1 MacSween R et al.: Developmental anatomy and normal structure. In MacSween R (ed): Pathology of the Liver, 4th ed. 2002.
2 Crawford JM. Development of the intrahepatic biliary tree. Semin Liver Dis. 2002;22:213.
3 Lim YS, Kim WR. The global impact of hepatic fibrosis and end-stage liver disease. Clin Liver Dis. 2008;12:733.
4 Lee WM. Etiologies of acute liver failure. Semin Liver Dis. 2008;28:142.
5 Fontana RJ. Acute liver failure due to drugs. Semin Liver Dis. 2008;28:175.
6 Haussinger D, Schliess F. Pathogenetic mechanisms of hepatic encephalopathy. Gut. 2008;57:1156.
7 Munoz SJ. The hepatorenal syndrome. Med Clin N Am. 2008;92:813.
8 Rodriguez-Rosin R, Krowka MJ. Hepatopulmonary syndrome—a liver induced lung vascular disease. N Engl J Med. 2008;358:2378.
9 Friedman SE. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655.
10 Sanyal AJ, et al. Portal hypertension and its complications. Gastroenterology. 2008;134:1715.
11 Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838.
12 Desmet VJ, Roskams T. Cirrhosis reversal: a duel between dogma and myth. J Hepatol. 2004;40:860.
13 Iwakiri Y, Groszmann RJ. Vascular endothelial dysfunction in cirrhosis. J Hepatol. 2007;46:927.
14 Bosch J. Vascular deterioration in cirrhosis: The big picture. J Clin Gastroenterol. 2007;41:S247.
15 Servedio V, et al. Spectrum of UGT1A1 mutations in Crigler-Najjar (CN) syndrome patients: identification of twelve novel alleles and genotype-phenotype correlation. Hum Mutat. 2005;25:325.
16 Kartenbeck J, et al. Absence of the canalicular isoform of the MRP gene-encoded conjugate export pump from the hepatocytes in Dubin-Johnson syndrome. Hepatology. 1996;23:1061.
17 O’Leary JG, Pratt DS. Cholestasis and cholestatic syndromes. Curr Opin Gastroenterol. 2007;23:232.
18 Paulusma CC, et al. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology. 2008;47:268.
19 Suchy FJ, Ananthanarayanan M. Bile salt excretory pump: biology and pathobiology. J Pediatr Gastroenterol Nutr. 2006;43(Suppl 1):S10.
20 Sundaram SS, Sokol RJ. The multiple facets of ABCB4 (MDR3) deficiency. Current Treat Options Gastroenterol. 2007;101:495.
21 Feigelstock D, et al. The human homolog of HAVcr-1 codes for a hepatitis A virus cellular receptor. J Virol. 1998;72:6621.
22 Martin A, Lemon SM. Hepatitis A virus: from discovery to vaccines. Hepatology. 2006;43:S164.
23 Andre FE. Universal mass vaccination against hepatitis A. Curr Top Microbiol Immunol. 2006;304:95.
24 Pungpapong S, et al. Natural history of hepatitis B infection: an update for clinicians. May Clin Proc. 2007;82:967.
25 Blumberg BS, et al. A “new” antigen in leukemia sera. JAMA. 1965;191:541.
26 Block TM, et al. Molecular virology of hepatitis B virus for clinicians. Clin Liver Dis. 2007;11:685.
27 Bertoletti A, Gehring AJ. The immune response during hepatitis B virus infection. J Gen Virol. 2006;87:1439.
28 Pawlotsky JM. Hepatitis C virus population dynamics during infection. Curr Top Microbiol Immunol. 2006;299:261.
29 Missiha SB, et al. Disease progression in chronic hepatitis C: modifiable and nonmodifiable factors. Gastroenterology. 2008;134:1699.
30 Koytak ES, Yurdaydin C, Glenn JS. Hepatitis D. Current Treat Options Gastroenterol. 2007;10:456.
31 Purcell RH, Emerson SU. Hepatitis E: an emerging awareness of an old disease. J Hepatol. 2008;48:494.
32 Williams CF, et al. Persistent GB virus C infection and survival in HIV-infected men. N Engl J Med. 2004;350:981.
33 Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology. 2006;43:S173.
34 Weber R, et al. Liver-related deaths in persons infected with the human immunodeficiency virus: the D:A:D study. Arch Intern Med. 2006;166:1632.
35 Goodman ZD. Grading and staging systems for inflammation and fibrosis in chronic liver diseases. J Hepatol. 2007;47:598.
36 Leifeld L, et al. Intrahepatic activation of caspases in human fulminant hepatic failure. Liver Int. 2006;26:872.
37 Krawitt EL. Autoimmune hepatitis. N Engl J Med. 2006;354:54.
38 Czaja AJ. Autoimmune liver disease. Curr Opin Gastroenterol. 2007;23:255.
39 Arundel C, Lewis JH. Drug-induced liver disease in 2006. Curr Opin Gastroenterol. 2007;23:244.
40 Bleibel W, et al. Drug-induced liver injury: review article. Dig Dis Sci. 2007;52:2463.
41 Aithal GP. Dangerous liaisons: drug, host and the environment. J Hepatol. 2007;46:995.
42 Gramenzi A, et al. Review article: alcoholic liver disease—pathophysiological aspects and risk factors. Aliment Pharmacol Ther. 2006;24:1151.
43 Ongi JP, Younossi ZM. Epidemiology and natural history of NAFLD and NASH. Clin Liv Dis. 2007;11:1.
44 Yeh MM, Brunt EM. Pathology of non-alcoholic fatty liver disease. Am J Clin Pathol. 2007;128:837.
45 Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99.
46 Greenfield V, Cheung O, Sanyal AJ. Recent advances in non-alcoholic fatty liver. Curr Opin Gastroenterol. 2008;24:320.
47 Pietrangelo A. Hereditary hemochromatosis. Annu Rev Nutr. 2006;26:251.
48 Du X, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320:1088.
49 Whitington PF. Neonatal hemochromatosis: a congenital alloimmune hepatitis. Semin Liver Dis. 2007;27:243.
50 Gordeuk VR, et al. Iron overload in Africans and African-Americans and a common mutation in the SCL40A1 (ferroportin 1) gene. Blood Cells Mol Dis. 2003;31:299.
51 Pfeiffer RF. Wilson’s disease. Semin Neurol. 2007;27:123.
52 Ioachimescu OC, Stoller JK. A review of alpha-1 antitrypsin deficiency. COPD. 2005;2:263.
53 Perlmutter DH. Pathogenesis of chronic liver injury and hepato-cellular carcinoma in alpha-1-antitrypsin deficiency. Pediatr Res. 2006;60:233.
54 Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261.
55 Jones DE. Pathogenesis of primary biliary cirrhosis. Gut. 2007;56:1615.
56 Abe M, Onji M. Natural history of primary biliary cirrhosis. Hepatology Res. 2008;38:639.
57 Weismüller TJ, et al. The challenges in primary sclerosing cholangitis—Aetiopathogenesis, autoimmunity, management and malignancy. J Hepatol. 2008;48:S38.
58 Maggs JL, Champamn RW. An update on primary sclerosing cholangitis. Curr Opin Gastroenter. 2008;24:377.
59 Nakanuma Y, Zen Y. Pathology and immunopathology of immunoglobulin G4-related sclerosing cholangitis: the latest addition to the sclerosing cholangitis family. Hepatol Res. 2007;37(Suppl 3):S478.
60 Everson GT, et al. Advances in the management of polycycstic liver disease. Expert Rev Gastroenterol Hepatol. 2008;2:563.
61 Masyuk T, LaRusso N. Polycystic liver disease: New insights into disease pathogenesis. Hepatology. 2006;43:906.
62 Gunay-Aygun M, et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: summary statement of a first National Institutes of Health/Office of Rare Diseases conference. J Pediatr. 2006;149:159.
63 Warthen DM, et al. Jagged1 (JAG1) mutations in Alagille syndrome: increasing the mutation detection rate. Hum Mutat. 2006;27:436.
64 Chawla Y, Dhiman RK. Intrahepatic portal venopathy and related disorders of the liver. Semin Liv Dis. 2008;208:270.
65 Pulliainen AT, Dehio C. Bartonella henselae: Subversion of vascular endothelial cell functions by translocated bacterial effector proteins. Int J Biochem Cell Biol. 2008. epub.
66 DeLeve LD. Hepatic microvasculature in liver injury. Semin Liver Dis. 2007;27:390.
67 Demetris A, et al. Update of the International Banff Schema for Liver Allograft Rejection: working recommendations for the histopathologic staging and reporting of chronic rejection. An International Panel. Hepatology. 2000;31:792.
68 Hay JE. Liver disease in pregnancy. Hepatology. 2008;47:1067.
69 Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv. 2004;59:838.
70 Browning MF, et al. Fetal fatty acid oxidation defects and maternal liver disease in pregnancy. Obstet Gynecol. 2006;107:115.
71 Mallet V, et al. Nodular regenerative hyperplasia is a new cause of chronic liver disease in HIV-infected patients. AIDS. 2007;21:187.
72 Zucman-Rossi J, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43:515.
73 Reznik Y, et al. Hepatocyte nuclear factor-1 alpha gene inactivation: cosegregation between liver adenomatosis and diabetes phenotypes in two maturity-onset diabetes of the young (MODY)3 families. J Clin Endocrinol Metab. 2004;89:1476.
74 Linabery AM, Ross JA. Trends in childhood cancer incidence in the U.S. (1992–2004). Cancer. 2008;112:416.
75 Cairo S, et al. Hepatic stem-like phenotype and interplay of Wnt/beta catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;9:14.
76 Adesina AM, et al. FOXG1 is overexpressed in hepatoblastoma. Hum Pathol. 2007;38:400.
77 Parkin DM, et al. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74.
78 Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25:3834.
79 Ni YH, et al. Two decades of universal hepatitis B vaccination in Taiwan: impact and implication for future strategies. Gastroenterology. 2007;132:1287.
80 Torbenson M. Review of the clinicopathologic features of fibrolamellar carcinoma. Adv Anat Pathol. 2007;14:217.
81 Lee JS, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410.
82 Llovet JM, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Nat Cancer Inst. 2008;100:698.
83 Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Eng J Med. 2008;359:378.
84 Gores GJ, Blechacz B. Cholangiocarcinoma: Advances in pathogenesis, diagnosis and treatment. Hepatology. 2008;48:308.
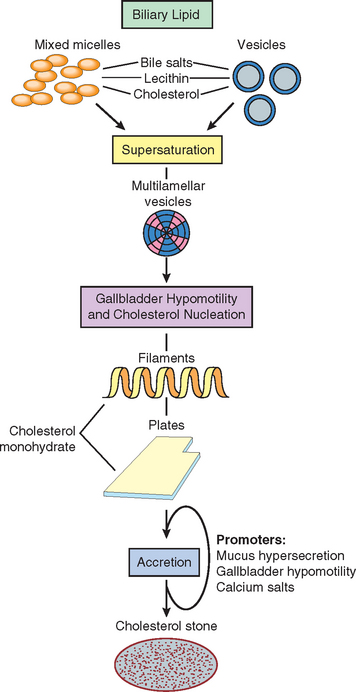
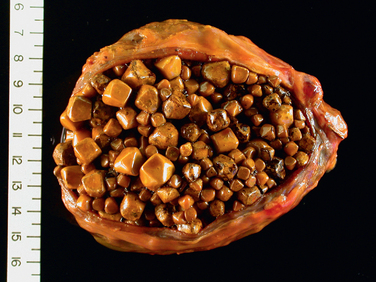
85 Lammert F, Miguel JF. Gallstone disease: from genes to evidence based therapy. J Hepatol. 2008;48:S124.
86 Strasberg SM. Acute calculous cholecystitis. New Engl J Med. 2008;358:2804.
87 Elwood DR. Cholecystitis. Surg Clin N Am. 2008;88:1241.
88 Petersen C. Pathogenesis and treatment opportunities for biliary atresia. Clin Liver Dis. 2006;10:73. vi,
89 Mack CL. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis. 2007;27:233.
90 Kerkar N, et al. The hepatic fibrocystic diseases. Clin Liver Dis. 2006;10:55. v–vi,
91 Randi G, et al. Gallbladder cancer worldwide: geographical distribution and risk factors. Int J Cancer. 2006;118:1591.