Chapter 3 Tissue Renewal, Regeneration, and Repair
Injury to cells and tissues sets in motion a series of events that contain the damage and initiate the healing process. This process can be broadly separated into regeneration and repair (Fig. 3-1). Regeneration results in the complete restitution of lost or damaged tissue; repair may restore some original structures but can cause structural derangements. In healthy tissues, healing, in the form of regeneration or repair, occurs after practically any insult that causes tissue destruction, and is essential for the survival of the organism.1

FIGURE 3-1 Overview of healing responses after injury. Healing after acute injury can occur by regeneration that restores normal tissue structure or by repair with scar formation. Healing in chronic injury involves scar formation and fibrosis (see text). GI, gastrointestinal.
Regeneration refers to the proliferation of cells and tissues to replace lost structures, such as the growth of an amputated limb in amphibians. In mammals, whole organs and complex tissues rarely regenerate after injury, and the term is usually applied to processes such as liver growth after partial resection or necrosis, but these processes consist of compensatory growth rather than true regeneration.2 Regardless, the term regeneration is well established and is used throughout this book. Tissues with high proliferative capacity, such as the hematopoietic system and the epithelia of the skin and gastrointestinal (GI) tract, renew themselves continuously and can regenerate after injury, as long as the stem cells of these tissues are not destroyed.3
Repair most often consists of a combination of regeneration and scar formation by the deposition of collagen. The relative contribution of regeneration and scarring in tissue repair depends on the ability of the tissue to regenerate and the extent of the injury. For instance, a superficial skin wound heals through the regeneration of the surface epithelium. However, as discussed later, scar formation is the predominant healing process that occurs when the extracellular matrix (ECM) framework is damaged by severe injury (Fig. 3-2). Chronic inflammation that accompanies persistent injury also stimulates scar formation because of local production of growth factors and cytokines that promote fibroblast proliferation and collagen synthesis. The term fibrosis is used to describe the extensive deposition of collagen that occurs under these situations. ECM components are essential for wound healing, because they provide the framework for cell migration, maintain the correct cell polarity for the re-assembly of multilayer structures,4 and participate in the formation of new blood vessels (angiogenesis). Furthermore, cells in the ECM (fibroblasts, macrophages, and other cell types) produce growth factors, cytokines, and chemokines that are critical for regeneration and repair. Although repair is a healing process, it may itself cause tissue dysfunction, as, for instance, in the development of atherosclerosis (Chapter 11).
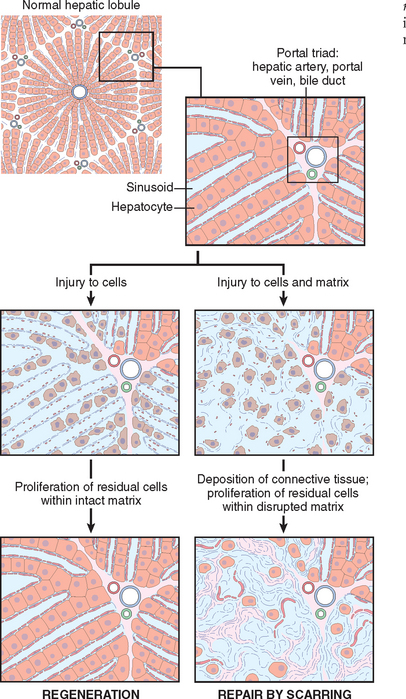
FIGURE 3-2 Role of the extracellular matrix in regeneration and repair. Liver regeneration with restoration of normal tissue after injury requires an intact cellular matrix. If the matrix is damaged the injury is repaired by fibrous tissue deposition and scar formation.
Understanding the mechanisms of regeneration and repair requires some knowledge of the control of cell proliferation and signal transduction pathways, and the many functions of ECM components.
In this chapter we first discuss the principles of cell proliferation, the proliferative capacity of tissues, and the role of stem cells in tissue homeostasis. This is followed by an overview of growth factors and cell signaling mechanisms relevant to healing processes. We then discuss regenerative processes with emphasis on liver regeneration, and examine the properties of the ECM and its components. These sections lay the foundation for the consideration of the main features of wound healing and fibrosis.
Control of Normal Cell Proliferation and Tissue Growth
In adult tissues the size of cell populations is determined by the rates of cell proliferation, differentiation, and death by apoptosis (Fig. 3-3), and increased cell numbers may result from either increased proliferation or decreased cell death.5 Apoptosis is a physiologic process required for tissue homeostasis, but it can also be induced by a variety of pathologic stimuli (see Chapter 1). Differentiated cells incapable of replication are referred to as terminally differentiated cells. The impact of differentiation depends on the tissue under which it occurs: in some tissues differentiated cells are not replaced, while in others they die but are continuously replaced by new cells generated from stem cells (discussed in the next section).
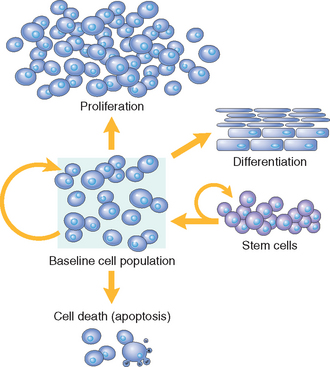
FIGURE 3-3 Mechanisms regulating cell populations. Cell numbers can be altered by increased or decreased rates of stem cell input, cell death due to apoptosis, or changes in the rates of proliferation or differentiation.
(Modified from McCarthy NJ et al: Apoptosis in the development of the immune system: growth factors, clonal selection and bcl-2. Cancer Metastasis Rev 11:157, 1992.)
Cell proliferation can be stimulated by physiologic and pathologic conditions. The proliferation of endometrial cells under estrogen stimulation during the menstrual cycle and the thyroid-stimulating hormone–mediated replication of cells of the thyroid that enlarges the gland during pregnancy are examples of physiologic proliferation. Physiologic stimuli may become excessive, creating pathologic conditions such as nodular prostatic hyperplasia resulting from dihydrotestosterone stimulation (Chapter 21) and the development of nodular goiters in the thyroid as a consequence of increased serum levels of thyroid-stimulating hormone (Chapter 24). Cell proliferation is largely controlled by signals (soluble or contact-dependent) from the microenvironment that either stimulate or inhibit proliferation. An excess of stimulators or a deficiency of inhibitors leads to net growth and, in the case of cancer, uncontrolled growth.
TISSUE PROLIFERATIVE ACTIVITY
The tissues of the body are divided into three groups on the basis of the proliferative activity of their cells: continuously dividing (labile tissues), quiescent (stable tissues), and nondividing (permanent tissues). This time-honored classification should be interpreted in the light of recent findings on stem cells and the reprogramming of cell differentiation.
STEM CELLS
Research on stem cells is at the forefront of modern-day biomedical investigation and stands at the core of a new field called regenerative medicine. The enthusiasm created by stem cell research derives from findings that challenge established views about cell differentiation, and from the hope that stem cells may one day be used to repair damaged human tissues, such as heart, brain, liver, and skeletal muscle.3,6,7
Stem cells are characterized by their self-renewal properties and by their capacity to generate differentiated cell lineages (Fig. 3-4). To give rise to these lineages, stem cells need to be maintained during the life of the organism. Such maintenance is achieved by two mechanisms8: (a) obligatory asymmetric replication, in which with each stem cell division, one of the daughter cells retains its self-renewing capacity while the other enters a differentiation pathway, and (b) stochastic differentiation, in which a stem cell population is maintained by the balance between stem cell divisions that generate either two self-renewing stem cells or two cells that will differentiate. In early stages of embryonic development, stem cells, known as embryonic stem cells or ES cells, are pluripotent, that is, they can generate all tissues of the body (Fig. 3-4). Pluripotent stem cells give rise to multipotent stem cells, which have more restricted developmental potential, and eventually produce differentiated cells from the three embryonic layers. The term transdifferentiation (discussed later) indicates a change in the lineage commitment of a stem cell.
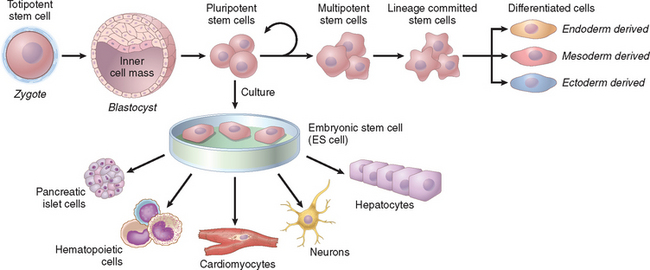
FIGURE 3-4 Stem cell generation and differentiation. The zygote, formed by the union of sperm and egg, divides to form blastocysts, and the inner cell mass of the blastocyst generates the embryo. The cells of the inner cell mass, known as embryonic stem (ES) cells, maintained in culture, can be induced to differentiate into cells of multiple lineages. In the embryo, pluripotent stem cells divide, but the pool of these cells is maintained (see text). As pluripotent cells differentiate, they give rise to cells with more restricted developmental capacity, and finally generate stem cells that are committed to specific lineages.
In adults, stem cells (often referred to as adult stem cells or somatic stem cells) with a more restricted capacity to generate different cell types have been identified in many tissues. They have been studied in detail in the skin, the lining of the gut, the cornea, and particularly in the hematopoietic tissue. An unexpected finding has been the discovery of stem cells and neurogenesis in areas of the central nervous system of adult animals and humans.9 Somatic stem cells for the most part reside in special microenvironments called niches (Fig. 3-5), composed of mesenchymal, endothelial, and other cell types.10,11 It is believed that niche cells generate or transmit stimuli that regulate stem cell self-renewal and the generation of progeny cells. Recent groundbreaking research has now demonstrated that differentiated cells of rodents and humans can be reprogrammed into pluripotent cells, similar to ES cells, by the transduction of genes encoding ES cell transcription factors.12,13 These reprogrammed cells have been named induced pluripotent stem cells (iPS cells). Their discovery has opened open an exciting new era in stem cell research and its applications.
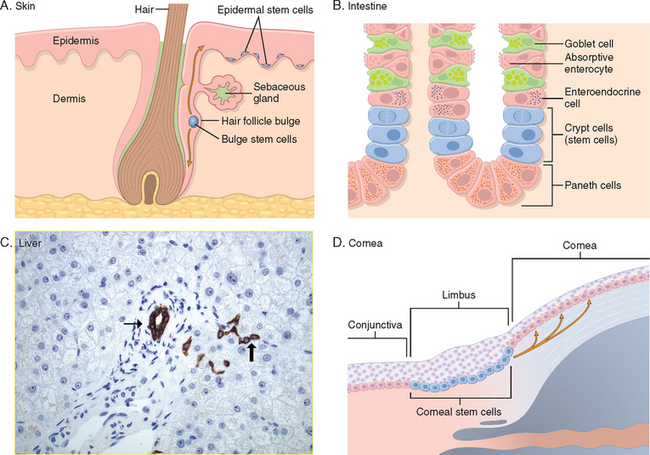
FIGURE 3-5 Stem cell niches in various tissues. A, Skin stem cells are located in the bulge area of the hair follicle, in sebaceous glands, and in the lower layer of the epidermis. B, Small intestine stem cells located near the base of a crypt, above Paneth cells (stem cells in the small intestine may also be located at the bottom of the crypt25). C, Liver stem (progenitor) cells, known as oval cells, are located in the canals of Hering (thick arrow), structures that connect bile ductules (thin arrow) with parenchymal hepatocytes (bile duct and Hering canals are stained for cytokeratin 7). D, Corneal stem cells are located in the limbus region, between the conjunctiva and the cornea.
(C, courtesy of Tania Roskams, MD, University of Leuven, Leuven, Belgium; D, Courtesy of T-T Sun, MD, New York University, New York, NY.)
We start our discussion of stem cells with a brief consideration of ES cells and the newly identified iPS cells. This is followed by a presentation on adult stem cells from a few selected tissues and the role they play in regeneration and repair.
Embryonic Stem Cells
The inner cell mass of blastocysts in early embryonic development contains pluripotent stem cells known as ES cells.14 Cells isolated from blastocysts can be maintained in culture as undifferentiated cell lines or be induced to differentiate into specific lineages (see Fig. 3-4) such as heart and liver cells.15
The study of ES cells has had an enormous impact on biology and medicine:
Reprogramming of Differentiated Cells: Induced Pluripotent Stem Cells
Differentiated cells of adult tissues can be reprogrammed to become pluripotent by transferring their nucleus to an enucleated oocyte. The oocytes implanted into a surrogate mother can generate cloned embryos that develop into complete animals. This procedure, known as reproductive cloning, was successfully demonstrated in 1997 by the cloning of Dolly the sheep.17 There has been great hope that the technique of nuclear transfer to oocytes may be used for therapeutic cloning in the treatment of human diseases (illustrated in Fig. 3-6). In this technique the nucleus of a skin fibroblast from a patient is introduced into an enucleated human oocyte to generate ES cells, which are kept in culture, and then induced to differentiate into various cell types. In principle, these cells can then be transplanted into the patient to repopulate damaged organs.18 In addition to the ethical issues associated with this technique, therapeutic as well as reproductive cloning are inefficient and often inaccurate. One of the main reasons for the inaccuracy is the deficiency in histone methylation in reprogrammed ES cells, which results in improper gene expression.
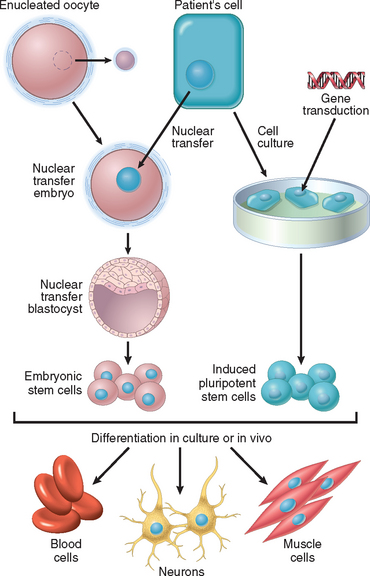
FIGURE 3-6 Steps involved in stem cell therapy, using embryonic stem (ES) cells or induced pluripotent stem (iPS) cells. Left side, Therapeutic cloning using ES cells. The diploid nucleus of an adult cell from a patient is introduced into an enucleated oocyte. The oocyte is activated, and the zygote divides to become a blastocyst that contains the donor DNA. The blastocyst is dissociated to obtain ES cells. Right side, Stem cell therapy using iPS cells. The cells of a patient are placed in culture and transduced with genes encoding transcription factors, to generate iPS cells. Both ES and iPS cells are capable of differentiating into various cell types. The goal of stem cell therapy is to repopulate damaged organs of a patient or to correct a genetic defect, using the cells of the same patient to avoid immunological rejection.
(Modified from Hochedlinger K, Jaenisch R: Nuclear transplantation, embryonic stem cells, and the potential for cell therapy. N Engl J Med 349:275–286, 2003.)
Until recently there were no clues about the mechanisms that maintain pluripotency of ES cells. A series of landmark experiments have now demonstrated that the pluripotency of mouse ES cells depends on the expression of four transcription factors, Oct3/4, Sox2, c-myc, and Klf4, while the homeobox protein Nanog (named after Tir na n’Og, the Celtic land of the ever-young) acts to prevent differentiation.19-22 Human fibroblasts from adults and newborns have been reprogrammed into pluripotent cells by the transduction of four genes encoding transcription factors (Oct3/4, Sox2, c-myc and Kfl4 in one laboratory; Oct3/4, Sox2, Nanog, and Lin28 in experiments from another laboratory).12,13 The reprogrammed cells, known as iPS cells, are able to generate cells from endodermal, mesodermal, and ectodermal origin. They have also been used to rescue mice with a model of sickle cell anemia, proving that they function in vivo even after genetic manipulation and transplantation.23 More recently, pluripotent iPS cells were generated by transfecting mouse hepatocytes, gastric cells, and terminally differentiated mature B lymphocytes with genes for the same four transcription factors.24,25 Thus, iPS cells may become a source of cells for patient-specific stem cell therapy, without the involvement of nuclear transfer into oocytes (see Fig. 3-6). To make the dream of using iPS cells in human regenerative medicine a reality (iPS cells have been called ES cells without embryos), much additional work is required, including the development of new methods for gene delivery and the replacement of c-myc and Kfl4, which are oncogenes.26 In any event, exciting new achievements can be expected in the near future from work on ES cells, iPS cells, and cell reprogramming.
Adult (Somatic) Stem Cells
In the adult organism, stem cells are present in tissues that continuously divide such as the bone marrow, the skin, and the lining of the GI tract. Stem cells may also be present in organs such as liver, pancreas, and adipose tissue, in which, under normal conditions, they do not actively produce differentiated cell lineages. Stem cells divide very slowly in most tissues, but there is evidence that they may be continuously cycling in the small intestine epithelium.27 Regardless of their proliferative activity, somatic stem cells generate rapidly dividing cells known as transit amplifying cells. These cells lose the capacity of self-perpetuation, and give rise to cells with restricted developmental potential known as progenitor cells. Regrettably, the terms stem cell and progenitor cell continue to be used interchangeably, despite the fact that cell lineage hierarchies have been well defined only for hemopoietic stem cells (HSC).
A change in the differentiation of a cell from one type to another is known as transdifferentiation, and the capacity of a cell to transdifferentiate into diverse lineages is referred to as developmental plasticity. HSCs maintained in culture have been shown to transdifferentiate into other cell types, such as hepatocytes and neurons. In addition, some studies indicated that when injected in appropriate sites, HSC could transdifferentiate in vivo into cells such as neurons, skeletal and cardiac myocytes, and hepatocytes. However, many of the findings attributed to HSC transdifferentiation in vivo have been difficult to reproduce, because cells assumed to be the product of transdifferentiation could not be detected or were present at a very low frequency.28 Moreover, the reported generation of neurons, skeletal myocytes, and hepatocytes from injected HSCs seems to be caused primarily by fusion of the hematopoietic cells or their progeny with differentiated or progenitor cells of the appropriate tissues.29,30 Hence, so far, there is little conclusive evidence that transdifferentiation of HSCs contributes to tissue renewal in normal homeostasis or tissue regeneration and repair after injury.31 On the other hand, it is possible that HSCs may migrate to sites of inflammation and injury, where they generate innate immune cells, or release growth factors and cytokines that promote repair and cell replication through a paracrine effect.32 The issue of transdifferentiation and developmental plasticity in tissue repopulation continues to be explored.
Stem Cells in Tissue Homeostasis
To illustrate the importance of stem cells in tissue maintenance and regeneration, we briefly discuss stem cells in the bone marrow, skin, gut, liver, brain, muscle, and cornea.
Cell Cycle and the Regulation of Cell Replication
Cell proliferation is a tightly regulated process that involves a large number of molecules and interrelated pathways. To understand how cells proliferate during regeneration and repair, it is useful to summarize the key features of the normal cell cycle and its regulation. We discuss the details of the cell cycle and its abnormalities in Chapter 7, in the context of cancer. Here we summarize some salient features of the process of cellular proliferation.
The replication of cells is stimulated by growth factors or by signaling from ECM components through integrins. To achieve DNA replication and division, the cell goes through a tightly controlled sequence of events known as the cell cycle. The cell cycle consists of G1 (presynthetic), S (DNA synthesis), G2 (premitotic), and M (mitotic) phases. Quiescent cells that have not entered the cell cycle are in the G0 state (Fig. 3-7). Each cell cycle phase is dependent on the proper activation and completion of the previous one, and the cycle stops at a place at which an essential gene function is deficient. Because of its central role in maintaining tissue homeostasis and regulating physiologic growth processes such as regeneration and repair, the cell cycle has multiple controls and redundancies, particularly during the transition between the G1 and S phases. These controls include activators and inhibitors, as well as sensors that are responsible for checkpoints, described below.52
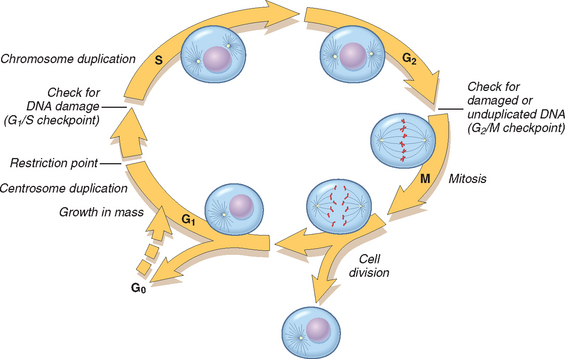
FIGURE 3-7 Cell cycle landmarks. The figure shows the cell cycle phases (G0, G1, G2, S, and M), the location of the G1 restriction point, and the G1/S and G2/M cell cycle checkpoints. Cells from labile tissues such as the epidermis and the GI tract may cycle continuously; stable cells such as hepatocytes are quiescent but can enter the cell cycle; permanent cells such as neurons and cardiac myocytes have lost the capacity to proliferate.
(Modified from Pollard TD, Earnshaw WC: Cell Biology. Philadelphia, Saunders, 2002.)
Cells can enter G1 either from G0 (quiescent cells) or after completing mitosis (continuously replicating cells). Quiescent cells first must go through the transition from G0 to G1, the first decision step, which functions as a gateway to the cell cycle. This transition involves the transcriptional activation of a large set of genes, including various proto-oncogenes and genes required for ribosome synthesis and protein translation. Cells in G1 progress through the cycle and reach a critical stage at the G1/S transition, known as a restriction point, a ratelimiting step for replication (see Fig. 3-7). Upon passing this restriction point, normal cells become irreversibly committed to DNA replication. Progression through the cell cycle, particularly at the G1/S transition, is tightly regulated by proteins called cyclins and associated enzymes called cyclin-dependent kinases (CDKs). CDKs acquire catalytic activity by binding to and forming complexes with the cyclins. Activated CDKs in these complexes drive the cell cycle by phosphorylating proteins that are critical for cell cycle transitions. One such protein is the retinoblastoma susceptibility (RB) protein, which normally prevents cells from replicating by forming a tight, inactive complex with the transcription factor E2F. Phosphorylation of RB causes its release, which activates E2F and allows it to stimulate transcription of genes whose products drive cells through the cycle. More details are provided in Chapter 7.
The activity of cyclin-CDK complexes is tightly regulated by CDK inhibitors. Some growth factors shut off production of these inhibitors. Embedded in the cell cycle are surveillance mechanisms that are geared primarily at sensing damage to DNA and chromosomes. These quality control checks are called checkpoints; they ensure that cells with damaged DNA or chromosomes do not complete replication.53 The G1/S checkpoint monitors the integrity of DNA before replication, whereas the G2/M checkpoint checks DNA after replication and monitors whether the cell can safely enter mitosis. When cells sense DNA damage, checkpoint activation delays the cell cycle and triggers DNA repair mechanisms. If DNA damage is too severe to be repaired, the cells are eliminated by apoptosis, or enter a nonreplicative state called senescence, primarily through p53-dependent mechanisms. Checkpoint defects that allow cells with DNA strand breaks and chromosome abnormalities to divide produce mutations in daughter cells that may lead to neoplasia (Chapter 7).54
GROWTH FACTORS
The proliferation of many cell types is driven by polypeptides known as growth factors. These factors, which can have restricted or multiple cell targets, may also promote cell survival, locomotion, contractility, differentiation, and angiogenesis, activities that may be as important as their growth-promoting effects. All growth factors function as ligands that bind to specific receptors, which deliver signals to the target cells. These signals stimulate the transcription of genes that may be silent in resting cells, including genes that control cell cycle entry and progression. Table 3-1 lists some of the most important growth factors involved in tissue regeneration and repair. Here we review only those that have major roles in these processes. Other growth factors are alluded to in various sections of the book.
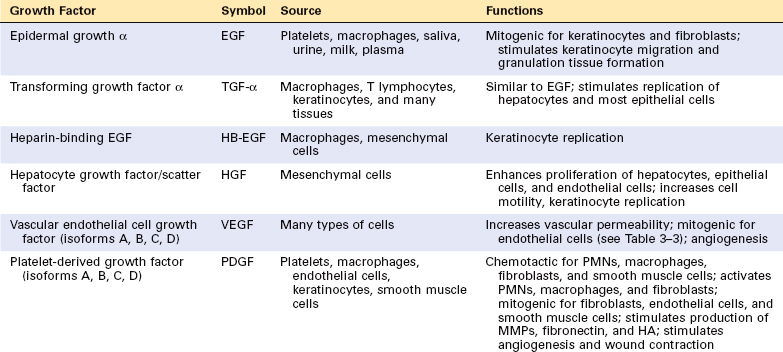
Epidermal Growth Factor (EGF) and Transforming Growth Factor α (TGF-α).
These two factors belong to the EGF family and share a common receptor (EGFR).55 EGF is mitogenic for a variety of epithelial cells, hepatocytes, and fibroblasts, and is widely distributed in tissue secretions and fluids. In healing wounds of the skin, EGF is produced by keratinocytes, macrophages, and other inflammatory cells that migrate into the area. TGF-α was originally extracted from sarcoma virus–transformed cells and is involved in epithelial cell proliferation in embryos and adults, and in malignant transformation of normal cells to cancer. TGF-α has homology with EGF, binds to EGFR, and shares most of the biologic activities of EGF. The “EGF receptor” is actually a family of four membrane receptors with intrinsic tyrosine kinase activity. The best-characterized EGFR is referred to as EGFR1, ERB B1, or simply EGFR. It responds to EGF, TGF-α, and other ligands of the EGF family, such as HB-EGF (heparin-binding EGF) and amphiregulin. EGFR1 mutations and amplification have been detected in cancers of the lung, head and neck, and breast, glioblastomas, and other cancers, leading to the development of new types of treatments for these conditions. The ERB B2 receptor (also known as HER-2 or HER2/Neu), whose main ligand has not been identified, has received great attention because it is overexpressed in a subset of breast cancers and is an important therapeutic target.
Hepatocyte Growth Factor (HGF).
HGF was originally isolated from platelets and serum. Subsequent studies demonstrated that it is identical to a previously identified growth factor isolated from fibroblasts known as scatter factor.56 The factor is often referred to as HGF/SF, but in this chapter we will use the simpler notation, HGF.
HGF has mitogenic effects on hepatocytes and most epithelial cells, including cells of the biliary epithelium, and epithelial cells of the lungs, kidney, mammary gland, and skin. HGF acts as a morphogen in embryonic development, promotes cell scattering and migration, and enhances survival of hepatocytes. It is produced by fibroblasts and most mesenchymal cells, endothelial cells, and liver nonparenchymal cells. It is produced as an inactive single-chain form (pro-HGF) that is activated by serine proteases released in damaged tissues. The receptor for HGF, c-MET, is often highly expressed or mutated in human tumors, especially in renal and thyroid papillary carcinomas. HGF signaling is required for survival during embryonic development, as demonstrated by defects in the development of muscles, kidney, liver, and brain, and the lethality of knockout mice that lack c-met. Several HGF and c-MET inhibitors are presently being evaluated in cancer therapy clinical trials.
Platelet-Derived Growth Factor (PDGF).
PDGF is a family of several closely related proteins, each consisting of two chains. Three isoforms of PDGF (AA, AB, and BB) are secreted as biologically active molecules. The more recently identified isoforms PDGF-CC and PDGF-DD require extracellular proteolytic cleavage to release the active growth factor.57 All PDGF isoforms exert their effects by binding to two cell surface receptors, designated PDGFR α and β, which have different ligand specificities. PDGF is stored in platelet granules and is released on platelet activation. It is produced by a variety of cells, including activated macrophages, endothelial cells, smooth muscle cells, and many tumor cells. PDGF causes migration and proliferation of fibroblasts, smooth muscle cells, and monocytes to areas of inflammation and healing skin wounds, as demonstrated by defects in these functions in mice deficient in either the A or the B chain of PDGF. PDGF-B and C participate in the activation of hepatic stellate cells in the initial steps of liver fibrosis (Chapter 18) and stimulate wound contraction.
Vascular Endothelial Growth Factor (VEGF).
VEGFs are a family of homodimeric proteins that include VEGF-A (referred throughout as VEGF), VEGF-B, VEGF-C, VEGF-D, and PIGF (placental growth factor).58 VEGF is a potent inducer of blood vessel formation in early development (vasculogenesis) and has a central role in the growth of new blood vessels (angiogenesis) in adults (see Table 3-3). It promotes angiogenesis in chronic inflammation, healing of wounds, and in tumors (discussed later in this chapter, in “Mechanisms of Angiogenesis”). Mice that lack a single VEGF allele (heterozygous VEGF knockout mice) die during embryonic development as a result of defective vasculogenesis and hematopoiesis. VEGF family members signal through three tyrosine kinase receptors: VEGFR-1, VEGFR-2, and VEGFR-3. VEGFR-2, located in endothelial cells and many other cell types, is the main receptor for the vasculogenic and angiogenic effects of VEGF. The role of VEGFR-1 is less well understood, but it may facilitate the mobilization of endothelial stem cells and has a role in inflammation. VEGF-C and VEGF-D bind to VEGFR-3 and act on lymphatic endothelial cells to induce the production of lymphatic vessels (lymphangiogenesis).
TABLE 3-3 Vascular Endothelial Growth Factor (VEGF)
| Proteins | Family members: VEGF (VEGF-A), VEGF-B, VEGF-C, VEGF-D |
| Dimeric glycoprotein with multiple isoforms | |
| Targeted mutations in VEGF result in defective vasculogenesis and angiogenesis. | |
| Production | Expressed at low levels in a variety of adult tissues and at higher levels in a few sites, such as podocytes in the glomerulus and cardiac myocytes |
| Inducing agents | Hypoxia |
| TGF-β | |
| PDGF | |
| TGF-α | |
| Receptors | VEGFR-1 |
| VEGFR-2 | |
| VEGFR-3 (lymphatic endothelial cells) | |
| Targeted mutations in the receptors result in lack of vasculogenesis | |
| Functions | Promotes angiogenesis |
| Increases vascular permeability | |
| Stimulates endothelial cell migration | |
| Stimulates endothelial cell proliferation | |
| VEGF-C selectively induces hyperplasia of lymphatic vasculature | |
| Up-regulates endothelial expression of plasminogen activator, plasminogen activator inhibitor 1, and collagenase |
PDGF, platelet-derived growth factor; TGF-β, -α, transforming growth factor beta, alpha.
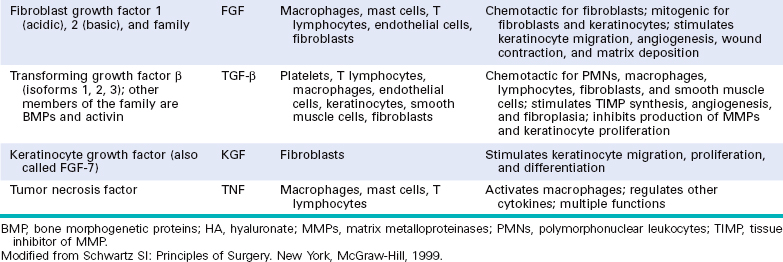
Fibroblast Growth Factor (FGF).
This is a family of growth factors containing more than 20 members, of which acidic FGF (aFGF, or FGF-1) and basic FGF (bFGF, or FGF-2) are the best characterized.59 FGFs transduce signals through four tyrosine kinase receptors (FGFRs 1–4). FGF-1 binds to all receptors; FGF-7 is referred to as keratinocyte growth factor or KGF. Released FGFs associate with heparan sulfate in the ECM, which can serve as a reservoir for the storage of inactive factors. FGFs contribute to wound healing responses, hematopoiesis, angiogenesis, development, and other processes through several functions:
Transforming Growth Factor β (TGF-β) and Related Growth Factors.
TGF-β belongs to a superfamily of about 30 members that includes three TGF-β isoforms (TGF-β1, TGF-β2, TGF-β3) and factors with wide-ranging functions, such as BMPs, activins, inhibins, and müllerian inhibiting substance.60 TGF-β1 has the most widespread distribution in mammals and will be referred to as TGF-β. It is a homodimeric protein produced by a variety of different cell types, including platelets, endothelial cells, lymphocytes, and macrophages. Native TGF-β is synthesized as a precursor protein, which is secreted and then proteolytically cleaved to yield the biologically active growth factor and a second latent component. Active TGF-β binds to two cell surface receptors (types I and II) with serine/threonine kinase activity and triggers the phosphorylation of cytoplasmic transcription factors called Smads (of which there are several forms, e.g., Smad 1, 2, 3, 5, and 8). These phosphorylated Smads in turn form heterodimers with Smad 4, which enter the nucleus and associate with other DNA-binding proteins to activate or inhibit gene transcription. TGF-β has multiple and often opposing effects depending on the tissue and the type of injury. Agents that have multiple effects are called pleiotropic; because of the large diversity of TGF-β effects, it has been said that TGF-β is pleiotropic with a vengeance.
Cytokines.
Cytokines have important functions as mediators of inflammation and immune responses (Chapter 6). Some of these proteins can also be considered as growth factors, because they have growth-promoting activities for a variety of cells. Cytokines are discussed in Chapters 2 and 6. Tumor necrosis factor (TNF) and IL-1 participate in wound healing reactions (see Table 3-1), and TNF and IL-6 are involved in the initiation of liver regeneration (discussed later).
SIGNALING MECHANISMS IN CELL GROWTH
In this section we review the process of receptor-mediated signal transduction, which is activated by the binding of ligands such as growth factors, and cytokines to specific receptors. Different classes of receptor molecules and pathways initiate a cascade of events by which receptor activation leads to expression of specific genes. Here we focus on the biochemical pathways and transcriptional regulation that mediate growth factor activity.
According to the source of the ligand and the location of its receptors (i.e., in the same, adjacent, or distant cells), three general modes of signaling, named autocrine, paracrine, and endocrine, can be distinguished (Fig. 3-8).
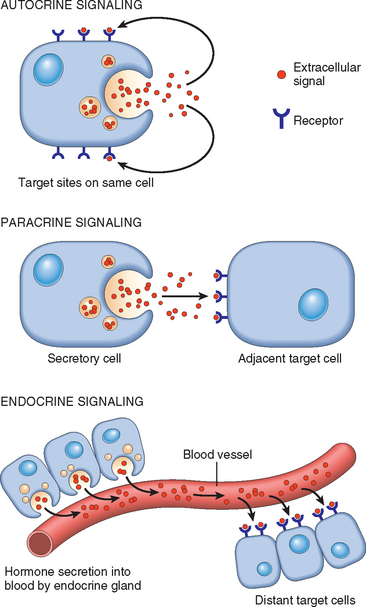
FIGURE 3-8 General patterns of intercellular signaling demonstrating autocrine, paracrine, and endocrine signaling (see text).
(Modified from Lodish H et al [eds]: Molecular Cell Biology, 3rd ed. New York, WH Freeman, 1995, p 855. © 1995 by Scientific American Books. Used with permission of WH Freeman and Company.)
Receptors and Signal Transduction Pathways
The binding of a ligand to its receptor triggers a series of events by which extracellular signals are transduced into the cell resulting in changes in gene expression. Although single receptor molecules can transduce some signals upon ligand binding, signaling typically involves clustering of two or more receptor molecules by the ligand. Receptors are generally located on the surface of the target cell but can also be found in the cytoplasm or nucleus.
It is useful at this point to summarize the properties of the major types of receptors and how they deliver signals to the cell interior (Fig. 3-9). This is pertinent to an understanding of normal and unregulated (neoplastic) cell growth (discussed in Chapter 7).
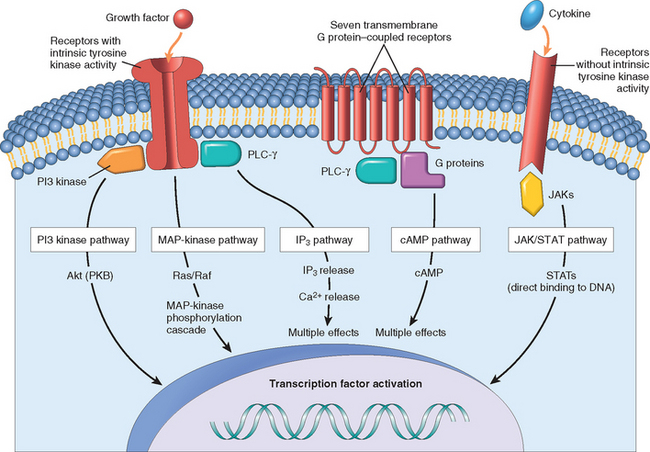
FIGURE 3-9 Overview of the main types of cell surface receptors and their principal signal transduction pathways (see text). Shown are receptors with intrinsic tyrosine kinase activity, seven transmembrane G protein–coupled receptors, and receptors without intrinsic tyrosine kinase activity. cAMP, cyclic adenosine monophosphate: IP3, inositol triphosphate; JAK, Janus kinase; MAP kinase, mitogen-activated protein kinase; PI3 kinase, phosphatidylinositol 3-kinase; PKB, protein kinase B, also known as Akt; PLC-γ, phospholipase C gamma; STATs, signal transducers and activators of transcription.

FIGURE 3-10 Signaling from tyrosine kinase receptors. Binding of the growth factor (ligand) causes receptor dimerization and autophosphorylation of tyrosine residues. Attachment of adapter (or bridging) proteins (e.g., GRB2 and SOS) couples the receptor to inactive RAS. Cycling of RAS between its inactive and active forms is regulated by GAP. Activated RAS interacts with and activates RAF (also known as MAP kinase kinase kinase). This kinase then phosphorylates a component of the MAP kinase signaling pathway, MEK (also known as MAP kinase kinase or MKK), which then phosphorylates ERK (MAP kinase or MK). Activated MAP kinase phosphorylates other cytoplasmic proteins and nuclear transcription factors, generating cellular responses. The phosphorylated tyrosine kinase receptor can also bind other components, such as phosphatidyl 3-kinase (PI3 kinase), which activates other signaling systems.
Transcription Factors
Many of the signal transduction systems used by growth factors transfer information to the nucleus and modulate gene transcription through the activity of transcription factors. Among the transcription factors that regulate cell proliferation are products of several growth-promoting genes, such as c-MYC and c-JUN, and of cell cycle–inhibiting genes, such as p53. Transcription factors have a modular design and contain domains for DNA binding and for transcriptional regulation. The DNA-binding domain permits binding to short sequence motifs of DNA, which may be unique to a particular target gene or may be present in many genes. The transactivating domain stimulates transcription of the adjacent gene.
Growth factors induce the synthesis or activity of transcription factors. Cellular events requiring rapid responses do not rely on new synthesis of transcription factors but depend on post-translational modifications that lead to their activation. These modifications include (a) heterodimerization, as for instance, the dimerization of the products of the protooncogenes c-FOS and c-JUN to form the transcription factor activator protein-1 (AP-1), which is activated by MAP kinase signaling pathways, (b) phosphorylation, as for STATs in the JAK/STAT pathway, (c) release of inhibition to permit migration into the nucleus, as for NF-κB, and (d) release from membranes by proteolytic cleavage, as for Notch receptors (see Fig. 3-16).

FIGURE 3-16 Notch signaling and angiogenesis. A, The Notch receptor binds a ligand (a delta-like ligand, Dll, is shown in the figure) located in an adjacent cell, and undergoes two proteolytic cleavages (the first cleavage by ADAM protease, the second by δ-secretase), releasing a C-terminal fragment known as Notch intracellular domain (Notch-ICD). B, Notch signaling in endothelial cells during angiogenesis, triggered by the binding of the Dll4 ligand in a tip cell to a Notch receptor in a stalk cell. Notch-ICD migrates into the nucleus and activates the transcription of target genes. C, Sprouting angiogenesis, showing a migrating tip cell and stalk cells connected to the endothelial cells of the main vessel.
(A, modified from Weinberg RA: The Biology of Cancer. New York, Garland Science, 2007, Fig. 5.22; B, modified from Kerbel RS: Tumor angiogenesis. N Engl J Med 358:2039, 2008.)
Mechanisms of Tissue and Organ Regeneration
Urodele amphibians such as the newt can regenerate their tails, limbs, lens, retina, jaws, and even a large portion of the heart, but the capacity for regeneration of whole tissues and organs has been lost in mammals.1 The inadequacy of true regeneration in mammals has been attributed to the absence of blastema formation (the source of cells for regeneration) and to the rapid fibroproliferative response after wounding. The Wnt/β-catenin is a highly conserved pathway that participates in the regeneration of planaria flatworms, fin and heart regeneration in zebra fish, and blastema and patterning formation in limb regeneration in newts. In mammals, Wnt/β-catenin modulates stem cell functions in the intestinal epithelium, bone marrow, and muscle, participates in liver regeneration after partial hepatectomy, and stimulates oval cell proliferation after liver injury.27,65,66
In this section we have chosen the liver to illustrate the mechanisms of regeneration, because it has been studied in detail and has important biologic and clinical aspects. Even this process is not one of true regeneration, because the resection of tissue does not cause new growth of liver but instead triggers a process of compensatory hyperplasia in the remaining parts of the organ (discussed below). Other organs, including kidney, pancreas, adrenal glands, thyroid, and the lungs of very young animals, are also capable of compensatory growth, although they display it in less dramatic form than the liver. Because new nephrons cannot be generated in the adult kidney, the growth of the contralateral kidney after unilateral nephrectomy involves nephron hypertrophy and some replication of proximal tubule cells. The pancreas has a limited capacity to regenerate its exocrine components and islets. Regeneration of pancreatic beta cells may involve beta-cell replication, transdifferentiation of ductal cells, or differentiation of putative stem cells that express the transcription factors Oct4 and Sox2.67 Recently, pancreatic exocrine cells have been reprogrammed into insulin-secreting β-cells.
LIVER REGENERATION
The human liver has a remarkable capacity to regenerate, as demonstrated by its growth after partial hepatectomy, which may be performed for tumor resection or for living-donor hepatic transplantation (Fig. 3-11). The popular image of liver regeneration is the daily regrowth of the liver of Prometheus, which was eaten every day by an eagle sent by Zeus (Zeus was angry at Prometheus for stealing the secret of fire, but did he know that Prometheus’s liver would regenerate?). The reality, although less dramatic, is still quite impressive. In humans, resection of approximately 60% of the liver in living donors results in the doubling of the liver remnant in about one month. The portions of the liver that remain after partial hepatectomy constitute an intact “mini-liver” that rapidly expands and reaches the mass of the original liver (see Fig. 3-11). Restoration of liver mass is achieved without the regrowth of the lobes that were resected at the operation. Instead, growth occurs by enlargement of the lobes that remain after the operation, a process known as compensatory growth or compensatory hyperplasia. In both humans and rodents, the end point of liver regeneration after partial hepatectomy is the restitution of functional mass rather than the reconstitution of the original form.69
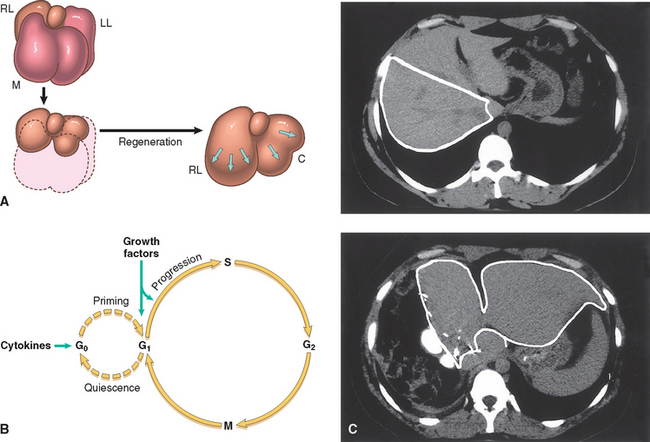
FIGURE 3-11 Liver regeneration after partial hepatectomy. A, The lobes of the liver of a rat (M, median; RL and LL, right and left lateral lobes; C, caudate lobe). Partial hepatectomy removes two thirds of the liver (median and left lateral lobes). After 3 weeks the right lateral and caudate lobes enlarge to reach a mass equivalent to that of the original liver without regrowth of the median and left lateral lobes. B, Entry and progression of hepatocytes in the cell cycle (see text for details). C, Regeneration of the human liver in living-donor transplantation. Computed tomography scans of the donor liver in living-donor hepatic transplantation. Upper panel is a scan of the liver of the donor before the operation. The right lobe, to be used as a transplant, is outlined. Lower panel is a scan of the liver 1 week after performance of partial hepatectomy. Note the great enlargement of the left lobe (outlined in the panel) without regrowth of the right lobe.
(A, From Goss RJ: Regeneration versus repair. In Cohen IK et al [eds]: Wound Healing. Biochemical and Clinical Aspects. Philadelphia, WB Saunders, 1992, pp 20–39; C, courtesy of R. Troisi, MD, Ghent University, Ghent, Belgium; reproduced in part from Fausto N: Liver regeneration. In Arias I, et al: The Liver: Biology and Pathobiology, 4th ed. Philadelphia, Lippincott Williams & Wilkins, 2001.)
Almost all hepatocytes replicate during liver regeneration after partial hepatectomy. Because hepatocytes are quiescent cells, it takes them several hours to enter the cell cycle, progress through G1, and reach the S phase of DNA replication. The wave of hepatocyte replication is synchronized and is followed by synchronous replication of nonparenchymal cells (Kupffer cells, endothelial cells, and stellate cells).
There is substantial evidence that hepatocyte proliferation in the regenerating liver is triggered by the combined actions of cytokines and polypeptide growth factors. With the exception of the autocrine activity of TGF-α, hepatocyte replication is strictly dependent on paracrine effects of growth factors and cytokines such as HGF and IL-6 produced by hepatic nonparenchymal cells. There are two major restriction points for hepatocyte replication: the G0/G1 transition that bring quiescent hepatocytes into the cell cycle, and the G1/S transition needed for passage through the late G1 restriction point. Gene expression in the regenerating liver proceeds in phases, starting with the immediate early gene response, which is a transient response that corresponds to the G0/G1 transition. More than 70 genes are activated during this response, including the proto-oncogenes c-FOS and c-JUN, whose products dimerize to form the transcription factor AP-1; c-MYC, which encodes a transcription factor that activates many different genes; and other transcription factors, such as NF-κB, STAT-3, and C/EBP.70 The immediate early gene response sets the stage for the sequential activation of multiple genes, as hepatocytes progress into the G1 phase. The G1 to S transition occurs as previously described (see Fig. 3-7).
Quiescent hepatocytes become competent to enter the cell cycle through a priming phase that is mostly mediated by the cytokines TNF and IL-6, and components of the complement system. Priming signals activate several signal transduction pathways as a necessary prelude to cell proliferation. Under the stimulation of HGF, TGFα, and HB-EGF, primed hepatocytes enter the cell cycle and undergo DNA replication (Fig. 3-11). Norepinephrine, serotonin, insulin, thyroid and growth hormone, act as adjuvants for liver regeneration, facilitating the entry of hepatocytes into the cell cycle.
Individual hepatocytes replicate once or twice during regeneration and then return to quiescence in a strictly regulated sequence of events, but the mechanisms of growth cessation have not been established. Growth inhibitors, such as TGF-β and activins, may be involved in terminating hepatocyte replication, but there is no clear understanding of their mode of action. Intrahepatic stem or progenitor cells do not play a role in the compensatory growth that occurs after partial hepatectomy, and there is no evidence for hepatocyte generation from bone marrow–derived cells during this process.28,37 However, endothelial cells and other nonparenchymal cells in the regenerating liver may originate from bone marrow precursors.
Extracellular Matrix and Cell-Matrix Interactions
Tissue repair and regeneration depend not only on the activity of soluble factors, but also on interactions between cells and the components of the extracellular matrix (ECM). The ECM regulates the growth, proliferation, movement, and differentiation of the cells living within it. It is constantly remodeling, and its synthesis and degradation accompanies morphogenesis, regeneration, wound healing, chronic fibrotic processes, tumor invasion, and metastasis. The ECM sequesters water, providing turgor to soft tissues, and minerals that give rigidity to bone, but it does much more than just fill the spaces around cells to maintain tissue structure. Its various functions include:
The ECM is composed of three groups of macromolecules: fibrous structural proteins, such as collagens and elastins that provide tensile strength and recoil; adhesive glycoproteins that connect the matrix elements to one another and to cells; and proteoglycans and hyaluronan that provide resilience and lubrication. These molecules assemble to form two basic forms of ECM: interstitial matrix and basement membranes. The interstitial matrix is found in spaces between epithelial, endothelial, and smooth muscle cells, as well as in connective tissue. It consists mostly of fibrillar and nonfibrillar collagen, elastin, fibronectin, proteoglycans, and hyaluronan. Basement membranes are closely associated with cell surfaces, and consist of nonfibrillar collagen (mostly type IV), laminin, heparin sulfate, and proteoglycans.71
We will now consider the main components of the ECM.
COLLAGEN
Collagen is the most common protein in the animal world, providing the extracellular framework for all multicellular organisms. Without collagen, a human being would be reduced to a clump of cells, like the “Blob” (the “gelatinous horror from outer space” of 1950s movie fame), interconnected by a few neurons. Currently, 27 different types of collagens encoded by 41 genes dispersed on at least 14 chromosomes are known72 (Table 3-2). Each collagen is composed of three chains that form a trimer in the shape of a triple helix. The polypeptide is characterized by a repeating sequence in which glycine is in every third position (Gly-X-Y, in which X and Y can be any amino acid other than cysteine or tryptophan), and it contains
TABLE 3-2 Main Types of Collagens, Tissue Distribution, and Genetic Disorders
| Collagen Type | Tissue Distribution | Genetic Disorders |
|---|---|---|
| FIBRILLAR COLLAGENS | ||
| I | Ubiquitous in hard and soft tissues | Osteogenesis imperfecta; Ehlers-Danlos syndrome—arthrochalasias type I |
| II | Cartilage, intervertebral disk, vitreous | Achondrogenesis type II, spondyloepiphysea dysplasia syndrome |
| III | Hollow organs, soft tissues | Vascular Ehlers-Danlos syndrome |
| V | Soft tissues, blood vessels | Classical Ehlers-Danlos syndrome |
| IX | Cartilage, vitreous | Stickler syndrome |
| BASEMENT MEMBRANE COLLAGENS | ||
| IV | Basement membranes | Alport syndrome |
| OTHER COLLAGENS | ||
| VI | Ubiquitous in microfibrils | Bethlem myopathy |
| VII | Anchoring fibrils at dermal-epidermal junctions | Dystrophic epidermolysis bullosa |
| IX | Cartilage, intervertebral disks | Multiple epiphyseal dysplasias |
| XVII | Transmembrane collagen in epidermal cells | Benign atrophic generalized epidermolysis bullosa |
| XV and XVIII | Endostatin-forming collagens, endothelial cells | Knobloch syndrome (type XVIII collagen) |
Courtesy of Dr. Peter H. Byers, Department of Pathology, University of Washington, Seattle, WA.
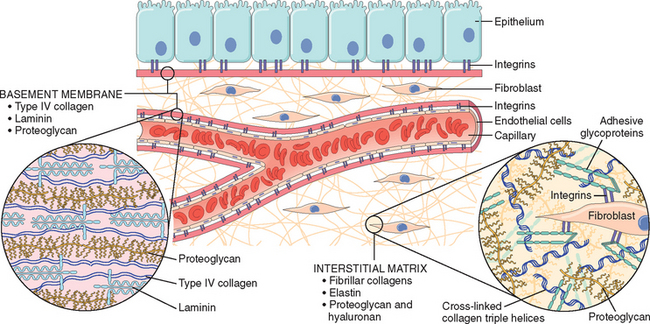
FIGURE 3-12 Main components of the extracellular matrix (ECM), including collagens, proteoglycans, and adhesive glycoproteins. Both epithelial and mesenchymal cells (e.g., fibroblasts) interact with ECM via integrins. Basement membranes and interstitial ECM have different architecture and general composition, although there is some overlap in their constituents. For the sake of simplification, many ECM components (e.g., elastin, fibrillin, hyaluronan, and syndecan) are not included.
the specialized amino acids 4-hydroxyproline and hydroxylysine. Prolyl residues in the Y-position are characteristically hydroxylated to produce hydroxyproline, which serves to stabilize the triple helix. Types I, II, III and V, and XI are the fibrillar collagens, in which the triple-helical domain is uninterrupted for more than 1000 residues; these proteins are found in extracellular fibrillar structures. Type IV collagens have long but interrupted triple-helical domains and form sheets instead of fibrils; they are the main components of the basement membrane, together with laminin. Another collagen with a long interrupted triple-helical domain (type VII) forms the anchoring fibrils between some epithelial and mesenchymal structures, such as epidermis and dermis. Still other collagens are transmembrane and may also help to anchor epidermal and dermal structures.
The messenger RNAs transcribed from fibrillar collagen genes are translated into pre-pro-α chains that assemble in a type-specific manner into trimers. Hydroxylation of proline and lysine residues and lysine glycosylation occur during translation. Three chains of a particular collagen type assemble to form the triple helix (Fig. 3-15). Procollagen is secreted from the cell and cleaved by proteases to form the basic unit of the fibrils. Collagen fibril formation is associated with the oxidation of lysine and hydroxylysine residues by the extracellular enzyme lysyl oxidase. This results in cross-linking between the chains of adjacent molecules, which stabilizes the array, and is a major contributor to the tensile strength of collagen. Vitamin C is required for the hydroxylation of procollagen, a requirement that explains the inadequate wound healing in scurvy (Chapter 9). Genetic defects in collagen production (see Table 3-2) cause many inherited syndromes, including various forms of the Ehlers-Danlos syndrome and osteogenesis imperfecta73 (Chapters 5 and 26.
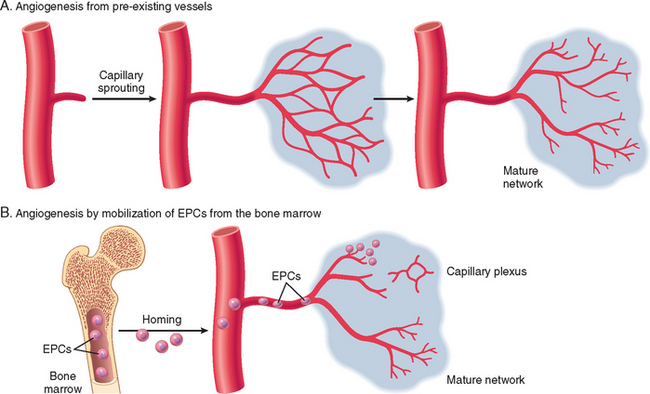
FIGURE 3-15 Angiogenesis by mobilization of endothelial precursor cells (EPCs) from the bone marrow and from preexisting vessels (capillary growth). A, In angiogenesis from preexisting vessels, endothelial cells from these vessels become motile and proliferate to form capillary sprouts. Regardless of the initiating mechanism, vessel maturation (stabilization) involves the recruitment of pericytes and smooth muscle cells to form the periendothelial layer. B, EPCs are mobilized from the bone marrow and may migrate to a site of injury or tumor growth. At these sites, EPCs differentiate and form a mature network by linking to existing vessels.
(Modified from Conway EM et al: Molecular mechanisms of blood vessel growth. Cardiovasc Res 49:507, 2001.)
ELASTIN, FIBRILLIN, AND ELASTIC FIBERS
Tissues such as blood vessels, skin, uterus, and lung require elasticity for their function. Proteins of the collagen family provide tensile strength, but the ability of these tissues to expand and recoil (compliance) depends on the elastic fibers. These fibers can stretch and then return to their original size after release of the tension. Morphologically, elastic fibers consist of a central core made of elastin, surrounded by a peripheral network of microfibrils. Substantial amounts of elastin are found in the walls of large blood vessels, such as the aorta, and in the uterus, skin, and ligaments. The peripheral microfibrillar network that surrounds the core consists largely of fibrillin, a 350-kD secreted glycoprotein, which associates either with itself or with other components of the ECM. The microfibrils serve, in part, as scaffolding for deposition of elastin and the assembly of elastic fibers. They also influence the availability of active TGFβ in the ECM. As already mentioned, inherited defects in fibrillin result in formation of abnormal elastic fibers in Marfan syndrome, manifested by changes in the cardiovascular system (aortic dissection) and the skeleton74 (Chapter 5).
CELL ADHESION PROTEINS
Most adhesion proteins, also called CAMs (cell adhesion molecules), can be classified into four main families: immunoglobulin family CAMs, cadherins, integrins, and selectins. These proteins function as transmembrane receptors but are sometimes stored in the cytoplasm.75 As receptors, CAMs can bind to similar or different molecules in other cells, providing for interaction between the same cells (homotypic interaction) or different cell types (heterotypic interaction). Selectins have been discussed in Chapter 2 in the context of leukocyte/endothelial interactions. Selected aspects of other cell adhesion proteins are described here. Integrins bind to ECM proteins such as fibronectin, laminin, and osteopontin providing a connection between cells and ECM, and also to adhesive proteins in other cells, establishing cell-to-cell contact. Fibronectin is a large protein that binds to many molecules, such as collagen, fibrin, proteoglycans, and cell surface receptors. It consists of two glycoprotein chains, held together by disulfide bonds. Fibronectin messenger RNA has two splice forms, giving rise to tissue fibronectin and plasma fibronectin. The plasma form binds to fibrin, helping to stabilize the blood clot that fills the gaps created by wounds, and serves as a substratum for ECM deposition and formation of the provisional matrix during wound healing (discussed later). Laminin is the most abundant glycoprotein in the basement membrane and has binding domains for both ECM and cell surface receptors. In the basement membrane, polymers of laminin and collagen type IV form tightly bound networks. Laminin can also mediate the attachment of cells to connective tissue substrates.
Cadherins and integrins link the cell surface with the cytoskeleton through binding to actin and intermediate filaments. These linkages, particularly for the integrins, provide a mechanism for the transmission of mechanical force and the activation of intracellular signal transduction pathways that respond to these forces. Ligand binding to integrins causes clustering of the receptors in the cell membrane and formation of focal adhesion complexes. The cytoskeletal proteins that co-localize with integrins at the cell focal adhesion complex include talin, vinculin, and paxillin. The integrin-cytoskeleton complexes function as activated receptors and trigger a number of signal transduction pathways, including the MAP kinase, PKC, and PI3K pathways, which are also activated by growth factors. Not only is there a functional overlap between integrin and growth factor receptors, but integrins and growth factor receptors interact (“cross-talk”) to transmit environmental signals to the cell that regulate proliferation, apoptosis, and differentiation (Fig. 3-13).
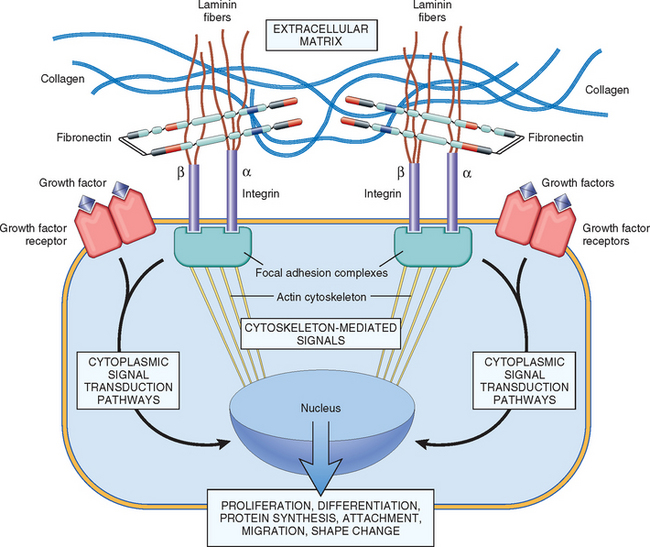
FIGURE 3-13 Mechanisms by which ECM components and growth factors interact and activate signaling pathways. Integrins bind ECM components and interact with the cytoskeleton at focal adhesion complexes (protein aggregates that include vinculin, α-actin, and talin). This can initiate the production of intracellular messengers or can directly mediate nuclear signals. Cell surface receptors for growth factors may activate signal transduction pathways that overlap with those activated by integrins. Signaling from ECM components and growth factors is integrated by the cell to produce various responses, including changes in cell proliferation, locomotion, and differentiation.
The name cadherin is derived from the term “calcium-dependent adherence protein.” This family contains almost 90 members, which participate in interactions between cells of the same type. These interactions connect the plasma membrane of adjacent cells, forming two types of cell junctions called (1) zonula adherens, small, spotlike junctions located near the apical surface of epithelial cells, and (2) desmosomes, stronger and more extensive junctions, present in epithelial and muscle cells. Migration of keratinocytes in the re-epithelialization of skin wounds is dependent on the formation of dermosomal junctions. Linkage of cadherins with the cytoskeleton occurs through two classes of catenins. β-catenin links cadherins with α-catenin, which, in turn, connects to actin, thus completing the connection with the cytoskeleton. Cell-to-cell interactions mediated by cadherins and catenins play a major role in regulating cell motility, proliferation, and differentiation and account for the inhibition of cell proliferation that occurs when cultured normal cells contact each other (“contact inhibition”). Diminished function of E-cadherin contributes to certain forms of breast and gastric cancer. As already mentioned, free β-catenin acts independently of cadherins in the Wnt signaling pathway, which participates in stem cell homeostasis and regeneration. Mutation and altered expression of the Wnt/β-catenin pathway is implicated in cancer development, particularly in gastrointestinal and liver cancers (Chapter 7).
In addition to the main families of adhesive proteins described earlier, some other secreted adhesion molecules are mentioned because of their potential role in disease processes: (1) SPARC (secreted protein acidic and rich in cysteine), also known as osteonectin, contributes to tissue remodeling in response to injury and functions as an angiogenesis inhibitor; (2) the thrombospondins, a family of large multifunctional proteins, some of which, similar to SPARC, also inhibit angiogenesis; (3) osteopontin (OPN) is a glycoprotein that regulates calcification, is a mediator of leukocyte migration involved in inflammation, vascular remodeling, and fibrosis in various organs76,77 (discussed later in this chapter); and (4) the tenascin family, which consist of large multimeric proteins involved in morphogenesis and cell adhesion.
GLYCOSAMINOGLYCANS (GAGS) AND PROTEOGLYCANS
GAGs make up the third type of component in the ECM, besides the fibrous structural proteins and cell adhesion proteins. GAGs consist of long repeating polymers of specific disaccharides. With the exception of hyaluronan (discussed later), GAGs are linked to a core protein, forming molecules called proteoglycans.78 Proteoglycans are remarkable in their diversity. At most sites, ECM may contain several different core proteins, each containing different GAGs. Proteoglycans were originally described as ground substances or mucopolysaccharides, whose main function was to organize the ECM, but it is now recognized that these molecules have diverse roles in regulating connective tissue structure and permeability (Fig. 3-14). Proteoglycans can be integral membrane proteins and, through their binding to other proteins and the activation of growth factors and chemokines, act as modulators of inflammation, immune responses, and cell growth and differentiation.
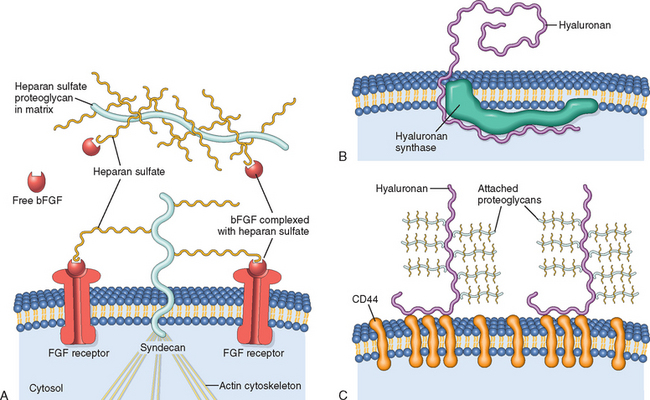
FIGURE 3-14 Proteoglycans, glycosaminoglycans (GAGs), and hyaluronan. A, Regulation of FGF-2 activity by ECM and cellular proteoglycans. Heparan sulfate binds FGF-2 (basic FGF) secreted into the ECM. Syndecan is a cell surface proteoglycan with a transmembrane core protein, extracellular GAG side chains that can bind FGF-2, and a cytoplasmic tail that binds to the actin cytoskeleton. Syndecan side chains bind FGF-2 released by damage to the ECM and facilitate the interaction with cell surface receptors. B, Synthesis of hyaluronan at the inner surface of the plasma membrane. The molecule extends to the extracellular space, while still attached to hyaluronan synthase. C, Hyaluronan chains in the extracellular space are bound to the plasma membrane through the CD44 receptor. Multiple proteoglycans may attach to hyaluronan chains in the ECM.
(B and C, modified from Toole KR: Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer 4:528, 2004.)
There are four structurally distinct families of GAGs: heparan sulfate, chondroitin/dermatan sulfate, keratan sulfate, and hyaluronan (HA). The first three of these families are synthesized and assembled in the Golgi apparatus and rough endoplasmic reticulum as proteoglycans. By contrast, HA is produced at the plasma membrane by enzymes called hyaluronan synthases and is not linked to a protein backbone.
HA is a polysaccharide of the GAG family found in the ECM of many tissues and is abundant in heart valves, skin and skeletal tissues, synovial fluid, the vitreous of the eye, and the umbilical cord.79 It is a huge molecule that consists of many repeats of a simple disaccharide stretched end-to-end. It binds a large amount of water (about 1000-fold its own weight), forming a viscous hydrated gel that gives connective tissue the ability to resist compression forces. HA helps provide resilience and lubrication to many types of connective tissue, notably for the cartilage in joints. Its concentration increases in inflammatory diseases such as rheumatoid arthritis, scleroderma, psoriasis, and osteoarthritis. Enzymes called hyaluronidases fragment HA into lower molecular weight molecules (LMW HA) that have different functions than the parent molecule. LMW HA produced by endothelial cells binds to the CD44 receptor on leukocytes, promoting the recruitment of leukocytes to the sites of inflammation. In addition, LMW HA molecules stimulate the production of inflammatory cytokines and chemokines by white cells recruited to the sites of injury. The leukocyte recruitment process and the production of pro-inflammatory molecules by LMW HA are strictly regulated processes; these activities are beneficial if short-lived, but their persistence may lead to prolonged inflammation.
Healing by Repair, Scar Formation and Fibrosis
If tissue injury is severe or chronic, and results in damage of both parenchymal cells and the stromal framework of the tissue, healing can not be accomplished by regeneration. Under these conditions, the main healing process is repair by deposition of collagen and other ECM components, causing the formation of a scar. In contrast to regeneration which involves the restitution of tissue components, repair is a fibroproliferative response that “patches” rather than restores the tissue. The term scar is most often used in connection to wound healing in the skin, but is also used to describe the replacement of parenchymal cells in any tissue by collagen, as in the heart after myocardial infarction. Repair by connective tissue deposition includes the following basic features:
We will examine all of these events under the context of cutaneous wound healing, as a prototype repair process. Suffice to say here that regardless of site, the inflammatory reaction elicited by the injury contains the damage, removes injured tissue, and promotes the deposition of ECM components in the area of injury, at the same time that angiogenesis is stimulated. However, if the damage persists, inflammation becomes chronic, leading to an excess deposition of connective tissue known as fibrosis. In most healing processes, a combination of repair and regeneration occurs. The relative contributions of repair and regeneration are influenced by: (1) the proliferative capacity of the cells of the tissue; (2) the integrity of the extracellular matrix; and (3) the resolution or chronicity of the injury and inflammation.
Because of the great importance of angiogenesis in processes other than wound healing, we start with a discussion of the mechanisms of angiogenesis before considering the steps of cutaneous wound healing.
MECHANISMS OF ANGIOGENESIS
Angiogenesis is a fundamental process that affects physiologic reactions (e.g. wound healing, regeneration, the vascularization of ischemic tissues, and menstruation), and pathologic processes, such as tumor development and metastasis, diabetic retinopathy, and chronic inflammation. Therefore great efforts have been made to understand the mechanisms of angiogenesis, and to develop agents that have pro- or anti-angiogenic activity.
Around 4000 BC, Egyptian physicians believed that there were “vessels for every part of the body, which are hollow, having a mouth which opens to absorb medications and eliminate waste”.80 Fortunately, our understanding of blood vessels has clearly improved since then.81,82 We know now that blood vessels are assembled during embryonic development by vasculogenesis, in which a primitive vascular network is established from endothelial cell precursors (angioblasts), or from dual hemopoietic/endothelial cell precursors called hemangioblasts. Blood vessel formation in adults, known as angiogenesis or neovascularization, involves the branching and extension of adjacent pre-existing vessels, but it can also occur by recruitment of endothelial progenitor cells (EPCs) from the bone marrow (Fig. 3-15).81
Angiogenesis from Preexisting Vessels.
In this type of angiogenesis there is vasodilation and increased permeability of the existing vessels, degradation of ECM, and migration of endothelial cells. The major steps are listed below.
Angiogenesis from Endothelial Precursor Cells (EPCs).
EPCs can be recruited from the bone marrow into tissues to initiate angiogenesis (Fig. 3-15). The nature of the homing mechanism is uncertain. These cells express some markers of hematopoietic stem cells as well as VEGFR-2, and vascular endothelial–cadherin (VE-cadherin). EPCs may contribute to the re-endothelization of vascular implants and the neovascularization of ischemic organs, cutaneous wounds, and tumors. The number of circulating EPCs increases greatly in patients with ischemic conditions, suggesting that EPCs may influence vascular function and determine the risk of cardiovascular diseases.
Growth Factors and Receptors Involved in Angiogenesis
Despite the diversity of factors that participate in angiogenesis, VEGF is the most important growth factor in adult tissues undergoing physiologic angiogenesis (e.g., proliferating endometrium) as well as angiogenesis occurring in chronic inflammation, wound healing, tumors, and diabetic retinopathy.81,82
As mentioned earlier,58 VEGF is secreted by many mesenchymal and stromal cells. Of the various receptors for VEGF, VEGFR-2, a tyrosine kinase receptor, is the most important in angiogenesis. It is expressed by endothelial cells and their precursors, by other cell types, and by many tumor cells. VEGF, or more specifically, its circulating isoforms VEGF121 and VEGF165, signal through VEGFR-2 (also known as KDR in humans and flk-1 in mice). VEGF induces the migration of EPCs in the bone marrow, and enhances the proliferation and differentiation of these cells at sites of angiogenesis. In angiogenesis originating from preexisting local vessels, VEGF signaling stimulates the survival of endothelial cells, their proliferation and their motility, initiating the sprouting of new capillaries. The main components of the VEGF/VEGFR system and their main actions are listed in Table 3-3. Endothelial cell proliferation, differentiation, and migration can also be stimulated by FGF-2.
Given the multiplicity of VEGF effects and the diverse mechanisms that regulate its expression, how do endothelial cells develop into a perfect pattern of vessels during angiogenesis? One recently identified mechanism for the modulation of vasculogenesis is the Notch pathway, which promotes the proper branching of new vessels and prevents excessive angiogenesis by decreasing the responsiveness to VEGF.83-85 Notch ligands and receptors are membrane-bound molecules conserved between species. In mammals there are five Notch ligands (Jagged 1 and 2, and Delta-like ligand [Dll] 1, 3, and 4) and four transmembrane receptors (Notch 1–4). The receptors contain EGF-like repeats on their extracellular surface that serve as ligand-binding sites (Fig. 3-16). The Delta-like ligand 4 (Dll4) ligand is endothelial cell–specific and is expressed in arteries and capillaries but not in veins; the importance of this ligand is demonstrated by the embryonic lethality of mice that lack a single Delta-like ligand 4 allele. During angiogenesis the leading cell, known as a tip cell, undergoes proliferation and migration, but stalk cells maintain their connection with the existing vessel. VEGF induces Delta-like ligand 4 in tip cells, while Notch1 and Notch4 are expressed in stalk cells (see Fig. 3-16C). The interaction between Delta-like ligand 4 and Notch receptors in adjacent tip and stalk cells leads to a two-step proteolytic cleavage of the receptor, releasing the Notch intracellular domain, which translocates to the nucleus and activates genes that dampen responsiveness to VEGF. Blockade of Delta-like ligand 4 causes increased proliferation of endothelial cells and capillary sprouting; VEGF blockade has the opposite effects and also decreases the survival of endothelial cells (Fig. 3-17).
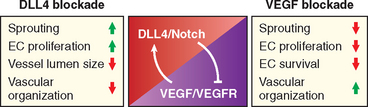
FIGURE 3-17 Interactions between Notch and VEGF during angiogenesis. VEGF stimulates delta-like ligand 4 (Dll4)/Notch, which inhibits VEGFR signaling. Compared with unperturbed angiogenesis, Dll4 blockade causes an increase in capillary sprouting and endothelial cell (EC) proliferation, creating vessels that are disorganized and have a small lumen size. VEGF blockade decreases capillary sprouting, and the proliferation and survival of ECs.
(Courtesy of Minhong Yan, Genentech, San Francisco, CA).
Regardless of the process that leads to capillary formation, newly formed vessels are fragile and need to become “stabilized.” Stabilization requires the recruitment of pericytes and smooth muscle cells (periendothelial cells) and the deposition of ECM proteins. Angiopoietins 1 and 2 (Ang1 and Ang2), PDGF, and TGF-β participate in the stabilization process. Ang1 interacts with a receptor on endothelial cells called Tie2 to recruit periendothelial cells. PDGF participates in the recruitment of smooth muscle cells, while TGF-β stabilizes newly formed vessels by enhancing the production of ECM proteins.58 The Ang1-Tie2 interaction mediates vessel maturation from simple endothelial tubes into more elaborate vascular structures and helps maintain endothelial quiescence. Ang2, in contrast, which also interacts with Tie2, has the opposite effect, making endothelial cells become either more responsive to stimulation by growth factors such as VEGF or, in the absence of VEGF, more responsive to inhibitors of angiogenesis. A telling proof of the importance of these molecules is the existence of a genetic disorder caused by mutations in Tie2 that is characterized by venous malformations. Agents or conditions that stimulate VEGF expression, such as certain cytokines and growth factors (e.g., TGF-β, PDGF, TGF-α), and notably, tissue hypoxia, can influence physiologic and pathologic angiogenesis. VEGF transcription is regulated by the transcription factor HIF, which is induced by hypoxia.
ECM Proteins as Regulators of Angiogenesis
A key component of angiogenesis is the motility and directed migration of endothelial cells, required for the formation of new blood vessels. These processes are controlled by several classes of proteins, including (1) integrins, especially αvβ3, which is critical for the formation and maintenance of newly formed blood vessels, (2) matricellular proteins, including thrombospondin 1, SPARC, and tenascin C, which destabilize cell-matrix interactions and therefore promote angiogenesis, and (3) proteinases, such as the plasminogen activators and MMPs, which are important in tissue remodeling during endothelial invasion. Additionally, these proteinases cleave extracellular proteins, releasing matrix-bound growth factors such as VEGF and FGF-2 that stimulate angiogenesis. Proteinases can also release inhibitors such as endostatin, a small fragment of collagen that inhibits endothelial proliferation and angiogenesis. αVβ3 Integrin expression in endothelial cells is stimulated by hypoxia and has multiple effects on angiogenesis: it interacts with a metalloproteinase (MMP-2, discussed below), it binds to and regulates the activity of VEGFR-2, and it mediates adhesion to ECM components such as fibronectin, thrombospondin, and OPN.72
The review of ECM components, cell-matrix interactions, and the mechanisms of angiogenesis sets the stage for the discussion of tissue healing that involves repair and scar formation, with special emphasis on the steps and main mechanisms of cutaneous wound healing.
CUTANEOUS WOUND HEALING
Cutaneous wound healing is divided into three phases: inflammation, proliferation, and maturation86 (Fig. 3-18). These phases overlap, and their separation is somewhat arbitrary, but they help to understand the sequence of events that take place in the healing of skin wounds. The initial injury causes platelet adhesion and aggregation and the formation of a clot in the surface of the wound, leading to inflammation. In the proliferative phase there is formation of granulation tissue, proliferation and migration of connective tissue cells, and re-epithelialization of the wound surface. Maturation involves ECM deposition, tissue remodeling, and wound contraction.
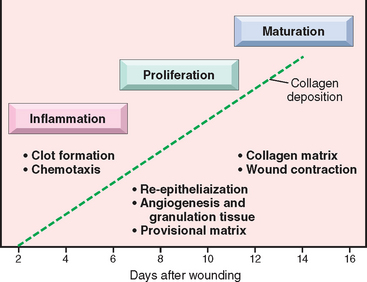
FIGURE 3-18 Phases of cutaneous wound healing: inflammation, proliferation, and maturation (see text for details).
(Modified from Broughton G et al: The basic science of wound healing. Plast Reconstr Surg 117:12S–34S, 2006.)
The simplest type of cutaneous wound repair is the healing of a clean, uninfected surgical incision approximated by surgical sutures (Fig. 3-19). Such healing is referred to as healing by primary union or by first intention.86-88 The incision causes death of a limited number of epithelial and connective tissue cells and disruption of epithelial basement membrane continuity. Re-epithelialization to close the wound occurs with formation of a relatively thin scar. The repair process is more complicated in excisional wounds that create large defects on the skin surface, causing extensive loss of cells and tissue. The healing of these wounds involves a more intense inflammatory reaction, the formation of abundant granulation tissue (described below), and extensive collagen deposition, leading to the formation of a substantial scar, which generally contracts. This form of healing is referred to as healing by secondary union or by second intention (see Figs. 3-19 and 3-20). Despite these differences, the basic mechanisms of healing by primary (first intention) and secondary (second intention) union are similar. They are described together and the differences will be indicated where appropriate.

FIGURE 3-19 Wound healing and scar formation. A, Healing of wound that caused little loss of tissue: note the small amount of granulation tissue, and formation of a thin scar with minimal contraction. B, Healing of large wound: note large amounts of granulation tissue and scar tissue, and wound contraction.
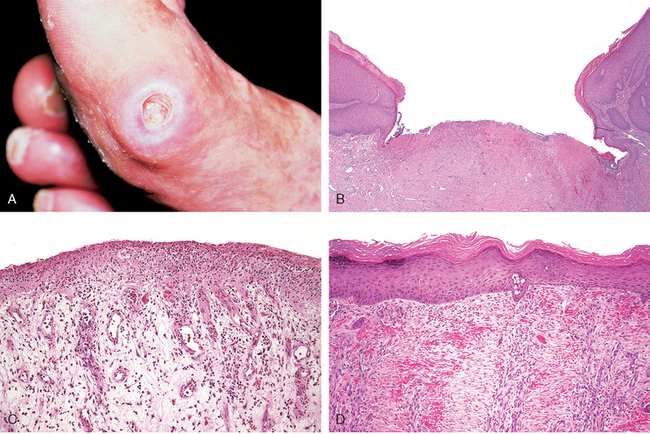
FIGURE 3-20 Healing of skin ulcers. A, Pressure ulcer of the skin, commonly found in diabetic patients. The histologic slides show: B, a skin ulcer with a large gap between the edges of the lesion; C, a thin layer of epidermal re-epithelialization and extensive granulation tissue formation in the dermis; and D, continuing re-epithelialization of the epidermis and wound contraction.
(Courtesy of Z. Argenyi, MD, University of Washington, Seattle, WA.)
A large number of growth factors and cytokines are involved in cutaneous wound healing.89 The main agents, and the steps at which they participate in the repair process, are listed in Table 3-4. We next discuss the sequence of events in wound healing.
TABLE 3-4 Growth Factors and Cytokines Affecting Various Steps in Wound Healing
| Monocyte chemotaxis | Chemokines, TNF, PDGF, FGF, TGF-β |
| Fibroblast migration/replication | PDGF, EGF, FGF, TGF-β, TNF, IL-1 |
| Keratinocyte replication | HB-EGF, FGF-7, HGF |
| Angiogenesis | VEGF, angiopoietins, FGF |
| Collagen synthesis | TGF-β, PDGF |
| Collagenase secretion | PDGF, FGF, TNF; TGF-β inhibits |
HB-EGF, heparin-binding EGF; IL-1, interleukin 1; TNF, tumor necrosis factor; other abbreviations as given in Table 3-1.
Formation of Blood Clot.
Wounding causes the rapid activation of coagulation pathways, which results in the formation of a blood clot on the wound surface (Chapter 4). In addition to entrapped red cells, the clot contains fibrin, fibronectin, and complement components. The clot serves to stop bleeding and also as a scaffold for migrating cells, which are attracted by growth factors, cytokines and chemokines released into the area.89 Release of VEGF leads to increased vessel permeability and edema. However, dehydration occurs at the external surface of the clot, forming a scab that covers the wound. In wounds causing large tissue deficits, the fibrin clot is larger, and there is more exudate and necrotic debris in the wounded area. Within 24 hours, neutrophils appear at the margins of the incision, and use the scaffold provided by the fibrin clot to march in. They release proteolytic enzymes that clean out debris and invading bacteria.
Formation of Granulation Tissue.
Fibroblasts and vascular endothelial cells proliferate in the first 24 to 72 hours of the repair process to form a specialized type of tissue called granulation tissue, which is a hallmark of tissue repair. The term derives from its pink, soft, granular appearance on the surface of wounds. Its characteristic histologic feature is the presence of new small blood vessels (angiogenesis) and the proliferation of fibroblasts (Fig. 3-21). These new vessels are leaky, allowing the passage of plasma proteins and fluid into the extravascular space. Thus, new granulation tissue is often edematous. Granulation tissue progressively invades the incision space; the amount of granulation tissue that is formed depends on the size of the tissue deficit created by the wound and the intensity of inflammation. Hence, it is much more prominent in healing by secondary union. By 5 to 7 days, granulation tissue fills the wound area and neovascularization is maximal. The mechanisms of angiogenesis in repair processes were discussed earlier in this chapter.
FIGURE 3-21 A, Granulation tissue showing numerous blood vessels, edema, and a loose ECM containing occasional inflammatory cells. Collagen is stained blue by the trichrome stain; minimal mature collagen can be seen at this point. B, Trichrome stain of mature scar, showing dense collagen, with only scattered vascular channels.
Cell Proliferation and Collagen Deposition.
Neutrophils are largely replaced by macrophages by 48 to 96 hours. Macrophages are key cellular constituents of tissue repair, clearing extracellular debris, fibrin, and other foreign material at the site of repair, and promoting angiogenesis and ECM deposition (Fig. 3-22).
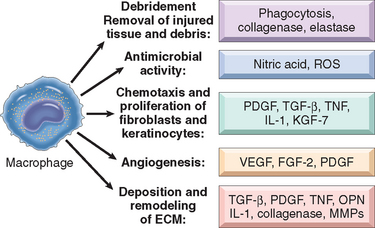
FIGURE 3-22 Multiple roles of macrophages in wound healing. Macrophages participate in wound debridement, have antimicrobial activity, stimulate chemotaxis and the activation of inflammatory cells and fibroblasts, promote angiogenesis, and stimulate matrix remodeling and synthesis. ROS, reactive oxygen species.
Migration of fibroblasts to the site of injury is driven by chemokines, TNF, PDGF, TGF-β, and FGF. Their subsequent proliferation is triggered by multiple growth factors, including PDGF, EGF, TGF-β, FGF, and the cytokines IL-1 and TNF (see Table 3-4). Macrophages are the main source for these factors, although other inflammatory cells and platelets may also produce them. Collagen fibers are now present at the margins of the incision, but at first these are vertically oriented and do not bridge the incision. In 24 to 48 hours, spurs of epithelial cells move from the wound edge (initially with little cell proliferation) along the cut margins of the dermis, depositing basement membrane components as they move. They fuse in the midline beneath the surface scab, producing a thin, continuous epithelial layer that closes the wound. Full epithelialization of the wound surface is much slower in healing by secondary union because the gap to be bridged is much greater. Subsequent epithelial cell proliferation thickens the epidermal layer. Macrophages stimulate fibroblasts to produce FGF-7 (keratinocyte growth factor) and IL-6, which enhance keratinocyte migration and proliferation. Other mediators of re-epithelialization are HGF and HB-EGF.89 Signaling through the chemokine receptor CXCR 3 also promotes skin re-epithelialization.
Concurrently with epithelialization, collagen fibrils become more abundant and begin to bridge the incision. At first a provisional matrix containing fibrin, plasma fibronectin, and type III collagen is formed, but this is replaced by a matrix composed primarily of type I collagen. TGF-β is the most important fibrogenic agent (see Table 3-4). It is produced by most of the cells in granulation tissue and causes fibroblast migration and proliferation, increased synthesis of collagen and fibronectin, and decreased degradation of ECM by metalloproteinases. The epidermis recovers its normal thickness and architecture, and surface keratinization.
Scar Formation.
The leukocytic infiltrate, edema, and increased vascularity largely disappear during the second week. Blanching begins, accomplished by the increased accumulation of collagen within the wound area and regression of vascular channels. Ultimately, the original granulation tissue scaffolding is converted into a pale, avascular scar, composed of spindle-shaped fibroblasts, dense collagen, fragments of elastic tissue, and other ECM components. The dermal appendages that have been destroyed in the line of the incision are permanently lost, although in rats new hair follicles may develop in large healing wounds under Wnt stimulation.90 This result suggests that, with appropriate treatment procedures, regrowth of skin appendages during wound healing might be achieved in humans. By the end of the first month, the scar is made up of acellular connective tissue devoid of inflammatory infiltrate, covered by intact epidermis.
Wound Contraction.
Wound contraction generally occurs in large surface wounds. The contraction helps to close the wound by decreasing the gap between its dermal edges and by reducing the wound surface area. Hence, it is an important feature in healing by secondary union. The initial steps of wound contraction involve the formation, at the edge of the wound, of a network of myofibroblasts that express smooth muscle α-actin and vimentin. These cells have ultrastructural characteristics of smooth muscle cells, contract in the wound tissue, and may produce large amounts of ECM components (see discussion of hypertrophic scars in this chapter), such as type I collagen, tenascin-C, SPARC, and extra-domain fibronectin.91 Myofibroblasts are formed from tissue fibroblasts through the effects of PDGF, TGF-β, and FGF-2 released by macrophages at the wound site, but they can also originate from bone marrow precursors known as fibrocytes, or from epithelial cells, through the process of epithelial-to-mesenchymal transition.
Connective Tissue Remodeling.
The replacement of granulation tissue with a scar involves changes in the composition of the ECM. The balance between ECM synthesis and degradation results in remodeling of the connective tissue framework – an important feature of tissue repair. Some of the growth factors that stimulate synthesis of collagen and other connective tissue molecules also modulate the synthesis and activation of metalloproteinases, enzymes that degrade these ECM components.
Degradation of collagen and other ECM proteins is achieved by matrix metalloproteinases (MMPs), a family of enzymes that includes more than 20 members that have in common a 180-residue zinc-protease domain (MMPs should be distinguished from neutrophil elastase, cathepsin G, kinins, plasmin, and other important proteolytic enzymes, which also degrade EMC components and which are serine proteinases, not metalloenzymes). Matrix metalloproteinases include interstitial collagenases (MMP-1, -2, and -3), which cleave the fibrillar collagen types I, II, and III; gelatinases (MMP-2 and 9), which degrade amorphous collagen as well as fibronectin; stromelysins (MMP-3, 10, and 11), which act on a variety of ECM components, including proteoglycans, laminin, fibronectin, and amorphous collagens; and the family of membrane-bound MMPs (ADAMs) described below. MMPs are produced by fibroblasts, macrophages, neutrophils, synovial cells, and some epithelial cells. Their secretion is induced by growth factors (PDGF, FGF), cytokines (IL-1, TNF), and phagocytosis in macrophages, and is inhibited by TGF-β and steroids. Collagenases cleave collagen under physiologic conditions. They are synthesized as a latent precursor (procollagenase) that is activated by chemicals, such as free radicals produced during the oxidative burst of leukocytes, and proteinases (plasmin). Once formed, activated collagenases are rapidly inhibited by a family of specific tissue inhibitors of metalloproteinases, which are produced by most mesenchymal cells, thus preventing uncontrolled action of these proteases. Collagenases and their inhibitors are essential in the debridement of injured sites and in the remodeling of connective tissue necessary to repair the defect.
A large and important family of enzymes related to MMPs is called ADAM (disintegrin and metalloproteinase-domain family). Most ADAMs are anchored by a single transmembrane domain to cell surface. ADAM-17 (also known as TACE, for TNF-converting enzyme) cleaves the membrane-bound precursor forms of TNF and TGF-α, releasing the active molecules. ADAM-17 deficiency in mice causes embryonic or neonatal lethality associated with pulmonary hypoplasia. Members of the ADAM family are also involved in the pathogenesis of bronchial asthma (Chapter 15), and thrombotic microangiopathies (Chapter 13).
Recovery of Tensile Strength.
Fibrillar collagens (mostly type I collagen) form a major portion of the connective tissue in repair sites and are essential for the development of strength in healing wounds. Net collagen accumulation, however, depends not only on increased collagen synthesis but also on decreased degradation. How long does it take for a skin wound to achieve its maximal strength? When sutures are removed from an incisional surgical wound, usually at the end of the first week, wound strength is approximately 10% that of unwounded skin. Wound strength increases rapidly over the next 4 weeks, slows down at approximately the third month after the original incision, and reaches a plateau at about 70% to 80% of the tensile strength of unwounded skin. Lower tensile strength in the healed wound area may persist for life. The recovery of tensile strength results from the excess of collagen synthesis over collagen degradation during the first 2 months of healing, and, at later times, from structural modifications of collagen fibers (cross-linking, increased fiber size) after collagen synthesis ceases.
LOCAL AND SYSTEMIC FACTORS THAT INFLUENCE WOUND HEALING
The adequacy of wound repair may be impaired by systemic and local host factors.
Systemic factors include those listed below:
Local factors that influence healing include those listed here:
PATHOLOGIC ASPECTS OF REPAIR
Complications in wound healing can arise from abnormalities in any of the basic components of the repair process. These aberrations can be grouped into three general categories: (1) deficient scar formation, (2) excessive formation of the repair components, and (3) formation of contractures.
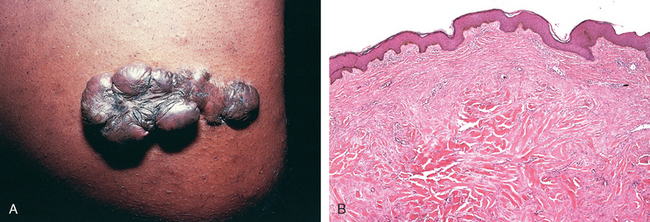
FIGURE 3-23 Keloid. A, Excess collagen deposition in the skin forming a raised scar known as keloid. B, Note the thick connective tissue deposition in the dermis.
(A, from Murphy GF, Herzberg AJ: Atlas of Dermatopathology. Philadelphia, WB Saunders, 1996, p 219; B, courtesy of Z. Argenyi, MD, University of Washington, Seattle, WA.)
Exuberant granulation is another deviation in wound healing consisting of the formation of excessive amounts of granulation tissue, which protrudes above the level of the surrounding skin and blocks re-epithelialization (this process has been called, with more literary fervor, proud flesh). Excessive granulation must be removed by cautery or surgical excision to permit restoration of the continuity of the epithelium. Finally (fortunately rarely), incisional scars or traumatic injuries may be followed by exuberant proliferation of fibroblasts and other connective tissue elements that may, in fact, recur after excision. Called desmoids, or aggressive fibromatoses, these lie in the interface between benign proliferations and malignant (though low-grade) tumors. The line between the benign hyperplasias characteristic of repair and neoplasia is frequently finely drawn (Chapter 7).
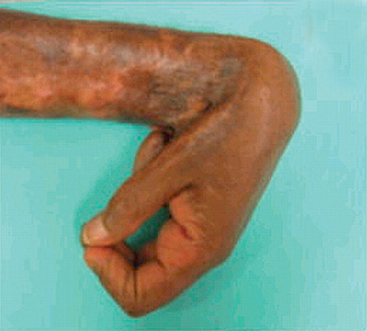
FIGURE 3-24 Wound contracture. Severe contracture of a wound after deep burn injury.
(From Aarabi S et al: Hypertrophic scar formation following burns and trauma: new approaches to treatment. PLOS Med 4:e234, 2007.)
Fibrosis
Deposition of collagen is part of normal wound healing. However, the term fibrosis is used more broadly to denote the excessive deposition of collagen and other ECM components in a tissue. As already mentioned, the terms scar and fibrosis are used interchangeably, but fibrosis most often indicates the deposition of collagen in chronic diseases. The basic mechanisms that occur in the development of fibrosis associated with chronic inflammatory diseases are generally similar to the mechanisms of skin wound healing that have been discussed in this chapter. However, in contrast to the short-lived stimulus that triggers the orderly steps of wound healing in the skin, the injurious stimulus caused by infections, autoimmune reactions, trauma, and other types of tissue injury persists in chronic diseases, causing organ dysfunction and often organ failure.
The persistence of injury leads to chronic inflammation, which is associated with the proliferation and activation of macrophages and lymphocytes, and the production of a plethora of inflammatory and fibrogenic growth factors and cytokines, mentioned earlier and summarized in Figure 3-25.
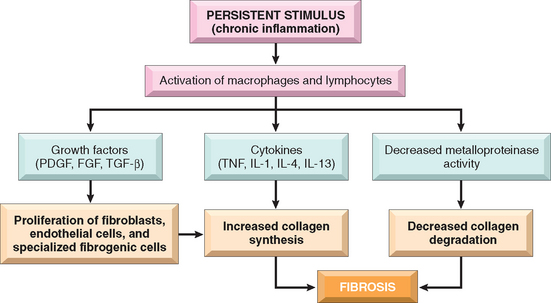
FIGURE 3-25 Development of fibrosis in chronic inflammation. The persistent stimulus of chronic inflammation activates macrophages and lymphocytes, leading to the production of growth factors and cytokines, which increase the synthesis of collagen. Deposition of collagen is enhanced by decreased activity of metalloproteinases.
The host response to harmful stimuli is orchestrated to first clear these stimuli and then heal the damage. As we discussed in Chapter 2 (see Fig. 2-10), the initial wave of the host response to external invaders and tissue injury generates “classically activated macrophages”, which are effective in ingesting and destroying microbes and dead tissues. This is followed by the accumulation of “alternatively activated macrophages”, which suppress the microbicidal activities and instead function to remodel tissues and promote angiogenesis and scar formation.93 The cytokines that induce classical macrophage activation are those produced by TH1 cells, notably IFN-γ and TNF, whereas alternative macrophage activation is best induced by IL-4 and IL-13, cytokines produced by TH2 cells and other cells including mast cells and eosinophils. Alternatively activated macrophages produce TGF-β and other growth factors that are involved in the repair process.
TGF-β is practically always involved as an important fibrogenic agent (see Table 3-4) in these diseases, regardless of the original cause. It is produced by most of the cells in granulation tissue and causes fibroblast migration and proliferation, increased synthesis of collagen and fibronectin, and decreased degradation of ECM due to inhibition of metalloproteinases. The levels of TGF-β in tissues are not primarily regulated by the transcription of the gene but depend on the post-transcriptional activation of latent TGF-β, the rate of secretion of the active molecule, and factors in the ECM that enhance or diminish TGF-β activity.
The mechanisms that lead to the activation of TGF-β in fibrosis are not precisely known, but cell death by necrosis or apoptosis and the production of reactive oxygen species seem to be important triggers of the activation, regardless of the tissue. Similarly, the cells that produce collagen under TGF-β stimulation may vary depending on the tissue. In most cases, such as in lung and kidney fibrosis, myofibroblasts (already discussed in this chapter) are the main source of collagen, but stellate cells are the major collagen producers in liver cirrhosis.
Recent studies provide evidence for an important role for osteopontin (OPN) in would healing and fibrosis.77 OPN is strongly expressed in fibrosis of the heart, lung, liver, kidney and some other tissues. In animal experiments, blockage of OPN expression during wound healing decreases the formation of granulation tissue and scarring.94 Although the mechanisms by which OPN promotes fibrosis are not completely understood, recent data show that OPN is a mediator of the differentiation of myofibroblasts induced by TGF-β.
In marked contrast to wounds in adults, fetal cutaneous wounds heal without scar formation.95,96 Several factors have been proposed to promote scarless healing, including the secretion of non-fibrogenic forms of TGF-β, the lack of osteopontin, and the absence of a TH2 response, but no definitive results have been obtained. Given the serious organ dysfunction produced by fibrosis, there is a major effort to develop clinically useful antifibrotic agents. Among agents being tested are inhibitors of TGF-β binding or signaling, angiogenesis inhibitors, Toll-like receptors antagonists, and the decoy receptor IL-13Rα2 which blocks IL-13.
Fibrotic disorders include diverse diseases such as liver cirrhosis, systemic sclerosis, fibrosing diseases of the lung (idiopathic pulmonary fibrosis, pneumoconioses, and drug-, radiation-induced pulmonay fibrosis), chronic pancreatitis, glomerulonephritis, and constrictive pericarditis. These conditions are discussed in the appropriate chapters throughout the book.
This concludes the discussion, begun in Chapter 1, of cellular and tissue injury, the inflammatory reaction to such injury (Chapter 2), and tissue healing by regeneration and repair. An overview of the relationships between these processes is illustrated in Fig. 3-26.
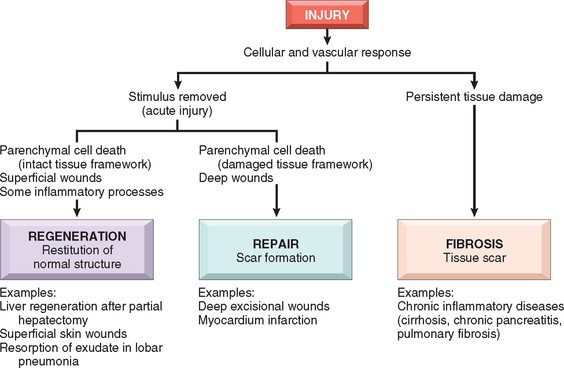
1 Goss RJ. Regeneration versus repair. In: Cohen IK, Diegelman RF, Lindblad WJ, editors. Wound Healing: Biochemical and Clinical Aspects. Philadelphia: WB Saunders; 1992:20.
2 Fausto N, Campbell JS. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev. 2003;120:117.
3 Mimeault M, et al. Stem cells: a revolution in therapeutics—recent advances in stem cell biology and their therapeutic applications in regenerative medicine and cancer therapies. Clin Pharmacol Ther. 2007;82:252.
4 Ott HC, et al. Perfusion-decellularized matrix: using nature’s platform to engineer a bioartificial heart. Nat Med. 2008;14:213.
5 Pellettieri J, Alvarado AS. Cell turnover and adult tissue homeostasis: from humans to planarians. Annu Rev Genet. 2007;41:83.
6 Park IH, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877.
7 Gardner RL. Stem cells and regenerative medicine: principles, prospects and problems. C R Biol. 2007;330:465.
8 Schroeder M. Asymmetric cell division in normal and malignant hematopoietic precursor cells. Cell Stem Cell. 2007;1:479.
9 Conover JC, Notti RQ. The neural stem cell niche. Cell Tissue Res. 2008;331:211.
10 Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880.
11 Xie T, Li L. Stem cells and their niche: an inseparable relationship. Development. 2007;134:2001.
12 Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861.
13 Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1916.
14 Wu DC, et al. Embryonic stem cell transplantation: potential applicability in cell replacement therapy and regenerative medicine. Front Biosci. 2007;12:4525.
15 Laflamme MA, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015.
16 Manis JP. Knock out, knock in, knock down—genetically manipulated mice and the Nobel Prize. N Engl J Med. 2007;357:2426.
17 Paterson L, et al. Application of reproductive biotechnology in animals: implications and potentials. Applications of reproductive cloning. Anim Reprod Sci. 2003;79:137.
18 Han Z, et al. Therapeutic cloning: status and prospects. Curr Opin Mol Ther. 2007;9:392.
19 Okita K, et al. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313.
20 Lowry WE, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci USA. 2008;105:2883.
21 Chambers I, et al. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230.
22 Chen L, Daley GQ. Molecular basis of pluripotency. Hum Mol Genet. 2008;17:R23.
23 Hanna J, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920.
24 Aoi T, et al. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 2008;321:699.
25 Hanna J, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133:250.
26 Nakagawa M, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101.
27 Barker N, et al. Identification of stem cells in the small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003.
28 Thorgeirsson SS, Grisham JW. Hematopoietic cells as hepatocyte stem cells: a critical review of the evidence. Hepatology. 2006;43:2.
29 Rizvi AZ, et al. Bone marrow–derived cells fuse with normal and transformed intestinal stem cells. Proc Natl Acad Sci U S A. 2006;103:6321.
30 Camargo FD, et al. Hematopoietic myelomonocytic cells are the major source of hepatocyte fusion partners. J Clin Invest. 2004;113:1266.
31 Vieyra DS, et al. Plasticity and tissue regenerative potential of bone marrow–derived cells. Stem Cell Rev. 2005;1:65.
32 Massberg S, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994.
33 Bryder D, et al. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am J Pathol. 2006;169:338.
34 Levesque JP, Winkler IG. Mobilization of hematopoietic stem cells: state of the art. Curr Opin Organ Transplant. 2008;13:53.
35 Fox JM, et al. Recent advances into the understanding of mesenchymal stem cell trafficking. Br J Haematol. 2007;137:491.
36 Caplan AI. Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J Cell Physiol. 2007;213:341.
37 Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology. 2004;39:1477.
38 Taupin P. Adult neural stem cells, neurogenic niches, and cellular therapy. Stem Cell Rev. 2006;2:213.
39 Okano H, et al. Regeneration of the central nervous system using endogenous repair mechanisms. J Neurochem. 2007;102:1459.
40 Sieber-Blum M, Hu Y. Epidermal neural crest stem cells (EPI-NCSC) and pluripotency. Stem Cell Rev. August 2008. Epub.
41 Fuchs E. Skin stem cells: rising to the surface. J Cell Biol. 2008;180:273.
42 Braun KM, Prowse DM. Distinct epidermal stem cell compartments are maintained by independent niche microenvironments. Stem Cell Rev. 2006;2:221.
43 Watt FM, et al. Epidermal stem cells: an update. Curr Opin Genet Dev. 2006;16:518.
44 Ohyama M, et al. Characterization and isolation of stem cell–enriched human hair follicle bulge cells. J Clin Invest. 2006;116:249.
45 Yen TH, Wright NA. The gastrointestinal tract stem cell niche. Stem Cell Rev. 2006;2:203.
46 Barker N, et al. The intestinal stem cell. Genes Dev. 2008;22:1856.
47 Shi X, Garry DJ. Muscle stem cells in development, regeneration, and disease. Genes Dev. 2006;20:1692.
48 Hsieh PCH, et al. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970.
49 Murry CE. Cardiac aid to the injured but not the elderly? Nat Med. 2007;13:901.
50 Daniels JT, et al. Corneal epithelial stem cells in health and disease. Stem Cell Rev. 2006;2:247.
51 Djojosubroto MW, Arsenijevic Y. Retinal stem cells: promising candidates for retina transplantation. Cell Tissue Res. 2008;331:347.
52 Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298.
53 Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19:238.
54 Ashwell S, Zabludoff S. DNA damage detection and repair pathways—recent advances with inhibitors of checkpoint kinases in cancer therapy. ClinCancer Res. 2008;14:4032.
55 Zandi R, et al. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal. 2007;19:2013.
56 Conway K, et al. The molecular and clinical impact of hepatocyte growth factor, its receptor, activators, and inhibitors in wound healing. Wound Repair Regen. 2006;14:2.
57 Andrae J, et al. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276.
58 Nagy JA, et al. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007;2:251.
59 Itoh N. The Fgf families in humans, mice, and zebrafish: their evolutional processes and roles in development, metabolism, and disease. Biol Pharm Bull. 2007;30:1819.
60 Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506.
61 Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911.
62 Rawlings JS, et al. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281.
63 Deupi X, Kobilka B. Activation of G protein–coupled receptors. Adv Protein Chem. 2007;74:137.
64 Ahmed W, et al. PPARs and their metabolic modulation: new mechanisms for transcriptional regulation? J Intern Med. 2007;262:184.
65 Thompson MD, Monga SP. WNT/beta-catenin signaling in liver health and disease. Hepatology. 2007;45:1298.
66 Stoick-Cooper CL, et al. Advances in signaling in vertebrate regeneration as a prelude to regenerative medicine. Genes Dev. 2007;21:1292.
67 Zhao M, et al. Evidence for the presence of stem cell-like progenitor cells in human adult pancreas. J Endocrinol. 2007;195:407.
68 Zhou Q, et al. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. Aug 2008. Epub.
69 Fausto N, et al. Liver regeneration. Hepatology. 2006;43:S45.
70 Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836.
71 LeBleu VS, et al. Structure and function of basement membranes. Exp Biol Med (Maywood). 2007;232:112.
72 Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33.
73 Byers PH, et al. Genetic evaluation of suspected osteogenesis imperfecta (OI). Genet Med. 2006;8:383.
74 Robinson PN, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006;43:769.
75 Morgan MR, et al. Synergistic control of cell adhesion by integrins and syndecans. Nat Rev Mol Cell Biol. 2007;8:957.
76 Scatena M, et al. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:2302.
77 Lenga Y, et al. Osteopontin expression is required for myofibroblast differentiation. Circ Res. 2008;102:319.
78 Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2006;20:9.
79 Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528.
80 Carmeliet P. Manipulating angiogenesis in medicine. J Intern Med. 2004;255:538.
81 Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932.
82 Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464.
83 Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039.
84 Holderfield MT, Hughes CCW. Crosstalk between vascular endothelial growth factor, Notch and transforming growth factor-β in vascular morphogenesis. Circ Res. 2008;102:637.
85 Siekmann AF, et al. Modulation of VEGF signaling output by the NOTCH pathway. BioEssays. 2008;30:303.
86 Broughton G, et al. The basic science of wound healing. Plast Reconstr Surg. 2006;117:12S.
87 Gurtner GC, et al. Wound repair and regeneration. Nature. 2008;453:314.
88 Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci. 2004;9:283.
89 Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83:835.
90 Ito M, et al. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature. 2007;447:316.
91 Darby IA, Hewitson TD. Fibroblast differentiation in wound healing and fibrosis. Int Rev Cytol. 2007;257:143.
92 Dabiri G, et al. Hic-5 promotes the hypertrophic scar myofibroblast phenotype by regulating the TGF-β1 autocrine loop. J Invest Dermatol [serial on the internet]. 10 April 2008. 10.1038/jid.2008.90.
93 Martinez FO, et al. Macrophage activation and polarization. Front Biosci. 2008;13:453.
94 Mori R, et al. Molecular mechanisms linking wound inflammation and fibrosis: knockdown of osteopontin leads to rapid repair and reduced scarring. J Exp Med. 2008;205:43.
95 Ferguson MW, O’Kane S. Scar-free healing: from embryonic mechanisms to adult therapeutic intervention. Philos Trans R Soc Lond B Biol Sci. 2004;359:839.
96 Hantash BM, et al. Adult and fetal wound healing. Front Biosci. 2008;13:51.