have cystic fibrosis associations, which offer encouragement and support for patients and their families. In spite of the frequently high demands of treatment, most patients with cystic fibrosis are leading fulfilling lives and many adults are in full-time employment, homeowners and parents (Walters et al 1993).
Abbott J, Bilton D, Dodd M, Webb AK. Treatment compliance in adults with cystic fibrosis. Thorax. 1994;59:115-120.
Abbott J, Dodd M, Webb AK. Health perceptions and treatment adherence in adults with cystic fibrosis. Thorax. 1996;51:1233-1238.
Abbott J, Dodd M, Gee L, Webb AK. Ways of coping with cystic fibrosis: implications for treatment adherence. Disability and Rehabilitation. 2001;23:315-324.
Accurso FJ, Sontag MK, Koenig JM, et al. Multi-centre airway secretion clearance study in cystic fibrosis. Pediatric Pulmonolology. 2004(suppl 27):314.
Albini S, Rath R, Renner S, et al. Additional inspiratory muscle training intensifies the beneficial effects of cycle ergometer training in patients with cystic fibrosis. Journal of Cystic Fibrosis. 2004;3(Suppl 1):63.
Alton E, Caplen N, Geddes D, Williamson R. New treatments for cystic fibrosis. British Medical Bulletin. 1992;48:785-804.
Anderson DH. Cystic fibrosis of the pancreas and its relation to celiac disease: clinical and pathological study. American Journal of Disease in Childhood. 1938;56:344-399.
Andreasson B, Jonson B, Kornfalt R, et al. Long-term effects of physical exercise on working capacity and pulmonary function in cystic fibrosis. Acta Paediatrica Scandinavica. 1987;76:70-75.
Armstrong DS, Grimwood K, Carzino R, et al. Lower respiratory tract infection and inflammation in infants with newly diagnosed cystic fibrosis. British Medical Journal. 1995;310:1571-1572.
Asher MI, Pardy RL, Coates AL. The effects of inspiratory muscle training in patients with cystic fibrosis. American Review of Respiratory Disease. 1982;126:855-859.
Ashleigh RJ, Webb AK. Radiological intervention for haemoptysis in cystic fibrosis. Journal Royal Society of Medicine. 2007;100(Suppl 47):38-45.
Astrand PO, Rodahl K. Text book of work physiology. London: McGraw-Hill, 1977.
Aurora P, Bush A, Gustafsson P, et al. Multiple-breath washout as a marker of lung disease in preschool children with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 2005;171(3):249-256.
Aurora P, Gustafsson P, Bush A, et al. Multiple breath inert gas washout as a measure of ventilation distribution in children with cystic fibrosis. Thorax. 2004;59(12):1068-1073.
Aurora P, Kozlowska W, Stocks J. Gas mixing efficiency from birth to adulthood measured by multiple breath washout. Respiratory Physiology and Neurobiology. 2005;148:125-134.
Bachrach LK, Loutit CW, Moss RB. Osteopenia in adults with cystic fibrosis. American Journal of Medicine. 1994;96:27-34.
Baldwin DR, Hill AL, Peckham KG, et al. Effect of addition of exercise to chest physiotherapy on sputum expectoration and lung function in adults with cystic fibrosis. Respiratory Medicine. 1994;88:49-53.
Balfour-Lynn IM, Klein NH, Dinwiddie R. Randomized controlled trial of inhaled corticosteroids (fluticasone propionate) in cystic fibrosis. Archives of Disease in Childhood. 1997;77:124-130.
Balfour-Lynn IM, Lees B, Hall B, et al. Multicentre randomized controlled trial of withdrawal of inhaled corticosteroids in cystic fibrosis. American Journal of Respiratory Critical Care Medicine. 2006;173:1356-1362.
Balfour-Lynn IM, Prasad SA, Laverty A. A step in the right direction: assessing exercise tolerance in cystic fibrosis. Pediatric Pulmonology. 1998;25:78-84.
Balfour-Lynn IM, Walters S, Dezateux C, 2000 Inhaled corticosteroids for cystic fibrosis. Cochrane Database of Systemic Reviews. Issue 1. Art. No.: CD001915
Bargon J, Viel K, Dauletbaev N, et al. Short-term effects of regular salmeterol treatment on adult cystic fibrosis patients. European Respiratory Journal. 1997;10:2307-2311.
Barker AF, Couch L, Fiel SB, et al. Tobramycin solution for inhalation reduces sputum Pseudomonas aeruginosa density in bronchiectasis. American Journal of Respiratory and Critical Care Medicine. 2000;162:481-485.
Bar-Or O, Blimkie CJ, Hay JA, et al. Voluntary dehydration and heat intolerance in cystic fibrosis. Lancet. 1992;339:696-699.
Bhudmkanok GS, Lim J, Marcus R, et al. Correlates of osteopenia in patients with cystic fibrosis. Journal of Pediatrics. 1996;97:103-111.
Bilton D, Dodd ME, Abbot J, et al. The benefits of exercise combined with physiotherapy in the treatment of adults with cystic fibrosis. Respiratory Medicine. 1992;86:507-511.
Bilton D, Webb AK, Foster H, et al. Life threatening haemoptysis in cystic fibrosis: an alternative therapeutic approach. Thorax. 1990;45:523-524.
Birrer P, McElvaney NG, Rüdeberg A, et al. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 1994;150:207-213.
Bisgaard H, Pederson SS, Nielsen KG, et al. Controlled trial of inhaled budesonide in patients with cystic fibrosis and chronic Pseudomonas aeruginosa infection. American Journal of Respiratory and Critical Care Medicine. 1997;156:1190-1196.
Boas SR, Danduran MJ, McColley SA. Parental attitudes about exercise regarding their children with cystic fibrosis. International Journal of Sports Medicine. 1999;20:334-338.
Bourke S, Rooney M, Fitzgerald M, et al. Episodic arthropathy in adult cystic fibrosis. Quarterly Journal of Medicine. 1987;64:651-659.
Boyd AC. Gene and stem cell therapy. In: Bush A, Alton EWFW, Davies J, Griesenbach J, Jaffe A, editors. Cystic fibrosis in the 21st century. Progress in Respiratory Research. Basel: Karger; 2006:221-229. 34
Boyle L, Bradley JM, McGregor K. Thoracic kyphosis in cystic fibrosis. The flexible ruler technique. Pediatric Pulmonology. 2003;36:331.
Bradley J, Howard J, Wallace E, et al. The validity of a modified shuttle test in adult cystic fibrosis. Thorax. 1999;54:437-439.
Bradley JM, Moran FM, Greenstone M. Physical training for bronchiectasis. Cochrane Database of Systematic Reviews. 3, 2002. : CD002166
Bradley JM, Moran FM, Elborn JS. Evidence for physical therapies (airway clearance and physical training) in cystic fibrosis: an overview of five Cochrane Systematic Reviews. Respiratory Medicine. 2006;100:191-201.
Bridges N, Spowart K. Diabetes in cystic fibrosis. In: Bush A, Alton EWFW, Davies J, Griesenbach J, Jaffe A, editors. Cystic fibrosis in the 21st century. Progress in Respiratory Research. Basel: Karger; 2006:278-283. 34
British Thoracic Society. Recommendations: Managing patients with lung disease planning air travel. London: British Thoracic Society, 2004.
British Thoracic Society. Guidelines: Bronchiectasis. London: British Thoracic Society, 2007.
Britto MT, Garrett JM, Konrad TR, et al. Comparison of physical activity in adolescents with cystic fibrosis versus age-matched controls. Pediatric Pulmonology. 2000;30:86-91.
Bureau MA, Lupien L, Begin R. Neural drive and ventilatory strategy of breathing in normal children and in patients with cystic fibrosis and asthma. Pediatrics. 1981;68:187-194.
Bush A, Ferkol T. The emerging genetics of primary ciliary dyskinesia. American Journal of Respiratory and Critical Care Medicine. 2006;174:109-111.
Bush A, Gotz M, 2006 Cystic fibrosis. Respiratory Monograph 11(37): 234-291
Button BM. Postural drainage techniques and gastro-oesophageal reflux in infants with cystic fibrosis. European Respiratory Journal. 1999;14:456-1457.
Button BM, Heine RG, Catto-Smith AG, et al. Postural drainage and gastro-oesophageal reflux in infants with cystic fibrosis. Archives of Disease in Childhood. 1997;76:148-150.
Button BM, Heine RG, Catto-Smith AG, et al. Chest physiotherapy in infants with cystic fibrosis: to tip or not? A five-year study. Pediatric Pulmonology. 2003;35:208-213.
Button BM, Heine RG, Catto-Smith AG, et al. Chest physiotherapy, gastro-oesophageal reflux, and arousal in infants with cystic fibrosis. Archives of Disease in Childhood. 2004;89(5):435-439.
Button BM, Roberts S, Kotsimbos TC, et al. Gastroesophageal reflux (symptomatic and silent): a potentially significant problem in patients with cystic fibrosis before and after lung transplantation. Journal of Heart and Lung Transplantation. 2005;24:1522-1529.
Button BM, Sherburn M, Chase J, et al. Pelvic floor muscle function in women with chronic lung disease (cystic fibrosis and COPD) versus controls: relationship to urinary incontinence. Pediatric Pulmonology. 2005;113(Suppl 28):a368.
Button BM, Sherburn M, Chase J, et al. Effect of a three-month physiotherapeutic intervention on incontinence in women with chronic cough related to cystic fibrosis and COPD. Pediatric Pulmonology. 2005;113(Suppl 28):a369.
Carr L, Smith RE, Pryor JA, et al. Cystic fibrosis patients’ views and beliefs about chest clearance and exercise - a pilot study. Physiotherapy. 1996;82:621-627.
Cecins NM, Jenkins SC, Pengelly J, et al. The active cycle of breathing techniques - to tip or not to tip? Respiratory Medicine. 1999;93:660-665.
Chatham K, Ionescu AA, Nixon LS, Shale DJ. A short-term comparison of two methods of sputum expectoration in cystic fibrosis. European Respiratory Journal. 2004;23(3):435-439.
Chen HC, Liu CY, Cheng HF, et al. Chest physiotherapy does not exacerbate gastroesophageal reflux in patients with chronic bronchitis and bronchiectasis. Respiratory Medicine. 1998;21:409-414.
Chen KY, Ko SC, Hsueh PR, et al. Pulmonary fungal infection: emphasis on microbiological spectra, patient outcome and prognostic factors. Chest. 2001;120:177-184.
Cheng K, Smyth RL, Motley J, et al. Randomized controlled trials in cystic fibrosis (1966-1997) categorised by time, design and intervention. Pediatric Pulmonology. 2000;29:1-7.
Chiarelli P, Brown W, McElduff P. Leaking urine: prevalence and associated factors in Australian women. Neurourology and Urodynamics. 1999;18:567-577.
Chua HL, Collis GG, Le-Souef PN. Bronchial response to nebulized antibiotics in children with cystic fibrosis. European Respiratory Journal. 1990;3:1114-1116.
Coakley RD, Boucher RC. Pathophysiology: epithelial cell biology, ASL channel functions including sweat gland and pancreas. In: Hodson M, Geddes D, Bush A, editors. Cystic fibrosis. 3rd edn. London: Hodder Arnold; 2007:59-68.
Cole P. Bronchiectasis. In Brewis RAL, Corrin B, Geddes DM, Gibson GJ, editors: Respiratory medicine, 2nd edn., London: WB Saunders, 1995., ch 39
Congleton J, Hodson M, Duncan-Skingle F. Quality of life in adults with cystic fibrosis. Thorax. 1997;52:397-400.
Constantini D, Brivio A, Brusa D, et al. PEP-mask versus postural drainage in CF infants: a long-term comparative trial. Pediatric Pulmonology Suppl. 2001;22:A400.
Conway JH, Fleming JS, Perring S, et al. Humidification as an adjunct to chest physiotherapy in aiding tracheo-bronchial clearance in patients with bronchiectasis. Respiratory Medicine. 1992;86:109-111.
Conway SP, Pond MN, Watson A, et al. Knowledge of adult patients with cystic fibrosis about their illness. Thorax. 1996;51:34-38.
Conway SP, Watson A. Nebulized bronchodilators, corticosteroids and rhDNase in adult patients with cystic fibrosis. Thorax. 1997;52(Suppl 2):S64-68.
Corless JA, Warburton CJ. Surgery vs non-surgical treatment for bronchiectasis. Cochrane Database Systematic Review. 4, 2000. : CD002180
Cornaccia M, Zenorini A, Braagion C, et al. Prevalence of urinary incontinence in women with cystic fibrosis. British Journal of Urology International. 2001;88:44-48.
Cropp GJA, Pullano TP, Cerny FJ, et al. Exercise tolerance and cardiorespiratory adjustments at peak work capacity in cystic fibrosis. American Review of Respiratory Disease. 1982;126:211-216.
Cunningham S, Prasad SA, Collyer L, et al. Bronchoconstriction following nebulized colistin in cystic fibrosis. Archives of Disease in Childhood. 2001;84:432-433.
Currie DC. Nebulizers for bronchiectasis. Thorax. 1997;52(Suppl 2):S72-74.
Currie DC, Cooke JC, Morgan AD, et al. Interpretation of bronchograms and chest radiographs in patients with chronic sputum production. Thorax. 1987;42:278-284.
Currie DC, Munro C, Gaskell D, et al. Practice, problems and compliance with postural drainage: a survey of chronic sputum producers. British Journal of Diseases of the Chest. 1986;80:249-253.
Cystic Fibrosis Foundation, 2006 www.cff.org
Cystic Fibrosis Trust. Antibiotic treatment for cystic fibrosis. Bromley: Cystic Fibrosis Trust, 2002.
Cystic Fibrosis Trust. Clinical guidelines for the physiotherapy management of cystic fibrosis. Bromley: Cystic Fibrosis Trust, 2002.
Cystic Fibrosis Trust. The Burkholderia cepacia complex. Bromley: Cystic Fibrosis Trust, 2004.
Cystic Fibrosis Trust. Pseudomonas aeruginosa infection in people with cystic fibrosis. Bromley: Cystic Fibrosis Trust, 2004.
Cystic Fibrosis Trust. Management of cystic fibrosis related diabetes mellitus. Bromley: Cystic Fibrosis Trust, 2004.
Cystic Fibrosis Trust. Bone mineralisation in cystic fibrosis. Bromley: Cystic Fibrosis Trust, 2007.
Davidson AGF. Gastrointestinal and pancreatic disease in cystic fibrosis. In: Hodson ME, Geddes DM, editors. Cystic fibrosis. London: Arnold; 2000:261-289.
Davis PB, Drumm M, Konstan MW. Cystic fibrosis: state of the art. American Journal of Respiratory and Critical Care Medicine. 1996;154:1229-1256.
De Boeck K. Improving standards of clinical care in cystic fibrosis. European Respiratory Journal. 2000;16:585-587.
De Jong W, Kaptein AA, Van Der Schans CP, et al. Quality of life in patients with cystic fibrosis. Pediatric Pulmonology. 1997;23:95-100.
Department of Health, 2004 Home oxygen therapy service: Service specifications. www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_4126268 (Accessed 19 July 2007)
Desai M, Weller PH, Spencer DA. Clinical benefit from nebulized human recombinent DNase in Kartagener’s syndrome. Pediatric Pulmonology. 1995;20:307-308.
Desmond KJ, Schwenk WF, Thomas E, et al. Immediate and long-term effects of chest physiotherapy in patients with cystic fibrosis. Journal of Pediatrics. 1983;103:538-542.
Di Sant’Agnese PA, Davis PB. Cystic fibrosis in adults. American Journal of Medicine. 1979;66:121-132.
Dodd ME, Langman H. Urinary incontinence in cystic fibrosis. Journal of Royal Society of Medicine. 2005;98(Suppl 45):28-36.
Dodd ME, Prasad SA. Physiotherapy management of cystic fibrosis. Chronic Respiratory Disease. 2005;2:139-149.
Dodd ME, Webb AK. Understanding non-compliance with treatment in adults with cystic fibrosis. Journal of the Royal Society of Medicine. 2000;93(Suppl 38):2-8.
Dodd ME, Abbott J, Maddison J, et al. The effect of the tonicity of nebulized colistin on chest tightness and lung function in adults with cystic fibrosis. Thorax. 1997;52:656-658.
Dodd ME, Haworth CS, Webb AK. Practical application of oxygen therapy in cystic fibrosis. Journal of the Royal Society of Medicine. 1998;91(Suppl 34):30-39.
Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947-2003. European Respiratory Journal. 2007;29:522-526.
Dodge JA, Morison S, Lewis PA, et al. Cystic fibrosis in the United Kingdom, 1968-1988: incidence, population and survival. Paediatric and Perinatal Epidemiology. 1993;7:157-166.
Donaldson SH, Bennett WD, Zeman KL, et al. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. New England Journal of Medicine. 2006;354:241-250.
Doring G, Conway SP, Heijerman HGM, et al. Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis. European Respiratory Journal. 2000;16:749-767.
Downs AM. ACT and the non-participating patient/family. Pediatric Pulmonology Suppl. 2006;29:S21.2.
Du Bois RM. Respiratory medicine - recent advances. British Medical Journal. 1995;310:1594-1597.
Dyer J, Morais JA. Supporting children with cystic fibrosis in school. Professional Nurse. 1996;11:518-520.
Eaton T, Young P, Zeng I, Kolbe J. A randomized evaluation of the acute efficacy, acceptability and tolerability of Flutter and active cycle of breathing with and without postural drainage in non-cystic fibrosis bronchiectasis. Chronic Respiratory Disease. 2007;4:23-30.
Edenborough FP, Stableforth DE, Webb AK, et al. Outcome of pregnancy in women with cystic fibrosis. Thorax. 1995;50:170-174.
Edlund LD, French RW, Herbst JJ, et al. Effects of a swimming program on children with cystic fibrosis. American Journal of Diseases of Childhood. 1986;140:80-83.
Elborn JS, Prescott RJ, Stack BH, et al. Elective versus symptomatic antibiotic treatment in cystic fibrosis patients with chronic Pseudomonas infection of the lungs. Thorax. 2000;55:355-358.
Elkin SL, Fairney A, Burnett S, et al. Vertebral deformities and low bone mineral density in adults with cystic fibrosis: a cross sectional study. Osteoporosis International. 2001;12:366-372.
Elkins MR, Alison JA, Bye PT. Effect of body position on maximal expiratory pressure and flow in adults with cystic fibrosis. Pediatric Pulmonology. 2005;40:385-391.
Elkins MR, Eberl S, Constable C, et al. The effect of manual chest physiotherapy, positive expiratory pressure (PEP) and oscillating PEP on mucociliary clearance in subjects with cystic fibrosis. Pediatric Pulmonology. 2005;40(Suppl 28):A377.
Elkins MR, Robinson M, Rose BR, et al. controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. New England Journal of Medicine. 2006;354:229-240.
Elkins MR, Jones A, Schans C, 2006b Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis. Cochrane Database of Systematic Reviews. Issue 2. Art. No.: CD003147
Elkus R, Popovich J. Respiratory physiology in pregnancy. Clinics in Chest Medicine. 1992;13:555-565.
Eng PA, Morton J, Douglass JA, et al. Short-term efficacy of ultrasonically nebulized hypertonic saline in cystic fibrosis. Pediatric Pulmonology. 1996;21:77-83.
Enright S, Chatham K, Ionescu AA, et al. Inspiratory muscle training improves lung function and exercise capacity in adults with cystic fibrosis. Chest. 2004;126:405-411.
Equi A, Balfour-Lynn IM, Bush A, Rosenthal M. Long-term azithromycin in children with cystic fibrosis: a randomized, placebo-controlled crossover trial. Lancet. 2002;360(9338):978-984.
Evans SA, Turner SM, Bosch BJ, Hardy MA, Woodhead MA. Lung function in bronchiectasis: the influence of Pseudomonas aeruginosa.. European Respiratory Journal. 1996;9:1601-1604.
Farrell PM, Kosorok MR, Rock MJ, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Paediatrics. 2001;107:1-13.
Fauroux B, Boule M, Lofaso F, et al. Chest physiotherapy in cystic fibrosis: improved tolerance with nasal pressure support ventilation. Pediatrics. 1999;103:1-9.
Ferkol T, Mitchison HM, O’Callaghan C et al. 2006 Current issues in the basic mechanisms, pathophysiology, diagnosis and management of primary ciliary dyskinesia. European Respiratory Journal, Monograph 37: 291-313
Finnegan MJ, Hughes DV, Hodson ME. Comparison of nebulized and intravenous terbutaline during acute exacerbations of pulmonary infection in patients with cystic fibrosis. European Respiratory Journal. 1992;5:1089-1091.
FitzSimmons SC. The changing epidemiology of cystic fibrosis. Journal of Pediatrics. 1993;122:1-9.
Freeman W, Stableforth DE, Cayton R, et al. Endurance exercise capacity in adults with cystic fibrosis. Respiratory Medicine. 1993;87:252-257.
Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. New England Journal of Medicine. 1994;331:637-642.
Gee L, Abbott J, Conway SP, et al. Development of a disease specific health related quality of life measure for adults and adolescents with cystic fibrosis. Thorax. 2000;55:946-954.
Gibson GJ. Spirometry: then and now. Breathe. 2005;1:206-216. ;
Gillet D, de Braekeleer M, Bellis G, Durieu I. French cystic fibrosis registry: cystic fibrosis and pregnancy. Report from French data (1980-1999). British Journal of Obstetrics and Gynaecology. 2002;109:912-918.
Godfrey S, Mearns M. Pulmonary function and response to exercise in cystic fibrosis. Archives of Disease in Childhood. 1971;46:144-151.
Govan J, Brown PH, Maddison J, et al. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet. 1993;342:15-18.
Gozal D. Nocturnal ventilatory support in patients with cystic fibrosis: comparison with supplemental oxygen. European Respiratory Journal. 1997;10:1999-2003.
Greenstone M, Rutman A, Dewar I, et al. Primary ciliary dyskinesia: cytological and clinical features. Quarterly Journal of Medicine. 1988;67(253):405-430.
Gremmo ML, Guenza MC. Positive expiratory pressure in the physiotherapeutic management of primary ciliary dyskinesia in paediatric age. Monaldi Archives of Chest Disease. 1999;54:255-257.
Gudas LJ, Koocher GP, Wypij D. Perceptions of medical compliance in children and adolescents with cystic fibrosis. Journal of Behaviour in Pediatrics. 1991;12:236-242.
Gumery L, Lee J, Whitehouse J, et al. The prevalence of urinary incontinence in adult cystic fibrosis males. Journal of Cystic Fibrosis. 2005;4:S97.
Hasani A, Pavia D, Agnew JE, Clarke SW. Regional lung clearance during cough and forced expiration technique (FET): effects of flow and viscoelasticity. Thorax. 1994;49:557-561.
Hassan JA, Saadiah S, Roslan H, et al. Bronchodilator response to inhaled beta 2-agonist and anticholinergic drugs in patients with bronchiectasis. Respirology. 1999;4:423-426.
Haworth CS, Dodd ME, Atkins M, et al. Pneumothorax in adults with cystic fibrosis dependent on nasal intermittent positive pressure ventilation (NIPPV): a management dilemma. Thorax. 2000;55:620-662.
Haworth CS, Selby PL, Webb AK, et al. Low bone mineral density in adults with cystic fibrosis. Thorax. 1999;54:961-967.
Heijerman HG, Bakker W, Sterk PJ, et al. Long-term effects of exercise training and hyperalimentation in adult cystic fibrosis patients with severe pulmonary dysfunction. International Journal of Rehabilitation and Respiration. 1992;15:252-257.
Hill AT, Edenborough FP, Cayton RM, et al. Long-term nasal intermittent positive pressure ventilation in patients with cystic fibrosis and hypercapnic respiratory failure (1991-1996). Respiratory Medicine. 1998;92:523-526.
Hill LS, Slater AL. A comparison of the performance of two modern multidose dry powder asthma inhalers. Respiratory Medicine. 1998;92:105-110.
Hodson ME. Adults. In: Hodson ME, Geddes DM, editors. Cystic fibrosis. London: Chapman & Hall; 1994:237-253.
Hodson ME, 1996 Principles of antibiotic management. In: Issues in cystic fibrosis: antibiotic therapy. Report of a meeting held at Royal College of Pathologists, November (Zeneca), pp 6-14
Hodson ME. The respiratory system: adults. In: Hodson ME, Geddes DM, editors. Cystic fibrosis. London: Arnold; 2000:218-242.
Hodson ME, Shah PL. DNase trials in cystic fibrosis. European Respiratory Journal. 1995;8:1786-1791.
Hodson ME, Madden BP, Steven MH, et al. Non-invasive mechanical ventilation for cystic fibrosis patients - a potential bridge to transplantation. European Respiratory Journal. 1991;4:524-527.
Høiby N, Koch C. Pseudomonas aeruginosa infection in cystic fibrosis and its management. Thorax. 1990;45:881-884.
Holland AE, Denehy L, Ntoumenopoulos G, et al. Non-invasive ventilation assists chest physiotherapy in adults with acute exacerbations of cystic fibrosis. Thorax. 2003;58:880-884.
Houtmeyers E, Gosselink R, Gayan-Ramirez G, et al. Effects of drugs on mucus clearance. European Respiratory Journal. 1999;14:452-467.
Hutchinson GR, Parker S, Pryor JA, et al. Home-nebulizers: a potential primary source of Burkholderia cepacia and other colistin-resistant, gram-negative bacteria in patients with cystic fibrosis. Journal of Clinical Microbiology. 1996;34:584-587.
Johnson S, Knox AJ. Arthropathy in cystic fibrosis. Respiratory Medicine. 1994;88:567-570.
Jones AM, Dodd ME, Govan JRW, et al. Burkholderia cenocepacia and Burkholderia multivorans: influence on survival in cystic fibrosis. Thorax. 2004;59:948-951.
Jones AM, Dodd ME, Govan JRW, et al. Prospective surveillance for Pseudomonas aeruginosa cross-infection at a cystic fibrosis centre. American Journal of Respiratory and Critical Care Medicine. 2005;171:257-260.
Jones AM, Govan JR, Doherty C, et al. Spread of a multiresistant strain of Pseudomonas aeruginosa in an adult cystic fibrosis clinic. Lancet. 2001;358:557-558.
Jones DK, Davis RJ. Massive haemoptysis. British Medical Journal. 1990;300:889-890.
Jones J, Hunter D. Consensus methods for medical and health services research. British Medical Journal. 1995;311:376-380.
Jones NL. Clinical exercise testing, 3rd edn., Philadelphia: WB Saunders; 1988:306-307.
Karadag B, Gultekin E, Wilson N, et al. Exhaled nitric oxide (NO) in children with primary ciliary dyskinesia (abstrct). European Respiratory Journal. 1997;10(Suppl 25):339s.
Kartagener M. Zur Pathogenese der Bronchiektasien. Beitrage zur Klinik der Tuberkulose. 1933;83:489-501. B
Keens TG, Krastins IRB, Wannamaker EM, et al. Ventilatory muscle endurance training in normal subjects and patients with cystic fibrosis. American Review of Respiratory Disease. 1977;116:853-860.
Kellett F, Redfern J, Niven RM. Evaluation of nebulized hypertonic saline (7%) as an adjunct to physiotherapy in patients with stable bronchiectasis. Respiratory Medicine. 2005;99:27-31.
Kerem E, Reisman J, Corey M, et al. Prediction of mortality in patients with cystic fibrosis. New England Journal of Medicine. 1992;326:1187-1191.
Khan TZ, Wagener JS, Bost T, et al. Early pulmonary inflammation in infants with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 1995;151:1075-1082.
King AD, Cumberland DC, Brennan SR. Management of severe haemoptysis by bronchial artery embolisation in a patient with cystic fibrosis. Thorax. 1989;44:523-524.
Klijn PH, Oudshoorn A, van der Ent CK, et al. Effects of anaerobic training in children with cystic fibrosis: a randomized controlled study. Chest. 2004;125:1299-1305.
Koch C, Lanng S. Other organ systems. In: Hodson ME, Geddes DM, editors. Cystic fibrosis. London: Arnold; 2000:314-328.
Koh YY, Park Y, Jeong JH, et al. The effect of regular salbutamol on lung function and bronchial responsiveness in patients with primary ciliary dyskinesia. Chest. 2000;117:427-433.
Kolbe J, Wells A. Inhaled steroids for bronchiectasis. Cochrane Database of Systematic Reviews. 2, 2000. : CD000996
Konstan MW, Hilliard KA, Norvell TM, et al. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. American Journal of Respiratory and Critical Care Medicine. 1994;150:448-454.
Koscik RL, Farrell PM, Kosorok MR, et al. Cognitive function of children with cystic fibrosis: deleterious effect of early malnutrition. Pediatrics. 2004;113:1549-1558.
Kotloff RM, Fitzsimmons SC, Fiel SB. Fertility and pregnancy in patients with cystic fibrosis. Clinics in Chest Medicine. 1992;13:623-635.
Kraemer R, Delosea N, Baliinari P, et al. Effects of allergic bronchopulmonary aspergillosis on lung function in children with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 2006;74:1211-1220.
Kuhn RJ, Nahata MC. Therapeutic management of cystic fibrosis. Clinical Pharmacology. 1985;4:555-565.
Kuo C, Lin S, Wang J. Aerosol, humidity and oxygenation. Chest. 1991;99:1352-1356.
Langlands J. The dynamics of cough in health and in chronic bronchitis. Thorax. 1967;22:88-96.
Langman HB, Webb AK, Jones AM, et al. Airway clearance therapy and urinary incontinence. Pediatric Pulmonology. 2006(Suppl 29):21.3. S
Lannefors L, Wollmer P. Mucus clearance with three chest physiotherapy regimes in cystic fibrosis: a comparison between postural drainage, PEP and physical exercise. European Respiratory Journal. 1992;5:748-753.
Lanng S, Thorsteinsson B, Nerup J, et al. Influence of the development of diabetes mellitus on clinical status in patients with cystic fibrosis. European Journal of Paediatrics. 1992;151:684-687.
Lapin CD. Mixing and matching airway clearance techniques to patients. Pediatric Pulmonology Supplement. 2000;20:S12.3.
Ledson MJ, Gallagher MJ, Corkhill JE, et al. Cross infection between cystic fibrosis patients colonized with Burkholderia cepacia. Thorax. 1998;53:432-436.
LiPuma JJ, Dasen SE, Nielson DW, et al. Person-to-person transmission of Pseudomonas cepacia between patients with cystic fibrosis. Lancet. 1990;336:1094-1096.
Long FR, Williams RS, Castille RG. Structural airway abnormalities in infants and young children with cystic fibrosis. Journal of Pediatrics. 2004;144(2):154-161.
Lum S, Gustafsson P, Ljungberg H, et al. Early detection of cystic fibrosis lung disease: multiple breath washout versus raised volume tests. Thorax. 2007;62(4):341-347.
Madden BP, Kariyawasam H, Siddiqi AJ, et al. Non-invasive ventilation in cystic fibrosis patients with acute or chronic respiratory failure. European Respiratory Journal. 2002;19:310-313.
Maddison J, Dodd M, Webb AK. Nebulized colistin causes chest tightness in adults with cystic fibrosis. Respiratory Medicine. 1994;88:145-147.
Mahadeva R, Webb AK, Westerbeek RC, et al. Clinical outcome in relation to care in centres specialising in cystic fibrosis: cross-sectional study. British Medical Journal. 1998;316:1771-1775.
Main E, Prasad A, Schans C. Conventional chest physiotherapy compared to other airway clearance techniques for cystic fibrosis. Cochrane Database of Systematic Reviews. 25(1), 2005. CD002011
Maral I, Ozkardes H, Peskircioglu L, Bumin MA. Prevalence of stress urinary incontinence in both sexes at or after age 15 years: a cross-sectional study. Journal of Urology. 2001;165(2):408-412.
Marcotte JE, Grisdale RK, Levison H, et al. Multiple factors limit exercise in cystic fibrosis. Pediatric Pulmonology. 1986;2:274-281.
Marcus CL, Bader D, Stabile M, et al. Supplemental oxygen and exercise performance in patients with cystic fibrosis with severe pulmonary disease. Chest. 1992;105:52-57.
Martinez TM, Llapur CJ, Williams TH, et al. High-resolution computed tomography imaging of airway disease in infants with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 2005;172:1133-1138.
Massery M. Musculoskeletal and neuromuscular interventions: a physical approach to cystic fibrosis. Journal of the Royal Society of Medicine. 2005;98(Suppl 45):55-66.
Mastella G, Rainisio M, Harms HK, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis: a European epidemiological study. European Respiratory Journal. 2000;16:464-471.
McBride G. More progress in cystic fibrosis. British Medical Journal. 1990;301:627.
McCallum SJ, Corkhill J, Gallagher M, et al. Superinfection with a transmissible strain of a Pseudomonas aeruginosa in adults with cystic fibrosis chronically colonized by P aeruginosa.. Lancet. 2001;358:558-560.
McIlwaine MP, Davidson GF. Airway clearance techniques in the treatment of cystic fibrosis. Current Opinion in Pulmonary Medicine. 1996;2:447-451.
McMullen AH, Pasta DJ, Frederick PD, et al. Impact of pregnancy on women with cystic fibrosis. Chest. 2006;129:706-711.
McVean R, Orr A, Webb AK, et al. Treatment of urinary incontinence in cystic fibrosis. Journal of Cystic Fibrosis. 2003;2:171-176.
Miller JM, Ashton-Miller JA, DeLancey JO. A pelvic muscle contraction can reduce cough-related urine loss in selected women with mild SUI. Journal of the American Geriatric Society. 1998;46(7):870-874.
Moorcroft AJ, Dodd ME, Webb AK. Individualised unsupervised exercise training in adults with cystic fibrosis: a one-year randomized controlled trial. Thorax. 2004;59:1074-1080.
Muhdi K, Edenborough FP, Gumery L, et al. Outcome for patients colonised with Burkholderia cepacia in a Birmingham adult cystic fibrosis clinic and the end of an epidemic. Thorax. 1996;51:374-377.
Mukhopadhyay S, Singh M, Cater JI, et al. Nebulized antipseudomonal antibiotic therapy in cystic fibrosis: a meta-analysis of benefits and risks. Thorax. 1996;51:364-368.
Mullinger B, Sommerer K, Herpich C, et al. Inhalation therapy can be improved in CF patients by controlling the breathing pattern during inspiration. Journal of Cystic Fibrosis. 2004;3:S65.
Nazer D, Abdulhamid I, Thomas R, Pendleton S. Home versus hospital intravenous antibiotic therapy for acute pulmonary exacerbations in children with cystic fibrosis. Pediatric Pulmonology. 2006;41(8):744-749.
Newall C, Stockley RA, Hill SL. Exercise training and inspiratory muscle training in patients with bronchiectasis. Thorax. 2005;60:943-948.
Nikolaizik WH, Jenni-Galovie V, Schoni MH. Bronchial constriction after nebulized tobramycin preparations and saline in patients with cystic fibrosis. European Journal of Pediatrics. 1996;155:608-611.
Noone PG, Bennett WD, Regnis JA, et al. Effect of aerolised uridine-5’-triphosphate on airway clearance with cough in patients with primary ciliary dyskinesia. American Journal of Respiratory and Critical Care Medicine. 1999;160:144-149.
Noone PG, Leigh MW, Sannuti A, et al. Primary ciliary dyskinesia; diagnostic and phenotypic features. American Journal of Respiratory and Critical Care Medicine. 2004;169:459-467.
O’Neill PA, Dodd M, Phillips B, et al. Regular exercise and reduction of breathlessness in cystic fibrosis. British Journal of Diseases of the Chest. 1987;81:62-66.
Orenstein DM, Franklin BA, Doershuk CF, et al. Exercise conditioning and cardiopulmonary fitness in cystic fibrosis. Chest. 1981;80:392-398.
Orenstein DM, Holt LS, Rebovich P, et al. Measuring ease of breathing in young patients with cystic fibrosis. Pediatric Pulmonology. 2002;34(6):473-477.
Orenstein DM, Hovell MF, Mulvihill M, et al. Strength vs aerobic training in children with cystic fibrosis. Chest. 2004;126:1204-1214.
Orenstein DM, Nixon PA, Ross EA, et al. The quality of well-being in cystic fibrosis. Chest. 1989;95:344-347.
Oriols R, Roig J, Ferrer J, et al. Inhaled antibiotic therapy in non-cystic fibrosis patients with bronchiectasis and chronic bronchial infection by Pseudomonas aeruginosa.. Respiratory Medicine. 1999;93:476-480.
Orr A, McVean R, Webb AK, et al. A questionnaire survey of the prevalence of urinary incontinence in females with cystic fibrosis: a marginalised and undertreated problem. British Medical Journal. 2001;322:1521.
Park RW, Grand RJ. Gastrointestinal manifestations of cystic fibrosis: a review. Gastroenterology. 1981;81:1143-1216.
Passero MA, Remor B, Salomon J. Patient-reported compliance with cystic fibrosis therapy. Clinical Pediatrics. 1981;20:264-268.
Pasteur MC, Heliwell SM, Houghton SJ, et al. An investigation into the causative factors in patients with bronchiectasis. American Journal of Respiratory and Critical Care Medicine. 2000;162:1277-1284.
Patterson JE, Bradley JM, Elborn JS. Airway clearance in bronchiectasis: a randomized crossover trial of active cycle of breathing techniques (incorporating postural drainage and vibration) versus test of incremental respiratory endurance. Chronic Respiratory Disease. 2004;1:127-130.
Patterson JE, Bradley JM, Hewitt O, et al. Airway clearance in bronchiectasis: a randomized crossover trial of active cycle of breathing techniques versus Acapella®. Respiration. 2005;72:239-242.
Patterson JE, Bradley JM, Hewitt O, Kent L, et al. Acapella vs ‘usual airway clearance’ during acute exacerbation in bronchiectasis: a randomized crossover trial. Chronic Respiratory Disease. 2007;4:67-74.
Penketh ARL, Knight RK, Hodson M, et al. Management of pneumothorax in adults with cystic fibrosis. Thorax. 1982;37:850-853.
Phillips G. To tip or not to tip? Physiotherapy Research International. 1996;1:1-6.
Phillips GE, Thomas S, Heather S, Bush A. Airway responsiveness in primary ciliary dyskinesia: intrasubject variability and the effects of exercise and bronchodilator therapy (abstract). European Respiratory Journal. 1996;9(Suppl 23):36s.
Phillipson GTM, Petrucco OM, Mathews CD. Congenital absence of the vas deferens, cystic fibrosis mutational analysis and intracytoplasmic sperm injection. Human Reproduction. 2000;15:431-435.
Piper AJ, Parker S, Torzillo PJ, et al. Nocturnal nasal IPPV stabilizes patients with cystic fibrosis and hypercapnic respiratory failure. Chest. 1992;102:846-850.
Poole S. Dietary treatment of cystic fibrosis. In: Hodson ME, Geddes DM, editors. Cystic fibrosis. London: Chapman & Hall; 1995:383-395.
Prasad SA, Cerny FJ. Factors that influence adherence to exercise and their effectiveness: application to cystic fibrosis. Pediatric Pulmonology. 2002;34(1):66-72.
Prasad SA, Main E. Finding evidence to support airway clearance techniques in cystic fibrosis. Disability and Rehabilitation. 1998;20:235-246.
Prasad SA, Main E. Routine airway clearance in asymptomatic infants and babies with cystic fibrosis in the UK: obligatory or obsolete? Physical Therapy Reviews. 2006;11(1):11-20.
Prasad SA, Balfour-Lynn IM, Carr S, et al. A comparison of the prevalence of urinary incontinence in girls with cystic fibrosis, asthma and healthy controls. Pediatric Pulmonology. 2006;41:1065-1068.
Prasad SA, Dodd M, Main E, 2008 Finding consensus on the physiotherapy management of asymptomatic infants with cystic fibrosis. Pediatric Pulmonology In Press
Prasad SA, Randall SD, Balfour-Lynn I. Fifteen-count breathlessness score: an objective measure for children. Pediatric Pulmonology. 2000;30:56-62.
Pringle MB. Swimming and grommets. British Medical Journal. 1992;304:198.
Pryor JA, Tannenbaum E, Cramer D, et al. comparison of five airway clearance techniques in the treatment of people with cystic fibrosis. Journal of Cystic Fibrosis. 2006;5:76. : S
Pryor JA, Main E, Agent P, et al. Physiotherapy. In: Bush A, Alton EWFW, Davies JC, Griesenbach U, Jaffe A, editors. Cystic fibrosis in the 21st century. Progress in Respiratory Research. Basel: Karger; 2006:303-305.
Quittner AL, Sweeny S, Watrous M, et al. Translation of a linguistic validation of a disease specific quality of life measure for cystic fibrosis. Journal of Pediatric Psychology. 2000;25:403-414.
Ramsey BW, Dorkin HL. Consensus conference: practical applications of Pulmozyme. Pediatric Pulmonology. 1994;17:404-408.
Ramsey BW, Pepe MS, Quan JM, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. New England Journal of Medicine. 1999;340:23030.
Ranganathan S. Recent advances in infant and pre-school lung function. In: Bush A, Alton EWFW, Davies JC, Griesenbach U, Jaffe A, editors. Cystic fibrosis in the 21st century. Progress in Respiratory Research. Basel: Karger, 2006.
Ranganathan SC, Dezateux C, Bush A, et al. London Collaborative Cystic Fibrosis Group: airway function in infants newly diagnosed with cystic fibrosis. Lancet. 2001;358:1964-1965.
Ranganathan SC, Stocks J, Dezateux C, et al. The evolution of airway function in early childhood following clinical diagnosis of cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 2004;169:928-933.
Range SP, Knox AJ. rhDNase in cystic fibrosis. Thorax. 1995;50:321-322.
Reid L, De Haller R. The bronchial mucous glands - their hypertrophy and changes in intracellular mucus. Bibliotheca Pediatrica. 1967;86:195-200.
Robinson M, Regnis JA, Bailey DL, et al. Effect of hypertonic saline, amiloride, and cough on mucociliary clearance in patients with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 1996;153:1503-1509.
Rodwell LT, Anderson SD. Airway responsiveness to hyperosmolar saline challenge in cystic fibrosis: a pilot study. Pediatric Pulmonology. 1996;21:282-289.
Rogers D, Goodchild MC. Role of a domiciliary physiotherapist in the treatment of children with cystic fibrosis. Physiotherapy. 1996;82:396-402.
Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059-1065.
Rosen MJ. Chronic cough due to bronchiectasis. ACCP evidence-based clinical practice guidelines. Chest. 2006;129:122S-131S.
Rubin BK. Immotile cilia syndrome (primary ciliary dyskinesia) and inflammatory lung disease. Clinics of Chest Medicine. 1988;9:657-668.
Rubin K. Emerging therapies for cystic fibrosis lung disease. Chest. 1998;115:1120-1126.
Rush PJ, Shore A, Coblentz C, et al. The musculoskeletal manifestations of C.F. Seminars in Arthritis and Rheumatism. 1986;15(3):213-225.
Rutland J, Cole PJ. Non-invasive sampling of nasal cilia for measurement of beat frequency and study of ultrastructure. Lancet. 1980;ii:564-565.
Sahl W, Bilton D, Dodd M, Webb AK. Effect of exercise and physiotherapy in aiding sputum expectoration in adults with cystic fibrosis. Thorax. 1989;44:1006-1008.
Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa. A randomized clinical controlled trial. Journal of the American Medical Association. 2003;290:1749-1756.
Sapsford RR, Hodges PW, Richardson CA, et al. Co-activation of the abdominal and pelvic floor muscles during voluntary exercises. Neurourology and Urodynamics. 2001;20:31-42.
Sawyer E, Clayton TL. Improved pulmonary function and exercise tolerance with inspiratory muscle conditioning in children with cystic fibrosis. Chest. 1993;104:1490-1497.
Scheinberg P, Shore E. A pilot study of the safety and the efficacy of tobramycin solution for inhalation in patients with severe bronchiectasis. Chest. 2005;127:1420-1426.
Schneiderman-Walker J, Pollack SL, Corey M, et al. A randomized controlled trial of a 3-year home exercise program in cystic fibrosis. Journal of Pediatrics. 2000;136:304-310.
Schöni MH. Autogenic drainage - a modern approach to chest physiotherapy in cystic fibrosis. Journal of the Royal Society of Medicine. 1989;82(Suppl 16):32-37.
Shah PL, Scott SF, Knight RA, et al. In vivo effects of recombinant human DNase I on sputum in patients with cystic fibrosis. Thorax. 1996;51:119-125.
Shah PL, Scott SF, Geddes DM, et al. An evaluation of two aerosol delivery systems for rhDNase. European Respiratory Journal. 1997;10:1261-1267.
Shale DJ. Predicting survival in cystic fibrosis. Thorax. 1997;52:309.
Shale DJ. Commentary. Thorax. 1997;52:95-96.
Sharp JT, Drutz WS, Moisan T, Forster J, Machnach W. Postural relief of dyspnea in severe chronic obstructive pulmonary disease. American Review of Respiratory Disease. 1980;122:201-211.
Shepherd SL, Hovell MF, Slymen DJ, et al. Functional status as an overall measure of health in adults with cystic fibrosis: a further validation of a generic health measure. Journal of Clinical Epidemiology. 1992;45:117-125.
Simmonds EJ, Littlewood JM, Evans EGV. Cystic fibrosis and allergic bronchopulmonary aspergillosis. Archives of Disease in Childhood. 1990;65:507-511.
Sindel BD, Maisels MJ, Ballantine TVN. Gastroesophageal reflux to the proximal esophagus in infants with bronchopulmonary dysplasia. American Journal of Disease in Childhood. 1989;143:1103-1106.
Skov M, McKay K, Koch C, Cooper PJ. Prevalence of allergic bronchopulmonary aspergillosis in cystic fibrosis in an area with a high frequency of atopy. Respiratory Medicine. 2005;99:887-893.
Smith IE, Flower CDR. Review article: imaging in bronchiectasis. British Journal of Radiology. 1996;69:589-593.
Speechly-Dick ME, Rimmer SJ, Hodson ME. Exacerbation of cystic fibrosis after holidays at high altitude: a cautionary tale. Respiratory Medicine. 1992;86:55-56.
Spier S, Rivlin J, Hughes D, et al. The effect of oxygen on sleep, blood gases and ventilation in cystic fibrosis. American Review of Respiratory Disease. 1984;129:712-718.
Stanley P, MacWilliam L, Greenstone M, et al. Efficacy of a saccharin test for screening to detect abnormal mucociliary clearance. British Journal of Diseases of the Chest. 1984;78:62-65.
Stead RJ, Davidson TI, Duncan FR, et al. Use of a totally implantable system for venous access in cystic fibrosis. Thorax. 1987;42:149-150.
Stern M, Geddes D. Gene therapy for cystic fibrosis. Respiratory Disease in Practice. 1994;Winter:18-23.
Strauss GD, Osher A, Wang C, et al. Variable weight training in cystic fibrosis. Chest. 1987;92:273-276.
Suri R. The use of human deoxyribonuclease (rhDNase) in the management of cystic fibrosis. BioDrugs. 2005;19(3):135-144.
Suri R, Grieve R, Normand C, et al. Effects of hypertonic saline, alternate day and daily rhDNase on healthcare use, costs and outcomes in children with cystic fibrosis. Thorax. 2002;57:841-846.
Tannenbaum E, Prasad SA, Dinwiddie R, Main E. Chest physiotherapy during anaesthesia for children with cystic fibrosis: effects on respiratory function. Pediatric Pulmonology. 2007;42(12):1152-1158.
Tattersall R, Callaghan H, Groves D, Walshaw MJ, 2000 Assessment of posture by Quantec scanning in adult cystic fibrosis (CF) patients. Proceedings of the XIIIth International Cystic Fibrosis Congress, Stockholm S237, p 148
Taylor CJ, Threlfall D. Postural drainage techniques and gastro-oesophageal reflux in cystic fibrosis. Lancet. 1997;349:1567-1568.
ten Berge M, Brinkhorst G, Kroon AA, et al. DNase treatment in primary ciliary dyskinesia - assessment by nocturnal pulse oximetry. Pediatric Pulmonology. 1999;27:59-61.
Thomas J, Cook DJ, Brooks D. Chest physical therapy management of patients with cystic fibrosis: a meta-analysis. American Journal of Respiratory and Critical Care Medicine. 1995;151:846-885.
Thomas TM, Plymat KR, Blannin J, Meade TW. Prevalence of urinary incontinence. British Medical Journal. 1980;281:1243-1324.
Thompson CS, Harrison S, Ashley J, et al. Randomized crossover study of the Flutter device and the active cycle of breathing technique in non-cystic fibrosis bronchiectasis. Thorax. 2002;57:446-448.
Thornton J, Elliot R, Tully MP, Dodd M, Webb AK. Long-term clinical outcome of home and hospital intravenous antibiotic treatment in adults with cystic fibrosis. Thorax. 2004;59:242-246.
Touw DJ, Brimicombe RW, Hodson ME, et al. Inhalation of antibiotics in cystic fibrosis. European Respiratory Journal. 1995;8:1594-1604.
Turner JA, Corkey CW, Lee JY, et al. Clinical expression of immotile cilia syndrome. Pediatrics. 1981;67:805-810.
Turner M, Baildam E, Patel L, et al. Joint disorders in CF. Journal of the Royal Society of Medicine. 1997;90(Suppl 31):13-20.
Unsworth R, Davis A, Dodd ME 1998 Does education improve patient understanding and adherence with airway clearance techniques? Proceedings of 22nd European Cystic Fibrosis Conference
Valerius NH, Koch C, Høiby N. Prevention of chronic Pseudomonas aeruginosa colonisation in cystic fibrosis by early treatment. Lancet. 1991;338:725-772.
van der Schans CP, Postma KS, Koeter GH, Rubin BK. Physiotherapy and bronchial mucus transport. European Respiratory Journal. 1999;13:1477-1486.
van der Schans C, Prasad A, Main E, 2000 Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis. Cochrane Database of Systematic Reviews 2000, Issue 2. Art. No.: CD001401. DOI: 10.1002/14651858.CD001401
Vibekk P. Chest mobilization and respiratory function. In: Pryor JA, editor. Respiratory care. Edinburgh: Churchill Livingstone; 1991:103-119.
Vidal V, Therasse E, Berthiaume Y, et al. Bronchial artery embolization in adults with cystic fibrosis: impact on the clinical course and survival. Journal of Vascular and Interventional Radiology. 2006;17(6):953-958.
Vizza CD, Yusen RD, Jynch JP, et al. Outcome of patients with cystic fibrosis awaiting lung transplantation. American Journal of Respiratory and Critical Care Medicine. 2000;162:819-825.
Walters S, Britton J, Hodson ME. Demographic and social characteristics of adults with cystic fibrosis in the United Kingdom. British Medical Journal. 1993;306:549-552.
Wanner A, Rao A. Clinical implications for and effects of bland, mucolytic and antibiotic aerosols. American Review of Respiratory Diseases. 1980;122:79-87.
Webb AK, Dodd ME. Nebulized antibiotics for adults with cystic fibrosis. Thorax. 1997;52(Suppl 2):S69-71.
Webb AK, Dodd ME. Exercise and sport in cystic fibrosis: benefits and risks. British Journal of Sports Medicine. 1999;33:77-78.
Webb AK, Dodd ME. Exercise and training for adults with cystic fibrosis. In: Hodson ME, Geddes DM, editors. Cystic fibrosis. London: Arnold; 2000:433-444.
Webb AK, Egan J, Dodd ME. Clinical management of cystic fibrosis patients awaiting and immediately following lung transplantation. In: Dodge A, Brock DJH, Widdecombe JH, editors. Cystic fibrosis: current topics, vol 3. Chichester: John Wiley; 1996:311-337.
Weinberger JBL, Weiss ST, Cohen WR, et al. Pregnancy and the lung. American Review of Respiratory Disease. 1980;121:559-581.
Westaby D. Liver and biliary disease. In: Hodson ME, Geddes DM, editors. Cystic fibrosis. London: Arnold; 2000:289-300.
Wills P, Greenstone M, 2006 Inhaled hyperosmolar agents for bronchiectasis. Cochrane Database of Systematic Reviews 2006, Issue 2. Art. No.: CD002996. DOI: 10.1002/14651858.CD002996.pub2
Wills PJ, Wodehouse T, Corkery K, et al. Short-term recombinant human DNase in bronchiectasis. American Journal of Respiratory and Critical Care Medicine. 1996;154:413-417.
Wilson R, Sykes DA, Chan KL, et al. Effect of head position on the efficacy of topical treatment of chronic mucopurulent rhinosinusitis. Thorax. 1987;42:631-663.
Wolfe S, Collins S. Nutritional aspects. In Hodson M, Geddes D, Bush A, editors: Cystic fibrosis, 3rd edn., London: Hodder Arnold, 2007.
Wolfgang K, Schwabe A, Kramer I. Inhalation solutions - which ones are allowed to be mixed? Physico-chemical compatibility of drug solutions in nebulizers. Journal of Cystic Fibrosis. 2006;5:205-213.
Wolter J, Seeney S, Bell S, et al. Effect of long-term treatment with azithromycin on disease parameters in cystic fibrosis: a randomized trial. Thorax. 2002;57:212-216.
Wood RE, Wanner A, Hirsch J, et al. Tracheal muco-ciliary transport in cystic fibrosis and its stimulation by terbutaline. American Review of Respiratory Disease. 1975;111:733-738.
World Health Organization Report, 2003 Adherence to long-term therapies. O3.01
Zabner JJJ, Smith PH, Karp J, Widdicombe JH, Welsh MJ. Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Molecular Cell. 1998;2(3):397-403.
Zach MS, Oberwaldner B, Forche G, et al. Bronchodilators increase airway instability in cystic fibrosis. American Review of Respiratory Disease. 1985;131:537-543.
Zihlif N, Paraskakis E, Lex C, Van de Pohl LA, Bush A. Correlation between cough frequency and airway inflammation in children with primary ciliary dyskinesia. Pediatric Pulmonology. 2005;39(6):551-557.
Bush A, Alton EWFW, Davies JC, Griesenbach U, Jaffe A, editors. Cystic fibrosis in the 21st century. Basel: Karger, 2006.
Bluebond-Langner M, Lask B, Angst D. Psychosocial aspects of cystic fibrosis. London: Arnold, 2001.
Hodson M, Geddes D, Bush A, editors. Cystic fibrosis, 3rd edn., London: Hodder Arnold, 2007.