Chapter 10 Diseases of Infancy and Childhood
Children are not merely little adults, and their diseases are not merely variants of adult diseases. Many childhood conditions are unique to, or at least take distinctive forms in, this stage of life and so are discussed separately in this chapter. Diseases originating in the perinatal period are important in that they account for significant morbidity and mortality. As would be expected, the chances for survival of live-born infants improve with each passing week. This progressrepresents, at least in part, a triumph of improved medical care. Better prenatal care, more effective methods of monitoring the condition of the fetus, and judicious resort to cesarean section before term when there is evidence of fetal distress, have all contributed toward bringing into this “mortal coil” live-born infants who in past years might have been stillborn. These infants represent an increased number of high-risk infants. Nonetheless, the infant mortality rate in the United States has shown a decline from a level of 20.0 deaths per 1000 live births in 1970 to about 6.8 deaths in 2004, the latest year for which these data are systematically available.1 Although the death rate has continued to decline for all infants, African Americans continue to have an infant mortality rate more than twice (13.6 deaths per 1000 live births) that of American whites (5.6 deaths). Worldwide, infant mortality rates vary widely, from as low as 2.3 deaths per 1000 live births in Singapore, to as high as 180 deaths in the African subcontinent.
Each stage of development of the infant and child is prey to a somewhat different group of disorders. The data available permit a survey of four time spans: (1) the neonatal period (the first 4 weeks of life), (2) infancy (the first year of life), (3) age 1 to 4 years, and (4) age 5 to 14 years.
The major causes of death in infancy and childhood are listed in Table 10-1. Congenital anomalies, disorders relating to short gestation (prematurity) and low birth weight, and sudden infant death syndrome (SIDS) represent the leading causes of death in the first 12 months of life. Once the infant survives the first year of life, the outlook brightens measurably. In the next two age groups—1 to 4 years and 5 to 14 years—injuries resulting from accidents have become the leading cause of death. Among the natural diseases, in order of importance, congenital anomalies and malignant neoplasms assume major significance. It would appear then that, in a sense, life is an obstacle course. For the great majority, the obstacles are surmounted or, even better, bypassed.
TABLE 10-1 Cause of Death Related with Age
| Causes* | Rate† |
|---|---|
| UNDER 1 YEAR | 685.2 |
| 1–4 YEARS | 29.9 |
| 5–14 YEARS | 16.8 |
| 15–24 YEARS | 80.1 |
* Causes are listed in decreasing order of frequency. All causes and rates are final 2004 statistics.
† Rates are expressed per 100,000 population from all causes within each age group.
‡ Excludes congenital heart disease.
From Minino AM et al.: Deaths: final data for 2004. National Vital Statistics Rep 55:19, 2007.
We now take a closer look at the specific conditions en-countered during the various stages of infant and child development.
Congenital Anomalies
Congenital anomalies are morphologic defects that are present at birth, but some, such as cardiac defects and renal anomalies, may not become clinically apparent until years later. The term congenital means “born with,” but it does not imply or exclude a genetic basis for the birth defect. It is estimated that about 120,000 (1 in 33) babies are born with a birth defect each year in the United States. They are the most common cause of mortality in the first year and contribute significantly to morbidity and mortality throughout the early years of life. In a sense, anomalies found in live-born infants represent the less serious developmental failures in embryogenesis that are compatible with live birth. Perhaps 20% of fertilized ova are so anomalous that they are blighted from the outset. Others may be compatible with early fetal development, only to lead to spontaneous abortion. Less severe anomalies allow more prolonged intrauterine survival, with some disorders terminating in stillbirth and those still less significant permitting live birth despite the handicaps imposed.
DEFINITIONS
Before proceeding, we define some of the terms used for various kinds of errors in morphogenesis—malformations, disruptions, deformations, sequences, and syndromes.
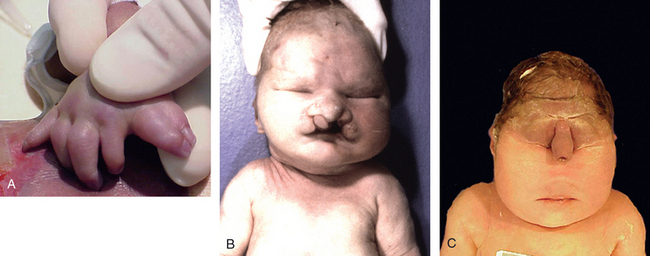
FIGURE 10-1 Examples of malformations. Polydactyly (one or more extra digits) and syndactyly (fusion of digits), both of which are illustrated in A, have little functional consequence when they occur in isolation. Similarly, cleft lip (B), with or without associated cleft palate, is compatible with life when it occurs as an isolated anomaly; in the present case, however, this neonate had an underlying malformation syndrome (trisomy 13) and expired because of severe cardiac defects. C, The stillbirth illustrated represents a severe and essentially lethal malformation, wherein the midface structures are fused or ill-formed; in almost all cases, this degree of external dysmorphogenesis is associated with severe internal anomalies such as maldevelopment of the brain and cardiac defects.
(A and C, courtesy of Dr. Reade Quinton; and B, courtesy of Dr. Beverly Rogers, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX.)
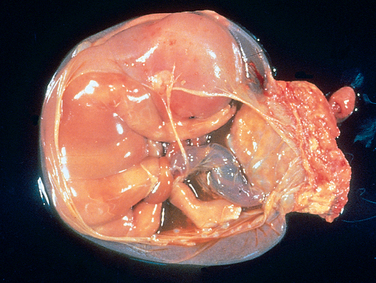
FIGURE 10-2 Disruption of morphogenesis by an amniotic band. Note the placenta at the right of the diagram and the band of amnion extending from the top portion of the amniotic sac to encircle the leg of the fetus.
(Courtesy of Dr. Theonia Boyd, Children’s Hospital of Boston, Boston, MA.)
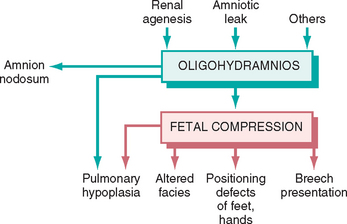
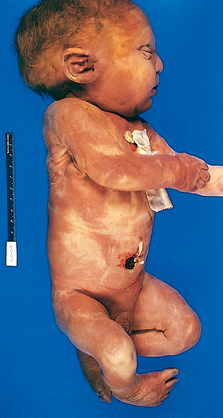
FIGURE 10-4 Infant with oligohydramnios sequence. Note the flattened facial features and deformed right foot
(talipes equinovarus).
In addition to the aforementioned general definitions, a few organ-specific terms should be defined. Agenesis refers to the complete absence of an organ and its associated primordium. A closely related term, aplasia, refers also to the absence of an organ but one due to failure of development of the primordium. Atresia describes the absence of an opening, usually of a hollow visceral organ, such as the trachea and intestine. Hypoplasia refers to incomplete development or decreased size of an organ with decreased numbers of cells, whereas hyperplasia refers to the converse, that is, the enlargement of an organ due to increased numbers of cells. An abnormality in an organ or a tissue as a result of an increase or a decrease in the size (rather than the number) of individual cells defines hypertrophy or hypotrophy, respectively. Finally, dysplasia, in the context of malformations (versus neoplasia) describes an abnormal organization of cells.
CAUSES OF ANOMALIES
At one time, it was believed that the presence of a visible, external anomaly was divine punishment for wickedness, a belief that occasionally jeopardized the mother’s life. Although we are learning a great deal about some of the molecular bases of congenital anomalies, the exact cause remains unknown in at least half to three quarters of the cases. The common known causes of congenital anomalies can be grouped into three major categories: genetic, environmental, and multifactorial (Table 10-2).
TABLE 10-2 Causes of Congenital Anomalies in Humans
| Cause | Frequency (%) |
|---|---|
| GENETIC | |
| Chromosomal aberrations | 10–15 |
| Mendelian inheritance | 2–10 |
| ENVIRONMENTAL | |
| Maternal/placental infections | 2–3 |
| Rubella | |
| Toxoplasmosis | |
| Syphilis | |
| Cytomegalovirus | |
| Human immunodeficiency virus | |
| Maternal disease states | 6–8 |
| Diabetes | |
| Phenylketonuria | |
| Endocrinopathies | |
| Drugs and chemicals | 1 |
| Alcohol | |
| Folic acid antagonists | |
| Androgens | |
| Phenytoin | |
| Thalidomide | |
| Warfarin | |
| 13-cis-retinoic acid | |
| Others | |
| Irradiations | 1 |
| MULTIFACTORIAL | 20–25 |
| UNKNOWN | 40–60 |
Adapted from Stevenson RE et al (eds): Human Malformations and related Anomalies. New York, Oxford University Press, 1993, p 115.
Genetic Causes
Anomalies that are known to be genetic in origin can be divided into two groups:
A third group is suspected of resulting from multifactorial inheritance, a term that implies the interaction of two or more genes of small effect with environmental factors, and is discussed separately.
Karyotypic abnormalities are present in approximately 10% to 15% of live-born infants with congenital anomalies, but trisomy 21 (Down syndrome) is the only one that approaches a birth frequency of greater than 10 in 10,000 total births. Next in order of frequency are trisomies 13 and 18 (Table 10-3). The remaining chromosomal syndromes associated with malformations are far rarer. The great preponderance of these cytogenetic aberrations arises as defects in gametogenesis and so are not familial. There are, however, several transmissible chromosomal abnormalities, for example, the form of Down syndrome associated with a robertsonian translocation in the parent, which is passed from one generation to the next, thus constituting a familial pattern of structural abnormalities (see Chapter 5). It should come as a sobering thought that 80% to 90% of fetuses with aneuploidy and other abnormalities of chromosome number die in utero, the majority in the earliest stages of gestation.
TABLE 10-3 National Prevalence Estimates for the Most Common Birth Defects in the United States, 1999–2001
| Birth Defect | Estimated National Prevalence (per 10,000 live births) |
|---|---|
| CHROMOSOMAL DEFECTS | |
| Down syndrome (Trisomy 21) | 12.8 |
| Trisomy 13 | 1.3 |
| Trisomy 18 | 2.3 |
| OROFACIAL DEFECTS | |
| Cleft palate | 6.4 |
| Cleft lip with and without cleft palate | 10.5 |
| CARDIOVASCULAR DEFECTS | |
| Atrioventricular septal defect (endocardial cushion defect) | 4.4 |
| Transposition of great arteries | 4.7 |
| Tetrology of Fallot | 3.9 |
| CENTRAL NERVOUS SYSTEM DEFECTS | |
| Spina bifida without anencephalus | 3.7 |
| Anencephalus | 2.5 |
| GASTROINTESTINAL DEFECTS | |
| Rectal and large intestinal atresia/stenosis | 4.8 |
| Esophageal atresia/tracheoesophageal fistula | 2.4 |
| MUSCULOSKELETAL DEFECTS | |
| Gastroschisis | 3.7 |
| Diaphragmatic hernia | 2.9 |
| Omphalocele | 2.1 |
Adapted from Canfield MA et al.: National estimates and race/ethnic-specific variation of selected birth defects in the United States, 1999–2001. Birth Defects Res A 76:747–756, 2006. The data have been adjusted for maternal race and ethnicity.
Single-gene mutations of large effect may underlie major congenital anomalies, which, as expected, follow mendelian patterns of inheritance. Of these, approximately 90% are inherited in an autosomal dominant or recessive pattern, while the remainder segregates in an X-linked pattern. Not surprisingly, many of the mutations that give rise to birth defects involve loss of function of genes involved in normal organogenesis and development. For example, holoprosencephaly is the most common developmental defect of the forebrain and midface in humans (see Chapter 28); the Hedgehog signaling pathway plays a critical role in the morphogenesis of these structures, and loss-of-function mutations of individual components within this pathway are reported in families with a history of recurrent holoprosencephaly.2 Similarly, achondroplasia, which is the most common form of short-limb dwarfism, is caused by gain-of-function mutations in fibroblast growth factor receptor 3 (FGFR3).3 The FGFR3 protein is a negative regulator of bone growth, and the activating FGFR3 mutations in achondroplasia are thought to exaggerate this physiologic inhibition, resulting in dwarfism.
Environmental Causes
Environmental influences, such as viral infections, drugs, and irradiation to which the mother was exposed during pregnancy, may cause fetal malformations (the appellation of “malformation” is loosely used in this context, since technically these anomalies represent disruptions).
Viruses.
Many viruses have been implicated in causing malformations, including the agents responsible for rubella, cytomegalic inclusion disease, herpes simplex, varicella-zoster infection, influenza, mumps, human immunodeficiency virus (HIV), and enterovirus infections. Among these, the rubella virus and cytomegalovirus are the most extensively investigated agents. With all viruses, the gestational age at which the infection occurs in the mother is critically important. The at-risk period for rubella infection extends from shortly before conception to the sixteenth week of gestation, the hazard being greater in the first 8 weeks than in the second 8 weeks. The incidence of malformations is reduced from 50% to 20% to 7% if infection occurs in the first, second, or third month of gestation. The fetal defects are varied, but the major tetrad comprises cataracts, heart defects (persistent ductus arteriosus, pulmonary artery hypoplasia or stenosis, ventricular septal defect, tetralogy of Fallot), deafness, and mental retardation, referred to as congenital rubella syndrome.
Intrauterine infection with cytomegalovirus, mostly asymptomatic, is the most common fetal viral infection. This viral disease is considered in detail in Chapter 8; the highest at-risk period is the second trimester of pregnancy. Because organogenesis is largely completed by the end of the first trimester, congenital malformations occur less frequently than in rubella; nevertheless, the effects of virus-induced injury on the formed organs are often severe. Involvement of the central nervous system is a major feature, and the most prominent clinical changes are mental retardation, microcephaly, deafness, and hepatosplenomegaly.
Drugs and Other Chemicals.
A variety of drugs and chemicals have been suspected to be teratogenic, but perhaps less than 1% of congenital malformations are caused by these agents. The list includes thalidomide, folate antagonists, androgenic hormones, alcohol, anticonvulsants, warfarin (oral anticoagulant), and 13-cis-retinoic acid used in the treatment of severe acne (see below). In many instances experimental studies in lower organisms (chick, zebrafish, etc.) have been instrumental in elucidating which developmental pathway(s) is affected by a given teratogen. For example, thalidomide, once used as a tranquilizer in Europe, caused an extremely high frequency (50% to 80%) of limb abnormalities in exposed fetuses. The mechanism of thalidomide teratogenicity involves downregulation of the developmentally important wingless (WNT) signaling pathway through upregulation of endogenous WNT repressors.4 Thalidomide and related drugs have staged a remarkable comeback as antineoplastic agents, with potent immunomodulatory and anti-angiogenic properties. Much caution must be taken when these drugs are given to cancer patients who are of reproductive age. Alcohol is probably the most widely used teratogen. Alcohol is responsible for several structural anomalies, as well as more subtle cognitive and behavioral defects in the fetus, collectively termed fetal alcohol spectrum disorders (FASDs). The most severely affected infants with FASDs have growth retardation, microcephaly, atrial septal defect, short palpebral fissures, and maxillary hypoplasia, and this classic teratogenic phenotype is labeled as fetal alcohol syndrome. Experiments performed in animals suggest that prenatal exposure to alcohol disrupts at least two seminal developmental signaling pathways—retinoic acid and Hedgehog—with critical roles during development.5,6 While cigarette smoke–derived nicotine has not been convincingly demonstrated to be a teratogen, there is a high incidence of spontaneous abortions, premature labor, and placental abnormalities in pregnant smokers; babies born to smoking mothers often have a low birth weight and may be prone to SIDS (see later). In light of these findings, it is best to avoid nicotine exposure altogether during pregnancy.
Radiation.
In addition to being mutagenic and carcinogenic, radiation is teratogenic. Exposure to heavy doses of radiation during the period of organogenesis leads to malformations, such as microcephaly, blindness, skull defects, spina bifida, and other deformities. Such exposure occurred in the past when radiation was used to treat cervical cancer during pregnancy.
Maternal Diabetes.
Diabetes mellitus is a common entity, and despite advances in antenatal obstetric monitoring and glucose control, the incidence of major malformations in infants of diabetic mothers stands between 6% and 10% in most series. Maternal hyperglycemia-induced fetal hyperinsulinemia results in increased body fat, muscle mass, and organomegaly (fetal macrosomia); cardiac anomalies, neural tube defects, and other central nervous system malformations are some of the major anomalies seen in diabetic embryopathy.
Multifactorial Causes
In contrast to monogenic disorders like achondroplasia, which are caused by functional perturbation of a single gene, congenital anomalies with a multifactorial basis arise as a result of inheritance of multiple genetic polymorphisms that confer a “susceptibility phenotype.” The interaction of this underlying phenotype with the environment is then required before the disorder becomes manifest. In the case of congenital dislocation of the hip, for example, a shallow acetabular socket and laxity of the supporting ligaments are believed to be genetically determined, whereas frank breech position in utero, with hips flexed and knees extended is a key environmental factor. Such complex gene-environment interactions might explain why the monozygotic concordance rate for some common congenital anomalies like cleft lip or cleft palate is only in the range of 25% to 50%. The importance of environmental contribution to multifactorial inheritance is further underscored by a marked reduction in the incidence of neural tube defects by periconceptional intake of folic acid in the diet.7
The estimated frequency of some common birth defects in the United States is presented in Table 10-3.
PATHOGENESIS OF CONGENITAL ANOMALIES
The pathogenesis of congenital anomalies is complex and still poorly understood, but two general principles of developmental pathology are relevant regardless of the etiologic agent.
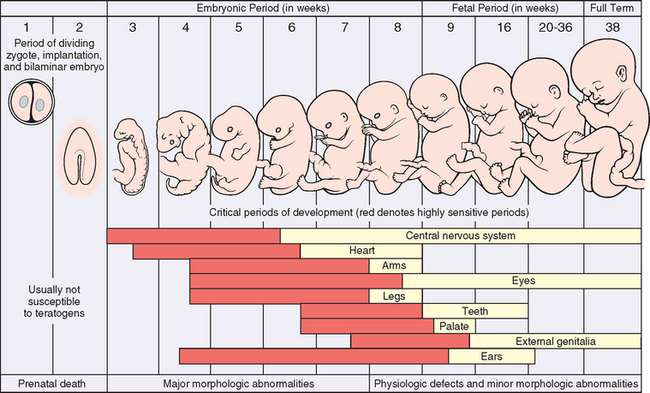
Disorders of Prematurity
Infants born before completion of the normal gestation period or who have failed to grow normally during gestation have higher morbidity and mortality rates than full-term infants. For example, an infant weighing 2300 gm and born at 34 weeks of gestation is likely to be physiologically immature and therefore at greater riskfor suffering the consequences of organ system immaturity (e.g., respiratory distress syndrome [RDS] or transient hyperbilirubinemia) than a full-term infant also weighing 2300 gm but with corresponding functional maturity of most organ systems. Therefore, a system of classification that takes into account both birth weight and gestational age has been adopted. Based on birth weight, infants are classified as being
Infants whose birth weight falls between the 10th and the 90th percentiles for a given gestational age are considered AGA, whereas those who fall above or below these norms are classified as LGA or SGA, respectively. With respect to gestational age, infants born before 37 weeks are considered preterm, whereas those delivered after the forty-second week are considered post-term. Such a classification is useful in risk stratification. For example, an AGA 1500-gm infant born at 32 weeks’ gestation has a much lower mortality risk than an SGA, 700-gm infant born at a similar gestational age. We briefly discuss the subgroups of infants who are SGA and/or preterm, since they account for a significant proportion of perinatal mortality.
CAUSES OF PREMATURITY AND FETAL GROWTH RESTRICTION
Prematurity, defined by a gestational age less than 37 weeks, is the second most common cause of neonatal mortality, behind only congenital anomalies. The American College of Obstetrics and Gynecology (ACOG) estimates that 12% of all births in the United States are preterm deliveries, and despite extensive research into this area, this rate has increased over the last two decades.10 The major risk factors for prematurity include:
The hazards of prematurity are manifold for the newborn and include one or more of the entities listed below:
Although preterm infants have low birth weights, it is often appropriate once adjusted for their gestational age. In contrast, at least one third of infants who weigh less than 2500 gm are born at term and therefore are undergrown rather than immature. Hence, fetal growth restriction (FGR) commonly underlies SGA. FGR has also been called intrauterine growth retardation; however, the term FGR probably better reflects the pathophysiology of this disorder.13 FGR can be detected before delivery by ultrasonographic measurement of various fetal dimensions, such as biparietal diameter, head circumference, abdominal circumference, femur length (as an indicator of fetal length), head-to-abdominal circumference ratio, femur length–to–abdominal circumference ratio, and total intrauterine volume. Influences known to result in FGR can be divided into three main groups: fetal, placental, and maternal.
Fetal.
Fetal influences are those that intrinsically reduce growth potential of the fetus despite an adequate supply of nutrients from the mother. Prominent among such fetal conditions are chromosomal disorders, congenital anomalies, and congenital infections. Chromosomal abnormalities may be detected in up to 17% of fetuses sampled for FGR and in up to 66% of fetuses with documented ultrasonographic malformations. Among the first group, the abnormalities include triploidy (7%), trisomy 18 (6%), trisomy 21 (1%), trisomy 13 (1%), and a variety of deletions and translocations (2%). Fetal infection should be considered in all infants with FGR. Those most commonly responsible for FGR are the TORCH group of infections (toxoplasmosis, rubella, cytomegalovirus, herpesvirus, and other viruses and bacteria, such as syphilis). Infants who are SGA because of fetal factors are usually characterized by symmetric growth restriction (also referred to as proportionate FGR), meaning that all organ systems are similarly affected.
Placental.
During the third trimester of pregnancy, vigorous fetal growth places particularly heavy demands on the uteroplacental supply line. Therefore, the adequacy of placental growth in the preceding midtrimester is extremely important, and uteroplacental insufficiency is an important cause of growth restriction. This insufficiency may result from umbilical-placental vascular anomalies (such as single umbilical artery, abnormal cord insertion, placental hemangioma), placental abruption, placenta previa, placental thrombosis and infarction, placental infection, or multiple gestations (Chapter 22). In some cases the placenta may be small without any detectable underlying cause. Placental causes of FGR tend to result in asymmetric (or disproportionate) growth retardation of the fetus with relative sparing of the brain. Physiologically, this general type of FGR is viewed as a down-regulation of growth in the latter half of gestation because of limited availability of nutrients or oxygen.
Genetic mosaicism confined to the placenta (confined placental mosaicism) is a more recently discovered cause of FGR and has been documented in up to 15% of pregnancies with FGR.14 Chromosomal mosaicism, in general, results from viable genetic mutations occurring after zygote formation. Depending on the developmental timing and cell of origin of the mutation, different forms of chromosomal mosaicism result. For example, genetic mutations occurring at the time of the first or second postzygotic division result in generalized constitutional mosaicism of the fetus and placenta. Conversely, if the mutation occurs later and within dividing trophoblast or extraembryonic progenitor cells of the inner cell mass (approximately 90% of the time), a genetic abnormality limited to the placenta results—confined placental mosaicism (Fig. 10-6). The phenotypic consequences of such placental mosaicism depend on both the specific cytogenetic abnormality and the percentage of cells involved. Chromosomal trisomies, in particular trisomy 7, are the abnormality most frequently documented.
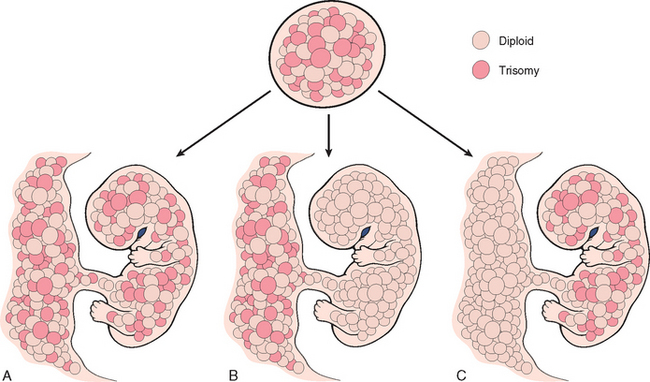
FIGURE 10-6 Diagrammatic representation of constitutional chromosomal mosaicism. A, Generalized. B, Confined to the placenta. C, Confined to the embryo. See text for details. See text for details.
(Modified and redrawn from Kalousek DK: Confined placental mosaicism and intrauterine development. Pediatr Pathol 10:69, 1990.)
Maternal.
By far the most common factors associated with SGA infants are maternal conditions that result in decreased placental blood flow. Vascular diseases, such as preeclampsia (toxemia of pregnancy) and chronic hypertension, are often the underlying cause. Another class of maternal diseases increasingly being recognized in the setting of FGR is inherited thrombophilias, such as the factor V Leiden mutation (Chapter 4).15 Inherited diseases of hypercoagulability are also associated with recurrent early pregnancy losses. The list of other maternal conditions associated with SGA infants is long, but some of the avoidable factors worth mentioning are maternal narcotic abuse, alcohol intake, and heavy cigarette smoking. Drugs causing FGR include both classic teratogens, such as antimetabolites, and some commonly administered therapeutic agents, such as phenytoin (Dilantin). Maternal malnutrition (in particular, prolonged hypoglycemia) may also affect fetal growth, but the association between SGA infants and the nutritional status of the mother is complex.
The SGA infant faces a difficult course, not only during the struggle for survival in the perinatal period, but also in childhood and adult life. Depending on the underlying cause of FGR and, to a lesser extent, the degree of prematurity, there is a significant risk of morbidity in the form of a major handicap, cerebral dysfunction, learning disability, or hearing and visual impairment.
Neonatal Respiratory Distress Syndrome (RDS)
There are many causes of respiratory distress in the newborn, including excessive sedation of the mother, fetal head injury during delivery, aspiration of blood or amniotic fluid, and intrauterine hypoxia brought about by coiling of the umbilical cord about the neck. The most common cause, however, is RDS, also known as hyaline membrane disease because of the deposition of a layer of hyaline proteinacecous material in the peripheral airspaces of infants who succumb to this condition. An estimated 24,000 cases of RDS are reported annually in the United States, and improvements in management of this condition have sharply decreased deaths due to respiratory insufficiency from as many as 5000 per year a decade earlier to less than 900 cases.16
In untreated infants (not receiving surfactant), RDS generally presents in a stereotyped fashion, with characteristic clinical findings. The infant is almost always preterm and AGA, and there are strong, but not invariable, associations with male gender, maternal diabetes, and delivery by cesarean section. Resuscitation may be necessary at birth, but usually within a few minutes rhythmic breathing and normal color are re-established. Soon afterward, often within 30 minutes, breathing becomes more difficult, and within a few hours cyanosis becomes evident. Fine rales can now be heard over both lung fields. A chest x-ray film at this time usually reveals uniform minute reticulogranular densities, producing a so-called ground-glass picture. In the full-blown condition the respiratory distress persists, cyanosis increases, and even the administration of 80% oxygen by a variety of ventilatory methods fails to improve the situation. If therapy staves off death for the first 3 or 4 days, however, the infant has an excellent chance of recovery.
Etiology and Pathogenesis.
Immaturity of the lungs is the most important substrate on which this condition develops. It may be encountered in full-term infants but is much less frequent than in those “born before their time into this breathing world.” The incidence of RDS is inversely proportional to gestational age. It occurs in about 60% of infants born at less than 28 weeks of gestation, 30% of those born between 28 to 34 weeks’ gestation, and less than 5% of those born after 34 weeks’ gestation.
The fundamental defect in RDS is a deficiency of pulmonary surfactant. As described in Chapter 15, surfactant consists predominantly of dipalmitoyl phosphatidylcholine (lecithin), smaller amounts of phosphatidylglycerol, and two groups of surfactant-associated proteins. The first group is composed of hydrophilic glycoproteins SP-A and SP-D, which play a role in pulmonary host defense (innate immunity). The second group consists of hydrophobic surfactant proteins SP-B and SP-C, which, in concert with the surfactant lipids, are involved in the reduction of surface tension at the air-liquid barrier in the alveoli of the lung. With reduced surface tension in the alveoli, less pressure is required to keep them patent and hence aerated. The importance of surfactant proteins in normal lung function can be gauged by the occurrence of severe respiratory failure in neonates with congenital deficiency of surfactant caused by mutations in the SFTPB or SFTBC genes.17
Surfactant production by type II alveolar cells is accelerated after the thirty-fifth week of gestation in the fetus. At birth, the first breath of life requires high inspiratory pressures to expand the lungs. With normal levels of surfactant, the lungs retain up to 40% of the residual air volume after the first breath; thus, subsequent breaths require far lower inspiratory pressures. With a deficiency of surfactant, the lungs collapse with each successive breath, and so infants must work as hard with each successive breath as they did with the first. The problem of stiff atelectatic lungs is compounded by the soft thoracic wall that is pulled in as the diaphragm descends. Progressive atelectasis and reduced lung compliance then lead to a train of events as depicted in Figure 10-7, resulting in a protein-rich, fibrin-rich exudation into the alveolar spaces with the formation of hyaline membranes. The fibrin-hyaline membranes constitute barriers to gas exchange, leading to carbon dioxide retention and hypoxemia. The hypoxemia itself further impairs surfactant synthesis, and a vicious cycle ensues.
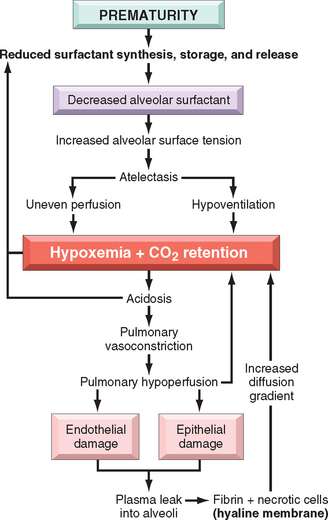
Surfactant synthesis is modulated by a variety of hormones and growth factors, including cortisol, insulin, prolactin, thyroxine, and TGF-β. The role of glucocorticoids is particularly important. Conditions associated with intrauterine stress and FGR that increase corticosteroid release lower the risk of developing RDS. Surfactant synthesis can be suppressed by the compensatory high blood levels of insulin in infants of diabetic mothers, which counteracts the effects of steroids. This may explain, in part, why infants of diabetic mothers have a higher risk of developing RDS. Labor is known to increase surfactant synthesis; hence, cesarean section before the onset of labor may increase the risk of RDS.
Morphology. The lungs are distinctive on gross examination. Though of normal size, they are solid, airless, and reddish purple, similar to the color of the liver, and they usually sink in water. Microscopically, alveoli are poorly developed, and those that are present are collapsed (Fig. 10-8). When the infant dies early in the course of the disease, necrotic cellular debris can be seen in the terminal bronchioles and alveolar ducts. The necrotic material becomes incorporated within eosinophilic hyaline membranes lining the respiratory bronchioles, alveolar ducts, and random alveoli. The membranes are largely made up of fibrin admixed with cell debris derived chiefly from necrotic type II pneumocytes. The sequence of events that leads to the formation of hyaline membranes is depicted in Figure 10-7. There is a remarkable paucity of neutrophilic inflammatory reaction associated with these membranes. The lesions of hyaline membrane disease are never seen in stillborn infants.
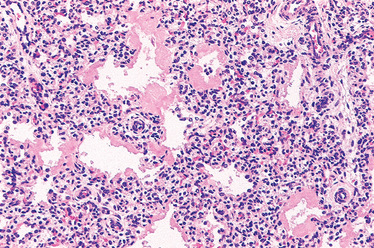
FIGURE 10-8 Hyaline membrane disease. There is alternating atelectasis and dilation of the alveoli. Note the eosinophilic thick hyaline membranes lining the dilated alveoli.
In infants who survive more than 48 hours, reparative changes occur in the lungs. The alveolar epithelium proliferates under the surface of the membrane, which may be desquamated into the airspace, where it may undergo partial digestion or phagocytosis by macrophages.
Clinical Course.
Although a classic clinical presentation before the era of treatment with exogenous surfactant was described earlier, the actual clinical course and prognosis for neonatal RDS vary, dependent on the maturity and birth weight of the infant and the promptness of institution of therapy. A major thrust in the control of RDS focuses on prevention, either by delaying labor until the fetal lung reaches maturity or by inducing maturation of the lung in the fetus at risk. Critical to these objectives is the ability to assess fetal lung maturity accurately. Because pulmonary secretions are discharged into the amniotic fluid, analysis of amniotic fluid phospholipids provides a good estimate of the level of surfactant in the alveolar lining. Prophylactic administration of exogenous surfactant at birth to extremely premature infants (gestational age ∼26 to 28 weeks) and administration of surfactant to older premature infants who are symptomatic have been shown to be extremely beneficial, such that it is now uncommon for infants to die of acute RDS. In addition, antenatal corticosteroids decrease neonatal morbidity and mortality when administered to mothers with threatened premature delivery at 24 to 34 weeks’ gestation. Once the infant is born, the cornerstone of treatment is the delivery of surfactant replacement therapy and oxygen, usually accomplished by a variety of ventilatory assistance methods, including high-frequency ventilation.
In uncomplicated cases, recovery begins to occur within 3 or 4 days. Therapy, however, carries with it the now well-recognized hazard of oxygen toxicity, caused by oxygen-derived free radicals. High concentrations of oxygen administered for prolonged periods cause two well-known complications: retrolental fibroplasia (also called retinopathy of prematurity) in the eyes (Chapter 29) and bronchopulmonary dysplasia. The retinopathy has been ascribed to changes in expression of vascular endothelial growth factor (VEGF), which is strongly induced by hypoxia, and also serves as a survival factor for endothelial cells and promotes angiogenesis (Chapter 3).18 During the initial hyperoxic phase of RDS therapy (phase I), VEGF is markedly decreased, causing endothelial cell apoptosis; VEGF increases after return to relatively hypoxic room air ventilation, inducing the retinal vessel proliferation (neovascularization) characteristic of the lesions in the retina (phase II).
Bronchopulmonary dysplasia (BPD), originally described in 1967, is now infrequent in infants of more than 1200 gm birth weight or with gestations exceeding 30 weeks. Gentler ventilation techniques, antenatal glucocorticoid therapy, and surfactant treatments have minimized severe lung injury in larger and more mature infants. The definition of BPD has evolved in recent years to reflect these trends, and at least 28 days of oxygen therapy in an infant who is beyond 36 weeks’ post-menstrual age is required to render a diagnosis of BPD.19 The original histopathologic descriptions of BPD reported airway epithelial hyperplasia and squamous metaplasia, alveolar wall thickening, and peribronchial as well as interstitial fibrosis. The major abnormalities in “new” BPD are a striking decrease in alveolar septation (manifested as large, simplified alveolar structures) and a dysmorphic capillary configuration. Thus, the current view is that BPD is caused by a potentially reversible impairment in the development of alveolar septation at the saccular stage.
Multiple factors—hyperoxemia, hyperventilation, prematurity, inflammatory cytokines, and vascular maldevelopment—contribute to BPD and probably act additively or synergistically to promote injury.20 Oxygen alone can arrest septation of lungs that are in the saccular stage of development, with infants receiving higher levels of supplemental oxygen having more persistent lung disease. Mechanical ventilation of preterm animals without simultaneous exposure to high levels of supplemental oxygen also results in the pathologic lesion of BPD. The levels of a variety of pro-inflammatory cytokines (TNF, interleukin-1β [IL-1β], IL-6, and IL-8) are increased in the alveoli of infants who develop BPD, and their deregulation in animal models can impair alveolar septation, suggesting a role for these cytokines in arresting pulmonary development.21 Recent studies in experimental models of lung development have also elucidated the requirement of appropriate vascularization within the pulmonary mesenchyme for allowing branching morphogenesis of the epithelium. In corroboration, infants who succumb to BPD often demonstrate dysmorphic capillaries and reduced levels of the angiogenic growth factor, VEGF.22
If oxygen toxicity is avoided, as is usually the case, and the infant can be kept alive for about 3 or 4 days, recovery of infants of 31 weeks’ gestation or more can be anticipated without permanent sequelae. Infants who recover from RDS are at increased risk for developing a variety of other complications associated with preterm birth; most important among these are patent ductus arteriosus, intraventricular hemorrhage, and necrotizing enterocolitis. Thus, although current high technology saves many infants with RDS, it also brings to the surface the exquisite fragility of the immature neonate.
NECROTIZING ENTEROCOLITIS
Necrotizing enterocolitis (NEC) is most common in premature infants, with the incidence of the disease being inversely proportional to the gestational age. It occurs in approximately 1 out of 10 very low birth weight infants (<1500 gm). Approximately 2500 cases occur annually in the United States.
The pathogenesis of NEC is uncertain, but is in all likelihood multifactorial. In addition to prematurity, most cases are associated with enteral feeding, suggesting that some postnatal insult (such as introduction of bacteria) sets in motion the cascade culminating in tissue destruction. While infectious agents likely play a role in NEC pathogenesis, no single bacterial pathogen has been linked to the disease. A large number of inflammatory mediators have been associated with NEC, and their discussion is beyond the scope of this book. One particular mediator, platelet activating factor (PAF), has been implicated in increasing mucosal permeability by promoting enterocyte apoptosis and compromising intercellular tight junctions, thus adding “fuel to the fire.”23 Stool and serum samples of infants with NEC demonstrate higher PAF levels than age-matched controls. Ultimately, breakdown of mucosal barrier functions permits transluminal migration of gut bacteria, leading to a vicious cycle of inflammation, mucosal necrosis, and further bacterial entry, eventually culminating in sepsis and shock (Chapter 4).
The clinical course is fairly typical, with the onset of bloody stools, abdominal distention, and development of circulatory collapse. Abdominal radiographs often demonstrate gas within the intestinal wall (pneumatosis intestinalis). NEC typically involves the terminal ileum, cecum, and right colon, although any part of the small or large intestines may be involved. The involved segment is distended, friable, and congested, or it can be frankly gangrenous; intestinal perforation with accompanying peritonitis may be seen. Microscopically, mucosal or transmural coagulative necrosis, ulceration, bacterial colonization, and submucosal gas bubbles may be seen (Fig. 10-9). Reparative changes, such as the formation of granulation tissue and fibrosis, may begin shortly after the acute episode. When detected early on, NEC can be often managed conservatively, but many cases (20% to 60%) require resection of the necrotic segments of bowel. NEC is associated with high perinatal mortality; those who survive often develop post-NEC strictures from fibrosis caused by the healing process.

FIGURE 10-9 Necrotizing enterocolitis (NEC). A, Postmortem examination in a severe case of NEC shows the entire small bowel is markedly distended with a perilously thin wall (usually this implies impending perforation). B, The congested portion of the ileum corresponds to areas of hemorrhagic infarction and transmural necrosis microscopically. Submucosal gas bubbles (pneumatosis intestinalis) can be seen in several areas (arrows).
Perinatal Infections
Infections of the embryo, fetus, and neonate are manifested in a variety of ways and are mentioned as etiologic factors in numerous other sections within this chapter. In general, fetal and perinatal infections are acquired through one of two primary routes—transcervically (also referred to as ascending) or transplacentally (hematologic). Occasionally, infections occur by a combination of the two routes in that an ascending microorganism infects the endometrium and then the fetal bloodstream via the chorionic villi.
TRANSCERVICAL (ASCENDING) INFECTIONS
Most bacterial and a few viral (e.g., herpes simplex II) infections are acquired by the cervicovaginal route. Such infections may be acquired in utero or around the time of birth. In general the fetus acquires the infection either by inhaling infected amniotic fluid into the lungs shortly before birth or by passing through an infected birth canal during delivery. As stated before, preterm birth is often an unfortunate consequence and may be related either to damage and rupture of the amniotic sac as a direct consequence of the inflammation or to the induction of labor associated with a release of prostaglandins by the infiltrating neutrophils. Inflammation of the placental membranes and cord are usually demonstrable, although the presence or absence and severity of chorioamnionitis do not necessarily correlate with the severity of the fetal infection. In the fetus infected by inhalation of amniotic fluid, pneumonia, sepsis, and meningitis are the most common sequelae.
TRANSPLACENTAL (HEMATOLOGIC) INFECTIONS
Most parasitic (e.g., toxoplasma, malaria) and viral infections and a few bacterial infections (i.e., Listeria, Treponema) gain access to the fetal bloodstream transplacentally via the chorionic villi. This hematogenous transmission may occur at any time during gestation or occasionally, as may be the case with hepatitis B and HIV, at the time of delivery via maternal-to-fetal transfusion. The clinical manifestations of these infections are highly variable, depending largely on the gestational timing and microorganism involved.
Parvovirus B19, which causes erythema infectiosum or “fifth disease of childhood” in immunocompetent older children, can infect 1% to 5% of pregnant women, and the vast majorityhave a normal pregnancy outcome. Adverse pregnancy outcomes in a minority of intrauterine infections include spontaneous abortion (particularly in the second trimester), stillbirth, hydrops fetalis (see below), and congenital anemia. Parvovirus B19 has a particular tropism for erythroid cells, and diagnostic viral inclusions can be seen in early erythroid progenitor cells in infected infants (Fig. 10-10).
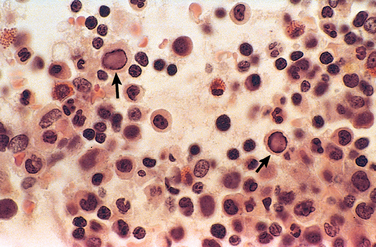
FIGURE 10-10 Bone marrow from an infant infected with parvovirus B19. The arrows indicate two erythroid precursors with large homogeneous intranuclear inclusions and a surrounding peripheral rim of residual chromatin.
The TORCH group of infections (see above) are grouped together because they may evoke similar clinical and pathologic manifestations, including fever, encephalitis, chorioretinitis, hepatosplenomegaly, pneumonitis, myocarditis, hemolytic anemia, and vesicular or hemorrhagic skin lesions. Such infections occurring early in gestation may also cause chronic sequelae in the child, including growth and mental retardation, cataracts, congenital cardiac anomalies, and bone defects.
SEPSIS
Perinatal sepsis can also be grouped clinically based on early onset (within the first 7 days of life) versus late onset (from 7 days to 3 months). Most cases of early-onset sepsis are acquired at or shortly before birth and tend to result in clinical signs and symptoms of pneumonia, sepsis, and occasionally meningitis within 4 or 5 days of life. Group B streptococcus is the most common organism isolated in early-onset sepsis and is also the most common cause of bacterial meningitis. Infections with Listeria and Candida follow a latent period between the time of microorganism inoculation and the appearance of clinical symptoms and present as late-onset sepsis.
Fetal Hydrops
Fetal hydrops refers to the accumulation of edema fluid in the fetus during intrauterine growth. Until recently, hemolytic anemia caused by Rh blood group incompatibility between mother and fetus (immune hydrops) was the most common cause, but with the successful prophylaxis of this disorder during pregnancy, causes of nonimmune hydrops have emerged as the principal culprits (Table 10-4). The intrauterine fluid accumulation can be quite variable, from progressive, generalized edema of the fetus (hydrops fetalis), a usually lethal condition, to more localized degrees of edema, such as isolated pleural and peritoneal effusions, or postnuchal fluid accumulation (cystic hygroma, see later) that are compatible with life.
TABLE 10-4 Selected Causes of Non-Immune Fetal Hydrops
| CARDIOVASCULAR |
| CHROMOSOMAL |
| THORACIC CAUSES |
| FETAL ANEMIA |
| TWIN GESTATION |
| Twin-to-twin transfusion |
| INFECTION (EXCLUDING PARVOVIRUS) |
| GENITOURINARY TRACT MALFORMATIONS |
| TUMORS |
| GENETIC/METABOLIC DISORDERS |
IMMUNE HYDROPS
Immune hydrops is a hemolytic disease caused by blood group incompatibility between mother and fetus. When the fetus inherits red cell antigenic determinants from the father that are foreign to the mother, a maternal immune reaction may occur, leading to hemolytic disease (in utero). The major antigens known to induce clinically significant immunological disease are certain of the Rh antigens the ABO and blood groups. The incidence of immune hydrops in urban populations has declined remarkably, mostly as a result of the current methods of preventing Rh immunization in at-risk women. Successful prophylaxis of this disorder has resulted from an understanding of its pathogenesis.
Etiology and Pathogenesis.
The underlying basis of immune hydrops is the immunization of the mother by blood group antigens on fetal red cells and the free passage of antibodies from the mother through the placenta to the fetus (Fig. 10-11). Fetal red cells may reach the maternal circulation during the last trimester of pregnancy, when the cytotrophoblast is no longer present as a barrier, or during childbirth itself. The mother thus becomes sensitized to the foreign antigen.
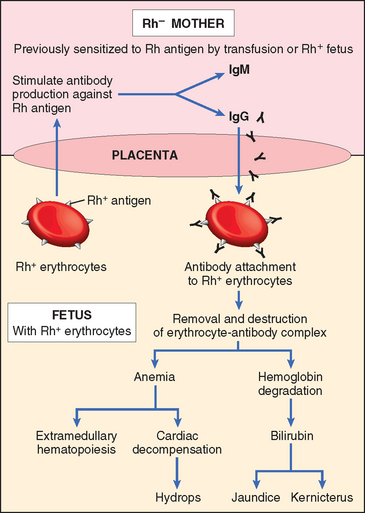
Of the numerous antigens included in the Rh system, only the D antigen is a major cause of Rh incompatibility. Several factors influence the immune response to Rh-positive fetal red cells that reach the maternal circulation.
The incidence of maternal Rh isoimmunization has decreased significantly since the use of Rhesus immune globulin (RhIg) containing anti-D antibodies. Administration of RhIg at 28 weeks and within 72 hours of delivery to Rhnegative mothers significantly decreases the risk for hemolytic disease in Rh-positive neonates and in subsequent pregnancies; RhIg is also administered following abortions, as thesetoo can lead to immunization. Antenatal identification and management of the at-risk fetus have been greatly facilitated by amniocentesis and the advent of chorionic villus and fetal blood sampling. In addition, cloning of the RHD gene has resulted in efforts to determine fetal Rh status using maternal blood. When identified, cases of severe intrauterine hemolysis may be treated by fetal intravascular transfusions via the umbilical cord and early delivery.
The pathogenesis of fetal hemolysis caused by maternal-fetal ABO incompatibility is slightly different from that caused by differences in the Rh antigens. ABO incompatibility occurs in approximately 20% to 25% of pregnancies, but laboratory evidence of hemolytic disease occurs in only 1 in 10 of such infants, and the hemolytic disease is severe enough to require treatment in only 1 in 200 cases. Several factors account for this. First, as mentioned, most anti-A and anti-B antibodies are of the IgM type and hence do not cross the placenta. Second, neonatal red cells express blood group antigens A and B poorly. Third, many cells other than red cells express A and B antigens and thus absorb some of the transferred antibody. ABO hemolytic disease occurs almost exclusively in infants of group A or B who are born of group O mothers. For reasons unknown, certain group O women possess IgG antibodies directed against group A or B antigens (or both) even without prior sensitization. Therefore, the firstborn may be affected. Fortunately, even with transplacentally acquired antibodies, lysis of the infant’s red cells is minimal. There is no effective protection against ABO reactions.
There are two consequences of excessive destruction of red cells in the neonate (see Fig. 10-11). The severity of these changes varies considerably, depending on the degree of hemolysis and the maturity of the infant.
NONIMMUNE HYDROPS
The three major causes of nonimmune hydrops include cardiovascular defects, chromosomal anomalies, and fetal anemia (Table 10-4).24 Both structural and functional cardiovascular defects, such as congenital cardiac defects and arrhythmias, may result in intrauterine cardiac failure and hydrops. Among the chromosomal anomalies, 45,X karyotype (Turner syndrome) and the trisomies 21 and 18 are associated with fetal hydrops. Most often, underlying structural cardiac anomalies associated with the chromosomal aberrations form the basis of fetal hydrops. In the Turner phenotype, however, abnormalities of lymphatic drainage from the neck may lead to postnuchal fluid accumulation (cystic hygromas). Fetal anemia, not caused by Rh- or ABO-associated antibodies, also results in hydrops. In fact, in some parts of the world (e.g., Southeast Asia), severe fetal anemia due to homozygous α-thalassemia is probably the most common cause of nonimmune hydrops. Transplacental infection by parvovirus B19 is rapidly emerging as an important cause of hydrops (see above). The virus gains preferential entry into erythroid precursors (normoblasts), where it replicates, leading to apoptosis of red cell progenitors and isolated red cell aplasia. Parvoviral intranuclear inclusions can be seen within circulating and marrow erythroid precursors (see Fig. 10-10). Approximately 10% of cases of nonimmune hydrops are related to monozygous twin pregnancies and twin-to-twin transfusion occurring through anastomoses between the two circulations.
Morphology of Hydrops Fetalis. The anatomic findings in fetuses with intrauterine fluid accumulation vary with both the severity of the disease and the underlying etiology. As previously noted, hydropsfetalis represents the most severe and generalized manifestation (Fig. 10-12), and lesser degrees of edema such as isolated pleural, peritoneal, or postnuchal fluid collections can occur. Accordingly, infants may be stillborn, die within the first few days, or recover completely. The presence of dysmorphic features suggests a chromosomal abnormality; postmortem examination may reveal an underlying cardiac anomaly.
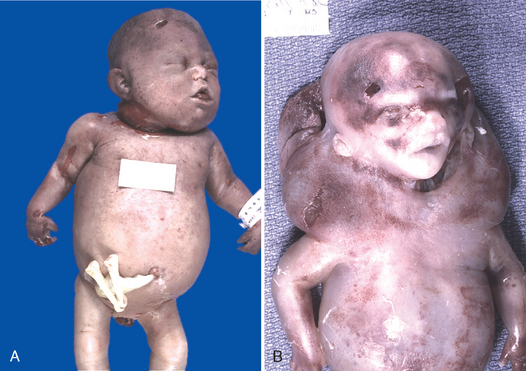
FIGURE 10-12 Hydrops fetalis. A, There is generalized accumulation of fluid in the fetus. B, Fluid accumulation is particularly prominent in the soft tissues of the neck, and this condition has been termed cystic hygroma. Cystic hygromas are characteristically seen, but not limited to, constitutional chromosomal anomalies such as 45,X0 karyotypes.
(Courtesy of Dr. Beverly Rogers, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX.)
In hydrops associated with fetal anemia, both fetus and placenta are characteristically pale; in most cases the liver and spleen are enlarged from cardiac failure and congestion. Additionally, the bone marrow demonstrates compensatory hyperplasia of erythroid precursors (parvovirus-associated red cell aplasia being a notable exception), and extramedullary hematopoiesis is present in the liver, spleen, and lymph nodes, and possibly other tissues such as the kidneys, lungs, and even the heart. The increased hematopoietic activity accounts for the presence in the peripheral circulation of large numbers of immature red cells, including reticulocytes, normoblasts, and erythroblasts (erythroblastosis fetalis) (Fig. 10-13).
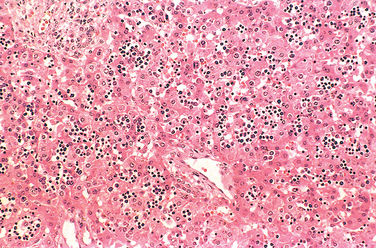
FIGURE 10-13 Numerous islands of extramedullary hematopoiesis (small blue cells) are scattered among mature hepatocytes in this infant with nonimmune hydrops fetalis.
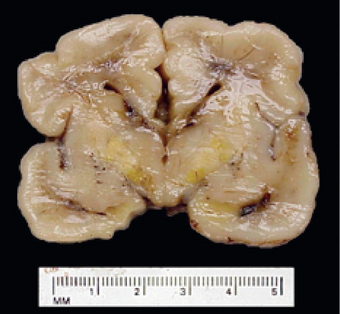
The most serious threat in fetal hydrops is central nervous system damage known as “kernicterus” (Fig. 10-14). The affected brain is enlarged and edematous and, when sectioned, has a bright yellow color, particularly the basal ganglia, thalamus, cerebellum, cerebral gray matter, and spinal cord. The precise level of bilirubin that induces kernicterus is unpredictable, but neural damage usually requires a blood bilirubin level greater than 20 mg/dL in term infants; in premature infants this threshold may be considerably lower.
Clinical Features.
The clinical manifestations of fetal hydrops vary with the severity of the disease and can be inferred from the preceding discussion. Minimally affected infants display pallor, possibly accompanied by hepatosplenomegaly (to which may be added jaundice with more severe hemolytic reactions), whereas the most gravely ill neonatespresent with intense jaundice, generalized edema, and signs of neurologic involvement. These infants may be supported by a variety of measures, including phototherapy (visual light oxidizes toxic unconjugated bilirubin to harmless, readily excreted, water-soluble dipyrroles) and, in severe cases, total exchange transfusion of the infant.
Inborn Errors of Metabolism and Other Genetic Disorders
Sir Archibald Garrod coined the term inborn errors of metabolism in 1908; since that time the number of well-characterized genetic disorders giving rise to congenital metabolic abnormalities has increased exponentially and is beyond the scope of this chapter. Most inborn errors of metabolism are rare, and some were discussed in Chapter 5. They are inherited, most commonly, as autosomal recessive or X-linked diseases; a few are inherited as dominant traits. Mitochondrial disorders (Chapter 5) form a distinct entity by themselves. Some of the clinical features that suggest an underlying metabolic disorder in a neonate are tabulated in Table 10-5. Three metabolic genetic defects, phenylketonuria (PKU), galactosemia, and cystic fibrosis, are selected for discussion here. PKU and galactosemia are reviewed because their early diagnosis (via neonatal screening programs) is particularly important, since appropriate dietary regimens can prevent early death or mental retardation. Cystic fibrosis is included because it is one of the most common, potentially lethal diseases occurring in individuals of Caucasian descent. Neonatal screening for cystic fibrosis remains a controversial topic, with the benefits and risks much less clear than in the other two diseases.
TABLE 10-5 Abnormalities Suggesting Inborn Errors of Metabolism
| GENERAL |
| NEUROLOGIC |
| GASTROINTESTINAL |
| EYES |
| MUSCLE, JOINTS |
Adapted from Barness LA, Gilbert-Barness E: Metabolic diseases. In Gilbert-Barness E, et al (eds): Potter’s Pathology of the Fetus, Infant, and Child. St. Louis, Mosby, 2007.
PHENYLKETONURIA (PKU)
PKU is characterized by abnormalities of phenylalanine metabolism, resulting in hyperphenylalaninemia. PKU is an autosomal recessive condition, and the vast majority of PKU is caused by bi-allelic mutations of the gene encoding the enzyme phenylalanine hydroxylase (PAH). Nevertheless, the great diversity in clinical presentation underscores the genetic complexities that underlie even classic “mendelian” diseases like PKU.25 At the molecular level, more than 500 diseaseassociated alleles of the PAH gene have been identified in populations worldwide. Each mutation induces a particular alteration in the enzyme resulting in a corresponding quantitative effect on residual enzyme activity ranging from complete absence to 50% of normal values. The degree of hyperphenylalaninemia and clinical phenotype is inversely related to the amount of residual enzyme activity. Infants with mutations resulting in a lack of PAH activity present with the classic features of PKU, while those with up to 6% residual activity present with milder disease. Moreover, some mutations result in only modest elevations of blood phenylalanine levels without associated neurologic damage. This latter condition, referred to as benign hyperphenylalaninemia, is important to recognize, because the individuals may well have positive screening tests but do not develop the stigmata of classic PKU. Measurement of serum phenylalanine differentiates benign hyperphenylalaninemia and classic PKU, with the concentrations being typically above 600 μM in PKU (normal phenylalanine concentrations, by contrast, are less than 120 μM).
The biochemical abnormality in PKU is an inability to convert phenylalanine into tyrosine. In normal children, less than 50% of the dietary intake of phenylalanine is necessary for protein synthesis. The rest is irreversibly converted to tyrosine by PAH in the liver as part of a complex metabolic pathway, the hepatic PAH system (Fig. 10-15), which, in addition to the enzyme PAH, has two other components: the cofactor tetrahydrobiopterin (BH4) and the enzyme dihydropteridine reductase, which regenerates BH4. Although neonatal hyperphenylalaninemia can be caused by deficiencies in any of these components, about 98% of cases are attributable to abnormalities in PAH and the remaining 2% to abnormalities in synthesis or recycling of BH4. BH4 is not only an essential cofactor for PAH but is also required for tyrosine and tryptophan hydroxylation. Concomitant defects in BH4 recycling disturb the synthesis of neurotransmitters. As a result, in patients with BH4 recycling defects neurologic damage is not arrested despite normalization of phenylalanine levels. Although they account for a small minority of patients with hyperphenylalaninemia, it is important to recognize these PKU variants because the ongoing neurologic disturbances cannot be treated by dietary control of phenylalanine levels alone.
Individuals with “classic” PKU have a severe deficiency of PAH, leading to hyperphenylalaninemia and its pathologic consequences. With a block in phenylalanine metabolism due to lack of PAH, minor shunt pathways come into play, yielding phenylpyruvic acid, phenyllactic acid, phenylacetic acid, and o-hydroxyphenylacetic acid, which are excreted in large amounts in the urine in PKU. Some of these abnormal metabolites are excreted in the sweat, and phenylacetic acid in particular imparts a strong musty or mousy odor to affected infants. It is believed that excess phenylalanine or its metabolites contribute to the brain damage in PKU. Affected infants are normal at birth but within a few weeks develop a rising plasma phenylalanine level, which in some way impairs brain development. Usually by 6 months of life severe mental retardation becomes evident; fewer than 4% of untreated PKU children have intelligence quotient values greater than 50 or 60. About one third of these children are never able to walk, and two thirds cannot talk. Seizures, other neurologic abnormalities, decreased pigmentation of hair and skin, and eczema often accompany the mental retardation in untreated children. Hyperphenylalaninemia and the resultant mental retardation can be avoided by restriction of phenylalanine intake early in life. Hence, several screening procedures are routinely used for detection of PKU in the immediate postnatal period.
Many clinically normal female PKU patients who are treated with dietary control early in life reach childbearing age. If such individuals were to discontinue dietary treatment, the result would be marked hyperphenylalaninemia. Between 75% and 90% of children born to such women are mentally retarded and microcephalic, and 15% have congenital heart disease, even though the infants themselves are heterozygotes. This syndrome, termed maternal PKU, results from the teratogenic effects of phenylalanine or its metabolites that cross the placenta and affect specific fetal organs during development. The presence and severity of the fetal anomalies directly correlate with the maternal phenylalanine level, so it is imperative that maternal dietary restriction of phenylalanine be initiated before conception and continue throughout pregnancy.
Although dietary restriction of phenylalanine is usually successful in reducing or preventing the mental retardation associated with PKU, there are problems with long-term compliance (resulting in a decline in mental or behavioral status) and nutritional imbalances involving trace minerals, fatty acids, and lipids. A subset of patients with PAH missense mutations are responsive to pharmacologic dosages of BH4; some recent analyses have predicted that as many as half of prevalent PAH mutations in some populations may be “BH4 responsive.”26 Since there are no primary abnormalities of BH4 in these patients, it is believed that this cofactor acts as a “molecular chaperone,” preventing the degradation of misfolded PAH protein. Permanent restitution of PAH activity through gene therapy remains the ultimate goal; recent studies in animal models of PKU have produced encouraging results.27
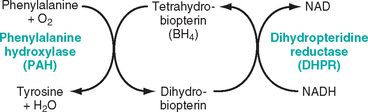
GALACTOSEMIA
Galactosemia is an autosomal recessive disorder of galactose metabolism. Normally, lactose, the major carbohydrate of mammalian milk, is split into glucose and galactose in the intestinal microvilli by lactase. Galactose is then converted to glucose in three steps (Fig. 10-16). Two variants of galactosemia have been identified. In the more common variant there is a total lack of galactose-1-phosphate uridyl transferase (also known as GALT) involved in reaction 2. The rare variant arises from a deficiency of galactokinase, involved in reaction 1. Because galactokinase deficiency leads to a milder form of the disease not associated with mental retardation, it is not considered in this discussion. As a result of the transferase lack, galactose-1-phosphate accumulates in many locations, including the liver, spleen, lens of the eye, kidneys, heart muscle, cerebral cortex, and erythrocytes. Alternative metabolic pathways are activated, leading to the production of galactitol (a polyol metabolite of galactose) and galactonate, an oxidized by-product of excess galactose, both of which also accumulate in the tissues. Long-term toxicity in galactosemics has been variously imputed to these metabolic intermediates.28 Heterozygotes may have a mild deficiency but are spared the clinical and pathologic consequences of the homozygous state.
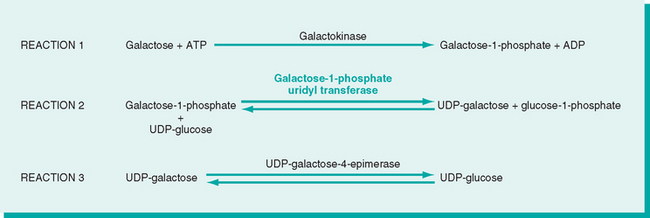
FIGURE 10-16 Pathways of galactose metabolism. ADP, adenosine diphosphate; ATP, adenosine triphosphate; UDP, uridine diphosphate.
The clinical picture is variable, probably reflecting the heterogeneity of mutations in the galactose-1-phosphate uridyl transferase gene leading to galactosemia. The liver, eyes, and brain bear the brunt of the damage. The early-to-develop hepatomegaly is due largely to fatty change, but in time widespread scarring that closely resembles the cirrhosis of alcohol abuse may supervene (Fig. 10-17). Opacification of the lens (cataract) develops, probably because the lens absorbs water and swells as galactitol, produced by alternative metabolic pathways, accumulates and increases its tonicity. Nonspecific alterations appear in the central nervous system, including loss of nerve cells, gliosis, and edema, particularly in the dentate nuclei of the cerebellum and the olivary nuclei of the medulla. Similar changes may occur in the cerebral cortex and white matter.
FIGURE 10-17 Galactosemia. The liver shows extensive fatty change and a delicate fibrosis.
(Courtesy of Dr. Wesley Tyson, The Children’s Hospital, Denver, CO.)
These infants fail to thrive almost from birth. Vomiting and diarrhea appear within a few days of milk ingestion. Jaundice and hepatomegaly usually become evident during the first week of life and may seem to be a continuation of the physiologic jaundice of the newborn. The cataracts develop within a few weeks, and within the first 6 to 12 months of life mental retardation may be detected. Even in untreated infants, however, the mental deficit is usually not as severe as that seen in PKU. Accumulation of galactose and galactose-1-phosphate in the kidney impairs amino acid transport, resulting in aminoaciduria. There is an increased frequency of fulminant Escherichia coli septicemia, possibly arising from depressed neutrophil bactericidal activity. Hemolysis and coagulopathy in the newborn period can occur as well.
The diagnosis of galactosemia can be suspected by the demonstration in the urine of a reducing sugar other than glucose, but tests that directly identify the deficiency of the transferase in leukocytes and erythrocytes are more reliable. Antenatal diagnosis is possible by the assay of GALT activity in cultured amniotic fluid cells or determination of galactitol level in amniotic fluid supernatant. More than 140 mutations have been documented in GALT; among these, a glutamine-to-arginine substitution at codon 188 (Gln188Arg) is the most prevalent mutation in non-Hispanic whites, while a serine-to-leucine substitution at codon 135 (Ser135Leu) is the most common mutation in African Americans.
Many of the clinical and morphologic changes of galactosemia can be prevented or ameliorated by early removal of galactose from the diet for at least the first 2 years of life. Control instituted soon after birth prevents the cataracts and liver damage and permits almost normal development. Even with dietary restrictions, however, it is now established that older patients are frequently affected by a speech disorder and gonadal failure (especially premature ovarian failure) and, less commonly, by an ataxic condition.
CYSTIC FIBROSIS (MUCOVISCIDOSIS)
Cystic fibrosis is a disorder of ion transport in epithelial cells that affects fluid secretion in exocrine glands and the epithelial lining of the respiratory, gastrointestinal, and reproductive tracts. In many infants this disorder leads to abnormally viscous secretions, which obstruct organ passages, resulting in most of the clinical features of this disorder, such as chronic lung disease secondary to recurrent infections, pancreatic insufficiency, steatorrhea, malnutrition, hepatic cirrhosis, intestinal obstruction, and male infertility. These manifestations may appear at any point in life from before birth to much later in childhood or even in adolescence.
With an incidence of 1 in 2500 live births, cystic fibrosis is the most common lethal genetic disease that affects Caucasian populations. The carrier frequency in the United States is 1 in 20 among Caucasians but significantly lower in African Americans, Asians, and Hispanics. Although cystic fibrosis follows an autosomal recessive transmission, recent data suggest that even heterozygote carriers have a higher incidence of respiratory and pancreatic diseases as compared with the general population.29,30 In addition, despite the classification of cystic fibrosis as a “mendelian” disorder, there is a wide degree of phenotypic variation that results from diverse mutations in the gene associated with cystic fibrosis, the tissue-specific effects of this gene, and the influence of newly recognized disease modifiers.31
The Cystic Fibrosis–Associated Gene: Normal Structure and Function.
In normal duct epithelia, chloride is transported by plasma membrane channels (chloride channels). The primary defect in cystic fibrosis results from abnormal function of an epithelial chloride channel protein encoded by the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome 7q31.2. The 1480–amino acid polypeptide encoded by CFTR has two transmembrane domains (each containing six α-helices), two cytoplasmic nucleotide-binding domains (NBDs), and a regulatory domain (R domain) that contains protein kinase A and C phosphorylation sites (Fig. 10-18). The two transmembrane domains form a channel through which chloride passes. Activation of the CFTR channel is mediated by agonist-induced increases in cyclic adenosine monophosphate (cAMP), followed by activation of a protein kinase A that phosphorylates the R domain. Adenosine triphosphate (ATP) binding and hydrolysis occurs at the NBD and is essential for the opening and closing of the channel pore in response to cAMP-mediated signaling. Several important facets of CFTR function have emerged in recent years:
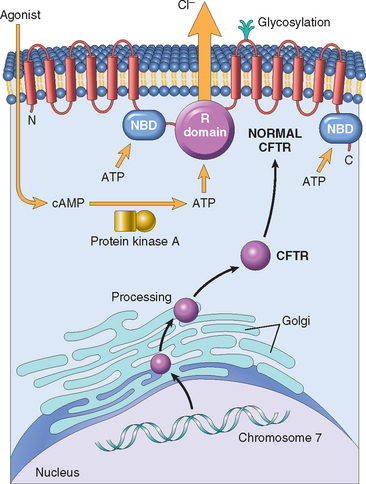
FIGURE 10-18 Top, Normal cystic fibrosis transmembrane conductance regulator (CFTR) structure and activation. CFTR consists of two transmembrane domains, two nucleotide-binding domains (NBDs), and a regulatory R domain. Agonists (e.g., acetylcholine) bind to epithelial cells and increase cyclic adenosine monophosphate (cAMP), which activates protein kinase A, the latter phosphorylating the CFTR at the R domain, resulting in opening of the chloride channel. Bottom, CFTR from gene to protein. The most common mutation in the CFTR gene results in defective protein folding in the Golgi/endoplasmic reticulum and degradation of CFTR before it reaches the cell surface. Other mutations affect synthesis of CFTR, NBDs and R domains, as well as membrane-spanning domains. (See text for details.)
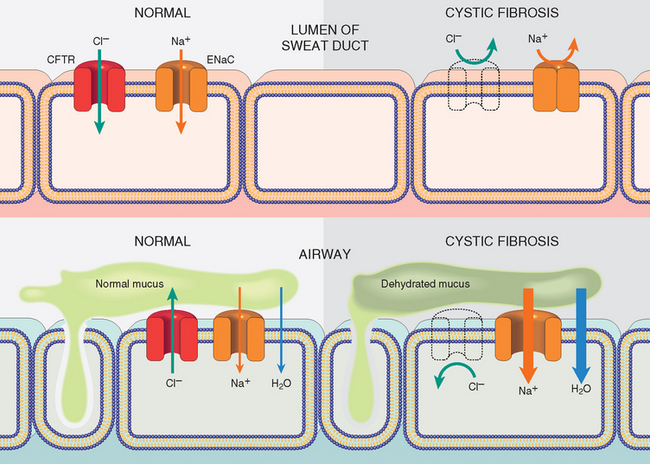
FIGURE 10-19 Chloride channel defect in the sweat duct (top) causes increased chloride and sodium concentration in sweat. In the airway (bottom), cystic fibrosis patients have decreased chloride secretion and increased sodium and water reabsorption leading to dehydration of the mucus layer coating epithelial cells, defective mucociliary action, and mucus plugging of airways. CFTR, Cystic fibrosis transmembrane conductance regulator; ENaC, epithelial sodium channel.
The Cystic Fibrosis Gene: Mutational Spectra and Genotype-Phenotype Correlation.
Since the CFTR gene was cloned in 1989, more than 1300 disease-associated mutations have been identified. Various mutations can be grouped into six “classes” based on their effect on the CFTR protein:
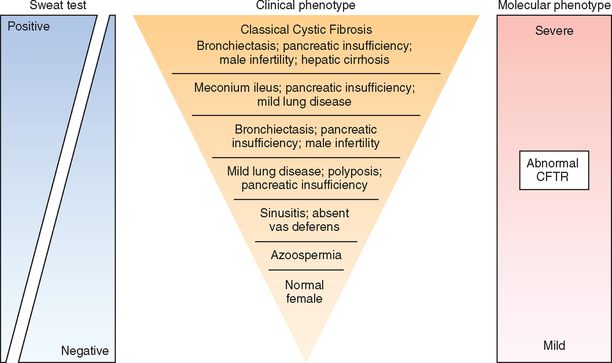
Since cystic fibrosis is an autosomal recessive disease, affected individuals harbor mutations on both alleles. However, the combination of mutations on the two alleles can have a remarkable effect on the overall phenotype, as well as on organ-specific manifestations (Fig. 10-20). Thus, two “severe” (class I, II, and III) mutations that produce virtual absence of membrane CFTR are associated with the classic cystic fibrosis phenotype (pancreatic insufficiency, sinopulmonary infections, and gastrointestinal symptoms), while the presence of a “mild” (class IV or V) mutation on one or both alleles results in a less severe phenotype. This general dictum of genotype-phenotype correlation is most consistent for pancreatic disease, wherein the presence of a “mild” mutation in one allele can revert to the pancreatic insufficiency phenotype conferred by homozygosity for “severe” mutations. By contrast, genotype-phenotype correlations are far less consistent in pulmonary disease, reflecting an effect of secondary modifiers (see below). As genetic testing for CFTR mutations has expanded, it has become increasingly evident that patients who present with a variety of apparently unrelated clinical phenotypes may also harbor CFTR mutations. These include individuals with idiopathic chronic pancreatitis, late-onset chronic pulmonary disease, idiopathic bronchiectasis, and obstructive azoospermia caused by bilateral absence of the vas deferens (see detailed discussion of individual phenotypes later). Most of these patients do not demonstrate other features of cystic fibrosis, despite the presence of bi-allelic CFTR mutations, and are classified as nonclassic or atypical cystic fibrosis.34 Identifying these individuals is important not only for subsequent management, but also for the purposes of genetic counseling.
Genetic and Environmental Modifiers.
Although cystic fibrosis remains one of the best-known examples of the “one gene, one disease” axiom, there is increasing evidence that genes other than CFTR modify the frequency and severity of organ-specific manifestations.35 The severity of pulmonary manifestations in cystic fibrosis is associated with polymorphic variants at several genes, the best known examples of which are mannose-binding lectin 2 (MBL2) and transforming growth factor β1 (TGFB1). MBL is a key effector of innate immunity involved in opsonization and phagocytosis of microorganisms, and polymorphisms in the MBL2 gene that are associated with lower circulating levels of the protein confer a threefold higher risk of end-stage lung disease. TGFβ is a direct inhibitor of CFTR function.36,37 A large multicenter study of patients homozygous for the ΔF508 CFTR mutation found two specific polymorphisms in the 5′ end of the TGFB1 gene to be associated with severe pulmonary phenotypes.38 Similarly, several putative genetic modifiers have been identified that influence the incidence of meconium ileus in cystic fibrosis, although the precise genes associated with the linked chromosomal regions have not yet been identified.39
Environmental modifiers may also cause significant phenotypic differences between individuals who share the same CFTR genotype. This is best exemplified in pulmonary disease, where CFTR genotype and phenotype correlations can be perplexing. As stated above, defective mucociliary action because of deficient hydration of the mucus results in an inability to clear bacteria from the airways. Pseudomonas aeruginosa species, in particular, colonize the lower respiratory tract, first intermittently and then chronically. Concurrent viral infections predispose to such colonization. The static mucus creates a hypoxic microenvironment in the airway surface fluid, which in turn favors the production of alginate, a mucoid polysaccharide capsule. Alginate production permits the formation of a biofilm that protects the bacteria from antibodies and antibiotics, allowing them to evade host defenses, and produce a chronic destructive lung disease. Antibody- and cell-mediated immune reactions induced by the organisms result in further pulmonary destruction, but are ineffective against the organism. It is evident, therefore, that in addition to genetic factors (e.g., class of mutation), a plethora of environmental modifiers (e.g., virulence of organisms, efficacy of therapy, intercurrent and concurrent infections by other organisms, exposure to smoking and allergens) can influence the severity and progression of lung disease in cystic fibrosis.
Morphology. The anatomic changes are highly variable in distribution and severity. In individuals with nonclassic cystic fibrosis, the disease is quite mild and does not seriously disturb their growth and development. In others, the pancreatic involvement is severe and impairs intestinal absorption because of the pancreatic achylia, and so malabsorption stunts development and post-natal growth. In others, the mucus secretion defect leads to defective mucociliary action, obstruction of bronchi and bronchioles, and crippling fatal pulmonary infections (Fig. 10-21). In all variants, the sweat glands are morphologically unaffected.
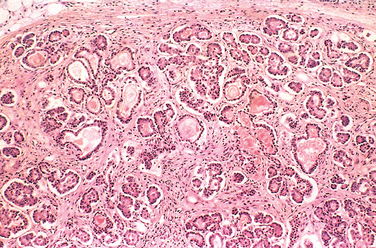
FIGURE 10-21 Mild to moderate cystic fibrosis changes in the pancreas. The ducts are dilated and plugged with eosinophilic mucin, and the parenchymal glands are atrophic and replaced by fibrous tissue.
Pancreatic abnormalities are present in approximately 85% to 90% of patients with cystic fibrosis. In the milder cases, there may be only accumulations of mucus in the small ducts with some dilation of the exocrine glands. In more severe cases, usually seen in older children or adolescents, the ducts are completely plugged, causing atrophy of the exocrine glands and progressive fibrosis (Fig. 10-21). Atrophy of the exocrine portion of the pancreas may occur, leaving only the islets within a fibrofatty stroma. The loss of pancreatic exocrine secretion impairs fat absorption, and the associated avitaminosis A may contribute to squamous metaplasia of the lining epithelium of the ducts in the pancreas, which are already injured by the inspissated mucus secretions. Thick viscid plugs of mucus may also be found in the small intestine of infants. Sometimes these cause small-bowel obstruction, known as meconium ileus.
The liver involvement follows the same basic pattern. Bile canaliculi are plugged by mucinous material, accompanied by ductular proliferation and portal inflammation. Hepatic steatosis is not an uncommon finding in liver biopsies. Over time, focal biliary cirrhosis develops in approximately a third of patients (Chapter 18), which can eventually involve the entire liver, resulting in diffuse hepatic nodularity. Such severe hepatic involvement is encountered in less than 10% of patients.
The salivary glands frequently show histologic changes similar to those described in the pancreas: progressive dilation of ducts, squamous metaplasia of the lining epithelium, and glandular atrophy followed by fibrosis.
The pulmonary changes are the most serious complications of this disease (Fig. 10-22). These stem from the viscous mucus secretions of the submucosal glands of the respiratory tree leading to secondary obstruction and infection of the air passages. The bronchioles are often distended with thick mucus associated with marked hyperplasia and hypertrophy of the mucus-secreting cells. Superimposed infections give rise to severe chronic bronchitis and bronchiectasis (Chapter 15). In many instances, lung abscesses develop. Staphylococcus aureus, Hemophilus influenzae, and Pseudomonas aeruginosa are the three most common organisms responsible for lung infections. As mentioned above, a mucoid form of P. aeruginosa (alginate-producing) is particularly frequent and causes chronic inflammation. Even more sinister is the increasing frequency of infection with another group of pseudomonads, the Burkholderia cepacia complex, which includes at least nine different species; of these, infections with B. cenocepacia are the most common in cystic fibrosis patients. This opportunistic bacterium is particularly hardy, and infection with this organism has been associated with fulminant illness (“cepacia syndrome”), longer hospital stays, and increased mortality.40 Other opportunistic bacterial pathogens include Stenotrophomonas maltophila and nontuberculous mycobacteria; allergic bronchopulmonary aspergillosis also occurs with increased frequency in cystic fibrosis.
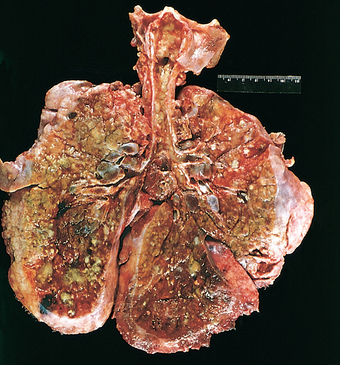
FIGURE 10-22 Lungs of a patient dying of cystic fibrosis. There is extensive mucus plugging and dilation of the tracheobronchial tree. The pulmonary parenchyma is consolidated by a combination of both secretions and pneumonia—the green color associated with Pseudomonas infections.
(Courtesy of Dr. Eduardo Yunis, Children’s Hospital of Pittsburgh, Pittsburgh, PA.)
Azoospermia and infertilityare found in 95% of the males who survive to adulthood; congenital bilateral absence of the vas deferens is a frequent finding in these patients. In some males, bilateral absence of the vas deferens may be the only feature suggesting an underlying CFTR mutation.
Clinical Features.
Few childhood diseases are as protean as cystic fibrosis in clinical manifestations (Table 10-6). The symptoms are extremely varied and may appear at birth to years later, and involve one organ system or many. Approximately 5% to 10% of the cases come to clinical attention at birth or soon after because of meconium ileus. Distal intestinal obstruction can also occur in older individuals, manifesting as recurrent episodes of right lower quadrant pain sometimes associated with a palpable mass in the right iliac fossa.
TABLE 10-6 Clinical Features and Diagnostic Criteria for Cystic Fibrosis
Adapted with permission from Rosenstein BJ, Cutting GR: The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 132:589, 1998.
Exocrine pancreatic insufficiency occurs in the majority (85% to 90%) of patients with cystic fibrosis and is associated with “severe” CFTR mutations on both alleles (e.g., ΔF508/ΔF508), whereas 10% to 15% of patients with one “severe” and one “mild” CFTR mutation (ΔF508/R117H) or two “mild” CFTR mutations retain enough pancreatic exocrine function so as not to require enzyme supplementation (pancreassufficient phenotype). Pancreatic insufficiency is associated with protein and fat malabsorption and increased fecal loss. Manifestations of malabsorption (e.g., large, foul-smelling stools, abdominal distention, and poor weight gain) appear during the first year of life. The faulty fat absorption may induce deficiency of the fat-soluble vitamins, resulting in manifestations of avitaminosis A, D, or K. Hypoproteinemia may be severe enough to cause generalized edema. Persistent diarrhea may result in rectal prolapse in up to 10% of children with cystic fibrosis. The pancreas-sufficient phenotype is usually not associated with other gastrointestinal complications, and in general, these individuals demonstrate excellent growth and development. “Idiopathic” chronic pancreatitis occurs in a subset of patients with pancreas-sufficient cystic fibrosis and is associated with recurrent abdominal pain with life-threatening complications. These patients have other features of cystic fibrosis, such as pulmonary disease. By contrast, “idiopathic” chronic pancreatitis can also occur as an isolated late-onset finding in the absence of other stigmata of cystic fibrosis (Chapter 19); bi-allelic CFTR mutations (usually one “mild,” one “severe”) are demonstrable in the majority of these individuals who have nonclassic or atypical cystic fibrosis. Endocrine pancreatic insufficiency (i.e., diabetes) is uncommon in cystic fibrosis and is usually accompanied by substantial destruction of pancreatic parenchyma.
Cardiorespiratory complications, such as persistent lung infections, obstructive pulmonary disease, and cor pulmonale, are the most common cause of death (∼80%) in patients in the United States. By age 18, 80% of patients with classic cystic fibrosis harbor P. aeruginosa. With the indiscriminate use of antibiotic prophylaxis against Staphylococcus, there has been an unfortunate resurgence of resistant strains of Pseudomonas in many patients. Individuals who carry one “severe” and one “mild” CFTR mutation may develop late-onset mild pulmonary disease, another example of nonclassic or atypical cystic fibrosis. Patients with mild pulmonary disease usually have little or no pancreatic disease. Adult-onset “idiopathic” bronchiectasis, has been linked to CFTR mutations in a subset of cases. Recurrent sinonasal polyps can occur in up to 25% of individuals with cystic fibrosis; hence, children who present with this finding should be tested for cystic fibrosis.
Significant liver disease occurs late in the natural history of cystic fibrosis and is gaining in clinical importance as life expectancies increase. In fact, after cardiopulmonary and transplantation-related complications, liver disease is the most common cause of death in cystic fibrosis. Most studies suggest that symptomatic or biochemical liver disease has its onset at or around puberty, with a prevalence of approximately 13% to 17%. However, asymptomatic hepatomegaly may be present in up to a third of individuals. Obstruction of the common bile duct may occur due to stones or sludge; it presents with abdominal pain and the acute onset of jaundice. As previously noted, diffuse biliary cirrhosis develops in less than 10% of individuals with cystic fibrosis.
Approximately 95% of males with cystic fibrosis are infertile, as a result of obstructive azoospermia. As mentioned earlier, this is most commonly due to congenital bilateral absence of the vas deferens, which is caused in 80% of cases by bi-allelic CFTR mutations.
In most cases, the diagnosis of cystic fibrosis is based on persistently elevated sweat electrolyte concentrations (often the mother makes the diagnosis by recognizing her infant’s abnormally salty sweat), characteristic clinical findings (sinopulmonary disease and gastrointestinal manifestations), an abnormal newborn screening test, or a family history. A minority of patients with cystic fibrosis, especially those with at least one “mild” CFTR mutation, may have a normal or near-normal sweat test (<60 mM/L). Measurement of nasal transepithelial potential difference in vivo can be a useful adjunct test under these circumstances; individuals with cystic fibrosis demonstrate a significantly more negative baseline nasal potential difference than controls. Sequencing the CFTR gene is, of course, the “gold standard” for diagnosis of cystic fibrosis. Therefore, in patients with suggestive clinical findings or family history (or both), genetic analysis may be warranted.
There have been major improvements in the management of acute and chronic complications for cystic fibrosis, including more potent antimicrobial therapies, pancreatic enzyme replacement, and bilateral lung transplantation. Newer modalities for restituting endogenous CFTR function have also emerged in recent years. In principle, cystic fibrosis, like other single-gene disorders, should be amenable to gene therapy, and several adenoviral gene therapy vectors are currently undergoing early-phase clinical trials. Improvement in management of cystic fibrosis has resulted in enhancement of median life expectancy to above 36 years as of 2006,41 and increasingly, a lethal disease of childhood is changing into a chronic disease of adults.
Sudden Infant Death Syndrome (SIDS)
The National Institute of Child Health and Human Development defines SIDS as “the sudden death of an infant under 1 year of age which remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of the clinical history.”42 Thus, SIDS is a disease of unknown cause. It is important to emphasize that many cases of sudden death in infancy may have an unexpected anatomic or biochemical basis discernible at autopsy (Table 10-7), and these should not be labeled as SIDS. Furthermore, instances of sudden death during infancy wherein evidence for excluding alternative diagnoses is equivocal, or a postmortem diagnosis cannot be performed, are best labeled as unclassified sudden infant death, in line with the current rigorous definition of SIDS.43 An aspect of SIDS that is not stressed in the definition is that the infant usually dies while asleep, mostly in the prone or side position, hence the pseudonyms of crib death or cot death.
TABLE 10-7 Risk Factors and Postmortem Findings Associated with Sudden Infant Death Syndrome
| PARENTAL |
| INFANT |
| ENVIRONMENT |
| POSTMORTEM ABNORMALITIES DETECTED IN CASES OF SUDDEN UNEXPECTED INFANT DEATH* |
| Infections |
| Unsuspected congenital anomaly |
| Traumatic child abuse |
| Genetic and metabolic defects |
* SIDS is not the only cause of sudden unexpected death in infancy but rather is a diagnosis of exclusion. Therefore, performance of an autopsy may often reveal findings that would explain the cause of sudden unexpected death. These cases should not, strictly speaking, be labeled as “SIDS.” SCN5A, sodium channel, voltage-gated, type V, alpha polypeptide; KCNQ1, potassium voltage-gated channel, KQT-like subfamily, member 1; MCAD, medium-chain acyl coenzyme A dehydrogenase; LCHAD, long-chain 3-hydroxyacyl coenzyme A dehydrogenase; SCHAD, short-chain 3-hydroxyacyl coenzyme A dehydrogenase; MTCYB, mitochondrial cytochrome b; C4, complement component 4.
Epidemiology.
As infantile deaths due to nutritional problems and infections have come under control in developed countries, SIDS has assumed greater importance in these countries, including the United States. SIDS is the leading cause of death between age 1 month and 1 year in this country and the third leading cause of death overall in infancy, after congenital anomalies and diseases of prematurity and low birth weight. Mostly because of nationwide SIDS awareness campaigns by organizations such as the American Academy of Pediatrics, there has been a significant drop in SIDS-related mortality in the past decade, from an estimated 120 deaths per 100,000 live births in 1992 to 57 per 100,000 in 2002. Worldwide, in countries where unexpected infant deaths are diagnosed as SIDS only after postmortem examination, the death rates from SIDS range from 10 per 100,000 live births in the Netherlands to 80 per 100,000 in New Zealand.44
Approximately 90% of all SIDS deaths occur during the first 6 months of life, most between ages 2 and 4 months. Thisnarrow window of peak susceptibility is a unique characteristic that is independent of other risk factors (to be described) and the geographic locale. Most infants who die of SIDS die at home, usually during the night after a period of sleep. For many years, prolonged apnea was considered to be a risk factor for SIDS. Infants who developed a so-called “apparent life-threatening event” (ALTE), characterized by some combination of apnea, marked change in color or muscle tone, choking or gagging, were considered at risk for subsequent SIDS. However, epidemiologic studies have demonstrated that these “life-threatening events” and SIDS have different risk factors and ages of onset, and are probably unrelated entities. Children experiencing ALTEs are often premature or have a mechanical basis for respiratory compromise. This distinction might explain why home apnea monitors, which have proliferated among American families for “SIDS prevention,” have had minimal impact on reducing the risk of SIDS.45
Morphology. In infants who have died of suspected SIDS, a variety of findings have been reported at postmortem examination. They are usually subtle and of uncertain significance and are not present in all cases. Multiple petechiae are the most common finding (∼80% of cases); these are usually present on the thymus, visceral and parietal pleura, and epicardium. Grossly, the lungs are usually congested, and vascular engorgement with or without pulmonary edema is demonstrable microscopically in the majority of cases. These changes possibly represent agonal events, since they are found with comparable frequencies in explained sudden deaths in infancy. Within the upper respiratory system (larynx and trachea), there may be some histologic evidence of recent infection (correlating with the clinical symptoms), although the changes are not sufficiently severe to account for death and should not detract from the diagnosis of SIDS. The central nervous system demonstrates astrogliosis of the brain stem and cerebellum. Sophisticated morphometric studies have revealed quantitative brain-stem abnormalities such as hypoplasia of the arcuate nucleus or a decrease in brain-stem neuronal populations in several cases; these observations are not uniform, however. Nonspecific findings include frequent persistence of hepatic extramedullary hematopoiesis and periadrenal brown fat; it is tempting to speculate that these latter findings relate to chronic hypoxemia, retardation of normal development, and chronic stress. Thus, autopsy usually fails to provide a clear cause of death, and this may well be related to the etiologic heterogeneity of SIDS. The importance of a postmortem examination rests in identifying other causes of sudden unexpected death in infancy, such as unsuspected infection, congenital anomaly, or a genetic disorder (see Table 10-7), the presence of any of which would exclude a diagnosis of SIDS; and in ruling out the unfortunate possibility of traumatic child abuse.
Pathogenesis.
The circumstances surrounding SIDS have been explored in great detail, and it is generally accepted that it is a multifactorial condition, with a variable mixture of contributing factors. A “triple-risk” model of SIDS has been proposed, which postulates the intersection of three overlapping factors: (1) a vulnerable infant, (2) a critical developmental period in homeostatic control, and (3) an exogenous stressor(s).46 According to this model, several factors make the infant vulnerable to sudden death during the critical developmental period (i.e., the first 6 months of life). These vulnerability factors may be attributable to the parents or the infant, while the exogenous stressor(s) is attributable to the environment (see Table 10-7).
While numerous factors have been proposed to account for a vulnerable infant, the most compelling hypothesis is that SIDS reflects a delayed development of “arousal” and cardiorespiratory control. The brain stem, and in particular the medulla oblongata, plays a critical role in the body’s “arousal” response to noxious stimuli such as episodic hypercarbia, hypoxia, and thermal stress encountered during sleep. The serotonergic (5-HT) system of the medulla is implicated in these “arousal” responses, as well as regulation of other critical homeostatic functions such as respiratory drive, blood pressure, and upper airway reflexes. Abnormalities in serotonin-dependent signaling in the brainstem may be the underlying basis for SIDS in some infants.47 A recently developed mouse model of disrupted somatostatin signaling in the medulla recapitulates many of the features likely to contribute to sudden death in SIDS infants, including lack of an appropriate “arousal” response to environmental stressors.48
Epidemiologic and genetic studies have identified additional vulnerability factors for SIDS in the “triple-risk” model. Infants who are born before term or who are low birth weight are at increased risk, and risk increases with decreasing gestational age or birth weight. As stated, male sex is associated with a slightly greater incidence of SIDS. SIDS in a prior sibling is associated with a fivefold relative risk of recurrence, underscoring the importance of a genetic predisposition (see below); traumatic child abuse must be carefully excluded under these circumstances. Most SIDS babies have an immediate prior history of a mild respiratory tract infection, but no single causative organism has been isolated. These infections may predispose an already vulnerable infant to even greater impairment of cardiorespiratory control and delayed arousal. In this context, laryngeal chemoreceptors have emerged as a putative “missing link” between upper respiratory tract infections, the prone position (see below), and SIDS. When stimulated, these laryngeal chemoreceptors typically elicit an inhibitory cardiorespiratory reflex. Stimulation of the chemoreceptors is augmented by respiratory tract infections, which increase the volume of secretions, and by the prone position, which impairs swallowing and clearing of the airways even in healthy infants. In a previously vulnerable infant with impaired arousal, the resulting inhibitory cardiorespiratory reflex may prove fatal. Genetic vulnerability factors in the infant include polymorphic variants in genes that may confer an increased risk of SIDS. These genes include those related to serotonergic signaling and autonomic innervation, underscoring the importance of these processes in the pathophysiology of SIDS.49
In addition to infant vulnerability factors, several maternal risk factors have also been identified. Maternal smoking during pregnancy has consistently emerged as a risk factor in epidemiologic studies of SIDS, with children exposed to in utero nicotine having more than double the risk of SIDS as compared with children born to nonsmokers.50 Young maternal age, frequent childbirths, and inadequate prenatal care are all risk factors associated with increased incidence of SIDS in the offspring.
Among the potential “environmental stressors,” prone or side sleeping positions, sleeping with parents in the first 3 months, sleeping on soft surfaces, and thermal stress arepossibly the most important modifiable risk factors for SIDS.42,44 The prone or side positions predispose an infant to one or more recognized noxious stimuli (hypoxia, hypercarbia, and thermal stress) during sleep. The side position was considered a reliable alternative to the prone sleeping position, but a number of studies have established a substantial risk for both positions vis-à-vis SIDS. The American Academy of Pediatrics now recognizes the supine sleeping position as the only safe position that reduces the risk of SIDS. This “Back to Sleep” campaign has resulted in substantial reductions in SIDS-related deaths since its inception in 1994.
As has been stated, SIDS is not the only cause of sudden unexpected deaths in infancy. In fact, SIDS is a diagnosis of exclusion, requiring careful examination of the death scene and a complete postmortem examination. The latter can reveal an unsuspected cause of sudden death in as many as 20% or more of “SIDS” babies. Infections (e.g., viral myocarditis or bronchopneumonia) are the most common causes of sudden “unexpected” death, followed by unsuspected congenital anomalies. In part as a result of advancements in molecular diagnostics and knowledge of the human genome, several genetic causes of sudden “unexpected” infant death have emerged. For example, fatty acid oxidation disorders, characterized by defects in mitochondrial fatty acid oxidative enzymes, may be responsible for as many as 5% of sudden deaths in infancy. Other newly emerging genetic causes of explained sudden death are listed in Table 10-7.
Tumors and Tumor-like Lesions of Infancy and Childhood
Only 2% of all malignant tumors occur in infancy and childhood; nonetheless, cancer (including leukemia) accounts for about 9% of deaths in the United States in children over age 4 and up to age 14, and only accidents cause significantly more deaths. Benign tumors are even more common than cancers. Most benign tumors are of little concern, but on occasion they cause serious complications by virtue of their location or rapid increase in size.
It is sometimes difficult to separate, on morphologic grounds, true tumors or neoplasms from tumor-like lesions in the infant and child. In this context, two special categories of tumor-like lesions should be distinguished from true tumors.
The term heterotopia (or choristoma) is applied to microscopically normal cells or tissues that are present in abnormal locations. Examples of heterotopias include a rest of pancreatic tissue found in the wall of the stomach or small intestine, or a small mass of adrenal cells found in the kidney, lungs, ovaries, or elsewhere. These heterotopic rests are usually of little significance, but they can be confused clinically with neoplasms. Rarely, they are sites of origin of true neoplasms, producing paradoxes such as an adrenal carcinoma arising in the ovary.
The term hamartoma refers to an excessive, focal overgrowth of cells and tissues native to the organ in which it occurs. Although the cellular elements are mature and identical to those found in the remainder of the organ, they do not reproduce the normal architecture of the surrounding tissue. The line of demarcation between a hamartoma and a benign neoplasm is often unclear, as both lesions can be clonal. Hemangiomas, lymphangiomas, rhabdomyomas of the heart, adenomas of the liver, and developmental cysts within the kidneys, lungs, or pancreas are interpreted by some as hamartomas and by others as true neoplasms. The frequency of these lesions in infancy and childhood and their clinical behavior give credence to the belief that many are developmental aberrations. Their unequivocally benign histology, however, does not preclude bothersome and rarely life-threatening clinical problems in some cases.
BENIGN TUMORS AND TUMOR-LIKE LESIONS
Virtually any tumor may be encountered in children, but within this wide array hemangiomas, lymphangiomas, fibrous lesions, and teratomas deserve special mention. You will notice that the most common neoplasms of childhood are so-called soft-tissue tumors of mesenchymal derivation. This contrasts with adults, in whom the most common tumors, benign or malignant, have an epithelial origin. Benign tumors of various tissues are described in greater detail in appropriate chapters; here a few comments are made about their special features in childhood.
Hemangioma.
Hemangiomas (Chapter 11) are the most common tumors of infancy. Architecturally, they do not differ from those occurring in adults. Both cavernous and capillary hemangiomas may be encountered, although the latter are often more cellular than in adults, a feature that is deceptively worrisome. In children, most are located in the skin, particularly on the face and scalp, where they produce flat to elevated, irregular, red-blue masses; some of the flat, larger lesions (considered by some to represent vascular ectasias) are referred to as port-wine stains. Hemangiomas may enlarge along with the growth of the child, but in many instances they spontaneously regress (Fig. 10-23). In addition to their cosmetic significance, they can represent one facet of the hereditary disorder von Hippel-Lindau disease (Chapter 20). A subset of central nervous system cavernous hemangiomas can occur in the familial setting; these families harbor mutations in one of three cerebral cavernous malformation (CCM) genes.
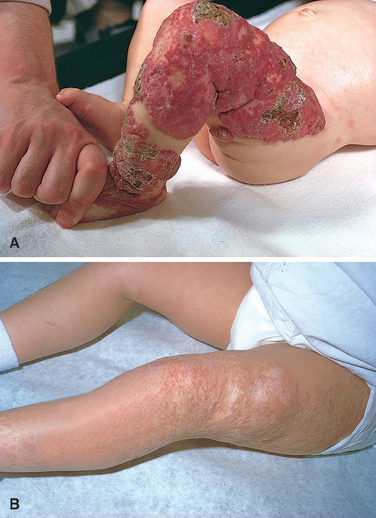
Lymphatic Tumors.
A wide variety of lesions are of lymphatic origin. Some of them—lymphangiomas—are hamartomatous or neoplastic, whereas others seem to represent abnormal dilations of preexisting lymph channels known as lymphangiectasis. The lymphangiomas are usually characterized by cystic and cavernous spaces. Lesions of this nature may occur in the skin but are more often encountered in the deeper regions of the neck, axilla, mediastinum, retroperitoneal tissue, and elsewhere. Although histologically benign, they tend to increase in size after birth, owing to the accumulation of fluid and the budding of preexisting spaces. In this manner they may encroach on vital structures, such as those in the mediastinum or nerve trunks in the axilla, and give rise to clinical problems. Lymphangiectasis, in contrast, usually presents as a diffuse swelling of part or all of an extremity; considerable distortion and deformation may occur as a consequence of the spongy, dilated subcutaneous and deeper lymphatics. The lesion is not progressive, however, and does not extend beyond its original location. Nonetheless, it creates cosmetic problems that are often difficult to correct surgically.
Fibrous Tumors.
Fibrous tumors occurring in infants and children range from sparsely cellular proliferations of spindle-shaped cells (designated as fibromatosis) to richly cellular lesions indistinguishable from fibrosarcomas occurring in adults (designated as congenital-infantile fibrosarcomas). Biologic behavior cannot be predicted based on histology alone, however, in that despite their histologic similarities with adult fibrosarcomas, the congenital-infantile variants have an excellent prognosis. Recently, a characteristic chromosomal translocation, t(12;15)(p13;q25), has been described in congenital-infantile fibrosarcomas, which results in generation of an ETV6-NTRK3 fusion transcript.51 The normal ETV6 gene product is a transcription factor, while the NTRK3 gene product (also known as TRKC, see below) is a tyrosine kinase. Like other tyrosine kinase fusion proteins found in human neoplasms, ETV6-TRKC is constitutively active and stimulates signaling through the oncogenic RAS and PI-3K/AKT pathways (Chapter 4). Among soft-tissue tumors, the ETV6-NTRK3 fusion transcript is unique to infantile fibrosarcomas, making it a useful diagnostic marker.
Teratomas.
Teratomas illustrate the relationship of histologic maturity to biologic behavior. They may occur as benign, well-differentiated cystic lesions (mature teratomas), as lesions of indeterminate potential (immature teratomas), or as unequivocally malignant teratomas (usually admixed with another germ cell tumor component such as endodermal sinus tumor) (Chapter 21). They exhibit two peaks in incidence: the first at approximately 2 years of age and the second in late adolescence or early adulthood. The first peak are congenital neoplasms; the later occurring lesions may also be of prenatal origin but are more slowly growing. Sacrococcygeal teratomas are the most common teratomas of childhood, accounting for 40% or more of cases (Fig. 10-24). They occur with a frequency of 1 in 20,000 to 40,000 live births, and are four times more common in girls than boys. In view of the overlap in the mechanisms underlying teratogenesis and oncogenesis, it is interesting that approximately 10% of sacrococcygeal teratomas are associated with congenital anomalies, primarily defects of the hindgut and cloacal region and other midline defects (e.g., meningocele, spina bifida) not believed to result from local effects of the tumor. Approximately 75% of these tumors are mature teratomas, and about 12% are unequivocally malignant and lethal. The remainder is immature teratomas; their malignant potential correlates with the amount of immature tissue, usually immature neuroepithelial elements, present. Most of the benign teratomas are encountered in younger infants (<4 months), whereas children with malignant lesions tend to be somewhat older. Other sites for teratomas in childhood include the testis (Chapter 21), ovaries (Chapter 22), and various midline locations, such as the mediastinum, retroperitoneum, and head and neck.
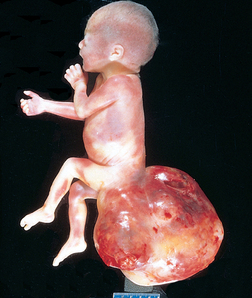
MALIGNANT TUMORS
Cancers of infancy and childhood differ biologically and histologically from their counterparts occurring later in life. The main differences, some of which have already been alluded to, include the following:
Incidence and Types
The most frequent childhood cancers arise in the hematopoietic system, nervous tissue (including the central and sympathetic nervous system, adrenal medulla, and retina), soft tissues, bone, and kidney. This is in sharp contrast to adults, in whom the skin, lung, breast, prostate, and colon are the most common sites of tumors.
Neoplasms that exhibit sharp peaks in incidence in children younger than age 10 years include (1) leukemia (principally acute lymphoblastic leukemia), (2) neuroblastoma, (3) Wilms tumor, (4) hepatoblastoma, (5) retinoblastoma, (6) rhabdomyosarcoma, (7) teratoma, (8) Ewing sarcoma, and finally, posterior fossa neoplasms—principally (9) juvenile astrocytoma, (10) medulloblastoma, and (11) ependymoma. Other forms of cancer are also common in childhood but do not have the same striking early peak. The approximate age distribution of these cancers is indicated in Table 10-8. Within this large array, leukemia alone accounts for more deaths in children younger than age 15 years than all of the other tumors combined.
TABLE 10-8 Common Malignant Neoplasms of Infancy and Childhood
| 0 to 4 Years | 5 to 9 Years | 10 to 14 Years |
|---|---|---|
| Leukemia | Leukemia | |
| Retinoblastoma | Retinoblastoma | |
| Neuroblastoma | Neuroblastoma | |
| Wilms tumor | ||
| Hepatoblastoma | Hepatocellular carcinoma | Hepatocellular carcinoma |
| Soft-tissue sarcoma (especially rhabdomyosarcoma) | Soft-tissue sarcoma | Soft-tissue sarcoma |
| Teratomas | ||
| Central nervous system tumors | Central nervous system tumors | Osteogenic sarcoma |
| Ewing sarcoma | Thyroid carcinoma | |
| Lymphoma | Hodgkin disease | |
Histologically, many of the malignant non-hematopoietic pediatric neoplasms are unique. In general, they tend to have a more primitive (embryonal) rather than pleomorphic-anaplastic microscopic appearance, are often characterized by sheets of cells with small, round nuclei, and frequently show features of organogenesis specific to the site of tumor origin. Because of this latter characteristic, these tumors are frequently designated by the suffix -blastoma, for example, nephroblastoma (Wilms tumor), hepatoblastoma, and neuroblastoma. Because of their primitive histologic appearance, many childhood tumors have been collectively referred to as small round blue cell tumors. The differential diagnosis of such tumors includes neuroblastoma, Wilms tumor, lymphoma (Chapter 14), rhabdomyosarcoma (Chapter 26), Ewing sarcoma/primitive neuroectodermal tumor (Chapter 26), medulloblastoma (Chapter 28), and retinoblastoma (Chapter 29). If the anatomic site of origin is known, diagnosis is usually possible on histologic grounds alone. Occasionally, a combination of chromosome analysis, immunoperoxidase stains, or electron microscopy is required. Two of these tumors are particularly illustrative and are discussed here: the neuroblastic tumors, specifically neuroblastoma, and Wilms tumor. The remaining tumors are discussed in their respective organspecific chapters.
The Neuroblastic Tumors
The term neuroblastic tumor includes tumors of the sympathetic ganglia and adrenal medulla that are derived from primordial neural crest cells populating these sites. As a family, neuroblastic tumors demonstrate certain characteristic features including spontaneous or therapy-induced differentiation of primitive neuroblasts into mature elements, spontaneous tumor regression, and a wide range of clinical behavior and prognosis, which often mirror the extent of histologic differentiation. Neuroblastoma is the most important member of this family. It is the most common extracranial solid tumor of childhood, and the most frequently diagnosed tumor of infancy. The prevalence is about one case in 7000 live births, and there are approximately 700 cases diagnosed each year in the United States. The median age at diagnosis is 18 months; approximately 40% of cases are diagnosed in infancy. Most neuroblastomas occur sporadically, but 1% to 2% are familial, and in such cases the neoplasms may involve both of the adrenals or multiple primary autonomic sites. Germline mutations in the anaplastic lymphoma kinase (ALK) gene (Chapter 14) have recently been identified as a major cause of familial predisposition to neuroblastoma.52 Somatic gain-of-function ALK mutations are also observed in a subset of sporadic neuroblastomas. It is envisioned that tumors harboring ALK mutations in either the germline or somatic setting will be amenable to treatment using drugs that target the activity of this kinase.
Despite the remarkable progress made in the therapy of this disease, long-term prognosis for the high-risk subsets remains modest, with a 5-year survival in the range of 40%. As will be evident later, age and stage have a remarkable effect on prognosis, and, in general, children younger than 18 months of age tend to have a significantly better prognosis than older individuals at comparable disease burdens.
Morphology In childhood about 40% of neuroblastomas arise in the adrenal medulla. The remainder occur anywhere along the sympathetic chain, with the most common locations being the paravertebral region of the abdomen (25%) and posterior mediastinum (15%). Tumors may arise in numerous other sites, including the pelvis, the neck, and within the brain (cerebral neuroblastomas).
Neuroblastomas range in size from minute nodules (so-called in situ lesions) to large masses more than 1 kg in weight (Fig. 10-25). In situ neuroblastomas are reported to occur 40 times more frequently than clinically overt tumors. The great majority of these silent lesions spontaneously regress, leaving only a focus of fibrosis or calcification in the adult; this has led some to question the neoplastic connotation for the in situ lesions, arguing instead in favor of labeling them as developmental anomalies (“rests”). Some neuroblastomas are often sharply demarcated by a fibrous pseudo-capsule, but others are far more infiltrative and invade surrounding structures, including the kidneys, renal vein, and vena cava, and envelop the aorta. On transection, they are composed of soft, gray-tan, tissue. Larger tumors have areas of necrosis, cystic softening, and hemorrhage. Occasionally, foci of punctate intra-tumoral calcification can be palpated.
FIGURE 10-25 Adrenal neuroblastoma in a 6-month-old child. The hemorrhagic, partially encapsulated tumor has displaced the opened left kidney and is impinging on the aorta and left renal artery.
(Courtesy of Dr. Arthur Weinberg, University of Texas Southwestern Medical School, Dallas, TX.)
Histologically, classic neuroblastomas are composed of small, primitive-appearing cells with dark nuclei, scant cytoplasm, and poorly defined cell borders growing in solid sheets. Such tumors may be difficult to differentiate morphologically from other small round blue cell tumors. Mitotic activity, nuclear breakdown (“karyorrhexis”), and pleomorphism may be prominent. The background often demonstrates a faintly eosinophilic fibrillary material (neuropil) that corresponds to neuritic processes of the primitive neuroblasts. Typically, rosettes (Homer-Wright pseudorosettes) can be found in which the tumor cells are concentrically arranged about a central space filled with neuropil (Fig. 10-26). Other helpful features include positive immunochemical reactions for neuron-specific enolase and ultrastructural demonstration of small, membrane-bound, cytoplasmic catecholamine-containing secretory granules; the latter contain characteristic central dense cores surrounded by a peripheral halo (dense core granules). Some neoplasms show signs of maturation that can be spontaneous or therapy-induced. Larger cells having more abundant cytoplasm, large vesicular nuclei, and a prominent nucleolus, representing ganglion cells in various stages of maturation, may be found in tumors admixed with primitive neuroblasts (ganglioneuroblastoma). Even better differentiated lesions contain many more large cells resembling mature ganglion cells with few if any residual neuroblasts; such neoplasms merit the designation ganglioneuroma (Fig. 10-27). Maturation of neuroblasts into ganglion cells is usually accompanied by the appearance of Schwann cells. In fact, the presence of a so-called schwannian stroma composed of organized fascicles of neuritic processes, mature Schwann cells, and fibroblasts is a histologic prerequisite for the designation of ganglioneuroblastoma and ganglioneuroma; ganglion cells in and of themselves do not fulfill the criteria for maturation. The origin of Schwann cells in neuroblastoma remains an issue of contention; some investigators believe they represent a reactive population recruited by the tumor cells. However, studies using microdissection techniques have demonstrated that the Schwann cells harbor at least a subset of the same genetic alterations found in neuroblasts, and therefore are a component of the malignant clone.53 Irrespective of histogenesis, documenting the presence of schwannian stroma is essential, since its presence is associated with a favorable outcome (Table 10-9).
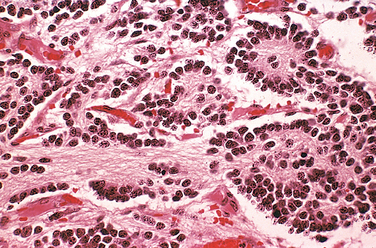
FIGURE 10-26 Adrenal neuroblastoma. This tumor is composed of small cells embedded in a finely fibrillar matrix.
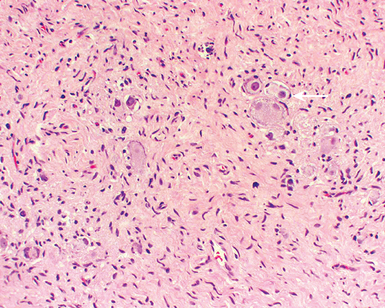
FIGURE 10-27 Ganglioneuromas, arising from spontaneous or therapy-induced maturation of neuroblastomas, are characterized by clusters of large cells with vesicular nuclei and abundant eosinophilic cytoplasm, representing neoplastic ganglion cells (arrow). Spindle-shaped Schwann cells are present in the background stroma.
TABLE 10-9 Prognostic Factors in Neuroblastomas
| Variable | Favorable | Unfavorable |
|---|---|---|
| Stage* | Stage 1, 2A, 2B, 4S | Stage 3, 4 |
| Age* | <18 months | >18 months |
| Histology* | ||
| Evidence of schwannian stroma and gangliocytic differentiation† | Present | Absent |
| Mitosis-karyorrhexis index‡ | <200/5000 cells | >200/5000 cells |
| DNA ploidy* | Hyperdiploid or near-triploid | Near-diploid |
| N-MYC* | Not amplified | Amplified |
| Chromosome 17q gain | Absent | Present |
| Chromosome 1p loss | Absent | Present |
| Chromosome 11q loss | Absent | Present |
| TRKA expression | Present | Absent |
| TRKB expression | Absent | Present |
| Telomerase expression | Low or absent | Highly expressed |
* Corresponds to the most commonly used parameters in clinical practice for assessment of prognosis and risk stratification.
† It is not only the presence but also the amount of schwannian stroma that confers the designation of a favorable histology. At least 50% or more schwannian stroma is required before a neoplasm can be classified as ganglioneuroblastoma or ganglioneuroma.
‡ Mitotic karyorrhexis index (MKI) is defined as the number of mitotic or karyorrhectic cells per 5000 tumor cells in random foci.
Metastases, when they develop, appear early and widely. In addition to local infiltration and lymph node spread, there is a pronounced tendency to spread through the bloodstream to involve the liver, lungs, bone marrow, and bones.
Staging.The International Neuroblastoma Staging System, which is the most widely used staging scheme worldwide, is detailed below:
Unfortunately, most (60% to 80%) children present with stage 3 or 4 tumors, and only 20% to 40% present with stage 1, 2A, 2B, or 4S neuroblastomas. The staging system is of paramount importance in determining prognosis.
Clinical Course and Prognostic Features.
In young children, under age 2 years, neuroblastomas generally present with large abdominal masses, fever, and possibly weight loss. In older children, they may not come to attention until metastases produce manifestations, such as bone pain, respiratory symptoms, or gastrointestinal complaints. Neuroblastomas may metastasize widely through the hematogenous and lymphatic systems, particularly to liver, lungs, bones, and bone marrow. Proptosis and ecchymosis may also be present, because the periorbital region is a common metastatic site. Bladder and bowel dysfunction may be caused by paraspinal neuroblastomas that impinge on nerves. In neonates, disseminated neuroblastomas may present with multiple cutaneous metastases that cause deep blue discoloration of the skin (earning the unfortunate designation of “blueberry muffin baby”). About 90% of neuroblastomas, regardless of location, produce catecholamines (similar to the catecholamines associated with pheochromocytomas), which are an important diagnostic feature (i.e., elevated blood levels of catecholamines and elevated urine levels of the metabolites vanillylmandelic acid [VMA] and homovanillic acid [HVA]). Despite the elaboration of catecholamines, hypertension is much less frequent with these neoplasms than with pheochromocytomas (Chapter 24). Ganglioneuromas, unlike their malignant counterparts, tend to produce either asymptomatic mass lesions or symptoms related to compression.
The course of neuroblastomas is extremely variable. Several clinical, histopathologic, molecular, and biochemical factors have been identified that have a bearing on prognosis (see Table 10-9)54; based on the collection of prognostic factors present in a given patient, they are classified as either “low,” “intermediate,” or “high” risk. With improvements in therapy, the first two categories result in long-term survival of 80% to 90% of patients, while less than 40% of patients in the high-risk category are long-term survivors. The most pertinent prognostic factors in neuroblastomas include the following:
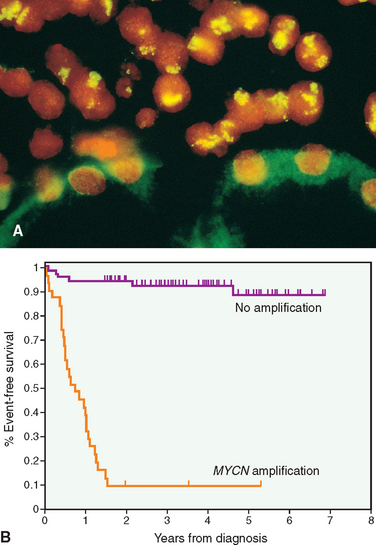
FIGURE 10-28 A, Fluorescence in situ hybridization using a fluorescein-labeled cosmid probe for N-myc on a tissue section. Note the neuroblastoma cells on the upper half of the photo with large areas of staining (yellow-green); this corresponds to amplified N-MYC in the form of homogeneously staining regions. Renal tubular epithelial cells in the lower half of the photograph show no nuclear staining and background (green) cytoplasmic staining.
(Courtesy of Dr. Timothy Triche, Children’s Hospital, Los Angeles, CA.) B, A Kaplan–Meier survival curve of infants younger than 1 year of age with metastatic neuroblastoma. The 3-year event-free survival (EFS) of infants whose tumors lacked MYCN amplification was 93%, whereas those with tumors that had MYCN amplification had only a 10% EFS. (Reproduced with permission from Brodeur GM: Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 3:203–216; 2003).
While age, stage, N-MYC status, histology, and DNA ploidy are currently the “core” criteria used for the purposes of formal risk stratification and therapeutic decision, several additional molecular variables have been described with prognostic implications. The most pertinent ones include the following:
Although discussion of the treatment modalities for neuroblastoma is beyond the scope of this book, we will mention in passing two promising experimental approaches that are being evaluated. The first involves the use of retinoids as an adjunct therapy for inducing the differentiation of neuroblastoma. Recall that the retinoic acid pathway plays a critical role in cellular differentiation during embryogenesis. Another emerging “targeted” therapy involves disrupting oncogenic TRKB signaling in neuroblastomas, by using small-molecule inhibitors of its tyrosine kinase activity. Finally, we should mention the current status of screening programs for neuroblastoma. Since the vast majority of neuroblastomas release catecholamines into the circulation, detection of catecholamine metabolites (VMA and HVA) in urine could, in principle, form the basis for screening for asymptomatic tumors in children. However, two large studies in Europe and North America have failed to demonstrate improved mortality rates with such population screening, because most tumors detected had favorable biologic characteristics, and the costs of screening failed to outweigh the benefits.60,61 Therefore, community-based screening programs for neuroblastomas are not currently advocated.
Wilms Tumor
Wilms tumor afflicts approximately 1 in every 10,000 children in the United States, and it is the most common primary renal tumor of childhood and the fourth most common pediatric malignancy in the United States. The peak incidence for Wilms tumor is between 2 and 5 years of age, and 95% of tumors occur before the age of 10 years. Approximately 5% to 10% of Wilms tumors involve both kidneys, either simultaneously (synchronous) or one after the other (metachronous). Bilateral Wilms tumors have a median age of onset approximately 10 months earlier than tumors restricted to one kidney, and these patients are presumed to harbor a germline mutation in one of the Wilms tumor–predisposing genes (see below). The biology of this tumor illustrates several important aspects of childhood neoplasms, such as the relationship between malformations and neoplasia, the histologic similarities between organogenesis and oncogenesis, the two-hit theory of recessive tumor suppressor genes (Chapter 7), the role of premalignant lesions, and perhaps most importantly, the potential for judicious treatment modalities to dramatically affect prognosis and outcome.62 Improvements in the cure rates for Wilms tumor (from as low as 30% a few decades ago, to 85% currently) represent one of the greatest successes of pediatric oncology.
Pathogenesis and Genetics.
The risk of Wilms tumor is increased in association with at least four recognizable groups of congenital malformations associated with distinct chromosomal loci. Although Wilms tumors arising in this setting account for no more than 10% of cases, these syndromic tumors have provided important insight into the biology of this neoplasm.
The first group of patients has the WAGR syndrome, characterized by aniridia, genital anomalies, and mental retardation and a 33% chance of developing Wilms tumor. Individuals with WAGR syndrome carry constitutional (germline) deletions of 11p13. Studies on these patients led to the identification of the first Wilms tumor–associated gene, WT1, and a contiguously deleted autosomal dominant gene for aniridia, PAX6, both located on chromosome 11p13. Patients with deletions restricted to PAX6, with normal WT1 function, develop sporadic aniridia, but they are not at increased risk for Wilms tumors. The presence of germline WT1 deletions in WAGR syndrome represents the “first hit”; the development of Wilms tumor in these patients frequently correlates with the occurrence of a nonsense or frameshift mutation in the second WT1 allele (“second hit”).
A second group of patients at a much higher risk for Wilms tumor (∼90%) have the Denys-Drash syndrome, which is characterized by gonadal dysgenesis (male pseudohermaphroditism) and early-onset nephropathy leading to renal failure. The characteristic glomerular lesion in these patients is a diffuse mesangial sclerosis (Chapter 20). As in patients with WAGR, these patients also demonstrate germline abnormalities in WT1. In patients with the Denys-Drash syndrome, however, the genetic abnormality is a dominant-negative missense mutation in the zinc-finger region of the WT1 gene that affects its DNA-binding properties. This mutation interferes with the function of the remaining wild-type allele, yet strangely, it is sufficient only in causing genitourinary abnormalities, but not tumorigenesis; Wilms tumors arising in Denys-Drash syndrome demonstrate bi-allelic inactivation of WT1. In addition to Wilms tumors, these individuals are also at increased risk for developing germ cell tumors called gonadoblastomas (Chapter 21), almost certainly a consequence of disruption in normal gonadal development.
WT1 encodes a DNA-binding transcription factor that is expressed within several tissues, including the kidney and gonads, during embryogenesis. The WT1 protein is critical for normal renal and gonadal development. WT1 has multiple binding partners, and the choice of this partner can affect whether WT1 functions as a transcriptional activator or repressor in a given cellular context.63 Numerous transcriptional targets of WT1 have been identified, including glomerular podocyte-specific proteins, and genes associated with inducing differentiation. Despite the importance of WT1 in nephrogenesis and its unequivocal role as a tumor suppressor gene, only about 10% of patients with sporadic (nonsyndromic) Wilms tumors demonstrate WT1 mutations, suggesting that the majority of these tumors arise by genetically distinct pathways.
Clinically distinct from these previous two groups of patients but also having an increased risk of developing Wilms tumor are children with Beckwith-Wiedemann syndrome (BWS), characterized by enlargement of body organs (organomegaly), macroglossia, hemihypertrophy, omphalocele, and abnormal large cells in the adrenal cortex (adrenal cytomegaly). BWS has served as a model for a nonclassical mechanism of tumorigenesis in humans—genomic imprinting (Chapter 5).64 The chromosomal region implicated in BWS has been localized to band 11p15.5 (“WT2”), distal to the WT1 locus. This region contains multiple genes that are normally expressed from only one of the two parental alleles, with transcriptional silencing (i.e., imprinting) of the other parental homologue by methylation of the promoter region. Unlike WAGR or Denys-Drash syndromes, the genetic basis for BWS is considerably more heterogeneous in that no single 11p15.5 gene is involved in all cases. Moreover, the phenotype of BWS, including the predisposition to tumorigenesis, is influenced by the specific “WT2” imprinting abnormalities present. One of the genes in this region—insulin-like growth factor-2 (IGF2)—is normally expressed solely from the paternal allele, while the maternal allele is silenced by imprinting. In some Wilms tumors, loss of imprinting (i.e., re-expression of the maternal IGF2 allele) can be demonstrated, leading to overexpression of the IGF-2 protein. In other instances there is a selective deletion of the imprinted maternal allele, combined with duplication of the transcriptionally active paternal allele in the tumor (uniparental paternal disomy), which has an identical functional effect in terms of overexpression of IGF-2. Since the IGF-2 protein is an embryonal growth factor, it could conceivably explain the features of overgrowth associated with BWS, as well as the increased risk for Wilms tumors in these patients. Of all the “WT2” genes, imprinting abnormalties of IGF2 have the strongest relationship to tumor predisposition in BWS.65 A subset of patients with BWS harbor mutations of the cell cycle regulator CDKN1C (also known as p57 or KIP2); however, these patients have a significantly lower risk for developing Wilms tumors. In addition to Wilms tumors, patients with BWS are also at increased risk for developing hepatoblastoma, pancreatoblastoma, adrenocortical tumors, and rhabdomyosarcomas.
Recent genetic studies have also elucidated the role of β-catenin in Wilms tumor. It will be recalled (Chapter 7) that β-catenin belongs to the developmentally important WNT (wingless) signaling pathway. Gain-of-function mutations of the gene encoding β-catenin have been demonstrated in approximately 10% of sporadic Wilms tumors; there is a significant overlap between the presence of WT1 and β-catenin mutations, suggesting a synergistic role for these events in the genesis of Wilms tumors.66
Nephrogenic Rests
Nephrogenic rests are putative precursor lesions of Wilms tumors and are seen in the renal parenchyma adjacent to approximately 25% to 40% of unilateral tumors; this frequency rises to nearly 100% in cases of bilateral Wilms tumors. In many instances the nephrogenic rests share genetic alterations with the adjacent Wilms tumor, underscoring their preneoplastic status. The appearance of nephrogenic rests varies from expansile masses that resemble Wilms tumors (hyperplastic rests) to sclerotic rests consisting predominantly of fibrous tissue and occasional admixed immature tubules or glomeruli. It is important to document the presence of nephrogenic rests in the resected specimen, since these patients are at an increased risk of developing Wilms tumors in the contralateral kidney and require frequent and regular surveillance for many years.
Morphology. Grossly, Wilms tumor tends to present as a large, solitary, well-circumscribed mass, although 10% are either bilateral or multicentric at the time of diagnosis. On cut section, the tumor is soft, homogeneous, and tan to gray with occasional foci of hemorrhage, cyst formation, and necrosis (Fig. 10-29).
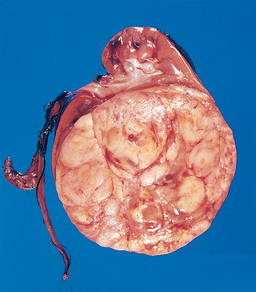
FIGURE 10-29 Wilms’ tumor in the lower pole of the kidney with the characteristic tan-to-gray color and well-circumscribed margins.
Microscopically, Wilms tumors are characterized by recognizable attempts to recapitulate different stages of nephrogenesis. The classic triphasic combination of blastemal, stromal, and epithelial cell types is observed in the vast majority of lesions, although the percentage of each component is variable (Fig. 10-30). Sheets of small blue cells with few distinctive features characterize the blastemal component. Epithelial differentiation is usually in the form of abortive tubules or glomeruli. Stromal cells are usually fibrocytic or myxoid in nature, although skeletal muscle differentiation is not uncommon. Rarely, other heterologous elements are identified, including squamous or mucinous epithelium, smooth muscle, adipose tissue, cartilage, and osteoid and neurogenic tissue. Approximately 5% of tumors reveal anaplasia, defined as the presence of cells with large, hyperchromatic, pleomorphic nuclei and abnormal mitoses. The presence of anaplasia correlates with the presence of p53 mutations and the emergence of resistance to chemotherapy.67 Recall that p53 elicits pro-apoptotic signals in response to DNA damage (Chapter 1). The loss of p53 function might explain the relative unresponsiveness of anaplastic cells to cytotoxic chemotherapy.
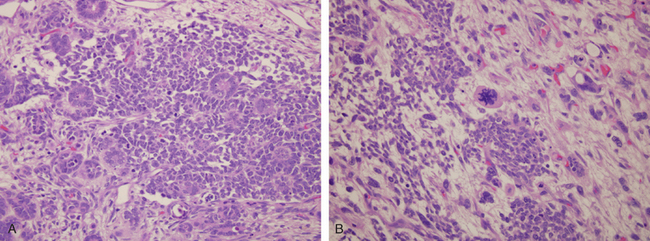
FIGURE 10-30 A, Wilms tumor with tightly packed blue cells consistent with the blastemal component and interspersed primitive tubules, representing the epithelial component. Although multiple mitotic figures are seen, none are atypical in this field. B, Focal anaplasia was present in this Wilms’ tumor in other areas, characterized by cells with hyperchromatic, pleomorphic nuclei and abnormal mitoses.
Clinical Features.
Most children with Wilms tumors present with a large abdominal mass that may be unilateral or, when very large, may extend across the midline and down into the pelvis. Hematuria, pain in the abdomen after some traumatic incident, intestinal obstruction, and appearance of hypertension are other patterns of presentation. In a considerable number of these patients, pulmonary metastases are present at the time of primary diagnosis.
As stated, most patients with Wilms tumor can expect to be cured of their malignancy. Anaplastic histology remains a critical determinant of adverse prognosis. Even anaplasia restricted to the kidney (i.e., without extra-renal spread) confers an increased risk of recurrence and death, underscoring the need for correctly identifying this histologic feature. Molecular parameters that correlate with adverse prognosis include loss of genetic material on chromosomes 11q and 16q, and gain of chromosome 1q in the tumor cells. Along with the increased survival of individuals with Wilms tumor have come reports of an increased relative risk of developing second primary tumors, including bone and soft-tissue sarcomas, leukemia and lymphomas, and breast cancers. While some of these neoplasms represent the presence of a germline mutation in a cancer predisposition gene, others are a consequence of therapy, most commonly radiation administered to the cancer field.68 This tragic, albeit uncommon, outcome has mandated that radiation therapy be used judiciously in the treatment of this and other childhood cancers.
1 Minino AM, Heron MP, Murphy SL, Kochanek KD. Deaths: final data for 2004. Natl Vital Stat Rep. 2007;55:1.
2 Roessler E, Muenke M. How a Hedgehog might see holoprosencephaly. Hum Mol Genet. 2003;12(Spec No 1):R15.
3 Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;370:162.
4 Knobloch J, Shaughnessy JDJr., Ruther U. Thalidomide induces limb deformities by perturbing the Bmp/Dkk1/Wnt signaling pathway. Faseb J. 2007;21:1410.
5 Yelin R, Schyr RB, Kot H, et al. Ethanol exposure affects gene expression in the embryonic organizer and reduces retinoic acid levels. Dev Biol. 2005;279:193.
6 Li YX, Yang HT, Zdanowicz M, et al. Fetal alcohol exposure impairs Hedgehog cholesterol modification and signaling. Lab Invest. 2007;87:231.
7 Blom HJ, Shaw GM, den Heijer M, Finnell RH. Neural tube defects and folate: case far from closed. Nat Rev Neurosci. 2006;7:724.
8 Faiella A, Wernig M, Consalez GG, et al. A mouse model for valproate teratogenicity: parental effects, homeotic transformations, and altered HOX expression. Hum Mol Genet. 2000;9:227.
9 Koo SH, Cunningham MC, Arabshahi B, Gruss JS, Grant JH3rd. The transforming growth factor-beta 3 knock-out mouse: an animal model for cleft palate. Plast Reconstr Surg. 2001;108:938.
10 ACOG Practice Bulletin No. 80: premature rupture of membranes. Clinical management guidelines for obstetrician-gynecologists. Obstet Gynecol. 2007;109:1007.
11 Nesin M. Genetic basis of preterm birth. Front Biosci. 2007;12:115.
12 Elovitz MA, Wang Z, Chien EK, Rychlik DF, Phillippe M. A new model for inflammation-induced preterm birth: the role of platelet-activating factor and Toll-like receptor-4. Am J Pathol. 2003;163:2103.
13 Kinzler WL, Kaminsky L. Fetal growth restriction and subsequent pregnancy risks. Semin Perinatol. 2007;31:126.
14 Miura K, Yoshiura K, Miura S, et al. Clinical outcome of infants with confined placental mosaicism and intrauterine growth restriction of unknown cause. Am J Med Genet A. 2006;140:1827.
15 Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol. 2006;132:171.
16 Hermansen CL, Lorah KN. Respiratory distress in the newborn. Am Fam Physician. 2007;76:987.
17 Hamvas A. Inherited surfactant protein-B deficiency and surfactant protein-C associated disease: clinical features and evaluation. Semin Perinatol. 2006;30:316.
18 Chen J, Smith LE. Retinopathy of prematurity. Angiogenesis. 2007;10:133.
19 Bancalari E, Claure N. Definitions and diagnostic criteria for bronchopulmonary dysplasia. Semin Perinatol. 2006;30:164.
20 Chess PR, D’Angio CT, Pryhuber GS, Maniscalco WM. Pathogenesis of bronchopulmonary dysplasia. Semin Perinatol. 2006;30:171.
21 Speer CP. Inflammation and bronchopulmonary dysplasia: a continuing story. Semin Fetal Neonatal Med. 2006;11:354.
22 Thebaud B. Angiogenesis in lung development, injury and repair: implications for chronic lung disease of prematurity. Neonatology. 2007;91:291.
23 Caplan MS, Simon D, Jilling T. The role of PAF, TLR, and the inflammatory response in neonatal necrotizing enterocolitis. Semin Pediatr Surg. 2005;14:145.
24 Abrams ME, Meredith KS, Kinnard P, Clark RH. Hydrops fetalis: a retrospective review of cases reported to a large national database and identification of risk factors associated with death. Pediatrics. 2007;120:84.
25 Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat. 2007;28:831.
26 Zurfluh MR, Zschocke J, Lindner M, et al. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum Mutat. 2008.
27 Harding CO, Gillingham MB, Hamman K, et al. Complete correction of hyperphenylalaninemia following liver-directed, recombinant AAV2/8 vector-mediated gene therapy in murine phenylketonuria. Gene Ther. 2006;13:457.
28 Fridovich-Keil JL. Galactosemia: the good, the bad, and the unknown. J Cell Physiol. 2006;209:701.
29 Wang X, Kim J, McWilliams R, Cutting GR. Increased prevalence of chronic rhinosinusitis in carriers of a cystic fibrosis mutation. Arch Otolaryngol Head Neck Surg. 2005;131:237.
30 Cohn JA, Neoptolemos JP, Feng J, et al. Increased risk of idiopathic chronic pancreatitis in cystic fibrosis carriers. Hum Mutat. 2005;26:303.
31 Castellani C, Cuppens H, Macek MJr, et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cyst Fibros. 2008;7:179.
32 Boucher RC. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends in molecular medicine. 2007;13:231.
33 Shcheynikov N, Ko SB, Zeng W, et al. Regulatory interaction between CFTR and the SLC26 transporters. Novartis Found Symp. 2006;273:177.
34 Farrell PM, Rosenstein BJ, White TB, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. The Journal of pediatrics. 2008;153:S4.
35 Cutting GR. Modifier genetics: cystic fibrosis. Annu Rev Genomics Hum Genet. 2005;6:237.
36 Howe KL, Wang A, Hunter MM, Stanton BA, McKay DM. TGFbeta down-regulation of the CFTR: a means to limit epithelial chloride secretion. Exp Cell Res. 2004;298:473.
37 Pruliere-Escabasse V, Fanen P, Dazy AC, et al. TGF-beta 1 downregulates CFTR expression and function in nasal polyps of non-CF patients. Am J Physiol Lung Cell Mol Physiol. 2005;288:L77.
38 Drumm ML, Konstan MW, Schluchter MD, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443.
39 Blackman SM, Deering-Brose R, McWilliams R, et al. Relative contribution of genetic and nongenetic modifiers to intestinal obstruction in cystic fibrosis. Gastroenterology. 2006;131:1030.
40 Mahenthiralingam E, Urban TA, Goldberg JB. The multifarious, multireplicon Burkholderia cepacia complex. Nat Rev Microbiol. 2005;3:144.
41 Boyle MP. Adult cystic fibrosis. JAMA. 2007;298:1787.
42 American Academy of Pediatrics Task Force on Sudden Infant Death Syndrome. The changing concept of sudden infant death syndrome: diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Pediatrics. 2005;116:1245.
43 Krous HF, Beckwith JB, Byard RW, et al. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics. 2004;114:234.
44 Moon RY, Horne RS, Hauck FR. Sudden infant death syndrome. Lancet. 2007;370:1578.
45 American Academy of Pediatrics, Committee on Fetus and Newborn. Apnea, sudden infant death syndrome, and home monitoring. Pediatrics. 2003;111:914.
46 Guntheroth WG, Spiers PS. The triple risk hypotheses in sudden infant death syndrome. Pediatrics. 2002;110:e64.
47 Paterson DS, Trachtenberg FL, Thompson EG, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA. 2006;296:2124.
48 Audero E, Coppi E, Mlinar B, et al. Sporadic autonomic dysregulation and death associated with excessive serotonin autoinhibition. Science, New York, NY. 2008;321:130.
49 Weese-Mayer DE, Ackerman MJ, Marazita ML, Berry-Kravis EM. Sudden Infant Death Syndrome: review of implicated genetic factors. Am J Med Genet A. 2007;143:771.
50 Adgent MA. Environmental tobacco smoke and sudden infant death syndrome: a review. Birth Defects Res B Dev Reprod Toxicol. 2006;77:69.
51 Lannon CL, Sorensen PH. ETV6-NTRK3: a chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin Cancer Biol. 2005;15:215.
52 Mosse YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:950.
53 Mora J, Cheung NK, Juan G, et al. Neuroblastic and Schwannian stromal cells of neuroblastoma are derived from a tumoral progenitor cell. Cancer research. 2001;61:6892.
54 Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106.
55 London WB, Castleberry RP, Matthay KK, et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. J Clin Oncol. 2005;23:6459.
56 Brodeur GM, Maris JM. Neuroblastoma. In: Pizzo PA, Poplack DG, editors. Principles and Practice of Pediatric Oncology. Philadelphia: JB Lippincott; 2006:933.
57 Attiyeh EF, London WB, Mosse YP, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005;353:2243.
58 George RE, Attiyeh EF, Li S, et al. Genome-wide analysis of neuroblastomas using high-density single nucleotide polymorphism arrays. PLoS ONE. 2007;2:e255.
59 Schleiermacher G, Michon J, Huon I, et al. Chromosomal CGH identifies patients with a higher risk of relapse in neuroblastoma without MYCN amplification. Br J Cancer. 2007;97:238.
60 Woods WG, Gao RN, Shuster JJ, et al. Screening of infants and mortality due to neuroblastoma. N Engl J Med. 2002;346:1041.
61 Schilling FH, Spix C, Berthold F, et al. Neuroblastoma screening at one year of age. N Engl J Med. 2002;346:1047.
62 Rivera MN, Haber DA. Wilms’ tumour: connecting tumorigenesis and organ development in the kidney. Nat Rev Cancer. 2005;5:699.
63 Hohenstein P, Hastie ND. The many facets of the Wilms’ tumour gene, WT1. Hum Mol Genet. 2006;15(Spec No 2):R196.
64 Feinberg AP. The epigenetics of cancer etiology. Semin Cancer Biol. 2004;14:427.
65 Bjornsson HT, Brown LJ, Fallin MD, et al. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst. 2007;99:1270.
66 Tycko B, Li CM, Buttyan R. The Wnt/beta-catenin pathway in Wilms tumors and prostate cancers. Curr Mol Med. 2007;7:479.
67 Dome JS, Cotton CA, Perlman EJ, et al. Treatment of anaplastic histology Wilms’ tumor: results from the fifth National Wilms’ Tumor Study. J Clin Oncol. 2006;24:2352.
68 Robison LL, Green DM, Hudson M, et al. Long-term outcomes of adult survivors of childhood cancer. Cancer. 2005;104:2557.