Chapter 7 Neoplasia
Cancer is the second leading cause of death in the United States; only cardiovascular diseases exact a higher toll. Even more agonizing than the mortality rate is the emotional and physical suffering inflicted by neoplasms. Patients and the public often ask, “When will there be a cure for cancer?” The answer to this simple question is difficult, because cancer is not one disease but many disorders that share a profound growth dysregulation. Some cancers, such as Hodgkin lymphoma, are curable, whereas others, such as pancreatic adenocarcinoma, have a high mortality. The only hope for controlling cancer lies in learning more about its cause and pathogenesis, and great strides have been made in understanding its molecular basis. Indeed, some good news has emerged: cancer mortality for both men and women in the United States declined during the last decade of the twentieth century and has continued its downward course in the 21st.1 The discussion that follows deals with both benign and malignant tumors, focusing on the basic morphologic and biologic properties of tumors and the molecular basis of carcinogenesis. We also discuss the interactions of the tumor with the host and the host response to tumors.
Nomenclature
Neoplasia means “new growth,” and a new growth is called a neoplasm. Tumor originally applied to the swelling caused by inflammation, but the non-neoplastic usage of tumor has almost vanished; thus, the term is now equated with neoplasm. Oncology (Greek oncos = tumor) is the study of tumors or neoplasms.
Although all physicians know what they mean when they use the term neoplasm, it has been surprisingly difficult to develop an accurate definition. The eminent British oncologist Willis2 has come closest: “A neoplasm is an abnormal mass of tissue, the growth of which exceeds and is uncoordinated with that of the normal tissues and persists in the same excessive manner after cessation of the stimuli which evoked the change.” We know that the persistence of tumors, even after the inciting stimulus is gone, results from genetic alterations that are passed down to the progeny of the tumor cells. These genetic changes allow excessive and unregulated proliferation that becomes autonomous (independent of physiologic growth stimuli), although tumors generally remain dependent on the host for their nutrition and blood supply. As we shall discuss later, the entire population of neoplastic cells within an individual tumor arises from a single cell that has incurred genetic change, and hence tumors are said to be clonal.
A tumor is said to be benign when its microscopic and gross characteristics are considered relatively innocent, implying that it will remain localized, it cannot spread to other sites, and it is generally amenable to local surgical removal; the patient generally survives. It should be noted, however, that benign tumors can produce more than localized lumps, and sometimes they are responsible for serious disease.
Malignant tumors are collectively referred to as cancers, derived from the Latin word for crab, because they adhere to any part that they seize on in an obstinate manner, similar to a crab. Malignant, as applied to a neoplasm, implies that the lesion can invade and destroy adjacent structures and spread to distant sites (metastasize) to cause death. Not all cancers pursue so deadly a course. Some are discovered early and are treated successfully, but the designation malignant always raises a red flag.
All tumors, benign and malignant, have two basic components: (1) clonal neoplastic cells that constitute their parenchyma and (2) reactive stroma made up of connective tissue, blood vessels, and variable numbers of macrophages and lymphocytes. Although the neoplastic cells largely determine a tumor’s behavior and pathologic consequences, their growth and evolution is critically dependent on their stroma. An adequate stromal blood supply is requisite for the tumor cells to live and divide, and the stromal connective tissue provides the structural framework essential for the growing cells. In addition, there is cross-talk between tumor cells and stromal cells that directly influences the growth of tumors. In some tumors, the stromal support is scant and so the neoplasm is soft and fleshy. In other cases the parenchymal cells stimulate the formation of an abundant collagenous stroma, referred to as desmoplasia. Some demoplastic tumors—for example, some cancers of the female breast—are stony hard or scirrhous. The nomenclature of tumors and their biologic behavior are based primarily on the parenchymal component.
Benign Tumors.
In general, benign tumors are designated by attaching the suffix -oma to the cell of origin. Tumors of mesenchymal cells generally follow this rule. For example, a benign tumor arising in fibrous tisssue is called a fibroma, whereas a benign cartilaginous tumor is a chondroma. In contrast, the nomenclature of benign epithelial tumors is more complex. These are variously classified, some based on their cells of origin, others on microscopic pattern, and still others on their macroscopic architecture.
Adenoma is applied to a benign epithelial neoplasm derived from glands, although they may or may not form glandular structures. On this basis, a benign epithelial neoplasm that arises from renal tubular cells growing in the form of numerous tightly clustered small glands would be termed an adenoma, as would a heterogeneous mass of adrenal cortical cells growing as a solid sheet. Benign epithelial neoplasms producing microscopically or macroscopically visible finger-like or warty projections from epithelial surfaces are referred to as papillomas. Those that form large cystic masses, as in the ovary, are referred to as cystadenomas. Some tumors produce papillary patterns that protrude into cystic spaces and are called papillary cystadenomas. When a neoplasm, benign or malignant, produces a macroscopically visible projection above a mucosal surface and projects, for example, into the gastric or colonic lumen, it is termed a polyp (Fig. 7-1).
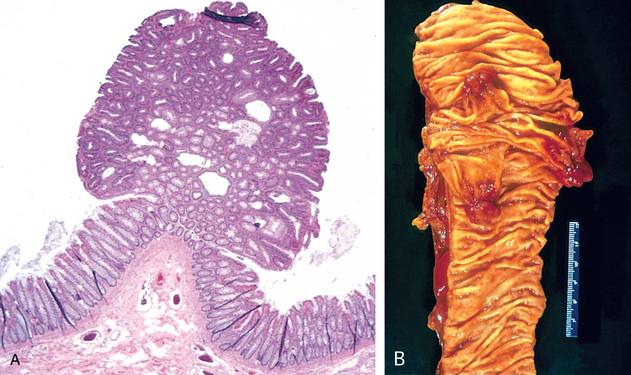
Malignant Tumors.
The nomenclature of malignant tumors essentially follows the same schema used for benign neoplasms, with certain additions. Malignant tumors arising in mesenchymal tissue are usually called sarcomas (Greek sar = fleshy), because they have little connective tissue stroma and so are fleshy (e.g., fibrosarcoma, chondrosarcoma, leiomyosarcoma, and rhabdomyosarcoma). Malignant neoplasms of epithelial cell origin, derived from any of the three germ layers, are called carcinomas. Thus, cancer arising in the epidermis of ectodermal origin is a carcinoma, as is a cancer arising in the mesodermally derived cells of the renal tubules and the endodermally derived cells of the lining of the gastrointestinal tract. Carcinomas may be further qualified. Squamous cell carcinoma would denote a cancer in which the tumor cells resemble stratified squamous epithelium, and adenocarcinoma denotes a lesion in which the neoplastic epithelial cells grow in glandular patterns. Sometimes the tissue or organ of origin can be identified, as in the designation of renal cell adenocarcinoma or bronchogenic squamous cell carcinoma. Not infrequently, however, a cancer is composed of undifferentiated cells of unknown tissue origin, and must be designated merely as an undifferentiated malignant tumor.
In many benign and malignant neoplasms, the parenchymal cells bear a close resemblance to each other, as though all were derived from a single cell. Indeed, neoplasms are of monoclonal origin, as is documented later. Infrequently, divergent differentiation of a single neoplastic clone along two lineages creates what are called mixed tumors. The best example of this is the mixed tumor of salivary gland origin. These tumors contain epithelial components scattered within a myxoid stroma that sometimes contains islands of cartilage or bone (Fig. 7-2). All these elements, it is believed, arise from a single clone capable of giving rise to epithelial and myoepithelial cells; thus, the preferred designation of these neoplasms is pleomorphic adenoma. The great majority of neoplasms, even mixed tumors, are composed of cells representative of a single germ layer. The multifaceted mixed tumors should not be confused with a teratoma, which contains recognizable mature or immature cells or tissues representative of more than one germ cell layer and sometimes all three. Teratomas originate from totipotential cells such as those normally present in the ovary and testis and sometimes abnormally present in sequestered midline embryonic rests. Such cells have the capacity to differentiate into any of the cell types found in the adult body and so, not surprisingly, may give rise to neoplasms that mimic, in a helter-skelter fashion, bits of bone, epithelium, muscle, fat, nerve, and other tissues. When all the component parts are well differentiated, it is a benign (mature) teratoma; when less well differentiated, it is an immature, potentially or overtly, malignant teratoma. A particularly common pattern is seen in the ovarian cystic teratoma (dermoid cyst), which differentiates principally along ectodermal lines to create a cystic tumor lined by skin replete with hair, sebaceous glands, and tooth structures (Fig. 7-3).
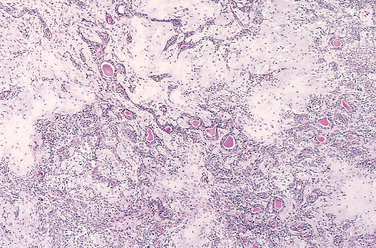
FIGURE 7-2 This mixed tumor of the parotid gland contains epithelial cells forming ducts and myxoid stroma that resembles cartilage.
(Courtesy of Dr. Trace Worrell, University of Texas Southwestern Medical School, Dallas, TX.)
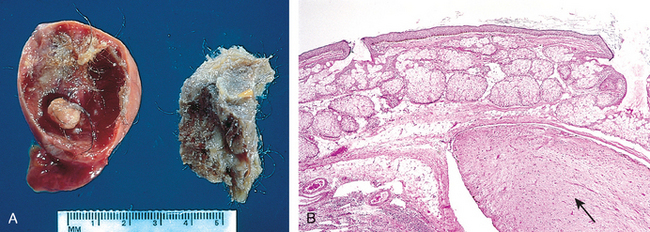
FIGURE 7-3 A, Gross appearance of an opened cystic teratoma of the ovary. Note the presence of hair, sebaceous material, and tooth. B, A microscopic view of a similar tumor shows skin, sebaceous glands, fat cells, and a tract of neural tissue (arrow).
The nomenclature of the more common forms of neoplasia is presented in Table 7-1. It is evident from this compilation that there are some inappropriate but deeply entrenched usages. For generations, benign-sounding designations such as lymphoma, melanoma, mesothelioma, and seminoma have been used for certain malignant neoplasms. The converse is also true; ominous terms may be applied to trivial lesions. Hamartomas present as disorganized but benign-appearing masses composed of cells indigenous to the particular site. They were once thought to be a developmental malformation, unworthy of the -oma designation. For example, pulmonary chondroid harmatoma contains islands of disorganized, but histologically normal cartilage, bronchi, and vessels. However, many hamartomas, including pulmonary chondroid hamartoma, have clonal, recurrent translocations involving genes encoding certain chromatin proteins.3 Thus, through molecular biology they have finally earned their -oma designation. Another misnomer is the term choristoma. This congenital anomaly is better described as a heterotopic rest of cells. For example, a small nodule of well-developed and normally organized pancreatic substance may be found in the submucosa of the stomach, duodenum, or small intestine. This heterotopic rest may be replete with islets of Langerhans and exocrine glands. The term choristoma, connoting a neoplasm, imparts to the heterotopic rest a gravity far beyond its usual trivial significance. Although regrettably the terminology of neoplasms is not simple, it is important because it is the language by which the nature and significance of tumors are categorized.
TABLE 7-1 Nomenclature of Tumors
| Tissue of Origin | Benign | Malignant |
|---|---|---|
| COMPOSED OF ONE PARENCHYMAL CELL TYPE | ||
| Tumors of Mesenchymal Origin | ||
| Connective tissue and derivatives | Fibroma | Fibrosarcoma |
| Lipoma | Liposarcoma | |
| Chondroma | Chondrosarcoma | |
| Osteoma | Osteogenic sarcoma | |
| Endothelial and Related Tissues | ||
| Blood vessels | Hemangioma | Angiosarcoma |
| Lymph vessels | Lymphangioma | Lymphangiosarcoma |
| Synovium | Synovial sarcoma | |
| Mesothelium | Mesothelioma | |
| Brain coverings | Meningioma | Invasive meningioma |
| Blood Cells and Related Cells | ||
| Hematopoietic cells | Leukemias | |
| Lymphoid tissue | Lymphomas | |
| Muscle | ||
| Smooth | Leiomyoma | Leiomyosarcoma |
| Striated | Rhabdomyoma | Rhabdomyosarcoma |
| Tumors of Epithelial Origin | ||
| Stratified squamous | Squamous cell papilloma | Squamous cell carcinoma |
| Basal cells of skin or adnexa | Basal cell carcinoma | |
| Epithelial lining of glands or ducts | Adenoma | Adenocarcinoma |
| Papilloma | Papillary carcinomas | |
| Cystadenoma | Cystadenocarcinoma | |
| Respiratory passages | Bronchial adenoma | Bronchogenic carcinoma |
| Renal epithelium | Renal tubular adenoma | Renal cell carcinoma |
| Liver cells | Liver cell adenoma | Hepatocellular carcinoma |
| Urinary tract epithelium (transitional) | Transitional-cell papilloma | Transitional-cell carcinoma |
| Placental epithelium | Hydatidiform mole | Choriocarcinoma |
| Testicular epithelium (germ cells) | Seminoma | |
| Embryonal carcinoma | ||
| Tumors of Melanocytes | Nevus | Malignant melanoma |
| MORE THAN ONE NEOPLASTIC CELL TYPE—MIXED TUMORS, USUALLY DERIVED FROM ONE GERM CELL LAYER | ||
| Salivary glands | Pleomorphic adenoma (mixed tumor of salivary origin) | Malignant mixed tumor of salivary gland origin |
| Renal anlage | Wilms tumor | |
| MORE THAN ONE NEOPLASTIC CELL TYPE DERIVED FROM MORE THAN ONE GERM CELL LAYER—TERATOGENOUS | ||
| Totipotential cells in gonads or in embryonic rests | Mature teratoma, dermoid cyst | Immature teratoma, teratocarcinoma |
Characteristics of Benign and Malignant Neoplasms
Nothing is more important to the individual with a tumor than being told “It is benign,” and so the differentiation between benign and malignant tumors is one of the most important distinctions a pathologist can make. In the great majority of instances, a benign tumor may be distinguished from a malignant tumor with considerable confidence on the basis of morphology. Occasionally, despite the pathologist’s best efforts, a neoplasm defies categorization. Certain anatomic features may suggest innocence, whereas others point toward cancerous potential. In a few instances there is not perfect concordance between the appearance of a neoplasm and its biologic behavior. In these cases molecular profiling (see below) or other molecular ancillary tests may provide useful information. Although an innocent face may mask an ugly nature, in general, benign and malignant tumors can be distinguished on the basis of differentiation and anaplasia, rate of growth, local invasion, and metastasis.
DIFFERENTIATION AND ANAPLASIA
Differentiation refers to the extent to which neoplastic parenchymal cells resemble the corresponding normal parenchymal cells, both morphologically and functionally; lack of differentiation is called anaplasia. In general, benign tumors are well differentiated (Figs. 7-4 and 7-5). The neoplastic cell in a benign adipocyte tumor—a lipoma—so closely resembles the normal cell that it may be impossible to recognize it as a tumor by microscopic examination of individual cells. Only the growth of these cells into a discrete mass discloses the neoplastic nature of the lesion. One may get so close to the tree that one loses sight of the forest. In well-differentiated benign tumors, mitoses are extremely scant in number and are of normal configuration.
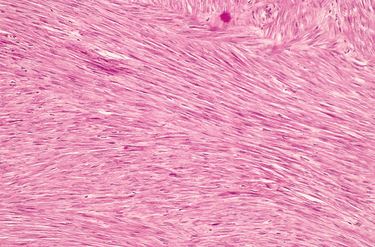
FIGURE 7-4 Leiomyoma of the uterus. This benign, well-differentiated tumor contains interlacing bundles of neoplastic smooth muscle cells that are virtually identical in appearance to normal smooth muscle cells in the myometrium.
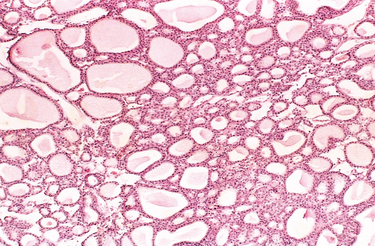
FIGURE 7-5 Benign tumor (adenoma) of the thyroid. Note the normal-looking (well-differentiated), colloid-filled thyroid follicles.
(Courtesy of Dr. Trace Worrell, University of Texas Southwestern Medical School, Dallas, TX.)
Malignant neoplasms are characterized by a wide range of parenchymal cell differentiation, from surprisingly well differentiated (Fig. 7-6) to completely undifferentiated. Certain well-differentiated adenocarcinomas of the thyroid, for example, may form normal-appearing follicles, and some squamous cell carcinomas contain cells that do not differ cytologically from normal squamous epithelial cells (Fig. 7-7). Thus, the morphologic diagnosis of malignancy in well-differentiated tumors may sometimes be quite difficult. In between the two extremes lie tumors that are loosely referred to as moderately well differentiated.
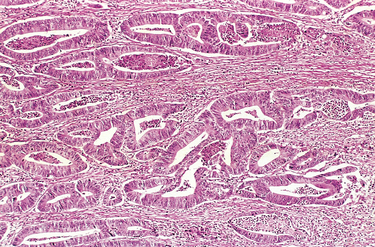
FIGURE 7-6 Malignant tumor (adenocarcinoma) of the colon. Note that compared with the well-formed and normal-looking glands characteristic of a benign tumor (see Fig. 7-5), the cancerous glands are irregular in shape and size and do not resemble the normal colonic glands. This tumor is considered differentiated because gland formation can be seen. The malignant glands have invaded the muscular layer of the colon.
(Courtesy of Dr. Trace Worrell, University of Texas Southwestern Medical School, Dallas, TX.)
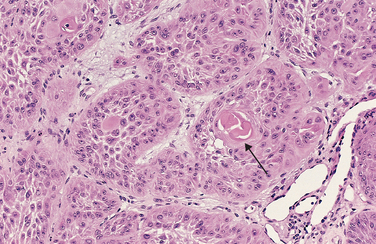
FIGURE 7-7 Well-differentiated squamous cell carcinoma of the skin. The tumor cells are strikingly similar to normal squamous epithelial cells, with intercellular bridges and nests of keratin pearls (arrow).
(Courtesy of Dr. Trace Worrell, University of Texas Southwestern Medical School, Dallas, TX.)
Malignant neoplasms that are composed of poorly differentiated cells are said to be anaplastic. Lack of differentiation, or anaplasia, is considered a hallmark of malignancy. The term anaplasia literally means “to form backward,” implying a reversal of differentiation to a more primitive level. It is believed, however, that most cancers do not represent “reverse differentiation” of mature normal cells but, in fact, arise from less mature cells with “stem-cell-like” properties, such as tissue stem cells (Chapter 3). In well-differentiated tumors (Fig. 7-7), daughter cells derived from these “cancer stem cells” retain the capacity for differentiation, whereas in poorly differentiated tumors that capacity is lost.
Lack of differentiation, or anaplasia, is often associated with many other morphologic changes.
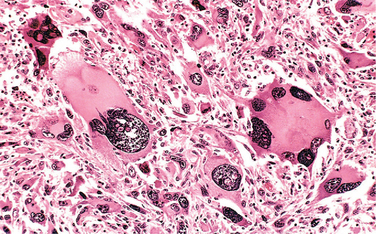
FIGURE 7-8 Anaplastic tumor of the skeletal muscle (rhabdomyosarcoma). Note the marked cellular and nuclear pleomorphism, hyperchromatic nuclei, and tumor giant cells.
(Courtesy of Dr. Trace Worrell, University of Texas Southwestern Medical School, Dallas, TX.)
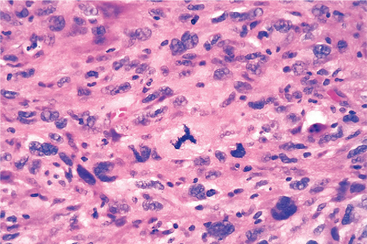
FIGURE 7-9 Anaplastic tumor showing cellular and nuclear variation in size and shape. The prominent cell in the center field has an abnormal tripolar spindle.
Before we leave the subject of differentiation and anaplasia, we should discuss metaplasia and dysplasia. Metaplasia is defined as the replacement of one type of cell with another type. Metaplasia is nearly always found in association with tissue damage, repair, and regeneration. Often the replacing cell type is more suited to a change in environment. For example, gastroesophageal reflux damages the squamous epithelium of the esophagus, leading to its replacement by glandular (gastric or intestinal) epithelium, more suited to the acidic environment. Dysplasia is a term that literally means disordered growth. Dysplasia often occurs in metaplastic epithelium, but not all metaplastic epithelium is also dysplastic. Dysplasia is encountered principally in epithelia, and it is characterized by a constellation of changes that include a loss in the uniformity of the individual cells as well as a loss in their architectural orientation. Dysplastic cells exhibit considerable pleomorphism and often contain large hyperchromatic nuclei with a high nuclearto-cytoplasmic ratio. The architecture of the tissue may be disorderly. For example, in squamous epithelium the usual progressive maturation of tall cells in the basal layer to flattened squames on the surface may be lost and replaced by a scrambling of dark basal-appearing cells throughout the epithelium. Mitotic figures are more abundant than usual, although almost invariably they have a normal configuration. Frequently, however, the mitoses appear in abnormal locations within the epithelium. For example, in dysplastic stratified squamous epithelium, mitoses are not confined to the basal layers but instead may appear at all levels, including surface cells. When dysplastic changes are marked and involve the entire thickness of the epithelium but the lesion remains confined by the basement membrane, it is considered a preinvasive neoplasm and is referred to as carcinoma in situ (Fig. 7-10). Once the tumor cells breach the basement membrane, the tumor is said to be invasive. Dysplastic changes are often found adjacent to foci of invasive carcinoma, and in some situations, such as in long-term cigarette smokers and persons with Barrett esophagus, severe epithelial dysplasia frequently antedates the appearance of cancer. However, dysplasia does not necessarily progress to cancer. Mild to moderate changes that do not involve the entire thickness of epithelium may be reversible, and with removal of the inciting causes the epithelium may revert to normal. Even carcinoma in situ may take years to become invasive.
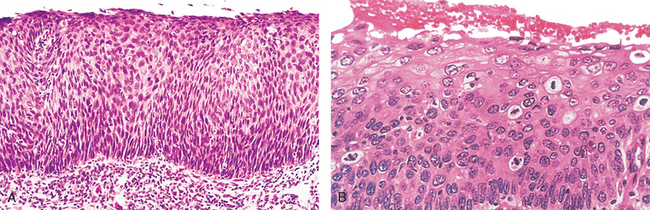
FIGURE 7-10 A, Carcinoma in situ. This low-power view shows that the entire thickness of the epithelium is replaced by atypical dysplastic cells. There is no orderly differentiation of squamous cells. The basement membrane is intact, and there is no tumor in the subepithelial stroma. B, A high-power view of another region shows failure of normal differentiation, marked nuclear and cellular pleomorphism, and numerous mitotic figures extending toward the surface. The basement membrane is not seen in this section.
As you might presume, the better the differentiation of the transformed cell, the more completely it retains the functional capabilities found in its normal counterparts. Thus, benign neoplasms and well-differentiated carcinomas of endocrine glands frequently elaborate the hormones characteristic of their origin. Increased levels of these hormones in the blood are used clinically to detect and follow such tumors. Welldifferentiated squamous cell carcinomas of the epidermis elaborate keratin, just as well-differentiated hepatocellular carcinomas elaborate bile. Highly anaplastic undifferentiated cells, whatever their tissue of origin, lose their resemblance to the normal cells from which they have arisen. In some instances, new and unanticipated functions emerge. Some tumors may elaborate fetal proteins not produced by comparable cells in the adult. Carcinomas of nonendocrine origin may produce a variety of hormones. For example, bronchogenic carcinomas may produce corticotropin, parathyroid-like hormone, insulin, and glucagon, as well as others. Despite exceptions, the more rapidly growing and the more anaplastic a tumor, the less likely it will have specialized functional activity. The cells in benign tumors are almost always well differentiated and resemble their normal cells of origin; the cells in cancer are more or less differentiated, but some derangement of differentiation is always present.
RATES OF GROWTH
A fundamental issue in tumor biology is to understand the factors that affect the growth rates of tumors and their influence on clinical outcome and therapeutic responses. One can begin the consideration of tumor cell kinetics by asking the question: How long does it take to produce a clinically overt tumor mass? It is a reasonable estimate the original transformed cell (approximately 10 μm in diameter) must undergo at least 30 population doublings to produce 109 cells (weighing approximately 1 gm), which is the smallest clinically detectable mass. In contrast, only 10 additional doubling cycles are required to produce a tumor containing 1012 cells (weighing −1kg), which is usually the maximal size compatible with life. These are minimal estimates, based on the assumption that all descendants of the transformed cell retain the ability to divide and that there is no loss of cells from the replicative pool. This concept of tumor as a “pathologic dynamo” is not entirely correct, as we discuss subsequently. Nevertheless, this calculation highlights an extremely important concept about tumor growth: By the time a solid tumor is clinically detected, it has already completed a major portion of its life span. This is a major impediment in the treatment of cancer and underscores the need to develop diagnostic markers to detect early cancers.
The rate of growth of a tumor is determined by three main factors: the doubling time of tumor cells, the fraction of tumor cells that are in the replicative pool, and the rate at which cells are shed or die. Because cell cycle controls are deranged in most tumors, tumor cells can be triggered to cycle without the usual restraints. The dividing cells, however, do not necessarily complete the cell cycle more rapidly than do normal cells. In reality, total cell cycle time for many tumors is equal to or longer than that of corresponding normal cells. Thus, it can be safely concluded that growth of tumors is not commonly associated with a shortening of cell cycle time.
The proportion of cells within the tumor population that are in the proliferative pool is referred to as the growth fraction. Clinical and experimental studies suggest that during the early, submicroscopic phase of tumor growth, the vast majority of transformed cells are in the proliferative pool (Fig. 7-11). As tumors continue to grow, cells leave the proliferative pool in ever-increasing numbers as a result of shedding, lack of nutrients, necrosis, apoptosis, differentiation, and reversion to the nonproliferative phase of the cell cycle (G0). Thus, by the time a tumor is clinically detectable, most cells are not in the replicative pool. Even in some rapidly growing tumors, the growth fraction is only about 20% or less.
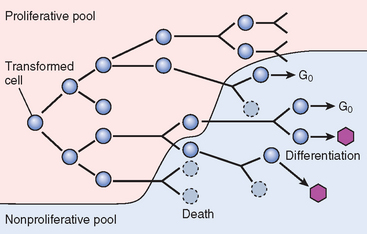
FIGURE 7-11 Schematic representation of tumor growth. As the cell population expands, a progressively higher percentage of tumor cells leaves the replicative pool by reversion to G0, differentiation, and death.
Ultimately the progressive growth of tumors and the rate at which they grow are determined by an excess of cell production over cell loss. In some tumors, especially those with a relatively high growth fraction, the imbalance is large, resulting in more rapid growth than in those in which cell production exceeds cell loss by only a small margin. Some leukemias and lymphomas and certain lung cancers (i.e., small-cell carcinoma) have a relatively high growth fraction, and their clinical course is rapid. By comparison, many common tumors, such as cancers of the colon and breast, have low growth fractions, and cell production exceeds cell loss by only about 10%; they tend to grow at a much slower pace.
Several important conceptual and practical lessons can be learned from studies of tumor cell kinetics:
We can now return to the question posed earlier: How long does it take for one transformed cell to produce a clinically detectable tumor containing 109 cells? If every one of the daughter cells remained in cell cycle and no cells were shed or lost, we could anticipate the answer to be 90 days (30 population doublings, with a cell cycle time of 3 days). In reality, the latent period before which a tumor becomes clinically detectable is unpredictable but typically much longer than 90 days, as long as many years for most solid tumors, emphasizing once again that human cancers are diagnosed only after they are fairly advanced in their life cycle. After they become clinically detectable, the average volume-doubling time for such common killers as cancer of the lung and colon is about 2 to 3 months. As might be anticipated from the discussion of the variables that affect growth rate, however, the range of doubling time values is extremely broad, varying from less than 1 month for some childhood cancers to more than 1 year for certain salivary gland tumors. Cancer is indeed an unpredictable group of disorders.
In general, the growth rate of tumors correlates with their level of differentiation, and thus most malignant tumors grow more rapidly than do benign lesions. There are, however, many exceptions to such an oversimplification. Some benign tumors have a higher growth rate than malignant tumors. Moreover, the rate of growth of benign as well as malignant neoplasms may not be constant over time. Factors such as hormonal stimulation, adequacy of blood supply, and unknown influences may affect their growth. For example, the growth of uterine leiomyomas (benign smooth muscle tumors) may change over time because of hormonal variations. Not infrequently, repeated clinical examination of women bearing such neoplasms over the span of decades discloses no significant increase in size. After menopause the neoplasms may atrophy and may be replaced largely by collagenous, sometimes calcified, tissue. During pregnancy leiomyomas frequently enter a growth spurt. Such changes reflect the responsiveness of the tumor cells to circulating levels of steroid hormones, particularly estrogens. Cancers show a wide range of growth. Some malignant tumors grow slowly for years and then suddenly increase in size, explosively disseminating to cause death within a few months of discovery. It is possible that such behavior results from the emergence of an aggressive subclone of transformed cells. At the other extreme are malignant neoplasms that grow more slowly than do benign tumors and may even enter periods of dormancy lasting for years. On occasion, cancers decrease in size and even spontaneously disappear, but such “miracles” are rare enough that they remain intriguing curiosities.
CANCER STEM CELLS AND CANCER CELL LINEAGES
The continued growth and maintenance of many tissues that contain short-lived cells, such as the formed elements of the blood and the epithelial cells of the gastrointestinal tract and skin, require a resident population of tissue stem cells that are long-lived and capable of self-renewal. Tissue stem cells are rare and exist in a niche created by support cells, which produce paracrine factors that sustain the stem cell.4 Recall from Chapter 3 that tissue stem cells divide asymmetrically to produce two types of daughter cells—those with limited proliferative potential, which undergo terminal differentiation and die, and those that retain stem cell potential.
Cancers are immortal and have limitless proliferative capacity, indicating that like normal tissues, they also must contain cells with “stemlike” properties.5,6 The concept of cancer stem cells has several important implications. Most notably, if cancer stem cells are essential for tumor persistence, it follows that these cells must be eliminated to cure the affected patient. It is hypothesized that like normal stem cells, cancer stem cells have a high intrinsic resistance to conventional therapies, because of their low rate of cell division and the expression of factors, such as multiple drug resistance-1 (MDR1), that counteract the effects of chemotherapeutic drugs.5,6 Thus, the limited success of current therapies may in part be explained by their failure to kill the malignant stem cells that lie at the root of cancer. Cancer stem cells could arise from normal tissue stem cells or from more differentiated cells that, as part of the transformation process, acquire the property of self-renewal. Studies of certain leukemias (Chapter 13) support both of these possibilities. For example, chronic myelogenous leukemia (CML) originates from the malignant counterpart of a normal hematopoietic stem cell, whereas certain acute myeloid leukemias (AMLs) are derived from more differentiated myeloid precursors that acquire an abnormal capacity for self-renewal. The identification of “leukemia stem cells” has spurred the search for cancer stem cells in solid tumors. Most such studies have focused on the identification of tumor-initiating cells (T-ICs), which are defined as cells that allow a human tumor to grow and maintain itself indefinitely when transplanted into an immunodeficient mouse. T-ICs have been identified in several human tumors, including breast carcinoma, glioblastoma multiforme, colon cancer, and AML,5-8 in which they constitute 0.1% to 2% of the total cellularity.
More recent studies have shown that in some cancers, T-ICs are very common, representing 25% of the total cellularity.9 Thus some tumors may have a small number of T-ICs that then “differentiate” to form the bulk of the tumor, while other tumors may be primarily composed of T-ICs. In the future, it will be important to identify the tumorigenic population in each tumor to direct therapy against tumor stem cells. An emerging theme is that the genes and pathways that maintain cancer stem cells are the same as those that regulate normal tissue stem cell homeostasis. Examples include BMI1, a component of the polycomb chromatin-remodeling complex that promotes “stem-ness” in both normal hematopoietic and leukemic stem cells; and the WNT pathway, a key regulator of normal colonic crypt stem cells that has been implicated in the maintenance of colonic adenocarcinoma “stem cells.”9,10 Important remaining questions revolve around whether T-ICs are an accurate measure of cancer stem cells, if cancer stem cells remain dependent on the “niche” that supports normal stem cells, and if it will be possible to selectively target cancer cell “stem-ness” factors.
LOCAL INVASION
Nearly all benign tumors grow as cohesive expansile masses that remain localized to their site of origin and do not have the capacity to infiltrate, invade, or metastasize to distant sites, as do malignant tumors. Because they grow and expand slowly, they usually develop a rim of compressed connective tissue, sometimes called a fibrous capsule, which separates them from the host tissue. This capsule is derived largely from the extracellular matrix of the native tissue due to atrophy of normal parenchymal cells under the pressure of an expanding tumor. Such encapsulation does not prevent tumor growth, but it keeps the benign neoplasm as a discrete, readily palpable, and easily movable mass that can be surgically enucleated (Figs. 7-12 and 7-13). Although a well-defined cleavage plane exists around most benign tumors, in some it is lacking. For example, hemangiomas (neoplasms composed of tangled blood vessels) are often unencapsulated and may appear to permeate the site in which they arise (commonly the dermis of the skin).
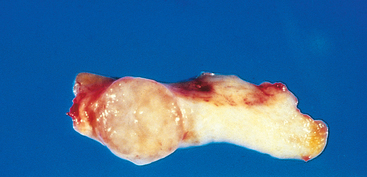
FIGURE 7-12 Fibroadenoma of the breast. The tan-colored, encapsulated small tumor is sharply demarcated from the whiter breast tissue.
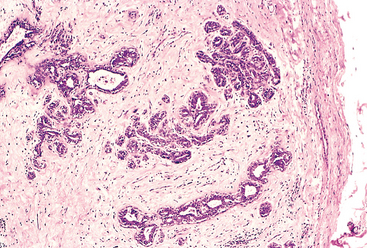
FIGURE 7-13 Microscopic view of fibroadenoma of the breast seen in Figure 7-12. The fibrous capsule (right) delimits the tumor from the surrounding tissue.
(Courtesy of Dr. Trace Worrell, University of Texas Southwestern Medical School, Dallas, TX.)
The growth of cancers is accompanied by progressive infiltration, invasion, and destruction of the surrounding tissue. In general, malignant tumors are poorly demarcated from the surrounding normal tissue, and a well-defined cleavage plane is lacking (Figs. 7-14 and 7-15). Slowly expanding malignant tumors, however, may develop an apparently enclosing fibrous capsule and may push along a broad front into adjacent normal structures. Histologic examination of such pseudo-encapsulated masses almost always shows rows of cells penetrating the margin and infiltrating the adjacent structures, a crablike pattern of growth that constitutes the popular image of cancer.
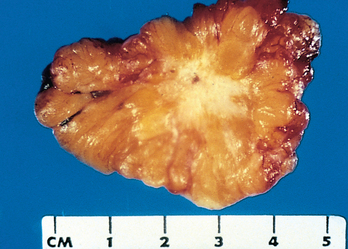
FIGURE 7-14 Cut section of an invasive ductal carcinoma of the breast. The lesion is retracted, infiltrating the surrounding breast substance, and would be stony hard on palpation.
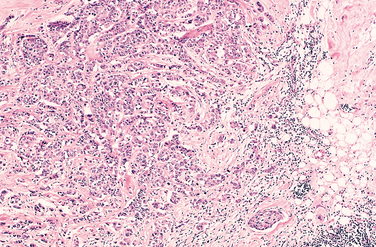
FIGURE 7-15 The microscopic view of the breast carcinoma seen in Figure 7-14 illustrates the invasion of breast stroma and fat by nests and cords of tumor cells (compare with fibroadenoma shown in Fig. 7-13). The absence of a well-defined capsule should be noted.
(Courtesy of Dr. Trace Worrell, University of Texas Southwestern Medical School, Dallas, TX.)
Most malignant tumors are obviously invasive and can be expected to penetrate the wall of the colon or uterus, for example, or fungate through the surface of the skin. They recognize no normal anatomic boundaries. Such invasiveness makes their surgical resection difficult or impossible, and even if the tumor appears well circumscribed it is necessary to remove a considerable margin of apparently normal tissues adjacent to the infiltrative neoplasm. Next to the development of metastases, invasiveness is the most reliable feature that differentiates malignant from benign tumors. We noted earlier that some cancers seem to evolve from a preinvasive stage referred to as carcinoma in situ. This commonly occurs in carcinomas of the skin, breast, and certain other sites and is best illustrated by carcinoma of the uterine cervix (Chapter 22). In situ epithelial cancers display the cytologic features of malignancy without invasion of the basement membrane. They may be considered one step removed from invasive cancer; with time, most penetrate the basement membrane and invade the subepithelial stroma.
METASTASIS
Metastases are tumor implants discontinuous with the primary tumor. Metastasis unequivocally marks a tumor as malignant because benign neoplasms do not metastasize. The invasiveness of cancers permits them to penetrate into blood vessels, lymphatics, and body cavities, providing the opportunity for spread. With few exceptions, all malignant tumors can metastasize. The major exceptions are most malignant neoplasms of the glial cells in the central nervous system, called gliomas, and basal cell carcinomas of the skin. Both are locally invasive forms of cancer, but they rarely metastasize. It is evident then that the properties of invasion and metastasis are separable.
In general, the more aggressive, the more rapidly growing, and the larger the primary neoplasm, the greater the likelihood that it will metastasize or already has metastasized. There are innumerable exceptions, however. Small, well-differentiated, slowly growing lesions sometimes metastasize widely; conversely, some rapidly growing, large lesions remain localized for years. Many factors relating to both invader and host are involved.
Approximately 30% of newly diagnosed individuals with solid tumors (excluding skin cancers other than melanomas) present with metastases. Metastatic spread strongly reduces the possibility of cure; hence, short of prevention of cancer, no achievement would be of greater benefit to patients than methods to block metastases.
Pathways of Spread
Dissemination of cancers may occur through one of three pathways: (1) direct seeding of body cavities or surfaces, (2) lymphatic spread, and (3) hematogenous spread. Although direct transplantation of tumor cells, as for example on surgical instruments, may theoretically occur, it is rare and we do not discuss this artificial mode of dissemination further. Each of the three major pathways is described separately.
Seeding of Body Cavities and Surfaces.
Seeding of body cavities and surfaces may occur whenever a malignant neoplasm penetrates into a natural “open field.” Most often involved is the peritoneal cavity (Fig. 7-16), but any other cavity—pleural, pericardial, subarachnoid, and joint space—may be affected. Such seeding is particularly characteristic of carcinomas arising in the ovaries, when, not infrequently, all peritoneal surfaces become coated with a heavy layer of cancerous glaze. Remarkably, the tumor cells may remain confined to the surface of the coated abdominal viscera without penetrating into the substance. Sometimes mucus-secreting appendiceal carcinomas fill the peritoneal cavity with a gelatinous neoplastic mass referred to as pseudomyxoma peritonei.
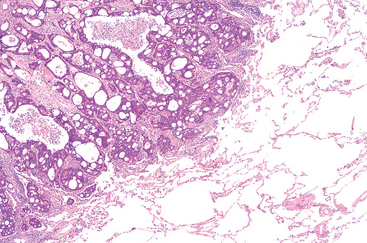
Lymphatic Spread.
Transport through lymphatics is the most common pathway for the initial dissemination of carcinomas (Fig. 7-17), and sarcomas may also use this route. Tumors do not contain functional lymphatics, but lymphatic vessels located at the tumor margins are apparently sufficient for the lymphatic spread of tumor cells.11 The emphasis on lymphatic spread for carcinomas and hematogenous spread for sarcomas is misleading, because ultimately there are numerous interconnections between the vascular and the lymphatic systems. The pattern of lymph node involvement follows the natural routes of lymphatic drainage. Because carcinomas of the breast usually arise in the upper outer quadrants, they generally disseminate first to the axillary lymph nodes. Cancers of the inner quadrants drain to the nodes along the internal mammary arteries. Thereafter the infraclavicular and supraclavicular nodes may become involved. Carcinomas of the lung arising in the major respiratory passages metastasize first to the perihilar tracheobronchial and mediastinal nodes. Local lymph nodes, however, may be bypassed—so-called “skip metastasis”—because of venous-lymphatic anastomoses or because inflammation or radiation has obliterated lymphatic channels.
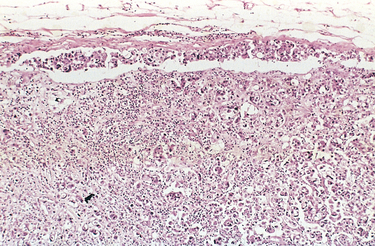
FIGURE 7-17 Axillary lymph node with metastatic breast carcinoma. The subcapsular sinus (top) is distended with tumor cells. Nests of tumor cells have also invaded the subcapsular cortex.
(Courtesy of Dr. Trace Worrell, University of Texas Southwestern Medical School, Dallas, TX.)
In breast cancer, determining the involvement of axillary lymph nodes is very important for assessing the future course of the disease and for selecting suitable therapeutic strategies. To avoid the considerable surgical morbidity associated with a full axillary lymph node dissection, biopsy of sentinel nodes is often used to assess the presence or absence of metastatic lesions in the lymph nodes. A sentinel lymph node is defined as “the first node in a regional lymphatic basin that receives lymph flow from the primary tumor.”12 Sentinel node mapping can be done by injection of radiolabeled tracers and blue dyes, and the use of frozen section upon the sentinel lymph node at the time of surgery can guide the surgeon to the appropriate therapy. Sentinel node biopsy has also been used for detecting the spread of melanomas, colon cancers, and other tumors.12,13
In many cases the regional nodes serve as effective barriers to further dissemination of the tumor, at least for a while. Conceivably the cells, after arrest within the node, may be destroyed by a tumor-specific immune response. Drainage of tumor cell debris or tumor antigens, or both, also induces reactive changes within nodes. Thus, enlargement of nodes may be caused by (1) the spread and growth of cancer cells or (2) reactive hyperplasia (Chapter 13). Therefore, nodal enlargement in proximity to a cancer, while it must arouse suspicion, does not necessarily mean dissemination of the primary lesion.
Hematogenous Spread.
Hematogenous spread is typical of sarcomas but is also seen with carcinomas. Arteries, with their thicker walls, are less readily penetrated than are veins. Arterial spread may occur, however, when tumor cells pass through the pulmonary capillary beds or pulmonary arteriovenous shunts or when pulmonary metastases themselves give rise to additional tumor emboli. In such vascular spread, several factors influence the patterns of distribution of the metastases. With venous invasion the blood-borne cells follow the venous flow draining the site of the neoplasm, and the tumor cells often come to rest in the first capillary bed they encounter. Understandably the liver and lungs are most frequently involved in such hematogenous dissemination (Figs. 7-18 and 7-19), because all portal area drainage flows to the liver and all caval blood flows to the lungs. Cancers arising in close proximity to the vertebral column often embolize through the paravertebral plexus, and this pathway is involved in the frequent vertebral metastases of carcinomas of the thyroid and prostate.
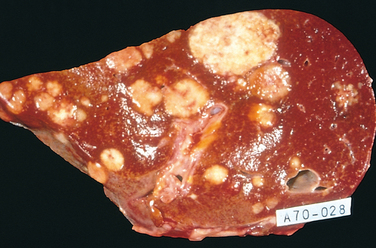
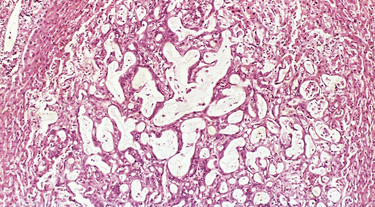
FIGURE 7-19 Microscopic view of liver metastasis. A pancreatic adenocarcinoma has formed a metastatic nodule in the liver.
(Courtesy of Dr. Trace Worrell, University of Texas Southwestern Medical School, Dallax, TX.)
Certain cancers have a propensity for invasion of veins. Renal cell carcinoma often invades the branches of the renal vein and then the renal vein itself to grow in a snakelike fashion up the inferior vena cava, sometimes reaching the right side of the heart. Hepatocellular carcinomas often penetrate portal and hepatic radicles to grow within them into the main venous channels. Remarkably, such intravenous growth may not be accompanied by widespread dissemination. Histologic evidence of penetration of small vessels at the site of the primary neoplasm is obviously an ominous feature. Such changes, however, must be viewed guardedly because, for reasons discussed later, they do not indicate the inevitable development of metastases.
Many observations suggest that mere anatomic localization of the neoplasm and natural pathways of venous drainage do not wholly explain the systemic distributions of metastases. For example, breast carcinoma preferentially spreads to bone, bronchogenic carcinomas tend to involve the adrenals and the brain, and neuroblastomas spread to the liver and bones. Conversely, skeletal muscles and the spleen, despite the large percentage of blood flow they receive and the enormous vascular beds present, are rarely the site of secondary deposits. The probable basis of such tissue-specific homing of tumor cells is discussed later.
The distinguishing features of benign and malignant tumors discussed in this overview are summarized in Table 7-2 and Figure 7-20. With this background on the structure and behavior of neoplasms, we now discuss the origin of tumors, starting with insights gained from the epidemiology of cancer and followed by the molecular basis of carcinogenesis.
TABLE 7-2 Comparisons between Benign and Malignant Tumors
| Characteristics | Benign | Malignant |
|---|---|---|
| Differentiation/anaplasia | Well differentiated; structure sometimes typical of tissue of origin | Some lack of differentiation with anaplasia; structure often atypical |
| Rate of growth | Usually progressive and slow; may come to a standstill or regress; mitotic figures rare and normal | Erratic and may be slow to rapid; mitotic figures may be numerous and abnormal |
| Local invasion | Usually cohesive expansile well-demarcated masses that do not invade or infiltrate surrounding normal tissues | Locally invasive, infiltrating surrounding tissue; sometimes may be seemingly cohesive and expansile |
| Metastasis | Absent | Frequently present; the larger and more undifferentiated the primary, the more likely are metastases |
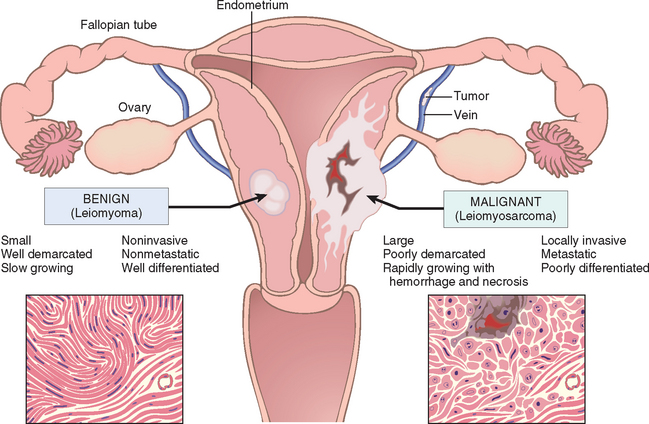
Epidemiology
Because cancer is a disorder of cell growth and behavior, its ultimate cause has to be defined at the cellular and subcellular levels. Study of cancer patterns in populations, however, can contribute substantially to knowledge about the origins of cancer. Epidemiologic studies have established the causative link between smoking and lung cancer, and comparison of diet and cancer rates in the Western world and Africa has implicated high dietary fat and low fiber in the development of colon cancer. Major insights into the causes of cancer can be obtained by epidemiologic studies that relate particular environmental, racial (possibly hereditary), and cultural influences to the occurrence of specific neoplasms. Certain diseases associated with an increased risk of developing cancer (preneoplastic disorders) also provide clues to the pathogenesis of cancer. In the following discussion we first summarize the overall incidence of cancer to gain an insight into the magnitude of the cancer problem, then we review some factors relating to the patient and environment that influence the predisposition to cancer.
CANCER INCIDENCE
In some measure, an individual’s likelihood of developing a cancer is expressed by national incidence and mortality rates. For example, residents of the United States have about a one in five chance of dying of cancer. There were, it is estimated, about 1,437,180 new cancer cases and 565,650 deaths from cancer in 2008, representing 23% of all mortality,1 a frequency surpassed only by deaths caused by cardiovascular diseases. These data do not include an additional 1 million, for the most part readily curable, non-melanoma cancers of the skin and 122,000 cases of carcinoma in situ, largely of the female breast and melanomas.1 The major organ sites affected and the estimated frequency of cancer deaths are shown in Figure 7-21. The most common tumors in men arise in the prostate, lung, and colorectum. In women, cancers of the breast, lung, and colon and rectum are the most frequent. Cancers of the lung, female breast, prostate, and colon/rectum constitute more than 50% of cancer diagnoses and cancer deaths in the U.S. population.1
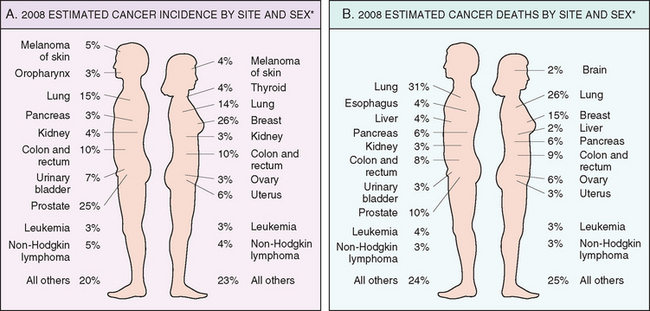
FIGURE 7-21 Cancer incidence and mortality by site and sex. Excludes basal cell and squamous cell skin cancers and in situ carcinomas, except urinary bladder.
(Adapted from Jemal A et al.: Cancer statistics, 2008. CA Cancer J Clin 58:2, 2008.)
The age-adjusted death rates (number of deaths per 100,000 population) for many forms of cancer have significantly changed over the years. Many of the long-term comparisons are noteworthy. Over the last 50 years of the twentieth century, the overall age-adjusted cancer death rate significantly increased in both men and women. However, since 1995 the cancer incidence rate in men has stabilized and since 1990 the cancer death rate in men has decreased 18.4%.1 In women the cancer incidence rate stabilized in 1995, and the cancer death rate has decreased 10.4% since 1991.1 Among men nearly 80% of the total decrease in cancer death rates is accounted for by decreases in death rates from lung, prostate, and colorectal cancers since 1990.1 Among women nearly 60% of the decrease in cancer death rates is due to reductions in death rates from breast and colorectal cancers.1 Nearly 40% of the sex-specific decreases in cancer death rates is accounted for by a reduction in lung cancer deaths in men and breast cancer deaths in women.1 Decreased use of tobacco products is responsible for the reduction in lung cancer deaths, while improved detection and treatment are responsible for the decrease in death rates for colorectal, female breast, and prostate cancer.1 The last half century has seen a decline in the number of deaths caused by cervical cancer that relates to earlier diagnosis made possible by the Papanicolaou (Pap) smear. The downward trend in deaths from stomach cancer has been attributed to a decrease in some dietary carcinogens, as a consequence of better food preservation or changes in dietary habits. Unfortunately, between 1990–1991 and 2004, lung cancer death rates in women, and liver and intrahepatic bile duct cancer death rates in men, increased substantially, offsetting some of the improvement in survival from other cancers.1 Indeed, although in women carcinomas of the breast occur about 2.5 times more frequently than those of the lung, lung cancer has become the leading cause of cancer deaths in women. Deaths from primary liver cancers, which declined between 1930 and 1970, have approximately doubled during the past 30 years. This number is expected to increase over the coming decades, as the large number of individuals infected with the hepatitis C virus (HCV) begin to develop hepatocellular carcinoma.
Although race is not a strict biologic category, it can define groups at risk for certain cancers.14,15 The disparity in cancer mortality rates between white and black Americans persists, but African Americans had the largest decline in cancer mortality during the past decade. Hispanics living in the United States have a lower frequency of the most common tumors than the white non-Hispanic population but a higher incidence of tumors of the stomach, liver, uterine cervix, and gallbladder, as well as certain childhood leukemias.
GEOGRAPHIC AND ENVIRONMENTAL FACTORS
Although genetics and environmental triggers both play a role in the pathogenesis of cancer, environmental factors are thought to be the more significant contributors in most common sporadic cancers. In one large study the proportion of risk from environmental causes was found to be 65%, whereas heritable factors contributed 26% to 42% of cancer risk. Remarkable differences found in the incidence and death rates of specific forms of cancer around the world also suggest a role for environmental factors.16,17 For example, the death rate for stomach carcinoma in both men and women is seven to eight times higher in Japan than in the United States. In contrast, the death rate from carcinoma of the lung is slightly more than twice as great in the United States as in Japan. Although racial predispositions cannot be ruled out, it is generally believed that most of these geographic differences are the consequence of environmental influences. Indeed, comparing mortality rates for Japanese immigrants to the United States and Japanese born in the United States of immigrant parents (Nisei) with those of long-term residents of both countries shows that cancer mortality rates for first-generation Japanese immigrants are intermediate between those of natives of Japan and natives of California, and the two rates come closer with each passing generation (Fig. 7-22). This points strongly to environmental and cultural factors rather than genetic predisposition.

FIGURE 7-22 The change in incidence of various cancers with migration from Japan to the United States provides evidence that the occurrence of cancers is related to components of the environment that differ in the two countries. The incidence of each kind of cancer is expressed as the ratio of the death rate in the population being considered to that in a hypothetical population of California whites with the same age distribution; the death rates for whites are thus defined as 1. The death rates among immigrants and immigrants’ sons tend consistently toward California norms.
(From Cairns J: The cancer problem. In Readings from Scientific American—Cancer Biology. New York, WH Freeman, 1986, p 13.)
There is no paucity of carcinogenic environmental factors: they lurk in the ambient environment, in the workplace, in food, and in personal practices. Individuals may be exposed to carcinogenic factors when they go outside (ultraviolet [UV] rays, smog), in their medication (methotrexate), at work (asbestos, vinyl chloride; Table 7-3), or at home (high-fat diet, alcohol). Overall, mortality data indicate that the most overweight individuals in the U.S. population have a 52% (men) and 62% (women) higher death rate from cancer than do their slimmer counterparts. Indeed, obesity is associated with approximately 14% of cancer deaths in men and 20% in women.18 Alcohol abuse alone increases the risk of carcinomas of the oropharynx (excluding lip), larynx, and esophagus and, by the development of alcoholic cirrhosis, hepatocellular carcinoma. Smoking, particularly of cigarettes, has been implicated in cancer of the mouth, pharynx, larynx, esophagus, pancreas, bladder, and most significantly, about 90% of lung cancer deaths (Chapter 9). Cigarette smoking has been called the single most important environmental factor contributing to premature death in the United States. Alcohol and tobacco together synergistically increase the danger of incurring cancers in the upper aerodigestive tract. The risk of cervical cancer is linked to age at first intercourse and the number of sex partners, and it is now known that infection by venereally transmitted human papillomavirus (HPV) contributes to cervical dysplasia and cancer. It appears that almost everything one does to gain a livelihood or for pleasure is fattening, immoral, illegal, or, even worse, oncogenic.
TABLE 7-3 Occupational Cancers
| Agents or Groups of Agents | Human Cancer Site for Which Reasonable Evidence Is Available | Typical Use or Occurrence |
|---|---|---|
| Arsenic and arsenic compounds | Lung, skin, hemangiosarcoma | Byproduct of metal smelting; component of alloys, electrical and semiconductor devices, medications and herbicides, fungicides, and animal dips |
| Asbestos | Lung, mesothelioma; gastrointestinal tract (esophagus, stomach, large intestine) | Formerly used for many applications because of fire, heat, and friction resistance; still found in existing construction as well as fire-resistant textiles, friction materials (i.e., brake linings), underlayment and roofing papers, and floor tiles |
| Benzene | Leukemia, Hodgkin lymphoma | Principal component of light oil; despite known risk, many applications exist in printing and lithography, paint, rubber, dry cleaning, adhesives and coatings, and detergents; formerly widely used as solvent and fumigant |
| Beryllium and beryllium compounds | Lung | Missile fuel and space vehicles; hardener for lightweight metal alloys, particularly in aerospace applications and nuclear reactors |
| Cadmium and cadmium compounds | Prostate | Uses include yellow pigments and phosphors; found in solders; used in batteries and as alloy and in metal platings and coatings |
| Chromium compounds | Lung | Component of metal alloys, paints, pigments, and preservatives |
| Nickel compounds | Nose, lung | Nickel plating; component of ferrous alloys, ceramics, and batteries; by-product of stainless-steel arc welding |
| Radon and its decay products | Lung | From decay of minerals containing uranium; potentially serious hazard in quarries and underground mines |
| Vinyl chloride | Angiosarcoma, liver | Refrigerant; monomer for vinyl polymers; adhesive for plastics; formerly inert aerosol propellant in pressurized containers |
Modified from Stellman JM, Stellman SD: Cancer and workplace. CA Cancer J Clin 46:70, 1996.
AGE
Age has an important influence on the likelihood of being afflicted with cancer. Most carcinomas occur in the later years of life (>55 years). Cancer is the main cause of death among women aged 40 to 79 and among men aged 60 to 79; the decline in deaths after age 80 is due to the lower number of individuals who reach this age. The rising incidence with age may be explained by the accumulation of somatic mutations associated with the emergence of malignant neoplasms (discussed later). The decline in immune competence that accompanies aging may also be a factor.
However, children are not spared; cancer accounts for slightly more than 10% of all deaths in children under age 15 in the United States, second only to accidents. However, the types of cancers that predominate in children are significantly different from those seen in adults. Carcinomas, the most common general category of tumor in adults, are extraordinarily rare among children. Instead, acute leukemia and primitive neoplasms of the central nervous system are responsible for approximately 60% of childhood cancer deaths. The common neoplasms of infancy and childhood include the so-called small round blue cell tumors such as neuroblastoma, Wilms tumor, retinoblastoma, acute leukemias, and rhabdomyosarcomas. These are discussed in Chapter 10 and elsewhere in the text.
GENETIC PREDISPOSITION TO CANCER
One frequently asked question is: “My mother and father both died of cancer. Does that mean I am doomed to get it?” Based on current knowledge, the answer must be carefully qualified.19,20 Evidence now indicates that for a large number of cancer types, including the most common forms, there exist not only environmental influences but also hereditary predispositions. For example, lung cancer is in most instances clearly related to cigarette smoking, yet mortality from lung cancer has been shown to be four times greater among nonsmoking relatives (parents and siblings) of lung cancer patients than among nonsmoking relatives of controls (the effects of second-hand smoke may confound some of these results). Less than 10% of cancer patients have inherited mutations that predispose to cancer, and the frequency is even lower (around 0.1%) for certain types of tumors. Despite the low frequency, the recognition of inherited predisposition to cancer has had a major impact on the understanding of cancer pathogenesis. Moreover, genes that are causally associated with cancers that have a strong hereditary component are generally also involved in the much more common sporadic forms of the same tumor. Genetic predisposition to cancer can be divided into three categories (Table 7-4).
TABLE 7-4 Examples of Inherited Predisposition to Cancer
| INHERITED CANCER SYNDROMES (AUTOSOMAL DOMINANT) | |
|---|---|
| Gene | Inherited Predisposition |
| RB | Retinoblastoma |
| p53 | Li-Fraumeni syndrome (various tumors) |
| p16/INK4A | Melanoma |
| APC | Familial adenomatous polyposis/colon cancer |
| NF1, NF2 | Neurofibromatosis 1 and 2 |
| BRCA1, BRCA2 | Breast and ovarian tumors |
| MEN1, RET | Multiple endocrine neoplasia 1 and 2 |
| MSH2, MLH1, MSH6 | Hereditary nonpolyposis colon cancer |
| PTCH | Nevoid basal cell carcinoma syndrome |
| PTEN | Cowden syndrome (epithelial cancers) |
| LKB1 | Peutz-Jegher syndrome (epithelial cancers) |
| VHL | Renal cell carcinomas |
| INHERITED AUTOSOMAL RECESSIVE SYNDROMES OF DEFECTIVE DNA REPAIR | |
| Xeroderma pigmentosum | |
| Ataxia-telangiectasia | |
| Bloom syndrome | |
| Fanconi anemia | |
| FAMILIAL CANCERS | |
| Familial clustering of cases, but role of inherited predisposition not clear for each individual | |
| Breast cancer | |
| Ovarian cancer | |
| Pancreatic cancer | |
Autosomal Dominant Inherited Cancer Syndromes.
Inherited cancer syndromes include several well-defined cancers in which inheritance of a single autosomal dominant mutant gene greatly increases the risk of developing a tumor. The inherited mutation is usually a point mutation occurring in a single allele of a tumor suppressor gene. The silencing of the second allele occurs in somatic cells, generally as a consequence of deletion or recombination. Childhood retinoblastoma is the most striking example in this category. Approximately 40% of retinoblastomas are inherited. Carriers of a mutant of the RB tumor suppressor gene have a 10,000-fold increased risk of developing retinoblastoma, usually bilateral. They also have a greatly increased risk of developing a second cancer, particularly osteosarcoma. Familial adenomatous polyposis is an autosomal dominant hereditary disorder caused by mutation of the adenomatous polyposis coli (APC) tumor suppressor gene. Other autosomal dominant cancer syndromes include Li-Fraumeni syndrome resulting from germ-line mutations of the p53 gene; multiple endocrine neoplasia types 1 and 2 (MEN-1 and MEN-2) caused by mutation in the genes that encode the menin transcription factor and the RET tyrosine kinase, respectively; hereditary nonpolyposis colon cancer (HNPCC), a condition caused by inactivation of a DNA mismatch repair gene (also listed below among repair defects); and several others listed in Table 7-4.
There are several features that characterize inherited cancer syndromes:
As in other autosomal dominant conditions, both incomplete penetrance and variable expressivity occur.
Defective DNA-Repair Syndromes.
Besides the dominantly inherited precancerous conditions, a group of cancer-predisposing conditions is collectively characterized by defects in DNA repair and resultant DNA instability. These conditions generally have an autosomal recessive pattern of inheritance. Included in this group are xeroderma pigmentosum, ataxia-telangiectasia, and Bloom syndrome, all rare diseases characterized by genetic instability resulting from defects in DNA-repair genes. Also included here is HNPCC, an autosomal dominant condition caused by inactivation of a DNA mismatch repair gene.21 HNPCC is the most common cancer predisposition syndrome, increasing the susceptibility of cancer of the colon, the small intestine, endometrium, and ovary (Chapter 17).
Familial Cancers.
Besides the inherited syndromes of cancer susceptibility, cancer may occur at higher frequency in certain families without a clearly defined pattern of transmission. Virtually all the common types of cancers that occur sporadically have also been reported to occur in familial forms. Examples include carcinomas of colon, breast, ovary, and brain, as well as melanomas and lymphomas. Features that characterize familial cancers include early age at onset, tumors arising in two or more close relatives of the index case, and sometimes, multiple or bilateral tumors. Familial cancers are not associated with specific marker phenotypes. For example, in contrast to the familial adenomatous polyp syndrome, familial colonic cancers do not arise in preexisting benign polyps. The transmission pattern of familial cancers is not clear. In general, siblings have a risk between two and three times greater than unrelated individuals. Segregation analyses of large families usually show that predisposition to the tumors is dominant, but multifactorial inheritance cannot be easily ruled out. It is likely that familial susceptibility to cancer may depend on multiple low-penetrance alleles, each contributing to only a small increase in the risk of tumor development. Genome-wide association studies show great promise in identifying such alleles (Chapter 5).22 It has been estimated that 10% to 20% of patients with breast or ovarian cancer have a first- or second-degree relative with one of these tumors. Although two breast cancer susceptibility genes, named BRCA1 and BRCA2, have been identified, mutation of these genes occurs in no more than 3% of breast cancers.20 A similar situation occurs in familial melanomas, in which a mutation of the p16 tumor suppressor gene has been identified. However, mutation in this gene accounts for only about 20% of familial melanoma kindreds, suggesting that other factors are involved in the familial predisposition.23
Interactions between Genetic and Nongenetic Factors.
What can be said about the influence of heredity on the majority of malignant neoplasms? It could be argued that they are largely of environmental origin, but lack of family history does not preclude an inherited component. It is generally difficult to sort out the hereditary and acquired basis of a tumor, because these factors often interact closely. The interaction between genetic and nongenetic factors is particularly complex when tumor development depends on the action of multiple contributory genes. Even in tumors with a well-defined inherited component, the risk of developing the tumor can be greatly influenced by nongenetic factors. For instance, breast cancer risk in female carriers of BRCA1 or BRCA2 mutations is almost threefold higher for women born after 1940, as compared with the risks for women born before that year.20 Furthermore, the genotype can significantly influence the likelihood of developing environmentally induced cancers. Inherited variations (polymorphisms) of enzymes that metabolize procarcinogens to their active carcinogenic forms (see “Initiation of Carcinogenesis”) can influence the susceptibility to cancer. Of interest in this regard are genes that encode the cytochrome P-450 enzymes. As discussed later under “Chemical Carcinogenesis,” polymorphism at one of the P-450 loci confers inherited susceptibility to lung cancers in cigarette smokers. More such associations are likely to be found.
NONHEREDITARY PREDISPOSING CONDITIONS
The only certain way of avoiding cancer is not to be born; to live is to incur the risk. Certain predisposing influences, such as environment, behaviors, and clinical conditions, can increase that risk, however. For example, regenerative, metaplastic, hyperplastic, and dysplastic proliferations are fertile soil for the origin of a malignant tumor, because cell replication is involved in neoplastic transformation. Indeed, proliferation may be required for neoplastic transformation in some settings, since it is proliferating cells that accumulate the genetic lesions required for carcinogenesis.
Chronic Inflammation and Cancer.
In 1863 Virchow proposed that cancer develops at sites of chronic inflammation, and the potential relationships between cancer and inflammation have been studied since then.24 This is exemplified by the increased risk of cancer in individuals affected by a variety of chronic inflammatory diseases of the gastrointestinal tract (Table 7-5). These include ulcerative colitis, Helicobacter pylori gastritis, viral hepatitis, and chronic pancreatitis. Although the precise mechanisms that link inflammation and cancer development have not been established, recent work has demonstrated that in the setting of unresolved chronic inflammation, as occurs in viral hepatitis or chronic gastritis, the immune response may become maladaptive, promoting tumorigenesis.24 As with any cause of tissue injury, there is a compensatory proliferation of cells so as to repair the damage. This regenerative process is aided and abetted by a plethora of growth factors, cytokines, chemokines, and other bioactive substances produced by activated immune cells that promote cell survival, tissue remodeling, and angiogenesis. In some cases, chronic inflammation may increase the pool of tissue stem cells, which become subject to the effect of mutagens. These mediators also cause genomic stress and mutations; additionally the activated immune cells produce reactive oxygen species that are directly genotoxic. To add insult to injury, many of these mediators promote cell survival, even in the face of genomic damage. In the short term this can be adaptive; the organism must survive, and the damaged cells can be repaired or eliminated later. However, in chronic inflammation such behavior is maladaptive, since it allows the creation and fixation of such mutations, eventually leading to cancer. Whatever the precise mechanism, the link between chronic inflammation and cancer has practical implications. For instance, expression of the enzyme cyclooxygenase-2 (COX-2), which brings about the conversion of arachidonic acid into prostaglandins (Chapter 2), is induced by inflammatory stimuli and is increased in colon cancers and other tumors.25 The development of COX-2 inhibitors for cancer treatment is an active area of research.26
TABLE 7-5 Chronic Inflammatory States and Cancer
| Pathologic Condition | Associated Neoplasm(s) | Etiologic Agent |
|---|---|---|
| Asbestosis, silicosis | Mesothelioma, lung carcinoma | Asbestos fibers, silica particles |
| Bronchitis | Lung carcinoma | Silica, asbestos, smoking (nitrosamines, peroxides) |
| Cystitis, bladder inflammation | Bladder carcinoma | Chronic indwelling urinary catheters |
| Gingivitis, lichen planus | Oral squamous cell carcinoma | |
| Inflammatory bowel disease | Colorectal carcinoma | |
| Lichen sclerosis | Vulvar squamous cell carcinoma | |
| Chronic pancreatitis | Pancreatic carcinoma | Alcoholism |
| Hereditary pancreatitis | Pancreatic carcinoma | Mutation in trypsinogen gene |
| Reflux esophagitis, Barrett esophagus | Esophageal carcinoma | Gastric acids |
| Sialadenitis | Salivary gland carcinoma | |
| Sjögren syndrome, Hashimoto thyroiditis | MALT lymphoma | |
| CANCERS ASSOCIATED WITH INFECTIOUS AGENTS | ||
| Opisthorchis, cholangitis | Cholangiosarcoma, colon carcinoma | Liver flukes (Opisthorchis viverrini) |
| Bile acids | ||
| Chronic cholecystitis | Gallbladder cancer | Bacteria, gallbladder stones |
| Gastritis/ulcers | Gastric adenocarcinoma, MALT | Helicobacter pylori |
| Hepatitis | Hepatocellular carcinoma | Hepatitis B and/or C virus |
| Mononucleosis | B-cell non-Hodgkin lymphoma and Hodgkin lymphoma | Epstein-Barr virus |
| AIDS | Non-Hodgkin lymphoma, squamous cell carcinoma, Kaposi sarcoma | Human immunodeficiency virus, human herpesvirus type 8 |
| Osteomyelitis | Carcinoma in draining sinuses | Bacterial infection |
| Pelvic inflammatory disease, chronic cervicitis | Ovrian carcinoma, cervical/anal carcinoma | Gonorrhea, chlamydia, human papillomavirus |
| Chronic cystitis | Bladder, liver, rectal carcinoma | Schistosomiasis |
Adapted from Tlsty TD, Coussens LM: Tumor stroma and regulation of cancer development. Ann Rev Pathol Mech Dis 1:119, 2006.
Precancerous Conditions.
Certain non-neoplastic disorders—the chronic atrophic gastritis of pernicious anemia, solar keratosis of the skin, chronic ulcerative colitis, and leukoplakia of the oral cavity, vulva, and penis—have such a well-defined association with cancer that they have been termed precancerous conditions. This designation is somewhat unfortunate, because in the great majority of these lesions no malignant neoplasm emerges. Nonetheless, the term persists because it calls attention to the increased risk. Certain forms of benign neoplasia also constitute precancerous conditions. The villous adenoma of the colon, as it increases in size, becomes malignant in up to 50% of cases. It might be asked: Is there not a risk with all benign neoplasms? Although some risk may be inherent, a large cumulative experience indicates that most benign neoplasms do not become cancerous. Nonetheless, numerous examples could be offered of cancers arising, albeit rarely, in benign tumors—for example, a leiomyosarcoma beginning in a leiomyoma, and carcinoma appearing in long-standing pleomorphic adenomas. Generalization is impossible, because each type of benign neoplasm is associated with a particular level of risk ranging from virtually never to frequently. Only follow-up studies of large series of each neoplasm can establish the level of risk, and always the question remains: Did the cancer arise from a nonmalignant cell in the benign tumor, or did the benign tumor contain, from the outset, a silent or indolent malignant focus?
Molecular Basis of Cancer
The literature on the molecular basis of cancer continues to proliferate at such a rapid pace that it is easy to get lost in the growing forest of information. We list some fundamental principles before delving into the details of the molecular basis of cancer.
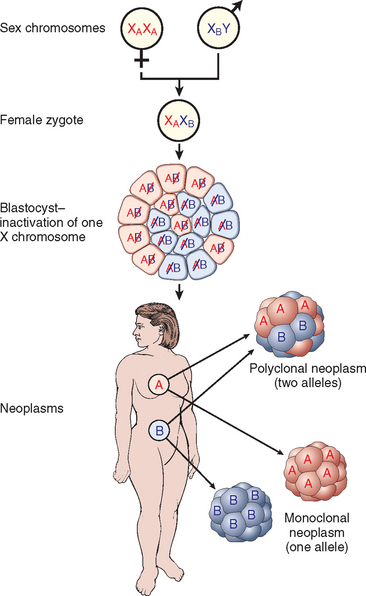
FIGURE 7-23 The use of X-linked markers as evidence of the monoclonality of neoplasms. Because of random X inactivation, all females are mosaics with two cell populations (with different alleles for the androgen receptor labeled A and B in this case). When neoplasms that arise in women who are heterozygous for X-linked markers are analyzed, they are made up of cells that contain the active maternal (XA) or the paternal (XB) X chromosome but not both.
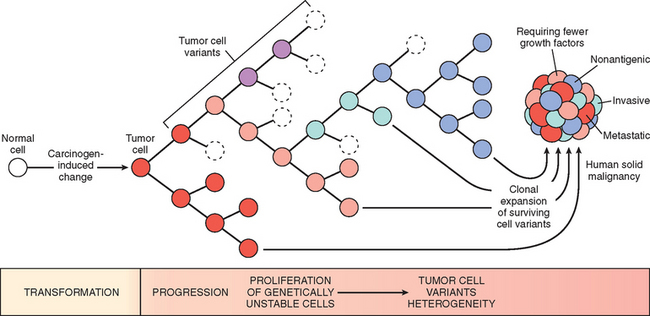
FIGURE 7-24 Tumor progression and generation of heterogeneity. New subclones arise from the descendants of the original transformed cell by multiple mutations. With progression the tumor mass becomes enriched for variants that are more adept at evading host defenses and are likely to be more aggressive.
ESSENTIAL ALTERATIONS FOR MALIGNANT TRANSFORMATION
With this overview we can now address in some detail the molecular pathogenesis of cancer and then discuss the carcinogenic agents that inflict genetic damage. Over the past two decades, hundreds of cancer-associated genes have been discovered. Some, such as p53, are mutated in many different cancers; others, such as ABL1, are affected only in one or few. Each of the cancer-associated genes has a specific function, the dysregulation of which contributes to the origin or progression of malignancy. It is traditional to describe cancer-associated genes on the basis of their presumed function. It is beneficial, however, to consider cancer-related genes in the context of seven fundamental changes in cell physiology that together determine malignant phenotype.32 (Another important change for tumor development is escape from immune attack. This property is discussed later in this chapter.) The seven key changes are the following:
Mutations in one or more genes that regulate these cellular traits are seen in every cancer. However, the precise genetic pathways that give rise to these attributes differ between individual cancers, even within the same organ. It is widely believed that the occurrence of mutations in cancer-related genes is conditioned by the robustness of the DNA-repair machinery, as well as protective mechanisms such as apoptosis and senescence that prevent the proliferation of cells with damaged DNA. Indeed, recent studies in a variety of human tumors, such as melanoma and prostate adenocarcinoma, have shown that oncogene-induced senescence, wherein mutation of a proto-oncogene drives cells into senescence rather than proliferation, is an important barrier to carcinogenesis.33 Some limits to neoplastic growth are even physical. If a tumor is to grow larger than 1 to 2 mm, mechanisms that allow the delivery of nutrients and the elimination of waste products must be provided (angiogenesis). Furthermore, epithelia are separated from the interstitial matrix by a basement membrane, composed of extracellular matrix molecules, that must be broken down by invasive carcinoma cells. These protective barriers, both intrinsic and extrinsic to the cell, must be breached, and feedback loops that normally prevent uncontrolled cell division must be disabled by mutations before a fully malignant tumor can emerge. The main principles of the molecular basis of cancer are summarized in a simplified form in Figure 7-25.
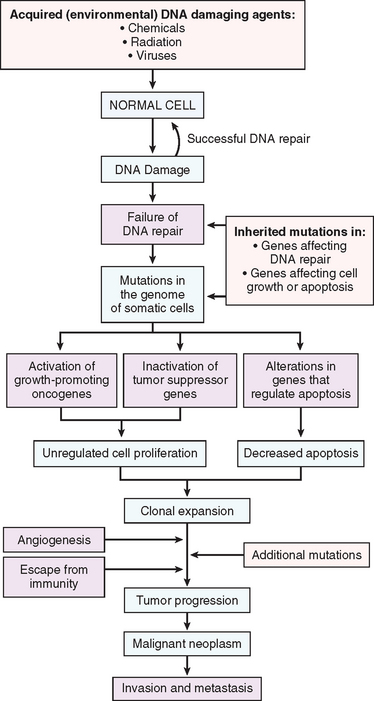
In the following sections we discuss the nature of the genes involved in each of the seven biologic alterations listed earlier. We end with a discussion of epigenetic changes and chromosomal abnormalities in cancer.
SELF-SUFFICIENCY IN GROWTH SIGNALS: ONCOGENES
Genes that promote autonomous cell growth in cancer cells are called oncogenes, and their unmutated cellular counterparts are called proto-oncogenes. Oncogenes are created by mutations in proto-oncogenes and are characterized by the ability to promote cell growth in the absence of normal growth-promoting signals. Their products, called oncoproteins, resemble the normal products of proto-oncogenes except that oncoproteins are often devoid of important internal regulatory elements, and their production in the transformed cells does not depend on growth factors or other external signals. In this way cell growth becomes autonomous, freed from checkpoints and dependence upon external signals. To aid in the understanding of the nature and functions of oncoproteins and their role in cancer, it is necessary to briefly mention the sequential steps that characterize normal cell proliferation. Under physiologic conditions cell proliferation can be readily resolved into the following steps:
With this background we can readily identify the strategies used by cancer cells to acquire self-sufficiency in growth signals. They can be grouped on the basis of their role in growth factor–mediated signal transduction cascades and cell cycle regulation.
Proto-oncogenes, Oncogenes, and Oncoproteins
Proto-oncogenes have multiple roles, participating in cellular functions related to growth and proliferation. Proteins encoded by proto-oncogenes may function as growth factors or their receptors, signal transducers, transcription factors, or cell cycle components. Oncoproteins encoded by oncogenes generally serve functions similar to their normal counterparts (Table 7-6). However, mutations convert proto-oncogenes into constitutively active cellular oncogenes that are involved in tumor development because the oncoproteins they encode endow the cell with self-sufficiency in growth.34
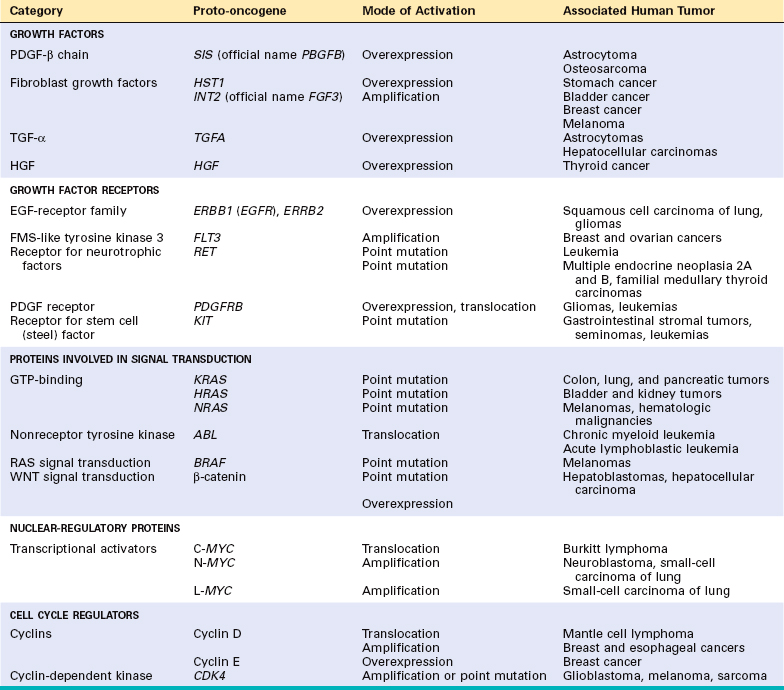
Two questions follow: (1) What are the functions of oncogene products, the oncoproteins? (2) How do the normally “civilized” proto-oncogenes turn into “enemies within”? These issues are discussed below.
Growth Factors.
Normal cells require stimulation by growth factors to undergo proliferation. Most soluble growth factors are made by one cell type and act on a neighboring cell to stimulate proliferation (paracrine action). Many cancer cells, however, acquire the ability to synthesize the same growth factors to which they are responsive, generating an autocrine loop. For example, many glioblastomas secrete platelet-derived growth factor (PDGF) and express the PDGF receptor, and many sarcomas make both transforming growth factor α (TGF-α) and its receptor. Although an autocrine loop is considered to be an important element in the pathogenesis of several tumors, in most instances the growth factor gene itself is not altered or mutated. More commonly, products of other oncogenes that lie along many signal transduction pathways, such as RAS, cause overexpression of growth factor genes, thus forcing the cells to secrete large amounts of growth factors, such as TGF-α. Nevertheless, increased growth factor production is not sufficient for neoplastic transformation. In all likelihood growth factor driven proliferation contributes to the malignant phenotype by increasing the risk of spontaneous or induced mutations in the proliferating cell population.
Growth Factor Receptors.
Several oncogenes that encode growth factor receptors have been found. To understand how mutations affect the function of these receptors, it should be recalled that one important class of growth factor receptors are transmembrane proteins with an external ligand-binding domain and a cytoplasmic tyrosine kinase domain (Chapter 3). In the normal forms of these receptors, the kinase is transiently activated by binding of the specific growth factors, followed rapidly by receptor dimerization and tyrosine phosphorylation of several substrates that are a part of the signaling cascade. The oncogenic versions of these receptors are associated with constitutive dimerization and activation without binding to the growth factor. Hence, the mutant receptors deliver continuous mitogenic signals to the cell, even in the absence of growth factor in the environment.
Growth factor receptors can be constitutively activated in tumors by multiple different mechanisms, including mutations, gene rearrangements, and overexpression. The RET proto-oncogene, a receptor tyrosine kinase, exemplifies oncogenic conversion via mutations and gene rearrangements.33 The RET protein is a receptor for the glial cell line–derived neurotrophic factor and structurally related proteins that promote cell survival during neural development. RET is normally expressed in neuroendocrine cells, such as parafollicular C cells of the thyroid, adrenal medulla, and parathyroid cell precursors. Point mutations in the RET proto-oncogene are associated with dominantly inherited MEN types 2A and 2B and familial medullary thyroid carcinoma (Chapter 24). In MEN-2A, point mutations in the RET extracellular domain cause constitutive dimerization and activation, leading to medullary thyroid carcinomas and adrenal and parathyroid tumors. In MEN-2B, point mutations in the RET cytoplasmic catalytic domain alter the substrate specificity of the tyrosine kinase and lead to thyroid and adrenal tumors without involvement of the parathyroid. In all these familial conditions, the affected individuals inherit the RET mutation in the germline. Sporadic medullary carcinomas of the thyroid are associated with somatic rearrangements of the RET gene, generally similar to those found in MEN-2B.35,36
Oncogenic conversions by mutations and rearrangements have been found in other growth factor receptor genes. Point mutations in FLT3, the gene encoding the FMS-like tyrosine kinase 3 receptor, that lead to constitutive signaling have been detected in myeloid leukemias. In certain chronic myelomonocytic leukemias with the (5;12) translocation, the entire cytoplasmic domain of the PDGF receptor is fused with a segment of an ETS family transcription factor, resulting in permanent dimerization of the PDGF receptor. Greater than 90% of gastrointestinal stromal tumors have a constitutively activating mutation in the receptor tyrosine kinase c-KIT or PDGFR, which are the receptors for stem cell factor and PDGF, respectively. These mutations are amenable to specific inhibition by the tyrosine kinase inhibitor imatinib mesylate. This type of therapy, directed to a specific alteration in the cancer cell, is called targeted therapy.37
Far more common than mutations of these proto-oncogenes is overexpression of normal forms of growth factor receptors. In some tumors increased receptor expression results from gene amplification, but in many cases the molecular basis of increased receptor expression is not fully known. Two members of the epidermal growth factor (EGF) receptor family are the best described. The normal form of ERBB1, the EGF receptor gene, is overexpressed in up to 80% of squamous cell carcinomas of the lung, in 50% or more of glioblastomas (Chapter 28), and in 80% to 100% of head and neck tumors.38,39 Likewise, the ERBB2 gene (also called HER-2/NEU), the second member of the EGF receptor family, is amplified in approximately 25% of breast cancers and in human adenocarcinomas arising within the ovary, lung, stomach, and salivary glands.36 Because the molecular alteration in ERBB2 is specific for the cancer cells, new therapeutic agents consisting of monoclonal antibodies specific to ERBB2 have been developed and are currently in use clinically, providing yet another example of targeted therapy.38,39
Signal-Transducing Proteins.
Several examples of oncoproteins that mimic the function of normal cytoplasmic signal-transducing proteins have been found. Most such proteins are strategically located on the inner leaflet of the plasma membrane, where they receive signals from outside the cell (e.g., by activation of growth factor receptors) and transmit them to the cell’s nucleus. Biochemically, the signal-transducing proteins are heterogeneous. The most well-studied example of a signal-transducing oncoprotein is the RAS family of guanine triphosphate (GTP)-binding proteins (G proteins).
The RAS Oncogene.
The RAS genes, of which there are three in the human genome (HRAS, KRAS, NRAS), were discovered initially in transforming retroviruses. Point mutation of RAS family genes is the single most common abnormality of proto-oncogenes in human tumors. Approximately 15% to 20% of all human tumors contain mutated versions of RAS proteins.40 The frequency of such mutations varies with different tumors, but in some types of cancers it is very high. For example, 90% of pancreatic adenocarcinomas and cholangiocarcinomas contain a RAS point mutation, as do about 50% of colon, endometrial, and thyroid cancers and 30% of lung adenocarcinomas and myeloid leukemias.41,42 In general, carcinomas (particularly from colon and pancreas) have mutations of KRAS, bladder tumors have HRAS mutations, and hematopoietic tumors bear NRAS mutations. RAS mutations are infrequent in certain other cancers, such as those arising in the uterine cervix or breast.
RAS plays an important role in signaling cascades downstream of growth factor receptors, resulting in mitogenesis. For example, abrogation of RAS function blocks the proliferative response to EGF, PDGF, and CSF-1. Normal RAS proteins are tethered to the cytoplasmic aspect of the plasma membrane, as well as the endoplasmic reticulum and Golgi membranes. They can be activated by growth factor binding to receptors at the plasma membrane.40 RAS is a member of a family of small G proteins that bind guanosine nucleotides (guanosine triphosphate, GTP and guanosine diphosphate, GDP), similar to the larger trimolecular G proteins. Normally RAS proteins flip back and forth between an excited signal-transmitting state and a quiescent state. In the inactive state, RAS proteins bind GDP. Stimulation of cells by growth factors leads to exchange of GDP for GTP and subsequent conformational changes that generates active RAS (Fig. 7-26). The activated RAS stimulates downstream regulators of proliferation, such as the mitogen-activated protein (MAP) kinase cascade, which floods the nucleus with signals for cell proliferation.
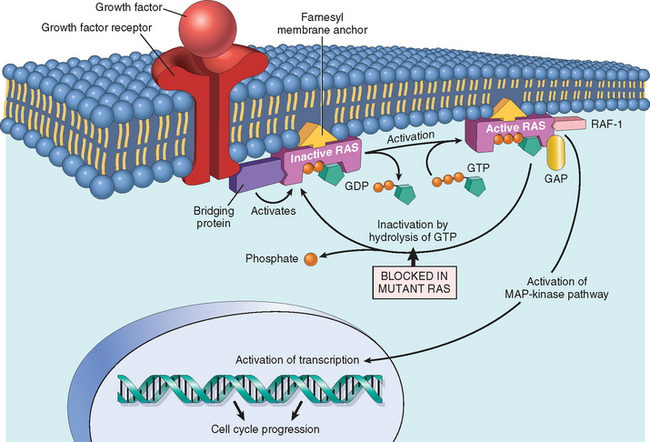
FIGURE 7-26 Model for action of RAS genes. When a normal cell is stimulated through a growth factor receptor, inactive (GDP-bound) RAS is activated to a GTP-bound state. Activated RAS recruits RAF and stimulates the MAP-kinase pathway to transmit growth-promoting signals to the nucleus. The mutated RAS protein is permanently activated because of inability to hydrolyze GTP, leading to continuous stimulation of cells without any external trigger. The anchoring of RAS to the cell membrane by the farnesyl moiety is essential for its action. See text for explanation of abbreviations.
The orderly cycling of the RAS protein depends on two reactions: (1) nucleotide exchange (GDP by GTP), which activates RAS protein, and (2) GTP hydrolysis, which converts the GTP-bound, active RAS to the GDP-bound, inactive form. Both these processes are extrinsically regulated by other proteins. The removal of GDP and its replacement by GTP during RAS activation are catalyzed by a family of guanine nucleotide–releasing proteins. Conversely, the GTPase activity intrinsic to normal RAS proteins is dramatically accelerated by GTPase-activating proteins (GAPs). These widely distributed proteins bind to the active RAS and augment its GTPase activity by more than 1000-fold, leading to termination of signal transduction. Thus, GAPs function as “brakes” that prevent uncontrolled RAS activity.
Several distinct point mutations of RAS have been identified in cancer cells. The affected residues lie within either the GTP-binding pocket or the enzymatic region essential for GTP hydrolysis, and thus markedly reduce the GTPase activity of the RAS protein. Mutated RAS is trapped in its activated GTP-bound form, and the cell is forced into a continuously proliferating state. It follows from this scenario that the consequences of mutations in RAS protein would be mimicked by mutations in the GAPs that fail to activate the GTPase activity and thus restrain normal RAS proteins. Indeed, disabling mutation of neurofibromin 1, a GAP, is associated with the inherited cancer syndrome familial neurofibromatosis type 1 (Chapter 27).
In addition to RAS, downstream members of the RAS signaling cascade (RAS/RAF/MAP kinase) may also be altered in cancer cells, resulting in a similar phenotype. Thus, mutations in BRAF, one of the members of the RAF family, have been detected in more than 60% of melanomas and in more than 80% of benign nevi.44,45 This suggests that dysregulation of the RAS/RAF/MAP kinase pathway may be one of the initiating events in the development of melanomas, although it is not sufficient by itself to cause tumorigenesis. Indeed, BRAF mutations alone lead to oncogene-induced senescence giving rise to benign nevi rather than malignant melanoma. Thus, oncogene-induced senescence is a barrier to carcinogenesis that must be overcome by mutation and disabling of key protective mechanisms, such as those provided by the p53 gene (discussed later).33
Because RAS is so frequently mutated in human cancers, much effort has been spent to develop anti-RAS modalities of targeted therapy. Unfortunately, none of these strategies has so far proven to be successful for clinical use.
Alterations in Nonreceptor Tyrosine Kinases
Mutations that unleash latent oncogenic activity occur in several non-receptor-associated tyrosine kinases, which normally function in signal transduction pathways that regulate cell growth (Chapter 3). As with receptor tyrosine kinases, in some instances the mutations take the form of chromosomal translocations or rearrangements that create fusion genes encoding constitutively active tyrosine kinases. An important example of this oncogenic mechanism involves the c-ABL tyrosine kinase. In CML and some acute lymphoblastic leukemias, the ABL gene is translocated from its normal abode on chromosome 9 to chromosome 22 (Fig. 7-27), where it fuses with the BCR gene (see discussion of chromosomal translocations, later in this chapter). The resultant chimeric gene encodes a constitutively active, oncogenic BCR-ABL tyrosine kinase. Several structural features of the BCR-ABL fusion protein contribute to the increased kinase activity, but the most important is that the BCR moiety promotes the self-association of BCR-ABL. This is a common theme, since many different oncogenic tyrosine kinases consist of fusion proteins in which the non–tyrosine kinase partner drives self-association.46 Treatment of CML has been revolutionized by the development of imatinib mesylate, a “designer” drug with low toxicity and high therapeutic efficacy that inhibits the BCR-ABL kinase.47-49 This is another example of rational drug design emerging from an understanding of the molecular basis of cancer. It is also an example of the concept of oncogene addiction.50 Despite accumulation of numerous mutations throughout the genome, signaling through the BCR-ABL gene is required for the tumor to persist, hence inhibition of its activity is effective therapy. BCR-ABL translocation is an early, perhaps initiating event, during leukemogenesis. The remaining mutations are selected for, and built around, the constant signaling through BCR-ABL. BCR-ABL signaling can be seen as the central lodgepole around which the structure is built. Remove the lodgepole by inhibition of the BCR-ABL kinase, and the structure collapses.

FIGURE 7-27 The chromosomal translocation and associated oncogenes in Burkitt lymphoma and chronic myelogenous leukemia.
In other instances, nonreceptor tyrosine kinases are activated by point mutations that abrogate the function of negative regulatory domains that normally hold enzyme activity in check. For example, several myeloproliferative disorders, particularly polycythemia vera and primary myelofibrosis, are highly associated with activating point mutations in the tyrosine kinase JAK2 (Chapter 13).51 The aberrant JAK2 kinase in turn activates transcription factors of the STAT family, which promote the growth factor–independent proliferation and survival of the tumor cells. Recognition of this molecular lesion has led to trials of JAK2 inhibitors in myeloproliferative disorders, and stimulated searches for activating mutations in other nonreceptor tyrosine kinases in a wide variety of human cancers.
Transcription Factors.
Just as all roads lead to Rome, all signal transduction pathways converge to the nucleus, where a large bank of responder genes that orchestrate the cell’s orderly advance through the mitotic cycle are activated. Indeed, the ultimate consequence of signaling through oncogenes like RAS or ABL is inappropriate and continuous stimulation of nuclear transcription factors that drive growth-promoting genes. Transcription factors contain specific amino acid sequences or motifs that allow them to bind DNA or to dimerize for DNA binding. Binding of these proteins to specific sequences in the genomic DNA activates transcription of genes. Growth autonomy may thus occur as a consequence of mutations affecting genes that regulate transcription. A host of oncoproteins, including products of the MYC, MYB, JUN, FOS, and REL oncogenes, are transcription factors that regulate the expression of growth-promoting genes, such as cyclins. Of these, MYC is most commonly involved in human tumors, and hence a brief overview of its function is warranted.
The MYC Oncogene.
The MYC proto-oncogene is expressed in virtually all eukaryotic cells and belongs to the immediate early response genes, which are rapidly induced when quiescent cells receive a signal to divide (see discussion of liver regeneration in Chapter 3). After a transient increase of MYC messenger RNA, the expression declines to a basal level. The molecular basis of MYC function in cell replication is not entirely clear. As with many transcription factors, it is thought that MYC is involved in carcinogenesis by activating genes that are involved in proliferation. Indeed, some of its target genes, such as ornithine decarboxylase and cyclin D2, are known to be associated with cell proliferation. However, the range of activities modulated by MYC is very broad and includes histone acetylation, reduced cell adhesion, increased cell motility, increased telomerase activity, increased protein synthesis, decreased proteinase activity, and other changes in cellular matbolism that enable a high rate of cell division.52 Genomic mapping of MYC binding sites has identified thousands of different sites and an equivalent number of genes that might be regulated.53 However, there is little overlap in the MYC target genes in different cancers, preventing identification of a canonical MYC carcinogenesis program. Interestingly, it has been recently suggested that MYC interacts with components of the DNA-replication machinery, and plays a role in the selection of origins of replication.54 Thus, overexpression of MYC may drive activation of more origins than needed for normal cell division, or bypass checkpoints involved in replication, leading to genomic damage and accumulation of mutations. Finally, MYC is one of a handful of transcription factors that can act in concert to reprogram somatic cells into pluripotent stem cells (Chapter 3); MYC may also enhance self-renewal, block differentiation, or both.
While on one hand MYC activation is linked to proliferation, on the other hand, cells in culture undergo apoptosis if MYC activation occurs in the absence of survival signals (growth factors).55 The MYC proto-oncogene contains separate domains that encode the growth-promoting and apoptotic activities, but it is not clear whether MYC-induced apoptosis occurs in vivo.
In contrast to the regulated expression of MYC during normal cell proliferation, persistent expression, and in some cases overexpression, of the MYC protein are commonly found in tumors. Dysregulation of MYC expression resulting from translocation of the gene occurs in Burkitt lymphoma, a B-cell tumor (see Fig. 7-27). MYC is amplified in some cases of breast, colon, lung, and many other carcinomas. The related N-MYC and L-MYC genes are amplified in neuroblastomas (Fig. 7-28) and small-cell cancers of the lung, respectively.
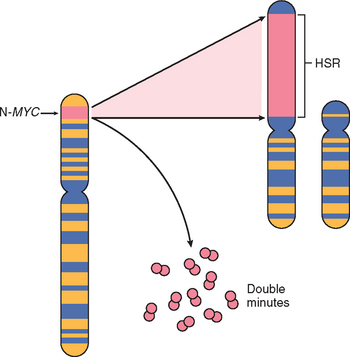
FIGURE 7-28 Amplification of the N-MYC gene in human neuroblastomas. The N-MYC gene, normally present on chromosome 2p, becomes amplified and is seen either as extra chromosomal double minutes or as a chromosomally integrated, homogeneous staining region (HSR). The integration involves other autosomes, such as 4, 9, or 13.
(Modified from Brodeur GM: Molecular correlates of cytogenetic abnormalities in human cancer cells: implications for oncogene activation. In Brown EB (ed): Progress in Hematology, Vol 14. Orlando, FL, Grune & Stratton, 1986, p 229–256.)
Cyclins and Cyclin-Dependent Kinases.
The ultimate outcome of all growth-promoting stimuli is the entry of quiescent cells into the cell cycle. Cancers may grow autonomously if the genes that drive the cell cycle become dysregulated by mutations or amplification. As described in Chapter 3, the orderly progression of cells through the various phases of the cell cycle is orchestrated by cyclin-dependent kinases (CDKs), which are activated by binding to cyclins, so called because of the cyclic nature of their production and degradation. The CDK-cyclin complexes phosphorylate crucial target proteins that drive the cell through the cell cycle. On completion of this task, cyclin levels decline rapidly. More than 15 cyclins have been identified; cyclins D, E, A, and B appear sequentially during the cell cycle and bind to one or more CDK. The cell cycle may thus be seen as a relay race in which each lap is regulated by a distinct set of cyclins, and as one set of cyclins leaves the track, the next set takes over (Fig. 7-29 and Table 7-7).
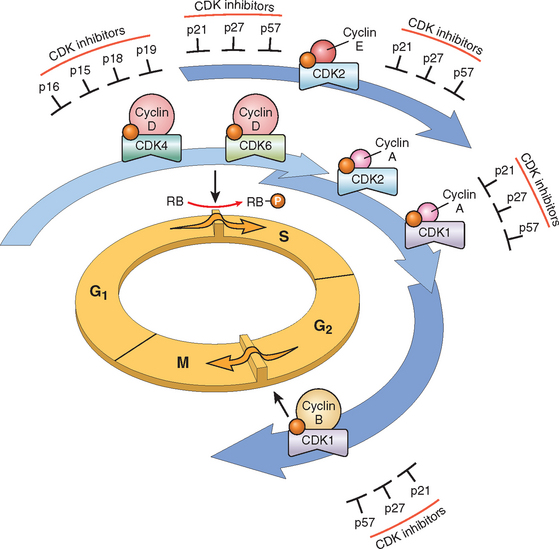
FIGURE 7-29 Schematic illustration of the role of cyclins, CDKs, and CDK inhibitors (CDKIs) in regulating the cell cycle. The shaded arrows represent the phases of the cell cycle during which specific cyclin-CDK complexes are active. As illustrated, cyclin D–CDK4, cyclin D–CDK6, and cyclin E–CDK2 regulate the G1-to-S transition by phosphorylation of the RB protein (pRB). Cyclin A–CDK2 and cyclin A–CDK1 are active in the S phase. Cyclin B–CDK1 is essential for the G2-to-M transition. Two families of CDKIs can block activity of CDKs and progression through the cell cycle. The so-called INK4 inhibitors, composed of p16, p15, p18, and p19, act on cyclin D–CDK4 and cyclin D–CDK6. The other family of three inhibitors, p21, p27, and p57, can inhibit all CDKs.
TABLE 7-7 Main Cell Cycle Components and Their Inhibitors
| Cell Cycle Component | Main Function |
|---|---|
| CYCLIN-DEPENDENT KINASES | |
| CDK4 | Forms a complex with cyclin D that phosphorylates RB, allowing the cell to progress through the G1 restriction point. |
| CDK2 | Forms a complex with cyclin E in late G1, which is involved in G1/S transition. Forms a complex with cyclin A at the S phase that facilitates G2/M transition. |
| CDK1 | Forms a complex with cyclin B that facilitates G2/M transition. |
| INHIBITORS | |
| CIP/KIP family: p21, p27 (CDKN2A-C) | Block the cell cycle by binding to cyclin-CDK complexes; p21 is induced by the tumor suppressor p53; p27 responds to growth suppressors such as TGF-β. |
| INK4/ARF family (CDKN1A-D) | p16/INK4a binds to cyclin D–CDK4 and promotes the inhibitory effects of RB; p14/ARF increases p53 levels by inhibiting MDM2 activity. |
| CHECKPOINT COMPONENTS | |
| p53 | Tumor suppressor gene altered in the majority of cancers; causes cell cycle arrest and apoptosis. Acts mainly through p21 to cause cell cycle arrest. Causes apoptosis by inducing the transcription of pro-apoptotic genes such as BAX. Levels of p53 are negatively regulated by MDM2 through a feedback loop. p53 is required for the G1/S checkpoint and is a main component of the G2/M checkpoint. |
| Ataxia-telangiectasia mutated | Activated by mechanisms that sense double-stranded DNA breaks. Transmits signals to arrest the cell cycle after DNA damage. Acts through p53 in the G1/S checkpoint. At the G2/M checkpoint, it acts both through p53-dependent mechanisms and through the inactivation of CDC25 phosphatase, which disrupts the cyclin B–CDK1 complex. Component of a network of genes that include BRCA1 and BRCA2, which link DNA damage with cell cycle arrest and apoptosis. |
With this background it is easy to appreciate that mutations that dysregulate the activity of cyclins and CDKs favor cell proliferation. Mishaps affecting the expression of cyclin D or CDK4 seem to be a common event in neoplastic transformation. The cyclin D genes are overexpressed in many cancers, including those affecting the breast, esophagus, liver, and a subset of lymphomas. Amplification of the CDK4 gene occurs in melanomas, sarcomas, and glioblastomas. Mutations affecting cyclin B and cyclin E and other CDKs also occur, but they are much less frequent.
While cyclins arouse the CDKs, their inhibitors (CDKIs), of which there are many, silence the CDKs and exert negative control over the cell cycle. The CIP/WAF family of CDKIs, composed of three proteins, called p21 (CDKN1A), p27 (CDKN1B), and p57 (CDKN1C), inhibits the CDKs broadly, whereas the INK4 family of CDK1s, made up of p15 (CDKN2B), p16 (CDKN2A), p18 (CDKN2C), and p19 (CDKN2D), has selective effects on cyclin D/CDK4 and cyclin D/CDK6. Expression of these inhibitors is down-regulated by mitogenic signaling pathways, thus promoting the progression of the cell cycle. For example, p27 (CDKN1B), a CDKI that inhibits cyclin E, is expressed throughout G1. Mitogenic signals dampen the activity of p27 in a variety of ways, relieving inhibition of cyclin E-CDK2 and thus allowing the cell cycle to proceed.56 The CDKIs are frequently mutated or otherwise silenced in many human malignancies. Germline mutations of p16 (CDKN2A) are associated with 25% of melanoma-prone kindreds.23 Somatically acquired deletion or inactivation of p16 is seen in 75% of pancreatic carcinomas, 40% to 70% of glioblastomas, 50% of esophageal cancers, 20% to 70% of acute lymphoblastic leukemias, and 20% of non-small-cell lung carcinomas, soft-tissue sarcomas, and bladder cancers.57
Before closing this discussion of the cell cycle and its regulation, we should briefly discuss the internal controls of the cell cycle called checkpoints, since later discussion of tumor suppressor genes will illustrate the importance of cell cycle checkpoints in maintaining genomic integrity. There are two main cell cycle checkpoints, one at the G1/S transition and the other at G2/M. The S phase is the point of no return in the cell cycle. Before a cell makes the final commitment to replicate, the G1/S checkpoint checks for DNA damage; if damage is present, the DNA-repair machinery and mechanisms that arrest the cell cycle are put in motion. The delay in cell cycle progression provides the time needed for DNA repair; if the damage is not repairable, apoptotic pathways are activated to kill the cell. Thus, the G1/S checkpoint prevents the replication of cells that have defects in DNA, which would be perpetuated as mutations or chromosomal breaks in the progeny of the cell. DNA damaged after its replication can still be repaired as long as the chromatids have not separated. The G2/M checkpoint monitors the completion of DNA replication and checks whether the cell can safely initiate mitosis and separate sister chromatids. This checkpoint is particularly important in cells exposed to ionizing radiation. Cells damaged by ionizing radiation activate the G2/M checkpoint and arrest in G2; defects in this checkpoint give rise to chromosomal abnormalities. To function properly, cell cycle checkpoints require sensors of DNA damage, signal transducers, and effector molecules.58 The sensors and transducers of DNA damage seem to be similar for the G1/S and G2/M checkpoints. They include, as sensors, proteins of the RAD family and ataxia telangiectasia mutated (ATM) and as transducers, the CHK kinase families.59 The checkpoint effector molecules differ, depending on the cell cycle stage at which they act. In the G1/S checkpoint, cell cycle arrest is mostly mediated through p53, which induces the cell cycle inhibitor p21. Arrest of the cell cycle by the G2/M checkpoint involves both p53-dependent and p53-independent mechanisms. Defects in cell cycle checkpoint components are a major cause of genetic instability in cancer cells.
INSENSITIVITY TO GROWTH INHIBITION AND ESCAPE FROM SENESCENCE: TUMOR SUPPRESSOR GENES
Failure of growth inhibition is one of the fundamental alterations in the process of carcinogenesis. Whereas oncogenes drive the proliferation of cells, the products of tumor suppressor genes apply brakes to cell proliferation (Table 7-8). It has become apparent that the tumor suppressor proteins form a network of checkpoints that prevent uncontrolled growth. Many tumor suppressors, such as RB and p53, are part of a regulatory network that recognizes genotoxic stress from any source, and responds by shutting down proliferation. Indeed, expression of an oncogene in an otherwise completely normal cell leads to quiescence, or to permanent cell cycle arrest (oncogene-induced senescence), rather than uncontrolled proliferation. Ultimately, the growth-inhibitory pathways may prod the cells into apoptosis. Another set of tumor suppressors seem to be involved in cell differentiation, causing cells to enter a postmitotic, differentiated pool without replicative potential. Similar to mitogenic signals, growth-inhibitory, pro-differentiation signals originate outside the cell and use receptors, signal transducers, and nuclear transcription regulators to accomplish their effects; tumor suppressors form a portion of these networks.
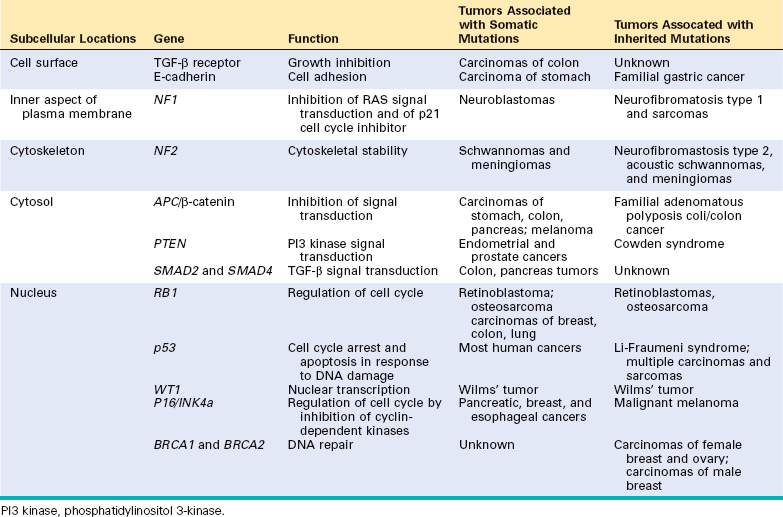
In this section we describe tumor suppressor genes, their products, and possible mechanisms by which loss of their function contributes to unregulated cell growth. The protein products of tumor suppressor genes may function as transcription factors, cell cycle inhibitors, signal transduction molecules, cell surface receptors, and regulators of cellular responses to DNA damage. In the following section we discuss the functions of the most important tumor suppressor genes, and how their defects contribute to carcinogenesis.
We begin our discussion with RB, the first, and prototypic, tumor suppressor gene discovered. Like many discoveries in medicine, RB was discovered by studying a rare disease, in this case retinoblastoma. Approximately 60% of retinoblastomas are sporadic, and the remaining are familial, with the predisposition to develop the tumor being transmitted as an autosomal dominant trait. Patients with familial retinoblastoma are also at greatly increased risk of developing osteosarcoma and other soft-tissue sarcomas. To explain the inherited and sporadic occurrence of an apparently identical tumor, Knudson proposed his now canonical “two-hit” hypothesis of oncogenesis.19,60 In molecular terms, Knudson’s hypothesis can be stated as follows (Fig. 7-30):
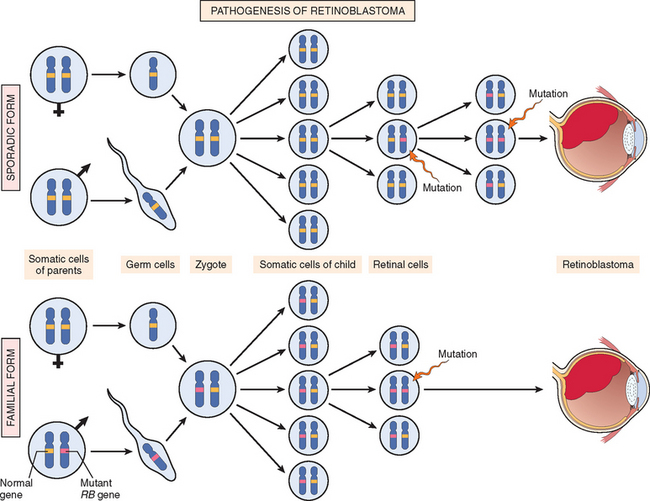
FIGURE 7-30 Pathogenesis of retinoblastoma. Two mutations of the RB locus on chromosome 13q14 lead to neoplastic proliferation of the retinal cells. In the sporadic form both mutations at the RB locus are acquired by the retinal cells after birth. In the familial form, all somatic cells inherit one mutant RB gene from a carrier parent. The second mutation affects the RB locus in one of the retinal cells after birth.
At this point we should clarify some terminology. A child carrying an inherited mutant RB allele in all somatic cells is perfectly normal (except for the increased risk of developing cancer). Because such a child is heterozygous at the RB locus, this implies that heterozygosity for the RB gene does not affect cell behavior. Cancer develops when the cell becomes homozygous for the mutant allele or, put another way, when the cell loses heterozygosity for the normal RB gene (a condition known as LOH, for loss of heterozygosity). The RB gene stands as a paradigm for several other genes that act similarly. For example, one or more genes on the short arm of chromosome 11 play a role in the formation of Wilms’ tumor, hepatoblastoma, and rhabdomyosarcoma. The von Hippel-Lindau (VHL) gene is a tumor suppressor gene that causes familial clear cell renal carcinomas and is also involved in sporadic forms of the same tumor.61 Consistent and nonrandom LOH has provided important clues to the location of several tumor suppressor genes.
RB.
RB protein, the product of the RB gene, is a ubiquitously expressed nuclear phosphoprotein that plays a key role in regulating the cell cycle. RB exists in an active hypophosphorylated state in quiescent cells and an inactive hyperphosphorylated state in the G1/S cell cycle transition (Fig. 7-31). The importance of RB lies in its enforcement of G1, or the gap between mitosis (M) and DNA replication (S). In embryos, cell divisions proceed at an amazing clip, with DNA replication beginning immediately after mitosis ends. However, as development proceeds, two gaps are incorporated into the cell cycle: Gap 1 (G1) between mitosis (M) and DNA replication (S), and Gap 2 (G2) between DNA replication (S) and mitosis (M) (see Fig. 7-29). Although each phase of the cell cycle circuitry is monitored carefully, the transition from G1 to S is believed to be an extremely important checkpoint in the cell cycle clock. Once cells cross the G1 checkpoint they can pause the cell cycle for a time, but they are obligated to complete mitosis. In G1, however, cells can exit the cell cycle, either temporarily, called quiescence, or permanently, called senescence. In G1, therefore, diverse signals are integrated to determine whether the cell should enter the cell cycle, exit the cell cycle and differentiate, or die. RB is a key node in this decision process. To understand why RB is such a crucial player, we must review the mechanisms that police the G1 phase.62
FIGURE 7-31 The role of RB in regulating the G1-S checkpoint of the cell cycle. Hypophosphorylated RB in complex with the E2F transcription factors binds to DNA, recruits chromatin-remodeling factors (histone deacetylases and histone methyltransferases), and inhibits transcription of genes whose products are required for the S phase of the cell cycle. When RB is phosphorylated by the cyclin D–CDK4, cyclin D–CDK6, and cyclin E–CDK2 complexes, it releases E2F. The latter then activates transcription of S-phase genes. The phosphorylation of RB is inhibited by CDKIs, because they inactivate cyclin-CDK complexes. Virtually all cancer cells show dysregulation of the G1-S checkpoint as a result of mutation in one of four genes that regulate the phosphorylation of RB; these genes are RB1, CDK4, the genes encoding cyclin D proteins, and CDKN2A (p16). EGF, epidermal growth factor; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-beta.
The initiation of DNA replication requires the activity of cyclin E–CDK2 complexes, and expression of cyclin E is dependent on the E2F family of transcription factors. Early in G1, RB is in its hypophosphorylated active form, and it binds to and inhibits the E2F family of transcription factors, preventing transcription of cyclin E. Hypophosphorylated RB blocks E2F-mediated transcription in at least two ways (see Fig. 7-31). First, it sequesters E2F, preventing it from interacting with other transcriptional activators. Second, RB recruits chromatin-remodeling proteins, such as histone deacetylases and histone methyltransferases, which bind to the promoters of E2F-responsive genes such as cyclin E. These enzymes modify chromatin so as to make promoters insensitive to transcription factors. Mitogenic signaling leads to cyclin D expression and activation of cyclin D–CDK4/6 complexes. These complexes phosphorylate RB, inactivating the protein and releasing E2F to induce target genes such as cyclin E. Expression of cyclin E then stimulates DNA replication and progression through the cell cycle. When the cells enter S phase, they are committed to divide without additional growth factor stimulation. During the ensuing M phase the phosphate groups are removed from RB by cellular phosphatases, regenerating the hypophosphorylated form of RB. E2Fs are not the sole effectors of Rb-mediated G1 arrest. Rb also controls the stability of the cell cycle inhibitor p27.63,64
If RB is absent (as a result of gene mutations) or its ability to regulate E2F transcription factors is derailed, the molecular brakes on the cell cycle are released, and the cells move through the cell cycle. The mutations of RB genes found in tumors are localized to a region of the RB protein, called the “RB pocket,” that is involved in binding to E2F. However, the versatile RB protein has also been shown to bind to a variety of other transcription factors that regulate cell differentiation.65 For example, RB stimulates myocyte-, adipocyte-, melanocyte-, and macrophage-specific transcription factors. Thus, the RB pathway couples control of cell cycle progression at G1 with differentiation, which may explain how differentiation is associated with exit from the cell cycle. In addition to these dual activities, RB can also induce senescence, discussed below.
It was mentioned previously that germline loss or mutations of the RB gene predispose to occurrence of retinoblastomas and to a lesser extent osteosarcomas. Furthermore, somatically acquired RB mutations have been described in glioblastomas, small-cell carcinomas of lung, breast cancers, and bladder carcinomas. Given the presence of RB in every cell and its importance in cell cycle control, two questions arise: (1) Why do patients with germline mutation of the RB locus develop mainly retinoblastomas? (2) Why are inactivating mutations of RB not much more common in human cancers? The reason for the occurrence of tumors restricted to the retina in persons who inherit one defective allele of RB is not fully understood, but some possible explanations have emerged from the study of mice with targeted disruption of the rb locus. For instance, RB family members may partially complement its function in cell types other than retinoblasts. Indeed, RB is a member of a small family of proteins, so-called pocket proteins, which also include p107 and p130.66 All three proteins bind to E2F transcription factors. The complexity grows; there are seven E2F proteins (named E2F1 through E2F7), which function as either transcriptional activators or repressors. The pocket proteins are all thought to regulate progression through the cell cycle as well as differentiation in a manner similar to that described for RB above. However, each member of this protein family binds a different set of E2F proteins and is also expressed at different times in the cell cycle. Thus, although there is some redundancy in the network, their functions are not completely overlapping. The complexity of the pocket protein–E2F network is just now being unraveled. For example, in a mouse model of retinoblastoma, it has been shown that mutation of different members of the network in various combinations generates retinoblastomas not just from retinoblasts, but also from differentiated cells in the retina, such as horizontal interneurons.67
With respect to the second question (i.e., why the loss of RB is not more common in human tumors), the answer is much simpler: Mutations in other genes that control RB phosphorylation can mimic the effect of RB loss, and such genes are mutated in many cancers that may have normal RB genes. Thus, for example, mutational activation of cyclin D or CDK4 would favor cell proliferation by facilitating RB phosphorylation. As previously discussed, cyclin D is overexpressed in many tumors because of gene amplification or translocation. Mutational inactivation of CDKIs would also drive the cell cycle by unregulated activation of cyclins and CDKs. Thus, the emerging paradigm is that loss of normal cell cycle control is central to malignant transformation and that at least one of four key regulators of the cell cycle (p16/INK4a, cyclin D, CDK4, RB) is dysregulated in the vast majority of human cancers.68 In cells that harbor mutations in any one of these other genes, the function of RB is disrupted even if the RB gene itself is not mutated.34
The transforming proteins of several oncogenic animal and human DNA viruses seem to act, in part, by neutralizing the growth-inhibitory activities of RB. In these cases, RB protein is functionally inactivated by the binding of a viral protein and no longer acts as a cell cycle inhibitor. Simian virus 40 and polyomavirus large T antigens, adenoviruses EIA protein, and HPV E7 protein all bind to the hypophosphorylated form of RB. The binding occurs in the same RB pocket that normally sequesters E2F transcription factors; in the case of HPV the binding is particularly strong for viral types, such as HPV type 16, that confer high risk for the development of cervical carcinomas. Thus, the RB protein, unable to bind the E2F transcription factors, is functionally inactivated, and the transcription factors are free to cause cell cycle progression.
Several other pathways of cell growth regulation, some to be discussed in more detail later, also converge on RB (see Fig. 7-31):
p53: Guardian of the Genome.
The p53 gene is located on chromosome 17p13.1, and it is the most common target for genetic alteration in human tumors.69 (The official name of the gene is TP53 and the protein is p53; for the sake of simplicity, we refer to both as “p53”.) A little over 50% of human tumors contain mutations in this gene. Homozygous loss of p53 occurs in virtually every type of cancer, including carcinomas of the lung, colon, and breast—the three leading causes of cancer death. In most cases, the inactivating mutations affect both p53 alleles and are acquired in somatic cells (not inherited in the germ line). Less commonly, some individuals inherit one mutant p53 allele. As with the RB gene, inheritance of one mutant allele predisposes individuals to develop malignant tumors because only one additional “hit” is needed to inactivate the second, normal allele. Such individuals, said to have the Li-Fraumeni syndrome, have a 25-fold greater chance of developing a malignant tumor by age 50 than the general population.70 In contrast to individuals who inherit a mutant RB allele, the spectrum of tumors that develop in persons with the Li-Fraumeni syndrome is quite varied; the most common types of tumors are sarcomas, breast cancer, leukemia, brain tumors, and carcinomas of the adrenal cortex. As compared with sporadic tumors, those that afflict patients with the Li-Fraumeni syndrome occur at a younger age, and a given individual may develop multiple primary tumors.71
The fact that p53 mutations are common in a variety of human tumors suggests that the p53 protein functions as a critical gatekeeper against the formation of cancer. Indeed, it is evident that p53 acts as a “molecular policeman” that prevents the propagation of genetically damaged cells. p53 is a transcription factor that is at the center of a large network of signals that sense cellular stress, such as DNA damage, shortened telomeres, and hypoxia. Many activities of the p53 protein are related to its function as a transcription factor. Several hundred genes have been shown to be regulated by p53 in numerous different contexts, but which genes are the key for the p53 response is not yet clear. Approximately 80% of the p53 point mutations present in human cancers are located in the DNA-binding domain of the protein. However, the effects of different point mutations vary considerably; in some cases there is complete abrogation of transcriptional capabilities, whereas other mutants retain the ability to bind to and activate a subset of genes. In addition to somatic and inherited mutations, p53 functions can be inactivated by other mechanisms. As with RB, the transforming proteins of several DNA viruses, including the E6 protein of HPV, can bind to and promote the degradation of p53. Also, analagous to RB, it is thought that in the majority of tumors without a p53 mutation, the function of the p53 pathway is blocked by mutation in another gene that regulates p53 function. For example, MDM2 and MDMX stimulate the degradation of p53; these proteins are frequently overexpressed in malignancies in which the gene encoding p53 is not mutated. Indeed, MDM2 is amplified in 33% of human sarcomas, thereby causing functional loss of p53 in these tumors.72,73
p53 thwarts neoplastic transformation by three interlocking mechanisms: activation of temporary cell cycle arrest (quiescence), induction of permanent cell cycle arrest (senescence), or triggering of programmed cell death (apoptosis).
In nonstressed, healthy cells, p53 has a short half-life (20 minutes), because of its association with MDM2, a protein that targets it for destruction. When the cell is stressed, for example by an assault on its DNA, p53 undergoes post-transcriptional modifications that release it from MDM2 and increase its half-life. Unshackled from MDM2, p53 also becomes activated as a transcription factor. Hundreds of genes whose transcription is triggered by p53 have been found.74,75 They can be grouped into two broad categories: those that cause cell cycle arrest and those that cause apoptosis. If DNA damage can be repaired during cell cycle arrest, the cell reverts to a normal state; if the repair fails, p53 induces apoptosis or senescence. Recently, however, the plot has thickened. It has been known that repression of a subset of pro-proliferative and anti-apoptotic genes is key to the p53 response, but it was not clear how p53 achieved repression, since in most assays it seemed to be an activator of transcription. At this point enter the recently famous miRNAs, the small guys with big clubs. It has been shown that p53 activates transcription of the mir34 family of miRNAs (mir34a–mir34c).76 miRNAs, as discussed in Chapter 5, bind to cognate sequences in the 3′ untranslated region of mRNAs, preventing translation (Fig. 7-32B). Interestingly, blocking mir34 severely hampered the p53 response in cells, while ectopic expression of mir34 without p53 activation is sufficient to induce growth arrest and apoptosis. Thus, mir34 microRNAs are able to recapitulate many of the functions of p53 and are necessary for these functions, demonstrating the importance of mir34 to the p53 response. Targets of mir34s include pro-proliferative genes such as cyclins, and anti-apoptotic genes such as BCL2. p53 regulation of mir34 explains, at least in part, how p53 is able to repress gene expression, and it seems that regulation of this miRNA is crucial for the p53 response.
FIGURE 7-32 A, The role of p53 in maintaining the integrity of the genome. Activation of normal p53 by DNA-damaging agents or by hypoxia leads to cell cycle arrest in G1 and induction of DNA repair, by transcriptional up-regulation of the cyclin-dependent kinase inhibitor CDKN1A (p21) and the GADD45 genes. Successful repair of DNA allows cells to proceed with the cell cycle; if DNA repair fails, p53 triggers either apoptosis or senescence. In cells with loss or mutations of p53, DNA damage does not induce cell cycle arrest or DNA repair, and genetically damaged cells proliferate, giving rise eventually to malignant neoplasms. B, p53 mediates gene repression by activating transcription of miRNAs. p53 activates transcription of the mir34 family of miRNAs. mir34s repress translation of both proliferative genes, such as cyclins, and anti-apoptotic genes, such as BCL2. Repression of these genes can promote either quiescence or senescence as well as apoptosis.
The manner in which p53 senses DNA damage and determines the adequacy of DNA repair is beginning to be understood. The key initiators of the DNA-damage pathway are two related protein kinases: ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3 related (ATR).77,78 As the name implies, the ATM gene was originally identified as the germ-line mutation in individuals with ataxia-telangiectasia. Persons with this disease, which is characterized by an inability to repair certain kinds of DNA damage, suffer from an increased incidence of cancer. The types of damage sensed by ATM and ATR are different, but the downstream pathways they activate are similar. Once triggered, both ATM and ATR phosphorylate a variety of targets, including p53 and DNA-repair proteins. Phosphorylation of these two targets leads to a pause in the cell cycle and stimulation of DNA-repair pathways, respectively.
p53-mediated cell cycle arrest may be considered the primordial response to DNA damage (Fig. 7-32). It occurs late in the G1 phase and is caused mainly by p53-dependent transcription of the CDK inhibitor CDKN1A (p21). As discussed, p21 inhibits cyclin-CDK complexes and phosphorylation of RB, thereby preventing cells from entering G1 phase. Such a pause in cell cycling is welcome, because it gives the cells “breathing time” to repair DNA damage. p53 also helps the process by inducing certain proteins, such as GADD45 (growth arrest and DNA damage), that help in DNA repair.75 p53 can stimulate DNA-repair pathways by transcription-independent mechanisms as well. If DNA damage is repaired successfully, p53 up-regulates transcription of MDM2, leading to its own destruction and thus releasing the cell cycle block. If the damage cannot be repaired, the cell may enter p53-induced senescence or undergo p53-directed apoptosis.
p53-induced senescence is a permanent cell cycle arrest characterized by specific changes in morphology and gene expression that differentiate it from quiescence or reversible cell cycle arrest. Senescence requires activation of p53 and/or RB and expression of their mediators, such as the CDK inhibitors, and is generally irreversible, although it may require the continued expression of p53. The mechanisms of senescence are unclear but involve epigenetic changes that result in the formation of heterochromatin at different loci throughout the genome.80 These senescence-associated heterochromatin foci include pro-proliferative genes regulated by E2F; this drastically and permanently alters expression of these E2F targets. Like all p53 responses, senescence may be stimulated in response to a variety of stresses, such as unopposed oncogene signaling, hypoxia, and shortened telomeres.
p53-induced apoptosis of cells with irreversible DNA damage is the ultimate protective mechanism against neoplastic transformation. p53 directs the transcription of several pro-apoptotic genes such as BAX and PUMA (approved name BBC3; described later). Exactly how a cell decides whether to repair its DNA or to enter apoptosis is unclear. It appears that the affinity of p53 for the promoters and enhancers of DNA-repair genes is stronger than its affinity for pro-apoptotic genes.80 Thus, the DNA-repair pathway is stimulated first, while p53 continues to accumulate. Eventually, if the DNA damage is not repaired, enough p53 accumulates to stimulate transcription of the pro-apoptotic genes and the cell dies. While this scheme is generally correct, there seem to be important cell type–specific responses as well, with some cell types succumbing to apoptosis early, while others opt for senescence.80 Such differential responses may be related to the functions of other p53 family members expressed in different cell types (see below).
To summarize, p53 links cell damage with DNA repair, cell cycle arrest, and apoptosis. In response to DNA damage, p53 is phosphorylated by genes that sense the damage and are involved in DNA repair. p53 assists in DNA repair by causing G1 arrest and inducing DNA-repair genes. A cell with damaged DNA that cannot be repaired is directed by p53 to undergo apoptosis (see Fig. 7-32). In view of these activities, p53 has been rightfully called a “guardian of the genome.” With loss of function of p53, DNA damage goes unrepaired, mutations accumulate in dividing cells, and the cell marches along a one-way street leading to malignant transformation.
The ability of p53 to control apoptosis in response to DNA damage has important practical therapeutic implications. Irradiation and chemotherapy, the two common modalities of cancer treatment, mediate their effects by inducing DNA damage and subsequent apoptosis. Tumors that retain normal p53 are more likely to respond to such therapy than tumors that carry mutated alleles of the gene. Such is the case with testicular teratocarcinomas and childhood acute lymphoblastic leukemias. By contrast, tumors such as lung cancers and colorectal cancers, which frequently carry p53 mutations, are relatively resistant to chemotherapy and irradiation. Various therapeutic modalities aimed at increasing normal p53 activity in tumor cells that retain this type of activity or selectively killing cells with defective p53 function are being investigated.
The discovery of p53 family members p63 and p73 has revealed that p53 has collaborators. Indeed, p53, p63, and p73 are players in a complex network with significant cross-talk that is only beginning to be unraveled.81,82 p53 is ubiquitously expressed, while p63 and p73 show more tissue specificity. For example, p63 is essential for the differentiation of stratified squamous epithelia, while p73 has strong pro-apoptotic effects after DNA damage induced by chemotheraputic agents. Furthermore, both p63 and p73, and probably p53 as well, are expressed as different isoforms, some of which act as transcriptional activators and others that function as dominant negatives. An illustrative example of the concerted actions of these three musketeers is seen in the so-called basal subset of breast cancers, which have a poor prognosis. These tumors have been shown to have mutations in p53 and additionally express a dominant-negative version of p63 that antagonizes the apoptotic activity of p73. This perturbation of the p53p63-p73 network contributes to the chemoresistance and poor prognosis of these tumors.83
APC/β-Catenin Pathway.
Adenomatous polyposis coli genes (APC) represents a class of tumor suppressors whose main function is to down-regulate growth-promoting signals. Germ-line mutations at the APC (5q21) loci are associated with familial adenomatous polyposis, in which all individuals born with one mutant allele develop thousands of adenomatous polyps in the colon during their teens or 20s (familial adenomatous polyposis; Chapter 17). Almost invariably, one or more of these polyps undergoes malignant transformation, giving rise to colon cancer. As with other tumor suppressor genes, both copies of the APC gene must be lost for a tumor to arise. This conclusion is supported by the development of colon adenomas in mice with targeted disruption of apc genes in the colonic mucosa.84 As discussed later, several additional mutations must occur if cancers are to develop in the adenomas. In addition to these tumors, which have a strong hereditary predisposition, 70% to 80% of nonfamilial colorectal carcinomas and sporadic adenomas also show homozygous loss of the APC gene, thus firmly implicating APC loss in the pathogenesis of colonic tumors.85
APC is a component of the WNT signaling pathway, which has a major role in controlling cell fate, adhesion, and cell polarity during embryonic development (Fig. 7-33). WNT signaling is also required for self-renewal of hematopoietic stem cells. WNT signals through a family of cell surface receptors called frizzled (FRZ), and stimulates several pathways, the central one involving β-catenin and APC.
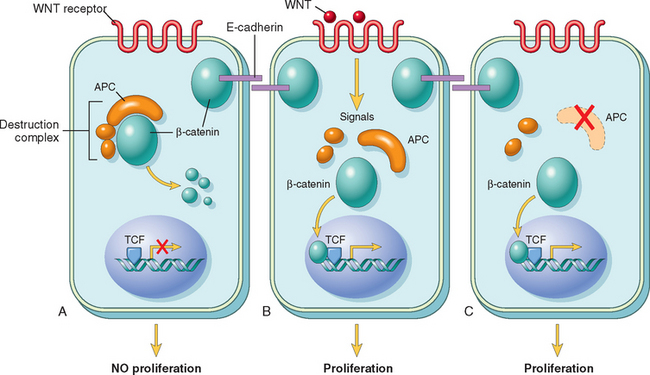
FIGURE 7-33 A, The role of APC in regulating the stability and function of β-catenin. APC and β-catenin are components of the WNT signaling pathway. In resting cells (not exposed to WNT), β-catenin forms a macromolecular complex containing the APC protein. This complex leads to the destruction of β-catenin, and intracellular levels of β-catenin are low. B, When cells are stimulated by WNT molecules, the destruction complex is deactivated, β-catenin degradation does not occur, and cytoplasmic levels increase. β-catenin translocates to the nucleus, where it binds to TCF, a transcription factor that activates genes involved in cell cycle progression. C, When APC is mutated or absent, the destruction of β-catenin cannot occur. β-catenin translocates to the nucleus and coactivates genes that promote entry into the cell cycle, and cells behave as if they are under constant stimulation by the WNT pathway.
An important function of the APC protein is to down-regulate β-catenin. In the absence of WNT signaling APC causes degradation of β-catenin, preventing its accumulation in the cytoplasm.85 It does so by forming a macromolecular complex with β-catenin, axin, and GSK3β, which leads to the phosphorylation and eventually ubiquitination of β-catenin and destruction by the proteasome. Signaling by WNT blocks the APC-AXIN-GSK3β destruction complex, allowing β-catenin to translocate from the cytoplasm to the nucleus. In the cell nucleus, β-catenin forms a complex with TCF, a transcription factor that up-regulates cellular proliferation by increasing the transcription of c-MYC, cyclin D1, and other genes. Since inactivation of the APC gene disrupts the destruction complex, β-catenin survives and translocates to the nucleus, where it can activate transcription in cooperation with TCF.85 Thus, cells with loss of APC behave as if they are under continuous WNT signaling. The importance of the APC/β-catenin signaling pathway in tumorigenesis is attested to by the fact that colon tumors that have normal APC genes harbor mutations in β-catenin that prevent its destruction by APC, allowing the mutant protein to accumulate in the nucleus. Dysregulation of the APC/β-catenin pathway is not restricted to colon cancers; mutations in the β-catenin gene are present in more than 50% of hepatoblastomas and in approximately 20% of hepatocellular carcinomas.86 As mentioned in Chapter 3, β-catenin binds to the cytoplasmic tail of E-cadherin, a cell surface protein that maintains intercellular adhesiveness. Loss of cell-cell contact, such as in a wound or injury to the epithelium, disrupts the interaction between E-cadherin and β-catenin, and allows β-catenin to travel to the nucleus and stimulate proliferation; this is an appropriate response to injury that can help repair the wound. Re-establishment of these E-cadherin contacts as the wound heals leads to β-catenin again being sequestered at the membrane and reduction in the proliferative signal; these cells are said to be “contact-inhibited.” Loss of contact inhibition, by mutation of the E-cadherin/β-catenin axis, or by other methods, is a key characteristic of carcinomas. Furthermore, loss of cadherins can favor the malignant phenotype by allowing easy disaggregation of cells, which can then invade locally or metastasize. Reduced cell surface expression of E-cadherin has been noted in many types of cancers, including those that arise in the esophagus, colon, breast, ovary, and prostate.87 Germline mutations of the E-cadherin gene can predispose to familial gastric carcinoma, and mutation of the gene and decreased E-cadherin expression are present in a variable proportion of gastric cancers of the diffuse type. The molecular basis of reduced E-cadherin expression is varied. In a small proportion of cases, there are mutations in the E-cadherin gene (located on 16q); in other cancers, E-cadherin expression is reduced as a secondary effect of mutations in β-catenin genes. Additionally, E-cadherin may be down-regulated by transcription repressors, such as SNAIL, which have been implicated in epithelial-to-mesenchymal transition and metastasis88 (discussed below).
Other Genes That Function as Tumor Suppressors.
There is little doubt that many more tumor suppressor genes remain to be discovered. Often, their location is suspected by the detection of consistent sites of chromosomal deletions or by analysis of LOH. Some of the tumor suppressor genes that are associated with well-defined clinical syndromes are briefly described below (see Table 7-8):
INK4a/ARF.
Also called the CDKN2A gene locus, the INK4a/ARF locus encodes two protein products; the p16/INK4a CDKI, which blocks cyclin D/CDK2-mediated phosphorylation of RB, keeping the RB checkpoint in place. The second gene product, p14/ARF, activates the p53 pathway by inhibiting MDM2 and preventing destruction of p53. Both protein products function as tumor suppressors, and thus mutation or silencing of this locus impacts both the RB and p53 pathways. p16 in particular is crucial for the induction of senescence. Mutations at this locus have been detected in bladder, head and neck tumors, acute lymphoblastic leukemias, and cholangiocarcinomas. In some tumors, such as cervical cancer, p16/INK4a is frequently silenced by hypermethylation of the gene, without the presence of a mutation (see discussion of epigenetic changes). The other CDKIs also function as tumor suppressors and are frequently mutated or otherwise silenced in many human malignancies, including 20% of familial melanomas, 50% of sporadic pancreatic adenocarcinomas, and squamous cell carcinomas of the esophagus.
The TGF-β Pathway.
In most normal epithelial, endothelial, and hematopoietic cells, TGF-β is a potent inhibitor of proliferation. It regulates cellular processes by binding to a serine-threonine kinase complex composed of TGF-β receptors I and II. Dimerization of the receptor upon ligand binding leads to activation of the kinase and phosphorylation of receptor SMADs (R-SMADs). Upon phosphorylation, R-SMADs can enter the nucleus, bind to SMAD-4, and activate transcription of genes, including the CDKIs p21 and p15/INK4b. In addition, TGF-β signaling leads to repression of c-MYC, CDK2, CDK4, and cyclins A and E. As can be inferred from our earlier discussion, these changes result in decreased phosphorylation of RB and cell cycle arrest.
In many forms of cancer the growth-inhibiting effects of TGF-β pathways are impaired by mutations in the TGF-β signaling pathway. These mutations may affect the type II TGF-β receptor or interfere with SMAD molecules that serve to transduce antiproliferative signals from the receptor to the nucleus. Mutations affecting the type II receptor are seen in cancers of the colon, stomach, and endometrium. Mutational inactivation of SMAD4 is common in pancreatic cancers. In 100% of pancreatic cancers and 83% of colon cancers, at least one component of the TGF-β pathway is mutated. However, in many cancers, loss of TGF-β-mediated growth inhibition occurs at a level downstream of the core signaling pathway, for example, loss of p21 and/or persistent expression of c-Myc. These tumor cells can then use other elements of the TGFβ–induced program, including immune system suppression/evasion or promotion of angiogenesis, to facilitate tumor progression.89 Thus TGF-β can function to prevent or promote tumor growth, depending on the state of other genes in the cell.
PTEN.
PTEN (Phosphatase and tensin homologue) is a membrane-associated phosphatase encoded by a gene on chromosome 10q23 that is mutated in Cowden syndrome, an autosomal dominant disorder marked by frequent benign growths, such as tumors of the skin appendages, and an increased incidence of epithelial cancers, particularly of the breast (Chapter 23), endometrium, and thyroid. PTEN acts as a tumor suppressor by serving as a brake on the pro-survival/pro-growth PI3K/AKT pathway.90,91 As you will recall from Chapter 3, this pathway is normally stimulated (along with the RAS and JAK/STAT pathways) when ligands bind to receptor tyrosine kinases and involves a cascade of phosphorylation events. First, PI3K (phosphoinositide 3-kinase) phosphorylates the lipid inositide-3-phosphate to give rise to inositide-3,4,5-triphosphate, which binds and activates the kinase PDK1. PDK1 and other factors in turn phosphorylate and activate the serine/threonine kinase AKT, which is a major node in the pathway with several important functions. By phosphorylating a number of substrates, including BAD and MDM2, AKT enhances cell survival. AKT also inactivates the TSC1/TSC2 complex. TSC1 and TSC2 are the products of two tumor suppressor genes that are mutated in tuberous sclerosis (Chapter 28), an autosomal dominant disorder associated with developmental malformations and unusual benign neoplasms such as cardiac rhabdomyomas (Chapter 12), renal angiomyolipomas, and giant cell astrocytomas. Inactivation of TSC1/TSC2 unleashes the activity of yet another kinase called mTOR (mammalian target of rapamycin, a potent immunosuppressive drug), which stimulates the uptake of nutrients such as glucose and amino acids that are needed for growth and augments the activity of several factors that are required for protein synthesis. Although acquired loss of PTEN function is one of the most common ways that PI3K/AKT signaling is upregulated in various cancers, many other components of the pathway, including PI3K itself, may also be mutated so as to increase signaling. Considering all of these molecular lesions collectively, it is said that this may be the most commonly mutated pathway in human cancer. As a result there is great interest in targeting the PI3K/AKT pathway with inhibitors of mTOR, AKT, and other kinases in the pathway.
NF1.
Individuals who inherit one mutant allele of the NF1 gene develop numerous benign neurofibromas and optic nerve gliomas as a result of inactivation of the second copy of the gene.92 This condition is called neurofibromatosis type 1 (Chapter 27). Some of the neurofibromas later develop into malignant peripheral nerve sheath tumors. Neurofibromin, the protein product of the NF1 gene, contains a GTPase-activating domain, which regulates signal transduction through RAS proteins. Recall that RAS transmits growth-promoting signals and flips back and forth between GDP-binding (inactive) and GTP-binding (active) states. Neurofibromin facilitates conversion of RAS from an active to an inactive state. With loss of neurofibromin function, RAS is trapped in an active, signal-emitting state.
NF2.
Germline mutations in the NF2 gene predispose to the development of neurofibromatosis type 2.93 As discussed in Chapter 27, individuals with mutations in NF2 develop benign bilateral schwannomas of the acoustic nerve. In addition, somatic mutations affecting both alleles of NF2 have also been found in sporadic meningiomas and ependymomas. The product of the NF2 gene, called neurofibromin 2 or merlin, shows a great deal of homology with the red cell membrane cytoskeletal protein 4.1 (Chapter 14), and is related to the ERM (ezrin, radixin, and moesin) family of membrane cytoskeleton-associated proteins. Although the mechanism by which merlin deficiency leads to carcinogenesis is not known, cells lacking this protein are not capable of establishing stable cell-to-cell junctions and are insensitive to normal growth arrest signals generated by cell-to-cell contact. Merlin is a key member of the Salvador-Warts-Hippo (SWH) tumor suppressor pathway, originally described in Drosophila. The signaling pathway controls organ size by modulating cell growth, proliferation, and apoptosis. Many human homologues of genes in the SWH pathway have been implicated in human cancers.94
VHL.
Germline mutations of the von Hippel-Lindau (VHL) gene on chromosome 3p are associated with hereditary renal cell cancers, pheochromocytomas, hemangioblastomas of the central nervous system, retinal angiomas, and renal cysts.60 Mutations of the VHL gene have also been noted in sporadic renal cell cancers (Chapter 20). The VHL protein is part of a ubiquitin ligase complex. A critical substrate for this activity is HIF1α (hypoxia-inducible transcription factor 1α). In the presence of oxygen, HIF1α is hydroxylated and binds to the VHL protein, leading to ubiquitination and proteasomal degradation. This hydroxylation reaction requires oxygen; in hypoxic environments the reaction cannot occur, and HIF1α escapes recognition by VHL and subsequent degradation. HIF1α can then translocate to the nucleus and turn on many genes, such as the growth/angiogenic factors vascular endothelial growth factor (VEGF) and PDGF. Lack of VHL activity prevents ubiquitination and degradation of HIF1α and is associated with increased levels of angiogenic growth factors.
WT1.
The WT1 gene, located on chromosome 11p13, is associated with the development of Wilms’ tumor, a pediatric kidney cancer.95 Both inherited and sporadic forms of Wilms’ tumor occur, and mutational inactivation of the WT1 locus has been seen in both forms. The WT1 protein is a transcriptional activator of genes involved in renal and gonadal differentiation. It regulates the mesenchymal-to-epithelial transition that occurs in kidney development. Though not precisely known, it is likely that the tumorigenic effect of WT1 deficiency is intimately connected with the role of the gene in the differentiation of genitourinary tissues. Interestingly, although WT1 is a tumor suppressor in Wilms’ tumor, a variety of adult cancers, including leukemias and breast carcinomas, have also been shown to overexpress WT1. Since these tissues do not normally express WT1 at all, it has been suggested that WT1 may function as an oncogene in these cancers. Another Wilms’ gene, WT2, located on 11p15, is associated with the Beckwith-Wiedemann syndrome (Chapter 10).
Patched (PTCH).
PTCH1 and PTCH2 are tumor suppressor genes that encode a cell membrane protein (PATCHED), which functions as a receptor for a family of proteins called Hedgehog.96 The Hedgehog/PATCHED pathway regulates several genes, including TGF-β and PDGFRA and PDGFRB. Mutations in PTCH are related to Gorlin syndrome, an inherited condition also known as nevoid basal cell carcinoma syndrome (see Chapter 26). PTCH mutations are present in 20% to 50% of sporadic cases of basal cell carcinoma. About one half of such mutations are of the type caused by UV exposure.
EVASION OF APOPTOSIS
Accumulation of neoplastic cells may result not only from activation of growth-promoting oncogenes or inactivation of growth-suppressing tumor suppressor genes, but also from mutations in the genes that regulate apoptosis.97-99 Thus, apoptosis represents a barrier that must be surmounted for cancer to occur. In the adult, cell death by apoptosis is a physiologic response to several pathologic conditions that might contribute to malignancy if the cells remained viable. A cell with genomic injury can be induced to die, preventing the accumulation of cells with mutations. A variety of signals, ranging from DNA damage to loss of adhesion to the basement membrane (termed anoikis), can trigger apoptosis. A large family of genes that regulate apoptosis has been identified. Before we can understand how tumor cells evade apoptosis, it is essential to review briefly the biochemical pathways to apoptosis.
As discussed in Chapter 1, there are two distinct programs that activate apoptosis, the extrinsic and intrinsic pathways. Figure 7-34 shows, in simplified form, the sequence of events that lead to apoptosis by signaling through the death receptor CD95/Fas (extrinsic pathway) and by DNA damage (intrinsic pathway). The extrinsic pathway is initiated when CD95/Fas binds to its ligand, CD95L/FasL, leading to trimerization of the receptor and its cytoplasmic death domains, which attract the intracellular adaptor protein FADD. This protein recruits procaspase 8 to form the death-inducing signaling complex. Procaspase 8 is activated by cleavage into smaller subunits, generating caspase 8. Caspase 8 then activates downstream caspases such as caspase 3, a typical executioner caspase that cleaves DNA and other substrates to cause cell death. Additionally, caspase 8 can cleave and activate the BH3-only protein BID, activating the intrinsic pathway as well. The intrinsic pathway of apoptosis is triggered by a variety of stimuli, including withdrawal of survival factors, stress, and injury. Activation of this pathway leads to permeabilization of the mitochondrial outer membrane, with resultant release of molecules, such as cytochrome c, that initiate apoptosis. The integrity of the mitochondrial outer membrane is regulated by pro-apoptotic and anti-apoptotic members of the BCL2 family of proteins.100 The pro-apoptotic proteins BAX and BAK are required for apoptosis and directly promote mitochondrial permeabilization. Their action is inhibited by the anti-apoptotic members of this family exemplified by BCL2 and BCL-XL. A third set of proteins (so-called BH3-only proteins), including BAD, BID, and PUMA, regulate the balance between the pro- and anti-apoptotic members of the BCL2 family. The BH3-only proteins sense death-inducing stimuli and promote apoptosis by neutralizing the actions of anti-apoptotic proteins like BCL2 and BCL-XL. When the sum total of all BH3 proteins expressed “overwhelms” the anti-apoptotic BCL2/BCL-XL protein barrier, BAX and BAK are activated and form pores in the mitochondrial membrane. Cytochrome c leaks into the cytosol, where it binds to APAF1, activating caspase 9. Like caspase 8 of the extrinsic pathway, caspase 9 can cleave and activate the executioner caspases. The caspases can be inhibited by a family of proteins called Inhibitors of Apoptosis Proteins (IAPs). Some tumors avoid apoptosis by upregulating these proteins, and there is interest in developing drugs that can block the interaction between IAPs and caspases. Because of the pro-apoptotic effect of BH3-only proteins, efforts are underway to develop BH3 mimetic drugs.

FIGURE 7-34 CD95 receptor–induced and DNA damage–triggered pathways of apoptosis and mechanisms used by tumor cells to evade cell death. (1) Reduced CD95 level. (2) Inactivation of death-induced signaling complex by FLICE protein (caspase 8; apoptosis-related cysteine peptidase). (3) Reduced egress of cytochrome c from mitochondrion as a result of up-regulation of BCL2. (4) Reduced levels of pro-apoptotic BAX resulting from loss of p53. (5) Loss of apoptotic peptidase activating factor 1 (APAF1). (6) Up-regulation of inhibitors of apoptosis (IAP). FADD, Fas-associated via death domain.
Within this framework it is possible to illustrate the multiple sites at which apoptosis is frustrated by cancer cells101 (see Fig. 7-34). Starting from the surface, reduced levels of CD95/Fas may render the tumor cells less susceptible to apoptosis by CD95L/FasL. Some tumors have high levels of FLIP, a protein that can bind death-inducing signaling complex and prevent activation of caspase 8. Of all these genes, perhaps best established is the role of BCL2 in protecting tumor cells from apoptosis. As discussed later, approximately 85% of B-cell lymphomas of the follicular type (Chapter 13) carry a characteristic t(14;18)(q32;q21) translocation. Recall that 14q32, the site where immunoglobulin heavy-chain (IgH) genes are found, is also involved in the pathogenesis of Burkitt lymphoma. Juxtaposition of this transcriptionally active locus with BCL2 (located at 18q21) causes overexpression of the BCL2 protein. This in turn increases the BCL2/BCL-XL buffer, protecting lymphocytes from apoptosis and allowing them to survive for long periods; there is therefore a steady accumulation of B lymphocytes, resulting in lymphadenopathy and marrow infiltration. Because BCL2-overexpressing lymphomas arise in large part from reduced cell death rather than explosive cell proliferation, they tend to be indolent (slow growing) compared with many other lymphomas.
As mentioned before, p53 is an important pro-apoptotic gene that induces apoptosis in cells that are unable to repair DNA damage. The actions of p53 are mediated in part by transcriptional activation of BAX, but there are other connections as well between p53 and the apoptotic machinery. Thus, the apoptotic machinery in cancer may be thwarted by mutations affecting the component proteins directly, as well as by loss of sensors of genomic integrity such as p53.
LIMITLESS REPLICATIVE POTENTIAL: TELOMERASE
As was discussed in the section on cellular aging (Chapter 1), most normal human cells have a capacity of 60 to 70 doublings. After this, the cells lose their ability to divide and become senescent. This phenomenon has been ascribed to progressive shortening of telomeres at the ends of chromosomes. Indeed, short telomeres seem to be recognized by the DNA-repair machinery as double-stranded DNA breaks, and this leads to cell cycle arrest mediated by p53 and RB.102 In cells in which the checkpoints are disabled by p53 or RB1 mutations, the nonhomologous end-joining pathway is activated as a last-ditch effort to save the cell, joining the shortened ends of two chromosomes.103 This inappropriately activated repair system results in dicentric chromosomes that are pulled apart at anaphase, resulting in new double-stranded DNA breaks. The resulting genomic instability from the repeated bridge-fusion-breakage cycles eventually produces mitotic catastrophe, characterized by massive cell death. It follows that for tumors to grow indefinitely, as they often do, loss of growth restraints is not enough. Tumor cells must also develop ways to avoid both cellular senescence and mitotic catastrophe (Fig. 7-35). If during crisis a cell manages to reactivate telomerase, the bridge-fusion-breakage cycles cease and the cell is able to avoid death. However, during the period of genomic instability that precedes telomerase activation, numerous mutations could accumulate, helping the cell march toward malignancy. Passage through a period of genomic instability may explain the complex karyotypes frequently seen in human carcinomas. Telomerase, active in normal stem cells, is normally absent, or expressed at very low levels in most somatic cells. By contrast, telomere maintenance is seen in virtually all types of cancers. In 85% to 95% of cancers, this is due to up-regulation of the enzyme telomerase. A few tumors use other mechanisms, termed alternative lengthening of telomeres, which probably depend on DNA recombination. Interestingly, in the progression from colonic adenoma to colonic adenocarcinoma, early lesions had a high degree of genomic instability with low telomerase expression, whereas malignant lesions had complex karyotypes with high levels of telomerase activity, consistent with a model of telomere-driven tumorigenesis in human cancer. Several other mechanisms of genomic instability are discussed later.
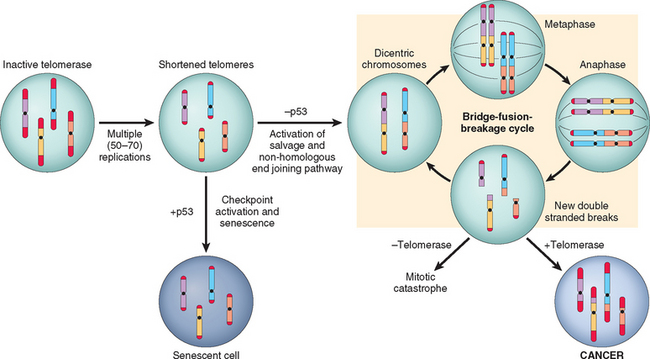
FIGURE 7-35 Sequence of events in the development of limitless replicative potential. Replication of somatic cells, which do not express telomerase, leads to shortened telomeres. In the presence of competent checkpoints, cells undergo arrest and enter nonreplicative senescence. In the absence of checkpoints, DNA-repair pathways are inappropriately activated, leading to the formation of dicentric chromosomes. At mitosis the dicentric chromosomes are pulled apart, generating random double-stranded breaks, which then activate DNA-repair pathways, leading to the random association of double-stranded ends and the formation, again, of dicentric chromosomes. Cells undergo numerous rounds of this bridge-fusion-breakage cycle, which generates massive chromosomal instability and numerous mutations. If cells fail to re-express telomerase, they eventually undergo mitotic catastrophe and death. Re-expression of telomerase allows the cells to escape the bridge-fusion-breakage cycle, thus promoting their survival and tumorigenesis.
ANGIOGENESIS
Even with all the genetic abnormalities discussed above, solid tumors cannot enlarge beyond 1 to 2 mm in diameter unless they are vascularized. Like normal tissues, tumors require delivery of oxygen and nutrients and removal of waste products; presumably the 1- to 2-mm zone represents the maximal distance across which oxygen, nutrients, and waste can diffuse from blood vessels. Cancer cells can stimulate neo-angiogenesis, during which new vessels sprout from previously existing capillaries, or, in some cases, vasculogenesis, in which endothelial cells are recruited from the bone marrow (Chapter 3). Tumor vasculature is abnormal, however. The vessels are leaky and dilated, and have a haphazard pattern of connection. Neovascularization has a dual effect on tumor growth: perfusion supplies needed nutrients and oxygen, and newly formed endothelial cells stimulate the growth of adjacent tumor cells by secreting growth factors, such as insulin-like growth factors (IGFs), PDGF, and granulocyte-macrophage colony-stimulating factor. Angiogenesis is required not only for continued tumor growth but also for access to the vasculature and hence for metastasis. Angiogenesis is thus a necessary biologic correlate of malignancy.104
How do growing tumors develop a blood supply? The emerging paradigm is that tumor angiogenesis is controlled by the balance between angiogenesis promoters and inhibitors. Early in their growth, most human tumors do not induce angiogenesis. They remain small or in situ, possibly for years, until the angiogenic switch terminates this stage of vascular quiescence.105 The molecular basis of the angiogenic switch involves increased production of angiogenic factors and/or loss of angiogenic inhibitors. These factors may be produced directly by the tumor cells themselves or by inflammatory cells (e.g., macrophages) or other stromal cells associated with the tumors. Proteases, either elaborated by the tumor cells directly or from stromal cells in response to the tumor, are also involved in regulating the balance between angiogenic and anti-angiogenic factors. Many proteases can release the proangiogenic basic fibroblast growth factors (bFGF) stored in the ECM; conversely, three potent angiogenesis inhibitors—angiostatin, endostatin, and vasculostatin—are produced by proteolytic cleavage of plasminogen, collagen, and transthyretin, respectively. The angiogenic switch is controlled by several physiologic stimuli, such as hypoxia. Relative lack of oxygen stimulates HIF1α, an oxygen-sensitive transcription factor mentioned above, which then activates transcription of a variety of pro-angiogenic cytokines, such as VEGF and bFGF. These factors create an angiogenic gradient that stimulates the proliferation of endothelial cells and guides the growth of new vessels toward the tumor. VEGF also increases the expression of ligands that activate the Notch signaling pathway, which plays a crucial role in regulating the branching and density of the new vessels (Chapter 3). Both pro- and anti-angiogenic factors are regulated by many other genes frequently mutated in cancer. For example, in normal cells, p53 can stimulate expression of anti-angiogenic molecules such as thrombospondin-1, and repress expression of pro-angiogenic molecules such as VEGF. Thus, loss of p53 in tumor cells not only removes the cell cycle checkpoints listed above but also provides a more permissive environment for angiogenesis. The transcription of VEGF is also influenced by signals from the RAS-MAP kinase pathway, and mutations of RAS or MYC up-regulate the production of VEGF. The mechanisms whereby bFGF, VEGF, and the Notch pathway work together to coordinate angiogenesis were discussed in Chapter 3. bFGF and VEGF are commonly expressed in a wide variety of tumor cells, and elevated levels can be detected in the serum and urine of a significant fraction of cancer patients. Indeed, an anti-VEGF monoclonal antibody, bevacizumab, has recently been approved for use in the treatment of multiple cancers.106 Another emerging strategy involves the use of antibodies that inhibit Notch activation. These antibodies cause new vessels to be so malformed that they cannot deliver blood to the tumor effectively.107,108
INVASION AND METASTASIS
Invasion and metastasis are biologic hallmarks of malignant tumors. They are the major cause of cancer-related morbidity and mortality and hence are the subjects of intense scrutiny. Studies in mice and humans reveal that although millions of cells are released into the circulation each day from a primary tumor, only a few metastases are produced. Indeed, tumor cells can be frequently detected in the blood and marrow of patients with breast cancer who have not, and do not ever, develop gross metastatic disease. Why is the metastatic process so inefficient? Each step in the process is subject to a multitude of controls; hence, at any point in the sequence the breakaway cell may not survive.109 For tumor cells to break loose from a primary mass, enter blood vessels or lymphatics, and produce a secondary growth at a distant site, they must go through a series of steps (summarized in Fig. 7-36). For the purpose of this discussion, the metastatic cascade will be divided into two phases: (1) invasion of the extracellular matrix (ECM); (2) vascular dissemination, homing of tumor cells, and colonization. Subsequently, the molecular genetics of the metastatic cascade, as currently understood, will be presented.
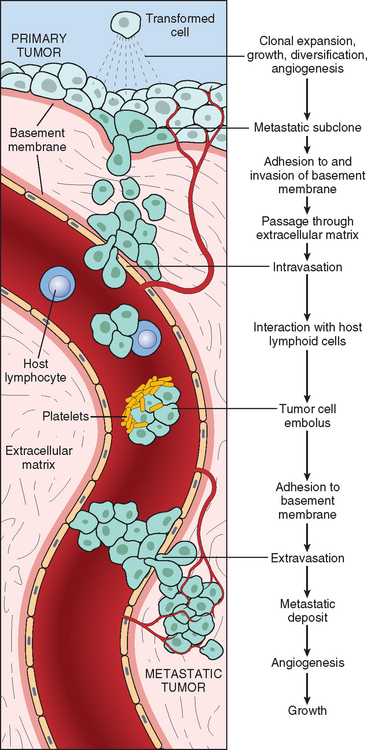
FIGURE 7-36 The metastatic cascade. Sequential steps involved in the hematogenous spread of a tumor.
Invasion of Extracellular Matrix
The structural organization and function of normal tissues is to a great extent determined by interactions between cells and the ECM.110 As we discussed in Chapter 3, tissues are organized into compartments separated from each other by two types of ECM: basement membrane and interstitial connective tissue. Though organized differently, each of these components of ECM is made up of collagens, glycoproteins, and proteoglycans. As shown in Figure 7-36, tumor cells must interact with the ECM at several stages in the metastatic cascade. A carcinoma must first breach the underlying basement membrane, then traverse the interstitial connective tissue, and ultimately gain access to the circulation by penetrating the vascular basement membrane. This process is repeated in reverse when tumor cell emboli extravasate at a distant site. Invasion of the ECM initiates the metastatic cascade and is an active process that can be resolved into several steps (Fig. 7-37):
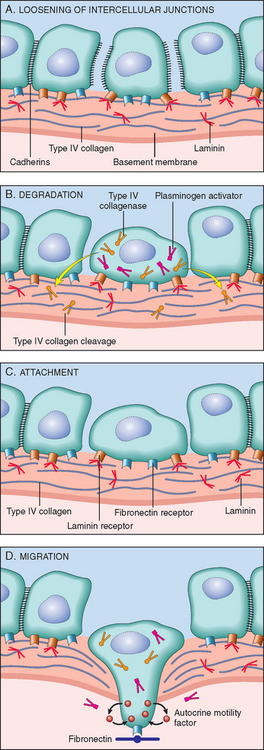
FIGURE 7-37 A–D, Sequence of events in the invasion of epithelial basement membranes by tumor cells. Tumor cells detach from each other because of reduced adhesiveness, then secrete proteolytic enzymes, degrading the basement membrane. Binding to proteolytically generated binding sites and tumor cell migration follow.
Dissociation of cells from one another is often the result of alterations in intercellular adhesion molecules. Normal cells are neatly glued to each other and their surroundings by a variety of adhesion molecules.111 Cell-cell interactions are mediated by the cadherin family of transmembrane glycoproteins. E-cadherins mediate homotypic adhesions in epithelial tissue, thus serving to keep the epithelial cells together and to relay signals between the cells; intracellularly the E-cadherins are connected to β-catenin and the actin cytoskeleton. In several epithelial tumors, including adenocarcinomas of the colon and breast, there is a down-regulation of E-cadherin expression. Presumably, this down-regulation reduces the ability of cells to adhere to each other and facilitates their detachment from the primary tumor and their advance into the surrounding tissues. E-cadherins are linked to the cytoskeleton by the catenins, proteins that lie under the plasma membrane (see Fig. 7-33). The normal function of E-cadherin is dependent on its linkage to catenins. In some tumors E-cadherin is normal, but its expression is reduced because of mutations in the gene for α catenin.
The second step in invasion is local degradation of the basement membrane and interstitial connective tissue. Tumor cells may either secrete proteolytic enzymes themselves or induce stromal cells (e.g., fibroblasts and inflammatory cells) to elaborate proteases. Many different families of proteases, such as matrix metalloproteinases (MMPs), cathepsin D, and urokinase plasminogen activator, have been implicated in tumor cell invasion. MMPs regulate tumor invasion not only by remodeling insoluble components of the basement membrane and interstitial matrix but also by releasing ECM-sequestered growth factors. Indeed, cleavage products of collagen and proteoglycans also have chemotactic, angiogenic, and growth-promoting effects.112 For example, MMP9 is a gelatinase that cleaves type IV collagen of the epithelial and vascular basement membrane and also stimulates release of VEGF from ECM-sequestered pools. Benign tumors of the breast, colon, and stomach show little type IV collagenase activity, whereas their malignant counterparts overexpress this enzyme. Concurrently, the concentrations of metalloproteinase inhibitors are reduced so that the balance is tilted greatly toward tissue degradation. Indeed, overexpression of MMPs and other proteases has been reported for many tumors. However, recent in vivo imaging experiments have shown that tumor cells can adopt a second mode of invasion, termed ameboid migration.113 In this type of migration the cell squeezes through spaces in the matrix instead of cutting its way through it. This ameboid migration is much quicker, and tumor cells seem to be able to use collagen fibers as high-speed railways in their travels. Tumor cells, in vitro at least, seem to be able to switch between the two forms of migration, perhaps explaining the disappointing performance of MMP inhibitors in clinical trials.
The third step in invasion involves changes in attachment of tumor cells to ECM proteins. Normal epithelial cells have receptors, such as integrins, for basement membrane laminin and collagens that are polarized at their basal surface; these receptors help to maintain the cells in a resting, differentiated state. Loss of adhesion in normal cells leads to induction of apoptosis, while, not surprisingly, tumor cells are resistant to this form of cell death. Additionally, the matrix itself is modified in ways that promote invasion and metastasis. For example, cleavage of the basement membrane proteins collagen IV and laminin by MMP2 or MMP9 generates novel sites that bind to receptors on tumor cells and stimulate migration.
Locomotion is the final step of invasion, propelling tumor cells through the degraded basement membranes and zones of matrix proteolysis. Migration is a complex, multistep process that involves many families of receptors and signaling proteins that eventually impinge on the actin cytoskeleton. Cells must attach to the matrix at the leading edge, detach from the matrix at the trailing edge, and contract the actin cytoskeleton to ratchet forward. Such movement seems to be potentiated and directed by tumor cell–derived cytokines, such as autocrine motility factors. In addition, cleavage products of matrix components (e.g., collagen, laminin) and some growth factors (e.g., IGFs I and II) have chemotactic activity for tumor cells. Furthermore, proteolytic cleavage liberates growth factors bound to matrix molecules. Stromal cells also produce paracrine effectors of cell motility, such as hepatocyte growth factor–scatter factor, which bind to receptors on tumor cells. Concentrations of hepatocyte growth factor–scatter factor are elevated at the advancing edges of the highly invasive brain tumor glioblastoma multiforme, supporting their role in motility.
It has become clear in recent years that the ECM and stromal cells surrounding tumor cells do not merely represent a static barrier for tumor cells to traverse but instead represent a varied environment in which reciprocal signaling between tumor cells and stromal cells may either promote or prevent tumorigenesis and/or tumor progression.24 Stromal cells that interact with tumors include innate and adaptive immune cells (discussed later), as well as fibroblasts. A variety of studies have demonstrated that tumor-associated fibroblasts exhibit altered expression of genes that encode ECM molecules, proteases, protease inhibitors, and various growth factors. Thus, tumor cells live in a complex and ever-changing milieu composed of ECM, growth factors, fibroblasts, and immune cells, with significant cross-talk among all the components. The most successful tumors may be those that can co-opt and adapt this environment to their own nefarious ends.
Vascular Dissemination and Homing of Tumor Cells
Once in the circulation, tumor cells are vulnerable to destruction by a variety of mechanisms, including mechanical shear stress, apoptosis stimulated by loss of adhesion, (which has been termed anoikis), and innate and adaptive immune defenses. The details of tumor immunity are considered later.
Within the circulation, tumor cells tend to aggregate in clumps. This is favored by homotypic adhesions among tumor cells as well as heterotypic adhesion between tumor cells and blood cells, particularly platelets (see Fig. 7-36). Formation of platelet-tumor aggregates may enhance tumor cell survival and implantability. Tumor cells may also bind and activate coagulation factors, resulting in the formation of emboli. Arrest and extravasation of tumor emboli at distant sites involves adhesion to the endothelium, followed by egress through the basement membrane. Involved in these processes are adhesion molecules (integrins, laminin receptors) and proteolytic enzymes, discussed earlier. Of particular interest is the CD44 adhesion molecule, which is expressed on normal T lymphocytes and is used by these cells to migrate to selective sites in the lymphoid tissue. Such migration is accomplished by the binding of CD44 to hyaluronate on high endothelial venules, and overexpression of CD44 may favor metastatic spread. At the new site, tumor cells must proliferate, develop a vascular supply, and evade the host defenses.109
The site at which circulating tumor cells leave the capillaries to form secondary deposits is related, in part, to the anatomic location of the primary tumor, with most metastases occurring in the first capillary bed available to the tumor. Many observations, however, suggest that natural pathways of drainage do not wholly explain the distribution of metastases. For example, prostatic carcinoma preferentially spreads to bone, bronchogenic carcinomas tend to involve the adrenals and the brain, and neuroblastomas spread to the liver and bones. Such organ tropism may be related to the following mechanisms:
Despite their “cleverness” in escaping their sites of origin, tumor cells are quite inefficient in colonizing distant organs. Millions of tumors cells are shed daily from even small tumors. These cells can be detected in the bloodstream and in small foci in the bone marrow, even in patients that never develop gross metastatic lesions. Indeed, the concept of dormancy, referring to the prolonged survival of micrometastases without progression, is well described in melanoma and in breast and prostate cancer. Although the molecular mechanisms of colonization are just beginning to be unraveled in mouse models, a constant pattern seems to be that tumor cells secrete cytokines, growth factors, and ECM molecules that act on the resident stromal cells, which in turn make the metastatic site habitable for the cancer cell.115 For example, breast cancer metastases to bone are osteolytic because of the activation of osteoclasts in the metastatic site. Breast cancer cells secrete parathyroid hormone–related protein (PTHRP), which stimulates osteoblasts to make RANK ligand (RANKL). RANKL then activates osteoclasts, which degrade the bone matrix and release growth factors embedded within it, like IGF and TGF-β. With a better molecular understanding of the mechanisms of metastasis our ability to target them therapeutically will be greatly enhanced.
Molecular Genetics of Metastasis Development
Why do only some tumors metastasize? What are the genetic changes that allow metastases? Why is the metastatic process so inefficient? Several competing theories have been proposed to explain how the metastatic phenotype arises. The clonal evolution model suggest that, as mutations accumulate in genetically unstable cancer cells and the tumor become heterogeneous (Fig. 7-38A), a subset of tumor cell subclones develop the right combination of gene products to complete all the steps involved in metastasis. Thus, metastatic subclones result from clonal evolution, and it is only the rare cell that acquires all the necessary genetic alterations and can complete all the steps. However, recent experiments, in which gene expression profiles of primary tumors and metastatic deposits have been compared, challenge this hypothesis. For example, a subset of breast cancers has a gene expression signature similar to that found in metastases, although no clinical evidence for metastasis is apparent. In these tumors it seems that most if not all cells develop a predilection for metastatic spread during early stages of carcinogenesis. Metastases, according to this view, are not dependent on the stochastic generation of metastatic subclones postulated above. The alternative hypothesis suggested by these data is that metastasis is the result of multiple abnormalities that occur in many, perhaps most, cells of a primary tumor, and perhaps early in the development of the tumor (Fig. 7-38B and C). Such abnormalities give most cells within the tumor a general predisposition for metastasis, often called the “metastasis signature.”116 This signature may involve not only properties intrinsic to the cancer cells but also the characteristics of their microenvironment, such as the components of the stroma, the presence of infiltrating immune cells, and angiogenesis (Fig. 7-38D). It should be noted, however, that gene expression analyses like those described above would not detect a small subset of metastatic subclones within a large tumor. Perhaps both mechanisms are operative, with aggressive tumors acquiring a metastases-permissive gene expression pattern early in tumorigenesis that requires some additional random mutations to complete the metastatic phenotype. A third hypothesis suggests that background genetic variation, and the resulting variation in gene expression, in the human population contributes to the generation of metastases. In mouse models, cancers induced with the same oncogenic mutations can have very different metastatic outcomes depending on the strain (i.e., background genetics) of the mouse used. Even very strong oncogenes can be significantly affected by background genetics. The fourth hypothesis is a corollary of the tumor stem cell hypothesis, which suggests that if tumors derive from rare tumor stem cells, metastases require the spread of the tumor stem cells themselves.
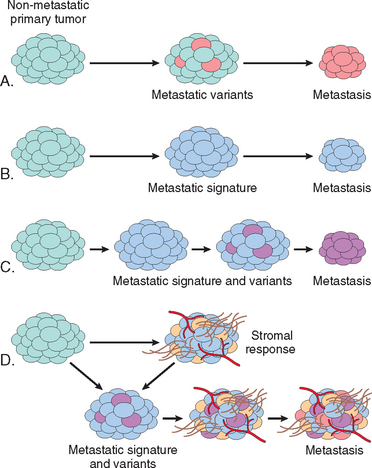
FIGURE 7-38 Mechanisms of metastasis development within a primary tumor. A nonmetastatic primary tumor is shown (light blue) on the left side of all diagrams. Four models are presented: A, Metastasis is caused by rare variant clones that develop in the primary tumor; B, Metastasis is caused by the gene expression pattern of most cells of the primary tumor, referred to as a metastatic signature; C, A combination of A and B, in which metastatic variants appear in a tumor with a metastatic gene signature; D, Metastasis development is greatly influenced by the tumor stroma, which may regulate angiogenesis, local invasiveness, and resistance to immune elimination, allowing cells of the primary tumor, as in C, to become metastatic.
One open question in the field is, are there genes whose principal or sole contribution to tumorigenesis is to control metastasis? This question is of more than academic interest, because if altered forms of certain genes promote or suppress the metastatic phenotype, their detection in a primary tumor would have both prognostic and therapeutic implications. Since metastasis is a complex phenomenon involving a variety of steps and pathways described above, it is thought that, unlike transformation, in which a subset of proteins like p53 and RB seem to play a key role, genes that function as “metastasis oncogenes” or “metastatic suppressors” are rare. A metastasis suppressor gene is defined as a gene whose loss promotes the development of metastasis without an effect on the primary tumor. Accordingly, expression of a metastasis oncogene favors the development of metastasis without effect upon the primary tumor. At least a dozen genes lost in metastatic lesions have been confirmed to function as “metastasis suppressors”.117,118 Their molecular functions are varied and not yet completely clear; however, most appear to affect various signaling pathways. Interestingly, recent work has suggested that two miRNAs, mir335 and mir126, suppress the metastasis of breast cancer, while a second set (mir10b) promotes metastasis.119,120
Among candidates for metastasis oncogenes are SNAIL and TWIST, which encode transcription factors whose primary function is to promote a process called epithelial-tomesenchymal transition (EMT).88 In EMT, carcinoma cells down-regulate certain epithelial markers (e.g., E-cadherin) and up-regulate certain mesenchymal markers (e.g., vimentin and smooth muscle actin). These changes are believed to favor the development of a promigratory phenotype that is essential for metastasis. Loss of E-cadherin expression seems to be a key event in EMT, and SNAIL and TWIST are transcriptional repressors that down-regulate E-cadherin expression.121 EMT has been documented mainly in breast cancers; whether this is a general phenomenon remains to be established.
GENOMIC INSTABILITY—ENABLER OF MALIGNANCY
Although humans literally swim in environmental agents that are mutagenic (e.g., chemicals, radiation, sunlight), cancers are relatively rare outcomes of these encounters. This state of affairs results from the ability of normal cells to repair DNA damage, the death of cells with unrepairable damage122 (see “Evasion of Apoptosis” above), and other mechanisms, such as oncogene-induced senescence and immune surveillance (discussed later). The importance of DNA repair in maintaining the integrity of the genome is highlighted by several inherited disorders in which genes that encode proteins involved in DNA repair are defective. Individuals born with such inherited defects in DNA-repair proteins are at a greatly increased risk of developing cancer. Moreover, defects in repair mechanisms are present in sporadic human cancers. DNA-repair genes themselves are not oncogenic, but their abnormalities allow mutations in other genes during the process of normal cell division. Typically, genomic instability occurs when both copies of the DNA repair gene are lost; however, recent work has suggested that at least a subset of these genes may promote cancer in a haploinsufficient manner. Defects in three types of DNA-repair systems—mismatch repair, nucleotide excision repair, and recombination repair—contribute to different types of cancers.
Hereditary Nonpolyposis Colon Cancer Syndrome.
HNPCC syndrome, characterized by familial carcinomas of the colon affecting predominantly the cecum and proximal colon (Chapter 17), results from defects in genes involved in DNA mismatch repair.123 When a strand of DNA is being replicated, these genes act as “spell checkers.” For example, if there is an erroneous pairing of G with T rather than the normal A with T, the mismatch-repair genes correct the defect. Without these “proofreaders,” errors gradually accumulate randomly in the genome, and some of these errors may involve proto-oncogenes and tumor suppressor genes. One of the hallmarks of patients with mismatch-repair defects is microsatellite instability.21 Microsatellites are tandem repeats of one to six nucleotides found throughout the genome. In normal people the length of these microsatellites remains constant. However, in people with HNPCC, these satellites are unstable and increase or decrease in length in tumor cells, creating alleles not found in normal cells of the same patient. Of the various DNA mismatch-repair genes, at least four are involved in the pathogenesis of HNPCC, but germline mutations in the MSH2 (2p16) and MLH1 (3p21) genes each account for approximately 30% of cases. The remaining cases have mutations in other mismatch repair genes. Each affected individual inherits one defective copy of a DNA mismatch-repair gene and acquires the second hit in colonic epithelial cells. Thus, DNA-repair genes behave like tumor suppressor genes in their mode of inheritance, but in contrast to tumor suppressor genes (and oncogenes), they affect cell growth only indirectly—by allowing mutations in other genes during the process of normal cell division. Although HNPCC accounts only for 2% to 4% of all colonic cancers, microsatellite instability can be detected in about 15% of sporadic colon cancers. The growth-regulating genes that are mutated in HNPCC tumors have not yet been fully characterized but include the genes encoding TGF-β receptor II, the TCF component of the β-catenin pathway, BAX, and other oncogenes and tumor suppressor genes.124
Xeroderma Pigmentosum.
Individuals with another inherited disorder of defective DNA repair, xeroderma pigmentosum, are at increased risk for the development of cancers of the skin particularly following exposure to the UV light contained in sun rays.125 UV radiation causes cross-linking of pyrimidine residues, preventing normal DNA replication. Such DNA damage is repaired by the nucleotide excision repair system. Several proteins are involved in nucleotide excision repair, and an inherited loss of any one can give rise to xeroderma pigmentosum.
Diseases with Defects in DNA Repair by Homologous Recombination.
A group of autosomal recessive disorders comprising Bloom syndrome, ataxia-telangiectasia, and Fanconi anemia is characterized by hypersensitivity to other DNA-damaging agents, such as ionizing radiation (Bloom syndrome and ataxia-telangiectasia), or DNA cross-linking agents, such as many chemotherapeutic agents (Fanconi anemia).126,127 Their phenotype is complex and includes, in addition to predisposition to cancer, features such as neural symptoms (ataxia-telangiectasia), bone marrow aplasia (Fanconi anemia), and developmental defects (Bloom syndrome). As mentioned earlier, the gene mutated in ataxia-telangiectasia, ATM, is important in recognizing and responding to DNA damage caused by ionizing radiation. Persons with Bloom syndrome have a predisposition to a very broad spectrum of tumors. The defective gene is located on chromosome 15 and encodes a helicase that participates in DNA repair by homologous recombination. There are 13 genes that make up the Fanconi anemia complex; mutation of any one of these genes can result in the phenotype.126,128 Interestingly, BRCA2, which is mutated in some individuals with familial breast cancer, is also mutated in a subset of persons with Fanconi anemia. Evidence for the role of DNA-repair genes in the origin of cancer also comes from the study of hereditary breast cancer. Mutations in two genes, BRCA1 (chromosome 17q21) and BRCA2 (chromosome 13q12–13), account for 25% of cases of familial breast cancer. In addition to breast cancer, women with BRCA1 mutations have a substantially higher risk of epithelial ovarian cancers, and men have a slightly higher risk of prostate cancer. Likewise, mutations in the BRCA2 gene increase the risk of breast cancer in both men and women as well as cancer of the ovary, prostate, pancreas, bile ducts, stomach, and melanocytes. Although the functions of these genes have not been elucidated fully, cells that lack these genes develop chromosomal breaks and severe aneuploidy. Indeed, both BRCA1 and BRCA2 have been shown to associate with a variety of proteins involved in the homologous recombination repair pathway. The Fanconi anemia proteins and the BRCA proteins form a DNA-damage response network whose purpose is to resolve and repair intrastrand and interstrand DNA cross-links induced by chemical cross-linking agents. Failure to resolve these cross-links before separation of the two strands would lead to chromosome breakage and exposed chromosome ends. Generation of such ends would, as with short telomeres (see above), lead to the activation of the salvage nonhomologous end joining pathway, formation of dicentric chromosomes, bridge-fusion-breakage cycles, and massive aneuploidy. Similar to other tumor suppressor genes, both copies of BRCA1 and BRCA2 must be inactivated for cancer to develop. Although linkage of BRCA1 and BRCA2 to familial breast cancers is established, these genes are rarely inactivated in sporadic cases of breast cancer. In this regard, BRCA1 and BRCA2 are different from other tumor suppressor genes, such as APC and p53, which are inactivated in both familial and sporadic cancers.
STROMAL MICROENVIRONMENT AND CARCINOGENESIS
Although we have mostly focused on the neoplastic parenchymal cells in our discussion, tumors are not composed of a single cell type. Indeed, tumors are comprised of a complex mixture of cells of numerous lineages, including the tumor cells themselves, innate and adaptive immune cells, fibroblasts, endothelial cells, and others. Additionally, numerous examples of cross-talk between the ECM and tumor cells have been described. For example, cleavage of matrix components such as type IV collagen releases angiogenic factors (VEGF), and enzymatic degradation of laminin-5 by MMP2 reveals a cryptic proteolytic fragment that favors cancer cell motility.112 The ECM also stores growth factors in inactive forms, which are released by active matrix proteases. Such factors include PDGF, TGF-β, and bFGF, which in turn affect the growth of tumor cells in a paracrine manner. Successful tumor cells must co-opt these and other interactions and use them to promote their growth and invasion. More interesting is whether tumor cells are dependent upon these interactions for proliferation, survival, or metastases. If so, these interactions, and the stromal cells themselves, become potential therapeutic targets.
Both the inflammatory cells and fibroblasts within the tumor have been shown to have a complex relationship to the cancer cells and to each other. The role of chronic inflammation in the development of cancer has already been described (see above). Various mechanisms, such as the expression of pro-survival and pro-proliferation cytokines by immune cells, not only promote the development of cancer, but also promote survival and progression of tumor cells. Additionally, it has been suggested that macrophages infiltrating the tumor can be induced by the tumor cells to secrete factors that promote metastasis.129 In a mouse model of breast cancer, genetic deletion of macrophages prevented metastases. Furthermore, in vivo imaging of tumors in animal models has shown that macrophages surrounding blood vessels secrete EGF, resulting in chemotactic migration of tumor cells toward the vasculature.113 Fibroblasts play an important role in tumors as well. Fibroblasts secrete the matrix that results in the desmoplastic response to tumors. Interestingly, in vitro experiments that altered the stiffness of the matrix alone could change the aggressiveness of a cancer cell line. Thus, the desmoplastic response to cancer may be stimulated by the cancer cells and may promote their growth. On the other hand, in a prostate cancer model, injection of immortalized, but nontumorigenic cells, together with fibroblasts derived from a tumor (cancer-associated fibroblasts) led to the development of poorly differentiated tumors in athymic mice.130 These carcinomas had multiple genetic abnormalities that were not present in the parent cell line, suggesting that the stroma can drive genetic changes that promote carcinogenesis. Indeed, some of the predictions of tumor behavior based on gene expression profiling are turning out to be based on genes highly expressed in stromal cells, rather than tumor cells. How such changes come about remains mysterious, as does their relevance to carcinogenesis in vivo. However, the results are sufficiently intriguing to merit attention, since they suggest a novel form of cancer therapy that could be targeted to stromal cells. The role of stromal cells in tumor growth and progression is highlighted by recent studies in which gene expression profiles of stroma cells predicted clinical outcome in human breast cancer.131
METABOLIC ALTERATIONS: THE WARBURG EFFECT
Even in the presence of ample oxygen, cancer cells shift their glucose metabolism away from the oxygen hungry, but efficient, mitochondria to glycolysis.132-134 This phenomenon, called the Warburg effect and also known as aerobic glycolysis, has been recognized for many years (indeed, Otto Warburg received the Nobel Prize for discovery of the effect that bears his name in 1931), but was largely neglected until recently. This metabolic alteration is so common to tumors that some would call it the eighth hallmark of cancer. Indeed, clinically, the “glucose-hunger” of tumors is used to visualize tumors via positron emission tomography (PET) scanning, in which patients are injected with 18F-fluorodeoxyglucose, a non-metabolizable derivative of glucose that is preferentially taken up into tumor cells (as well as normal, actively dividing tissues such as the bone marrow). Most tumors are PETpositive, and rapidly growing ones are markedly so. However, the causal relationship between aerobic glycolysis and tumor progression is not entirely clear, nor is the initial insult that drives these metabolic changes.
Indeed, as is well known, glycolysis generates 2 ATP molecules per molecule of glucose, while oxidative phosphorylation in the mitochondria generates more than 20. How does a switch to the less efficient glycolysis lead to a growth advantage for a tumor? Several mutually non-exclusive hypotheses have been offered. One attractive hypothesis to explain the Warburg effect is that altered metabolism confers a growth advantage in the hypoxic tumor microenvironment.133-134 Although angiogenesis generates increased vasculature, the vessels are poorly formed, and tumors are still relatively hypoxic compared to normal tissues. Indeed, the activation of HIF1α by hypoxia not only stimulates angiogenesis, but also increases the expression of numerous metabolic enzymes in the glycolytic pathway as well as downregulates genes involved in oxidative phosphorylation. So the simplest explanation is basic economics: supply and demand. Decreased demand by individual tumor cells increases the oxygen supply, thus increasing the number of tumor cells that can be supported by the vasculature and increasing the size of the tumor.
However, the Warburg effect refers to aerobic glycolysis; glycolysis that occurs in the face of adequate oxygen for oxidative phosphorylation. Thus, the changes that promote the switch in metabolism during hypoxia must become fixed in the tumor cell. It may be that continuous rounds of hypoxia followed by normoxia, as is frequently seen in tumors, select for tumor cells that constitutively upregulate glycolysis. Additionally, or perhaps alternatively, mutations in oncogenes and tumor suppressors that favor growth, such as RAS, p53 and PTEN, also stimulate metabolic changes in the cell. Which brings us to the second part of the supply and demand equation that may help explain why tumor cells opt for a less efficient energy production pipeline.
In addition to doubling its DNA content prior to division, an actively dividing cell (whether normal or transformed) must also double all of its other components, including membranes, proteins, and organelles. This task requires increased uptake of nutrients, particularly glucose (which produces the energy needed for biosynthesis of these components) and amino acids (which provide the building blocks used for protein synthesis) as well as increased synthesis of the necessary building blocks. Halting the breakdown of glucose at pyruvate allows these carbons to be shunted to anabolic pathways, such as lipid and nucleotide production; additionally, tumor cells are able to shunt glutamine into both the glycolytic as well as anabolic pathways.134-135 Thus, the metabolic changes that tumor cells undergo increase their ability to synthesize the building blocks they need for cell division. Indeed, alterations in signaling pathways involved in cancer also stimulate the uptake of glucose and other nutrients, favor glycolysis over oxidative phosphorylation, and increase anabolic pathways in the cell. Normally, growth factors stimulate glucose and amino acid uptake through the PI3K/AKT/mTOR pathway, which is downstream of receptor tyrosine kinases and other growth factor receptors; in tumors, these signals are cell autonomous. Thus, mutation of tumor suppressors and oncogenes not only leads to constitutive activation of pathways that favor survival and proliferation, but they also make glycolysis and anabolic biosynthesis a permanent fixture of the tumor cell.135-136
Now that the Warburg effect has been “rediscovered,” other fascinating connections between metabolism and neoplasia are emerging that involve both tumor suppressors and oncoproteins. One example of the former involves LKB1, a tumor suppressor gene encoding a threonine kinase that is mutated in Peutz-Jegher syndrome (Chapter 17), which is associated with benign and malignant epithelial proliferations of the gastrointestinal tract. At least one aspect of LKB1’s tumor suppressive activity is mediated through its ability to activate AMP-dependent protein kinase (AMPK), a conserved sensor of cellular energy status that is an important negative regulator of mTOR. Thus LKB1 suppresses tumor formation, at least in part, by putting the brakes on anabolic metabolism. Of note, two other tumor suppressors that are mutated in tuberous sclerosis, TSC1 and TSC2 (Chapter 28), also negatively regulate mTOR. On the other side of the ledger, it has been claimed that the transforming effects of many oncoproteins, including mutated receptor tyrosine kinases and the notorious oncogenic transcription factor c-MYC, are mediated in part through induction of the “Warburg effect.” These kinds of insights have spurred many recent attempts to target signaling pathways that drive anabolic metabolism in cancer cells, such as the PI3K/AKT/mTOR pathway.
As we have discussed earlier, cells have many regulatory barriers to prevent inappropriate growth. One adaptive response of normal cells to oxygen and glucose deprivation is autophagy, a state in which cells arrest their growth and cannibalize their own organelles, proteins, and membranes as carbon sources for energy production (Chapter 1). If this adaptation fails, the cells die. Tumor cells often seem to be able to grow under marginal environmental conditions without triggering autophagy, suggesting that the pathways that induce autophagy are deranged. In keeping with this, several genes that promote autophagy are tumor suppressors, most notable PTEN (a negative regulator of the PI3K/AKT pathway), which is mutated or epigenetically silenced in a wide variety of human cancers. Whether autophagy is always bad from the vantage point of the tumor, however, is a matter of active investigation and debate. For example, under conditions of severe nutrient deprivation tumor cells may use autophagy to become “dormant,” a state of metabolic hibernation that allows cells to survive hard times for long periods. Such cells are believed to be resistant to therapies that kill actively dividing cells, and could therefore be responsible for therapeutic failures. Thus, autophagy may be a tumor’s friend or foe depending on how the signaling pathways that regulate it are “wired” in a given tumor.