Chapter 9 Environmental and Nutritional Diseases
The term “environment” encompasses the outdoor, indoor, and occupational environments shared by small and large populations, and our own personal environment. In each of these environments, the air we collectively breathe, the food and water we consume, and exposure to toxic agents are major determinants of our health. Our personal environment is greatly influenced by tobacco use, alcohol ingestion, therapeutic and nontherapeutic drug consumption, and diet. Factors in the personal environment may have a larger effect on human health than the ambient environment. The term environmental diseases refers to conditions caused by exposure to chemical or physical agents in the ambient, workplace, and personal environment, including diseases of nutritional origin. Environmental diseases mostly come to the public’s attention after major disasters, such as the methyl mercury contamination of Minamata Bay in Japan in the 1960s, the exposure to dioxin in Seveso, Italy, in 1976, the leakage of methyl isocyanate gas in Bhopal, India, in 1984, the Chernobyl nuclear accident in 1986, and the contamination of Tokyo subways by the organophosphate pesticide sarin. Fortunately, these are unusual and infrequent occurrences, but environmental diseases caused by chronic exposure to relatively low levels of contaminants, occupational injuries, and nutritional deficiencies are widespread. The International Labor Organization has estimated that work-related injuries and illnesses kill approximately 2 million people per year globally (more deaths than are caused by road accidents and wars combined). A comprehensive report from the Disease Control Priorities Project (http://www.dcp2.org) estimated that there are 130 million undernourished children worldwide, and that malnutrition alone is responsible for 2.67 million deaths per year. Estimating the burden of disease in the general population caused by nonoccupational exposures to toxic agents is complicated by the diversity of agents and difficulties in determining the extent and duration of exposures. Whatever the precise numbers, environmental (including nutritional) diseases are major causes of disability and suffering, and constitute a heavy financial burden, particularly in developing countries. During the last few years new concerns have been raised about air and water quality, and the potential health effects of climate change.
In this chapter, we first consider two key issues in global health: the global burden of disease, and the emerging problem of the health effects of climate change. We then discuss the mechanisms of toxicity of chemical and physical agents, and address specific environmental disorders, including those of nutritional origin.
The Global Burden of Disease
Until about 1990 global health data were fragmented and lacked a uniform standard of measurement.1 Since then, a project entitled The Global Burden of Disease (GBD) has set the standard for reporting health information. The GBD approach is now applied to the measurement of the burden imposed by environmental disease, including those caused by communicable and nutritional diseases. In addition, a unit of measurement (“metric”) called DALY (disability-adjusted life year, a time based measure that adds the years of life lost to premature mortality with the years lived with illness and disability), has been used to assess both premature mortality and disease morbidity. DALY reporting provides a high degree of uniformity for health information gathered about acute and chronic diseases in different parts of the world and at multiple locations in a single country. The new methodology has revealed important trends in the worldwide morbidity and mortality of disease.
Health Effects of Climate Change
There is general agreement that the earth has been warming at an accelerating pace during the last 40 years, and that the rate of warming is more rapid than at any other period in perhaps 1000 years.5 Since 1960 the global average surface temperature increased by 0.6°C; the increase is not uniform, being greatest at latitudes between 40° N and 70° N.6 Glacier melting has accelerated, and in polar regions, snow cover and ice thickness have diminished. At the same time, the sea level has risen 1 to 2 mm/year as a result of thermal expansion.6 The importance of climate change was highlighted by the awarding of the 2007 Nobel Peace Prize to individuals and organizations concerned with the impact of these changes on human health.
The causes of global climate change are the subject of debate, but human activity is a major contributor, through increases of carbon dioxide (CO2), methane, and ozone (discussed later), the main agents of the greenhouse effect. These gases (along with water vapor) act like a blanket by absorbing energy radiated from the earth’s surface that would otherwise be lost into space. Recent increases in levels of greenhouse gases, particularly CO2 and ozone produced by the combustion of hydrocarbons in automobiles and energy plants, are strongly correlated with warming of the earth (Fig. 9-2). The present concentration of atmospheric CO2, estimated to be 370 ppm (highest in about 1 million years), is expected to increase to 500 to 1200 ppm at the end of this century. Also contributing to the increase in atmospheric CO2 is large-scale deforestation (present estimates are that the Amazon forest will lose 50% of the original area by 2050), which decreases carbon sequestration by trees. Beyond certain levels of warming of the land and seas, it is predicted that positive-feedback loops will amplify the process further. Examples include increases in heat absorption due to the loss of reflective snow and ice; increases in water vapor in the atmosphere due to greater evaporation from bodies of water and transpiration from trees; large releases of stored CO2 and methane from thawing arctic tundra; and decreased sequestration of CO2 in the oceans, due to diminished growth of diatoms, which serve as an important CO2 sink. Depending on the model used, these changes are predicted to cause the global temperature to rise 2° to 5°C by the year 2100 (see Fig. 9-2).

FIGURE 9-2 Sources and consequences of increased greenhouse gases. A, Predicted temperature increases during the twenty-first century. Different computer models plot anticipated rises in temperature of 2° to 5°C by the year 2100. B, Release of carbon dioxide (CO2) from combustion sources in China, 1970 to 2005. China has now surpassed the United States as the world’s largest producer of CO2. C, Regions of the United States in which ozone levels are above existing accepted standards (80 ppb during an 8-hour period). These areas include about 500 counties located predominantly in the East Coast corridor, the Los Angeles basin, and areas with large coal-burning plants.
The future impact of global warming on health will depend on the extent and rapidity of climate change, the severity of the ensuing consequences, and humankind’s ability to adapt to or otherwise mitigate the damaging effects. Even in the best-case scenario, however, it is expected that climate change will seriously impact human health by increasing the incidence of several diseases.7
Despite recognition of these dangers, climate change is just one of multiple factors that contribute to the incidence of a disease at a particular geographic location, making it difficult to establish precise risk estimates for effects which are specifically caused by global warming.8
Both developed and developing countries will suffer the consequences of climate change, but the burden will be heaviest in developing nations. Wealthy countries are the main producers of the emissions that cause global warming, but rapidly developing countries such as China and India are using increasingly large amounts of energy to sustain their growth. The urgent challenge ahead is to develop new methods of energy production that do not harm the environment and do not contribute to global warming.
Toxicity of Chemical and Physical Agents
Toxicology is defined as the science of poisons. It studies the distribution, effects, and mechanisms of action of toxic agents. More broadly, it also includes the study of the effects of physical agents such as radiation and heat. Approximately 4 billion pounds of toxic chemicals, including 72 million pounds of recognized carcinogens, are released per year in the United States. Of about 100,000 chemicals in commercial use in the United States, only a very small proportion has been tested experimentally for health effects. Several agencies in the United States set permissible levels of exposure to known environmental hazards (e.g., the maximum level of carbon monoxide in air that is noninjurious or the tolerable levels of radiation that are harmless or “safe”). But factors such as the complex interaction between various pollutants, and the age, genetic predisposition, and the different tissue sensitivities of exposed persons, create wide variations in individual sensitivity to toxic agents, limiting the value of establishing rigid “safe levels” for entire populations. Nevertheless, such levels are useful for comparative studies of the effects of harmful agents between specific populations, and for estimating risk of disease in heavily exposed individuals.
We now consider some basic principles relevant to the effects of toxic chemicals and drugs.
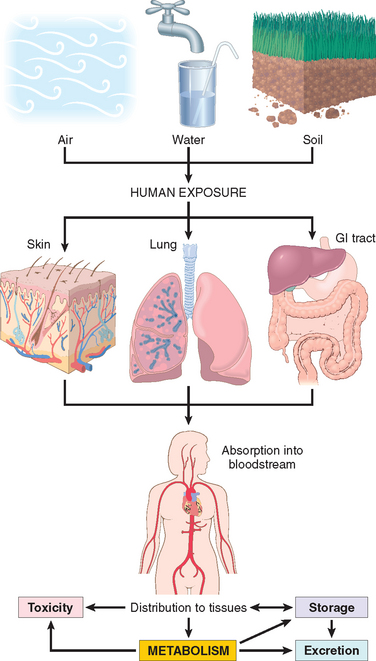
FIGURE 9-3 Human exposure to pollutants. Pollutants contained in air, water, and soil are absorbed through the lungs, gastrointestinal tract, and skin. In the body they may act at the site of absorption but are generally transported through the bloodstream to various organs where they may be stored or metabolized. Metabolism of xenobiotics may result in the formation of water-soluble compounds that are excreted, or in activation of the agent, creating a toxic metabolite.
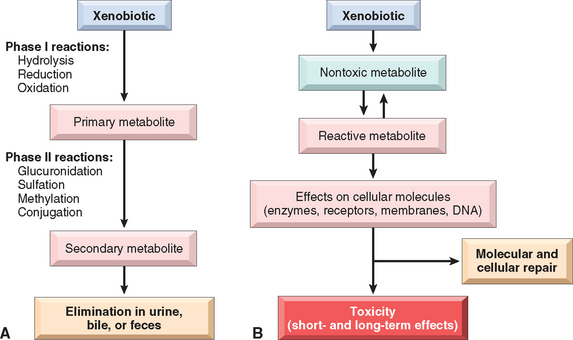
FIGURE 9-4 Xenobiotic metabolism. A, Xenobiotics can be metabolized to nontoxic metabolites and eliminated from the body (detoxification). B, Xenobiotic metabolism may also result in the formation of a reactive metabolite that is toxic to cellular components. If repair is not effective, short- and long-term effects develop.
(Based on Hodgson E: A Textbook of Modern Toxicology, 3rd ed. Hoboken, NJ, Wiley, 2004.)
This brief overview of the general mechanisms of toxicity provides the background for the discussion of environmental diseases presented in this chapter.
Environmental Pollution
AIR POLLUTION
Precious as air is—especially to those deprived of it—it is often loaded with many potential causes of disease. Airborne microorganisms contaminating food and water have long been major causes of morbidity and mortality, especially in developing countries. More widespread are the chemical and particulate pollutants found in the air, especially in industrialized nations. Here, we consider these hazards in outdoor and indoor air.
Outdoor Air Pollution
The ambient air in industrialized nations is contaminated with an unsavory mixture of gaseous and particulate pollutants, more heavily in cities and in proximity to heavy industry. In the United States the Environmental Protection Agency monitors and sets allowable upper limits for six pollutants: sulfur dioxide, carbon monoxide, ozone, nitrogen dioxide, lead, and particulate matter. Collectively, these agents produce the well-known smog (smoke and fog) that sometimes stifles large cities such as Beijing, Los Angeles, Houston, Cairo, New Delhi, Mexico City, and São Paulo. It may seem that air pollution is a modern phenomenon. This is not so, since John Evelyn wrote in 1661 that inhabitants of London suffered from “Catharrs, Phthisicks and Consumptions” (bronchitis, pneumonia, and tuberculosis) and breathed “nothing but an impure and thick mist, accompanied by a fuliginous and filthy vapour, which renders them obnoxious to a thousand inconveniences, corrupting the lungs, and disordering the entire habit of their bodies.” The first environmental control law, proclaimed by Edward I in 1306, was straightforward in its simplicity: “whoever should be found guilty of burning coal shall suffer the loss of his head.” Thus, what has changed in modern times is the nature and sources of air pollutants, and the types of regulations that control their emission.
Although the lungs bear the brunt of the adverse consequences, air pollutants can affect many organ systems (see, for instance, the discussion of lead poisoning and carbon monoxide effects in this chapter). Except for some comments on smoking, pollutant-caused lung diseases are discussed in Chapter 15. Major health effects of outdoor pollutants are described in Table 9-1. Here we discuss ozone, sulfur dioxide, particulates, and carbon monoxide.
TABLE 9-1 Health Effects of Outdoor Air Pollutants
| Pollutant | Populations at Risk | Effects |
|---|---|---|
| Ozone | Healthy adults and children | Decreased lung function |
| Increased airway reactivity | ||
| Lung inflammation | ||
| Athletes, outdoor workers | Decreased exercise capacity | |
| Asthmatics | Increased hospitalizations | |
| Nitrogen dioxide | Healthy adults | Increased airway reactivity |
| Asthmatics | Decreased lung function | |
| Children | Increased respiratory infections | |
| Sulfur dioxide | Healthy adults | Increased respiratory symptoms |
| Individuals with chronic lung disease | Increased mortality | |
| Asthmatics | Increased hospitalization | |
| Decreased lung function | ||
| Acid aerosols | Healthy adults | Altered mucociliary clearance |
| Children | Increased respiratory infections | |
| Asthmatics | Decreased lung function | |
| Increased hospitalizations | ||
| Particulates | Children | Increased respiratory infections |
| Individuals with chronic lung or heart disease | Decreased lung function | |
| Asthmatics | Excess mortality | |
| Increased attacks |
Data from Bascom R, et al.: Health effects of outdoor air pollution. Am J Respir Crit Care Med 153:477, 1996.
Ozone. The interaction of ultraviolet (UV) radiation and oxygen (O2) in the stratosphere leads to the formation of ozone (O3), which accumulates in the so-called ozone layer 10 to 30 miles above the earth’s surface. This layer protects life on earth by absorbing the most dangerous UV radiation emitted by the sun. During the last 30 years, the stratospheric ozone layer decreased in both thickness and extent due to the widespread use of aerosols, which drift up into the upper atmosphere and participate in chemical reactions that destroy ozone. The resulting depletion has been most profound over polar regions, particularly Antarctica, during the winter months. Recognition of the problem led to the ban of chlorofluorocarbons as aerosol propellants and their replacement by hydrofluoroalkanes, resulting in a decrease in the extent of stratospheric ozone “holes.”
In contrast to the “good” ozone in the stratosphere, ozone that accumulates in the lower atmosphere (ground-level ozone) is one of the most pernicious air pollutants (see Fig. 9-2). Ground-level ozone is a gas formed by the reaction of nitrogen oxides and volatile organic compounds in the presence of sunlight. These chemicals are released by industrial emissions and motor vehicle exhaust. Ozone toxicity is in large part mediated by the production of free radicals, which injure epithelial cells along the respiratory tract and type I alveolar cells, and cause the release of inflammatory mediators. Healthy individuals exposed to ozone experience upper respiratory tract inflammation and mild symptoms (decreased lung function and chest discomfort), but exposure is much more dangerous for people with asthma or emphysema. Ozone-induced asthma is associated with airway hyper-reactivity and neutrophilia.11
Even low levels of ozone may be detrimental to the lung function of normal individuals when combined with other air pollutants. Unfortunately, air pollutants often combine to create a veritable “witches’ brew” of ozone and other agents such as sulfur dioxide and particulates. Sulfur dioxide is produced by power plants burning coal and oil, from copper smelting, and as a byproduct of paper mills. Released into the air, it may be converted into sulfuric acid and sulfuric trioxide, which cause a burning sensation in the nose and throat, difficulty in breathing, and asthma attacks in susceptible individuals.
Particulate matter (known as “soot”) is emitted by coal- and oil-fired power plants, by industrial processes burning these fuels, and by diesel exhaust. Exposure to particulates was the main cause of morbidity and mortality in the air pollution episodes that occurred in London in 1952 and 1962. Although the particles have not been well characterized chemically or physically, fine or ultrafine particles that are less than 10 μm in diameter are the most harmful. They are readily inhaled into the alveoli, where they are phagocytosed by macrophages and neutrophils, which release inflammatory mediators such as macrophage inflammatory protein 1α and endothelin. Acute exposure to diesel exhaust that contains fine particles may cause irritation to the eyes, throat, and lungs, induce asthma attacks,12 and promote myocardial ischemia.13 In contrast, exposure to particles that are greater than 10 μm in diameter is of lesser consequence, because these particles are generally removed in the nose, or trapped by the mucociliary epithelium of the airways.
Carbon monoxide (CO). CO is a nonirritating, colorless, tasteless, odorless gas produced by the incomplete oxidation of carbonaceous materials. Its sources include automotive engines, industrial processes using fossil fuels, wood and charcoal burning with an inadequate supply of oxygen, and cigarette smoke. The low levels often found in ambient air may contribute to impaired respiratory function, but of themselves they are not life-threatening. However, chronic poisoning can occur in individuals working in confined environments with high exposure to fumes, such as tunnels, underground garages, and in highway toll workers. CO is included here as an air pollutant, but it is also an important cause of accidental and suicidal death. In a small, closed garage, the average car exhaust can induce lethal coma within 5 minutes. CO is a systemic asphyxiant that kills by inducing central nervous system (CNS) depression, which appears so insidiously that victims are often unaware of their plight and fail to help themselves. Hemoglobin has 200-fold greater affinity for CO than for oxygen, and the resultant carboxyhemoglobin does not carry oxygen. Systemic hypoxia develops when the hemoglobin is 20% to 30% saturated with CO; unconsciousness and death are likely with 60% to 70% saturation.
Morphology. Chronic poisoning by CO develops because carboxyhemoglobin, once formed, is remarkably stable. Even with low-level, but persistent, exposure to CO, carboxyhemoglobin may rise to life-threatening levels in the blood. The slowly developing hypoxia can insidiously evoke widespread ischemic changes in the central nervous system; these are particularly marked in the basal ganglia and lenticular nuclei. With cessation of exposure to CO, the patient usually recovers, but often there are permanent neurologic sequelae such as impairment of memory, vision, hearing, and speech. The diagnosis is made by measuring carboxyhemoglobin levels in the blood.
Acute poisoning by CO is generally a consequence of accidental exposure or suicide attempt. In light-skinned individuals, acute poisoning is marked by a characteristic generalized cherry-red color of the skin and mucous membranes, which result from high levels of carboxyhemoglobin. If death occurs rapidly morphologic changes may not be present; with longer survival the brain may be slightly edematous, with punctate hemorrhages and hypoxia-induced neuronal changes. The morphologic changes are not specific and stem from systemic hypoxia.
Indoor Air Pollution
As we increasingly “button up” our homes to exclude the environment, the potential for pollution of the indoor air increases. The commonest pollutant is tobacco smoke (discussed later), but additional offenders are CO, nitrogen dioxide (both already mentioned as outdoor pollutants), and asbestos (discussed in Chapter 15). Volatile substances containing polycyclic aromatic hydrocarbons generated by cooking oils and coal burning are important indoor pollutants in some regions of China. Only a few comments about other agents will be made here.
Wood smoke, containing various oxides of nitrogen and carbon particulates, may not only be an irritant but also predisposes to lung infections and may contain the far more dangerous carcinogenic polycyclic hydrocarbons. Bioaerosols range from microbiologic agents capable of causing infectious diseases such as Legionnaires’ disease, viral pneumonia, and the common cold, to less threatening but nonetheless distressing allergens derived from pet dander, dust mites, and fungi and molds responsible for rhinitis, eye irritation, and asthma. Radon, a radioactive gas derived from uranium widely present in soil and in homes, can cause lung cancer in uranium miners. However, it does not seem that low-level chronic exposures in the home increase lung cancer risk, at least for nonsmokers. Exposure to formaldehyde, used in the manufacture of building materials (cabinetry, furniture, adhesives, etc.) has become a common health problem in refugees from environmental disasters living in poorly ventilated trailers. Many of these cases occurred in trailers occupied by families displaced from their homes after Hurricane Katrina, which hit the southeastern United States in 2005. At concentrations of 0.1 ppm or higher, it causes breathing difficulties and a burning sensation in the eyes and throat, and can trigger asthma attacks. Formaldehyde is classified as a carcinogen for humans and animals. Finally, the so-called sick building syndrome remains an elusive problem, since it may be a consequence of exposure to one or more of the indoor pollutants already mentioned or be caused by poor ventilation.
METALS AS ENVIRONMENTAL POLLUTANTS
Lead, mercury, arsenic, and cadmium are the heavy metals most commonly associated with harmful effects in humans.
Lead
Lead exposure occurs through contaminated air and food and water. For most of the twentieth century the major sources of lead in the environment were lead-containing house paints and gasoline. Although limits have been set for the amounts of lead contained in residential paints, and leaded gasoline has practically disappeared in the United States, lead contamination remains an important health hazard, particularly for children. The large-scale recall of toys containing lead in 2007 alerted the general public to the dangers of lead exposures. There are many sources of lead in the environment, such as from mining, foundries, batteries, and spray painting, which constitute occupational hazards. However, flaking lead paint in older houses and soil contamination pose major hazards to youngsters, and ingestion of up to 200 mg/day can occur. During the last 30 years the median blood level of lead in preschool children in the United States decreased from 15 μg/dL to the present level of less than 2 μg/dL. However, lead blood levels in children living in older homes containing lead-based paint or lead-contaminated dust, often exceed the maximal allowed level of 10 μg/dL. Subclinical lead poisoning may occur in children exposed to levels of lead below 10 μg/dL, causing low intellectual capacity, behavioral problems such as hyperactivity, and poor organizational skills.14,15 Lead poisoning, although less common in adults, occurs mainly as an occupational hazard in those involved in the manufacturing of batteries, pigments, car radiators, and tin cans. The main clinical features of lead poisoning in children and adults are shown in Figures 9-5 and 9-6.
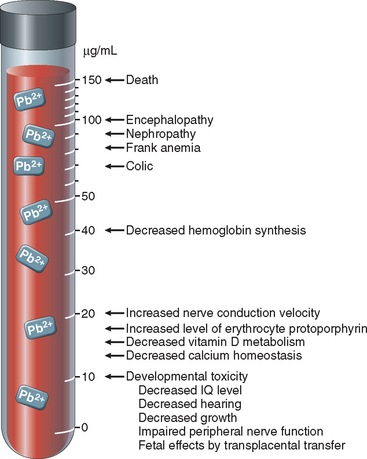
FIGURE 9-5 Effects of lead poisoning in children related to blood levels.
(Modified from Bellinger DC, Bellinger AM: Childhood lead poisoning: the tortuous path from science to policy. J Clin Invest 116:853, 2006.)
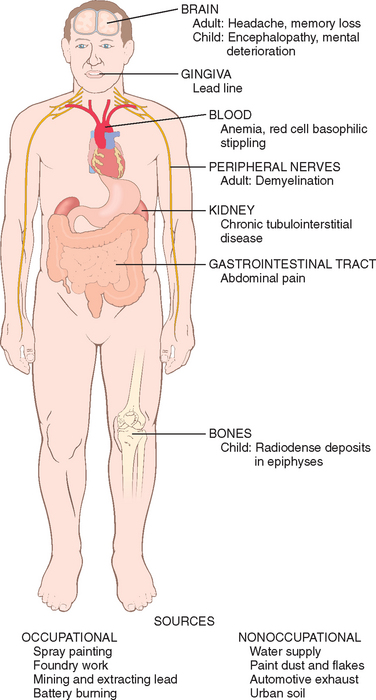
Most of the absorbed lead (80% to 85%) is incorporated into bone and developing teeth, where it competes with calcium; its half-life in bone is 20 to 30 years. High levels of lead cause disturbances in the CNS in adults and children, but peripheral neuropathies predominate in adults. Children absorb more than 50% of ingested lead (as compared with ≤15% in adults); the higher intestinal absorption and the more permeable blood-brain barrier of children create a high susceptibility to brain damage. The neurotoxic effects of lead are attributed to the inhibition of neurotransmitters caused by the disruption of calcium homeostasis. Other effects of lead exposure are listed below.
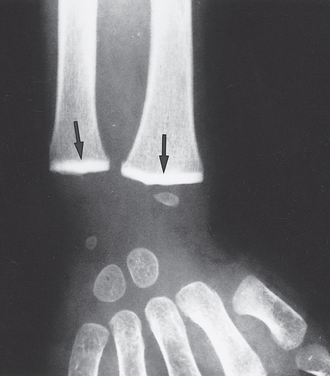
FIGURE 9-7 Lead poisoning. Impaired remodeling of calcified cartilage in the epiphyses (arrows) of the wrist has caused a marked increase in their radiodensity, so that they are as radiopaque as the cortical bone.
(Courtesy of Dr. G.W. Dietz, Department of Radiology, University of Texas Southwestern Medical School, Dallas, TX.)
The diagnosis of lead poisoning requires constant awareness of its prevalence. In children it may be suspected on the basis of neurologic and behavioral changes, or by unexplained anemia with basophilic stippling in red cells. Definitive diagnosis requires the detection of elevated blood levels of lead and free (or zinc-bound) red cell protoporphyrin.
Morphology. The major anatomic targets of lead toxicity are the bone marrow and blood, nervous system, gastrointestinal tract, and kidneys (see Fig. 9-6).
Blood and marrow changes occur fairly early and are characteristic. The inhibition of ferrochelatase by lead results in the appearance of scattered ringed sideroblasts, red cell precursors with iron-laden mitochondria that are detected with a Prussian blue stain. In the peripheral blood the defect in hemoglobin synthesis appears as a microcytic, hypochromic anemia that is often accompanied by mild hemolysis. Even more distinctive is a punctate basophilic stippling of the red cells.
Brain damage is prone to occur in children. It can be very subtle, producing mild dysfunction, or it can be massive and lethal. In young children, sensory, motor, intellectual, and psychologic impairments have been described, including reduced IQ, learning disabilities, retarded psychomotor development, blindness, and, in more severe cases, psychoses, seizures, and coma (see Fig. 9-5). Lead toxicity in the mother may impair brain development in the prenatal infant. The anatomic changes underlying the more subtle functional deficits are ill-defined, but there is concern that some of the defects may be permanent. At the more severe end of the spectrum are marked brain edema, demyelination of the cerebral and cerebellar white matter, and necrosis of cortical neurons accompanied by diffuse astrocytic proliferation. In adults the CNS is less often affected, but frequently a peripheral demyelinating neuropathy appears, typically involving the motor nerves of the most commonly used muscles. Thus, the extensor muscles of the wrist and fingers are often the first to be affected (causing wristdrop), followed by paralysis of the peroneal muscles (causing footdrop).
The gastrointestinal tract is also a major source of clinical manifestations. Lead “colic” is characterized by extremely severe, poorly localized abdominal pain.
Kidneys may develop proximal tubular damage with intranuclear lead inclusions. Chronic renal damage leads eventually to interstitial fibrosis and possibly renal failure. Decreases in uric acid excretion can lead to gout (“saturnine gout”).
Mercury
Mercury has had many uses throughout history such as a pigment in cave paintings, a cosmetic, a remedy for syphilis, and a component of diuretics. Alchemists tried (without much success) to produce gold from mercury. Poisoning from inhalation of mercury vapors has long been recognized and is associated with tremor, gingivitis, and bizarre behavior, such as that displayed by the Mad Hatter in Alice in Wonderland. There are three forms of mercury: metallic mercury (also referred to as elemental mercury), inorganic mercury compounds (mostly mercuric chloride), and organic mercury (mostly methyl mercury). Today, the main sources of exposure to mercury are contaminated fish (methyl mercury) and mercury vapors released from metallic mercury in dental amalgams, a possible occupational hazard for dental workers. In some areas of the world, mercury used in gold mining has contaminated rivers and streams.
Inorganic mercury from the natural degassing of the earth’s crust or from industrial contamination is converted to organic compounds such as methyl mercury by bacteria. Methyl mercury enters the food chain, and in carnivorous fish such as swordfish, shark, and bluefish, mercury may be concentrated to levels a million-fold higher than in the surrounding water. Disasters caused by the consumption of fish contaminated by the release of methyl mercury from industrial sources in Minamata Bay and the Agano River in Japan caused widespread mortality and morbidity. Acute exposure through consumption of bread made from grain treated with a methyl mercury–based fungicide in Iraq in 1971 resulted in hundreds of deaths and thousands of hospitalizations. The medical disorders associated with the Minamata episode became known as “Minamata disease” and include cerebral palsy, deafness, blindness, mental retardation, and major CNS defects in children exposed in utero. For unclear reasons, the developing brain is extremely sensitive to methyl mercury. The lipid solubility of methyl mercury and metallic mercury facilitate their accumulation in the brain, disturbing neuromotor, cognitive, and behavioral functions.16 Mercury binds with high affinity to thiol groups, a property that contributes to its toxicity. Intracellular glutathione, acting as thiol donor, is the main protective mechanism against mercury-induced CNS and kidney damage.
Mercury continues to be released into the environment by power plants and other industrial sources, and there are serious concerns about the effects of chronic low-level exposure to methyl mercury in the food supply. To protect against potential fetal brain damage, the Centers for Disease Control and Prevention has recommended that pregnant women reduce their consumption of fish known to contain mercury to a minimum. There has been much publicity about a possible relationship between thimerosal (a compound that contains ethyl mercury, used until recently as a preservative in some vaccines) and the development of autism, but multiple studies have failed to find evidence of a causal relationship.17
Arsenic
Arsenic was the poison of choice in Renaissance Italy, with members of the Borgia and Medici families being highly skilled practitioners of the art. Because of its favored use as a murder weapon among royal families, arsenic has been called “the poison of kings and the king of poisons.”18 Deliberate poisoning by arsenic is exceedingly rare today, but exposure to arsenic is an important health problem in many areas of the world. Arsenic is found naturally in soils and water and is used in products such as wood preservers, as well as herbicides and other agricultural products. It may be released into the environment from mines and smelting industries. Arsenic is present in Chinese and Indian herbal medicine, and arsenic trioxide is used in the treatment of relapsing acute promyelocytic leukemia. Large concentrations of inorganic arsenic are present in ground water used for drinking in countries such as Bangladesh, Chile, and China. Between 35 and 77 million people in Bangladesh drink water contaminated by arsenic, constituting the highest environmental cancer risk ever found.
The most toxic forms of arsenic are the trivalent compounds arsenic trioxide, sodium arsenite, and arsenic trichloride.19 If ingested in large quantities, arsenic causes acute toxic effects consisting of severe disturbances of the gastrointestinal, cardiovascular, and central nervous systems that are often fatal. These effects may be attributed to interference with mitochondrial oxidative phosphorylation, since trivalent arsenic can replace the phosphates in adenosine triphosphate. Neurologic effects usually occur 2 to 8 weeks after exposure and consist of a sensorimotor neuropathy that causes paresthesias, numbness, and pain. The most serious consequence of chronic exposure is the increased risk for the development of cancers in almost all tissues, but particularly in the lungs and skin. Chronic exposure to arsenic causes skin changes consisting of hyperpigmentation and hyperkeratosis, which may be followed by the development of basal and squamous cell carcinomas. Arsenic-induced skin tumors differ from those induced by sunlight; they are often multiple and usually appear on the palms and soles. The mechanisms of arsenic carcinogenesis in skin and lung have not been elucidated but may involve defects in nucleotide excision repair mechanisms that protect against DNA damage.18 Recent studies suggest that chronic exposure to arsenic in drinking water can also cause non-malignant respiratory disease.20
Cadmium
In contrast to the other metals discussed in this section, cadmium toxicity is a relatively modern problem. It is an occupational and environmental pollutant generated by mining, electroplating, and production of nickel-cadmium batteries, which are usually disposed of as household waste. Cadmium can contaminate the soil and plants directly or through fertilizers and irrigation water. Food is the most important source of cadmium exposure for the general population. The toxic effects of excess cadmium consist of obstructive lung disease caused by necrosis of alveolar macrophages, and kidney damage, initially consisting of tubular damage that may progress to end-stage renal disease. Cadmium exposure can also cause skeletal abnormalities associated with calcium loss. Cadmium-containing water used to irrigate rice fields in Japan caused a disease in postmenopausal women known as “Itai-Itai” (ouch-ouch), a combination of osteoporosis and osteomalacia associated with renal disease. Cadmium exposure is also associated with elevated risk of lung cancer, which has been demonstrated in workers exposed occupationally and in populations living near zinc smelters.21 Cadmium is not directly genotoxic and most likely produces DNA damage through the generation of reactive oxygen species (see Chapter 1). A recent survey showed that 5% of the US population age 20 years and older have urinary cadmium levels that may produce subtle kidney injury and calcium loss.
Occupational Health Risks: Industrial and Agricultural Exposures
More than 10 million injuries and about 100,000 deaths occur yearly in the United States as a consequence of work-related accidents and illnesses. Work-related accidents are the biggest problem in developing countries, while work-related diseases are more frequent in industrialized countries. The fraction of global disease attributed to occupational exposures includes 13% of all cases of chronic obstructive pulmonary disease, 9% of lung cancers, and 2 % of leukemias. Industrial exposures to toxic agents are as varied as the industries themselves. They range from mere irritation of the respiratory mucosa by formaldehyde or ammonia fumes; to lung cancer induced by exposure to asbestos, arsenic, or uranium mining; to leukemia caused by chronic exposure to benzene. Human diseases associated with occupational exposures are listed in Table 9-2. Here we provide a few examples of important agents that contribute to occupational diseases. Toxicity caused by metals has already been discussed in this chapter.
TABLE 9-2 Human Diseases Associated with Occupational Exposures
| Organ/System | Effect | Toxicant |
|---|---|---|
| Cardiovascular system | Heart disease | Carbon monoxide, lead, solvents, cobalt, cadmium |
| Respiratory system | Nasal cancer | Isopropyl alcohol, wood dust |
| Lung cancer | Radon, asbestos, silica, bis(chloromethyl)ether, nickel, arsenic, chromium, mustard gas, uranium | |
| Chronic obstructive lung disease | Grain dust, coal dust, cadmium | |
| Hypersensitivity | Beryllium, isocyanates | |
| Irritation | Ammonia, sulfur oxides, formaldehyde | |
| Fibrosis | Silica, asbestos, cobalt | |
| Nervous system | Peripheral neuropathies | Solvents, acrylamide, methyl chloride, mercury, lead, arsenic, DDT |
| Ataxic gait | ||
| Central nervous system depression | Chlordane, toluene, acrylamide, mercury | |
| Cataracts | Alcohols, ketones, aldehydes, solvents | |
| Ultraviolet radiation | ||
| Urinary system | Toxicity | Mercury, lead, glycol ethers, solvents |
| Bladder cancer | Naphthylamines, 4-aminobiphenyl, benzidine, rubber products | |
| Reproductive system | Male infertility | Lead, phthalate plasticizers, cadmium |
| Female infertility/stillbirths | Lead, mercury | |
| Teratogenesis | Mercury, polychlorinated biphenyls | |
| Hematopoietic system | Leukemia | Benzene |
| Skin | Folliculitis and acneiform dermatosis | Polychlorinated biphenyls, dioxins, herbicides |
| Cancer | Ultraviolet radiation | |
| Gastrointestinal tract | Liver angiosarcoma | Vinyl chloride |
Data from Leigh JP, et al.: Occupational injury and illness in the United States. Estimates of costs, morbidity, and mortality, Arch Intern Med 157:1557, 1997; Mitchell FL: Hazardous waste. In Rom WN (ed): Environmental and Occupational Medicine, 2nd ed. Boston, Little, Brown, 1992, p 1275; and Levi PE: Classes of toxic chemicals. In Hodgson E, Levi PE (eds): A Textbook of Modern Toxicology. Stamford, CT, Appleton & Lange, 1997, p 229.
Effects of Tobacco
Tobacco is the most common exogenous cause of human cancers, being responsible for 90% of lung cancers. The main culprit is cigarette smoking, but smokeless tobacco (snuff, chewing tobacco, etc.) is also harmful to health and an important cause of oral cancer. The use of tobacco products not only creates personal risks, but passive tobacco inhalation from the environment (“second-hand smoke”) can cause lung cancer in nonsmokers.22 Cigarette smoking causes, worldwide, more than 5 million deaths annually, mostly from cardiovascular disease, various types of cancers, and chronic respiratory problems, that result in a total of more than 35 million years of life lost. These figures are expected to rise to 8 million tobacco-related deaths by 2020, the major increase occurring in developing countries. It has been estimated that of people alive today, approximately 500 million will die from tobacco-related illnesses. In the United States alone, tobacco is responsible for over 400,000 deaths annually, one third of these attributable to lung cancer. Two thirds of smokers live in 10 countries, led by China, which accounts for nearly 30%, and India with about 10%, followed by Indonesia, Russia, the United States, Japan, Brazil, Bangladesh, Germany, and Turkey.
Smoking is the most preventable cause of human death. It reduces overall survival through dose-dependent effects. For instance, while 80% of a population of nonsmokers is alive at age 70, only about 50% of smokers survive to that age (Fig. 9-8). The prevalence of smoking has decreased in US teenagers, a hopeful trend. However, recent surveys estimate that 7%, 14%, and 22% of students in grades 8, 10, and 12, respectively, had used tobacco products during the month before the survey. Delaying the age at which smoking is initiated reduces the future risk of lung and other types of cancers, but, unfortunately, initiation seems to be occurring at younger ages. Cessation of smoking greatly reduces, within 5 years, the overall mortality and the risk of death from cardiovascular diseases. Lung cancer mortality decreases by 21% within 5 years, but the excess risk lasts for 30 years.22

FIGURE 9-8 The effects of smoking on survival. The study compared age-specific death rates for current cigarette smokers with that of individuals who never smoked regularly (British Doctors Study). Measured at age 75, the difference in survival between smokers and nonsmokers is 7.5 years.
(Modified from Stewart BW, Kleihues P (eds): World Cancer Report. Lyon, IARC Press, 2003.)
The number of potentially noxious chemicals in tobacco smoke is extraordinary. Tobacco contains between 2000 and 4000 substances, more than 60 of which have been identified as carcinogens. Table 9-3 provides only a partial list and includes various types of injuries produced by these agents. Nicotine, an alkaloid present in tobacco leaves, is not a direct cause of tobacco-related diseases, but is addictive. Without it, it would be easy for smokers to stop the habit. Nicotine binds to receptors in the brain, and through the release of catecholamines, is responsible for the acute effects of smoking, such as the increase in heart rate and blood pressure, and the elevation in cardiac contractility and output. The most common diseases caused by cigarette smoking involve the lung and include emphysema, chronic bronchitis, chronic obstructive pulmonary disease, and lung cancer, conditions that are discussed in Chapter 15. Cigarette smoking is also strongly associated with the development of atherosclerosis, myocardial infarcts, and cancers of the lip, mouth, pharynx, esophagus, pancreas, bladder, kidney, and cervix. Adverse effects of smoking in various organs systems are shown in Figure 9-9.
TABLE 9-3 Effects of Selected Tobacco Smoke Constituents
| Substance | Effect |
|---|---|
| Tar | Carcinogenesis |
| Polycyclic aromatic hydrocarbons | Carcinogenesis |
| Nicotine | Ganglionic stimulation and depression; tumor promotion |
| Phenol | Tumor promotion; mucosal irritation |
| Benzopyrene | Carcinogenesis |
| Carbon monoxide | Impaired oxygen transport and utilization |
| Formaldehyde | Toxicity to cilia; mucosal irritation |
| Oxides of nitrogen | Toxicity to cilia; mucosal irritation |
| Nitrosamine | Carcinogenesis |

Smoking and Lung Cancer.
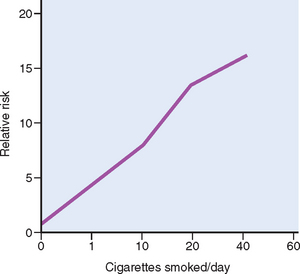
Agents in smoke have a direct irritant effect on the tracheobronchial mucosa, producing inflammation and increased mucus production (bronchitis). Cigarette smoke also causes the recruitment of leukocytes to the lung, with increased local elastase production and subsequent injury to lung tissue, leading to emphysema. Components of cigarette smoke, particularly polycyclic hydrocarbons and nitrosamines (Table 9-4), are potent carcinogens in animals and likely to be directly involved in the development of lung cancer in humans (see Chapter 15). CYPs (cytochrome P-450 phase I enzymes) and phase II enzymes increase the water solubility of the carcinogens, facilitating their excretion. However, some intermediates produced by CYPs are electrophilic and form DNA adducts. If such adducts persist, they can cause mutations in oncogenes and tumor suppressors such as K-Ras and p53,23 respectively. The risk of developing lung cancer is related to the intensity of exposure, frequently expressed in terms of “pack years” (e.g., one pack smoked daily for 20 years equals 20 pack years) or in cigarettes smoked per day (Fig. 9-10). Moreover, smoking multiplies the risk of other carcinogenic influences. Witness the ten-fold higher incidence of lung carcinomas in asbestos workers and uranium miners who smoke over those who do not smoke, and the interaction between tobacco consumption and alcohol in the development of oral cancers (mentioned below).
TABLE 9-4 Organ-Specific Carcinogens in Tobacco Smoke
| Organ | Carcinogen |
|---|---|
| Lung, larynx | Polycyclic aromatic hydrocarbons 4-(Methylnitrosoamino)-1-(3-pyridyl)-1-buta-none (NNK), polonium 210 |
| Esophagus | N′-Nitrosonornicotine (NNN) |
| Pancreas | NNK (?) |
| Bladder | 4-Aminobiphenyl, 2-naphthylamine |
| Oral cavity (smoking) | Polycyclic aromatic hydrocarbons, NNK, NNN |
| Oral cavity (snuff) | NNK, NNN, polonium 210 |
Data from Szczesny LB, Holbrook JH: Cigarette smoking. In Rom WH (ed): Environmental and Occupational Medicine, 2nd ed. Boston, Little, Brown, 1992, p 1211.
Smoking and Other Diseases.
In addition to lung cancers, tobacco contributes to the development of cancers of the oral cavity, esophagus, pancreas, and bladder. Smoke and smokeless tobacco interact with alcohol in the development of laryngeal cancer. The combination of these agents has a multiplicative effect on the risk of developing this tumor (Fig. 9-11).
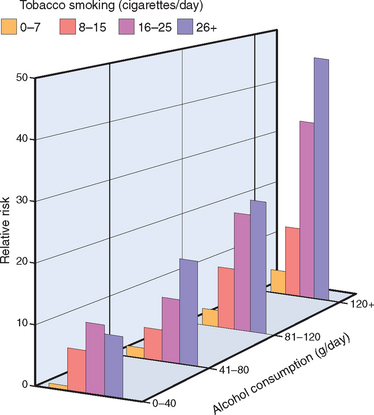
FIGURE 9-11 Multiplicative increase in the risk of laryngeal cancer from the interaction between cigarette smoking and alcohol consumption.
(Modified from Stewart BW, Kleihues P (eds): World Cancer Report. Lyon, IARC Press, 2003.)
Cigarette smoking is strongly linked to the development of atherosclerosis and its major complication, myocardial infarction. The causal mechanisms probably relate to several factors, including increased platelet aggregation, decreased myocardial oxygen supply (because of significant lung disease coupled with the hypoxia related to the CO content of cigarette smoke) accompanied by an increased oxygen demand, and a decreased threshold for ventricular fibrillation. Smoking has a multiplicative effect on the incidence of myocardial infarction when combined with hypertension and hypercholesterolemia.
Maternal smoking increases the risk of spontaneous abortions and preterm births and results in intrauterine growth retardation (Chapter 10). Birth weights of infants born to mothers who stopped smoking before pregnancy are, however, normal.
Exposure to environmental tobacco smoke (passive smoke inhalation) is also associated with some of the same detrimental effects that result from active smoking. It is estimated that the relative risk of lung cancer in nonsmokers exposed to environmental smoke is about 1.3 times higher than that of nonsmokers who are not exposed to smoke. In the United States, approximately 3000 lung cancer deaths in nonsmokers over the age of 35 years can be attributed each year to environmental tobacco smoke. Even more striking is the increased risk of coronary atherosclerosis and fatal myocardial infarction. Studies report that every year 30,000 to 60,000 cardiac deaths in the United States are associated with exposure to passive smoke. Passive smoke inhalation in nonsmokers can be estimated by measuring the blood levels of cotinine, a metabolite of nicotine. Median cotinine levels in nonsmokers have decreased by more than 60% during the last 10 years, but exposure to environmental tobacco smoke in the home remains a major public health concern, particularly for children who may develop respiratory illnesses and asthma. It is clear that the transient pleasure a puff may give comes with a heavy long-term price.
Effects of Alcohol
Ethanol consumption in moderate amounts is generally not injurious, but in excessive amounts alcohol causes serious physical and psychologic damage. In this section we describe the steps of alcohol metabolism and the major health consequences associated with alcohol abuse.
Despite all the attention given to illicit drugs such as cocaine and heroin, alcohol abuse is a more widespread hazard and claims many more lives. Fifty percent of adults in the Western world drink alcohol, and about 5% to 10% have chronic alcoholism. It is estimated that there are more than 10 million chronic alcoholics in the United States and that alcohol consumption is responsible for more than 100,000 deaths annually. More than 50% of these deaths result from accidents caused by drunken driving and alcohol-related homicides and suicides, and about 15,000 annual deaths are a consequence of cirrhosis of the liver. Worldwide, alcohol accounts for approximately 1.8 million deaths per year (3.2% of all deaths). After consumption, ethanol is absorbed unaltered in the stomach and small intestine. It is then distributed to all the tissues and fluids of the body in direct proportion to the blood level. Less than 10% is excreted unchanged in the urine, sweat, and breath. The amount exhaled is proportional to the blood level and forms the basis of the breath test used by law enforcement agencies. A concentration of 80 mg/dL in the blood constitutes the legal definition of drunk driving in the United States. For an average individual, this alcohol concentration may be reached after consumption of three standard drinks, contained in about 3 (12 ounce) bottles of beer, 15 ounces of wine, or 4–5 ounces of 80 proof distilled spirits. Drowsiness occurs at 200 mg/dL, stupor at 300 mg/dL, and coma, with possible respiratory arrest, at higher levels. The rate of metabolism affects the blood alcohol level. Chronic alcoholics can tolerate levels of up to 700 mg/dL, a situation that is partially explained by accelerated ethanol metabolism caused by a five- to ten-fold induction of liver CYPs discussed below. The effects of alcohol also vary by age, sex, and body fat.
Most of the alcohol in the blood is biotransformed to acetaldehyde in the liver by three enzyme systems consisting of alcohol dehydrogenase (ADH), the microsomal ethanol-oxidizing system (MEOS), and catalase (Fig. 9-12). The main enzyme system involved in alcohol metabolism is ADH, located in the cytosol of hepatocytes. At high blood alcohol levels, the microsomal ethanol-oxidizing system participates in its metabolism. Catalase, which uses hydrogen peroxide as substrate, is of minor importance, since it metabolizes no more than 5% of ethanol in the liver. Acetaldehyde produced by alcohol metabolism through ADH or MEOS is converted to acetate by acetaldehyde dehydrogenase (ALDH), which is then utilized in the mitochondrial respiratory chain.
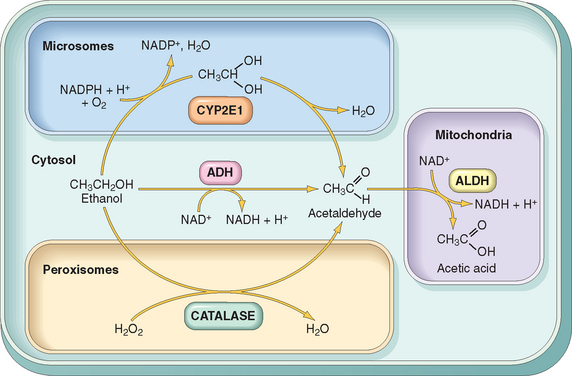
FIGURE 9-12 Metabolism of ethanol: oxidation of ethanol to acetaldehyde by three different routes, and the generation of acetic acid. Note that oxidation by ADH (alcohol dehydrogenase) takes place in the cytosol; the cytochrome P-450 system and its CYP2E1 isoform are located in the endoplasmic reticulum (microsomes), and catalase is located in peroxisomes. Oxidation of acetaldehyde by ALDH (aldehyde dehydrogenase) oc-curs in mitochondria. ADH oxidation is the most important route; catalase is involved in only 5% of ethanol metabolism. Oxidation through CYPs may also generate reactive oxygen species (not shown).
(From Parkinson A: Biotransformation of xenobiotics. In Klassen CD [ed]: Casarett and Doull’s Toxicology: The Basic Science of Poisons, 6th ed. New York, McGraw-Hill, 2001, p 133.)
The microsomal oxidation system involves CYPs, particularly CYP2E1 located in the smooth endoplasmic reticulum. Induction of CYPs by alcohol explains the increased susceptibility of alcoholics to other compounds metabolized by the same enzyme system, which include drugs, anesthetics, carcinogens, and industrial solvents. Note, however, that when alcohol is present in the blood at high concentrations, it competes with other CYP2E1 substrates and delays drug catabolism, potentiating the depressant effects of narcotic, sedative, and psychoactive drugs in the central nervous system. The oxidation of ethanol produces toxic agents and disrupts metabolic pathways. Here we mention only the most important of these changes.
The adverse effects of ethanol can be classified as acute or chronic.
Acute alcoholism exerts its effects mainly on the CNS, but it may induce hepatic and gastric changes that are reversible if alcohol consumption is discontinued. Even with moderate intake of alcohol, multiple fat droplets accumulate in the cytoplasm of hepatocytes (fatty change or hepatic steatosis). The gastric changes are acute gastritis and ulceration. In the CNS, alcohol is a depressant, first affecting subcortical structures (probably the high brain stem reticular formation) that modulate cerebral cortical activity. Consequently, there is stimulation and disordered cortical, motor, and intellectual behavior. At progressively higher blood levels, cortical neurons and then lower medullary centers are depressed, including those that regulate respiration. Respiratory arrest may follow.
Chronic alcoholism affects not only the liver and stomach, but virtually all other organs and tissues as well. Chronic alcoholics suffer significant morbidity and have a shortened life span, related principally to damage to the liver, gastrointestinal tract, CNS, cardiovascular system, and pancreas.
TABLE 9-9 Vitamins: Major Functions and Deficiency Syndromes
| Vitamin | Functions | Deficiency Syndromes |
|---|---|---|
| FAT-SOLUBLE | ||
| Vitamin A | A component of visual pigment | Night blindness, xerophthalmia, blindness |
| Maintenance of specialized epithelia | Squamous metaplasia | |
| Maintenance of resistance to infection | Vulnerability to infection, particularly measles | |
| Vitamin D | Facilitates intestinal absorption of calcium and phosphorus and mineralization of bone | Riskets in children |
| Osteomalacia in adults | ||
| Vitamin E | Major antioxidant; scavenges free radicals | Spinocerebellar degeneration |
| Vitamin K | Cofactor in hepatic carboxylation of procoagulants—factors II (prothrombin), VII, IX, and X; and protein C and protein S | Bleeding diathesis (Chapter 14) |
| WATER-SOLUBLE | ||
| Vitamin B1 (thiamine) | As pyrophosphate, is coenzyme in decarboxylation reactions | Dry and wet beriberi, Wernicke syndrome, Korsakoff syndrome (Chapter 28) |
| Vitamin B2 (riboflavin) | Converted to coenzymes flavin mononucleotide and flavin adenine dinucleotide, cofactors for many enzymes in intermediary metabolism | Ariboflavinosis, cheilosis, stomatitis, glossitis, dermatitis, corneal vascularization |
| Niacin | Incorporated into nicotinamide adenine dinucleotide (NAD) and NAD phosphate, involved in a variety of redox reactions | Pellagra—“three Ds”: dementia, dermatitis, diarrhea |
| Vitamin B6 (pyridoxine) | Derivatives serve as coenzymes in many intermediary reactions | Cheilosis, glossitis, dermatitis, peripheral neuropathy (Chapter 28) |
| Vitamin B12 | Required for normal folate metabolism and DNA synthesis | Megaloblastic pernicious anemia and degeneration of posterolateral spinal cord tracts (Chapter 14) |
| Maintenance of myelinization of spinal cord tracts | ||
| Vitamin C | Serves in many oxidation-reduction (redox) reactions and hydroxylation of collagen | Scurvy |
| Folate | Essential for transfer and use of one-carbon units in DNA synthesis | Megaloblastic anemia, neural tube defects (Chapter 14) |
| Pantothenic acid | Incorporated in coenzyme A | No nonexperimental syndrome recognized |
| Biotin | Cofactor in carboxylation reactions | No clearly defined clinical syndrome |
And now, a bit of good news: red wine contains resveratrol, a polyphenolic compound that increases life span in worms and flies, promotes longevity in mice, and protects mice against diet-induced obesity and insulin resistance. Resveratrol contributes to the protective effect against cardiovascular disease in moderate wine drinkers and possibly provides the clue to the “French paradox,” a wine- and food-loving population with a low incidence of obesity and cardiovascular disease. The effects of resveratrol on longevity have been attributed to its activation of protein deacetylases of the Sir2 (sirtuin) family of enzymes, which include histone deacetylases (Chapter 1). However, because resveratrol also interacts with various other proteins, ongoing studies seek to identify the precise mechanisms of its protective effects.26,27
Injury by Therapeutic Drugs and Drugs of Abuse
INJURY BY THERAPEUTIC DRUGS (ADVERSE DRUG REACTIONS)
Adverse drug reactions (ADRs) refer to untoward effects of drugs that are given in conventional therapeutic settings. These reactions are extremely common in the practice of medicine (Fig. 9-13) and affect almost 10% of patients admitted to a hospital. It is estimated that in about 10% of these patients, ADRs are fatal. Table 9-5 lists common pathologic findings in ADRs and the drugs most frequently involved. As can be seen in the table, many of the drugs that produce ADRs, such as antineoplastic agents, are highly potent, and the adverse reactions are expected risks of the treatment. In this section, we examine the adverse reactions to some commonly used drugs. We first discuss the adverse effects of hormonal replacement therapy (HRT), oral contraceptives (OCs), and anabolic steroids. This is followed by a discussion of the effects of the drugs acetaminophen and aspirin, because all of these are used very commonly.
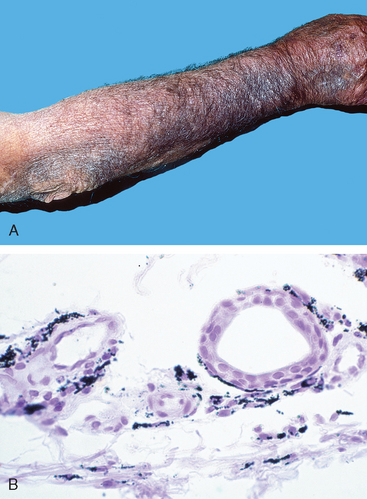
FIGURE 9-13 Adverse drug reaction. Skin pigmentation caused by minocycline, a long-acting tetracycline derivative. A, Diffuse blue-gray pigmentation of the forearm; B, Deposition of drug metabolite/iron/melanin pigment particles in the dermis.
(Courtesy of Dr. Zsolt Argenyi, Department of Pathology, University of Washington, Seattle, WA.)
TABLE 9-5 Some Common Adverse Drug Reactions and Their Agents
| Reaction | Major Offenders |
|---|---|
| BONE MARROW AND BLOOD CELLS* | |
| Granulocytopenia, aplastic anemia, pancytopenia | Antineoplastic agents, immunosuppressives, and chloramphenicol |
| Hemolytic anemia, thrombocytopenia | Penicillin, methyldopa, quinidine, heparin |
| CUTANEOUS | |
| Urticaria, macules, papules, vesicles, petechiae, exfoliative dermatitis, fixed drug eruptions, abnormal pigmentation | Antineoplastic agents, sulfonamides, hydantoins, some antibiotics, and many other agents |
| CARDIAC | |
| Arrhythmias | Theophylline, hydantoins, digoxin |
| Cardiomyopathy | Doxorubicin, daunorubicin |
| RENAL | |
| Glomerulonephritis | Penicillamine |
| Acute tubular necrosis | Aminoglycoside antibiotics, cyclosporin, amphotericin B |
| Tubulointerstitial disease with papillary necrosis | Phenacetin, salicylates |
| PULMONARY | |
| Asthma | Salicylates |
| Acute pneumonitis | Nitrofurantoin |
| Interstitial fibrosis | Busulfan, nitrofurantoin, bleomycin |
| HEPATIC | |
| Fatty change | Tetracycline |
| Diffuse hepatocellular damage | Halothane, isoniazid, acetominophen |
| Cholestasis | Chlorpromazine, estrogens, contraceptive agents |
| SYSTEMIC | |
| Anaphylaxis | Penicillin |
| Lupus erythematosus syndrome (drug-induced lupus) | Hydralazine, procainamide |
| CENTRAL NERVOUS SYSTEM | |
| Tinnitus and dizziness | Salicylates |
| Acute dystonic reactions and parkinsonian syndrome | Phenothiazine antipsychotics |
| Respiratory depression | Sedatives |
* Affected in almost half of all drug-related deaths.
Hormonal Replacement Therapy (HRT)
The most common type of HRT consists of the administration of estrogens together with progesterone. Because of the risk of uterine cancer, estrogen therapy alone is used only in hysterectomized women. Once prescribed primarily for distressing menopausal symptoms (e.g., hot flashes), HRT had been widely used in postmenopausal women to prevent or slow the progression of osteoporosis (Chapter 26) and to reduce the likelihood of myocardial infarction. However, the results of the Women’s Health Initiative published in 2002, stunned the scientific community by failing to find support for some of the presumed beneficial effects of the therapy. This large epidemiologic study involved approximately 17,000 women who were taking a combination of estrogen (equine estrogen) and progesterone (medroxyprogesterone acetate). Although the study found that HRT caused a reduction in the number of fractures, it also reported that after 5 years of treatment, HRT increased the risk of breast cancer (as discussed in Chapter 23) and thromboembolism, and had no effect on preventing cardiovascular disease. The wide dissemination of these findings led to a drastic decrease in the use of HRT, from 16 million prescriptions in 2001 to 6 million in 2006, which was accompanied by an apparent drop in the incidence of newly diagnosed breast cancers. During the last few years there has been a reappraisal of the risks and benefits of HRT.28 The new analyses showed that HRT effects depend on the type of estrogen/progesterone used, the mode of drug administration, the age of the person at the start of treatment, the duration of the treatment, and the presence of associated diseases.
Oral Contraceptives (OCs)
Worldwide, more than 100 million women use hormonal contraception. OCs nearly always contain a synthetic estradiol and a variable amount of a progestin, but some preparations contain only progestins. They act by inhibiting ovulation or preventing implantation. Currently prescribed OCs contain a much smaller amount of estrogens (as little as 20 μg of ethinyl estradiol) than the earliest formulations approved for use in the United States in 1960, and are associated with fewer side effects. Transdermal and implantable formulations have also become available. Hence, the results of epidemiologic studies should be interpreted in the context of the dosage and the delivery system. Nevertheless, there is good evidence that the use of OCs is associated with the following conditions31:
Anabolic Steroids
The use of steroids to increase performance by baseball players, track-and-field athletes, and wrestlers has received wide publicity during the last few years. Anabolic steroids are synthetic versions of testosterone, and for performance enhancement they are used at doses that are about 10 to 100 times higher than therapeutic indications. The high concentration of testosterone and its derivatives inhibits production and release of luteinizing hormone and follicle-stimulating hormone by a feedback mechanism, and increases the amount of estrogens, which are produced from anabolic steroids. Anabolic steroids have multiple adverse effects including stunted growth in adolescents, acne, gynecomastia and testicular atrophy in males, and growth of facial hair and menstrual changes in women. Other effects include psychiatric problems and premature heart attacks. Hepatic cholestasis may develop in individuals receiving orally administered anabolic steroids.
Acetaminophen
Acetaminophen is the most commonly used analgesic in the United States. It is present in over 300 products, alone or in combination with other agents. Hence, acetaminophen toxicity is common, being responsible for more than 50,000 emergency room visits per year. In the United States, it is the cause of about 50% of cases of acute liver failure, with 30% mortality. Intentional overdosage (suicide attempts) is the most common cause of acetaminophen toxicity in Great Britain, but unintentional overdosage is the most frequent cause in the United States, representing almost 50% of the total intoxication cases.
At therapeutic doses about 95% of acetaminophen undergoes detoxification in the liver by phase II enzymes and is excreted in the urine as glucuronate or sulfate conjugates (Fig. 9-14). About 5% or less is metabolized through the activity of CYPs (primarily CYP2E) to NAPQI (N-acetyl-p-benzoquinoneimine), a highly reactive metabolite.32,33 NAPQI is normally conjugated with glutathione (GSH), but when taken in larger doses unconjugated NAPQI accumulates and causes hepatocellular injury leading to centrilobular necrosis and liver failure. The injury produced by NAPQI involves two mechanisms: (1) covalent binding to hepatic proteins, which causes damage to cellular membranes and mitochondrial dysfunction, and (2) depletion of GSH, making hepatocytes more susceptible to reactive oxygen species–induced injury. It should be noted that because alcohol induces CYP2E in the liver, toxicity can occur at lower doses in chronic alcoholics.
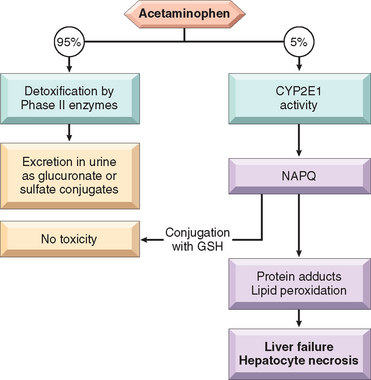
FIGURE 9-14 Acetaminophen metabolism and toxicity. (See text for details.)
(Courtesy of Dr. Xavier Vaquero, Department of Pathology, University of Washington, Seattle, WA.)
The window between the usual dose (0.5 gm) and the toxic dose (15 to 25 gm) is large, and the drug is ordinarily very safe. Toxicity begins with nausea, vomiting, diarrhea, and sometimes shock, followed in a few days by evidence of jaundice. Overdoses of acetaminophen can be treated at its early stages (within 12 hours) by administration of N-acetylcysteine, which restores GSH. In serious overdose liver failure ensues, starting with centrilobular necrosis that may extend to entire lobules, requiring liver transplantation for survival. Some patients show evidence of concurrent renal damage.
Aspirin (Acetylsalicylic Acid)
Overdose may result from accidental ingestion of a large number of tablets by young children; in adults overdose is frequently suicidal. A source of salicylate poisoning is the excessive use of ointments containing oil of wintergreen (methyl salicylate). Acute salicylate overdose causes alkalosis as a consequence of the stimulation of the respiratory center in the medulla. This is followed by metabolic acidosis and accumulation of pyruvate and lactate, caused by uncoupling of oxidative phosphorylation and inhibition of the Krebs cycle. Metabolic acidosis enhances the formation of non-ionized forms of salicylates, which diffuse into the brain and produce effects from nausea to coma. Ingestion of 2 to 4 gm by children or 10 to 30 gm by adults may be fatal, but survival has been reported after ingestion of doses five times larger.
Chronic aspirin toxicity (salicylism) may develop in persons who take 3 gm or more daily for long periods of time for treatment of chronic pain or inflammatory conditions. Chronic salicylism is manifested by headaches, dizziness, ringing in the ears (tinnitus), hearing impairment, mental confusion, drowsiness, nausea, vomiting, and diarrhea. The CNS changes may progress to convulsions and coma. The morphologic consequences of chronic salicylism are varied. Most often there is an acute erosive gastritis (Chapter 17), which may produce overt or covert gastrointestinal bleeding and lead to gastric ulceration. A bleeding tendency may appear concurrently with chronic toxicity, because aspirin acetylates platelet cyclooxygenase and irreversibly blocks the production of thromboxane A2, an activator of platelet aggregation. Petechial hemorrhages may appear in the skin and internal viscera, and bleeding from gastric ulcerations may be exaggerated. With the recognition of gastric ulceration and bleeding as an important complication of ingestion of large doses of aspirin, its chronic toxicity is now quite uncommon.
Proprietary analgesic mixtures of aspirin and phenacetin or its active metabolite, acetaminophen, when taken over several years, can cause tubulointerstitial nephritis with renal papillary necrosis, referred to as analgesic nephropathy (Chapter 20).
INJURY BY NONTHERAPEUTIC AGENTS (DRUG ABUSE)
Drug abuse generally involves the use of mind-altering substances, beyond therapeutic or social norms. Drug addiction and overdose are serious public health problems. Common drugs of abuse are listed in Table 9-6. Here we consider cocaine, heroin, amphetamines, and marijuana, and briefly mention a few others.
TABLE 9-6 Common Drugs of Abuse
| Class | Molecular Target | Example |
|---|---|---|
| Opioid narcotics | Mu opioid receptor (agonist) | Heroin, hydromorphone (Dilaudid) |
| Oxycodone (Percodan, Percocet, Oxycontin) | ||
| Methadone (Dolophine) | ||
| Meperidine (Demerol) | ||
| Sedative-hypnotics | GABAA receptor (agonist) | Barbiturates |
| Ethanol | ||
| Methaqualone (Quaalude) | ||
| Glutethimide (Doriden) | ||
| Ethchlorvynol (Placidyl) | ||
| Psychomotor stimulants | Dopamine transporter (antagonist) | Cocaine |
| Serotonin receptors (toxicity) | Amphetamines | |
| 3,4-methylenedioxymethamphetamine (MDMA, ecstasy) | ||
| Phencyclidine-like drugs | NMDA glutamate receptor channel (antagonist) | Phencyclidine (PCP, angel dust) |
| Ketamine | ||
| Cannabinoids | CBI cannabinoid receptors (agonist) | Marijuana |
| Hashish | ||
| Hallucinogens | Serotonin 5-HT2 receptors (agonist) | Lysergic acid diethylamide (LSD) |
| Mescaline | ||
| Psilocybin |
GABA, γ-aminobutyric acid; 5-HT2, 5-hydroxytryptamine; NMDA, N-methyl D-aspartate.
Data from Hyman SE: A 28-year-old man addicted to cocaine. JAMA 286:2586, 2001.
Cocaine
The use of cocaine and crack continues to increase. According to a 2006 survey, approximately 35.3 million Americans aged 12 or older have tried cocaine, with 6.1 million having used cocaine in the past year. Cocaine is extracted from the leaves of the coca plant, and is usually prepared as a water-soluble powder, cocaine hydrochloride. Sold on the street, it is liberally diluted with talcum powder, lactose, or other look-alikes. Cocaine can be snorted or dissolved in water and injected subcutaneously or intravenously. Crystallization of the pure alkaloid yields nuggets of crack, so called because of the cracking or popping sound it makes when heated to produce vapors that are inhaled. The pharmacologic actions of cocaine and crack are identical, but crack is far more potent.
Cocaine produces an intense euphoria and stimulation, making it one of the most addictive drugs. Experimental animals will press a lever more than 1000 times and forgo food and drink to obtain it. In the cocaine user, although physical dependence generally does not occur, the psychologic withdrawal is profound and can be extremely difficult to treat. Intense cravings are particularly severe in the first several months after abstinence and can recur for years. Acute overdose can produce seizures, cardiac arrhythmias, and respiratory arrest.
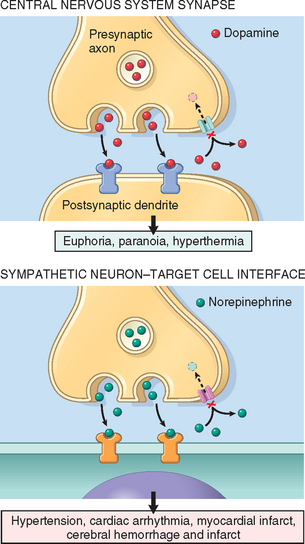
Heroin
Heroin is an addictive opioid derived from the poppy plant that is closely related to morphine. Its use is even more harmful than that of cocaine. As sold on the street, it is cut (diluted) with an agent (often talc or quinine); thus, the size of the dose is not only variable but also usually unknown to the buyer. Heroin, along with any contaminating substances, is usually self-administered intravenously or subcutaneously. Effects are varied and include euphoria, hallucinations, somnolence, and sedation. Heroin has a wide range of adverse physical effects related to (1) the pharmacologic action of the agent, (2) reactions to the cutting agents or contaminants, (3) hypersensitivity reactions to the drug or its adulterants (quinine itself has neurologic, renal, and auditory toxicity), and (4) diseases contracted incident to the use of infected needles. Some of the most important adverse effects of heroin are the following:
Methadone, originally used in the treatment of heroin addiction, is increasingly being prescribed as a painkiller. Unfortunately, its careless use has contributed to more than 800 deaths per year in the United States.
Amphetamines
Methamphetamine.
This addictive drug, known as “speed” or “meth, ” is closely related to amphetamine but has stronger effects in the CNS. It is estimated that there are approximately 500,000 current users in the United States. Approximately 2.5% of youths in grade 8 and 6.5% in grade 12 have tried methamphetamine at least once. It acts by releasing dopamine in the brain, which inhibits presynaptic neurotransmission at corticostriatal synapses, slowing glutamate release.34 Metamphetamine produces a feeling of euphoria, which is followed by a “crash.” Long-term use leads to violent behaviors, confusion, and psychotic features that include paranoia and hallucinations.
MDMA.
MDMA (3,4 methylenedioxymethamphetamine) is popularly known as ecstasy. MDMA is generally taken orally. Its effects, which include euphoria and hallucinogen-like feelings that last for 4 to 6 hours, are mainly due to an increase in serotonin release in the CNS. This is coupled with interference in serotonin synthesis, causing a reduction in serotonin that is only slowly replenished. MDMA use also reduces the number of serotonergic axon terminals in the striatum and the cortex, and it may increase the peripheral effects of dopamine and adrenergic agents. MDMA tablets may be spiked with other drugs, including methamphetamine and cocaine, which greatly enhance the effects on the CNS.
Marijuana
Marijuana, or “pot,” is made from the leaves of the Cannabis sativa plant, which contain the psychoactive substance Δ9-tetrahydrocannabinol (THC). About 5% to 10% of THC is absorbed when it is smoked in a hand-rolled cigarette (“joint”). Despite numerous studies, the central question of whether the drug has persistent adverse physical and functional effects remains unresolved.35 Some of the untoward anecdotal effects may be allergic or idiosyncratic reactions or possibly related to contaminants in the preparations rather than to the pharmacologic effects of marijuana. Among the beneficial effects of marijuana is its potential use to treat nausea secondary to cancer chemotherapy, and as an agent capable of decreasing pain in some chronic conditions that are otherwise difficult to treat. The functional and organic CNS consequences of marijuana smoking have received most scrutiny. Its use distorts sensory perception and impairs motor coordination, but these acute effects generally clear in 4 to 5 hours. With continued use these changes may progress to cognitive and psychomotor impairments, such as inability to judge time, speed, and distance, a frequent cause of automobile accidents. Marijuana increases the heart rate and sometimes blood pressure, and it may cause angina in a person with coronary artery disease.
The respiratory system is also affected by chronic marijuana smoking; laryngitis, pharyngitis, bronchitis, cough and hoarseness, and asthma-like symptoms have all been described, along with mild but significant airway obstruction. Marijuana cigarettes contain a large number of carcinogens that are also present in tobacco. Smoking a marijuana cigarette, compared with a tobacco cigarette, is associated with a three-fold increase in the amount of tar inhaled and retained in the lungs, presumably because of the larger puff volume, deeper inhalation, and longer breath holding.
Regardless of the use of THC as a recreational drug, a large number of studies have characterized the endogenous cannabinoid system, which consists of the cannabinoid receptors CB1 and CB2, and the endogenous lipid ligands known as endocannabinoids.36 This system participates in the regulation of the hypothalamic-pituitary-adrenal axis, and modulates the control of appetite, food intake, and energy balance, as well as fertility and sexual behavior.37
Other Drugs
The variety of drugs that have been tried by those seeking “new experiences” (e.g., “highs,” “lows,” “out-of-the-body experiences”) defies belief. Overall, there has been a decrease in the use of most illegal drugs, but large increases have occurred in prescription and nonprescription drug abuse, and in the inhalation of potentially toxic household products. These drugs include various stimulants, depressants, analgesics, and hallucinogens (see Table 9-6). Among these are PCP (phenylcyclidine, an anesthetic agent), analgesics such as oxycontin and vicodin, and ketamine, an anesthetic agent used in animal surgery. Chronic inhalation of vapors of spray paints, paint thinners, and some glues that contain toluene (“glue sniffing”) can cause cognitive abnormalities and magnetic resonance imaging–detectable brain damage that ranges from mild to severe dementia. Because they are used haphazardly and in various combinations, not much is known about the long-time deleterious effects of most of these agents. However, their acute effects are clear: they cause bizarre and often aggressive behavior that leads to violence, or depressed mood and suicidal ideation.
Injury by Physical Agents
Injury induced by physical agents is divided into the following categories: mechanical trauma, thermal injury, electrical injury, and injury produced by ionizing radiation. Each type is considered separately.
MECHANICAL TRAUMA
Mechanical forces may inflict a variety of forms of damage. The type of injury depends on the shape of the colliding object, the amount of energy discharged at impact, and the tissues or organs that bear the impact. Bone and head injuries result in unique damage and are discussed elsewhere (Chapter 28). All soft tissues react similarly to mechanical forces, and the patterns of injury can be divided into abrasions, contusions, lacerations, incised wounds, and puncture wounds. This is just a small sampling of the various forms of trauma encountered by forensic pathologists, who deal with wounds produced by shooting, stabbing, blunt force, traffic accidents, and other causes. In addition to morphologic analyses, forensic pathology now includes molecular methods for identity testing and sophisticated methods to detect the presence of foreign substances. Details about the practice of forensic pathology can be found in specialized textbooks.
Morphology. An abrasion is a wound produced by scraping or rubbing, resulting in removal of the superficial layer. Skin abrasions may remove only the epidermal layer. A contusion, or bruise, is an injury usually produced by a blunt object characterized by damage to blood vessels and extravasation of blood into tissues (Fig. 9-16A). A laceration is a tear or disruptive stretching of tissue caused by the application of force by a blunt object (Fig. 9-16B). In contrast to an incision, most lacerations have intact bridging blood vessels and jagged, irregular edges. An incised wound is one inflicted by a sharp instrument. The bridging blood vessels are severed. A puncture wound is caused by a long, narrow instrument and is termed penetrating when the instrument pierces the tissue and perforating when it traverses a tissue to also create an exit wound. Gunshot wounds are special forms of puncture wounds that demonstrate distinctive features important to the forensic pathologist. For example, a wound from a bullet fired at close range leaves powder burns, whereas one fired from more than 4 or 5 feet away does not.
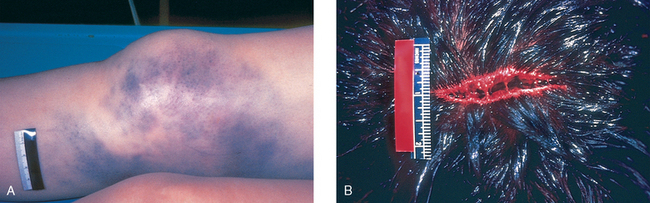
FIGURE 9-16 A, Contusion resulting from blunt trauma. The skin is intact, but there is hemorrhage of subcutaneous vessels, producing extensive discoloration. B, Laceration of the scalp; the bridging strands of fibrous tissues are evident.
(From the Department of Pathology, Southwestern Medical School, Dallas, TX.)
One of the most common causes of mechanical injury is vehicular accident. The typical injuries result from (1) hitting a part of the interior of the vehicle or being hit by an object that enters the passenger compartment during the crash, such as the motor; (2) being thrown from the vehicle; or (3) being trapped in a burning vehicle. The pattern of injury relates to whether one or all three of these mechanisms are operative. For example, in a head-on collision, a common pattern of injury sustained by a driver who is not wearing a seat belt includes trauma to the head (windshield impact), chest (steering column impact), and knees (dashboard impact). Under these conditions, common chest injuries include sternal and rib fractures, heart contusions, aortic lacerations, and (less commonly) lacerations of the spleen and liver. Thus, in caring for an automobile injury victim, it is essential to remember that internal wounds often accompany superficial abrasions, contusions, and lacerations. Indeed, in many cases external evidence of serious internal damage is completely absent.
THERMAL INJURY
Both excessive heat and excessive cold are important causes of injury. Burns are the most common cause of thermal injury and are discussed first; a brief discussion of hyperthermia and hypothermia follows.
Thermal Burns
In the United States, approximately 500,000 persons per year receive medical treatment for burn injuries. It is estimated that approximately 4000 persons per year die as a consequence of injuries caused by fire and smoke inhalation, mostly originating in homes. Fortunately, since the 1970s, marked decreases have been seen in both mortality rates and the length of hospitalizations of burn patients. In 2007 there were 40,000 hospitalizations in specialized burn centers, with a 90% survival. Eighty percent of the burns were caused by fire or scalding, the latter being a major cause of injury in children. Improvements in burn treatment have been achieved by a better understanding of the systemic effects of massive burns, the prevention of wound infection, and improvements in treatments that promote the healing of skin surfaces.
The clinical significance of a burn injury depends on the following factors:
Burns used to be classified as first to fourth degree, according to the depth of the injury (first-degree burns being the most superficial). This classification has been replaced by the terms superficial, partial thickness, and full-thickness burns.
Shock, sepsis, and respiratory insufficiency are the greatest threats to life in burn patients. Particularly in burns of more than 20% of the body surface, there is a rapid (within hours) shift of body fluids into the interstitial compartments, both at the burn site and systemically, which can result in hypovolemic shock (Chapter 4). Because protein from the blood is lost into interstitial tissue, generalized edema, including pulmonary edema, can be severe. An important pathophysiologic effect of burns is the development of a hypermetabolic state associated with excess heat loss and an increased need for nutritional support. It is estimated that when more than 40% of the body surface is burned, the resting metabolic rate may double.
The burn site is ideal for the growth of microorganisms; the serum and debris provide nutrients, and the burn injury compromises blood flow, blocking effective inflammatory responses. The most common offender is the opportunist Pseudomonas aeruginosa, but antibiotic-resistant strains of other common hospital-acquired bacteria, such as S. aureus, and fungi, particularly Candida species, may also be involved. Furthermore, cellular and humoral defenses against infections are compromised, and both lymphocyte and phagocyte functions are impaired. Direct bacteremic spread and release of toxic substances such as endotoxin from the local site have dire consequences. Pneumonia or septic shock with renal failure and/or the acute respiratory distress syndrome (Chapter 15) are the most common serious sequelae.
Organ system failure resulting from burn sepsis has greatly diminished during the last 30 years, because of the introduction of techniques for early excision and grafting of the burn wound. Removal of the burn wound decreases infection and reduces the need for reconstructive surgery.38 Grafting is done with split-thickness skin grafts; dermal substitutes, which serve as a bed for cell repopulation, may be used in large full-thickness burns.
Injury to the airways and lungs may develop within 24 to 48 hours after the burn and may result from the direct effect of heat on the mouth, nose, and upper airways or from the inhalation of heated air and noxious gases in the smoke. Water-soluble gases, such as chlorine, sulfur oxides, and ammonia, may react with water to form acids or alkalis, particularly in the upper airways, producing inflammation and swelling, which may lead to partial or complete airway obstruction. Lipid-soluble gases, such as nitrous oxide and products of burning plastics, are more likely to reach deeper airways, producing pneumonitis.
In burn survivors the development of hypertrophic scars, both at the site of the original burn and at donor graft sites, and itching may become long-term, difficult-to-treat problems. Hypertrophic scars after burn injury may be a consequence of continuous angiogenesis in the wound caused by excess neuropeptides, such as substance P, released from injured nerve endings.39
Morphology. Grossly, full-thickness burns are white or charred, dry, and anesthetic (because of destruction of nerve endings), whereas, depending on the depth, partial-thickness burns are pink or mottled with blisters and are painful. Histologically, devitalized tissue reveals coagulative necrosis, adjacent to vital tissue that quickly accumulates inflammatory cells and marked exudation.
Hyperthermia
Prolonged exposure to elevated ambient temperatures can result in heat cramps, heat exhaustion, and heat stroke.
Hypothermia
Prolonged exposure to low ambient temperature leads to hypothermia, a condition seen all too frequently in homeless persons. High humidity, wet clothing, and dilation of superficial blood vessels resulting from the ingestion of alcohol hasten the lowering of body temperature. At a body temperature of about 90°F, loss of consciousness occurs, followed by bradycardia and atrial fibrillation at lower core temperatures.
Hypothermia causes injury by two mechanisms:
ELECTRICAL INJURY
Electrical injuries, which are often fatal, can arise from contact with low-voltage currents (i.e., in the home and workplace) or high-voltage currents carried by high-power lines or lightning. Injuries are of two types: (1) burns and (2) ventricular fibrillation or cardiac and respiratory center failure, resulting from disruption of normal electrical impulses. The type of injury and the severity and extent of burns depend on the strength (amperage), duration, and path of the electric current within the body.
Voltage in the household and workplace (120 or 220 V) is high enough that with low resistance at the site of contact (as when the skin is wet), sufficient current can pass through the body to cause serious injury, including ventricular fibrillation. If current flow continues long enough, it generates enough heat to produce burns at the site of entry and exit as well as in internal organs. An important characteristic of alternating current, the type available in most homes, is that it induces tetanic muscle spasm, so that when a live wire or switch is grasped, irreversible clutching is likely to occur, prolonging the period of current flow. This results in a greater likelihood of developing extensive electrical burns and, in some cases, spasm of the chest wall muscles, producing death from asphyxia. Currents generated from high-voltage sources cause similar damage; however, because of the large current flows generated, these are more likely to produce paralysis of medullary centers and extensive burns. Lightning is a classic cause of high-voltage electrical injury.
Earlier studies had linked exposure to high-voltage magnetic fields to an increased risk of cancer, mainly leukemias, among workers on electric high-power lines and children living near power transmission lines. However, further analyses have failed to find a consistent association between these exposures and cancer development. Electric and magnetic fields and microwave radiation, when sufficiently intense, may produce burns, usually of the skin and subjacent connective tissue, and both forms of radiation can interfere with cardiac pacemakers.
INJURY PRODUCED BY IONIZING RADIATION
Radiation is energy that travels in the form of waves or high-speed particles. Radiation has a wide range of energies that span the electromagnetic spectrum; it can be divided into non-ionizing and ionizing radiation. The energy of non-ionizing radiation such as UV and infrared light, microwave, and sound waves, can move atoms in a molecule or cause them to vibrate, but is not sufficient to displace bound electrons from atoms. By contrast, ionizing radiation has sufficient energy to remove tightly bound electrons. Collision of electrons with other molecules releases electrons in a reaction cascade, referred to as ionization. The main sources of ionizing radiation are x-rays and gamma rays (electromagnetic waves of very high frequencies), high-energy neutrons, alpha particles (composed of two protons and two neutrons), and beta particles, which are essentially electrons. At equivalent amounts of energy, alpha particles induce heavy damage in a restricted area, whereas x-rays and gamma rays dissipate energy over a longer, deeper course, and produce considerably less damage per unit of tissue. About 25% of the total dose of ionizing radiation received by the US population is human-made, mostly originated in medical devices and radioisotopes.
Ionizing radiation is a double-edged sword. It is indispensable in medical practice, being used in the treatment of cancer, in diagnostic imaging, and in therapeutic or diagnostic radioisotopes, but it also produces adverse short- and long-term effects such as fibrosis, mutagenesis, carcinogenesis, and teratogenesis.41
Radiation Units.
Several somewhat confusing terms are used to describe the radiation doses. This is because radiation is measured in three different ways. These are, the amount of radiation emitted by a source, the radiation dose absorbed by a person, and the biologic effect of the radiation. These are described below:
Main Determinants of the Biologic Effects of Ionizing Radiation.
In addition to the physical properties of the radiation, its biologic effects depend heavily on the following factors.
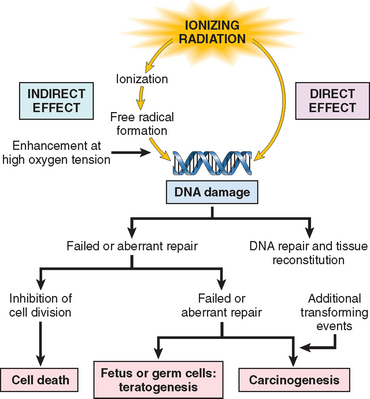
FIGURE 9-17 Effects of ionizing radiation on DNA and its consequences. The effects on DNA can be direct, or most importantly, indirect, through free radical formation.
FIGURE 9-18 Chronic vascular injury with subintimal fibrosis occluding the lumen.
(American Registry of Pathology © 1990.)
Figure 9-19 shows the overall consequences of radiation exposure. These consequences may vary according to the dose of radiation and the type of exposure. Table 9-7 lists the estimated threshold doses for acute effects of radiation aimed at specific organs; Table 9-8 shows the syndromes caused by exposure to various doses of total-body radiation.
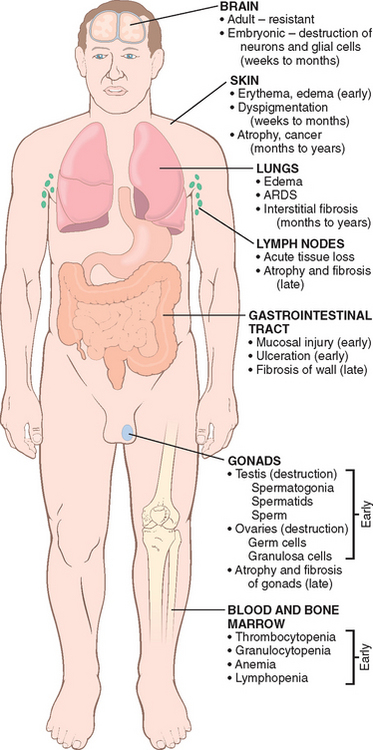
FIGURE 9-19 Overview of the major morphologic consequences of radiation injury. Early changes occur in hours to weeks; late changes occur in months to years. ARDS, acute respiratory distress syndrome.
TABLE 9-7 Estimated Threshold Doses for Acute Radiation Effects on Specific Organs
| Health Effect | Organ | Dose (Sv) |
|---|---|---|
| Temporary sterility | Testes | 0.15 |
| Depression of hematopoiesis | Bone marrow | 0.50 |
| Reversible skin effects (e.g., erythema) | Skin | 1.0–2.0 |
| Permanent sterility | Ovaries | 2.5–6.0 |
| Temporary hair loss | Skin | 3.0–5.0 |
| Permanent sterility | Testis | 3.5 |
| Cataract | Lens of eye | 5.0 |
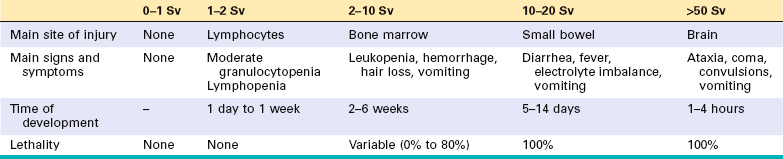
Morphology. Cells surviving radiant energy damage show a wide range of structural changes in chromosomes, including deletions, breaks, translocations, and fragmentation. The mitotic spindle often becomes disorderly, and polyploidy and aneuploidy may be encountered. Nuclear swelling and condensation and clumping of chromatin may appear; sometimes the nuclear membrane breaks down. Apoptosis may occur. All forms of abnormal nuclear morphology may be seen. Giant cells with pleomorphic nuclei or more than one nucleus may appear and persist for years after exposure. At extremely high doses of radiant energy, markers of cell death, such as nuclear pyknosis, and lysis appear quickly.
In addition to affecting DNA and nuclei, radiant energy may induce a variety of cytoplasmic changes, including cytoplasmic swelling, mitochondrial distortion, and degeneration of the endoplasmic reticulum. Plasma membrane breaks and focal defects may be seen. The histologic constellation of cellular pleomorphism, giant-cell formation, conformational changes in nuclei, and abnormal mitotic figures creates a more than passing similarity between radiation-injured cells and cancer cells, a problem that plagues the pathologist when evaluating post-irradiation tissues for the possible persistence of tumor cells.
At the light microscopic level, vascular changes and interstitial fibrosis are prominent in irradiated tissues (Fig. 9-20). During the immediate post-irradiation period, vessels may show only dilation. With time, or with higher doses, a variety of degenerative changes appear, including endothelial cell swelling and vacuolation, or even dissolution with total necrosis of the walls of small vessels such as capillaries and venules. Affected vessels may rupture or thrombose. Still later, endothelial cell proliferation and collagenous hyalinization with thickening of the media are seen in irradiated vessels, resulting in marked narrowing or even obliteration of the vascular lumens. At this time, an increase in interstitial collagen in the irradiated field usually becomes evident, leading to scarring and contractions.
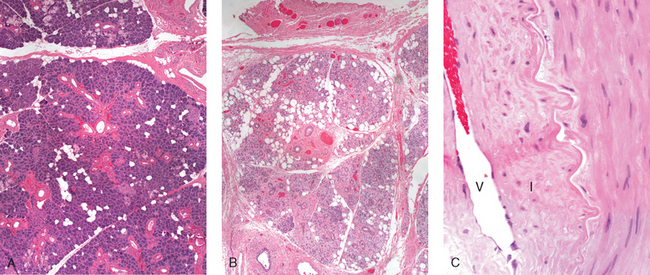
FIGURE 9-20 Fibrosis and vascular changes in salivary glands produced by radiation therapy of the neck region. A, Normal salivary gland; B, fibrosis caused by radiation; C, fibrosis and vascular changes consisting of fibrointimal thickening and arteriolar sclerosis. V, vessel lumen; I, thickened intima.
(Courtesy of Dr. Melissa Upton, Department of Pathology, University of Washington, Seattle, WA.)
Total-Body Irradiation.
Exposure of large areas of the body to even very small doses of radiation may have devastating effects. Dosages below 1 Sv produce minimal or no symptoms. However, higher levels of exposure cause health effects known as acute radiation syndromes, which at progressively higher doses involve the hematopoietic, gastrointestinal, and central nervous systems. The syndromes associated with total-body exposure to ionizing radiation are presented in Table 9-8.
Acute Effects on Hematopoietic and Lymphoid Systems.
The hematopoietic and lymphoid systems are extremely susceptible to radiation injury and deserve special mention. With high dose levels and large exposure fields, severe lymphopenia may appear within hours of irradiation, along with shrinkage of the lymph nodes and spleen. Radiation directly destroys lymphocytes, both in the circulating blood and in tissues (nodes, spleen, thymus, gut). With sublethal doses of radiation, regeneration from viable precursors is prompt, leading to restoration of a normal lymphocyte count in the blood within weeks to months. Hematopoietic precursors in the bone marrow are also quite sensitive to radiant energy, which produces a dose-dependent marrow aplasia. Very high doses of radiation kill marrow stem cells and induce permanent aplasia (aplastic anemia), whereas with lower doses the aplasia is transient. The circulating granulocyte count may first rise but begins to fall toward the end of the first week. Levels near zero may be reached during the second week. If the patient survives, recovery of the normal granulocyte count may require 2 to 3 months. Platelets are similarly affected, with the nadir of the count occurring somewhat later than that of granulocytes; recovery is similarly delayed. Red cell counts fall and anemia appears after 2 to 3 weeks and may persist for months.
Fibrosis.
A common consequence of radiation therapy for cancer is the development of fibrosis in the tissues included in the irradiated field (see Fig. 9-20). Fibrosis may occur weeks or months after irradiation as a consequence of the replacement of dead parenchymal cells by connective tissue, leading to the formation of scars and adhesions (see Chapter 3). Vascular damage, the killing of tissue stem cells, and the release of cytokines and chemokines that promote an inflammatory reaction and fibroblast activation are the main contributors to the development of radiation-induced fibrosis (Figs. 9-21 and 9-22). Common sites of fibrosis after radiation treatment are the lungs, the salivary glands after radiation therapy for head and neck cancers, and colorectal and pelvic areas after treatment for prostate cancer.
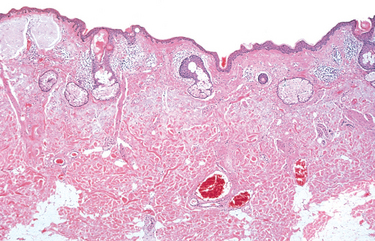
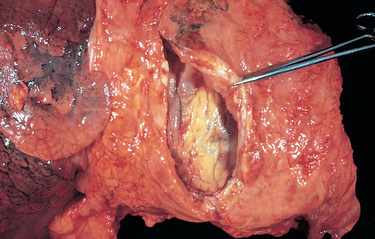
DNA Damage and Carcinogenesis.
Ionizing radiation can cause multiple types of damage in DNA, including single-base damage, single- and double-stranded breaks, and DNA-protein cross-links. In surviving cells, simple defects may be repaired by various enzyme systems present in most mammalian cells (see Chapter 7). However, the most serious damage to DNA is caused by double-stranded breaks (DSBs). Two types of mechanisms can repair DSBs in mammalian cells: homologous recombination and nonhomologous end joining (NHEJ), with NHEJ being the most common repair pathway. DNA repair through NHEJ often produces mutations, including short deletions or duplications, or gross chromosomal aberrations such as translocations and inversions. If the replication of cells containing DSBs is not stopped by cell cycle checkpoint controls (Chapter 3), cells with chromosomal damage persist and may initiate carcinogenesis many years later. More recently it has been recognized that these abnormal cells may also have a “bystander effect,” that is, they may promote growth of non-irradiated surrounding cells through the production of growth factors and cytokines.42,43 Bystander effects are referred to as non-target effects of radiation.
Cancer Risks from Exposures to Low-level Radiation.
Any cell capable of division that has sustained a mutation has the potential to become cancerous. Thus, an increased incidence of neoplasms may occur in any organ after exposure to ionizing radiation. The level of radiation required to increase the risk of cancer development is difficult to determine, but there is little doubt that acute or prolonged exposures that result in doses of greater than 100 mSv cause serious consequences including cancer.44 This is documented by the increased incidence of leukemias and tumors at various sites (such as thyroid, breast, and lungs) in survivors of the atomic bombings of Hiroshima and Nagasaki; the high number of thyroid cancers in survivors of the Chernobyl accident; the high incidence of thyroid tumors, and the elevated frequency of leukemias and birth defects in inhabitants of the Marshall Islands exposed to nuclear fallout; and the development of “second cancers,” such as acute myeloid leukemia, myelodysplastic syndrome, Hodgkin lymphoma and solid tumors, in individuals who received radiation therapy for childhood cancers. The long-term cancer risks caused by radiation exposures in the range of 5 to 100 mSv are much more difficult to establish, because accurate measurements of risks require large population groups ranging from 50,000 to 5 million people.
Estimation of cancer risks at low levels of exposure to ionizing radiation relies in part on models that extrapolate from higher doses. Nevertheless, for x-rays and gamma rays there is good evidence for a statistically significant increase in the risk of cancer at acute doses of greater than 50 mSv and “reasonable” evidence for acute doses of greater than 5 mSv. For protracted exposures, the approximate values suggested are greater than 100 mSv (good evidence for a statistically significant increase of risk) and 50 mSv (reasonable evidence for increased risk). As a comparison, a single posterior-anterior chest radiograph, a lateral chest film chest radiograph, and a computed tomography of the chest deliver effective dosages to the lungs of 0.01, 0.15, and 10 mSv, respectively.45
Increased risk of cancer development may also be associated with occupational exposures. Radon gas is a ubiquitous product of the spontaneous decay of uranium. Its carcinogenic effects are largely attributable to two decay products, polonium 214 and 218 (or “radon daughters”), which emit alpha particles. Polonium 214 and 218 produced from inhaled radon tend to deposit in the lung, and chronic exposure in uranium miners may give rise to lung carcinomas. Risks are also present in homes in which the levels of radon are very high, comparable to those found in mines. However, there is little or no evidence to suggest that radon contributes to the risk of lung cancer in the average household. Among other polonium isotopes, polonium 210 came to the public attention in November 2006 with the highly publicized use of this isotope to kill an individual in England. For historical reasons, we also mention here the development of osteogenic sarcomas after radium exposure in radium dial painters, chemists, radiologists, and patients exposed to radium as a treatment for various ailments, during the first part of the twentieth century.
Nutritional Diseases
Malnutrition, also referred to as protein energy malnutrition or PEM, is a consequence of inadequate intake of proteins and calories, or deficiencies in the digestion or absorption of proteins, resulting in the loss of fat and muscle tissue, weight loss, lethargy, and generalized weakness. Millions of people in developing nations are malnourished and starving, or living on the cruel edge of starvation. In the industrial world and, more recently, also in developing countries, obesity has become a major public health problem, associated with the development of diseases such as diabetes and atherosclerosis.
The sections that follow barely skim the surface of nutritional disorders. Particular attention is devoted to PEM, anorexia nervosa and bulimia, deficiencies of vitamins and trace minerals, obesity, and a brief overview of the relationships of diet to atherosclerosis and cancer. Other nutrients and nutritional issues are discussed in the context of specific diseases.
DIETARY INSUFFICIENCY
An appropriate diet should provide (1) sufficient energy, in the form of carbohydrates, fats, and proteins, for the body’s daily metabolic needs; (2) amino acids and fatty acids to be used as building blocks for synthesis of structural and functional proteins and lipids; and (3) vitamins and minerals, which function as coenzymes or hormones in vital metabolic pathways or, as in the case of calcium and phosphate, as important structural components. In primary malnutrition, one or all of these components are missing from the diet. By contrast, in secondary malnutrition, the supply of nutrients is adequate, but malnutrition results from insufficient intake, malabsorption, impaired utilization or storage, excess loss, or increased need for nutrients.
There are several conditions that may lead to dietary insufficiencies.
PROTEIN-ENERGY MALNUTRITION (PEM)
Severe PEM is a serious, often lethal disease affecting children. It is common in low-income countries, where up to 25% of children may be affected, and where it is a major factor in the high death rates among children younger than 5 years. In the West Africa country of Niger, which suffered a severe famine in 2005, United Nations reports estimate that there were, respectively, 150,000 and 650,000 children with severe and moderate malnutrition. In that country, malnutrition was a direct or indirect cause of mortality in 60% of children under age 5. Decreased food intake can also occur due to sharp increases in prices, as was seen in the first half of 2008. In developed countries, PEM occurs in elderly and debilitated patients in nursing homes and hospitals.
Malnutrition is determined according to the body mass index (BMI, weight in kilograms divided by height in meters squared). A BMI less than 16 kg/m2 is considered malnutrition (normal range 18.5 to 25 kg/m2). In more practical ways, a child whose weight falls to less than 80% of normal (provided in standard tables) is considered malnourished. However, loss of weight may be masked by generalized edema, as discussed later. Other helpful parameters are the evaluation of fat stores (thickness of skin folds), muscle mass (reduced circumference of mid-arm), and serum proteins (albumin and transferrin measurements provide a measure of the adequacy of the visceral protein compartment).
Marasmus and Kwashiorkor.
In malnourished children, PEM presents as a range of clinical syndromes, all characterized by a dietary intake of protein and calories inadequate to meet the body’s needs. The two ends of the spectrum of PEM syndromes are known as marasmus and kwashiorkor. From a functional standpoint, there are two differentially regulated protein compartments in the body: the somatic compartment, represented by proteins in skeletal muscles, and the visceral compartment, represented by protein stores in the visceral organs, primarily the liver. As we shall see, the somatic compartment is affected more severely in marasmus, and the visceral compartment is depleted more severely in kwashiorkor.
A child is considered to have marasmus when weight falls to 60% of normal for sex, height, and age. A marasmic child suffers growth retardation and loss of muscle, the latter resulting from catabolism and depletion of the somatic protein compartment. This seems to be an adaptive response that provides the body with amino acids as a source of energy. The visceral protein compartment, which is presumably more precious and critical for survival, is only marginally depleted, and hence serum albumin levels are either normal or only slightly reduced. In addition to muscle proteins, subcutaneous fat is also mobilized and used as fuel. The production of leptin (discussed in “Obesity”) is low, which may stimulate the hypothalamic-pituitary-adrenal axis to produce high levels of cortisol that contribute to lipolysis. With such losses of muscle and subcutaneous fat, the extremities are emaciated; by comparison, the head appears too large for the body (Fig. 9-23A). Anemia and manifestations of multiple vitamin deficiencies are present, and there is evidence of immune deficiency, particularly T cell–mediated immunity. Hence, concurrent infections are usually present, which impose additional nutritional demands. Unfortunately, images of children dead or near death with marasmus, have become almost commonplace in television and newspaper reports of famine and disasters in various areas of the world.
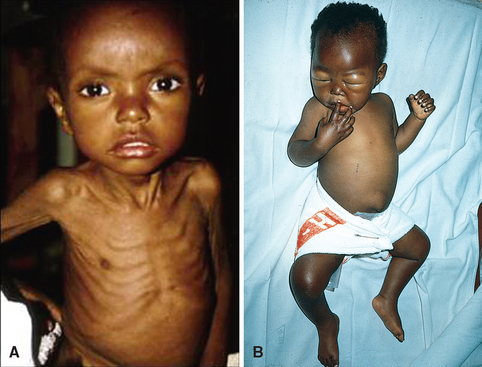
FIGURE 9-23 Childhood malnutrition. A, Marasmus. Note the loss of muscle mass and subcutaneous fat; the head appears to be too large for the emaciated body. B, Kwashiorkor. The infant shows generalized edema, seen as ascites and puffiness of the face, hands, and legs.
(A, From Clinic Barak, Reisebericht Kenya.)
Kwashiorkor occurs when protein deprivation is relatively greater than the reduction in total calories (Fig. 9-23B). This is the most common form of PEM seen in African children who have been weaned too early and subsequently fed, almost exclusively, a carbohydrate diet (the name kwashiorkor is from the Ga language in Ghana describing a disease of a baby due to the arrival of another child). The prevalence of kwashiorkor is also high in impoverished countries of Southeast Asia. Less severe forms may occur worldwide in persons with chronic diarrheal states in which protein is not absorbed or in those with chronic protein loss due to conditions such as protein-losing enteropathies, the nephrotic syndrome, or after extensive burns. Cases of kwashiorkor resulting from fad diets or replacement of milk by rice-based beverages have been reported in the United States.
In kwashiorkor, marked protein deprivation is associated with severe loss of the visceral protein compartment, and the resultant hypoalbuminemia gives rise to generalized or dependent edema (Fig. 9-23B). The loss of weight in these patients is masked by the increased fluid retention. In further contrast to marasmus, there is relative sparing of subcutaneous fat and muscle mass. Children with kwashiorkor have characteristic skin lesions, with alternating zones of hyperpigmentation, areas of desquamation, and hypopigmentation, giving a “flaky paint” appearance. Hair changes include overall loss of color or alternating bands of pale and darker hair. Other features that differentiate kwashiorkor from marasmus include an enlarged, fatty liver (resulting from reduced synthesis of the carrier protein component of lipoproteins), and the development of apathy, listlessness, and loss of appetite. Vitamin deficiencies are likely to be present, as are defects in immunity and secondary infections. As already stated, marasmus and kwashiorkor are two ends of a spectrum, and considerable overlap exists between these conditions.
Secondary PEM often develops in chronically ill, elderly, and bedridden patients. An 18-item questionnaire known as the Mininutritional Assessment (MNA) is often used to measure the nutritional status of elderly persons. It is estimated that more than 50% of elderly residents in nursing homes in the United States are malnourished. Weight loss of more than 5% associated with PEM increases the risk of mortality in nursing home patients by almost five-fold. The most obvious signs of secondary PEM include: (1) depletion of subcutaneous fat in the arms, chest wall, shoulders, or metacarpal regions; (2) wasting of the quadriceps femoris and deltoid muscles; and (3) ankle or sacral edema. Bedridden or hospitalized malnourished patients have an increased risk of infection, sepsis, impaired wound healing, and death after surgery.
Morphology. The central anatomic changes in PEM are (1) growth failure, (2) peripheral edema in kwashiorkor, and (3) loss of body fat and atrophy of muscle, more marked in marasmus.
The liver in kwashiorkor, but not in marasmus, is enlarged and fatty; superimposed cirrhosis is rare.
In kwashiorkor (rarely in marasmus) the small bowel shows a decrease in the mitotic index in the crypts of the glands, associated with mucosal atrophy and loss of villi and microvilli. In such cases concurrent loss of small intestinal enzymes occurs, most often manifested as disaccharidase deficiency. Hence, infants with kwashiorkor initially may not respond well to full-strength, milk-based diets. With treatment, the mucosal changes are reversible.
The bone marrow in both kwashiorkor and marasmus may be hypoplastic, mainly as a result of decreased numbers of red cell precursors. The peripheral blood commonly reveals mild to moderate anemia, which often has a multifactorial origin; nutritional deficiencies of iron, folate, and protein, as well as the suppressive effects of infection (anemia of chronic disease) may all contribute. Depending on the predominant factor, the red cells may be microcytic, normocytic, or macrocytic.
The brain in infants who are born to malnourished mothers and who suffer PEM during the first 1 or 2 years of life has been reported by some to show cerebral atrophy, a reduced number of neurons, and impaired myelinization of white matter.
Many other changes may be present, including (1) thymic and lymphoid atrophy (more marked in kwashiorkor than in marasmus), (2) anatomic alterations induced by intercurrent infections, particularly with all manner of endemic worms and other parasites, and (3) deficiencies of other required nutrients such as iodine and vitamins.
Cachexia.
PEM is a common complication in patients with AIDS or advanced cancers, and in these settings it is known as cachexia. Cachexia occurs in about 50% of cancer patients, most commonly in individuals with gastrointestinal, pancreatic, and lung cancers, and is responsible for about 30% of cancer deaths. It is a highly debilitating condition characterized by extreme weight loss, fatigue, muscle atrophy, anemia, anorexia, and edema. Mortality is generally the consequence of atrophy of the diaphragm and other respiratory muscles. The precise causes of cachexia are not known, but it is clear that agents secreted by tumors and host responses contribute to its development (Fig. 9-24). Cachetic agents produced by tumors include:
FIGURE 9-24 Mechanisms of cancer cachexia. The figure illustrates three mechanisms that cause muscle atrophy and muscle degradation leading to cachexia. (1) Proteolysis-inducing factor (PIF) produced by tumors degrades myosin heavy chain through the proteasome, causing muscle atrophy; (2) TNF and other cytokines produced by tumors and the host activate NF-κB and initiate the transcription of the ubiquitin ligases MAFBx and MuRF1, contributing to protein breakdown; (3) alterations in the dystrophin-glycoprotein complex leading to dystrophin-degradation by the proteasome also participate in the muscle atrophy of cachexia.
Proteolysis-inducing factor (PIF) and pro-inflammatory cytokines cause skeletal muscle breakdown through the NFκB-induced activation of the ubiquitin proteasome pathway, leading to the degradation of myosin heavy chain.47 The induction of the ubiquitin proteasome pathway involves the production of two muscle-specific ubiquitin ligases, MuRF1 (muscle RING finger-1) and MAFBx (muscle atrophy F-box, or atroglin-1). More recent data also implicate alterations in the myofibrillar membrane of skeletal muscle with loss of dystrophin caused by alterations in the dystrophin-glycoprotein complex (Fig. 9-24) as contributors to muscle atrophy, through a mechanism similar to that which occurs in some muscular dystrophies.48
ANOREXIA NERVOSA AND BULIMIA
Anorexia nervosa is self-induced starvation, resulting in marked weight loss; bulimia is a condition in which the patient binges on food and then induces vomiting. Anorexia nervosa has the highest death rate of any psychiatric disorder. Bulimia is more common than anorexia nervosa, and generally has a better prognosis; it is estimated to occur in 1% to 2% of women and 0.1% of men, with an average onset at 20 years of age. These eating disorders occur primarily in previously healthy young women who have developed an obsession with body image and thinness. The neurobiologic underpinnings of these diseases are unknown, but it has been suggested that altered serotonin metabolism may be an important component.49
The clinical findings in anorexia nervosa are generally similar to those in severe PEM. In addition, effects on the endocrine system are prominent. Amenorrhea, resulting from decreased secretion of gonadotropin-releasing hormone, and subsequent decreased secretion of luteinizing hormone and follicle-stimulating hormone, is so common that its presence is a diagnostic feature for the disorder. Other common findings, related to decreased thyroid hormone release, include cold intolerance, bradycardia, constipation, and changes in the skin and hair. In addition, dehydration and electrolyte abnormalities are frequently present. The skin becomes dry and scaly. Bone density is decreased, most likely because of low estrogen levels, which mimics the postmenopausal acceleration of osteoporosis. Anemia, lymphopenia, and hypoalbuminemia may be present. A major complication of anorexia nervosa (and also bulimia) is an increased susceptibility to cardiac arrhythmia and sudden death, resulting from hypokalemia.
In bulimia, binge eating is the norm. Large amounts of food, principally carbohydrates, are ingested, only to be followed by induced vomiting. Although menstrual irregularities are common, amenorrhea occurs in less than 50% of bulimic patients, probably because weight and gonadotropin levels are maintained near normal. The major medical complications relate to continual induced vomiting, and the chronic use of laxatives and diuretics. They include (1) electrolyte imbalances (hypokalemia), which predispose the patient to cardiac arrhythmias; (2) pulmonary aspiration of gastric contents; and (3) esophageal and gastric cardiac rupture. Nevertheless, there are no signs and symptoms that are specific for bulimia; the diagnosis must rely on a comprehensive psychologic assessment of the person. A recent trend in bulimic patients has been the combination of binge eating with high ingestion of alcohol. Needless to say, the combined effects of bulimia and alcoholism are devastating.
VITAMIN DEFICIENCIES
Thirteen vitamins are necessary for health; vitamins A, D, E, and K are fat-soluble, and all others are water-soluble. The distinction between fat- and water-soluble vitamins is important. Fat-soluble vitamins are more readily stored in the body, but they may be poorly absorbed in fat malabsorption disorders, caused by disturbances of digestive functions (discussed in Chapter 17). Certain vitamins can be synthesized endogenously—vitamin D from precursor steroids, vitamin K and biotin by the intestinal microflora, and niacin from tryptophan, an essential amino acid. Notwithstanding this endogenous synthesis, a dietary supply of all vitamins is essential for health.
A deficiency of vitamins may be primary (dietary in origin) or secondary because of disturbances in intestinal absorption, transport in the blood, tissue storage, or metabolic conversion. In the following sections, vitamins A, D, and C are presented in some detail because of their wide-ranging activities and the morphologic changes of deficient states. This is followed by presentation in tabular form of the main consequences of deficiencies of the remaining vitamins (E, K, and the B complex) and some essential minerals. However, it should be emphasized that deficiency of a single vitamin is uncommon, and that single or multiple vitamin deficiencies may be associated with PEM.
Vitamin A
Vitamin A is the name given to a group of related compounds that include retinol (vitamin A alcohol), retinal (vitamin A aldehyde), and retinoic acid (vitamin A acid), which have similar biologic activities. Retinol is the chemical name given to vitamin A. It is the transport form and, as retinol ester, also the storage form. The generic term retinoids encompasses vitamin A in its various forms and both natural and synthetic chemicals that are structurally related to vitamin A, but may not necessarily have vitamin A–like biologic activity.50 Animal-derived foods such as liver, fish, eggs, milk, and butter are important dietary sources of preformed vitamin A. Yellow and leafy green vegetables such as carrots, squash, and spinach supply large amounts of carotenoids, which are provitamins that can be metabolized to active vitamin A in the body. Carotenoids contribute approximately 30% of the vitamin A in human diets; the most important of these is β-carotene, which is efficiently converted to vitamin A. The Recommended Dietary Allowance for vitamin A is expressed in retinol equivalents, to take into account both preformed vitamin A and β-carotene.
Vitamin A is a fat-soluble vitamin, and its absorption requires bile, pancreatic enzymes, and some level of antioxidant activity in the food. Retinol (generally ingested as retinol ester) and β-carotene are absorbed in the intestine, where β-carotene is converted to retinol (Fig. 9-25). Retinol is then transported in chylomicrons to the liver for esterification and storage. Uptake in liver cells takes place through the apolipoprotein E receptor. More than 90% of the body’s vitamin A reserves are stored in the liver, predominantly in the perisinusoidal stellate (Ito) cells. In healthy persons who consume an adequate diet, these reserves are sufficient to meet the body’s demands for at least 6 months. Retinol esters stored in the liver can be mobilized; before release, retinol binds to a specific retinol-binding protein (RBP), synthesized in the liver. The uptake of retinol/RBP in peripheral tissues is dependent on cell surface receptors specific for RBP.51 After uptake, retinol binds to a cellular RBP, and the RBP is released back into the blood. Retinol may be stored in peripheral tissues as retinol ester or be oxidized to form retinoic acid. Retinoic acid has important effects in epithelial differentiation and growth.
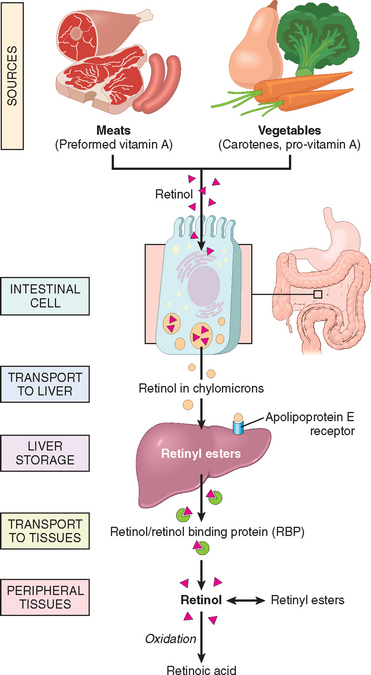
Function.
In humans the main functions of vitamin A are the following:
In addition, the retinoids, β-carotene, and some related carotenoids can function as photoprotective and antioxidant agents.
Retinoids are used clinically for the treatment of skin disorders such as severe acne and certain forms of psoriasis, and also in the treatment of acute promyelocytic leukemia. In this leukemia, a (15 : 17) translocation (Chapter 13) results in the fusion of a truncated RARα gene on chromosome 17 with the PML gene on chromosome 15. The fusion gene encodes an abnormal RAR that blocks myeloid cell differentiation. Pharmacologic doses of all-trans retinoic acid overcome the block, causing leukemia cells to differentiate into neutrophils, which subsequently die by apoptosis. This “differentiation therapy” induces remission in most individuals with acute promyelocytic leukemia and in combination with other chemotherapeutic agents can be curative. A different isomer, 13-cis retinoic acid, has been used with some success in the treatment of neuroblastomas in children.
Vitamin A Deficiency.
Vitamin A deficiency occurs worldwide either as a consequence of general undernutrition or as a secondary deficiency in individuals with conditions that cause malabsorption of fats. In children, stores of vitamin A are depleted by infections, and the absorption of the vitamin is poor in newborn infants. Adult patients with malaborption syndromes, such as celiac disease, Crohn’s disease, and colitis, may develop vitamin A deficiency, in conjunction with depletion of other fat-soluble vitamins. Bariatric surgery and, in elderly persons, continuous use of mineral oil as a laxative may lead to deficiency. The pathologic effects of vitamin A deficiency are summarized in Figure 9-26.
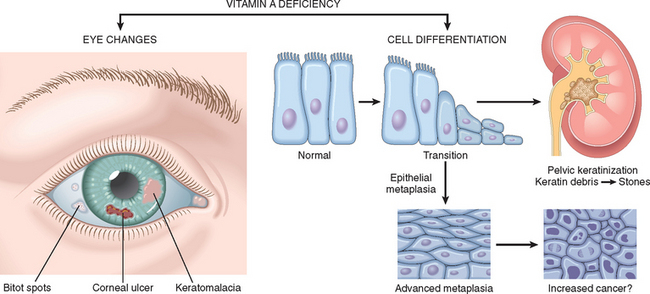
FIGURE 9-26 Vitamin A deficiency: its major consequences in the eye and in the production of keratinizing metaplasia of specialized epithelial surfaces, and its possible role in epithelial metaplasia. Not depicted are night blindness and immune deficiency.
As already discussed, vitamin A is a component of rhodopsin and other visual pigments. Not surprisingly, one of the earliest manifestations of vitamin A deficiency is impaired vision, particularly in reduced light (night blindness). Other effects of deficiency are related to the role of vitamin A in maintaining the differentiation of epithelial cells. Persistent deficiency gives rise to a series of changes involving epithelial metaplasia and keratinization. The most devastating changes occur in the eyes and are referred to as xerophthalmia (dry eye). First, there is dryness of the conjunctiva (xerosis conjunctivae) as the normal lacrimal and mucus-secreting epithelium is replaced by keratinized epithelium. This is followed by buildup of keratin debris in small opaque plaques (Bitot spots) and, eventually, erosion of the roughened corneal surface with softening and destruction of the cornea (keratomalacia) and total blindness.
In addition to the ocular epithelium, the epithelium lining the upper respiratory passage and urinary tract is replaced by keratinizing squamous cells (squamous metaplasia). Loss of the mucociliary epithelium of the airways predisposes to secondary pulmonary infections, and desquamation of keratin debris in the urinary tract predisposes to renal and urinary bladder stones. Hyperplasia and hyperkeratinization of the epidermis with plugging of the ducts of the adnexal glands may produce follicular or papular dermatosis. Another very serious consequence is immune deficiency, which is responsible for higher mortality rates from common infections such as measles, pneumonia, and infectious diarrhea. In parts of the world where a deficiency of vitamin A is prevalent, dietary supplements reduce mortality by 20% to 30%.
Vitamin A Toxicity.
Both short- and long-term excesses of vitamin A may produce toxic manifestations, a point of concern because of the megadoses touted by certain sellers of supplements. The consequences of acute hypervitaminosis A were first described by Gerrit de Veer in 1597, a ship’s carpenter stranded in the Arctic, who recounted in his diary the serious symptoms that he and other members of the crew developed after eating polar bear liver. With this cautionary tale in mind, the adventurous eater should be aware that acute vitamin A toxicity has also been described in individuals who ingested the livers of whales, sharks, and even tuna! The symptoms of acute vitamin A toxicity include headache, dizziness, vomiting, stupor, and blurred vision, symptoms that may be confused with those of a brain tumor (pseudotumor cerebri). Chronic toxicity is associated with weight loss, anorexia, nausea, vomiting, and bone and joint pain. Retinoic acid stimulates osteoclast production and activity, which lead to increased bone resorption and high risk of fractures. Although synthetic retinoids used for the treatment of acne are not associated with these types of conditions, their use in pregnancy should be avoided because of the well-established teratogenic effects of retinoids.
Vitamin D
The major function of the fat-soluble vitamin D is the maintenance of adequate plasma levels of calcium and phosphorus to support metabolic functions, bone mineralization, and neuromuscular transmission.53 Vitamin D is required for the prevention of bone diseases known as rickets (in children whose epiphyses have not already closed), osteomalacia (in adults), and hypocalcemic tetany. This latter condition is a convulsive state caused by an insufficient extracellular concentration of ionized calcium, which is required for normal neural excitation and the relaxation of muscles. Rickets was nearly endemic in large European cities and poor areas of New York and Boston at the end of the nineteenth century. Although cod liver oil was recognized for its anti-rachitic properties in the early part of that century, it took almost 100 years for it to be accepted by the medical profession as an effective preventive agent (it did not help that cod liver oil consumed in fishing villages in Northern Europe, Scandinavia, and Iceland was a dark, foul-smelling liquid).54 In addition to its effects on calcium and phosphorus homeostasis, vitamin D has effects in non-skeletal tissues (so called “nonclassical” effects).
Metabolism of Vitamin D.
The major source of vitamin D for humans is its endogenous synthesis in the skin by photochemical conversion of a precursor, 7-dehydrocholesterol, via the energy of solar or artificial UV light in the range of 290 to 315 nm (UVB radiation). Irradiation of 7-dehydrocholesterol forms cholecalciferol, known as vitamin D3. For the sake of simplicity we will use the term vitamin D to refer to this compound. Under usual conditions of sun exposure, about 90% of the vitamin D requirement is endogenously derived from 7-dehydrocholesterol present in the skin. However, individuals with dark skin generally have a lower level of vitamin D production because of melanin pigmentation. Dietary sources, such as deep-sea fish, plants, and grains, contribute about 10% of required vitamin D and depend on adequate intestinal fat absorption. In plants, vitamin D is present in its precursor form (ergosterol), which is converted to vitamin D in the body.
The main steps of vitamin D metabolism are summarized below53 and shown in Figure 9-27.
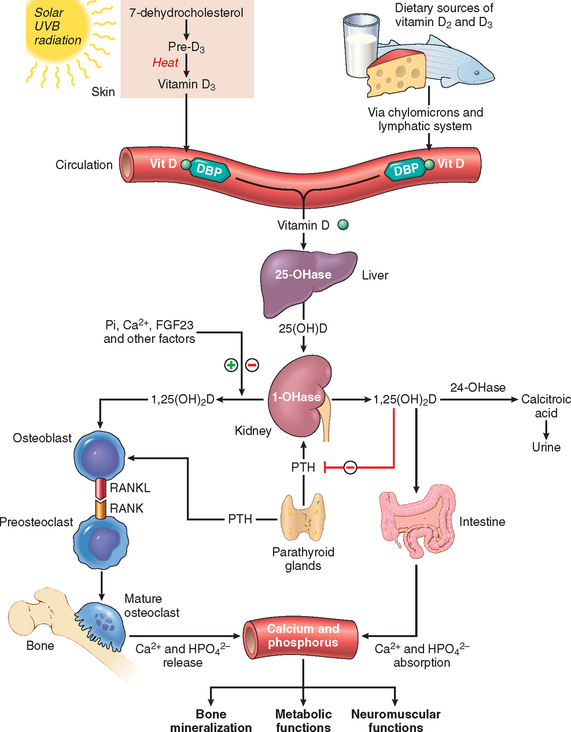
FIGURE 9-27 Vitamin D metabolism. Vitamin D is produced from 7-dehydrocholesterol in the skin or is ingested in the diet. It is converted in the liver into 25(OH)D, and in kidney into 1,25(OH)2D (1,25-dihydroxyvitamin D), the active form of the vitamin. 1,25(OH)2D stimulates the expression of RANKL, an important regulator of osteoclast maturation and function, on osteoblasts, and enhances the intestinal absorption of calcium and phosphorus in the intestine. See text for further details. DBP, vitamin d-binding protein (α1-globulin).
The production of 1,25-dihydroxyvitamin D in the kidney is regulated by three main mechanisms (Fig. 9-27): (a) hypocalcemia stimulates secretion of parathyroid hormone (PTH), which in turn augments the conversion of 25-OH-D into 1,25-dihydroxyvitamin D by activating 1α-hydroxylase; (b) hypophosphatemia directly activates α1-hydroxylase, increasing the production of 1,25-dihydroxyvitamin D; (c) through a feedback mechanism, increased levels of 1,25dihydroxyvitamin D down-regulate its own synthesis through inhibition of 1α-hydroxylase activity.
Mechanisms of Action.
1,25-dihydroxyvitamin D, the biologically active form of vitamin D, is best regarded as a steroid hormone. It binds to the high-affinity vitamin D receptor (VDR), which associates with the already mentioned RXR. This heterodimeric complex binds to vitamin D response elements located in the promoter of vitamin D target genes. The receptors for 1,25-dihydroxyvitamin D are present in most cells of the body and transduce signals that regulate plasma levels of calcium and phosphorus, through action on the small intestine, bones, and kidneys. Beyond its role on skeletal homeostasis, vitamin D also has immunomodulatory and antiproliferative effects. More recently it has been proposed that 1,25-dihydroxyvitamin D may also act through nongenomic mechanisms, which do not require the transcription of target genes. Nongenomic mechanisms may involve the binding of 1,25-dihydroxyvitamin D to a membrane vitamin D receptor, leading to the activation of protein kinase C and opening of calcium channels.55
Effects of Vitamin D on Calcium and Phosphorus Homeostasis.
The main functions of 1,25-dihydroxyvitamin D on calcium and phosphorus homeostasis are the following:
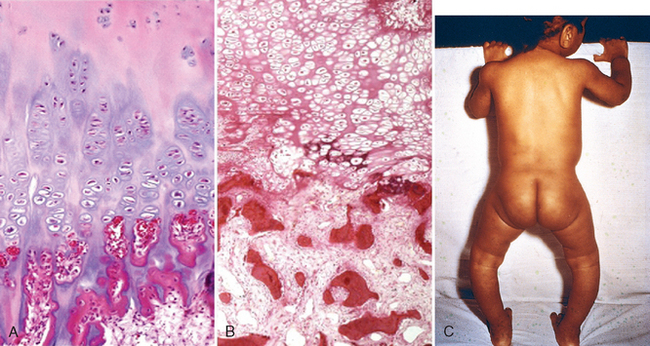
FIGURE 9-28 Rickets. A, Normal costochondral junction of a young child, illustrating formation of cartilage palisades and orderly transition from cartilage to new bone. B, Detail of a rachitic costochondral junction in which the palisades of cartilage is lost. Darker trabeculae are well-formed bone; paler trabeculae consist of uncalcified osteoid. C, Rickets, note bowing of legs due to formation of poorly mineralized bones.
(B, Courtesy of Dr. Andrew E. Rosenberg, Massachusetts General Hospital, Boston, MA.)
When hypocalcemia occurs in vitamin D deficiency (Fig. 9-29), PTH production is elevated, causing (1) activation of renal 1α-hydroxylase, increasing the amount of active vitamin D and calcium absorption; (2) increased resorption of calcium from bone by osteoclasts; (3) decreased renal calcium excretion; and (4) increased renal excretion of phosphate. Fibroblast growth factor 23, which is produced by bone, is one of a group of agents known as phosphatonins, which block the absorption of phosphate in the intestine, and phosphate reabsorption in the kidney, causing increased urinary excretion of phosphate. Although a normal serum level of calcium may be restored, hypophosphatemia persists, impairing the mineralization of bone. Increased production of fibroblast growth factor 23 may be responsible for tumor-induced osteomalacia and some forms of hypophosphatemic rickets.58

FIGURE 9-29 Vitamin D deficiency. There is inadequate substrate for the renal 1α-hydroxylase (1), yielding a deficiency of 1,25(OH)2D (2), and deficient absorption of calcium and phosphorus from the gut (3), with consequently depressed serum levels of both (4). The hypocalcemia activates the parathyroid glands (5), causing mobilization of calcium and phosphorus from bone (6a). Simultaneously, the parathyroid hormone (PTH) induces wasting of phosphate in the urine (6b) and calcium retention. As a result, the serum levels of calcium are normal or nearly normal, but phosphate levels are low; hence, mineralization is impaired (7).
Deficiency States.
The normal reference range for circulating 25-(OH)-D is 20 to 100 ng/mL; concentrations of less than 20 ng/mL constitute vitamin D deficiency.
Rickets in growing children (see Fig. 9-28C) and osteomalacia in adults are skeletal diseases with worldwide distribution. They may result from diets deficient in calcium and vitamin D, but an equally important cause of vitamin D deficiency is limited exposure to sunlight. This most often affects inhabitants of northern latitudes, but can even be a problem in tropical countries, in heavily veiled women, and in children born to mothers who have frequent pregnancies followed by lactation. In all of these situations, vitamin D deficiency can be prevented by a diet high in fish oils. Other, less common causes of rickets and osteomalacia include renal disorders causing decreased synthesis of 1,25-dihydroxyvitamin D, phosphate depletion, malabsorption disorders, and some rare inherited disorders.53 Although rickets and osteomalacia rarely occur outside high-risk groups, milder forms of vitamin D deficiency (also called vitamin D insufficiency), leading to an increase risk of bone loss and hip fractures, are quite common in the elderly in the United States and Europe.59 Some genetically determined variants of the vitamin D receptors are associated with an accelerated loss of bone minerals with aging and in certain familial forms of osteoporosis (Chapter 26).
Morphology. The basic derangement in both rickets and osteomalacia is an excess of unmineralized matrix. The following sequence ensues in rickets:
Rickets is most common during the first year of life. The gross skeletal changes depend on the severity and duration of the process and, in particular, the stresses to which individual bones are subjected. During the nonambulatory stage of infancy, the head and chest sustain the greatest stresses. The softened occipital bones may become flattened, and the parietal bones can be buckled inward by pressure; with the release of the pressure, elastic recoil snaps the bones back into their original positions (craniotabes). An excess of osteoid produces frontal bossing and a squared appearance to the head. Deformation of the chest results from overgrowth of cartilage or osteoid tissue at the costochondral junction, producing the “rachitic rosary.” The weakened metaphyseal areas of the ribs are subject to the pull of the respiratory muscles and thus bend inward, creating anterior protrusion of the sternum (pigeon breast deformity). When an ambulating child develops rickets, deformities are likely to affect the spine, pelvis, and tibia, causing lumbar lordosis and bowing of the legs (see Fig. 9-28C).
In adults, the lack of vitamin D deranges the normal bone remodeling that occurs throughout life. The newly formed osteoid matrix laid down by osteoblasts is inadequately mineralized, thus producing the excess of persistent osteoid that is characteristic of osteomalacia. Although the contours of the bone are not affected, the bone is weak and vulnerable to gross fractures or microfractures, which are most likely to affect vertebral bodies and femoral necks.
Histologically, the unmineralized osteoid can be visualized as a thickened layer of matrix (which stains pink in hematoxylin and eosin preparations) arranged about the more basophilic, normally mineralized trabeculae.
Non-Skeletal Effects of Vitamin D.
It was mentioned earlier that the vitamin D receptor is present in various cells and tissues that do not participate in calcium and phophorus homeostasis. Macrophages, keratinocytes, and tissues such as breast, prostate, and colon can produce 1,25-dihydroxyvitamin D.60 Within macrophages, synthesis of 1,25-dihydroxyvitamin D occurs through the activity of CYP27B located in the mitochondria. It has been proposed that pathogen-induced activation of Toll-like receptors in macrophages causes a transcription-induced increase in vitamin D receptor and CYP27B (Fig. 9-30). The resultant production of 1,25-dihydroxyvitamin D then stimulates the synthesis of cathelicidin, an antimicrobial peptide from the defensin family, which is effective against infection by Mycobacterium tuberculosis. Other effects of vitamin D in the innate and adaptive immune system have been reported,61 but the data are often contradictory. Vitamin D regulates the expression of more than 200 genes, including genes that participate in cell proliferation, differentiation, apoptosis, and angiogenesis. It has been reported that levels of 1,25-dihydroxyvitamin D below 20 ng/mL are associated with a 30% to 50% increase in the incidence of colon, prostate, and breast cancers.
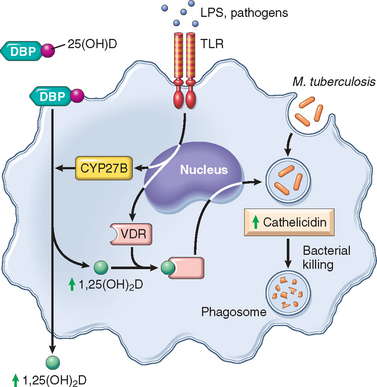
FIGURE 9-30 Anti-microbial effect of vitamin D. Pathogens and lipopolysaccharides (LPS) stimulate Toll-like receptors (TLRs) in macrophages, causing the transcription of vitamin D receptor (VDR) and an increase in CYP27B activity in mitochondria. This causes the production of 1,25(OH)2D (1,25-dihydroxyvitamin D), which stimulates the synthesis of cathelicidin, an antimicrobial peptide that is particularly active against Mycobacterium tuberculosis.
Vitamin D Toxicity.
Prolonged exposure to normal sunlight does not produce an excess of vitamin D, but megadoses of orally administered vitamin can lead to hypervitaminosis. In children, hypervitaminosis D may take the form of metastatic calcifications of soft tissues such as the kidney; in adults it causes bone pain and hypercalcemia. In passing, we might point out that the toxic potential of this vitamin is so great that in sufficiently large doses it is a potent rodenticide!
Vitamin C (Ascorbic Acid)
A deficiency of water-soluble vitamin C leads to the development of scurvy, characterized principally by bone disease in growing children and by hemorrhages and healing defects in both children and adults. Sailors of the British Royal Navy were nicknamed “limeys,” because at the end of the eighteenth century the Navy began to provide lime and lemon juice (rich sources of vitamin C) to sailors to prevent scurvy during their long sojourn at sea. It was not until 1932 that ascorbic acid was identified and synthesized. Ascorbic acid is not synthesized endogenously in humans; therefore, we are entirely dependent on the diet for this nutrient. Vitamin C is present in milk and some animal products (liver, fish) and is abundant in a variety of fruits and vegetables. All but the most restricted diets provide adequate amounts of vitamin C.
Function.
Ascorbic acid functions in a variety of biosynthetic pathways by accelerating hydroxylation and amidation reactions. The best-established function of vitamin C is the activation of prolyl and lysyl hydroxylases from inactive precursors, providing for hydroxylation of procollagen. Inadequately hydroxylated procollagen cannot acquire a stable helical configuration or be adequately cross-linked, so it is poorly secreted from the fibroblast. Those molecules that are secreted lack tensile strength and are more soluble and vulnerable to enzymatic degradation. Collagen, which normally has the highest content, of hydroxyproline, of any polypeptide is most affected, particularly in blood vessels, accounting for the predisposition to hemorrhages in scurvy. In addition, a deficiency of vitamin C suppresses the rate of synthesis of procollagen, independent of an effect on proline hydroxylation.
While the role of vitamin C in collagen synthesis has been known for many decades, it is only in relatively recent years that its antioxidant properties have been recognized. Vitamin C can scavenge free radicals directly and can act indirectly by regenerating the antioxidant form of vitamin E.
Deficiency States.
Consequences of vitamin C deficiency (scurvy) are illustrated in Figure 9-31. Fortunately, because of the abundance of ascorbic acid in many foods, scurvy has ceased to be a global problem. It is sometimes encountered even in affluent populations as a secondary deficiency, particularly among elderly individuals, persons who live alone, and chronic alcoholics, groups that often have erratic and inadequate eating patterns. Occasionally, scurvy appears in patients undergoing peritoneal dialysis and hemodialysis and among food faddists. Tragically, the condition sometimes appears in infants who are maintained on formulas of evaporated milk without supplementation of vitamin C.
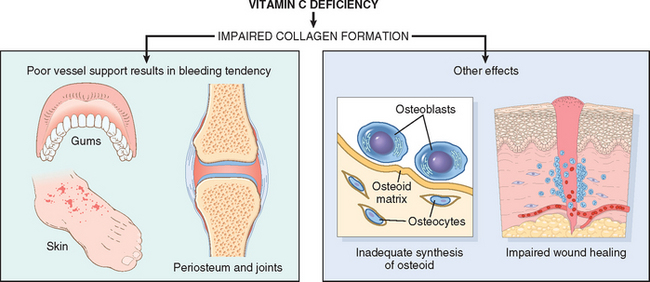
Vitamin C Toxicity.
The popular notion that megadoses of vitamin C protect against the common cold, or at least allay the symptoms, has not been borne out by controlled clinical studies. Such slight relief as may be experienced is probably due to the mild antihistamine action of ascorbic acid. Similarly there is little support that large doses of vitamin C protect against cancer development. The physiologic availability of vitamin C is limited. It is unstable, poorly absorbed in the intestine, and promptly excreted in the urine.
Other vitamins and some essential minerals are listed and briefly described in Tables 9-9 and 9-10. Some vitamins are discussed in other chapters, as indicated in the tables.

OBESITY
Excess adiposity (known as obesity) and excess body weight are associated with the increased incidence of several of the most important diseases of humans, including type 2 diabetes, dyslipidemias, cardiovascular disease, hypertension, and cancer. Obesity is defined as an accumulation of adipose tissue that is of sufficient magnitude to impair health. As with weight loss, excess weight is best assessed by the body mass index or BMI. For practical reasons, body weight, which generally correlates well with BMI, is often used as a surrogate for BMI measurements. The normal BMI range is 18.5 to 25 kg/m2, although the range may differ for different countries. Individuals with BMI above 30 kg/m2 are classified as obese; those with BMI between 25 kg/m2 and 30 kg/m2 are considered to be overweight. For the sake of simplicity, unless otherwise noted, the term obesity will be applied to both the truly obese and the overweight.
Accumulation of body fat may also be measured by triceps skinfold thickness, mid-arm circumference, and the ratio between waist and hip circumferences. Not only the total body weight but also the distribution of the stored fat is of importance in obesity. Central, or visceral, obesity, in which fat accumulates in the trunk and in the abdominal cavity (in the mesentery and around viscera), is associated with a much higher risk for several diseases than is excess accumulation of fat diffusely in subcutaneous tissue.
Obesity is a major public health problem, which, until about a dozen years ago, was confined to developed countries. Since then, it has also become an important health problem in developing nations, and in certain countries obesity coexists with malnutrition in individual families. In the United States obesity has reached epidemic proportions. The prevalence of obesity increased from 13% to 32% between 1960 and 2004; currently 66% of adults in the United States are overweight or obese, and 16% of children are overweight. If current trends continue, it is projected that by the year 2015, 41% of adults will be obese.62 The increase in obesity in the United States has been associated with the higher caloric content of the diet, mostly caused by increased consumption of refined sugars, sweetened beverages, and vegetable oils.
At its simplest level, obesity is a disease of caloric imbalance that results from an excess intake of calories above their consumption by the body. However, the pathogenesis of obesity is exceedingly complex and not yet completely understood. Ongoing research has identified complex humoral and neural mechanisms that control appetite and satiety. These neurohumoral mechanisms respond to genetic, nutritional, environmental, and psychologic signals, and trigger a metabolic response through the stimulation of centers located in the hypothalamus. There is little doubt that genetic influences play an important role in weight control, but obesity is a disease that depends on the interaction between multiple factors. After all, regardless of genetic makeup, obesity would not occur without intake of food!
In a simplified way the neurohumoral mechanisms that regulate energy balance can be subdivided into three components (illustrated in Figs. 9-29 and 9-30):
POMC/CART neurons enhance energy expenditure and weight loss through the production of the anorexigenic α-melanocyte-stimulating hormone (MSH), and the activation of the melanocortin receptors 3 and 4 (MC3/4R) in second-order neurons. NPY/AgRP neurons promote food intake (orexigenic effect) and weight gain, through the activation of Y1/5 receptors in secondary neurons.
We will now discuss three important components of the afferent system that regulates appetite and satiety: leptin, adiponectin, and gut hormones.
Leptin.
The name leptin is derived from the Greek term leptos, meaning “thin.” Leptin, a 16-kD hormone synthesized by fat cells, is the product of the ob gene. The leptin receptor (OB-R) is the product of the diabetes (db) gene and belongs to the type I cytokine receptor superfamily that includes the gp130, granulocyte-colony-stimulating factor, IL-2, and IL-6 receptors. Mice genetically deficient in leptin (ob/ob mice) or leptin receptors (db/db mice) fail to sense the adequacy of fat stores, overeat, and gain weight, behaving as if they are undernourished. Thus, the obesity of these animals is a consequence of the lack of the signal for energy sufficiency that is normally provided by leptin.63
Although in a general sense leptin levels are regulated by the adequacy of fat stores, the precise mechanisms that regulate the output of leptin from adipose tissue have not been completely defined, but it has been established that leptin secretion is stimulated when fat stores are abundant. It is believed that insulin-stimulated glucose metabolism is an important factor in the regulation of leptin levels. Leptin levels are regulated by multiple post-transcriptional mechanisms that affect its synthesis, secretion, and turnover. In the hypothalamus, leptin stimulates POMC/CART neurons that produce anorexigenic neuropeptides (primarily melanocyte-stimulating hormone) and inhibits NPY/AgRP neurons that produce feeding-inducing (orexigenic) neuropeptides (see Figs. 9-32 and 9-33). In individuals with stable weight, the activities of the opposing POMC/CART and NPY/AgRP pathways are properly balanced. However, when there are inadequate stores of body fat, leptin secretion is diminished and food intake is increased.
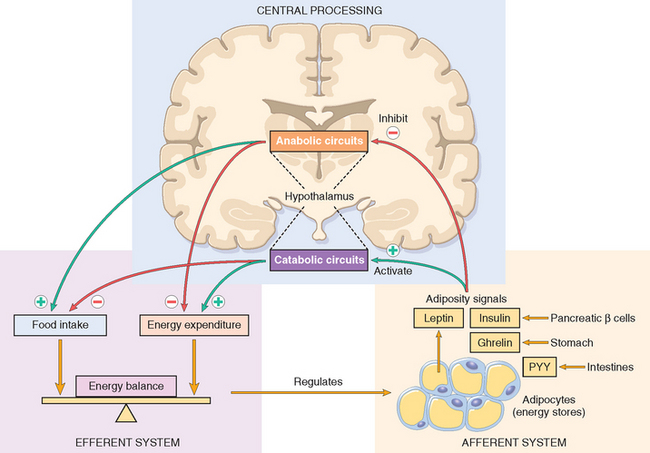
FIGURE 9-32 Regulation of energy balance. Adipose tissues generate afferent signals that influence the activity of the hypothalamus, which is the central regulator of appetite and satiety. These signals decrease food intake by inhibiting anabolic circuits, and enhance energy expenditure through the activation of catabolic circuits. PYY, peptide YY. See text for details.
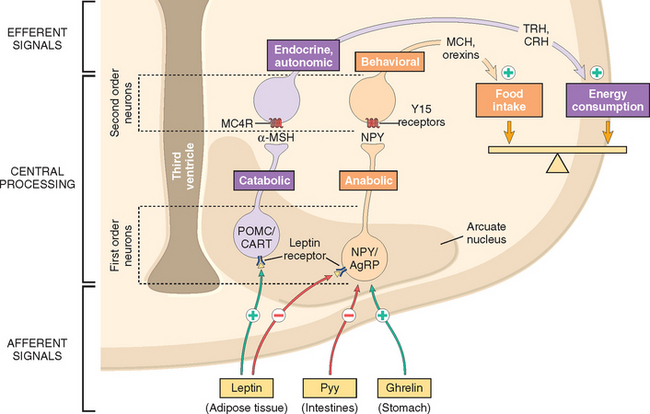
FIGURE 9-33 Neurohumoral circuits in the hypothalamus that regulate energy balance. Shown are POMC/CART anorexigenic neurons and NPY/AgRP orexigenic neurons in the arcuate nucleus of the hypothalamus, and their pathways. See text for details.
Humans with loss-of-function mutations in the leptin system develop early-onset severe obesity, but this is a rare condition. Mutations of melanocortin receptor 4 (MC4R) and its downstream pathways are more frequent, being responsible for about 5% of massive obesity. In these individuals, sensing of satiety (anorexigenic signal) is not generated, and hence they behave as if they are undernourished. It has recently been reported64 that haplo-insufficiency of brain-derived neurotrophic factor (BDNF), an important component of MC4R downstream signaling in the hypothalamus, is associated with obesity in patients with the WAGR syndrome (this is a very rare condition that includes Wilms tumor, aniria, genito-urinary defects, and mental retardation in addition to obesity Chapter 10). Although the defects in leptin and MC4R detected so far are uncommon, they underscore the importance of these systems in the control of energy balance and body weight. Perhaps other defects in these pathways may have pathogenic effects in more common forms of obesity. For instance, it has been proposed that leptin resistance rather than leptin deficiency may be prevalent in humans.
Leptin regulates not only food intake but also energy expenditure, through a distinct set of pathways. Thus, an abundance of leptin stimulates physical activity, heat production, and energy expenditure. The neurohumoral mediators of leptin-induced energy expenditure are less well defined. Thermogenesis, an important catabolic effect mediated by leptin, is controlled in part by hypothalamic signals that increase the release of norepinephrine from sympathetic nerve endings in adipose tissue. In addition to these effects, leptin can function as a pro-inflammatory cytokine and participates in the regulation of hematopoiesis and lymphopoiesis.65 The OB-R receptor is highly similar structurally to the IL-6 receptor and activates the JAK/STAT pathway.
Adiponectin.
Injections of adiponectin in mice stimulate fatty acid oxidation in muscle, causing a decrease in fat mass. This hormone is produced mainly by adipocytes. Its levels in the blood are very high, about 1000 times higher than those of other polypeptide hormones, and are lower in obese than in lean individuals.66 Adiponectin, which has been called a “fat-burning molecule” and the “guardian angel against obesity,” directs fatty acids to muscle for their oxidation. It decreases the influx of fatty acids to the liver and the total hepatic triglyceride content, and also decreases the glucose production in the liver, causing an increase in insulin sensitivity and a protection against the metabolic syndrome (described later).67 Adiponectin circulates as a complex of three, six, or even more aggregates of the monomeric form, and binds to two receptors, AdipoR1 and AdipoR2. These receptors are found in many tissues, including the brain, but AdipoR1 and AdipoR2 are most highly expressed in skeletal muscle and liver, respectively. Binding of adiponectin to its receptors triggers signals that activate cyclic adenosine monophosphate–activated protein kinase, which in turn phosphorylates and inactivates acetyl coenzyme A carboxylase, a key enzyme required for fatty acid synthesis.
Adipose Tissue.
In addition to leptin and adiponectin, adipose tissue produces cytokines such as TNF, IL-6, IL-1, and IL-18, chemokines, and steroid hormones. The increased production of cytokines and chemokines by adipose tissue in obese patients creates a chronic sub-clinical (asymptomatic) inflammatory state that includes high levels of circulating C-reactive protein. Through its multiple activities, adipose tissue participates in the control of energy balance and energy metabolism, functioning as a link between lipid metabolism, nutrition, and inflammatory responses. Thus, the adipocyte that was relegated to an obscure and passive role as the “Cinderella of cells of metabolism,” is now “the Belle of the Ball” at the forefront of metabolic research.68
The total number of adipocytes is established during childhood and adolescence, and it is higher in obese than in lean individuals.69 In adults the number of adipocytes remains constant, even after losses or weight gains, but there is a continuous turnover of the cell population. It is estimated that approximately 10% of adipocytes are renewed annually, regardless of the level of the individual’s body mass. Thus, although the fat mass in an adult person can increase through the enlargement of existing adipocytes, their number is tightly controlled, and is predetermined in childhood and adolescence. In individuals who lose weight after dietary regimens, the well-known difficulties in maintaining weight losses are, in part, a consequence of the lack of a decrease in the number of adipocytes, and the enhanced appetite caused by leptin deficiency.
Gut Hormones.
Gut peptides act as short-term meal initiators and terminators. They include ghrelin, PYY, pancreatic polypeptide, insulin, and amylin among others.70 Ghrelin is produced in the stomach and in the arcuate nucleus of the hypothalamus. It is the only known gut hormone that increases food intake (orexigenic effect). Its injection in rodents elicits voracious feeding, even after repeated administration. Long-term injections cause weight gain, by increasing caloric intake and reducing energy utilization. Ghrelin acts by binding the growth hormone secretagogue receptor, which is abundant in the hypothalamus and the pituitary. Although the precise mechanisms of ghrelin action have not been identified, it most likely stimulates NPY/AgRP neurons to increase food intake. Ghrelin levels rise before meals and fall between 1 and 2 hours after eating. However, in obese individuals the postprandial suppression of ghrelin is attenuated, leading to maintenance of the obesity.
PYY is secreted from endocrine cells in the ileum and colon. Plasma levels of PYY are low during fasting and increase shortly after food intake. Intravenous administration of PYY reduces energy intake, and its levels generally increase after gastric bypass surgery. By contrast, levels of PYY generally decrease in individuals with the Prader-Willi syndrome (caused by loss of imprinted genes on chromosome 15q11–q13),71 and may contribute to the development of hyperphagia and obesity in these persons. These observations have led to ongoing work to produce PYYs for the treatment of obesity. Amylin, a peptide secreted with insulin from pancreatic β-cells that reduces food intake and weight gain, is also being evaluated for the treatment of obesity and diabetes. Both PYY and amylin act centrally by stimulating POMC/CART neurons in the hypothalamus, causing a decrease in food intake.
General Consequences of Obesity
Obesity, particularly central obesity, increases the risk for a number of conditions, including type 2 diabetes and cardiovascular disease (Fig. 9-34). Obesity is the main driver of a cluster of alterations known as the metabolic syndrome characterized by visceral or intra-abdominal adiposity, insulin resistance, hyperinsulinemia, glucose intolerance, hypertension, hypertriglyceridemia, and low HDL cholesterol (Chapter 11).
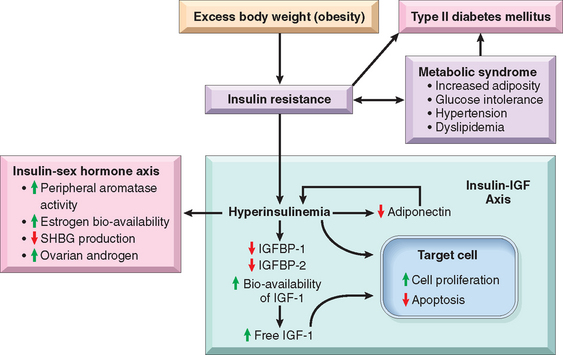
FIGURE 9-34 Obesity, metabolic syndrome, and cancer. Obesity and excessive weight are precursors of the metabolic syndrome, which is associated with insulin resistance, type 2 diabetes, and hormonal changes. Increases in insulin and IGF-1 (insulin-like growth factor-1) stimulate cell proliferation and inhibit apoptosis and may contribute to tumor development. IGF, insulin-like growth factor; IGFBP, insulin-like growth factor–binding protein; SHBG, sex hormone–binding globulin.
(Modified from Renehan AG et al.: Obesity and cancer risk: the role of the insulin-I6F axis. Trends Endocrinol Metab 17:328, 2006.)
Obesity and Cancer
Approximately 4% of cancers in men and 7% in women are associated with obesity.72 Data on the relationships between obesity and cancer have been obtained from the Million Women Study that examined the relationship between BMI and cancer in women aged 50 to 64 years in the United Kingdom, and from a systematic analysis of published data sets involving more than 280,000 cases of cancer in men and women.73,74
The mechanisms by which obesity is associated with these specific types of cancers are unknown, but a proposed hypothesis is that the increased cancer risk in obese individuals is a consequence of hyperinsulinemia and insulin resistance (Fig. 9-34). Insulin at high concentrations has multiple effects on cell growth, including the activation of phosphatidylinositol 3-kinase, extracellular-signal-regulated kinases 1 and 2, β-catenin, and Ras. All of these are important components of pathways that are dysregulated during cancer development. Hyperinsulinemia also causes an increase in insulin-like growth factor-1 (IGF-1) concentrations, because insulin inhibits the production of the IGF-binding proteins IGFBP-1 and IGFBP-2. IGF-1 is a mitogenic and anti-apoptotic agent that is highly expressed in many human cancers.75 It binds with high affinity to the IGF-1R receptor, and with low affinity to the insulin receptor. IGF-1 activates many of the cell growth pathways that are also activated by insulin, and increases the production of vascular endothelial growth factor, by inducing the expression of hypoxia-inducible factor 1.
In addition to the obesity-associated effects of insulin and IGF-1 in cell growth pathways, obesity and hyperinsulimia have an effect on steroid hormones that regulate cell growth and differentiation in the breast, uterus, and other tissues: (1) obesity increases the synthesis of estrogen from androgen precursors through an effect of adipose tissue aromatases; (2) insulin increases androgen synthesis in ovaries and adrenals, and enhances estrogen availability in obese persons by inhibiting the production of sex-hormone-binding globulin (SHBG) in the liver (see Fig. 9-34).
As already discussed in this chapter, adiponectin, secreted mostly from adipose tissue, is an abundant hormone that is inversely correlated with obesity and acts as an insulinsensitizing agent. Thus, the decreased levels of adiponectin in obese persons contribute to hyperinsulinemia and the impairment of insulin sensitivity.
DIETS, CANCER, AND ATHEROSCLEROSIS
Diet and Cancer
The incidence of specific cancers varies widely throughout the world. The frequency of some tumors varies as much as 100-fold in different geographic areas. It is also well known that differences in incidence of various cancers is not fixed and can be modified by nongenetic factors, including changes in diet. For instance, the incidence of colon cancer in Japanese men and women 55 to 60 years of age was negligible about 50 years ago, but it is now higher than that in men of the same age in the United Kingdom.76 Studies have also shown a progressive increase in colon cancers in Japanese populations as they moved from Japan to Hawaii and from there to the continental United States. Nevertheless, despite the very large amount of experimental and epidemiologic research, relatively few mechanisms that link diets and specific types of cancer have been established.
With respect to carcinogenesis, three aspects of the diet are of major concern: (1) the content of exogenous carcinogens, (2) the endogenous synthesis of carcinogens from dietary components, and (3) the lack of protective factors.
Thus, we must conclude that despite many tantalizing trends and proclamations by “diet gurus,” thus far there is no definitive proof that a particular diet can cause or prevent cancer. On the other hand, given the relationships between obesity and cancer development, prevention of obesity through the consumption of a healthy diet is a commonsense measure that goes a long way in preserving good health. Concern persists that carcinogens lurk in things as pleasurable as a juicy steak, a rich ice cream, and in nuts contaminated with aflatoxin.
Diet and Atherosclerosis
A most important and controversial issue is the contribution of diet to atherogenesis. The central question is “can dietary modification—specifically, reduction in the consumption of cholesterol and saturated animal fats (e.g., eggs, butter, beef)—reduce serum cholesterol levels and prevent or retard the development of atherosclerosis (most importantly, coronary heart disease)?” The average adult in the United States consumes a large amount of fat and cholesterol daily, with a ratio of saturated fatty acids to polyunsaturated fatty acids of about 3 : 1. Lowering this ratio to 1 : 1 causes a 10% to 15% reduction in the serum cholesterol level within a few weeks. Vegetable oils (e.g., corn and safflower oils) and fish oil contain polyunsaturated fatty acids and are good sources of such cholesterol-lowering lipids. Fish oil fatty acids belonging to the omega-3 family have more double bonds than do the omega-6 fatty acids present in vegetable oils. A study of Dutch men whose usual daily diet contained 30 g of fish revealed a substantially lower frequency of death from coronary heart disease than that among comparable controls.
There is much talk about the role that caloric restriction and special diets may play in the control of body weight and prevention of cardiovascular disease. We offer just a few general observations on these topics.
1 Stein C, et al. The global burden of disease assessments—WHO is responsible? PLoS Negl Trop Dis. 2007;1:e161.
2 Mathers CD, et al. Measuring the burden of neglected tropical diseases: the global burden of disease framework. PLoS Negl Trop Dis. 2007;1:e114.
3 Murray CJ, et al. Can we achieve Millennium Development Goal 4? New analysis of country trends and forecasts of under-5 mortality to 2015. Lancet. 2007;370:1040.
4 Jones KE, et al. Global trends in emerging infectious diseases. Nature. 2008;451:990.
5 Patz JA, et al. Impact of regional climate change on human health. Nature. 2005;438:310.
6 Shea KM. Global climate change and children’s health. Pediatrics. 2007;120:e1359.
7 Patz JA, Kovats RS. Hotspots in climate change and human health. BMJ. 2002;325:1094.
8 McMichael AJ, et al. Climate change and human health: present and future risks. Lancet. 2006;367:859.
9 Iyanagi T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: implications for detoxification. Int Rev Cytol. 2007;260:35.
10 Tompkins LM, Wallace AD. Mechanisms of cytochrome P450 induction. J Biochem Mol Toxicol. 2007;21:176.
11 Pichavant M, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med. 2008;205:385.
12 McCreanor J, et al. Respiratory effects of exposure to diesel traffic in persons with asthma. N Engl J Med. 2007;357:2348.
13 Mills NL, et al. Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. N Engl J Med. 2007;357:1075.
14 Bellinger DC, Bellinger AM. Childhood lead poisoning: the torturous path from science to policy. J Clin Invest. 2006;116:853.
15 Bellinger DC. Very low lead exposures and children’s neurodevelopment. Curr Opin Pediatr. 2008;20:172.
16 Guzzi G, La Porta CA. Molecular mechanisms triggered by mercury. Toxicology. 2008;244:1.
17 Thompson WW, et al. Early thimerosal exposure and neuropsychological outcomes at 7 to 10 years. N Engl J Med. 2007;357:1281.
18 Vahidnia A, et al. Arsenic neurotoxicity—a review. Hum Exp Toxicol. 2007;26:823.
19 Ratnaike RN. Acute and chronic arsenic toxicity. Postgrad Med J. 2003;79:391.
20 Parvez F, et al. Non-malignant respiratory effects of chronic arsenic exposure from drinking water in never-smokers in Bangladesh. Environ Health Perspect. 2008;116:190.
21 Nawrot T, et al. Environmental exposure to cadmium and risk of cancer: a prospective population-based study. Lancet Oncol. 2006;7:119.
22 Kenfield SA, et al. Smoking and smoking cessation in relation to mortality in women. JAMA. 2008;299:2037.
23 Sun S, et al. Lung cancer in never smokers—a different disease. Nat Rev Cancer. 2007;7:778.
24 Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599.
25 Bailey BA, Sokol RJ. Pregnancy and alcohol use: evidence and recommendations for prenatal care. Clin Obst Gyn. 2008;51:436.
26 Baur JA, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337.
27 Lagouge M, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109.
28 MacLennan AH. HRT: a reappraisal of the risks and benefits. Med J Aust. 2007;186:643.
29 Li CI, et al. Relationship between menopausal hormone therapy and risk of ductal, lobular, and ductal-lobular breast carcinomas. Cancer Epidemiol Biomarkers Prev. 2008;17:43.
30 Mendelsohn ME, Karas RH. HRT and the young at heart. N Engl J Med. 2007;356:2639.
31 American Society for Reproductive Medicine. Hormonal contraception: recent advances and controversies. Fertil Steril. 2006;86:S229.
32 Bessems JG, Vermeulen NP. Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues and protective approaches. Crit Rev Toxicol. 2001;31:55.
33 Liu ZX, Kaplowitz N. Role of innate immunity in acetaminophen-induced hepatotoxicity. Expert Opin Drug Metab Toxicol. 2006;2:493.
34 Bamford NS, et al. Repeated exposure to methamphetamine causes long-lasting presynaptic corticostriatal depression that is renormalized with drug readministration. Neuron. 2008;58:89.
35 Wilkins MR. Cannabis and cannabis-based medicines: potential benefits and risks to health. Clin Med. 2006;6:16.
36 Pagotto U, et al. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev. 2006;27:73.
37 Kunos G, Osei-Hyiaman D. Endocannabinoid involvement in obesity and hepatic steatosis. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1101.
38 Gibran NS, et al. Cutaneous wound healing. J Burn Care Res. 2007;28:577.
39 Scott JR, et al. Making sense of hypertrophic scar: a role for nerves. Wound Repair Regen. 2007;15(Suppl 1):S27.
40 Durham WJ, et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53.
41 Stone HB, et al. Effects of radiation on normal tissue: consequences and mechanisms. Lancet Oncol. 2003;4:529.
42 Wright EG, Coates PJ. Untargeted effects of ionizing radiation: implications for radiation pathology. Mutat Res. 2006;597:119.
43 Hagelstrom RT, et al. DNA-PKcs and ATM influence the generation of ionizing radiation-induced bystander effects. Oncogene Epub. 2008.
44 Brenner DJ, et al. Cancer risks attributable to low doses of ionizing radiation: assessing what we really know. Proc Natl Acad Sci U S A. 2003;100:13761.
45 Brenner DJ, Hall EJ. Computed tomography—an increasing source of radiation exposure. N Engl J Med. 2007;357:2277.
46 Schaible UE, Kaufmann SH. Malnutrition and infection: complex mechanisms and global impacts. PLoS Med. 2007;4:e115.
47 Acharyya S, Guttridge DC. Cancer cachexia signaling pathways continue to emerge yet much still points to the proteasome. Clin Cancer Res. 2007;13:1356.
48 Acharyya S, et al. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell. 2005;8:421.
49 Kaye W. Neurobiology of anorexia and bulimia nervosa. Physiol Behav. 2008;94:121.
50 Ziouzenkova O, Plutzky J. Retinoid metabolism and nuclear receptor responses: new insights into coordinated regulation of the PPAR-RXR complex. FEBS Lett. 2008;582:32.
51 Germain P, et al. International Union of Pharmacology. LX. Retinoic acid receptors. Pharmacol Rev. 2006;58:712.
52 Ziouzenkova O, Plutsky J. Retinoid metabolism and nuclear receptor responses: new insights into coordinated regulation of the PPAR-RXR complex. FEBS Lett. 2008;9:582.
53 Holick MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest. 2006;116:2062.
54 Rajakumar K, et al. Solar ultraviolet radiation and vitamin D: a historical perspective. Am J Public Health. 2007;97:1746.
55 Deeb KK, et al. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007;7:684.
56 Mensenkamp AR, et al. TRPV5, the gateway to Ca2+ homeostasis. Handb Exp Pharmacol. 2007;179:207.
57 Hoenderop JG, et al. Calcium absorption across epithelia. Physiol Rev. 2005;85:373.
58 Berndt T, Kumar R. Phosphatonins and the regulation of phosphate homeostasis. Annu Rev Physiol. 2007;69:341.
59 Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266.
60 Schauber J, et al. Histone acetylation in keratinocytes enables control of the expression of cathelicidin and CD14 by 1,25-dihydroxyvitamin D3. J Invest Dermatol. 2008;128:816.
61 Adams JS, Hewison M. Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat Clin Pract Endocrinol Metab. 2008;4:80.
62 Wang Y, Beydoun MA. The obesity epidemic in the United States—gender, age, socioeconomic, racial/ethnic, and geographic characteristics: a systematic review and meta-regression analysis. Epidemiol Rev. 2007;29:6.
63 Badman MK, Flier JS. The adipocyte as an active participant in energy balance and metabolism. Gastroenterology. 2007;132:2103.
64 Froguel P, Blakemore AIF. The power of the extreme in elucidating obesity. N Engl J Med. 2008;359:891.
65 Lam QL, Lu L. Role of leptin in immunity. Cell Mol Immunol. 2007;4:1.
66 Guerre-Millo M. Adiponectin: an update. Diabetes Metab. 2008;34:12.
67 Garaulet M, et al. Adiponectin, the controversial hormone. Public Health Nutr. 2007;10:1145.
68 O’Rahilly S. Human obesity and insulin resistance: lessons from experiments of nature. Novartis Found Symp. 2007;286:13.
69 Spalding KL, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783.
70 Huda MS, et al. Gut peptides and the regulation of appetite. Obes Rev. 2006;7:163.
71 Davies W, et al. Imprinted genes and neuroendocrine function. Front Neuroendocrinol. 2007;29:413.
72 Polednak AP. Estimating the number of U.S. incidence cancers attributable to obesity and the impact on temporal trends in incidence rates for obesity-related cancers. Cancer Detect Prev Epub. 2008.
73 Reeves GK, et al. Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ. 2007;335:1134.
74 Renehan AG, et al. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569.
75 Renehan AG, et al. Obesity and cancer risk: the role of the insulin-IGF axis. Trends Endocrinol Metab. 2006;17:328.
76 Bingham S, Riboli E. Diet and cancer—the European Prospective Investigation into Cancer and Nutrition. Nat Rev Cancer. 2004;4:206.