Chapter 6 Diseases of the Immune System
The immune system is vital for survival, because our environment is teeming with potentially deadly microbes and the immune system protects us from infectious pathogens. Predictably, immune deficiencies render individuals easy prey to infections. But the immune system is similar to the proverbial double-edged sword. Although it normally defends us against infections, a hyperactive immune system may cause diseases that can sometimes be fatal. Examples of disorders caused by immune responses include allergic reactions and reactions against an individual’s own tissues and cells (autoimmunity).
This chapter is devoted to diseases caused by too little immunity or too much immunologic reactivity. We also consider amyloidosis, a disease in which an abnormal protein, derived in some cases from fragments of immunoglobulins, is deposited in tissues. First, we review some of the important features of normal immune responses, to provide a foundation for understanding the abnormalities that give rise to immunological diseases.
The Normal Immune Response
The normal immune response is best understood in the context of defense against infectious pathogens, the classical definition of immunity. The mechanisms of protection against infections fall into two broad categories. Innate immunity (also called natural, or native, immunity) refers to defense mechanisms that are present even before infection and that have evolved to specifically recognize microbes and protect individuals against infections. Adaptive immunity (also called acquired, or specific, immunity) consists of mechanisms that are stimulated by (“adapt to”) microbes and are capable of recognizing microbial and nonmicrobial substances. Innate immunity is the first line of defense, because it is always ready to prevent and eradicate infections. Adaptive immunity develops later, after exposure to microbes, and is even more powerful than innate immunity in combating infections. By convention, the term “immune response” refers to adaptive immunity.
INNATE IMMUNITY
The major components of innate immunity are epithelial barriers that block entry of microbes, phagocytic cells (mainly neutrophils and macrophages), dendritic cells, natural killer (NK) cells, and several plasma proteins, including the proteins of the complement system. The two most important cellular reactions of innate immunity are: inflammation, the process in which phagocytic leukocytes are recruited and activated to kill microbes, and anti-viral defense, mediated by dendritic cells and NK cells. Leukocytes and epithelial cells that participate in innate immunity are capable of recognizing components of microbes that are shared among related microbes and are often essential for the infectivity of these pathogens (and thus cannot be mutated to allow the microbes to evade the defense mechanisms). These microbial structures are called pathogenassociated molecular patterns. Leukocytes also recognize molecules released by injured and necrotic cells, which are sometimes called danger-associated molecular patterns. The cellular receptors that recognize these molecules are often called pattern recognition receptors. The best-defined pattern recognition receptors are a family of proteins called Toll-like receptors (TLRs)1 that are homologous to the Drosophila protein Toll. Different TLRs are specific for components of different bacteria and viruses. TLRs are located on the cell surface and in endosomes, so they are able to recognize and initiate cellular responses to extracellular and ingested microbes. Other microbial sensors are located in the cytoplasm, where they recognize bacteria and viruses that may have colonized cells. Upon recognition of microbes, the TLRs and other sensors signal by a common pathway that leads to the activation of transcription factors, notably NF-κB (nuclear factor κB). NF-κB turns on the production of cytokines and proteins that stimulate the microbicidal activities of various cells, notably the phagocytes. Other cellular receptors bind microbes for phagocytosis; these include receptors for mannose residues, which are typical of microbial but not host glycoproteins, and receptors for opsonins such as antibodies and complement proteins that coat microbes.
Epithelia of the skin and gastrointestinal and respiratory tracts provide mechanical barriers to the entry of microbes from the external environment. Epithelial cells also produce anti-microbial molecules such as defensins, and lymphocytes located in the epithelia combat microbes at these sites. If microbes do breach epithelial boundaries, other defense mechanisms are called in.
Monocytes and neutrophils are phagocytes in the blood that can rapidly be recruited to any site of infection; monocytes that enter the tissues and mature are called macrophages (Chapter 2). Dendritic cells produce type I interferons, anti-viral cytokines that inhibit viral infection and replication; these cells are described below, in the context of antigen display to lymphocytes. Natural killer cells provide early protection against many viruses and intracellular bacteria; their properties and functions are also described below.
The proteins of the complement system, which were described in Chapter 2, are some of the most important plasma proteins of the innate immune system. Recall that in innate immunity the complement system is activated by microbes using the alternative and lectin pathways; in adaptive immunity it is activated by antibodies using the classical pathway. Other circulating proteins of innate immunity are mannose-binding lectin and C-reactive protein, both of which coat microbes for phagocytosis. Lung surfactant is also a component of innate immunity, providing protection against inhaled microbes.
The early innate immune response not only provides the initial defense against infections but is also involved in triggering the subsequent, more powerful adaptive immune response.
ADAPTIVE IMMUNITY
The adaptive immune system consists of lymphocytes and their products, including antibodies. The receptors of lymphocytes are much more diverse than those of the innate immune system, but lymphocytes are not inherently specific for microbes, and they are capable of recognizing a vast array of foreign substances. In the remainder of this introductory section we focus on lymphocytes and the reactions of the adaptive immune system.
There are two types of adaptive immunity: humoral immunity, which protects against extracellular microbes and their toxins, and cell-mediated (or cellular) immunity, which is responsible for defense against intracellular microbes. Humoral immunity is mediated by B (bone marrow–derived) lymphocytes and their secreted products, antibodies (also called immunoglobulins, Ig), and cellular immunity is mediated by T (thymus-derived) lymphocytes. Both classes of lymphocytes express highly specific receptors for a wide variety of substances, called antigens.
COMPONENTS OF THE IMMUNE SYSTEM: CELLS, TISSUES, AND SELECTED MOLECULES
Before describing normal and pathologic immune responses, it is important to summarize the salient characteristics of some of the important participants in these responses.
Cells of the Immune System
Although lymphocytes appear morphologically unimpressive and similar to one another, they are actually remarkably heterogeneous and specialized in molecular properties and functions. The major classes of lymphocytes and their functions in adaptive immunity are illustrated in Figure 6-1. Lymphocytes and other cells involved in immune responses are not fixed in particular tissues (as are cells in most of the organs of the body) but are capable of migrating among lymphoid and other tissues and the vascular and lymphatic circulations. This feature permits lymphocytes to home to any site of infection. In lymphoid organs, different classes of lymphocytes are anatomically segregated in such a way that they interact with one another only when stimulated to do so by encounter with antigens and other stimuli. Mature lymphocytes that have not encountered the antigen for which they are specific are said to be naive (immunologically inexperienced). After they are activated by recognition of antigens and other signals described later, lymphocytes differentiate into effector cells, which perform the function of eliminating microbes, and memory cells, which live in a state of heightened awareness and are better able to combat the microbe in case it returns. The process of lymphocyte differentiation into effector and memory cells is summarized below.
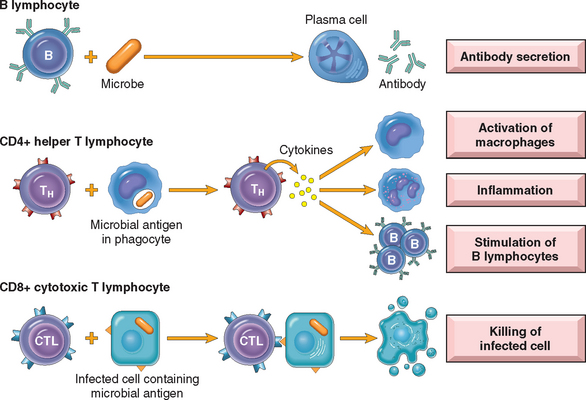
T Lymphocytes
T lymphocytes develop from precursors in the thymus. Mature T cells are found in the blood, where they constitute 60% to 70% of lymphocytes, and in T-cell zones of peripheral lymphoid organs (described below). Each T cell recognizes a specific cell-bound antigen by means of an antigen-specific T-cell receptor (TCR).2 In approximately 95% of T cells the TCR consists of a disulfide-linked heterodimer made up of an α and a β polypeptide chain (Fig. 6-2), each having a variable (antigen-binding) region and a constant region. The αβ TCR recognizes peptide antigens that are displayed by major histocompatibility complex (MHC) molecules on the surfaces of antigen-presenting cells (APCs). (The function of MHC proteins is described later.) By limiting the specificity of T cells for peptides displayed by cell surface MHC molecules, called MHC restriction, the immune system ensures that T cells see only cell-associated antigens (e.g., those derived from microbes in cells).
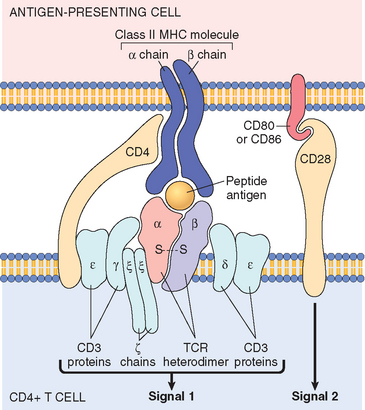
FIGURE 6-2 The T-cell receptor (TCR) complex and other molecules involved in T-cell activation. The TCR heterodimer, consisting of an α and a β chain, recognizes antigen (in the form of peptide-MHC complexes expressed on antigen-presenting cells, or APCs), and the linked CD3 complex and ζ chains initiate activating signals. CD4 and CD28 are also involved in T-cell activation. (Note that some T cells express CD8 and not CD4; these molecules serve analogous roles.) The sizes of the molecules are not drawn to scale. MHC, major histocompatibility complex.
TCR diversity is generated by somatic rearrangement of the genes that encode the TCR α and β chains.3 All cells of the body, including lymphocyte progenitors, contain TCR genes in the germ-line configuration, which cannot be expressed as TCR proteins. During T cell development in the thymus, the TCR genes rearrange to form many different combinations that can be transcribed and translated into functional antigen receptors. The enzyme in developing lymphocytes that mediates rearrangement of antigen receptor genes is the product of RAG-1 and RAG-2 (recombination activating genes); inherited defects in RAG proteins result in a failure to generate mature lymphocytes. Whereas each T cell expresses TCR molecules of one specificity, collectively, the full complement of T cells in an individual is capable of recognizing a very large number of antigens. It is important to note that unrearranged (germ-line) TCR genes are present in all non-T cells in the body, but only T cells contain rearranged TCR genes. Hence, the presence of rearranged TCR genes, which can be demonstrated by molecular analysis, is a marker of T-lineage cells. Furthermore, because each T cell and its clonal progeny have a unique DNA rearrangement (and hence a unique TCR), it is possible to distinguish polyclonal (non-neoplastic) T-cell proliferations from monoclonal (neoplastic) T-cell proliferations. Thus, analysis of antigen receptor gene rearrangements is a valuable assay for detecting lymphoid tumors (Chapter 13).
Each TCR is noncovalently linked to five polypeptide chains, which form the CD3 complex and the ζ chain dimer (see Fig. 6-2).4 The CD3 and ζ proteins are invariant (i.e., identical) in all T cells. They are involved in the transduction of signals into the T cell after the TCR has bound the antigen. Together with the TCR, these proteins form the “TCR complex.”
A small population of mature T cells expresses another type of TCR composed of γ and δ polypeptide chains.5 The γδ TCR recognizes peptides, lipids, and small molecules, without a requirement for display by MHC proteins. γδ T cells tend to aggregate at epithelial surfaces, such as the skin and mucosa of the gastrointestinal and urogenital tracts, suggesting that these cells are sentinels that protect against microbes that try to enter through epithelia. However, the functions of γδ T cells are not known. Another small subset of T cells expresses markers that are found on NK cells; these cells are called NK-T cells.6 NK-T cells express a very limited diversity of TCRs, and they recognize glycolipids that are displayed by the MHC-like molecule CD1. The functions of NK-T cells are also not well defined.
In addition to CD3 and ζ proteins, T cells express several other proteins that assist the TCR complex in functional responses. These include CD4, CD8, CD2, integrins, and CD28.7 CD4 and CD8 are expressed on two mutually exclusive subsets of αβ T cells. CD4 is expressed on approximately 60% of mature CD3+ T cells, which function as cytokine-secreting helper cells that help macrophages and B lymphocytes to combat infections, whereas CD8 is expressed on about 30% of T cells, which function as cytotoxic (killer) T lymphocytes (CTLs) to destroy host cells harboring microbes. CD4 and CD8 serve as “coreceptors” in T-cell activation, so called because they work with the antigen receptor in responses to antigen. During antigen presentation, CD4 molecules bind to class II MHC molecules that are displaying antigen (see Fig. 6-2), and CD8 molecules bind to class I MHC molecules. When the antigen receptor of a T cell recognizes antigen, the CD4 or CD8 coreceptor initiates signals that are necessary for activation of the T cells. Because of this requirement for co-receptors, CD4+ helper T cells can recognize and respond to antigen displayed only by class II MHC molecules, whereas CD8+ cytotoxic T cells recognize cell-bound antigens only in association with class I MHC molecules; this segregation is described below.
To respond, T cells have to recognize not only antigen-MHC complexes but additional signals provided by APCs. We will describe these later, when we summarize the steps in cell-mediated immune responses.
B Lymphocytes
B lymphocytes develop from precursors in the bone marrow. Mature B cells constitute 10% to 20% of the circulating peripheral lymphocyte population and are also present in peripheral lymphoid tissues such as lymph nodes, spleen, and mucosa-associated lymphoid tissues. B cells recognize antigen via the B-cell antigen receptor complex. Membrane-bound antibodies called IgM and IgD, present on the surface of all mature, naive B cells, are the antigen-binding component of the B-cell receptor complex (Fig. 6-3). As with T cells, each B-cell receptor has a unique antigen specificity, derived from RAG-mediated rearrangements of Ig genes. Thus, as in T cells, analysis of Ig gene rearrangements is useful for identifying monoclonal B-cell tumors. After stimulation by antigen and other signals (described later), B cells develop into plasma cells that secrete antibodies, the mediators of humoral immunity. In addition to membrane Ig, the B-cell antigen receptor complex contains a heterodimer of two invariant proteins called Igα and Igβ. Similar to the CD3 and ζ proteins of the TCR complex, Igα and Igβ are essential for signal transduction through the antigen receptor. B cells also express several other molecules that are essential for their responses. These include complement receptors, Fc receptors, and CD40. The type 2 complement receptor (CR2, or CD21) is also the receptor for the Epstein-Barr virus (EBV), and hence EBV readily infects B cells.
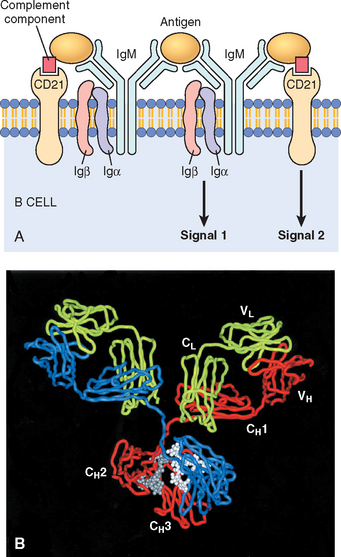
FIGURE 6-3 Structure of antibodies and the B-cell antigen receptor. A, The B-cell receptor complex is composed of membrane immunoglobulin M (IgM; or IgD, not shown), which recognize antigens, and the associated signaling proteins Igα and Igβ. CD21 is a receptor for a complement component that also promotes B-cell activation. B, Crystal structure of a secreted IgG molecule, showing the arrangement of the variable (V) and constant (C) regions of the heavy (H) and light (L) chains.
(Courtesy of Dr. Alex McPherson, University of California, Irvine, CA.)
Dendritic Cells
There are two types of cells with dendritic morphology that are functionally quite different. Both have numerous fine cytoplasmic processes that resemble dendrites, from which they derive their name. One type is called interdigitating dendritic cells, or just dendritic cells (Fig. 6-4).8 These cells are the most important antigen-presenting cells (APCs) for initiating primary T-cell responses against protein antigens (described later). Several features of dendritic cells account for their key role in antigen presentation. First, these cells are located at the right place to capture antigens—under epithelia, the common site of entry of microbes and foreign antigens, and in the interstitia of all tissues, where antigens may be produced. Immature dendritic cells within the epidermis are called Langerhans cells. Second, dendritic cells express many receptors for capturing and responding to microbes (and other antigens), including TLRs and mannose receptors. Third, in response to microbes, dendritic cells are recruited to the T-cell zones of lymphoid organs, where they are ideally located to present antigens to T cells. Fourth, dendritic cells express high levels of the molecules needed for presenting antigens to and activating CD4+ T cells.
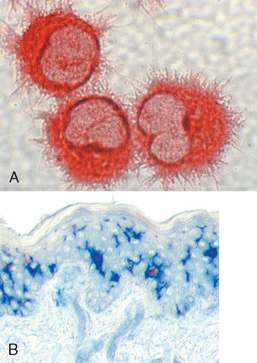
FIGURE 6-4 Dendritic cells. A, Cultured dendritic cells showing the prominent surface projections. B, The location of dendritic cells (Langerhans cells) in the epidermis (stained blue using an immunohistochemical method).
(Courtesy of Dr. Y-J. Liu, M.D. Anderson Cancer Center, Houston, TX.)
The other type of cell with dendritic morphology is present in the germinal centers of lymphoid follicles in the spleen and lymph nodes and is hence called follicular dendritic cell.9 These cells bear Fc receptors for IgG and receptors for C3b and can trap antigen bound to antibodies or complement proteins. Such cells play a role in humoral immune responses by presenting antigens to B cells and selecting the B cells that have the highest affinity for the antigen, thus improving the quality of the antibody produced.
Macrophages
Macrophages are a part of the mononuclear phagocyte system; their origin, differentiation, and role in inflammation are discussed in Chapter 2. Here we need only to emphasize their important functions in the induction and effector phases of adaptive immune responses.
Natural Killer Cells
NK cells make up approximately 10% to 15% of peripheral blood lymphocytes. They do not express TCRs or Ig. Morphologically, NK cells are somewhat larger than small lymphocytes, and they contain abundant azurophilic granules; because of these characteristics, they are also called large granular lymphocytes. NK cells are endowed with the ability to kill a variety of infected and tumor cells, without prior exposure to or activation by these microbes or tumors. This ability makes NK cells an early line of defense against viral infections and, perhaps, some tumors. Two cell surface molecules, CD16 and CD56, are commonly used to identify NK cells. CD16 is an Fc receptor for IgG, and it confers on NK cells the ability to lyse IgG-coated target cells. This phenomenon is known as antibody-dependent cell-mediated cytotoxicity (ADCC).
The functional activity of NK cells is regulated by a balance between signals from activating and inhibitory receptors10 (Fig. 6-5). There are many types of activating receptors, of which the NKG2D family is the best characterized. The NKG2D receptors recognize surface molecules that are induced by various kinds of stress, such as infection and DNA damage. NK cell inhibitory receptors recognize self–class I MHC molecules, which are expressed on all healthy cells. These receptors belong to two major families: killer cell Ig-like receptors and the CD94 family of lectins (carbohydrate-recognizing proteins). The inhibitory receptors prevent NK cells from killing normal cells. Virus infection or neoplastic transformation often induces expression of ligands for activating receptors and at the same time reduces the expression of class I MHC molecules. As a result the balance is tilted toward activation, and the infected or tumor cell is killed.
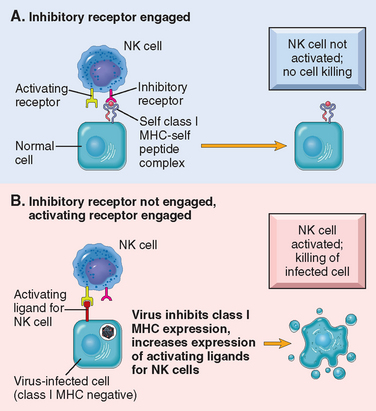
FIGURE 6-5 Activating and inhibitory receptors of natural killer (NK) cells. A, Healthy cells express self–class I MHC molecules, which are recognized by inhibitory receptors, thus ensuring that NK cells do not attack normal cells. Note that healthy cells may express ligands for activating receptors (not shown) or may not express such ligands (as shown), but they do not activate NK cells because they engage the inhibitory receptors. B, In infected and stressed cells, class I MHC expression is reduced so that the inhibitory receptors are not engaged, and ligands for activating receptors are expressed. The result is that NK cells are activated and the infected cells are killed.
NK cells also secrete cytokines, such as interferon-γ (IFN-γ), which activates macrophages to destroy ingested microbes, and thus NK cells provide early defense against intracellular microbial infections. The activity of NK cells is regulated by many cytokines, including the interleukins IL-2, IL-15, and IL-12. IL-2 and IL-15 stimulate proliferation of NK cells, whereas IL-12 activates killing and secretion of IFN-γ.
Tissues of the Immune System
The tissues of the immune system consist of the generative (also called primary, or central) lymphoid organs, in which T and B lymphocytes mature and become competent to respond to antigens, and the peripheral (or secondary) lymphoid organs, in which adaptive immune responses to microbes are initiated.
Generative Lymphoid Organs
The principal generative lymphoid organs are the thymus, where T cells develop, and the bone marrow, the site of production of all blood cells and where B lymphocytes mature. These organs are described in Chapter 13.
Peripheral Lymphoid Organs
The peripheral lymphoid organs consist of the lymph nodes, spleen, and the mucosal and cutaneous lymphoid tissues. These tissues are organized to concentrate antigens, APCs, and lymphocytes in a way that optimizes interactions among these cells and the development of adaptive immune responses.
Lymph nodes are nodular aggregates of lymphoid tissues located along lymphatic channels throughout the body (Fig. 6-6). As lymph passes through lymph nodes, APCs in the nodes are able to sample the antigens of microbes that may enter through epithelia into tissues and are carried in the lymph. In addition, dendritic cells pick up and transport antigens of microbes from epithelia via the lymphatic vessels to the lymph nodes. Thus, the antigens of microbes that enter through epithelia or colonize tissues become concentrated in draining lymph nodes.
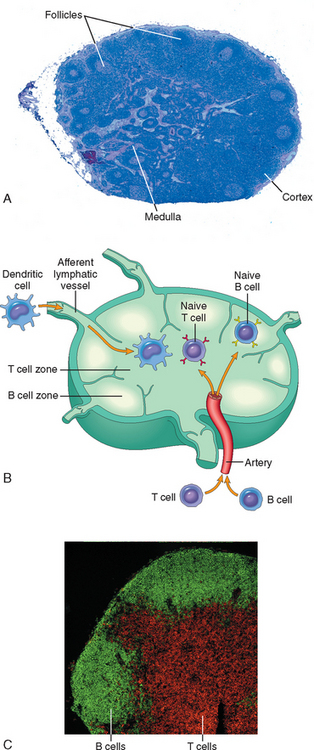
FIGURE 6-6 Morphology of a lymph node. A, The histology of a lymph node, with an outer cortex containing follicles and an inner medulla. B, The segregation of B cells and T cells in different regions of the lymph node, illustrated schematically. C, The location of B cells (stained green, using the immunofluorescence technique) and T cells (stained red) in a lymph node.
(Courtesy of Drs. Kathryn Pape and Jennifer Walter, University of Minnesota School of Medicine, Minneapolis, MN.)
The spleen is an abdominal organ that serves the same role in immune responses to blood-borne antigen as that of lymph nodes in responses to lymph-borne antigens. Blood entering the spleen flows through a network of sinusoids. Blood-borne antigens are trapped by dendritic cells and macrophages in the spleen.
The cutaneous and mucosal lymphoid systems are located under the epithelia of the skin and the gastrointestinal and respiratory tracts, respectively. They respond to antigens that enter by breaches in the epithelium. Pharyngeal tonsils and Peyer’s patches of the intestine are two anatomically defined mucosal lymphoid tissues. At any time, more than half the body’s lymphocytes are in the mucosal tissues (reflecting the large size of these tissues), and many of these are memory cells.
Within the peripheral lymphoid organs, T lymphocytes and B lymphocytes are segregated into different regions (see Fig. 6-6). In lymph nodes the B cells are concentrated in discrete structures, called follicles, located around the periphery, or cortex, of each node. If the B cells in a follicle have recently responded to an antigen, this follicle may contain a central region called a germinal center. The T lymphocytes are concentrated in the paracortex, adjacent to the follicles. The follicles contain the follicular dendritic cells that are involved in the activation of B cells, and the paracortex contains the dendritic cells that present antigens to T lymphocytes. In the spleen, T lymphocytes are concentrated in periarteriolar lymphoid sheaths surrounding small arterioles, and B cells reside in the follicles.
The anatomic organization of peripheral lymphoid organs is tightly regulated to allow immune responses to develop.11-13 The location of B cells and T cells in the lymphoid follicles and paracortical areas, respectively, is dictated by chemokines produced in these anatomic locales. When the lymphocytes are activated by antigens they alter their expression of chemokine receptors. As a result, the B cells and T cells leave their homes, migrate toward each other and meet at the edge of follicles, where helper T cells interact with and help B cells to differentiate into antibody-producing cells.
Lymphocyte Recirculation
Lymphocytes constantly recirculate between tissues and home to particular sites; naive lymphocytes traverse the peripheral lymphoid organs where immune responses are initiated, and effector lymphocytes migrate to sites of infection and inflammation14 (Fig. 6-7). This process of lymphocyte recirculation is most relevant for T cells, because effector T cells have to locate and eliminate microbes at any site of infection. By contrast, plasma cells remain in lymphoid organs and do not need to migrate to sites of infection because they secrete antibodies that are carried to distant tissues. Therefore, we will limit our discussion of lymphocyte recirculation to T lymphocytes.
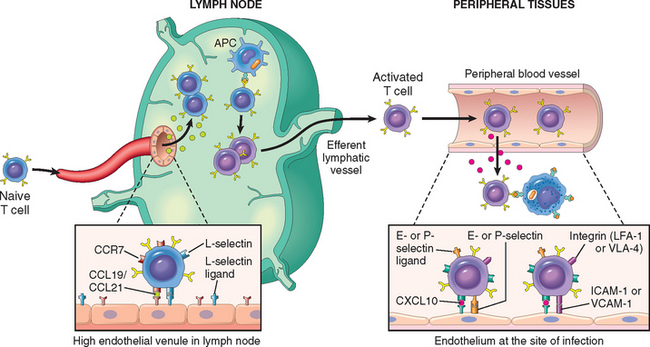
FIGURE 6-7 Migration of naive and effector T lymphocytes. Naive T lymphocytes home to lymph nodes as a result of L-selectin and integrin binding to their ligands on high endothelial venules (HEVs). Chemokines expressed in lymph nodes (called CCL19 and CCL21) bind to receptors (CCR7) on naive T cells, enhancing integrin-dependent adhesion and inducing migration of the cells through the HEV wall. Activated T lymphocytes, including effector and memory cells, home to sites of infection in peripheral tissues, and this migration is mediated by E-selectin and P-selectin, integrins, and chemokines secreted at inflammatory sites (e.g., CXCL10) that are recognized by chemokine receptors (e.g., CXCR3) that are expressed on activated T cells. APC, antigen-presenting cell; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1.
Naive T lymphocytes that have exited the thymus migrate to lymph nodes and enter the T-cell zones through specialized postcapillary venules, called high endothelial venules (HEVs) (see Fig. 6-7). In the lymph node, a naive T cell may encounter the antigen that it specifically recognizes on the surface of an APC and is activated. During this process, the cells alter their expression of adhesion molecules and chemokine receptors. Differentiated effector T cells ultimately leave the lymph nodes, enter the circulation, and migrate into the tissues that harbor the microbes.
Major Histocompatibility Complex (MHC) Molecules: Peptide Display System of Adaptive Immunity
Because MHC molecules are fundamental to the recognition of antigens by T cells and are linked to many autoimmune diseases, it is important to briefly review the structure and function of these molecules.15 MHC molecules were discovered as products of genes that evoke rejection of transplanted organs, and their name derives from the recognition that they are responsible for tissue compatibility between individuals. The physiologic function of MHC molecules is to display peptide fragments of proteins for recognition by antigen-specific T cells.16 In humans the genes encoding the major histocompatibility molecules are clustered on a small segment of chromosome 6, the major histocompatibility complex, or the human leukocyte antigen (HLA) complex (Fig. 6-8), so named because in humans MHC-encoded proteins were initially detected on leukocytes by the binding of antibodies. The HLA system is highly polymorphic, meaning that there are many alleles of each MHC gene in the population and each individual inherits one set of these alleles that is different from the alleles in most other individuals. This, as we see subsequently, constitutes a formidable barrier in organ transplantation.
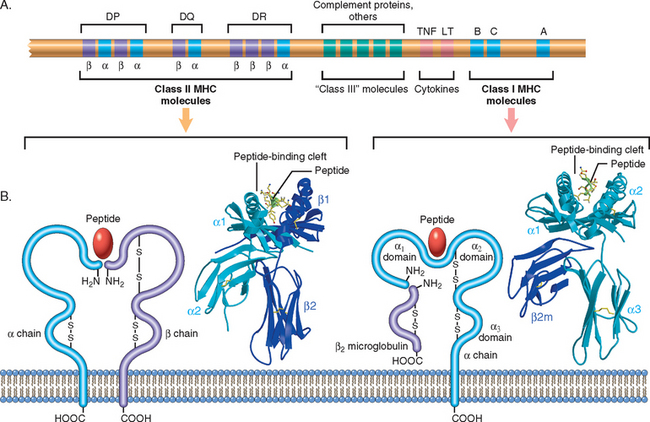
FIGURE 6-8 The human leukocyte antigen (HLA) complex and the structure of HLA molecules. A, The location of genes in the HLA complex. The relative locations, sizes, and distances between genes are not to scale. Genes that encode several proteins involved in antigen processing (the TAP transporter, components of the proteasome, and HLA-DM) are located in the class II region (not shown). B, Schematic diagrams and crystal structures of class I and class II HLA molecules.
(Courtesy of Drs. Kathryn Pape and Jennifer Walter, University of Minnesota School of Medicine, Minneapolis, MN.)
On the basis of their structure, cellular distribution, and function, MHC gene products are classified into three groups.
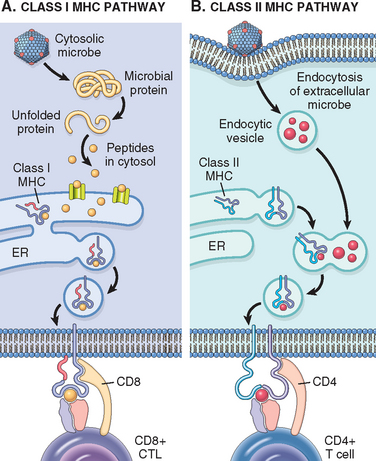
FIGURE 6-9 Antigen processing and display by major histocompatibility complex (MHC) molecules. A, In the class I MHC pathway, peptides are produced from proteins in the cytosol and transported to the endoplasmic reticulum (ER), where they bind to class I MHC molecules. The peptide-MHC complexes are transported to the cell surface and displayed for recognition by CD8+ T cells. B, In the class II MHC pathway, proteins are ingested into vesicles and degraded into peptides, which bind to class II MHC molecules being transported in the same vesicles. The class II–peptide complexes are expressed on the cell surface and recognized by CD4+ T cells.
Class I MHC molecules display peptides that are derived from proteins, such as viral antigens, that are located in the cytoplasm and usually produced in the cell, and class I–associated peptides are recognized by CD8+ T lymphocytes (Fig. 6-9A). Cytoplasmic proteins are degraded in proteasomes and peptides are transported into the endoplasmic reticulum (ER) where the peptides bind to newly synthesized class I molecules.17 Peptide-loaded MHC molecules associate with β2-microglobulin to form a stable trimer that is transported to the cell surface. The nonpolymorphic α3 domain of class I MHC molecules has a binding site for CD8, and therefore the peptide–class I complexes are recognized by CD8+ T cells, which function as CTLs. In this interaction, the TCR recognizes the MHC-peptide complex, and the CD8 molecule, acting as a coreceptor, binds to the class I heavy chain. Thus, CD8+ cytotoxic T cells recognize peptides that are produced by cytoplasmic microbes (typically viruses) or in tumors and kill cells harboring these infections or the tumor cells. Since CD8+ T cells recognize peptides only if presented as a complex with self–class I MHC molecules, CD8+ T cells are said to be class I MHC–restricted. Because one of the important functions of CD8+ CTLs is to eliminate viruses, which may infect any nucleated cell, it makes good sense that all nucleated cells express class I HLA molecules and can be surveyed by CD8+ T cells.
Class II MHC molecules present antigens that are internalized into vesicles, and are typically derived from extracellular microbes and soluble proteins (Fig. 6-9B). The internalized proteins are proteolytically digested in endosomes or lysosomes. Peptides resulting from proteolytic cleavage then associate with class II heterodimers in the vesicles, and the stable peptide-MHC complexes are transported to the cell surface. The class II β2 domain has a binding site for CD4, and therefore, the class II–peptide complex is recognized by CD4+ T cells, which function as helper cells. In this interaction, the CD4 molecule acts as the co-receptor. Because CD4+ T cells can recognize antigens only in the context of self–class II molecules, they are referred to as class II MHC–restricted. In contrast to class I molecules, class II MHC molecules are mainly expressed on cells that present ingested antigens and respond to T-cell help (macrophages, B lymphocytes, and dendritic cells).
The combination of HLA alleles in each individual is called the HLA haplotype. Any given individual inherits one set of HLA genes from each parent and thus typically expresses two different molecules for every locus. Because of the polymorphism of the HLA loci, virtually innumerable combinations of molecules exist in the population, and each individual expresses an MHC profile on his or her cell surface that is different from the haplotypes of most other individuals. It is believed that this polymorphism evolved to ensure that at least some individuals in a species would be able to display any microbial peptide and thus provide protection against any infection. The same polymorphism means that no two individuals (other than identical twins) are likely to express the same MHC molecules, and therefore grafts exchanged between these individuals are recognized as foreign and attacked by the immune system.
MHC molecules play key roles in regulating T cell–mediated immune responses in several ways. First, because different antigenic peptides bind to different MHC molecules, it follows that an individual mounts an immune response against a protein antigen only if he or she inherits the gene(s) for those MHC molecule(s) that can bind peptides derived from the antigen and present it to T cells. The consequences of inheriting a given MHC (e.g., class II) gene depend on the nature of the antigen bound by the class II molecule. For example, if the antigen is a peptide from ragweed pollen, the individual who expresses class II molecules capable of binding the antigen would be genetically prone to allergic reactions against pollen. In contrast, an inherited capacity to bind a bacterial peptide may provide resistance to the infection by evoking a protective antibody response. Second, by segregating cytoplasmic and internalized antigens, MHC molecules ensure that the correct immune response is mounted against different microbes—CTLs against cytoplasmic microbes, and antibodies and macrophages (both of which are activated by helper T cells) against extracellular microbes.
HLA and Disease Association
A variety of diseases are associated with the inheritance of certain HLA alleles (Table 6-1).18 The most striking of these is the association between ankylosing spondylitis and HLA-B27; individuals who inherit this class I HLA allele have a 90-fold greater chance (relative risk) of developing the disease as compared with those who do not carry HLA-B27. The diseases that show association with the HLA locus can be broadly grouped into the following categories:
TABLE 6-1 Association of HLA Alleles and Inflammatory Diseases
| Disease | HLA Allele | Relative Risk (%) |
|---|---|---|
| Ankylosing spondylitis | B27 | 90–100 |
| Postgonococcal arthritis | B27 | 14 |
| Acute anterior uveitis | B27 | 14 |
| Rheumatoid arthritis | DR4 | 4 |
| Chronic active hepatitis | DR3 | 13 |
| Primary Sjögren syndrome | DR3 | 9 |
| Type 1 diabetes | DR3 | 5 |
| DR4 | 6 | |
| DR3/DR4 | 20 |
The mechanisms underlying these associations are not fully understood. In the immunological and inflammatory diseases, the inheritance of particular HLA alleles likely influences the T-cell response, but it has proved difficult to define precisely how. In some cases (e.g., 21-hydroxylase deficiency), the linkage results because the relevant disease-associated gene, in this case the gene for 21-hydroxylase, maps within the HLA complex. Similarly, in hereditary hemochromatosis, a gene that is mutated, called HFE, maps within the HLA locus. The HFE protein resembles MHC molecules structurally, but its function is in the regulation of iron transport (Chapter 18).
Cytokines: Messenger Molecules of the Immune System
The induction and regulation of immune responses involve multiple interactions among lymphocytes, dendritic cells, macrophages, other inflammatory cells (e.g., neutrophils), and endothelial cells. Some of these interactions depend on cell-to-cell contact; however, many interactions and effector functions of leukocytes are mediated by short-acting secreted mediators called cytokines. Molecularly defined cytokines are called interleukins, because they mediate communications between leukocytes. Most cytokines have a wide spectrum of effects, and some are produced by several different cell types.
It is convenient to classify cytokines into distinct functional classes, although many belong to multiple categories.
The knowledge gained about cytokines has numerous practical therapeutic applications. Inhibiting cytokine production or actions is an approach for controlling the harmful effects of inflammation and tissue-damaging immune reactions. Patients with rheumatoid arthritis often show dramatic responses to TNF antagonists, an elegant example of rationally designed and molecularly targeted therapy. Conversely, recombinant cytokines can be administered to enhance immunity against cancer or microbial infections (immunotherapy).
OVERVIEW OF LYMPHOCYTE ACTIVATION AND IMMUNE RESPONSES
All adaptive immune responses develop in steps, consisting of: antigen recognition, activation of specific lymphocytes to proliferate and differentiate into effector and memory cells, elimination of the antigen, and decline of the response, with memory cells being the long-lived survivors. The major events in each step are summarized below; these general principles apply to protective responses against microbes as well as pathologic responses that injure the host.
The Display and Recognition of Antigens
Lymphocytes specific for a large number of antigens exist before exposure to the antigen, and when an antigen enters, it selects the specific cells and activates them. This fundamental concept is called the clonal selection hypothesis. According to this hypothesis, antigen-specific clones of lymphocytes develop before and independent of exposure to antigen. The cells constituting each clone have identical antigen receptors, which are different from the receptors on the cells of all other clones. It is estimated that there are about 107 to 109 different specificities in the total pool of about 1012 lymphocytes in an adult, and therefore, at least this many antigens can be recognized by the adaptive immune system. It follows that the number of lymphocytes specific for any one antigen is very small, probably less than 1 in 100,000 to 1 in 1 million cells. To permit a small number of lymphocytes to find antigen anywhere in the body, the immune system has specialized mechanisms for capturing antigens and displaying them to lymphocytes. Microbes and their protein antigens are captured by dendritic cells that are resident in epithelia and tissues. These cells carry their antigenic cargo to draining lymph nodes (Fig. 6-10).19 Here the antigens are processed and displayed complexed with MHC molecules on the cell surface (see Fig. 6-9).
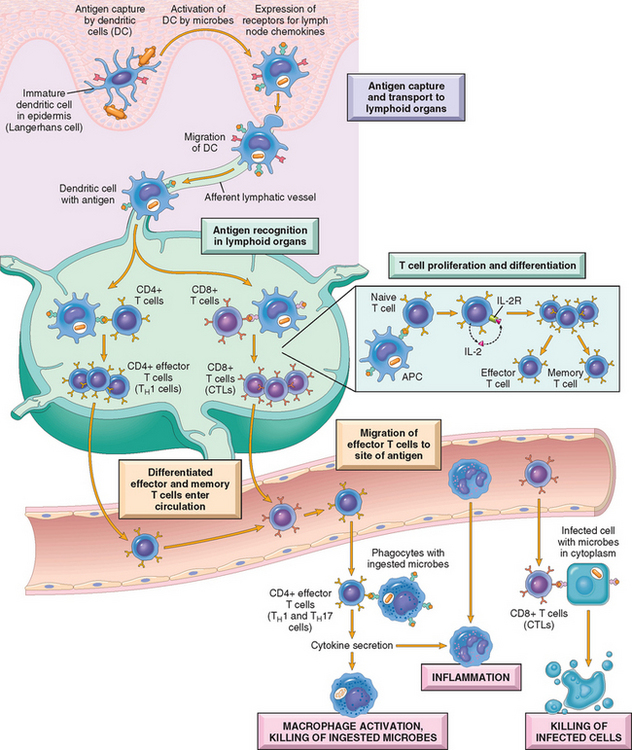
FIGURE 6-10 Cell-mediated immunity. Dendritic cells (DCs) capture microbial antigens from epithelia and tissues and transport the antigens to lymph nodes. During this process, the DCs mature, and express high levels of MHC molecules and costimulators. Naive T cells recognize MHC-associated peptide antigens displayed on DCs. The T cells are activated to proliferate and to differentiate into effector and memory cells, which migrate to sites of infection and serve various functions in cell-mediated immunity. CD4+ effector T cells of the TH1 subset recognize the antigens of microbes ingested by phagocytes, and activate the phagocytes to kill the microbes. CD4+ T cells also induce inflammation. CD8+ cytotoxic T lymphocytes (CTLs) kill infected cells harboring microbes in the cytoplasm. Not shown are TH2 cells, which are especially important in defense against helminthic infections. Some activated T cells differentiate into long-lived memory cells. APC, antigen-presenting cell.
B lymphocytes use their antigen receptors (membrane-bound antibody molecules) to recognize antigens of many different chemical types, including proteins, polysaccharides, and lipids.
At the same time as the antigens of a microbe are recognized by T and B lymphocytes, the microbe elicits an innate immune response; in the case of immunization with a protein antigen, the innate response is induced by the adjuvant given with the antigen. During this innate response the microbe activates APCs to express molecules called costimulators and to secrete cytokines that stimulate the proliferation and differentiation of T lymphocytes. The principal costimulators for T cells are the B7 proteins (CD80 and CD86) that are expressed on APCs and are recognized by the CD28 receptor on naive T cells.20 Thus, antigen (“signal 1”) and costimulatory molecules produced during innate immune responses to microbes (“signal 2”) function cooperatively to activate antigen-specific lymphocytes (see Fig. 6-3). The requirement for microbetriggered signal 2 ensures that the adaptive immune response is induced by microbes and not by harmless substances. In immune responses to tumors and transplants, “signal 2” may be provided by substances released from necrotic cells (the “danger-associated molecular patterns” mentioned earlier).
The reactions and functions of T and B lymphocytes differ in important ways and are best considered separately.
Cell-Mediated Immunity: Activation of T Lymphocytes and Elimination of Intracellular Microbes
Naive T lymphocytes are activated by antigen and costimulators in peripheral lymphoid organs, and proliferate and differentiate into effector cells that migrate to any site where the antigen (microbe) is present (see Fig. 6-10). One of the earliest responses of CD4+ helper T cells is secretion of the cytokine IL-2 and expression of high-affinity receptors for IL-2. IL-2 is a growth factor that acts on these T lymphocytes and stimulates their proliferation, leading to an increase in the number of antigen-specific lymphocytes. The functions of helper T cells are mediated by the combined actions of CD40-ligand (CD40L) and cytokines. When CD4+ helper T cells recognize antigens being displayed by macrophages or B lymphocytes, the T cells express CD40L, which engages CD40 on the macrophages or B cells and activates these cells.
Some of the progeny of the expanded T cells differentiate into effector cells that can secrete different sets of cytokines, and thus perform different functions (Fig. 6-11).21 The best defined subsets of differentiated CD4+ helper cells are the TH1 and TH2 subsets. Cells of the TH1 subset secrete the cytokine IFN-γ, which is a potent macrophage activator. The combination of CD40- and IFN-γ–mediated activation results in the induction of microbicidal substances in macrophages, leading to the destruction of ingested microbes. TH2 cells produce IL-4, which stimulates B cells to differentiate into IgE-secreting plasma cells, and IL-5, which activates eosinophils. Eosinophils and mast cells bind to IgE-coated microbes such as helminthic parasites, and function to eliminate helminths. A third subset of CD4+ T cells that has been discovered recently is called the TH17 subset because the signature cytokine of these cells is IL-17.22,23 TH17 cells are powerful recruiters of neutrophils and monocytes, and thus play major roles in several inflammatory diseases. They may also be important for defense against some bacterial and fungal infections in which neutrophilic inflammation is a prominent feature. We will return to the generation and functions of these subsets when we discuss hypersensitivity reactions.
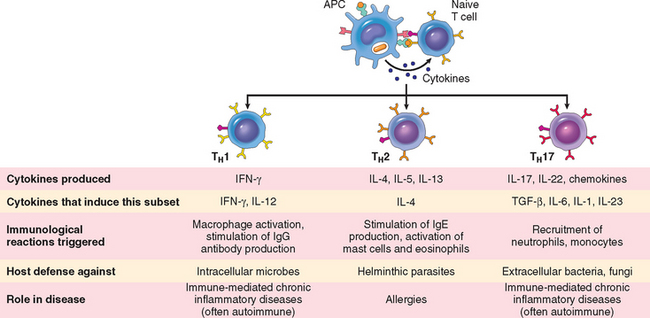
FIGURE 6-11 Subsets of helper T (TH) cells. In response to stimuli (mainly cytokines) present at the time of antigen recognition, naive CD4+ TH cells may differentiate into populations of effector cells that produce distinct sets of cytokines and perform different functions. The dominant immune reactions elicited by each subset, and its role in host defense and immunological diseases, are summarized.
Activated CD8+ lymphocytes differentiate into CTLs that kill cells harboring microbes in the cytoplasm. By destroying the infected cells, CTLs eliminate the reservoirs of infection.
Humoral Immunity: Activation of B Lymphocytes and Elimination of Extracellular Microbes
Upon activation, B lymphocytes proliferate and then differentiate into plasma cells that secrete different classes of antibodies with distinct functions (Fig. 6-12). Many polysaccharide and lipid antigens have multiple identical antigenic determinants (epitopes) that are able to engage many antigen receptor molecules on each B cell and initiate the process of B-cell activation. Typical globular protein antigens are not able to bind to many antigen receptors, and the full response of B cells to protein antigens requires help from CD4+ T cells.24 B cells ingest protein antigens into vesicles, degrade them, and display peptides bound to MHC molecules for recognition by helper T cells. The helper T cells express CD40L and secrete cytokines, which work together to activate the B cells.

FIGURE 6-12 Humoral immunity. Naive B lymphocytes recognize antigens, and under the influence of TH cells and other stimuli (not shown), the B cells are activated to proliferate and to differentiate into antibody-secreting plasma cells. Some of the activated B cells undergo heavy-chain class switching and affinity maturation, and some become long-lived memory cells. Antibodies of different heavy-chain classes (isotypes) perform different effector functions, shown on the right. See text for abbreviations.
Each plasma cell secretes antibodies that have the same antigen binding site as the cell surface antibodies (B-cell receptors) that first recognized the antigen. Polysaccharides and lipids stimulate secretion mainly of IgM antibody. Protein antigens, by virtue of CD40L- and cytokine-mediated helper T-cell actions, induce the production of antibodies of different classes, or isotypes (IgG, IgA, IgE). Cytokines that induce isotype switching include IFN-γ and IL-4. Helper T cells also stimulate the production of antibodies with high affinities for the antigen. This process, called affinity maturation, improves the quality of the humoral immune response. Isotype switching and affinity maturation occur mainly in germinal centers, which are formed by proliferating B cells, especially in helper T cell-dependent responses to protein antigens.
The humoral immune response combats microbes in many ways (see Fig. 6-12). Antibodies bind to microbes and prevent them from infecting cells, thus “neutralizing” the microbes. IgG antibodies coat (“opsonize”) microbes and target them for phagocytosis, since phagocytes (neutrophils and macrophages) express receptors for the Fc tails of IgG. IgG and IgM activate the complement system by the classical pathway, and complement products promote phagocytosis and destruction of microbes. The production of most opsonizing and complement-fixing IgG antibodies is stimulated by TH1 helper cells, which respond to many bacteria and viruses; thus, the protective response to most bacteria and viruses is driven by TH1 cells. Some antibodies serve special roles at particular anatomic sites. IgA is secreted from mucosal epithelia and neutralizes microbes in the lumens of the respiratory and gastrointestinal tracts (and other mucosal tissues). IgG is actively transported across the placenta and protects the newborn until the immune system becomes mature. IgE and eosinophils cooperate to kill parasites, mainly by release of eosinophil granule contents that are toxic to the worms. As mentioned above, TH2 cells secrete cytokines that stimulate the production of IgE and activate eosinophils, and thus the response to helminths is orchestrated by TH2 cells.
Most circulating IgG antibodies have half-lives of about 3 weeks. Some antibody-secreting plasma cells migrate to the bone marrow and live for years, continuing to produce low levels of antibodies.
Decline of Immune Responses and Immunological Memory
The majority of effector lymphocytes induced by an infectious pathogen die by apoptosis after the microbe is eliminated, thus returning the immune system to its basal resting state, called homeostasis. The initial activation of lymphocytes also generates long-lived memory cells, which may survive for years after the infection. Memory cells are an expanded pool of antigen-specific lymphocytes (more numerous than the naive cells specific for any antigen that are present before encounter with that antigen), and that respond faster and more effectively when re-exposed to the antigen than do naive cells.25 This is why the generation of memory cells is an important goal of vaccination.
The brief outline of basic immunology presented above provides a foundation for considering the diseases of the immune system. Our subsequent discussion will be divided into disorders caused by an abnormally active immune system, called hypersensitivity disorders, and the rejection of transplants, followed by diseases caused by a defective immune system, called immunodeficiency diseases. We close with a consideration of amyloidosis, a disorder that is often associated with immune and inflammatory diseases.
Hypersensitivity and Autoimmune Disorders
Before discussing specific immunological diseases, we start with a summary of the general mechanisms of hypersensitivity.
MECHANISMS OF HYPERSENSITIVITY REACTIONS
Individuals who have been previously exposed to an antigen are said to be sensitized. Sometimes, repeat exposures to the same antigen trigger a pathologic reaction; such reactions are described as hypersensitivity, implying an excessive response to antigen. There are several important general features of hypersensitivity disorders.
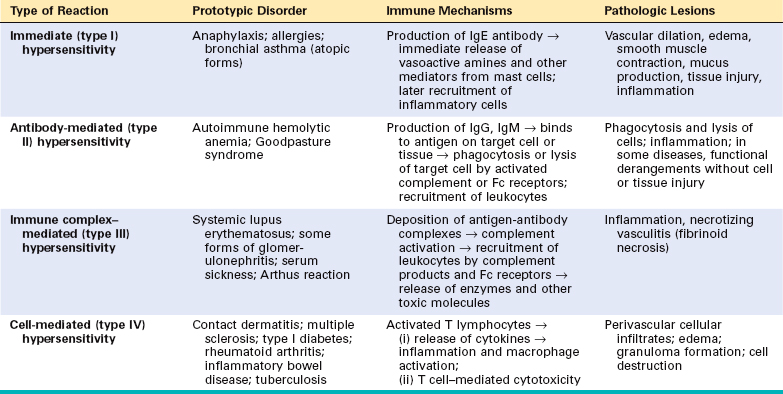
Hypersensitivity diseases can be classified on the basis of the immunologic mechanism that mediates the disease (Table 6-2). This classification is of value in distinguishing the manner in which the immune response causes tissue injury and disease, and the accompanying pathologic and clinical manifestations. However, it is now increasingly recognized that multiple mechanisms may be operative in any one hypersensitivity disease. The main types of hypersensitivity reactions are the following:
Immediate (Type I) Hypersensitivity
Immediate, or type I, hypersensitivity is a rapid immunologic reaction occurring within minutes after the combination of an antigen with antibody bound to mast cells in individuals previously sensitized to the antigen.26 These reactions are often called allergy, and the antigens that elicit them are allergens. Immediate hypersensitivity may occur as a systemic disorder or as a local reaction. The systemic reaction usually follows injection of an antigen into a sensitized individual. Sometimes, within minutes the patient goes into a state of shock, which may be fatal. Local reactions are diverse and vary depending on the portal of entry of the allergen. They may take the form of localized cutaneous swellings (skin allergy, hives), nasal and conjunctival discharge (allergic rhinitis and conjunctivitis), hay fever, bronchial asthma, or allergic gastroenteritis (food allergy). Many local type I hypersensitivity reactions have two well-defined phases (Fig. 6-13). The immediate or initial reaction is characterized by vasodilation, vascular leakage, and depending on the location, smooth muscle spasm or glandular secretions. These changes usually become evident within 5 to 30 minutes after exposure to an allergen and tend to subside in 60 minutes. In many instances (e.g., allergic rhinitis and bronchial asthma), a second, late-phase reaction sets in 2 to 24 hours later without additional exposure to antigen and may last for several days. This late-phase reaction is characterized by infiltration of tissues with eosinophils, neutrophils, basophils, monocytes, and CD4+ T cells as well as tissue destruction, typically in the form of mucosal epithelial cell damage.
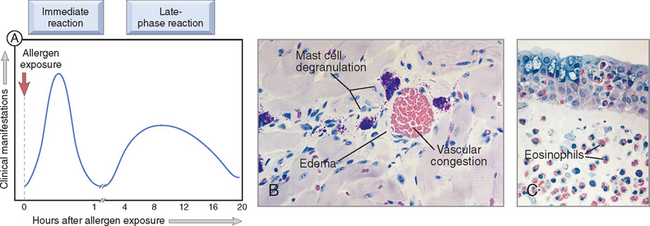
FIGURE 6-13 Immediate hypersensitivity. A, Kinetics of the immediate and late-phase reactions. The immediate vascular and smooth muscle reaction to allergen develops within minutes after challenge (allergen exposure in a previously sensitized individual), and the late-phase reaction develops 2 to 24 hours later. B, C, Morphology: The immediate reaction (B) is characterized by vasodilation, congestion, and edema, and the late-phase reaction (C) is characterized by an inflammatory infiltrate rich in eosinophils, neutrophils, and T cells.
(Courtesy of Dr. Daniel Friend, Department of Pathology, Brigham and Women’s Hospital, Boston, MA.)
Most immediate hypersensitivity reactions are mediated by IgE antibody–dependent activation of mast cells and other leukocytes (Fig. 6-14). Because mast cells are central to the development of immediate hypersensitivity, we first review some of their salient characteristics.27 Mast cells are bone marrow–derived cells that are widely distributed in the tissues. They are abundant near blood vessels and nerves and in subepithelial tissues, which explains why local immediate hypersensitivity reactions often occur at these sites. Mast cells have cytoplasmic membrane-bound granules that contain a variety of biologically active mediators. The granules also contain acidic proteoglycans that bind basic dyes such as toluidine blue. As is detailed next, mast cells (and basophils) are activated by the cross-linking of high-affinity IgE Fc receptors; in addition, mast cells may also be triggered by several other stimuli, such as complement components C5a and C3a (called anaphylatoxins because they elicit reactions that mimic anaphylaxis), both of which act by binding to receptors on the mast cell membrane. Other mast cell secretagogues include some chemokines (e.g., IL-8), drugs such as codeine and morphine, adenosine, mellitin (present in bee venom), and physical stimuli (e.g., heat, cold, sunlight). Basophils are similar to mast cells in many respects, including the presence of cell surface IgE Fc receptors as well as cytoplasmic granules. In contrast to mast cells, however, basophils are not normally present in tissues but rather circulate in the blood in extremely small numbers. (Most allergic reactions occur in tissues, and the role of basophils in these reactions is not as well established as that of mast cells.) Similar to other granulocytes, basophils can be recruited to inflammatory sites.
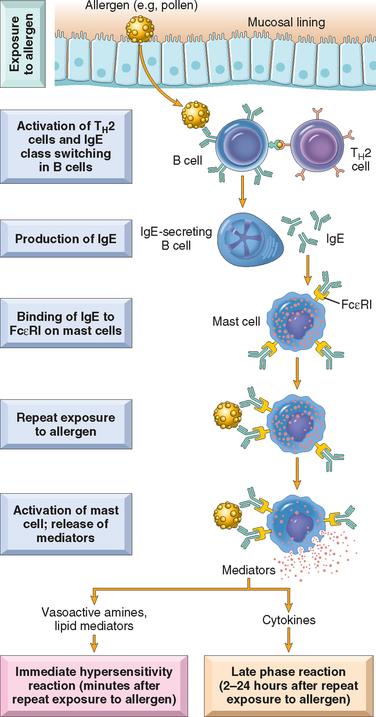
FIGURE 6-14 Sequence of events in immediate (type I) hypersensitivity. Immediate hypersensitivity reactions are initiated by the introduction of an allergen, which stimulates TH2 responses and IgE production in genetically susceptible individuals. IgE binds to Fc receptors (FcεRI) on mast cells, and subsequent exposure to the allergen activates the mast cells to secrete the mediators that are responsible for the pathologic manifestations of immediate hypersensitivity. See text for abbreviations.
TH2 cells play a central role in the initiation and propagation of immediate hypersensitivity reactions by stimulating IgE production and promoting inflammation.28,29 The first step in the generation of TH2 cells is the presentation of the antigen to naive CD4+ helper T cells, probably by dendritic cells that capture the antigen from its site of entry. In response to antigen and other stimuli, including cytokines such as IL-4 produced at the local site, the T cells differentiate into TH2 cells. The newly minted TH2 cells produce a number of cytokines upon subsequent encounter with the antigen; as we mentioned earlier, the signature cytokines of this subset are IL-4, IL-5, and IL-13. IL-4 acts on B cells to stimulate class switching to IgE, and promotes the development of additional TH2 cells. IL-5 is involved in the development and activation of eosinophils, which, as we discuss subsequently, are important effectors of type I hypersensitivity. IL-13 enhances IgE production and acts on epithelial cells to stimulate mucus secretion. In addition, TH2 cells (as well as mast cells and epithelial cells) produce chemokines that attract more TH2 cells, as well as other leukocytes, to the reaction site.28
Mast cells and basophils express a high-affinity receptor, called FcεRI, that is specific for the Fc portion of IgE, and therefore avidly binds IgE antibodies. When a mast cell, armed with IgE antibodies, is exposed to the specific allergen, a series of reactions takes place, leading eventually to the release of an arsenal of powerful mediators responsible for the clinical expression of immediate hypersensitivity reactions. In the first step in this sequence, antigen (allergen) binds to the IgE antibodies previously attached to the mast cells. Multivalent antigens bind to and cross-link adjacent IgE antibodies and the underlying IgE Fc receptors. The bridging of the Fcε receptors activates signal transduction pathways from the cytoplasmic portion of the receptors. These signals lead to mast cell degranulation with the discharge of preformed (primary) mediators that are stored in the granules, and de novo synthesis and release of secondary mediators, including lipid products and cytokines (Fig. 6-15). These mediators are responsible for the initial, sometimes explosive, symptoms of immediate hypersensitivity, and they also set into motion the events that lead to the late-phase reaction.26
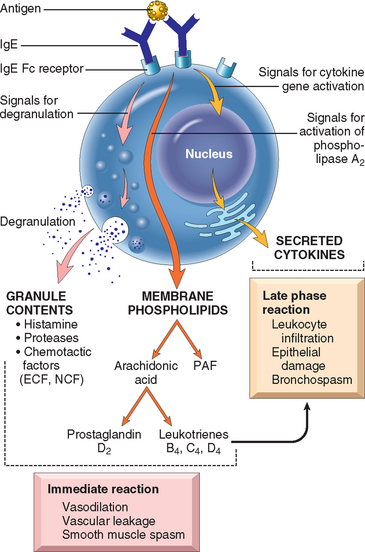
FIGURE 6-15 Mast cell mediators. Upon activation, mast cells release various classes of mediators that are responsible for the immediate and late-phase reactions. ECF, eosinophil chemotactic factor; NCF, neutrophil chemotactic factor (neither of these is biochemically defined); PAF, platelet-activating factor.
Preformed Mediators.
Mediators contained within mast cell granules are the first to be released, and can be divided into three categories:
Lipid Mediators.
The major lipid mediators are synthesized by sequential reactions in the mast cell membranes that lead to activation of phospholipase A2, an enzyme that acts on membrane phospholipids to yield arachidonic acid. This is the parent compound from which leukotrienes and prostaglandins are derived by the 5-lipoxygenase and cyclooxygenase pathways (Chapter 2).
Cytokines.
Mast cells are sources of many cytokines, which may play an important role at several stages of immediate hypersensitivity reactions. The cytokines include: TNF, IL-1, and chemokines, which promote leukocyte recruitment (typical of the late-phase reaction); IL-4, which amplifies the TH2 response; and numerous others. The inflammatory cells that are recruited by mast cell–derived TNF and chemokines are additional sources of cytokines and of histamine-releasing factors that cause further mast cell degranulation.
The development of immediate hypersensitivity reactions is dependent on the coordinated actions of a variety of chemotactic, vasoactive, and spasmogenic compounds (Table 6-3). Some, such as histamine and leukotrienes, are released rapidly from sensitized mast cells and are responsible for the intense immediate reactions characterized by edema, mucus secretion, and smooth muscle spasm; others, exemplified by cytokines, set the stage for the late-phase response by recruiting additional leukocytes. Not only do these inflammatory cells release additional waves of mediators (including cytokines), but they also cause epithelial cell damage. Epithelial cells themselves are not passive bystanders in this reaction; they can also produce soluble mediators, such as chemokines.
TABLE 6-3 Summary of the Action of Mast Cell Mediators in Immediate (Type I) Hypersensitivity
| Action | Mediators |
|---|---|
| Vasodilation, increased vascular permeability | Histamine |
| PAF | |
| Leukotrienes C4, D4, E4 | |
| Neutral proteases that activate complement and kinins | |
| Prostaglandin D2 | |
| Smooth muscle spasm | Leukotrienes C4, D4, E4 |
| Histamine | |
| Prostaglandins | |
| PAF | |
| Cellular infiltration | Cytokines (e.g., chemokines, TNF) |
| Leukotriene B4 | |
| Eosinophil and neutrophil chemotactic factors (not defined biochemically) |
PAF, platelet-activating factor; TNF, tumor necrosis factor.
Among the cells that are recruited in the late-phase reaction, eosinophils are particularly important.30 They are recruited to sites of immediate hypersensitivity reactions by chemokines, such as eotaxin and others, that may be produced by epithelial cells, TH2 cells, and mast cells. The survival of eosinophils in tissues is favored by IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-5 is the most potent eosinophil-activating cytokine known. Eosinophils liberate proteolytic enzymes as well as two unique proteins called major basic protein and eosinophil cationic protein, which are toxic to epithelial cells. Activated eosinophils and other leukocytes also produce leukotriene C4 and PAF and directly activate mast cells to release mediators. Thus, the recruited cells amplify and sustain the inflammatory response without additional exposure to the triggering antigen. It is now believed that this late-phase reaction is a major cause of symptoms in some type I hypersensitivity disorders, such as allergic asthma. Therefore, treatment of these diseases requires the use of broad-spectrum anti-inflammatory drugs, such as steroids.
Susceptibility to immediate hypersensitivity reactions is genetically determined. The term atopy refers to a predisposition to develop localized immediate hypersensitivity reactions to a variety of inhaled and ingested allergens. Atopic individuals tend to have higher serum IgE levels, and more IL-4–producing TH2 cells, compared with the general population. A positive family history of allergy is found in 50% of atopic individuals. The basis of familial predisposition is not clear, but studies in patients with asthma reveal linkage to several gene loci.31 Candidate genes have been mapped to 5q31, where genes encoding the cytokines IL-3, IL-4, IL-5, IL-9, IL-13, and GM-CSF are located. This locus has attracted great attention because of the known roles of many of these cytokines in the reaction, but how the disease-associated polymorphisms influence the biology of the cytokines is not known. Linkage has also been noted to 6p, close to the HLA complex, suggesting that the inheritance of certain HLA alleles permits reactivity to certain allergens.
A significant proportion of immediate hypersensitivity reactions are triggered by temperature extremes and exercise, and do not involve TH2 cells or IgE; such reactions are sometimes called “non-atopic allergy.” It is believed that in these cases mast cells are abnormally sensitive to activation by various non-immune stimuli.
A final point that should be mentioned in this general discussion of immediate hypersensitivity disorders is that the incidence of many of these diseases is increasing in developed countries, and seems to be related to a decrease in infections during early life. These observations have led to an idea, sometimes called the hygiene hypothesis, that reduced exposure to microbes resets the immune system in such a way that TH2 responses develop more readily against common environmental antigens. This hypothesis, however, is controversial, and the underlying mechanisms are not defined.
To summarize, immediate (type I) hypersensitivity is a complex disorder resulting from an IgE-mediated triggering of mast cells and subsequent accumulation of inflammatory cells at sites of antigen deposition. These events are regulated mainly by the induction of TH2 helper T cells that stimulate production of IgE (which promotes mast cell activation), cause accumulation of inflammatory cells (particularly eosinophils), and trigger secretion of mucus. The clinical features result from release of mast cell mediators as well as the eosinophil-rich inflammation.
With this consideration of the basic mechanisms of type I hypersensitivity, we turn to some conditions that are important examples of IgE-mediated disease.
Systemic Anaphylaxis
Systemic anaphylaxis is characterized by vascular shock, widespread edema, and difficulty in breathing. It may occur in sensitized individuals in hospital settings after administration of foreign proteins (e.g., antisera), hormones, enzymes, polysaccharides, and drugs (such as the antibiotic penicillin), or in the community setting following exposure to food allergens (e.g. peanuts, shellfish) or insect toxins (e.g. those in bee venom).32 Extremely small doses of antigen may trigger anaphylaxis, for example, the tiny amounts used in skin testing for various forms of allergies. Because of the risk of severe allergic reactions to minute quantities of peanuts, the U.S. Congress is considering a bill to ban peanut snacks from the confined quarters of commercial airplanes. Within minutes after exposure, itching, hives, and skin erythema appear, followed shortly thereafter by a striking contraction of respiratory bronchioles and respiratory distress. Laryngeal edema results in hoarseness and further compromises breathing. Vomiting, abdominal cramps, diarrhea, and laryngeal obstruction follow, and the patient may go into shock and even die within the hour. The risk of anaphylaxis must be borne in mind when certain therapeutic agents are administered. Although patients at risk can generally be identified by a previous history of some form of allergy, the absence of such a history does not preclude the possibility of an anaphylactic reaction.
Local Immediate Hypersensitivity Reactions
About 10% to 20% of the population suffers from allergies involving localized reactions to common environmental allergens, such as pollen, animal dander, house dust, foods, and the like. Specific diseases include urticaria, angioedema, allergic rhinitis (hay fever), and bronchial asthma; these are discussed elsewhere in the book.
Antibody-Mediated (Type II) Hypersensitivity
This type of hypersensitivity is caused by antibodies that react with antigens present on cell surfaces or in the extracellular matrix. The antigenic determinants may be intrinsic to the cell membrane or matrix, or they may take the form of an exogenous antigen, such as a drug metabolite, that is adsorbed on a cell surface or matrix. In either case the hypersensitivity reaction results from the binding of antibodies to normal or altered cell surface antigens. The antibody-dependent mechanisms that cause tissue injury and disease are illustrated in Figure 6-16 and described next.
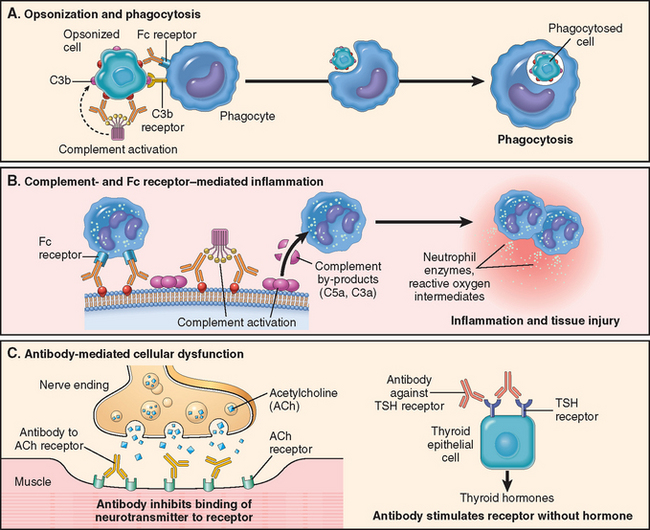
FIGURE 6-16 Mechanisms of antibody-mediated injury. A, Opsonization of cells by antibodies and complement components and ingestion by phagocytes. B, Inflammation induced by antibody binding to Fc receptors of leukocytes and by complement breakdown products. C, Anti-receptor antibodies disturb the normal function of receptors. In these examples, antibodies to the acetylcholine (ACh) receptor impair neuromuscular transmission in myasthenia gravis, and antibodies against the thyroid-stimulating hormone (TSH) receptor activate thyroid cells in Graves disease.
Opsonization and Phagocytosis
Phagocytosis is largely responsible for depletion of cells coated with antibodies. Cells opsonized by IgG antibodies are recognized by phagocyte Fc receptors, which are specific for the Fc portions of some IgG subclasses. In addition, when IgM or IgG antibodies are deposited on the surfaces of cells, they may activate the complement system by the classical pathway. Complement activation generates by-products, mainly C3b and C4b, which are deposited on the surfaces of the cells and recognized by phagocytes that express receptors for these proteins. The net result is phagocytosis of the opsonized cells and their destruction (Fig. 6-16A). Complement activation on cells also leads to the formation of the membrane attack complex, which disrupts membrane integrity by “drilling holes” through the lipid bilayer, thereby causing osmotic lysis of the cells. This mechanism of depletion is probably effective only with cells that have thin cell walls, such as Neisseria bacteria.
Antibody-mediated destruction of cells may occur by another process called antibody-dependent cellular cytotoxicity (ADCC). Cells that are coated with low concentrations of IgG antibody are killed by a variety of effector cells, which bind to the target by their receptors for the Fc fragment of IgG, and cell lysis proceeds without phagocytosis. ADCC may be mediated by monocytes, neutrophils, eosinophils, and NK cells. The role of ADCC in particular hypersensitivity diseases is uncertain.
Clinically, antibody-mediated cell destruction and phagocytosis occur in the following situations: (1) transfusion reactions, in which cells from an incompatible donor react with and are opsonized by preformed antibody in the host; (2) hemolytic disease of the newborn (erythroblastosis fetalis), in which there is an antigenic difference between the mother and the fetus, and antibodies (of the IgG class) from the mother cross the placenta and cause destruction of fetal red cells; (3) autoimmune hemolytic anemia, agranulocytosis, and thrombocytopenia, in which individuals produce antibodies to their own blood cells, which are then destroyed; and (4) certain drug reactions, in which a drug acts as a “hapten” by attaching to surface molecules of red cells and antibodies are produced against the drug–membrane protein complex.
Inflammation
When antibodies deposit in fixed tissues, such as basement membranes and extracellular matrix, the resultant injury is due to inflammation. The deposited antibodies activate complement, generating by-products, including chemotactic agents (mainly C5a), which direct the migration of polymorphonuclear leukocytes and monocytes, and anaphylatoxins (C3a and C5a), which increase vascular permeability (Fig. 6-16B). The leukocytes are activated by engagement of their C3b and Fc receptors. This results in the release or generation of a variety of pro-inflammatory substances, including prostaglandins, vasodilator peptides, and chemotactic substances. Leukocyte activation leads to the production of other substances that damage tissues, such as lysosomal enzymes, including proteases capable of digesting basement membrane, collagen, elastin, and cartilage, and reactive oxygen species. It was once thought that complement was the major mediator of antibody-induced inflammation, but knockout mice lacking Fc receptors also show striking reduction in these reactions. It is now believed that inflammation in antibody-mediated (and immune complex–mediated) diseases is due to both complement- and Fc receptor–dependent reactions.33
Antibody-mediated inflammation is the mechanism responsible for tissue injury in some forms of glomerulonephritis, vascular rejection in organ grafts, and other disorders (Table 6-4).
Cellular Dysfunction
In some cases, antibodies directed against cell surface receptors impair or dysregulate function without causing cell injury or inflammation (Fig. 6-16C). For example, in myasthenia gravis, antibodies reactive with acetylcholine receptors in the motor end plates of skeletal muscles block neuromuscular transmission and therefore cause muscle weakness. The converse (i.e., antibody-mediated stimulation of cell function) is the basis of Graves disease. In this disorder, antibodies against the thyroid-stimulating hormone receptor on thyroid epithelial cells stimulate the cells, resulting in hyperthyroidism.
Immune Complex–Mediated (Type III) Hypersensitivity
Antigen-antibody complexes produce tissue damage mainly by eliciting inflammation at the sites of deposition. The pathologic reaction is initiated when antigen combines with antibody within the circulation (circulating immune complexes), and these are deposited typically in vessel walls.34 Sometimes the complexes are formed at extravascular sites where antigen may have been “planted” previously (called in situ immune complexes). The antigens that form immune complexes may be exogenous, such as a foreign protein that is injected or produced by an infectious microbe, or endogenous, if the individual produces antibody against self-components (autoimmunity). Examples of immune complex disorders and the antigens involved are listed in Table 6-5. Immune complex–mediated diseases can be systemic, if immune complexes are formed in the circulation and are deposited in many organs, or localized to particular organs, such as the kidney (glomerulonephritis), joints (arthritis), or the small blood vessels of the skin if the complexes are deposited or formed in these tissues.
TABLE 6-5 Examples of Immune Complex–Mediated Diseases
| Disease | Antigen Involved | Clinicopathologic Manifestations |
|---|---|---|
| Systemic lupus erythematosus | Nuclear antigens | Nephritis, skin lesions, arthritis, others |
| Poststreptococcal glomerulonephritis | Streptococcal cell wall antigen(s); may be “planted” in glomerular basement membrane | Nephritis |
| Polyarteritis nodosa | Hepatitis B virus antigens in some cases | Systemic vasculitis |
| Reactive arthritis | Bacterial antigens (e.g., Yersinia) | Acute arthritis |
| Serum sickness | Various proteins, e.g., foreign serum protein (horse anti-thymocyte globulin) | Arthritis, vasculitis, nephritis |
| Arthus reaction (experimental) | Various foreign proteins | Cutaneous vasculitis |
Systemic Immune Complex Disease
Acute serum sickness is the prototype of a systemic immune complex disease; it was once a frequent sequela to the administration of large amounts of foreign serum (e.g., serum from immunized horses used for protection against diphtheria). In modern times the disease is infrequent, but it is an informative model that has taught us a great deal about systemic immune complex disorders.
The pathogenesis of systemic immune complex disease can be divided into three phases: (1) formation of antigen-antibody complexes in the circulation; (2) deposition of the immune complexes in various tissues, thus initiating (3) an inflammatory reaction at the sites of immune complex deposition (Fig. 6-17).
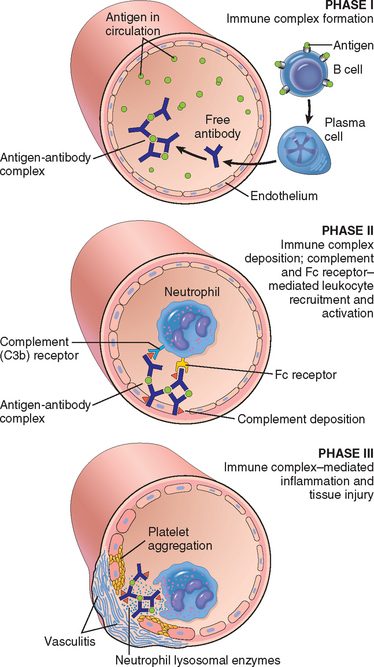
FIGURE 6-17 Pathogenesis of systemic immune complex–mediated disease (type III hypersensitivity). The three sequential phases in the development of immune complex diseases are shown.
Formation of Immune Complexes.
The introduction of a protein antigen triggers an immune response that results in the formation of antibodies, typically about a week after the injection of the protein. These antibodies are secreted into the blood, where they react with the antigen still present in the circulation and form antigen-antibody complexes.
Deposition of Immune Complexes.
In the next phase the circulating antigen-antibody complexes are deposited in various tissues. The factors that determine whether immune complex formation will lead to tissue deposition and disease are not fully understood, but the major influences seem to be the characteristics of the complexes and local vascular alterations.
In general, complexes that are of medium size, formed in slight antigen excess, are the most pathogenic. Organs where blood is filtered at high pressure to form other fluids, like urine and synovial fluid, are favored; hence, immune complexes frequently deposit in glomeruli and joints.35
Tissue Injury Caused by Immune Complexes.
Once complexes are deposited in the tissues, they initiate an acute inflammatory reaction (the third phase). During this phase (approximately 10 days after antigen administration), clinical features such as fever, urticaria, joint pains (arthralgias), lymph node enlargement, and proteinuria appear. Wherever complexes deposit the tissue damage is similar. The mechanisms of inflammation and injury were discussed above, in the discussion of antibody-mediated injury. The resultant inflammatory lesion is termed vasculitis if it occurs in blood vessels, glomerulonephritis if it occurs in renal glomeruli, arthritis if it occurs in the joints, and so on.
It is clear that complement-fixing antibodies (i.e., IgG and IgM) and antibodies that bind to leukocyte Fc receptors (some subclasses of IgG) induce the pathologic lesions of immune complex disorders. The important role of complement in the pathogenesis of the tissue injury is supported by the observations that during the active phase of the disease, consumption of complement leads to a decrease in serum levels of C3. In fact, serum C3 levels can, in some cases, be used to monitor disease activity.
Morphology. The principal morphologic manifestation of immune complex injury is acute necrotizing vasculitis, with necrosis of the vessel wall and intense neutrophilic infiltration. The necrotic tissue and deposits of immune complexes, complement, and plasma protein produce a smudgy eosinophilic deposit that obscures the underlying cellular detail, an appearance termed fibrinoid necrosis (Fig. 6-18). When deposited in the kidney, the complexes can be seen on immunofluorescence microscopy as granular lumpy deposits of immunoglobulin and complement and on electron microscopy as electron-dense deposits along the glomerular basement membrane (see Figs. 6-30 and 6-31).
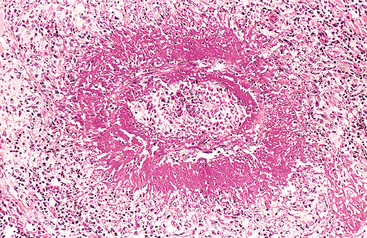
FIGURE 6-18 Immune complex vasculitis. The necrotic vessel wall is replaced by smudgy, pink “fibrinoid” material.
(Courtesy of Dr. Trace Worrell, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
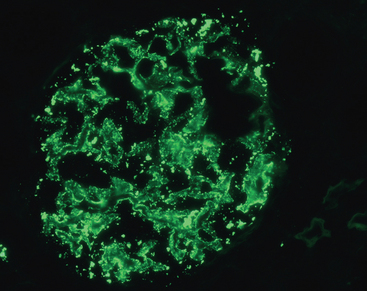
FIGURE 6-30 Immune complex deposition in systemic lupus erythematosus. Immunofluorescence micrograph of a glomerulus stained with fluorescent anti-IgG from a patient with diffuse proliferative lupus nephritis. Note the mesangial and capillary wall deposits of IgG.
(Courtesy of Dr. Jean Olson, Department of Pathology, University of California San Francisco, San Francisco, CA.)
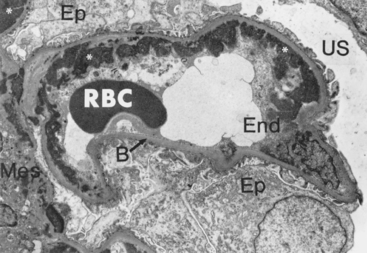
FIGURE 6-31 Immune complex deposition in systemic lupus erythematosus (SLE). Electron micrograph of a renal glomerular capillary loop from a patient with SLE nephritis shows subendothelial dense deposits corresponding to “wire loops” seen by light microscopy. Deposits are also present in the mesangium. B, basement membrane; End, endothelium; Ep, epithelium; RBC, red blood cell; US, urinary space
(Courtesy of Dr. Edwin Eigenbrodt, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
If the disease results from a single large exposure to antigen (e.g., acute serum sickness and perhaps acute poststreptococcal glomerulonephritis), the lesions tend to resolve, as a result of catabolism of the immune complexes. A chronic form of serum sickness results from repeated or prolonged exposure to an antigen. This occurs in several human diseases, such as systemic lupus erythematosus (SLE), which is associated with persistent antibody responses to autoantigens. In many diseases, however, the morphologic changes and other findings suggest immune complex deposition but the inciting antigens are unknown. Included in this category are membranous glomerulonephritis, many cases of polyarteritis nodosa, and several other vasculitides.
Local Immune Complex Disease (Arthus Reaction)
The Arthus reaction is a localized area of tissue necrosis resulting from acute immune complex vasculitis, usually elicited in the skin. The reaction can be produced experimentally by intracutaneous injection of antigen in a previously immunized animal that contains circulating antibodies against the antigen. As the antigen diffuses into the vascular wall, it binds the preformed antibody, and large immune complexes are formed locally. These complexes precipitate in the vessel walls and cause fibrinoid necrosis, and superimposed thrombosis worsens the ischemic injury.
T Cell–Mediated (Type IV) Hypersensitivity
The cell-mediated type of hypersensitivity is initiated by antigen-activated (sensitized) T lymphocytes, including CD4+ and CD8+ T cells (Fig. 6-19). CD4+ T cell–mediated hypersensitivity induced by environmental and self-antigens can be a cause of chronic inflammatory disease. Many autoimmune diseases are now known to be caused by inflammatory reactions driven by CD4+ T cells (Table 6-6). In some of these T cell–mediated autoimmune diseases, CD8+ cells may also be involved. In fact, in certain forms of T cell–mediated reactions, especially those that follow viral infections, CD8+ cells may be the dominant effector cells.
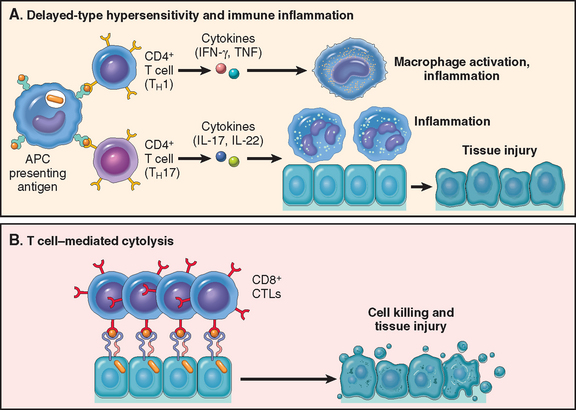
FIGURE 6-19 Mechanisms of T cell–mediated (type IV) hypersensitivity reactions. A, In delayed-type hypersensitivity reactions, CD4+ TH1 cells (and sometimes CD8+ T cells, not shown) respond to tissue antigens by secreting cytokines that stimulate inflammation and activate phagocytes, leading to tissue injury. CD4+ TH17 cells contribute to inflammation by recruiting neutrophils (and, to a lesser extent, monocytes). B, In some diseases, CD8+ cytotoxic T lymphocytes (CTLs) directly kill tissue cells. APC, antigen-presenting cell. See text for other abbreviations.
TABLE 6-6 Examples of T Cell–Mediated (Type IV) Hypersensitivity
| Disease | Specificity of Pathogenic T Cells | Clinicopathologic Manifestations |
|---|---|---|
| Type 1 diabetes mellitus | Antigens of pancreatic islet β cells (insulin, glutamic acid decarboxylase, others) | Insulitis (chronic inflammation in islets), destruction of β cells; diabetes |
| Multiple sclerosis | Protein antigens in CNS myelin (myelin basic protein, proteolipid protein) | Demyelination in CNS with perivascular inflammation; paralysis, ocular lesions |
| Rheumatoid arthritis | Unknown antigen in joint synovium (type II collagen?); role of antibodies? | Chronic arthritis with inflammation, destruction of articular cartilage and bone |
| Crohn disease | Unknown antigen; role for commensal bacteria | Chronic intestinal inflammation, obstruction |
| Peripheral neuropathy; Guillain-Barré syndrome? | Protein antigens of peripheral nerve myelin | Neuritis, paralysis |
| Contact sensitivity (dermatitis) | Various environmental antigens (e.g., poison ivy) | Skin inflammation with blisters |
CNS, central nervous system.
Reactions of CD4+ T Cells: Delayed-Type Hypersensitivity and Immune Inflammation
Inflammatory reactions caused by CD4+ T cells were initially characterized on the basis of delayed-type hypersensitivity (DTH) to exogenously administered antigens. The same immunological events are responsible for chronic inflammatory reactions against self-tissues. Because of the central role of the adaptive immune system in such inflammation, it is sometimes referred to as immune inflammation. Both TH1 and TH17 cells contribute to organ-specific diseases in which inflammation is a prominent aspect of the pathology.36 The inflammatory reaction associated with TH1 cells is dominated by activated macrophages, and that triggered by TH17 cells has a greater neutrophil component.
The cellular events in T cell–mediated hypersensitivity consist of a series of reactions in which cytokines play important roles. The reactions can be divided into the following stages.
Proliferation and Differentiation of CD4+ T Cells.
Naive CD4+ T cells recognize peptides displayed by dendritic cells and secrete IL-2, which functions as an autocrine growth factor to stimulate proliferation of the antigen-responsive T cells. The subsequent differentiation of antigen-stimulated T cells to TH1 or TH17 cells is driven by the cytokines produced by APCs at the time of T-cell activation (see Fig. 6-13).36 In some situations the APCs (dendritic cells and macrophages) produce IL-12, which induces differentiation of CD4+ T cells to the TH1 subset. IFN-γ produced by these effector cells promotes further TH1 development, thus amplifying the reaction. If the APCs produce inflammatory cytokines such as IL-1, IL-6, and a close relative of IL-12 called IL-23, these work in collaboration with transforming growth factor-β (TGF-β) (made by many cell types) to stimulate differentiation of T cells to the TH17 subset. Some of the differentiated effector cells enter the circulation and may remain in the memory pool of T cells for long periods, sometimes years.
Responses of Differentiated Effector T Cells.
Upon repeat exposure to an antigen, previously activated T cells recognize the antigen displayed by APCs and respond. TH1 cells secrete cytokines, mainly IFN-γ, which are responsible for many of the manifestations of delayed-type hypersensitivity. IFN-γ–activated macrophages are altered in several ways: their ability to phagocytose and kill microorganisms is markedly augmented; they express more class II MHC molecules on the surface, thus facilitating further antigen presentation; they secrete TNF, IL-1, and chemokines, which promote inflammation (Chapter 2); and they produce more IL-12, thereby amplifying the TH1 response. Thus, activated macrophages serve to eliminate the offending antigen; if the activation is sustained, continued inflammation and tissue injury result.
TH17 cells are activated by some microbial antigens and by self-antigens in autoimmune diseases. Activated TH17 cells secrete IL-17, IL-22, chemokines, and several other cytokines. Collectively, these cytokines recruit neutrophils and monocytes to the reaction, thus promoting inflammation. TH17 cells also produce IL-21, which amplifies the TH17 response.
The classic example of DTH is the tuberculin reaction, which is produced by the intracutaneous injection of purified protein derivative (PPD, also called tuberculin), a protein-containing antigen of the tubercle bacillus. In a previously sensitized individual, reddening and induration of the site appear in 8 to 12 hours, reach a peak in 24 to 72 hours, and thereafter slowly subside. Morphologically, delayed-type hypersensitivity is characterized by the accumulation of mononuclear cells, mainly CD4+ T cells and macrophages, around venules, producing perivascular “cuffing” (Fig. 6-20). In fully developed lesions, the venules show marked endothelial hypertrophy, reflecting cytokine-mediated endothelial activation.
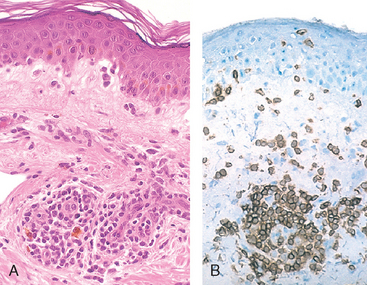
FIGURE 6-20 Delayed hypersensitivity reaction in the skin. A, Perivascular infiltration by T cells and mononuclear phagocytes. B, Immunoperoxidase staining reveals a predominantly perivascular cellular infiltrate that marks positively with antibodies specific to CD4.
(Courtesy of Dr. Louis Picker, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
With certain persistent or nondegradable antigens, such as tubercle bacilli colonizing the lungs or other tissues, the perivascular infiltrate is dominated by macrophages over a period of 2 or 3 weeks. The activated macrophages often undergo a morphologic transformation into epithelium-like cells and are then referred to as epithelioid cells. A microscopic aggregation of epithelioid cells, usually surrounded by a collar of lymphocytes, is referred to as a granuloma (Fig. 6-21). This pattern of inflammation, called granulomatous inflammation (Chapter 2), is typically associated with strong T-cell activation with cytokine production (Fig. 6-22). It can also be caused by foreign bodies that activate macrophages without eliciting an adaptive immune response.
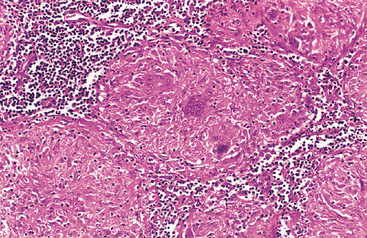
FIGURE 6-21 Granulomatous inflammation. A section of a lymph node shows several granulomas, each made up of an aggregate of epithelioid cells and surrounded by lymphocytes. The granuloma in the center shows several multinucleate giant cells.
(Courtesy of Dr. Trace Worrell, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
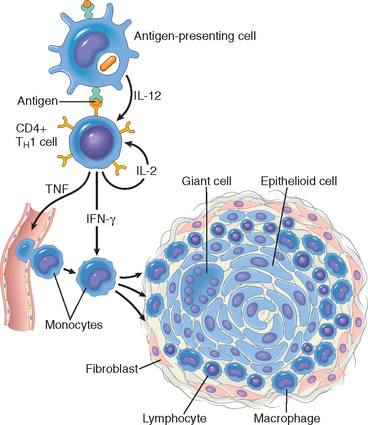
FIGURE 6-22 Mechanisms of granuloma formation. Schematic illustration of the events that give rise to the formation of granulomas in cell-mediated (type IV) hypersensitivity reactions. Note the role played by cytokines. See text for abbreviations.
Contact dermatitis is a common example of tissue injury resulting from DTH reactions. It may be evoked by contact with urushiol, the antigenic component of poison ivy or poison oak, and presents as a vesicular dermatitis (Fig. 6-23).
Reactions of CD8+ T Cells: Cell-Mediated Cytotoxicity
In this type of T cell–mediated reaction, CD8+ CTLs kill antigen-bearing target cells. Tissue destruction by CTLs may be an important component of many T cell–mediated diseases, such as type 1 diabetes. CTLs directed against cell surface histocompatibility antigens play an important role in graft rejection, to be discussed later. They also play a role in reactions against viruses. In a virus-infected cell, viral peptides are displayed by class I MHC molecules and the complex is recognized by the TCR of CD8+ T lymphocytes. The killing of infected cells leads to the elimination of the infection, and is responsible for cell damage that accompanies the infection (e.g., in viral hepatitis). Tumor-associated antigens are also presented on the cell surface, and CTLs are involved in tumor rejection (Chapter 7).
The principal mechanism of T cell–mediated killing of targets involves perforins and granzymes, preformed mediators contained in the lysosome-like granules of CTLs.37 CTLs that recognize the target cells secrete a complex consisting of perforin, granzymes, and a protein called serglycin, which enters target cells by endocytosis. In the target cell cytoplasm, perforin facilitates the release of the granzymes from the complex. Granzymes are proteases that cleave and activate caspases, which induce apoptosis of the target cells (Chapter 1). Activated CTLs also express Fas ligand, a molecule with homology to TNF, which can bind to Fas expressed on target cells and trigger apoptosis.
CD8+ T cells also produce cytokines, notably IFN-γ, and are involved in inflammatory reactions resembling DTH, especially following virus infections and exposure to some contact sensitizing agents.
AUTOIMMUNE DISEASES
Immune reactions against self-antigens—autoimmunity—are an important cause of certain diseases in humans, estimated to affect at least 1% to 2% of the US population. A growing number of diseases have been attributed to autoimmunity (Table 6-7). Autoantibodies can be found in the serum of apparently normal individuals, particularly in older age groups. Furthermore, innocuous autoantibodies are also formed after damage to tissue and may serve a physiologic role in the removal of tissue breakdown products. How, then, does one define pathologic autoimmunity? Ideally, at least three requirements should be met before a disorder is categorized as truly due to autoimmunity: (1) the presence of an immune reaction specific for some self-antigen or self-tissue; (2) evidence that such a reaction is not secondary to tissue damage but is of primary pathogenic significance; and (3) the absence of another well-defined cause of the disease. Similarity with experimental models of proven autoimmunity is also often used to support this mechanism in human diseases. Because of the uncertainty about the target antigens and the contribution of “true” autoimmunity, these disorders are often grouped under immune-mediated inflammatory diseases. This term also emphasizes the important contribution of chronic inflammation to the pathogenesis of these diseases.
TABLE 6-7 Immune-Mediated Inflammatory Diseases
* Antibodies may also play a role in these diseases.
The clinical manifestations of autoimmune disorders are extremely varied. On one end are conditions in which the immune responses are directed against a single organ or tissue, resulting in organ-specific disease, and on the other end are diseases in which the autoimmune reactions are against widespread antigens, resulting in systemic or generalized disease. Examples of organ-specific autoimmunity are type 1 diabetes mellitus, in which the autoreactive T cells and antibodies are specific for β cells of the pancreatic islets, and multiple sclerosis, in which autoreactive T cells react against central nervous system myelin. The best example of systemic autoimmune disease is SLE, in which a diversity of antibodies directed against DNA, platelets, red cells, and protein-phospholipid complexes result in widespread lesions throughout the body. In the middle of the spectrum falls Goodpasture’s syndrome, in which antibodies to basement membranes of lung and kidney induce lesions in these organs.
It is obvious that autoimmunity results from the loss of self-tolerance, and the question arises as to how this happens. Before we look for answers to this question, we review the mechanisms of immunological tolerance to self-antigens.
Immunological Tolerance
Immunological tolerance is the phenomenon of unresponsiveness to an antigen as a result of exposure of lymphocytes to that antigen. Self-tolerance refers to lack of responsiveness to an individual’s own antigens, and it underlies our ability to live in harmony with our cells and tissues. Lymphocytes with receptors capable of recognizing self-antigens are being generated constantly, and these cells have to be eliminated or inactivated as soon as they recognize the antigens, to prevent them from causing harm. The mechanisms of self-tolerance can be broadly classified into two groups: central tolerance and peripheral tolerance (Fig. 6-24).38-40 Each of these is considered briefly.
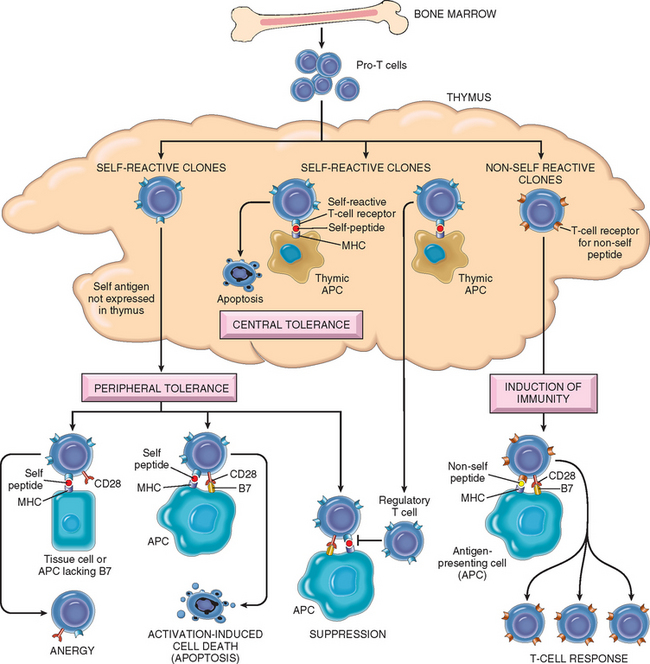
FIGURE 6-24 Mechanisms of immunological tolerance. Schematic illustration of the mechanisms of central and peripheral tolerance to self-antigens, shown for CD4+ T cells. APC, antigen-presenting cell. See text for other abbreviations.
Central Tolerance.
In this process, immature self-reactive T- and B-lymphocyte clones that recognize self-antigens during their maturation in the central (or generative) lymphoid organs (the thymus for T cells and the bone marrow for B cells) are killed or rendered harmless.41 The mechanisms of central tolerance in T and B cells show some similarities and differences.
Central tolerance, however, is far from perfect. Not all self-antigens may be present in the thymus, and hence T cells bearing receptors for such autoantigens escape into the periphery. There is similar “slippage” in the B-cell system. Self-reactive lymphocytes that escape negative selection can inflict tissue injury unless they are deleted or muzzled in the peripheral tissues.
Peripheral Tolerance.
Several mechanisms silence potentially autoreactive T and B cells in peripheral tissues; these are best defined for T cells.40 These mechanisms include the following:
Anergy also affects mature B cells in peripheral tissues. It is believed that if B cells encounter self-antigen in peripheral tissues, especially in the absence of specific helper T cells, the B cells become unable to respond to subsequent antigenic stimulation and may be excluded from lymphoid follicles, resulting in their death.
Some antigens are hidden (sequestered) from the immune system, because the tissues in which these antigens are located do not communicate with the blood and lymph. Thus, self-antigens in these tissues do not induce tolerance but fail to elicit immune responses and are essentially ignored by the immune system. This is believed to be the case for the testis, eye, and brain, all of which are called immune-privileged sites because it is difficult to induce immune responses to antigens introduced into these sites. If the antigens of these tissues are released, for example, as a consequence of trauma or infection, the result may be an immune response that leads to prolonged tissue inflammation and injury. This is the postulated mechanism for post-traumatic orchitis and uveitis.
Mechanisms of Autoimmunity: General Principles
Autoimmunity arises from a combination of the inheritance of susceptibility genes, which may contribute to the breakdown of self-tolerance, and environmental triggers, such as infections and tissue damage, which promote the activation of self-reactive lymphocytes (Fig. 6-25).51,52 In general, these genetic and environmental influences conspire to create an imbalance between control mechanisms that normally function to prevent self-reactivity and pathways that lead to the generation and activation of pathogenic effector lymphocytes. In the following section we discuss how genetic and other factors contribute to the development of autoimmunity.
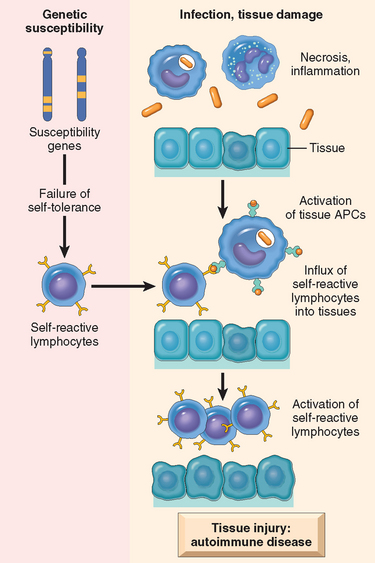
FIGURE 6-25 Pathogenesis of autoimmunity. Autoimmunity results from multiple factors, including susceptibility genes that may interfere with self-tolerance and environmental triggers (tissue injury, inflammation) that promote lymphocyte entry into tissues, activation of self-reactive lymphocytes, and tissue damage.
Role of Susceptibility Genes.
It has been known for decades that autoimmunity has a genetic component. The incidence of the disease is greater in twins of affected individuals than in the general population, and greater in monozygotic than in dizygotic twins. Most autoimmune diseases are complex multigenic disorders.53-55 Among the genes known to be associated with autoimmunity, the greatest contribution is that of HLA genes. The concept of HLA association with diseases was mentioned earlier (see Table 6-1). Although this association has been well established for many years, the underlying mechanisms remain obscure. It is postulated that the presence of particular MHC alleles affects the negative selection of T cells in the thymus or the development of regulatory T cells, but there is little proof for either possibility. It should be pointed out that many normal individuals inherit the MHC alleles that are disease-associated in patient populations, and normal MHC molecules are capable of presenting self-antigens. Therefore, the presence of particular MHC alleles is not, by itself, the cause of autoimmunity.
Genome-wide association studies (Chapter 5) have shown that multiple non-MHC genes are associated with various autoimmune diseases. Some of these genes are disease-specific, but many of the associations are seen in multiple disorders, suggesting that the products of these genes affect general mechanisms of immune regulation and self-tolerance. Three recently described genetic associations are especially interesting. Polymorphisms in a gene called PTPN-22, which encodes a protein tyrosine phosphatase, are associated with rheumatoid arthritis, type 1 diabetes, and several other autoimmune diseases.56 Because these disorders have a fairly high prevalence (especially rheumatoid arthritis), PTPN-22 is said to be the gene that is most frequently implicated in autoimmunity. It is postulated that the disease-associated variants encode a phosphatase that is functionally defective and is thus unable to fully control the activity of tyrosine kinases, which are involved in many lymphocyte responses. The net result is excessive lymphocyte activation. Polymorphisms in the gene for NOD-2 are associated with Crohn disease, a form of inflammatory bowel disease, especially in certain ethnic populations.57 NOD-2 is a cytoplasmic sensor of microbes, expressed in epithelial and many other cells. According to one hypothesis, the disease-associated variant is ineffective at sensing intestinal microbes, resulting in entry of and chronic inflammatory responses against normally well-tolerated commensal bacteria. The genes encoding the IL-2 receptor (CD25) and IL-7 receptor α chains are associated with multiple sclerosis and other autoimmune diseases. These cytokines may control the maintenance of regulatory T cells. Although these genetic associations are beginning to reveal interesting clues about pathogenesis, the links between the genes, functions of their encoded proteins, and the diseases remain to be established.
We have already mentioned that in mice and humans, knockouts and natural mutations affecting several individual genes result in autoimmunity. These genes include AIRE, CTLA4, PD1, Fas, FasL, and IL2 and its receptor CD25. In addition, B cells express an Fc receptor that recognizes IgG antibodies bound to antigens and switches off further antibody production (a normal negative-feedback mechanism). Knockout of this receptor results in autoimmunity, pre-sumably because the B cells can no longer be controlled. These examples are very informative about pathways of self-tolerance and immune regulation, but the diseases caused by these single gene mutations are rare and not representative of the common autoimmune disorders.
Role of Infections.
Many autoimmune diseases are associated with infections, and clinical flare-ups are often preceded by infectious prodromes. Two mechanisms have been postulated to explain the link between infections and autoimmunity (Fig. 6-26). First, infections may up-regulate the expression of costimulators on APCs. If these cells are presenting self-antigens, the result may be a breakdown of anergy and activation of T cells specific for the self-antigens. Second, some microbes may express antigens that have the same amino acid sequences as self-antigens. Immune responses against the microbial antigens may result in the activation of self-reactive lymphocytes. This phenomenon is called molecular mimicry. A clear example of such mimicry is rheumatic heart disease, in which antibodies against streptococcal proteins cross-react with myocardial proteins and cause myocarditis (Chapter 12). But more subtle molecular mimicry may be involved in classical autoimmune diseases as well.
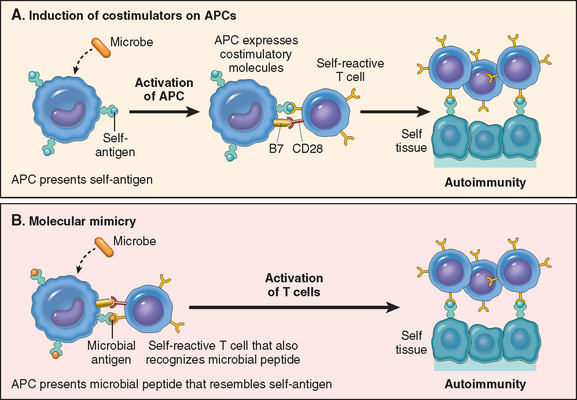
FIGURE 6-26 Postulated role of infections in autoimmunity. Infections may promote activation of self-reactive lymphocytes by inducing the expression of costimulators (A), or microbial antigens may mimic self-antigens and activate self-reactive lymphocytes as a cross-reaction (B).
Microbes may induce other abnormalities that promote autoimmune reactions. Some viruses, such as Epstein-Barr virus (EBV) and HIV, cause polyclonal B-cell activation, which may result in production of autoantibodies. The tissue injury that is common in infections may release self-antigens and structurally alter self-antigens so that they are able to activate T cells that are not tolerant to these new, modified antigens. Infections may induce the production of cytokines that recruit lymphocytes, including potentially self-reactive lymphocytes, to sites of self-antigens.
Although the role of infections in triggering autoimmunity has received a great deal of attention, recent epidemiologic studies suggest that the incidence of autoimmune diseases is increasing in developed countries as infections are better controlled. In some animal models (e.g., of type 1 diabetes) infections greatly reduce the incidence of disease. Thus, paradoxically, infections may protect against some autoimmune diseases.58 The underlying mechanisms are unclear; an intriguing possibility is that infections promote low-level IL-2 production, and this is essential for maintaining regulatory T cells.
General Features of Autoimmune Diseases
Diseases caused by autoimmunity have some important general features.
With this background we can proceed to a discussion of specific autoimmune diseases. Table 6-7 lists both systemic and organ-specific autoimmune disorders. The systemic diseases tend to involve blood vessels and connective tissues, and therefore, they are often classified as collagen vascular diseases. Our focus here is on prototypic systemic autoimmune diseases; organ-specific disorders are covered elsewhere in the book.
SYSTEMIC LUPUS ERYTHEMATOSUS (SLE)
SLE is the prototype of a multisystem disease of autoimmune origin, characterized by a vast array of autoantibodies, particularly antinuclear antibodies (ANAs). Acute or insidious in its onset, it is a chronic, remitting and relapsing, often febrile illness characterized principally by injury to the skin, joints, kidney, and serosal membranes. Virtually every other organ in the body, however, may also be affected. The clinical presentation of SLE is so variable that the American College of Rheumatology has established a complex set of criteria for this disorder (Table 6-8). SLE is a fairly common disease, with a prevalence that may be as high as 1 in 2500 in certain populations.60 Similar to many autoimmune diseases, SLE predominantly affects women, with a frequency of 1 in 700 among women of childbearing age and a female-to-male ratio of 9 : 1. By comparison, the female-to-male ratio is only 2 : 1 for disease developing during childhood or after the age of 65. The prevalence of the disease is 2–3 fold higher in blacks and Hispanics than in whites. Although SLE usually arises in the 20s and 30s, it may manifest at any age, even in early childhood.
TABLE 6-8 1997 Revised Criteria for Classification of Systemic Lupus Erythematosus*
| Criterion | Definition |
|---|---|
| Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds | |
| Erythematous raised patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in older lesions | |
| Rash as a result of unusual reaction to sunlight, by patient history or physician observation | |
| Oral or nasopharyngeal ulceration, usually painless, observed by a physician | |
| Nonerosive arthritis involving two or more peripheral joints, characterized by tenderness, swelling, or effusion | |
| Pleuritis—convincing history of pleuritic pain or rub heard by a physician or evidence of pleural effusion, or | |
| Pericarditis—documented by electrocardiogram or rub or evidence of pericardial effusion | |
| Persistent proteinuria >0.5 gm/dL or >3 if quantitation not performed or | |
| Cellular casts—may be red blood cell, hemoglobin, granular, tubular, or mixed | |
| Seizures—in the absence of offending drugs or known metabolic derangements (e.g., uremia, ketoacidosis, or electrolyte imbalance), or | |
| Psychosis—in the absence of offending drugs or known metabolic derangements (e.g., uremia, ketoacidosis, or electrolyte imbalance) | |
| Hemolytic anemia—with reticulocytosis, or | |
| Leukopenia—<4.0 × 109 cells/L (4000 cells/mm3) total on two or more occasions, or | |
| Lymphopenia—<1.5 × 109 cells/L (1500 cells/mm3) on two or more occasions, or | |
| Thrombocytopenia—<100 × 109 cells/L (100 × 103 cells/mm3) in the absence of offending drugs | |
| Anti-DNA antibody to native DNA in abnormal titer, or | |
| Anti-Sm—presence of antibody to Sm nuclear antigen, or | |
| Positive finding of antiphospholipid antibodies based on (1) an abnormal serum level of IgG or IgM anticardiolipin antibodies, (2) a positive test for lupus anticoagulant using a standard test, or (3) a false-positive serologic test for syphilis known to be positive for at least 6 months and confirmed by negative Treponema pallidum immobilization or fluorescent treponemal antibody absorption test | |
| An abnormal titer of antinuclear antibody by immunofluorescence or an equivalent assay at any point in time and in the absence of drugs known to be associated with drug-induced lupus syndrome |
* This classification, based on 11 criteria, was proposed for the purpose of identifying patients in clinical studies. A person is said to have systemic lupus erythematosus if any 4 or more of the 11 criteria are present, serially or simultaneously, during any period of observation.
From Tan EM et al: The revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25:1271, 1982; and Hochberg MC: Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40:1725, 1997.
Spectrum of Autoantibodies in SLE
The hallmark of the disease is the production of autoantibodies. Some antibodies recognize diverse nuclear and cytoplasmic components of the cell that are neither organ- nor species-specific, and others are directed against cell surface antigens of blood cells. Apart from their value in the diagnosis and management of patients with SLE, these antibodies are of major pathogenetic significance, as, for example, in the immune complex–mediated glomerulonephritis so typical of this disease.61,62
Antinuclear antibodies (ANAs) are directed against nuclear antigens and can be grouped into four categories63: (1) antibodies to DNA, (2) antibodies to histones, (3) antibodies to nonhistone proteins bound to RNA, and (4) antibodies to nucleolar antigens. Table 6-9 lists several ANAs and their association with SLE as well as with other autoimmune diseases to be discussed later. The most widely used method for detecting ANAs is indirect immunofluorescence, which can identify antibodies that bind to a variety of nuclear antigens, including DNA, RNA, and proteins (collectively called generic ANAs). The pattern of nuclear fluorescence suggests the type of antibody present in the patient’s serum. Four basic patterns are recognized:
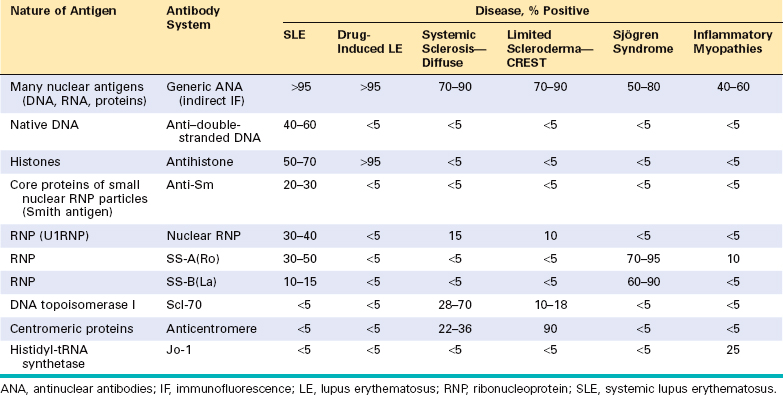
The fluorescence patterns are not absolutely specific for the type of antibody, and because many autoantibodies may be present, combinations of patterns are frequent. The immunofluorescence test for ANAs is sensitive because it is positive in virtually every patient with SLE, but it is not specific because patients with other autoimmune diseases also frequently score positive (see Table 6-9). Furthermore, approximately 5% to 15% of normal individuals have low titers of these antibodies, and the incidence increases with age. Antibodies to double -stranded DNA and the so-called Smith (Sm) antigen are virtually diagnostic of SLE.64
In addition to ANAs, lupus patients have a host of other autoantibodies. Some are directed against blood cells, such as red cells, platelets, and lymphocytes; others react with proteins in complex with phospholipids. In recent years there has been much interest in these so-called antiphospholipid antibodies. They are present in 40% to 50% of lupus patients. They are actually directed against epitopes of plasma proteins that are revealed when the proteins are in complex with phospholipids. Included among these proteins are prothrombin, annexin V, β2-glycoprotein I, protein S, and protein C.65 Antibodies against the phospholipid–β2-glycoprotein complex also bind to cardiolipin antigen, used in syphilis serology, and therefore lupus patients may have a false-positive test result for syphilis. Some of these antibodies interfere with in vitro clotting tests, such as partial thromboplastin time. Therefore, these antibodies are sometimes referred to as lupus anticoagulant. Despite having a circulating anticoagulant that delays clotting in vitro, these patients have complications associated with a hypercoagulable state.66 They have venous and arterial thromboses, which may be associated with recurrent spontaneous miscarriages and focal cerebral or ocular ischemia. This constellation of clinical features, in association with lupus, is referred to as the secondary antiphospholipid antibody syndrome. The pathogenesis of thrombosis in these patients is unknown; possible mechanisms are discussed in Chapter 4. Some patients develop these autoantibodies and the clinical syndrome without associated SLE. They are said to have the primary antiphospholipid syndrome (Chapter 4).
Etiology and Pathogenesis of SLE
The cause of SLE remains unknown, but the existence in these patients of a seemingly limitless number of antibodies against self-constituents indicates that the fundamental defect in SLE is a failure of the mechanisms that maintain self-tolerance. As is true of most autoimmune diseases, both genetic and environmental factors play a role in the pathogenesis of SLE.67
Genetic Factors
SLE is a genetically complex disease with contributions from MHC and multiple non-MHC genes. Many lines of evidence support a genetic predisposition.68,69
Immunological Factors
Recent studies in animal models and patients are revealing several immunological aberrations that collectively may result in the persistence and uncontrolled activation of self-reactive lymphocytes.
Environmental Factors.
There are many indications that environmental or nongenetic factors must also be involved in the pathogenesis of SLE. Exposure to ultraviolet (UV) light exacerbates the disease in many individuals. UV irradiation may induce apoptosis in cells and may alter the DNA in such a way that it becomes immunogenic, perhaps because of enhanced recognition by TLRs.77 In addition, UV light may modulate the immune response, for example, by stimulating keratinocytes to produce IL-1, a cytokine known to promote inflammation. Sex hormones seem to exert an important influence on the occurrence and manifestations of SLE. During the reproductive years the frequency of SLE is 10 times greater in women than in men in the age group of 17 through 55 years, and exacerbation has been noted during normal menses and pregnancy. Drugs such as hydralazine, procainamide, and d-penicillamine can induce an SLE-like response in humans.78
A Model for the Pathogenesis of SLE.
It is clear from this discussion that the immunological abnormalities in SLE—both documented and postulated—are as varied and complex as is the clinical presentation (discussed later). Nevertheless, an attempt can be made to synthesize the new results into a hypothetical model of the pathogenesis of SLE (Fig. 6-27). UV irradiation and other environmental insults lead to the apoptosis of cells. Inadequate clearance of the nuclei of these cells results in a large burden of nuclear antigens.79 An underlying abnormality in B and T lymphocytes is responsible for defective tolerance, because of which self-reactive lymphocytes survive and remain functional. These lymphocytes are stimulated by self nuclear antigens, and antibodies are produced against the antigens. Complexes of the antigens and antibodies bind to Fc receptors on B cells and dendritic cells, and may be internalized. The nucleic acid components engage TLRs and stimulate B cells to produce autoantibodies and activate dendritic cells to produce interferons and other cytokines, which further enhance the immune response and cause more apoptosis. The net result is a cycle of antigen release and immune activation resulting in the production of high-affinity autoantibodies.
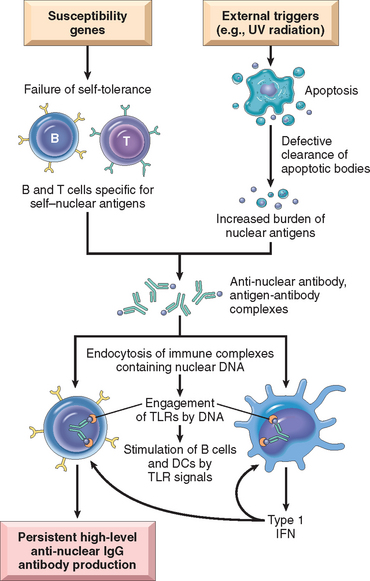
FIGURE 6-27 Model for the pathogenesis of systemic lupus erythematosus. In this hypothetical model, susceptibility genes interfere with the maintenance of self-tolerance and external triggers lead to persistence of nuclear antigens. The result is an antibody response against self–nuclear antigens, which is amplified by the action of nucleic acids on dendritic cells (DCs) and B cells, and the production of type 1 interferons. TLRs, Toll-like receptors.
Mechanisms of Tissue Injury.
Regardless of the exact mechanisms by which autoantibodies are formed, they are clearly the mediators of tissue injury. Most of the visceral lesions are caused by immune complexes (type III hypersensitivity). DNA–anti-DNA complexes can be detected in the glomeruli and small blood vessels. Low levels of serum complement (secondary to consumption of complement proteins) and granular deposits of complement and immunoglobulins in the glomeruli further support the immune complex nature of the disease. Autoantibodies specific for red cells, white cells, and platelets opsonize these cells and promote their phagocytosis and lysis. There is no evidence that ANAs, which are involved in immune complex formation, can penetrate intact cells. If cell nuclei are exposed, however, the ANAs can bind to them. In tissues, nuclei of damaged cells react with ANAs, lose their chromatin pattern, and become homogeneous, to produce so-called LE bodies or hematoxylin bodies. Related to this phenomenon is the LE cell, which is readily seen when blood is agitated in vitro. The LE cell is any phagocytic leukocyte (blood neutrophil or macrophage) that has engulfed the denatured nucleus of an injured cell. The demonstration of LE cells in vitro was used in the past as a test for SLE. With new techniques for detection of ANAs, however, this test is now largely of historical interest. Sometimes, LE cells are found in pericardial or pleural effusions in patients.
To summarize, SLE is a complex disorder of multifactorial origin resulting from interactions among genetic, immunological, and environmental factors that act in concert to cause activation of helper T cells and B cells and result in the production of several species of pathogenic autoantibodies.
Morphology. The morphologic changes in SLE are extremely variable, as are the clinical manifestations and course of disease. The constellation of clinical, serologic, and morphologic changes is essential for diagnosis (see Table 6-8). The frequency of individual organ involvement is shown in Table 6-10. The most characteristic lesions result from immune complexes depositing in blood vessels, kidneys, connective tissue, and skin.
TABLE 6-10 Clinical and Pathologic Manifestations of Systemic Lupus Erythematosus
| Clinical Manifestation | Prevalence in Patients (%)* |
|---|---|
| Hematologic | 100 |
| Arthritis | 80–90 |
| Skin | 85 |
| Fever | 55–85 |
| Fatigue | 80–100 |
| Weight loss | 60 |
| Renal | 50–70 |
| Neuropsychiatric | 25–35 |
| Pleuritis | 45 |
| Myalgia | 35 |
| Pericarditis | 25 |
| Gastrointestinal | 20 |
| Raynaud phenomenon | 15–40 |
| Ocular | 15 |
| Peripheral neuropathy | 15 |
* The percentages are approximate and may vary with age, ethnicity, and other factors.
An acute necrotizing vasculitis involving capillaries, small arteries and arterioles may be present in any tissue.80 The arteritis is characterized by fibrinoid deposits in the vessel walls. In chronic stages, vessels undergo fibrous thickening with luminal narrowing.
Kidney. Lupus nephritis affects up to 50% of SLE patients. The principal mechanism of injury is immune complex deposition in the glomeruli, tubular or peritubular capillary basement membranes, or larger blood vessels. Other injuries may include thrombi in glomerular capillaries, arterioles, or arteries, often associated with antiphospholipid antibodies.
All of the glomerular lesions described below are the result of deposition of immune complexes that are regularly present in the mesangium or along the entire basement membrane and sometimes throughout the glomerulus. The immune complexes consist of DNA and anti-DNA antibodies, but other antigens such as histones have also been implicated. Both in situ formation and deposition of preformed circulating immune complexes may contribute to the injury, but the reason for the wide spectrum of histopathologic lesions (and clinical manifestations) in lupus nephritis patients remains uncertain.
A morphologic classification of lupus nephritis has proven to be clinically useful.81 Five patterns are recognized: minimal mesangial (class I); mesangial proliferative (class II); focal proliferative (class III); diffuse proliferative (class IV); and membranous (class V). None of these patterns is specific for lupus.
Mesangial lupus glomerulonephritis is seen in 10% to 25% of patients and is characterized by mesangial cell proliferation and immune complex deposition without involvement of glomerular capillaries. There is no or slight (class I) to moderate (class II) increase in both mesangial matrix and number of mesangial cells. Granular mesangial deposits of immunoglobulin and complement are always present. Classes III to V nephritis, described below, are usually superimposed on some degree of mesangial changes.
Focal proliferative glomerulonephritis (class III) is seen in 20% to 35% of patients, and is defined by fewer than 50% involvement of all glomeruli. The lesions may be segmental (affecting only a portion of the glomerulus) or global (involving the entire glomerulus). Affected glomeruli may exhibit crescent formation, fibrinoid necrosis, proliferation of endothelial and mesangial cells, infiltrating leukocytes, and eosinophilic deposits or intracapillary thrombi (Fig. 6-28), which often correlate with hematuria and proteinuria. Some patients may progress to diffuse proliferative glomerulonephritis. The active (or proliferative) inflammatory lesions can heal completely or lead to chronic global or segmental glomerular scarring.
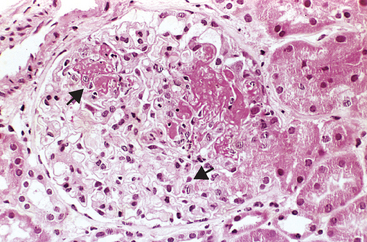
FIGURE 6-28 Lupus nephritis, focal proliferative type. There are two focal necrotizing lesions in the glomerulus (arrows).
(Courtesy of Dr. Helmut Rennke, Department of Pathology, Brigham and Women’s Hospital, Boston, MA.)
Diffuse proliferative glomerulonephritis (class IV) is the most severe form of lupus nephritis, occurring in 35% to 60% of patients. Pathologic glomerular changes may be identical to focal (class III) lupus nephritis, including proliferation of endothelial, mesangial and, sometimes, epithelial cells (Fig. 6-29), with the latter producing cellular crescents that fill Bowman’s space (Chapter 20). The entire glomerulus is frequently affected but segmental lesions also may occur. Both acutely injured and chronically scarred glomeruli in focal or diffuse lupus nephritis are qualitatively indistinguishable from one another; the distinction is based solely on the percentage of glomerular involvement (<50% for class III vs >50% for class IV). Patients with diffuse glomerulonephritis are usually symptomatic, showing hematuria as well as proteinuria. Hypertension and mild to severe renal insufficiency are also common.
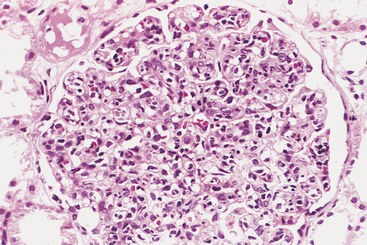
FIGURE 6-29 Lupus nephritis, diffuse proliferative type. Note the marked increase in cellularity throughout the glomerulus.
(Courtesy of Dr. Helmut Rennke, Department of Pathology, Brigham and Women’s Hospital, Boston, MA.)
Membranous glomerulonephritis (class V) is characterized by diffuse thickening of the capillary walls, which is similar to idiopathic membranous glomerulonephritis, described in Chapter 20. This lesion is seen in 10% to 15% of lupus nephritis patients, is usually accompanied by severe proteinuria or nephrotic syndrome, and may occur concurrently with focal or diffuse lupus nephritis.
Granular deposits of antibody and complement can be detected by immunofluorescence (Fig. 6-30). Electron microscopy demonstrates electron-dense deposits that represent immune complexes in mesangial, intramembranous, subepithelial, or subendothelial locations. All classes show variable amounts of mesangial deposits. In membranous lupus nephritis, the deposits are predominantly subepithelial (between the basement membrane and visceral epithelial cells). Subendothelial deposits (between the endothelium and the basement membrane) are seen in the proliferative types (classes III and IV) but may be encountered rarely in class I, II, and V lupus nephritis (Fig. 6-31). When prominent, subendothelial deposits create a homogeneous thickening of the capillary wall, which are seen by light microscopy as a “wire-loop” lesion (Fig. 6-32). Such wire loops are often found in both focal and diffuse proliferative (class III or IV) lupus nephritis, which reflects active disease.
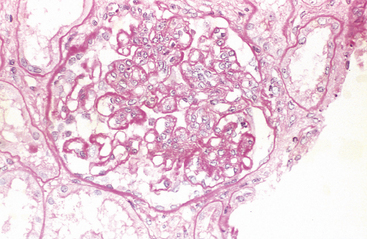
FIGURE 6-32 Lupus nephritis. A glomerulus with several “wire loop” lesions representing extensive subendothelial deposits of immune complexes is seen. (Periodic acid-Schiff [PAS] stain.)
(Courtesy of Dr. Helmut Rennke, Department of Pathology, Brigham and Women’s Hospital, Boston, MA.)
Changes in the interstitium and tubules are frequently present in lupus nephritis patients. Rarely, tubulointerstitial lesions may be the dominant abnormality. Discrete immune complexes similar to those in glomeruli are present in the tubular or peritubular capillary basement membranes in many lupus nephritis patients.
Skin. Characteristic erythema affects the facial butterfly (malar) area (bridge of the nose and cheeks) in approximately 50% of patients, but a similar rash may also be seen on the extremities and trunk. Urticaria, bullae, maculopapular lesions, and ulcerations also occur. Exposure to sunlight incites or accentuates the erythema. Histologically the involved areas show vacuolar degeneration of the basal layer of the epidermis (Fig. 6-33A). In the dermis, there is variable edema and perivascular inflammation. Vasculitis with fibrinoid necrosis may be prominent. Immunofluorescence microscopy shows deposition of immunoglobulin and complement along the dermoepidermal junction (Fig. 6-33B), which may also be present in uninvolved skin. This finding is not diagnostic of SLE and is sometimes seen in scleroderma or dermatomyositis.
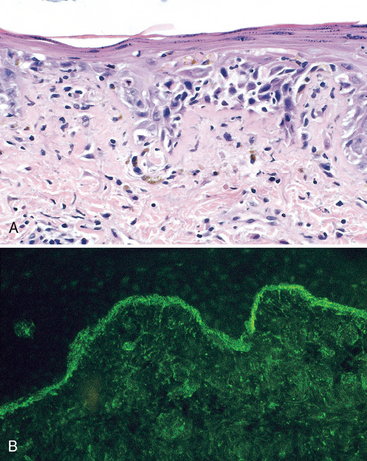
FIGURE 6-33 Systemic lupus erythematosus involving the skin. A, An H&E-stained section shows liquefactive degeneration of the basal layer of the epidermis and edema at the dermoepidermal junction.
(Courtesy of Dr. Jag Bhawan, Boston University School of Medicine, Boston, MA.) B, An immunofluorescence micrograph stained for IgG reveals deposits of Ig along the dermoepidermal junction. (Courtesy of Dr. Richard Sontheimer, Department of Dermatology, University of Texas Southwestern Medical School, Dallas, TX.)
Joints. Joint involvement is typically a nonerosive synovitis with little deformity, which contrasts with rheumatoid arthritis.
Central Nervous System. The pathologic basis of central nervous system symptoms is not entirely clear, but antibodies against a synaptic membrane protein have been implicated.82,83 Neuropsychiatric symptoms of SLE have often been ascribed to acute vasculitis, but in histologic studies of the nervous system in such patients significant vasculitis is rarely present. Instead, noninflammatory occlusion of small vessels by intimal proliferation is sometimes noted, which may be due to endothelial damage by antiphospholipid antibodies.
Pericarditis and Other Serosal Cavity Involvement. Inflammation of the serosal lining membranes may be acute, subacute, or chronic. During the acute phases, the mesothelial surfaces are sometimes covered with fibrinous exudate. Later they become thickened, opaque, and coated with a shaggy fibrous tissue that may lead to partial or total obliteration of the serosal cavity.
Cardiovascular system involvement may manifest as damage to any layer of the heart.84 Symptomatic or asymptomatic pericardial involvement is present in up to 50% of patients. Myocarditis, or mononuclear cell infiltration, is less common and may cause resting tachycardia and electrocardiographic abnormalities. Valvular abnormalities primarily of the mitral and aortic valves manifest as diffuse leaflet thickening that may be associated with dysfunction (stenosis and/or regurgitation). Valvular (or so-called Libman-Sacks) endocarditis was more common prior to the widespread use of steroids. This nonbacterial verrucous endocarditis takes the form of single or multiple 1- to 3-mm warty deposits on any heart valve, distinctively on either surface of the leaflets (Fig. 6-34). By comparison, the vegetations in infective endocarditis are considerably larger, and those in rheumatic heart disease (Chapter 12) are smaller and confined to the lines of closure of the valve leaflets.
FIGURE 6-34 Libman-Sacks endocarditis of the mitral valve in lupus erythematosus. The vegetations attached to the margin of the thickened valve leaflet are indicated by arrows.
(Courtesy of Dr. Fred Schoen, Department of Pathology, Brigham and Women’s Hospital, Boston, MA.)
An increasing number of patients have clinical evidence of coronary artery disease (angina, myocardial infarction) owing to coronary atherosclerosis. This complication is noted particularly in young patients with long-standing disease and especially in those who have been treated with corticosteroids. The pathogenesis of accelerated coronary atherosclerosis is unclear but is probably multifactorial. The traditional risk factors, including hypertension, obesity, and hyperlipidemia, are more common in SLE patients than in control populations. In addition, immune complexes and antiphospholipid antibodies may cause endothelial damage and promote atherosclerosis.
Spleen. Splenomegaly, capsular thickening, and follicular hyperplasia are common features. Central penicilliary arteries may show concentric intimal and smooth muscle cell hyperplasia, producing so-called onion-skin lesions.
Lungs. Pleuritis and pleural effusions are the most common pulmonary manifestations, affecting almost 50% of patients. Alveolar injury with edema and hemorrhage is less common. In some cases, there is chronic interstitial fibrosis and secondary pulmonary hypertension. None of these changes is specific for SLE.
Other Organs and Tissues. LE, or hematoxylin, bodies in the bone marrow or other organs are strongly indicative of SLE. Lymph nodes may be enlarged with hyperplastic follicles or even demonstrate necrotizing lymphadenitis.
Clinical Features.
SLE is a multisystem disease that is highly variable in its clinical presentation. Typically, the patient is a young woman with some, but not necessarily all, of the following features: a butterfly rash over the face, fever, pain but no deformity in one or more peripheral joints (feet, ankles, knees, hips, fingers, wrists, elbows, shoulders), pleuritic chest pain, and photosensitivity. In many patients, however, the presentation of SLE is subtle and puzzling, taking forms such as a febrile illness of unknown origin, abnormal urinary findings, or joint disease masquerading as rheumatoid arthritis or rheumatic fever. ANAs are found in virtually 100% of patients, but it must be remembered that ANAs are not specific (see Table 6-9). A variety of clinical findings may point toward renal involvement, including hematuria, red cell casts, proteinuria, and in some cases the classic nephrotic syndrome (Chapter 20). Laboratory evidence of some hematologic derangement is seen in virtually every case, but in some patients anemia or thrombocytopenia may be the presenting manifestation as well as the dominant clinical problem. In still others, mental aberrations, including psychosis or convulsions, or coronary artery disease may be prominent clinical problems. Patients with SLE are also prone to infections, presumably because of their underlying immune dysfunction and treatment with immunosuppressive drugs.
The course of the disease is variable and unpredictable. Rare acute cases result in death within weeks to months. More often, with appropriate therapy, the disease is characterized by flare-ups and remissions spanning a period of years or even decades. During acute flare-ups, increased formation of immune complexes and the accompanying complement activation often result in hypocomplementemia. Disease exacerbations are usually treated by corticosteroids or other immunosuppressive drugs. Even without therapy, in some patients the disease may run a benign course with skin manifestations and mild hematuria for years. The outcome has improved significantly, and an approximately 90% 5-year and 80% 10-year survival can be expected. The most common causes of death are renal failure and intercurrent infections. Coronary artery disease is also becoming an important cause of death. Patients treated with steroids and immunosuppressive drugs incur the usual risks associated with such therapy.
As mentioned earlier, involvement of skin along with multisystem disease is fairly common in SLE. The following sections describe two syndromes in which the cutaneous involvement is the exclusive or most prominent feature.
Chronic Discoid Lupus Erythematosus.
Chronic discoid lupus erythematosus is a disease in which the skin manifestations may mimic SLE, but systemic manifestations are rare.85 It is characterized by the presence of skin plaques showing varying degrees of edema, erythema, scaliness, follicular plugging, and skin atrophy surrounded by an elevated erythematous border. The face and scalp are usually affected, but widely disseminated lesions occasionally occur. The disease is usually confined to the skin, but 5% to 10% of patients with discoid lupus erythematosus develop multisystem manifestations after many years. Conversely, some patients with SLE may have prominent discoid lesions in the skin. Approximately 35% of patients show a positive ANA test, but antibodies to double-stranded DNA are rarely present. Immunofluorescence studies of skin biopsy specimens show deposition of immunoglobulin and C3 at the dermoepidermal junction similar to that in SLE.
Subacute Cutaneous Lupus Erythematosus.
This condition also presents with predominant skin involvement and can be distinguished from chronic discoid lupus erythematosus by several criteria. The skin rash in this disease tends to be widespread, superficial, and nonscarring, although scarring lesions may occur in some patients. Most patients have mild systemic symptoms consistent with SLE. Furthermore, there is a strong association with antibodies to the SS-A antigen and with the HLA-DR3 genotype. Thus, the term subacute cutaneous lupus erythematosus seems to define a group intermediate between SLE and lupus erythematosus localized only to skin.85
Drug-Induced Lupus Erythematosus
A lupus erythematosus–like syndrome may develop in patients receiving a variety of drugs, including hydralazine, procainamide, isoniazid, and d-penicillamine, to name only a few.78 Many of these drugs are associated with the development of ANAs, but most patients do not have symptoms of lupus erythematosus. For example, 80% of patients receiving procainamide test positive for ANAs, but only one third of these manifest clinical symptoms, such as arthralgias, fever, and serositis. Although multiple organs are affected, renal and central nervous system involvement is distinctly uncommon. There are serologic and genetic differences from classical SLE, as well. Antibodies specific for double-stranded DNA are rare, but there is an extremely high frequency of antibodies specific for histone. Persons with the HLA-DR4 allele are at a greater risk of developing lupus erythematosus after administration of hydralazine. The disease remits after withdrawal of the offending drug.
RHEUMATOID ARTHRITIS
Rheumatoid arthritis is a chronic inflammatory disease that affects primarily the joints but may involve extra-articular tissues such as the skin, blood vessels, lungs, and heart. Abundant evidence supports the autoimmune nature of the disease. Because the principal manifestations of the disease are in the joints, it is discussed in Chapter 26.
SJÖGREN SYNDROME
Sjögren syndrome is a chronic disease characterized by dry eyes (keratoconjunctivitis sicca) and dry mouth (xerostomia) resulting from immunologically mediated destruction of the lacrimal and salivary glands. It occurs as an isolated disorder (primary form), also known as the sicca syndrome, or more often in association with another autoimmune disease (secondary form). Among the associated disorders, rheumatoid arthritis is the most common, but some patients have SLE, polymyositis, scleroderma, vasculitis, mixed connective tissue disease, or thyroiditis.
Etiology and Pathogenesis
The characteristic decrease in tears and saliva (sicca syndrome) is the result of lymphocytic infiltration and fibrosis of the lacrimal and salivary glands.86,87 The infiltrate contains predominantly activated CD4+ helper T cells and some B cells, including plasma cells. About 75% of patients have rheumatoid factor (an antibody reactive with self-IgG) whether or not coexisting rheumatoid arthritis is present. ANAs are detected in 50% to 80% of patients. A host of other organ-specific and non–organ-specific antibodies have also been identified. Most important, however, are antibodies directed against two ribonucleoprotein antigens, SS-A (Ro) and SS-B (La) (see Table 6-9), which can be detected in as many as 90% of patients by sensitive techniques. These antibodies are thus considered serologic markers of the disease. Patients with high titers of antibodies to SS-A are more likely to have early disease onset, longer disease duration, and extraglandular manifestations, such as cutaneous vasculitis and nephritis.62 These autoantibodies are also present in a smaller percentage of patients with SLE and hence are not diagnostic of Sjögren syndrome.
As with other autoimmune diseases, Sjögren syndrome shows some association, albeit weak, with certain HLA alleles. Studies of whites and blacks suggest linkage of the primary form with HLA-B8, HLA-DR3, and DRW52 as well as HLA-DQA1 and HLA-DQB1 loci; in patients with anti-SS-A or anti-SS-B antibodies, specific alleles of HLA-DQA1 and HLA-DQB1 are frequent. This suggests that, as in SLE, inheritance of certain class II molecules predisposes to the development of particular autoantibodies.
Although the pathogenesis of Sjögren syndrome remains obscure, aberrant T-cell and B-cell activation are both implicated. The initiating trigger may be a viral infection of the salivary glands, which causes local cell death and release of tissue self-antigens. In genetically susceptible individuals, CD4+ T cells and B cells specific for these self-antigens may have escaped tolerance and are able to react. The result is inflammation, tissue damage, and, eventually, fibrosis. The nature of the autoantigen(s) recognized by these lymphocytes is still mysterious. A cytoskeletal protein called α-fodrin is a candidate autoantigen, but its role in disease development has not been established yet.88 The viruses that may serve as the initiating stimuli are also unknown but may include the perennial culprit in chronic inflammatory diseases, Epstein-Barr virus, and hepatitis C virus.89 In addition, a small proportion of individuals infected with the human retrovirus human T-cell lymphotropic virus type 1 develop a clinical picture and pathologic changes virtually identical to those seen in Sjögren syndrome.
Morphology. As mentioned earlier, lacrimal and salivary glands are the major targets of the disease, although other exocrine glands, including those lining the respiratory and gastrointestinal tracts and the vagina, may also be involved. The earliest histologic finding in both the major and the minor salivary glands is periductal and perivascular lymphocytic infiltration. Eventually the lymphocytic infiltrate becomes extensive (Fig. 6-35), and in the larger salivary glands lymphoid follicles with germinal centers may be seen. The ductal lining epithelial cells may show hyperplasia, thus obstructing the ducts. Later there is atrophy of the acini, fibrosis, and hyalinization; still later in the course atrophy and replacement of parenchyma with fat are seen. In some cases the lymphoid infiltrate may be so intense as to give the appearance of a lymphoma. Indeed, these patients are at high risk for development of B-cell lymphomas, and molecular assessments of clonality may be necessary to distinguish intense reactive chronic inflammation from early involvement by lymphoma.
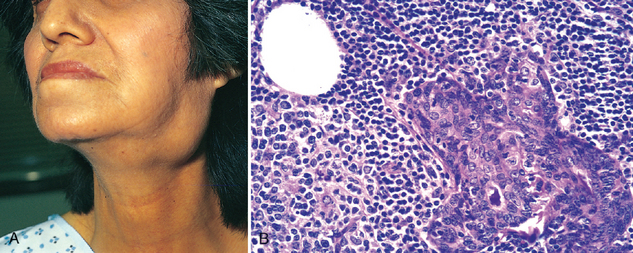
FIGURE 6-35 Sjögren syndrome. A, Enlargement of the salivary gland.
(Courtesy of Dr. Richard Sontheimer, Department of Dermatology, University of Texas Southwestern Medical School, Dallas, TX.) B, Intense lymphocytic and plasma cell infiltration with ductal epithelial hyperplasia in a salivary gland. (Courtesy of Dr. Dennis Burns, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
The lack of tears leads to drying of the corneal epithelium, which becomes inflamed, eroded, and ulcerated; the oral mucosa may atrophy, with inflammatory fissuring and ulceration; and dryness and crusting of the nose may lead to ulcerations and even perforation of the nasal septum.
Clinical Features.
Sjögren syndrome occurs most commonly in women between the ages of 50 and 60. As might be expected, symptoms result from inflammatory destruction of the exocrine glands. The keratoconjunctivitis produces blurring of vision, burning, and itching, and thick secretions accumulate in the conjunctival sac. The xerostomia results in difficulty in swallowing solid foods, a decrease in the ability to taste, cracks and fissures in the mouth, and dryness of the buccal mucosa. Parotid gland enlargement is present in half the patients; dryness of the nasal mucosa, epistaxis, recurrent bronchitis, and pneumonitis are other symptoms. Manifestations of extraglandular disease are seen in one third of patients and include synovitis, diffuse pulmonary fibrosis, and peripheral neuropathy. These are more common in patients with high titers of antibodies specific for SS-A. In contrast to SLE, glomerular lesions are extremely rare in Sjögren syndrome. Defects of tubular function, however, including renal tubular acidosis, uricosuria, and phosphaturia, are often seen and are associated histologically with tubulointerstitial nephritis (Chapter 20). About 60% of patients have another accompanying autoimmune disorder, such as rheumatoid arthritis, and these patients also have the symptoms and signs of that disorder.
The combination of lacrimal and salivary gland inflammatory involvement was once called Mikulicz disease. The name has now been replaced, however, by Mikulicz syndrome, broadened to include lacrimal and salivary gland enlargement from any cause, including sarcoidosis, leukemia, lymphoma, and other tumors. Biopsy of the lip (to examine minor salivary glands) is essential for the diagnosis of Sjögren syndrome.
The lymph nodes of patients with Sjögren syndrome are often hyperplastic, but the most intense lymphocytic response is seen in the tissues that are the focal point of the autoimmune response, particularly the salivary and lacrimal glands. In early stages of the disease, this immune infiltrate consists of a mixture of polyclonal T and B cells. However, if the reaction continues unabated there is a strong tendency over time for individual clones within the population of B cells to gain a growth advantage, presumably because of the acquisition of somatic mutations. Emergence of a dominant B cell clone is usually indicative of the development of a marginal zone lymphoma, a specific type of B cell malignancy that often arises in the setting of chronic lymphocytic inflammation. About 5% of Sjögren patients develop lymphoma, an incidence that is 40-fold greater than normal. Certain other autoimmune disorders (e.g., Hashimoto thyroiditis) are also associated with a high risk of marginal zone lymphoma (Chapter 13).
SYSTEMIC SCLEROSIS (SCLERODERMA)
Systemic sclerosis is a chronic disease characterized by: (1) chronic inflammation thought to be the result of autoimmunity, (2) widespread damage to small blood vessels, and (3) progressive interstitial and perivascular fibrosis in the skin and multiple organs.90 Although the term scleroderma is ingrained in clinical medicine, this disease is better named systemic sclerosis because it is characterized by excessive fibrosis throughout the body. The skin is most commonly affected, but the gastrointestinal tract, kidneys, heart, muscles, and lungs also are frequently involved. In some patients the disease seems to remain confined to the skin for many years, but in the majority it progresses to visceral involvement with death from renal failure, cardiac failure, pulmonary insufficiency, or intestinal malabsorption. The clinical heterogeneity of systemic sclerosis has been recognized by classifying the disease into two major categories: diffuse scleroderma, characterized by widespread skin involvement at onset, with rapid progression and early visceral involvement; and limited scleroderma, in which the skin involvement is often confined to fingers, forearms, and face. Visceral involvement occurs late; hence, the clinical course is relatively benign. Some patents with the limited disease also develop a combination of calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia, called the CREST syndrome. Several other variants and related conditions, such as eosinophilic fasciitis, occur far less frequently and are not described here.
Etiology and Pathogenesis
The cause of systemic sclerosis is not known. Autoimmune responses, vascular damage, and collagen deposition all contribute to the ultimate tissue injury (Fig. 6-36).90,91
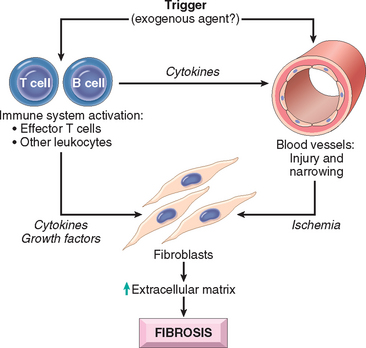
Abnormal Immune Responses
It is proposed that CD4+ T cells responding to an as yet unidentified antigen accumulate in the skin and release cytokines that activate inflammatory cells and fibroblasts.92 Although inflammatory infiltrates are typically sparse in the skin of patients with systemic sclerosis, activated CD4+ T cells can be found in many patients, and TH2 cells have been isolated from the skin. Several cytokines produced by these T cells, including TGF-β and IL-13, can stimulate transcription of genes that encode collagen and other extracellular matrix proteins (e.g., fibronectin) in fibroblasts. Other cytokines recruit leukocytes and propagate the chronic inflammation.
There is also evidence for inappropriate activation of humoral immunity, and the presence of various autoantibodies provides diagnostic and prognostic information.93 Virtually all patients have ANAs that react with a variety of nuclear antigens. Two ANAs strongly associated with systemic sclerosis have been described. One of these, directed against DNA topoisomerase I (anti-Scl 70), is highly specific. Depending on the ethnic group and the assay, it is present in 10% to 20% of patients with diffuse systemic sclerosis. Patients who have this antibody are more likely to have pulmonary fibrosis and peripheral vascular disease. The other, an anticentromere antibody, is found in 20% to 30% of patients, who tend to have the CREST syndrome or limited cutaneous systemic sclerosis. Only rarely does the same patient have both antibodies. The role of these ANAs in the pathogenesis of the disease is unclear; it has been postulated that some of these antibodies may stimulate fibrosis, but the evidence in support of this idea is not convincing.
Vascular Damage
Microvascular disease is consistently present early in the course of systemic sclerosis and may be the initial lesion. Intimal proliferation is evident in 100% of digital arteries of patients with systemic sclerosis. Capillary dilation with leaking, as well as destruction, is also common. Nailfold capillary loops are distorted early in the course of disease, and later they disappear. Thus, there is unmistakable morphologic evidence of microvascular injury. Telltale signs of endothelial activation and injury (e.g., increased levels of von Willebrand factor) and increased platelet activation (increased percentage of circulating platelet aggregates) have also been noted. However, what causes the vascular injury is not known; it could be the initiating event or the result of chronic inflammation, with mediators released by inflammatory cells inflicting damage on microvascular endothelium. Repeated cycles of endothelial injury followed by platelet aggregation lead to release of platelet and endothelial factors (e.g., PDGF, TGF-β) that trigger perivascular fibrosis. Activated or injured endothelial cells themselves may release PDGF and factors chemotactic for fibroblasts. Vascular smooth muscle cells also show abnormalities, such as increased expression of adrenergic receptors. Eventually, widespread narrowing of the microvasculature leads to ischemic injury and scarring. Whether endothelial injury can also be initiated by toxic effects of environmental triggers remains uncertain but cannot be definitively excluded.
Fibrosis
The progressive fibrosis characteristic of the disease may be the culmination of multiple abnormalities, including the actions of fibrogenic cytokines produced by infiltrating leukocytes, hyperresponsiveness of fibroblasts to these cytokines, and scarring following upon ischemic damage caused by the vascular lesions. There is also evidence for a primary abnormality in collagen production. Consistent with this notion is the finding that a polymorphism in the gene encoding connective tissue growth factor is associated with systemic sclerosis.94 In mouse models of Marfan syndrome caused by mutations in the fibrillin-1 gene, some features of systemic sclerosis are also seen,95 suggesting again that connective tissue abnormalities may contribute to this disease.
Morphology. Virtually all organs can be involved in systemic sclerosis. Prominent changes occur in the skin, alimentary tract, musculoskeletal system, and kidney, but lesions also are often present in the blood vessels, heart, lungs, and peripheral nerves.
Skin. A great majority of patients have diffuse, sclerotic atrophy of the skin, which usually begins in the fingers and distal regions of the upper extremities and extends proximally to involve the upper arms, shoulders, neck, and face. Histologically, there are edema and perivascular infiltrates containing CD4+ T cells, together with swelling and degeneration of collagen fibers, which become eosinophilic. Capillaries and small arteries (150 to 500 μm in diameter) may show thickening of the basal lamina, endothelial cell damage, and partial occlusion. With progression of the disease, there is increasing fibrosis of the dermis, which becomes tightly bound to the subcutaneous structures. There is marked increase of compact collagen in the dermis, usually with thinning of the epidermis, loss of rete pegs, atrophy of the dermal appendages, and hyaline thickening of the walls of dermal arterioles and capillaries (Fig. 6-37). Focal and sometimes diffuse subcutaneous calcifications may develop, especially in patients with the CREST syndrome. In advanced stages the fingers take on a tapered, clawlike appearance with limitation of motion in the joints, and the face becomes a drawn mask. Loss of blood supply may lead to cutaneous ulcerations and to atrophic changes in the terminal phalanges (Fig. 6-38). Sometimes the tips of the fingers undergo autoamputation.
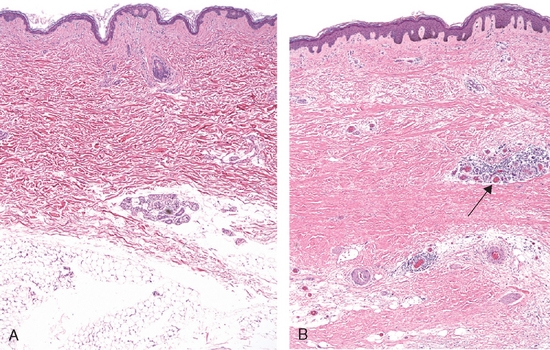
FIGURE 6-37 Systemic sclerosis. A, Normal skin. B, Skin biopsy from a patient with systemic sclerosis. Note the extensive deposition of dense collagen in the dermis with virtual absence of appendages (e.g., hair follicles) and foci of inflammation (arrow).
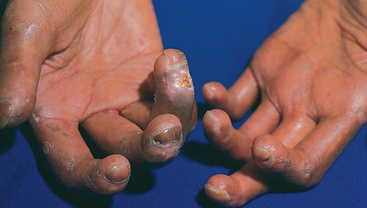
FIGURE 6-38 Advanced systemic sclerosis. The extensive subcutaneous fibrosis has virtually immobilized the fingers, creating a clawlike flexion deformity. Loss of blood supply has led to cutaneous ulcerations.
(Courtesy of Dr. Richard Sontheimer, Department of Dermatology, University of Texas Southwestern Medical School, Dallas, TX.)
Alimentary Tract. The alimentary tract is affected in approximately 90% of patients. Progressive atrophy and collagenous fibrous replacement of the muscularis may develop at any level of the gut but are most severe in the esophagus. The lower two thirds of the esophagus often develops a rubber-hose inflexibility. The associated dysfunction of the lower esophageal sphincter gives rise to gastroesophageal reflux and its complications, including Barrett metaplasia (Chapter 17) and strictures. The mucosa is thinned and may be ulcerated, and there is excessive collagenization of the lamina propria and submucosa. Loss of villi and microvilli in the small bowel is the anatomic basis for the malabsorption syndrome sometimes encountered.
Musculoskeletal System. Inflammation of the synovium, associated with hypertrophy and hyperplasia of the synovial soft tissues, is common in the early stages; fibrosis later ensues. These changes are reminiscent of rheumatoid arthritis, but joint destruction is not common in systemic sclerosis. In a small subset of patients (approximately 10%), inflammatory myositis indistinguishable from polymyositis may develop.
Kidneys. Renal abnormalities occur in two thirds of patients with systemic sclerosis. The most prominent are the vascular lesions. Interlobular arteries show intimal thickening as a result of deposition of mucinous or finely collagenous material, which stains histochemically for glycoprotein and acid mucopolysaccharides. There is also concentric proliferation of intimal cells. These changes may resemble those seen in malignant hypertension, but in scleroderma the alterations are restricted to vessels 150 to 500 μm in diameter and are not always associated with hypertension. Hypertension, however, does occur in 30% of patients with scleroderma, and in 20% it takes an ominously rapid, downhill course (malignant hypertension). In hypertensive patients, vascular alterations are more pronounced and are often associated with fibrinoid necrosis involving the arterioles together with thrombosis and infarction. Such patients often die of renal failure, which accounts for about 50% of deaths in persons with this disease. There are no specific glomerular changes.
Lungs. The lungs are involved in more than 50% of individuals with systemic sclerosis. This involvement may manifest as pulmonary hypertension and interstitial fibrosis. Pulmonary vasospasm, secondary to pulmonary vascular endothelial dysfunction, is considered important in the pathogenesis of pulmonary hypertension. Pulmonary fibrosis, when present, is indistinguishable from that seen in idiopathic pulmonary fibrosis (Chapter 15).
Heart. Pericarditis with effusion and myocardial fibrosis, along with thickening of intramyocardial arterioles, occurs in one third of the patients. Clinical myocardial involvement, however, is less common.
Clinical Features.
Systemic sclerosis has a female-to-male ratio of 3 : 1, with a peak incidence in the 50- to 60-year age group. Although systemic sclerosis shares many features with SLE, rheumatoid arthritis (Chapter 26), and polymyositis (Chapter 27), its distinctive features are the striking cutaneous changes, notably skin thickening. Raynaud’s phenomenon, manifested as episodic vasoconstriction of the arteries and arterioles of the extremities, is seen in virtually all patients and precedes other symptoms in 70% of cases. Dysphagia attributable to esophageal fibrosis and its resultant hypomotility are present in more than 50% of patients. Eventually, destruction of the esophageal wall leads to atony and dilation, especially at its lower end. Abdominal pain, intestinal obstruction, or malabsorption syndrome with weight loss and anemia reflect involvement of the small intestine. Respiratory difficulties caused by the pulmonary fibrosis may result in right-sided cardiac dysfunction, and myocardial fibrosis may cause either arrhythmias or cardiac failure. Mild proteinuria occurs in as many as 30% of patients, but rarely is the proteinuria severe enough to cause a nephrotic syndrome. The most ominous manifestation is malignant hypertension, with the subsequent development of fatal renal failure, but in its absence progression of the disease may be slow. The disease tends to be more severe in blacks, especially black women. As treatment of the renal crises has improved, pulmonary disease has become the major cause of death in systemic sclerosis.
As mentioned earlier, the CREST syndrome is seen in some patients with limited systemic sclerosis. It is characterized by calcinosis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly, telangiectasia, and the presence of anticentromere antibodies. Patients with the CREST syndrome have relatively limited involvement of skin, often confined to fingers, forearms, and face, and calcification of the subcutaneous tissues. Involvement of the viscera, including esophageal lesions, pulmonary hypertension, and biliary cirrhosis, may not occur at all or occur late. In general the patients live longer than those with systemic sclerosis with diffuse visceral involvement at the outset.
INFLAMMATORY MYOPATHIES
Inflammatory myopathies comprise an uncommon, heterogeneous group of disorders characterized by injury and inflammation of mainly the skeletal muscles, which are probably immunologically mediated. Three distinct disorders, dermatomyositis, polymyositis, and inclusion-body myositis, are included in this category. These may occur alone or with other immune-mediated diseases, particularly systemic sclerosis. These diseases are described in Chapter 27.
MIXED CONNECTIVE TISSUE DISEASE
The term mixed connective tissue disease is used to describe a disease with clinical features that are a mixture of the features of SLE, systemic sclerosis, and polymyositis.96 The disease is characterized serologically by high titers of antibodies to ribonucleoprotein particle–containing U1 ribonucleoprotein. Typically, mixed connective tissue disease shows modest renal involvement and a good response to corticosteroids, at least in the short term. Because the clinical features overlap with other diseases, it has been suggested that mixed connective tissue disease is not a distinct entity but that different patients represent subsets of SLE, systemic sclerosis, and polymyositis. The disease can also, over time, evolve into classical SLE or systemic sclerosis. Two of the more serious complications of mixed connective tissue disease are pulmonary hypertension and renal disease resembling that associated with systemic sclerosis.
POLYARTERITIS NODOSA AND OTHER VASCULITIDES
Polyarteritis nodosa belongs to a group of diseases characterized by necrotizing inflammation of the walls of blood vessels and showing strong evidence of an immunological pathogenetic mechanism.97,98 The general term noninfectious vasculitis differentiates these conditions from those due to direct infection of the blood vessel wall (such as occurs in the wall of an abscess) and serves to emphasize that any type of vessel may be involved—arteries, arterioles, veins, or capillaries.
Noninfectious vasculitis is encountered in many clinical settings. A detailed classification and description of vasculitides is presented in Chapter 11 on blood vessels, where the immunological mechanisms are also discussed.