Chapter 4 Hemodynamic Disorders, Thromboembolic Disease, and Shock
As a group, cardiovascular diseases are the most important cause of morbidity and mortality in Western society. In the year 2005, it was estimated that 81 million people in the United States had one or more forms of cardiovascular disease, which were responsible for 35% to 40% of deaths. Included in this category are diseases that primarily affect one of the three major components of the cardiovascular system: the heart; the blood vessels; and the blood itself, which is composed of water, salts, a wide variety of proteins, elements that regulate clotting (the coagulation factors and platelets), and other formed elements (red cells and white cells). For the sake of simplicity we will consider disorders affecting each component of the cardiovascular system separately, recognizing that disturbances affecting one component often lead to adaptations and abnormalities involving others. Here, we focus on disorders of hemodynamics (edema, congestion, and shock) and hemostasis (hemorrhage and thrombosis), as well as various forms of embolism. Diseases that primarily affect the blood vessels and the heart will be discussed in Chapters 11 and 12, respectively.
Edema
Approximately 60% of lean body weight is water. Two thirds of the body’s water is intracellular, and the remainder is in extracellular compartments, mostly the interstitium (or third space) that lies between cells; only about 5% of total body water is in blood plasma. The movement of water and low molecular weight solutes such as salts between the intravascular and interstitial spaces is controlled primarily by the opposing effect of vascular hydrostatic pressure and plasma colloid osmotic pressure. Normally the outflow of fluid from the arteriolar end of the microcirculation into the interstitium is nearly balanced by inflow at the venular end; a small residual amount of fluid may be left in the interstitium and is drained by the lymphatic vessels, ultimately returning to the bloodstream via the thoracic duct. Either increased capillary pressure or diminished colloid osmotic pressure can result in increased interstitial fluid (Fig. 4-1). If the movement of water into tissues (or body cavities) exceeds lymphatic drainage, fluid accumulates. An abnormal increase in interstitial fluid within tissues is called edema, while fluid collections in the different body cavities are variously designated hydrothorax, hydropericardium, and hydroperitoneum (the last is more commonly called ascites). Anasarca is a severe and generalized edema with widespread subcutaneous tissue swelling.

FIGURE 4-1 Factors influencing fluid transit across capillary walls. Capillary hydrostatic and osmotic forces are normally balanced so that there is no net loss or gain of fluid across the capillary bed. However, increased hydrostatic pressure or diminished plasma osmotic pressure will cause extravascular fluid to accumulate. Tissue lymphatics remove much of the excess volume, eventually returning it to the circulation via the thoracic duct; however, if the capacity for lymphatic drainage is exceeded, tissue edema results.
There are several pathophysiologic categories of edema (Table 4-1). Edema caused by increased hydrostatic pressure or reduced plasma protein is typically a protein-poor fluid called a transudate. Edema fluid of this type is seen in patients suffering from heart failure, renal failure, hepatic failure, and certain forms of malnutrition, as described below and outlined in Figure 4-2. In contrast, inflammatory edema is a protein-rich exudate that is a result of increased vascular permeability. Edema in inflamed tissues is discussed in Chapter 2; noninflammatory causes of edema (see Fig. 4-2) are described below.
TABLE 4-1 Pathophysiologic Categories of Edema
| INCREASED HYDROSTATIC PRESSURE |
| REDUCED PLASMA OSMOTIC PRESSURE (HYPOPROTEINEMIA) |
| LYMPHATIC OBSTRUCTION |
| SODIUM RETENTION |
| INFLAMMATION |
Modified from Leaf A, Cotran RS: Renal Pathophysiology, 3rd ed. New York, Oxford University Press, 1985, p 146.
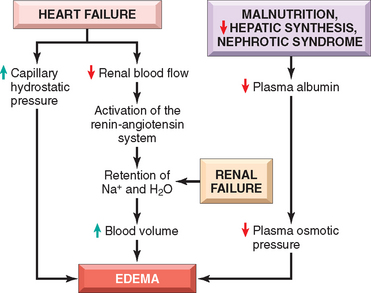
FIGURE 4-2 Pathways leading to systemic edema from primary heart failure, primary renal failure, or reduced plasma osmotic pressure (e.g., from malnutrition, diminished hepatic synthesis, or protein loss from nephrotic syndrome).
Increased Hydrostatic Pressure.
Regional increases in hydrostatic pressure can result from a focal impairment in venous return. Thus, deep venous thrombosis in a lower extremity may cause localized edema in the affected leg. On the other hand, generalized increases in venous pressure, with resulting systemic edema, occur most commonly in congestive heart failure (Chapter 12), where compromised right ventricular function leads to pooling of blood on the venous side of the circulation.
Reduced Plasma Osmotic Pressure.
Reduced plasma osmotic pressure occurs when albumin, the major plasma protein, is not synthesized in adequate amounts or is lost from the circulation. An important cause of albumin loss is the nephrotic syndrome (Chapter 20), in which glomerular capillaries become leaky; patients typically present with generalized edema. Reduced albumin synthesis occurs in the setting of severe liver diseases (e.g., cirrhosis, Chapter 18) or protein malnutrition (Chapter 9). In each case, reduced plasma osmotic pressure leads to a net movement of fluid into the interstitial tissues with subsequent plasma volume contraction. The reduced intravascular volume leads to decreased renal perfusion. This triggers increased production of renin, angiotensin, and aldosterone, but the resulting salt and water retention cannot correct the plasma volume deficit because the primary defect of low serum protein persists.
Sodium and Water Retention.
Salt and water retention can also be a primary cause of edema. Increased salt retention—with obligate associated water—causes both increased hydrostatic pressure (due to intravascular fluid volume expansion) and diminished vascular colloid osmotic pressure (due to dilution). Salt retention occurs whenever renal function is compromised, such as in primary disorders of the kidney and disorders that decrease renal perfusion. One of the most important causes of renal hypoperfusion is congestive heart failure, which (like hypoproteinemia) results in the activation of the renin-angiotensin-aldosterone axis. In early heart failure, this response tends to be beneficial, as the retention of sodium and water and other adaptations, including increased vascular tone and elevated levels of antidiuretic hormone (ADH), improve cardiac output and restore normal renal perfusion.1,2 However, as heart failure worsens and cardiac output diminishes, the retained fluid merely increases the venous pressure, which (as already mentioned) is a major cause of edema in this disorder. Unless cardiac output is restored or renal sodium and water retention is reduced (e.g., by salt restriction, diuretics, or aldosterone antagonists), a downward spiral of fluid retention and worsening edema ensues. Salt restriction, diuretics, and aldosterone antagonists are also of value in managing generalized edema arising from other causes. Primary retention of water (and modest vasoconstriction) is produced by the release of ADH from the posterior pituitary, which normally occurs in the setting of reduced plasma volumes or increased plasma osmolarity.2 Inappropriate increases in ADH are seen in association with certain malignancies and lung and pituitary disorders and can lead to hyponatremia and cerebral edema (but interestingly not to peripheral edema).
Lymphatic Obstruction.
Impaired lymphatic drainage results in lymphedema that is typically localized; causes include chronic inflammation with fibrosis, invasive malignant tumors, physical disruption, radiation damage, and certain infectious agents. One dramatic example is seen in parasitic filariasis, in which lymphatic obstruction due to extensive inguinal lymphatic and lymph node fibrosis can result in edema of the external genitalia and lower limbs that is so massive as to earn the appellation elephantiasis. Severe edema of the upper extremity may also complicate surgical removal and/or irradiation of the breast and associated axillary lymph nodes in patients with breast cancer.
Morphology. Edema is easily recognized grossly; microscopically, it is appreciated as clearing and separation of the extracellular matrix and subtle cell swelling. Any organ or tissue can be involved, but edema is most commonly seen in subcutaneous tissues, the lungs, and the brain. Subcutaneous edema can be diffuse or more conspicuous in regions with high hydrostatic pressures. In most cases the distribution is influenced by gravity and is termed dependent edema (e.g., the legs when standing, the sacrum when recumbent). Finger pressure over substantially edematous subcutaneous tissue displaces the interstitial fluid and leaves a depression, a sign called pitting edema.
Edema as a result of renal dysfunction can affect all parts of the body. It often initially manifests in tissues with loose connective tissue matrix, such as the eyelids; periorbital edema is thus a characteristic finding in severe renal disease. With pulmonary edema, the lungs are often two to three times their normal weight, and sectioning yields frothy, blood-tinged fluid—a mixture of air, edema, and extravasated red cells. Brain edema can be localized or generalized depending on the nature and extent of the pathologic process or injury. With generalized edema the brain is grossly swollen with narrowed sulci; distended gyri show evidence of compression against the unyielding skull (Chapter 28).
Clinical Consequences.
The consequences of edema range from merely annoying to rapidly fatal. Subcutaneous tissue edema is important primarily because it signals potential underlying cardiac or renal disease; however, when significant, it can also impair wound healing or the clearance of infection. Pulmonary edema is a common clinical problem that is most frequently seen in the setting of left ventricular failure; it can also occur with renal failure, acute respiratory distress syndrome (Chapter 15), and pulmonary inflammation or infection. Not only does fluid collect in the alveolar septa around capillaries and impede oxygen diffusion, but edema fluid in the alveolar spaces also creates a favorable environment for bacterial infection. Brain edema is life-threatening; if severe, brain substance can herniate (extrude) through the foramen magnum, or the brain stem vascular supply can be compressed. Either condition can injure the medullary centers and cause death (Chapter 28).
Hyperemia and Congestion
Hyperemia and congestion both stem from locally increased blood volumes. Hyperemia is an active process in which arteriolar dilation (e.g., at sites of inflammation or in skeletal muscle during exercise) leads to increased blood flow. Affected tissues turn red (erythema) because of the engorgement of vessels with oxygenated blood. Congestion is a passive process resulting from reduced outflow of blood from a tissue. It can be systemic, as in cardiac failure, or local, as in isolated venous obstruction. Congested tissues take on a dusky reddish-blue color (cyanosis) due to red cell stasis and the accumulation of deoxygenated hemoglobin.
As a result of the increased volumes and pressures, congestion commonly leads to edema. In long-standing chronic passive congestion, the lack of blood flow causes chronic hypoxia, potentially resulting in ischemic tissue injury and scarring. Capillary rupture in chronic congestion can also cause small hemorrhagic foci; subsequent catabolism of extravasated red cells can leave residual telltale clusters of hemosiderin-laden macrophages.
Morphology. The cut surfaces of congested tissues are often discolored due to the presence of high levels of poorly oxygenated blood. Microscopically, acute pulmonary congestion exhibits engorged alveolar capillaries often with alveolar septal edema and focal intra-alveolar hemorrhage. In chronic pulmonary congestion the septa are thickened and fibrotic, and the alveoli often contain numerous hemosiderin-laden macrophages called heart failure cells. In acute hepatic congestion, the central vein and sinusoids are distended; centrilobular hepatocytes can be frankly ischemic while the periportal hepatocytes—better oxygenated because of proximity to hepatic arterioles—may only develop fatty change. In chronic passive hepatic congestion the centrilobular regions are grossly red-brown and slightly depressed (because of cell death) and are accentuated against the surrounding zones of uncongested tan liver (nutmeg liver) (Fig. 4-3A). Microscopically, there is centrilobular hemorrhage, hemosiderin-laden macrophages, and degeneration of hepatocytes (Fig. 4-3B). Because the centrilobular area is at the distal end of the blood supply to the liver, it is prone to undergo necrosis whenever the blood supply is compromised.
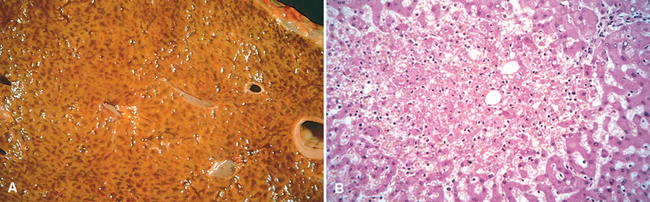
FIGURE 4-3 Liver with chronic passive congestion and hemorrhagic necrosis. A, Central areas are red and slightly depressed compared with the surrounding tan viable parenchyma, forming a “nutmeg liver” pattern (so-called because it resembles the cut surface of a nutmeg. B, Centrilobular necrosis with degenerating hepatocytes and hemorrhage.
(Courtesy of Dr. James Crawford, Department of Pathology, University of Florida, Gainesville, FL.)
Hemorrhage
Hemorrhage is defined as the extravasation of blood into the extravascular space. As described above, capillary bleeding can occur under conditions of chronic congestion; an increased tendency to hemorrhage (usually with insignificant injury) also occurs in a variety of clinical disorders that are collectively called hemorrhagic diatheses. Rupture of a large artery or vein results in severe hemorrhage and is almost always due to vascular injury, including trauma, atherosclerosis, or inflammatory or neoplastic erosion of the vessel wall.
Tissue hemorrhage can occur in distinct patterns, each with its own clinical implications:
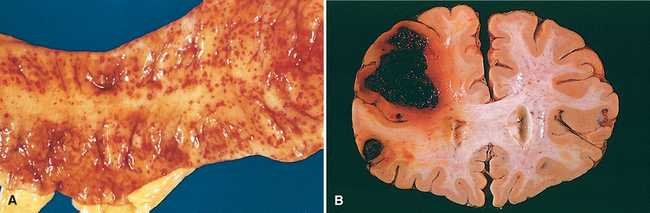
FIGURE 4-4 A, Punctate petechial hemorrhages of the colonic mucosa, a consequence of thrombocytopenia. B, Fatal intracerebral bleed.
The clinical significance of hemorrhage depends on the volume and rate of bleeding. Rapid loss of up to 20% of the blood volume or slow losses of even larger amounts may have little impact in healthy adults; greater losses, however, can cause hemorrhagic (hypovolemic) shock (discussed later). The site of hemorrhage is also important. For example, bleeding that is trivial in the subcutaneous tissues can cause death if located in the brain (Fig. 4-4B); because the skull is unyielding, intracranial hemorrhage can result in an increase in pressure that is sufficient to compromise the blood supply or to cause the herniation of the brainstem (Chapter 28). Finally, chronic or recurrent external blood loss (e.g., peptic ulcer or menstrual bleeding) causes a net loss in iron and can lead to an iron deficiency anemia. In contrast, when red cells are retained (e.g., hemorrhage into body cavities or tissues), iron is recovered and recycled for use in the synthesis of hemoglobin.
Hemostasis and Thrombosis
Normal hemostasis is a consequence of tightly regulated processes that maintain blood in a fluid state in normal vessels, yet also permit the rapid formation of a hemostatic clot at the site of a vascular injury. The pathologic counterpart of hemostasis is thrombosis; it involves blood clot (thrombus) formation within intact vessels. Both hemostasis and thrombosis involve three components: the vascular wall (particularly the endothelium), platelets, and the coagulation cascade. We begin the discussion with the normal hemostatic pathway and how it is regulated.
NORMAL HEMOSTASIS
The general sequence of events in hemostasis at a site of vascular injury is shown in Figure 4-5.3,4
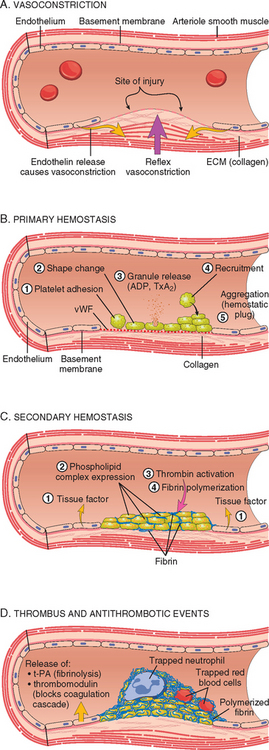
FIGURE 4-5 Normal hemostasis. A, After vascular injury local neurohumoral factors induce a transient vasoconstriction. B, Platelets bind via glycoprotein Ib (GpIb) receptors to von Willebrand factor (vWF) on exposed extracellular matrix (ECM) and are activated, undergoing a shape change and granule release. Released adenosine diphosphate (ADP) and thromboxane A2 (TxA2) induce additional platelet aggregation through platelet GpIIb-IIIa receptor binding to fibrinogen, and form the primary hemostatic plug. C, Local activation of the coagulation cascade (involving tissue factor and platelet phospholipids) results in fibrin polymerization, “cementing” the platelets into a definitive secondary hemostatic plug. D, Counter-regulatory mechanisms, mediated by tissue plasminogen activator (t-PA, a fibrinolytic product) and thrombomodulin, confine the hemostatic process to the site of injury.
The following sections discuss the roles of the endothelium, platelets, and the coagulation cascade in greater detail.
Endothelium
Endothelial cells are key players in the regulation of homeostasis, as the balance between the anti- and prothrombotic activities of endothelium determines whether thrombus formation, propagation, or dissolution occurs.5-7 Normally, endothelial cells exhibit antiplatelet, anticoagulant, and fibrinolytic properties; however, after injury or activation they acquire numerous procoagulant activities (Fig. 4-6). Besides trauma, endothelium can be activated by infectious agents, hemodynamic forces, plasma mediators, and cytokines.
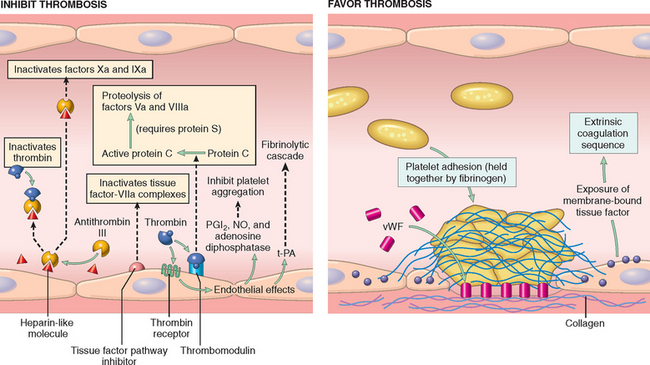
FIGURE 4-6 Anti- and procoagulant activities of endothelium. NO, nitric oxide; PGI2, prostacyclin; t-PA, tissue plasminogen activator; vWF, von Willebrand factor. The thrombin receptor is also called a protease-activated receptor (PAR).
Antithrombotic Properties
Under normal circumstances endothelial cells actively prevent thrombosis by producing factors that variously block platelet adhesion and aggregation, inhibit coagulation, and lyse clots.
Prothrombotic Properties
While normal endothelial cells limit clotting, trauma and inflammation of endothelial cells induce a prothrombotic state that alters the activities of platelets, coagulation proteins, and the fibrinolytic system.
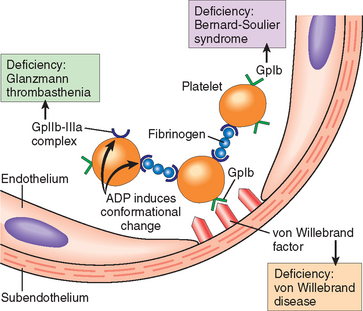
FIGURE 4-7 Platelet adhesion and aggregation. Von Willebrand factor functions as an adhesion bridge between subendothelial collagen and the glycoprotein Ib (GpIb) platelet receptor. Aggregation is accomplished by fibrinogen bridging GpIIb-IIIa receptors on different platelets. Congenital deficiencies in the various receptors or bridging molecules lead to the diseases indicated in the colored boxes. ADP, adenosine diphosphate.
In summary, intact, nonactivated endothelial cells inhibit platelet adhesion and blood clotting. Endothelial injury or activation, however, results in a procoagulant phenotype that enhances thrombus formation.
Platelets
Platelets are disc-shaped, anucleate cell fragments that are shed from megakaryocytes in the bone marrow into the blood stream. They play a critical role in normal hemostasis,13 by forming the hemostatic plug that initially seals vascular defects, and by providing a surface that recruits and concentrates activated coagulation factors. Their function depends on several glycoprotein receptors, a contractile cytoskeleton, and two types of cytoplasmic granules. α-Granules have the adhesion molecule P-selectin on their membranes (Chapter 2) and contain fibrinogen, fibronectin, factors V and VIII, platelet factor 4 (a heparin-binding chemokine), platelet-derived growth factor (PDGF), and transforming growth factor-β (TGF-β). Dense (or δ) granules contain ADP and ATP, ionized calcium, histamine, serotonin, and epinephrine.
After vascular injury, platelets encounter ECM constituents such as collagen and the adhesive glycoprotein vWF. On contact with these proteins, platelets undergo: (1) adhesion and shape change, (2) secretion (release reaction), and (3) aggregation (see Fig. 4-5B).
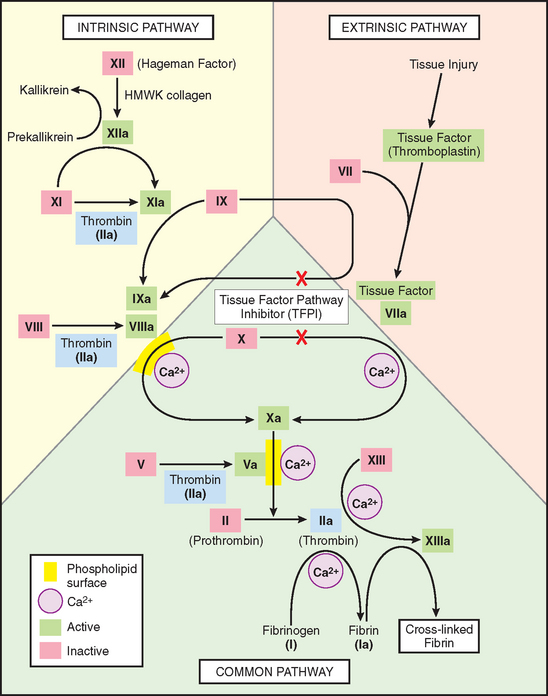
FIGURE 4-8 The coagulation cascade. Factor IX can be activated either by factor XIa or factor VIIa; in lab tests, activation is predominantly dependent on factor XIa of the intrinsic pathway. Factors in red boxes represent inactive molecules; activated factors are indicated with a lower case “a” and a green box. Note also the multiple points where thrombin (factor IIa; light blue boxes) contributes to coagulation through positive feedback loops. The red “X”s denote points of action of tissue factor pathway inhibitor (TFPI), which inhibits the activation of factors X and IX by factor VIIa. PL, phospholipid; HMWK, high-molecular-weight kininogen.
Noncleaved fibrinogen is also an important component of platelet aggregation. Platelet activation by ADP triggers a conformational change in the platelet GpIIb-IIIa receptors that induces binding to fibrinogen, a large protein that forms bridging interactions between platelets that promote platelet aggregation (see Fig. 4-7). Predictably, inherited deficiency of GpIIb-IIIa results in a bleeding disorder (Glanzmann thrombasthenia).16 The recognition of the central role of the various receptors and mediators in platelet cross-linking has led to the development of therapeutic agents that block platelet aggregation—for example, by interfering with thrombin activity,17 by blocking ADP binding (clopidogrel), or by binding to the GpIIb-IIIa receptors (synthetic antagonists or monoclonal antibodies).18 Antibodies against GpIb are on the horizon.
Red cells and leukocytes are also found in hemostatic plugs. Leukocytes adhere to platelets via P-selectin and to endothelium using several adhesion receptors (Chapter 2); they contribute to the inflammation that accompanies thrombosis. Thrombin also drives thrombus-associated inflammation by directly stimulating neutrophil and monocyte adhesion and by generating chemotactic fibrin split products during fibrinogen cleavage.
Platelet-Endothelial Cell Interactions.
The interplay of platelets and endothelium has a profound impact on clot formation. The endothelial cell-derived prostaglandin PGI2 (prostacyclin) inhibits platelet aggregation and is a potent vasodilator; conversely, the platelet-derived prostaglandin TxA2 activates platelet aggregation and is a vasoconstrictor (Chapter 2). Effects mediated by PGI2 and TxA2 are exquisitely balanced to effectively modulate platelet and vascular wall function: at baseline, platelet aggregation is prevented, whereas endothelial injury promotes hemostatic plug formation. The clinical utility of aspirin (an irreversible cyclooxygenase inhibitor) in persons at risk for coronary thrombosis resides in its ability to permanently block platelet TxA2 synthesis. Although endothelial PGI2 production is also inhibited by aspirin, endothelial cells can resynthesize active cyclooxygenase and thereby overcome the blockade. In a manner similar to PGI2, endothelial-derived nitric oxide also acts as a vasodilator and inhibitor of platelet aggregation (see Fig. 4-6).
Coagulation Cascade
The coagulation cascade is the third arm of the hemostatic process. The pathways are schematically presented in Figure 4-8; only general principles are discussed here.4,19
The coagulation cascade is essentially an amplifying series of enzymatic conversions; each step proteolytically cleaves an inactive proenzyme into an activated enzyme, culminating in thrombin formation. Thrombin is the most important coagulation factor, and indeed can act at numerous stages in the process (see blue boxes in Fig. 4-8).20 At the conclusion of the proteolytic cascade, thrombin converts the soluble plasma protein fibrinogen into fibrin monomers that polymerize into an insoluble gel. The fibrin gel encases platelets and other circulating cells in the definitive secondary hemostatic plug, and the fibrin polymers are covalently cross-linked and stabilized by factor XIIIa (which itself is activated by thrombin).
Each reaction in the pathway results from the assembly of a complex composed of an enzyme (activated coagulation factor), a substrate (proenzyme form of coagulation factor), and a cofactor (reaction accelerator). These components are typically assembled on a phospholipid surface and held together by calcium ions (as an aside, the clotting of blood is prevented by the presence of calcium chelators). The requirement that coagulation factors be brought close together ensures that clotting is normally localized to the surface of activated platelets or endothelium;4 as shown in Figure 4-9, it can be likened to a “dance” of complexes, in which coagulation factors are passed successfully from one partner to the next. Parenthetically, the binding of coagulation factors II, XII, IX, and X to calcium depends on the addition of γ-carboxyl groups to certain glutamic acid residues on these proteins. This reaction uses vitamin K as a cofactor and is antagonized by drugs such as coumadin, which is a widely used anticoagulant.
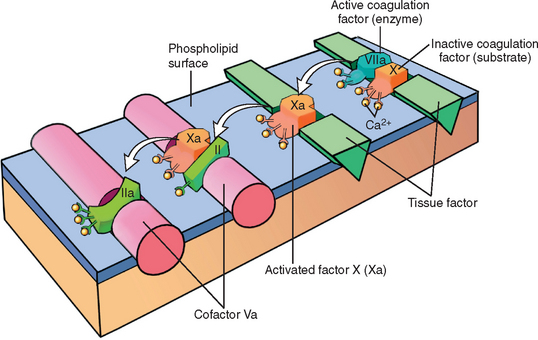
FIGURE 4-9 Schematic illustration of the conversion of factor X to factor Xa via the extrinsic pathway, which in turn converts factor II (prothrombin) to factor IIa (thrombin). The initial reaction complex consists of a proteolytic enzyme (factor VIIa), a substrate (factor X), and a reaction accelerator (tissue factor), all assembled on a platelet phospholipid surface. Calcium ions hold the assembled components together and are essential for the reaction. Activated factor Xa becomes the protease for the second adjacent complex in the coagulation cascade, converting prothrombin substrate (II) to thrombin (IIa) using factor Va as the reaction accelerator.
Blood coagulation is traditionally classified into extrinsic and intrinsic pathways that converge on the activation of factor X (see Fig. 4-8). The extrinsic pathway was so designated because it required the addition of an exogenous trigger (originally provided by tissue extracts); the intrinsic pathway only required exposing factor XII (Hageman factor) to thrombogenic surfaces (even glass would suffice). However, such a division is largely an artifact of in vitro testing; there are, in fact, several interconnections between the two pathways. Moreover, the extrinsic pathway is the most physiologically relevant pathway for coagulation occurring when vascular damage has occurred; it is activated by tissue factor (also known as thromboplastin or factor III), a membrane-bound lipoprotein expressed at sites of injury (see Fig. 4-8).12
Clinical laboratories assess the function of the two arms of the coagulation pathway through two standard assays: prothrombin time (PT) and partial thromboplastin time (PTT). The PT assay assesses the function of the proteins in the extrinsic pathway (factors VII, X, II, V, and fibrinogen). This is accomplished by adding tissue factor and phospholipids to citrated plasma (sodium citrate chelates calcium and prevents spontaneous clotting). Coagulation is initiated by the addition of exogenous calcium and the time for a fibrin clot to form is recorded. The partial thromboplastin time (PTT) screens for the function of the proteins in the intrinsic pathway (factors XII, XI, IX, VIII, X, V, II, and fibrinogen). In this assay, clotting is initiated through the addition of negative charged particles (e.g., ground glass), which you will recall activates factor XII (Hageman factor), phospholipids, and calcium, and the time to fibrin clot formation is recorded.
In addition to catalyzing the final steps in the coagulation cascade, thrombin exerts a wide variety of proinflammatery effects (Fig. 4-10). Most of these effects of thrombin occur through its activation of a family of protease activated receptors (PARs) that belong to the seven-transmembrane G protein–coupled receptor family21,22 (see also Fig. 4-6). PARs are expressed on endothelium, monocytes, dendritic cells, T lymphocytes, and other cell types. Receptor activation is initiated by cleavage of the extracellular end of the PAR; this generates a tethered peptide that binds to the “clipped” receptor, causing a conformational change that triggers signaling.
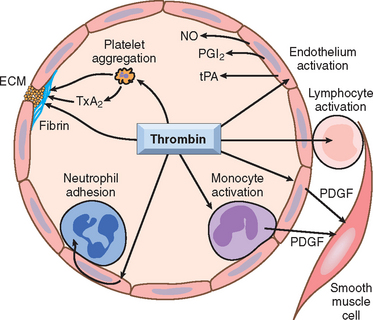
FIGURE 4-10 Role of thrombin in hemostasis and cellular activation. Thrombin plays a critical role in generating cross-linked fibrin (by cleaving fibrinogen to fibrin, and by activating factor XIII), as well as activating several other coagulation factors (see Fig. 4-8). Through protease-activated receptors (PARs, see text), thrombin also modulates several cellular activities. It directly induces platelet aggregation and TxA2 production, and activates ECs to express adhesion molecules, and a variety of fibrinolytic (t-PA), vasoactive (NO, PGI2), and cytokine mediators (e.g., PDGF). Thrombin also directly activates leukocytes. ECM, extracellular matrix; NO, nitric oxide; PDGF, platelet-derived growth factor; PGI2, prostacyclin; TxA2, thromboxane A2; t-PA, tissue plasminogen activator. See Figure 4-7 for additional anticoagulant activities mediated by thrombin, including via thrombomodulin.
(Courtesy of Shaun Coughlin, MD, PhD, Cardiovascular Research Institute, University of California at San Francisco; modified with permission.)
Once activated, the coagulation cascade must be restricted to the site of vascular injury to prevent runaway clotting of the entire vascular tree. Besides restricting factor activation to sites of exposed phospholipids, three categories of endogenous anticoagulants also control clotting. (1) Antithrombins (e.g., antithrombin III) inhibit the activity of thrombin and other serine proteases, including factors IXa, Xa, XIa, and XIIa. Antithrombin III is activated by binding to heparin-like molecules on endothelial cells; hence the clinical usefulness of administering heparin to minimize thrombosis (see Fig. 4-6). (2) Proteins C and S are vitamin K–dependent proteins that act in a complex that proteolytically inactivates factors Va and VIIIa. Protein C activation by thrombomodulin was described earlier. (3) TFPI is a protein produced by endothelium (and other cell types) that inactivates tissue factor–factor VIIa complexes (see Figs. 4-6 and 4-8).10
Activation of the coagulation cascade also sets into motion a fibrinolytic cascade that moderates the size of the ultimate clot. Fibrinolysis is largely accomplished through the enzymatic activity of plasmin, which breaks down fibrin and interferes with its polymerization (Fig. 4-11).23 The resulting fibrin split products (FSPs or fibrin degradation products) can also act as weak anticoagulants. Elevated levels of FSPs (most notably fibrin-derived D-dimers) can be used in diagnosing abnormal thrombotic states including disseminated intravascular coagulation (DIC), deep venous thrombosis, or pulmonary embolism (described later). Plasmin is generated by enzymatic catabolism of the inactive circulating precursor plasminogen, either by a factor XII–dependent pathway or by plasminogen activators (PAs; see Fig. 4-11). The most important of the PAs is t-PA; it is synthesized principally by endothelium and is most active when bound to fibrin. The affinity for fibrin makes t-PA a useful therapeutic agent, since it largely confines fibrinolytic activity to sites of recent thrombosis. Urokinase-like PA (u-PA) is another PA present in plasma and in various tissues; it can activate plasmin in the fluid phase. Finally, plasminogen can be cleaved to plasmin by the bacterial enzyme streptokinase, an activity that may be clinically significant in certain bacterial infections. As with any potent regulator, plasmin activity is tightly restricted. To prevent excess plasmin from lysing thrombi indiscriminately elsewhere in the body, free plasmin is rapidly inactivated by α2-plasmin inhibitor (see Fig. 4-11).
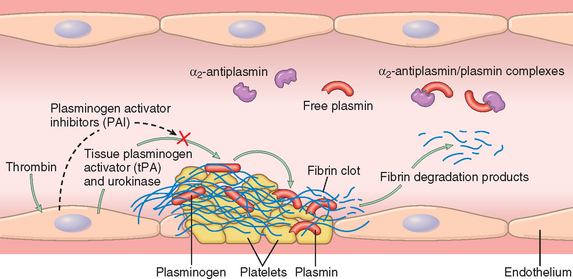
FIGURE 4-11 The fibrinolytic system, illustrating various plasminogen activators and inhibitors (see text).
Endothelial cells also fine-tune the coagulation/anticoagulation balance by releasing plasminogen activator inhibitor (PAI); it blocks fibrinolysis by inhibiting t-PA binding to fibrin and confers an overall procoagulant effect (see Fig. 4-11). PAI production is increased by thrombin as well as certain cytokines, and probably plays a role in the intravascular thrombosis accompanying severe inflammation.24
THROMBOSIS
Having discussed the components of normal hemostasis, we now turn our attention to the three primary abnormalities that lead to thrombus formation (called Virchow’s triad): (1) endothelial injury, (2) stasis or turbulent blood flow, and (3) hypercoagulability of the blood (Fig. 4-12).
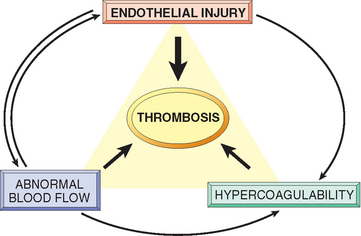
FIGURE 4-12 Virchow’s triad in thrombosis. Endothelial integrity is the most important factor. Injury to endothelial cells can alter local blood flow and affect coagulability. Abnormal blood flow (stasis or turbulence), in turn, can cause endothelial injury. The factors promote thrombosis independently or in combination.
Endothelial Injury.
Endothelial injury is particularly important for thrombus formation in the heart or the arterial circulation, where the normally high flow rates might otherwise impede clotting by preventing platelet adhesion and washing out activated coagulation factors. Thus, thrombus formation within cardiac chambers (e.g., after endocardial injury due to myocardial infarction), over ulcerated plaques in atherosclerotic arteries, or at sites of traumatic or inflammatory vascular injury (vasculitis) is largely a consequence of endothelial cell injury. Clearly, physical loss of endothelium can lead to exposure of the subendothelial ECM, adhesion of platelets, release of tissue factor, and local depletion of PGI2 and plasminogen activators. However, it should be emphasized that endothelium need not be denuded or physically disrupted to contribute to the development of thrombosis; any perturbation in the dynamic balance of the prothombotic and antithrombotic activities of endothelium can influence local clotting events (see Fig. 4-6). Thus, dysfunctional endothelial cells can produce more procoagulant factors (e.g., platelet adhesion molecules, tissue factor, PAIs) or may synthesize less anticoagulant effectors (e.g., thrombomodulin, PGI2, t-PA). Endothelial dysfunction can be induced by a wide variety of insults, including hypertension, turbulent blood flow, bacterial endotoxins, radiation injury, metabolic abnormalities such as homocystinemia or hypercholesterolemia, and toxins absorbed from cigarette smoke.
Alterations in Normal Blood Flow.
Turbulence contributes to arterial and cardiac thrombosis by causing endothelial injury or dysfunction, as well as by forming countercurrents and local pockets of stasis; stasis is a major contributor in the development of venous thrombi.25 Normal blood flow is laminar such that the platelets (and other blood cellular elements) flow centrally in the vessel lumen, separated from endothelium by a slower moving layer of plasma. Stasis and turbulence therefore:
Turbulence and stasis contribute to thrombosis in several clinical settings. Ulcerated atherosclerotic plaques not only expose subendothelial ECM but also cause turbulence. Aortic and arterial dilations called aneurysms result in local stasis and are therefore fertile sites for thrombosis (Chapter 11). Acute myocardial infarctions result in areas of noncontractile myocardium and sometimes cardiac aneurysms; both are associated with stasis and flow abnormalities that promote the formation of cardiac mural thrombi (Chapter 12). Rheumatic mitral valve stenosis results in left atrial dilation; in conjunction with atrial fibrillation, a dilated atrium is a site of profound stasis and a prime location for developing thrombi (Chapter 12). Hyperviscosity (such as is seen with polycythemia vera; Chapter 13) increases resistance to flow and causes small vessel stasis; the deformed red cells in sickle cell anemia (Chapter 14) cause vascular occlusions, with the resulting stasis also predisposing to thrombosis.
Hypercoagulability.
Hypercoagulability (also called thrombophilia) is a less frequent contributor to thrombotic states but is nevertheless an important component in the equation, and in some situations can predominate. It is loosely defined as any alteration of the coagulation pathways that predisposes to thrombosis; it can be divided into primary (genetic) and secondary (acquired) disorders (Table 4-2).27-29 Of the inherited causes of hypercoagulability, point mutations in the factor V gene and prothrombin gene are the most common.
TABLE 4-2 Hypercoagulable States
| PRIMARY (GENETIC) |
| Common |
| Rare |
| Very Rare |
| SECONDARY (ACQUIRED) |
| High Risk for Thrombosis |
| Lower Risk for Thrombosis |
The most common thrombophilic genotypes found in various populations (heterozygosity for factor V Leiden and heterozygosity for prothrombin) impart only a moderately increased risk of thrombosis; most individuals with these genotypes, when otherwise healthy, are free of thrombotic complications. However, mutations in factor V and prothrombin are frequent enough that homozygosity and compound heterozygosity are not rare, and such genotypes are associated with greater risk.35 Moreover, individuals with such mutations have a significantly increased frequency of venous thrombosis in the setting of other acquired risk factors (e.g., pregnancy or prolonged bedrest). Thus, factor V Leiden heterozygosity (which by itself has only a modest effect) may trigger deep venous thrombosis when combined with enforced inactivity, such as during prolonged plane travel. Consequently, inherited causes of hypercoagulability must be considered in patients under the age of 50 who present with thrombosis—even when acquired risk factors are present.36,37
Unlike hereditary disorders, the pathogenesis of acquired thrombophilia is frequently multifactorial (see Table 4-2). In some cases (e.g., cardiac failure or trauma), stasis or vascular injury may be most important. Hypercoagulability due to oral contraceptive use or the hyperestrogenic state of pregnancy is probably caused by increased hepatic synthesis of coagulation factors and reduced anticoagulant synthesis.38 In disseminated cancers, release of procoagulant tumor products predisposes to thrombosis.39 The hypercoagulability seen with advancing age may be due to reduced endothelial PGI2. Smoking and obesity promote hypercoagulability by unknown mechanisms.
Among the acquired thrombophilic states, two that are particularly important clinical problems deserve special mention.
Heparin-induced thrombocytopenia (HIT) syndrome.
HIT occurs following the administration of unfractionated heparin, which may induce the appearance of antibodies that recognize complexes of heparin and platelet factor 4 on the surface of platelets (Chapter 14), as well as complexes of heparin-like molecules and platelet factor 4-like proteins on endothelial cells.40-42 Binding of these antibodies to platelets results in their activation, aggregation, and consumption (hence the thrombocytopenia in the syndrome name). This effect on platelets and endothelial damage combine to produce a prothrombotic state, even in the face of heparin administration and low platelet counts. Newer low-molecular weight heparin preparations induce antibody formation less frequently, but still cause thrombosis if antibodies have already formed.41 Other anticoagulants such as fondaparinux (a pentasaccharide inhibitor of factor X) also cause a HIT-like syndrome on rare occasions.42
Antiphospholipid antibody syndrome43
(previously called the lupus anticoagulant syndrome). This syndrome has protean clinical manifestations, including recurrent thromboses, repeated miscarriages, cardiac valve vegetations, and thrombocytopenia. Depending on the vascular bed involved, the clinical presentations can include pulmonary embolism (following lower extremity venous thrombosis), pulmonary hypertension (from recurrent subclinical pulmonary emboli), stroke, bowel infarction, or renovascular hypertension. Fetal loss is attributable to antibody-mediated inhibition of t-PA activity necessary for trophoblastic invasion of the uterus. Antiphospholipid antibody syndrome is also a cause of renal microangiopathy, resulting in renal failure associated with multiple capillary and arterial thromboses (Chapter 20).
The name antiphospholipid antibody syndrome is a bit of a misnomer, as it is believed that the most important pathologic effects are mediated through binding of the antibodies to epitopes on plasma proteins (e.g., prothrombin) that are somehow induced or “unveiled” by phospholipids. In vivo, these autoantibodies induce a hypercoagulable state by causing endothelial injury, by activating platelets and complement directly, and through interaction with the catalytic domains of certain coagulation factors.43 However, in vitro (in the absence of platelets and endothelial cells), the autoantibodies interfere with phospholipids and thus inhibit coagulation. The antibodies also frequently give a false-positive serologic test for syphilis because the antigen in the standard assay is embedded in cardiolipin.
Antiphospholipid antibody syndrome has primary and secondary forms. Individuals with a well-defined autoimmune disease, such as systemic lupus erythematosus (Chapter 6), are designated as having secondary antiphospholipid syndrome (hence the earlier term lupus anticoagulant syndrome). In primary antiphospholipid syndrome, patients exhibit only the manifestations of a hypercoagulable state and lack evidence of other autoimmune disorders; occasionally this happens in association with certain drugs or infections. A particularly aggressive form (catastrophic antiphospholipid syndrome) characterized by widespread small-vessel thrombi and multi-organ failure has a 50% mortality.44 The antibodies also make surgical procedures more difficult; for example, nearly 90% of patients with anti-phospholipid antibodies undergoing cardiovascular surgery have complications related to the antibodies.45 Therapy involves anticoagulation and immunosuppression. Although antiphospholipid antibodies are clearly associated with thrombotic diatheses, they have also been identified in 5% to 15% of apparently normal individuals, implying that they are necessary but not sufficient to cause the full-blown syndrome.
Morphology. Thrombi can develop anywhere in the cardiovascular system (e.g., in cardiac chambers, on valves, or in arteries, veins, or capillaries). The size and shape of thrombi depend on the site of origin and the cause. Arterial or cardiac thrombi usually begin at sites of turbulence or endothelial injury; venous thrombi characteristically occur at sites of stasis. Thrombi are focally attached to the underlying vascular surface; arterial thrombi tend to grow retrograde from the point of attachment, while venous thrombi extend in the direction of blood flow (thus both propagate toward the heart). The propagating portion of a thrombus is often poorly attached and therefore prone to fragmentation and embolization.
Thrombi often have grossly and microscopically apparent laminations called lines of Zahn; these represent pale platelet and fibrin deposits alternating with darker red cell–rich layers. Such laminations signify that a thrombus has formed in flowing blood; their presence can therefore distinguish antemortem thrombosis from the bland nonlaminated clots that occur postmortem (see below).
Thrombi occurring in heart chambers or in the aortic lumen are designated mural thrombi. Abnormal myocardial contraction (arrhythmias, dilated cardiomyopathy, or myocardial infarction) or endomyocardial injury (myocarditis or catheter trauma) promotes cardiac mural thrombi (Fig. 4-13A), while ulcerated atherosclerotic plaque and aneurysmal dilation are the precursors of aortic thrombus (Fig. 4-13B).
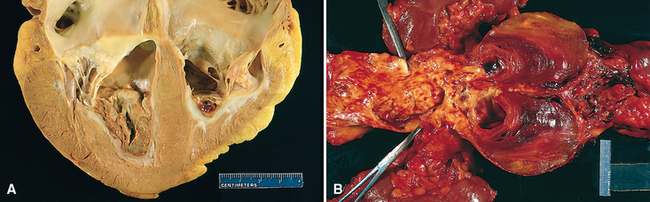
FIGURE 4-13 Mural thrombi. A, Thrombus in the left and right ventricular apices, overlying white fibrous scar. B, Laminated thrombus in a dilated abdominal aortic aneurysm. Numerous friable mural thrombi are also superimposed on advanced atherosclerotic lesions of the more proximal aorta (left side of picture).
Arterial thrombi are frequently occlusive; the most common sites in decreasing order of frequency are the coronary, cerebral, and femoral arteries. They typically cosist of a friable meshwork of platelets, fibrin, red cells, and degenerating leukocytes. Although these are usually superimposed on a ruptured atherosclerotic plaque, other vascular injuries (vasculitis, trauma) may be the underlying cause.
Venous thrombosis (phlebothrombosis) is almost invariably occlusive, with the thrombus forming a long cast of the lumen. Because these thrombi form in the sluggish venous circulation, they tend to contain more enmeshed red cells (and relatively few platelets) and are therefore known as red, or stasis, thrombi. The veins of the lower extremities are most commonly involved (90% of cases); however, upper extremities, periprostatic plexus, or the ovarian and periuterine veins can also develop venous thrombi. Under special circumstances, they can also occur in the dural sinuses, portal vein, or hepatic vein.
Postmortem clots can sometimes be mistaken for antemortem venous thrombi. However, postmortem clots are gelatinous with a dark red dependent portion where red cells have settled by gravity and a yellow “chicken fat” upper portion; they are usually not attached to the underlying wall. In comparison, red thrombi are firmer and are focally attached, and sectioning typically reveals gross and/or microscopic lines of Zahn.
Thrombi on heart valves are called vegetations. Blood-borne bacteria or fungi can adhere to previously damaged valves (e.g., due to rheumatic heart disease) or can directly cause valve damage; in both cases, endothelial injury and disturbed blood flow can induce the formation of large thrombotic masses (infective endocarditis; Chapter 12). Sterile vegetations can also develop on noninfected valves in persons with hypercoagulable states, so-called nonbacterial thrombotic endocarditis (Chapter 12). Less commonly, sterile, verrucous endocarditis (Libman-Sacks endocarditis) can occur in the setting of systemic lupus erythematosus (Chapter 6).
Fate of the Thrombus.
If a patient survives the initial thrombosis, in the ensuing days to weeks thrombi undergo some combination of the following four events:
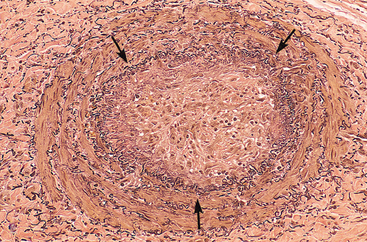
FIGURE 4-14 Low-power view of a thrombosed artery stained for elastic tissue. The original lumen is delineated by the internal elastic lamina (arrows) and is totally filled with organized thrombus, now punctuated by several recanalized endothelium-lined channels (white spaces).
Although the earliest capillary channels may not restore significant flow to obstructed vessels, continued recanalization may convert a thrombus into a smaller mass of connective tissue that becomes incorporated into the vessel wall. Eventually, with remodeling and contraction of the mesenchymal elements, only a fibrous lump may remain to mark the original thrombus. Occasionally the centers of thrombi undergo enzymatic digestion, presumably as a result of the release of lysosomal enzymes from trapped leukocytes and platelets. In the setting of bacteremia such thrombi may become infected, producing an inflammatory mass that erodes and weakens the vessel wall. If unchecked, this may result in a mycotic aneurysm (Chapter 11).
Clinical Consequences.
Thrombi are significant because they cause obstruction of arteries and veins, and are sources of emboli. Which effect predominates depends on the site of the thrombosis. Venous thrombi can cause congestion and edema in vascular beds distal to an obstruction, but they are far more worrisome for their capacity to embolize to the lungs and cause death (see below). Conversely, although arterial thrombi can embolize and cause downstream infarctions, a thrombotic occlusion at a critical site (e.g., a coronary artery) can have more serious clinical consequences.
Venous Thrombosis (Phlebothrombosis).
Most venous thrombi occur in the superficial or deep veins of the leg.25 Superficial venous thrombi typically occur in the saphenous veins in the setting of varicosities. Although such thrombi can cause local congestion, swelling, pain, and tenderness, they rarely embolize. Nevertheless, the local edema and impaired venous drainage do predispose the overlying skin to infections from slight trauma and to the development of varicose ulcers. Deep venous thrombosis (DVT) in the larger leg veins—at or above the knee (e.g., popliteal, femoral, and iliac veins)—is more serious because such thrombi more often embolize to the lungs and give rise to pulmonary infarction (see below and Chapter 15). Although they can cause local pain and edema, venous obstructions from DVTs can be rapidly offset by collateral channels. Consequently, DVTs are asymptomatic in approximately 50% of affected individuals and are recognized only in retrospect after embolization.
Lower extremity DVTs are associated with hypercoagulable states, as described earlier (see Table 4-2). Common predisposing factors include bed rest and immobilization (because they reduce the milking action of the leg muscles, resulting in reduced venous return), and congestive heart failure (also a cause of impaired venous return). Trauma, surgery, and burns not only immobilize a person but are also associated with vascular insults, procoagulant release from injured tissues, increased hepatic synthesis of coagulation factors, and altered t-PA production. Many elements contribute to the thrombotic diathesis of pregnancy; besides the potential for amniotic fluid infusion into the circulation at the time of delivery, late pregnancy and the postpartum period are also associated with systemic hypercoagulability. Tumor-associated inflammation and coagulation factors (tissue factor, factor VIII) and procoagulants (e.g., mucin) released from tumor cells all contribute to the increased risk of thromboembolism in disseminated cancers, so-called migratory thrombophlebitis or Trousseau syndrome.39,46 Regardless of the specific clinical setting, advanced age also increases the risk of DVT.
Arterial and Cardiac Thrombosis.
Atherosclerosis is a major cause of arterial thromboses, because it is associated with loss of endothelial integrity and with abnormal vascular flow (see Fig. 4-13B). Myocardial infarction can predispose to cardiac mural thrombi by causing dyskinetic myocardial contraction as well as damage to the adjacent endocardium (see Fig. 4-13A), and rheumatic heart disease may engender atrial mural thrombi as discussed above. Besides local obstructive consequences, cardiac and aortic mural thrombi can also embolize peripherally. Although any tissue can be affected, the brain, kidneys, and spleen are particularly likely targets because of their rich blood supply.
DISSEMINATED INTRAVASCULAR COAGULATION (DIC)
Disorders ranging from obstetric complications to advanced malignancy can be complicated by DIC, the sudden or insidious onset of widespread fibrin thrombi in the microcirculation. Although these thrombi are not grossly visible, they are readily apparent microscopically and can cause diffuse circulatory insufficiency, particularly in the brain, lungs, heart, and kidneys. To complicate matters, the widespread microvascular thrombosis results in platelet and coagulation protein consumption (hence the synonym consumption coagulopathy), and at the same time, fibrinolytic mechanisms are activated. Thus, an initially thrombotic disorder can evolve into a bleeding catastrophe. It should be emphasized that DIC is not a primary disease but rather a potential complication of any condition associated with widespread activation of thrombin.47 It is discussed in greater detail along with other bleeding diatheses in Chapter 14.
Embolism
An embolus is a detached intravascular solid, liquid, or gaseous mass that is carried by the blood to a site distant from its point of origin. The term embolus was coined by Rudolf Virchow in 1848 to describe objects that lodge in blood vessels and obstruct the flow of blood. Almost all emboli represent some part of a dislodged thrombus, hence the term thromboembolism. Rare forms of emboli include fat droplets, nitrogen bubbles, atherosclerotic debris (cholesterol emboli), tumor fragments, bone marrow, or even foreign bodies. However, unless otherwise specified, emboli should be considered thrombotic in origin. Inevitably, emboli lodge in vessels too small to permit further passage, causing partial or complete vascular occlusion; a major consequence is ischemic necrosis (infarction) of the downstream tissue. Depending on where they originate, emboli can lodge anywhere in the vascular tree; the clinical outcomes are best understood based on whether emboli lodge in the pulmonary or systemic circulations.
PULMONARY EMBOLISM
Pulmonary embolism (PE) has had a fairly stable incidence since the 1970s of roughly 2 to 4 per 1000 hospitalized patients in the United States, although the numbers vary depending on the mix of patient age and diagnosis (i.e., surgery, pregnancy, and malignancy all increase the risk).48 While the rate of fatal PEs (as assessed at autopsy) has declined from 6% to 2% over the last quarter century, PE still causes about 200,000 deaths per year in the United States.49 In more than 95% of cases, PEs originate from leg deep vein thromboses (DVTs), although it is important to realize that DVTs occur roughly two to three times more frequently than PEs.48
Fragmented thrombi from DVTs are carried through progressively larger channels and the right side of the heart before slamming into the pulmonary arterial vasculature. Depending on the size of the embolus, it can occlude the main pulmonary artery, straddle the pulmonary artery bifurcation (saddle embolus), or pass out into the smaller, branching arteries (Fig. 4-15). Frequently there are multiple emboli, perhaps sequentially or as a shower of smaller emboli from a single large mass; in general, the patient who has had one PE is at high risk of having more. Rarely, an embolus can pass through an interatrial or interventricular defect and gain access to the systemic circulation (paradoxical embolism). A more complete discussion of PEs is presented in Chapter 15; an overview is offered here.49-51
SYSTEMIC THROMBOEMBOLISM
Systemic thromboembolism refers to emboli in the arterial circulation. Most (80%) arise from intracardiac mural thrombi, two thirds of which are associated with left ventricular wall infarcts and another quarter with left atrial dilation and fibrillation. The remainder originate from aortic aneurysms, thrombi on ulcerated atherosclerotic plaques, or fragmentation of a valvular vegetation, with a small fraction due to paradoxical emboli; 10% to 15% of systemic emboli are of unknown origin. In contrast to venous emboli, which tend to lodge primarily in one vascular bed (the lung), arterial emboli can travel to a wide variety of sites; the point of arrest depends on the source and the relative amount of blood flow that downstream tissues receive. Major sites for arteriolar embolization are the lower extremities (75%) and the brain (10%), with the intestines, kidneys, spleen, and upper extremities involved to a lesser extent. The consequences of embolization in a tissue depend on its vulnerability to ischemia, the caliber of the occluded vessel, and whether there is a collateral blood supply; in general, arterial emboli cause infarction of the affected tissues.
FAT AND MARROW EMBOLISM
Microscopic fat globules—with or without associated hematopoietic marrow elements—can be found in the circulation and impacted in the pulmonary vasculature after fractures of long bones (which have fatty marrow) or, rarely, in the setting of soft tissue trauma and burns. Fat and associated cells released by marrow or adipose tissue injury may enter the circulation after the rupture of the marrow vascular sinusoids or venules. Fat and marrow PEs are very common incidental findings after vigorous cardiopulmonary resuscitation and are probably of no clinical consequence. Indeed, fat embolism occurs in some 90% of individuals with severe skeletal injuries (Fig. 4-16), but less than 10% of such patients have any clinical findings.
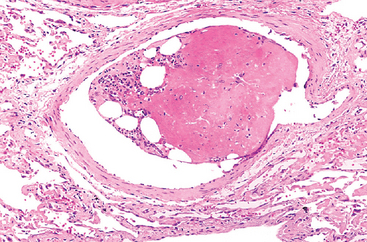
FIGURE 4-16 Bone marrow embolus in the pulmonary circulation. The cellular elements on the left side of the embolus are hematopoietic precursors, while the cleared vacuoles represent marrow fat. The relatively uniform red area on the right of the embolus is an early organizing thrombus.
Fat embolism syndrome is the term applied to the minority of patients who become symptomatic. It is characterized by pulmonary insufficiency, neurologic symptoms, anemia, and thrombocytopenia, and is fatal in about 5% to 15% of cases.52,53 Typically, 1 to 3 days after injury there is a sudden onset of tachypnea, dyspnea, and tachycardia; irritability and restlessness can progress to delirium or coma. Thrombocytopenia is attributed to platelet adhesion to fat globules and subsequent aggregation or splenic sequestration; anemia can result from similar red cell aggregation and/or hemolysis. A diffuse petechial rash (seen in 20% to 50% of cases) is related to rapid onset of thrombocytopenia and can be a useful diagnostic feature.
The pathogenesis of fat emboli syndrome probably involves both mechanical obstruction and biochemical injury.52 Fat microemboli and associated red cell and platelet aggregates can occlude the pulmonary and cerebral microvasculature. Release of free fatty acids from the fat globules exacerbates the situation by causing local toxic injury to endothelium, and platelet activation and granulocyte recruitment (with free radical, protease, and eicosanoid release) complete the vascular assault. Because lipids are dissolved out of tissue preparations by the solvents routinely used in paraffin embedding, the microscopic demonstration of fat microglobules (in the absence of accompanying marrow) typically requires specialized techniques, including frozen sections and stains for fat.
AIR EMBOLISM
Gas bubbles within the circulation can coalesce to form frothy masses that obstruct vascular flow (and cause distal ischemic injury). For example, a very small volume of air trapped in a coronary artery during bypass surgery, or introduced into the cerebral circulation by neurosurgery in the “sitting position,” can occlude flow with dire consequences. Generally, more than 100 cc of air are required to have a clinical effect in the pulmonary circulation; however, this volume of air can be inadvertently introduced during obstetric or laparoscopic procedures, or as a consequence of chest wall injury.54
A particular form of gas embolism, called decompression sickness, occurs when individuals experience sudden decreases in atmospheric pressure.55 Scuba and deep sea divers, underwater construction workers, and individuals in unpressurized aircraft in rapid ascent are all at risk. When air is breathed at high pressure (e.g., during a deep sea dive), increased amounts of gas (particularly nitrogen) are dissolved in the blood and tissues. If the diver then ascends (depressurizes) too rapidly, the nitrogen comes out of solution in the tissues and the blood.
The rapid formation of gas bubbles within skeletal muscles and supporting tissues in and about joints is responsible for the painful condition called the bends. In the lungs, gas bubbles in the vasculature cause edema, hemorrhage, and focal atelectasis or emphysema, leading to a form of respiratory distress called the chokes. A more chronic form of decompression sickness is called caisson disease (named for the pressurized vessels used in the bridge construction; workers in these vessels suffered both acute and chronic forms of decompression sickness). In caisson disease, persistence of gas emboli in the skeletal system leads to multiple foci of ischemic necrosis; the more common sites are the femoral heads, tibia, and humeri.
Acute decompression sickness is treated by placing the individual in a high pressure chamber, which serves to force the gas bubbles back into solution. Subsequent slow decompression theoretically permits gradual resorption and exhalation of the gases so that obstructive bubbles do not re-form.
AMNIOTIC FLUID EMBOLISM
Amniotic fluid embolism is an ominous complication of labor and the immediate postpartum period. Although the incidence is only approximately 1 in 40,000 deliveries, the mortality rate is up to 80%, making amniotic fluid embolism the fifth most common cause of maternal mortality worldwide; it accounts for roughly 10% of maternal deaths in the United States and results in permanent neurologic deficit in as many as 85% of survivors.56 The onset is characterized by sudden severe dyspnea, cyanosis, and shock, followed by neurologic impairment ranging from headache to seizures and coma. If the patient survives the initial crisis, pulmonary edema typically develops, along with (in half the patients) DIC, as a result of release of thrombogenic substances from the amniotic fluid.56
The underlying cause is the infusion of amniotic fluid or fetal tissue into the maternal circulation via a tear in the placental membranes or rupture of uterine veins. Classic findings include the presence of squamous cells shed from fetal skin, lanugo hair, fat from vernix caseosa, and mucin derived from the fetal respiratory or gastrointestinal tract in the maternal pulmonary microvasculature (Fig. 4-17). Other findings include marked pulmonary edema, diffuse alveolar damage (Chapter 15), and the presence of fibrin thrombi in many vascular beds due to DIC.
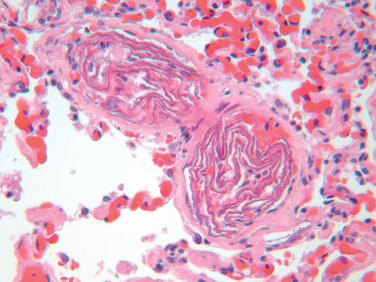
FIGURE 4-17 Amniotic fluid embolism. Two small pulmonary arterioles are packed with laminated swirls of fetal squamous cells. There is marked edema and congestion, and elsewhere in the lung were small organizing thrombi consistent with disseminated intravascular coagulation.
(Courtesy of Dr. Beth Schwartz, Baltimore, MD.)
Infarction
An infarct is an area of ischemic necrosis caused by occlusion of either the arterial supply or the venous drainage. Tissue infarction is a common and extremely important cause of clinical illness. Roughly 40% of all deaths in the United States are caused by cardiovascular disease, and most of these are attributable to myocardial or cerebral infarction. Pulmonary infarction is also a common complication in many clinical settings, bowel infarction is frequently fatal, and ischemic necrosis of the extremities (gangrene) is a serious problem in the diabetic population.
Nearly all infarcts result from thrombotic or embolic arterial occlusions. Occasionally infarctions are caused by other mechanisms, including local vasospasm, hemorrhage into an atheromatous plaque, or extrinsic vessel compression (e.g., by tumor). Rarer causes include torsion of a vessel (e.g., in testicular torsion or bowel volvulus), traumatic rupture, or vascular compromise by edema (e.g., anterior compartment syndrome) or by entrapment in a hernia sac. Although venous thrombosis can cause infarction, the more common outcome is just congestion; in this setting, bypass channels rapidly open and permit vascular outflow, which then improves arterial inflow. Infarcts caused by venous thrombosis are thus more likely in organs with a single efferent vein (e.g., testis and ovary).
Morphology. Infarcts are classified according to color and the presence or absence of infection; they are either red (hemorrhagic) or white (anemic) and may be septic or bland.
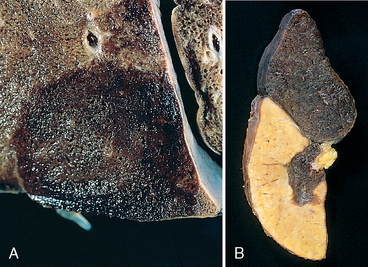
FIGURE 4-18 Red and white infarcts. A, Hemorrhagic, roughly wedge-shaped pulmonary red infarct. B, Sharply demarcated white infarct in the spleen.
Infarcts tend to be wedge-shaped, with the occluded vessel at the apex and the periphery of the organ forming the base (see Fig. 4-18); when the base is a serosal surface there can be an overlying fibrinous exudate. Acute infarcts are poorly defined and slightly hemorrhagic. With time the margins tend to become better defined by a narrow rim of congestion attributable to inflammation.
Infarcts resulting from arterial occlusions in organs without a dual blood supply typically become progressively paler and more sharply defined with time (see Fig. 4-18B). By comparison, in the lung hemorrhagic infarcts are the rule (see Fig. 4-18A). Extravasated red cells in hemorrhagic infarcts are phagocytosed by macrophages, which convert heme iron into hemosiderin; small amounts do not grossly impart any appreciable color to the tissue, but extensive hemorrhage can leave a firm, brown residuum.
The dominant histologic characteristic of infarction is ischemic coagulative necrosis (Chapter 1). It is important to recall that if the vascular occlusion has occurred shortly (minutes to hours) before the death of the person, no demonstrable histologic changes may be evident; it takes 4 to 12 hours for the tissue to show frank necrosis. Acute inflammation is present along the margins of infarcts within a few hours and is usually well defined within 1 to 2 days. Eventually the inflammatory response is followed by a reparative response beginning in the preserved margins (Chapter 2). In stable or labile tissues, parenchymal regeneration can occur at the periphery where underlying stromal architecture is preserved. However, most infarcts are ultimately replaced by scar (Fig. 4-19). The brain is an exception to these generalizations, as central nervous system infarction results in liquefactive necrosis (Chapter 1).
Septic infarctions occur when infected cardiac valve vegetations embolize or when microbes seed necrotic tissue. In these cases the infarct is converted into an abscess, with a correspondingly greater inflammatory response (Chapter 2). The eventual sequence of organization, however, follows the pattern already described.
Factors That Influence Development of an Infarct.
The effects of vascular occlusion can range from no or minimal effect to causing the death of a tissue or person. The major determinants of the eventual outcome are: (1) the nature of the vascular supply, (2) the rate at which an occlusion develops, (3) vulnerability to hypoxia, and (4) the oxygen content of the blood.
Shock
Shock is the final common pathway for several potentially lethal clinical events, including severe hemorrhage, extensive trauma or burns, large myocardial infarction, massive pulmonary embolism, and microbial sepsis. Shock is characterized by systemic hypotension due either to reduced cardiac output or to reduced effective circulating blood volume. The consequences are impaired tissue perfusion and cellular hypoxia. At the outset the cellular injury is reversible; however, prolonged shock eventually leads to irreversible tissue injury that often proves fatal.
The causes of shock fall into three general categories (Table 4-3):
TABLE 4-3 Three Major Types of Shock
| Type of Shock | Clinical Example | Principal Mechanisms | |
|---|---|---|---|
| CARDIOGENIC | |||
| Failure of myocardial pump resulting from intrinsic myocardial damage, extrinsic pressure, or obstruction to outflow | |||
| HYPOVOLEMIC | |||
| Fluid loss (e.g., hemorrhage, vomiting, diarrhea, burns, or trauma) | Inadequate blood or plasma volume | ||
| SEPTIC | |||
| Peripheral vasodilation and pooling of blood; endothelial activation/injury; leukocyte-induced damage, disseminated intravascular coagulation; activation of cytokine cascades | |||
Less commonly, shock can occur in the setting of anesthetic accident or a spinal cord injury (neurogenic shock), as a result of loss of vascular tone and peripheral pooling of blood. Anaphylactic shock denotes systemic vasodilation and increased vascular permeability caused by an IgE–mediated hypersensitivity reaction (Chapter 6). In these situations, acute widespread vasodilation results in tissue hypoperfusion and hypoxia.
PATHOGENESIS OF SEPTIC SHOCK
Septic shock is associated with severe hemodynamic and hemostatic derangements, and therefore merits more detailed consideration here. With a mortality rate near 20%, septic shock ranks first among the causes of death in intensive care units and accounts for over 200,000 lost lives each year in the United States.57 Its incidence is rising, ironically due to improvements in life support for critically ill patients and the growing ranks of immunocompromised hosts (due to chemotherapy, immunosuppression, or HIV infection). Currently, septic shock is most frequently triggered by gram-positive bacterial infections, followed by gram-negative bacteria and fungi.57 Hence, the older synonym of “endotoxic shock” is not appropriate.
In septic shock, systemic vasodilation and pooling of blood in the periphery leads to tissue hypoperfusion, even though cardiac output may be preserved or even increased early in the course. This is accompanied by widespread endothelial cell activation and injury, often leading to a hypercoagulable state that can manifest as DIC. In addition, septic shock is associated with changes in metabolism that directly suppress cellular function. The net effect of these abnormalities is hypoperfusion and dysfunction of multiple organs—culminating in the extraordinary morbidity and mortality associated with sepsis.
The ability of diverse microorganisms to cause septic shock (sometimes even when the infection is localized to one area of the body)58 is consistent with the idea that several microbial constituents can initiate the process. As you will recall from Chapter 2, macrophages, neutrophils, and other cells of the innate immune system express a number of receptors that respond to a variety of substances derived from microorganisms. Once activated, these cells release inflammatory mediators, as well as a variety of immunosuppressive factors that modify the host response. In addition, microbial constituents also activate humoral elements of innate immunity, particularly the complement and coagulation pathways. These mediators combine with the direct effects of microbial constituents on endothelium in a complex, incompletely understood fashion to produce septic shock (Fig. 4-20).59-61 The major factors contributing to its pathophysiology include the following:
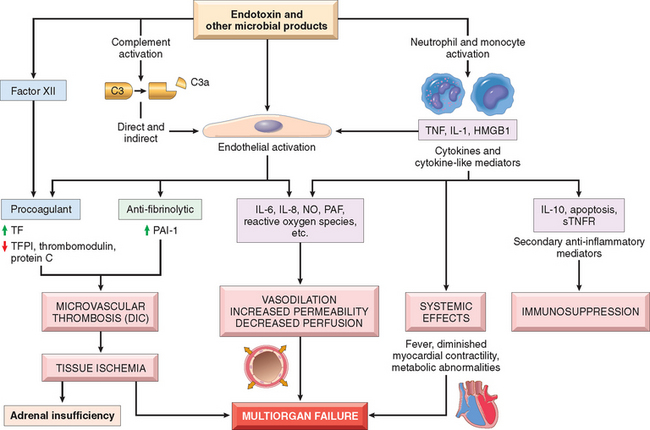
FIGURE 4-20 Major pathogenic pathways in septic shock. Microbial products activate endothelial cells and cellular and humoral elements of the innate immune system, initiating a cascade of events that lead to end-stage multiorgan failure. Additional details are given in the text. DIC, disseminated vascular coagulation; HMGB1, high mobility group box 1 protein; NO, nitric oxide; PAF, platelet activating factor; PAI-1, plasminogen activator inhibitor 1; STNFR, soluble TNF receptor; TF, tissue factor; TFPI, tissue factor pathway inhibitor.
The severity and outcome of septic shock are likely dependent upon the extent and virulence of the infection; the immune status of the host; the presence of other co-morbid conditions; and the pattern and level of mediator production. The multiplicity of factors and the complexity of the interactions that underlie sepsis explain why most attempts to intervene therapeutically with antagonists of specific mediators have been of very modest benefit at best, and may even have had deleterious effects in some cases.59 The standard of care remains treatment with appropriate antibiotics, intensive insulin therapy for hyperglycemia, fluid resuscitation to maintain systemic pressures, and “physiologic doses” of corticosteroids to correct relative adrenal insufficiency.59 Administration of activated protein C (to prevent thrombin generation and thereby reduce coagulation and inflammation) may have some benefit in cases of severe sepsis, but this remains controversial. Suffice it to say, even in the best of clinical centers, septic shock remains an obstinate clinical challenge.58
It is worth mentioning here that an additional group of secreted bacterial proteins called superantigens also cause a syndrome similar to septic shock (e.g., toxic shock syndrome). Superantigens are polyclonal T-lymphocyte activators that induce the release of high levels of cytokines that result in a variety of clinical manifestations, ranging from a diffuse rash to vasodilation, hypotension, and death.66
STAGES OF SHOCK
Shock is a progressive disorder that, if uncorrected, leads to death. The exact mechanism(s) of death from sepsis are still unclear; aside from increased lymphocyte and enterocyte apoptosis there is only minimal cell death, and patients rarely have refractory hypotension.61 For hypovolemic and cardiogenic shock, however, the pathways to death are reasonably well understood. Unless the insult is massive and rapidly lethal (e.g., a massive hemorrhage from a ruptured aortic aneurysm), shock in those settings tends to evolve through three general (albeit somewhat artificial) phases:
In the early nonprogressive phase of shock, a variety of neurohumoral mechanisms help to maintain cardiac output and blood pressure. These include baroreceptor reflexes, catecholamine release, activation of the renin-angiotensin axis, ADH release, and generalized sympathetic stimulation. The net effect is tachycardia, peripheral vasoconstriction, and renal conservation of fluid. Cutaneous vasoconstriction, for example, is responsible for the characteristic coolness and pallor of the skin in well-developed shock (although septic shock can initially cause cutaneous vasodilation and thus present with warm, flushed skin). Coronary and cerebral vessels are less sensitive to the sympathetic response and thus maintain relatively normal caliber, blood flow, and oxygen delivery.
If the underlying causes are not corrected, shock passes imperceptibly to the progressive phase, during which there is widespread tissue hypoxia. In the setting of persistent oxygen deficit, intracellular aerobic respiration is replaced by anaerobic glycolysis with excessive production of lactic acid. The resultant metabolic lactic acidosis lowers the tissue pH and blunts the vasomotor response; arterioles dilate, and blood begins to pool in the microcirculation. Peripheral pooling not only worsens the cardiac output, but also puts EC at risk for developing anoxic injury with subsequent DIC. With widespread tissue hypoxia, vital organs are affected and begin to fail.
Without intervention, the process eventually enters an irreversible stage. Widespread cell injury is reflected in lysosomal enzyme leakage, further aggravating the shock state. Myocardial contractile function worsens in part because of nitric oxide synthesis. If ischemic bowel allows intestinal flora to enter the circulation, bacteremic shock may be superimposed. At this point the patient has complete renal shutdown as a result of acute tubular necrosis (Chapter 20), and despite heroic measures the downward clinical spiral almost inevitably culminates in death.
Morphology. The cellular and tissue changes induced by cardiogenic or hypovolemic shock are essentially those of hypoxic injury (Chapter 1); changes can manifest in any tissue although they are particularly evident in brain, heart, lungs, kidneys, adrenals, and gastrointestinal tract. The adrenal changes in shock are those seen in all forms of stress; essentially there is cortical cell lipid depletion. This does not reflect adrenal exhaustion but rather conversion of the relatively inactive vacuolated cells to metabolically active cells that utilize stored lipids for the synthesis of steroids. The kidneys typically exhibit acute tubular necrosis (Chapter 20). The lungs are seldom affected in pure hypovolemic shock, because they are somewhat resistant to hypoxic injury. However, when shock is caused by bacterial sepsis or trauma, changes of diffuse alveolar damage (Chapter 15) may develop, the so-called shock lung. In septic shock, the development of DIC leads to widespread deposition of fibrin-rich microthrombi, particularly in the brain, heart, lungs, kidney, adrenal glands, and gastrointestinal tract. The consumption of platelets and coagulation factors also often leads to the appearance of petechial hemorrhages on serosal surface and the skin.
With the exception of neuronal and myocyte ischemic loss, virtually all of these tissues may revert to normal if the individual survives. Unfortunately, most patients with irreversible changes due to severe shock die before the tissues can recover.
Clinical Consequences.
The clinical manifestations of shock depend on the precipitating insult. In hypovolemic and cardiogenic shock the patient presents with hypotension; a weak, rapid pulse; tachypnea; and cool, clammy, cyanotic skin. In septic shock the skin may initially be warm and flushed because of peripheral vasodilation. The initial threat to life stems from the underlying catastrophe that precipitated the shock (e.g., myocardial infarct, severe hemorrhage, or sepsis). Rapidly, however, the cardiac, cerebral, and pulmonary changes secondary to shock worsen the problem. Eventually, electrolyte disturbances and metabolic acidosis also exacerbate the situation. Individuals who survive the initial complications may enter a second phase dominated by renal insufficiency and marked by a progressive fall in urine output as well as severe fluid and electrolyte imbalances.
The prognosis varies with the origin of shock and its duration. Thus, greater than 90% of young, otherwise healthy patients with hypovolemic shock survive with appropriate management; in comparison, septic shock, or cardiogenic shock associated with extensive myocardial infarction, can have substantially worse mortality rates, even with optimal care.
1 Schrier R, Abraham W. Hormones and hemodynamics in heart failure. N Engl J Med. 1999;341:57.
2 Chen H, Schrier R. Pathophysiology of volume overload in acute heart failure syndromes. Am J Med. 2006;119:S11.
3 Arnout J, et al. Haemostasis. Handb Exp Pharmacol. 2006;176(Pt 2):1.
4 Hoffman M, Monroe D. Coagulation 2006: a modern view of hemostasis. Hematol Oncol Clin North Am. 2007;21:1.
5 Michiels C. Endothelial cell functions. J Cell Physiol. 2003;196:430.
6 Galley H, Webster N. Physiology of the endothelium. Br J Anaesth. 2004;93:105.
7 Pries A, Kuebler W. Normal endothelium. Handb Exp Pharmacol. 2006;176(Pt 1):1.
8 Mackman N. Tissue-specific hemostasis in mice. Arterioscler Thromb Vasc Biol. 2005;25:2273.
9 Esmon C. Inflammation and the activated protein C anticoagulant pathway. Semin Thromb Hemost. 2006;32(Suppl 1):49.
10 Crawley J, Lane D. The haemostatic role of tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol. 2007.
11 Ruggeri Z. Von Willebrand factor: looking back and looking forward. Thromb Haemost. 2007;98:55.
12 Monroe D, Key N. The tissue factor–factor VIIa complex: procoagulant activity, regulation, and multitasking. J Thromb Haemost. 2007;5:1097.
13 Andrews R, Berndt M. Platelet physiology and thrombosis. Thromb Res. 2004;114:447.
14 Briedé J, et al. von Willebrand factor stimulates thrombin-induced exposure of procoagulant phospholipids on the surface of fibrin-adherent platelets. J Thromb Haemost. 2003;1:559.
15 Lentz B. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res. 2003;42:423.
16 Salles I, et al. Inherited traits affecting platelet function. Blood Rev. 2008.
17 Husmann M, Barton M. Therapeutical potential of direct thrombin inhibitors for atherosclerotic vascular disease. Expert Opin Investig Drugs. 2007;16:563.
18 Schneider D, Aggarwal A. Development of glycoprotein IIb–IIIa antagonists: translation of pharmacodynamic effects into clinical benefit. Expert Rev Cardiovasc Ther. 2004;2:903.
19 Mackman N, et al. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687.
20 Crawley J, et al. The central role of thrombin in hemostasis. J Thromb Haemost. 2007;5(Suppl 1):95.
21 Coughlin S. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800.
22 Landis R. Protease activated receptors: clinical relevance to hemostasis and inflammation. Hematol Oncol Clin North Am. 2007;21:103.
23 Cesarman-Maus G, Hajjar K. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005;129:307.
24 Cale J, Lawrence D. Structure-function relationships of plasminogen activator inhibitor-1 and its potential as a therapeutic agent. Curr Drug Targets. 2007;8:971.
25 Cushman M. Epidemiology and risk factors for venous thrombosis. Semin Hematol. 2007;44:62.
26 Nesbitt W, et al. The impact of blood rheology on the molecular and cellular events underlying arterial thrombosis. J Mol Med. 2006;84:989.
27 Seligsohn U, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med. 2001;344:1222.
28 Feero W. Genetic thrombophilia. Prim Care. 2004;31:685.
29 Middeldorp S, Levi M. Thrombophilia: an update. Semin Thromb Hemost. 2007;33:563.
30 Rosendorff A, Dorfman D. Activated protein C resistance and factor V Leiden. Arch Pathol Lab Med. 2007;131:866.
31 Danckwardt S, et al. 3′ end processing of the prothrombin mRNA in thrombophilia. Acta Haematol. 2006;115:192.
32 Jakubowski H. The molecular basis of homocysteine thiolactone-mediated vascular disease. Clin Chem Lab Med. 2007;45:1704.
33 Gatt A, Makris M. Hyperhomocysteinemia and venous thrombosis. Semin Hematol. 2007;44:70.
34 Kottke-Marchant K. Genetic polymorphisms associated with venous and arterial thrombosis: an overview. Arch Pathol Lab Med. 2002;126:295.
35 Emmerich J, et al. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism—pooled analysis of 8 case-control studies including 2310 cases and 3204 controls. Study Group for Pooled-Analysis in Venous Thromboembolism. Thromb Haemost. 2001;86:809.
36 Gallus A. Travel, venous thromboembolism, and thrombophilia. Semin Thromb Hemost. 2005;31:90.
37 Kuipers S, et al. Travel and venous thrombosis: a systematic review. J Intern Med. 2007;262:615.
38 Rosendaal F, et al. Estrogens, progestogens and thrombosis. J Thromb Haemost. 2003;1:1371.
39 Zwicker J, et al. Cancer-associated thrombosis. Crit Rev Oncol Hematol. 2007;62:126.
40 Castelli R, et al. Heparin induced thrombocytopenia: pathogenetic, clinical, diagnostic and therapeutic aspects. Cardiovasc Hematol Disord Drug Targets. 2007;7:153.
41 Warkentin T. Heparin-induced thrombocytopenia. Hematol Oncol Clin North Am. 2007;21:589.
42 Warkentin T, et al. Heparin-induced thrombocytopenia associated with fondaparinux. N Engl J Med. 2007;356:2653.
43 Pierangeli S, et al. Antiphospholipid antibodies and the antiphospholipid syndrome: an update on treatment and pathogenic mechanisms. Curr Opin Hematol. 2006;13:366.
44 Merrill J, Asherson R. Catastrophic antiphospholipid syndrome. Nat Clin Pract Rheumatol. 2006;2:81.
45 Hegde V, et al. Cardiovascular surgical outcomes in patients with the antiphospholipid syndrome—a case-series. Heart Lung Circ. 2007;16:4237.
46 Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood. 2007;110:1723.
47 Levi M. Disseminated intravascular coagulation. Crit Care Med. 2007;35:21915.
48 Stein P, et al. Trends in the incidence of pulmonary embolism and deep venous thrombosis in hospitalized patients. Am J Cardiol. 2005;95:1525.
49 Heit J. Venous thromboembolism epidemiology: implications for prevention and management. Semin Thromb Hemost. 2002;28(Suppl 2):3.
50 Goldhaber S. Pulmonary embolism. Lancet. 2004;363:1295.
51 Rahimtoola A, Bergin J. Acute pulmonary embolism: an update on diagnosis and management. Curr Probl Cardiol. 2005;30:61.
52 Parisi D, et al. Fat embolism syndrome. Am J Orthop. 2002;31:507.
53 Habashi N, et al. Therapeutic aspects of fat embolism syndrome. Injury. 2006;37(Suppl 4):S68.
54 Mirski M, et al. Diagnosis and treatment of vascular air embolism. Anesthesiology. 2007;106:164.
55 Tetzlaff K, Thorsen E. Breathing at depth: physiologic and clinical aspects of diving while breathing compressed gas. Clin Chest Med. 2005;26:355.
56 Moore J, Baldisseri M. Amniotic fluid embolism. Crit Care Med. 2005;33(10 Suppl):S279.
57 Martin G, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546.
58 Munford R. Severe sepsis and septic shock: the role of gram-negative bacteremia. Annu Rev Pathol. 2006;1:467.
59 Hotchkiss R, Karl I. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138.
60 Remick D. Pathophysiology of sepsis. Am J Pathol. 2007;170:1435.
61 Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885.
62 Fink M. Neuropetide modulators of high mobility group box 1 secretion as potential therapeutic agents for severe sepsis. Am J Pathol. 2008;172:1171.
63 Albrecht E, Ward P. Complement-induced impairment of the innate immune system during sepsis. Curr Infect Dis Rep. 2005;7:349.
64 vanAmersfoort E, et al. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin Microbiol Rev. 2003;16:379.
65 Marik P, Raghaven M. Stress-hyperglycemia, insulin and immunomodulation in sepsis. Int Care Med. 2004;30:748.
66 Zamoyska R. Superantigens: supersignalers? Sci STKE. 2006;358:45.