Chapter 2 Acute and Chronic Inflammation
Overview of Inflammation
Essential to the survival of organisms is their ability to get rid of damaged or necrotic tissues and foreign invaders, such as microbes. The host response that accomplishes these goals is called inflammation. This is fundamentally a protective response, designed to rid the organism of both the initial cause of cell injury (e.g., microbes, toxins) and the consequences of such injury (e.g., necrotic cells and tissues). Without inflammation infections would go unchecked, wounds would never heal, and injured tissues might remain permanent festering sores. In the practice of medicine the importance of inflammation is that it can sometimes be inappropriately triggered or poorly controlled, and is thus the cause of tissue injury in many disorders.
Inflammation is a complex reaction in tissues that consists mainly of responses of blood vessels and leukocytes. The body’s principal defenders against foreign invaders are plasma proteins and circulating leukocytes (white blood cells), as well as tissue phagocytes that are derived from circulating cells. The presence of proteins and leukocytes in the blood gives them the ability to home to any site where they may be needed. Because invaders such as microbes and necrotic cells are typically present in tissues, outside the circulation, it follows that the circulating cells and proteins have to be rapidly recruited to these extravascular sites. The inflammatory response coordinates the reactions of vessels, leukocytes, and plasma proteins to achieve this goal.
The vascular and cellular reactions of inflammation are triggered by soluble factors that are produced by various cells or derived from plasma proteins and are generated or activated in response to the inflammatory stimulus. Microbes, necrotic cells (whatever the cause of cell death) and even hypoxia can trigger the elaboration of inflammatory mediators, and thus elicit inflammation. Such mediators initiate and amplify the inflammatory response and determine its pattern, severity, and clinical and pathologic manifestations.
Inflammation may be acute or chronic, depending on the nature of the stimulus and the effectiveness of the initial reaction in eliminating the stimulus or the damaged tissues. Acute inflammation is rapid in onset (typically minutes) and is of short duration, lasting for hours or a few days; its main characteristics are the exudation of fluid and plasma proteins (edema) and the emigration of leukocytes, predominantly neutrophils (also called polymorphonuclear leukocytes). When acute inflammation is successful in eliminating the offenders the reaction subsides, but if the response fails to clear the invaders it can progress to a chronic phase. Chronic inflammation may follow acute inflammation or be insidious in onset. It is of longer duration and is associated with the presence of lymphocytes and macrophages, the proliferation of blood vessels, fibrosis, and tissue destruction.
Inflammation is terminated when the offending agent is eliminated. The reaction resolves rapidly, because the mediators are broken down and dissipated and the leukocytes have short life spans in tissues. In addition, anti-inflammatory mechanisms are activated that serve to control the response and prevent it from causing excessive damage to the host.
The inflammatory response is closely intertwined with the process of repair. At the same time as inflammation destroys, dilutes, and walls off the injurious agent, it sets into motion a series of events that try to heal the damaged tissue. Repair begins during inflammation but reaches completion usually after the injurious influence has been neutralized. In the process of repair the injured tissue is replaced through regeneration of native parenchymal cells, by filling of the defect with fibrous tissue (scarring) or, most commonly, by a combination of these two processes (Chapter 3).
Inflammation may be harmful in some situations. Mechanisms designed to destroy foreign invaders and necrotic tissues have an intrinsic ability to injure normal tissues. When inflammation is inappropriately directed against self tissues or is not adequately controlled, it becomes the cause of injury and disease. In fact, in clinical medicine, great attention is given to the damaging consequences of inflammation. Inflammatory reactions underlie common chronic diseases, such as rheumatoid arthritis, atherosclerosis, and lung fibrosis, as well as life-threatening hypersensitivity reactions to insect bites, drugs, and toxins. For this reason our pharmacies abound with anti-inflammatory drugs, which ideally would control the harmful sequelae of inflammation yet not interfere with its beneficial effects.
Inflammation may contribute to a variety of diseases that are not thought to be primarily due to abnormal host responses. For instance, chronic inflammation may play a role in atherosclerosis, type 2 diabetes, degenerative disorders like Alzheimer disease, and cancer. In recognition of the wide-ranging harmful consequences of inflammation, the lay press has rather melodramatically referred to it as “the silent killer.”
This chapter describes the sequence of events and mediators of acute inflammation, and then its morphologic patterns. This is followed by a discussion of the major features of chronic inflammation. Inflammation has a rich history, and we first touch on some of the historical highlights in our consideration of this fascinating process.
Historical Highlights
Although clinical features of inflammation were described in an Egyptian papyrus dated around 3000 bc, Celsus, a Roman writer of the first century ad, first listed the four cardinal signs of inflammation: rubor (redness), tumor (swelling), calor (heat), and dolor (pain).1 These signs are typically more prominent in acute inflammation than in chronic inflammation. A fifth clinical sign, loss of function (functio laesa), was added by Rudolf Virchow in the 19th century. In 1793 the Scottish surgeon John Hunter noted what is now considered an obvious fact: that inflammation is not a disease but a nonspecific response that has a salutary effect on its host.2 In the 1880s the Russian biologist Elie Metchnikoff discovered the process of phagocytosis by observing the ingestion of rose thorns by amebocytes of starfish larvae and of bacteria by mammalian leukocytes.3 He concluded that the purpose of inflammation was to bring phagocytic cells to the injured area to engulf invading bacteria. This concept was elegantly satirized by George Bernard Shaw in his play “The Doctor’s Dilemma,” in which one physician’s cure-all is to “stimulate the phagocytes”! Sir Thomas Lewis, studying the inflammatory response in skin, established the concept that chemical substances, such as histamine (produced locally in response to injury), mediate the vascular changes of inflammation. This fundamental concept underlies the important discoveries of chemical mediators of inflammation and the use of anti-inflammatory drugs in clinical medicine.
Acute Inflammation
Acute inflammation is a rapid host response that serves to deliver leukocytes and plasma proteins, such as antibodies, to sites of infection or tissue injury. Acute inflammation has three major components: (1) alterations in vascular caliber that lead to an increase in blood flow, (2) structural changes in the microvasculature that permit plasma proteins and leukocytes to leave the circulation, and (3) emigration of the leukocytes from the microcirculation, their accumulation in the focus of injury, and their activation to eliminate the offending agent (Fig. 2-1).
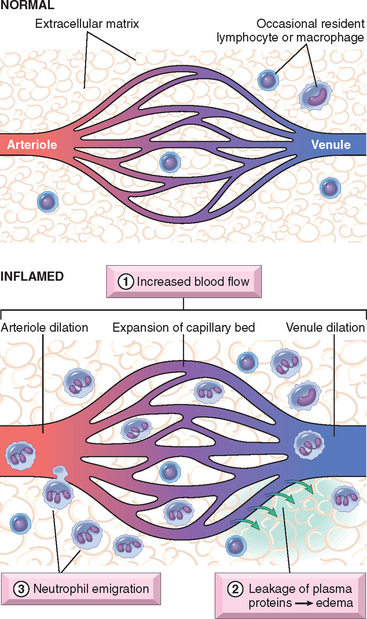
FIGURE 2-1 The major local manifestations of acute inflammation, compared to normal. (1) Vascular dilation and increased blood flow (causing erythema and warmth); (2) extravasation and extravascular deposition of plasma fluid and proteins (edema); (3) leukocyte emigration and accumulation in the site of injury.
STIMULI FOR ACUTE INFLAMMATION
Acute inflammatory reactions may be triggered by a variety of stimuli:
All inflammatory reactions share the same basic features, although different stimuli may induce reactions with some distinctive characteristics. We first describe the typical sequence of events in acute inflammation, and then the chemical mediators responsible for inflammation and the morphologic appearance of these reactions.
REACTIONS OF BLOOD VESSELS IN ACUTE INFLAMMATION
In inflammation, blood vessels undergo a series of changes that are designed to maximize the movement of plasma proteins and circulating cells out of the circulation and into the site of infection or injury. The escape of fluid, proteins, and blood cells from the vascular system into the interstitial tissue or body cavities is known as exudation. An exudate is an extravascular fluid that has a high protein concentration, contains cellular debris, and has a high specific gravity. Its presence implies an increase in the normal permeability of small blood vessels in an area of injury and, therefore, an inflammatory reaction (Fig. 2-2). In contrast, a transudate is a fluid with low protein content (most of which is albumin), little or no cellular material, and low specific gravity. It is essentially an ultrafiltrate of blood plasma that results from osmotic or hydrostatic imbalance across the vessel wall without an increase in vascular permeability (Chapter 4). Edema denotes an excess of fluid in the interstitial tissue or serous cavities; it can be either an exudate or a transudate. Pus, a purulent exudate, is an inflammatory exudate rich in leukocytes (mostly neutrophils), the debris of dead cells and, in many cases, microbes.
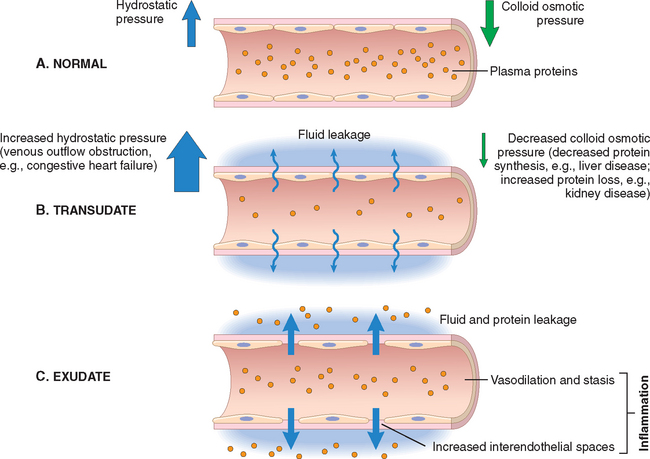
FIGURE 2-2 Formation of transudates and exudates. A, Normal hydrostatic pressure (blue arrows) is about 32 mm Hg at the arterial end of a capillary bed and 12 mm Hg at the venous end; the mean colloid osmotic pressure of tissues is approximately 25 mm Hg (green arrows), which is equal to the mean capillary pressure. Therefore, the net flow of fluid across the vascular bed is almost nil. B, A transudate is formed when fluid leaks out because of increased hydrostatic pressure or decreased osmotic pressure. C, An exudate is formed in inflammation, because vascular permeability increases as a result of increased interendothelial spaces.
The vascular reactions of acute inflammation consist of changes in the flow of blood and the permeability of vessels. Proliferation of blood vessels (angiogenesis) is prominent during repair and in chronic inflammation; this process is discussed in Chapter 3.
Changes in Vascular Flow and Caliber
Changes in vascular flow and caliber begin early after injury and consist of the following.
Increased Vascular Permeability (Vascular Leakage)
A hallmark of acute inflammation is increased vascular permeability leading to the escape of a protein-rich exudate into the extravascular tissue, causing edema. Several mechanisms are responsible for the increased vascular permeability (Fig. 2-3):

FIGURE 2-3 Principal mechanisms of increased vascular permeability in inflammation, and their features and underlying causes. NO, nitric oxide; VEGF, vascular endothelial growth factor.
Although these mechanisms of increased vascular permeability are described separately, all probably contribute in varying degrees in responses to most stimuli. For example, at different stages of a thermal burn, leakage results from chemically mediated endothelial contraction and direct and leukocyte-dependent endothelial injury. The vascular leakage induced by all these mechanisms can cause life-threatening loss of fluid in severely burned patients.
Responses of Lymphatic Vessels
Although much of the emphasis in our discussion of inflammation is on the reactions of blood vessels, lymphatic vessels also participate in the response. The system of lymphatics and lymph nodes filters and polices the extravascular fluids. Recall that lymphatics normally drain the small amount of extravascular fluid that has seeped out of capillaries. In inflammation, lymph flow is increased and helps drain edema fluid that accumulates due to increased vascular permeability. In addition to fluid, leukocytes and cell debris, as well as microbes, may find their way into lymph. Lymphatic vessels, like blood vessels, proliferate during inflammatory reactions to handle the increased load.11,12 The lymphatics may become secondarily inflamed (lymphangitis), as may the draining lymph nodes (lymphadenitis). Inflamed lymph nodes are often enlarged because of hyperplasia of the lymphoid follicles and increased numbers of lymphocytes and macrophages. This constellation of pathologic changes is termed reactive, or inflammatory, lymphadenitis (Chapter 13). For clinicians the presence of red streaks near a skin wound is a telltale sign of an infection in the wound. This streaking follows the course of the lymphatic channels and is diagnostic of lymphangitis; it may be accompanied by painful enlargement of the draining lymph nodes, indicating lymphadenitis.
REACTIONS OF LEUKOCYTES IN INFLAMMATION
As mentioned earlier, a critical function of inflammation is to deliver leukocytes to the site of injury and to activate the leukocytes to eliminate the offending agents. The most important leukocytes in typical inflammatory reactions are the ones capable of phagocytosis, namely neutrophils and macrophages. These leukocytes ingest and kill bacteria and other microbes, and eliminate necrotic tissue and foreign substances. Leukocytes also produce growth factors that aid in repair. A price that is paid for the defensive potency of leukocytes is that, when strongly activated, they may induce tissue damage and prolong inflammation, because the leukocyte products that destroy microbes and necrotic tissues can also injure normal host tissues.
The processes involving leukocytes in inflammation consist of: their recruitment from the blood into extravascular tissues, recognition of microbes and necrotic tissues, and removal of the offending agent.
Recruitment of Leukocytes to Sites of Infection and Injury
The journey of leukocytes from the vessel lumen to the interstitial tissue, called extravasation, can be divided into the following steps13 (Fig. 2-4):
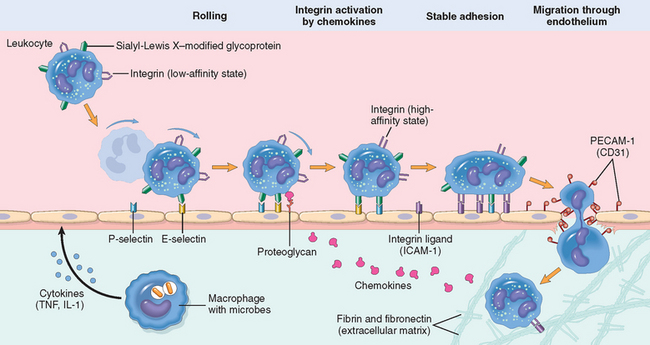
FIGURE 2-4 The multistep process of leukocyte migration through blood vessels, shown here for neutrophils. The leukocytes first roll, then become activated and adhere to endothelium, then transmigrate across the endothelium, pierce the basement membrane, and migrate toward chemoattractants emanating from the source of injury. Different molecules play predominant roles in different steps of this process—selectins in rolling; chemokines (usually displayed bound to proteoglycans) in activating the neutrophils to increase avidity of integrins; integrins in firm adhesion; and CD31 (PECAM-1) in transmigration. Neutrophils express low levels of L-selectin; they bind to endothelial cells predom in antly via P- and E-selectins. ICAM-1, intercellular adhesion molecule 1; TNF, tumor necrosis factor.
Leukocyte Adhesion to Endothelium.
In normally flowing blood in venules, red cells are confined to a central axial column, displacing the leukocytes toward the wall of the vessel. Because blood flow slows early in inflammation (stasis), hemodynamic conditions change (wall shear stress decreases), and more white cells assume a peripheral position along the endothelial surface. This process of leukocyte redistribution is called margination. Subsequently, individual and then rows of leukocytes adhere transiently to the endothelium, detach and bind again, thus rolling on the vessel wall. The cells finally come to rest at some point where they adhere firmly (resembling pebbles over which a stream runs without disturbing them).
The adhesion of leukocytes to endothelial cells is mediated by complementary adhesion molecules on the two cell types whose expression is enhanced by secreted proteins called cytokines.13,14 Cytokines are secreted by cells in tissues in response to microbes and other injurious agents, thus ensuring that leukocytes are recruited to the tissues where these stimuli are present. The initial rolling interactions are mediated by a family of proteins called selectins15,16 (Table 2-1). There are three types of selectins: one expressed on leukocytes (L-selectin), one on endothelium (E-selectin), and one in platelets and on endothelium (P-selectin). The ligands for selectins are sialylated oligosaccharides bound to mucin-like glycoprotein backbones. The expression of selectins and their ligands is regulated by cytokines produced in response to infection and injury. Tissue macrophages, mast cells, and endothelial cells that encounter microbes and dead tissues respond by secreting several cytokines, including tumor necrosis factor (TNF),17 interleukin-1 (IL-1),18 and chemokines (chemoattractant cytokines).19,20 (Cytokines are described in more detail below and in Chapter 6.) TNF and IL-1 act on the endothelial cells of post-capillary venules adjacent to the infection and induce the coordinate expression of numerous adhesion molecules (Fig. 2-5). Within 1 to 2 hours the endothelial cells begin to express E-selectin and the ligands for L-selectin. Other mediators such as histamine, thrombin, and platelet-activating factor (PAF), described later, stimulate the redistribution of P-selectin from its normal intracellular stores in endothelial cell granules (called Weibel-Palade bodies) to the cell surface. Leukocytes express L-selectin at the tips of their microvilli and also express ligands for E- and P-selectins, all of which bind to the complementary molecules on the endothelial cells. These are low-affinity interactions with a fast off-rate, and they are easily disrupted by the flowing blood. As a result, the bound leukocytes bind, detach, and bind again, and thus begin to roll along the endothelial surface.
TABLE 2-1 Endothelial-Leukocyte Adhesion Molecules
| Endothelial Molecule | Leukocyte Molecule | Major Role |
|---|---|---|
| P-selectin | Sialyl-Lewis X–modified proteins | Rolling (neutrophils, monocytes, T lymphocytes) |
| E-selectin | Sialyl-Lewis X–modified proteins | Rolling and adhesion (neutrophils, monocytes, T lymphocytes) |
| GlyCam-1, CD34 | L-selectin* | Rolling (neutrophils, monocytes) |
| ICAM-1 (immunoglobulin family) | CD11/CD18 (β2) integrins (LFA-1, Mac-1) | Adhesion, arrest, transmigration (neutrophils, monocytes, lymphocytes) |
| VCAM-1 (immunoglobulin family) | VLA-4 (β1) integrin | Adhesion (eosinophils, monocytes, lymphocytes) |
* L-selectin is expressed weakly on neutrophils. It is involved in the binding of circulating T-lymphocytes to the high endothelial venules in lymph nodes and mucosal lymphoid tissues, and subsequent “homing” of lymphocytes to these tissues.
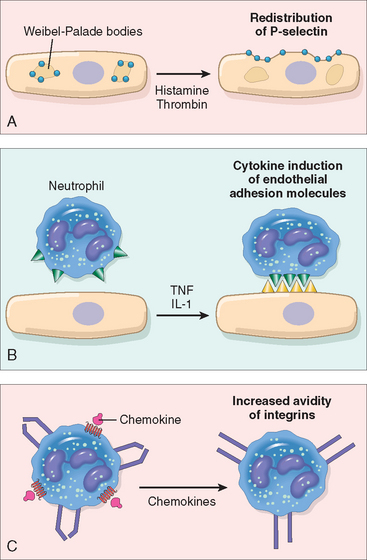
FIGURE 2-5 Regulation of expression of endothelial and leukocyte adhesion molecules. A, Redistribution of P-selectin from intracellular stores to the cell surface. B, Increased surface expression of selectins and ligands for integrins upon cytokine activation of endothelium. C, Increased binding avidity of integrins induced by chemokines. Clustering of integrins contributes to their increased binding avidity (not shown). IL-1, interleukin-1; TNF, tumor necrosis factor.
These weak rolling interactions slow down the leukocytes and give them the opportunity to bind more firmly to the endothelium. Firm adhesion is mediated by a family of heterodimeric leukocyte surface proteins called integrins21 (see Table 2-1). TNF and IL-1 induce endothelial expression of ligands for integrins, mainly vascular cell adhesion molecule 1 (VCAM-1, the ligand for the VLA-4 integrin) and intercellular adhesion molecule-1 (ICAM-1, the ligand for the LFA-1 and Mac-1 integrins). Leukocytes normally express integrins in a low-affinity state. Meanwhile, chemokines that were produced at the site of injury enter the blood vessel, bind to endothelial cell proteoglycans, and are displayed at high concentrations on the endothelial surface. These chemokines bind to and activate the rolling leukocytes. One of the consequences of activation is the conversion of VLA-4 and LFA-1 integrins on the leukocytes to a high-affinity state.22 The combination of cytokine-induced expression of integrin ligands on the endothelium and activation of integrins on the leukocytes results in firm integrin-mediated binding of the leukocytes to the endothelium at the site of inflammation. The leukocytes stop rolling, their cytoskeleton is reorganized, and they spread out on the endothelial surface.
Leukocyte Migration through Endothelium.
The next step in the process of leukocyte recruitment is migration of the leukocytes through the endothelium, called transmigration or diapedesis. Transmigration of leukocytes occurs mainly in post-capillary venules. Chemokines act on the adherent leukocytes and stimulate the cells to migrate through interendothelial spaces toward the chemical concentration gradient, that is, toward the site of injury or infection where the chemokines are being produced.23 Several adhesion molecules present in the intercellular junctions between endothelial cells are involved in the migration of leukocytes. These molecules include a member of the immunoglobulin superfamily called PECAM-1 (platelet endothelial cell adhesion molecule) or CD3124 and several junctional adhesion molecules.25 After traversing the endothelium, leukocytes pierce the basement membrane, probably by secreting collagenases, and enter the extravascular tissue. The cells then migrate toward the chemotactic gradient created by chemokines and accumulate in the extravascular site. In the connective tissue, the leukocytes are able to adhere to the extracellular matrix by virtue of integrins and CD44 binding to matrix proteins. Thus, leukocytes are retained at the site where they are needed.
The most telling proof of the importance of leukocyte adhesion molecules is the existence of genetic deficiencies in these molecules, which result in recurrent bacterial infections as a consequence of impaired leukocyte adhesion and defective inflammation.26 Individuals with leukocyte adhesion deficiency type 1 have a defect in the biosynthesis of the β2 chain shared by the LFA-1 and Mac-1 integrins. Leukocyte adhesion deficiency type 2 is caused by the absence of sialyl-Lewis X, the fucose-containing ligand for E- and P-selectins, as a result of a defect in a fucosyl transferase, the enzyme that attaches fucose moieties to protein backbones.
Chemotaxis of Leukocytes.
After exiting the circulation, leukocytes emigrate in tissues toward the site of injury by a process called chemotaxis, which is defined as locomotion oriented along a chemical gradient. Both exogenous and endogenous substances can act as chemoattractants. The most common exogenous agents are bacterial products, including peptides that possess an N-formylmethionine terminal amino acid, and some lipids. Endogenous chemoattractants include several chemical mediators (described later): (1) cytokines, particularly those of the chemokine family (e.g., IL-8); (2) components of the complement system, particularly C5a; and (3) arachidonic acid (AA) metabolites, mainly leukotriene B4 (LTB4). All these chemotactic agents bind to specific seven-transmembrane G protein–coupled receptors on the surface of leukocytes.27 Signals initiated from these receptors result in activation of second messengers that increase cytosolic calcium and activate small guanosine triphosphatases of the Rac/Rho/cdc42 family as well as numerous kinases. These signals induce polymerization of actin, resulting in increased amounts of polymerized actin at the leading edge of the cell and localization of myosin filaments at the back. The leukocyte moves by extending filopodia that pull the back of the cell in the direction of extension, much as an automobile with front-wheel drive is pulled by the wheels in front (Fig. 2-6). The net result is that leukocytes migrate toward the inflammatory stimulus in the direction of the gradient of locally produced chemoattractants.
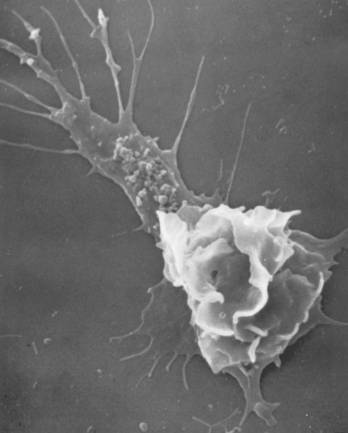
FIGURE 2-6 Scanning electron micrograph of a moving leukocyte in culture showing a filopodium (upper left) and a trailing tail.
(Courtesy of Dr. Morris J. Karnovsky, Harvard Medical School, Boston, MA.)
The nature of the leukocyte infiltrate varies with the age of the inflammatory response and the type of stimulus. In most forms of acute inflammation neutrophils predominate in the inflammatory infiltrate during the first 6 to 24 hours and are replaced by monocytes in 24 to 48 hours (Fig. 2-7). Several reasons account for the early appearance of neutrophils: they are more numerous in the blood, they respond more rapidly to chemokines, and they may attach more firmly to the adhesion molecules that are rapidly induced on endothelial cells, such as P- and E-selectins. After entering tissues, neutrophils are short-lived; they undergo apoptosis and disappear after 24 to 48 hours. Monocytes not only survive longer but may proliferate in the tissues, and thus become the dominant population in chronic inflammatory reactions. There are, however, exceptions to this pattern of cellular infiltration. In certain infections—for example, those produced by Pseudomonas bacteria—the cellular infiltrate is dominated by continuously recruited neutrophils for several days; in viral infections, lymphocytes may be the first cells to arrive; in some hypersensitivity reactions, eosinophils may be the main cell type.
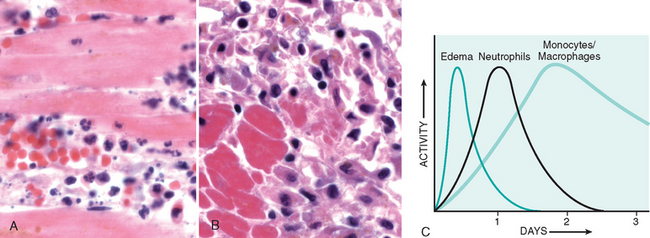
FIGURE 2-7 Nature of leukocyte infiltrates in inflammatory reactions. The photomicrographs are representative of the early (neutrophilic) (A) and later (mononuclear) cellular infiltrates (B) seen in an inflammatory reaction in the myocardium following ischemic necrosis (infarction). The kinetics of edema and cellular infiltration (C) are approximations.
The molecular understanding of leukocyte recruitment and migration has provided a large number of potential therapeutic targets for controlling harmful inflammation.14 Agents that block TNF, one of the major cytokines in leukocyte recruitment, are among the most successful therapeutics ever developed for chronic inflammatory diseases, and antagonists of leukocyte integrins (e.g. VLA-4), selectins, and chemokines are approved for inflammatory diseases or in clinical trials. Predictably, these antagonists not only have the desired effect of controlling the inflammation but can compromise the ability of treated patients to defend themselves against microbes, which, of course, is the physiologic function of the inflammatory response.
Recognition of Microbes and Dead Tissues
Once leukocytes (neutrophils and monocytes) have been recruited to a site of infection or cell death, they must be activated to perform their functions. The responses of leukocytes consist of two sequential sets of events: (1) recognition of the offending agents, which deliver signals that (2) activate the leukocytes to ingest and destroy the offending agents and amplify the inflammatory reaction.
Leukocytes express several receptors that recognize external stimuli and deliver activating signals (Fig. 2-8).
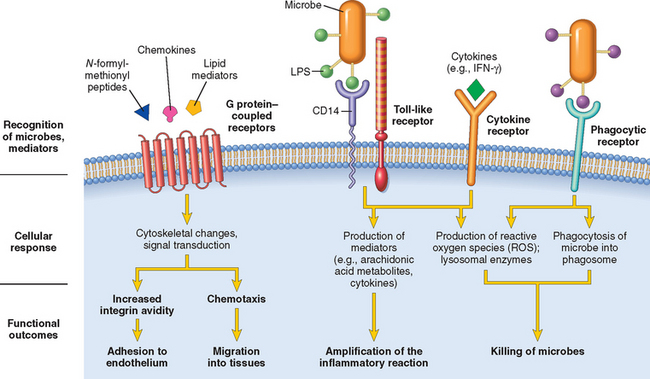
FIGURE 2-8 Leukocyte receptors and responses. Different classes of cell surface receptors of leukocytes recognize different stimuli. The receptors initiate responses that mediate the functions of the leukocytes. Only some receptors are depicted (see text for details). IFN-γ, interferon-γ; LPS, lipopolysaccharide(s).
Removal of the Offending Agents
Recognition of microbes or dead cells by the receptors described above induces several responses in leukocytes that are referred to under the rubric of leukocyte activation (see Fig. 2-8). Activation results from signaling pathways that are triggered in leukocytes, resulting in increases in cytosolic Ca2+ and activation of enzymes such as protein kinase C and phospholipase A2. The functional responses that are most important for destruction of microbes and other offenders are phagocytosis and intracellular killing. Several other responses aid in the defensive functions of inflammation and may contribute to its injurious consequences.
Phagocytosis.
Phagocytosis involves three sequential steps (Fig. 2-9): (1) recognition and attachment of the particle to be ingested by the leukocyte; (2) its engulfment, with subsequent formation of a phagocytic vacuole; and (3) killing or degradation of the ingested material.30
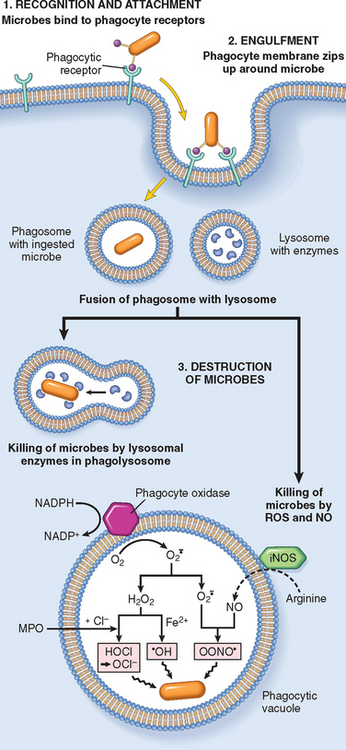
FIGURE 2-9 Phagocytosis and intracellular destruction of microbes. Phagocytosis of a particle (e.g., bacterium) involves binding to receptors on the leukocyte membrane, engulfment, and fusion of lysosomes with phagocytic vacuoles. This is followed by destruction of ingested particles within the phagolysosomes by lysosomal enzymes and by reactive oxygen and nitrogen species. The microbicidal products generated from superoxide ( ) are hypochlorite (HOCl•) and hydroxyl radical (•OH), and from nitric oxide (NO) it is peroxynitrite (OONO•). During phagocytosis, granule contents may be released into extracellular tissues (not shown). MPO, myeloperoxidase; iNOS, inducible NO synthase.
) are hypochlorite (HOCl•) and hydroxyl radical (•OH), and from nitric oxide (NO) it is peroxynitrite (OONO•). During phagocytosis, granule contents may be released into extracellular tissues (not shown). MPO, myeloperoxidase; iNOS, inducible NO synthase.
Mannose receptors, scavenger receptors, and receptors for various opsonins all function to bind and ingest microbes. The macrophage mannose receptor is a lectin that binds terminal mannose and fucose residues of glycoproteins and glycolipids. These sugars are typically part of molecules found on microbial cell walls, whereas mammalian glycoproteins and glycolipids contain terminal sialic acid or N-acetylgalactosamine. Therefore, the mannose receptor recognizes microbes and not host cells. Scavenger receptors were originally defined as molecules that bind and mediate endocytosis of oxidized or acetylated low-density lipoprotein (LDL) particles that can no longer interact with the conventional LDL receptor. Macrophage scavenger receptors bind a variety of microbes in addition to modified LDL particles. Macrophage integrins, notably Mac-1 (CD11b/CD18), may also bind microbes for phagocytosis.
The efficiency of phagocytosis is greatly enhanced when microbes are opsonized by specific proteins (opsonins) for which the phagocytes express high-affinity receptors. As described above, the major opsonins are IgG antibodies, the C3b breakdown product of complement, and certain plasma lectins, notably mannan-binding lectin, all of which are recognized by specific receptors on leukocytes.
Engulfment.
After a particle is bound to phagocyte receptors, extensions of the cytoplasm (pseudopods) flow around it, and the plasma membrane pinches off to form a vesicle (phagosome) that encloses the particle. The phagosome then fuses with a lysosomal granule, resulting in discharge of the granule’s contents into the phagolysosome (see Fig. 2-9). During this process the phagocyte may also release granule contents into the extracellular space.
The process of phagocytosis is complex and involves the integration of many receptor-initiated signals to lead to membrane remodeling and cytoskeletal changes.30 Phagocytosis is dependent on polymerization of actin filaments; it is, therefore, not surprising that the signals that trigger phagocytosis are many of the same that are involved in chemotaxis. (In contrast, fluid-phase pinocytosis and receptor-mediated endocytosis of small particles involve internalization into clathrin-coated pits and vesicles and are not dependent on the actin cytoskeleton.)
Killing and Degradation.
The final step in the elimination of infectious agents and necrotic cells is their killing and degradation within neutrophils and macrophages, which occur most efficiently after activation of the phagocytes. Microbial killing is accomplished largely by reactive oxygen species (ROS, also called reactive oxygen intermediates) and reactive nitrogen species, mainly derived from NO (see Fig. 2-9).31,32 The generation of ROS is due to the rapid assembly and activation of a multicomponent oxidase (NADPH oxidase, also called phagocyte oxidase), which oxidizes NADPH (reduced nicotinamide-adenine dinucleotide phosphate) and, in the process, reduces oxygen to superoxide anion (). In neutrophils, this rapid oxidative reaction is triggered by activating signals and accompanies phagocytosis, and is called the respiratory burst. Phagocyte oxidase is an enzyme complex consisting of at least seven proteins.33 In resting neutrophils, different components of the enzyme are located in the plasma membrane and the cytoplasm. In response to activating stimuli, the cytosolic protein components translocate to the phagosomal membrane, where they assemble and form the functional enzyme complex. Thus, the ROS are produced within the lysosome where the ingested substances are segregated, and the cell’s own organelles are protected from the harmful effects of the ROS. is then converted into hydrogen peroxide (H2O2), mostly by spontaneous dismutation. H2O2 is not able to efficiently kill microbes by itself. However, the azurophilic granules of neutrophils contain the enzyme myeloperoxidase (MPO), which, in the presence of a halide such as Cl−, converts H2O2 to hypochlorite (OCl•, the active ingredient in household bleach). The latter is a potent antimicrobial agent that destroys microbes by halogenation (in which the halide is bound covalently to cellular constituents) or by oxidation of proteins and lipids (lipid peroxidation). The H2O2-MPO-halide system is the most efficient bactericidal system of neutrophils. H2O2 is also converted to hydroxyl radical (•OH), another powerful destructive agent.
NO, produced from arginine by the action of nitric oxide synthase (NOS), also participates in microbial killing.34 NO reacts with superoxide () to generate the highly reactive free radical peroxynitrite (ONOO•). These oxygen- and nitrogen-derived free radicals attack and damage the lipids, proteins, and nucleic acids of microbes as they do with host macromolecules (Chapter 1). Reactive oxygen and nitrogen species have overlapping actions, as shown by the observation that knockout mice lacking either phagocyte oxidase or inducible nitric oxide synthase (iNOS) are only mildly susceptible to infections, but mice lacking both succumb rapidly to disseminated infections by normally harmless commensal bacteria. The roles of ROS and NO as mediators of inflammation are described later in the chapter.
Microbial killing can also occur through the action of other substances in leukocyte granules. Neutrophil granules contain many enzymes, such as elastase, that contribute to microbial killing.35 Other microbicidal granule contents include defensins, cationic arginine-rich granule peptides that are toxic to microbes36; cathelicidins, antimicrobial proteins found in neutrophils and other cells37; lysozyme, which hydrolyzes the muramic acid–N-acetylglucosamine bond, found in the glycopeptide coat of all bacteria; lactoferrin, an iron-binding protein present in specific granules; major basic protein, a cationic protein of eosinophils, which has limited bactericidal activity but is cytotoxic to many parasites; and bactericidal/permeability increasing protein, which binds bacterial endotoxin and is believed to be important in defense against some gram-negative bacteria.
Other Functional Responses of Activated Leukocytes
In addition to eliminating microbes and dead cells, activated leukocytes play several other roles in host defense. Importantly, these cells, especially macrophages, produce a number of growth factors that stimulate the proliferation of endothelial cells and fibroblasts and the synthesis of collagen, and enzymes that remodel connective tissues. These products drive the process of repair after tissue injury (Chapter 3). An emerging concept is that macrophages can be activated to perform different functions—“classically activated” macrophages respond to microbial products and T-cell cytokines such as IFN-γ and have strong microbicidal activity, whereas “alternatively activated” macrophages respond to cytokines such as IL-4 and IL-13 (typically, the products of the TH2 subset of T-cells, see Chapter 6) and are mainly involved in tissue repair and fibrosis (Fig. 2-10).38 Different stimuli activate leukocytes to secrete mediators of inflammation as well as inhibitors of the inflammatory response, and thus serve to both amplify and control the reaction. This may be another distinction between classically and alternatively activated macrophages—the former trigger inflammation and the latter function to limit inflammatory reactions.
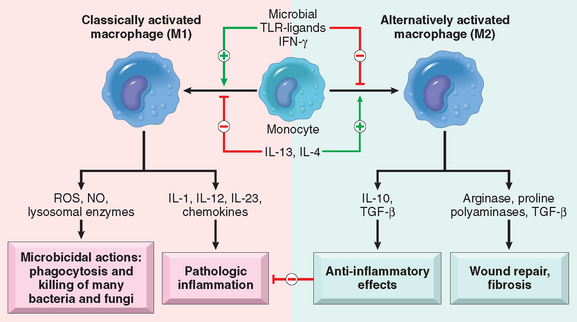
FIGURE 2-10 Subsets of activated macrophages. Different stimuli activate monocytes/macrophages to develop into functionally distinct populations. Classically activated macrophages are induced by microbial products and cytokines, particularly IFN-γ, and are microbicidal and involved in potentially harmful inflammation. Alternatively activated macrophages are induced by other cytokines and in response to helminths (not shown), and are important in tissue repair and the resolution of inflammation (and may play a role in defense against helminthic parasites, also not shown).
Release of Leukocyte Products and Leukocyte-Mediated Tissue Injury
Leukocytes are important causes of injury to normal cells and tissues under several circumstances:
In all these situations, the mechanisms by which leukocytes damage normal tissues are the same as the mechanisms involved in antimicrobial defense, because once the leukocytes are activated, their effector mechanisms do not distinguish between offender and host. During activation and phagocytosis, neutrophils and macrophages release microbicidal and other products not only within the phagolysosome but also into the extracellular space. The most important of these substances are lysosomal enzymes, present in the granules, and reactive oxygen and nitrogen species. These released substances are capable of damaging normal cells and vascular endothelium, and may thus amplify the effects of the initial injurious agent. In fact, if unchecked or inappropriately directed against host tissues, the leukocyte infiltrate itself becomes the offender,39 and indeed leukocyte-dependent tissue injury underlies many acute and chronic human diseases (Table 2-2). This fact becomes evident in the discussion of specific disorders throughout the book.
TABLE 2-2 Clinical Examples of Leukocyte-Induced Injury*
| Disorders | Cells and Molecules Involved in Injury |
|---|---|
| ACUTE | |
| Acute respiratory distress syndrome | Neutrophils |
| Acute transplant rejection | Lymphocytes; antibodies and complement |
| Asthma | Eosinophils; IgE antibodies |
| Glomerulonephritis | Neutrophils, monocytes; antibodies and complement |
| Septic shock | Cytokines |
| Lung abscess | Neutrophils (and bacteria) |
| CHRONIC | |
| Arthritis | Lymphocytes, macrophages; antibodies? |
| Asthma | Eosinophils; IgE antibodies |
| Atherosclerosis | Macrophages; lymphocytes? |
| Chronic transplant rejection | Lymphocytes; cytokines |
| Pulmonary fibrosis | Macrophages; fibroblasts |
* Listed are selected examples of diseases in which the host response plays a significant role in tissue injury, and the principal cells and molecules that cause the injury. These diseases and their pathogenesis will be discussed in detail in relevant chapters.
The contents of lysosomal granules are secreted by leukocytes into the extracellular milieu by several mechanisms.40 Controlled secretion of granule contents is a normal response of activated leukocytes. If phagocytes encounter materials that cannot be easily ingested, such as immune complexes deposited on immovable flat surfaces (e.g., glomerular basement membrane), the inability of the leukocytes to surround and ingest these substances (frustrated phagocytosis) triggers strong activation, and the release of large amounts of lysosomal enzymes into the extracellular environment. Phagocytosis of membrane-damaging substances, such as urate crystals, may injure the membrane of the phagolysosome and also lead to the release of lysosomal granule contents.
Defects in Leukocyte Function
Because leukocytes play a central role in host defense, defects in leukocyte function, both inherited and acquired, lead to increased vulnerability to infections (Table 2-3). Impairments of virtually every phase of leukocyte function have been identified—from adherence to vascular endothelium to microbicidal activity. These include the following:
. The most common variants are an X-linked defect in one of the membrane-bound components (gp91phox) and autosomal recessive defects in the genes encoding two of the cytoplasmic components (p47phox and p67phox).42 The name of this disease comes from the macrophage-rich chronic inflammatory reaction that tries to control the infection when the initial neutrophil defense is inadequate. This often leads to collections of activated macrophages that wall off the microbes, forming aggregates called granulomas (described in more detail later in the chapter).TABLE 2-3 Defects in Leukocyte Functions
| Disease | Defect |
|---|---|
| GENETIC | |
| Leukocyte adhesion deficiency 1 | Defective leukocyte adhesion because of mutations in β chain of CD11/CD18 integrins |
| Leukocyte adhesion deficiency 2 | Defective leukocyte adhesion because of mutations in fucosyl transferase required for synthesis of sialylated oligosaccharide (ligand for selectins) |
| MPO deficiency | Decreased microbial killing because of defective MPO—H2O2 system |
| Chédiak-Higashi syndrome | Decreased leukocyte functions because of mutations affecting protein involved in lysosomal membrane traffic |
| ACQUIRED | |
| Bone marrow suppression: tumors, radiation, and chemotherapy | Production of leukocytes |
| Diabetes, malignancy, sepsis, chronic dialysis | Adhesion and chemotaxis |
| Leukemia, anemia, sepsis, diabetes, malnutrition | Phagocytosis and microbicidal activity |
MPO, myeloperoxidase.
Modified from Gallin JI: Disorders of phagocytic cells. In Gallin JI, et al (eds): Inflammation: Basic Principles and Clinical Correlates, 2nd ed. New York, Raven Press, 1992, pp 860, 861.
Although we have emphasized the role of leukocytes recruited from the circulation in the acute inflammatory response, cells resident in tissues also serve important functions in initiating acute inflammation. The two most important of these cell types are mast cells and tissue macrophages. These “sentinel” cells are stationed in tissues to rapidly recognize potentially injurious stimuli and initiate the host defense reaction. Mast cells react to physical trauma, breakdown products of complement, microbial products, and neuropeptides. These cells release histamine, leukotrienes, enzymes, and many cytokines (including TNF, IL-1, and chemokines), all of which contribute to inflammation. The functions of mast cells are discussed in more detail in Chapter 6. Macrophages recognize microbial products and secrete most of the cytokines important in acute inflammation. We will return to a discussion of the role of macrophages in inflammation later in the chapter.
TERMINATION OF THE ACUTE INFLAMMATORY RESPONSE
It is predictable that such a powerful system of host defense, with its inherent capacity to cause tissue damage, needs tight controls to minimize the damage. In part, inflammation declines simply because the mediators of inflammation are produced in rapid bursts, only as long as the stimulus persists, have short half-lives, and are degraded after their release. Neutrophils also have short half-lives in tissues and die by apoptosis within a few hours after leaving the blood. In addition, as inflammation develops the process also triggers a variety of stop signals that serve to actively terminate the reaction.43,44 These active termination mechanisms include a switch in the type of arachidonic acid metabolite produced, from pro-inflammatory leukotrienes to anti-inflammatory lipoxins (described below); the liberation of anti-inflammatory cytokines, including transforming growth factor-β (TGF-β) and IL-10, from macrophages and other cells; the production of anti-inflammatory lipid mediators, called resolvins and protectins, derived from polyunsaturated fatty acids45; and neural impulses (cholinergic discharge) that inhibit the production of TNF in macrophages.46
Mediators of Inflammation
Having described the sequence of events in acute inflammation, we can now turn to a discussion of the chemical mediators that are responsible for these reactions. Many mediators have been identified, and how they function in a coordinated manner is still not fully understood. The sources of the principal mediators and their roles in the inflammatory reaction are summarized in Table 2-4. We start our discussion of the mediators of inflammation by reviewing some of their shared properties and the general principles of their production and actions.
TABLE 2-4 The Actions of the Principal Mediators of Inflammation
| Mediator | Principal Sources | Actions |
|---|---|---|
| CELL-DERIVED | ||
| Histamine | Mast cells, basophils, platelets | Vasodilation, increased vascular permeability, endothelial activation |
| Serotonin | Platelets | Vasodilation, increased vascular permeability |
| Prostaglandins | Mast cells, leukocytes | Vasodilation, pain, fever |
| Leukotrienes | Mast cells, leukocytes | Increased vascular permeability, chemotaxis, leukocyte adhesion and activation |
| Platelet-activating factor | Leukocytes, mast cells | Vasodilation, increased vascular permeability, leukocyte adhesion, chemotaxis, degranulation, oxidative burst |
| Reactive oxygen species | Leukocytes | Killing of microbes, tissue damage |
| Nitric oxide | Endothelium, macrophages | Vascular smooth muscle relaxation, killing of microbes |
| Cytokines (TNF, IL-1) | Macrophages, endothelial cells, mast cells | Local endothelial activation (expression of adhesion molecules), fever/pain/anorexia/hypotension, decreased vascular resistance (shock) |
| Chemokines | Leukocytes, activated macrophages | Chemotaxis, leukocyte activation |
| PLASMA PROTEIN–DERIVED | ||
| Complement products (C5a, C3a, C4a) | Plasma (produced in liver) | |
| Kinins | Plasma (produced in liver) | |
| Proteases activated during coagulation | Plasma (produced in liver) | |
IL-1, interleukin-1; MAC, membrane attack complex; TNF, tumor necrosis factor.
We now discuss some of the more important mediators of acute inflammation, starting with the cell-derived mediators and moving on to those derived from plasma proteins.
CELL-DERIVED MEDIATORS
Vasoactive Amines: Histamine and Serotonin
The two major vasoactive amines, so named because they have important actions on blood vessels, are histamine and serotonin. They are stored as preformed molecules in cells and are therefore among the first mediators to be released during inflammation. The richest sources of histamine are the mast cells that are normally present in the connective tissue adjacent to blood vessels. It is also found in blood basophils and platelets. Histamine is present in mast cell granules and is released by mast cell degranulation in response to a variety of stimuli, including (1) physical injury such as trauma, cold, or heat; (2) binding of antibodies to mast cells, which underlies allergic reactions (Chapter 6); (3) fragments of complement called anaphylatoxins (C3a and C5a); (4) histamine-releasing proteins derived from leukocytes; (5) neuropeptides (e.g., substance P); and (6) cytokines (IL-1, IL-8).
Histamine causes dilation of arterioles and increases the permeability of venules. It is considered to be the principal mediator of the immediate transient phase of increased vascular permeability, producing interendothelial gaps in venules, as we have seen. Its vasoactive effects are mediated mainly via binding to H1 receptors on microvascular endothelial cells.47
Serotonin (5-hydroxytryptamine) is a preformed vasoactive mediator with actions similar to those of histamine. It is present in platelets and certain neuroendocrine cells, e.g. in the gastrointestinal tract, and in mast cells in rodents but not humans. Release of serotonin (and histamine) from platelets is stimulated when platelets aggregate after contact with collagen, thrombin, adenosine diphosphate, and antigenantibody complexes. Thus, the platelet release reaction, which is a key component of coagulation, also results in increased vascular permeability. This is one of several links between clotting and inflammation.
Arachidonic Acid (AA) Metabolites: Prostaglandins, Leukotrienes, and Lipoxins
When cells are activated by diverse stimuli, such as microbial products and various mediators of inflammation, membrane AA is rapidly converted by the actions of enzymes to produce prostaglandins and leukotrienes. These biologically active lipid mediators serve as intracellular or extracellular signals to affect a variety of biologic processes, including inflammation and hemostasis.48-50
AA is a 20-carbon polyunsaturated fatty acid (5,8,11,14-eicosatetraenoic acid) that is derived from dietary sources or by conversion from the essential fatty acid linoleic acid. It does not occur free in the cell but is normally esterified in membrane phospholipids. Mechanical, chemical, and physical stimuli or other mediators (e.g., C5a) release AA from membrane phospholipids through the action of cellular phospholipases, mainly phospholipase A2. The biochemical signals involved in the activation of phospholipase A2 include an increase in cytoplasmic Ca2+ and activation of various kinases in response to external stimuli.51 AA-derived mediators, also called eicosanoids, are synthesized by two major classes of enzymes: cyclooxygenases (which generate prostaglandins) and lipoxygenases (which produce leukotrienes and lipoxins) (Fig. 2-11). Eicosanoids bind to G protein–coupled receptors on many cell types and can mediate virtually every step of inflammation (Table 2-5).
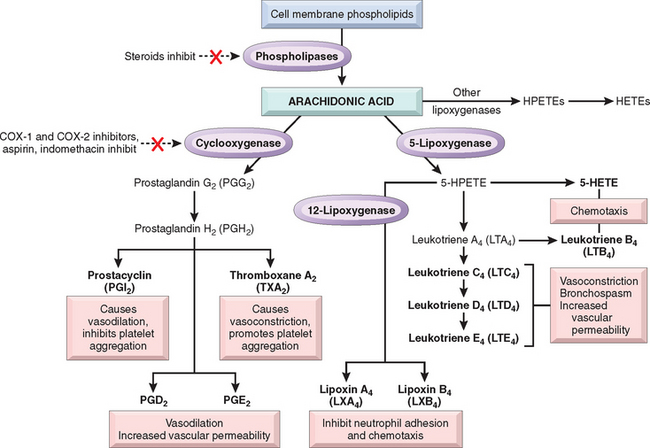
FIGURE 2-11 Generation of arachidonic acid metabolites and their roles in inflammation. The molecular targets of action of some anti-inflammatory drugs are indicated by a red X. Not shown are agents that inhibit leukotriene production by inhibition of 5-lipoxygenase (e.g., Zileuton) or block leukotriene receptors (e.g., Monteleukast). COX, cyclooxygenase; HETE, hydroxyeicosatetraenoic acid; HPETE, hydroperoxyeicosatetraenoic acid.
TABLE 2-5 Principal Inflammatory Actions of Arachidonic Acid Metabolites (Eicosanoids)
| Action | Eicosanoid |
|---|---|
| Vasodilation | PGI2 (prostacyclin), PGE1, PGE2, PGD2 |
| Vasoconstriction | Thromboxane A2, leukotrienes C4, D4, E4 |
| Increased vascular permeability | Leukotrienes C4, D4, E4 |
| Chemotaxis, leukocyte adhesion | Leukotriene B4, HETE |
HETE, hydroxyeicosatetraenoic acid; PGI2, etc., prostaglandin I2, etc.
In addition to their local effects, the prostaglandins are involved in the pathogenesis of pain and fever in inflammation. PGE2 is hyperalgesic and makes the skin hypersensitive to painful stimuli, such as intradermal injection of suboptimal concentrations of histamine and bradykinin. It is involved in cytokine-induced fever during infections (described later).
Many anti-inflammatory drugs work by inhibiting the synthesis of eicosanoids:
Platelet-Activating Factor (PAF)
PAF is another phospholipid-derived mediator.54 Its name comes from its discovery as a factor that causes platelet aggregation, but it is now known to have multiple inflammatory effects. A variety of cell types, including platelets themselves, basophils, mast cells, neutrophils, macrophages, and endothelial cells, can elaborate PAF, in both secreted and cell-bound forms. In addition to platelet aggregation, PAF causes vasoconstriction and bronchoconstriction, and at extremely low concentrations it induces vasodilation and increased venular permeability with a potency 100 to 10,000 times greater than that of histamine. PAF also causes increased leukocyte adhesion to endothelium (by enhancing integrin-mediated leukocyte binding), chemotaxis, degranulation, and the oxidative burst. Thus, PAF can elicit most of the vascular and cellular reactions of inflammation. PAF also boosts the synthesis of other mediators, particularly eicosanoids, by leukocytes and other cells. A role for PAF in vivo is supported by the ability of synthetic PAF receptor antagonists to inhibit inflammation in some experimental models.
Reactive Oxygen Species
Oxygen-derived free radicals may be released extracellularly from leukocytes after exposure to microbes, chemokines, and immune complexes, or following a phagocytic challenge.55 Their production is dependent, as we have seen, on the activation of the NADPH oxidase system. Superoxide anion (), hydrogen peroxide (H2O2), and hydroxyl radical (•OH) are the major species produced within cells, and O can combine with NO to form reactive nitrogen species. Extracellular release of low levels of these potent mediators can increase the expression of chemokines (e.g., IL-8), cytokines, and endothelial leukocyte adhesion molecules, amplifying the inflammatory response. As mentioned earlier, the physiologic function of these ROS in leukocytes is to destroy phagocytosed microbes, but release of these potent mediators can be damaging to the host (Chapter 1). They are implicated in the following responses in inflammation:
Serum, tissue fluids, and host cells possess antioxidant mechanisms that protect against these potentially harmful oxygen-derived radicals. These antioxidants were discussed in Chapter 1; they include (1) the enzyme superoxide dismutase, which is found in or can be activated in a variety of cell types; (2) the enzyme catalase, which detoxifies H2O2; (3) glutathione peroxidase, another powerful H2O2 detoxifier; (4) the copper-containing serum protein ceruloplasmin; and (5) the iron-free fraction of serum transferrin. Thus, the influence of oxygen-derived free radicals in any given inflammatory reaction depends on the balance between production and inactivation of these metabolites by cells and tissues.
Nitric Oxide (NO)
NO was discovered as a factor released from endothelial cells that caused vasodilation and was therefore called endothelium-derived relaxing factor. NO is a soluble gas that is produced not only by endothelial cells but also by macrophages and some neurons in the brain. It acts in a paracrine manner on target cells through induction of cyclic guanosine monophosphate, which, in turn, initiates a series of intracellular events leading to a response, such as the relaxation of vascular smooth muscle cells. Because the in vivo half-life of NO is only seconds, the gas acts only on cells in close proximity to where it is produced.
NO is synthesized from l-arginine by the enzyme nitric oxide synthase (NOS). There are three different types of NOS: endothelial (eNOS), neuronal (nNOS), and inducible (iNOS) (Fig. 2-12). eNOS and nNOS are constitutively expressed at low levels and can be activated rapidly by an increase in cytoplasmic Ca2+. iNOS, in contrast, is induced when macrophages and other cells are activated by cytokines (e.g., TNF, IFN-γ) or microbial products.
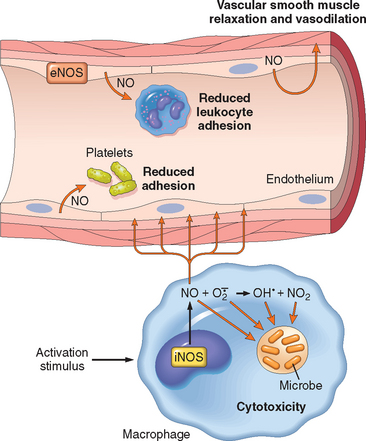
FIGURE 2-12 Functions of nitric oxide (NO) in blood vessels and macrophages. NO is produced by two NO synthase (NOS) enzymes. It causes vasodilation, and NO-derived free radicals are toxic to microbial and mammalian cells.
NO has dual actions in inflammation: it relaxes vascular smooth muscle and promotes vasodilation, thus contributing to the vascular reaction, but it is also an inhibitor of the cellular component of inflammatory responses.56,57 NO reduces platelet aggregation and adhesion (Chapter 4), inhibits several features of mast cell–induced inflammation, and inhibits leukocyte recruitment. Because of these inhibitory actions, production of NO is thought to be an endogenous mechanism for controlling inflammatory responses.
NO and its derivatives are microbicidal, and thus NO is a mediator of host defense against infection (discussed earlier). High levels of iNOS-induced NO are produced by leukocytes, mainly neutrophils and macrophages, in response to microbes.
Cytokines and Chemokines
Cytokines are proteins produced by many cell types (principally activated lymphocytes and macrophages, but also endothelial, epithelial, and connective tissue cells) that modulate the functions of other cell types. Long known to be involved in cellular immune responses, these products have additional effects that play important roles in both acute and chronic inflammation. Their general properties and functions are discussed in Chapter 6. Here we review the pro-perties of cytokines that are involved in acute inflammation (Table 2-6).
TABLE 2-6 Cytokines in Inflammation
| Cytokine | Principal Sources | Principal Actions in Inflammation |
|---|---|---|
| IN ACUTE INFLAMMATION | ||
| TNF | Macrophages, mast cells, T lymphocytes | Stimulates expression of endothelial adhesion molecules and secretion of other cytokines; systemic effects |
| IL-1 | Macrophages, endothelial cells, some epithelial cells | Similar to TNF; greater role in fever |
| IL-6 | Macrophages, other cells | Systemic effects (acute-phase response) |
| Chemokines | Macrophages, endothelial cells, T lymphocytes, mast cells, other cell types | Recruitment of leukocytes to sites of inflammation; migration of cells to normal tissues |
| IN CHRONIC INFLAMMATION | ||
| IL-12 | Dendritic cells, macrophages | Increased production of IFN-γ |
| IFN-γ | T lymphocytes, NK cells | Activation of macrophages (increased ability to kill microbes and tumor cells) |
| IL-17 | T lymphocytes | Recruitment of neutrophils and monocytes |
IFN-γ, interferon-γ; IL-1, interleukin-1; NK cells, natural killer cells; TNF, tumor necrosis factor.
The most important cytokines involved in inflammatory reactions are listed. Many other cytokines may play lesser roles in inflammation. There is also considerable overlap between the cytokines involved in acute and chronic inflammation. Specifically, all the cytokines listed under acute inflammation may also contribute to chronic inflammatory reactions.
Tumor Necrosis Factor and Interleukin-1.
TNF and IL-1 are two of the major cytokines that mediate inflammation. They are produced mainly by activated macrophages. The secretion of TNF and IL-1 can be stimulated by endotoxin and other microbial products, immune complexes, physical injury, and a variety of inflammatory stimuli. Their most important actions in inflammation are their effects on endothelium, leukocytes, and fibroblasts, and induction of systemic acute-phase reactions (Fig. 2-13). In endothelium they induce a spectrum of changes referred to as endothelial activation.58 In particular, they induce the expression of endothelial adhesion molecules; synthesis of chemical mediators, including other cytokines, chemokines, growth factors, eicosanoids, and NO; production of enzymes associated with matrix remodeling; and increases in the surface thrombogenicity of the endothelium.59 TNF also augments responses of neutrophils to other stimuli such as bacterial endotoxin.
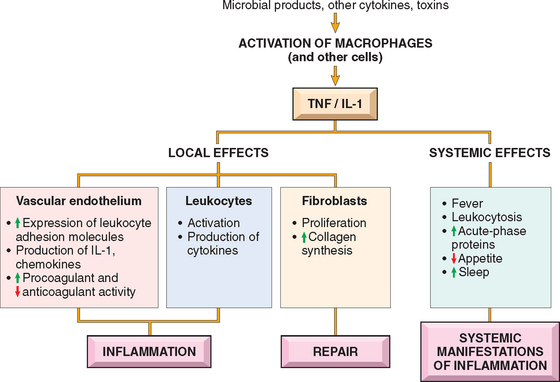
FIGURE 2-13 Principal local and systemic actions of tumor necrosis factor (TNF) and interleukin-1 (IL-1).
The production of IL-1 is controlled by a multi-protein cellular complex, sometimes called the “inflammasome,” that responds to stimuli from microbes and dead cells. This complex activates proteases that are members of the caspase family, which function to cleave the newly synthesized inactive precursor of IL-1 into the biologically active cytokine. Mutations in genes encoding members of this protein complex are the cause of inherited autoinflammatory syndromes, the best known of which is familial Mediterranean fever.60 The mutant proteins either constitutively activate the inflammatory caspases or interfere with the negative regulation of this enzymatic process. The net result is unregulated IL-1 production.61,62 Affected patients present with fever and other systemic manifestations of inflammation without overt provocation. Over time, some of these patients also develop amyloidosis, a disease of extracellular protein deposition that is often the result of persistent inflammation (Chapter 6). IL-1 antagonists are effective for treating these disorders, an excellent example of molecularly targeted rational therapy. The same inflammasome complex may be activated by urate crystals in the disease called gout, and the inflammation in this disease also seems to be, at least partly, mediated by IL-1 (Chapter 26).
IL-1 and TNF (as well as IL-6) induce the systemic acute-phase responses associated with infection or injury (described later in the chapter). TNF also regulates energy balance by promoting lipid and protein mobilization and by suppressing appetite. Therefore, sustained production of TNF contributes to cachexia, a pathologic state characterized by weight loss and anorexia that accompanies some chronic infections and neoplastic diseases (Chapter 9).
Chemokines.
Chemokines are a family of small (8 to 10 kD) proteins that act primarily as chemoattractants for specific types of leukocytes.63 About 40 different chemokines and 20 different receptors for chemokines have been identified. They are classified into four major groups, according to the arrangement of the conserved cysteine (C) residues in the mature proteins64,65:
Chemokines mediate their activities by binding to seven-transmembrane G protein–coupled receptors. These receptors (called CXCR or CCR, for C-X-C or C-C chemokine receptors) usually exhibit overlapping ligand specificities, and leukocytes generally express more than one receptor type. As discussed in Chapter 6, certain chemokine receptors (CXCR-4, CCR-5) act as coreceptors for a viral envelope glycoprotein of human immunodeficiency virus 1 and are thus involved in binding and entry of the virus into cells.
Chemokines have two main functions: they stimulate leukocyte recruitment in inflammation and control the normal migration of cells through various tissues.20,65 Some chemokines are produced transiently in response to inflammatory stimuli and promote the recruitment of leukocytes to the sites of inflammation. Other chemokines are produced constitutively in tissues and function to organize different cell types in different anatomic regions of the tissues. In both situations, chemokines may be displayed at high concentrations attached to proteoglycans on the surface of endothelial cells and in the extracellular matrix.
Other Cytokines in Acute Inflammation.
The list of cytokines implicated in inflammation is huge and constantly growing.66 Two that have received considerable recent interest are: IL-6, made by macrophages and other cells, which is involved in local and systemic reactions67; and IL-17, produced mainly by T lymphocytes, which promotes neutrophil recruitment.68 Antagonists against both are in clinical trials for inflammatory diseases. Cytokines also play central roles in chronic inflammation; these are described later in the chapter.
Lysosomal Constituents of Leukocytes
Neutrophils and monocytes contain lysosomal granules, which, when released, may contribute to the inflammatory response. Neutrophils have two main types of granules. The smaller specific (or secondary) granules contain lysozyme, collagenase, gelatinase, lactoferrin, plasminogen activator, histaminase, and alkaline phosphatase. The larger azurophil (or primary) granules contain myeloperoxidase, bactericidal factors (lysozyme, defensins), acid hydrolases, and a variety of neutral proteases (elastase, cathepsin G, nonspecific collagenases, proteinase 3).40 Both types of granules can fuse with phagocytic vacuoles containing engulfed material, or the granule contents can be released into the extracellular space.
Different granule enzymes serve different functions. Acid proteases degrade bacteria and debris within the phagolysosomes, in which an acid pH is readily reached. Neutral proteases are capable of degrading various extracellular components, such as collagen, basement membrane, fibrin, elastin, and cartilage, resulting in the tissue destruction that accompanies inflammatory processes. Neutral proteases can also cleave C3 and C5 complement proteins directly, releasing anaphylatoxins, and release a kinin-like peptide from kininogen. Neutrophil elastase has been shown to degrade virulence factors of bacteria and thus combat bacterial infections. Monocytes and macrophages also contain acid hydrolases, collagenase, elastase, phospholipase, and plasminogen activator. These may be particularly active in chronic inflammatory reactions.
Because of the destructive effects of lysosomal enzymes, the initial leukocytic infiltration, if unchecked, can potentiate further inflammation and tissue damage. These harmful proteases, however, are held in check by a system of antiproteases in the serum and tissue fluids. Foremost among these is α1-antitrypsin, which is the major inhibitor of neutrophil elastase. A deficiency of these inhibitors may lead to sustained action of leukocyte proteases, as is the case in patients with α1-antitrypsin deficiency (Chapter 15). α2-Macroglobulin is another antiprotease found in serum and various secretions.
Neuropeptides
Neuropeptides are secreted by sensory nerves and various leukocytes, and play a role in the initiation and propagation of an inflammatory response. The small peptides, such as substance P and neurokinin A, belong to a family of tachykinin neuropeptides produced in the central and peripheral nervous systems.69 Nerve fibers containing substance P are prominent in the lung and gastrointestinal tract. Substance P has many biologic functions, including the transmission of pain signals, regulation of blood pressure, stimulation of secretion by endocrine cells, and increasing vascular permeability. Sensory neurons can also produce other pro-inflammatory molecules, such as calcitonin-related gene product, which are thought to link the sensing of painful stimuli to the development of protective host responses.70
PLASMA PROTEIN–DERIVED MEDIATORS
A variety of phenomena in the inflammatory response are mediated by plasma proteins that belong to three interrelated systems: the complement, kinin, and clotting systems.
Complement System
The complement system consists of more than 20 proteins, some of which are numbered C1 through C9. This system functions in both innate and adaptive immunity for defense against microbial pathogens.71-73 In the process of complement activation several cleavage products of complement proteins are elaborated that cause increased vascular permeability, chemotaxis, and opsonization. The activation and functions of the complement system are outlined in Figure 2-14.
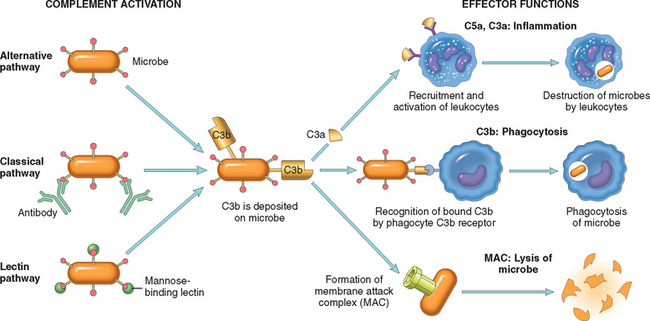
FIGURE 2-14 The activation and functions of the complement system. Activation of complement by different pathways leads to cleavage of C3. The functions of the complement system are mediated by breakdown products of C3 and other complement proteins, and by the membrane attack complex (MAC).
Complement proteins are present in inactive forms in the plasma, and many of them are activated to become proteolytic enzymes that degrade other complement proteins, thus forming an enzymatic cascade capable of tremendous amplification. The critical step in complement activation is the proteolysis of the third (and most abundant) component, C3. Cleavage of C3 can occur by one of three pathways: the classical pathway, which is triggered by fixation of C1 to antibody (IgM or IgG) that has combined with antigen; the alternative pathway, which can be triggered by microbial surface molecules (e.g., endotoxin, or LPS), complex polysaccharides, cobra venom, and other substances, in the absence of antibody; and the lectin pathway, in which plasma mannose-binding lectin binds to carbohydrates on microbes and directly activates C1. Whichever pathway is involved in the early steps of complement activation, they all lead to the formation of an active enzyme called the C3 convertase, which splits C3 into two functionally distinct fragments, C3a and C3b. C3a is released, and C3b becomes covalently attached to the cell or molecule where complement is being activated. More C3b then binds to the previously generated fragments to form C5 convertase, which cleaves C5 to release C5a and leave C5b attached to the cell surface. C5b binds the late components (C6–C9), culminating in the formation of the membrane attack complex (MAC, composed of multiple C9 molecules).
The biologic functions of the complement system fall into three general categories (see Fig. 2-14):
Among the complement components, C3a and C5a are the most important inflammatory mediators. In addition to the mechanisms already discussed, C3 and C5 can be cleaved by several proteolytic enzymes present within the inflammatory exudate. These include plasmin and lysosomal enzymes released from neutrophils (discussed earlier). Thus, the chemotactic actions of complement and the complement-activating effects of neutrophils can initiate a self-perpetuating cycle of neutrophil recruitment.
The activation of complement is tightly controlled by cell-associated and circulating regulatory proteins. Different regulatory proteins inhibit the production of active complement fragments or remove fragments that deposit on cells. These regulators are expressed on normal host cells and are thus designed to prevent healthy tissues from being injured at sites of complement activation. Regulatory proteins can be overwhelmed when large amounts of complement are deposited on host cells and tissues, as happens in autoimmune diseases, in which individuals produce complement-fixing antibodies against their own tissue antigens (Chapter 6).
Coagulation and Kinin Systems
Inflammation and blood clotting are often intertwined, with each promoting the other.74 The clotting system is divided into two pathways that converge, culminating in the activation of thrombin and the formation of fibrin (Fig. 2-15) (Chapter 4). The intrinsic clotting pathway is a series of plasma proteins that can be activated by Hageman factor (factor XII), a protein synthesized by the liver that circulates in an inactive form. Factor XII is activated upon contact with negatively charged surfaces, for instance when vascular permeability increases and plasma proteins leak into the extravascular space and come into contact with collagen, or when it comes into contact with basement membranes exposed as a result of endothelial damage. Factor XII then undergoes a conformational change (becoming factor XIIa), exposing an active serine center that can subsequently cleave protein substrates and activate a variety of mediator systems (see later). Inflammation increases the production of several coagulation factors, makes the endothelial surface pro-thrombogenic, and inhibits anticoagulation mechanisms, thus promoting clotting. Conversely, thrombin, a product of clotting, promotes inflammation by engaging receptors that are called protease-activated receptors (PARs) because they bind multiple trypsin-like serine proteases in addition to thrombin.75 These receptors are seven-transmembrane G protein–coupled receptors that are expressed on platelets, endothelial and smooth muscle cells, and many other cell types. Engagement of the so-called type 1 receptor (PAR-1) by proteases, particularly thrombin, triggers several responses that induce inflammation. They include mobilization of P-selectin; production of chemokines and other cytokines; expression of endothelial adhesion molecules for leukocyte integrins; induction of cyclooxygenase-2 and production of prostaglandins; production of PAF and NO; and changes in endothelial shape.75 As we have seen, these responses promote the recruitment of leukocytes and many other reactions of inflammation. Because coagulation and inflammation can initiate a vicious cycle of amplification, interfering with clotting is a potential therapeutic strategy for the systemic inflammatory disease seen with severe, disseminated bacterial infections. This is the rationale for treating this disorder with the anticoagulant, activated protein C, which may benefit a subset of the patients (Chapter 4).76
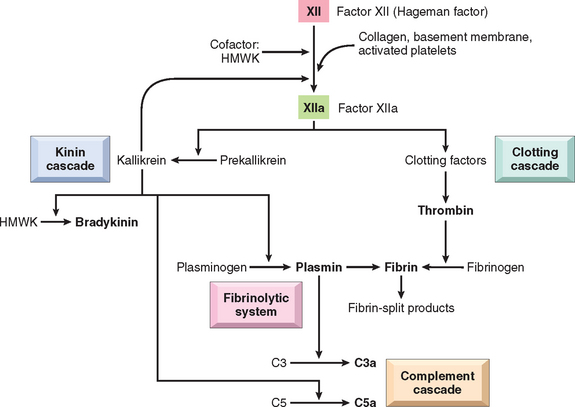
FIGURE 2-15 Interrelationships between the four plasma mediator systems triggered by activation of factor XII (Hageman factor). Note that thrombin induces inflammation by binding to protease-activated receptors (principally PAR-1) on platelets, endothelium, smooth muscle cells, and other cells. HMWK, high molecular weight kininogen.
Kinins are vasoactive peptides derived from plasma proteins, called kininogens, by the action of specific proteases called kallikreins. The kinin and coagulation systems are also intimately connected. The active form of factor XII, factor XIIa, converts plasma prekallikrein into an active proteolytic form, the enzyme kallikrein, which cleaves a plasma glycoprotein precursor, high-molecular-weight kininogen, to produce bradykinin (see Fig. 2-15).77 Bradykinin increases vascular permeability and causes contraction of smooth muscle, dilation of blood vessels, and pain when injected into the skin. These effects are similar to those of histamine. The action of bradykinin is short-lived, because it is quickly inactivated by an enzyme called kininase. Any remaining kinin is inactivated during passage of plasma through the lung by angiotensin-converting enzyme. Kallikrein itself is a potent activator of Hageman factor, allowing for autocatalytic amplification of the initial stimulus. Kallikrein has chemotactic activity, and it also directly converts C5 to the chemoattractant product C5a.
At the same time that factor XIIa is inducing fibrin clot formation, it activates the fibrinolytic system. This cascade counterbalances clotting by cleaving fibrin, thereby solubilizing the clot. Kallikrein, as well as plasminogen activator (released from endothelium, leukocytes, and other tissues), cleaves plasminogen, a plasma protein that binds to the evolving fibrin clot to generate plasmin, a multifunctional protease. The fibrinolytic system contributes to the vascular phenomena of inflammation in several ways. Although the primary function of plasmin is to lyse fibrin clots, during inflammation it also cleaves the complement protein C3 to produce C3 fragments, and it degrades fibrin to form fibrin split products, which may have permeability-inducing properties. Plasmin can also activate Hageman factor, which can trigger multiple cascades (see Fig. 2-15), amplifying the response.
From this discussion of the plasma proteases activated by the complement, kinin, and clotting systems, a few general conclusions can be drawn:
When Lewis discovered the role of histamine in inflammation, one mediator was thought to be enough. Now, we are wallowing in them! Yet, from this large compendium, it is likely that a few mediators are most important for the reactions of acute inflammation in vivo, and these are summarized in Table 2-7. The redundancy of the mediators and their actions ensures that this protective response remains robust and is not easy to perturb.
TABLE 2-7 Role of Mediators in Different Reactions of Inflammation
| Role in Inflammation | Mediators |
|---|---|
| Vasodilation | Prostaglandins |
| Nitric oxide | |
| Histamine | |
| Increased vascular permeability | Histamine and serotonin |
| C3a and C5a (by liberating vasoactive amines from mast cells, other cells) | |
| Bradykinin | |
| Leukotrienes C4, D4, E4 | |
| PAF | |
| Substance P | |
| Chemotaxis, leukocyte recruitment and activation | TNF, IL-1 |
| Chemokines | |
| C3a, C5a | |
| Leukotriene B4 | |
| (Bacterial products, e.g., N-formyl methyl peptides) | |
| Fever | IL-1, TNF |
| Prostaglandins | |
| Pain | Prostaglandins |
| Bradykinin | |
| Tissue damage | Lysosomal enzymes of leukocytes |
| Reactive oxygen species | |
| Nitric oxide |
IL-1, interleukin-1; PAF, platelet-activating factor; TNF, tumor necrosis factor.
Outcomes of Acute Inflammation
Although, as might be expected, many variables may modify the basic process of inflammation, including the nature and intensity of the injury, the site and tissue affected, and the responsiveness of the host, all acute inflammatory reactions may have one of three outcomes (Fig. 2-16):

Morphologic Patterns of Acute Inflammation
The morphologic hallmarks of all acute inflammatory reactions are dilation of small blood vessels, slowing of blood flow, and accumulation of leukocytes and fluid in the extravascular tissue (Fig. 2-17). However, special morphologic patterns are often superimposed on these general features, depending on the severity of the reaction, its specific cause, and the particular tissue and site involved. The importance of recognizing the gross and microscopic patterns is that they often provide valuable clues about the underlying cause.
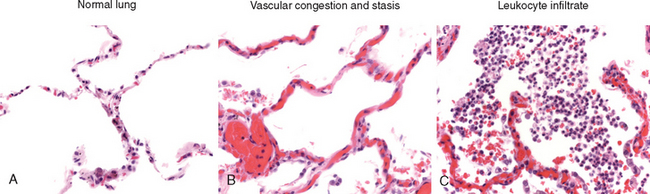
FIGURE 2-17 The characteristic histopathology of acute inflammation. A, Normal lung shows thin (virtually invisible) blood vessels in the alveolar walls and no cells in the alveoli. B, The vascular component of acute inflammation is manifested by congested blood vessels (packed with erythrocytes), resulting from stasis. C, The cellular component of the response is manifested by large numbers of leukocytes (neutrophils) in the alveoli.
SEROUS INFLAMMATION
Serous inflammation is marked by the outpouring of a thin fluid that may be derived from the plasma or from the secretions of mesothelial cells lining the peritoneal, pleural, and pericardial cavities. Accumulation of fluid in these cavities is called an effusion. The skin blister resulting from a burn or viral infection represents a large accumulation of serous fluid, either within or immediately beneath the epidermis of the skin (Fig. 2-18).
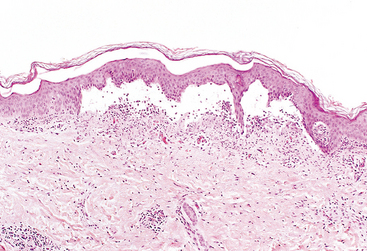
FIBRINOUS INFLAMMATION
With greater increase in vascular permeability, large molecules such as fibrinogen pass the vascular barrier, and fibrin is formed and deposited in the extracellular space. A fibrinous exudate develops when the vascular leaks are large or there is a local procoagulant stimulus (e.g., cancer cells). A fibrinous exudate is characteristic of inflammation in the lining of body cavities, such as the meninges, pericardium (Fig. 2-19A) and pleura. Histologically, fibrin appears as an eosinophilic meshwork of threads or sometimes as an amorphous coagulum (Fig. 2-19B). Fibrinous exudates may be removed by fibrinolysis and clearing of other debris by macrophages. If the fibrin is not removed, over time it may stimulate the ingrowth of fibroblasts and blood vessels and thus lead to scarring. Conversion of the fibrinous exudate to scar tissue (organization) within the pericardial sac leads to opaque fibrous thickening of the pericardium and epicardium in the area of exudation and, if the fibrosis is extensive, obliteration of the pericardial space.
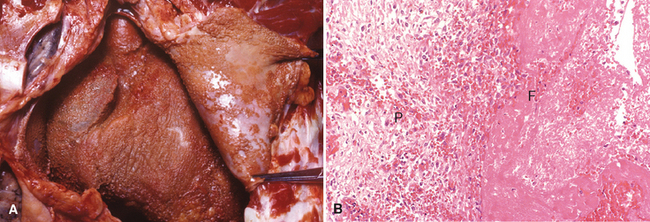
SUPPURATIVE OR PURULENT INFLAMMATION; ABSCESS
This type of inflammation is characterized by the production of large amounts of pus or purulent exudate consisting of neutrophils, liquefactive necrosis, and edema fluid. Certain bacteria (e.g., staphylococci) produce this localized suppuration and are therefore referred to as pyogenic (pus-producing) bacteria. A common example of an acute suppurative inflammation is acute appendicitis. Abscesses are localized collections of purulent inflammatory tissue caused by suppuration buried in a tissue, an organ, or a confined space. They are produced by deep seeding of pyogenic bacteria into a tissue (Fig. 2-20). Abscesses have a central region that appears as a mass of necrotic leukocytes and tissue cells. There is usually a zone of preserved neutrophils around this necrotic focus, and outside this region vascular dilation and parenchymal and fibroblastic proliferation occur, indicating chronic inflammation and repair. In time the abscess may become walled off and ultimately replaced by connective tissue.
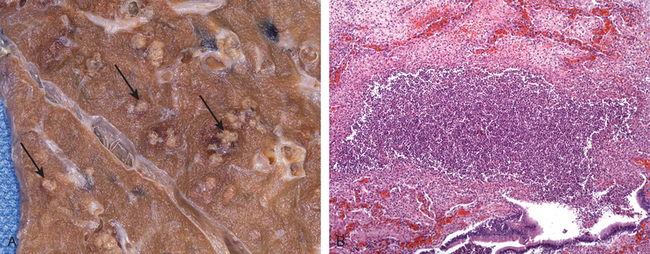
ULCERS
An ulcer is a local defect, or excavation, of the surface of an organ or tissue that is produced by the sloughing (shedding) of inflamed necrotic tissue (Fig. 2-21). Ulceration can occur only when tissue necrosis and resultant inflammation exist on or near a surface. It is most commonly encountered in (1) the mucosa of the mouth, stomach, intestines, or genitourinary tract; and (2) the skin and subcutaneous tissue of the lower extremities in older persons who have circulatory disturbances that predispose to extensive ischemic necrosis.
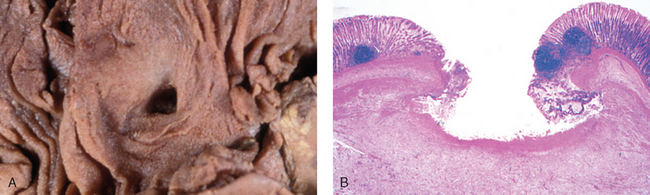
FIGURE 2-21 The morphology of an ulcer. A, A chronic duodenal ulcer. B, Low-power cross-section of a duodenal ulcer crater with an acute inflammatory exudate in the base.
Ulcerations are best exemplified by peptic ulcer of the stomach or duodenum, in which acute and chronic inflammation coexist. During the acute stage there is intense polymorphonuclear infiltration and vascular dilation in the margins of the defect. With chronicity, the margins and base of the ulcer develop fibroblastic proliferation, scarring, and the accumulation of lymphocytes, macrophages, and plasma cells.
Summary of Acute Inflammation
Now that we have described the components, mediators, and pathologic manifestations of acute inflammatory responses, it is useful to summarize the sequence of events in a typical response of this type.78 When a host encounters an injurious agent, such as an infectious microbe or dead cells, phagocytes that reside in all tissues try to eliminate these agents. At the same time phagocytes and other host cells react to the presence of the foreign or abnormal substance by liberating cytokines, lipid messengers, and other mediators of inflammation. Some of these mediators act on small blood vessels in the vicinity and promote the efflux of plasma and the recruitment of circulating leukocytes to the site where the offending agent is located. The recruited leukocytes are activated by the injurious agent and by locally produced mediators, and the activated leukocytes try to remove the offending agent by phagocytosis. As the injurious agent is eliminated and anti-inflammatory mechanisms become active, the process subsides and the host returns to a normal state of health. If the injurious agent cannot be quickly eliminated, the result may be chronic inflammation.
The clinical and pathologic manifestations of the inflammatory response are caused by several reactions. The vascular phenomena of acute inflammation are characterized by increased blood flow to the injured area, resulting mainly from arteriolar dilation and opening of capillary beds induced by mediators such as histamine. Increased vascular permeability results in the accumulation of protein-rich extravascular fluid, which forms the exudate. Plasma proteins leave the vessels, most commonly through widened interendothelial cell junctions of the venules. The redness (rubor), warmth (calor), and swelling (tumor) of acute inflammation are caused by the increased blood flow and edema. Circulating leukocytes, initially predominantly neutrophils, adhere to the endothelium via adhesion molecules, traverse the endothelium, and migrate to the site of injury under the influence of chemotactic agents. Leukocytes that are activated by the offending agent and by endogenous mediators may release toxic metabolites and proteases extracellularly, causing tissue damage. During the damage, and in part as a result of the liberation of prostaglandins, neuropeptides, and cytokines, one of the local symptoms is pain (dolor).
In clinical practice the underlying cause determines whether the therapeutic goal is to promote or reduce inflammation. In infections the treatment is intended to increase the host response and eliminate the infection—hence the practice of warm compresses and gargles in case of pharyngitis (sore throat). On the other hand, in traumatic injuries and chronic inflammatory diseases, inflammation serves no useful purpose and the goal is to reduce it with application of cold (in trauma) and anti-inflammatory drugs. At certain locations, such as the cornea, it may be desirable to suppress even acute inflammation so that corneal transparency can be maintained.
Chronic Inflammation
Chronic inflammation is inflammation of prolonged duration (weeks or months) in which inflammation, tissue injury, and attempts at repair coexist, in varying combinations. It may follow acute inflammation, as described earlier, or chronic inflammation may begin insidiously, as a low-grade, smoldering response without any manifestations of an acute reaction. This latter type of chronic inflammation is the cause of tissue damage in some of the most common and disabling human diseases, such as rheumatoid arthritis, atherosclerosis, tuberculosis, and pulmonary fibrosis. It has also been implicated in the progression of cancer and in diseases once thought to be purely degenerative, such as Alzheimer disease.
CAUSES OF CHRONIC INFLAMMATION
Chronic inflammation arises in the following settings:
MORPHOLOGIC FEATURES
In contrast to acute inflammation, which is manifested by vascular changes, edema, and predominantly neutrophilic infiltration, chronic inflammation is characterized by:
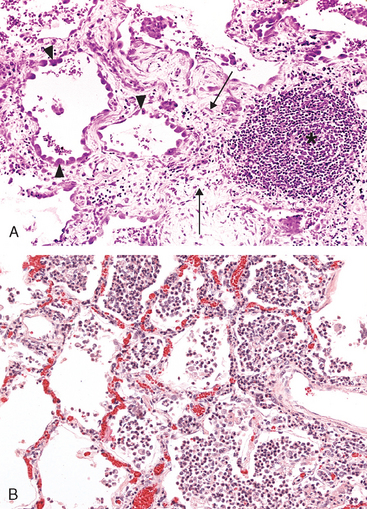
FIGURE 2-22 A, Chronic inflammation in the lung, showing all three characteristic histologic features: (1) collection of chronic inflammatory cells (*), (2) destruction of parenchyma (normal alveoli are replaced by spaces lined by cuboidal epithelium, arrowheads), and (3) replacement by connective tissue (fibrosis, arrows). B, By contrast, in acute inflammation of the lung (acute bronchopneumonia), neutrophils fill the alveolar spaces and blood vessels are congested.
Because angiogenesis and fibrosis are also components of wound healing and repair, they are discussed more fully in Chapter 3.
ROLE OF MACROPHAGES IN CHRONIC INFLAMMATION
The macrophage is the dominant cellular player in chronic inflammation, and we begin our discussion with a brief review of its biology. Macrophages are one component of the mononuclear phagocyte system (Fig. 2-23). The mononuclear phagocyte system (sometimes called reticuloendothelial system) consists of closely related cells of bone marrow origin, including blood monocytes and tissue macrophages. The latter are diffusely scattered in the connective tissue or located in organs such as the liver (Kupffer cells), spleen and lymph nodes (sinus histiocytes), lungs (alveolar macrophages), and central nervous system (microglia). Mononuclear phagocytes arise from a common precursor in the bone marrow, which gives rise to blood monocytes. From the blood, monocytes migrate into various tissues and differentiate into macrophages. The half-life of blood monocytes is about 1 day, whereas the life span of tissue macrophages is several months or years. The journey from bone marrow stem cell to tissue macrophage is regulated by a variety of growth and differentiation factors, cytokines, adhesion molecules, and cellular interactions.

FIGURE 2-23 Maturation of mononuclear phagocytes.
(From Abbas AK et al: Cellular and Molecular Immunology, 5th ed. Philadelphia, WB Saunders, 2003.)
As discussed earlier, monocytes begin to emigrate into extravascular tissues quite early in acute inflammation, and within 48 hours they may constitute the predominant cell type. Extravasation of monocytes is governed by the same factors that are involved in neutrophil emigration, that is, adhesion molecules and chemical mediators with chemotactic and activating properties.81 When a monocyte reaches the extravascular tissue, it undergoes transformation into a larger phagocytic cell, the macrophage. Macrophages may be activated by a variety of stimuli, including microbial products that engage TLRs and other cellular receptors, cytokines (e.g., IFN-γ) secreted by sensitized T lymphocytes and by natural killer cells, and other chemical mediators (Fig. 2-24).
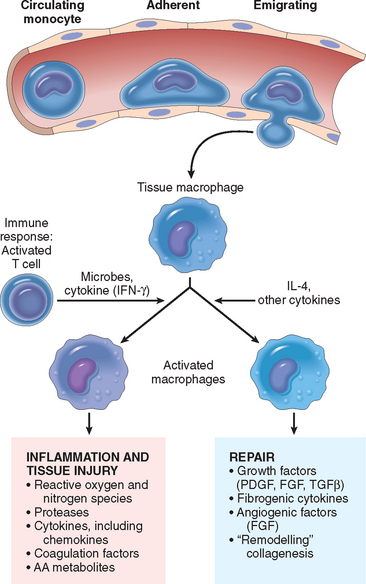
FIGURE 2-24 The roles of activated macrophages in chronic inflammation. Macrophages are activated by nonimmunologic stimuli such as endotoxin or by cytokines from immune-activated T cells (particularly IFN-γ). The products made by activated macrophages that cause tissue injury and fibrosis are indicated. AA, arachidonic acid; PDGF, platelet-derived growth factor; FGF, fibroblast growth factor; TGFβ, transforming growth factor β.
The products of activated macrophages serve to eliminate injurious agents such as microbes and to initiate the process of repair, and are responsible for much of the tissue injury in chronic inflammation. Activation of macrophages results in increased levels of lysosomal enzymes and reactive oxygen and nitrogen species, and production of cytokines, growth factors, and other mediators of inflammation. Some of these products are toxic to microbes and host cells (e.g., reactive oxygen and nitrogen species) or to extracellular matrix (proteases); some cause influx of other cell types (e.g., cytokines, chemotactic factors); and still others cause fibroblast proliferation, collagen deposition, and angiogenesis (e.g., growth factors). As illustrated in Fig. 2-10, different macrophage populations may serve distinct functions—some may be important for microbial killing and inflammation, and others for repair.38 Their impressive arsenal of mediators makes macrophages powerful allies in the body’s defense against unwanted invaders, but the same weaponry can also induce considerable tissue destruction when macrophages are inappropriately activated. It is because of the activities of these macrophages that tissue destruction is one of the hallmarks of chronic inflammation. The ongoing tissue destruction can itself activate the inflammatory cascade, so that features of both acute and chronic inflammation may coexist in certain circumstances.
In short-lived inflammation, if the irritant is eliminated, macrophages eventually disappear (either dying off or making their way into the lymphatics and lymph nodes). In chronic inflammation, macrophage accumulation persists, as a result of continuous recruitment from the circulation and local proliferation at the site of inflammation.
OTHER CELLS IN CHRONIC INFLAMMATION
Other cell types involved in chronic inflammation include lymphocytes, plasma cells, eosinophils, and mast cells:
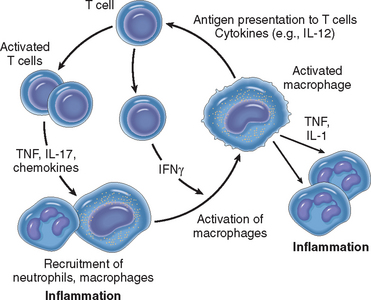
FIGURE 2-25 Macrophage-lymphocyte interactions in chronic inflammation. Activated T cells produce cytokines that recruit macrophages (TNF, IL-17, chemokines) and others that activate macrophages (IFNγ). Different subsets of T cells (called TH1 and TH17) may produce different sets of cytokines; these are described in Chapter 6. Activated macrophages in turn stimulate T cells by presenting antigens and via cytokines (such as IL-12).
Although neutrophils are characteristic of acute inflammation, many forms of chronic inflammation, lasting for months, continue to show large numbers of neutrophils, induced either by persistent microbes or by mediators produced by activated macrophages and T lymphocytes. In chronic bacterial infection of bone (osteomyelitis), a neutrophilic exudate can persist for many months. Neutrophils are also important in the chronic damage induced in lungs by smoking and other irritant stimuli (Chapter 15).
In addition to cellular infiltrates, growth of blood vessels and lymphatic vessels is often prominent in chronic inflammatory reactions. This growth of vessels is stimulated by growth factors, such as VEGF, produced by macrophages and endothelial cells (Chapter 3).
GRANULOMATOUS INFLAMMATION
Granulomatous inflammation is a distinctive pattern of chronic inflammation that is encountered in a limited number of infectious and some noninfectious conditions. Immune reactions are usually involved in the development of granulomas, and therefore this process is described in more detail in Chapter 6. Briefly, a granuloma is a cellular attempt to contain an offending agent that is difficult to eradicate. In this attempt there is often strong activation of T lymphocytes leading to macrophage activation, which can cause injury to normal tissues. Tuberculosis is the prototype of the granulomatous diseases, but sarcoidosis, cat-scratch disease, lymphogranuloma inguinale, leprosy, brucellosis, syphilis, some mycotic infections, berylliosis, reactions of irritant lipids, and some autoimmune diseases are also included (Table 2-8). Recognition of the granulomatous pattern in a biopsy specimen is important because of the limited number of possible conditions that cause it and the significance of the diagnoses associated with the lesions.
TABLE 2-8 Examples of Diseases with Granulomatous Inflammation
| Disease | Cause | Tissue Reaction |
|---|---|---|
| Tuberculosis | Mycobacterium tuberculosis | Caseating granuloma (tubercle): focus of activated macrophages (epithelioid cells), rimmed by fibroblasts, lymphocytes, histiocytes, occasional Langhans giant cells; central necrosis with amorphous granular debris; acid-fast bacilli |
| Leprosy | Mycobacterium leprae | Acid-fast bacilli in macrophages; noncaseating granulomas |
| Syphilis | Treponema pallidum | Gumma: microscopic to grossly visible lesion, enclosing wall of histiocytes; plasma cell infiltrate; central cells necrotic without loss of cellular outline |
| Cat-scratch disease | Gram-negative bacillus | Rounded or stellate granuloma containing central granular debris and recognizable neutrophils; giant cells uncommon |
| Sarcoidosis | Unknown etiology | Noncaseating granulomas with abundant activated macrophages |
| Crohn disease (inflammatory bowel disease) | Immune reaction against intestinal bacteria, self-antigens | Occasional noncaseating granulomas in the wall of the intestine, with dense chronic inflammatory infiltrate |
A granuloma is a focus of chronic inflammation consisting of a microscopic aggregation of macrophages that are transformed into epithelium-like cells, surrounded by a collar of mononuclear leukocytes, principally lymphocytes and occasionally plasma cells. In the usual hematoxylin and eosin–stained tissue sections, the epithelioid cells have a pale pink granular cytoplasm with indistinct cell boundaries, often appearing to merge into one another. The nucleus is less dense than that of a lymphocyte, is oval or elongate, and may show folding of the nuclear membrane. Older granulomas develop an enclosing rim of fibroblasts and connective tissue. Frequently, epithelioid cells fuse to form giant cells in the periphery or sometimes in the center of granulomas. These giant cells may attain diameters of 40 to 50 μm. They have a large mass of cytoplasm containing 20 or more small nuclei arranged either peripherally (Langhans-type giant cell) or haphazardly (foreign body–type giant cell) (Fig. 2-27). There is no known functional difference between these two types of giant cells, yet some pathologists persist in describing them—perhaps because they make nice exam questions!
FIGURE 2-27 Typical tuberculous granuloma showing an area of central necrosis surrounded by multiple Langhans-type giant cells, epithelioid cells, and lymphocytes.
There are two types of granulomas, which differ in their pathogenesis. Foreign body granulomas are incited by relatively inert foreign bodies. Typically, foreign body granulomas form around material such as talc (associated with intravenous drug abuse) (Chapter 9), sutures, or other fibers that are large enough to preclude phagocytosis by a single macrophage and do not incite any specific inflammatory or immune response. Epithelioid cells and giant cells are apposed to the surface of the foreign body. The foreign material can usually be identified in the center of the granuloma, particularly if viewed with polarized light, in which it appears refractile.
Immune granulomas are caused by a variety of agents that are capable of inducing a cell-mediated immune response (Chapter 6). This type of immune response produces granulomas usually when the inciting agent is poorly degradable or particulate. In such responses macrophages engulf foreign protein antigen, process it, and present peptides to antigen-specific T lymphocytes, causing their activation (Chapter 6). The responding T cells produce cytokines, such as IL-2, which activates other T cells, perpetuating the response, and IFN-γ, which is important in activating macrophages and transforming them into epithelioid cells and multinucleate giant cells.
The prototype of the immune granuloma is that caused by infection with Mycobacterium tuberculosis. In this disease the granuloma is referred to as a tubercle. It is often characterized by the presence of central caseous necrosis (see Fig. 2-27). In contrast, caseous necrosis is rare in other granulomatous diseases. The morphologic patterns in the various granulomatous diseases may be sufficiently different to allow reasonably accurate diagnosis by an experienced pathologist (see Table 2-8); however, there are so many atypical presentations that it is always necessary to identify the specific etiologic agent by special stains for organisms (e.g., acid-fast stains for tubercle bacilli), by culture methods (e.g., in tuberculosis and fungal diseases), by molecular techniques (e.g., the polymerase chain reaction in tuberculosis), and by serologic studies (e.g., in syphilis).
Systemic Effects of Inflammation
Anyone who has suffered a severe sore throat or a respiratory infection has experienced the systemic manifestations of acute inflammation. The systemic changes associated with acute inflammation are collectively called the acute-phase response, or the systemic inflammatory response syndrome. These changes are reactions to cytokines whose production is stimulated by bacterial products such as LPS and by other inflammatory stimuli. The acute-phase response consists of several clinical and pathologic changes:
Consequences of Defective or Excessive Inflammation
Now that we have described the process of inflammation and its outcomes, it is helpful to summarize the clinical and pathologic consequences of too much or too little inflammation.
Now that our discussion of the molecular and cellular events in acute and chronic inflammation is concluded, in Chapter 3 we consider the body’s attempts to heal the damage, the process of repair. Repair begins almost as soon as the inflammatory reaction has started and involves several processes, including cell proliferation, angiogenesis, and collagen synthesis and deposition. Many aspects of repair were mentioned in this chapter, but the process is sufficiently complex and important to deserve its own chapter!
1 Weissman G, editor. Inflammation: Historical Perspectives. New York: Raven Press, 1992.
2 Hunter J. A Treatise of the Blood, Inflammation, and Gunshot Wounds. London: J. Nicoli, 1794.
3 Heifets L. Centennial of Metchnikoff’s discovery. J Reticuloendothel Soc. 1982;31:381.
4 Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol Mech Dis. 2008;3:99.
5 Hellwig-Burgel T, et al. Review: hypoxia-inducible factor-1 (HIF-1): a novel transcription factor in immune reactions. J Interferon Cytokine Res. 2005;25:297.
6 Lampugnani MG, Dejana E. Interendothelial junctions: structure, signalling and functional roles. Curr Opin Cell Biol. 1997;9:674.
7 Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279.
8 Lentsch AB, Ward PA. Regulation of inflammatory vascular damage. J Pathol. 2000;190:343.
9 Valbuena G, Walker DH. Endothelium as a target of infections. Annu Rev Pathol Mech Dis. 2006;1:151.
10 Dvorak AM, Feng D. The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle. J Histochem Cytochem. 2001;49:419.
11 Oliver G, Alitalo K. The lymphatic vasculature: recent progress and paradigms. Annu Rev Cell Dev Biol. 2005;21:457.
12 Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464.
13 Muller WA. Leukocyte-endothelial cell interactions in the inflammatory response. Lab Invest. 2002;82:521.
14 Luster AD, et al. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182.
15 McEver RP. Selectins: lectins that initiate cell adhesion under flow. Curr Opin Cell Biol. 2002;14:581.
16 Sperandio M. Selectins and glycosyltransferases in leukocyte rolling in vivo. FEBS J. 2006;273:4377.
17 Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115:1.
18 Dinarello CA. Interleukin-1 β. Crit Care Med. 2005;33:S460.
19 Johnston B, Butcher EC. Chemokines in rapid leukocyte adhesion triggering and migration. Semin Immunol. 2002;14:83.
20 Sallusto F, Mackay CR. Chemoattractants and their receptors in homeostasis and inflammation. Curr Opin Immunol. 2004;16:724.
21 Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673.
22 Cook-Mills JM, Deem TL. Active participation of endothelial cells in inflammation. J Leukoc Biol. 2005;77:487.
23 Petri B, Bixel MG. Molecular events during leukocyte diapedesis. FEBS J. 2006;273:4399.
24 Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24:327.
25 Weber C, et al. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. 2007;7:467.
26 Bunting M, et al. Leukocyte adhesion deficiency syndromes: adhesion and tethering defects involving beta 2 integrins and selectin ligands. Curr Opin Hematol. 2002;9:30.
27 Van Haastert PJ, Devreotes PN. Chemotaxis: signalling the way forward. Nat Rev Mol Cell Biol. 2004;5:626.
28 Akira S, et al. Pathogen recognition and innate immunity. Cell. 2006;124:783.
29 Meylan E, et al. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39.
30 Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. 2002;20:825.
31 Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197.
32 Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol. 2004;2:820.
33 Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42.
34 Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A. 2000;97:8841.
35 Belaaouaj A. Neutrophil elastase-mediated killing of bacteria: lessons from targeted mutagenesis. Microbes Infect. 2002;4:1259.
36 Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol. 2005;6:551.
37 Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. 2004;75:39.
38 Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953.
39 Jaeschke H, Smith CW. Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol. 1997;61:647.
40 Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5:1317.
41 Ward DM, et al. Chediak-Higashi syndrome: a clinical and molecular view of a rare lysosomal storage disorder. Curr Mol Med. 2002;2:469.
42 Heyworth PG, et al. Chronic granulomatous disease. Curr Opin Immunol. 2003;15:578.
43 Nathan C. Points of control in inflammation. Nature. 2002;420:846.
44 Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191.
45 Serhan CN, Chang N, van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349.
46 Tracey KJ. The inflammatory reflex. Nature. 2002;420:853.
47 Repka-Ramirez MS, Baraniuk JN. Histamine in health and disease. Clin Allergy Immunol. 2002;17:1.
48 Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871.
49 Miller SB. Prostaglandins in health and disease: an overview. Semin Arthritis Rheum. 2006;36:37.
50 Khanapure SP, et al. Eicosanoids in inflammation: biosynthesis, pharmacology, and therapeutic frontiers. Curr Top Med Chem. 2007;7:311.
51 Murakami M, Kudo I. Cellular arachidonate-releasing functions of various phospholipase A2s. Adv Exp Med Biol. 2003;525:87.
52 Flower RJ. The development of COX2 inhibitors. Nat Rev Drug Discov. 2003;2:179.
53 Krotz F, et al. Selective COX-2 inhibitors and risk of myocardial infarction. J Vasc Res. 2005;42:312.
54 Stafforini DM, et al. Platelet-activating factor, a pleiotrophic mediator of physiological and pathological processes. Crit Rev Clin Lab Sci. 2003;40:643.
55 Salvemini D, et al. Superoxide, peroxynitrite and oxidative/nitrative stress in inflammation. Biochem Soc Trans. 2006;34:965.
56 Laroux FS, et al. Role of nitric oxide in inflammation. Acta Physiol Scand. 2001;173:113.
57 Cirino G, et al. Nitric oxide and inflammation. Inflamm Allergy Drug Targets. 2006;5:115.
58 Mantovani A, et al. Endothelial activation by cytokines. Ann N Y Acad Sci. 1997;832:93.
59 Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol. 2001;70:317.
60 Stojanov S, Kastner DL. Familial autoinflammatory diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol. 2005;17:586.
61 Ting JP, et al. CATERPILLERs, pyrin and hereditary immunological disorders. Nat Rev Immunol. 2006;6:183.
62 Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10.
63 Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610.
64 Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121.
65 Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891.
66 Conti P, et al. Modulation of autoimmunity by the latest interleukins (with special emphasis on IL-32). Autoimmun Rev. 2007;6:131.
67 Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nat Clin Pract Rheumatol. 2006;2:619.
68 Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467.
69 O’Connor TM, et al. The role of substance P in inflammatory disease. J Cell Physiol. 2004;201:167.
70 Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839.
71 Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058.
72 Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140.
73 Barrington R, et al. The role of complement in inflammation and adaptive immunity. Immunol Rev. 2001;180:5.
74 Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131:417.
75 Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest. 2003;111:25.
76 Esmon CT. Inflammation and the activated protein C anticoagulant pathway. Semin Thromb Hemost. 2006;32(Suppl 1):49.
77 Joseph K, Kaplan AP. Formation of bradykinin: a major contributor to the innate inflammatory response. Adv Immunol. 2005;86:159.
78 Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428.
79 Lawrence T, Gilroy DW. Chronic inflammation: a failure of resolution? Int J Exp Pathol. 2007;88:85.
80 Majno G. Chronic inflammation: links with angiogenesis and wound healing. Am J Pathol. 1998;153:1035.
81 Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol. 2004;4:432.
82 Drayton DL, et al. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. 2006;7:344.
83 Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol. 2006;24:147.
84 Dinarello CA. Cytokines as endogenous pyrogens. J Infect Dis. 1999;179(Suppl 2):S294.
85 Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448.
86 Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007;49:2129.
87 Ganz T. Molecular control of iron transport. J Am Soc Nephrol. 2007;18:394.
88 Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138.
89 Munford RS. Severe sepsis and septic shock: the role of Gram-negative bacteremia. Annu Rev Pathology Mech Dis. 2006;1:467.