Chapter 11 Blood Vessels
Vascular disorders—and their downstream sequelae—are responsible for more morbidity and mortality than any other category of human disease. Although the most clinically significant lesions typically involve arteries, venous diseases also occur. Vascular pathology results in disease via two principal mechanisms: (1) Narrowing (stenosis) or complete obstruction of vessel lumens, either progressively (e.g., by atherosclerosis) or precipitously (e.g., by thrombosis or embolism); and (2) weakening of vessel walls, leading to dilation or rupture.
We will first describe the important structural and functional characteristics of blood vessels to better appreciate how pathologic changes can result in disease states.
The Structure and Function of Blood Vessels
The general architecture and cellular composition of blood vessels are the same throughout the cardiovascular system. However, certain features of the vasculature vary with and reflect distinct functional requirements at different locations (Fig. 11-1). To withstand the pulsatile flow and higher blood pressures in arteries, arterial walls are generally thicker than the walls of veins. Arterial wall thickness gradually diminishes as the vessels become smaller, but the ratio of wall thickness to lumen diameter becomes greater.
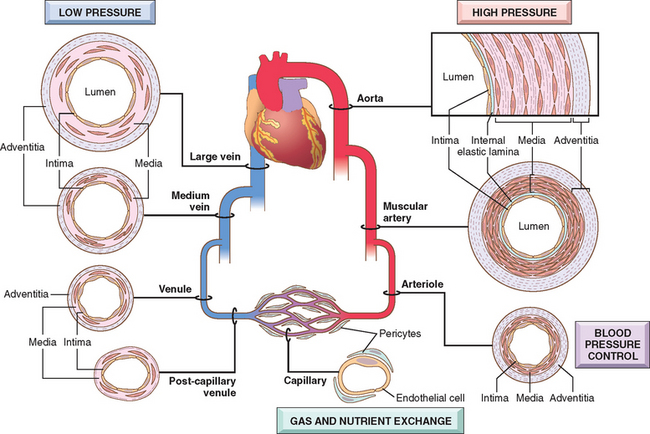
FIGURE 11-1 Regional specializations of the vasculature. Although the basic organization of the vasculature is constant, the thickness and composition of the various layers differ according to hemodynamic forces and tissue requirements.
The basic constituents of the walls of blood vessels are endothelial cells and smooth muscle cells, and extracellular matrix (ECM), including elastin, collagen, and glycosoaminoglycans. The three concentric layers—intima, media, and adventitia—are most clearly defined in the larger vessels, particularly arteries. In normal arteries, the intima consists of a single layer of endothelial cells with minimal underlying subendothelial connective tissue. It is separated from the media by a dense elastic membrane called the internal elastic lamina. The smooth muscle cell layers of the media near the vessel lumen receive oxygen and nutrients by direct diffusion from the vessel lumen, facilitated by holes in the internal elastic membrane. However, diffusion from the lumen is inadequate for the outer portions of the media in large and medium-sized vessels, therefore these areas are nourished by small arterioles arising from outside the vessel (called vasa vasorum, literally “vessels of the vessels”) coursing into the outer one half to two thirds of the media. The outer limit of the media of most arteries is a well-defined external elastic lamina. External to the media is the adventitia, consisting of connective tissue with nerve fibers and the vasa vasorum.
Based on their size and structural features, arteries are divided into three types: (1) large or elastic arteries, including the aorta, its large branches (particularly the innominate, subclavian, common carotid, and iliac), and pulmonary arteries; (2) medium-sized or muscular arteries, comprising other branches of the aorta (e.g., coronary and renal arteries); and (3) small arteries (less than approximately 2 mm in diameter) and arterioles (20 to 100 μm in diameter), within the substance of tissues and organs.
The relative amount and configuration of the basic constituents differ along the arterial system owing to local adaptations to mechanical or metabolic needs. These structural variations, from location to location, are principally in the media and in the ECM. In the elastic arteries the media is rich in elastic fibers. This allows vessels such as the aorta to expand during systole and recoil during diastole, thus propelling blood through the peripheral vascular system. With aging, the aorta loses elasticity, and large vessels expand less readily, particularly when blood pressure is increased. Thus, the arteries of older in-dividuals often become progressively tortuous and dilated (ectatic). In muscular arteries the media is composed predominantly of circularly or spirally arranged smooth muscle cells. In the muscular arteries and arterioles (see below), regional blood flow and blood pressure are regulated by changes in lumen size through smooth muscle cell contraction (vasoconstriction) or relaxation (vasodilation), controlled in part by the autonomic nervous system and in part by local metabolic factors and cellular interactions. Since the resistance of a tube to fluid flow is inversely proportional to the fourth power of the diameter (i.e., halving the diameter increases resistance 16-fold), small changes in the lumen size of small arteries caused by structural change or vasoconstriction can have a profound effect. Thus, arterioles are the principal points of physiologic resistance to blood flow.
Capillaries, approximately the diameter of a red blood cell (7 to 8 μm), have an endothelial cell lining but no media. Collectively, capillaries have a very large total cross-sectional area; within the capillaries, the flow rate slows dramatically. With thin walls only and slow flow, capillaries are ideally suited to the rapid exchange of diffusible substances between blood and tissues. As normal tissue function depends on an adequate supply of oxygen through blood vessels, and since diffusion of oxygen in solid tissues is inefficient over distances of greater than approximately 100 μm,1 the capillary network of most tissues is very rich. Metabolically highly active tissues, such as the myocardium, have the highest density of capillaries.
Blood from capillary beds flows initially into the postcapillary venules and then sequentially through collecting venules and small, medium, and large veins. In many types of inflammation, vascular leakage and leukocyte exudation occur preferentially in postcapillary venules (Chapter 2).
Relative to arteries, veins have larger diameters, larger lumens, and thinner and less well organized walls (see Fig. 11-1). Thus, because of their poor support, veins are predisposed to irregular dilation, compression, and easy penetration by tumors and inflammatory processes. The venous system collectively has a large capacity; approximately two thirds of all the blood is in veins. Reverse flow is prevented by venous valves in the extremities, where blood flows against gravity.
Lymphatics are thin-walled, endothelium-lined channels that serve as a drainage system for returning interstitial tissue fluid and inflammatory cells to the blood. Lymphatics constitute an important pathway for disease dissemination through transport of bacteria and tumor cells to distant sites.
As will be discussed in detail in this chapter, pathologic lesions involve vessels of a characteristic size, range, and/or type. Atherosclerosis, for example, affects elastic and muscular arteries, hypertension affects small muscular arteries and arterioles, and specific types of vasculitis involve different vascular segments.
Vessel Development, Growth, and Remodeling
Three major processes characterize blood vessel formation and remodeling (covered in detail in Chapter 3): vasculogenesis, angiogenesis, and arteriogenesis.1
Congenital Anomalies
Though rarely symptomatic, variants of the usual anatomic pattern of vascular supply can become important during surgery when a vessel in an unexpected location is injured. Variations in the normal coronary artery anatomy are also extremely important to the cardiac surgeon or interventional cardiologist.5,6 Among the other congenital vascular anomalies, three are particularly significant, though not necessarily common:
Vascular Wall Cells and Their Response to Injury
As the main cellular components of the blood vessels, endothelial cells and smooth muscle cells play central roles in vascular biology and pathology. Therefore, we will describe their functions and dysfunctions briefly before we discuss specific vascular disorders.
Endothelial Cells.
Endothelium is critical for maintaining vessel wall homeostasis and circulatory function. Endothelial cells contain Weibel-Palade bodies, intracellular membrane-bound storage organelles for von Willebrand’s factor (Chapter 4). Antibodies to von Willebrand’s factor and/or platelet-endothelial cell adhesion molecule-1 (PECAM-1 or CD31, a protein localized to interendothelial junctions) can be used to identify endothelial cells immunohistochemically.
Vascular endothelium is a multifunctional tissue with a wealth of synthetic and metabolic properties; at baseline it has several constitutive activities critical for normal vessel homeostasis (Table 11-1). Thus, endothelial cells maintain a nonthrombogenic blood-tissue interface (until clotting is necessitated by local injury, Chapter 4), modulate vascular resistance, metabolize hormones, regulate inflammation, and affect the growth of other cell types, particularly smooth muscle cells. In most regions the interendothelial junctions are substantially impermeable. However, tight endothelial cell junctions can loosen under the influence of hemodynamic factors (e.g., high blood pressure) and/or vasoactive agents (e.g., histamine in inflammation), resulting in the flooding of adjacent tissues by electrolytes and protein; in inflammatory states, even leukocytes can slip between adjacent endothelial cells (Chapter 2).
TABLE 11-1 Endothelial Cell Properties and Functions
| MAINTENANCE OF PERMEABILITY BARRIER |
| ELABORATION OF ANTICOAGULANT, ANTITHROMBOTIC, FIBRINOLYTIC REGULATORS |
| ELABORATION OF PROTHROMBOTIC MOLECULES |
| EXTRACELLULAR MATRIX PRODUCTION (COLLAGEN, PROTEOGLYCANS) |
| MODULATION OF BLOOD FLOW AND VASCULAR REACTIVITY |
| REGULATION OF INFLAMMATION AND IMMUNITY |
| REGULATION OF CELL GROWTH |
| OXIDATION OF LDL |
ACE, angiotensin-converting enzyme; CSF, colony-stimulating factor; FGF, fibroblast growth factor; IL, interleukin; LDL, low-density lipoprotein; NO, nitric oxide; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-β.
Although endothelial cells share many general attributes, endothelial cell populations that line different portions of the vascular tree (large vessels vs. capillaries, arterial vs. venous) have distinct transcriptional repertoires and behavior.9 There is also substantial phenotypic variability depending on specific anatomic site. Thus, endothelial cells in liver sinusoids or in renal glomeruli are fenestrated (they have holes, presumably to facilitate filtration), while the endothelial cells of the central nervous system (with the associated perivascular cells) create an impermeable blood-brain barrier.
Structurally intact endothelial cells can respond to various pathophysiologic stimuli by adjusting their usual (constitutive) functions and by expressing newly acquired (inducible) properties—a process termed endothelial activation (Fig. 11-2).10,11 Inducers of endothelial activation include cytokines and bacterial products, which cause inflammation and septic shock (Chapter 2); hemodynamic stresses and lipid products, critical to the pathogenesis of atherosclerosis (see later); advanced glycosylation end products (important in diabetes, Chapter 24); as well as viruses, complement components, and hypoxia. Activated endothelial cells, in turn, express adhesion molecules (Chapter 2), and produce cytokines and chemokines, growth factors, vasoactive molecules that result either in vasoconstriction or in vasodilation, major histocompatibility complex molecules, procoagulant and anticoagulant moieties, and a variety of other biologically active products. Endothelial cells influence the vasoreactivity of the underlying smooth muscle cells through the production of both relaxing factors (e.g., nitric oxide [NO]) and contracting factors (e.g., endothelin).12 Normal endothelial function is characterized by a balance of these responses.
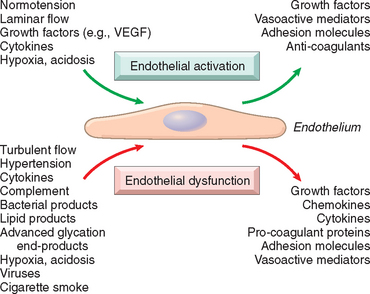
FIGURE 11-2 Endothelial cell responses to environmental stimuli. Certain cues (e.g., laminar flow and constant growth factor levels) lead to stable endothelial cell activation that maintains a nonthrombotic interface with appropriate smooth muscle cell tone. Pathologic mediators or excessive stimulation by normal physiologic pathways (e.g., increased inflammatory cytokines) can result in endothelial cell dysfunction. VEGF, vascular endothelial growth factor.
Endothelial dysfunction is defined as an altered phenotype that impairs vasoreactivity or induces a surface that is thrombogenic or abnormally adhesive to inflammatory cells. It is responsible, at least in part, for the initiation of thrombus formation, atherosclerosis, and the vascular lesions of hypertension and other disorders. Certain forms of endothelial cell dysfunction are rapid in onset (within minutes), reversible, and independent of new protein synthesis (e.g., endothelial cell contraction induced by histamine and other vasoactive mediators that cause gaps in venular endothelium, Chapter 2). Other changes involve alterations in gene expression and protein synthesis and may require hours or even days to develop.
Vascular Smooth Muscle Cells.
As the predominant cellular element of the vascular media, smooth muscle cells play important roles in normal vascular repair and pathologic processes such as atherosclerosis. Smooth muscle cells have the capacity to proliferate when appropriately stimulated; they can also synthesize ECM collagen, elastin, and proteoglycans and elaborate growth factors and cytokines. Smooth muscle cells are also responsible for the vasoconstriction or dilation that occurs in response to physiologic or pharmacologic stimuli.
The migratory and proliferative activities of smooth muscle cells are regulated by growth promoters and inhibitors. Promoters include PDGF, as well as endothelin-1, thrombin, fibroblast growth factor (FGF), interferon-γ (IFN-γ), and interleukin-1(IL-1). Inhibitors include heparan sulfates, nitric oxide, and TGF-β. Other regulators include the renin-angiotensin system (e.g., angiotensin II), catecholamines, the estrogen receptor, and osteopontin, a component of the ECM.13
Intimal Thickening—a Stereotypic Response To Vascular Injury.
Vascular injury—with endothelial cell loss or even just dysfunction—stimulates smooth muscle cell growth and associated matrix synthesis that thickens the intima. Healing of injured vessels is analogous to the healing process that occurs in other damaged tissues (Chapter 3); in vessels, it results in the formation of a neointima. During the healing process, endothelial cells that fill areas of denudation may migrate from adjacent uninjured areas or may be derived from circulating precursors.14 Medial smooth muscle cells or smooth muscle precursor cells also migrate into the intima, proliferate, and synthesize ECM in much the same way that fibroblasts fill in a wound (Fig. 11-3). The resulting neointima is typically completely covered by endothelial cells. This neointimal response occurs with any form of vascular damage or dysfunction, regardless of cause. Thus, intimal thickening is the stereotypical response of the vessel wall to any insult.
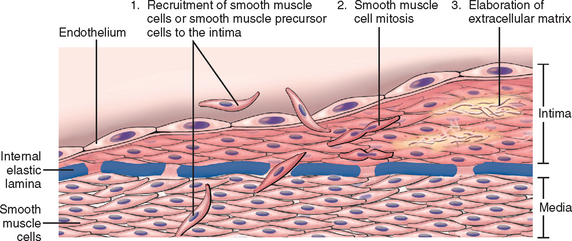
FIGURE 11-3 Schematic of intimal thickening, emphasizing smooth muscle cell migration and proliferation within the intima, with associated ECM synthesis. Intimal smooth muscle cells may derive from the underlying media or may be recruited from circulating precursors; they are shown in a different color from the medial cells to emphasize that they have a proliferative, synthetic, and noncontractile phenotype distinct from medial smooth muscle cells.
(Modified and redrawn from Schoen FJ: Interventional and Surgical Cardiovascular Pathology: Clinical Correlations and Basic Principles. Philadelphia, WB Saunders, 1989, p 254.)
It should be emphasized that the phenotype of neointimal smooth muscle cells is distinct from that of medial smooth muscle cells; neointimal smooth muscle cells do not contract like medial smooth muscle cells but have the capacity to divide. Although these neointimal cells have long been thought to be derived from de-differentiation of smooth muscle cells migrating from the underlying media, there is increasing evidence that the intimal smooth muscle cells are at least in part derived from circulating precursor cells.14-17 The migratory, proliferative, and synthetic activities of the intimal smooth muscle cells are physiologically regulated by products derived from platelets, endothelial cells, and macrophages, as well as by activated coagulation and complement factors. PDGF, endothelin-1, thrombin, FGF, IFN-γ, and IL-1 stimulate neointimal smooth muscle cells, while heparan sulfates, nitric oxide, and TGF-β antagonize their growth.
With time and restoration and/or normalization of the endothelial layer, the intimal smooth muscle cells can return to a nonproliferative state. However, the healing response results in permanent intimal thickening. With persistent or recurrent insults, excessive thickening can cause narrowing or stenosis of small and medium-sized blood vessels (e.g., atherosclerosis, see below), impeding downstream tissue perfusion. As a final note, it is important to remember that intimal thickening also occurs in otherwise normal arteries as a result of maturation and aging. In adult coronaries, for example, the intima and the media are frequently of approximately equal thickness. Such age-related intimal change is typically of no consequence, in part because a compensatory outward remodeling of the vessel results in little net change in the luminal diameter18; it also suggests that not all intimal thickening is a harbinger of disease.
Hypertensive Vascular Disease
Systemic and local tissue blood pressures must be maintained within a narrow range to prevent untoward consequences. Low pressures (hypotension) result in inadequate organ perfusion and can lead to dysfunction or tissue death. Conversely, high pressures (hypertension) can cause vessel and end-organ damage.
Like height and weight, blood pressure is a continuously distributed variable, and detrimental effects of blood pressure increase continuously as the pressure rises; no rigidly defined threshold level of blood pressure distinguishes risk from safety. Nevertheless, according to the National Heart, Lung, and Blood Institute of the U.S.A., a sustained diastolic pressure greater than 89 mm Hg, or a sustained systolic pressure in excess of 139 mm Hg, are associated with a measurably increased risk of atherosclerosis, and are therefore felt to represent clinically significant hypertension. Both the systolic and diastolic blood pressure are important in determining cardiovascular risk.19 By either criterion, some 25% of individuals in the general population are hypertensive. However, it must be emphasized that these cut-offs are somewhat arbitrary, and in patients with other risk factors for vascular disease such as diabetes, lower thresholds are applicable.
Although we have an improved understanding of the molecular pathways that regulate normal blood pressure,20,21 the mechanisms that result in hypertension remain largely unknown in most individuals. Typically, for individuals with such “essential hypertension,” the best we can say is that the disorder is multifactorial, resulting from the combined effects of multiple genetic polymorphisms and interacting environmental factors.22,23
The prevalence and vulnerability to complications of hypertension increase with age; they are also higher in African Americans. As we will see below, hypertension is one of the major risk factors for atherosclerosis and underlies numerous other diseases. It can cause—among other things—cardiac hypertrophy and heart failure (hypertensive heart disease, Chapter 12), multi-infarct dementia (Chapter 28), aortic dissection, and renal failure. Unfortunately, hypertension typically remains asymptomatic until late in its course and even severely elevated pressures can be clinically silent for years. Left untreated, roughly half of hypertensive patients die of ischemic heart disease (IHD) or congestive heart failure, and another third die of stroke. Prophylactic blood pressure reduction dramatically reduces the incidence and death rates from all forms of hypertension-related pathology.
Table 11-2 lists the major causes of hypertension. A small number of patients (approximately 5%) have underlying renal or adrenal disease (such as primary aldosteronism, Cushing syndrome, pheochromocytoma), narrowing of the renal artery, usually by an atheromatous plaque (renovascular hypertension) or other identifiable cause (secondary hypertension). However, about 95% of hypertension is idiopathic (called essential hypertension). This form of hypertension generally does not cause short-term problems. When controlled, it is compatible with long life and is asymptomatic, unless a myocardial infarction, cerebrovascular accident, or other complication supervenes.
TABLE 11-2 Types and Causes of Hypertension (Systolic and Diastolic)
A small percentage, perhaps 5%, of hypertensive persons show a rapidly rising blood pressure that, if untreated, leads to death within a year or two. Called accelerated or malignant hypertension, this clinical syndrome is characterized by severe hypertension (i.e., systolic pressure over 200 mm Hg, diastolic pressure over 120 mm Hg), renal failure, and retinal hemorrhages and exudates, with or without papilledema. It may develop in previously normotensive persons but more often is superimposed on pre-existing benign hypertension, either essential or secondary.24,25
Regulation of Normal Blood Pressure.
Blood pressure is a function of cardiac output and peripheral vascular resistance (Fig. 11-4A), two hemodynamic variables that are influenced by multiple genetic, environmental, and demographic factors. The major factors that determine blood pressure variation within and between populations include age, gender, body mass index, and diet, particularly sodium intake.
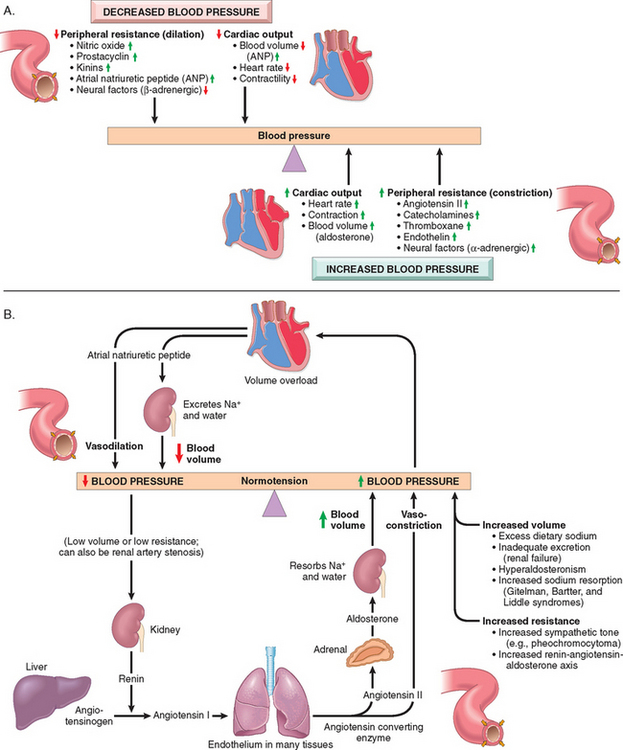
FIGURE 11-4 Blood pressure regulation. A, The critical roles played by cardiac output and peripheral resistance in modulating blood pressure. B, Interplay of renin-angiotensin-aldosterone and atrial natriuretic peptide in maintaining blood pressure homeostasis.
Cardiac output is highly dependent on blood volume, itself greatly influenced by the sodium homeostasis. Peripheral vascular resistance is determined mainly at the level of the arterioles and is affected by neural and hormonal factors. Normal vascular tone reflects the balance between humoral vasoconstricting influences (including angiotensin II, catecholamines, and endothelin) and vasodilators (including kinins, prostaglandins, and NO). Resistance vessels also exhibit autoregulation, whereby increased blood flow induces vasoconstriction to protect against tissue hyperperfusion. Other local factors such as pH and hypoxia, and the α- and β-adrenergic systems, which influence heart rate, cardiac contraction, and vascular tone, may also be important in regulating blood pressure. The integrated function of these systems ensures adequate perfusion of all tissues, despite regional differences in demand.
The kidneys play an important role in blood pressure regulation as follows (Fig. 11-4B):
Mechanisms of Essential Hypertension
Genetic factors play a definite role in determining blood pressure levels, as shown by studies comparing blood pressure in monozygotic and dizygotic twins, and other types of family studies, including comparisons of genetically related and adopted family members. Moreover, several single-gene disorders cause relatively rare forms of hypertension (and hypotension) by altering net sodium reabsorption in the kidney. The importance of sodium balance is emphasized by considering that the kidneys filter 170 liters of plasma containing 23 moles of salt daily; on a typical 100-mEq sodium diet, this means that 99.5% of the filtered salt must be reabsorbed. About 98% of the filtered sodium is reabsorbed by a number of ion channels, exchangers, and transporters that are constitutively active and not subject to regulation. Absorption of the remaining 2% of sodium occurs via the epithelial Na+ channel (ENaC), which is tightly regulated by the renin-angiotensin system in the cortical collecting tubule; it is this resorption pathway that determines net sodium balance.26
Single-gene disorders cause severe but rare forms of hypertension through several mechanisms. These include:
Inherited variations in blood pressure may also depend on the cumulative effects of polymorphisms in several genes that affect blood pressure. For example, predisposition to essential hypertension has been associated with variations in the genes encoding components of the renin-angiotensin system: there is an association of hypertension with polymorphisms in both the angiotensinogen locus and the angiotensin receptor locus. Genetic variants in the renin-angiotensin system may contribute to the known racial differences in blood pressure regulation.
Reduced renal sodium excretion in the presence of normal arterial pressure may be a key initiating event in essential hypertension and, indeed, a final common pathway for the pathogenesis of hypertension. Decreased sodium excretion may lead sequentially to an increase in fluid volume, increased cardiac output, and peripheral vasoconstriction, thereby elevating blood pressure. At the higher setting of blood pressure, enough additional sodium would be excreted by the kidneys to equal intake and prevent further fluid retention. Thus, an altered but steady state of sodium excretion would be achieved (“resetting of pressure natriuresis”), but at the expense of an increase in blood pressure.
Vasoconstrictive influences, such as factors that induce vasoconstriction or stimuli that cause structural changes in the vessel wall, can lead to an increase in peripheral resistance and may also play a role in primary hypertension. Moreover, chronic or repeated vasoconstrictive influences could cause thickening and rigidity of the involved vessels.
Environmental factors can modify the impact of genetic determinants. Stress, obesity, smoking, physical inactivity, and heavy consumption of salt have all been implicated as exogenous factors in hypertension. Indeed, evidence linking the level of dietary sodium intake with the prevalence of hypertension in different population groups is particularly impressive. Moreover, in both essential and secondary hypertension, heavy sodium intake augments the condition.
To summarize, essential hypertension is a complex, multifactorial disorder. Although single gene disorders can be responsible for hypertension in rare cases, it is unlikely that such mutations are a major cause of essential hypertension. It is more likely that essential hypertension results from interactions of mutations or polymorphisms at several loci that influence blood pressure, with a variety of environmental factors (e.g., stress, salt intake). Mendelian forms of hypertension and hypotension are rare but yield insights into pathways and mechanisms of blood pressure regulation, and they may help define rational targets for therapeutic intervention. Sustained hypertension requires participation of the kidney, which normally responds to hypertension by eliminating salt and water. Susceptibility genes for essential hypertension in the larger population are currently unknown but may well include genes that govern responses to an increased renal sodium load, levels of pressor substances, reactivity of vascular smooth muscle cells to vasoconstrictive agents, or smooth muscle cell growth. In established hypertension, both increased blood volume and increased peripheral resistance contribute to the increased pressure.
Pathogenesis of Secondary Hypertension.
For many of the secondary forms of hypertension, the underlying pathways are reasonably well understood. For example, in renovascular hypertension, renal artery stenosis causes decreased glomerular flow and pressure in the afferent arteriole of the glomerulus. This (1) induces renin secretion, initiating angiotensin II–mediated vasoconstriction and increased peripheral resistance, and (2) increases sodium reabsorption and therefore blood volume through the aldosterone mechanism. Primary hyperaldosteronism is one of the most common causes of secondary hypertension (Chapter 24).
VASCULAR PATHOLOGY IN HYPERTENSION
Hypertension not only accelerates atherogenesis (see below) but also causes degenerative changes in the walls of large and medium arteries that can lead to aortic dissection and cerebrovascular hemorrhage.
Hypertension is associated with two forms of small blood vessel disease: hyaline arteriolosclerosis and hyperplastic arteriolosclerosis.
Hyaline Arteriolosclerosis. Arterioles show homogeneous, pink hyaline thickening with associated luminal narrowing (Fig. 11-5A). These changes stem from plasma protein leakage across injured endothelial cells, and increased smooth muscle cell matrix synthesis in response to chronic hemodynamic stress. Although the vessels of elderly persons (either normo- or hypertensive) also frequently show hyaline arteriosclerosis, it is more generalized and severe in individuals with hypertension. The same lesions are also a common feature of diabetic microangiography; in that case the underlying etiology is hyperglycemia-induced endothelial cell dysfunction (Chapter 24). In nephrosclerosis due to chronic hypertension, the arteriolar narrowing of hyaline arteriosclerosis causes diffuse impairment of renal blood supply and causes glomerular scarring (Chapter 20).
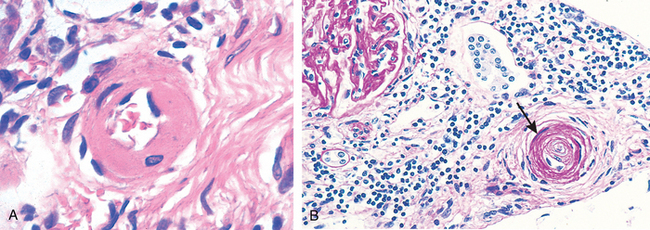
FIGURE 11-5 Vascular pathology in hypertension. A, Hyaline arteriolosclerosis. The arteriolar wall is thickened with increased protein deposition (hyalinized), and the lumen is markedly narrowed. B, Hyperplastic arteriolosclerosis (onion-skinning; arrow) causing lumenal obliteration (arrow; periodic acid–Schiff stain).
(Courtesy of Helmut Rennke, M.D., Brigham and Women’s Hospital, Boston, MA.)
Hyperplastic Arteriolosclerosis. This lesion occurs in severe (malignant) hypertension; vessels exhibit “onion-skin lesions,” characterized by concentric, laminated thickening of the walls and luminal narrowing (Fig. 11-5B). The laminations consist of smooth muscle cells with thickened, reduplicated basement membranes; in malignant hypertension they are accompanied by fibrinoid deposits and vessel wall necrosis (necrotizing arteriolitis), particularly in the kidney.
Arteriosclerosis
Arteriosclerosis literally means “hardening of the arteries”; it is a generic term reflecting arterial wall thickening and loss of elasticity. There are three general patterns, with differing clinical and pathologic consequences:
Atherosclerosis
Atherosclerosis is characterized by intimal lesions called atheromas (also called atheromatous or atherosclerotic plaques) that protrude into vessel lumens. An atheromatous plaque consists of a raised lesion with a soft, yellow, grumous core of lipid (mainly cholesterol and cholesterol esters) covered by a white fibrous cap (Fig. 11-6). Besides mechanically obstructing blood flow, atherosclerotic plaques can rupture, leading to catastrophic vessel thrombosis; plaques also weaken the underlying media and thereby lead to aneurysm formation. Atherosclerosis causes far more morbidity and mortality (roughly half of all deaths) in the Western world than any other disorder. Because coronary artery disease is an important manifestation of the disease, epidemiologic data related to atherosclerosis mortality typically reflect deaths caused by heart disease (Chapter 12); indeed, myocardial infarction is responsible for almost a quarter of all deaths in the United States. Significant morbidity and mortality are also caused by aortic and carotid atherosclerotic disease and stroke.
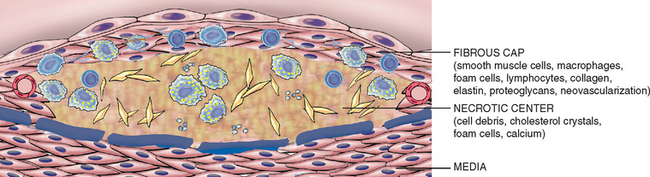
FIGURE 11-6 The major components of a well-developed intimal atheromatous plaque overlying an intact media.
EPIDEMIOLOGY
Virtually ubiquitous among most developed nations, atherosclerosis is much less prevalent in Central and South America, Africa, and parts of Asia. The mortality rate for ischemic heart disease (IHD) in the United States is among the highest in the world and is approximately five times higher than that in Japan. Nevertheless, IHD has been increasing in Japan and is now the second leading cause of death there. Moreover, Japanese immigrants who adopt American life styles and dietary customs acquire the same predisposition to atherosclerosis as the indigenous population.
The prevalence and severity of atherosclerosis and IHD among individuals and groups are related to several risk factors, some constitutional (and therefore less controllable), others acquired or related to behaviors that are potentially amenable to intervention (Table 11-3). Risk factors have been identified through several prospective studies in well-defined populations, most notably the Framingham Heart Study and Atherosclerosis Risk in Communities Study (Fig. 11-7).27,28 Risk factors have a multiplicative effect; two risk factors increase the risk approximately fourfold. When three risk factors are present (e.g., hyperlipidemia, hypertension, and smoking), the rate of myocardial infarction is increased seven times.
TABLE 11-3 Major Risk Factors for Atherosclerosis
| NONMODIFIABLE | |
| MODIFIABLE | |
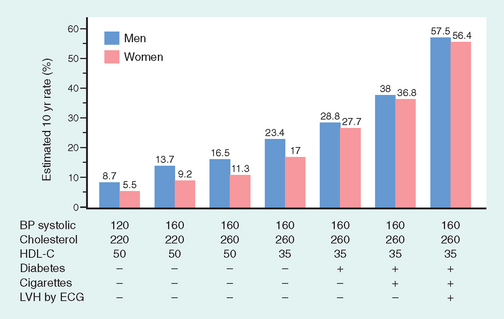
FIGURE 11-7 Estimated 10-year risk of coronary artery disease in hypothetical 55-year-old men and women as a function of traditional risk factors (hyperlipidemia, hypertension, smoking, and diabetes). BP, blood pressure; ECG, electrocardiogram; HDL-C, high-density lipoprotein cholesterol; LVH, left ventricular hypertrophy.
(From O’Donnell CJ, Kannel WB: Cardiovascular risks of hypertension: lessons from observational studies. J Hypertension 16 [Suppl. 6]:3, 1998, with permission from Lippincott Williams & Wilkins.)
Constitutional risk factors in IHD.
These include age, gender, and genetics.
Modifiable risk factors in IHD.
These include hyperlipidemia, hypertension, cigarette smoking, and diabetes.
Additional risk factors.
As many as 20% of all cardiovascular events occur in the absence of hypertension, hyperlipidemia, smoking, or diabetes. Indeed, more than 75% of cardiovascular events in previously healthy women occurred with LDL cholesterol levels below 160 mg/dL (a cutoff generally considered to connote low risk).32 Clearly, other factors contribute to risk; the assessment of some of these has entered clinical practice.
CRP is an acute-phase reactant synthesized primarily by the liver. It is downstream of a number of inflammatory triggers and plays a role in the innate immune response by opsonizing bacteria and activating complement. When CRP is secreted from cells within the atherosclerotic intima, it can activate local endothelial cells and induce a prothrombotic state and also increase the adhesiveness of endothelium for leukocytes. Most importantly, it strongly and independently predicts the risk of myocardial infarction, stroke, peripheral arterial disease, and sudden cardiac death, even among apparently healthy individuals (Fig. 11-8). Indeed, CRP levels have recently been incorporated into risk stratification algorithms.34 Interestingly, although there is as yet no direct evidence that lowering CRP directly reduces cardiovascular risk, smoking cessation, weight loss, and exercise all reduce CRP; moreover, statins reduce CRP levels largely independent of their effects on LDL cholesterol.
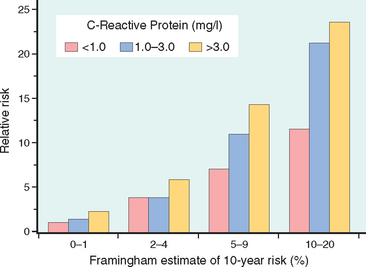
FIGURE 11-8 C-reactive protein (CRP) adds prognostic information at all levels of traditional risk identified from the Framingham Heart Study. Relative risk (y-axis) refers to the risk of a cardiovascular event (e.g., myocardial infarction). The x-axis is the 10-year risk of a cardiovascular event derived from the traditional risk factors identified in the Framingham Study. In each group of Framingham “risk”, CRP values further stratify the patients.
(Adapted from Ridker PM et al: Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med 347:1557, 2002.)
PATHOGENESIS OF ATHEROSCLEROSIS
The clinical importance of atherosclerosis has stimulated enormous interest in understanding the mechanisms that underlie this disease and its complications. Historically, there have been two dominant hypotheses: one emphasizes intimal cellular proliferation, while the other focuses on the repetitive formation and organization of thrombi. The contemporary view of atherogenesis incorporates elements of both theories and also integrates the risk factors previously discussed.40,41 Called the response-to-injury hypothesis,42 the model views atherosclerosis as a chronic inflammatory and healing response of the arterial wall to endothelial injury. Lesion progression occurs through the interaction of modified lipoproteins, monocyte-derived macrophages, and T lymphocytes with the normal cellular constituents of the arterial wall (Fig. 11-9). According to this model, atherosclerosis is produced by the following pathogenic events:
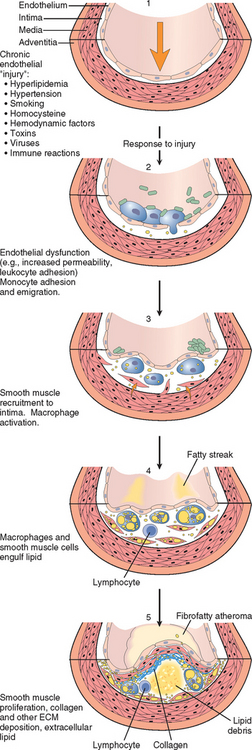
FIGURE 11-9 Evolution of arterial wall changes in the response to injury hypothesis. 1, Normal. 2, Endothelial injury with adhesion of monocytes and platelets (the latter to sites where endothelium has been lost). 3, Migration of monocytes and smooth muscle cells into the intima. 4, Smooth muscle cell proliferation in the intima with ECM production. 5, Well-developed plaque.
The major mechanisms of atherogenesis will now be considered in detail.
Endothelial Injury
Endothelial cell injury is the cornerstone of the responseto-injury hypothesis. Endothelial loss due to any kind of injury—induced experimentally by mechanical denudation, hemodynamic forces, immune complex deposition, irradiation, or chemicals—results in intimal thickening; in the presence of high-lipid diets, typical atheromas ensue. However, early human lesions begin at sites of morphologically intact endothelium. Thus, endothelial dysfunction underlies human atherosclerosis; in this setting, dysfunctional endothelial cells show increased endothelial permeability, enhanced leukocyte adhesion, and altered gene expression.
The specific pathways and factors contributing to endothelial cell dysfunction in early atherosclerosis are not completely understood; etiologic culprits include hypertension, hyperlipidemia, toxins from cigarette smoke, homocysteine, and even infectious agents. Inflammatory cytokines (e.g., tumor necrosis factor [TNF]) can also stimulate pro-atherogenic patterns of endothelial cell gene expression. However, the two most important causes of endothelial dysfunction are hemodynamic disturbances and hypercholesterolemia.
Hemodynamic Disturbances.
The importance of hemodynamic turbulence in atherogenesis is illustrated by the observation that plaques tend to occur at ostia of exiting vessels, branch points, and along the posterior wall of the abdominal aorta, where there are disturbed flow patterns.43 In vitro studies further demonstrate that nonturbulent laminar flow in the normal vasculature leads to the induction of endothelial genes whose products (e.g., the antioxidant superoxide dismutase) protect against atherosclerosis. Such “atheroprotective” genes could explain the nonrandom localization of early atherosclerotic lesions.11
Lipids.
Lipids are typically transported in the bloodstream bound to specific apoproteins (forming lipoprotein complexes). Dyslipoproteinemias can result from mutations that alter the apoproteins or the lipoprotein receptors on cells,44 or from other disorders that affect the circulating levels of lipids (e.g., nephrotic syndrome, alcoholism, hypothyroidism, or diabetes mellitus).45 Common lipoprotein abnormalities in the general population (indeed, present in many myocardial infarction survivors) include (1) increased LDL cholesterol levels, (2) decreased HDL cholesterol levels, and (3) increased levels of the abnormal lipoprotein (a) (see above).
The evidence implicating hypercholesterolemia in atherogenesis includes the following observations:
The mechanisms by which hyperlipidemia contributes to atherogenesis include the following:44
Inflammation.
Inflammatory cells and pathways contribute to the initiation, progression, and complications of atherosclerotic lesions.41,46 Although normal vessels do not bind inflammatory cells, early in atherogenesis, dysfunctional arterial endothelial cells express adhesion molecules that encourage leukocyte adhesion; vascular cell adhesion molecule 1 (VCAM-1), in particular, binds monocytes and T cells. After these cells adhere to the endothelium, they migrate into the intima under the influence of locally produced chemokines.
Infection.
Although there is tantalizing evidence that infections may drive the local inflammatory process that underlies atherosclerosis, this hypothesis has yet to be conclusively proven. Herpesvirus, cytomegalovirus, and Chlamydia pneumoniae have all been detected in atherosclerotic plaques but not in normal arteries, and seroepidemiologic studies find increased antibody titers to C. pneumoniae in patients with more severe atherosclerosis. Of course, some of these observations are confounded by the fact that C. pneumoniae bronchitis is also associated with smoking, a well-established IHD risk factor. Moreover, infections with these organisms are exceedingly common (as is atherosclerosis), so that distinguishing coincidence from causality is difficult. Nevertheless, it is certainly possible that such organisms could infect sites of early atheroma formation; their foreign antigens could potentiate atherogenesis by driving local immune responses, or infectious agents could contribute to the local prothrombotic state.47
Smooth Muscle Proliferation
Intimal smooth muscle cell proliferation and ECM deposition convert a fatty streak, the earliest lesion, into a mature atheroma and contribute to the progressive growth of atherosclerotic lesions (see Fig. 11-9, steps 4 and 5). (Recall that the intimal smooth muscle cells may be recruited from circulating precursors and they have a proliferative and synthetic phenotype distinct from the underlying medial smooth muscle cells.) Several growth factors are implicated in smooth muscle cell proliferation and ECM synthesis, including PDGF (released by locally adherent platelets, as well as macrophages, endothelial cells, and smooth muscle cells), FGF, and TGF-α. The recruited smooth muscle cells synthesize ECM (notably collagen) that stabilizes atherosclerotic plaques. However, activated inflammatory cells in atheromas can cause intimal smooth muscle cell apoptosis, and also increase ECM catabolism resulting in unstable plaques (see below).
Overview
Figure 11-10 highlights the concept of atherosclerosis as a chronic inflammatory response—and ultimately an attempt at vascular “healing”—driven by a variety of insults, including endothelial cell injury, lipid accumulation and oxidation, and thrombosis. Atheromas are dynamic lesions consisting of dysfunctional endothelial cells, recruited and proliferating smooth muscle cells, and admixed lymphocytes and macrophages. All four cell types are capable of liberating mediators that can influence atherogenesis. Thus, at early stages, intimal plaques are little more than smooth muscle cell and macrophage foam cell aggregates. With progression, the atheroma is modified by ECM synthesized by smooth muscle cells; connective tissue is particularly prominent in the intima, where it forms a fibrous cap, although lesions also typically retain a central core of lipid-laden cells and fatty debris that can become calcified. The intimal plaque may progressively encroach on the vessel lumen, or compress and cause degeneration of the underlying media; disruption of the fibrous cap can lead to thrombosis and acute vascular occlusion.
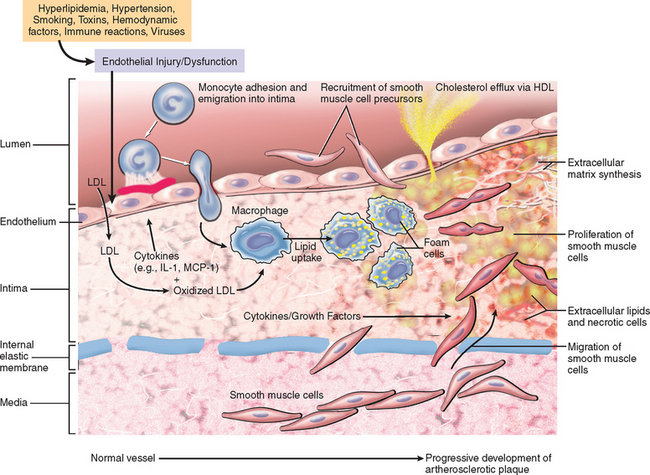
FIGURE 11-10 Hypothetical sequence of cellular interactions in atherosclerosis. Hyperlipidemia and other risk factors are thought to cause endothelial injury, resulting in adhesion of platelets and monocytes and release of growth factors, including platelet-derived growth factor (PDGF), which lead to smooth muscle cell migration and proliferation. Foam cells of atheromatous plaques are derived from both macrophages and smooth muscle cells—from macrophages via the very-low-density lipoprotein (VLDL) receptor and low-density lipoprotein (LDL) modifications recognized by scavenger receptors (e.g., oxidized LDL), and from smooth muscle cells by less certain mechanisms. Extracellular lipid is derived from insudation from the vessel lumen, particularly in the presence of hypercholesterolemia, and also from degenerating foam cells. Cholesterol accumulation in the plaque reflects an imbalance between influx and efflux, and high-density lipoprotein (HDL) probably helps clear cholesterol from these accumulations. Smooth muscle cells migrate to the intima, proliferate, and produce ECM, including collagen and proteoglycans. IL-1, interleukin-1; MCP-1, monocyte chemoattractant protein 1.
With this overview of pathogenesis, we will now discuss the morphologic features and evolution of atherosclerosis.
Fatty Streaks. Fatty streaks are the earliest lesions in atherosclerosis. They are composed of lipid-filled foamy macrophages. Beginning as multiple minute flat yellow spots, they eventually coalesce into elongated streaks 1 cm or more in length. These lesions are not significantly raised and do not cause any flow disturbance (Fig. 11-11). Aortas of infants less than 1 year old can exhibit fatty streaks, and such lesions are seen in virtually all children older than 10 years, regardless of geography, race, sex, or environment. The relationship of fatty streaks to atherosclerotic plaques is uncertain; although they may evolve into precursors of plaques, not all fatty streaks are destined to become advanced lesions. Nevertheless, coronary fatty streaks begin to form in adolescence, at the same anatomic sites that later tend to develop plaques.
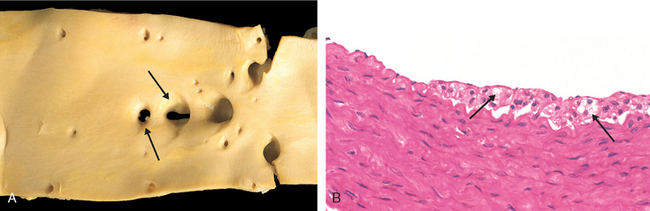
FIGURE 11-11 Fatty streak, a collection of foamy macrophages in the intima. A, Aorta with fatty streaks (arrows), associated largely with the ostia of branch vessels. B, Photomicrograph of fatty streak in an experimental hypercholesterolemic rabbit, demonstrating intimal, macrophage-derived foam cells (arrows).
(B, Courtesy of Myron I. Cybulsky, M.D., University of Toronto, Toronto, ON, Canada.)
Atherosclerotic Plaque. The key processes in atherosclerosis are intimal thickening and lipid accumulation (see Fig. 11-10). Atheromatous plaques impinge on the lumen of the artery and grossly appear white to yellow; superimposed thrombus over ulcerated plaques is red-brown. Plaques vary from 0.3 to 1.5 cm in diameter but can coalesce to form larger masses (Fig. 11-12).
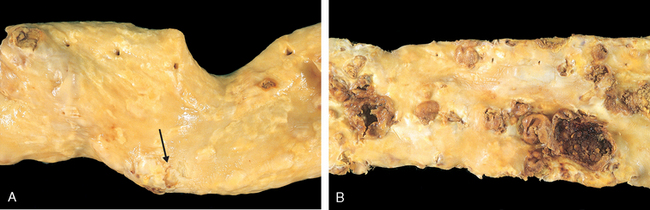
FIGURE 11-12 Gross views of atherosclerosis in the aorta. A, Mild atherosclerosis composed of fibrous plaques, one of which is denoted by the arrow. B, Severe disease with diffuse and complicated lesions (with plaque rupture and superimposed thrombosis), some of which have coalesced.
Atherosclerotic lesions are patchy, usually involving only a portion of any given arterial wall, and are rarely circumferential; on cross-section, the lesions therefore appear “eccentric” (Fig. 11-13A). The focality of atherosclerotic lesions—despite the uniform exposure of vessel walls to such factors as cigarette smoke toxins, elevated LDL, hyperglycemia, etc.—is attributable to the vagaries of vascular hemodynamics. Local flow disturbances (e.g., turbulence at branch points) leads to increased susceptibility of certain portions of a vessel wall to plaque formation. Though focal and sparsely distributed at first, atherosclerotic lesions can become more numerous and more diffuse with time.
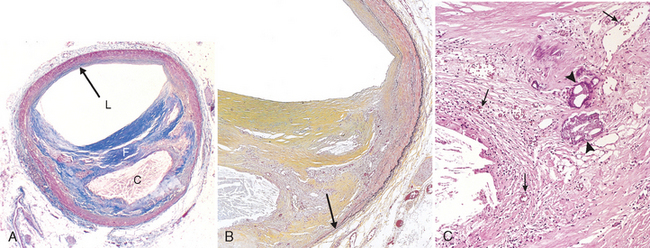
FIGURE 11-13 Histologic features of atheromatous plaque in the coronary artery. A, Overall architecture demonstrating fibrous cap (F) and a central necrotic (largely lipid) core (C). The lumen (L) has been moderately compromised. Note that a segment of the wall is free of plaque (arrow); the lesion is therefore “eccentric”. In this section, collagen has been stained blue (Masson’s trichrome stain). B, Higher power photograph of a section of the plaque shown in A, stained for elastin (black), demonstrating that the internal and external elastic membranes are attenuated and the media of the artery is thinned under the most advanced plaque (arrow). C, Higher magnification photomicrograph at the junction of the fibrous cap and core, showing scattered inflammatory cells, calcification (arrowhead) and neovascularization (small arrows).
In humans, the abdominal aorta is typically involved to a much greater degree than the thoracic aorta. In descending order, the most extensively involved vessels are the lower abdominal aorta, the coronary arteries, the popliteal arteries, the internal carotid arteries, and the vessels of the circle of Willis. Vessels of the upper extremities are usually spared, as are the mesenteric and renal arteries, except at their ostia. Nevertheless, in an individual case, the severity of atherosclerosis in one artery does not predict its severity in another. Moreover, in any given vessel, lesions at various stages often coexist.
Atherosclerotic plaques have three principal components: (1) cells, including smooth muscle cells, macrophages, and T cells; (2) ECM, including collagen, elastic fibers, and proteoglycans; and (3) intracellular and extracellular lipid (Fig. 11-13). These components occur in varying proportions and configurations in different lesions. Typically, there is a superficial fibrous cap composed of smooth muscle cells and relatively dense collagen. Beneath and to the side of the cap (the “shoulder”) is a more cellular area containing macrophages, T cells, and smooth muscle cells. Deep to the fibrous cap is a necrotic core, containing lipid (primarily cholesterol and cholesterol esters), debris from dead cells, foam cells (lipid-laden macrophages and smooth muscle cells), fibrin, variably organized thrombus, and other plasma proteins; the cholesterol is frequently present as crystalline aggregates that are washed out during routine tissue processing and leave behind only empty “clefts.” The periphery of the lesions show neovascularization (proliferating small blood vessels; Fig. 11-13C). Typical atheromas contain abundant lipid, but some plaques (“fibrous plaques”) are composed almost exclusively of smooth muscle cells and fibrous tissue.
Plaques generally continue to change and progressively enlarge due to cell death and degeneration, synthesis and degradation (remodeling) of ECM, and organization of thrombus. Moreover, atheromas often undergo calcification (see Fig. 11-13C).
Atherosclerotic plaques are susceptible to the following clinically important changes (see also subsequent discussion):
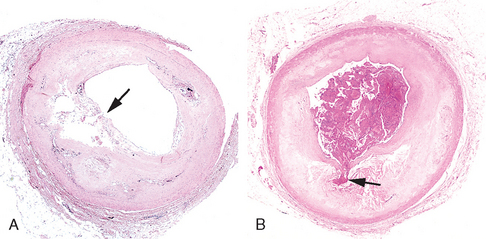
FIGURE 11-14 Atherosclerotic plaque rupture. A, Plaque rupture without superimposed thrombus, in a patient who died suddenly. B, Acute coronary thrombosis superimposed on an atherosclerotic plaque with focal disruption of the fibrous cap, triggering fatal myocardial infarction. In both A and B, an arrow points to the site of plaque rupture.
(B, Reproduced from Schoen FJ: Interventional and Surgical Cardiovascular pathology: Clinical Correlations and Basic Principles. Philadelphia, WB Saunders, 1989, p 61.)
CONSEQUENCES OF ATHEROSCLEROTIC DISEASE
Large elastic arteries (e.g., the aorta, carotid, and iliac arteries) and large and medium-sized muscular arteries (e.g., coronary and popliteal arteries) are the major targets of atherosclerosis. Symptomatic atherosclerotic disease most often involves the arteries supplying the heart, brain, kidneys, and lower extremities. Myocardial infarction (heart attack), cerebral infarction (stroke), aortic aneurysms, and peripheral vascular disease (gangrene of the legs) are the major consequences of atherosclerosis. The natural history, principal morphologic features, and main pathogenic events are schematized in Figure 11-15. The principal outcomes depend on the size of the involved vessels, the relative stability of the plaque itself, and the degree of degeneration of the underlying arterial wall:
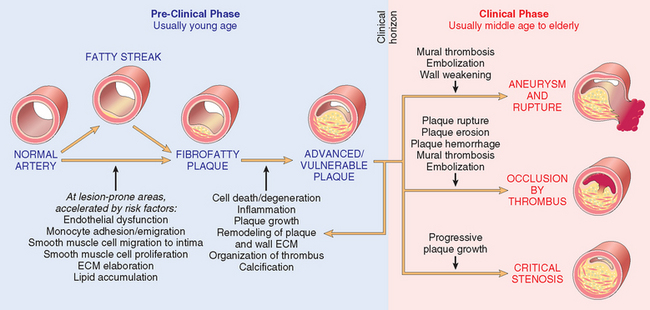
FIGURE 11-15 The natural history, morphologic features, main pathogenic events, and clinical complications of atherosclerosis.
Chronic stenosis and plaque rupture will be covered next, followed by a discussion of aneurysms.
Atherosclerotic Stenosis.
In small arteries, atherosclerotic plaques can gradually occlude vessel lumens, compromising blood flow and causing ischemic injury. At early stages of stenosis, outward remodeling of the vessel media tends to preserve luminal diameter as the total circumference expands.18 However, there are limits on this outward remodeling, and eventually the expanding atheroma impinges on blood flow. Critical stenosis is the Rubicon at which chronic occlusion significantly limits flow, and demand begins exceeding supply. In the coronary (and other) circulations, this typically occurs at approximately 70% fixed occlusion (i.e., loss of area through which blood can flow); at this degree of stenosis, patients classically develop chest pain (angina) on exertion (so-called stable angina; see Chapter 12). Although acute plaque rupture (below) is the most dangerous complication, atherosclerosis also takes a toll through chronically diminished arterial perfusion: mesenteric occlusion and bowel ischemia, chronic IHD, ischemic encephalopathy, and intermittent claudication (diminished extremity perfusion) are all consequences of flowlimiting stenoses. The effects of vascular occlusion ultimately depend on arterial supply and the metabolic demand of the affected tissue.
Acute Plaque Change.
Plaque erosion or rupture is typically promptly followed by partial or complete vascular thrombosis (see Fig. 11-14), resulting in acute tissue infarction (e.g., myocardial or cerebral infarction).40,48 Plaque changes fall into three general categories:
It is now recognized that the precipitating lesion in patients who develop myocardial infarction and other acute coronary syndromes is not necessarily a severely stenotic and hemodynamically significant lesion before its acute change. Pathologic and clinical studies show that the majority of plaques that undergo abrupt disruption and coronary occlusion previously showed only mild to moderate luminal stenosis.49 The worrisome conclusion is that a rather large number of now asymptomatic adults may well have a real but unpredictable risk of a catastrophic coronary event. Regrettably, it is presently impossible to reliably detect individuals who will have plaque disruption or subsequent thrombosis.
The events that trigger abrupt changes in plaque configuration and superimposed thrombosis are complex and include both intrinsic factors (e.g., plaque structure and composition) and extrinsic factors (e.g., blood pressure, platelet reactivity)40,50; rupture of a plaque indicates that it was unable to withstand the mechanical stresses of vascular shear forces. We next discuss the intrinsic and extrinsic factors that influence the risk of plaque rupture.
It is important to remember that the composition of plaques is dynamic and can materially contribute to risk of rupture. Thus, plaques that contain large areas of foam cells and extracellular lipid, and those in which the fibrous caps are thin or contain few smooth muscle cells or have clusters of inflammatory cells, are more likely to rupture, and are therefore called “vulnerable plaques”48 (Fig. 11-16).
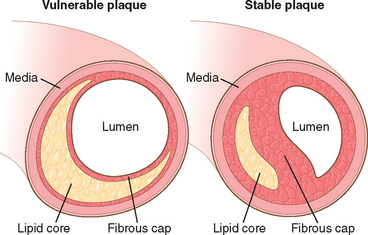
FIGURE 11-16 Schematic comparing vulnerable and stable atherosclerotic plaque. Whereas stable plaques have densely collagenous and thickened fibrous caps with minimal inflammation and negligible underlying atheromatous core, vulnerable plaques
(prone to rupture) are characterized by thin fibrous caps, large lipid cores, and increased inflammation. (Adapted from Libby P: Circulation 91:2844, 1995.)
It is also established that the fibrous cap undergoes continuous remodeling that may make the plaque susceptible to acute alterations. Collagen represents the major structural component of the fibrous cap, and accounts for its mechanical strength and stability. Thus, the balance of collagen synthesis versus degradation affects cap stability. Collagen in atherosclerotic plaque is produced primarily by smooth muscle cells, so that loss of these cellular elements results in a weaker cap. Moreover, collagen turnover is controlled by matrix metalloproteinases (MMPs), enzymes elaborated largely by macrophages within the atheromatous plaque; conversely, tissue inhibitors of metalloproteinases (TIMPs), produced by endothelial cells, smooth muscle cells, and macrophages, modulate MMP activity. In general, plaque inflammation results in a net increase in collagen degradation and reduces collagen synthesis, thereby destabilizing the mechanical integrity of the fibrous cap (see below). Interestingly, statins may have a beneficial therapeutic effect not only by reducing circulating cholesterol levels but also by stabilizing plaques through a reduction in plaque inflammation.51
Influences extrinsic to plaques are also important. Thus, adrenergic stimulation can increase systemic blood pressure or induce local vasoconstriction, thereby increasing the physical stresses on a given plaque. Indeed, the adrenergic stimulation associated with waking and rising can cause blood pressure spikes (followed by heightened platelet reactivity) that have been causally linked to the pronounced circadian periodicity for the peak time of onset of acute myocardial infarction (between 6 AM and 12 noon).52 Intense emotional stress can also contribute to plaque disruption; this is most dramatically illustrated by the uptick in the incidence of sudden death associated with disasters such as earthquakes and the September 11, 2001 attacks.53
It is also important to note that not all plaque ruptures result in occlusive thromboses with catastrophic consequences. Indeed, plaque disruption and ensuing platelet aggregation and thrombosis are probably common, repetitive, and often clinically silent complications of atheroma. Healing of these subclinical plaque disruptions—with their overlying thromboses—is an important mechanism in the growth of atherosclerotic lesions.
Thrombosis.
As mentioned above, partial or total thrombosis associated with a disrupted plaque is critical to the pathogenesis of the acute coronary syndromes. In the most serious form, thrombus superimposed on a disrupted but previously only partially stenotic plaque converts it to a total occlusion. In contrast, in other coronary syndromes (Chapter 12), luminal obstruction by thrombosis is usually incomplete, and can even wax and wane with time.
Mural thrombus in a coronary artery can also embolize. Indeed, small fragments of thrombotic material in the distal intra-myocardial circulation or microinfarcts can be found at autopsy in patients after sudden death or in rapidly accelerating anginal syndromes. Finally, thrombus is a potent activator of multiple growth-related signals in smooth muscle cells, which can contribute to the growth of atherosclerotic lesions.
Vasoconstriction.
Vasoconstriction compromises lumen size, and, by increasing the local mechanical forces can potentiate plaque disruption. Vasoconstriction at sites of atheroma is stimulated by (1) circulating adrenergic agonists, (2) locally released platelet contents, (3) impaired secretion of endothelial cell relaxing factors (nitric oxide) relative to contracting factors (endothelin) as a result of endothelial cell dysfunction, and possibly (4) mediators released from perivascular inflammatory cells.
Aneurysms and Dissection
An aneurysm is a localized abnormal dilation of a blood vessel or the heart (Fig. 11-17); it can be congenital or acquired. When an aneurysm involves an intact attenuated arterial wall or thinned ventricular wall of the heart, it is called a true aneurysm. Atherosclerotic, syphilitic, and congenital vascular aneurysms, and ventricular aneurysms that follow transmural myocardial infarctions are of this type. In contrast, a false aneurysm (also called pseudo-aneurysm) is a defect in the vascular wall leading to an extravascular hematoma that freely communicates with the intravascular space (“pulsating hematoma”). Examples include a ventricular rupture after myocardial infarction that is contained by a pericardial adhesion, or a leak at the sutured junction of a vascular graft with a natural artery. An arterial dissection arises when blood enters the arterial wall itself, as a hematoma dissecting between its layers. Dissections are often but not always aneurysmal (see also below). Both true and false aneurysms as well as dissections can rupture, often with catastrophic consequences.
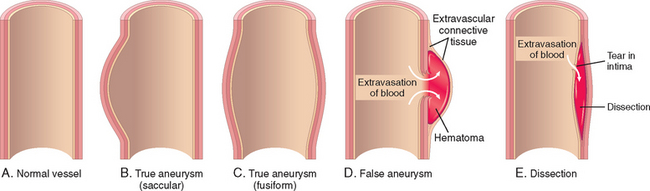
FIGURE 11-17 Aneurysms. A, Normal vessel. B, True aneurysm, saccular type. The wall focally bulges outward and may be attenuated but is otherwise intact. C, True aneurysm, fusiform type. There is circumferential dilation of the vessel, without rupture. D, False aneurysm. The wall is ruptured, and there is a collection of blood (hematoma) that is bounded externally by adherent extravascular tissues. E, Dissection. Blood has entered (dissected) the wall of the vessel and separated the layers. Although this is shown as occurring through a tear in the lumen, dissections can also occur by rupture of the vessels of the vaso vasorum within the media.
Aneurysms are generally classified by shape and size (see Fig. 11-17). Saccular aneurysms are spherical outpouchings (involving only a portion of the vessel wall); they vary from 5 to 20 cm in diameter and often contain thrombus. Fusiform aneurysms involve diffuse, circumferential dilation of a long vascular segment; they vary in diameter (up to 20 cm) and in length, and can involve extensive portions of the aortic arch, abdominal aorta, or even the iliac arteries. These types are not specific for any disease or clinical manifestations.
Pathogenesis of Aneurysms.
Arteries are dynamically remodeling tissues that maintain their integrity by constantly synthesizing, degrading, and repairing damage to their ECM constituents. Aneurysms can occur when the structure or function of the connective tissue within the vascular wall is compromised. Although we cite here examples of inherited defects in connective tissues, weakening of vessel walls is important in the common, sporadic forms of aneurysms as well.
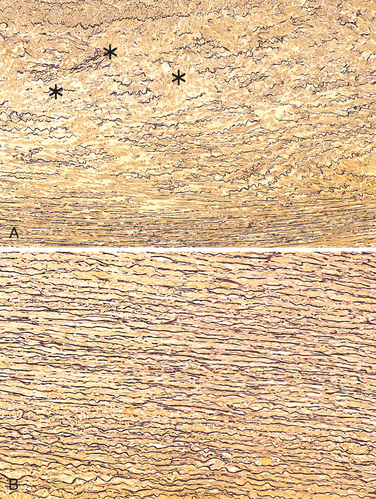
FIGURE 11-18 Cystic medial degeneration. A, Cross-section of aortic media from a patient with Marfan syndrome, showing marked elastin fragmentation and formation of areas devoid of elastin that resemble cystic spaces (asterisks). B, Normal media for comparison, showing the regular layered pattern of elastic tissue. In both A and B, elastin is stained black.
The two most important disorders that predispose to aortic aneurysms are atherosclerosis and hypertension; atherosclerosis is a greater factor in abdominal aortic aneurysms, while hypertension is the most common condition associated with aneurysms of the ascending aorta.58 Other conditions that weaken vessel walls and lead to aneurysms include trauma, vasculitis (see below), congenital defects (e.g., berry aneurysms typically in the circle of Willis; Chapter 28), and infections (mycotic aneurysms). Mycotic aneurysms can originate (1) from embolization of a septic embolus, usually as a complication of infective endocarditis; (2) as an extension of an adjacent suppurative process; or (3) by circulating organisms directly infecting the arterial wall. Tertiary syphilis is now a rare cause of aorticaneurysms. The obliterative endarteritis characteristic of late-stage syphilis shows a predilection for small vessels, including those of the vasa vasorum of the thoracic aorta. This leads to ischemic injury of the aortic media and aneurysmal dilation, which sometimes involves the aortic valve annulus.59
ABDOMINAL AORTIC ANEURYSM (AAA)
Aneurysms associated with atherosclerosis occur most commonly in the abdominal aorta. Atherosclerotic plaque in the intima compresses the underlying media and compromises nutrient and waste diffusion from the vascular lumen into the arterial wall. The media therefore undergoes degeneration and necrosis that results in arterial wall weakness and consequent thinning. Nevertheless, as described above, the major influence that leads to aneurysm formation is the production of MMP by inflammatory cell infiltrates.56
Abdominal aortic aneurysms occur more frequently in men and in smokers, and rarely develop before age 50. Atherosclerosis is a major cause of AAAs, but other factors clearly contribute since the incidence is less than 5% in men older than 60 years of age, despite almost universal abdominal aortic atherosclerosis in that population.
Morphology. Usually positioned below the renal arteries and above the bifurcation of the aorta, AAAs can be saccular or fusiform, up to 15 cm in diameter, and up to 25 cm in length (Fig. 11-19). Typically the intimal surface of the aneurysm shows severe complicated atherosclerosis with destruction and thinning of the underlying aortic media; the aneurysm frequently contains a bland, laminated, poorly organized mural thrombus that may fill some or all of the dilated segment. Occasionally the aneurysm can affect the renal and superior or inferior mesenteric arteries, either by producing direct pressure or by narrowing or occluding vessel ostia with mural thrombi. Not infrequently, AAAs are accompanied by smaller aneurysms of the iliac arteries.
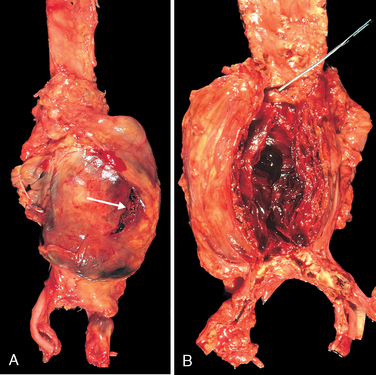
FIGURE 11-19 Abdominal aortic aneurysm. A, External view, gross photograph of a large aortic aneurysm that ruptured; the rupture site is indicated by the arrow. B, Opened view, with the location of the rupture tract indicated by a probe. The wall of the aneurysm is exceedingly thin, and the lumen is filled by a large quantity of layered but largely unorganized thrombus.
Clinical Features.
The clinical consequences of AAA include
The risk of rupture is directly related to the size of the aneurysm,60 varying from nil for AAAs of 4 cm or less in diameter, to 1% per year for AAAs between 4 and 5 cm, to 11% per year for AAAs between 5 and 6 cm, and 25% per year for aneurysms greater than 6 cm in diameter. Most aneurysms expand at a rate of 0.2 to 0.3 cm/yr, but 20% expand more rapidly. In general, aneurysms of 5 cm and larger are managed aggressively, usually by surgical bypass involving prosthetic grafts. Currently, aneurysm treatment is evolving toward endoluminal approaches using stent grafts (expandable wire frames covered by a cloth sleeve) in selected patients.61 Timely surgery is critical; operative mortality for unruptured aneurysms is approximately 5%, whereas emergency surgery after rupture carries a mortality rate of more than 50%. It is worth reiterating that because atherosclerosis is a systemic disease, a person with AAA is also very likely to have atherosclerosis in other vascular beds and is at a significantly increased risk of IHD and stroke.
THORACIC AORTIC ANEURYSMS
Thoracic aortic aneurysms are most commonly associated with hypertension, although other causes such as Marfan and Loeys-Dietz syndromes are increasingly recognized.58 Regardless of etiology, these give rise to signs and symptoms referable to (1) encroachment on mediastinal structures, (2) respiratory difficulties due to encroachment on the lungs and airways, (3) difficulty in swallowing due to compression of the esophagus, (4) persistent cough due to irritation of or pressure on the recurrent laryngeal nerves, (5) pain caused by erosion of bone (i.e., ribs and vertebral bodies), (6) cardiac disease as the aortic aneurysm leads to aortic valve dilation with valvular insufficiency or narrowing of the coronary ostia causing myocardial ischemia, and (7) rupture. Most patients with syphilitic aneurysms die of heart failure induced by aortic valvular incompetence.
AORTIC DISSECTION
Aortic dissection occurs when blood splays apart the laminar planes of the media to form a blood-filled channel within the aortic wall (Fig. 11-20); this can be catastrophic if the dissection then ruptures through the adventitia and hemorrhages into adjacent spaces.62 In contrast to atherosclerotic and syphilitic aneurysms, aortic dissection may or may not be associated with aortic dilation. Consequently, the older term “dissecting aneurysm” should be avoided.
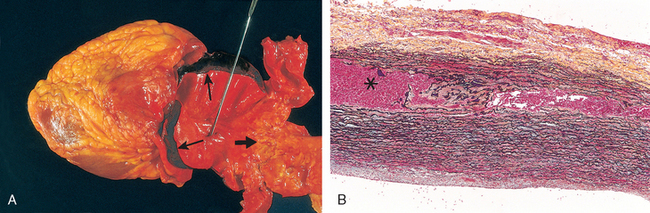
FIGURE 11-20 Aortic dissection. A, An opened aorta with proximal dissection originating from a small, oblique intimal tear (identified by the probe), allowing blood to enter the media and creating an intramural hematoma (narrow arrows). Note that the intimal tear has occurred in a region largely free of atherosclerotic plaque and that propagation of the intramural hematoma is arrested at a site more distally where atherosclerosis begins (broad arrow). B, Histologic view of the dissection demonstrating an aortic intramural hematoma (asterisk). Aortic elastic layers are black and blood is red in this section, stained with the Movat stain.
Aortic dissection occurs principally in two groups: (1) men aged 40 to 60, with antecedent hypertension (more than 90% of cases of dissection); and (2) younger patients with systemic or localized abnormalities of connective tissue affecting the aorta (e.g., Marfan syndrome). Dissections can also be iatrogenic (e.g., complicating arterial cannulations during diagnostic catheterization or cardiopulmonary bypass). Rarely, for unknown reasons, dissection of the aorta or other branches, including the coronary arteries, occurs during or after pregnancy. Dissection is unusual in the presence of substantial atherosclerosis or other cause of medial scarring such as syphilis, presumably because the medial fibrosis inhibits propagation of the dissecting hematoma.
Pathogenesis.
Hypertension is the major risk factor in aortic dissection. Aortas of hypertensive patients have medial hypertrophy of the vasa vasorum associated with degenerative changes in the aortic media and variable loss of medial smooth muscle cells, suggesting that pressure-related mechanical injury and/or ischemic injury (due to diminished flow through the vasa vasorum) is contributory. A considerably smaller number of dissections are related to inherited or acquired connective tissue disorders causing abnormal vascular ECM (e.g., Marfan syndrome, Ehlers-Danlos syndrome, vitamin C deficiency, copper metabolic defects). However, recognizable medial damage seems to be neither a prerequisite for dissection nor a guarantee that dissection is imminent. Regardless of the underlying etiology causing medial weakness, the trigger for the intimal tear and initial intramural aortic hemorrhage is not known in most cases. Nevertheless, once the tear has occurred, blood flow under systemic pressure dissects through the media, fostering progression of the medial hematoma. Accordingly, aggressive pressure-reducing therapy may be effective in limiting an evolving dissection. In some cases disruption of penetrating vessels of the vasa vasorum can give rise to an intramural hematoma without an intimal tear.
Morphology. In most cases, no specific underlying causal pathology is identified in the aortic wall. The most frequent preexisting histologically detectable lesion is cystic medial degeneration (see Fig. 11-18); inflammation is characteristically absent. However, dissections can occur in the setting of rather trivial medial degeneration, and the relationship of the structural changes to the pathogenesis of dissection is uncertain.
An aortic dissection usually initiates with an intimal tear. In the vast majority of spontaneous dissections, the tear is found in the ascending aorta, usually within 10 cm of the aortic valve (Fig. 11-20A). Such tears are typically transverse or oblique and 1 to 5 cm in length, with sharp, jagged edges. The dissection can extend along the aorta retrograde toward the heart as well as distally, sometimes into the iliac and femoral arteries. The dissecting hematoma spreads characteristically along the laminar planes of the aorta, usually between the middle and outer thirds (Fig. 11-20B). It often ruptures out through the adventitia causing massive hemorrhage (e.g., in the thoracic or abdominal cavities) or cardiac tamponade (hemorrhage into the pericardial sac).62 In some (lucky) instances, the dissecting hematoma reenters the lumen of the aorta through a second distal intimal tear, creating a new vascular channel and forming a “double-barreled aorta” with a false channel.62 This averts a fatal extra-aortic hemorrhage. In the course of time, false channels may be endothelialized and become chronic dissections.
Clinical Features.
The risk and nature of complications of aorta dissection depend strongly on the region(s) affected; the most serious complications occur with dissections that involve the aorta from the aortic valve to the arch. Thus, aortic dissections are generally classified into two types (Fig. 11-21). They are named after Dr. Michael DeBakey, a pioneer in vascular surgery.
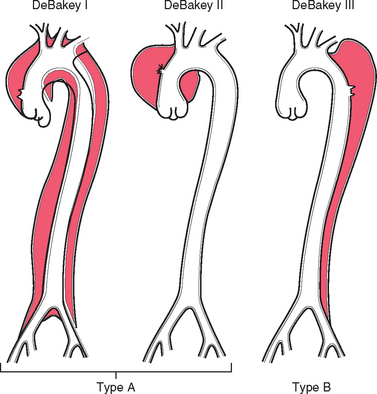
FIGURE 11-21 Classification of dissections. Type A (proximal) involves the ascending aorta, either as part of a more extensive dissection (DeBakey I) or in isolation (DeBakey II). Type B (distal or DeBakey III) dissections arise beyond the takeoff of the great vessels. The serious complications predominantly occur in type A dissections.
The classic clinical symptoms of aortic dissection are the sudden onset of excruciating pain, usually beginning in the anterior chest, radiating to the back between the scapulae, and moving downward as the dissection progresses; the pain can be confused with that of myocardial infarction.
The most common cause of death is rupture of the dissection outward into the pericardial, pleural, or peritoneal cavities. Retrograde dissection into the aortic root can cause disruption of the aortic valvular apparatus. Thus, common clinical manifestations include cardiac tamponade, aortic insufficiency, and myocardial infarction or extension of the dissection into the great arteries of the neck or into the coronary, renal, mesenteric, or iliac arteries, causing critical vascular obstruction and associated ischemic consequences; compression of spinal arteries may cause transverse myelitis.
In the past aortic dissection was typically fatal, but the prognosis has markedly improved. Rapid diagnosis and institution of intensive antihypertensive therapy, coupled with surgical procedures involving plication of the aortic wall, permit 65% to 75% of stricken individuals to be saved.
Vasculitis
Vasculitis is a general term for vessel wall inflammation. The clinical features of the various vasculitides are diverse and largely depend on the vascular bed affected (e.g., central nervous system vs. heart vs. small bowel). Besides the findings referable to the specific tissue(s) involved, the clinical manifestations typically include constitutional signs and symptoms such as fever, myalgias, arthralgias, and malaise.
Vessels of any type in virtually any organ can be affected; most vasculitides involve small vessels, from arterioles to capillaries to venules.63 Several of the vasculitides tend to affect only vessels of a particular size or particular vessel beds. There are vasculitic entities that primarily affect the aorta and medium-sized arteries, while others principally affect only smaller arterioles. Some 20 primary forms of vasculitis are recognized, and classification schemes attempt (with variable success) to group them according to vessel size, role of immune complexes, presence of specific autoantibodies, granuloma formation, organ specificity, and even population demographics! Though a subject of ongoing evolution,64 the so-called Chapel Hill nomenclature remains the most widely accepted approach to organizing this diverse group of entities65 (Table 11-4 and Fig. 11-22). As we will see, there is considerable clinical and pathologic overlap among many of them.
TABLE 11-4 Classification and Characteristics of Selected Immune-Mediated Vasculitides
| Vasculitis Type* | Examples | Description |
|---|---|---|
| LARGE-VESSEL VASCULITIS | Giant-cell (temporal) arteritis | Granulomatous inflammation; frequently involves the temporal artery. Usually occurs in patients older than age 50 and is associated with polymyalgia rheumatica. |
| Aorta and large branches to extremities, head, and neck | Takayasu arteritis | Granulomatous inflammation usually occurring in patients younger than age 50 |
| MEDIUM-VESSEL VASCULITIS | Polyarteritis nodosa | Necrotizing inflammation typically involving renal arteries but sparing pulmonary vessels |
| Main visceral arteries and their branches | Kawasaki disease | Arteritis with mucocutaneous lymph node syndrome; usually occurs in children. Coronary arteries can be involved with aneurysm formation and/or thrombosis. |
| SMALL-VESSEL VASCULITIS | Wegener granulomatosis | Granulomatous inflammation involving the respiratory tract and necrotizing vasculitis affecting small vessels, including glomerular vessels. Associated with PR3-ANCAs. |
| Arterioles, venules, capillaries, and occasionally small arteries | Churg-Strauss syndrome | Eosinophil-rich granulomatous inflammation involving the respiratory tract and necrotizing vasculitis affecting small vessels. Associated with asthma and blood eosinophilia. Associated with MPO-ANCAs. |
| Microscopic polyangiitis | Necrotizing small-vessel vasculitis with few or no immune deposits; necrotizing arteritis of small and medium-sized arteries can occur. Necrotizing glomerulonephritis and pulmonary capillaritis are common. Associated with MPO-ANCAs. |
MPO-ANCAs, antineutrophil cytoplasmic antibodies, directed against myeloperoxidase (p-ANCA); PR3-ANCAs, antineutrophil cytoplasmic antibodies, directed against proteinase 3 (c-ANCA).
* Note that some small- and large-vessel vasculitides may involve medium-sized arteries, but large- and medium-sized vessel vasculitides do not involve vessels smaller than arteries.
Modified from Jennette JC, et al. Nomenclature of systemic vasculitides: The proposal of an international consensus conference. Arthritis Rheum 37:187, 1994.
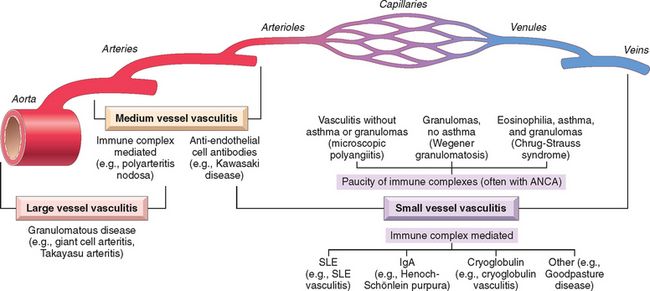
FIGURE 11-22 Diagrammatic representation of the typical vascular sites involved with the more common forms of vasculitis, as well as the presumptive etiologies. Note that there is a substantial overlap in distributions. ANCA, antineutrophil cytoplasmic antibody; SLE, systemic lupus erythematosus.
(Modified from Jennette JC, Falk RJ: Nosology of primary vasculitis. Curr Opin Rheumatol 19:10, 2007.)
The two most common pathogenic mechanisms of vasculitis are immune-mediated inflammation and direct invasion of vascular walls by infectious pathogens. Predictably, infections can also indirectly induce a noninfectious vasculitis, for example, by generating immune complexes or triggering cross-reactivity. In any given patient, it is critical to distinguish between infectious and immunological mechanisms, because immunosuppressive therapy is appropriate for immunemediated vasculitis but could very well worsen infectious vasculitis. Physical and chemical injury, such as from irradiation, mechanical trauma, and toxins, can also cause vasculitis.
NONINFECTIOUS VASCULITIS
The main immunological mechanisms that initiate noninfectious vasculitis are (1) immune complex deposition, (2) antineutrophil cytoplasmic antibodies, and (3) anti–endothelial cell antibodies.
Immune Complex–Associated Vasculitis.
The lesions resemble those found in experimental immune complex–mediated conditions such as the Arthus reaction and serum sickness (Chapter 6). Many systemic immunological diseases, such as systemic lupus erythematosus (SLE) and polyarteritis nodosa, manifest as immune complex-mediated vasculitis. Antibody and complement are typically detected in vasculitic lesions, although the nature of the antigens responsible for their deposition cannot usually be determined. Circulating antigen-antibody complexes may also be seen (e.g., DNA–anti-DNA complexes in SLE–associated vasculitis [Chapter 6]), but the sensitivity and specificity of circulating immune complex assays in such diseases are low. In addition, immune complexes are implicated in the following vasculitides:
In many cases of immune complex vasculitis, it is not clear whether the antigen-antibody complexes form elsewhere and then deposit in a particular vascular bed, or if they form in situ from the seeding of antigen in a vessel wall followed by antibody binding (Chapter 6). Moreover, in many cases of presumed immune complex vasculitis, antigen-antibody deposits are scarce. Either the immune complexes have been largely cleared at the time that the tissue diagnosis is made, or else other mechanisms may apply in such “pauci-immune” cases.
Antineutrophil Cytoplasmic Antibodies.
Many patients with vasculitis have circulating antibodies that react with neutrophil cytoplasmic antigens, so-called antineutrophil cytoplasmic antibodies (ANCAs). ANCAs are a heterogeneous group of autoantibodies directed against constituents (mainly enzymes) of neutrophil primary granules, monocyte lysosomes, and endothelial cells. These were previously classified according to their intracellular distribution, either cytoplasmic (c-ANCA) or perinuclear (p-ANCA). More commonly now, they are discriminated based on their target antigens:
Although not entirely specific, PR3-ANCAs are typical of Wegener granulomatosis and MPO-ANCAs are characteristic of microscopic polyangiitis and Churg-Strauss syndrome (see below); racial and geographic variables also influence the association of particular ANCAs and disease entities.
ANCAs serve as useful diagnostic markers for the ANCA-associated vasculitides, and their titers may reflect the degree of inflammatory activity. ANCA titers also rise with recurrent disease and are therefore useful in clinical management. The close association between ANCA titers and disease activity suggests a pathogenic role. Although the precise mechanisms are unknown, ANCA can directly activate neutrophils and may thereby stimulate neutrophils to release reactive oxygen species and proteolytic enzymes; within the vasculature, this also leads to endothelial cell-neutrophil interactions and subsequent endothelial cell damage.67 Moreover, the antigenic targets of ANCAs are primarily intracellular and therefore might not be expected to be accessible to circulating antibodies, but there is now abundant evidence that ANCA antigens (in particular PR3) are either constitutively present at low levels on the plasma membrane or are translocated to the cell surface in activated and apoptotic neutrophils.66,68
A plausible mechanism for ANCA vasculitis is the following66,68:
Interestingly, ANCAs directed against constituents other than PR3 and MPO are also found in some patients with inflammatory disorders that do not involve vasculitis (e.g., inflammatory bowel disease, primary sclerosing cholangitis, rheumatoid arthritis).
Anti-Endothelial Cell Antibodies.
Antibodies to endothelial cells may predispose to certain vasculitides, for example, Kawasaki disease69,70 (see below).
We will now briefly present several of the best-characterized vasculitides, again emphasizing that there is substantial overlap among the different entities. Moreover, it should be kept in mind that some patients with vasculitis do not have a classic constellation of findings that allows them to be neatly pigeonholed into one specific diagnosis.
GIANT-CELL (TEMPORAL) ARTERITIS
Giant-cell (temporal) arteritis is the most common form of vasculitis among elderly individuals in the United States and Europe. It is a chronic, typically granulomatous inflammation of large to small-sized arteries that affects principally the arteries in the head—especially the temporal arteries—but also the vertebral and ophthalmic arteries.71 Ophthalmic arterial involvement can lead to permanent blindness; consequently, giant-cell arteritis is a medical emergency requiring prompt recognition and treatment. Lesions also occur in other arteries, including the aorta (giant-cell aortitis).
Pathogenesis.
The cause of giant-cell arteritis remains elusive, although most evidence supports an initial T cell–mediated immune response against an unknown, possibly vessel wall, antigen. Pro-inflammatory cytokines (in particular TNF), and anti–endothelial cell humoral immune responses also probably contribute.72 An immune etiology is supported by the characteristic granulomatous reaction, a correlation with certain HLA class II haplotypes, and a therapeutic response to steroids. The extraordinary predilection for a single vascular site (temporal artery) remains unexplained.
Morphology. Involved arterial segments develop nodular intimal thickening (with occasional thromboses) that reduces the lumenal diameter. Classic lesions exhibit medial granulomatous inflammation that leads to elastic lamina fragmentation; there is an infiltrate of T cells (CD4+> CD8+) and macrophages. Multinucleated giant cells are found in upwards of 75% of adequately biopsied specimens (Fig. 11-23). Occasionally, granulomas and giant cells are rare or absent, and lesions show only a nonspecific panarteritis composed predominantly of lymphocytes and macrophages. Inflammatory lesions are not continuous along the vessel, and long segments of relatively normal artery may be interposed. The healed stage is marked by medial scarring and intimal thickening, typically with residual elastic tissue fragmentation.
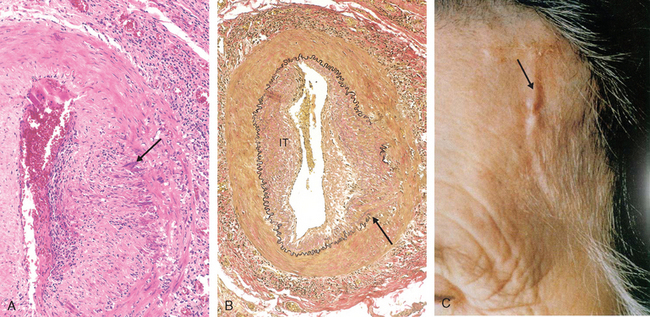
FIGURE 11-23 Giant-cell (temporal) arteritis. A, H&E stain of section of temporal artery showing giant cells at the degenerated internal elastic lamina in active arteritis (arrow). B, Elastic tissue stain demonstrating focal destruction of internal elastic lamina (arrow) and intimal thickening (IT) characteristic of long-standing or healed arteritis. C, Examination of the temporal artery of a patient with giant-cell arteritis shows a thickened, nodular, and tender segment of a vessel on the surface of head (arrow).
(C from Salvarani C et al.: Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med 347:261, 2002.)
Clinical Features.
Temporal arteritis is rare before the age of 50. Symptoms may be vague and constitutional—fever, fatigue, weight loss—or include facial pain or headache that is most intense along the course of the superficial temporal artery, which can be painful to palpation. Ocular symptoms (associated with involvement of the ophthalmic artery) appear abruptly in about 50% of patients; these range from diplopia to complete vision loss. Diagnosis depends on biopsy and histologic confirmation. However, because giant-cell arteritis is extremely segmental, adequate biopsy requires at least a 2- to 3-cm length of artery; even then, a negative biopsy result does not exclude the diagnosis. Treatment with corticosteroids is generally effective, with anti-TNF therapy showing promise in refractory cases.71
TAKAYASU ARTERITIS
This is a granulomatous vasculitis of medium and larger arteries characterized principally by ocular disturbances and marked weakening of the pulses in the upper extremities (hence, its other name, pulseless disease). Takayasu arteritis manifests with transmural fibrous thickening of the aorta—particularly the aortic arch and great vessels—and severe luminal narrowing of the major branch vessels (Fig. 11-24). Aortic lesions share many attributes with giant-cell aortitis, including clinical features and histology; indeed, the distinction is typically made only on the basis of the age of the patient. Those over 50 years of age are designated as having giant-cell aortitis, while those under 50 have Takayasu aortitis.64 Though traditionally associated with the Japanese population and a subset of HLA haplotypes, Takayasu aortitis has a global distribution. The cause and pathogenesis are unknown, although immune mechanisms are suspected.71,72
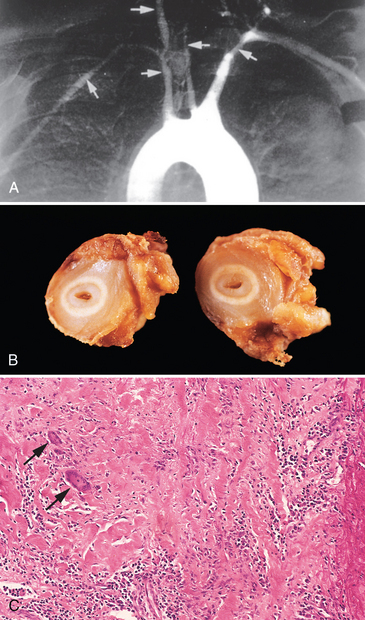
FIGURE 11-24 Takayasu arteritis. A, Aortic arch angiogram showing narrowing of brachiocephalic, carotid, and subclavian arteries (arrows). B, Gross photograph of two cross-sections of the right carotid artery taken at autopsy of the patient shown in A, demonstrating marked intimal thickening with minimal residual lumen. C, Histologic view of active Takayasu aortitis, illustrating destruction of the arterial media by mononuclear inflammation with giant cells (arrows).
Morphology. Takayasu arteritis classically involves the aortic arch. In a third of patients it also affects the remainder of the aorta and its branches. The pulmonary artery is involved in half of cases; coronary and renal arteries may be similarly affected. There is irregular thickening of the vessel wall with intimal hyperplasia; when the aortic arch is involved the great vessel lumens can be markedly narrowed or even obliterated (Fig. 11-24A and B). Such narrowing explains the weakness of the peripheral pulses. Histological changes range from adventitial mononuclear infiltrates with perivascular cuffing of the vasa vasorum, to intense mononuclear inflammation in the media, to granulomatous inflammation, replete with giant cells and patchy medial necrosis. The histologic appearance (Fig. 11-24C) is indistinguishable from that of giant-cell (temporal) arteritis. As the disease progresses, collagenous scarring, with admixed chronic inflammatory infiltrates, occurs in all three layers of the vessel wall. Occasionally, aortic root involvement causes aortic insufficiency.
Clinical Features.
Initial symptoms are usually nonspecific, including fatigue, weight loss, and fever. With progression, vascular symptoms appear and dominate the clinical picture, including reduced blood pressure and weaker pulses in the upper extremities; ocular disturbances, including visual defects, retinal hemorrhages, and total blindness; and neurologic deficits. Involvement of the more distal aorta may lead to claudication of the legs; pulmonary artery involvement may cause pulmonary hypertension. Narrowing of the coronary ostia may lead to myocardial infarction, and involvement of the renal arteries leads to systemic hypertension in roughly half of patients. The course of the disease is variable. In some there is rapid progression, while others enter a quiescent stage at 1 to 2 years, permitting long-term survival, albeit sometimes with visual or neurologic deficits.
POLYARTERITIS NODOSA
Polyarteritis nodosa (PAN) is a systemic vasculitis of small or medium-sized muscular arteries (but not arterioles, capillaries, or venules), typically involving renal and visceral vessels but sparing the pulmonary circulation. There is no association with ANCAs, but about 30% of patients with PAN have chronic hepatitis B with HBsAg-HbsAb complexes in affected vessels, indicating an immune complex–mediated etiology (see Chapter 6) in that subset. Nevertheless, the cause remains unknown in the majority of cases; there may be etiologic and important clinical distinctions between classic idiopathic PAN, the cutaneous forms of PAN, and the PAN associated with chronic hepatitis.73,74 Clinical manifestations result from ischemia and infarction of affected tissues and organs.73
Morphology. Classic PAN is characterized by segmental transmural necrotizing inflammation ofsmall to medium-sized arteries. Vessels of the kidneys, heart, liver, and gastrointestinal tract are involved in descending order of frequency. Lesions usually affect only part of the vessel circumference and show a predilection for branch points. The inflammatory process weakens the arterial wall and can lead to aneurysms or even rupture. Impaired perfusion resulting in ulcerations, infarcts, ischemic atrophy, or hemorrhages in the distribution of affected vessels may be the first sign of disease.
During the acute phase there is transmural inflammation of the arterial wall with a mixed infiltrate of neutrophils, eosinophils, and mononuclear cells, frequently accompanied by fibrinoid necrosis (Fig. 11-25). Luminal thrombosis can occur. Later, the acute inflammatory infiltrate is replaced by fibrous (occasionally nodular) thickening of the vessel wall that can extend into the adventitia. Characteristically, all stages of activity (from early to late) coexist in different vessels or even within the same vessel, suggesting ongoing and recurrent insults.
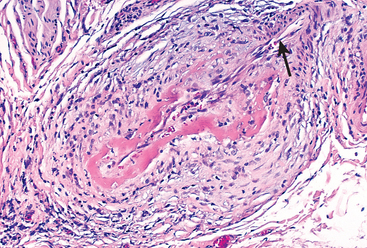
FIGURE 11-25 Polyarteritis nodosa. There is segmental fibrinoid necrosis and thrombotic occlusion of the lumen of this small artery. Note that part of the vessel wall at the upper right (arrow) is uninvolved.
(Courtesy of Sidney Murphree, M.D., Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
Clinical Features.
Though typically a disease of young adults, PAN can also occur in pediatric and geriatric populations. The course may be acute, subacute, or chronic, and is frequently remitting and episodic, with long symptom-free intervals. Because the vascular involvement is widely scattered, the clinical signs and symptoms of PAN may be varied, puzzling, and not always referable to a vascular source. The most common manifestations are malaise, fever, and weight loss; hypertension, usually developing rapidly due to renal involvement; abdominal pain and melena (bloody stool) due to vascular lesions in the gastrointestinal tract; diffuse muscular aches and pains; and peripheral neuritis. Renal arterial involvement is often prominent and a major cause of death. Untreated, the disease is fatal in most cases, either during an acute fulminant attack or following a protracted course. However, therapy with corticosteroids and cyclophosphamide results in remissions or cures in 90% of cases.
KAWASAKI DISEASE
The leading cause of acquired heart disease in children, Kawasaki disease, is an acute febrile, usually self-limited illness of infancy and childhood (80% are younger than 4 years) associated with an arteritis affecting large to medium-sized, and even small, vessels. Its clinical significance stems primarily from a predilection for coronary artery involvement; such coronary arteritis can cause aneurysms that rupture or thrombose, resulting in acute myocardial infarctions. Originally described in Japan, the disease is now increasingly reported in the United States and other countries. The etiology is uncertain, but the vasculitis is thought to result from a delayed-type hypersensitivity reaction of T cells to an as yet uncharacterized antigen. This leads to cytokine production and macrophage activation, and is accompanied by polyclonal B-cell activation. This results in formation of autoantibodies to endothelial cells and smooth muscle cells, which precipitate the acute vasculitis. It is currently speculated that a variety of infectious agents (most likely viral) can trigger the disease in genetically susceptible persons.69,70
Morphology. As with polyarteritis nodosa, lesions exhibit pronounced inflammation affecting the entire thickness of the vessel wall; however, fibrinoid necrosis is usually less prominent. Although the acute vasculitis subsides spontaneously or in response to treatment, aneurysm formation with thrombosis can supervene. As with other causes of arteritis, healed lesions may have obstructive intimal thickening. Pathologic changes outside the cardiovascular system are rarely significant.
Clinical Features.
Kawasaki disease is also known as mucocutaneous lymph node syndrome, because it presents with conjunctival and oral erythema and erosion, edema of the hands and feet, erythema of the palms and soles, a desquamative rash, and cervical lymph node enlargement. Approximately 20% of untreated patients develop cardiovascular sequelae, ranging from asymptomatic coronary arteritis, to coronary artery ectasia and aneurysm formation, to giant coronary artery aneurysms (7–8 mm) with rupture or thrombosis, myocardial infarction, and sudden death. With intravenous immunoglobulin therapy and aspirin, the rate of coronary artery disease is reduced to about 4%.69,70
MICROSCOPIC POLYANGIITIS
This is a necrotizing vasculitis that generally affects capillaries, as well as arterioles and venules of a size smaller than those involved in polyarteritis nodosa; rarely, larger arteries may be involved. It is also called hypersensitivity vasculitis or leukocytoclastic vasculitis. Unlike polyarteritis nodosa, all lesions of microscopic polyangiitis tend to be of the same age in any given patient. The skin, mucous membranes, lungs, brain, heart, gastrointestinal tract, kidneys, and muscle can all be involved; necrotizing glomerulonephritis (90% of patients) and pulmonary capillaritis are particularly common. Disseminated vascular lesions of hypersensitivity angiitis can also occur as a presentation of other disorders (e.g., Henoch-Schönlein purpura, essential mixed cryoglobulinemia, and vasculitis associated with connective tissue disorders).66,75
Pathogenesis.
In some cases, an antibody response to antigens such as drugs (e.g., penicillin), microorganisms (e.g., streptococci), heterologous proteins, or tumor proteins has been implicated; this can either result in immune complex deposition or may trigger secondary immune responses (e.g., the development of p-ANCAs) that are ultimately pathogenic. However, most lesions are pauci-immune (devoid of immune complexes), and increasingly, MPO-ANCAs are causally im-plicated.68 Recruitment and activation of neutrophils within a particular vascular bed may be responsible for the disease manifestations.
Morphology. Microscopic polyangiitis is characterized by segmental fibrinoid necrosis of the media with focal transmural necrotizing lesions; granulomatous inflammation is absent. These lesions morphologically resemble polyarteritis nodosa but typically spare medium-sized and larger arteries; consequently, macroscopic infarcts are uncommon. In some areas (typically post-capillary venules), only infiltrating and fragmenting neutrophils are seen, giving rise to the term leukocytoclastic vasculitis (Fig. 11-26A). Although immunoglobulins and complement components can be demonstrated in early skin lesions, little or no immunoglobulin can be seen in most lesions (so-called pauci-immune injury).
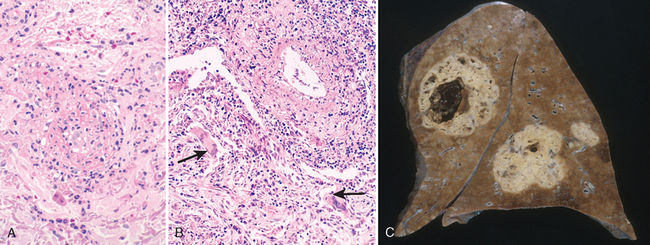
FIGURE 11-26 Representative forms of ANCA-associated small-vessel vasculitis. A, Leukocytoclastic vasculitis (microscopic polyangiitis) with fragmentation of neutrophils in and around blood vessel walls. B and C, Wegener granulomatosis. B, Vasculitis of a small artery with adjacent granulomatous inflammation including epithelioid cells and giant cells (arrows). C, Gross photo from the lung of a patient with fatal Wegener granulomatosis, demonstrating large nodular centrally cavitating lesions.
(A, Courtesy of Scott Granter, M.D., Brigham and Women’s Hospital, Boston, MA; C, courtesy of Sidney Murphree, M.D., Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
Clinical Features.
Depending on the vascular bed involved, major clinical features include hemoptysis; hematuria, and proteinuria; bowel pain or bleeding; muscle pain or weakness; and palpable cutaneous purpura. With the exception of those who develop widespread renal or brain involvement, cyclophosphamide and steroid immunosuppression induces remission and markedly improves long-term survival.76
CHURG-STRAUSS SYNDROME
Churg-Strauss syndrome (also called allergic granulomatosis and angiitis) is a relatively rare (roughly one in a million people) small-vessel necrotizing vasculitis classically associated with asthma, allergic rhinitis, lung infiltrates, peripheral hypereosinophilia, and extravascular necrotizing granulomas. Vascular lesions can be histologically similar to polyarteritis nodosa or microscopic polyangiitis but are also characteristically accompanied by granulomas and eosinophils.77 ANCAs (mostly MPO-ANCAs) are present in less than half the cases and raise the possibility that there are distinct subsets of patients with the syndrome. Nevertheless, when present, the ANCAs are probably responsible for the vascular manifestations of the disease. Cutaneous involvement (palpable purpura), gastrointestinal tract bleeding, and renal disease (primarily as focal and segmental glomerulosclerosis) are the major associations. Myocardial infiltrates of eosinophils and cytotoxicity caused by them are implicated in the cardiomyopathy seen in Churg-Strauss syndrome; the heart is involved in 60% of patients, and accounts for almost half of the deaths in the syndrome.77 The etiology remains obscure, but has been suggested to result from hyper-responsiveness to an allergic stimulus; in asthmatics, leukotriene receptor antagonists are reported as a trigger.78
WEGENER GRANULOMATOSIS
Wegener granulomatosis is a necrotizing vasculitis characterized by a triad of
“Limited” forms of Wegener granulomatosis may be restricted to the respiratory tract. Conversely, a widespread form of the disease can affect eyes, skin, and other organs, notably the heart; clinically, this resembles polyarteritis nodosa except that there is also respiratory involvement.
Pathogenesis.
Wegener granulomatosis probably represents a form of T cell–mediated hypersensitivity reaction, possibly to an inhaled infectious or other environmental agent; such a pathogenesis is supported by the presence of granulomas and a dramatic response to immunosuppressive therapy. PR3-ANCAs are present in up to 95% of cases; they are a useful marker of disease activity and may participate in disease pathogenesis. After immunosuppressive treatment, a rising PR3-ANCA titer suggests a relapse; most patients in remission have a negative test or falling titers.79
Morphology. Upper respiratory tract lesions range from inflammatory sinusitis with mucosal granulomas to ulcerative lesions of the nose, palate, or pharynx, rimmed by granulomas with geographic patterns of central necrosis and accompanying vasculitis (Fig. 11-26B). The necrotizing granulomas are surrounded by a zone of fibroblastic proliferation with giant cells and leukocyte infiltrate, reminiscent of mycobacterial or fungal infections. Multiple granulomas can coalesce to produce radiographically visible nodules that can also cavitate; late-stage disease may be marked by extensive necrotizing granulomatous involvement of the parenchyma (Fig. 11-26C); alveolar hemorrhage may be prominent. Lesions may ultimately undergo progressive fibrosis and organization.
A spectrum of renal lesions may be seen (Chapter 20). In early stages, glomeruli exhibit only focal necrosis with thrombosis of isolated glomerular capillary loops (focal and segmental necrotizing glomerulonephritis); there is minimal parietal cell proliferation in Bowman’s capsule. More advanced glomerular lesions are characterized by diffuse necrosis and parietal cell proliferation to form crescents (crescentic glomerulonephritis).
Clinical Features.
Males are affected more often than females, at an average age of about 40 years. Classic features include persistent pneumonitis with bilateral nodular and cavitary infiltrates (95%), chronic sinusitis (90%), mucosal ulcerations of the nasopharynx (75%), and evidence of renal disease (80%). Other features include rashes, muscle pains, articular involvement, mononeuritis or polyneuritis, and fever. Left untreated, the disease is usually rapidly fatal; 80% of patients die within 1 year. Treatment with steroids, cyclophosphamide, and more recently TNF-antagonists, have turned Wegener granulomatosis into a chronic remitting and relapsing disease.80
THROMBOANGIITIS OBLITERANS (BUERGER DISEASE)
Thromboangiitis obliterans (Buerger disease) is a distinctive disease that often leads to vascular insufficiency; it is characterized by segmental, thrombosing, acute and chronic inflammation of medium-sized and small arteries, principally the tibial and radial arteries, with occasional secondary extension into the veins and nerves of the extremities. Buerger disease is a condition that occurs almost exclusively in heavy cigarette smokers, usually before the age of 35.
Pathogenesis.
The strong relationship to cigarette smoking is thought to be due to direct endothelial cell toxicity by some component of tobacco, or an idiosyncratic immune response to the same agents. Most patients have hypersensitivity to intradermally injected tobacco extracts, and their vessels exhibit impaired endothelium-dependent vasodilation when challenged with acetylcholine. Genetic influences are suggested by an increased prevalence in certain ethnic groups (Israeli, Indian subcontinent, Japanese) and an association with certain HLA haplotypes.81
Morphology. Thromboangiitis obliterans is characterized by a sharply segmental acute and chronic vasculitis of medium-sized and small arteries, predominantly of the extremities. Microscopically, there is acute and chronic inflammation, accompanied by luminal thrombosis. Typically, the thrombus contains small microabscesses composed of neutrophils surrounded by granulomatous inflammation (Fig. 11-27); the thrombus may eventually organize and recanalize. The inflammatory process extends into contiguous veins and nerves (rare with other forms ofvasculitis), and in time all three structures become encased in fibrous tissue.
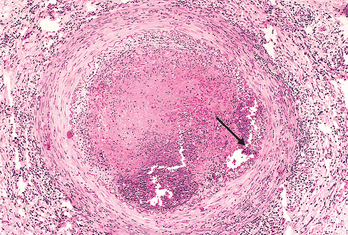
Clinical Features.
The early manifestations are a superficial nodular phlebitis, cold sensitivity of the Raynaud type (see below) in the hands, and pain in the instep of the foot induced by exercise (so-called instep claudication). In contrast to the insufficiency caused by atherosclerosis, in Buerger disease there tends to be severe pain, even at rest, related undoubtedly to the neural involvement. Chronic ulcerations of the toes, feet, or fingers may appear, which can be followed in time by frank gangrene. Abstinence from cigarette smoking in the early stages of the disease often brings dramatic relief from further attacks.
VASCULITIS ASSOCIATED WITH OTHER DISORDERS
Vasculitis resembling hypersensitivity angiitis or classic polyarteritis nodosa may sometimes be associated with other disorders, such as rheumatoid arthritis, SLE, cancer, or systemic illnesses such as mixed cryoglobulinemia, antiphospholipid antibody syndrome, and Henoch-Schönlein purpura. Rheumatoid vasculitis occurs predominantly after longstanding, severe rheumatoid arthritis and usually affects small and medium-sized arteries. It can lead to visceral infarction and sometimes causes a clinically significant aortitis. Identifying the underlying pathology may be therapeutically important. For example, although classic immune complex lupus vasculitis and antiphospholipid antibody syndrome are morphologically similar, anti-inflammatory therapy is required in the former while anticoagulant therapy is indicated in the latter.
INFECTIOUS VASCULITIS
Localized arteritis may be caused by the direct invasion of infectious agents, usually bacteria or fungi, and in particular Aspergillus and Mucor species. Vascular invasion can be part of a localized tissue infection (e.g., bacterial pneumonia or adjacent to abscesses), or—less commonly—it can arise from hematogenous seeding of bacteria during septicemia or embolization from sepsis of infective endocarditis.
Vascular infections can weaken arterial walls and culminate in mycotic aneurysms (see earlier), or can induce thrombosis and infarction. Thus, inflammation-induced thrombosis of meningeal vessels in bacterial meningitis can cause infarction of the underlying brain.
Raynaud Phenomenon
Raynaud phenomenon results from an exaggerated vasoconstriction of digital arteries and arterioles. These vascular changes induce paroxysmal pallor or cyanosis of the digits of the hands or feet; infrequently, the nose, earlobes, or lips can also be involved. Characteristically, the involved digits show red, white, and blue color changes from most proximal to most distal, correlating with proximal vasodilation, central vasoconstriction, and more distal cyanosis (Fig. 11-28). Raynaud phenomenon may be a primary disease entity or be secondary to a variety of conditions.82
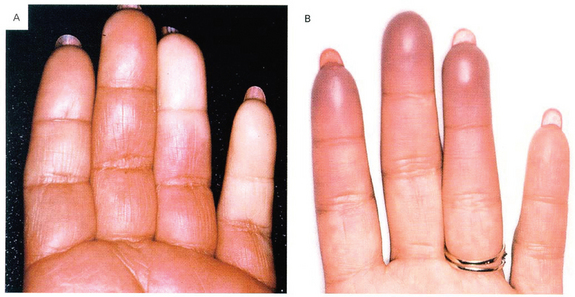
FIGURE 11-28 Raynaud’s phenomenon. A, Sharply demarcated pallor of the distal fingers resulting from the closure of digital arteries. B, Cyanosis of the fingertips.
(Reproduced from Salvarani C, et al.: Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med 347:261, 2002.)
Primary Raynaud phenomenon (previously called Raynaud disease) reflects an exaggeration of central and local vasomotor responses to cold or emotional stresses. It affects 3% to 5% of the general population and shows a predilection for young women. Structural changes in the arterial walls are absent except late in the course, when intimal thickening can appear. The course of Raynaud phenomenon is usually benign, but when long-standing can result in atrophy of the skin, subcutaneous tissues, and muscles. Ulceration and ischemic gangrene are rare.83
In contrast, secondary Raynaud phenomenon refers to vascular insufficiency of the extremities secondary to arterial disease caused by other entities including SLE, scleroderma, Buerger disease, or even atherosclerosis. Since Raynaud phenomenon may be the first manifestation of such conditions, any patient with new symptoms should be evaluated. Of these individuals, some 10% will eventually manifest an underlying disease.
Veins and Lymphatics
Varicose veins and phlebothrombosis/thrombophlebitis together account for at least 90% of clinical venous disease.
VARICOSE VEINS
Varicose veins are abnormally dilated, tortuous veins produced by prolonged, increased intraluminal pressure and loss of vessel wall support. The superficial veins of the upper and lower leg are typically involved (Fig. 11-29). When legs are dependent for prolonged periods, venous pressures in these sites can be markedly elevated (up to 10 times normal) and can lead to venous stasis and pedal edema, even in essentially normal veins (simple orthostatic edema). Some 10% to 20% of adult males and 25% to 33% of adult females develop lower extremity varicose veins; obesity increases risk, and the higher incidence in women is a reflection of the elevated venous pressure in lower legs caused by pregnancy. A familial tendency toward premature varicosities has been noted.
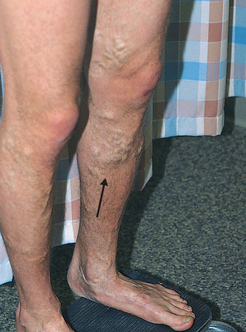
FIGURE 11-29 Varicose veins of the leg (arrow).
(Courtesy of Magruder C. Donaldson, M.D., Brigham and Women’s Hospital, Boston, MA.)
Clinical Features.
Varicose dilation renders the venous valves incompetent and leads to stasis, congestion, edema, pain, and thrombosis. The most disabling sequelae include persistent edema in the extremity and ischemic skin changes, including stasis dermatitis and ulcerations; poor wound healing and superimposed infections can lead to chronic varicose ulcers. Notably, embolism from these superficial veins is very rare. This is in sharp contrast to the relatively frequent thromboembolism that arises from thrombosed deep veins (see below and Chapter 4).
Varicosities that occur in two other sites deserve special mention:
THROMBOPHLEBITIS AND PHLEBOTHROMBOSIS
Deep leg veins are the site of more than 90% of cases of thrombophlebitis and phlebothrombosis; the two terms are largely interchangeable designations for venous thrombosis and inflammation. The periprostatic venous plexus in males and the pelvic venous plexus in females are additional sites, as are the large veins in the skull and the dural sinuses (especially in the setting of infection or inflammation). Peritoneal infections, including peritonitis, appendicitis, salpingitis, and pelvic abscesses, as well as certain thrombophilic conditions associated with platelet hyperactivity (e.g., polycythemia vera, Chapter 13), can lead to portal vein thrombosis. For deep venous thrombosis (DVT) of the legs, prolonged immobilization resulting in decreased blood flow through the veins is the most important predisposing condition. This can occur with extended bed rest or from just sitting during extended travel in an airplane or automobile; the postoperative state is another independent risk factor in DVT formation. Clearly, other mechanical factors that slow venous return also promote the development of DVT; these include congestive heart failure, pregnancy, and obesity.
Systemic hypercoagulability (Chapter 4) often predisposes to thrombophlebitis. In patients with cancer, particularly adenocarcinomas, hypercoagulability occurs as a paraneoplastic syndrome related to elaboration of pro-coagulant factors by the tumor cells (Chapter 7). In this setting, venous thromboses classically appear in one site, disappear, and then reoccur in other veins; this is referred to as migratory thrombophlebitis (Trousseau sign).
Thrombi in the legs tend to produce few, if any, reliable signs or symptoms. Indeed, local manifestations, including distal edema, cyanosis, superficial vein dilation, heat, tenderness, redness, swelling, and pain may be entirely absent, especially in bedridden patients. In some cases pain can be elicited by pressure over affected veins, squeezing the calf muscles, or forced dorsiflexion of the foot (Homan sign); absence of these findings does not exclude a diagnosis of DVT.
Pulmonary embolism is a serious complication of DVT (Chapter 4), resulting from fragmentation or detachment of the whole venous thrombus. In many cases the first manifestation of thrombophlebitis is a pulmonary embolus. Depending on the size and number of emboli, the outcome can range from no symptoms to death.
SUPERIOR AND INFERIOR VENA CAVAL SYNDROMES
The superior vena caval syndrome is usually caused by neoplasms that compress or invade the superior vena cava, such as bronchogenic carcinoma or mediastinal lymphoma. The resulting obstruction produces a characteristic clinical complex that includes marked dilation of the veins of the head, neck, and arms and cyanosis. Pulmonary vessels can also be compressed, causing respiratory distress.
The inferior vena caval syndrome can be caused by neoplasms that compress or invade the inferior vena cava (IVC) or by a thrombus from the hepatic, renal, or lower extremity veins that propagates upward. Certain neoplasms—particularly hepatocellular carcinoma and renal cell carcinoma—show a striking tendency to grow within veins, and these may ultimately occlude the IVC. IVC obstruction induces marked lower extremity edema, distention of the superficial collateral veins of the lower abdomen, and—with renal vein involvement—massive proteinuria.
LYMPHANGITIS AND LYMPHEDEMA
Primary disorders of lymphatic vessels are extremely uncommon; secondary processes are much more frequent and develop in association with inflammation or malignancies.
Lymphangitis is the acute inflammation elicited when bacterial infections spread into lymphatics; the most common agents are group A β-hemolytic streptococci, although any microbe may be associated. The affected lymphatics are dilated and filled with an exudate of neutrophils and monocytes; these infiltrates can extend through the vessel wall into the perilymphatic tissues and, in severe cases, produce cellulitis or focal abscesses. Clinically, lymphangitis is recognized by red, painful subcutaneous streaks (the inflamed lymphatics), and painful enlargement of draining lymph nodes (acute lymphadenitis). If bacteria are not successfully contained within the lymph nodes, subsequent passage into the venous circulation can result in bacteremia or sepsis.
Primary lymphedema can occur due to an isolated congenital defect (simple congenital lymphedema) or as the familial Milroy disease (heredofamilial congenital lymphedema), which causes lymphatic agenesis or hypoplasia. Secondary or obstructive lymphedema stems from blockage of a previously normal lymphatic; such obstruction can result from
Regardless of the cause, lymphedema increases the hydrostatic pressure in the lymphatics distal to the obstruction and causes increased interstitial fluid accumulation. Persistence of this edema leads to increased deposition of interstitial connective tissue, brawny induration or peau d’orange (orange peel) appearance of the overlying skin, and eventually ulcers due to inadequate tissue perfusion. Milky accumulations of lymph in various spaces are designated chylous ascites (abdomen), chylothorax, and chylopericardium; these are caused by rupture of dilated lymphatics, typically obstructed secondary to an infiltrating tumor mass.
Tumors
Tumors of blood vessels and lymphatics range from benign hemangiomas to lesions that are locally aggressive but infrequently metastasize, to relatively rare, highly malignant angiosarcomas (Table 11-5). Primary tumors of large vessels (aorta, pulmonary artery, and vena cava) are extremely rare and are mostly connective tissue sarcomas. Congenital or developmental malformations and reactive vascular proliferations (e.g., bacillary angiomatosis) can also present as tumor-like lesions.
TABLE 11-5 Classification of Vascular Tumors and Tumor-like Conditions
| BENIGN NEOPLASMS, DEVELOPMENTAL AND ACQUIRED CONDITIONS |
| INTERMEDIATE-GRADE NEOPLASMS |
| MALIGNANT NEOPLASMS |
Vascular neoplasms can be endothelium-derived (e.g., hemangioma, lymphangioma, angiosarcoma) or arise from cells that support and/or surround blood vessels (e.g., glomus tumor, hemangiopericytoma). Although a benign, welldifferentiated hemangioma can usually be readily discriminated from an anaplastic high-grade angiosarcoma, the distinction between benign and malignant can occasionally be difficult. Two general rules of thumb are as follows:
Because vascular tumors result from dysregulated vascular proliferation, the possibility of controlling such growth by inhibitors of blood vessel formation (anti-angiogenic factors) is being explored.
BENIGN TUMORS AND TUMOR-LIKE CONDITIONS
Hemangioma
Hemangiomas are very common tumors characterized by increased numbers of normal or abnormal vessels filled with blood (Fig. 11-30); they may be difficult to distinguish from vascular malformations. These lesions constitute 7% of all benign tumors of infancy and childhood; most are present from birth and expand along with the growth of the child. Nevertheless, many of the capillary lesions eventually regress spontaneously. Although some hemangiomas can involve large portions of the body (angiomatosis), most are localized. The majority are superficial lesions, often of the head or neck, but they can occur internally, with nearly one third being found in the liver. Malignant transformation occurs rarely, if ever. There are several histologic and clinical variants:
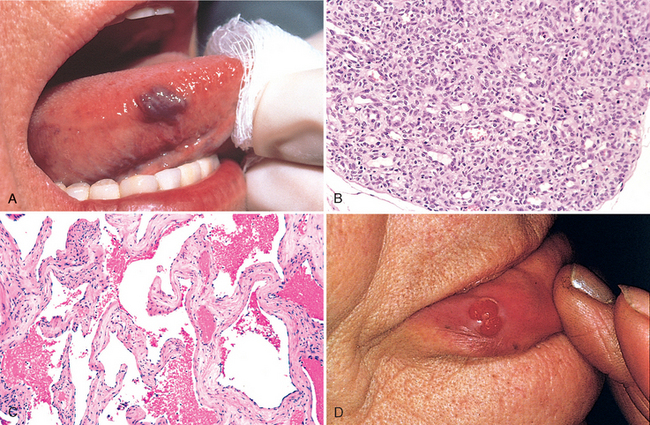
FIGURE 11-30 Hemangiomas. A, Hemangioma of the tongue. B and C, Histologic appearance of (B) juvenile capillary hemangioma and (C) cavernous hemangioma. D, Pyogenic granuloma of the lip.
(A and D, Courtesy of John Sexton, M.D., Beth Israel Hospital, Boston, MA; B, courtesy of Christopher D.M. Fletcher, M.D., Brigham and Women’s Hospital, Boston; C, courtesy of Thomas Rogers, M.D., University of Texas Southwestern Medical School, Dallas, TX.)
Capillary Hemangioma.
The most common variant, capillary hemangiomas, occur in the skin, subcutaneous tissues, and mucous membranes of the oral cavities and lips, as well as in the liver, spleen, and kidneys. The “strawberry type” or juvenile hemangioma of the skin of newborns is extremely common (1 in 200 births) and may be multiple. It grows rapidly in the first few months but then fades at 1 to 3 years of age and completely regresses by age 7 in 75% to 90% of cases.
Morphology. Capillary hemangiomas are bright red to blue and vary from a few millimeters to several centimeters in diameter; hemangiomas can be level with the surface of the skin or slightly elevated, and have an intact overlying epithelium (Fig. 11-30A). Histologically, these are unencapsulated aggregates of closely packed, thin-walled capillaries, usually blood-filled and lined by flattened endothelium; vessels are separated by scant connective tissue stroma (Fig. 11-30B). The lumens may be partially or completely thrombosed and organized. Vessel rupture accounts for hemosiderin pigment in these lesions as well as focal scarring.
Cavernous Hemangioma.
These exhibit large, dilated vascular channels; compared with capillary hemangiomas, cavernous hemangiomas are less well circumscribed and more frequently involve deep structures. Since they may be locally destructive and show no spontaneous tendency to regress, some may require surgery. In most cases, the tumors are of little clinical significance; however, they can be a cosmetic disturbance and are vulnerable to traumatic ulceration and bleeding. Moreover, visceral hemangiomas detected by imaging studies may have to be distinguished from more ominous (e.g., malignant) lesions. Brain hemangiomas are most problematic, because they can cause pressure symptoms or rupture. Cavernous hemangiomas are a component of von Hippel-Lindau disease (Chapter 28), occurring within the cerebellum, brain stem, or retina, along with similar angiomatous lesions or cystic neoplasms in the pancreas and liver; von Hippel-Lindau disease is also associated with renal neoplasms.
Morphology. Cavernous hemangiomas are red-blue, soft, spongy masses 1 to 2 cm in diameter; rare giant forms can affect large subcutaneous areas of the face, extremities, or other body regions. Histologically, the mass is sharply defined but not encapsulated, and composed of large, cavernous blood-filled vascular spaces, separated by a modest connective tissue stroma (Fig. 11-30C). Intravascular thrombosis with associated dystrophic calcification is common.
Pyogenic Granuloma.
This form of capillary hemangioma is a rapidly growing pedunculated red nodule on the skin, or gingival or oral mucosa; it bleeds easily and is often ulcerated (Fig. 11-30D). Roughly a third of the lesions develop after trauma, reaching a size of 1 to 2 cm within a few weeks. The proliferating capillaries are often accompanied by extensive edema and an acute and chronic inflammatory infiltrate, strikingly similar to exuberant granulation tissue. Pregnancy tumor (granuloma gravidarum) is a pyogenic granuloma that occurs infrequently (1% of patients) in the gingiva of pregnant women. These lesions can spontaneously regress (e.g., after pregnancy) or undergo fibrosis; in some cases surgical excision is required. Recurrence is rare.
Lymphangiomas
Lymphangiomas are the benign lymphatic analogues of blood vessel hemangiomas.
Simple (Capillary) Lymphangioma.
These are composed of small lymphatic channels predominantly occurring in the head, neck, and axillary subcutaneous tissues. They are slightly elevated or sometimes pedunculated lesions up to 1 to 2 cm in diameter. Histologically, lymphangiomas exhibit networks of endothelium-lined spaces that can be distinguished from capillary channels only by the absence of erythrocytes.
Cavernous Lymphangioma (Cystic Hygroma).
These lesions are typically found in the neck or axilla of children, and rarely occur in the retroperitoneum; cavernous lymphangiomas of the neck are common in Turner syndrome (Chapter 10). Cavernous lymphangiomas can occasionally be enormous (up to 15 cm in diameter) and may fill the axilla or produce gross deformities about the neck. Tumors are composed of massively dilated lymphatic spaces lined by endothelial cells and separated by intervening connective tissue stroma containing lymphoid aggregates. The tumor margins are not discrete and the lesions are not encapsulated, making definitive resection difficult.
Glomus Tumor (Glomangioma)
Glomus tumors are benign, exquisitely painful tumors arising from modified smooth muscle cells of the glomus body, a specialized arteriovenous structure involved in thermoregulation. Although they can resemble cavernous hemangiomas, glomus tumors constitute a distinct entity by virtue of their constituent cells. They are most commonly found in the distal portion of the digits, especially under the fingernails. Excision is curative.
Morphology. Glomus tumor lesions are round, slightly elevated, red-blue, firm nodules (usually <1 cm in diameter) that initially resemble a minute focus of hemorrhage. Histologically, these are aggregates, nests, and masses of specialized glomus cells intimately associated with branching vascular channels, all within a connective tissue stroma. Individual tumor cells are small, uniform, and round or cuboidal, with scant cytoplasm and ultrastructural features akin to smooth muscle cells.
Vascular Ectasias
Vascular ectasias are common lesions characterized by local dilation of preexisting vessels; they are not true neoplasms. Telangiectasia is a term used for a congenital anomaly or acquired exaggeration of preformed vessels—usually in the skin or mucous membranes—composed of capillaries, venules, and arterioles that creates a discrete red lesion.
Nevus Flammeus.
This lesion is the ordinary “birthmark” and is the most common form of ectasia; it is characteristically a flat lesion on the head or neck, ranging in color from light pink to deep purple. Histologically, there is only vascular dilation; most ultimately regress.
The so-called port wine stain is a special form of nevus flammeus; these lesions tend to grow with a child, thicken the skin surface, and demonstrate no tendency to fade. Such lesions in a trigeminal nerve distribution are occasionally associated with the Sturge-Weber syndrome (also called encephalotrigeminal angiomatosis). Sturge-Weber syndrome is a rare congenital disorder associated with venous angiomatous masses in the cortical leptomeninges and ipsilateral facial port wine nevi; mental retardation, seizures, hemiplegia, and skull radioopacities also occur. Thus, a large facial vascular malformation in a child with mental deficiency may indicate the presence of more extensive vascular malformations.84
Spider Telangiectasia.
This non-neoplastic vascular lesion grossly resembles a spider; there is a radial, often pulsatile array of dilated subcutaneous arteries or arterioles (resembling legs) about a central core (resembling a body) that blanches when pressure is applied to its center. It is commonly seen on the face, neck, or upper chest and is most frequently associated with hyper-estrogenic states such as pregnancy or patients with cirrhosis; how elevated estrogen levels contribute to “spider” formation is not known.
Hereditary Hemorrhagic Telangiectasia (OslerWeber-Rendu Disease).
In this autosomal dominant disorder the telangiectasias are malformations composed of dilated capillaries and veins. Present from birth, they are widely distributed over the skin and oral mucous membranes, as well as in the respiratory, gastrointestinal, and urinary tracts. Occasionally, these lesions rupture, causing serious epistaxis (nosebleeds), GI bleeding, or hematuria.
Bacillary Angiomatosis
Bacillary angiomatosis is a vascular proliferation resulting from an opportunistic infection in immunocompromised individuals; lesions can involve skin, bone, brain, and other organs. First described in patients with acquired immunodeficiency syndrome (AIDS), bacillary angiomatosis is caused by infection with gram-negative bacilli of the Bartonella family. Two species are implicated: Bartonella henselae, the organism responsible for cat-scratch disease (the domestic cat is the principal reservoir), and B. quintana, the cause of “trench fever” in World War I (the organism is transmitted by human body lice).85
Morphology. Skin lesions are red papules and nodules, or rounded subcutaneous masses; histologically, there is capillary proliferation with prominent epithelioid endothelial cells exhibiting nuclear atypia and mitoses (Fig. 11-31). Lesions contain neutrophils, nuclear dust, and the causal bacteria.
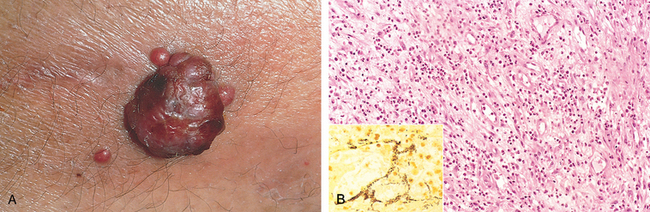
FIGURE 11-31 Bacillary angiomatosis. A, Photograph of a cutaneous lesion. B, Histologic appearance with acute neutrophilic inflammation and vascular (capillary) proliferation. (Inset) Demonstration by modified silver (Warthin-Starry) stain of clusters of tangled bacilli (black).
(A, Courtesy of Richard Johnson, M.D., Beth Israel Deaconess Medical Center, Boston, MA; B and inset, courtesy of Scott Granter, M.D., Brigham and Women’s Hospital, Boston.)
Though difficult to cultivate in the laboratory, the bacteria can be unequivocally demonstrated using molecular methods such as polymerase chain reaction with species-specific primers. The vascular proliferation results from induction of host HIF-1α by the bacteria; HIF-1α in turn drives VEGF production.86 The infections (and lesions) are cleared by macrolide antibiotics (including erythromycin).
INTERMEDIATE-GRADE (BORDERLINE) TUMORS
Kaposi Sarcoma
Though rare in other populations, Kaposi sarcoma (KS) is common in patients with AIDS; indeed, its presence is used as a criterion for diagnosing AIDS (Chapter 6). Four forms of the disease are recognized (based primarily on population demographics and risks), although all share the same underlying viral pathogenesis87 (see below):
Pathogenesis.
In 1994, a previously unrecognized herpesvirus—human herpesvirus-8 (HHV-8) or KS-associated herpesvirus (KSHV) was identified in a cutaneous KS lesion in an AIDS patient (Chapter 6). Indeed, regardless of the clinical subtype (described above), 95% of KS lesions have subsequently been shown to be KSHV-infected.88 Like Epstein-Barr virus, KSHV is a member of the γ-herpesvirus subfamily; it is transmitted sexually and by poorly understood nonsexual routes—perhaps including saliva.
KSHV is accepted as a necessary requirement for KS development, but tumor progression also requires a cofactor; HIV clearly can provide this, but the identity of the cofactor in non-HIV-associated KS is controversial. KSHV induces a lytic as well as a latent infection in endothelial cells, both of which are probably important in KS pathogenesis. Cytokines derived from HIV-infected T cells, or inflammatory cells recruited in response to the lytic infection, create a local proliferative milieu; a virally encoded G protein also induces local VEGF production. In latently infected cells, KSHV proteins disrupt normal cellular proliferation controls and prevent apoptosis by viral production of p53 inhibitors and a viral homologue of cyclin D. Thus, latently infected cells have a growth advantage; the local environment also favors cellular proliferation. The increasing recognition of the various viral gene products has nevertheless opened a number of new avenues for therapeutic interventions against affected intracellular kinase pathways and downstream targets. In its early stages, only a few cells are infected; with time virtually all spindle cells of late-stage lesions carry KSHV; these spindle cells express both endothelial cell and smooth muscle cell markers.
Morphology. In the indolent, classic disease of older men (and sometimes in other variants), three stages are recognized: patch, plaque, and nodule.
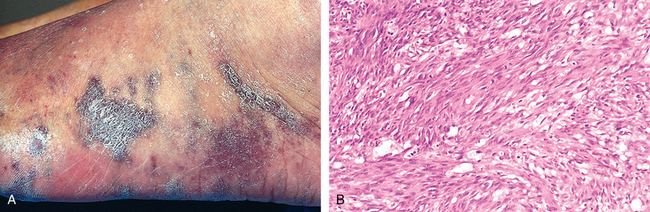
FIGURE 11-32 Kaposi sarcoma. A, Gross photograph, illustrating coalescent red-purple macules and plaques of the skin. B, Histologic appearance of nodular form, demonstrating sheets of plump, proliferating spindle cells.
(B, Courtesy of Christopher D.M. Fletcher, M.D., Brigham and Women’s Hospital, Boston, MA.)
Clinical Features.
The course of KS varies widely and is significantly affected by the clinical setting. Most primary KSHV infections are asymptomatic. Classic KS is—at least initially—largely restricted to the surface of the body, and surgical resection is usually adequate for an excellent prognosis. Radiation can be used for multiple lesions in a restricted area, and chemotherapy yields satisfactory results for more disseminated disease. Lymphadenopathic KS can also be treated with chemotherapy or radiation therapy with good results. In immunosuppression-associated KS, withdrawal of immunosuppression (perhaps with adjunct chemotherapy or radiation therapy) is often effective. For AIDS-associated KS, antiretroviral therapy for HIV is usually helpful, with or without therapy targeted to the KS lesions. IFN-α and angiogenesis inhibitors are variably effective, while newer strategies aimed at specific intracellular kinase pathways or the downstream mammalian target of rapamycin are showing promise.89,90
Hemangioendothelioma
Hemangioendothelioma denotes a wide spectrum of vascular neoplasms with clinical behaviors intermediate between benign, well-differentiated hemangiomas and highly malignant angiosarcomas, described below.
Epithelioid hemangioendothelioma is an example; it is a vascular tumor of adults occurring around medium-sized and large veins. The tumor cells are plump and often cuboidal (resembling epithelial cells); well-defined vascular channels are inconspicuous. Clinical behavior is variable; most are cured by excision, but up to 40% recur, 20% to 30% eventually metastasize, and perhaps 15% of patients die of the tumors.
MALIGNANT TUMORS
Angiosarcoma
Angiosarcomas are malignant endothelial neoplasms (Fig. 11-33) with histology varying from well-differentiated tumors that resemble hemangiomas (hemangiosarcoma) to anaplastic lesions difficult to distinguish from carcinomas or melanomas. Older adults are more commonly affected, with equal gender predilections; they occur at any site but most often involve skin, soft tissue, breast, and liver.

FIGURE 11-33 Angiosarcoma. A, Gross photograph of angiosarcoma of the heart (right ventricle). B, Photomicrograph of moderately well-differentiated angiosarcoma with dense clumps of irregular, moderate anaplastic cells and distinct vascular lumens. C, Immunohistochemical staining for the endothelial cell marker CD31, demonstrating the endothelial nature of the tumor cells.
Hepatic angiosarcomas are associated with carcinogen exposures, including arsenic (arsenical pesticides), Thorotrast (a radioactive contrast agent formerly used for radiologic imaging), and polyvinyl chloride (a widely used plastic). All of these agents have long latent periods between initial exposure and eventual tumor development. The increased frequency of angiosarcomas among polyvinyl chloride workers is one of the well-documented instances of human chemical carcinogenesis.
Angiosarcomas can also arise in the setting of lymphedema, classically in the ipsilateral upper extremity several years after radical mastectomy (i.e., with lymph node resection) for breast cancer; the tumor presumably arises from lymphatic vessels (lymphangiosarcoma). Angiosarcomas have also been induced by radiation and are rarely associated with foreign material introduced into the body either iatrogenically or accidentally.
Morphology. Cutaneous angiosarcomas can begin as deceptively small, sharply demarcated, asymptomatic, often multiple red nodules; most eventually become large, fleshy masses of red-tan to gray-white tissue (Fig. 11-33A). The margins blend imperceptibly with surrounding structures. Central areas of necrosis and hemorrhage are frequent.
Microscopically, all degrees of differentiation can be seen, from plump, anaplastic but recognizable endothelial cells producing vascular channels (Fig. 11-33B) to wildly undifferentiated tumors with a solid spindle cell appearance and without definite blood vessels. The endothelial cell origin of these tumors can be demonstrated by staining for CD31 or von Willebrand factor (Fig. 11-33C).
Clinically, angiosarcomas are locally invasive and can metastasize readily. Angiosarcomas are aggressive tumors with current 5-year survival rates approaching 30%.
Hemangiopericytoma
Hemangiopericytomas are rare tumors derived from pericytes—myofibroblast-like cells that are normally arranged around capillaries and venules. Hemangiopericytomas can occur as slowly enlarging, painless masses at any anatomic site, but are most common on the lower extremities (especially the thigh) and in the retroperitoneum. They consist of numerous branching capillary channels and gaping sinusoidal spaces enclosed within nests of spindle-shaped to round cells. Special stains confirm that these cells are outside the endothelial cell basement membrane and are therefore pericytes. The tumors may recur after excision, and roughly half will metastasize, usually hematogenously to lungs, bone, or liver.
Pathology of Vascular Interventions
The morphologic changes that occur in vessels after therapeutic intervention—balloon angioplasty, stenting, or bypass surgery—typically recapitulate many of the changes that occur in the setting of any vascular insult. Local endothelial cell trauma (e.g., due to a stent), vascular thrombosis (after angioplasty), or abnormal mechanical forces (e.g., a saphenous vein inserted into the arterial circulation as a coronary artery bypass graft), all induce similar responses characteristic of vessel wall healing. Thus, in the same way that various injuries can induce an intimal hyperplastic response that we recognize as atherosclerosis (see above), the trauma caused by vascular interventions tends to induce a concentric intimal thickening composed of recruited smooth muscle cells and their associated matrix deposition.
ANGIOPLASTY AND ENDOVASCULAR STENTS
Balloon angioplasty (dilation of a stenotic artery by inserting an intravascular catheter), with or without endovascular stenting, is used extensively to restore flow at sites of focal vascular stenosis—especially in the coronary circulation (percutaneous transluminal coronary angioplasty). The morphologic outcomes of angioplasty, as well as the now more commonly used endovascular stents, are demonstrated in Figure 11-34.
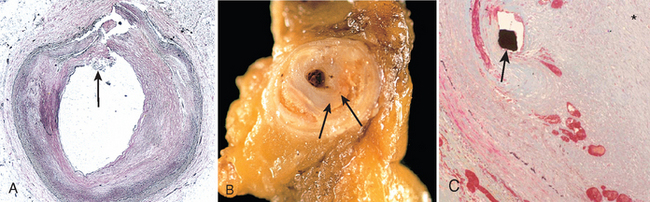
FIGURE 11-34 Balloon angioplasty and endovascular stents. A, Coronary artery after balloon angioplasty, showing the dissection encompassing the intima and media (arrow). B, Gross photograph of restenosis following balloon angioplasty, demonstrating residual atherosclerotic plaque (left arrow) and a new, glistening proliferative lesion (right arrow). C, Coronary arterial stent implanted long term, demonstrating thickened neointima separating the stent wires (black spot shown by arrow) from the lumen (asterisk).
(C, Reproduced from Schoen FJ, Edwards WD: Pathology of cardiovascular interventions, including endovascular therapies, revascularization, vascular replacement, cardiac assist/replacement, arrhythmia control, and repaired congenital heart disease. In Silver MD et al. (eds): Cardiovascular Pathology, 3rd ed. Philadelphia, Churchill Livingstone, 2001.)
Simple balloon dilation (without stenting) of an atherosclerotic vessel induces medial stretching and causes plaque fracture, often with accompanying localized hemorrhagic dissection of the adjacent arterial wall (Fig. 11-34A); in this manner vascular flow is restored, albeit with the attendant risks of more extensive dissection and luminal thrombosis (to prevent the latter, anticoagulation is required for a period of time after the procedure). Most patients improve symptomatically, at least in the short term. Abrupt reclosure may occur as a result of compression of the lumen by an extensive circumferential or longitudinal dissection or by thrombosis. The long-term success of angioplasty is primarily limited by the development of proliferative restenosis, due to intimal thickening; this occurs in approximately 30% to 50% of patients within the first 4 to 6 months after the procedure (Fig. 11-34B). The conditions causing restenosis are undoubtedly the same as those that occur in response to vascular damage of any type (e.g., atherosclerosis); in this case the injury is mechanical and is also compounded by the resolution of any associated thrombosis. The end result is an occlusive, progressive fibrous lesion that contains abundant smooth muscle cells and extracellular matrix.
Coronary stents are expandable tubes of metallic mesh that are inserted to preserve lumenal patency during angioplasty; they are now used in over 90% of angioplasty procedures. Stents provide a larger and more regular lumen, “tack down” the intimal flaps and dissections that occur during angioplasty, and mechanically limit vascular spasm. Nevertheless, by also causing focal plaque erosion and/or endothelial cell disruption, stents can cause acute thrombosis, and a successful procedure also requires at least transient anticoagulation with potent anti-thrombotic agents (platelet antagonists).91 A late complication of bare metal stents involves proliferative intimal thickening leading to proliferative restenosis much like that seen with percutaneous transluminal coronary angioplasty alone (Fig. 11-34C). The newest generation of stents are coated with anti-proliferative drugs (e.g., paclitaxel or rapamycin) that limit smooth muscle cell hyperplasia; this results in markedly diminished intimal thickening, although there is evidence that the anti-proliferative agents also slow the process of re-endothelialization of the coated stents and prolong the required period of anticoagulation therapy.92
VASCULAR REPLACEMENT
Synthetic or autologous vascular grafts are increasingly used to replace damaged vessels or bypass diseased arteries. With synthetic grafts (usually expanded polytetrafluoroethylene, a spongy Teflon fabric), large-bore (12- to 18-mm) conduits function well in high-flow locations such as the aorta; unfortunately, small-diameter artificial grafts (≤8 mm in diameter) generally fail because of early thrombosis or late intimal hyperplasia, primarily at the junction of the graft with the native vasculature (Fig. 11-35).
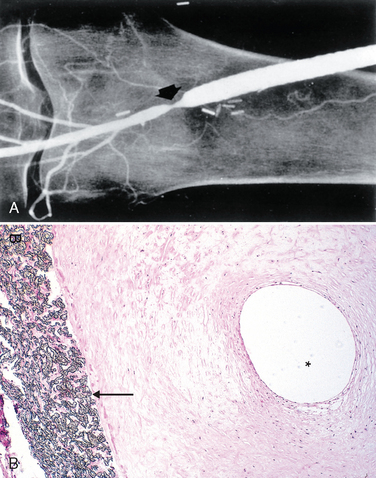
FIGURE 11-35 Anastomotic hyperplasia at the distal anastomosis of synthetic femoropopliteal graft. A, Angiogram demonstrating constriction (arrow). B, Photomicrograph demonstrating Gore-Tex graft (arrow) with prominent intimal proliferation and very small residual lumen (asterisk).
(A, Courtesy of Anthony D. Whittemore, M.D., Brigham and Women’s Hospital, Boston, MA.)
Consequently, where small-bore vessels are needed (e.g., for coronary artery bypass surgery performed in more than 400,000 U.S. patients per year), grafts are most commonly composed of either reversed autologous saphenous vein (taken from the patient’s own leg) or left internal mammary artery (because of its proximity to the heart). The long-term patency of saphenous vein grafts is only 50% at 10 years; grafts occlude because of thrombosis (typically early), intimal thickening (months to years postoperatively), and vein graft atherosclerosis—sometimes with superimposed plaque rupture, thrombi, or aneurysms (usually >2–3 years). In contrast, greater than 90% of internal mammary artery grafts are patent at 10 years.93
1 Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932.
2 Carmeliet P. Manipulating angiogenesis in medicine. J Intern Med. 2004;255:538.
3 Semenza G. Vasculogenesis, angiogenesis, and arteriogenesis: mechanisms of blood vessel formation and remodeling. J Cell Biochem. 2007;102:840.
4 Heil M, et al. Arteriogenesis versus angiogenesis: similarities and differences. J Cell Mol Med. 2006;10:45.
5 Angelini P, et al. Coronary anomalies: incidence, pathophysiology, and clinical relevance. Circulation. 2002;105:2449.
6 Angelini P. Coronary artery anomalies: an entity in search of an identity. Circulation. 2007;115:1296.
7 Friedlander R. Clinical practice. Arteriovenous malformations of the brain. N Engl J Med. 2007;356:2704.
8 Slovut D, Olin J. Fibromuscular dysplasia. N Engl J Med. 2004;350:1862.
9 Shin D, et al. Expression of ephrinB2 identifies a stable genetic difference between arterial and venous vascular smooth muscle as well as endothelial cells, and marks subsets of microvessels at sites of adult neovascularization. Dev Biol. 2001;230:139.
10 Garcia-Cardena G, Gimbrone M. Biomechanical modulation of endothelial phenotype: implications for health and disease. Handb Exp Pharmacol. 2006;176:79.
11 Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol Mech Dis. 2009;4:71.
12 Stevens T, et al. NHLBI workshop report: endothelial cell phenotypes in heart, lung, and blood diseases. Am J Physiol Cell Physiol. 2001;281:C1422.
13 Berk B. Vascular smooth muscle growth: autocrine growth mechanisms. Physiol Rev. 2001;81:999.
14 Sata M. Role of circulating vascular progenitors in angiogenesis, vascular healing, and pulmonary hypertension: lessons from animal models. Arterioscler Thromb Vasc Biol. 2006;26:1008.
15 Shimizu K, Mitchell R. Stem cell origins of intimal cells in graft arterial disease. Curr Athero Rep. 2003;5:230.
16 Hillebrands J, et al. Origin of vascular smooth muscle cells and the role of circulating stem cells in transplant arteriosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:380.
17 Caplice N, Doyle B. Vascular progenitor cells: origin and mechanisms of mobilization, differentiation, integration, and vasculogenesis. Stem Cells Dev. 2005;14:122.
18 Korshunov V, et al. Vascular remodeling: hemodynamic and biochemical mechanisms underlying Glagov’s phenomenon. Arterioscler Thromb Vasc Biol. 2007;27:1722.
19 Kannel W, et al. Concept and usefulness of cardiovascular risk profiles. Am Heart J. 2004;148:16.
20 Kaplan N. Systemic hypertension: mechanisms and diagnosis. In: Zipes D, et al, editors. Braunwald’s Heart Disease. 7th ed. Philadelphia: Elsevier Saunders; 2005:959.
21 Lifton R, et al. Molecular mechanisms of human hypertension. Cell. 2001;104:545.
22 Puddu P, et al. The genetic basis of essential hypertension. Acta Cardiol. 2007;62:281.
23 Messerli F, et al. Essential hypertension. Lancet. 2007;370:591.
24 Scarpelli P, et al. Continuing follow-up of malignant hypertension. J Nephrol. 2002;15:431.
25 Aggarwal M, Khan I. Hypertensive crisis: hypertensive emergencies and urgencies. Cardiol Clin. 2006;24:135.
26 Rodriguez-Iturbe B, et al. Pathophysiological mechanisms of salt-dependent hypertension. Am J Kidney Dis. 2007;50:655.
27 Chambless L, et al. Coronary heart disease risk prediction in the Atherosclerosis Risk in Communities (ARIC) study. J Clin Epidemiol. 2003;56:880.
28 Ridker P, Libby P. Risk factors for atherothrombotic disease. In: Zipes D, et al, editors. Braunwald’s Heart Disease. 7th ed. Philadelphia: Elsevier Saunders; 2005:939.
29 Sweitzer N, Douglas P. Cardiovascular disease in women. In: Zipes D, et al, editors. Braunwald’s Heart Disease. 7th ed. Philadelphia: Elsevier Saunders; 2005:1951.
30 Miller D, et al. Atherosclerosis: the path from genomics to therapeutics. J Am Coll Cardiol. 2007;49:1589.
31 Glassberg H, Rader D. Management of lipids in the prevention of cardiovascular events. Annu Rev Med. 2008;59:79-94.
32 Ridker P, et al. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557.
33 Ridker P. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007;49:2129.
34 Ridker P, et al. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. JAMA. 2007;297:611.
35 Guthikonda S, Haynes W. Homocysteine: role and implications in atherosclerosis. Curr Atheroscler Rep. 2006;8:100.
36 Meerarani P, et al. Metabolic syndrome and diabetic atherothrombosis: implications in vascular complications. Curr Mol Med. 2006;6:501.
37 Anuurad E, et al. Lipoprotein(a): a unique risk factor for cardiovascular disease. Clin Lab Med. 2006;26:751.
38 Croce K, Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr Opin Hematol. 2007;14:55.
39 Meadows T, Bhatt D. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res. 2007;100:1261.
40 Libby P. The vascular biology of atherosclerosis. In: Zipes D, et al, editors. Braunwald’s Heart Disease. 7th ed. Philadelphia: Elsevier Saunders; 2005:921.
41 Hansson G, et al. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297.
42 Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115.
43 Chatzizisis Y, et al. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol. 2007;49:2379.
44 Gau G, Wright R. Pathophysiology, diagnosis, and management of dyslipidemia. Curr Probl Cardiol. 2006;31:445.
45 Schiffrin E, et al. Chronic kidney disease: effects on the cardiovascular system. Circulation. 2007;116:85.
46 Hansson G. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685.
47 Mussa F, et al. Chlamydia pneumoniae and vascular disease: an update. J Vasc Surg. 2006;43:1301.
48 Libby P. Atherosclerosis: disease biology affecting the coronary vasculature. Am J Cardiol. 2006;98:3Q.
49 Davies M. A macro and micro view of coronary vascular insult in ischemic heart disease. Circulation. 1990;82(3 Suppl):II38.
50 Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481.
51 Libby P, Sasiela W. Plaque stabilization: can we turn theory into evidence? Am J Cardiol. 2006;98(11A):26P.
52 Curtis A, Fitzgerald G. Central and peripheral clocks in cardiovascular and metabolic function. Ann Med. 2006;38:552.
53 Brotman D, et al. The cardiovascular toll of stress. Lancet. 2007;370:1089.
54 Ramirez F, Dietz H. Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr Opin Genet Dev. 2007;17:252.
55 Loeys B, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788.
56 Hobeika M, et al. Matrix metalloproteinases in peripheral vascular disease. J Vasc Surg. 2007;45:849.
57 Shimizu K, et al. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006;26:987.
58 Homme J, et al. Surgical pathology of the ascending aorta: a clinicopathologic study of 513 cases. Am J Surg Pathol. 2006;30:1159.
59 Pagnoux C, et al. Vasculitides secondary to infections. Clin Exp Rheumatol. 2006;24(2 Suppl 41):S71.
60 Fillinger M. Who should we operate on and how do we decide: predicting rupture and survival in patients with aortic aneurysm. Semin Vasc Surg. 2007;20:121.
61 Eliason J, Clouse W. Current management of infrarenal abdominal aortic aneurysms. Surg Clin North Am. 2007;87:1017.
62 Kamalakannan D, et al. Acute aortic dissection. Crit Care Clin. 2007;23:779.
63 Iglesias-Gamarra A, et al. Small-vessel vasculitis. Curr Rheumatol Rep. 2007;9:304.
64 Jennette J, Falk R. Nosology of primary vasculitis. Curr Opin Rheumatol. 2007;19:10.
65 Jennette J, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187.
66 Kallenberg C. Antineutrophil cytoplasmic autoantibody–associated small-vessel vasculitis. Curr Opin Rheumatol. 2007;19:17.
67 Heeringa P, et al. Anti-neutrophil cytoplasmic autoantibodies and leukocyte-endothelial interactions: a sticky connection? Trends Immunol. 2005;26:561.
68 Jennette J, et al. Pathogenesis of vascular inflammation by anti-neutrophil cytoplasmic antibodies. J Am Soc Nephrol. 2006;17:1235.
69 Falcini F. Kawasaki disease. Curr Opin Rheumatol. 2006;18:33.
70 Gedalia A. Kawasaki disease: 40 years after the original report. Curr Rheumatol Rep. 2007;9:336.
71 Seko Y. Giant cell and Takayasu arteritis. Curr Opin Rheumatol. 2007;19:39.
72 Arnaud L, et al. Takayasu’s arteritis: an update on physiopathology. Eur J Intern Med. 2006;17:241.
73 Colmegna I, Maldonado-Cocco J. Polyarteritis nodosa revisited. Curr Rheumatol Rep. 2005;7:288.
74 Segelmark M, Selga D. The challenge of managing patients with polyarteritis nodosa. Curr Opin Rheumatol. 2007;19:33.
75 Kallenberg C, et al. Mechanisms of disease: pathogenesis and treatment of ANCA-associated vasculitides. Nat Clin Pract Rheumatol. 2006;2:661.
76 Langford C. Small-vessel vasculitis: therapeutic management. Curr Rheumatol Rep. 2007;9:328.
77 Pagnoux C, et al. Churg-Strauss syndrome. Curr Opin Rheumatol. 2007;19:25.
78 Cuchacovich R, et al. Churg-Strauss syndrome associated with leukotriene receptor antagonists (LTRA). Clin Rheumatol. 2007;26:1769.
79 Sarraf P, Sneller M. Pathogenesis of Wegener’s granulomatosis: current concepts. Expert Rev Mol Med. 2005;7:1.
80 Erickson V, Hwang P. Wegener’s granulomatosis: current trends in diagnosis and management. Curr Opin Otolaryngol Head Neck Surg. 2007;15:170.
81 Olin J, Shih A. Thromboangiitis obliterans (Buerger’s disease). Curr Opin Rheumatol. 2006;18:18.
82 Cooke J, Marshall J. Mechanisms of Raynaud’s disease. Vasc Med. 2005;10:293.
83 Boin F, Wigley F. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol. 2005;17:752.
84 Garzon M, et al. Vascular malformations. Part II: associated syndromes. J Am Acad Dermatol. 2007;56:541.
85 Chian C, et al. Skin manifestations of Bartonella infections. Int J Dermatol. 2002;41:461.
86 Kempf V, et al. Activation of hypoxia-inducible factor-1 in bacillary angiomatosis: evidence for a role of hypoxia-inducible factor-1 in bacterial infections. Circulation. 2005;111:1054.
87 Geraminejad P, et al. Kaposi’s sarcoma and other manifestations of human herpesvirus 8. J Am Acad Dermatol. 2002;47:641.
88 Ganem D. KSHV infection and pathogenesis of Kaposi’s sarcoma. Annu Rev Pathol. 2006;1:273.
89 Dittmer D, Krown S. Targeted therapy for Kaposi’s sarcoma and Kaposi’s sarcoma-associated herpesvirus. Curr Opin Oncol. 2007;19:452.
90 Lambert P, et al. Targeting the PI3K and MAPK pathways to treat Kaposi’s-sarcoma-associated herpes virus infection and pathogenesis. Expert Opin Ther Targets. 2007;11:589.
91 Jaffe R, Strauss B. Late and very late thrombosis of drug-eluting stents: evolving concepts and perspectives. J Am Coll Cardiol. 2007;50:119.
92 VanBelle E, et al. Drug-eluting stents: trading restenosis for thrombosis? J Thromb Haemost. 2007;5(Suppl 1):238.
93 Wallitt E, et al. Therapeutics of vein graft intimal hyperplasia: 100 years on. Ann Thorac Surg. 2007;84:317.