Chapter 1 Cellular Responses to Stress and Toxic Insults: Adaptation, Injury, and Death
Introduction to Pathology
Pathology is the study (logos) of disease (pathos). More specifically, it is devoted to the study of the structural, biochemical, and functional changes in cells, tissues, and organs that underlie disease. By the use of molecular, microbiologic, immunologic, and morphologic techniques, pathology attempts to explain the whys and wherefores of the signs and symptoms manifested by patients while providing a rational basis for clinical care and therapy. It thus serves as the bridge between the basic sciences and clinical medicine, and is the scientific foundation for all of medicine.
Traditionally the study of pathology is divided into general pathology and systemic pathology. The former is concerned with the reactions of cells and tissues to abnormal stimuli and to inherited defects, which are the main causes of disease. The latter examines the alterations in specialized organs and tissues that are responsible for disorders that involve these organs. In this book we first cover the principles of general pathology and then proceed to specific disease processes as they affect particular organs or systems.
The four aspects of a disease process that form the core of pathology are its cause (etiology), the mechanisms of its development (pathogenesis), the biochemical and structural alterations induced in the cells and organs of the body (molecular and morphologic changes), and the functional consequences of these changes (clinical manifestations).
Etiology or Cause.
The concept that certain abnormal symptoms or diseases are “caused” is as ancient as recorded history. For the Arcadians (2500 bc), if someone became ill it was the patient’s own fault (for having sinned) or the effects of outside agents, such as bad smells, cold, evil spirits, or gods.1 We now recognize that there are two major classes of etiologic factors: genetic (e.g., inherited mutations and disease-associated gene variants, or polymorphisms) and acquired (e.g., infectious, nutritional, chemical, physical). The idea that one etiologic agent is the cause of one disease—developed from the study of infections and single-gene disorders—is not applicable to the majority of diseases. In fact, most of our common afflictions, such as atherosclerosis and cancer, are multifactorial and arise from the effects of various external triggers on a genetically susceptible individual. The relative contribution of inherited susceptibility and external influences varies in different diseases.
Pathogenesis.
Pathogenesis refers to the sequence of events in the response of cells or tissues to the etiologic agent, from the initial stimulus to the ultimate expression of the disease. The study of pathogenesis remains one of the main domains of pathology. Even when the initial cause is known (e.g., infection or mutation), it is many steps removed from the expression of the disease. For example, to understand cystic fibrosis is to know not only the defective gene and gene product, but also the biochemical and morphologic events leading to the formation of cysts and fibrosis in the lungs, pancreas, and other organs. Indeed, as we shall see throughout the book, the molecular revolution has already identified mutant genes underlying a great number of diseases, and the entire human genome has been mapped. Nevertheless, the functions of the encoded proteins and how mutations induce disease—the pathogenesis—are still often obscure. Technologic advances are making it increasingly feasible to link specific molecular abnormalities to disease manifestations and to use this knowledge to design new therapeutic approaches. For these reasons, the study of pathogenesis has never been more exciting scientifically or more relevant to medicine.
Molecular and Morphologic Changes.
Morphologic changes refer to the structural alterations in cells or tissues that are either characteristic of a disease or diagnostic of an etiologic process. The practice of diagnostic pathology is devoted to identifying the nature and progression of disease by studying morphologic changes in tissues and chemical alterations in patients. More recently the limitations of morphology for diagnosing diseases have become increasingly evident, and the field of diagnostic pathology has expanded to encompass molecular biologic and immunologic approaches for analyzing disease states. Nowhere is this more striking than in the study of tumors; breast cancers that look morphologically identical may have widely different courses, therapeutic responses, and prognosis. Molecular analysis by techniques such as DNA microarrays (Chapter 5) has begun to reveal genetic differences that predict the behavior of the tumors as well as their responsiveness to different therapies. Increasingly, such techniques are being used to extend and even supplant traditional morphologic analyses.
Functional Derangements and Clinical Manifestations.
The end results of genetic, biochemical, and structural changes in cells and tissues are functional abnormalities, which lead to the clinical manifestations (symptoms and signs) of disease, as well as its progress (clinical course and outcome).
Virtually all forms of disease start with molecular or structural alterations in cells, a concept first put forth in the nineteenth century by Rudolf Virchow, known as the father of modern pathology. We therefore begin our consideration of pathology with the study of the causes, mechanisms, and morphologic and biochemical correlates of cell injury. Injury to cells and to extracellular matrix ultimately leads to tissue and organ injury, which determine the morphologic and clinical patterns of disease.
Overview: Cellular Responses to Stress and Noxious Stimuli
The normal cell is confined to a fairly narrow range of function and structure by its state of metabolism, differentiation, and specialization; by constraints of neighboring cells; and by the availability of metabolic substrates. It is nevertheless able to handle physiologic demands, maintaining a steady state called homeostasis. Adaptations are reversible functional and structural responses to more severe physiologic stresses and some pathologic stimuli, during which new but altered steady states are achieved, allowing the cell to survive and continue to function (Fig. 1-1 and Table 1-1). The adaptive response may consist of an increase in the size of cells (hypertrophy) and functional activity, an increase in their number (hyperplasia), a decrease in the size and metabolic activity of cells (atrophy), or a change in the phenotype of cells (metaplasia). When the stress is eliminated the cell can recover to its original state without having suffered any harmful consequences.
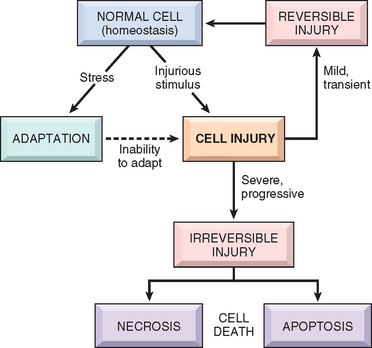
TABLE 1-1 Cellular Responses to Injury
| Nature of Injurious Stimulus | Cellular Response |
|---|---|
| ALTERED PHYSIOLOGICAL STIMULI; SOME NONLETHAL INJURIOUS STIMULI | CELLULAR ADAPTATIONS |
| REDUCED OXYGEN SUPPLY; CHEMICAL INJURY; MICROBIAL INFECTION | CELL INJURY |
| METABOLIC ALTERATIONS, GENETIC OR ACQUIRED; CHRONIC INJURY | INTRACELLULAR ACCUMULATIONS; CALCIFICATION |
| CUMULATIVE SUBLETHAL INJURY OVER LONG LIFE SPAN | CELLULAR AGING |
If the limits of adaptive responses are exceeded or if cells are exposed to injurious agents or stress, deprived of essential nutrients, or become compromised by mutations that affect essential cellular constituents, a sequence of events follows that is termed cell injury (see Fig. 1-1) Cell injury is reversible up to a certain point, but if the stimulus persists or is severe enough from the beginning, the cell suffers irreversible injury and ultimately cell death. Adaptation, reversible injury, and cell death may be stages of progressive impairment following different types of insults. For instance, in response to increased hemodynamic loads, the heart muscle becomes enlarged, a form of adaptation, and can even undergo injury. If the blood supply to the myocardium is compromised or inadequate, the muscle first suffers reversible injury, manifested by certain cytoplasmic changes (described later). Eventually, the cells suffer irreversible injury and die (Fig. 1-2).
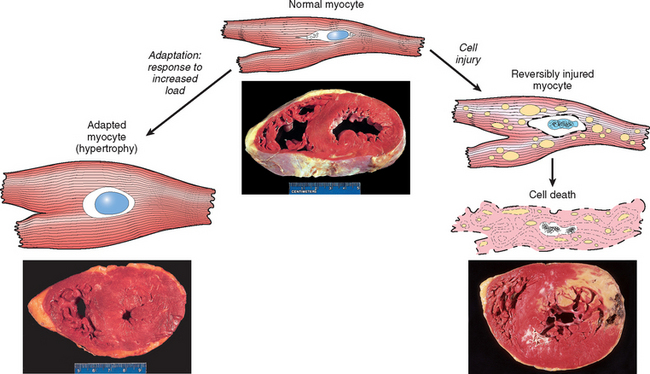
FIGURE 1-2 The relationship between normal, adapted, reversibly injured, and dead myocardial cells. The cellular adaptation is myocardial hypertrophy (lower left), caused by increased blood flow requiring greater mechanical effort by myocardial cells. This adaptation leads to thickening of the left ventricular wall to over 2 cm (normal, 1–1.5 cm). In reversibly injured myocardium (illustrated schematically, right) there are generally only functional effects, without any readily apparent gross or even microscopic changes. In the specimen showing necrosis, a form of cell death (lower right), the light area in the posterolateral left ventricle represents an acute myocardial infarction caused by reduced blood flow (ischemia). All three transverse sections of the heart have been stained with triphenyltetrazolium chloride, an enzyme substrate that colors viable myocardium magenta. Failure to stain is due to enzyme loss following cell death.
Cell death, the end result of progressive cell injury, is one of the most crucial events in the evolution of disease in any tissue or organ. It results from diverse causes, including ischemia (reduced blood flow), infection, and toxins. Cell death is also a normal and essential process in embryogenesis, the development of organs, and the maintenance of homeostasis. There are two principal pathways of cell death, necrosis and apoptosis. Nutrient deprivation triggers an adaptive cellular response called autophagy that may also culminate in cell death. We will return to a detailed discussion of these pathways of cell death later in the chapter.
Stresses of different types may induce changes in cells and tissues other than typical adaptations, cell injury, and death (see Table 1-1). Metabolic derangements in cells and sublethal, chronic injury may be associated with intracellular accumulations of a number of substances, including proteins, lipids, and carbohydrates. Calcium is often deposited at sites of cell death, resulting in pathologic calcification. Finally, the normal process of aging itself is accompanied by characteristic morphologic and functional changes in cells.
In this chapter we discuss first how cells adapt to stresses, and then the causes, mechanisms, and consequences of the various forms of acute cell damage, including reversible cell injury, and cell death. We conclude with three other processes that affect cells and tissues: intracellular accumulations, pathologic calcification, and cell aging.
Adaptations of Cellular Growth and Differentiation
Adaptations are reversible changes in the size, number, phenotype, metabolic activity, or functions of cells in response to changes in their environment. Such adaptations may take several distinct forms.
HYPERTROPHY
Hypertrophy refers to an increase in the size of cells, resulting in an increase in the size of the organ. The hypertrophied organ has no new cells, just larger cells. The increased size of the cells is due to the synthesis of more structural components of the cells. Cells capable of division may respond to stress by undergoing both hyperplasia (described below) and hypertrophy, whereas in nondividing cells (e.g., myocardial fibers) increased tissue mass is due to hypertrophy. In many organs hypertrophy and hyperplasia may coexist and contribute to increased size.
Hypertrophy can be physiologic or pathologic and is caused by increased functional demand or by stimulation by hormones and growth factors. The striated muscle cells in the heart and skeletal muscles have only a limited capacity for division, and respond to increased metabolic demands mainly by undergoing hypertrophy. The most common stimulus for hypertrophy of muscle is increased workload. For example, the bulging muscles of bodybuilders engaged in “pumping iron” result from an increase in size of the individual muscle fibers in response to increased demand. In the heart, the stimulus for hypertrophy is usually chronic hemodynamic overload, resulting from either hypertension or faulty valves (see Fig. 1-2). In both tissue types the muscle cells synthesize more proteins and the number of myofilaments increases. This increases the amount of force each myocyte can generate, and thus increases the strength and work capacity of the muscle as a whole.
The massive physiologic growth of the uterus during pregnancy is a good example of hormone-induced increase in the size of an organ that results mainly from hypertrophy of muscle fibers (Fig. 1-3). The cellular enlargement is stimulated by estrogenic hormones acting on smooth muscle estrogen receptors, eventually resulting in increased synthesis of smooth muscle proteins and an increase in cell size.
FIGURE 1-3 Physiologic hypertrophy of the uterus during pregnancy. A, Gross appearance of a normal uterus (right) and a gravid uterus (removed for postpartum bleeding) (left). B, Small spindle-shaped uterine smooth muscle cells from a normal uterus, compared with C, large plump cells from the gravid uterus, at the same magnification.
Although the traditional view of cardiac and skeletal muscle is that in adults these tissues are incapable of proliferation and, therefore, their enlargement is entirely a result of hypertrophy, there is now accumulating evidence that even these cell types are capable of some proliferation as well as repopulation from precursors, in addition to hypertrophy (Chapter 3).2
Mechanisms of Hypertrophy
Hypertrophy is the result of increased production of cellular proteins. Much of our understanding of hypertrophy is based on studies of the heart. Hypertrophy can be induced by the linked actions of mechanical sensors (that are triggered by increased work load), growth factors (including TGF-β, insulin-like growth factor-1 [IGF-1], fibroblast growth factor), and vasoactive agents (such as α-adrenergic agonists, endothelin-1, and angiotensin II). Indeed, mechanical sensors themselves induce production of growth factors and agonists (Fig. 1-4).3-5 These stimuli work coordinately to increase the synthesis of muscle proteins that are responsible for the hypertrophy. The two main biochemical pathways involved in muscle hypertrophy seem to be the phosphoinositide 3-kinase/Akt pathway (postulated to be most important in physiologic, e.g., exercise-induced, hypertrophy) and signaling downstream of G protein-coupled receptors (induced by many growth factors and vasoactive agents, and thought to be more important in pathologic hypertrophy). Hypertrophy may also be associated with a switch of contractile proteins from adult to fetal or neonatal forms. For example, during muscle hypertrophy the α isoform of myosin heavy chain is replaced by the β isoform, which has a slower, more energetically economical contraction. In addition, some genes that are expressed only during early development are re-expressed in hypertrophic cells, and the products of these genes participate in the cellular response to stress. For example, the gene for atrial natriuretic factor (ANF) is expressed in both the atrium and the ventricle in the embryonic heart, but it is down-regulated after birth. Cardiac hypertrophy, however, is associated with reinduction of ANF gene expression. ANF is a peptide hormone that causes salt secretion by the kidney, decreases blood volume and pressure, and therefore serves to reduce hemodynamic load.
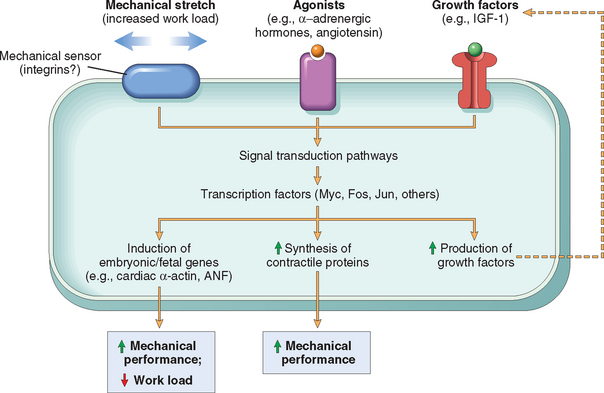
FIGURE 1-4 Biochemical mechanisms of myocardial hypertrophy. The major known signaling pathways and their functional effects are shown. Mechanical sensors appear to be the major triggers for physiologic hypertrophy, and agonists and growth factors may be more important in pathologic states. ANF, atrial natriuretic factor; IGF-1, insulin-like growth factor.
Whatever the exact cause and mechanism of cardiac hypertrophy, it eventually reaches a limit beyond which enlargement of muscle mass is no longer able to compensate for the increased burden. At this stage several regressive changes occur in the myocardial fibers, of which the most important are lysis and loss of myofibrillar contractile elements. In extreme cases myocyte death can occur by either apoptosis or necrosis.5,6 The net result of these changes is cardiac failure, a sequence of events that illustrates how an adaptation to stress can progress to functionally significant cell injury if the stress is not relieved.
Although hypertrophy usually refers to increase in size of cells or tissues, sometimes a subcellular organelle may undergo selective hypertrophy. For instance, individuals treated with drugs such as barbiturates show hypertrophy of the smooth endoplamic reticulum (ER) in hepatocytes, which is an adaptive response that increases the amount of enzymes (cytochrome P-450 mixed function oxidases) available to detoxify the drugs. Over time, the patients respond less to the drug because of this adaptation. Adaptation to one drug may result in an increased capacity to metabolize other drugs. For instance, alcohol intake causes hypertrophy of the smooth ER and may lead to reduced levels of available barbiturates that are being taken at the same time. Although P-450-mediated modification is often thought of as “detoxification,” many compounds are rendered more injurious by this process. In addition, the products formed by this oxidative metabolism include reactive oxygen species, which can injure the cell. Normal genetic variations (polymorphisms) influence the activity of P-450, and thus the sensitivity of different individuals to various drugs.7
HYPERPLASIA
Hyperplasia is an increase in the number of cells in an organ or tissue, usually resulting in increased mass of the organ or tissue. Although hyperplasia and hypertrophy are distinct processes, frequently they occur together, and they may be triggered by the same external stimulus. Hyperplasia takes place if the cell population is capable of dividing, and thus increasing the number of cells. Hyperplasia can be physiologic or pathologic.
Physiologic Hyperplasia
Physiologic hyperplasia can be divided into: (1) hormonal hyperplasia, which increases the functional capacity of a tissue when needed, and (2) compensatory hyperplasia, which increases tissue mass after damage or partial resection. Hormonal hyperplasia is well illustrated by the proliferation of the glandular epithelium of the female breast at puberty and during pregnancy, usually accompanied by enlargement (hypertrophy) of the glandular epithelial cells. The classical illustration of compensatory hyperplasia comes from the myth of Prometheus, which shows that the ancient Greeks recognized the capacity of the liver to regenerate. As punishment for having stolen the secret of fire from the gods, Prometheus was chained to a mountain, and his liver was devoured daily by an eagle, only to regenerate anew every night.1 In individuals who donate one lobe of the liver for transplantation, the remaining cells proliferate so that the organ soon grows back to its original size. Experimental models of partial hepatectomy have been very useful for defining the mechanisms that stimulate regeneration of the liver7 (Chapter 3).
Pathologic Hyperplasia
Most forms of pathologic hyperplasia are caused by excesses of hormones or growth factors acting on target cells. Endometrial hyperplasia is an example of abnormal hormone-induced hyperplasia. Normally, after a menstrual period there is a rapid burst of proliferative activity in the epithelium that is stimulated by pituitary hormones and ovarian estrogen. It is brought to a halt by the rising levels of progesterone, usually about 10 to 14 days before the end of the menstrual period. In some instances, however, the balance between estrogen and progesterone is disturbed. This results in absolute or relative increases in the amount of estrogen, with consequent hyperplasia of the endometrial glands. This form of pathologic hyperplasia is a common cause of abnormal menstrual bleeding. Benign prostatic hyperplasia is another common example of pathologic hyperplasia induced by responses to hormones, in this case, androgens. Although these forms of hyperplasia are abnormal, the process remains controlled because there are no mutations in genes that regulate cell division, and the hyperplasia regresses if the hormonal stimulation is eliminated. As is discussed in Chapter 7, in cancer the growth control mechanisms become dysregulated or ineffective because of genetic aberrations, resulting in unrestrained proliferation. Thus, hyperplasia is distinct from cancer, but pathologic hyperplasia constitutes a fertile soil in which cancerous proliferation may eventually arise. For instance, patients with hyperplasia of the endometrium are at increased risk for developing endometrial cancer (Chapter 22).
Hyperplasia is a characteristic response to certain viral infections, such as papillomaviruses, which cause skin warts and several mucosal lesions composed of masses of hyperplastic epithelium. Here, growth factors produced by viral genes or by infected cells may stimulate cellular proliferation (Chapter 7).
Mechanisms of Hyperplasia
Hyperplasia is the result of growth factor–driven proliferation of mature cells and, in some cases, by increased output of new cells from tissue stem cells. For instance, after partial hepatectomy growth factors are produced in the liver that engage receptors on the surviving cells and activate signaling pathways that stimulate cell proliferation. But if the proliferative capacity of the liver cells is compromised, as in some forms of hepatitis causing cell injury, hepatocytes can instead regenerate from intrahepatic stem cells.8 The roles of growth factors and stem cells in cellular replication and tissue hyperplasia are discussed in more detail in Chapter 3.
ATROPHY
Atrophy is reduced size of an organ or tissue resulting from a decrease in cell size and number. Atrophy can be physiologic or pathologic. Physiologic atrophy is common during normal development. Some embryonic structures, such as the notochord and thyroglossal duct, undergo atrophy during fetal development. The uterus decreases in size shortly after parturition, and this is a form of physiologic atrophy.
Pathologic atrophy depends on the underlying cause and can be local or generalized. The common causes of atrophy are the following:
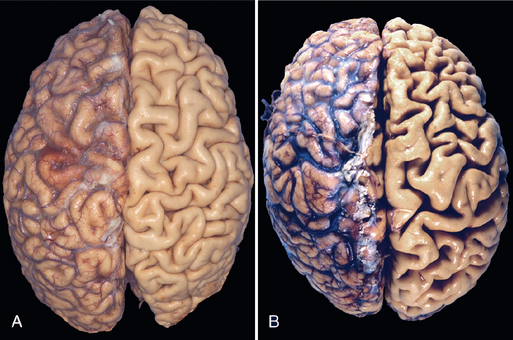
FIGURE 1-5 Atrophy. A, Normal brain of a young adult. B, Atrophy of the brain in an 82-year-old male with atherosclerotic cerebrovascular disease, resulting in reduced blood supply. Note that loss of brain substance narrows the gyri and widens the sulci. The meninges have been stripped from the right half of each specimen to reveal the surface of the brain.
The fundamental cellular changes associated with atrophy are identical in all of these settings. The initial response is a decrease in cell size and organelles, which may reduce the metabolic needs of the cell sufficiently to permit its survival. In atrophic muscle, the cells contain fewer mitochondria and myofilaments and a reduced amount of rough ER. By bringing into balance the cell’s metabolic demand and the lower levels of blood supply, nutrition, or trophic stimulation, a new equilibrium is achieved. Early in the process atrophic cells may have diminished function, but they are not dead. However, atrophy caused by gradually reduced blood supply may progress to the point at which cells are irreversibly injured and die, often by apoptosis. Cell death by apoptosis also contributes to the atrophy of endocrine organs after hormone withdrawal.
Mechanisms of Atrophy
Atrophy results from decreased protein synthesis and increased protein degradation in cells. Protein synthesis decreases because of reduced metabolic activity. The degradation of cellular proteins occurs mainly by the ubiquitin-proteasome pathway. Nutrient deficiency and disuse may activate ubiquitin ligases, which attach the small peptide ubiquitin to cellular proteins and target these proteins for degradation in proteasomes.3,9,10 This pathway is also thought to be responsible for the accelerated proteolysis seen in a variety of catabolic conditions, including cancer cachexia.
In many situations, atrophy is also accompanied by increased autophagy, with resulting increases in the number of autophagic vacuoles. Autophagy (“self eating”) is the process in which the starved cell eats its own components in an attempt to find nutrients and survive. Autophagic vacuoles are membrane-bound vacuoles that contain fragments of cell components. The vacuoles ultimately fuse with lysosomes, and their contents are digested by lysosomal enzymes. Some of the cell debris within the autophagic vacuoles may resist digestion and persist as membrane-bound residual bodies that may remain as a sarcophagus in the cytoplasm. An example of such residual bodies is the lipofuscin granules, discussed later in the chapter. When present in sufficient amounts, they impart a brown discoloration to the tissue (brown atrophy). Autophagy is associated with various types of cell injury, and we will discuss it in more detail later.
METAPLASIA
Metaplasia is a reversible change in which one differentiated cell type (epithelial or mesenchymal) is replaced by another cell type. It may represent an adaptive substitution of cells that are sensitive to stress by cell types better able to withstand the adverse environment.
The most common epithelial metaplasia is columnar to squamous (Fig. 1-6), as occurs in the respiratory tract in response to chronic irritation. In the habitual cigarette smoker, the normal ciliated columnar epithelial cells of the trachea and bronchi are often replaced by stratified squamous epithelial cells. Stones in the excretory ducts of the salivary glands, pancreas, or bile ducts may also cause replacement of the normal secretory columnar epithelium by stratified squamous epithelium. A deficiency of vitamin A (retinoic acid) induces squamous metaplasia in the respiratory epithelium (Chapter 9). In all these instances the more rugged stratified squamous epithelium is able to survive under circumstances in which the more fragile specialized columnar epithelium might have succumbed. However, the change to metaplastic squamous cells comes with a price. In the respiratory tract, for example, although the epithelial lining becomes tough, important mechanisms of protection against infection—mucus secretion and the ciliary action of the columnar epithelium—are lost. Thus, epithelial metaplasia is a double-edged sword and, in most circumstances, represents an undesirable change. Moreover, the influences that predispose to metaplasia, if persistent, may initiate malignant transformation in metaplastic epithelium. Thus, a common form of cancer in the respiratory tract is composed of squamous cells, which arise in areas of metaplasia of the normal columnar epithelium into squamous epithelium.
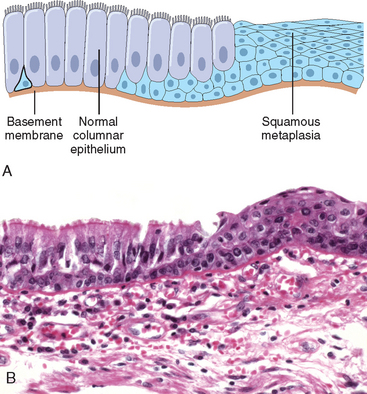
FIGURE 1-6 Metaplasia of columnar to squamous epithelium. A, Schematic diagram. B, Metaplasia of columnar epithelium (left) to squamous epithelium (right) in a bronchus.
Metaplasia from squamous to columnar type may also occur, as in Barrett esophagus, in which the esophageal squamous epithelium is replaced by intestinal-like columnar cells under the influence of refluxed gastric acid. Cancers may arise in these areas; these are typically glandular (adeno)carcinomas (Chapter 17).
Connective tissue metaplasia is the formation of cartilage, bone, or adipose tissue (mesenchymal tissues) in tissues that normally do not contain these elements. For example, bone formation in muscle, designated myositis ossificans, occasionally occurs after intramuscular hemorrhage. This type of metaplasia is less clearly seen as an adaptive response, and may be a result of cell or tissue injury.
Mechanisms of Metaplasia
Metaplasia does not result from a change in the phenotype of an already differentiated cell type; instead it is the result of a reprogramming of stem cells that are known to exist in normal tissues, or of undifferentiated mesenchymal cells present in connective tissue. In a metaplastic change, these precursor cells differentiate along a new pathway. The differentiation of stem cells to a particular lineage is brought about by signals generated by cytokines, growth factors, and extracellular matrix components in the cells’ environment.11,12 These external stimuli promote the expression of genes that drive cells toward a specific differentiation pathway. In the case of vitamin A deficiency or excess, it is known that retinoic acid regulates gene transcription directly through nuclear retinoid receptors (Chapter 9), which can influence the differentiation of progenitors derived from tissue stem cells. How other external stimuli cause metaplasia is unknown, but it is clear that they too somehow alter the activity of transcription factors that regulate differentiation.
Overview of Cell Injury and Cell Death
As stated at the beginning of the chapter, cell injury results when cells are stressed so severely that they are no longer able to adapt or when cells are exposed to inherently damaging agents or suffer from intrinsic abnormalities. Injury may progress through a reversible stage and culminate in cell death (see Fig. 1-1).
In the following sections we discuss the causes, morphologic features, and mechanisms of cell injury and its common end point, necrosis, with selected illustrative examples. We conclude with a discussion of the unique pattern of cell death represented by apoptosis, and then a brief description of the process of autophagy and how it may progress to cell death.
Causes of Cell Injury
The causes of cell injury range from the external gross physical violence of an automobile accident to subtle internal abnormalities, such as a genetic mutation causing lack of a vital enzyme that impairs normal metabolic function. Most injurious stimuli can be grouped into the following broad categories.
Oxygen Deprivation.
Hypoxia is a deficiency of oxygen, which causes cell injury by reducing aerobic oxidative respiration. Hypoxia is an extremely important and common cause of cell injury and cell death. Causes of hypoxia include reduced blood flow (celled ischemia), inadequate oxygenation of the blood due to cardiorespiratory failure, and decreased oxygen-carrying capacity of the blood, as in anemia or carbon monoxide poisoning (producing a stable carbon monoxyhemoglobin that blocks oxygen carriage) or after severe blood loss. Depending on the severity of the hypoxic state, cells may adapt, undergo injury, or die. For example, if an artery is narrowed, the tissue supplied by that vessel may initially shrink in size (atrophy), whereas more severe or sudden hypoxia induces injury and cell death.
Physical Agents.
Physical agents capable of causing cell injury include mechanical trauma, extremes of temperature (burns and deep cold), sudden changes in atmospheric pressure, radiation, and electric shock (Chapter 9).
Chemical Agents and Drugs.
The list of chemicals that may produce cell injury defies compilation. Simple chemicals such as glucose or salt in hypertonic concentrations may cause cell injury directly or by deranging electrolyte balance in cells. Even oxygen at high concentrations is toxic. Trace amounts of poisons, such as arsenic, cyanide, or mercuric salts, may destroy sufficient numbers of cells within minutes or hours to cause death. Other potentially injurious substances are our daily companions: environmental and air pollutants, insecticides, and herbicides; industrial and occupational hazards, such as carbon monoxide and asbestos; recreational drugs such as alcohol; and the ever-increasing variety of therapeutic drugs.
Infectious Agents.
These agents range from the submicroscopic viruses to the large tapeworms. In between are the rickettsiae, bacteria, fungi, and higher forms of parasites. The ways by which these biologic agents cause injury are diverse and are discussed in Chapter 8.
Immunologic Reactions.
The immune system serves an essential function in defense against infectious pathogens, but immune reactions may also cause cell injury. Injurious reactions to endogenous self-antigens are responsible for several autoimmune diseases (Chapter 6). Immune reactions to many external agents, such as microbes and environmental substances, are also important causes of cell and tissue injury (Chapters 2 and 6.
Genetic Derangements.
As described in Chapter 5, genetic abnormalities may result in a defect as severe as the congenital malformations associated with Down syndrome, caused by a chromosomal anomaly, or as subtle as the decreased life span of red blood cells caused by a single amino acid substitution in hemoglobin in sickle cell anemia. Genetic defects may cause cell injury because of deficiency of functional proteins, such as enzyme defects in inborn errors of metabolism, or accumulation of damaged DNA or misfolded proteins, both of which trigger cell death when they are beyond repair. Variations in the genetic makeup can also influence the susceptibility of cells to injury by chemicals and other environmental insults.
Nutritional Imbalances.
Nutritional imbalances continue to be major causes of cell injury. Protein-calorie deficiencies cause an appalling number of deaths, chiefly among underprivileged populations. Deficiencies of specific vitamins are found throughout the world (Chapter 9). Nutritional problems can be self-imposed, as in anorexia nervosa (self-induced starvation). Ironically, nutritional excesses have also become important causes of cell injury. Excess of cholesterol predisposes to atherosclerosis; obesity is associated with increased incidence of several important diseases, such as diabetes and cancer. Atherosclerosis is virtually endemic in the United States, and obesity is rampant. In addition to the problems of undernutrition and overnutrition, the composition of the diet makes a significant contribution to a number of diseases.
Morphologic Alterations in Cell Injury
It is useful to describe the basic alterations that occur in damaged cells before we discuss the biochemical mechanisms that bring about these changes. All stresses and noxious influences exert their effects first at the molecular or biochemical level. There is a time lag between the stress and the morphologic changes of cell injury or death; the duration of this delay may vary with the sensitivity of the methods used to detect these changes (Fig. 1-7). With histochemical or ultrastructural techniques, changes may be seen in minutes to hours after injury; however, it may take considerably longer (hours to days) before changes can be seen by light microscopy or on gross examination. As would be expected, the morphologic manifestations of necrosis take more time to develop than those of reversible damage. For example, in ischemia of the myocardium, cell swelling is a reversible morphologic change that may occur in a matter of minutes, and may progress to irreversibility within an hour or two. Unmistakable light microscopic changes of cell death, however, may not be seen until 4 to 12 hours after total ischemia.
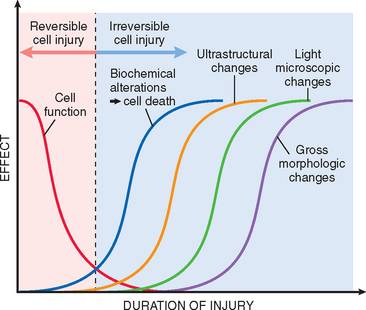
FIGURE 1-7 Sequential development of biochemical and morphologic changes in cell injury. Cells may become rapidly nonfunctional after the onset of injury, although they are still viable, with potentially reversible damage; a longer duration of injury may eventually lead to irreversible injury and cell death. Note that irreversible biochemical alterations may cause cell death, and typically this precedes ultrastructural, light microscopic, and grossly visible morphologic changes.
The sequential morphologic changes in cell injury progressing to cell death are illustrated in Figure 1-8. Reversible injury is characterized by generalized swelling of the cell and its organelles; blebbing of the plasma membrane; detachment of ribosomes from the ER; and clumping of nuclear chromatin. These morphologic changes are associated with decreased generation of ATP, loss of cell membrane integrity, defects in protein synthesis, cytoskeletal damage, and DNA damage. Within limits, the cell can repair these derangements and, if the injurious stimulus abates, will return to normalcy. Persistent or excessive injury, however, causes cells to pass the rather nebulous “point of no return” into irreversible injury and cell death. Different injurious stimuli may induce death by necrosis or apoptosis (Fig. 1-8 and Table 1-2). Severe mitochondrial damage with depletion of ATP and rupture of lysosomal and plasma membranes are typically associated with necrosis. Necrosis is the principal outcome in many commonly encountered injuries, such as those following ischemia, exposure to toxins, various infections, and trauma. Apoptosis has many unique features, and we will describe it later in the chapter.
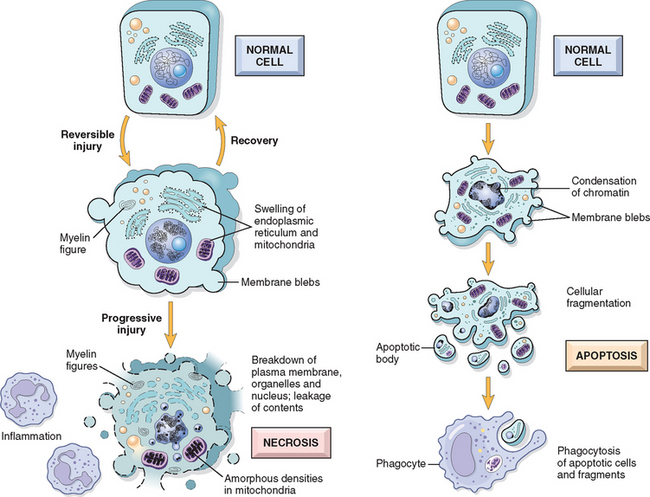
FIGURE 1-8 Schematic illustration of the morphologic changes in cell injury culminating in necrosis or apoptosis.
TABLE 1-2 Features of Necrosis and Apoptosis
| Feature | Necrosis | Apoptosis |
|---|---|---|
| Cell size | Enlarged (swelling) | Reduced (shrinkage) |
| Nucleus | Pyknosis → karyorrhexis → karyolysis | Fragmentation into nucleosome-size fragments |
| Plasma membrane | Disrupted | Intact; altered structure, especially orientation of lipids |
| Cellular contents | Enzymatic digestion; may leak out of cell | Intact; may be released in apoptotic bodies |
| Adjacent inflammation | Frequent | No |
| Physiologic or pathologic role | Invariably pathologic (culmination of irreversible cell injury) | Often physiologic, means of eliminating unwanted cells; may be pathologic after some forms of cell injury, especially DNA damage |
REVERSIBLE INJURY
Two features of reversible cell injury can be recognized under the light microscope: cellular swelling and fatty change. Cellular swelling appears whenever cells are incapable of maintaining ionic and fluid homeostasis and is the result of failure of energy-dependent ion pumps in the plasma membrane. Fatty change occurs in hypoxic injury and various forms of toxic or metabolic injury. It is manifested by the appearance of lipid vacuoles in the cytoplasm. It is seen mainly in cells involved in and dependent on fat metabolism, such as hepatocytes and myocardial cells. The mechanisms of fatty change are discussed later in the chapter.
Morphology. Cellular swelling is the first manifestation of almost all forms of injury to cells (Fig. 1-9B). It is a difficult morphologic change to appreciate with the light microscope; it may be more apparent at the level of the whole organ. When it affects many cells, it causes some pallor, increased turgor, and increase in weight of the organ. On microscopic examination, small clear vacuoles may be seen within the cytoplasm; these represent distended and pinched-off segments of the ER. This pattern of nonlethal injury is sometimes called hydropic change or vacuolar degeneration. Swelling of cells is reversible. Cells may also show increased eosinophilic staining, which becomes much more pronounced with progression to necrosis (described below).
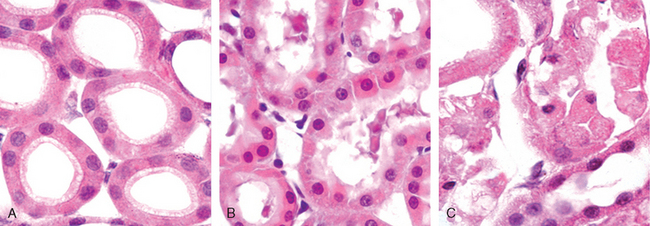
FIGURE 1-9 Morphologic changes in reversible cell injury and necrosis. A, Normal kidney tubules with viable epithelial cells. B, Early (reversible) ischemic injury showing surface blebs, increased eosinophilia of cytoplasm, and swelling of occasional cells. C, Necrosis (irreversible injury) of epithelial cells, with loss of nuclei, fragmentation of cells, and leakage of contents. The ultrastructural features of these stages of cell injury are shown in Figure 1-10.
(Courtesy of Drs. Neal Pinckard and M.A. Venkatachalam, University of Texas Health Sciences Center, San Antonio, TX.)
The ultrastructural changes of reversible cell injury (Fig. 1-10B) include:
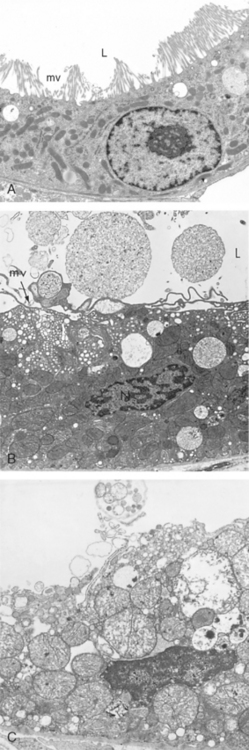
FIGURE 1-10 Ultrastructural features of reversible and irreversible cell injury (necrosis) in a rabbit kidney. A, Electron micrograph of a normal epithelial cell of the proximal kidney tubule. Note abundant microvilli (mv) lining the luminal surface (L). B, Epithelial cell of the proximal tubule showing early cell injury resulting from reperfusion following ischemia. The microvilli are lost and have been incorporated in apical cytoplasm; blebs have formed and are extruded in the lumen. Mitochondria would have been swollen during ischemia; with reperfusion, they rapidly undergo condensation and become electron-dense. C, Proximal tubular cell showing late injury, expected to be irreversible. Note the markedly swollen mitochondria containing electron-dense deposits, expected to contain precipitated calcium and proteins. Higher magnification micrographs of the cell would show disrupted plasma membrane and swelling and fragmentation of organelles.
(A, Courtesy of Dr. Brigitte Kaisslin, Institute of Anatomy, University of Zurich, Switzerland. B, C, Courtesy of Dr. M.A. Venkatachalam, University of Texas Health Sciences Center, San Antonio, TX.)
NECROSIS
The morphologic appearance of necrosis is the result of denaturation of intracellular proteins and enzymatic digestion of the lethally injured cell (cells placed immediately in fixative are dead but not necrotic). Necrotic cells are unable to maintain membrane integrity and their contents often leak out, a process that may elicit inflammation in the surrounding tissue. The enzymes that digest the necrotic cell are derived from the lysosomes of the dying cells themselves and from the lysosomes of leukocytes that are called in as part of the inflammatory reaction. Digestion of cellular contents and the host response may take hours to develop, and so there would be no detectable changes in cells if, for example, a myocardial infarct caused sudden death. The only circumstantial evidence might be occlusion of a coronary artery. The earliest histologic evidence of myocardial necrosis does not become apparent until 4 to 12 hours later. However, because of the loss of plasma membrane integrity, cardiac-specific enzymes and proteins are rapidly released from necrotic muscle and can be detected in the blood as early as 2 hours after myocardial cell necrosis.
Morphology. Necrotic cells show increased eosinophilia in hematoxylin and eosin (H & E) stains, attributable in part to the loss of cytoplasmic RNA (which binds the blue dye, hematoxylin) and in part to denatured cytoplasmic proteins (which bind the red dye, eosin). The necrotic cell may have a more glassy homogeneous appearance than do normal cells, mainly as a result of the loss of glycogen particles (Fig. 1-9C). When enzymes have digested the cytoplasmic organelles, the cytoplasm becomes vacuolated and appears moth-eaten. Dead cells may be replaced by large, whorled phospholipid masses called myelin figures that are derived from damaged cell membranes. These phospholipid precipitates are then either phagocytosed by other cells or further degraded into fatty acids; calcification of such fatty acid residues results in the generation of calcium soaps. Thus, the dead cells may ultimately become calcified. By electron microscopy, necrotic cells are characterized by discontinuities in plasma and organelle membranes, marked dilation of mitochondria with the appearance of large amorphous densities, intracytoplasmic myelin figures, amorphous debris, and aggregates of fluffy material probably representing denatured protein (see Fig. 1-10C).
Nuclear changes appear in one of three patterns, all due to nonspecific breakdown of DNA (see Fig. 1-9C). The basophilia of the chromatin may fade (karyolysis), a change that presumably reflects loss of DNA because of enzymatic degradation by endonucleases. A second pattern (which is also seen in apoptotic cell death) is pyknosis, characterized by nuclear shrinkage and increased basophilia. Here the chromatin condenses into a solid, shrunken basophilic mass. In the third pattern, known as karyorrhexis, the pyknotic nucleus undergoes fragmentation. With the passage of time (a day or two), the nucleus in the necrotic cell totally disappears.
Patterns of Tissue Necrosis
The discussion of necrosis has focused so far on changes in individual cells. When large numbers of cells die the tissue or organ is said to be necrotic; thus, a myocardial infarct is necrosis of a portion of the heart caused by death of many myocardial cells. Necrosis of tissues has several morphologically distinct patterns, which are important to recognize because they may provide clues about the underlying cause. Although the terms that describe these patterns are somewhat outmoded, they are used often and their implications are understood by pathologists and clinicians.
Morphology. Coagulative necrosis is a form of necrosis in which the architecture of dead tissues is preserved for a span of at least some days (Fig. 1-11). The affected tissues exhibit a firm texture. Presumably, the injury denatures not only structural proteins but also enzymes and so blocks the proteolysis of the dead cells; as a result, eosinophilic, anucleate cells may persist for days or weeks. Ultimately the necrotic cells are removed by phagocytosis of the cellular debris by infiltrating leukocytes and by digestion of the dead cells by the action of lysosomal enzymes of the leukocytes. Ischemia caused by obstruction in a vessel may lead to coagulative necrosis of the supplied tissue in all organs except the brain. A localized area of coagulative necrosis is called an infarct.
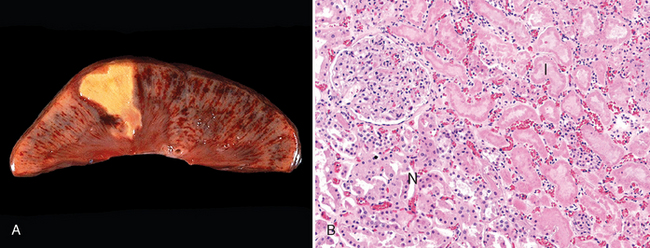
FIGURE 1-11 Coagulative necrosis. A, A wedge-shaped kidney infarct (yellow). B, Microscopic view of the edge of the infarct, with normal kidney (N) and necrotic cells in the infarct (I) showing preserved cellular outlines with loss of nuclei and an inflammatory infiltrate (which is difficult to discern at this magnification).
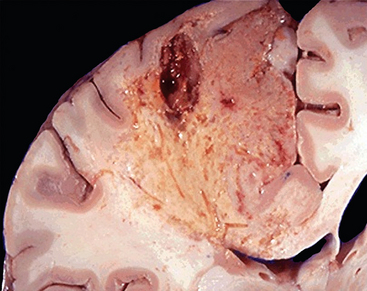
Liquefactive necrosis, in contrast to coagulative necrosis, is characterized by digestion of the dead cells, resulting in transformation of the tissue into a liquid viscous mass. It is seen in focal bacterial or, occasionally, fungal infections, because microbes stimulate the accumulation of leukocytes and the liberation of enzymes from these cells. The necrotic material is frequently creamy yellow because of the presence of dead leukocytes and is called pus. For unknown reasons, hypoxic death of cells within the central nervous system often manifests as liquefactive necrosis (Fig. 1-12).
Gangrenous necrosis is not a specific pattern of cell death, but the term is commonly used in clinical practice. It is usually applied to a limb, generally the lower leg, that has lost its blood supply and has undergone necrosis (typically coagulative necrosis) involving multiple tissue planes. When bacterial infection is superimposed there is more liquefactive necrosis because of the actions of degradative enzymes in the bacteria and the attracted leukocytes (giving rise to so-called wet gangrene).
Caseous necrosis is encountered most often in foci of tuberculous infection (Chapter 8). The term “caseous” (cheeselike) is derived from the friable white appearance of the area of necrosis (Fig. 1-13). On microscopic examination, the necrotic area appears as a collection of fragmented or lysed cells and amorphous granular debris enclosed within a distinctive inflammatory border; this appearance is characteristic of a focus of inflammation known as a granuloma (Chapter 2).
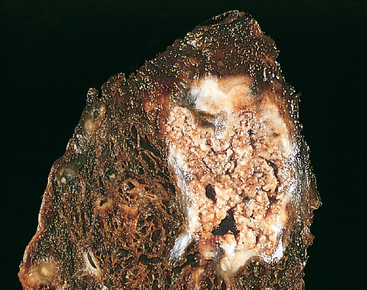
FIGURE 1-13 Caseous necrosis. Tuberculosis of the lung, with a large area of caseous necrosis containing yellow-white and cheesy debris.
Fat necrosis is a term that is well fixed in medical parlance but does not in reality denote a specific pattern of necrosis. Rather, it refers to focal areas of fat destruction, typically resulting from release of activated pancreatic lipases into the substance of the pancreas and the peritoneal cavity. This occurs in the calamitous abdominal emergency known as acute pancreatitis (Chapter 19). In this disorder pancreatic enzymes leak out of acinar cells and liquefy the membranes of fat cells in the peritoneum. The released lipases split the triglyceride esters contained within fat cells. The fatty acids, so derived, combine with calcium to produce grossly visible chalky-white areas (fat saponification), which enable the surgeon and the pathologist to identify the lesions (Fig. 1-14). On histologic examination the necrosis takes the form of foci of shadowy outlines of necrotic fat cells, with basophilic calcium deposits, surrounded by an inflammatory reaction.
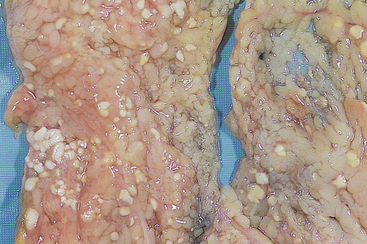
FIGURE 1-14 Fat necrosis. The areas of white chalky deposits represent foci of fat necrosis with calcium soap formation (saponification) at sites of lipid breakdown in the mesentery.
Fibrinoid necrosis is a special form of necrosis usually seen in immune reactions involving blood vessels. This pattern of necrosis typically occurs when complexes of antigens and antibodies are deposited in the walls of arteries. Deposits of these “immune complexes,” together with fibrin that has leaked out of vessels, result in a bright pink and amorphous appearance in H&E stains, called “fibrinoid” (fibrin-like) by pathologists (Fig. 1-15). The immunologically mediated vasculitis syndromes in which this type of necrosis is seen are described in Chapter 6.

Ultimately, in the living patient most necrotic cells and their contents disappear by phagocytosis of the debris and enzymatic digestion by leukocytes. If necrotic cells and cellular debris are not promptly destroyed and reabsorbed, they tend to attract calcium salts and other minerals and to become calcified. This phenomenon, called dystrophic calcification, is considered later in the chapter.
Mechanisms of Cell Injury
The discussion of the cellular pathology of cell injury and necrosis sets the stage for a consideration of the mechanisms and biochemical pathways of cell injury. The mechanisms responsible for cell injury are complex. There are, however, several principles that are relevant to most forms of cell injury:
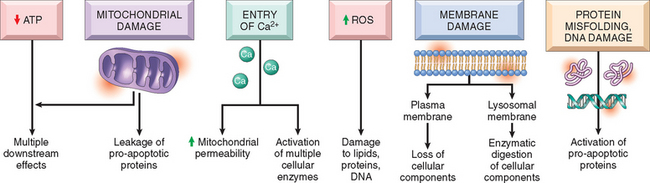
FIGURE 1-16 The principal mechanisms of cell injury, and their biochemical and functional effects, are shown. These are described in detail in the text.
In the following section we describe the biochemical mechanisms that may be activated by different injurious stimuli and contribute to cell injury.16 Our focus here is on reversible injury and necrosis, and the special cases of apoptosis and autophagy are best discussed separately.
DEPLETION OF ATP
ATP depletion and decreased ATP synthesis are frequently associated with both hypoxic and chemical (toxic) injury (Fig. 1-17). ATP is produced in two ways. The major pathway in mammalian cells is oxidative phosphorylation of adenosine diphosphate, in a reaction that results in reduction of oxygen by the electron transfer system of mitochondria. The second is the glycolytic pathway, which can generate ATP in the absence of oxygen using glucose derived either from body fluids or from the hydrolysis of glycogen. The major causes of ATP depletion are reduced supply of oxygen and nutrients, mitochondrial damage, and the actions of some toxins (e.g., cyanide). Tissues with a greater glycolytic capacity (e.g., the liver) are able to survive loss of oxygen and decreased oxidative phosphorylation better than are tissues with limited capacity for glycolysis (e.g., the brain).
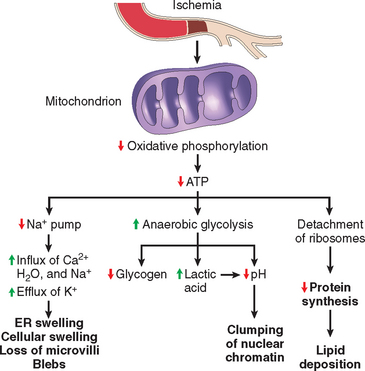
FIGURE 1-17 Functional and morphologic consequences of decreased intracellular ATP during cell injury. The morphologic changes shown here are indicative of reversible cell injury. Further depletion of ATP results in cell death, typically by necrosis. ER, endoplasmic reticulum.
High-energy phosphate in the form of ATP is required for virtually all synthetic and degradative processes within the cell. These include membrane transport, protein synthesis, lipogenesis, and the deacylation-reacylation reactions necessary for phospholipid turnover. Depletion of ATP to 5% to 10% of normal levels has widespread effects on many critical cellular systems:
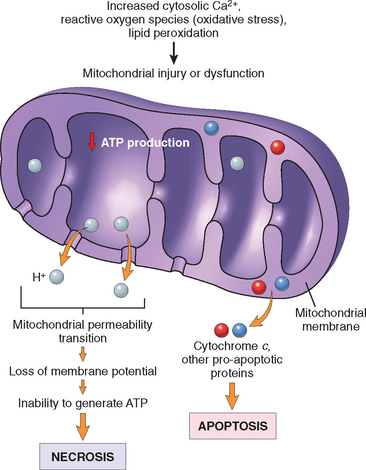
MITOCHONDRIAL DAMAGE
Mitochondria are the cell’s suppliers of life-sustaining energy in the form of ATP, but they are also critical players in cell injury and death.17 Mitochondria can be damaged by increases of cytosolic Ca2+, reactive oxygen species (discussed below), and oxygen deprivation, and so they are sensitive to virtually all types of injurious stimuli, including hypoxia and toxins. In addition, mutations in mitochondrial genes are the cause of some inherited diseases (Chapter 5).
There are two major consequences of mitochondrial damage.
INFLUX OF CALCIUM AND LOSS OF CALCIUM HOMEOSTASIS
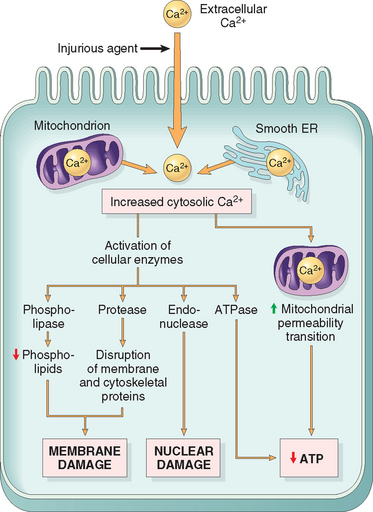
The finding that depleting calcium protects cells from injury induced by a variety of harmful stimuli indicates that calcium ions are important mediators of cell injury.19 Cytosolic free calcium is normally maintained at very low concentrations (−0.1 μmol) compared with extracellular levels of 1.3 mmol, and most intracellular calcium is sequestered in mitochondria and the ER. Ischemia and certain toxins cause an increase in cytosolic calcium concentration, initially because of release of Ca2+ from intracellular stores, and later resulting from increased influx across the plasma membrane (Fig. 1-19). Increased intracellular Ca2+ causes cell injury by several mechanisms.
ACCUMULATION OF OXYGEN-DERIVED FREE RADICALS (OXIDATIVE STRESS)
Cell injury induced by free radicals, particularly reactive oxygen species, is an important mechanism of cell damage in many pathologic conditions, such as chemical and radiation injury, ischemia-reperfusion injury (induced by restoration of blood flow in ischemic tissue), cellular aging, and microbial killing by phagocytes.21 Free radicals are chemical species that have a single unpaired electron in an outer orbit. Energy created by this unstable configuration is released through reactions with adjacent molecules, such as inorganic or organic chemicals—proteins, lipids, carbohydrates, nucleic acids—many of which are key components of cell membranes and nuclei. Moreover, free radicals initiate autocatalytic reactions, whereby molecules with which they react are themselves converted into free radicals, thus propagating the chain of damage. Reactive oxygen species (ROS) are a type of oxygen-derived free radical whose role in cell injury is well established. ROS are produced normally in cells during mitochondrial respiration and energy generation, but they are degraded and removed by cellular defense systems. Thus, cells are able to maintain a steady state in which free radicals may be present transiently at low concentrations but do not cause damage. When the production of ROS increases or the scavenging systems are ineffective, the result is an excess of these free radicals, leading to a condition called oxidative stress. Oxidative stress has been implicated in a wide variety of pathologic processes, including cell injury, cancer, aging, and some degenerative diseases such as Alzheimer disease. ROS are also produced in large amounts by leukocytes, particularly neutrophils and macrophages, as mediators for destroying microbes, dead tissue, and other unwanted substances. Therefore, injury caused by these reactive compounds often accompanies inflammatory reactions, during which leukocytes are recruited and activated (Chapter 2).
In the following section we discuss the generation and removal of ROS, and how they contribute to cell injury. The properties of some of the most important free radicals are summarized in Table 1-3.
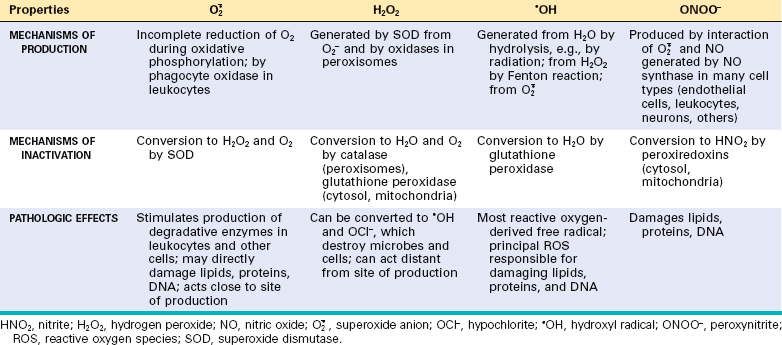
Generation of Free Radicals.
Free radicals may be generated within cells in several ways (Fig. 1-20):
 , one electron), hydrogen peroxide (H2O2, two electrons), and hydroxyl ions (
, one electron), hydrogen peroxide (H2O2, two electrons), and hydroxyl ions (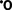 H, three electrons).H and hydrogen (H) free radicals.., and thus sources of iron and may cooperate in oxidative cell damage.
H, three electrons).H and hydrogen (H) free radicals.., and thus sources of iron and may cooperate in oxidative cell damage.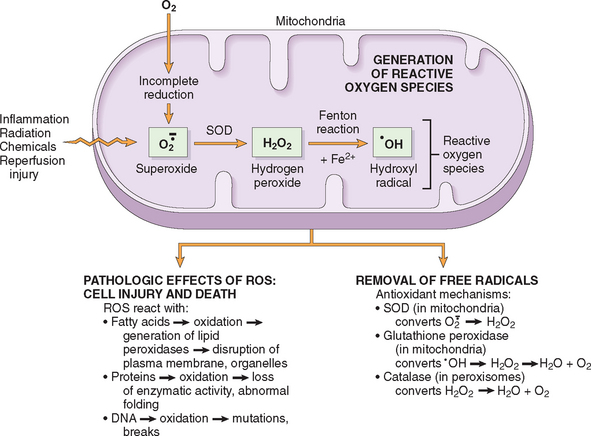
FIGURE 1-20 The role of reactive oxygen species (ROS) in cell injury. O2 is converted to superoxide () by oxidative enzymes in the endoplasmic reticulum (ER), mitochondria, plasma membrane, peroxisomes, and cytosol. is converted to H2O2 by dismutation and thence to H by the Cu2+/Fe2+-catalyzed Fenton reaction. H2O2 is also derived directly from oxidases in peroxisomes (not shown). Resultant free radical damage to lipids (by peroxidation), proteins, and DNA leads to injury to numerous cellular components. The major antioxidant enzymes are superoxide dismutase (SOD), glutathione peroxidase, and catalase.
Removal of Free Radicals.
Free radicals are inherently unstable and generally decay spontaneously. , for example, is unstable and decays (dismutates) spontaneously into O2 and H2O2 in the presence of water. In addition, cells have developed multiple nonenzymatic and enzymatic mechanisms to remove free radicals and thereby minimize injury (see Fig. 1-20). These include the following:
.21,23 These enzymes are lo-cated near the sites of generation of the oxidants and include the following:
to H2O2 (2 + 2H → H2O2 + O2). This group includes both manganese–SOD, which is localized in mitochondria, and copper-zinc–SOD, which is found in the cytosol.Pathologic Effects of Free Radicals.
The effects of ROS and other free radicals are wide-ranging, but three reactions are particularly relevant to cell injury (see Fig. 1-20):
H. The lipid–free radical interactions yield peroxides, which are themselves unstable and reactive, and an autocatalytic chain reaction ensues (called propagation), which can result in extensive membrane damage.The traditional thinking about free radicals was that they cause cell injury and death by necrosis, and, in fact, the production of ROS is a frequent prelude to necrosis. However, it is now clear that free radicals can trigger apoptosis as well.24 Recent studies have also revealed a role of ROS in signaling by a variety of cellular receptors and biochemical intermediates.25 In fact, according to one hypothesis, the major actions of stem from its ability to stimulate the production of degradative enzymes rather than direct damage of macromolecules. It is also possible that these potentially deadly molecules serve important physiologic functions.26
DEFECTS IN MEMBRANE PERMEABILITY
Early loss of selective membrane permeability leading ultimately to overt membrane damage is a consistent feature of most forms of cell injury (except apoptosis). Membrane damage may affect the functions and integrity of all cellular membranes. Below we discuss the mechanisms and pathologic consequences of membrane damage.
Mechanisms of Membrane Damage.
In ischemic cells membrane defects may be the result of ATP depletion and calcium-mediated activation of phospholipases (see below). The plasma membrane can also be damaged directly by various bacterial toxins, viral proteins, lytic complement components, and a variety of physical and chemical agents. Several biochemical mechanisms may contribute to membrane damage (Fig. 1-21):
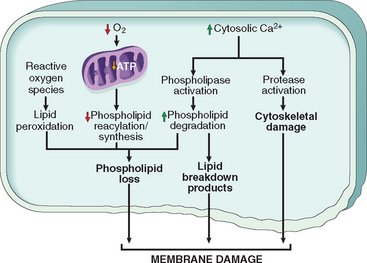
FIGURE 1-21 Mechanisms of membrane damage in cell injury. Decreased O2 and increased cytosolic Ca2+ are typically seen in ischemia but may accompany other forms of cell injury. Reactive oxygen species, which are often produced on reperfusion of ischemic tissues, also cause membrane damage (not shown).
Consequences of Membrane Damage.
The most important sites of membrane damage during cell injury are the mitochondrial membrane, the plasma membrane, and membranes of lysosomes.
DAMAGE TO DNA AND PROTEINS
Cells have mechanisms that repair damage to DNA, but if this damage is too severe to be corrected (e.g., after exposure to DNA damaging drugs, radiation, or oxidative stress), the cell initiates a suicide program that results in death by apoptosis. A similar reaction is triggered by improperly folded proteins, which may be the result of inherited mutations or external triggers such as free radicals. Because these mechanisms of cell injury typically cause apoptosis, they are discussed later in the chapter.
Before concluding our discussion of the mechanisms of cell injury, it is useful to consider the possible events that determine when reversible injury becomes irreversible and progresses to cell death. The clinical relevance of this question is obvious—if we can answer it we may be able to devise strategies for preventing cell injury from having permanent deleterious consequences. However, the molecular mechanisms connecting most forms of cell injury to ultimate cell death have proved elusive, for several reasons. The “point of no return,” at which the damage becomes irreversible, is still largely undefined, and there are no reliable morphologic or biochemical correlates of irreversibility. Two phenomena consistently characterize irreversibility—the inability to reverse mitochondrial dysfunction (lack of oxidative phosphorylation and ATP generation) even after resolution of the original injury, and profound disturbances in membrane function. As mentioned earlier, injury to lysosomal membranes results in the enzymatic dissolution of the injured cell that is characteristic of necrosis.
Leakage of intracellular proteins through the damaged cell membrane and ultimately into the circulation provides a means of detecting tissue-specific cellular injury and necrosis using blood serum samples. Cardiac muscle, for example, contains a specific isoform of the enzyme creatine kinase and of the contractile protein troponin; liver (and specifically bile duct epithelium) contains an isoform of the enzyme alkaline phosphatase; and hepatocytes contain transaminases. Irreversible injury and cell death in these tissues are reflected in increased levels of such proteins in the blood, and measurement of these biomarkers is used clinically to assess damage to these tissues.
Clinico-Pathologic Correlations: Selected Examples of Cell Injury and Necrosis
Having briefly reviewed the causes, morphology, and mechanisms of cell injury and necrotic cell death, we now describe some common and clinically significant forms of cell injury that typically culminate in necrosis. These examples illustrate many of the mechanisms and sequence of events in cell injury that were described above.
ISCHEMIC AND HYPOXIC INJURY
This is the most common type of cell injury in clinical medicine and has been studied extensively in humans, in experimental animals, and in culture systems. Hypoxia, referring to reduced oxygen availability, may occur in a variety of clinical settings, described earlier. In ischemia, on the other hand, the supply of oxygen and nutrients is decreased most often because of reduced blood flow as a consequence of a mechanical obstruction in the arterial system. It can also be caused by reduced venous drainage. In contrast to hypoxia, during which energy production by anaerobic glycolysis can continue, ischemia compromises the delivery of substrates for glycolysis. Thus, in ischemic tissues, not only is aerobic metabolism compromised but anaerobic energy generation also stops after glycolytic substrates are exhausted, or glycolysis is inhibited by the accumulation of metabolites that would have been removed otherwise by blood flow. For this reason, ischemia tends to cause more rapid and severe cell and tissue injury than does hypoxia in the absence of ischemia.
Mechanisms of Ischemic Cell Injury
The sequence of events following hypoxia or ischemia reflects many of the biochemical alterations in cell injury that have been described above. As the oxygen tension within the cell decreases, there is loss of oxidative phosphorylation and decreased generation of ATP. The depletion of ATP results in failure of the sodium pump, with loss of potassium, influx of sodium and water, and cell swelling. There is also influx of Ca2+, with its many deleterious effects. There is progressive loss of glycogen and decreased protein synthesis. The functional consequences may be severe at this stage. For instance, heart muscle ceases to contract within 60 seconds of coronary occlusion. Note, however, that loss of contractility does not mean cell death. If hypoxia continues, worsening ATP depletion causes further deterioration. The cytoskeleton disperses, resulting in the loss of ultrastructural features such as microvilli and the formation of “blebs” at the cell surface (see Figs. 1-9 and 1-10). “Myelin figures,” derived from degenerating cellular membranes, may be seen within the cytoplasm (in autophagic vacuoles) or extracellularly. They are thought to result from unmasking of phosphatide groups, promoting the uptake and intercalation of water between the lamellar stacks of membranes. At this time the mitochondria are usually swollen, as a result of loss of volume control in these organelles; the ER remains dilated; and the entire cell is markedly swollen, with increased concentrations of water, sodium, and chloride and a decreased concentration of potassium. If oxygen is restored, all of these disturbances are reversible.
If ischemia persists, irreversible injury and necrosis ensue. Irreversible injury is associated morphologically with severe swelling of mitochondria, extensive damage to plasma membranes (giving rise to myelin figures) and swelling of lysosomes (see Fig. 1-10C). Large, flocculent, amorphous densities develop in the mitochondrial matrix. In the myocardium, these are indications of irreversible injury and can be seen as early as 30 to 40 minutes after ischemia. Massive influx of calcium into the cell then occurs, particularly if the ischemic zone is reperfused. Death is mainly by necrosis, but apoptosis also contributes; the apoptotic pathway is probably activated by release of pro-apoptotic molecules from leaky mitochondria. The cell’s components are progressively degraded, and there is widespread leakage of cellular enzymes into the extracellular space and, conversely, entry of extracellular macromolecules from the interstitial space into the dying cells. Finally, the dead cells may become replaced by large masses composed of phospholipids in the form of myelin figures. These are then either phagocytosed by leukocytes or degraded further into fatty acids. Calcification of such fatty acid residues may occur, with the formation of calcium soaps.
As mentioned before, leakage of intracellular enzymes and other proteins across the abnormally permeable plasma membrane and into the blood provides important clinical indicators of cell death. For example, elevated serum levels of cardiac muscle creatine kinase MB and troponin are early signs of myocardial infarction, and may be seen before the infarct is detectable morphologically (Chapter 12).
Mammalian cells have developed protective responses to hypoxic stress. The best-defined of these is induction of a transcription factor called hypoxia-inducible factor-1, which promotes new blood vessel formation, stimulates cell survival pathways, and enhances anaerobic glycolysis.27 It remains to be seen if understanding of such oxygen-sensing mechanisms will lead to new strategies for preventing or treating ischemic and hypoxic cell injury.
Despite many investigations in experimental models there are still no reliable therapeutic approaches for reducing the injurious consequences of ischemia in clinical situations. The strategy that is perhaps the most useful in ischemic (and traumatic) brain and spinal cord injury is the transient induction of hypothermia (reducing the core body temperature to 92°F). This treatment reduces the metabolic demands of the stressed cells, decreases cell swelling, suppresses the formation of free radicals, and inhibits the host inflammatory response. All of these may contribute to decreased cell and tissue injury.28
ISCHEMIA-REPERFUSION INJURY
Restoration of blood flow to ischemic tissues can promote recovery of cells if they are reversibly injured. However, under certain circumstances, when blood flow is restored to cells that have been ischemic but have not died, injury is paradoxically exacerbated and proceeds at an accelerated pace. As a consequence, reperfused tissues may sustain loss of cells in addition to the cells that are irreversibly damaged at the end of ischemia. This process, called ischemia-reperfusion injury, is clinically important because it contributes to tissue damage during myocardial and cerebral infarction and following therapies to restore blood flow (Chapters 12 and 28.
How does reperfusion injury occur? The likely answer is that new damaging processes are set in motion during reperfusion, causing the death of cells that might have recovered otherwise.29 Several mechanisms have been proposed:
CHEMICAL (TOXIC) INJURY
Chemical injury remains a frequent problem in clinical medicine and is a major limitation to drug therapy. Because many drugs are metabolized in the liver, this organ is a frequent target of drug toxicity. In fact, toxic liver injury is perhaps the most frequent reason for terminating the therapeutic use or development of a drug.35 The mechanisms by which chemicals, certain drugs, and toxins produce injury are described in greater detail in Chapter 9 in the discussion of environmental diseases. Here we will describe the major pathways of chemically induced injury with selected examples.
Chemicals induce cell injury by one of two general mechanisms36:
Apoptosis
Apoptosis is a pathway of cell death that is induced by a tightly regulated suicide program in which cells destined to die activate enzymes that degrade the cells’ own nuclear DNA and nuclear and cytoplasmic proteins. Apoptotic cells break up into fragments, called apoptotic bodies, which contain portions of the cytoplasm and nucleus. The plasma membrane of the apoptotic cell and bodies remains intact, but its structure is altered in such a way that these become “tasty” targets for phagocytes. The dead cell and its fragments are rapidly devoured, before the contents have leaked out, and therefore cell death by this pathway does not elicit an inflammatory reaction in the host. The process was recognized in 1972 by the distinctive morphologic appearance of membrane-bound fragments derived from cells, and named after the Greek designation for “falling off.”37 It was quickly appreciated that apoptosis was a unique mechanism of cell death, distinct from necrosis, which is characterized by loss of membrane integrity, enzymatic digestion of cells, leakage of cellular contents, and frequently a host reaction (see Fig. 1-8 and Table 1-2). However, apoptosis and necrosis sometimes coexist, and apoptosis induced by some pathologic stimuli may progress to necrosis.
CAUSES OF APOPTOSIS
Apoptosis occurs normally both during development and throughout adulthood, and serves to eliminate unwanted, aged or potentially harmful cells. It is also a pathologic event when diseased cells become damaged beyond repair and are eliminated.
Apoptosis in Physiologic Situations
Death by apoptosis is a normal phenomenon that serves to eliminate cells that are no longer needed, and to maintain a steady number of various cell populations in tissues. It is important in the following physiologic situations:
Apoptosis in Pathologic Conditions
Apoptosis eliminates cells that are injured beyond repair without eliciting a host reaction, thus limiting collateral tissue damage. Death by apoptosis is responsible for loss of cells in a variety of pathologic states:
MORPHOLOGIC AND BIOCHEMICAL CHANGES IN APOPTOSIS
Before discussing the mechanisms of apoptosis, we describe the morphologic and biochemical characteristics of this process.
Morphology. The following morphologic features, some best seen with the electron microscope, characterize cells undergoing apoptosis (Fig. 1-22, and see Fig. 1-8).
FIGURE 1-22 Morphologic features of apoptosis. A, Apoptosis of an epidermal cell in an immune reaction. The cell is reduced in size and contains brightly eosinophilic cytoplasm and a condensed nucleus. B, This electron micrograph of cultured cells undergoing apoptosis shows some nuclei with peripheral crescents of compacted chromatin, and others that are uniformly dense or fragmented. C, These images of cultured cells undergoing apoptosis show blebbing and formation of apoptotic bodies (left panel, phase contrast micrograph), a stain for DNA showing nuclear fragmentation (middle panel), and activation of caspase-3 (right panel, immunofluorescence stain with an antibody specific for the active form of caspase-3, revealed as red color).
(B, From Kerr JFR, Harmon BV: Definition and incidence of apoptosis: a historical perspective. In Tomei LD, Cope FO (eds): Apoptosis: The Molecular Basis of Cell Death. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory Press, 1991, pp 5–29; C, Courtesy of Dr. Zheng Dong, Medical College of Georgia, Augusta, GA.)
Cell shrinkage. The cell is smaller in size; the cytoplasm is dense (Fig. 1-22A); and the organelles, though relatively normal, are more tightly packed. (Recall that in other forms of cell injury, an early feature is cell swelling, not shrinkage.)
Chromatin condensation. This is the most characteristic feature of apoptosis. The chromatin aggregates peripherally, under the nuclear membrane, into dense masses of various shapes and sizes (Fig. 1-22B). The nucleus itself may break up, producing two or more fragments.
Formation of cytoplasmic blebs and apoptotic bodies. The apoptotic cell first shows extensive surface blebbing, then undergoes fragmentation into membrane-bound apoptotic bodies composed of cytoplasm and tightly packed organelles, with or without nuclear fragments (Fig. 1-22C).
Phagocytosis of apoptotic cells or cell bodies, usually by macrophages. The apoptotic bodies are rapidly ingested by phagocytes and degraded by the phagocyte’s lysosomal enzymes.
Plasma membranes are thought to remain intact during apoptosis, until the last stages, when they become permeable to normally retained solutes. This classical description is accurate with respect to apoptosis during physiologic conditions such as embryogenesis and deletion of immune cells. However, forms of cell death with features of necrosis as well as of apoptosis are not uncommon after many injurious stimuli.39 Under such conditions the severity rather than the nature of the stimulus determines the pathway of cell death, necrosis being the major pathway when there is advanced ATP depletion and membrane damage.
On histologic examination, in tissues stained with hematoxylin and eosin, the apoptotic cell appears as a round or oval mass of intensely eosinophilic cytoplasm with fragments of dense nuclear chromatin (Fig. 1-22A). Because the cell shrinkage and formation of apoptotic bodies are rapid and the pieces are quickly phagocytosed, considerable apoptosis may occur in tissues before it becomes apparent in histologic sections. In addition, apoptosis—in contrast to necrosis—does not elicit inflammation, making it more difficult to detect histologically.
Biochemical Features of Apoptosis
Apoptotic cells usually exhibit a distinctive constellation of biochemical alterations that underlie the structural changes described above.
Activation of Caspases.
A specific feature of apoptosis is the activation of several members of a family of cysteine proteases named caspases.40 The term caspase is based on two properties of this family of enzymes: the “c” refers to a cysteine protease (i.e., an enzyme with cysteine in its active site), and “aspase” refers to the unique ability of these enzymes to cleave after aspartic acid residues. The caspase family, now including more than 10 members, can be divided functionally into two groups—initiator and executioner—depending on the order in which they are activated during apoptosis. Initiator caspases include caspase-8 and caspase-9. Several other caspases, including caspase-3 and caspase-6, serve as executioners. Like many proteases, caspases exist as inactive pro-enzymes, or zymogens, and must undergo an enzymatic cleavage to become active. The presence of cleaved, active caspases is a marker for cells undergoing apoptosis (Fig. 1-22C). We will discuss the roles of these enzymes in apoptosis later in this section.
DNA and Protein Breakdown.
Apoptotic cells exhibit a characteristic breakdown of DNA into large 50- to 300-kilobase pieces.41 Subsequently, there is cleavage of DNA by Ca2+- and Mg2+-dependent endonucleases into fragments whose sizes are multiples of 180 to 200 base pairs, reflecting cleavage between nucleosomal subunits. The fragments may be visualized by electrophoresis as DNA “ladders” (Fig. 1-23). Endonuclease activity also forms the basis for detecting cell death by cytochemical techniques that recognize double-stranded breaks of DNA.41 A “smeared” pattern of DNA fragmentation is thought to be indicative of necrosis, but this may be a late autolytic phenomenon, and typical DNA ladders are sometimes seen in necrotic cells as well.
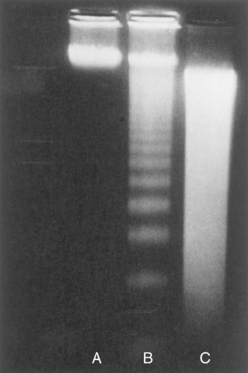
FIGURE 1-23 Agarose gel electrophoresis of DNA extracted from culture cells. Ethidium bromide stain; photographed under ultraviolet illumination. Lane A, Viable cells in culture. Lane B, Culture of cells exposed to heat showing extensive apoptosis; note ladder pattern of DNA fragments, which represent multiples of oligonucleosomes. Lane C, Culture showing cell necrosis; note diffuse smearing of DNA.
(From Kerr JFR, Harmon BV: Definition and incidence of apoptosis: a historical perspective. In Tomei LD, Cope FO: Apoptosis: The Molecular Basis of Cell Death. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory Press, 1991, p 13.)
Membrane Alterations and Recognition by Phagocytes.
The plasma membrane of apoptotic cells changes in ways that promote the recognition of the dead cells by phagocytes. One of these changes is the movement of some phospholipids (notably phosphatidylserine) from the inner leaflet to the outer leaflet of the membrane, where they are recognized by a number of receptors on phagocytes. These lipids are also detectable by binding of a protein called annexin V; thus, annexin V staining is commonly used to identify apoptotic cells. The clearance of apoptotic cells by phagocytes is described later.
MECHANISMS OF APOPTOSIS
All cells contain intrinsic mechanisms that signal death or survival, and apoptosis results from an imbalance in these signals. Because too much or too little apoptosis is thought to underlie many diseases, such as degenerative diseases and cancer, there is great interest in elucidating the mechanisms of this form of cell death. One of the remarkable facts to emerge is that the basic mechanisms of apoptosis—the genes and proteins that control the process and the sequence of events—are conserved in all multicellular organisms.38 In fact, some of the major breakthroughs came from observations made in the nematode Caenorhabditis elegans, whose development proceeds by a highly reproducible, programmed pattern of cell growth followed by cell death. Studies of mutant worms have allowed the identification of specific genes (called ced genes, for cell death abnormal) that initiate or inhibit apoptosis and for which there are defined mammalian homologues.38
The process of apoptosis may be divided into an initiation phase, during which some caspases become catalytically active, and an execution phase, during which other caspases trigger the degradation of critical cellular components. Initiation of apoptosis occurs principally by signals from two distinct pathways: the intrinsic, or mitochondrial, pathway, and the extrinsic, or death receptor–initiated, pathway (Fig. 1-24).42 These pathways are induced by distinct stimuli and involve different sets of proteins, although there is some cross-talk between them. Both pathways converge to activate caspases, which are the actual mediators of cell death.
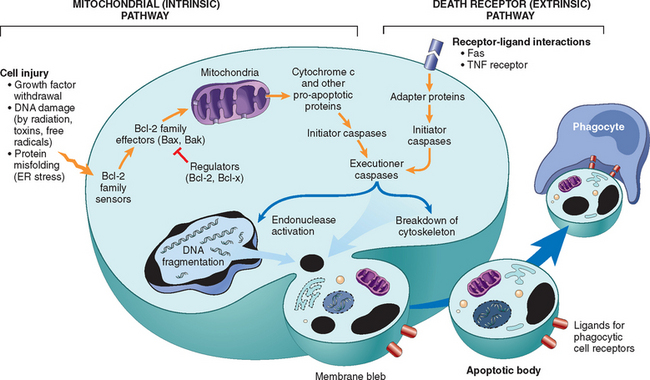
FIGURE 1-24 Mechanisms of apoptosis. The two pathways of apoptosis differ in their induction and regulation, and both culminate in the activation of “executioner” caspases. The induction of apoptosis by the mitochondrial pathway involves the action of sensors and effectors of the Bcl-2 family, which induce leakage of mitochondrial proteins. Also shown are some of the anti-apoptotic proteins (“regulators”) that inhibit mitochondrial leakiness and cytochrome c–dependent caspase activation in the mitochondrial pathway. In the death receptor pathway engagement of death receptors leads directly to caspase activation. The regulators of death receptor–mediated caspase activation are not shown. ER, endoplasmic reticulum; TNF, tumor necrosis factor.
The Intrinsic (Mitochondrial) Pathway of Apoptosis
The mitochondrial pathway is the major mechanism of apoptosis in all mammalian cells, and its role in a variety of physiologic and pathologic processes is well established. This pathway of apoptosis is the result of increased mitochondrial permeability and release of pro-apoptotic molecules (death inducers) into the cytoplasm (Fig. 1-25).42 Mitochondria are remarkable organelles in that they contain proteins such as cytochrome c that are essential for life, but some of the same proteins, when released into the cytoplasm (an indication that the cell is not healthy), initiate the suicide program of apoptosis. The release of these mitochondrial proteins is controlled by a finely orchestrated balance between pro- and anti-apoptotic members of the Bcl family of proteins.43 This family is named after Bcl-2, which was identified as an oncogene in a B-cell lymphoma and is homologous to the C. elegans protein Ced-9. There are more than 20 members of the Bcl family, and most of them function to regulate apoptosis. Growth factors and other survival signals stimulate production of anti-apoptotic proteins, the main ones being Bcl-2, Bcl-x, and Mcl-1. These proteins normally reside in the cytoplasm and in mitochondrial membranes, where they control mitochondrial permeability and prevent leakage of mitochondrial proteins that have the ability to trigger cell death (Fig. 1-25A). When cells are deprived of survival signals or their DNA is damaged, or misfolded proteins induce ER stress, sensors of damage or stress are activated. These sensors are also members of the Bcl family, and they include proteins called Bim, Bid, and Bad that contain a single “Bcl-2 homology domain” (the third of the four such domains present in Bcl-2) and are called “BH3-only proteins.” The sensors in turn activate two critical (proapoptotic) effectors, Bax and Bak, which form oligomers that insert into the mitochondrial membrane and create channels that allow proteins from the inner mitochondrial membrane to leak out into the cytoplasm. BH3-only proteins may also bind to and block the function of Bcl-2 and Bcl-x. At the same time, the synthesis of Bcl-2 and Bcl-x may decline. The net result of Bax-Bak activation coupled with loss of the protective functions of the anti-apoptotic Bcl family members is the release into the cytoplasm of several mitochondrial proteins that can activate the caspase cascade (Fig. 1-25B). One of these proteins is cytochrome c, well known for its role in mitochondrial respiration. Once released into the cytosol, cytochrome c binds to a protein called Apaf-1 (apoptosis-activating factor-1, homologous to Ced-4 in C. elegans), which forms a wheel-like hexamer that has been called the apoptosome.44 This complex is able to bind caspase-9, the critical initiator caspase of the mitochondrial pathway, and the enzyme cleaves adjacent caspase-9 molecules, thus setting up an auto-amplification process. Other mitochondrial proteins, with arcane names like Smac/DIABLO, enter the cytoplasm, where they bind to and neutralize cytoplasmic proteins that function as physiologic inhibitors of apoptosis (called IAPs). The normal function of the IAPs is to block the activation of caspases, including executioners like caspase-3, and keep cells alive.45,46 Thus, the neutralization of these IAPs permits the initiation of a caspase cascade.
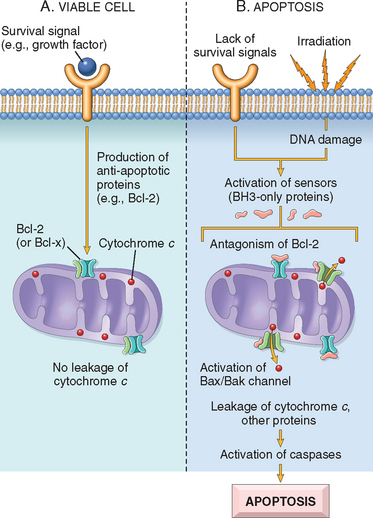
FIGURE 1-25 The intrinsic (mitochondrial) pathway of apoptosis. A, Cell viability is maintained by the induction of anti-apoptotic proteins such as Bcl-2 by survival signals. These proteins maintain the integrity of mitochondrial membranes and prevent leakage of mitochondrial proteins. B, Loss of survival signals, DNA damage, and other insults activate sensors that antagonize the anti-apoptotic proteins and activate the pro-apoptotic proteins Bax and Bak, which form channels in the mitochondrial membrane. The subsequent leakage of cytochrome c (and other proteins, not shown) leads to caspase activation and apoptosis.
There is some evidence that the intrinsic pathway of apoptosis can be triggered without a role for mitochondria.47 Apoptosis may be initiated by caspase activation upstream of mitochondria, and the subsequent increase in mitochondrial permeability and release of pro-apoptotic molecules serve to amplify the death signal. However, mechanisms of apoptosis involving mitochondria-independent initiation are not well defined.
The Extrinsic (Death Receptor–Initiated) Pathway of Apoptosis
This pathway is initiated by engagement of plasma membrane death receptors on a variety of cells.48-50 Death receptors are members of the TNF receptor family that contain a cytoplasmic domain involved in protein-protein interactions that is called the death domain because it is essential for delivering apoptotic signals. (Some TNF receptor family members do not contain cytoplasmic death domains; their function is to activate inflammatory cascades [Chapter 2], and their role in triggering apoptosis is much less established.) The best-known death receptors are the type 1 TNF receptor (TNFR1) and a related protein called Fas (CD95), but several others have been described. The mechanism of apoptosis induced by these death receptors is well illustrated by Fas, a death receptor expressed on many cell types (Fig. 1-26). The ligand for Fas is called Fas ligand (FasL). FasL is expressed on T cells that recognize self antigens (and functions to eliminate self-reactive lymphocytes), and on some cytotoxic T lymphocytes (which kill virus-infected and tumor cells). When FasL binds to Fas, three or more molecules of Fas are brought together, and their cytoplasmic death domains form a binding site for an adapter protein that also contains a death domain and is called FADD (Fas-associated death domain). FADD that is attached to the death receptors in turn binds an inactive form of caspase-8 (and, in humans, caspase-10), again via a death domain. Multiple pro-caspase-8 molecules are thus brought into proximity, and they cleave one another to generate active caspase-8. The enzyme then triggers a cascade of caspase activation by cleaving and thereby activating other pro-caspases, and the active enzymes mediate the execution phase of apoptosis (discussed below). This pathway of apoptosis can be inhibited by a protein called FLIP, which binds to pro-caspase-8 but cannot cleave and activate the caspase because it lacks a protease domain.51 Some viruses and normal cells produce FLIP and use this inhibitor to protect themselves from Fas-mediated apoptosis.

FIGURE 1-26 The extrinsic (death receptor–initiated) pathway of apoptosis, illustrated by the events following Fas engagement. FAAD, Fas-associated death domain; FasL, Fas ligand.
We have described the extrinsic and intrinsic pathways for initiating apoptosis as distinct because they involve fundamentally different molecules for their initiation, but there may be interconnections between them. For instance, in hepatocytes and several other cell types, Fas signaling activates a BH3-only protein called Bid, which then activates the mitochondrial pathway.
The Execution Phase of Apoptosis
The two initiating pathways converge to a cascade of caspase activation, which mediates the final phase of apoptosis. As we have seen, the mitochondrial pathway leads to activation of the initiator caspase-9, and the death receptor pathway to the initiators caspase-8 and -10. After an initiator caspase is cleaved to generate its active form, the enzymatic death program is set in motion by rapid and sequential activation of the executioner caspases. Executioner caspases, such as caspase-3 and -6, act on many cellular components. For instance, these caspases, once activated, cleave an inhibitor of a cytoplasmic DNase and thus make the DNase enzymatically active; this enzyme induces the characteristic cleavage of DNA into nucleosome-sized pieces, described earlier. Caspases also degrade structural components of the nuclear matrix, and thus promote fragmentation of nuclei. Some of the steps in apoptosis are not fully defined. For instance, we do not know how the structure of the plasma membrane is changed in apoptotic cells, or how membrane blebs and apoptotic bodies are formed.
Removal of Dead Cells
The formation of apoptotic bodies breaks cells up into “bite-sized” fragments that are edible for phagocytes. Apoptotic cells and their fragments also undergo several changes in their membranes that actively promote their phagocytosis so they are cleared before they undergo secondary necrosis and release their cellular contents (which can result in injurious inflammation). In healthy cells phosphatidylserine is present on the inner leaflet of the plasma membrane, but in apoptotic cells this phospholipid “flips” out and is expressed on the outer layer of the membrane, where it is recognized by several macrophage receptors. Cells that are dying by apoptosis secrete soluble factors that recruit phagocytes.52 Some apoptotic bodies express thrombospondin, an adhesive glycoprotein that is recognized by phagocytes, and macrophages themselves may produce proteins that bind to apoptotic cells (but not to live cells) and thus target the dead cells for engulfment. Apoptotic bodies may also become coated with natural antibodies and proteins of the complement system, notably C1q, which are recognized by phagocytes.53 Thus, numerous receptors on phagocytes and ligands induced on apoptotic cells are involved in the binding and engulfment of these cells. This process of phagocytosis of apoptotic cells is so efficient that dead cells disappear, often within minutes, without leaving a trace, and inflammation is absent even in the face of extensive apoptosis.
CLINICO-PATHOLOGIC CORRELATIONS: APOPTOSIS IN HEALTH AND DISEASE
Examples of Apoptosis
Cell death in many situations is known to be caused by apoptosis, and the selected examples listed below illustrate the role of this death pathway in normal physiology and in disease.54
Growth Factor Deprivation.
Hormone-sensitive cells deprived of the relevant hormone, lymphocytes that are not stimulated by antigens and cytokines, and neurons deprived of nerve growth factor die by apoptosis. In all these situations, apoptosis is triggered by the intrinsic (mitochondrial) pathway and is attributable to decreased synthesis of Bcl-2 and Bcl-x and activation of Bim and other pro-apoptotic members of the Bcl family.
DNA Damage.
Exposure of cells to radiation or chemotherapeutic agents induces apoptosis by a mechanism that is initiated by DNA damage (genotoxic stress) and that involves the tumor-suppressor gene p53.55 p53 protein accumulates in cells when DNA is damaged, and it arrests the cell cycle (at the G1 phase) to allow time for repair (Chapter 7). However, if the damage is too great to be repaired successfully, p53 triggers apoptosis. When p53 is mutated or absent (as it is in certain cancers), it is incapable of inducing apoptosis, so that cells with damaged DNA are allowed to survive. In such cells the DNA damage may result in mutations or translocations that lead to neoplastic transformation (Chapter 7). Thus, p53 serves as a critical “life or death” switch following genotoxic stress. The mechanism by which p53 triggers the distal death effector machinery—the caspases—is complex but seems to involve its function in transcriptional activation. Among the proteins whose production is stimulated by p53 are several pro-apoptotic members of the Bcl family, notably Bax, Bak, and some BH3-only proteins, mentioned earlier.
Protein Misfolding.
Chaperones in the ER control the proper folding of newly synthesized proteins, and misfolded polypeptides are ubiquitinated and targeted for proteolysis in proteasomes. If, however, unfolded or misfolded proteins accumulate in the ER, because of inherited mutations or stresses, they trigger a number of cellular responses, collectively called the unfolded protein response.56,57 This unfolded protein response activates signaling pathways that increase the production of chaperones, enhance proteasomal degradation of abnormal proteins, and slow protein translation, thus reducing the load of misfolded proteins in the cell (Fig. 1-27). However, if this cytoprotective response is unable to cope with the accumulation of misfolded proteins, the cell activates caspases and induces apoptosis.58-60 This process is called ER stress. Intracellular accumulation of abnormally folded proteins, caused by genetic mutations, aging, or unknown environmental factors, is now recognized as a feature of a number of neurodegenerative diseases, including Alzheimer, Huntington, and Parkinson diseases (Chapter 28), and possibly type 2 diabetes.61 Deprivation of glucose and oxygen, and stress such as heat, also result in protein misfolding, culminating in cell injury and death.
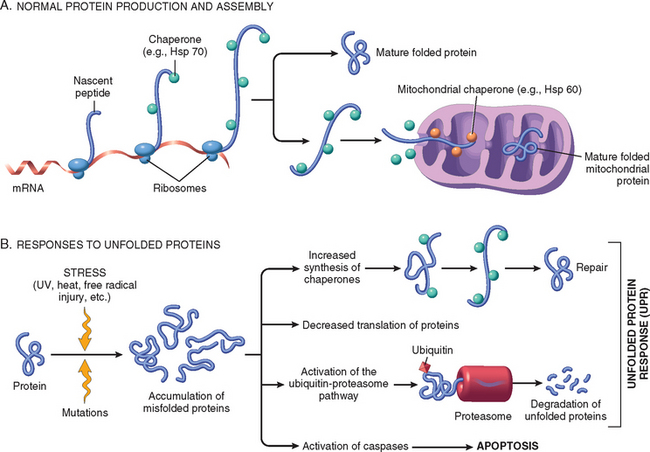
FIGURE 1-27 Mechanisms of protein folding and the unfolded protein response. A, Chaperones, such as heat shock proteins (Hsp), protect unfolded or partially folded proteins from degradation and guide proteins into organelles. B, Misfolded proteins trigger a protective unfolded protein response (UPR). If this response is inadequate to cope with the level of misfolded proteins, it induces apoptosis.
Apoptosis Induced By the TNF Receptor Family.
FasL on T cells binds to Fas on the same or neighboring lymphocytes. This interaction plays a role in the elimination of lymphocytes that recognize self-antigens, and mutations affecting Fas or FasL result in autoimmune diseases in humans and mice (Chapter 6).62 The cytokine TNF is an important mediator of the inflammatory reaction (Chapter 2), but it is also capable of inducing apoptosis. (The name “tumor necrosis factor” arose not because the cytokine kills tumor cells directly but because it induces thrombosis of tumor blood vessels, resulting in ischemic death of the tumor.) TNF-mediated death is readily demonstrated in cell cultures, but its physiologic or pathologic significance in vivo is not known. In fact, the major physiologic functions of TNF are mediated not by inducing apoptosis but by activating the important transcription factor NF-κB (nuclear factor-κB), which promotes cell survival by stimulating synthesis of anti-apoptotic members of the Bcl-2 family and, as we shall see in Chapter 2, activates a number of inflammatory responses. Since TNF can induce cell death and promote cell survival, what determines this yin and yang of its action? The answer is unclear, but it probably depends on which signaling proteins attach to the TNF receptor after binding of the cytokine.
Cytotoxic T Lymphocyte–Mediated Apoptosis.
Cytotoxic T lymphocytes (CTLs) recognize foreign antigens presented on the surface of infected host cells (Chapter 6). Upon activation, CTLs secrete perforin, a transmembrane pore-forming molecule, which promotes entry of the CTL granule serine proteases called granzymes. Granzymes have the ability to cleave proteins at aspartate residues and thus activate a variety of cellular caspases.63 In this way the CTL kills target cells by directly inducing the effector phase of apoptosis. CTLs also express FasL on their surface and may kill target cells by ligation of Fas receptors.
Disorders Associated with Dysregulated Apoptosis
Dysregulated apoptosis (“too little or too much”) has been postulated to explain aspects of a wide range of diseases.56
Autophagy
Autophagy is a process in which a cell eats its own contents. It is a survival mechanism in times of nutrient deprivation, when the starved cell lives by cannibalizing itself and recycling the digested contents. In this process intracellular organelles and portions of cytosol are first sequestered from the cytoplasm in an autophagic vacuole, which subsequently fuses with lysosomes to form an autophagolysosome, and the cellular components are digested by lysosomal enzymes (Fig. 1-28).64,65 Interest in autophagy has been spurred by the finding that it is regulated by a defined set of “autophagy genes” (called Atgs) in single-celled organisms and mammalian cells. The products of many of these genes function in the creation of the autophagic vacuole, but how they do so is unknown. It has also been suggested that autophagy triggers cell death that is distinct from necrosis and apoptosis.66 However, the mechanism of this type of cell death is not known, nor is it clear that the cell death is caused by autophagy rather than by the stress that triggers autophagy. Nevertheless, autophagy has been invoked as a mechanism of cell loss in various diseases, including degenerative diseases of the nervous system and muscle; in many of these disorders, the damaged cells contain abundant autophagic vacuoles.67
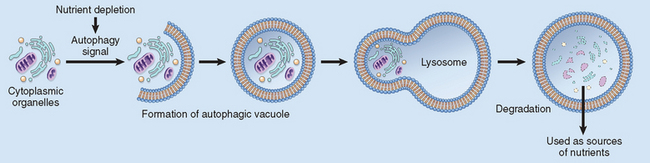
FIGURE 1-28 Autophagy. Cellular stresses, such as nutrient deprivation, activate autophagy genes that create vacuoles in which cellular organelles are sequestered and then degraded following fusion of the vesicles with lysosomes. The digested materials are recycled to provide nutrients for the cell.
Intracellular Accumulations
One of the manifestations of metabolic derangements in cells is the intracellular accumulation of abnormal amounts of various substances. The stockpiled substances fall into two categories: (1) a normal cellular constituent, such as water, lipids, proteins, and carbohydrates, that accumulates in excess; or (2) an abnormal substance, either exogenous, such as a mineral or products of infectious agents, or endogenous, such as a product of abnormal synthesis or metabolism. These substances may accumulate either transiently or permanently, and they may be harmless to the cells, but on occasion they are severely toxic. The substance may be located in either the cytoplasm (frequently within phagolysosomes) or the nucleus. In some instances the cell may be producing the abnormal substance, and in others it may be merely storing products of pathologic processes occurring elsewhere in the body.
Many processes result in abnormal intracellular accumulations, but most accumulations are attributable to four types of abnormalities (Fig. 1-29).
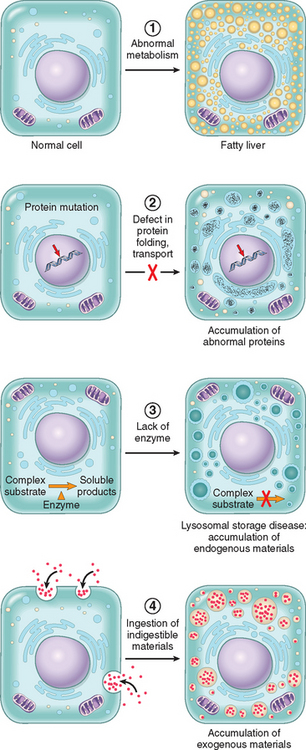
In many cases, if the overload can be controlled or stopped, the accumulation is reversible. In inherited storage diseases accumulation is progressive, and the overload may cause cellular injury, leading in some instances to death of the tissue and the patient.
LIPIDS
All major classes of lipids can accumulate in cells: triglycerides, cholesterol/cholesterol esters, and phospholipids. Phospholipids are components of the myelin figures found in necrotic cells. In addition, abnormal complexes of lipids and carbohydrates accumulate in the lysosomal storage diseases (Chapter 5). Here we concentrate on triglyceride and cholesterol accumulations.
Steatosis (Fatty Change)
The terms steatosis and fatty change describe abnormal accumulations of triglycerides within parenchymal cells. Fatty change is often seen in the liver because it is the major organ involved in fat metabolism, but it also occurs in heart, muscle, and kidney. The causes of steatosis include toxins, protein malnutrition, diabetes mellitus, obesity, and anoxia. In developed nations the most common causes of significant fatty change in the liver (fatty liver) are alcohol abuse and nonalcoholic fatty liver disease, which is often associated with diabetes and obesity (Chapter 18).
Different mechanisms account for triglyceride accumulation in the liver. Free fatty acids from adipose tissue or ingested food are normally transported into hepatocytes. In the liver they are esterified to triglycerides, converted into cholesterol or phospholipids, or oxidized to ketone bodies. Some fatty acids are synthesized from acetate as well. Release of triglycerides from the hepatocytes requires association with apoproteins to form lipoproteins, which may then be transported from the blood into the tissues (Chapter 4). Excess accumulation of triglycerides within the liver may result from excessive entry or defective metabolism and export of lipids (Fig. 1-30A). Several such defects are induced by alcohol, a hepatotoxin that alters mitochondrial and microsomal functions, leading to increased synthesis and reduced breakdown of lipids (Chapter 18). CCl4 and protein malnutrition cause fatty change by reducing synthesis of apoproteins, hypoxia inhibits fatty acid oxidation, and starvation increases fatty acid mobilization from peripheral stores.
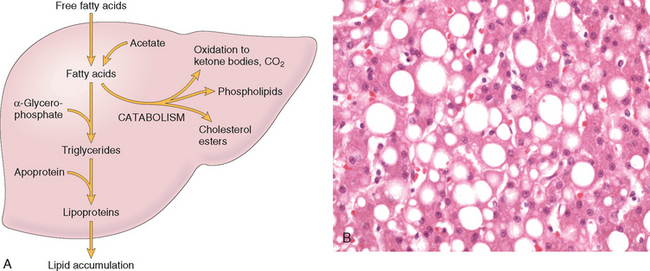
FIGURE 1-30 Fatty liver. A, Schematic diagram of the possible mechanisms leading to accumulation of triglycerides in fatty liver. Defects in any of the steps of uptake, catabolism, or secretion can result in lipid accumulation. B, High-power detail of fatty change of the liver. In most cells the well-preserved nucleus is squeezed into the displaced rim of cytoplasm about the fat vacuole.
(B, Courtesy of Dr. James Crawford, Department of Pathology, University of Florida School of Medicine, Gainesville, FL.)
The significance of fatty change depends on the cause and severity of the accumulation. When mild it may have no effect on cellular function. More severe fatty change may impair cellular function and may be a harbinger of cell death.
Morphology. Fatty change is most often seen in the liver and heart. In all organs fatty change appears as clear vacuoles within parenchymal cells. Intracellular accumulations of water or polysaccharides (e.g., glycogen) may also produce clear vacuoles. The identification of lipids requires the avoidance of fat solvents commonly used in paraffin embedding for routine hematoxylin and eosin stains. To identify the fat, it is necessary to prepare frozen tissue sections of either fresh or aqueous formalin-fixed tissues. The sections may then be stained with Sudan IV or Oil Red-O, both of which impart an orange-red color to the contained lipids. The periodic acid-Schiff (PAS) reaction, coupled with digestion by the enzyme diastase, is used to identify glycogen, although it is not specific. When neither fat nor polysaccharide can be demonstrated within a clear vacuole, it is presumed to contain water or fluid with a low protein content.
Liver. In the liver, mild fatty change may not affect the gross appearance. With progressive accumulation, the organ enlarges and becomes increasingly yellow until, in extreme instances, the liver may weigh two to four times normal and be transformed into a bright yellow, soft, greasy organ.
Fatty change begins with the development of minute, membrane-bound inclusions (liposomes) closely applied to the ER. Accumulation of fat is first seen by light microscopy as small vacuoles in the cytoplasm around the nucleus. As the process progresses the vacuoles coalesce, creating cleared spaces that displace the nucleus to the periphery of the cell (Fig. 1-30B). Occasionally contiguous cells rupture and the enclosed fat globules coalesce, producing so-called fatty cysts.
Heart. Lipid is found in cardiac muscle in the form of small droplets, occurring in two patterns. In one, prolonged moderate hypoxia, such as that produced by profound anemia, causes intracellular deposits of fat, which create grossly apparent bands of yellowed myocardium alternating with bands of darker, red-brown, uninvolved myocardium (tigered effect). The other pattern of fatty change is produced by more profound hypoxia or by some forms of myocarditis (e.g., diphtheria infection) and shows more uniformly affected myocytes.
Cholesterol and Cholesterol Esters
The cellular metabolism of cholesterol (discussed in detail in Chapter 5) is tightly regulated such that most cells use cholesterol for the synthesis of cell membranes without intracellular accumulation of cholesterol or cholesterol esters. Accumulations manifested histologically by intracellular vacuoles are seen in several pathologic processes.
PROTEINS
Intracellular accumulations of proteins usually appear as rounded, eosinophilic droplets, vacuoles, or aggregates in the cytoplasm. By electron microscopy they can be amorphous, fibrillar, or crystalline in appearance. In some disorders, such as certain forms of amyloidosis, abnormal proteins deposit primarily in extracellular spaces (Chapter 6).
Excesses of proteins within the cells sufficient to cause morphologically visible accumulation have diverse causes.
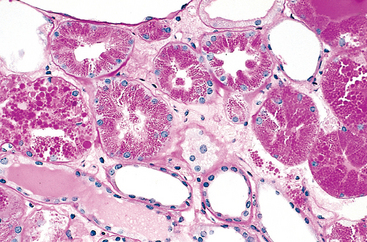
HYALINE CHANGE
The term hyaline usually refers to an alteration within cells or in the extracellular space that gives a homogeneous, glassy, pink appearance in routine histologic sections stained with hematoxylin and eosin. It is widely used as a descriptive histologic term rather than a specific marker for cell injury. This morphologic change is produced by a variety of alterations and does not represent a specific pattern of accumulation. Intracellular accumulations of protein, described earlier (reabsorption droplets, Russell bodies, alcoholic hyaline), are examples of intracellular hyaline deposits.
Extracellular hyaline has been more difficult to analyze. Collagenous fibrous tissue in old scars may appear hyalinized, but the biochemical basis of this change is not clear. In long-standing hypertension and diabetes mellitus, the walls of arterioles, especially in the kidney, become hyalinized, resulting from extravasated plasma protein and deposition of basement membrane material.
GLYCOGEN
Glycogen is a readily available energy source stored in the cytoplasm of healthy cells. Excessive intracellular deposits of glycogen are seen in patients with an abnormality in either glucose or glycogen metabolism. Whatever the clinical setting, the glycogen masses appear as clear vacuoles within the cytoplasm. Glycogen dissolves in aqueous fixatives; for its localization, tissues are best fixed in absolute alcohol. Staining with Best carmine or the PAS reaction imparts a rose-to-violet color to the glycogen, and diastase digestion of a parallel section before staining serves as a further control by hydrolyzing the glycogen.
Diabetes mellitus is the prime example of a disorder of glucose metabolism. In this disease glycogen is found in renal tubular epithelial cells, as well as within liver cells, β cells of the islets of Langerhans, and heart muscle cells.
Glycogen accumulates within the cells in a group of related genetic disorders that are collectively referred to as the glycogen storage diseases, or glycogenoses (Chapter 5). In these diseases enzymatic defects in the synthesis or breakdown of glycogen result in massive accumulation, causing cell injury and cell death.
PIGMENTS
Pigments are colored substances, some of which are normal constituents of cells (e.g., melanin), whereas others are abnormal and accumulate in cells only under special circumstances. Pigments can be exogenous, coming from outside the body, or endogenous, synthesized within the body itself.
Exogenous Pigments
The most common exogenous pigment is carbon (coal dust), a ubiquitous air pollutant of urban life. When inhaled it is picked up by macrophages within the alveoli and is then transported through lymphatic channels to the regional lymph nodes in the tracheobronchial region. Accumulations of this pigment blacken the tissues of the lungs (anthracosis) and the involved lymph nodes. In coal miners the aggregates of carbon dust may induce a fibroblastic reaction or even emphysema and thus cause a serious lung disease known as coal worker’s pneumoconiosis (Chapter 15). Tattooing is a form of localized, exogenous pigmentation of the skin. The pigments inoculated are phagocytosed by dermal macrophages, in which they reside for the remainder of the life of the embellished (sometimes with embarrassing consequences for the bearer of the tattoo!). The pigments do not usually evoke any inflammatory response.
Endogenous Pigments
Lipofuscin is an insoluble pigment, also known as lipochrome or wear-and-tear pigment. Lipofuscin is composed of polymers of lipids and phospholipids in complex with protein, suggesting that it is derived through lipid peroxidation of polyunsaturated lipids of subcellular membranes. Lipofuscin is not injurious to the cell or its functions. Its importance lies in its being a telltale sign of free radical injury and lipid peroxidation. The term is derived from the Latin (fuscus, brown), referring to brown lipid. In tissue sections it appears as a yellow-brown, finely granular cytoplasmic, often perinuclear, pigment (Fig. 1-33). It is seen in cells undergoing slow, regressive changes and is particularly prominent in the liver and heart of aging patients or patients with severe malnutrition and cancer cachexia.
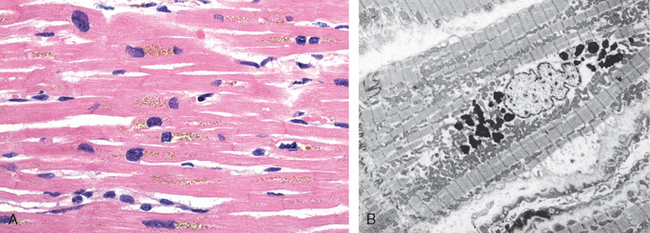
FIGURE 1-33 Lipofuscin granules in a cardiac myocyte shown by (A) light microscopy (deposits indicated by arrows), and (B) electron microscopy (note the perinuclear, intralysosomal location).
Melanin, derived from the Greek (melas, black), is an en-dogenous, non-hemoglobin-derived, brown-black pigment formed when the enzyme tyrosinase catalyzes the oxidation of tyrosine to dihydroxyphenylalanine in melanocytes. It is discussed further in Chapter 25. For practical purposes melanin is the only endogenous brown-black pigment. The only other that could be considered in this category is homogentisic acid, a black pigment that occurs in patients with alkaptonuria, a rare metabolic disease. Here the pigment is deposited in the skin, connective tissue, and cartilage, and the pigmentation is known as ochronosis (Chapter 5).
Hemosiderin is a hemoglobin-derived, golden yellow-to-brown, granular or crystalline pigment that serves as one of the major storage forms of iron. Iron metabolism and hemosiderin are considered in detail in Chapters 14 and 18. Iron is normally carried by specific transport proteins, transferrins. In cells, it is stored in association with a protein, apoferritin, to form ferritin micelles. Ferritin is a constituent of most cell types. When there is a local or systemic excess of iron, ferritin forms hemosiderin granules, which are easily seen with the light microscope (Fig. 1-34). Hemosiderin pigment represents aggregates of ferritin micelles. Under normal conditions small amounts of hemosiderin can be seen in the mononuclear phagocytes of the bone marrow, spleen, and liver, which are actively engaged in red cell breakdown.
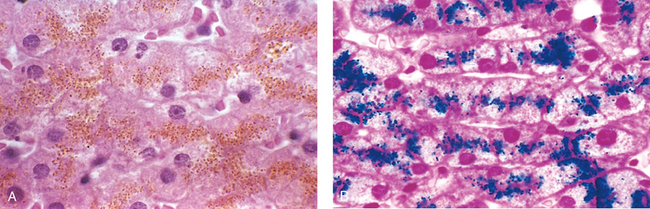
FIGURE 1-34 Hemosiderin granules in liver cells. A, H+E stain showing golden-brown, finely granular pigment. B, Prussian blue stain, specific for iron (seen as blue granules).
Local or systemic excesses of iron cause hemosiderin to accumulate within cells. Local excesses result from hemorrhages in tissues. The best example of localized hemosiderosis is the common bruise. Extravasated red blood cells at the site of injury are phagocytosed over several days by macrophages, which break down the hemoglobin and recover the iron. After removal of iron, the heme moiety is converted first to biliverdin (“green bile”) and then to bilirubin (“red bile”). In parallel, the iron released from heme is incorporated into ferritin and eventually hemosiderin. These conversions account for the often dramatic play of colors seen in a healing bruise, which typically changes from red-blue to green-blue to golden-yellow before it is resolved.
When there is systemic overload of iron hemosiderin may be deposited in many organs and tissues, a condition called hemosiderosis. The main causes of hemosiderosis are (1) increased absorption of dietary iron, (2) hemolytic anemias, in which abnormal quantities of iron are released from erythrocytes, and (3) repeated blood transfusions because the transfused red cells constitute an exogenous load of iron. These conditions are discussed in Chapter 18.
Morphology. Iron pigment appears as a coarse, golden, granular pigment lying within the cell’s cytoplasm (Fig. 1-34A). It can be visualized in tissues by the Prussian blue histochemical reaction, in which colorless potassium ferrocyanide is converted by iron to blue-black ferric ferrocyanide (Fig. 1-34B). When the underlying cause is the localized breakdown of red cells, the hemosiderin is found initially in the phagocytes in the area. In systemic hemosiderosis it is found at first in the mononuclear phagocytes of the liver, bone marrow, spleen, and lymph nodes and in scattered macrophages throughout other organs such as the skin, pancreas, and kidneys. With progressive accumulation, parenchymal cells throughout the body (principally in the liver, pancreas, heart, and endocrine organs) become pigmented.
In most instances of systemic hemosiderosis the pigment does not damage the parenchymal cells or impair organ function. The more extreme accumulation of iron, however, in an inherited disease called hemochromatosis, is associated with liver, heart, and pancreatic damage, resulting in liver fibrosis, heart failure, and diabetes mellitus (Chapter 18).
Bilirubin is the normal major pigment found in bile. It is derived from hemoglobin but contains no iron. Its normal formation and excretion are vital to health, and jaundice is a common clinical disorder caused by excesses of this pigment within cells and tissues. Bilirubin metabolism and jaundice are discussed in Chapter 18.
Pathologic Calcification
Pathologic calcification is the abnormal tissue deposition of calcium salts, together with smaller amounts of iron, magnesium, and other mineral salts. There are two forms of pathologic calcification. When the deposition occurs locally in dying tissues it is known as dystrophic calcification; it occurs despite normal serum levels of calcium and in the absence of derangements in calcium metabolism. In contrast, the deposition of calcium salts in otherwise normal tissues is known as metastatic calcification, and it almost always results from hypercalcemia secondary to some disturbance in calcium metabolism.
DYSTROPHIC CALCIFICATION
Dystrophic calcification is encountered in areas of necrosis, whether they are of coagulative, caseous, or liquefactive type, and in foci of enzymatic necrosis of fat. Calcification is almost always present in the atheromas of advanced atherosclerosis. It also commonly develops in aging or damaged heart valves, further hampering their function (Fig. 1-35). Whatever the site of deposition, the calcium salts appear macroscopically as fine, white granules or clumps, often felt as gritty deposits. Sometimes a tuberculous lymph node is virtually converted to stone.
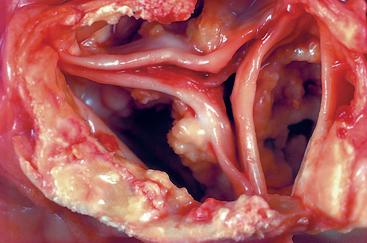
FIGURE 1-35 Dystrophic calcification of the aortic valve. View looking down onto the unopened aortic valve in a heart with calcific aortic stenosis. It is markedly narrowed (stenosis). The semilunar cusps are thickened and fibrotic, and behind each cusp are irregular masses of piled-up dystrophic calcification.
Morphology. Histologically, with the usual hematoxylin and eosin stain, calcium salts have a basophilic, amorphous granular, sometimes clumped appearance. They can be intracellular, extracellular, or in both locations. In the course of time, heterotopic bone may be formed in the focus of calcification. On occasion single necrotic cells may constitute seed crystals that become encrusted by the mineral deposits. The progressive acquisition of outer layers may create lamellated configurations, called psammoma bodies because of their resemblance to grains of sand. Some types of papillary cancers (e.g., thyroid) are apt to develop psammoma bodies. In asbestosis, calcium and iron salts gather about long slender spicules of asbestos in the lung, creating exotic, beaded dumbbell forms (Chapter 15).
Pathogenesis.
In the pathogenesis of dystrophic calcification, the final common pathway is the formation of crystalline calcium phosphate mineral in the form of an apatite similar to the hydroxyapatite of bone. It is thought that calcium is concentrated in membrane-bound vesicles in cells by a process that is initiated by membrane damage and has several steps: (1) calcium ion binds to the phospholipids present in the vesicle membrane; (2) phosphatases associated with the membrane generate phosphate groups, which bind to the calcium; (3) the cycle of calcium and phosphate binding is repeated, raising the local concentrations and producing a deposit near the membrane; and (4) a structural change occurs in the arrangement of calcium and phosphate groups, generating a microcrystal, which can then propagate and lead to more calcium deposition.
Although dystrophic calcification may simply be a telltale sign of previous cell injury, it is often a cause of organ dysfunction. Such is the case in calcific valvular disease and atherosclerosis, as will become clear in further discussion of these diseases.
METASTATIC CALCIFICATION
Metastatic calcification may occur in normal tissues whenever there is hypercalcemia. Hypercalcemia also accentuates dystrophic calcification. There are four principal causes of hypercalcemia: (1) increased secretion of parathyroid hormone (PTH) with subsequent bone resorption, as in hyperparathyroidism due to parathyroid tumors, and ectopic secretion of PTH-related protein by malignant tumors (Chapter 7); (2) destruction of bone tissue, secondary to primary tumors of bone marrow (e.g., multiple myeloma, leukemia) or diffuse skeletal metastasis (e.g., breast cancer), accelerated bone turnover (e.g., Paget disease), or immobilization; (3) vitamin D–related disorders, including vitamin D intoxication, sarcoidosis (in which macrophages activate a vitamin D precursor), and idiopathic hypercalcemia of infancy (Williams syndrome), characterized by abnormal sensitivity to vitamin D; and (4) renal failure, which causes retention of phosphate, leading to secondary hyperparathyroidism. Less common causes include aluminum intoxication, which occurs in patients on chronic renal dialysis, and milk-alkali syndrome, which is due to excessive ingestion of calcium and absorbable antacids such as milk or calcium carbonate.
Metastatic calcification may occur widely throughout the body but principally affects the interstitial tissues of the gastric mucosa, kidneys, lungs, systemic arteries, and pulmonary veins. Though quite different in location, all of these tissues excrete acid and therefore have an internal alkaline compartment that predisposes them to metastatic calcification. In all these sites the calcium salts morphologically resemble those described in dystrophic calcification. Thus, they may occur as noncrystalline amorphous deposits or, at other times, as hydroxyapatite crystals.
Usually the mineral salts cause no clinical dysfunction, but on occasion massive involvement of the lungs produces remarkable x-ray films and respiratory deficits. Massive deposits in the kidney (nephrocalcinosis) may in time cause renal damage (Chapter 20).
Cellular Aging
Shakespeare probably characterized aging best in his elegant description of the seven ages of man. It begins at the moment of conception, involves the differentiation and maturation of the organism and its cells, at some variable point in time leads to the progressive loss of functional capacity characteristic of senescence, and ends in death. With age there are physiologic and structural alterations in almost all organ systems. Aging in individuals is affected to a great extent by genetic factors, diet, social conditions, and occurrence of age-related diseases, such as atherosclerosis, diabetes, and osteoarthritis. In addition, there is good evidence that aging-induced alterations in cells are an important component of the aging of the organism. Here we discuss cellular aging because it could represent the progressive accumulation over the years of sublethal injury that may lead to cell death or to a diminished capacity of the cell to respond to injury.
Cellular aging is the result of a progressive decline in cellular function and viability caused by genetic abnormalities and the accumulation of cellular and molecular damage due to the effects of exposure to exogenous influences (Fig. 1-36). Studies in model systems have clearly established that aging is a regulated process that is influenced by a limited number of genes,69 and genetic anomalies underlie syndromes resembling premature aging in humans as well.70 Such findings suggest that aging is associated with definable mechanistic alterations. The known changes that contribute to cellular aging include the following.
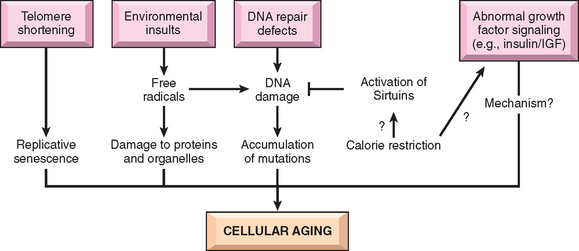
FIGURE 1-36 Mechanisms of cellular aging. Genetic factors and environmental insults combine to produce the cellular abnormalities characteristic of aging. How calorie restrictions prolong life span is net established. IGF, insulin-like growth factor.
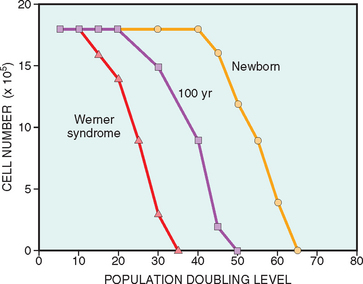
FIGURE 1-37 Finite population doublings of primary human fibroblasts derived from a newborn, a 100-year-old person, and a 20-year-old patient with Werner syndrome. The ability of cells to grow to a confluent monolayer decreases with increasing population-doubling levels.
(From Dice JF: Cellular and molecular mechanisms of aging. Physiol Rev 73:150, 1993.)
It is still not known why aging is associated with progressive senescence of cells.72 One probable mechanism in human cells is that with each cell division there is incomplete replication of chromosome ends (telomere shortening), which ultimately results in cell cycle arrest. Telomeres are short repeated sequences of DNA (TTAGGG) present at the linear ends of chromosomes that are important for ensuring the complete replication of chromosomal ends and for protecting chromosomal termini from fusion and degradation.73 When somatic cells replicate, a small section of the telomere is not duplicated and telomeres become progressively shortened. As the telomeres become shorter the ends of chromosomes cannot be protected and are seen as broken DNA, which activates the DNA damage response and signals cell cycle arrest. Telomere length is normally maintained by nucleotide addition mediated by an enzyme called telomerase. Telomerase is a specialized RNA-protein complex that uses its own RNA as a template for adding nucleotides to the ends of chromosomes (Fig. 1-38A). The activity of telomerase is repressed by regulatory proteins, which provide a mechanism for sensing telomere length and restrict unnecessary elongation. Telomerase activity is highest in germ cells and present at lower levels in stem cells, but it is usually undetectable in most somatic tissues (Fig. 1-38B). Therefore, as somatic cells divide, their telomeres become shorter, and they exit the cell cycle, resulting in an inability to generate new cells to replace damaged ones. Thus, both accumulation of senescent cells and depletion of stem cell pools via senescence contribute to aging. Conversely, in immortal cancer cells telomerase is reactivated and telomeres are stable, suggesting that maintenance of telomere length might be an important—possibly essential—step in tumor formation (Chapter 7). Despite such alluring observations, however, the relationship of telomerase activity and telomeric length to aging and cancer still must be fully established.74
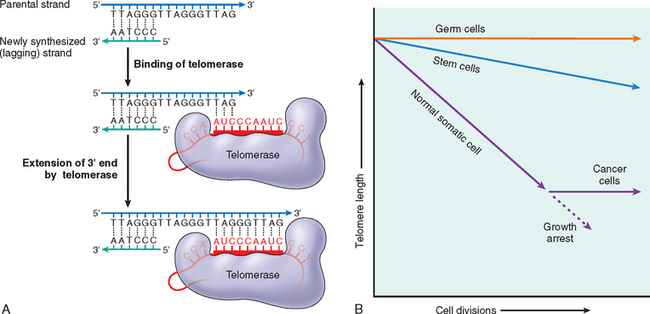
FIGURE 1-38 The role of telomeres and telomerase in replicative senescence of cells. A, Telomerase directs RNA template-dependent DNA synthesis, in which nucleotides are added to one strand at the end of a chromosome. The lagging strand is filled in by DNA polymerase. B, Telomere-telomerase hypothesis and proliferative capacity of cells. Telomere length is plotted against the number of cell divisions. Germ cells and stem cells both contain active telomerase, but only the germ cells have sufficient levels of the enzyme to stabilize telomere length completely. In normal somatic cells there is no telomerase activity, and telomeres progressively shorten with successive cell divisions until growth arrest, or senescence, occurs. Telomerase activation in cancer cells counteracts the telomere shortening that limits the proliferative capacity of normal somatic cells.
(A, Data from Alberts BR, et al: Molecular Biology of the Cell. New York, Garland Science, 2002. B, Modified and redrawn with permission from Holt SE, et al: Refining the telomere-telomerase hypothesis of aging and cancer. Nat Biotechnol 14:836, 1996. Copyright 1996, Macmillan Magazines Limited.)
Replicative senescence can also be induced by increased expression of the cell cycle inhibitor p16INK4a and by DNA damage (discussed further below). How these factors contribute to normal aging is not known.75
anion radical, and (2) overexpression of the antioxidative enzymes SOD and catalase extends life span in transgenic forms of Drosophila. Free radicals may have deleterious effects on DNA, leading to breaks and genome instability, thus affecting all cellular functions.77
Several protective responses counterbalance progressive damage in cells, and an important one is the recognition and repair of damaged DNA. Although most DNA damage is repaired by endogenous DNA repair enzymes, some persists and accumulates as cells age. Several lines of evidence point to the importance of DNA repair in the aging process. Patients with Werner syndrome show premature aging, and the defective gene product is a DNA helicase—a protein involved in DNA replication and repair and other functions requiring DNA unwinding.78 A defect in this enzyme causes rapid accumulation of chromosomal damage that may mimic the injury that normally accumulates during cellular aging. Genetic instability in somatic cells is also characteristic of other disorders in which patients display some of the manifestations of aging at an increased rate, such as ataxia-telangiectasia, in which the mutated gene encodes a protein involved in repairing double-strand breaks in DNA (Chapter 7). Thus, the balance between cumulative metabolic damage and the response to that damage could determine the rate at which we age. In this scenario aging can be delayed by decreasing the accumulation of damage or by increasing the response to that damage.
Not only damaged DNA but damaged cellular organelles also accumulate as cells age. In part this may be the result of declining function of the proteasome, the proteolytic machine that serves to eliminate abnormal and unwanted intracellular proteins.79
Studies in model organisms, from yeast to mammals, have shown that the most effective way of prolonging life span is calorie restriction. How this works is still not established, but the effect of calorie restriction on longevity appears to be mediated by a family of proteins called sirtuins.80 Sirtuins have histone deacetylase activity, and are thought to promote the expression of several genes whose products increase longevity. These products include proteins that increase metabolic activity, reduce apoptosis, stimulate protein folding, and inhibit the harmful effects of oxygen free radicals.81 Sirtuins also increase insulin sensitivity and glucose metabolism, and may be targets for the treatment of diabetes. Not surprisingly, optimistic wine-lovers have been delighted to hear that a constituent of red wine may activate sirtuins and thus increase life span! Other studies have shown that growth factors, such as insulin-like growth factor, and intracellular signaling pathways triggered by these hormones also influence life span.69 Transcription factors activated by insulin receptor signaling may induce genes that reduce longevity, and insulin receptor mutations are associated with increased life span. The relevance of these findings to aging in humans is an area of active investigation.
It should be apparent that the various forms of cellular derangements and adaptations described in this chapter cover a wide spectrum, ranging from adaptations in cell size, growth, and function; to the reversible and irreversible forms of acute cell injury; to the regulated type of cell death represented by apoptosis; to the pathologic alterations in cell organelles; and to the less ominous forms of intracellular accumulations, including pigmentations. Reference is made to all these alterations throughout this book, because all organ injury and ultimately all clinical disease arise from derangements in cell structure and function.
1 Majno G. The Healing Hand: Man and Wound in the Ancient World. Cambridge: Harvard University Press, 1975;43.
2 Anversa P, Nadal-Ginard B. Myocyte renewal and ventricular remodeling. Nature. 2002;415:240.
3 Glass DJ. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol. 2003;5:87.
4 Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45.
5 Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589.
6 Dorn GW. The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962.
7 Roots I, et al. Genotype and phenotype relationship in drug metabolism. Ernst Schering Res Found Workshop. 2007;59:81.
8 Tanimizu N, Miyajima A. Molecular mechanism of liver development and regeneration. Int Rev Cytol. 2007;259:1.
9 Kandarian SC, Jackman RW. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve. 2006;33:155.
10 Sacheck JM, et al. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007;21:140.
11 Tosh D, Slack JM. How cells change their phenotype. Nat Rev Mol Cell Biol. 2002;3:187.
12 Slack JM. Metaplasia and transdifferentiation: from pure biology to the clinic. Nat Rev Mol Cell Biol. 2007;8:369.
13 Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663.
14 Kroemer G, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;12(Suppl 2):1463.
15 Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37.
16 Vanlangenakker N, et al. Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med. 2008;8:207.
17 Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481.
18 Bernardi P, et al. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077.
19 Deng Z, et al. Calcium in cell injury and death. Annu Rev Pathol. 2006;1:405.
20 Orrenius S, et al. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552.
21 Valko M, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44.
22 Szabo C, et al. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662.
23 Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181.
24 Ryter SW, et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal. 2007;9:49.
25 D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813.
26 Afanas’ev IB. Signaling functions of free radicals superoxide and nitric oxide under physiological and pathological conditions. Mol Biotechnol. 2007;37:2.
27 Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol. 2006;70:1469.
28 Rincon F, Mayer SA. Therapeutic hypothermia for brain injury after cardiac arrest. Semin Neurol. 2006;26:387.
29 de Groot H, Rauen U. Ischemia-reperfusion injury: processes in pathogenetic networks: a review. Transplant Proc. 2007;39:481.
30 Kaminski KA, et al. Oxidative stress and neutrophil activation—the two keystones of ischemia/reperfusion injury. Int J Cardiol. 2002;86:41.
31 Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res. 2006;70:181.
32 Frangogiannis NG, et al. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31.
33 Riedemann NC, Ward PA. Complement in ischemia reperfusion injury. Am J Pathol. 2003;162:363.
34 Zhang M, et al. The role of natural IgM in myocardial ischemia-reperfusion injury. J Mol Cell Cardiol. 2006;41:62.
35 Bjornsson E. Drug-induced liver injury: Hy’s rule revisited. Clin Pharmacol Ther. 2006;79:521.
36 Kaplowitz N. Biochemical and cellular mechanisms of toxic liver injury. Semin Liver Dis. 2002;22:137.
37 Kerr JF, et al. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239.
38 Metzstein MM, et al. Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet. 1998;14:410.
39 Wyllie AH. Apoptosis: an overview. Br Med Bull. 1997;53:451.
40 Lavrik IN, et al. Caspases: pharmacological manipulation of cell death. J Clin Invest. 2005;115:2665.
41 McCarthy NJ, Evan GI. Methods for detecting and quantifying apoptosis. Curr Top Dev Biol. 1998;36:259.
42 Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205.
43 Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647.
44 Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405.
45 Vaux DL, Silke J. Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun. 2003;304:499.
46 Shiozaki EN, Shi Y. Caspases, IAPs and Smac/DIABLO: mechanisms from structural biology. Trends Biochem Sci. 2004;29:486.
47 Joza N, et al. Genetic analysis of the mammalian cell death machinery. Trends Genet. 2002;18:142.
48 Wallach D, et al. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999;17:331.
49 Nagata S. Fas ligand-induced apoptosis. Annu Rev Genet. 1999;33:29.
50 Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26.
51 Callus BA, Vaux DL. Caspase inhibitors: viral, cellular and chemical. Cell Death Differ. 2007;14:73.
52 Ravichandran KS. “Recruitment signals” from apoptotic cells: invitation to a quiet meal. Cell. 2003;113:817.
53 Ogden CA, Elkon KB. Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Curr Dir Autoimmun. 2006;9:120.
54 Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258:479.
55 Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12:440.
56 Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol. 2001;13:349.
57 Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739.
58 Xu C, et al. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656.
59 Macario AJ, Conway de Macario E. Sick chaperones, cellular stress, and disease. N Engl J Med. 2005;353:1489.
60 Marx J. Cell biology. A stressful situation. Science. 2006;313:1564.
61 Lin JH, et al. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399.
62 Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 2002;109(Suppl):S97.
63 Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20:323.
64 Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159.
65 Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427.
66 Maiuri MC, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741.
67 Huang J, Klionsky DJ. Autophagy and human disease. Cell Cycle. 2007;6:1837.
68 Omary MB, et al. “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem Sci. 2006;31:383.
69 Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449.
70 Martin GM, Oshima J. Lessons from human progeroid syndromes. Nature. 2000;408:263.
71 Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585.
72 Patil CK, et al. The thorny path linking cellular senescence to organismal aging. Mech Ageing Dev. 2005;126:1040.
73 Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106:661.
74 Stewart SA, Weinberg RA. Telomeres: cancer to human aging. Annu Rev Cell Dev Biol. 2006;22:531.
75 Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223.
76 Balaban RS, et al. Mitochondria, oxidants, and aging. Cell. 2005;120:483.
77 Lombard DB, et al. DNA repair, genome stability, and aging. Cell. 2005;120:497.
78 Kyng KJ, Bohr VA. Gene expression and DNA repair in progeroid syndromes and human aging. Ageing Res Rev. 2005;4:579.
79 Carrard G, et al. Impairment of proteasome structure and function in aging. Int J Biochem Cell Biol. 2002;34:1461.
80 Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1.
81 Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298.