Chapter 19 The Pancreas
The adult pancreas is a transversely oriented retroperitoneal organ extending from the “C” loop of the duodenum to the hilum of the spleen (Fig. 19-1). On average, the pancreas measures 20 cm in length and weighs 90 gm in men and 85 gm in women.1 The vasculature adjacent to the pancreas can be used to separate the pancreas into four parts: the head, neck, body, and tail.
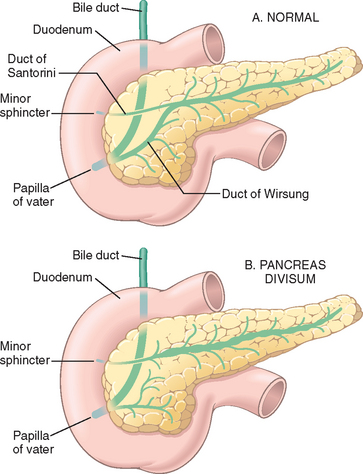
FIGURE 19-1 Pancreatic ductal anatomy. A, The normal ductal anatomy. B, The ductal anatomy in pancreatic divisum.
(Adapted from Gregg JA et al.: Pancreas divisum: results of surgical intervention. Am J Surg 145:488–492, 1983.)
The pancreatic duct system is highly variable. The main pancreatic duct, also known as the duct of Wirsung, most commonly drains into the duodenum at the papilla of Vater, whereas the accessory pancreatic duct, also known as the duct of Santorini, most often drains into the duodenum through a separate minor papilla approximately 2 cm cephalad (proximal) to the major papilla of Vater (Fig. 19-1A). In most adults the main pancreatic duct joins the common bile duct proximal to the papilla of Vater, thus creating the ampulla of Vater, a common channel for biliary and pancreatic drainage. This ductal architecture can differ significantly from person to person.
The pancreas arises from the fusion of dorsal and ventral outpouchings of the foregut, which fuse to form a single organ.2,3 The majority of the gland, including the body, the tail, the superior/anterior aspect of the head, and the accessory duct of Santorini, is derived from the dorsal primordium. The ventral primordium gives rise to the posterior/inferior part of the head of the pancreas, and drains into the papilla of Vater.
Although the organ gets its name from the Greek pankreas, meaning “all flesh,” the pancreas is, in fact, a complex lobulated organ with distinct exocrine and endocrine components. The exocrine portion, which produces digestive enzymes, constitutes 80% to 85% of the pancreas. The endocrine portion is composed of about 1 million clusters of cells, the islets of Langerhans. The islet cells secrete insulin, glucagon, and somatostatin and constitute only 1% to 2% of the organ. Diseases of the endocrine pancreas are described in detail in Chapter 24.
The exocrine pancreas is composed of acinar cells, which produce the enzymes needed for digestion, and a series of ductules and ducts that convey secretions to the duodenum.1 Acinar cells are pyramidally shaped epithelial cells that are radially oriented around a central lumen (Fig. 19-2). Acinar cells contain membrane-bound zymogen granules rich in digestive enzymes.
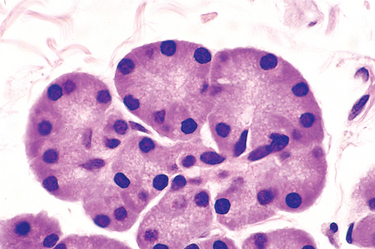
FIGURE 19-2 Pancreatic acini, showing the radial orientation of the pyramidal exocrine acinar cells. The cytoplasm is devoted to the synthesis and packaging of digestive enzymes for secretion into a central lumen.
The pancreas secretes its exocrine products as enzymatically inert proenzymes. They include trypsinogen, chymotrypsinogen, procarboxypeptidase, proelastase, kallikreinogen, and prophospholipase A and B.1 Self-digestion of pancreatic tissue is prevented by several mechanisms:
The most significant disorders of the exocrine pancreas include cystic fibrosis, congenital anomalies, acute and chronic pancreatitis, pseudocysts, and neoplasms. Cystic fibrosis is discussed in detail in Chapter 10.
Congenital Anomalies
The complex process by which the dorsal and ventral pancreatic primordia fuse during pancreatic development frequently gives rise to congenital variations in pancreatic anatomy.3 Most of these do not directly cause disease; however, such variations, especially in ductal anatomy, may present particular problems to endoscopists and surgeons. For example, failure to recognize aberrant ductal anatomy may lead to the inadvertent ligation of a pancreatic duct during surgery, causing serious sequelae such as pancreatitis.
AGENESIS
Very rarely the pancreas may be totally absent (agenesis), a condition associated with other severe malformations that are usually incompatible with life. PDX1 (pancreatic and duodenal homeobox-1 gene) encodes a transcription factor critical for the development of the pancreas.3 Homozygous PDX1 mutations on chromosome 13q12.1 have been reported in a person with pancreatic agenesis.3,4
PANCREAS DIVISUM
Pancreas divisum is the most common congenital anomaly of the pancreas, with an incidence of 3% to 10%.4 This anomaly is caused by a failure of fusion of the fetal duct systems of the dorsal and ventral pancreatic primordia.4 As a result, the bulk of the pancreas (formed by the dorsal pancreatic primordium) drains through the dorsal pancreatic duct and the small-caliber minor papilla (see Fig. 19-1B).4 The duct of Wirsung in persons with divisum, normally the main pancreatic duct, is very short (1 to 2 cm) and drains only a small portion of the head of the gland through the larger caliber major papilla of Vater. Although controversy exists about the clinical significance of pancreatic divisum, it has been suggested that the relative stenosis caused by the bulk of the pancreatic secretions passing through the minor papilla predisposes individuals to the development of chronic pancreatitis.4,5
ANNULAR PANCREAS
Annular pancreas is a band-like ring of normal pancreatic tissue that completely encircles the second portion of the duodenum. Annular pancreas is often associated with other congenital anomalies and may present early in life or in adults with signs and symptoms of duodenal obstruction such as gastric distention and vomiting.4,6
ECTOPIC PANCREAS
Aberrantly situated, or ectopic, pancreatic tissue is found in about 2% of careful routine postmortem examinations. The favored sites for ectopia are the stomach and duodenum, followed by the jejunum, Meckel diverticula, and ileum.4 These embryologic rests are a few millimeters to centimeters in size and are located in the submucosa. Histologic examination reveals that they are composed of normal-appearing pancreatic acini, glands, and sometimes islets of Langerhans. Though usually incidental, ectopic pancreas may cause pain from localized inflammation, or, rarely, may incite mucosal bleeding. Approximately 2% of islet cell neoplasms (Chapter 24) arise in ectopic pancreatic tissue. The pathogenesis of ectopic pancreas has not been established.
Pancreatitis
Pancreatitis is inflammation in the pancreas associated with injury to the exocrine parenchyma. The clinical manifestations range in severity from a mild, self-limited disease to a life-threatening acute inflammatory process, and the duration of the disease can range from a transient attack to a permanent loss of function.7,8 In acute pancreatitis the gland can return to normal if the underlying cause of the pancreatitis is removed.9,10 By contrast, chronic pancreatitis is defined by the irreversible loss of exocrine pancreatic parenchyma.7,11
ACUTE PANCREATITIS
Acute pancreatitis is reversible pancreatic parenchymal injury associated with inflammation. Acute pancreatitis is relatively common, with an annual incidence rate in Western countries of 10 to 20 cases per 100,000 people. Biliary tract disease and alcoholism account for approximately 80% of cases in Western countries (Table 19-1).8-10,12 Gallstones are present in 35% to 60% of cases of acute pancreatitis, and about 5% of patients with gallstones develop pancreatitis. The proportion of cases of acute pancreatitis caused by excessive alcohol intake varies from 65% in the United States to 20% in Sweden to 5% or less in southern France and the United Kingdom.13 The male-to-female ratio is 1 : 3 in the group with biliary tract disease and 6 : 1 in those with alcoholism.
TABLE 19-1 Etiologic Factors in Acute Pancreatitis
| METABOLIC |
| GENETIC |
| Mutations in the cationic trypsinogen (PRSS1) and trypsin inhibitor (SPINK1) genes |
| MECHANICAL |
| VASCULAR |
| INFECTIOUS |
| Mumps |
Less common causes of acute pancreatitis include the following:
Hereditary Pancreatitis.
Notably, 10% to 20% of individuals with acute pancreatitis have no known associated processes. Although this condition is currently termed idiopathic, a growing body of evidence suggests that some of these cases actually have a genetic basis. The genetic alterations associated with the development of pancreatitis therefore deserve special note.17 Hereditary pancreatitis is characterized by recurrent attacks of severe pancreatitis usually beginning in childhood.16,17 Most cases are caused by germline (inherited) mutations in the cationic trypsinogen gene (also known as PRSS1).16 These mutations abrogate a critical fail-safe mechanism by altering a site on the cationic trypsinogen molecule that is essential for the cleavage (inactivation) of trypsin by trypsin itself.17 When this site is mutated, trypsin becomes resistant to cleavage by another trypsin molecule, and if a small amount of this tryspin is inappropriately activated in the pancreas, it can activate other digestive proenzymes, resulting in the development of pancreatitis. Only one mutated allele is required for cleavage-resistant trypsin to be produced; thus, this form of hereditary pancreatitis has an autosomal dominant mode of inheritance.
The serine protease inhibitor Kazal type 1 (SPINK1) gene codes for a pancreatic secretory trypsin inhibitor that, as the name suggests, inhibits trypsin activity, helping to prevent the autodigestion of the pancreas by activated trypsin.17 As one might suspect, inherited inactivating mutations in the SPINK1 gene can also lead to the development of pancreatitis. This form of hereditary pancreatitis has an autosomal recessive mode of inheritance, as both alleles must be inactivated.
Morphology. The morphology of acute pancreatitis ranges from trivial inflammation and edema to severe extensive necrosis and hemorrhage. The basic alterations are (1) microvascular leakage causing edema, (2) necrosis of fat by lipolytic enzymes, (3) acute inflammation, (4) proteolytic destruction of pancreatic parenchyma, and (5) destruction of blood vessels and subsequent interstitial hemorrhage. The extent of each of these alterations depends on the duration and severity of the process.
In the milder form, acute interstitial pancreatitis, histologic alterations are limited to mild inflammation, interstitial edema, and focal areas of fat necrosis in the substance of the pancreas and in peripancreatic fat (Fig. 19-3). Fat necrosis, as we have seen, results from enzymatic activity of lipase. The released fatty acids combine with calcium to form insoluble salts that impart a granular blue microscopic appearance to the fat cells (Chapter 1).
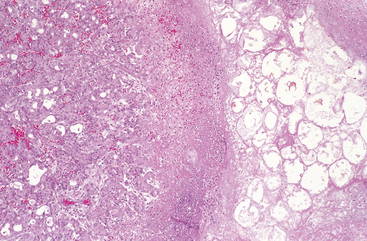
FIGURE 19-3 Acute pancreatitis. The microscopic field shows a region of fat necrosis on the right and focal pancreatic parenchymal necrosis (center).
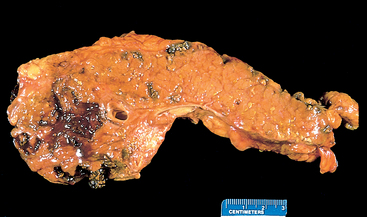
In the more severe form, acute necrotizing pancreatitis, the acinar and ductal tissues as well as the islets of Langerhans are necrotic. Vascular injury can lead to hemorrhage into the parenchyma of the pancreas. Macroscopically, the pancreatic substance shows areas of red-black hemorrhage interspersed with foci of yellow-white, chalky fat necrosis (Fig. 19-4). Foci of fat necrosis may also be found in extra-pancreatic collections of fat, such as the omentum and the mesentery of the bowel, and even outside the abdominal cavity, such as in the subcutaneous fat. In the majority of cases the peritoneal cavity contains a serous, slightly turbid, brown-tinged fluid in which globules of fat (derived from the action of enzymes on adipose tissue) can be identified. In its most severe form, hemorrhagic pancreatitis, extensive parenchymal necrosis is accompanied by dramatic hemorrhage within the substance of the gland.20,21
Pathogenesis.
The anatomic changes of acute pancreatitis strongly suggest autodigestion of the pancreatic substance by inappropriately activated pancreatic enzymes. This hypothesis is supported by the hereditary forms of pancreatitis described above. Here we focus on the more common, acquired forms of acute pancreatitis.
As has been discussed, pancreatic enzymes, including trypsin, are synthesized in an inactive proenzyme form. If trypsin is inappropriately activated it can in turn activate other proenzymes such as prophospholipase and proelastase, which then degrade fat cells and damage the elastic fibers of blood vessels, respectively.8-10,12 Trypsin also converts prekallikrein to its activated form, thus bringing into play the kinin system and, by activation of Hageman factor (factor XII), the clotting and complement systems as well (Chapters 2 and 4. In this way inflammation and small-vessel thromboses (which may lead to congestion and rupture of already weakened vessels) are amplified. Thus, the inappropriate activation of trypsinogen is an important triggering event in acute pancreatitis.
The mechanisms by which the activation of pancreatic enzymes is initiated are not entirely clear, but there is evidence for three possible events (Fig. 19-5):
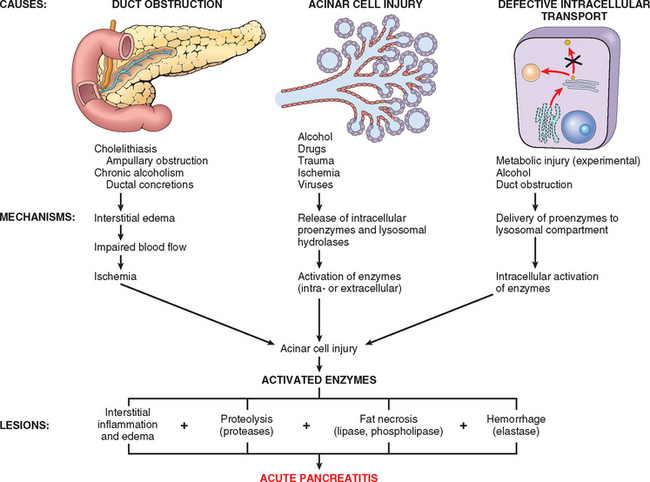
Alcohol consumption may cause pancreatitis by several mechanisms. Chronic alcohol ingestion results in the secretion of protein-rich pancreatic fluid, which leads to the deposition of inspissated protein plugs and obstruction of small pancreatic ducts. Alcohol also transiently increases pancreatic exocrine secretion and contraction of the sphincter of Oddi (the muscle at the ampulla of Vater), and it has direct toxic effects on acinar cells.29
Clinical Features.
Abdominal pain is the cardinal manifestation of acute pancreatitis.8-10,12 Characteristically, the pain is constant and intense and is often referred to the upper back and occasionally can be associated with referred pain to the left shoulder. Its severity varies from mild and uncomfortable to severe and incapacitating. Anorexia, nausea, and vomiting frequently accompany the pain. Suspected acute pancreatitis is primarily diagnosed by the presence of elevated plasma levels of amylase and lipase and the exclusion of other causes of abdominal pain.
Full-blown acute pancreatitis is a medical emergency. These patients usually have the sudden calamitous onset of an “acute abdomen.” Many of the systemic features of severe acute pancreatitis can be attributed to release of toxic enzymes, cytokines, and other mediators into the circulation and explosive activation of the systemic inflammatory response, resulting in leukocytosis, hemolysis, disseminated intravascular coagulation, fluid sequestration, acute respiratory distress syndrome, and diffuse fat necrosis. Peripheral vascular collapse and shock with acute renal tubular necrosis may occur.8-10,12
Laboratory findings include marked elevation of serum amylase levels during the first 24 hours, followed within 72 to 96 hours by a rising serum lipase level. Glycosuria occurs in 10% of cases. Hypocalcemia may result from precipitation of calcium soaps in necrotic fat; if persistent, it is a poor prognostic sign. Direct visualization of the enlarged inflamed pancreas by radiography is useful in the diagnosis of pancreatitis.
The key to the management of acute pancreatitis is “resting” the pancreas by total restriction of oral intake and by supportive therapy with intravenous fluids and analgesia. Although most individuals with acute pancreatitis recover fully, about 5% with severe acute pancreatitis die from shock during the first week of illness. Acute respiratory distress syndrome and acute renal failure are ominous complications.8-10,12 Sequelae can include a sterile pancreatic abscess and a pancreatic pseudocyst (discussed later). In 40% to 60% of patients with acute necrotizing pancreatitis the necrotic debris becomes infected, usually by gram-negative organisms from the alimentary tract, further complicating the clinical course.
CHRONIC PANCREATITIS
Chronic pancreatitis is defined as inflammation of the pancreas with irreversible destruction of exocrine parenchyma, fibrosis, and, in the late stages, the destruction of endocrine parenchyma.11,30 Although chronic pancreatitis may present as repeated bouts of acute pancreatitis, the chief distinction between acute and chronic pancreatitis is the irreversible impairment in pancreatic function that is characteristic of chronic pancreatitis. The prevalence of chronic pancreatitis ranges between 0.04% and 5%.7 There is significant overlap in the causes of acute and chronic pancreatitis. By far the most common cause of chronic pancreatitis is long-term alcohol abuse, and these patients are usually middle-aged males.
Less common causes of chronic pancreatitis include the following:
As many as 40% of individuals with chronic pancreatitis have no recognizable predisposing factor, but as is true for acute pancreatitis, a growing number of these “idiopathic” cases can now be shown to be caused by inherited mutations in pancreatitis-associated genes.19
Pathogenesis.
The pathogenesis of chronic pancreatitis is not well understood. Almost all individuals with repeated episodes of acute pancreatitis later develop chronic pancreatitis. It has been proposed that acute pancreatitis initiates a sequence of perilobular fibrosis, duct distortion, and altered pancreatic secretions. Over time and with multiple episodes, this can lead to loss of pancreatic parenchyma and fibrosis.32 The events that have been proposed to account for the development of chronic pancreatitis include:32,33
A variety of chemokines have been identified in chronic pancreatitis, including IL-8 and monocyte chemoattractant protein.35 In addition, transforming growth factor β (TGF-β) and platelet-derived growth factor induce the activation and proliferation of periacinar myofibroblasts (pancreatic stellate cells), resulting in the deposition of collagen and ultimately fibrosis (Fig. 19-6).36-38 While the chemokines produced during chronic pancreatitis are similar to those produced in acute pancreatitis, the profibrogenic chemokines tend to predominate in chronic pancreatitis.39
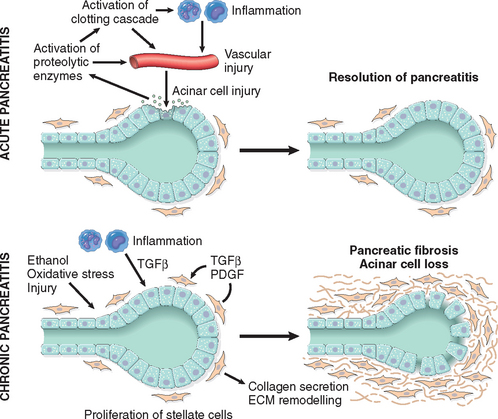
FIGURE 19-6 Comparison of the mediators in acute and chronic pancreatitis. In acute pancreatitis acinar injury results in release of proteolytic enzymes, leading to a cascade of events including activation of the clotting cascade, acute and chronic inflammation, vascular injury, and edema. In most patients, complete resolution of the acute injury occurs with restoration of acinar cell mass. In chronic pancreatitis, repeated episodes of acinar cell injury lead to the production of profibrogenic cytokines such as transforming growth factor β (TGF-β) and platelet-derived growth factor (PDGF), resulting in the proliferation of myofibroblasts, the secretion of collagen, and remodeling of the extracellular matrix (ECM). Repeated injury produces irreversible loss of acinar cell mass, fibrosis, and pancreatic insufficiency.
Morphology. Chronic pancreatitis is characterized by parenchymal fibrosis, reduced number and size of acini with relative sparing of the islets of Langerhans, and variable dilation of the pancreatic ducts (Fig. 19-7A). These changes are usually accompanied by a chronic inflammatory infiltrate around lobules and ducts. The interlobular and intralobular ducts are frequently dilated and contain protein plugs in their lumens. The ductal epithelium may be atrophied or hyperplastic or may show squamous metaplasia, and ductal concretions may be evident (Fig. 19-7B). Acinar loss is a constant feature. The remaining islets of Langerhans become embedded in the sclerotic tissue and may fuse and appear enlarged. Eventually, they too disappear. Grossly, the gland is hard, sometimes with extremely dilated ducts and visible calcified concretions. Lymphoplasmacytic sclerosing pancreatitis (autoimmune pancreatitis) is a distinct form of chronic pancreatitis characterized by a duct-centric mixed inflammatory cell infiltrate, venulitis, and increased numbers of IgG4-producing plasma cells.40 It is important to recognize lymphoplasmacytic sclerosing pancreatitis, since it can clinically mimic pancreatic cancer and also because it responds to steroid therapy.
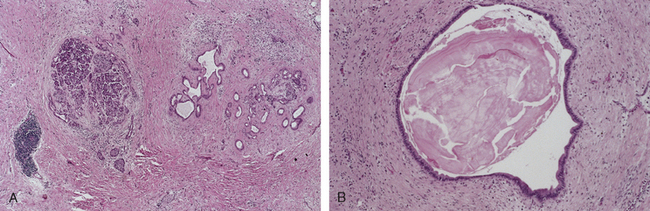
FIGURE 19-7 Chronic pancreatitis. A, Extensive fibrosis and atrophy has left only residual islets (left) and ducts (right), with a sprinkling of chronic inflammatory cells and acinar tissue. B, A higher power view demonstrating dilated ducts with inspissated eosinophilic ductal concretions in a person with alcoholic chronic pancreatitis.
Clinical Features.
Chronic pancreatitis may present in many different forms. It may be associated with repeated attacks of moderately severe abdominal pain, recurrent attacks of mild pain, or persistent abdominal and back pain. The disease may be entirely silent until pancreatic insufficiency and diabetes mellitus develop, the latter from associated destruction of islets of Langerhans. In still other instances, recurrent attacks of jaundice or vague attacks of indigestion may hint at pancreatic disease. Attacks may be precipitated by alcohol abuse, overeating (which increases demand on the pancreas), or the use of opiates and other drugs that increase the tone of the sphincter of Oddi.
The diagnosis of chronic pancreatitis requires a high degree of suspicion. During an attack of abdominal pain there may be mild fever and mild-to-moderate elevations of serum amylase. When the disease has been present for a long time, however, the destruction of acinar cells may preclude such diagnostic clues. Gallstone-induced obstruction may be evident as jaundice or elevations in serum levels of alkaline phosphatase. A very helpful finding is visualization of calcifications within the pancreas by computed tomography and ultrasonography. Weight loss and hypoalbuminemic edema from malabsorption caused by pancreatic exocrine insufficiency may point toward the disease.
Although chronic pancreatitis is usually not an immediately life-threatening condition, the long-term outlook for individuals with chronic pancreatitis is poor, with a 20- to 25-year mortality rate of 50%. Severe pancreatic exocrine insufficiency and chronic malabsorption may develop, as can diabetes mellitus. In other patients severe chronic pain may become the dominant problem. Pancreatic pseudocysts (described below) develop in about 10% of patients. While patients with hereditary pancreatitis have a 40% lifetime risk of developing pancreatic cancer, the degree to which other forms of chronic pancreatitis predispose to the development of pancreatic cancer is unclear.41,42
Non-Neoplastic Cysts
A variety of cysts can arise in the pancreas. Most are non-neoplastic pseudocysts (discussed later), but congenital cysts and neoplastic cysts also occur.
CONGENITAL CYSTS
Congenital cysts are believed to result from anomalous development of the pancreatic ducts. Cysts in the kidney, liver, and pancreas frequently coexist in polycystic disease (discussed in Chapter 20). Congenital cysts are usually unilocular, thin-walled, and range from microscopic lesions to 5 cm in diameter. They are lined by a glistening, uniform cuboidal epithelium or, if the intracystic pressure is high, by a flattened and attenuated cell layer; they are enclosed in a thin, fibrous capsule and are filled with a clear serous fluid. Congenital cysts may be sporadic, or part of autosomal-dominant polycystic kidney disease and von Hippel-Lindau disease.4 In von Hippel-Lindau disease (Chapter 20) vascular neoplasms are found in the retina and cerebellum or brain stem in association with congenital cysts (and also neoplasms) in the pancreas, liver, and kidney.
PSEUDOCYSTS
Pseudocysts are localized collections of necrotic-hemorrhagic material rich in pancreatic enzymes.43 Such cysts lack an epithelial lining (hence the prefix “pseudo”), and account for approximately 75% of cysts in the pancreas.43 Pseudocysts usually arise after an episode of acute pancreatitis, often in the setting of chronic alcoholic pancreatitis. Traumatic injury to the pancreas can also give rise to pseudocysts.
Morphology. Pseudocysts are usually solitary and may be situated within the substance of the pancreas, or, more commonly, involve the lesser omental sac or lie in the retroperitoneum between the stomach and transverse colon or between the stomach and liver. They can even be subdiaphragmatic43 (Fig. 19-8A). Pseudocysts are formed by the walling off of areas of peripancreatic hemorrhagic fat necrosis with fibrous tissue. As such, they usually are composed of central necrotic-hemorrhagic material rich in pancreatic enzymes surrounded by non-epithelial-lined fibrous walls of granulation tissue (Fig. 19-8B).43 They can range in size from 2 to 30 cm in diameter.
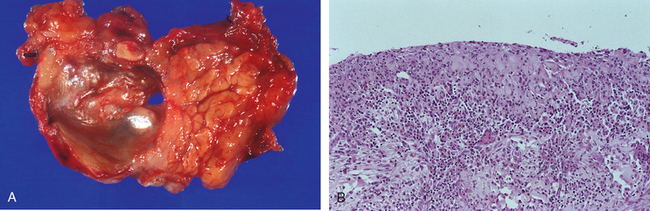
While many pseudocysts spontaneously resolve, they may become secondarily infected, and larger pseudocysts may compress or even perforate into adjacent structures.
Neoplasms
A broad spectrum of exocrine neoplasms can arise in the pancreas. They may be cystic or solid; some are benign, while others are among the most lethal of all malignancies.
CYSTIC NEOPLASMS
Only 5% to 15% of all pancreatic cysts are neoplastic (most cysts are pseudocysts; see the previous section), and cystic neoplasms make up fewer than 5% of all pancreatic neoplasms. While some, such as the serous cystadenoma, are entirely benign, others, such as mucinous cystic neoplasms, can be benign or malignant.
Serous cystadenomas are benign cystic neoplasms composed of glycogen-rich cuboidal cells surrounding small (1- to 3-mm) cysts containing clear, thin, straw-colored fluid (Fig. 19-9).1 They account for about 25% of all cystic neoplasms of the pancreas. These neoplasms arise twice as often in women as in men and typically present in the seventh decade of life with nonspecific symptoms such as abdominal pain. They may also present as palpable abdominal masses. Serous cystadenomas are almost always benign, and surgical resection is curative in the vast majority of patients.44
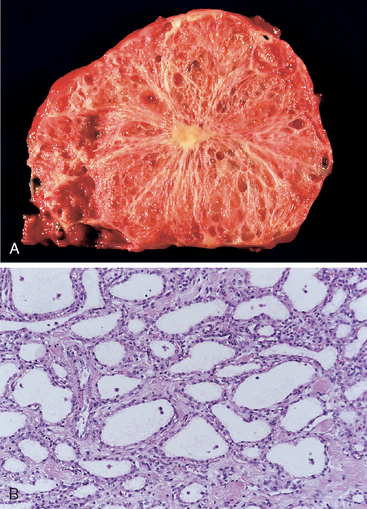
FIGURE 19-9 Serous cystadenoma. A, Cross-section through a serous cystadenoma. Only a thin rim of normal pancreatic parenchyma remains. The cysts are relatively small and contain clear, straw-colored fluid. B, The cysts are lined by cuboidal epithelium without atypia.
Close to 95% of mucinous cystic neoplasms arise in women, and, in contrast to serous cystadenomas, they can be associated with an invasive carcinoma.1,45,46 Mucinous cystic neoplasms usually arise in the body or tail of the pancreas and present as painless, slow-growing masses. The cysts are larger than those formed in serous cystadenomas; they are filled with thick, tenacious mucin and lined by a columnar mucin-producing epithelium with an associated dense stroma similar to ovarian stroma (Fig. 19-10).1 One third of surgically resected mucinous cystic neoplasms harbor an associated invasive adenocarcinoma. The best way to distinguish the entirely benign form (mucinous cystadenoma) from its malignant counterpart (invasive adenocarcinoma arising in association with a mucinous cystic neoplasm) is pathologic assessment after complete surgical removal, usually by distal pancreatectomy.45
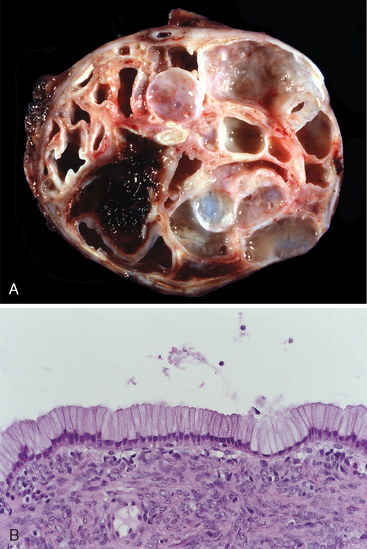
FIGURE 19-10 Pancreatic mucinous cystadenoma. A, Cross-section through a mucinous multiloculated cyst in the tail of the pancreas. The cysts are large and filled with tenacious mucin. B, The cysts are lined by columnar mucinous epithelium, and a dense “ovarian” stroma is noted.
Intraductal papillary mucinous neoplasms (IPMNs) are mucin-producing intraductal neoplasms.1,47,48 In contrast to mucinous cystic neoplasms, IPMNs arise more frequently in men than in women, and they involve the head of the pancreas more often than the tail. Ten to twenty percent are multifocal. Two features are useful in distinguishing IPMNs from mucinous cystic neoplasms: IPMNs lack the dense “ovarian” stroma seen in mucinous cystic neoplasms, and involve a larger pancreatic duct (Fig. 19-11), while mucinous cystic neoplasms do not connect to the pancreatic duct system. Just as with mucinous cystic neoplasms, benign IPMNs are distinguished from malignant IPMNs by the lack of tissue invasion.
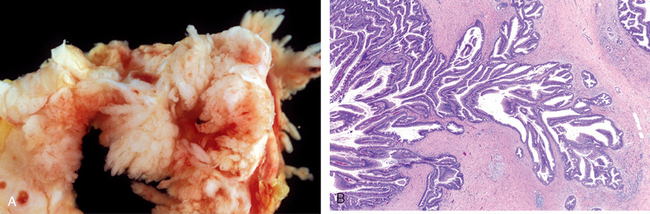
FIGURE 19-11 Intraductal papillary mucinous neoplasm. A, Cross-section through the head of the pancreas showing a prominent papillary neoplasm distending the main pancreatic duct. B, The papillary mucinous neoplasm involved the main pancreatic duct (left) and extending down into the smaller ducts and ductules (right).
The unusual solid-pseudopapillary neoplasm is seen mainly in young women.1,49 These large, well-circumscribed masses have solid and cystic components. The cystic areas are filled with hemorrhagic debris, and on histologic examination the neoplastic cells can be seen to grow in solid sheets or, as the name suggests, as papillary projections. These neoplasms often cause abdominal discomfort because of their large size. Of note, the β-catenin/adenomatous polyposis coli genetic pathway (Chapter 7) seems to be almost universally altered in these neoplasms often due to the presence of activating mutations of β-catenin.49 Surgical resection is the treatment of choice. Although some solid-pseudopapillary neoplasms are locally aggressive, most patients are cured following complete surgical resection of the neoplasm.
PANCREATIC CARCINOMA
Infiltrating ductal adenocarcinoma of the pancreas, more commonly known as “pancreatic cancer,” is the fourth leading cause of cancer deaths in the United States, preceded only by lung, colon, and breast cancers.50 Pancreatic cancer has one of the highest mortality rates of any cancer. It is estimated that in 2008 approximately 37,000 Americans were diagnosed with pancreatic cancer, and that virtually all of them will die from their disease. The 5-year survival rate is dismal, less than 5%.
Precursors to Pancreatic Cancer
Just as there is a progression in the colorectum from non-neoplastic epithelium to adenoma to invasive carcinoma (Chapters 7 and 17, there is a progression in the pancreas from non-neoplastic epithelium to histologically well-defined noninvasive lesions in small ducts and ductules to invasive carcinoma.51 These precursor lesions are called “pancreatic intraepithelial neoplasias” (PanINs). The PanIN-invasive carcinoma sequence is supported by the following observations:
The genetic and epigenetic alterations identified in PanINs are similar to those present in invasive cancers. The epithelial cells in PanINs show dramatic telomere shortening. A critical shortening of telomere length in PanINs may predispose these lesions to accumulate progressive chromosomal abnormalities and to develop invasive carcinoma.52
Based on these observations, a model for progression of PanINs has been proposed (Fig. 19-12).51
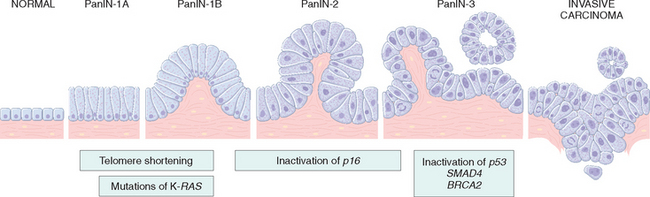
FIGURE 19-12 Progression model for the development of pancreatic cancer. It is postulated that telomere shortening and mutations of the oncogene KRAS occur at early stages, that inactivation of the p16 tumor suppressor gene occurs at intermediate stages, and the inactivation of the TP53, SMAD4 (DPC4), and BRCA2 tumor suppressor genes occur at late stages. It is important to note that while there is a general temporal sequence of changes, the accumulation of multiple mutations is more important than their occurrence in a specific order.
(Adapted from Wilentz RE et al.: Loss of expression of DPC4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res 60:2002, 2000.)
Molecular Carcinogenesis
Multiple genes are often altered in a single pancreatic cancer, and the patterns of genetic alterations differ from those seen in other malignancies.53 Molecular alterations in pancreatic carcinogenesis are summarized in Table 19-2 and include the following:
TABLE 19-2 Molecular Alterations in Invasive Pancreatic Adenocarcinoma
| Gene | Chromosomal Region | Percentage of Carcinoma with Genetic Alteration |
|---|---|---|
| KRAS | 12p | 90 |
| p16/CDKN2A | 9p | 95 |
| TP53 | 17p | 50–70 |
| SMAD4 | 18q | 55 |
| AKT2 | 19q | 10–20 |
| MYB | 6q | 10 |
| NCOA3/AIB1 | 20q | 10 |
| BRCA2 | 13q | 7–10 |
| GATA-6 | 18q | 10 |
| STK11 | 19p | 5 |
| MAP2K4/MKK4 | 17p | 5 |
| TGFβ-R1 | 9q | 2 |
| TGFβ-R2 | 3p | 2 |
| RB1 | 13q | 5 |
KRAS.
The KRAS gene (chromosome 12p) is the most frequently altered oncogene in pancreatic cancer. This oncogene is activated by point mutation in 80% to 90% of cases. These point mutations impair the intrinsic guanosine triphosphatase activity of the K-ras protein, resulting in a protein that is constitutively active. Ras in turn activates several intracellular signal transduction pathways that, among other effects, culminate in the activation of the transcription factors Fos and Jun.
CDKN2A (p16).
The p16/CDKN2A gene (chromosome 9p) is inactivated in 95% of the cases, making p16/CDKN2A the most frequently inactivated tumor suppressor gene in pancreatic cancer.54 The p16 protein plays a critical role in the control of the cell cycle, and inactivation of p16 abrogates an important cell cycle checkpoint.
SMAD4.
The SMAD4 tumor suppressor gene (chromosome 18q) is inactivated in 55% of pancreatic cancers.55 SMAD4 encodes a protein that plays an important role in signal transduction from the TGF-β family of cell surface receptors. SMAD4 is only rarely inactivated in other cancer types.
p53.
Inactivation of the p53 tumor suppressor gene (chromosome 17p) is seen in 50% to 70% of pancreatic cancers.56 As you recall, the p53 protein is a nuclear DNA-binding protein that acts both as a cell cycle checkpoint, as an inducer of cell death (apoptosis), and cellular senescence (Chapter 7).
Other Genes.
A growing number of less common, but nonetheless important, genetic loci have been reported to be damaged in pancreatic cancer (see Table 19-2). For example, the AKT2 gene (chromosome 19q) is amplified in 10% to 20%, the MYB gene (6q) in 10%, the GATA-6 gene (chromosome 18q) in 10%, and the NCOA3/AIB1 gene (chromosome 20q) in 10%.58 The BRCA2 (chromosome 13q), LKB1/STK11 (chromosome 19p), MAP2K4/MKK4 (chromosome 17p), TGFβ-R1 (chromosome 9q), TGFβ-R2 (chromosome 3p), and RB1 (chromosome 13q) tumor suppressor genes are inactivated in fewer than 10% of pancreatic cancers.
Methylation Abnormalities.
Several methylation abnormalities also occur in pancreatic cancer. Hypermethylation of the promoter of several tumor suppressor genes is associated with transcriptional silencing of the genes.
Gene Expression.
In addition to DNA alterations, global analyses of gene expression have identified several genes that are highly expressed in pancreatic cancers.54,59 These genes are potential targets for novel therapeutics and may form the basis of future screening tests. For example, the hedgehog signaling pathway has been shown to be activated in pancreatic cancer, and inhibition of this pathway with the drug cyclopamine blocks growth of pancreatic cancers in experimental systems.60
Epidemiology, Etiology, and Pathogenesis.
Pancreatic cancer is primarily a disease in the elderly, 80% of cases occurring between the ages of 60 and 80 years.61 It is more common in blacks than in whites, and it is slightly more common in individuals of Ashkenazi Jewish descent.
The strongest environmental influence is cigarette smoking, which is believed to double the risk of pancreatic cancer.59 Even though the magnitude of this increased risk is not great, the impact of smoking on pancreatic cancer is significant because of the large number of people who smoke. Consumption of a diet rich in fats has also been implicated, but less consistently. Chronic pancreatitis and diabetes mellitus have both been associated with an increased risk of pancreatic cancer. Pancreatic cancer arises with greater frequency in patients with chronic pancreatitis,42 but a causal role for pancreatitis, with the exception of hereditary pancreatitis, is not well established. Smoking and alcohol use in individuals with chronic pancreatitis may underlie some of the association.42 In an individual patient it can be difficult to sort out whether chronic pancreatitis is the cause of pancreatic cancer or an effect of the disease, since small pancreatic cancers may block the pancreatic duct and produce chronic pancreatitis. A similar argument applies to the association of diabetes mellitus with pancreatic cancer, since diabetes may develop as a consequence of pancreatic cancer. New-onset diabetes mellitus in an elderly patient may be the first sign that the patient has pancreatic cancer.62
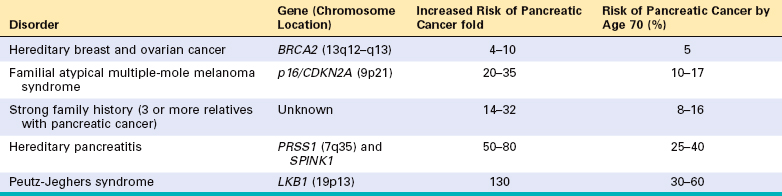
Familial clustering of pancreatic cancer has been reported, and a growing number of inherited genetic defects are recognized to increase pancreatic cancer risk (Table 19-3).63 BRCA2 mutations account for approximately 10% of pancreatic cancer cases in Ashkenazi Jews. Patients with these mutations may not have a family history of breast or ovarian cancers. Mutations in CDKN2A (p16) in pancreatic cancer almost always occur in individuals from melanoma-prone families.
A mutation in the PALLD gene, which encodes the extracellular matrix protein palladin, was reported in one family with high incidence of pancreatic cancer. The mutation was not found in other families, but palladin is highly expressed in the surrounding stroma of pancreatic cancers.
Morphology. Approximately 60% of cancers of the pancreas arise in the head of the gland, 15% in the body, and 5% in the tail; in 20% the neoplasm diffusely involves the entire gland. Carcinomas of the pancreas are usually hard, stellate, gray-white, poorly defined masses (Fig. 19-13A).
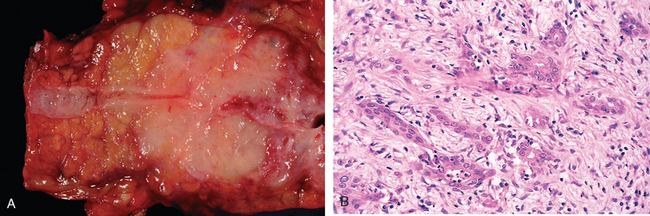
FIGURE 19-13 Carcinoma of the pancreas. A, A cross-section through the tail of the pancreas showing normal pancreatic parenchyma and a normal pancreatic duct (left), an ill-defined mass in the pancreatic substance (center) with narrowing of the pancreatic duct, and dilatation of the pancreatic duct upstream (right) from the mass. B, Poorly formed glands are present in densely fibrotic stroma within the pancreatic substance; there are some inflammatory cells.
The vast majority of carcinomas are ductal adenocarcinomas that recapitulate to some degree normal ductal epithelium by forming glands and secreting mucin. Two features are characteristic of pancreatic cancer: It is highly invasive (even “early” invasive pancreatic cancers extensively invade peripancreatic tissues), and elicits an intense non-neoplastic host reaction composed of fibroblasts, lymphocytes, and extracellular matrix (called a “desmoplastic response”).
Most carcinomas of the head of the pancreas obstruct the distal common bile duct as it courses through the head of the pancreas. As a consequence there is marked distention of the biliary tree in about 50% of patients with carcinoma of the head of the pancreas, and most develop jaundice. In marked contrast, carcinomas of the body and tail of the pancreas do not impinge on the biliary tract and hence remain silent for some time. They may be quite large and most are widely disseminated by the time they are discovered. Pancreatic cancers often grow along nerves and invade into the retroperitoneum. They can directly invade the spleen, adrenals, vertebral column, transverse colon, and stomach. Peripancreatic, gastric, mesenteric, omental, and portahepatic lymph nodes are frequently involved. Distant metastases occur, principally to the liver, lungs, and bones.
Microscopically, there is no difference between carcinomas of the head of the pancreas and those of the body and tail of the pancreas. The appearance is usually that of a moderately to poorly differentiated adenocarcinoma forming abortive tubular structures or cell clusters and showing an aggressive, deeply infiltrative growth pattern (Fig. 19-13B). Dense stromal fibrosis accompanies the invasive cancer, and there is a proclivity for perineural invasion within and beyond the organ. Lymphatic and large vessel invasion are also commonly seen. The malignant glands are poorly formed and are usually lined by pleomorphic cuboidal-to-columnar epithelial cells. Well-differentiated carcinomas are the exception.
Less common variants of pancreatic cancer include adenosquamous carcinomas, colloid carcinoma, hepatoid carcinoma, medullary carcinoma, signet-ring cell carcinoma, undifferentiated carcinoma, and undifferentiated carcinomas with osteoclast-like giant cells.1 Adenosquamous carcinomas have focal squamous differentiation in addition to glandular differentiation, and undifferentiated carcinomas may contain large multinucleated osteoclast-like giant cells.
Clinical Features.
From the preceding discussion it should be evident that carcinomas of the pancreas remain silent until they invade into adjacent structures. Pain is usually the first symptom, but by the time pain appears these cancers are usually beyond cure. Obstructive jaundice is associated with most cases of carcinoma of the head of the pancreas, but it rarely draws attention to the invasive cancer soon enough. Weight loss, anorexia, and generalized malaise and weakness tend to be signs of advanced disease. Migratory thrombophlebitis, known as the Trousseau sign, occurs in about 10% of patients and is attributable to the elaboration of platelet-aggregating factors and procoagulants from the carcinoma or its necrotic products (Chapter 4). On a sad note, Armand Trousseau (1801–1867, physician at Hotel Dieu, Paris) suspected that he had cancer when he developed spontaneously appearing and disappearing (migratory) thrombosis, and his autopsy revealed that he had pancreatic cancer.
The course of pancreatic carcinoma is typically brief and progressive. Despite the tendency of lesions of the head of the pancreas to obstruct the biliary system, fewer than 20% of pancreatic cancers overall are resectable at the time of diagnosis. There has long been a search for tests that could be useful in the early detection of pancreatic cancer. Serum levels of many enzymes and antigens (e.g., carcinoembryonic antigen and CA19–9 antigen) are often elevated in individuals with pancreatic cancer. These markers, while useful in following a patient’s response to treatment, are too nonspecific and lack the sensitivity needed to be used as screening tests. Several imaging techniques, such as endoscopic ultrasonography and computed tomography, have proved of great value in establishing the diagnosis once it is suspected, but are not useful as screening tests.
ACINAR CELL CARCINOMA
Acinar cell carcinomas, by definition, show prominent acinar cell differentiation, including the formation of zymogen granules and the production of exocrine enzymes including trypsin and lipase.64 Fifteen percent of individuals with acinar cell carcinoma develop the syndrome of metastatic fat necrosis caused by the release of lipase into the circulation.
PANCREATOBLASTOMA
Pancreatoblastomas are rare neoplasms that occur primarily in children aged 1 to 15 years.65 They have a distinct microscopic appearance with squamous islands admixed with acinar cells. These are fully malignant neoplasms, although survival may be better than that for pancreatic ductal adenocarcinomas.
1 Hruban RH, et al. Tumors of the pancreas. Atlas of tumor pathology. Fourth Series, Fascicle, 6 ed. Washington, DC: American Registry of Pathology and Armed Forces Institute of Pathology, 2007.
2 Oertel JE. The pancreas. Nonneoplastic alterations. Am J Surg Pathol. 1989;13:50.
3 Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science. 2008;322:1490.
4 Cano DA, et al. Pancreatic development and disease. Gastroenterology. 2007;132:745.
5 Spicak J, et al. Pancreas divisum does not modify the natural course of chronic pancreatitis. J Gastroenterol. 2007;42:135.
6 Jimenez JC, et al. Annular pancreas in children: a recent decade’s experience. J Pediatr Surg. 2004;39:1654.
7 Mitchell RM, et al. Pancreatitis. Lancet. 2003;361:1447.
8 Frossard JL, et al. Acure pancreatitis. Lancet. 2008;371:143.
9 Cappell MS. Acute pancreatitis: etiology, clinical presentation, diagnosis and therapies. Med Clin North Am. 2008;92:889.
10 Carroll JK, et al. Acute pancreatitis: diagnosis, prognosis, and treatment. Am Fam Physician. 2007;75:1513.
11 Witt H, et al. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology. 2007;132:1557.
12 Granger J, Remick D. Acute pancreatitis: models, markers, and mediators. Shock. 2005;24(Suppl 1):45.
13 Sand J, et al. Alcohol consumption in patients with acute or chronic pancreatitis. Pancreatology. 2007;7:147.
14 Pazzi P, et al. Biliary sludge: the sluggish gallbladder. Dig Liver Dis. 2003;35:S39.
15 Scarpelli DG. Toxicology of the pancreas. Toxicol Appl Pharmacol. 1989;101:543.
16 Whitcomb DC, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141.
17 Grendell JH. Genetic factors in pancreatitis. Curr Gastroenterol Rep. 2003;5:105.
18 Witt H, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213.
19 Noone PG, et al. Cystic fibrosis gene mutations and pancreatitis risk: relation to epithelial ion transport and trypsin inhibitor gene mutations. Gastroenterology. 2001;121:1310.
20 Phat VN, et al. Early histological changes in acute necrotizing hemorrhagic pancreatitis. Pathol Res Pract. 1984;178:273.
21 Pandol SJ, Raraty M. Pathobiology of alcoholic pancreatitis. Pancreatology. 2007;7:105.
22 Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg. 1998;175:76.
23 Saluja AK, Steer MLP. Pathophysiology of pancreatitis. Role of cytokines and other mediators of inflammation. Digestion. 1999;60(Suppl 1):27.
24 Rau B, et al. Differential effects of caspase-1/interleukin-1beta-converting enzyme on acinar cell necrosis and apoptosis in severe acute experimental pancreatitis. Lab Invest. 2001;81:1001.
25 Shimada M, et al. IL-6 secretion by human pancreatic periacinar myofibroblasts in response to inflammatory mediators. J Immunol. 2002;168:861.
26 Blackstone MO. Hypothesis: vascular compromise is the central pathogenic mechanism for acute hemorrhagic pancreatitis. Perspect Biol Med. 1995;39:56.
27 Steer ML. Pathogenesis of acute pancreatitis. Digestion. 1997;58(Suppl 1):46.
28 Whitcomb DC. Early trypsinogen activation in acute pancreatitis. Gastroenterology. 1999;116:770.
29 Pitchumoni CS, Bordalo O. Evaluation of hypotheses on pathogenesis of alcoholic pancreatitis. Am J Gastroenterol. 1996;91:637.
30 Vonlaufen A, et al. Molecular mechanisms of pancreatitis: current opinion. J Gastroenterol Hepatol. 2008;23:1339.
31 Witt H, Bhatia E. Genetic aspects of tropical calcific pancreatitis. Rev Endocr Metab Disord. 2008;9:213.
32 Klopel G. Chronic pancreatitis, pseudotumors and tumor-like lesions. Mod Pathol. 2007;20:S113.
33 Pitchumoni CS. Pathogenesis of alcohol-induced chronic pancreatitis: facts, perceptions, and misperceptions. Surg Clin North Am. 2001;81:379.
34 Tattersal SJN, et al. A fire inside: current concepts in chronic pancreatitis. Int Med J. 2008;38:592.
35 Saurer L, et al. Differential expression of chemokines in normal pancreas and in chronic pancreatitis. Gastroenterology. 2000;118:356.
36 Whitcomb DC. Hereditary pancreatitis: new insights into acute and chronic pancreatitis. Gut. 1999;45:317.
37 Luttenberger T, et al. Platelet-derived growth factors stimulate proliferation and extracellular matrix synthesis of pancreatic stellate cells: implications in pathogenesis of pancreas fibrosis. Lab Invest. 2000;80:47.
38 Van Laethem JL, et al. Localization of transforming growth factor beta 1 and its latent binding protein in human chronic pancreatitis. Gastroenterology. 1995;108:1873.
39 Detlefsen S, et al. Fibrogenesis in alcoholic chronic pancreatitis: the role of tissue necrosis, macrophages, myofibroblasts and cytokines. Mod Pathol. 2006;19:1019.
40 Hamano H, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732.
41 Treiber M, et al. Genetics of pancreatitis: a guide for clinicians. Curr Gastroenterol Rep. 2008;10:122.
42 Rebours V, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am J Gastroenterol. 2008;103:111.
43 Klöppel G. Pseudocysts and other non-neoplastic cysts of the pancreas. Semin Diagn Pathol. 2000;17:7.
44 Galanis C, et al. Resected serous cystic neoplasms of the pancreas: a review of 158 patients with recommendations for treatment. J Gastrointest Surg. 2007;11:820.
45 Wilentz RE, et al. Pathologic examination accurately predicts prognosis in mucinous cystic neoplasms of the pancreas. Am J Surg Pathol. 1999;23:1320.
46 Zamboni G, et al. Mucinous cystic tumors of the pancreas: clinicopathological features, prognosis, and relationship to other mucinous cystic tumors. Am J Surg Pathol. 1999;23:410.
47 Hruban RH, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28:977.
48 Chari ST, et al. Study of recurrence after surgical resection of intraductal papillary mucinous neoplasm of the pancreas. Gastroenterology. 2002;123:1500.
49 Abraham SC, et al. Solid-pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor beta-catenin mutations. Am J Pathol. 2002;160:1361.
50 American Cancer Society. Cancer Facts & Figures. Cancer 1–68. New York: American Cancer Society, 2008.
51 Hruban RH, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579.
52 van Heek NT, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol. 2002;161:1541.
53 Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897.
54 Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global analyses. Science. 2008;321:1801.
55 Caldas C, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27.
56 Hahn SA, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350.
57 Redston MS, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994;54:3025.
58 Fu B, et al. Frequent genomic copy number gain and overexpression of GATA-6 in parcreatic caroinoma. Cancer Biol Ther. 7, 2008. [epub ahead of print].
59 Iacobuzio-Donahue CA, et al. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003;63:8614.
60 Berman DM, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846.
61 Gold EB. Epidemiology of and risk factors for pancreatic cancer. Surg Clin North Am. 1995;75:819.
62 Chari ST, et al. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology. 2005;129:504.
63 Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med. 2008;359:2143.
64 Klimstra DS, et al. Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am J Surg Pathol. 1992;16:815.
65 Klimstra DS, et al. Pancreatoblastoma. A clinicopathologic study and review of the literature. Am J Surg Pathol. 1995;19:1371.