Chapter 17 The Gastrointestinal Tract

The gastrointestinal (GI) tract is a hollow tube extending from the oral cavity to the anus that consists of anatomically distinct segments, including the esophagus, stomach, small intestine, colon, rectum, and anus. Each of these segments has unique, complementary, and highly integrated functions, which together serve to regulate the intake, processing, and absorption of ingested nutrients and the disposal of waste products. The regional variations in structure and function are reflected in diseases of the GI tract, which often affect one or another segment preferentially. Accordingly, following consideration of several important congenital abnormalities, the discussion will be organized anatomically. Disorders affecting more than one segment of the GI tract, such as Crohn disease, will be discussed with the region that is involved most frequently.
CONGENITAL ABNORMALITIES
Depending on both the nature and timing of the insult, a variety of developmental anomalies can affect the GI tract. Importantly, because many organs develop simultaneously during embryogenesis, the presence of congenital GI disorders should prompt evaluation of other organs. Some defects are commonly associated with GI lesions.
Atresia, Fistulae, and Duplications
Atresia, fistulae, and duplications may occur in any part of the GI tract. When present within the esophagus they are discovered shortly after birth, usually because they cause regurgitation during feeding. These must be corrected promptly, since they are incompatible with life. Absence, or agenesis, of the esophagus is extremely rare, but atresia, in which development is incomplete, is more common. In esophageal atresia a thin, noncanalized cord replaces a segment of esophagus, causing a mechanical obstruction (Fig. 17-1A). Proximal and distal blind pouches connect to the pharynx and stomach, respectively. Atresia occurs most commonly at or near the tracheal bifurcation and is usually associated with a fistula connecting the upper or lower esophageal pouches to a bronchus or the trachea (17–1B). Fistulae can lead to aspiration, suffocation, pneumonia, and severe fluid and electrolyte imbalances (Fig. 17-1B,C). Esophageal atresia is associated with congenital heart defects, genitourinary malformations, and neurologic disease. Intestinal atresia is less common than esophageal atresia but frequently involves the duodenum and is characterized by a segment of bowel lacking a lumen.
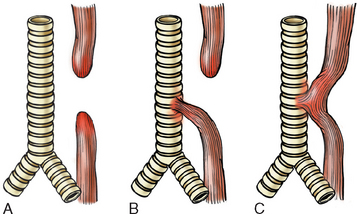
FIGURE 17-1 Esophageal atresia and tracheoesophageal fistula. A, Blind upper and lower esophageal segments. B, Blind upper segment with fistula between lower segment and trachea. C, Fistula between patent esophagus and trachea. Type B is the most common.
(Adapted from Morson BC, Dawson IMP, eds: Gastrointestinal Pathology. Oxford, Blackwell Scientific Publications, 1972, p 8.)
Stenosis is an incomplete form of atresia in which the lumen is markedly reduced in caliber as a result of fibrous thickening of the wall, resulting in partial or complete obstruction. Stenosis may involve any part of the GI tract, although the esophagus and small intestine are affected most often. Imperforate anus, the most common form of congenital intestinal atresia, is due to a failure of the cloacal diaphragm to involute. Stenosis can also be caused by inflammatory scarring, as may occur with chronic gastroesophageal reflux, irradiation, scleroderma, or caustic injury.
Congenital duplication cysts are saccular or elongated cystic masses that contain redundant smooth muscle layers. These may be present in the esophagus, small intestine, or colon.
Diaphragmatic Hernia, Omphalocele, and Gastroschisis
Diaphragmatic hernia occurs when incomplete formation of the diaphragm allows the abdominal viscera to herniate into the thoracic cavity. When severe, the space-filling effect of the displaced viscera can cause pulmonary hypoplasia that is incompatible with life after birth. Omphalocele occurs when closure of the abdominal musculature is incomplete and the abdominal viscera herniate into a ventral membranous sac. This may be repaired surgically, but as many as 40% of infants with an omphalocele have other birth defects, including diaphragmatic hernia and cardiac anomalies. Gastroschisis is another ventral wall defect similar to omphalocele except that it involves all of the layers of the abdominal wall, from the peritoneum to the skin.
Ectopia
Ectopic tissues (developmental rests) are common in the GI tract. The most frequent site of ectopic gastric mucosa is the upper third of the esophagus, where it is referred to as an inlet patch. While generally asymptomatic, acid released by gastric mucosa within the esophagus can result in dysphagia, esophagitis, Barrett esophagus, or, rarely, adenocarcinoma. Ectopic pancreatic tissue occurs less frequently and can be found in the esophagus or stomach. Like inlet patches, these nodules are most often asymptomatic but can produce damage and local inflammation. When ectopic pancreatic tissue is present in the pylorus, inflammation and scarring may lead to obstruction. Because the rests may be present within any layer of the gastric wall, they can mimic invasive cancer. Gastric heterotopia, small patches of ectopic gastric mucosa in the small bowel or colon, may present with occult blood loss due to peptic ulceration of adjacent mucosa.1
Meckel Diverticulum
A true diverticulum is a blind outpouching of the alimentary tract that is lined by mucosa, communicates with the lumen, and includes all three layers of the bowel wall. The most common type is the Meckel diverticulum, which occurs in the ileum.
The Meckel diverticulum occurs as a result of failed involution of the vitelline duct, which connects the lumen of the developing gut to the yolk sac. This solitary diverticulum is a small pouch extending from the antimesenteric side of the bowel (Fig. 17-2). It is a true diverticulum with a wall that includes mucosa, submucosa, and muscularis propria. Meckel diverticulae occur in approximately 2% of the population, are generally present within 2 feet (85 cm) of the ileocecal valve, are approximately 2 inches (5 cm) long, are twice as common in males as in females, and are most often symptomatic by age 2 (although only ∼4% of Meckel diverticulae are symptomatic). These facts comprise the “rule of 2s” that is often used to help remember characteristics of Meckel diverticulae. The mucosal lining of Meckel diverticulae may resemble that of normal small intestine, but ectopic pancreatic or gastric tissue may also be present. The latter may result in peptic ulceration of adjacent small intestinal mucosa and present with occult bleeding or abdominal pain resembling acute appendicitis or intestinal obstruction.
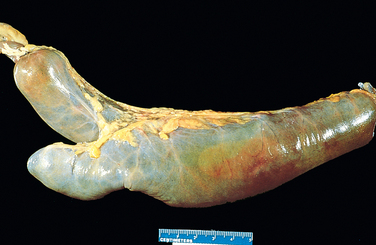
FIGURE 17-2 Meckel diverticulum. The blind pouch is located on the antimesenteric side of the small bowel.
Less commonly, congenital diverticulae occur in other parts of the small intestine and ascending colon. Virtually all other diverticulae are acquired and either lack muscularis entirely or have an attenuated muscularis propria. Although acquired diverticulae may occur in the esophagus, stomach, and duodenum, the most common site is the sigmoid colon (discussed later).
Pyloric Stenosis
Congenital hypertrophic pyloric stenosis is three to four times more common in males and occurs once in 300 to 900 live births. Monozygotic twins have a high rate of corcordance, suggesting a genetic basis. Family studies suggest a complex polygenic inheritance. Turner syndrome and trisomy 18 are also associated with the disease. Congenital hypertrophic pyloric stenosis generally presents in the second or third week of life as new-onset regurgitation and persistent, projectile, nonbilious vomiting. Physical examination reveals hyperperistalsis and a firm, ovoid abdominal mass. These findings stem from hyperplasia of the pyloric muscularis propria, which obstructs the gastric outflow tract. Edema and inflammatory changes in the mucosa and submucosa may aggravate the narrowing. Surgical splitting of the muscularis (myotomy) is curative. Acquired pyloric stenosis occurs in adults as a consequence of antral gastritis or peptic ulcers close to the pylorus. Carcinomas of the distal stomach and pancreas may also narrow the pyloric channel due to fibrosis or malignant infiltration.
Hirschsprung Disease
Hirschsprung disease occurs in approximately 1 of 5000 live births. It may be isolated or occur in combination with other developmental abnormalities; 10% of all cases occur in children with Down syndrome and serious neurologic abnormalities are present in another 5%.
Pathogenesis.
You may recall that the enteric neuronal plexus develops from neural crest cells that migrate into the bowel wall during embryogenesis. Hirschsprung disease, also known as congenital aganglionic megacolon, results when the normal migration of neural crest cells from cecum to rectum is arrested prematurely or when the ganglion cells undergo premature death. This produces a distal intestinal segment that lacks both the Meissner submucosal and the Auerbach myenteric plexus (“aganglionosis”). Coordinated peristaltic contractions are absent and functional obstruction occurs, resulting in dilation proximal to the affected segment.
The mechanisms underlying defective neural crest cell migration in Hirschsprung disease are unknown, but a genetic component is present in nearly all cases and 4% of patients’ siblings are affected.2 However, simple Mendelian inheritance is not involved in most cases. Heterozygous loss-of-function mutations in the receptor tyrosine kinase RET account for the majority of familial cases and approximately 15% of sporadic cases.3 Mutations also occur in at least seven other genes encoding proteins involved in enteric neurodevelopment, including the RET ligand glial-derived neurotrophic factor, endothelin, and the endothelin receptor, but, in aggregate, these account for fewer than 30% of patients, suggesting that other defects are yet to be discovered. Because penetrance is incomplete, modifying genes or environmental factors must also be important. In addition, it is clear that sex-linked factors exist, since males are affected preferentially while disease tends to be more extensive in females.
Morphology. Diagnosis of Hirschsprung disease requires documenting the absence of ganglion cells within the affected segment. Because migration of neural crest cells in the Meissner and Auerbach plexi are linked, it is possible to establish the diagnosis preoperatively by examining suction biopsy specimens. In addition to their characteristic morphology in hematoxylin and eosin (H&E)-stained sections, ganglion cells can be identified using immunohistochemical stains for acetylcholinesterase.
The rectum is always affected, but the length of the additional involved segments varies widely. Most cases are limited to the rectum and sigmoid colon, but severe cases can involve the entire colon. The aganglionic region may have a grossly normal or contracted appearance, while the normally innervated proximal colon may undergo progressive dilation (Fig. 17-3). With time the proximal colon may become massively distended (megacolon), reaching diameters of as much as 20 cm. Dilation may stretch and thin the colonic wall to the point of rupture, which occurs most frequently near the cecum. Mucosal inflammation or shallow ulcers may also be present. These changes proximal to the diseased segment can make gross identification of the extent of aganglionosis difficult. Hence, intraoperative frozen-section analysis of transmural sections is commonly used to confirm the presence of ganglion cells at the anastamotic margin.
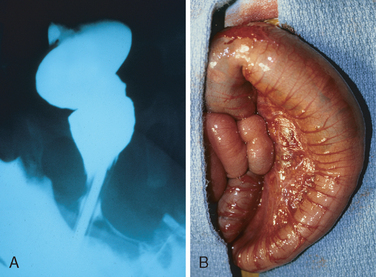
FIGURE 17-3 Hirschsprung disease. A, Preoperative barium enema study showing constricted rectum (bottom of the image) and dilated sigmoid colon. B, Corresponding intraoperative photograph showing constricted rectum and dilation of the sigmoid colon.
(Courtesy of Dr. Aliya Husain, The University of Chicago, Chicago, IL.)
Clinical Features.
Patients typically present neonatally, often with a failure to pass meconium in the immediate postnatal period. Obstructive constipation follows, although in cases where only a few centimeters of the rectum are involved there may be occasional passage of stool. The major threats to life are enterocolitis, fluid and electrolyte disturbances, perforation, and peritonitis. The primary mode of treatment is surgical resection of the aganglionic segment and anastamosis of the normal colon to the rectum. Even after successful surgery, it may take years for patients to attain normal bowel function and continence.
In contrast to the congenital megacolon of Hirschsprung disease, acquired megacolon may occur at any age as a result of Chagas disease, obstruction by a neoplasm or inflammatory stricture, toxic megacolon complicating ulcerative colitis, visceral myopathy, or in association with functional psychosomatic disorders. Of these, only Chagas disease (discussed later) is associated with loss of ganglia.
ESOPHAGUS
The esophagus develops from the cranial portion of the foregut and is recognizable by the third week of gestation. It is a hollow, highly distensible muscular tube that extends from the epiglottis in the pharynx to the gastroesophageal junction. Acquired diseases of the esophagus run the gamut from highly lethal cancers to the persistent “heartburn” that may be chronic and incapacitating or merely an occasional annoyance.
Esophageal Obstruction
For food and fluids to be delivered efficiently from the esophagus to the stomach, swallowing must be accompanied by a coordinated wave of peristaltic contractions. Esophageal dysmotility interferes with this process and can take several forms. High-amplitude esophageal contractions in which the outer longitudinal layer of smooth muscle contracts before the inner circular layer occur in some patients. Such lack of coordination results in a syndrome termed nutcracker esophagus that can cause periodic short-lived esophageal obstruction.4 Other motor disorders of the esophagus include diffuse esophageal spasm, which can also result in functional obstruction. Because it increases esophageal wall stress, diffuse esophageal spasm can cause small diverticulae to form. These small mucosal outpouchings, which are more accurately described as pseudo-diverticulae because they lack a true muscularis, are uncommon, probably because of the dense and continuous esophageal musculature. The Zenker diverticulum (pharyngoesophageal diverticulum) is located immediately above the upper esophageal sphincter; the traction diverticulum occurs near the midpoint of the esophagus; and the epiphrenic diverticulum is immediately above the lower esophageal sphincter. Zenker diverticulae may reach several centimeters in size and accumulate significant amounts of food, producing a mass and symptoms that include regurgitation.
Passage of food can also be impeded by esophageal stenosis, or narrowing of the lumen. This is generally caused by fibrous thickening of the submucosa and is associated with atrophy of the muscularis propria as well as secondary epithelial damage. Although occasionally congenital, stenosis is most often due to inflammation and scarring that may be caused by chronic gastroesophageal reflux, irradiation, or caustic injury. The dysphagia associated with stenosis is usually progressive, first affecting the ability to eat solids and only later interfering with ingestion of liquids. Because obstruction develops slowly, patients may subconsciously modify their diet to favor soft foods and liquids and be unaware of their condition until the obstruction is nearly complete.
Esophageal mucosal webs are uncommon ledge-like protrusions of mucosa that may cause obstruction. The pathogenesis is unknown, but webs are encountered most frequently in women over age 40. Webs are often associated with gastroesophageal reflux, chronic graft-versus-host disease, or blistering skin diseases. Upper esophageal webs accompanied by iron-deficiency anemia, glossitis, and cheilosis are part of the Paterson-Brown-Kelly or Plummer-Vinson syndrome. Esophageal webs are most common in the upper esophagus, where they are generally semicircumferential, eccentric lesions that protrude less than 5 mm and have a thickness of 2 to 4 mm. Microscopically, webs are composed of a fibrovascular connective tissue and overlying epithelium. The main symptom of webs is dysphagia associated with incompletely chewed food.
Esophageal rings, or Schatzki rings, are similar to webs, but are circumferential and thicker. Rings include mucosa, submucosa, and, in some cases, hypertrophic muscularis propria. When present in the distal esophagus, above the gastroesophageal junction, they are termed A rings and are covered by squamous mucosa; in contrast those located at the squamocolumnar junction of the lower esophagus are designated B rings and may have gastric cardia-type mucosa on their undersurface.
ACHALASIA
Increased tone of the lower esophageal sphincter (LES), as a result of impaired smooth muscle relaxation, is an important cause of esophageal obstruction. Release of nitric oxide and vasoactive intestinal polypeptide from inhibitory neurons, along with interruption of normal cholinergic signaling, allows the LES to relax during swallowing. Achalasia is characterized by the triad of incomplete LES relaxation, increased LES tone, and aperistalsis of the esophagus. Primary achalasia is caused by failure of distal esophageal inhibitory neurons and is, by definition, idiopathic.5 Degenerative changes in neural innervation, either intrinsic to the esophagus or within the extraesophageal vagus nerve or the dorsal motor nucleus of the vagus, may also occur. Secondary achalasia may arise in Chagas disease, in which Trypanosoma cruzi infection causes destruction of the myenteric plexus, failure of peristalsis, and esophageal dilatation. Duodenal, colonic, and ureteric myenteric plexi can also be affected in Chagas disease. Achalasia-like disease may be caused by diabetic autonomic neuropathy; infiltrative disorders such as malignancy, amyloidosis, or sarcoidosis; and lesions of dorsal motor nuclei, particularly polio or surgical ablation. Treatment options for primary and secondary achalasia include laparoscopic myotomy and pneumatic balloon dilatation. Botulinum neurotoxin (Botox) injection, to inhibit LES cholinergic neurons, can also be effective.
Esophagitis
LACERATIONS
Longitudinal tears in the esophagus near the gastroesophageal junction are termed Mallory-Weiss tears, and are most often associated with severe retching or vomiting secondary to acute alcohol intoxication. Normally, a reflex relaxation of the gastroesophageal musculature precedes the antiperistaltic contractile wave associated with vomiting. It is speculated that this relaxation fails during prolonged vomiting, with the result that refluxing gastric contents overwhelm the gastric inlet and cause the esophageal wall to stretch and tear. The roughly linear lacerations of Mallory-Weiss syndrome are longitudinally oriented and range in length from millimeters to several centimeters. These tears usually cross the gastroesophageal junction but may also be located in the proximal gastric mucosa. Up to 10% of upper GI bleeding, which often presents as hematemesis (Table 17-1), is due to superficial esophageal lacerations such as those associated with Mallory-Weiss syndrome. These do not generally require surgical intervention, and healing tends to be rapid and complete. In contrast, Boerhaave syndrome, characterized by distal esophageal rupture and mediastinitis, occurs rarely and is a catastrophic event.
TABLE 17-1 Esophageal Causes of Hematemesis
CHEMICAL AND INFECTIOUS ESOPHAGITIS
The stratified squamous mucosa of the esophagus may be damaged by a variety of irritants including alcohol, corrosive acids or alkalis, excessively hot fluids, and heavy smoking. The esophageal mucosa may also be injured when medicinal pills lodge and dissolve in the esophagus rather than passing into the stomach intact, a condition termed pill-induced esophagitis. Esophagitis due to chemical injury generally only causes self-limited pain, particularly dysphagia (pain with swallowing). Hemorrhage, stricture, or perforation may occur in severe cases. Iatrogenic esophageal injury may be caused by cytotoxic chemotherapy, radiation therapy, or graft-versus-host disease.
Infections may occur in otherwise healthy individuals but are most frequent in those who are debilitated or immunosuppressed as a result of disease or therapy. In these patients, esophageal infection by Herpes simplex viruses, cytomegalovirus (CMV), or fungal organisms is common. Among fungi, candidiasis is most common, although mucormycosis and aspergillosis may occur. The esophagus may also be involved by the desquamative skin diseases bullous pemphigoid and epidermolysis bullosa and, rarely, Crohn disease.
Morphology. The morphology of chemical and infectious esophagitis varies with etiology. Dense infiltrates of neutrophils are present in most cases but may be absent following injury induced by chemicals (lye, acids, or detergent), which may result in outright necrosis of the esophageal wall. Pill-induced esophagitis frequently occurs at the site of strictures that impede passage of luminal contents. When present, ulceration is accompanied by superficial necrosis with granulation tissue and eventual fibrosis.
Esophageal irradiation causes damage similar to that seen in other tissues and includes intimal proliferation and luminal narrowing of submucosal and mural blood vessels. The mucosal damage is, in part, often secondary to radiation-induced vascular injury as discussed in Chapter 9.
Infection by fungi or bacteria can either cause damage or complicate a preexisting ulcer. Nonpathogenic oral bacteria are frequently found in ulcer beds, while pathogenic organisms, which account for about 10% of infectious esophagitis, may invade the lamina propria and cause necrosis of overlying mucosa. Candidiasis, in its most advanced form, is characterized by adherent, gray-white pseudomembranes composed of densely matted fungal hyphae and inflammatory cells covering the esophageal mucosa.
The endoscopic appearance often provides a clue as to the infectious agent in viral esophagitis. Herpesviruses typically cause punched-out ulcers (Fig. 17-4A). Biopsy specimens demonstrate nuclear viral inclusions within a rim of degenerating epithelial cells at the margin of the ulcer (Fig. 17-4B). In contrast, CMV causes shallower ulcerations and characteristic nuclear and cytoplasmic inclusions within capillary endothelium and stromal cells (Fig. 17-4C). Although the histologic appearance is characteristic, immunohistochemical stains for virus-specific antigens are a sensitive and specific ancillary diagnostic tool.
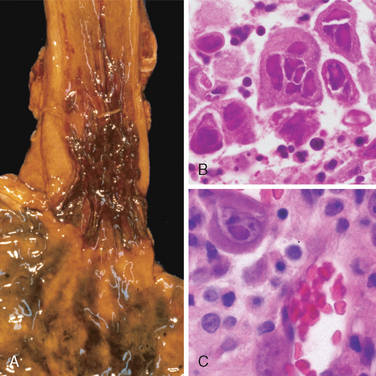
FIGURE 17-4 Viral esophagitis. A, Postmortem specimen with multiple herpetic ulcers in the distal esophagus. B, Multinucleate squamous cells containing Herpesvirus nuclear inclusions. C, Cytomegalovirus-infected endothelial cells with nuclear and cytoplasmic inclusions.
Histologic features of esophageal graft-versus-host disease are similar to those in the skin and include basal epithelial cell apoptosis, mucosal atrophy, and submucosal fibrosis without significant acute inflammatory infiltrates. The microscopic appearances of esophageal involvement in bullous pemphigoid, epidermolysis bullosa, and Crohn disease are also similar to those in the skin (Chapter 25).
REFLUX ESOPHAGITIS
The stratified squamous epithelium of the esophagus is resistant to abrasion from foods but is sensitive to acid. Submucosal glands, which are most abundant in the proximal and distal esophagus, contribute to mucosal protection by secreting mucin and bicarbonate. More importantly, constant lower esophageal sphincter tone prevents reflux of acidic gastric contents, which are under positive pressure and would otherwise enter the esophagus. Reflux of gastric contents into the lower esophagus is the most frequent cause of esophagitis and the most common outpatient GI diagnosis in the United States.6 The associated clinical condition is termed gastroesophageal reflux disease (GERD).
Pathogenesis.
Reflux of gastric juices is central to the development of mucosal injury in GERD. In severe cases, reflux of bile from the duodenum may exacerbate the damage. Conditions that decrease lower esophageal sphincter tone or increase abdominal pressure contribute to GERD and include alcohol and tobacco use, obesity, central nervous system depressants, pregnancy, hiatal hernia (discussed below), delayed gastric emptying, and increased gastric volume. In many cases, no definitive cause is identified.
Morphology. Simple hyperemia, evident to the endoscopist as redness, may be the only alteration. In mild GERD the mucosal histology is often unremarkable. With more significant disease, eosinophils are recruited into the squamous mucosa followed by neutrophils, which are usually associated with more severe injury (Fig. 17-5A). Basal zone hyperplasia exceeding 20% of the total epithelial thickness and elongation of lamina propria papillae, such that they extend into the upper third of the epithelium, may also be present.
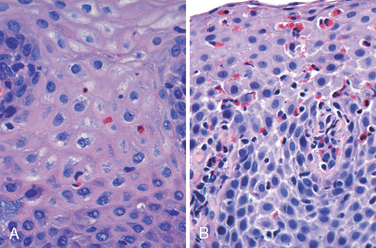
FIGURE 17-5 Esophagitis. A, Reflux esophagitis with scattered intraepithelial eosinophils. Although mild basal zone expansion can be appreciated, squamous cell maturation is relatively normal. B, Eosinophilic esophagitis is characterized by numerous intraepithelial eosinophils. Abnormal squamous maturation is also apparent.
Clinical Features.
GERD is most common in adults over age 40 but also occurs in infants and children. The most common clinical symptoms are dysphagia, heartburn, and, less frequently, noticeable regurgitation of sour-tasting gastric contents. Rarely, chronic GERD is punctuated by attacks of severe chest pain that may be mistaken for heart disease. Treatment with proton pump inhibitors or H2 histamine receptor antagonists, which reduce gastric acidity, typically provides symptomatic relief. While the severity of symptoms is not closely related to the degree of histologic damage, the latter tends to increase with disease duration. Complications of reflux esophagitis include esophageal ulceration, hematemesis, melena, stricture development, and Barrett esophagus.
Hiatal hernia is characterized by separation of the diaphragmatic crura and protrusion of the stomach into the thorax through the resulting gap. Congenital hiatal hernias are recognized in infants and children, but many are acquired in later life. Hiatal hernia is symptomatic in fewer than 10% of adults, and these cases are generally associated with other causes of LES incompetence. Symptoms, including heartburn and regurgitation of gastric juices, are similar to GERD.
EOSINOPHILIC ESOPHAGITIS
The incidence of eosinophilic esophagitis is increasing markedly.7 Symptoms include food impaction and dysphagia in adults and feeding intolerance or GERD-like symptoms in children. The cardinal histologic feature is large numbers of intraepithelial eosinophils, particularly superficially (Fig. 17-5B). Their abundance can help to differentiate eosinophilic esophagitis from GERD, Crohn disease, and other causes of esophagitis. Clinical characteristics, particularly failure of high-dose proton pump inhibitor treatment and the absence of acid reflux, are also necessary for diagnosis. The majority of individuals with eosinophilic esophagitis are atopic and many have atopic dermatitis, allergic rhinitis, asthma, or modest peripheral eosinophilia. Treatments include dietary restrictions to prevent exposure to food allergens, such as cow’s milk and soy products, and topical or systemic corticosteroids.
Barrett Esophagus
Barrett esophagus is a complication of chronic GERD that is characterized by intestinal metaplasia within the esophageal squamous mucosa. The incidence of Barrett esophagus is rising, and it is estimated to occur in as many as 10% of individuals with symptomatic GERD. Barrett esophagus is most common in white males and it typically presents between 40 and 60 years of age. The greatest concern in Barrett esophagus is that it confers an increased risk of esophageal adenocarcinoma. Molecular studies suggest that Barrett epithelium may be more similar to adenocarcinoma than to normal esophageal epithelium, consistent with the view that Barrett esophagus is a pre-malignant condition. In keeping with this, epithelial dysplasia, considered to be a pre-invasive lesion, is detected in 0.2% to 2.0% of persons with Barrett esophagus each year and is associated with prolonged symptoms and increased patient age. Although the vast majority of esophageal adenocarcinomas are associated with Barrett esophagus, it is important to remember that most individuals with Barrett esophagus do not develop esophageal tumors.
Morphology. Barrett esophagus can be recognized as one or several tongues or patches of red, velvety mucosa extending upward from the gastroesophageal junction. This metaplastic mucosa alternates with residual smooth, pale squamous (esophageal) mucosa and interfaces with light-brown columnar (gastric) mucosa distally (Fig. 17-6A, B). High-resolution endoscopes have increased the sensitivity of Barrett esophagus detection. This has led to subclassification of Barrett esophagus as long segment, in which 3 cm or more of esophagus is involved, or short segment, in which less than 3 cm is involved. It is not yet clear if the risk of dysplasia in short segment disease is less than in long segment Barrett esophagus.
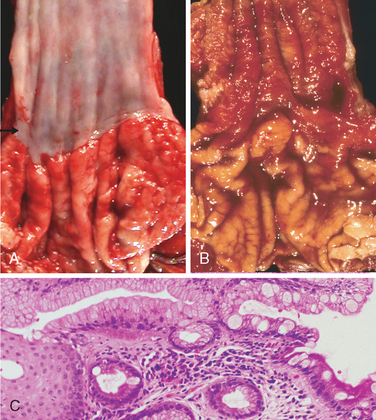
FIGURE 17-6 Barrett esophagus. A, Normal gastroesophageal junction. B, Barrett esophagus. Note the small islands of paler squamous mucosa within the Barrett mucosa. C, Histologic appearance of the gastroesophageal junction in Barrett esophagus. Note the transition between esophageal squamous mucosa (left) and Barrett metaplasia, with abundant metaplastic goblet cells (right).
Diagnosis of Barrett esophagus requires both endoscopic evidence of abnormal mucosa above the gastroesophageal junction and histologically documented intestinal metaplasia. Goblet cells, which have distinct mucous vacuoles that stain pale blue by H&E and impart the shape of a wine goblet to the remaining cytoplasm, define intestinal metaplasia and are necessary for diagnosis of Barrett esophagus (Fig. 17-6C). The requirement for intestinal metaplasia reflects the fact that this feature correlates with neoplastic risk. Foveolar mucus cells, which do not have distinct mucous vacuoles are insufficient for diagnosis. The requirement for an endoscopic abnormality helps to prevent misdiagnosis if metaplastic goblet cells within the cardia are included in the biopsy.
When dysplasia is present, it is classified as low grade or high grade. Increased epithelial proliferation, often with atypical mitoses, nuclear hyperchromasia and stratification, irregularly clumped chromatin, increased nuclear-to-cytoplasmic ratio, and a failure of epithelial cells to mature as they migrate to the esophageal surface are present in both grades of dysplasia (Fig. 17-7A). Gland architecture is frequently abnormal and is characterized by budding, irregular shapes, and cellular crowding (Fig. 17-7B). High-grade dysplasia exhibits more severe cytologic and architectural changes. Intramucosal carcinoma is characterized by invasion of neoplastic epithelial cells into the lamina propria.
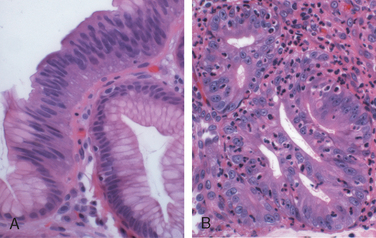
Clinical Features.
Barrett esophagus can only be identified thorough endoscopy and biopsy, which are usually prompted by GERD symptoms. Once diagnosed, the best course of management in Barrett esophagus is a matter of debate. However, most agree that periodic endoscopy with biopsy, for detection of dysplasia, has an important role. Nevertheless, uncertainties about the potential of dysplasia to regress, either spontaneously or in response to therapy, complicate clinical decisions when dysplasia is identified. In contrast, intramucosal carcinoma requires therapeutic intervention. Treatment options include surgical resection, or esophagectomy, as well as newer modalities such as photodynamic therapy, laser ablation, and endoscopic mucosectomy. Multifocal high-grade dysplasia, which carries a significant risk of progression to intramucosal or invasive carcinoma, is treated similarly to intramucosal carcinoma. Many physicians follow low-grade dysplasia or a single focus of high-grade dysplasia with endoscopy and biopsy at frequent intervals. However, management of esophageal dysplasia is evolving, and it is hoped that improved molecular understanding of neoplastic progression may allow development of chemopreventive approaches that reduce incidence of esophageal adenocarcinoma.8
ESOPHAGEAL VARICES
Instead of returning directly to the heart, venous blood from the GI tract is delivered to the liver via the portal vein before reaching the inferior vena cava. This circulatory pattern is responsible for the first-pass effect in which drugs and other materials absorbed in the intestines are processed by the liver before entering the systemic circulation. Diseases that impede this flow cause portal hypertension and can lead to the development of esophageal varices, an important cause of esophageal bleeding.
Pathogenesis.
Portal hypertension results in the development of collateral channels at sites where the portal and caval systems communicate. Although these collateral veins allow some drainage to occur, they lead to development of a congested subepithelial and submucosal venous plexus within the distal esophagus. These vessels, termed varices, develop in 90% of cirrhotic patients, most commonly in association with alcoholic liver disease. Worldwide, hepatic schistosomiasis is the second most common cause of varices. A more detailed consideration of portal hypertension is given in Chapter 18.
Morphology. Varices can be detected by venogram (Fig. 17-8A) and appear as tortuous dilated veins lying primarily within the submucosa of the distal esophagus and proximal stomach. Venous channels directly beneath the esophageal epithelium may also become massively dilated. Varices may not be grossly obvious in surgical or postmortem specimens, because they collapse in the absence of blood flow (Fig. 17-8B) and, when they are not ruptured, the overlying mucosa is intact (Fig. 17-8C). Variceal rupture results in hemorrhage into the lumen or esophageal wall, in which case the overlying mucosa appears ulcerated and necrotic. If rupture has occurred in the past, venous thrombosis, inflammation, and evidence of prior therapy may also be present.
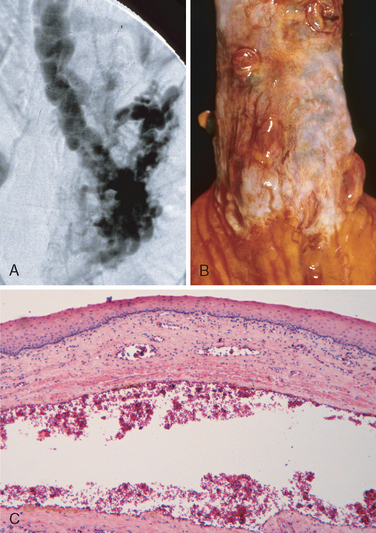
FIGURE 17-8 Esophageal varices. A, This angiogram shows several tortuous esophageal varices. B, Collapsed varices are present in this postmortem specimen corresponding to the angiogram in A. The polypoid areas represent previous sites of variceal hemorrhage that have been ligated with bands. C, Dilated varice beneath intact squamous mucosa.
Clinical Features.
While varices are often asymptomatic, they may rupture, causing massive hematemesis. The factors that lead to rupture are not well defined, but inflammatory erosion of thinned overlying mucosa, increased tension in progressively dilated veins, and increased vascular hydrostatic pressure associated with vomiting are likely to contribute. In any case, hemorrhage due to variceal rupture is a medical emergency that is treated by any of several methods: sclerotherapy by endoscopic injection of thrombotic agents; endoscopic balloon tamponade; or endoscopic rubber band ligation. Despite these interventions, as many as half of patients die from the first bleeding episode either as a direct consequence of hemorrhage or following hepatic coma triggered by hypovolemic shock. Among those who survive, additional instances of hemorrhage occur in over 50% within 1 year. Each episode has a similar rate of mortality. Thus, over half of deaths among individuals with advanced cirrhosis result from variceal rupture. It must be remembered, however, that even when varices are present, they are only one of several causes of hematemesis.
Esophageal Tumors
Two morphologic variants comprise the majority of esophageal cancers: adenocarcinoma and squamous cell carcinoma. Worldwide, squamous cell carcinoma is more common, but in the United States and other Western countries adenocarcinoma is on the rise. The potential reasons for these increases are discussed below.
ADENOCARCINOMA
Adenocarcinoma of the esophagus typically arises in a background of Barrett esophagus and long-standing GERD. Risk of adenocarcinoma is greater in those with documented dysplasia and is further increased by tobacco use, obesity, and prior radiation therapy.9 Conversely, risk of adenocarcinoma is reduced by diets rich in fresh fruits and vegetables. Some Helicobacter pylori serotypes are associated with a decreased risk of adenocarcinoma, perhaps by causing gastric atrophy and reducing acid reflux.
Esophageal adenocarcinoma occurs most frequently in Caucasians and shows a strong gender bias, being sevenfold more common in men. However, the incidence varies 60-fold worldwide, with rates being highest in certain developed Western countries, including the United States, the United Kingdom, Canada, Australia, the Netherlands, and Brazil and lowest in Korea, Thailand, Japan, and Ecuador. In countries where esophageal adenocarcinoma is more common, the incidence has increased markedly since 1970, more rapidly than almost any other cancer. As a result, esophageal adenocarcinoma, which represented less than 5% of esophageal cancers before 1970, now accounts for half of all esophageal cancers in the United States.
Pathogenesis.
Molecular studies suggest that the progression of Barrett esophagus to adenocarcinoma occurs over an extended period through the stepwise acquisition of genetic and epigenetic changes. This model is supported by the observation that epithelial clones identified in nondysplastic Barrett metaplasia persist and accumulate mutations during progression to dysplasia and invasive carcinoma. Chromosomal abnormalities and mutation or overexpression of p53 are present at early stages of esophageal adenocarcinoma. Additional genetic changes include amplification of c-ERB-B2, cyclin D1, and cyclin E genes; mutation of the retinoblastoma tumor suppressor gene; and allelic loss of the cyclindependent kinase inhibitor p16/INK4a. In other instances p16/INK4a is epigenetically silenced by hypermethylation. Increased epithelial expression of tumor necrosis factor (TNF)- and nuclear factor (NF)-κB–dependent genes suggests that inflammation may also contribute to neoplastic progression.
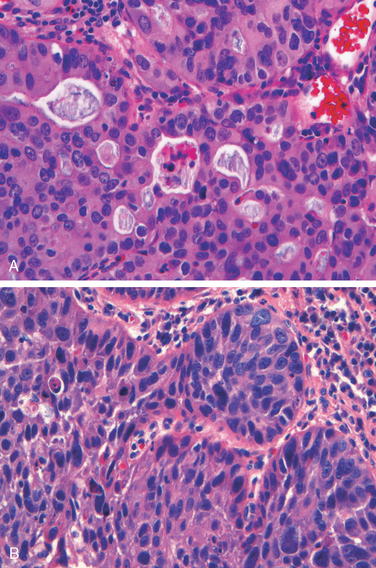
Morphology. Esophageal adenocarcinoma usually occurs in the distal third of the esophagus and may invade the adjacent gastric cardia (Fig. 17-9A). Initially appearing as flat or raised patches in otherwise intact mucosa, large masses of 5 cm or more in diameter may develop. Alternatively, tumors may infiltrate diffusely or ulcerate and invade deeply. Microscopically, Barrett esophagus is frequently present adjacent to the tumor. Tumors most commonly produce mucin and form glands (Fig. 17-10A), often with intestinal-type morphology; less frequently tumors are composed of diffusely infiltrative signet-ring cells (similar to those seen in diffuse gastric cancers) or, in rare cases, small poorly differentiated cells (similar to small-cell carcinoma of the lung).
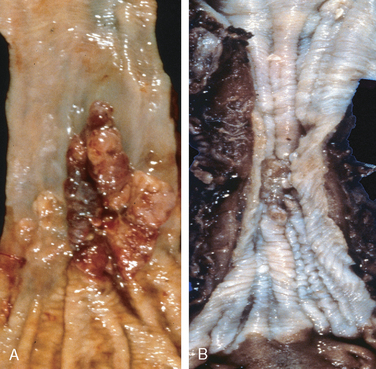
Clinical Features.
Although esophageal adenocarcinomas are occasionally discovered in evaluation of GERD or surveillance of Barrett esophagus, they more commonly present with pain or difficulty in swallowing, progressive weight loss, hematemesis, chest pain, or vomiting. By the time symptoms appear, the tumor has usually spread to submucosal lymphatic vessels. As a result of the advanced stage at diagnosis, overall 5-year survival is less than 25%. In contrast, 5-year survival approximates 80% in the few patients with adenocarcinoma limited to the mucosa or submucosa.
SQUAMOUS CELL CARCINOMA
In the United States, esophageal squamous cell carcinoma occurs in adults over age 45 and affects males four times more frequently than females.10 Risk factors include alcohol and tobacco use, poverty, caustic esophageal injury, achalasia, tylosis, Plummer-Vinson syndrome, and frequent consumption of very hot beverages.9 It is nearly sixfold more common in African-Americans than Caucasians, a striking risk disparity that reflects differences in rates of alcohol and tobacco use as well as other poorly understood factors.11 Previous radiation therapy to the mediastinum also predisposes individuals to esophageal carcinoma, typically 10 or more years after exposure.9
Esophageal squamous cell carcinoma incidence varies up to 180-fold between and within countries, being more common in rural and underdeveloped areas. The regions with highest incidences are Iran, central China, Hong Kong, Brazil, and South Africa.
Pathogenesis.
The majority of esophageal squamous cell carcinomas in Europe and the United States are at least partially attributable to the use of alcohol and tobacco, which synergize to increase risk. However, esophageal squamous cell carcinoma is also common in some regions where alcohol and tobacco use is uncommon. Thus, nutritional deficiencies, as well as polycyclic hydrocarbons, nitrosamines, and other mutagenic compounds, such as those found in fungus-contaminated foods, must be considered. Human papillomavirus (HPV) infection has also been implicated in esophageal squamous cell carcinoma in high-risk areas but not in low-risk regions.12 The molecular pathogenesis of esophageal squamous cell carcinoma remains incompletely defined, but loss of several tumor suppressor genes, including p53 and p16/INK4a, is involved.
Morphology. In contrast to adenocarcinoma, half of squamous cell carcinomas occur in the middle third of the esophagus (see Fig. 17-9B). Squamous cell carcinoma begins as an in situ lesion termed squamous dysplasia (this lesion is referred to as intraepithelial neoplasia or carcinoma in situ at other sites). Early lesions appear as small, gray-white, plaque-like thickenings. Over months to years they grow into tumor masses that may be polypoid or exophytic and protrude into and obstruct the lumen. Other tumors are either ulcerated or diffusely infiltrative lesions that spread within the esophageal wall and cause thickening, rigidity, and luminal narrowing. These may invade surrounding structures including the respiratory tree, causing pneumonia; the aorta, causing catastrophic exsanguination; or the mediastinum and pericardium.
Most squamous cell carcinomas are moderately to well-differentiated (see Fig. 17-10B). Less common histologic variants include verrucous squamous cell carcinoma, spindle cell carcinoma, and basaloid squamous cell carcinoma. Regardless of histology, symptomatic tumors are generally very large at diagnosis and have already invaded the esophageal wall. The rich submucosal lymphatic network promotes circumferential and longitudinal spread, and intramural tumor nodules may be present several centimeters away from the principal mass. The sites of lymph node metastases vary with tumor location: cancers in the upper third of the esophagus favor cervical lymph nodes; those in the middle third favor mediastinal, paratracheal, and tracheobronchial nodes; and those in the lower third spread to gastric and celiac nodes.
Clinical Features.
The onset of esophageal squamous cell carcinoma is insidious and ultimately produces dysphagia, odynophagia (pain on swallowing), and obstruction. Patients subconsciously adjust to the progressively increasing obstruction by altering their diet from solid to liquid foods. Extreme weight loss and debilitation result from both impaired nutrition and effects of the tumor itself. Hemorrhage and sepsis may accompany tumor ulceration. Occasionally, the first symptoms are caused by aspiration of food via a tracheoesophageal fistula.
Increased prevalence of endoscopic screening has led to earlier detection of esophageal squamous cell carcinoma. This is critical, because 5-year survival rates are 75% in individuals with superficial esophageal carcinoma but much lower in patients with more advanced tumors. Lymph node metastases, which are common, are associated with poor prognosis. The overall 5-year survival remains a dismal 9%.
UNCOMMON ESOPHAGEAL TUMORS
Other malignancies of the esophagus include unusual forms of adenocarcinoma, undifferentiated carcinoma, carcinoid tumor, melanoma, lymphoma, and sarcoma.
Benign tumors of the esophagus are generally mesenchymal in origin and arise within the esophageal wall. Tumors of smooth muscle origin, leiomyomas, are most common; fibromas, lipomas, hemangiomas, neurofibromas, and lymphangiomas also occur. Some benign tumors take the form of mucosal polyps. These are usually composed of fibrous and vascular tissue, or adipose tissue, and are known as fibrovascular polyps or pedunculated lipomas, respectively. Squamous papillomas are sessile lesions with a central core of connective tissue and a hyperplastic papilliform squamous mucosa. Uncommonly, papillomas are associated with HPV infection, in which case the term condyloma applies. In rare instances a mass of inflamed granulation tissue, growing either as an inflammatory polyp or an infiltrative mass in the wall of the esophagus, may resemble a malignant lesion. These benign lesions are called inflammatory pseudotumors.
STOMACH
Disorders of the stomach are a frequent cause of clinical disease, with inflammatory and neoplastic lesions being particularly common. In the United States, diseases related to gastric acid account for nearly one third of all health care spending on GI disease. In addition, despite a decreasing incidence in certain locales such as the United States, gastric cancer remains a leading cause of death worldwide.
The stomach is divided into four major anatomic regions: the cardia, fundus, body, and antrum. The cardia and antrum are lined mainly by mucin-secreting foveolar cells that form small glands. The antral glands are similar but also contain endocrine cells, such as G cells, that release gastrin to stimulate luminal acid secretion by parietal cells within the gastric fundus and body. The well-developed glands of the body and fundus also contain chief cells that produce and secrete digestive enzymes such as pepsin.
Acute Gastritis
Acute gastritis is a transient mucosal inflammatory process that may be asymptomatic or cause variable degrees of epigastric pain, nausea, and vomiting. In more severe cases there may be mucosal erosion, ulceration, hemorrhage, hematemesis, melena, or, rarely, massive blood loss.
Pathogenesis.
The gastric lumen is strongly acidic with pH close to 1, more than a million times more acidic than the blood. This harsh environment contributes to digestion but also has the potential to damage the gastric mucosa. Multiple mechanisms have evolved to protect the gastric mucosa (Fig. 17-11). Mucin secreted by surface foveolar cells forms a thin layer of mucus that prevents large food particles from directly touching the epithelium. The mucus layer also promotes formation of an “unstirred” layer of fluid over the epithelium that protects the mucosa and has a neutral pH as a result of bicarbonate ion secretion by surface epithelial cells. Finally, the rich vascular supply to the gastric mucosa delivers oxygen, bicarbonate, and nutrients while washing away acid that has back-diffused into the lamina propria. Acute or chronic gastritis can occur following disruption of any of these protective mechanisms. For example, reduced mucin synthesis in the elderly has been suggested as one factor that may explain their increased susceptibility to gastritis. Nonsteroidal anti-inflammatory drugs (NSAIDs) may interfere with cytoprotection normally provided by prostaglandins or reduce bicarbonate secretion, either of which increases the susceptibility of the gastric mucosa to injury. Similarly, the gastric injury that occurs in uremic patients and those infected with urease-secreting H. pylori may be due to inhibition of gastric bicarbonate transporters by ammonium ions. Ingestion of harsh chemicals, particularly acids or bases, either accidentally or as a suicide attempt, also results in severe gastric injury, predominantly as a result of direct injury to mucosal epithelial and stromal cells. Direct cellular injury is also implicated in gastritis due to excessive alcohol consumption, NSAIDs, radiation therapy, and chemotherapy. Since the entire gastric mucosal surface is replaced every 2 to 6 days, mitotic inhibitors, including those used in cancer chemotherapy, cause generalized mucosal damage due to insufficient epithelial regeneration. Finally, decreased oxygen delivery may explain increased incidence of acute gastritis at high altitudes.
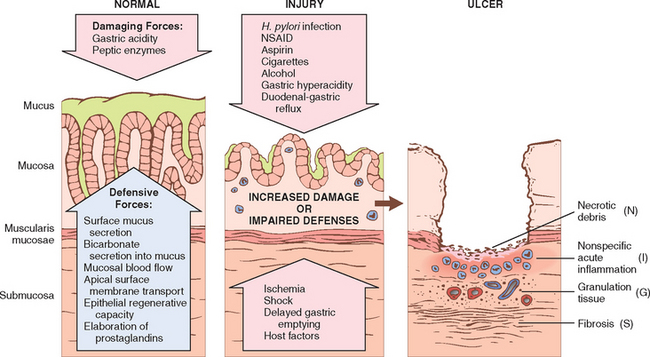
FIGURE 17-11 Mechanisms of gastric injury and protection. This diagram illustrates the progression from more mild forms of injury to ulceration that may occur with acute or chronic gastritis. Ulcers include layers of necrosis (N), inflammation (I), and granulation tissue (G), but a fibrotic scar (S), which takes time to develop, is only present in chronic lesions.
Morphology. Histologically, mild acute gastritis may be difficult to recognize, since the lamina propria shows only moderate edema and slight vascular congestion. The surface epithelium is intact, although scattered neutrophils may be present among the epithelial cells or within mucosal glands. In contrast, an abundance of lymphocytes or plasma cells suggests chronic disease. The presence of neutrophils above the basement membrane in direct contact with epithelial cells is abnormal in all parts of the GI tract and signifies active inflammation. This term is preferred over acute inflammation, since active inflammation may be present in both acute and chronic disease states. With more severe mucosal damage, erosions and hemorrhage develop. An erosion denotes loss of the superficial epithelium, generating a defect in the mucosa that is limited to the lamina propria. It is accompanied by a pronounced neutrophilic infiltrate within the mucosa and a fibrin-containing purulent exudate in the lumen. Hemorrhage may occur and cause dark punctae in an otherwise hyperemic mucosa. Concurrent erosion and hemorrhage is termed acute erosive hemorrhagic gastritis. Large areas of the gastric surface may be denuded, although the involvement is typically superficial. When erosions extend deeply, they may progress to ulcers, as described below.
ACUTE GASTRIC ULCERATION
Focal, acutely developing gastric mucosal defects are a well-known complication of therapy with NSAIDs. They may also appear after severe physiologic stress. Some of these are given specific names, based on location and clinical associations. For example:
Pathogenesis.
The pathogenesis of acute ulceration is complex and incompletely understood. NSAID-induced ulcers are related to cyclooxygenase inhibition. This prevents synthesis of prostaglandins, which enhance bicarbonate secretion, inhibit acid secretion, promote mucin synthesis, and increase vascular perfusion. Lesions associated with intracranial injury are thought to be caused by direct stimulation of vagal nuclei, which causes hypersecretion of gastric acid. Systemic acidosis, a frequent finding in these settings, may also contribute to mucosal injury by lowering the intracellular pH of mucosal cells. Hypoxia and reduced blood flow caused by stress-induced splanchnic vasoconstriction also contribute to the pathogenesis of acute ulcers.
Morphology. Lesions described as acute gastric ulcers range in depth from shallow erosions caused by superficial epithelial damage to deeper lesions that penetrate the depth of the mucosa. Acute ulcers are rounded and less than 1 cm in diameter. The ulcer base is frequently stained brown to black by acid digestion of extravasated blood and may be associated with transmural inflammation and local serositis. Unlike peptic ulcers, which arise in the setting of chronic injury, acute stress ulcers are found anywhere in the stomach. The gastric rugal folds are essentially normal, and the margins and base of the ulcers are not indurated. While they may occur singly, more often there are multiple ulcers throughout the stomach and duodenum. Microscopically, acute stress ulcers are sharply demarcated, with essentially normal adjacent mucosa. Depending on the duration of the ulceration, there may be a suffusion of blood into the mucosa and submucosa and some inflammatory reaction. Conspicuously absent are the scarring and thickening of blood vessels that characterize chronic peptic ulcers. Healing with complete re-epithelialization occurs after the injurious factors are removed. The time required for healing varies from days to several weeks.
Clinical Features.
Most critically ill patients admitted to hospital intensive care units have histologic evidence of gastric mucosal damage. Bleeding from superficial gastric erosions or ulcers that may require transfusion develops in 1% to 4% of these patients. Other complications, including perforation, can also occur (Table 17-2). Prophylactic H2 histamine receptor antagonists or proton pump inhibitors may blunt the impact of stress ulceration, but the most important determinant of clinical outcome is the ability to correct the underlying conditions. The gastric mucosa can recover completely if the patient does not succumb to their primary disease.
TABLE 17-2 Complications of Gastric Ulcers
| Bleeding |
| Perforation |
| Obstruction |
Chronic Gastritis
In contrast to acute gastritis, the symptoms associated with chronic gastritis are typically less severe but more persistent. Nausea and upper abdominal discomfort may occur, sometimes with vomiting, but hematemesis is uncommon. The most common cause of chronic gastritis is infection with the bacillus Helicobacter pylori. Before the acceptance of the central role of H. pylori infection in chronic gastritis, other chronic irritants, including psychologic stress, caffeine, alcohol, and tobacco use were considered the primary causes of gastritis. Autoimmune gastritis, the most common cause of atrophic gastritis, represents less than 10% of cases of chronic gastritis and is the most common form of chronic gastritis in patients without H. pylori infection. Less common etiologies include radiation injury, chronic bile reflux, mechanical injury, and involvement by systemic disease such as Crohn disease, amyloidosis, or graft-versus-host disease.
HELICOBACTER PYLORI GASTRITIS
The discovery of H. pylori has revolutionized our understanding of chronic gastritis.13 These spiral-shaped or curved bacilli are present in gastric biopsy specimens of almost all patients with duodenal ulcers and the majority of individuals with gastric ulcers or chronic gastritis.13 In a now-famous experiment, the Nobel laureate Barry Marshall ingested H. pylori cultures and developed mild gastritis. While not a recommended approach to infectious disease investigation, this experiment did demonstrate the pathogenicity of H. pylori. Acute H. pylori infection does not produce sufficient symptoms to require medical attention in most cases; it is the chronic gastritis that ultimately causes the individual to seek treatment. H. pylori organisms are present in 90% of individuals with chronic gastritis affecting the antrum. In addition, H. pylori has important roles in other gastric and duodenal diseases. For example, the increased acid secretion that occurs in H. pylori gastritis may result in peptic ulcer disease, and H. pylori infection also confers increased risk of gastric cancer.
Epidemiology.
In the United States, H. pylori infection is associated with poverty, household crowding, limited education, African-American or Mexican-American ethnicity, residence in rural areas, and birth outside of the United States. Colonization rates exceed 70% in some groups and vary from less than 10% to more than 80% worldwide. In high-prevalence areas infection is often acquired in childhood and then persists for decades, explaining the direct correlation between colonization rate and patient age.
The mode of H. pylori transmission is not well defined, but humans are the only known host, making oral-oral, fecal-oral, and environmental spread the most likely routes of infection. The related organism Helicobacter heilmannii causes similar disease and has reservoirs in cats, dogs, pigs, and nonhuman primates. While the morphologic differences between H. pylori and H. heilmannii organisms are subtle, recognition of H. heilmannii infection can be important since it may prompt treatment of household pets to prevent re-infection of the human companion.
Pathogenesis.
H. pylori infection is the most common cause of chronic gastritis. The disease most often presents as a predominantly antral gastritis with high acid production, despite hypogastrinemia. The risk of duodenal ulcer is increased in these patients and, in most, gastritis is limited to the antrum with occasional involvement of the cardia. In a subset of patients the gastritis progresses to involve the gastric body and fundus. This pangastritis is associated with multifocal mucosal atrophy, reduced acid secretion, intestinal metaplasia, and increased risk of gastric adenocarcinoma.
H. pylori organisms have adapted to the ecologic niche provided by gastric mucus. Although H. pylori may invade the gastric mucosa, this is not evident histologically and the contribution of invasion to disease is not known. Four features are linked to H. pylori virulence:
Although the mechanisms by which H. pylori cause gastritis are incompletely defined, it is clear that infection results in increased acid production and disruption of normal gastric and duodenal protective mechanisms, as described earlier (see Fig. 17-11). H. pylori gastritis is, therefore, the result of an imbalance between gastroduodenal mucosal defenses and damaging forces that overcome those defenses.
Over time chronic antral H. pylori gastritis may progress to pangastritis, resulting in multifocal atrophic gastritis. The underlying mechanisms contributing to this progression are not clear, but interactions between the host and bacterium seem to be critical. For example, particular polymorphisms in the gene encoding the pro-inflammatory cytokine interleukin-1β (IL-1β) correlate with the development of pangastritis after H. pylori infection. Polymorphisms in TNF and a variety of other genes associated with the inflammatory response also influence the clinical outcome in H. pylori infection.14 Severity of disease may also be influenced by genetic variation among H. pylori strains. For example, the CagA gene, a marker for a pathogenicity island of approximately 20 genes, is present in 50% of H. pylori isolates overall but in 90% of H. pylori isolates found in populations with elevated gastric cancer risk.
Morphology. Gastric biopsy specimens generally demonstrate H. pylori in infected individuals. The organism is concentrated within the superficial mucus overlying epithelial cells in the surface and neck regions. The distribution can be irregular, with areas of heavy colonization adjacent to those with few organisms. In extreme cases, the organisms carpet the luminal surfaces of foveolar and mucous neck cells, and can even extend into the gastric pits. Organisms are most easily demonstrated with a variety of special stains (Fig. 17-12A). H. pylori shows tropism for gastric epithelia and is generally not found in association with gastric intestinal metaplasia or duodenal epithelium. However, H. pylori may be present in foci of pyloric metaplasia within chronically injured duodenum or gastric-type mucosa within Barrett esophagus.
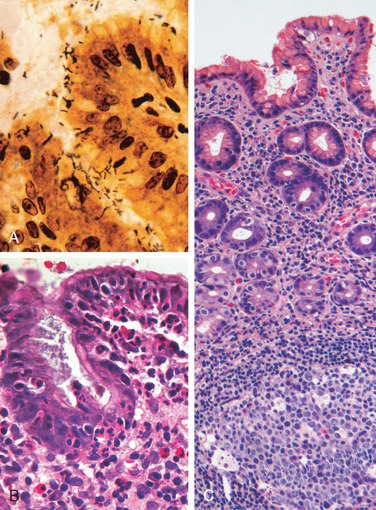
FIGURE 17-12 Helicobacter pylori gastritis. A, Spiral-shaped H. pylori are highlighted in this Warthin-Starry silver stain. Organisms are abundant within surface mucus. B, Intraepithelial and lamina propria neutrophils are prominent. C, Lymphoid aggregates with germinal centers and abundant subepithelial plasma cells within the superficial lamina propria are characteristic of H. pylori gastritis.
Within the stomach, H. pylori are typically found in the antrum (Table 17-3). Although there is a good concordance between colonization of the antrum and cardia, infection of the cardia occurs at somewhat lower rates. H. pylori are uncommon in oxyntic (acid-producing) mucosa of the fundus and body except in heavy colonization. Thus, an antral biopsy is preferred for evaluation of H. pylori gastritis. When viewed endoscopically, H. pylori–infected antral mucosa is usually erythematous and has a coarse or even nodular appearance. The inflammatory infiltrate generally includes variable numbers of neutrophils within the lamina propria, including some that cross the basement membrane to assume an intraepithelial location (Fig. 17-12B) and accumulate in the lumen of gastric pits to create pit abscesses. In addition, the superficial lamina propria includes large numbers of plasma cells, often in clusters or sheets, and increased numbers of lymphocytes and macrophages. Intraepithelial neutrophils and subepithelial plasma cells are characteristic of H. pylori gastritis. When intense, inflammatory infiltrates may create thickened rugal folds, mimicking early infiltrative lesions. Long-standing H. pylori gastritis may extend to involve the body and fundus, and the mucosa can become atrophic. Lymphoid aggregates, some with germinal centers, are frequently present (Fig. 17-12C) and represent an induced form of mucosa-associated lymphoid tissue, or MALT, that has the potential to transform into lymphoma.
TABLE 17-3 Characteristics of Helicobacter pylori–Associated and Autoimmune Gastritis
| H. pylori–Associated | Autoimmune | |
|---|---|---|
| Location | Antrum | Body |
| Inflammatory infiltrate | Neutrophils, subepithelial plasma cells | Lymphocytes, macrophages |
| Acid production | Increased to slightly decreased | Decreased |
| Gastrin | Normal to decreased | Increased |
| Other lesions | Hyperplastic/inflammatory polyps | Neuroendocrine hyperplasia |
| Serology | Antibodies to H. pylori | Antibodies to parietal cells (H+, K+-ATPase, intrinsic factor) |
| Sequelae | Peptic ulcer, adenocarcinoma | Atrophy, pernicious anemia, adenocarcinoma, carcinoid tumor |
| Associations | Low socioeconomic status, poverty, residence in rural areas | Autoimmune disease; thyroiditis, diabetes mellitus, Graves disease |
Clinical Features.
In addition to histologic identification of the organism, several diagnostic tests have been developed including a noninvasive serologic test for antibodies to H. pylori, fecal bacterial detection, and the urea breath test based on the generation of ammonia by the bacterial urease. Gastric biopsy specimens can also be analyzed by the rapid urease test, bacterial culture, or bacterial DNA detection by PCR.
Effective treatments for H. pylori infection include combinations of antibiotics and proton pump inhibitors. Individuals with H. pylori gastritis usually improve after treatment, although relapses can occur after incomplete eradication or re-infection. Prophylactic and therapeutic vaccine development is still at an early stage of development. Peptic ulcer disease, a complication of chronic H. pylori gastritis, is described later.
AUTOIMMUNE GASTRITIS
Autoimmune gastritis accounts for less than 10% of cases of chronic gastritis. In contrast to that caused by H. pylori, autoimmune gastritis typically spares the antrum and includes hypergastrinemia (see Table 17-3). Autoimmune gastritis is characterized by:
Pathogenesis.
Autoimmune gastritis is associated with loss of parietal cells, which are responsible for secretion of gastric acid and intrinsic factor. The absence of acid production stimulates gastrin release, resulting in hypergastrinemia and hyperplasia of antral gastrin-producing G cells. Lack of intrinsic factor disables ileal vitamin B12 absorption, leading to B12 deficiency and a slow-onset megaloblastic anemia (pernicious anemia). The reduced serum pepsinogen I concentration results from chief cell destruction. In contrast, although H. pylori can cause hypochlorhydria, it is not associated with achlorhydria or pernicious anemia because the parietal and chief cell damage is not as severe as in autoimmune gastritis.
It was initially thought that the autoantibodies to parietal cell components, most prominently the H+,K+-ATPase, or proton pump, and intrinsic factor were involved in the pathogenesis of autoimmune gastritis. However, this is unlikely because neither secreted intrinsic factor nor the luminally oriented proton pump are accessible to circulating antibodies, and passive transfer of these antibodies does not produce gastritis in experimental animals. It is more likely that CD4+ T cells directed against parietal cell components, including the H+,K+-ATPase, are the principal agents of injury. This is supported by the observation that transfer of H+,K+-ATPasereactive CD4+ T cells into naive mice results in gastritis and production of H+,K+-ATPase autoantibodies. There is no evidence of an autoimmune reaction to chief cells, suggesting that these are lost through gastric gland destruction during autoimmune attack on parietal cells. If autoimmune destruction is controlled by immunosuppression, the glands can repopulate, demonstrating that gastric stem cells survive and are able to differentiate into parietal and chief cells.
Morphology. Autoimmune gastritis is characterized by diffuse mucosal damage of the oxyntic (acid-producing) mucosa within the body and fundus. Damage to the antrum and cardia is typically absent or mild. With diffuse atrophy, the oxyntic mucosa of the body and fundus appears markedly thinned, and rugal folds are lost. If vitamin B12 deficiency is severe, nuclear enlargement (megaloblastic change) occurs within epithelial cells. Neutrophils may be present, but the inflammatory infiltrate is more often composed of lymphocytes, macrophages, and plasma cells. Lymphoid aggregates may be present. The superficial lamina propria plasma cells of H. pylori gastritis are typically absent, and the inflammatory reaction is most often deep and centered on the gastric glands (Fig. 17-13A). Loss of parietal and chief cells can be extensive. When atrophy is incomplete residual islands of oxyntic mucosa may give the appearance of multiple small polyps or nodules. Small surface elevations may be apparent, and these correlate with areas of intestinal metaplasia, characterized by the presence of goblet cells and columnar absorptive cells (Fig. 17-13B). The antral endocrine cell hyperplasia that develops in most patients can be difficult to appreciate on H&E-stained sections, since the endocrine cells, which are also referred to as enterochromaffin-like (ECL) cells, are not easily recognized. This hyperplasia parallels the degree of mucosal atrophy and is a physiologic response to decreased acid production. Over time, hypergastrinemia can stimulate endocrine cell hyperplasia in the fundus and body. Rarely, this may progress to form small, multicentric, low-grade neuroendocrine, or carcinoid, tumors.
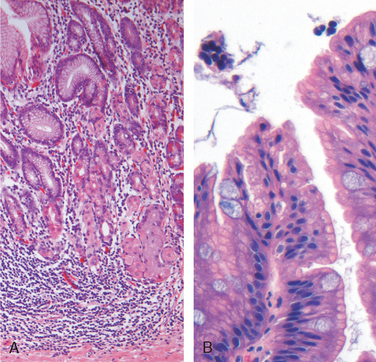
Clinical Features.
Antibodies to parietal cells and to intrinsic factor are present early in the disease course. Progression to gastric atrophy probably occurs over 2 to 3 decades, and anemia is seen in only a few patients. Because of the slow onset and variable progression, patients are generally diagnosed only after being affected for many years; the median age at diagnosis is 60 years. Slightly more women than men are affected. Pernicious anemia and autoimmune gastritis are often associated with other autoimmune diseases including Hashimoto thyroiditis, insulin-dependent (type I) diabetes mellitus, Addison disease, primary ovarian failure, primary hypoparathyroidism, Graves disease, vitiligo, myasthenia gravis, and Lambert-Eaton syndrome. These associations, along with concordance in some monozygotic twins and clustering of disease in families, support a genetic predisposition. In general, about 20% of relatives of individuals with pernicious anemia also have autoimmune gastritis, although they may be asymptomatic. Despite this strong genetic influence, autoimmune gastritis stands apart from other autoimmune diseases in that there is little evidence of linkage to specific HLA alleles.
Clinical presentation may be linked to symptoms of anemia.15 In addition, vitamin B12 deficiency may cause atrophic glossitis, in which the tongue becomes smooth and beefy red, epithelial megaloblastosis, and malabsorptive diarrhea. Vitamin B12 deficiency may also cause peripheral neuropathy, spinal cord lesions, and cerebral dysfunction. Neuropathic changes include demyelination, axonal degeneration, and neuronal death. The most frequent manifestations of peripheral neuropathy are paresthesias and numbness. The spinal lesions may be associated with a mixture of loss of vibration and position sense, sensory ataxia with positive Romberg sign, limb weakness, spasticity, and extensor plantar responses. Cerebral manifestations range from mild personality changes and memory loss to psychosis. In contrast to anemia, neurologic changes are not reversed by vitamin B12 replacement therapy.
UNCOMMON FORMS OF GASTRITIS
Reactive Gastropathy.
This group of disorders is marked by foveolar hyperplasia, glandular regenerative changes, and mucosal edema. Neutrophils are not abundant. Causes of reactive gastropathy include chemical injury, NSAID use, bile reflux, and mucosal trauma secondary to prolapse. Notably, reactive gastropathy and bile reflux are common after gastric surgeries that bypass the pylorus. Gastric antral trauma induces a grossly characteristic lesion referred to as gastric antral vascular ectasia (GAVE). Endoscopy shows longitudinal stripes of edematous erythematous mucosa alternating with less severely injured mucosa that is sometimes referred to as watermelon stomach. Histologically, the antral mucosa shows reactive gastropathy with dilated capillaries containing fibrin thrombi.16
Eosinophilic Gastritis.
As indicated by the name, this form of gastritis is characterized by tissue damage associated with dense infiltrates of eosinophils in the mucosa and muscularis, usually in the antral or pyloric region. The lesion is often present in other areas of the GI tract as well and is associated with peripheral eosinophilia and increased serum IgE levels. Allergic reactions are one cause of eosinophilic gastritis. In children, the allergens include cow’s milk and soy protein, while drugs are common allergens in children and adults. Eosinophilic gastritis can also occur in association with systemic collagen-vascular disease, such as systemic sclerosis and polymyositis. Parasitic infections and H. pylori infection are other causes of eosinophilic gastritis.
Lymphocytic Gastritis.
This disease preferentially affects women and produces nonspecific symptoms such as abdominal pain, anorexia, nausea, and vomiting. It is idiopathic, but approximately 40% of cases are associated with celiac disease, suggesting an immune-mediated pathogenesis. Lymphocytic gastritis is also referred to as varioliform gastritis based on the distinctive endoscopic appearance (thickened folds covered by small nodules with central aphthous ulceration).17 The entire stomach is affected in most cases, but disease is occasionally limited to the body. Histologically there is a marked increase in the number of intraepithelial T lymphocytes, mostly CD8+ cells, within surface and pit regions.
Granulomatous Gastritis.
This descriptive term is applied to any gastritis that contains granulomas, or aggregates of epithelioid histiocytes (tissue macrophages). It encompasses a diverse group of diseases with widely varying clinical and pathologic features. Correlation with clinical, endoscopic, radiologic, and serologic data is generally necessary for diagnosis. In Western populations, gastric involvement by Crohn disease is the most common specific cause of granulomatous gastritis.18 Sarcoidosis is the second most common cause, followed by a variety of infections including mycobacteria, fungi, CMV, and H. pylori. In addition to the presence of histologically evident granulomas, narrowing and rigidity of the gastric antrum may occur secondary to transmural granulomatous inflammation.
Complications of Chronic Gastritis
PEPTIC ULCER DISEASE
Peptic ulcer disease (PUD) is most often associated with H. pylori–induced hyperchlorhydric chronic gastritis, which is present in 85% to 100% of individuals with duodenal ulcers and in 65% with gastric ulcers. The presence of chronic gastritis can help to distinguish peptic ulcers from acute erosive gastritis or stress ulcers, since the mucosa adjacent to the ulcer is generally normal in the latter two conditions. PUD may occur in any portion of the GI tract exposed to acidic gastric juices, but is most common in the gastric antrum and first portion of the duodenum. PUD may also occur in the esophagus as a result of GERD or acid secretion by ectopic gastric mucosa. Gastric mucosa within a Meckel diverticulum can result in peptic ulceration of adjacent mucosa.
Epidemiology.
PUD is common and ranks fourth in both annual physician visits and costs among all GI diseases.19 In the United States, the lifetime risk of developing an ulcer is approximately 10% for males and 4% for females; the latter are typically affected during or after menopause. PUD affects more than 300 million people and is responsible for treatment and ongoing care of over 3 million people, 190,000 hospitalizations, and 5000 deaths in the United States each year.19
Pathogenesis.
The imbalances of mucosal defenses and damaging forces that cause chronic gastritis are also responsible for PUD. Thus, PUD generally develops on a background of chronic gastritis. The reasons why some people develop only chronic gastritis while others develop PUD are poorly understood.
H. pylori infection and NSAID use are the primary underlying causes of PUD, and both compromise mucosal defense while causing mucosal damage. Although more than 70% of individuals with PUD are infected by H. pylori, fewer than 20% of H. pylori–infected individuals develop peptic ulcer. It is probable that host factors as well as variation among H. pylori strains determine the clinical outcomes.
The gastric hyperacidity that drives PUD may be caused by H. pylori infection, parietal cell hyperplasia, excessive secretory responses, or impaired inhibition of stimulatory mechanisms such as gastrin release. For example, Zollinger-Ellison syndrome, in which there are multiple peptic ulcerations in the stomach, duodenum, and even jejunum, is caused by uncontrolled release of gastrin by a tumor and the resulting massive acid production. More common cofactors in peptic ulcerogenesis include chronic NSAID use, which causes direct chemical irritation while suppressing prostaglandin synthesis necessary for mucosal protection; cigarette smoking, which impairs mucosal blood flow and healing; and high-dose corticosteroids that suppress prostaglandin synthesis and impair healing. Duodenal ulcers are more frequent in individuals with alcoholic cirrhosis, chronic obstructive pulmonary disease, chronic renal failure, and hyperparathyroidism. In the latter two conditions, hypercalcemia stimulates gastrin production and therefore increases acid secretion. Finally, self-imposed or exogenous psychologic stress may increase gastric acid production.
Morphology. Peptic ulcers are four times more common in the proximal duodenum than in the stomach. Duodenal ulcers usually occur within a few centimeters of the pyloric valve and involve the anterior duodenal wall. Gastric peptic ulcers are predominantly located along the lesser curvature near the interface of the body and antrum.
Peptic ulcers are solitary in more than 80% of patients. Lesions less than 0.3 cm in diameter tend to be shallow while those over 0.6 cm are likely to be deeper ulcers. The classic peptic ulcer is a round to oval, sharply punched-out defect (Fig. 17-14A). The mucosal margin may overhang the base slightly, particularly on the upstream side, but is usually level with the surrounding mucosa. In contrast, heaped-up margins are more characteristic of cancers. The depth of ulcers may be limited by the thick gastric muscularis propria or by adherent pancreas, omental fat, or the liver. Hemorrhage and fibrin deposition are often present on the gastric serosa. Perforation into the peritoneal cavity is a surgical emergency that may be identified by the presence of free air under the diaphragm on upright radiographs of the abdomen.
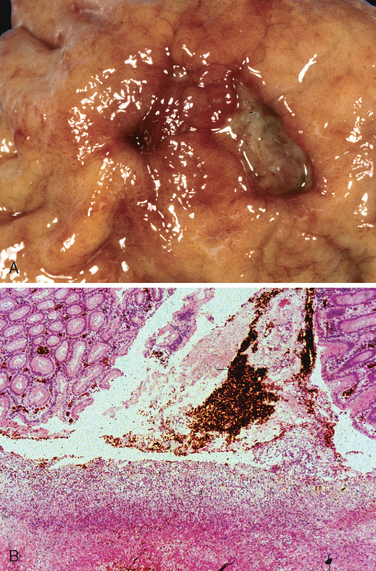
FIGURE 17-14 Acute gastric perforation in a patient presenting with free air under the diaphragm. A, Mucosal defect with clean edges. B, The necrotic ulcer base is composed of granulation tissue.
The base of peptic ulcers is smooth and clean as a result of peptic digestion of exudate, and blood vessels may be evident. In active ulcers the base may have a thin layer of fibrinoid debris underlaid by a predominantly neutrophilic inflammatory infiltrate. Beneath this, active granulation tissue infiltrated with mononuclear leukocytes and a fibrous or collagenous scar forms the ulcer base (Fig. 17-14B). Vessel walls within the scarred area are typically thickened and are occasionally thrombosed. Ongoing bleeding within the ulcer base may cause life-threatening hemorrhage. Scarring may involve the entire thickness of the wall and pucker the surrounding mucosa into folds that radiate outward.
Size and location do not differentiate benign and malignant ulcers. However, the gross appearance of chronic peptic ulcers is virtually diagnostic. Malignant transformation of peptic ulcers is very rare, and reports of transformation probably represent cases wherein a lesion thought to be benign was actually an ulcerated carcinoma from the start.
Clinical Features.
Peptic ulcers are notoriously chronic, recurring lesions with much greater morbidity than mortality. They may present in young adults but are most often diagnosed in middle-aged to older adults without obvious precipitating conditions. After a period of weeks to months of active disease, healing may occur with or without therapy, but the tendency to develop peptic ulcers remains. The majority of peptic ulcers come to clinical attention because of epigastric burning or aching pain, although a significant fraction present with complications such as iron deficiency anemia, frank hemorrhage, or perforation. The pain tends to occur 1 to 3 hours after meals during the day, is worse at night, and is relieved by alkali or food. Nausea, vomiting, bloating, belching, and significant weight loss are additional manifestations. With penetrating ulcers the pain is occasionally referred to the back, the left upper quadrant, or the chest, where it may be misinterpreted as cardiac in origin.
Current therapies for PUD are aimed at H. pylori eradication and neutralization of gastric acid, primarily with proton pump inhibitors or H2 histamine receptor antagonists.20 A variety of surgical approaches were formerly used, including antrectomy to remove gastrin-producing cells and vagotomy to remove the acid-stimulatory effects mediated by the vagus nerve. Proton pump inhibitors and H. pylori eradication have markedly reduced the need for surgical intervention, which is primarily reserved for treatment of bleeding or perforated peptic ulcers.
MUCOSAL ATROPHY AND INTESTINAL METAPLASIA
Long-standing chronic gastritis that involves the body and fundus may ultimately lead to significant loss of parietal cell mass. This oxyntic atrophy may be associated with intestinal metaplasia, recognized by the presence of goblet cells, and is strongly associated with increased risk of gastric adenocarcinoma. The risk of adenocarcinoma is greatest in autoimmune gastritis. This may be because achlorhydria of gastric mucosal atrophy permits overgrowth of bacteria that produce carcinogenic nitrosamines. Intestinal metaplasia also occurs in chronic H. pylori gastritis and may regress after clearance of the organism.
DYSPLASIA
Chronic gastritis exposes the epithelium to inflammation-related free radical damage and proliferative stimuli. Over time this combination of stressors can lead to the accumulation of genetic alterations that result in carcinoma. Pre-invasive in situ lesions can be recognized histologically as dysplasia. The morphologic hallmarks of dysplasia are variations in epithelial size, shape, and orientation along with coarse chromatin texture, hyperchromasia, and nuclear enlargement. The distinction between dysplasia and regenerative epithelial changes induced by active inflammation can be a challenge for the pathologist, since increased epithelial proliferation and mitotic figures may be prominent in both. However, reactive epithelial cells mature as they reach the mucosal surface, while dysplastic lesions remain cytologically immature.
GASTRITIS CYSTICA
Gastritis cystica refers to an exuberant reactive epithelial proliferation associated with entrapment of epithelial-lined cysts. These may be found within the submucosa (gastritis cystica polyposa) or deeper layers of the gastric wall (gastritis cystica profunda). Because of the association with chronic gastritis and partial gastrectomy, it is presumed that gastritis cystica is trauma-induced, but the reasons for the development of epithelial cysts within deeper portions of the gastric wall are not clear. Regenerative epithelial changes can be prominent in the entrapped epithelium, and gastritis cystica can therefore mimic invasive adenocarcinoma.
Hypertrophic Gastropathies
Hypertrophic gastropathies are uncommon diseases characterized by giant cerebriform enlargement of the rugal folds due to epithelial hyperplasia without inflammation. As might be expected, the hypertrophic gastropathies are linked to excessive growth factor release. The two most well-understood examples are Ménétrier disease and Zollinger-Ellison syndrome, the morphologic features of which are compared with other gastric proliferations in Table 17-4.
MÉNÉTRIER DISEASE
Ménétrier disease is a rare disorder caused by excessive secretion of transforming growth factor α (TGF-α)21 The disease is characterized by diffuse hyperplasia of the foveolar epithelium of the body and fundus and hypoproteinemia due to protein-losing enteropathy. Secondary symptoms, such as weight loss, diarrhea, and peripheral edema, are commonly present. Symptoms and pathologic features of Ménétrier disease in children are similar to those in adults, but pediatric disease is usually self-limited and often follows respiratory infection. Risk of gastric adenocarcinoma is increased in adults with Ménétrier disease.
Morphology. Ménétrier disease is characterized by irregular enlargement of the gastric rugae. Some areas may appear polypoid. Enlarged rugae are present in the body and fundus (Fig. 17-15A), but the antrum is generally spared. Histologically, the most characteristic feature is hyperplasia of foveolar mucous cells. The glands are elongated with a corkscrew-like appearance and cystic dilation is common (Fig. 17-15B). Inflammation is usually only modest, although some cases show marked intraepithelial lymphocytosis. Diffuse or patchy glandular atrophy, evident as hypoplasia of parietal and chief cells, is typical.
Treatment of Ménétrier disease is supportive, with intravenous albumin and parenteral nutritional supplementation. In severe cases gastrectomy may be performed. More recently, agents that block TGF-α–mediated activation of the epidermal growth factor receptor have shown promise.22
ZOLLINGER-ELLISON SYNDROME
Zollinger-Ellison syndrome is caused by gastrin-secreting tumors, gastrinomas, that are most commonly found in the small intestine or pancreas. Patients often present with duodenal ulcers or chronic diarrhea. Within the stomach, the most remarkable feature is a doubling of oxyntic mucosal thickness due to a fivefold increase in the number of parietal cells. Gastrin also induces hyperplasia of mucous neck cells, mucin hyperproduction, and proliferation of endocrine cells within oxyntic mucosa. In some cases these endocrine cells can form small dysplastic nodules or, rarely, true carcinoid tumors.
Treatment of individuals with Zollinger-Ellison syndrome includes blockade of acid hypersecretion, which is accomplished in almost all patients with proton pump inhibitors or high-dose H2 histamine receptor antagonists. Acid suppression allows peptic ulcers to heal and prevents gastric perforation, allowing treatment to focus on the gastrinoma, which becomes the main determinant of long-term survival.
Although they grow slowly, 60% to 90% of gastrinomas are malignant. Tumors are sporadic in 75% of patients. These tend to be solitary tumors and can be surgically resected. The remaining 25% of patients with gastrinomas have multiple endocrine neoplasia type I. These individuals often have multiple tumors or metastatic disease and may benefit from treatment with somatostatin analogues.23 Clinical identification of tumors may be enhanced by somatostatin receptor scintigraphy and endoscopic ultrasonography.
Gastric Polyps and Tumors
Polyps, nodules or masses that project above the level of the surrounding mucosa, are identified in up to 5% of upper GI endoscopies. Polyps may develop as a result of epithelial or stromal cell hyperplasia, inflammation, ectopia, or neoplasia. Only the most common types of polyps will be discussed here (Peutz-Jeghers and juvenile polyps are discussed with intestinal polyps). This is followed by a presentation of gastric tumors, including adenocarcinomas, lymphomas, carcinoid tumors, and stromal tumors.
INFLAMMATORY AND HYPERPLASTIC POLYPS
Approximately 75% of all gastric polyps are inflammatory or hyperplastic polyps. They are most common in individuals between 50 and 60 years of age. These polyps usually develop in association with chronic gastritis, which initiates the injury and reactive hyperplasia that leads to polyp growth. Inflammatory or hyperplastic polyps are most common in individuals between 50 and 60 years of age. Among individuals with H. pylori gastritis, polyps may regress after bacterial eradication. Because the risk of dysplasia correlates with size, polyps larger than 1.5 cm should be resected and examined histologically.
Morphology. The majority of inflammatory or hyperplastic polyps are smaller than 1 cm in diameter and are frequently multiple, particularly in individuals with atrophic gastritis. These polyps are ovoid in shape and have a smooth surface, though superficial erosions are common. Microscopically, polyps have irregular, cystically dilated, and elongated foveolar glands (Fig. 17-16A). The lamina propria is typically edematous with variable degrees of acute and chronic inflammation, and surface ulceration may be present (Fig. 17-16B).
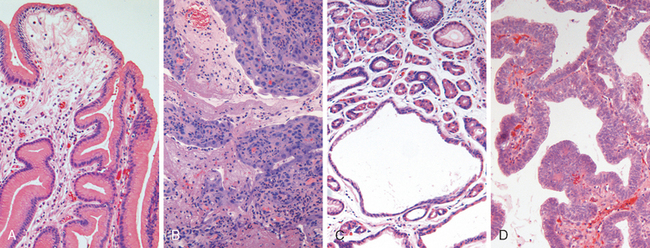
FIGURE 17-16 Gastric polyps. A, Hyperplastic polyp containing corkscrew-shaped foveolar glands. B, Hyperplastic polyp with ulceration. C, Fundic gland polyp composed of cystically dilated glands lined by parietal, chief, and foveolar cells. D, Gastric adenoma recognized by the presence of epithelial dysplasia.
FUNDIC GLAND POLYPS
Fundic gland polyps occur sporadically and in individuals with familial adenomatous polyposis (FAP). The prevalence of fundic gland polyps has increased markedly in recent years as a result of proton pump inhibitor therapy. This likely reflects increased gastrin secretion, in response to reduced gastric acidity, and the resulting glandular hyperplasia. These polyps are five times more common in women and are discovered at an average age of 50 years. Fundic gland polyps may be asymptomatic or associated with nausea, vomiting, or epigastric pain.
Morphology. Fundic gland polyps occur in the gastric body and fundus and are well-circumscribed lesions with a smooth surface. They may be single or multiple and are composed of cystically dilated, irregular glands lined by flattened parietal and chief cells. Inflammation is typically absent or minimal (Fig. 17-16C).
GASTRIC ADENOMA
Gastric adenomas represent as many as 10% of all gastric polyps (Table 17-4). Their incidence increases progressively with age,24 and there is a marked variation in rate among different populations that parallels the incidence of gastric adenocarcinoma. Patients are usually between 50 and 60 years of age, and males are affected three times more often than females. Like fundic gland polyps, the incidence of adenomas is increased in individuals with FAP. Similar to other forms of gastric dysplasia, adenomas almost always occur on a background of chronic gastritis with atrophy and intestinal metaplasia. The risk of adenocarcinoma in gastric adenomas is related to the size of the lesion and is particularly elevated in lesions greater than 2 cm in diameter. Overall, carcinoma may be present in up to 30% of gastric adenomas.24
Morphology. Gastric adenomas are usually solitary lesions less than 2 cm in diameter, most commonly located in the antrum. The majority of adenomas are composed of intestinal-type columnar epithelium. By definition, all GI adenomas have epithelial dysplasia (Fig. 17-16D) that can be classified as low or high grade. Both grades may include enlargement, elongation, and hyperchromasia of epithelial cell nuclei, epithelial crowding, and pseudostratification. High-grade dysplasia is characterized by more severe cytologic atypia and irregular architecture, including glandular budding and gland-within-gland, or cribriform, structures.25
GASTRIC ADENOCARCINOMA
Adenocarcinoma is the most common malignancy of the stomach, comprising over 90% of all gastric cancers. Early symptoms resemble those of chronic gastritis, including dyspepsia, dysphagia, and nausea. As a result, these tumors are often discovered at advanced stages, when symptoms such as weight loss, anorexia, altered bowel habits, anemia, and hemorrhage trigger further diagnostic evaluation.
Epidemiology.
Gastric cancer incidence varies markedly with geography. In Japan, Chile, Costa Rica, and Eastern Europe the incidence is up to 20-fold higher than in North America, northern Europe, Africa, and Southeast Asia. Mass endoscopic screening programs can be successful in regions where the incidence is high, such as Japan, where 35% of newly detected cases are early gastric cancer, tumors limited to the mucosa and submucosa. Unfortunately, mass screening programs are not cost-effective in regions where the incidence is low, and fewer than 20% of cases are detected at an early stage in North America and northern Europe.
In the United States, gastric cancer rates dropped by over 85% during the twentieth century.26 Adenocarcinoma of the stomach was the most common cause of cancer death in the United States in 1930 and remains a leading cause of cancer death worldwide, but now accounts for fewer than 2.5% of cancer deaths in the United States. Similar declines have been reported in many other Western countries, suggesting that environmental and dietary factors are responsible.26 Consistent with this conclusion, studies of migrants from high-risk to low-risk regions have shown that gastric cancer rates in second-generation immigrants are similar to those in their new country of residence.
The cause of the overall reduction in gastric cancer is unknown. One possible explanation is the decreased consumption of dietary carcinogens, such as N-nitroso compounds and benzo[a]pyrene, because of reduced use of salt and smoking for food preservation and the widespread availability of food refrigeration. Conversely, intake of green, leafy vegetables and citrus fruits, which contain antioxidants such as vitamin C, vitamin E, and beta-carotene, and is correlated with reduced risk of gastric cancers, may have increased as a result of improved food transportation networks.
Gastric cancer is more common in lower socioeconomic groups and in individuals with multifocal mucosal atrophy and intestinal metaplasia. PUD does not impart an increased risk of gastric cancer, but patients who have had partial gastrectomies for PUD have a slightly higher risk of developing cancer in the residual gastric stump as a result of hypochlorhydria, bile reflux, and chronic gastritis.
Although overall incidence of gastric adenocarcinoma is falling, cancer of the gastric cardia is on the rise. This is probably related to Barrett esophagus and may reflect the increasing incidence of chronic GERD and obesity.10 Consistent with this presumed common pathogenesis, distal esophageal adenocarcinomas and gastric cardia adenocarcinomas are similar in morphology, clinical behavior, and therapeutic response.27-29
Pathogenesis.
While the majority of gastric cancers are not hereditary, the mutations identified in familial gastric cancer have provided important insights into mechanisms of carcinogenesis in sporadic cases. Germline mutations in CDH1, which encodes E-cadherin, a protein that contributes to epithelial intercellular adhesion, are associated with familial gastric cancers, which are usually of the diffuse type. Mutations in CDH1 are present in about 50% of sporadic cases of diffuse gastric tumors, while E-cadherin expression is drastically decreased in the rest, often by methylation of the CDH1 promoter. Thus, the loss of E-cadherin function seems to be a key step in the development of diffuse gastric cancer. Notably, CDH1 mutations are also common in sporadic and familial lobular carcinoma of the breast, which also tends to infiltrate as single cells, and individuals with BRCA2 mutations are at increased risk of developing diffuse gastric cancer.
In contrast to diffuse gastric tumors, there is an increased risk of intestinal-type gastric cancer in individuals with FAP, particularly in Japan. This implies an interaction between host genetic background and environmental factors, since gastric cancer risk is less markedly elevated in individuals with FAP residing in areas of low gastric cancer incidence. Mutations in β-catenin, a protein that binds to both E-cadherin and adenomatous polyposis coli (APC), as well as microsatellite instability and hypermethylation of several genes including TGFβRII, BAX, IGFRII, and p16/INK4a have also been described in sporadic intestinal-type gastric cancer.
Genetic variants of pro-inflammatory and immune response genes, including those that encode IL-1β, TNF, IL-10, IL-8, and Toll-like receptor 4 (TLR4), are associated with elevated risk of gastric cancer when accompanied by H. pylori infection, and p53 mutations are present in the majority of sporadic gastric cancers of both histologic types. Thus, although specific sequences of events have not been defined, it is clear that chronic inflammation promotes neoplastic progression. Other associations between chronic inflammation and cancer were discussed in Chapter 7.
Morphology. Gastric adenocarcinomas are classified according to their location in the stomach, and most importantly, according to gross and histologic morphology. Most gastric adenocarcinomas involve the gastric antrum; the lesser curvature is involved more often than the greater curvature.28 Gastric tumors with an intestinal morphology tend to form bulky tumors (Fig. 17-17A) composed of glandular structures (Fig. 17-18A), while cancers with a diffuse infiltrative growth pattern (see Fig. 17-17B) are more often composed of signet-ring cells (see Fig. 17-18B). Although intestinal-type adenocarcinomas may penetrate the gastric wall, they typically grow along broad cohesive fronts to form either an exophytic mass or an ulcerated tumor. The neoplastic cells often contain apical mucin vacuoles, and abundant mucin may be present in gland lumens. In contrast, diffuse gastric cancer is generally composed of discohesive cells that do not form glands but instead have large mucin vacuoles that expand the cytoplasm and push the nucleus to the periphery, creating a signet-ring cell morphology. These cells permeate the mucosa and stomach wall individually or in small clusters, which makes tumor cells easy to confuse with inflammatory cells, such as macrophages, at low magnification. Extracellular mucin release in either type of gastric cancer can result in formation of large mucin lakes that dissect tissue planes.
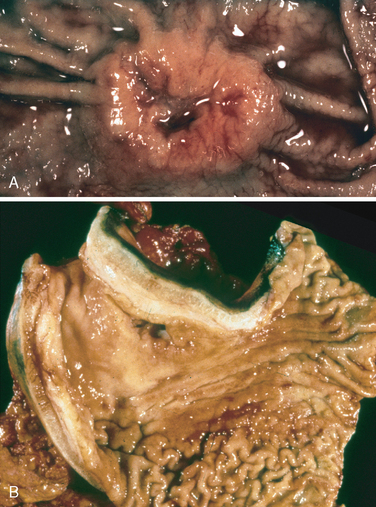
FIGURE 17-17 Gastric adenocarcinoma. A, Intestinal-type adenocarcinoma consisting of an elevated mass with heaped-up borders and central ulceration. Compare to the peptic ulcer in Figure 17-14A. B, Linitis plastica. The gastric wall is markedly thickened, and rugal folds are partially lost.
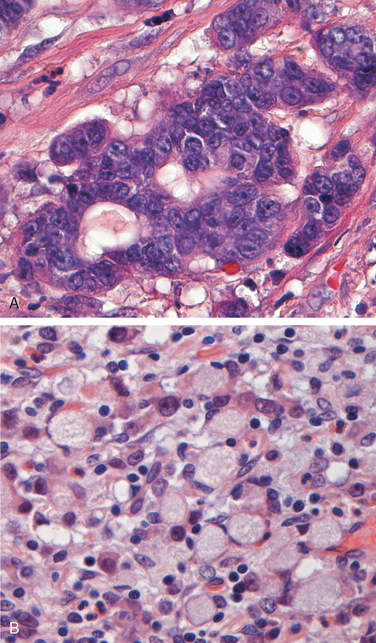
FIGURE 17-18 Gastric adenocarcinoma. A, Intestinal-type adenocarcinoma composed of columnar, gland-forming cells infiltrating through desmoplastic stroma. B, Signet-ring cells can be recognized by their large cytoplasmic mucin vacuoles and peripherally displaced, crescent-shaped nuclei.
A mass may be difficult to appreciate in diffuse gastric cancer, but these infiltrative tumors often evoke a desmoplastic reaction that stiffens the gastric wall and may provide a valuable diagnostic clue. When there are large areas of infitration, diffuse rugal flattening and a rigid, thickened wall may impart a leather bottle appearance termed linitis plastica (see Fig. 17-17B). Breast and lung cancers that metastasize to the stomach may also create a linitis plastica–like appearance.
Clinical Features.
Intestinal-type gastric cancer predominates in high-risk areas and develops from precursor lesions including flat dysplasia and adenomas. The mean age of presentation is 55 years, and the male-to-female ratio is 2 : 1. In contrast, the incidence of diffuse gastric cancer is relatively uniform across countries, there are no identified precursor lesions, and the disease occurs at similar frequencies in males and females. Notably, the remarkable decrease in gastric cancer incidence applies only to the intestinal type, which is most closely associated with atrophic gastritis and intestinal metaplasia. As a result, the incidences of intestinal and diffuse types of gastric cancers are now similar.
The depth of invasion and the extent of nodal and distant metastasis at the time of diagnosis remain the most powerful prognostic indicators for gastric cancer.30 In advanced cases gastric carcinoma may first be detected as metastases to the supraclavicular sentinel lymph node, also called Virchow’s node. Gastric tumors can also metastasize to the periumbilical region to form a subcutaneous nodule, termed a Sister Mary Joseph nodule, after the nurse who first noted this lesion as a marker of metastatic carcinoma. Local invasion into the duodenum, pancreas, and retroperitoneum is also characteristic. In such cases efforts are usually focused on chemotherapy or radiation therapy and palliative care. However, when possible, surgical resection remains the preferred treatment for gastric adenocarcinoma. After surgical resection, the 5-year survival rate of early gastric cancer can exceed 90%, even if lymph node metastases are present. In contrast, the 5-year survival rate for advanced gastric cancer remains below 20%.28 Because of the advanced stage at which most gastric cancers are discovered in the United States, the overall 5-year survival is less than 30%.28,31
LYMPHOMA
Although extra-nodal lymphomas can arise in virtually any tissue, they do so most commonly in the GI tract, particularly the stomach. In allogeneic bone marrow transplant and organ transplant recipients, the bowel is also the most frequent site for Epstein-Barr virus–positive B-cell lymphoproliferations,32 because the deficits in T-cell function caused by oral immunosuppressive agents (e.g., cyclosporine) are greatest at intestinal sites of drug absorption. Nearly 5% of all gastric malignancies are primary lymphomas, the most common of which are indolent extra-nodal marginal zone B-cell lymphomas. In the gut these tumors are often referred to as lymphomas of mucosa-associated lymphoid tissue (MALT), or MALTomas.33 This entity and the second most common primary lymphoma of the gut, diffuse large B-cell lymphoma, are also discussed in Chapter 13.
Pathogenesis.
Extra-nodal marginal zone B-cell lymphomas usually arise at sites of chronic inflammation. They can originate in the GI tract at sites of preexisting MALT, such as the Peyer’s patches of the small intestine, but more commonly arise within tissues that are normally devoid of organized lymphoid tissue. The most common cause of “pro-lymphomatous” inflammation in the stomach is chronic H. pylori infection, which is found in association with most cases of gastric MALToma.34 As with other low-grade lymphomas, MALTomas can transform into more aggressive tumors that are histologically identical to diffuse large B-cell lymphomas.
The most striking evidence linking H. pylori gastritis to MALToma is that eradication of the infection with antibiotics induces durable remissions with low rates of recurrence in most patients.35 Histologic features that predict antibiotic treatment failures include transformation to large-cell lymphoma, tumor invasion to the muscularis propria or beyond, and lymph node involvement.
Three translocations are associated with gastric MALToma, the t(11;18)(q21;q21) and the less common t(1;14)(p22;q32) and t(14;18)(q32;q21). They are also highly predictive of response failure.36,37 The t(11;18)(q21;q21) translocation brings together the apoptosis inhibitor 2 (API2) gene on chromosome 11 with the “mutated in MALT lymphoma,” or MLT, gene on chromosome 18. This creates a chimeric API2-MLT fusion gene that encodes an API2-MLT fusion protein. The t(14;18)(q32;q21) and t(1;14)(p22;q32) translocations cause increased expression of intact MLT and BCL-10 proteins, respectively.
Although some details remain uncertain, each of the three translocations has the same net effect, the constitutive activation of NF-κB, a transcription factor that promotes B-cell growth and survival. Remarkably, antigen-dependent activation of NF-κB in normal B and T cells requires both BCL-10 and MLT, which work together in a pathway down-stream of the B- and T-cell antigen receptors. In MALTomas that lack these translocations, H. pylori–induced inflammation may trigger NF-κB activation through the MLT/BCL-10 pathway. In these tumors elimination of the immune stimulus (H. pylori) down-regulates NF-κB, resulting in tumor regression. In contrast, NF-κB is constitutively active in tumors bearing translocations involving MLT or BCL10, and as a result the elimination of H. pylori has no effect. Additional genetic changes, such as inactivation of the tumor suppressor genes that encode p53 and p16, may lead to transformation of gastric MALToma into aggressive diffuse large B-cell lymphoma.38
Morphology. Histologically, gastric MALToma takes the form of a dense lymphocytic infiltrate in the lamina propria (Fig. 17-19A). Characteristically, the neoplastic lymphocytes infiltrate the gastric glands focally to create diagnostic lymphoepithelial lesions (Fig. 17-19A, inset). Reactive-appearing B-cell follicles may be present, and, in about 40% of tumors, plasmacytic differentiation is observed. Occasionally the tumor cells accumulate large amounts of pale cytoplasm, a feature referred to as “monocytoid” change.
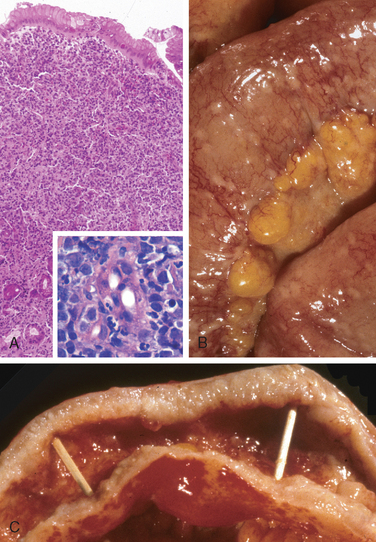
FIGURE 17-19 GI lymphoma. A, Gastric MALT lymphoma replacing much of the gastric epithelium. Inset shows lymphoepithelial lesions with neoplastic lymphocytes surrounding and infiltrating gastric glands. B, Disseminated lymphoma within the small intestine with numerous small serosal nodules. C, Large B-cell lymphoma infiltrating the small intestinal wall and producing diffuse thickening.
Like other tumors of mature B cells, MALTomas express the B-cell markers CD19 and CD20. They do not express CD5 and CD10, and are positive for CD43 in about 25% of cases, an unusual feature that can be diagnostically helpful. In cases lacking lymphoepithelial lesions, monoclonality may be demonstrated by restricted expression of either κ or γ immunoglobulin light chain or by molecular detection of clonal IgH rearrangements. Molecular cytogenetic analysis (e.g., fluorescent in situ hybridization) is being used increasingly to identify tumors with translocations that predict resistance to therapy.
Clinical Features.
The most common presenting symptoms are dyspepsia and epigastric pain. Hematemesis, melena, and constitutional symptoms such as weight loss can also be present. Because gastric MALTomas and H. pylori gastritis often coexist and have overlapping clinical symptoms and endoscopic appearances, diagnostic difficulties sometimes arise, particularly in small biopsy specimens. GI lymphomas may also disseminate as discrete small nodules (Fig. 17-19B) or infiltrate the wall diffusely (Fig. 17-19C).
CARCINOID TUMOR
Carcinoid tumors arise from the diffuse components of the endocrine system. The majority are found in the GI tract, and more than 40% occur in the small intestine (Table 17-5).39 The tracheobronchial tree and lungs are the next most commonly sites involved. Gastric carcinoids may be associated with endocrine cell hyperplasia, chronic atrophic gastritis, and Zollinger-Ellison syndrome. The term carcinoid, or “carcinoma-like,” was applied because these tumors tend to have a more indolent clinical course than GI carcinomas. Carcinoid tumors are best considered to be well-differentiated neuroendocrine carcinomas. Carcinoids within the GI tract arise from the endocrine cells that release peptide and nonpeptide hormones to coordinate gut function.
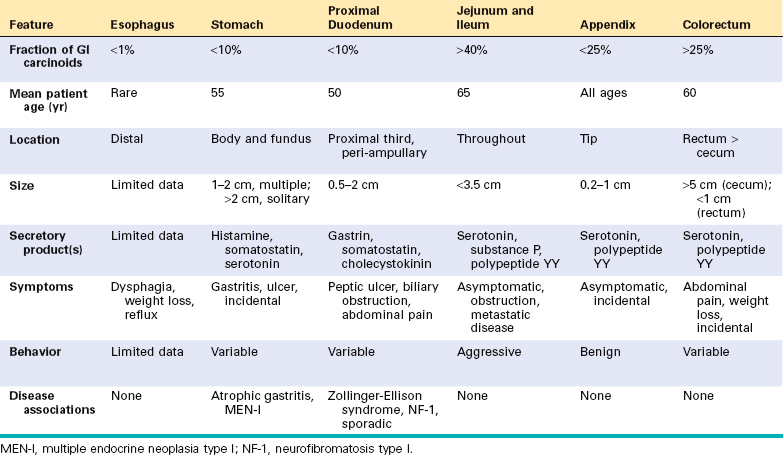
Morphology. Grossly, carcinoids are intramural or submucosal masses that create small polypoid lesions (Fig. 17-20A). The overlying mucosa may be intact or ulcerated, and the tumors may invade deeply to involve the mesentery. Carcinoids tend to be yellow or tan in color and are very firm as a consequence of an intense desmoplastic reaction, which may cause kinking of the bowel and obstruction. Histologically, carcinoid tumors are composed of islands, trabeculae, strands, glands, or sheets of uniform cells with scant, pink granular cytoplasm and a round to oval stippled nucleus (Fig. 17-20). In most tumors there is minimal pleomorphism, but anaplasia, mitotic activity, and necrosis may be present in rare cases. Immunohistochemical stains are typically positive for endocrine granule markers, such as synaptophysin and chromogranin A.
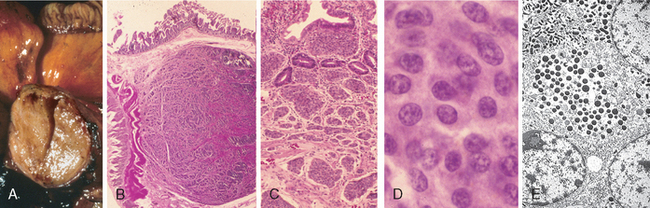
FIGURE 17-20 GI carcinoid tumor (neuroendocrine carcinoma). A, Gross cross-section of a submucosal tumor nodule. B, Microscopically the nodule is composed of tumor cells embedded in dense fibrous tissue. C, In other areas, the tumor has spread extensively within mucosal lymphatic channels. D, High magnification shows the bland cytology of carcinoid tumors. The chromatin texture, with fine and coarse clumps, is frequently described as a “salt and pepper” pattern. Despite their innocuous appearance, carcinoids can be extremely aggressive clinically. E, Electron microscopy reveals cytoplasmic dense core neurosecretory granules.
Clinical Features.
The peak incidence of carcinoid tumors is in the sixth decade, but they may appear at any age. Symptoms are determined by the hormones produced. For example, tumors that produce gastrin may cause Zollinger-Ellison syndrome, while ileal tumors may cause carcinoid syndrome, which is characterized by cutaneous flushing, sweating, bronchospasm, colicky abdominal pain, diarrhea, and right-sided cardiac valvular fibrosis. Carcinoid syndrome occurs in fewer than 10% of patients and is caused by vasoactive substances secreted by the tumor into the systemic circulation. When tumors are confined to the intestine, the vasoactive substances released are metabolized to inactive forms by the liver, a “first-pass” effect similar to that exerted on oral drugs. Carcinoid syndrome generally requires tumors to secrete hormones into a non-portal venous circulation and therefore is strongly associated with metastatic disease.
The most important prognostic factor for GI carcinoid tumors is location.
GASTROINTESTINAL STROMAL TUMOR
A wide variety of mesenchymal neoplasms may arise in the stomach. Many are named according to the cell type they most resemble; for example, smooth muscle tumors are called leiomyomas or leiomyosarcomas, nerve sheath tumors are termed schwannomas, and those resembling glomus bodies in the nailbeds and at other sites are termed glomus tumors. These are all rare and are discussed in greater detail in Chapter 26. GI stromal tumor (GIST) is the most common mesenchymal tumor of the abdomen, and more than half of these tumors occur in the stomach. As will be discussed below, the term stromal reflects historical confusion about the origin of this tumor.
Epidemiology.
Overall, GISTs are slightly more common in males. The peak age of GIST diagnosis in the stomach is approximately 60 years, with fewer than 10% occurring in individuals under 40 years of age. Of the uncommon GISTs in children, some are related to the Carney triad, a nonhereditary syndrome seen primarily in young females that includes gastric GIST, paraganglioma, and pulmonary chondroma. There is also an increased incidence of GIST in individuals with neurofibromatosis type 1.40
Pathogenesis.
Approximately 75% to 80% of all GISTs have oncogenic, gain-of-function mutations of the gene encoding the tyrosine kinase c-KIT, which is the receptor for stem cell factor. Approximately 8% of GISTs have mutations that activate a related tyrosine kinase, platelet-derived growth factor receptor α (PDGFRA).41 In sporadic GISTs, c-KIT and PDGFRA gene mutations are mutually exclusive.21 GISTs appear to arise from, or share a common stem cell with, the interstitial cells of Cajal, which are located in the muscularis propria and serve as pacemaker cells for gut peristalsis. Like GISTs, Cajal cells express c-KIT (also known as CD117) and CD34. Interestingly, familial GIST, which is rare, is associated with germline c-KIT or PDGFRA mutations; these patients, who develop multiple GISTs, may also have diffuse hyperplasia of Cajal cells.42 Mutation of c-KIT or PDGFRA is an early event in sporadic GISTs and is detectable in lesions as small as 3 mm. The constitutively active c-KIT and PDGFRA receptor tyrosine kinases produce intracellular signals that activate the RAS and PI3K/AKT pathways and thereby promote tumor cell proliferation and survival.21
Morphology. Primary gastric GISTs can be quite large, as much as 30 cm in diameter. They usually form a solitary, well-circumscribed, fleshy mass (Fig. 17-21A) covered by ulcerated or intact mucosa (Fig. 17-21B), but can also project outward toward the serosa. Metastases may take the form of multiple serosal nodules throughout the peritoneal cavity or as one or more nodules in the liver; spread outside of the abdomen is uncommon. GISTs composed of thin elongated cells are classified as spindle cell type (Fig. 17-21C), whereas tumors dominated by epithelial-appearing cells are termed epithelioid type; mixtures of the two patterns also occur. The most useful diagnostic marker is c-KIT, which is immunohistochemically detectable in 95% of gastric GISTs.
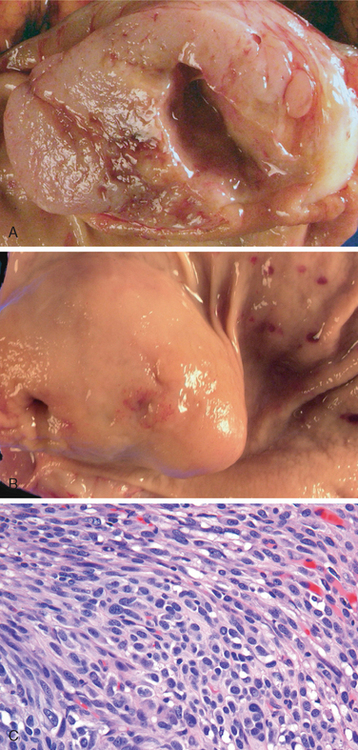
FIGURE 17-21 GI stromal tumor. A, On cross-section a whorled texture is evident within the white, fleshy tumor. B, The mass is covered by intact mucosa. C, Histologically the tumor is primarily composed of bundles, or fascicles, of spindle-shaped tumor cells.
(Courtesy of Dr. Christopher Weber, The University of Chicago, Chicago, IL.)
Clinical Features.
Symptoms of GISTs at presentation may be related to mass effects. Mucosal ulceration can cause blood loss, and approximately half of individuals with GIST present with anemia or related symptoms. GISTs may also be discovered as an incidental finding during radiologic imaging, endoscopy, or abdominal surgery performed for other reasons. Complete surgical resection is the primary treatment for localized gastric GIST. The prognosis correlates with tumor size, mitotic index, and location, with gastric GISTs being somewhat less aggressive than those arising in the small intestine. Recurrence or metastasis is rare for gastric GISTs under 5 cm but common for mitotically active tumors larger than 10 cm.
Patients with unresectable, recurrent, or metastatic disease often respond to imatinib, a tyrosine kinase inhibitor that inhibits c-KIT and PDGFRA, and is also effective in suppressing BCR-ABL kinase activity in chronic myeloid leukemia (Chapter 13).43 Development of resistance to imatinib is most often related to secondary c-KIT mutations that limit drug efficacy.
SMALL INTESTINE AND COLON
The small intestine and colon account for the majority of GI tract length and are the sites of a broad array of diseases. Some of these relate to nutrient and water transport. Perturbation of these processes can cause malabsorption and diarrhea. The intestines are also the principal site where the immune system interfaces with a diverse array of antigens present in food and gut microbes. Indeed, intestinal bacteria outnumber eukaryotic cells in our bodies by tenfold. Thus, it is not surprising that the small intestine and colon are frequently involved by infectious and inflammatory processes. Finally, the colon is the most common site of GI neoplasia in Western populations.
Intestinal Obstruction
Obstruction of the GI tract may occur at any level, but the small intestine is most often involved because of its relatively narrow lumen. Collectively, hernias, intestinal adhesions, intussusception, and volvulus account for 80% of mechanical obstructions (Fig. 17-22), while tumors and infarction account for only about 10% to 15% of small bowel obstructions. The clinical manifestations of intestinal obstruction include abdominal pain and distention, vomiting, and constipation. Surgical intervention is usually required in cases of mechanical obstruction or severe infarction.
FIGURE 17-22 Intestinal obstruction. The four major causes of intestinal obstruction are (1) herniation of a segment in the umbilical or inguinal regions, (2) adhesion between loops of intestine, (3) volvulus, and (4) intussusception.
HERNIAS
Any weakness or defect in the wall of the peritoneal cavity may permit protrusion of a serosa-lined pouch of peritoneum called a hernia sac. Acquired hernias most commonly occur anteriorly, via the inguinal and femoral canals or umbilicus, or at sites of surgical scars. These are of concern because of visceral protrusion (external herniation). This is particularly true of inguinal hernias, which tend to have narrow orifices and large sacs. Small bowel loops are involved most often, but portions of omentum or large bowel also protrude, and any of these may become entrapped. Pressure at the neck of the pouch may impair venous drainage of the entrapped viscus. The resultant stasis and edema increase the bulk of the herniated loop, leading to permanent entrapment, or incarceration, and, over time, arterial and venous compromise (strangulation) develops that can result in infarction (Fig. 17-23A).
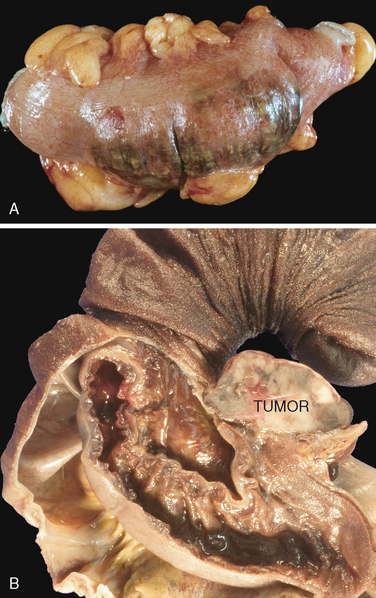
FIGURE 17-23 Intestinal obstruction. A, Portion of bowel incarcerated within an inguinal hernia. Note dusky serosa and hemorrhage that indicate ischemic damage. B, Intussusception caused by a tumor. The outermost layer of intestine with external serosa has been removed, leaving the mucosa of the second layer exposed. The serosa of the second layer is apposed to the serosa of the intussuscepted intestine. A tumor mass (right, labelled tumor) is present at the leading edge of the intussusception. Compare to Figure 17-22.
(B, Courtesy of Dr. Christopher Weber, The University of Chicago, Chicago, IL.)
ADHESIONS
Surgical procedures, infection, or other causes of peritoneal inflammation, such as endometriosis, may result in development of adhesions between bowel segments, the abdominal wall, and operative sites. These fibrous bridges can create closed loops through which other viscera may slide and become entrapped, resulting in internal herniation. Sequelae, including obstruction and strangulation, are much the same as with external hernias. Though rare, fibrous adhesions may be congenital, therefore, internal herniation must be considered even in the absence of a history of peritonitis or surgery.
VOLVULUS
Complete twisting of a loop of bowel about its mesenteric base of attachment is called volvulus and produces both luminal and vascular compromise. Thus, presentation includes features of obstruction and infarction. Volvulus occurs most often in large redundant loops of sigmoid colon, followed in frequency by the cecum, small bowel, stomach, or, rarely, transverse colon. Because it is rare, volvulus is often missed clinically.
INTUSSUSCEPTION
Intussusception occurs when a segment of the intestine, constricted by a wave of peristalsis, telescopes into the immediately distal segment. Once trapped, the invaginated segment is propelled by peristalsis and pulls the mesentery along. Untreated intussusception may progress to intestinal obstruction, compression of mesenteric vessels, and infarction.
When encountered in infants and children, there is usually no underlying anatomic defect and the patient is otherwise healthy, although some cases are associated with rotavirus infection. In older children and adults an intraluminal mass or tumor generally serves as the point of traction that causes intussusception (Fig. 17-23B). Barium enema may effectively reduce the intussusception in infants and young children, but surgical intervention is usually necessary in older patients.
Ischemic Bowel Disease
The majority of the GI tract is supplied by the celiac, superior mesenteric, and inferior mesenteric arteries. As they approach the intestinal wall the superior and inferior mesenteric arteries ramify into the mesenteric arcades. Interconnections between arcades, as well as collateral supplies from the proximal celiac and distal pudendal and iliac circulations, make it possible for the small intestine and colon to tolerate slowly progressive loss of the blood supply from one artery. In contrast, acute compromise of any major vessel can lead to infarction of several meters of intestine. Damage can range from mucosal infarction, extending no deeper than the muscularis mucosa; to mural infarction of mucosa and submucosa; to transmural infarction involving all three wall layers. While mucosal or mural infarctions are often secondary to acute or chronic hypoperfusion, transmural infarction is generally caused by acute vascular obstruction. Important causes of acute arterial obstruction include severe atherosclerosis (which is often prominent at the origin of mesenteric vessels), aortic aneurysm, hypercoagulable states, oral contraceptive use, and embolization of cardiac vegetations or aortic atheromas. Intestinal hypoperfusion can also be associated with cardiac failure, shock, dehydration, or vasoconstrictive drugs. Systemic vasculitides, such as polyarteritis nodosum, Henoch-Schönlein purpura, or Wegener granulomatosis, may also damage intestinal arteries. Mesenteric venous thrombosis, which can also lead to ischemic disease, is uncommon but can result from inherited or acquired hypercoagulable states, invasive neoplasms, cirrhosis, trauma, or abdominal masses that compress the portal drainage.
Pathogenesis.
Intestinal responses to ischemia occur in two phases. The initial hypoxic injury occurs at the onset of vascular compromise. While some damage occurs during this phase, the epithelial cells lining the intestine are relatively resistant to transient hypoxia. The second phase, reperfusion injury, is initiated by restoration of the blood supply and it is at this time that the greatest damage occurs. In severe cases this may trigger multiorgan failure. While the underlying mechanisms of reperfusion injury are incompletely understood, they involve free radical production, neutrophil infiltration, and release of inflammatory mediators, such as complement proteins and TNF (Chapter 1). Activation of intracellular signaling molecules and transcription factors, including hypoxia-inducible factor 1 (HIF-1) and NF-κB, also contribute to intestinal ischemia-reperfusion injury.44,45
The severity of vascular compromise, the time frame during which it develops, and the vessels affected are the major variables in ischemic bowel disease. Two aspects of intestinal vascular anatomy also contribute to the distribution of ischemic damage.
Morphology. Despite the increased susceptibility of watershed zones, mucosal and mural infarction may involve any level of the gut from stomach to anus. The lesions may be continuous but are more often segmental and patchy (Fig. 17-24A). The mucosa is hemorrhagic and may be ulcerated and dark red or purple as a result of luminal hemorrhage (Fig. 17-24B). The bowel wall is also thickened by edema that may involve the mucosa or extend into the submucosa and muscularis propria. When severe, there is extensive mucosal and submucosal hemorrhage and necrosis, but serosal hemorrhage and serositis are generally absent.
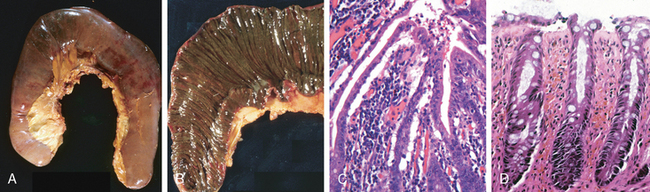
FIGURE 17-24 Ischemia. A, Jejunal resection with dusky serosa. B, Mucosa is stained with blood after hemorrhage. C, Characteristic attenuated villous epithelium in this case of acute jejunal ischemia. D, Chronic colonic ischemia with atrophic surface epithelium and fibrotic lamina propria.
Substantial portions of the bowel are generally involved in transmural infarction due to acute arterial obstruction. For reasons described above, the splenic flexure is the site at greatest risk. The demarcation between normal and ischemic bowel is sharply defined and the infarcted bowel is initially intensely congested and dusky to purple-red. Later, blood-tinged mucus or frank blood accumulates in the lumen and the wall becomes edematous, thickened, and rubbery. There is coagulative necrosis of the muscularis propria within 1 to 4 days, and perforation may occur. Serositis, with purulent exudates and fibrin deposition, may be prominent.
In mesenteric venous thrombosis arterial blood continues to flow for a time, resulting in a less abrupt transition from affected to normal bowel. However, propagation of the thrombus may lead to secondary involvement of the splanchnic bed. The ultimate result is similar to that produced by acute arterial obstruction because impaired venous drainage eventually prevents oxygenated arterial blood from entering the capillaries.
Microscopic examination of ischemic intestine demonstrates atrophy or sloughing of surface epithelium (Fig. 17-24C). In contrast, crypts may be hyperproliferative. Inflammatory infiltrates are initially absent in acute ischemia, but neutrophils are recruited within hours of reperfusion. Chronic ischemia is accompanied by fibrous scarring of the lamina propria (Fig. 17-24D) and, uncommonly, stricture formation. In acute phases of ischemic damage bacterial superinfection and enterotoxin release may induce pseudomembrane formation that can resemble Clostridium difficile–associated pseudomembranous colitis (discussed later).
Clinical Features.
Ischemic bowel disease tends to occur in older individuals with coexisting cardiac or vascular disease. Acute transmural infarction typically presents with sudden, severe abdominal pain and tenderness, sometimes accompanied by nausea, vomiting, bloody diarrhea, or grossly melanotic stool. Patients may progress to shock and vascular collapse within hours as a result of blood loss. Peristaltic sounds diminish or disappear, and muscular spasm creates board-like rigidity of the abdominal wall. Because these physical signs overlap with those of other abdominal emergencies, including acute appendicitis, perforated ulcer, and acute cholecystitis, the diagnosis of intestinal necrosis may be delayed or missed, with disastrous consequences. As the mucosal barrier breaks down, bacteria enter the circulation and sepsis can develop; mortality may exceed 50%. The overall progression of ischemic enteritis depends on the underlying cause and severity of injury.
Angiodysplasia
Angiodysplasia is characterized by malformed submucosal and mucosal blood vessels. It occurs most often in the cecum or right colon, usually after the sixth decade of life. Although the prevalence of angiodysplasia is less than 1% in the adult population, it accounts for 20% of major episodes of lower intestinal bleeding; intestinal hemorrhage may be chronic and intermittent or acute and massive.
The pathogenesis of angiodysplasia remains undefined but has been attributed to mechanical and congenital factors. Normal distention and contraction may intermittently occlude the submucosal veins that penetrate through the muscularis propria and can lead to focal dilation and tortuosity of overlying submucosal and mucosal vessels. Because the cecum has the largest diameter of any colonic segment, it develops the greatest wall tension. This may explain the preferential distribution of angiodysplastic lesions in the cecum and right colon. Degenerative vascular changes related to aging may also have some role. Finally, some data link angiodysplasia with aortic stenosis and Meckel diverticulum, suggesting the possibility of a developmental component.
Lesions of angiodysplasia are ectatic nests of tortuous veins, venules, and capillaries. The vascular channels may be separated from the intestinal lumen by only the vascular wall and a layer of attenuated epithelial cells; limited injury may therefore result in significant hemorrhage.
Malabsorption and Diarrhea
Malabsorption, which presents most commonly as chronic diarrhea, is characterized by defective absorption of fats, fat- and water-soluble vitamins, proteins, carbohydrates, electrolytes and minerals, and water. Chronic malabsorption can be accompanied by weight loss, anorexia, abdominal distention, borborygmi, and muscle wasting. A hallmark of malabsorption is steatorrhea, characterized by excessive fecal fat and bulky, frothy, greasy, yellow or clay-colored stools. The chronic malabsorptive disorders most commonly encountered in the United States are pancreatic insufficiency, celiac disease, and Crohn disease (Table 17-6). Intestinal graft-versus-host disease is an important cause of malabsorption and diarrhea after allogeneic bone marrow transplantation.
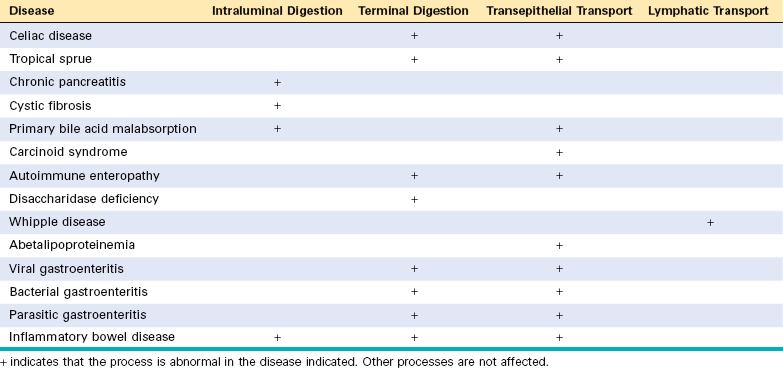
Malabsorption results from disturbance in at least one of the four phases of nutrient absorption: (1) intraluminal digestion, in which proteins, carbohydrates, and fats are broken down into forms suitable for absorption; (2) terminal digestion, which involves the hydrolysis of carbohydrates and peptides by disaccharidases and peptidases, respectively, in the brush border of the small intestinal mucosa; (3) transepithelial transport, in which nutrients, fluid, and electrolytes are transported across and processed within the small intestinal epithelium; and (4) lymphatic transport of absorbed lipids.
In many malabsorptive disorders a defect in one of these processes predominates, but more than one usually contributes. As a result, malabsorption syndromes resemble each other more than they differ. General symptoms include diarrhea (from nutrient malabsorption and excessive intestinal secretion), flatus, abdominal pain, and weight loss. Inadequate absorption of vitamins and minerals can result in anemia and mucositis due to pyridoxine, folate, or vitamin B12 deficiency; bleeding, due to vitamin K deficiency; osteopenia and tetany due to calcium, magnesium, or vitamin D deficiencies; or peripheral neuropathy due to vitamin A or B12 deficiencies. A variety of endocrine and skin disturbances may also occur.
Diarrhea is defined as an increase in stool mass, frequency, or fluidity, typically greater than 200 g per day. In severe cases stool volume can exceed 14 L per day and, without fluid resuscitation, result in death. Painful, bloody, small-volume diarrhea is known as dysentery. Diarrhea can be classified according to four major categories:
CYSTIC FIBROSIS
Cystic fibrosis is discussed in greater detail elsewhere (Chapter 10). Only the malabsorption associated with cystic fibrosis is considered here. Due to the absence of the epithelial cystic fibrosis transmembrane conductance regulator (CFTR), individuals with cystic fibrosis have defects in intestinal chloride ion secretion. This interferes with bicarbonate, sodium, and water secretion, ultimately resulting in defective luminal hydration. Formation of intraductal concretions can begin in utero, leading to duct obstruction, low-grade chronic autodigestion of the pancreas, and eventual exocrine pancreatic insufficiency in more than 80% of patients. The result is failure of the intraluminal phase of nutrient absorption, which can be effectively treated in most patients with oral enzyme supplementation.
CELIAC DISEASE
Celiac disease is also known as celiac sprue or gluten-sensitive enteropathy. It is an immune-mediated enteropathy triggered by the ingestion of gluten-containing cereals, such as wheat, rye, or barley, in genetically predisposed individuals. In countries where most people are Caucasians of European ancestry, celiac disease is a common disorder, with an estimated prevalence of 0.5% to 1%.
Pathogenesis.
Celiac disease is a unique intestinal immune disorder because the environmental precipitant, gluten, is known. Gluten is the major storage protein of wheat and similar grains, and the alcohol-soluble fraction of gluten, gliadin, contains most of the disease-producing components. Gluten is digested by luminal and brush-border enzymes into amino acids and peptides, including a 33–amino acid α-gliadin peptide that is resistant to degradation by gastric, pancreatic, and small intestinal proteases (Fig. 17-25). The network of immune reactions to gliadin that are thought to result in celiac disease is illustrated below. Some gliadin peptides induce epithelial cells to express IL-15, which in turn triggers activation and proliferation of CD8+ intraepithelial lymphocytes that are induced to express NKG2D, a natural killer cell marker. These lymphocytes become cytotoxic and kill enterocytes with surface MIC-A, an HLA class I–like protein expressed in response to stress. NKG2D is the receptor for MIC-A. Thus, unlike the CD4+ T cells, these NKG2D+ CD8+ T cells do not recognize gliadin. The resulting epithelial damage may contribute to the process by which other gliadin peptides cross the epithelium to be deamidated by tissue transglutaminase. Deamidated gliadin peptides are then able to interact with HLA-DQ2 or HLA-DQ8 on antigen-presenting cells and be presented to CD4+ T cells. These T cells produce cytokines that contribute to tissue damage and the characteristic mucosal pathology.
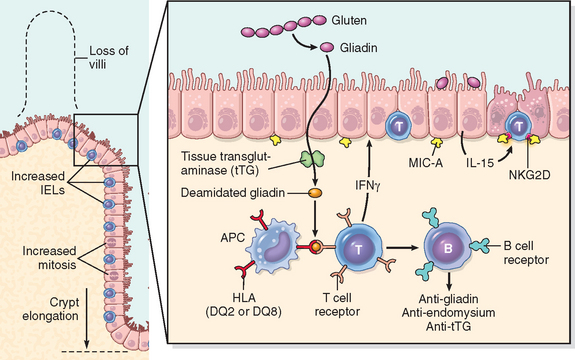
FIGURE 17-25 The left panel illustrates the morphologic alterations that may be present celiac disease, including villous atrophy, increased numbers of intraepithelial lymphocytes (IELs), and epithelial proliferation with crypt elongation (compare to Fig. 17-26). The right panel depicts a model for the pathogenesis of celiac disease. Note that both innate and adaptive immune mechanisms are involved in the tissue responses to gliadin.
While nearly all people eat grain and are exposed to gluten and gliadin, most do not develop celiac disease. Thus, host factors determine whether disease develops. Among these, HLA proteins seem to be critical, since almost all people with celiac disease carry the class II HLA-DQ2 or HLA-DQ8 allele. However, the HLA locus accounts for less than half of the genetic component of celiac disease. Remaining genetic factors may include polymorphisms of immune-regulatory genes, such as those encoding IL-2, IL-21, CCR3, and SH2B3,46 and genes that determine epithelial polarity.47,48 There is also an association of celiac disease with other immune diseases including type 1 diabetes, thyroiditis, and Sjögren syndrome, as well as ataxia, autism, depression, some forms of epilepsy, IgA nephropathy, Down syndrome, and Turner syndrome.
Morphology. Biopsy specimens from the second portion of the duodenum or proximal jejunum, which are exposed to the highest concentrations of dietary gluten, are generally diagnostic in celiac disease. The histopathology is characterized by increased numbers of intraepithelial CD8+ T lymphocytes (intraepithelial lymphocytosis), crypt hyperplasia, and villous atrophy (Fig. 17-26). This loss of mucosal and brush-border surface area probably accounts for the malabsorption. In addition, increased rates of epithelial turnover, reflected in increased crypt mitotic activity, may limit the ability of absorptive enterocytes to fully differentiate and contribute to defects in terminal digestion and transepithelial transport. Other features of fully developed celiac disease include increased numbers of plasma cells, mast cells, and eosinophils, especially within the upper part of the lamina propria. With increased frequency of serologic screening and early detection of disease-associated antibodies, it is now appreciated that an increase in the number of intraepithelial lymphocytes, particularly within the villus, is a marker of less advanced celiac disease.49 However, intraepithelial lymphocytosis and villous atrophy are not specific for celiac disease and can be present in other diseases, including viral enteritis. The combination of histology and serology is most specific for diagnosis of celiac disease.
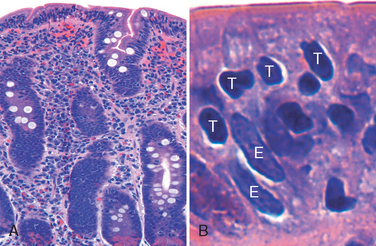
FIGURE 17-26 Celiac disease. A, Advanced cases of celiac disease show complete loss of villi, or total villous atrophy. Note the dense plasma cell infiltrates in the lamina propria. B, Infiltration of the surface epithelium by T lymphocytes, which can be recognized by their densely stained nuclei (labelled T). Compare to elongated, pale-staining epithelial nuclei (labelled E).
Clinical Features.
In adults, celiac disease presents most commonly between the ages of 30 and 60. However, many cases escape clinical attention for extended periods because of atypical presentations. Some patients may have silent celiac disease, defined as positive serology and villous atrophy without symptoms, or latent celiac disease, in which positive serology is not accompanied by villous atrophy. Symptomatic adult celiac disease is often associated with anemia, chronic diarrhea, bloating, or chronic fatigue. Although there is no gender preference, celiac disease is detected two- to threefold more commonly in women, perhaps because monthly menstrual bleeding increases the demand for iron and vitamins and accentuates the effects of impaired absorption.
Pediatric celiac disease, which affects males and females equally, may present with malabsorption or atypical symptoms affecting almost any organ.50 In those with classic symptoms, disease typically begins between ages of 6 and 24 months, after introduction of gluten to the diet, and includes irritability, abdominal distention, anorexia, chronic diarrhea, failure to thrive, weight loss, or muscle wasting.50 Children with nonclassic symptoms tend to present at older ages with complaints of abdominal pain, nausea, vomiting, bloating, or constipation. Common extra-intestinal complaints include arthritis or joint pain, seizure disorders, aphthous stomatitis, iron deficiency anemia, pubertal delay, and short stature.
A characteristic itchy, blistering skin lesion, dermatitis herpetiformis, can be present in as many as 10% of patients, and the incidence of lymphocytic gastritis and lymphocytic colitis is also increased. Unfortunately, the only treatment currently available for celiac disease is a gluten-free diet, but, despite the challenges of adhering to this diet, it does result in symptomatic improvement for most patients. A gluten-free diet may also reduce the risk of long-term complications including anemia, female infertility, osteoporosis, and cancer.
Noninvasive serologic tests are generally performed prior to biopsy.51 The most sensitive tests are the presence of IgA antibodies to tissue transglutaminase or IgA or IgG antibodies to deamidated gliadin. Anti-endomysial antibodies are highly specific but less sensitive than other antibodies. In cases with negative IgA tests, IgA deficiency, which is more common in celiac patients, should be ruled out. If IgA deficiency is present, titers of IgG antibodies to tissue transglutaminase and deamidated gliadin should be measured. The absence of HLA-DQ2 or HLA-DQ8 is useful for its high negative predictive value, but the presence of these alleles is not helpful in confirming the diagnosis.
Individuals with celiac disease have a higher than normal rate of malignancy. The most common celiac disease–associated cancer is enteropathy-associated T-cell lymphoma, an aggressive lymphoma of intraepithelial T lymphocytes. Small intestinal adenocarcinoma is also more frequent in individuals with celiac disease. Thus, when symptoms such as abdominal pain, diarrhea, and weight loss develop despite a strict gluten-free diet, cancer or refractory sprue, in which the response to a gluten-free diet is lost, must be considered. It is, however, important to remember that failure to adhere to a gluten-free diet is the most common cause of recurrent symptoms, and that most people with celiac disease do well with dietary restrictions and die of unrelated causes.
TROPICAL SPRUE
Tropical sprue is a malabsorption syndrome that occurs almost exclusively in people living in or visiting the tropics, including Puerto Rico, the Caribbean, northern South America, West Africa, India, and Southeast Asia. Inexplicably, it is uncommon in Jamaica. The disease is generally endemic, although epidemics have occurred.
Histologic changes of tropical sprue are similar to celiac disease, but total villous atrophy is uncommon, and tropical sprue tends to involve the distal small bowel. The latter probably explains the frequency of folate or vitamin B12 deficiencies with enlarged (megaloblastic) nuclei within epithelial cells that are reminiscent of those seen in pernicious anemia.
Malabsorption usually becomes apparent within days or a few weeks of an acute diarrheal enteric infection in visitors. Although no definite causal organism has been identified, overgrowth of aerobic enteric bacteria has been documented, and broad-spectrum antibiotics usually effect rapid recovery. Various infections, including Cyclospora or enterotoxigenic bacteria, have been suggested as etiologic factors.
AUTOIMMUNE ENTEROPATHY
Autoimmune enteropathy is an X-linked disorder characterized by severe persistent diarrhea and autoimmune disease that occurs most often in young children. A particularly severe familial form, termed IPEX, an acronym denoting immune dysregulation, polyendocrinopathy, enteropathy, and X-linkage, is due to a germline mutation in the FOXP3 gene, which is located on the X chromosome.52 FOXP3 is a transcription factor expressed in CD4+ regulatory T cells,53 and individuals with IPEX and FOXP3 mutations have defective T-regulatory function. Other defects in regulatory T cell function have also been linked to less severe forms of autoimmune enteropathy. Autoantibodies to enterocytes and goblet cells are common, and some patients may have antibodies to parietal or islet cells. Within the small intestine intraepithelial lymphocytes may be increased, but not to the extent seen in celiac disease, and neutrophils are often present. Therapy includes immunosuppressive drugs such as cyclosporine and, in rare cases, bone marrow transplantation.54
LACTASE (DISACCHARIDASE) DEFICIENCY
The disaccharidases, including lactase, are located in the apical brush-border membrane of the villus absorptive epithelial cells. Because the defect is biochemical, biopsy histology is generally unremarkable. Lactase deficiency is of two types:
ABETALIPOPROTEINEMIA
Abetalipoproteinemia is a rare autosomal recessive disease characterized by an inability to secrete triglyceride-rich lipoproteins. It is caused by a mutation in the microsomal triglyceride transfer protein (MTP) that catalyzes transport of triglycerides, cholesterol esters, and phospholipids. MTP-deficient enterocytes are unable to export lipoproteins and free fatty acids. As a result, monoglycerides cannot be assembled into chylomicrons and triglycerides accumulate within the epithelial cells. The malabsorption of abetalipoproteinemia is therefore a failure of transepithelial transport. Lipid vacuolization of small intestinal epithelial cells is evident under the light microscope and can be highlighted by special stains, such as oil red-O, particularly after a fatty meal.
Abetalipoproteinemia presents in infancy and the clinical picture is dominated by failure to thrive, diarrhea, and steatorrhea. Patients also have a complete absence of all plasma lipoproteins containing apolipoprotein B, although the gene that encodes apolipoprotein B itself is not affected. Failure to absorb essential fatty acids leads to deficiencies of fat-soluble vitamins as well as lipid membrane defects that can be recognized by the presence of acanthocytic red cells (burr cells) in peripheral blood smears.