Chapter 20 The Kidney
What is a human but an ingenious machine designed to turn, with “infinite artfulness, the red wine of Shiraz into urine”? So said the storyteller in Isak Dinesen’s Seven Gothic Tales.1 More accurately but less poetically, human kidneys serve to convert more than 1700 liters of blood per day into about 1 liter of a highly specialized concentrated fluid called urine. In so doing the kidney excretes the waste products of metabolism, precisely regulates the body’s concentration of water and salt, maintains the appropriate acid balance of plasma, and serves as an endocrine organ, secreting such hormones as erythropoietin, renin, and prostaglandins. The physiologic mechanisms that the kidney has developed to carry out these functions require a high degree of structural complexity.
Renal diseases are responsible for a great deal of morbidity but, fortunately, are not equally major causes of mortality. To place the problem in some perspective, approximately 45,000 deaths are attributed yearly to renal disease in the United States, in contrast to about 650,000 to heart disease, 560,000 to cancer, and 145,000 to stroke.2 Morbidity, however, is by no means insignificant. Millions of people are affected annually by nonfatal kidney diseases, most notably infections of the kidney or lower urinary tract, kidney stones, and urinary obstruction. Twenty percent of all women suffer from infection of the urinary tract or kidney at some time in their lives, and as many as 5% of the U.S. population develops renal stones. Similarly, modern treatments, notably dialysis and transplantation, keep many patients alive who earlier would have died of renal failure, adding to the pool of renal morbidity. Further, people with even mild chronic kidney disease have greatly enhanced risk for cardiovascular disease.
The study of kidney diseases is facilitated by dividing them into those that affect the four basic morphologic components: glomeruli, tubules, interstitium, and blood vessels. This traditional approach is useful, since the early manifestations of disease affecting each of these components tend to be distinct. Further, some components seem to be more vulnerable to specific forms of renal injury; for example, most glomerular diseases are immunologically mediated, whereas tubular and interstitial disorders are frequently caused by toxic or infectious agents. Nevertheless, some agents affect more than one structure. In addition, the anatomic and functional interdependence of the components of the kidney implies that damage to one almost always secondarily affects the others. Disease primarily in the blood vessels, for example, inevitably affects all the structures that depend on this blood supply. Severe glomerular damage impairs the flow through the peritubular vascular system and also delivers potentially toxic products to tubules; conversely, tubular destruction, by increasing intraglomerular pressure, may induce glomerular injury. Thus, whatever the origin, there is a tendency for all forms of chronic kidney disease ultimately to destroy all four components of the kidney, culminating in chronic renal failure and what has been called end-stage kidneys. The functional reserve of the kidney is large, and much damage may occur before there is evident functional impairment. For these reasons the early signs and symptoms are particularly important clinically.
Clinical Manifestations of Renal Diseases
The clinical manifestations of renal disease can be grouped into reasonably well-defined syndromes. Some are peculiar to glomerular diseases, and others are present in diseases that affect any one of the components. Before we list the syndromes, a few terms must be clarified.
Azotemia is a biochemical abnormality that refers to an elevation of the blood urea nitrogen (BUN) and creatinine levels, and is related largely to a decreased glomerular filtration rate (GFR). Azotemia is a consequence of many renal disorders, but it also arises from extrarenal disorders. Prerenal azotemia is encountered when there is hypoperfusion of the kidneys (e.g., in hemorrhage, shock, volume depletion, and congestive heart failure) that impairs renal function in the absence of parenchymal damage. Postrenal azotemia is seen whenever urine flow is obstructed beyond the level of the kidney. Relief of the obstruction is followed by correction of the azotemia.
When azotemia becomes associated with a constellation of clinical signs and symptoms and biochemical abnormalities, it is termed uremia. Uremia is characterized not only by failure of renal excretory function but also by a host of metabolic and endocrine alterations resulting from renal damage. Uremic patients frequently manifest secondary involvement of the gastrointestinal system (e.g., uremic gastroenteritis), peripheral nerves (e.g., peripheral neuropathy), and heart (e.g., uremic fibrinous pericarditis).
We can now turn to a brief description of the clinical presentations of renal disease:
Renal Failure.
Acute renal failure implies a rapid and frequently reversible deterioration of renal function. It is discussed in the section on “Acute Kidney Injury (Acute Tubular Necrosis),” because it commonly occurs in this disorder. Here, the discussion is limited to chronic renal failure, which is the end result of a variety of renal diseases and the major cause of death from renal disease.
Although exceptions abound, the evolution from normal renal function to symptomatic chronic renal failure broadly progresses through a series of four stages that merge into one another.
The details of the pathophysiology of chronic renal failure are beyond the scope of this book and are well covered in various nephrology texts. Table 20-1 lists the major systemic abnormalities in chronic kidney failure.
TABLE 20-1 Principal Systemic Manifestations of Chronic Kidney Disease and Uremia
| FLUID AND ELECTROLYTES |
| CALCIUM PHOSPHATE AND BONE |
| HEMATOLOGIC |
| CARDIOPULMONARY |
| GASTROINTESTINAL |
| NEUROMUSCULAR |
| DERMATOLOGIC |
Glomerular Diseases
Glomerular diseases constitute some of the major problems in nephrology; indeed, chronic glomerulonephritis is one of the most common causes of chronic kidney disease in humans. Glomeruli may be injured by a variety of factors and in the course of several systemic diseases. Systemic immunological diseases such as systemic lupus erythematosus (SLE), vascular disorders such as hypertension, metabolic diseases such as diabetes mellitus, and some hereditary conditions such as Fabry disease often affect the glomerulus. These are termed secondary glomerular diseases to differentiate them from disorders in which the kidney is the only or predominant organ involved. The latter constitute the various types of primary glomerulonephritis or, because some do not have a cellular inflammatory component, glomerulopathy. However, both the clinical manifestations and glomerular histologic changes in primary and secondary forms can be similar.
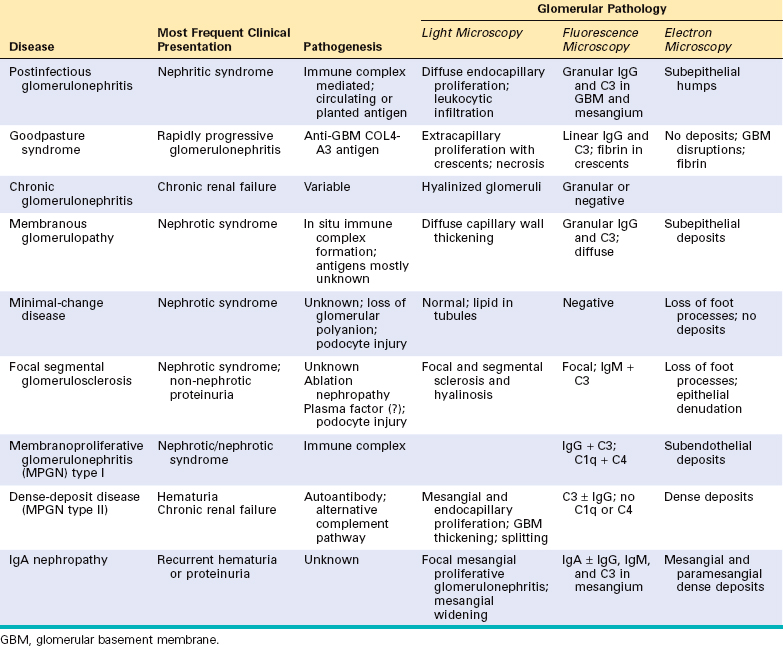
Here we discuss the various types of primary glomerulopathies and briefly review the secondary forms covered in other parts of this book. Table 20-2 lists the most common forms of glomerulonephritis that have reasonably well defined morphologic and clinical characteristics.
TABLE 20-2 Glomerular Diseases
| PRIMARY GLOMERULOPATHIES |
| SYSTEMIC DISEASES WITH GLOMERULAR INVOLVEMENT |
| HEREDITARY DISORDERS |
CLINICAL MANIFESTATIONS
The clinical manifestations of glomerular disease are clustered into the five major glomerular syndromes summarized in Table 20-3. Both the primary glomerulopathies and the systemic diseases affecting the glomerulus can result in these syndromes. Because glomerular diseases are often associated with systemic disorders, mainly diabetes mellitus, SLE, vasculitis, and amyloidosis, in any patient with manifestations of glomerular disease it is essential to consider these systemic conditions.
TABLE 20-3 The Glomerular Syndromes
| Syndrome | Manifestations |
|---|---|
| Nephritic syndrome | Hematuria, azotemia, variable proteinuria, oliguria, edema, and hypertension |
| Rapidly progressive glomerulonephritis | Acute nephritis, proteinuria, and acute renal failure |
| Nephrotic syndrome | >3.5 gm/day proteinuria, hypoalbuminemia, hyperlipidemia, lipiduria |
| Chronic renal failure | Azotemia → uremia progressing for months to years |
| Isolated urinary abnormalities | Glomerular hematuria and/or subnephrotic proteinuria |
Many clinical manifestations of glomerular disease result from perturbations of specific components of the glomerular tuft, so we present key anatomic structures that are subject to alteration in disease. The glomerulus consists of an anastomosing network of capillaries lined by fenestrated endothelium invested by two layers of epithelium (Fig. 20-1). The visceral epithelium is incorporated into and becomes an intrinsic part of the capillary wall, separated from endothelial cells by a basement membrane. The parietal epithelium, situated on the Bowman capsule, lines the urinary space, the cavity in which plasma filtrate first collects.
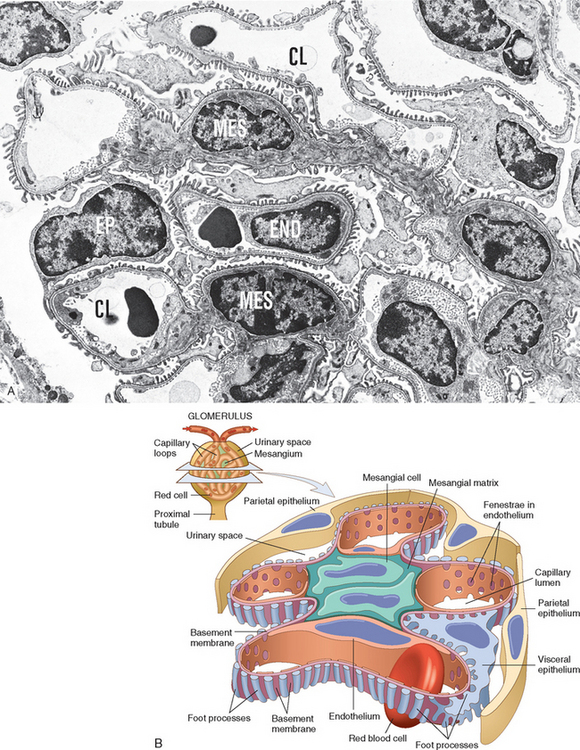
FIGURE 20-1 A, Low-power electron micrograph of renal glomerulus. CL, capillary lumen; EP, visceral epithelial cells with foot processes; END, endothelium; MES, mesangium. B, Schematic representation of a glomerular lobe.
(A, Courtesy of Dr. Vicki Kelley, Brigham and Women’s Hospital, Boston, MA.)
The glomerular capillary wall is the filtering membrane and consists of the following structures3,4 (Fig. 20-2):
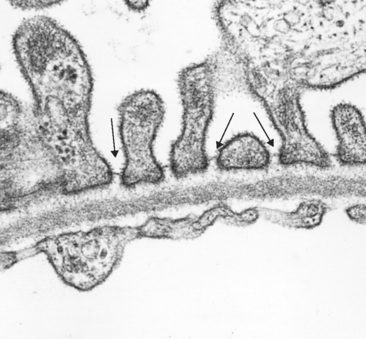
FIGURE 20-2 Glomerular filter consisting, from bottom to top, of fenestrated endothelium, basement membrane, and foot processes of epithelial cells. Note the filtration slits (arrows) and diaphragm situated between the foot processes. Note also that the basement membrane consists of a central lamina densa, sandwiched between two looser layers, the lamina rara interna and lamina rara externa.
(Courtesy of Dr. Helmut Rennke, Brigham and Women’s Hospital, Boston, MA.)
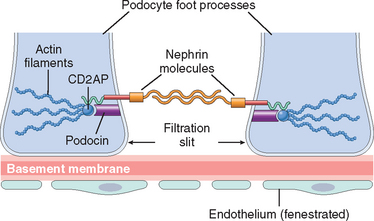
The major characteristics of normal glomerular filtration are an extraordinarily high permeability to water and small solutes, because of the highly fenestrated nature of the endothelium, and impermeability to proteins, such as molecules of the size of albumin (∼3.6-nm radius; 70 kilodaltons [kD] molecular weight) or larger. The latter property of the glomerular filtration barrier allows discrimination among various protein molecules, depending on their size (the larger, the less permeable) and charge (the more cationic, the more permeable). This size- and charge-dependent barrier function is accounted for by the complex structure of the capillary wall, the collagenous porous and charged structure of the GBM, and the many anionic moieties present within the wall, including the acidic proteoglycans of the GBM and the sialoglycoproteins of epithelial and endothelial cell coats (also called glycocalyx). The charge-dependent restriction is important in the virtually complete exclusion of albumin from the filtrate, because albumin is an anionic molecule of a pI 4.5. The visceral epithelial cell, also known as a podocyte, is important for the maintenance of glomerular barrier function; its slit diaphragm presents a size-selective distal diffusion barrier to the filtration of proteins, and it is the cell type that is largely responsible for synthesis of GBM components. Proteins located in the slit diaphragm control glomerular permeability. Three of the most important slit diaphragm proteins are depicted in Figure 20-3. Nephrin is a transmembrane protein with a large extracellular portion made up of immunoglobulin (Ig)-like domains. Nephrin molecules extend toward each other from neighboring foot processes and dimerize across the slit diaphragm. Within the cytoplasm of the foot processes, nephrin forms molecular connections with podocin, CD2-associated protein, and ultimately the actin cytoskeleton. The number of identified slit diaphragm proteins continues to grow rapidly, and more comprehensive descriptions of their complex localization and interactions have been published.6,7 The importance of these proteins in maintaining glomerular permeability is demonstrated by the observation that mutations in the genes encoding them give rise to nephrotic syndrome (discussed later). This has resulted in renewed appreciation of the importance of the slit diaphragm in glomerular barrier function and its contribution to protein leakage in disease states.8
HISTOLOGIC ALTERATIONS
Various types of glomerulopathies are characterized by one or more of four basic tissue reactions.
Hypercellularity.
Some inflammatory diseases of the glomerulus are characterized by an increase in the number of cells in the glomerular tufts. This hypercellularity is characterized by one or more combinations of the following:
Basement Membrane Thickening.
By light microscopy, this change appears as thickening of the capillary walls, best seen in sections stained with periodic acid–Schiff (PAS). By electron microscopy such thickening takes one of two forms:
Hyalinosis and Sclerosis.
Hyalinosis, as applied to the glomerulus, denotes the accumulation of material that is homogeneous and eosinophilic by light microscopy. By electron microscopy the hyalin is extracellular and amorphous. It is made up of plasma proteins that have insudated from the circulation into glomerular structures. When extensive, this change contributes to obliteration of the capillary lumens of the glomerular tuft. Hyalinosis is usually a consequence of endothelial or capillary wall injury and typically the end result of various forms of glomerular damage. It is a common feature of focal segmental glomerulosclerosis.
Sclerosis is characterized by accumulations of extracellular collagenous matrix, either confined to mesangial areas as is often the case in diabetic glomerulosclerosis, or involving the capillary loops, or both. The sclerosing process may also result in obliteration of some or all of the capillary lumens in affected glomeruli, which in turn can result in formation of fibrous adhesions between the sclerotic portions of glomeruli and the nearby parietal epithelium and Bowman capsules.
Because many of the primary glomerulopathies are of unknown cause, they are often classified by their histology, as can be seen in Table 20-2. The histologic changes can be further subdivided by their distribution into diffuse, involving all glomeruli; global, involving the entire glomerulus; focal, involving only a proportion of the glomeruli; segmental, affecting a part of each glomerulus; and by either capillary loop or mesangial, affecting predominantly capillary or mesangial regions. These terms are sometimes appended to the histologic classifications.
PATHOGENESIS OF GLOMERULAR INJURY
Although much is not known about etiologic agents and triggering events, it is clear that immune mechanisms underlie most forms of primary glomerulopathy and many of the secondary glomerular disorders10,11 (Table 20-4). Glomerulonephritis can be readily induced experimentally by antigen-antibody reactions. Furthermore, glomerular deposits of immunoglobulins, often with components of complement, are found in the majority of individuals with glomerulonephritis. Cell-mediated immune reactions also may play a role, usually in concert with antibody-mediated events. We begin this discussion with a review of antibody-instigated injury.
TABLE 20-4 Immune Mechanisms of Glomerular Injury
| ANTIBODY-MEDIATED INJURY |
| IN SITU IMMUNE COMPLEX DEPOSITION |
| CIRCULATING IMMUNE COMPLEX DEPOSITION |
| CYTOTOXIC ANTIBODIES |
| CELL-MEDIATED IMMUNE INJURY |
| ACTIVATION OF ALTERNATIVE COMPLEMENT PATHWAY |
GBM, glomerular basement membrane.
Two forms of antibody-associated injury have been established: (1) injury by antibodies reacting in situ within the glomerulus, either binding to insoluble fixed (intrinsic) glomerular antigens or to molecules planted within the glomerulus, and (2) injury resulting from deposition of circulating antigen-antibody complexes in the glomerulus. In addition, there is experimental evidence that cytotoxic antibodies directed against glomerular cell components may cause glomerular injury. These pathways are not mutually exclusive, and in humans, all may contribute to injury.
Immune Complex Deposition Involving Intrinsic and in Situ Renal Antigens
In these forms of injury, antibodies react directly with intrinsic tissue antigen, or antigens “planted” in the glomerulus from the circulation. The best established experimental models for anti–glomerular antibody–mediated glomerular injury, for which there are counterparts in human disease, are anti–glomerular basement membrane (antiGBM) antibody–induced glomerulonephritis and Heymann nephritis.
Heymann Nephritis
The Heymann model of rat glomerulonephritis is induced by immunizing animals with an antigen contained within preparations of proximal tubular brush border (Fig. 20-4C). The rats develop antibodies to this antigen, and a membranous nephropathy, resembling human membranous nephropathy, develops (discussed later; see also Fig. 20-13). On electron microscopy the glomerulopathy is characterized by the presence of numerous discrete electron-dense deposits (made up largely of immune reactants) along the subepithelial aspect of the basement membrane. The pattern of immune deposition by immunofluorescence microscopy is granular rather than linear. It is now clear that this type of disease results largely from the reaction of antibody with an antigen complex located on the basal surface of visceral epithelial cells and cross-reacting with the brush-border antigen used in the original experiments. This so-called Heymann antigen in rats is a large 330-kD protein called megalin, which has homology to the low-density lipoprotein receptor (Chapter 5); the corresponding antigen in human membranous nephropathy has not yet been identified.12 Antibody binding to glomerular epithelial cell membrane is followed by complement activation and then shedding of the immune aggregates from the cell surface to form the characteristic subepithelial deposits (see Fig. 20-4C).
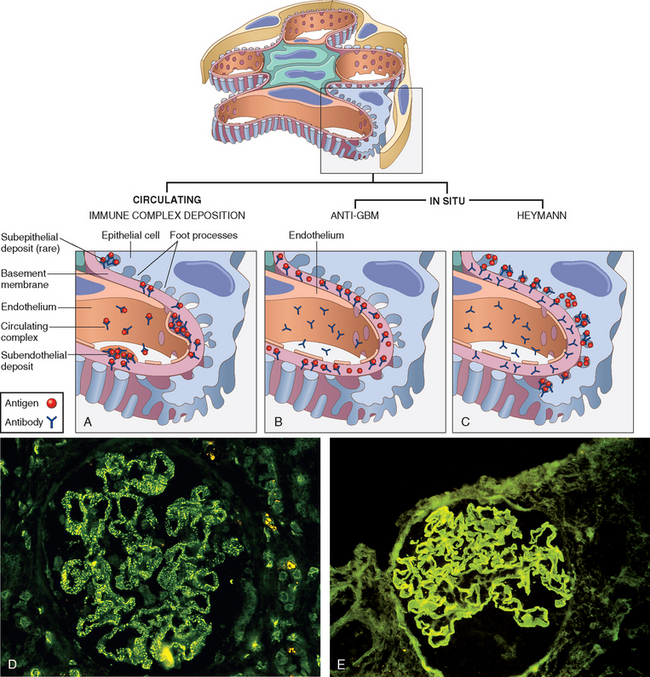
FIGURE 20-4 Antibody-mediated glomerular injury can result either from the deposition of circulating immune complexes (A) or, more commonly, from in situ formation of complexes exemplified by anti-GBM disease (B) or Heymann nephritis (C). D and E, Two patterns of deposition of immune complexes as seen by immunofluorescence microscopy: granular, characteristic of circulating and in situ immune complex nephritis (D), and linear, characteristic of classic anti-GBM disease (E).
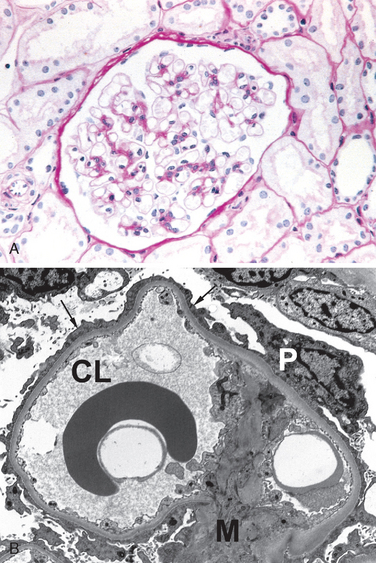
FIGURE 20-13 Minimal-change disease. A, Glomerulus stained with PAS. Note normal basement membranes and absence of proliferation. B, Ultrastructural characteristics of minimal-change disease include effacement of foot processes (arrows) and absence of deposits. CL, capillary lumen; M, mesangium; P, podocyte cell body.
In humans, anti-GBM antibody–induced disease and membranous nephropathy are autoimmune diseases, caused by antibodies to endogenous tissue components. What triggers these autoantibodies is unclear, but any one of the several mechanisms responsible for autoimmunity, discussed in Chapter 6, may be involved. Several forms of autoimmune glomerulonephritis can be experimentally induced by drugs (e.g., mercuric chloride), infectious products (endotoxin), and the graft-versus-host reaction (Chapter 6). In such models there is an alteration of immune regulation associated with B-cell activation and the induction of an array of autoantibodies that react with renal antigens.
Antibodies against Planted Antigens
Antibodies can react in situ with antigens that are not normally present in the glomerulus but are “planted” there. There is increasing experimental support for such a mechanism of glomerulonephritis. Such antigens may localize in the kidney by interacting with various intrinsic components of the glomerulus. Planted antigens include cationic molecules that bind to anionic components of the glomerulus; DNA, nucleosomes, and other nuclear proteins, which have an affinity for GBM components; bacterial products; large aggregated proteins (e.g., aggregated immunoglobulins, which deposit in the mesangium because of their size; and immune complexes themselves, since they continue to have reactive sites for further interactions with free antibody, free antigen, or complement. There is no dearth of other possible planted antigens, including viral, bacterial, and parasitic products and drugs. Antibodies that bind to most of these planted antigens induce a discrete pattern of Ig deposition detected as granular staining by immunofluorescence microscopy, similar to the pattern found in circulating immune complex nephritis.
Anti-GBM Antibody–Induced Glomerulonephritis
In this type of injury antibodies are directed against intrinsic fixed antigens that are normal components of the GBM proper. It has its experimental counterpart in so-called Masugi or nephrotoxic nephritis, produced in rats by injections of anti-rat kidney antibodies prepared in rabbits by immunization with rat kidney tissue. The injected antibodies bind along the entire length of the GBM, resulting in a diffuse linear pattern of staining for the antibodies by immunofluorescent techniques (see Fig. 20-4B and E). This is contrasted with the granular lumpy pattern of immunofluorescent staining seen in other in situ models, such as the Heymann model of membranous glomerulopathy, or after deposition of circulating immune complexes.
In the Masugi model the injected anti-GBM antibody is rabbit Ig, which is foreign to the host and thus acts as an antigen eliciting anti-Ig antibody in the rat. The rat antibodies then react with the rabbit Ig deposited in the basement membrane, leading to further glomerular injury. Often the anti-GBM antibodies cross-react with other basement membranes, especially those in the lung alveoli, resulting in simultaneous lung and kidney lesions (Goodpasture syndrome). The GBM antigen that is responsible for classic anti-GBM antibody–induced glomerulonephritis and Goodpasture syndrome is a component of the noncollagenous domain (NC1) of the α3 chain of collagen type IV that is critical for maintenance of GBM suprastructure.5 Anti-GBM antibody–induced glomerulonephritis accounts for fewer than 5% of cases of human glomerulonephritis. It is solidly established as the cause of injury in Goodpasture syndrome, discussed later. Most instances of anti-GBM antibody–induced glomerulonephritis are characterized by severe crescentic glomerular damage and the clinical syndrome of rapidly progressive glomerulonephritis.
Circulating Immune Complex Glomerulonephritis
In this type of nephritis glomerular injury is caused by the trapping of circulating antigen-antibody complexes within glomeruli. The antibodies have no immunological specificity for glomerular constituents, and the complexes localize within the glomeruli because of their physicochemical properties and the hemodynamic factors peculiar to the glomerulus (see Fig. 20-4A).
The pathogenesis of immune complex diseases was discussed in Chapter 6. Here we briefly review the salient features that relate to glomerular injury.
The antigens that trigger the formation of circulating immune complexes may be of endogenous origin, as in the glomerulonephritis associated with SLE, or they may be exogenous, as is likely in the glomerulonephritis that follows certain infections. Microbial antigens that are implicated include bacterial products (streptococci), the surface antigen of hepatitis B virus, hepatitis C virus antigens, and antigens of Treponema pallidum, Plasmodium falciparum, and several viruses. Some tumor antigens are also thought to cause immune complex–mediated nephritis. In many cases the inciting antigen is unknown.
Whatever the antigen may be, antigen-antibody complexes are formed in the circulation and then trapped in the glomeruli, where they produce injury. It has long been thought that this injury is mediated and amplified by the binding of complement, but recent studies in knockout mice also point to the importance of engagement of Fc receptors on leukocytes and perhaps intrinsic renal cells as mediators of the injury process.13 The glomerular lesions usually exhibit leukocytic infiltration and proliferation of mesangial and endothelial cells. Electron microscopy reveals the immune complexes as electron-dense deposits that lie in the mesangium, between the endothelial cells and the GBM (subendothelial deposits), or between the outer surface of the GBM and the podocytes (subepithelial deposits). Deposits may be located at more than one site in a given case. By immunofluorescence microscopy the immune complexes are seen as granular deposits along the basement membrane, in the mesangium, or in both locations (see Fig. 20-4D). Once deposited in the kidney, immune complexes may eventually be degraded, mostly by infiltrating neutrophils and monocytes/macrophages, mesangial cells, and endogenous proteases, and the inflammatory reaction may then subside. Such a course occurs when the exposure to the inciting antigen is short-lived and limited, as in most cases of poststreptococcal glomerulonephritis. However, if a continuous shower of antigens develops, as may be seen in SLE or viral hepatitis, repeated cycles of immune complex formation, deposition, and injury may occur, leading to a more chronic membranous or membranoproliferative type of glomerulonephritis.
Several factors affect glomerular localization of antigen, antibody, or complexes of both. The molecular charge and size of these reactants are clearly important. Highly cationic immunogens tend to cross the GBM, and the resultant complexes eventually reside in a subepithelial location. Highly anionic macromolecules are excluded from the GBM and either are trapped subendothelially or are not nephritogenic at all. Molecules of neutral charge and immune complexes containing these molecules tend to accumulate in the mesangium. Large circulating complexes are not usually nephritogenic, because they are cleared by the mononuclear phagocyte system and do not enter the GBM in sufficient quantities. The pattern of localization is also affected by changes in glomerular hemodynamics, mesangial function, and integrity of the charge-selective barrier in the glomerulus. These influences may underlie the variable pattern of immune reactant deposition in various forms of glomerulonephritis, as shown in Figure 20-5. In turn, the distinct patterns of localization of immune complexes is a key determinant of the injury response and the histologic features that subsequently develop.
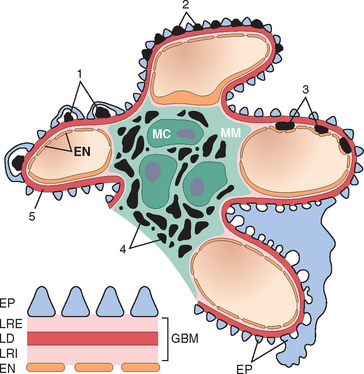
FIGURE 20-5 Localization of immune complexes in the glomerulus: (1) subepithelial humps, as in acute glomerulonephritis; (2) epimembranous deposits, as in membranous nephropathy and Heymann glomerulonephritis; (3) subendothelial deposits, as in lupus nephritis and membranoproliferative glomerulonephritis; (4) mesangial deposits, as in IgA nephropathy; (5) basement membrane. EN, endothelium; EP, epithelium; LD, lamina densa; LRE, lamina rara externa; LRI, lamina rara interna; MC, mesangial cell; MM, mesangial matrix.
(Modified from Couser WG: Mediation of immune glomerular injury. J Am Soc Nephrol 1:13, 1990.)
Antibodies to Glomerular Cells
In addition to causing immune deposits, antibodies against glomerular cell antigens may react with cellular components and cause injury by cytotoxic or other mechanisms. Antibodies to mesangial cell antigens, for example, can cause mesangiolysis followed by mesangial cell proliferation; antibodies to endothelial cell antigens cause endothelial injury and intravascular thrombosis; and antibodies to certain visceral epithelial cell components cause proteinuria in experimental animals. This mechanism may play a role in certain human immune disorders that are without demonstrable immune deposits.
In summary, most cases of human glomerulonephritis are a consequence of deposits of discrete immune complexes, which are visualized by granular immunofluorescence staining along the basement membranes or in the mesangium. However, it may be difficult to determine whether the deposition has occurred in situ, by circulating complexes, or by both mechanisms because, as was discussed earlier, trapping of circulating immune complexes can initiate further in situ complex formation. Single etiologic agents, such as hepatitis B and C viruses, can cause either a membranous pattern of glomerulonephritis, suggesting in situ deposition, or a membranoproliferative pattern, more indicative of circulating complexes. It is best then to consider that antigen-antibody deposition in the glomerulus is a major pathway of glomerular injury and that in situ immune reactions, trapping of circulating complexes, interactions between these two events, and local hemodynamic and structural determinants in the glomerulus all contribute to the diverse morphologic and functional alterations in glomerulonephritis.
Cell-Mediated Immunity in Glomerulonephritis
Although antibody-mediated mechanisms may initiate many forms of glomerulonephritis, there is now considerable evidence that sensitized T cells cause some forms of glomerular injury and are involved in the progression of many glomerulonephritides.14 Clues to the role of cellular immunity include the presence of activated macrophages and T cells and their products in the glomerulus in some forms of human and experimental glomerulonephritis;15 in vitro and in vivo evidence of lymphocyte activation on exposure to antigen in human and experimental glomerulonephritis; abrogation of glomerular injury by lymphocyte depletion; and successful attempts to induce glomerular injury by transfer of T cells from nephritic animals to normal recipients. The evidence is most compelling for certain types of experimental crescentic glomerulonephritis in which antibodies to GBM may initiate glomerular injury but activated T lymphocytes may propogate the inflammation.15
Activation of Alternative Complement Pathway
Alternative complement pathway activation occurs in the clinicopathologic entity called dense-deposit disease, also referred to as membranoproliferative glomerulonephritis (MPGN type II). It may also occur in some forms of proliferative glomerulonephritis. This mechanism is discussed later.
Epithelial Cell Injury
This can be induced by antibodies to visceral epithelial cell antigens; by toxins, as in an experimental model of proteinuria induced by puromycin aminonucleoside; conceivably by certain cytokines; or by still poorly characterized factors, as in the case of human minimal-change disease and focal segmental glomerulosclerosis, discussed later. Such injury is reflected morphologically by changes in the visceral epithelial cells, which include effacement of foot processes, vacuolization, retraction, and detachment of cells from the GBM, and functionally by proteinuria. It is hypothesized that the detachment of visceral epithelial cells is caused by loss of adhesive interactions with the basement membrane and that this detachment contributes to protein leakage (Fig. 20-6).
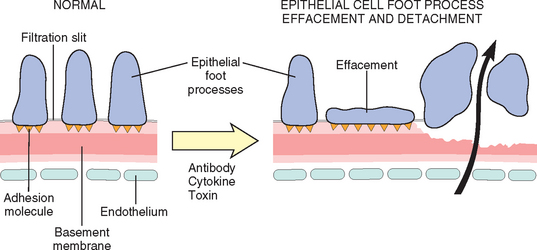
FIGURE 20-6 Epithelial cell injury. The postulated sequence is a consequence of antibodies specific to epithelial cell antigens, toxins, cytokines, or other factors causing injury; this results in foot process effacement and sometimes detachment of epithelial cells and protein leakage through defective GBM and filtration slits.
Mediators of Glomerular Injury
Once immune reactants or sensitized T cells have localized in the glomerulus, how does the glomerular damage ensue? The mediators—both cells and molecules—are the usual suspects involved in acute and chronic inflammation, described in Chapter 2, and only a few are highlighted here (Fig. 20-7).
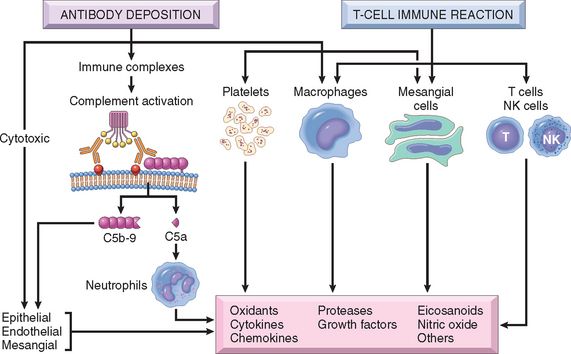
Cells
Soluble Mediators
Virtually all the known inflammatory chemical mediators have been implicated in glomerular injury.
MECHANISMS OF PROGRESSION IN GLOMERULAR DISEASES
Thus far we have discussed the immunological mechanisms and mediators that initiate glomerular injury. The outcome of such injury depends on several factors, including the initial severity of renal damage, the nature and persistence of the antigens, and the immune status, age, and genetic predisposition of the host.
It has long been known that once any renal disease, glomerular or otherwise, destroys functioning nephrons and reduces the GFR to about 30% to 50% of normal, progression to end-stage renal failure proceeds at a relatively constant rate, independent of the original stimulus or activity of the underlying disease. The secondary factors that lead to progression are of great clinical interest, since they can be targets of therapy that delays or even prevents the inexorable journey to dialysis or transplantation.
The two major histologic characteristics of such progressive renal damage are focal segmental glomerulosclerosis and tubulointerstitial fibrosis; we discuss these separately.18-20
Focal Segmental Glomerulosclerosis (FSGS).
Patients with this secondary change develop proteinuria, even if the primary disease was nonglomerular. The glomerulosclerosis seems to be initiated by the adaptive change that occurs in the relatively unaffected glomeruli of diseased kidneys.19,21 Such a mechanism is suggested by experiments in rats subjected to ablation of renal mass by subtotal nephrectomy. Compensatory hypertrophy of the remaining glomeruli serves to maintain renal function in these animals, but proteinuria and segmental glomerulosclerosis soon develop, leading eventually to total glomerular sclerosis and uremia. The glomerular hypertrophy is associated with hemodynamic changes, including increases in glomerular blood flow, filtration, and transcapillary pressure (glomerular hypertension), and often with systemic hypertension. The sequence of events (Fig. 20-8) that is thought to lead to sclerosis in this setting entails endothelial and epithelial cell injury, increased glomerular permeability to proteins, and accumulation of proteins in the mesangial matrix. This is followed by proliferation of mesangial cells, infiltration by macrophages, increased accumulation of extracellular matrix (ECM), and segmental and eventually global sclerosis of glomeruli. This results in further reductions in nephron mass, ongoing activation of these compensatory changes, and a vicious circle of continuing glomerulosclerosis. Most of the mediators of chronic inflammation and fibrosis, particularly TGF-β, play a role in the induction of sclerosis. Currently, the most successful interventions to interrupt these mechanisms of progressive glomerulosclerosis involve treatment with inhibitors of the renin-angiotensin system, which not only reduce intraglomerular hypertension, but also have direct effects on each of the mechanisms identified above.21 Importantly, these agents have been shown to ameliorate progression of sclerosis in both animal and human studies.20
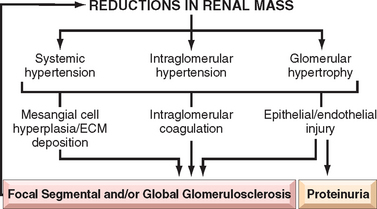
FIGURE 20-8 Focal segmental glomerulosclerosis associated with loss of renal mass. The adaptive changes in glomeruli (hypertrophy and glomerular capillary hypertension), as well as systemic hypertension, cause epithelial and endothelial injury and resultant proteinuria. The mesangial response, involving mesangial cell proliferation and ECM production together with intraglomerular coagulation, causes the glomerulosclerosis. This results in further loss of functioning nephrons and a vicious circle of progressive glomerulosclerosis.
Contributing to the progressive injury of focal and segmental glomerulosclerosis is the inability of mature visceral epithelial cells (podocytes) to proliferate after injury. This can lead to a decrease in glomerular podocyte number after a severe injury resulting in loss of some of these cells, leading in turn to a process whereby remaining podocytes are either abnormally stretched to maintain an appropriate filtration barrier or unable to cover portions of the GBM, which become denuded of the overlying podocyte foot processes. These alterations lead to abnormal protein filtration as well as loss of structural support for the glomerular capillary walls. This latter alteration in turn may lead to segmental loop dilation because of now incompletely opposed intracapillary pressures, with subsequent formation of a fibrous attachment to Bowman capsule by the bulging capillary segment, and eventual sclerosis of this segment.22
Tubulointerstitial Fibrosis.
Tubulointerstitial injury, manifested by tubular damage and interstitial inflammation, is a component of many acute and chronic glomerulonephritides. Tubulointerstitial fibrosis contributes to progression in both immune and nonimmune glomerular diseases, for example, diabetic nephropathy. Indeed, there is often a much better correlation of decline in renal function with the extent of tubulointerstitial damage than with the severity of glomerular injury.18 Many factors may lead to such tubulointerstitial injury, including ischemia of tubule segments downstream from sclerotic glomeruli, acute and chronic inflammation in the adjacent interstitium, and damage or loss of the peritubular capillary blood supply. Current work also points to the effects of proteinuria on tubular cell structure and function23. On the basis of in vitro and animal studies, proteinuria is thought to cause direct injury to and activation of tubular cells. Activated tubular cells in turn express adhesion molecules and elaborate pro-inflammatory cytokines, chemokines, and growth factors that contribute to interstitial fibrosis. Filtered proteins that may produce these tubular effects include cytokines, complement products, the iron in transferrin, immunoglobulins, lipid moieties, and oxidatively modified plasma proteins.
Having discussed factors in the initiation and progression of glomerular injury, we now turn to a discussion of individual glomerular diseases. Table 20-5 summarizes the main clinical and pathologic features of the major forms of primary glomerulopathies.
NEPHRITIC SYNDROME
Glomerular diseases presenting with a nephritic syndrome are often characterized by inflammation in the glomeruli. The nephritic patient usually presents with hematuria, red cell casts in the urine, azotemia, oliguria, and mild to moderate hypertension. Proteinuria and edema are common, but these are not as severe as those encountered in the nephrotic syndrome, discussed later. The acute nephritic syndrome may occur in such multisystem diseases as SLE and microscopic polyangiitis. Typically, however, it is characteristic of acute proliferative glomerulonephritis and is an important component of crescentic glomerulonephritis, which is described later.
Acute Proliferative (Poststreptococcal, Postinfectious) Glomerulonephritis
As the name implies, this cluster of diseases is characterized histologically by diffuse proliferation of glomerular cells, associated with influx of leukocytes. These lesions are typically caused by immune complexes. The inciting antigen may be exogenous or endogenous. The prototypic exogenous antigen-induced disease pattern is postinfectious glomerulonephritis, whereas an example of an endogenous antigen-induced disease is the nephritis of SLE, described in Chapter 6. The most common underlying infections are streptococcal, but the disorder also has been associated with other infections.
Poststreptococcal Glomerulonephritis
This glomerular disease is decreasing in frequency in the United States but continues to be a fairly common disorder worldwide.24 It usually appears 1 to 4 weeks after a streptococcal infection of the pharynx or skin (impetigo). Skin infections are commonly associated with overcrowding and poor hygiene. Poststreptococcal glomerulonephritis occurs most frequently in children 6 to 10 years of age, but adults of any age can also be affected.
Etiology and Pathogenesis.
Only certain strains of group A β-hemolytic streptococci are nephritogenic, more than 90% of cases being traced to types 12, 4, and 1, which can be identified by typing of M protein of the cell wall.
Poststreptococcal glomerulonephritis is an immunologically mediated disease. The latent period between infection and onset of nephritis is compatible with the time required for the production of antibodies and the formation of immune complexes. Elevated titers of antibodies against one or more streptococcal antigens are present in a great majority of patients. Serum complement levels are low, compatible with activation of the complement system and consumption of complement components. There are granular immune deposits in the glomeruli, supporting an immune complex–mediated mechanism. The streptococcal antigenic com-ponent responsible for the immune reaction has eluded identification for years. Several cationic antigens, including a nephritis-associated streptococcal plasmin receptor (NAPlr), unique to nephritogenic strains of streptococci, can be found in affected glomeruli. Other evidence suggests that streptococcal pyogenic exotoxin B (SpeB) and its zymogen precursor (zSpeB), another protein that functions as a plasmin receptor, are the principal antigenic determinants in most cases of poststreptococcal glomerulonephritis.25 It is not known if these represent antigens planted in the GBM, or parts of circulating immune complexes, or both. GBM proteins altered by streptococcal enzymes have also been implicated as antigens.
Morphology. The classic diagnostic picture is one of enlarged, hypercellular glomeruli (Fig. 20-9). The hypercellularity is caused by (1) infiltration by leukocytes, both neutrophils and monocytes; (2) proliferation of endothelial and mesangial cells; and (3) in severe cases by crescent formation. The proliferation and leukocyte infiltration are diffuse, that is, involving all lobules of all glomeruli. There is also swelling of endothelial cells, and the combination of proliferation, swelling, and leukocyte infiltration obliterates the capillary lumens. There may be interstitial edema and inflammation, and the tubules often contain red cell casts.
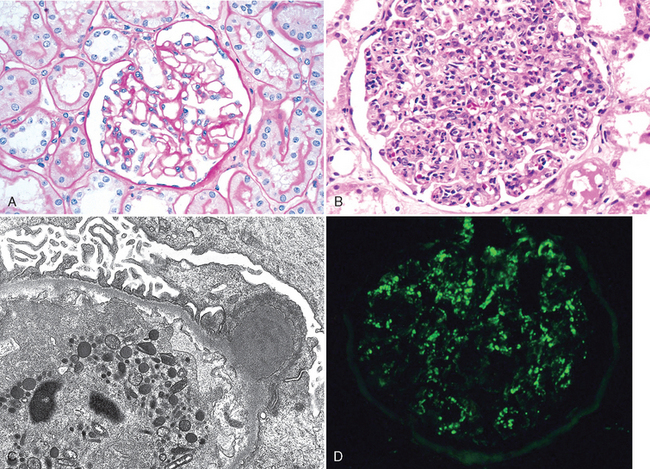
FIGURE 20-9 Acute proliferative glomerulonephritis. A, Normal glomerulus. B, Glomerular hypercellularity is due to intracapillary leukocytes and proliferation of intrinsic glomerular cells. C, Typical electron-dense subepithelial “hump” and a neutrophil in the lumen. D, Immunofluorescent stain demonstrates discrete, coarsly granular deposits of complement protein C3, corresponding to “humps” illustrated in part C.
(A–C, courtesy of Dr. H. Rennke, Brigham and Women’s Hospital, Boston, MA. D, courtesy of D. J. Kowaleska, University of Washington, Seattle, WA.)
By immunofluorescence microscopy, there are granular deposits of IgG, IgM, and C3 in the mesangium and along the GBM (Fig. 20-9D). Although immune complex deposits are almost universally present, they are often focal and sparse. The characteristic electron microscopic findings are discrete, amorphous, electron-dense deposits on the epithelial side of the membrane, often having the appearance of “humps” (Fig. 20-9C), presumably representing the antigen-antibody complexes at the epithelial cell surface. Subendothelial and intramembranous deposits are also commonly seen, and mesangial deposits may be present.
Clinical Course.
In the classic case, a young child abruptly develops malaise, fever, nausea, oliguria, and hematuria (smoky or cola-colored urine) 1 to 2 weeks after recovery from a sore throat. The patients have red cell casts in the urine, mild proteinuria (usually less than 1 gm/day), periorbital edema, and mild to moderate hypertension. In adults the onset is more likely to be atypical, such as the sudden appearance of hypertension or edema, frequently with elevation of BUN. During epidemics caused by nephritogenic streptococcal infections, glomerulonephritis may be asymptomatic, discovered only on screening for microscopic hematuria. Important laboratory findings include elevations of antistreptococcal antibody titers and a decline in the serum concentration of C3 and other components of the complement cascade.
More than 95% of affected children eventually recover totally with conservative therapy aimed at maintaining sodium and water balance. A small minority of children (perhaps fewer than 1%) do not improve, become severely oliguric, and develop a rapidly progressive form of glomerulonephritis (described later). Some of the remaining patients may undergo slow progression to chronic glomerulonephritis with or without recurrence of an active nephritic picture. Prolonged and persistent heavy proteinuria and abnormal GFR mark patients with an unfavorable prognosis.
In adults the disease is less benign. Although the overall prognosis in epidemics is good, in only about 60% of sporadic cases do the patients recover promptly. In the remainder the glomerular lesions fail to resolve quickly, as manifested by persistent proteinuria, hematuria, and hypertension. In some of these patients, the lesions eventually clear totally, but others develop chronic glomerulonephritis. Some patients will develop a syndrome of rapidly progressive glomerulonephritis.
Nonstreptococcal Acute Glomerulonephritis (Postinfectious Glomerulonephritis)
A similar form of glomerulonephritis occurs sporadically in association with other infections, including bacterial (e.g., staphylococcal endocarditis, pneumococcal pneumonia, and meningococcemia), viral (e.g., hepatitis B, hepatitis C, mumps, human immunodeficiency virus [HIV] infection, varicella, and infectious mononucleosis), and parasitic (malaria, toxoplasmosis). In these settings, granular immunofluorescent deposits and subepithelial humps characteristic of immune complex nephritis are present.
RAPIDLY PROGRESSIVE (CRESCENTIC) GLOMERULONEPHRITIS
Rapidly progressive glomerulonephritis (RPGN) is a syndrome associated with severe glomerular injury and does not denote a specific etiologic form of glomerulonephritis. It is characterized clinically by rapid and progressive loss of renal function associated with severe oliguria and signs of nephritic syndrome; if untreated, death from renal failure occurs within weeks to months. The most common histologic picture is the presence of crescents in most of the glomeruli (crescentic glomerulonephritis). As discussed earlier, these are produced by the proliferation of the parietal epithelial cells lining Bowman capsule and by the infiltration of monocytes and macrophages.
Classification and Pathogenesis.
RPGN may be caused by a number of different diseases, some restricted to the kidney and others systemic. Although no single mechanism can explain all cases, there is little doubt that in most cases the glomerular injury is immunologically mediated. A practical classification divides RPGN into three groups on the basis of immunological findings (Table 20-6). In each group the disease may be associated with a known disorder, or it may be idiopathic.
TABLE 20-6 Rapidly Progressive Glomerulonephritides
| TYPE I (ANTI-GBM ANTIBODY) |
| TYPE II (IMMUNE COMPLEX) |
| TYPE III (PAUCI-IMMUNE) |
ANCA, antineutrophil cytoplasmic antibodies; GBM, glomerular basement membrane.
The first type of RPGN is anti-GBM antibody–induced disease, characterized by linear deposits of IgG and, in many cases, C3 in the GBM that are visualized by immunofluorescence.26 In some of these patients, the anti-GBM antibodies cross-react with pulmonary alveolar basement membranes to produce the clinical picture of pulmonary hemorrhage associated with renal failure (Goodpasture syndrome). Plasmapheresis to remove the pathogenic circulating antibodies is usually part of the treatment, which also includes therapy to suppress the underlying immune response.
The Goodpasture antigen is a peptide within the noncollagenous portion of the α3 chain of collagen type IV.5 What triggers the formation of these antibodies is unclear in most patients. Exposure to viruses or hydrocarbon solvents (found in paints and dyes) has been implicated in some patients, as have various drugs and cancers. There is a high prevalence of certain HLA subtypes and haplotypes (e.g., HLA-DRB1) in affected patients, a finding consistent with the genetic predisposition to autoimmunity.27
The second type of RPGN is the result of immune complex deposition. It can be a complication of any of the immune complex nephritides, including postinfectious glomerulonephritis, lupus nephritis, IgA nephropathy, and HenochSchönlein purpura. In all these cases, immunofluorescence studies reveal the granular pattern of staining characteristic of immune complex deposition. This type of RPGN frequently demonstrates cellular proliferation within the glomerular tuft, in addition to crescent formation. These patients cannot usually be helped by plasmapheresis, and they require treatment for the underlying disease.
The third type of RPGN, also called pauci-immune type, is defined by the lack of anti-GBM antibodies or immune complexes by immunofluorescence and electron microscopy. Most patients with this type of RPGN have circulating antineutrophil cytoplasmic antibodies (ANCAs) that produce cytoplasmic (c) or perinuclear (p) staining patterns and, as we have seen (Chapter 11), play a role in some vasculitides. Hence, in some cases this type of RPGN is a component of a systemic vasculitis such as Wegener granulomatosis or microscopic polyangiitis. In many cases, however, pauci-immune crescentic glomerulonephritis is isolated and hence idiopathic. More than 90% of such idiopathic cases have c-ANCAs or p-ANCAs in the sera.26 The presence of circulating ANCAs in both idiopathic crescentic glomerulonephritis and cases of crescentic glomerulonephritis that occur as a component of systemic vasculitis, and the similar pathologic features in either setting, have led to the idea that these disorders are pathogenetically related. According to this concept, all cases of crescentic glomerulonephritis of the pauci-immune type are manifestations of small-vessel vasculitis or polyangiitis, which is limited to glomerular and perhaps peritubular capillaries in cases of idiopathic crescentic glomerulonephritis. The clinical distinction between systemic vasculitis with pauci-immune renal involvement and idiopathic crescentic glomerulonephritis accordingly has become de-emphasized, since these entities are viewed as part of a spectrum of vasculitic disease. ANCAs have proved to be invaluable as a highly sensitive diagnostic marker for pauci-immune crescentic glomerulonephritis, but proof of their role as a direct cause of this glomerulonephritis has been elusive. Recent strong evidence of their pathogenic potential has been obtained by studies in mice showing that transferring antibodies against myeloperoxidase (the target antigen of most p-ANCAs) induces a form of RPGN.28
To summarize, all three types of RPGN may be associated with a well-defined renal or extrarenal disease, but in many cases (∼50%), the disorder is idiopathic. Of the patients with this syndrome, about one fifth have anti–GBM antibody–mediated glomerulonephritis without lung involvement; another one fourth have immune complex–mediated crescentic glomerulonephritis; and the remainder are of the pauci-immune type. The common denominator in all types of RPGN is severe glomerular injury.
Morphology. The kidneys are enlarged and pale, often with petechial hemorrhages on the cortical surfaces. Depending on the underlying cause, the glomeruli may show focal necrosis, diffuse or focal endothelial proliferation, and mesangial proliferation. The histologic picture, however, is dominated by distinctive crescents (Fig. 20-10). Crescents are formed by proliferation of parietal cells and by migration of monocytes and macrophages into the urinary space. Neutrophils and lymphocytes may be present. The crescents eventually obliterate Bowman space and compress the glomerular tuft. Fibrin strands are frequently prominent between the cellular layers in the crescents; indeed, as discussed earlier, the escape of fibrinogen into Bowman space and its conversion to fibrin are an important contributor to crescent formation. By immunofluorescence microscopy, immune complex–mediated cases show granular immune deposits; Goodpasture syndrome cases show linear GBM fluorescence for Ig and complement, and pauci-immune cases have little or no deposition of immune reactants. Electron microscopy discloses deposits in those cases due to immune complex deposition (type II). Regardless of type, electron microscopy may show distinct ruptures in the GBM, the severe injury that allows leukocytes, proteins, and inflammatory mediators to reach the urinary space, where they trigger the crescent formation (Fig. 20-11). In time, most crescents undergo sclerosis, but restoration of normal glomerular architecture may be achieved with early aggressive therapy.
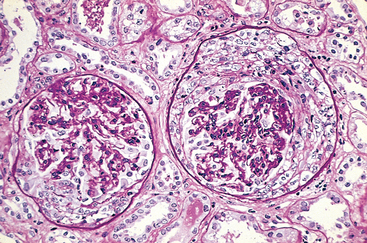
FIGURE 20-10 Crescentic glomerulonephritis (PAS stain). Note the collapsed glomerular tufts and the crescent-shaped mass of proliferating parietal epithelial cells and leukocytes internal to Bowman capsule.
(Courtesy of Dr. M.A. Venkatachalam, University of Texas Health Sciences Center, San Antonio, TX.)
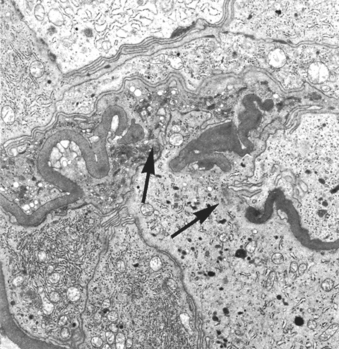
Clinical Course.
The renal manifestations of all forms of crescentic glomerulonephritis include hematuria with red blood cell casts in the urine, moderate proteinuria occasionally reaching the nephrotic range, and variable hypertension and edema. In Goodpasture syndrome the course may be dominated by recurrent hemoptysis or even life-threatening pulmonary hemorrhage. Serum analyses for anti-GBM antibodies, antinuclear antibodies, and ANCAs are helpful in the diagnosis of specific subtypes. Although milder forms of glomerular injury may subside, the renal involvement is usually progressive over a matter of weeks and culminates in severe oliguria. Recovery of renal function may follow early intensive plasmapheresis (plasma exchange) combined with steroids and cytotoxic agents in Goodpasture syndrome. This therapy can reverse both pulmonary hemorrhage and renal failure. Other forms of RPGN also respond well to steroids and cytotoxic agents. However, despite therapy, some patients may eventually require chronic dialysis or transplantation, particularly if the disease is discovered at a late stage.
NEPHROTIC SYNDROME
Certain glomerular diseases virtually always produce the nephrotic syndrome. In addition, many other forms of primary and secondary glomerulopathies discussed in this chapter may underlie the syndrome. Before the major diseases associated with nephrotic syndrome are presented, the causes and pathophysiology of this clinical complex are briefly discussed.
Pathophysiology.
The manifestations of the nephrotic syndrome include:
The various components of nephrotic syndrome bear a logical relationship to one another. The initial event is a derangement in glomerular capillary walls resulting in increased permeability to plasma proteins. The glomerular capillary wall, with its endothelium, GBM, and visceral epithelial cells, acts as a size and charge barrier through which the plasma filtrate passes. Increased permeability resulting from either structural or physicochemical alterations allows protein to escape from the plasma into the urinary space. Massive proteinuria results.
The heavy proteinuria depletes serum albumin levels at a rate beyond the compensatory synthetic capacity of the liver, resulting in hypoalbuminemia and a reversed albumin-to-globulin ratio. Increased renal catabolism of filtered albumin also contributes to the hypoalbuminemia. The generalized edema is, in turn, the consequence of decreased colloid osmotic pressure of the blood with subsequent accumulation of fluid in the interstitial tissues. There is also sodium and water retention, which aggravates the edema (Chapter 4). This seems to be due to several factors, including compensatory secretion of aldosterone, mediated by the hypovolemia-enhanced renin secretion; stimulation of the sympathetic system; and a reduction in the secretion of natriuretic factors such as atrial peptides. Edema is characteristically soft and pitting, most marked in the periorbital regions and dependent portions of the body. It may be massive, with pleural effusions and ascites.
The largest proportion of protein lost in the urine is albumin, but globulins are also excreted in some diseases. The ratio of low- to high-molecular-weight proteins in the urine in various cases of nephrotic syndrome is a manifestation of the selectivity of proteinuria. A highly selective proteinuria consists mostly of low-molecular-weight proteins (albumin, 70 kD; transferrin, 76 kD molecular weight), whereas a poorly selective proteinuria consists of higher molecular-weight globulins in addition to albumin.
The genesis of the hyperlipidemia is complex. Most patients with nephritic syndrome have increased blood levels of cholesterol, triglyceride, very-low-density lipoprotein, low-density lipoprotein, Lp(a) lipoprotein, and apoprotein, and there is a decrease in high-density lipoprotein concentration in some patients. These defects seem to be due in part to increased synthesis of lipoproteins in the liver, abnormal transport of circulating lipid particles, and decreased catabolism. Lipiduria follows the hyperlipidemia, because lipoproteins also leak across the glomerular capillary wall. The lipid appears in the urine either as free fat or as oval fat bodies, representing lipoprotein resorbed by tubular epithelial cells and then shed along with the degenerated cells.
Nephrotic patients are particularly vulnerable to infection, especially staphylococcal and pneumococcal, probably related to loss of immunoglobulins in the urine. Thrombotic and thromboembolic complications are also common in nephrotic syndrome, due in part to loss of endogenous anticoagulants (e.g., antithrombin III) and antiplasmins in the urine. Renal vein thrombosis, once thought to be a cause of nephrotic syndrome, is most often a consequence of this hypercoagulable state, particularly in patients with membranous nephropathy (see below).
Causes.
The relative frequencies of the several causes of the nephrotic syndrome vary according to age and geography. In children younger than 17 years in North America, for example, the nephrotic syndrome is almost always caused by a lesion primary to the kidney; among adults, in contrast, it may often be associated with a systemic disease. Table 20-7 represents a composite derived from several studies of the causes of the nephrotic syndrome and is therefore only approximate. The most frequent systemic causes of the nephrotic syndrome are diabetes, amyloidosis, and SLE. The most important of the primary glomerular lesions are minimal-change disease, membranous glomerulopathy, and focal segmental glomerulosclerosis. The first is most common in children in North America, the second is most common in older adults, and focal segmental glomerulosclerosis occurs at all ages.29 These three lesions are discussed individually in the following sections. Other primary causes, the various proliferative glomerulonephritides including MPGN, frequently present as a mixed syndrome with nephrotic and nephritic features.
TABLE 20-7 Cause of Nephrotic Syndrome
| Prevalence (%)* | ||
|---|---|---|
| Causes | Children | Adults |
| PRIMARY GLOMERULAR DISEASE | ||
| Membranous glomerulopathy | 5 | 30 |
| Minimal-change disease | 65 | 10 |
| Focal segmental glomerulosclerosis | 10 | 35 |
| Membranoproliferative | 10 | 10 |
| glomerulonephritis† | 10 | 15 |
| Other proliferative glomerulonephritides (focal, “pure mesangial,” IgA nephropathy)† | ||
| SYSTEMIC DISEASES | ||
* Approximate prevalence of primary disease = 95% of nephrotic syndrome in children, 60% in adults. Approximate prevalence of systemic disease = 5% in children, 40% in adults.
† Membranoproliferative and other proliferative glomerulonephritides may have mixed nephrotic/nephritic syndromes.
Membranous Nephropathy
Membranous nephropathy is a common cause of the nephrotic syndrome in adults. It is characterized by diffuse thickening of the glomerular capillary wall due to the accumulation of electron-dense, Ig-containing deposits along the subepithelial side of the basement membrane.30
Membranous glomerulopathy occurring in association with other systemic diseases and a variety of identifiable etiologic agents is referred to as secondary membranous glomerulopathy. The most notable such associations are as follows:
In about 85% of patients no associated condition can be uncovered, and the disease is considered idiopathic.
Pathogenesis.
Membranous glomerulopathy is a form of chronic immune complex–mediated disease. In secondary membranous glomerulopathy, the inciting antigens can sometimes be identified in the immune complexes. For example, membranous glomerulopathy in SLE is associated with deposition of autoantigen-antibody complexes. Antigens that have been identified in the deposits in some patients include exogenous antigens (e.g., hepatitis B, Treponema), endogenous nonrenal antigens (e.g., thyroglobulin), and endogenous renal antigens (e.g., the membrane protein neutral endopeptidase recognized by placental transfer of maternal antibodies in cases of neonatal membranous nephropathy and possibly the phospholipase A2 receptor in adult cases).12
The lesions bear a striking resemblance to those of experimental Heymann nephritis, which, as you might recall, is induced by antibodies to a megalin antigenic complex. It is still not known if a similar antigen is present in most cases of idiopathic membranous glomerulopathy in humans. Susceptibility to Heymann nephritis in rats and membranous glomerulopathy in humans is linked to the major histocompatibility complex locus, which can influence the ability to produce antibodies to the nephritogenic antigen. Thus, idiopathic membranous glomerulopathy, like Heymann nephritis, is considered an autoimmune disease linked to susceptibility genes and caused most likely by antibodies to a renal autoantigen.
How does the glomerular capillary wall become leaky in membranous glomerulopathy? There is a paucity of neutrophils, monocytes, or platelets in glomeruli. The virtually uniform presence of complement and corroborating experimental work suggest a direct action of C5b-C9, the pathway leading to the formation of the membrane attack complex. It is postulated that C5b-C9 activates glomerular epithelial and mesangial cells, inducing them to liberate proteases and oxidants, which cause capillary wall injury and increased protein leakage.32
Morphology. By light microscopy the glomeruli either appear normal in the early stages of the disease or exhibit uniform, diffuse thickening of the glomerular capillary wall (Fig. 20-12A). By electron microscopy the thickening is seen to be caused by irregular dense deposits of immune complexes between the basement membrane and the overlying epithelial cells, the latter having effaced foot processes (Fig. 20-12B and D). Basement membrane material is laid down between these deposits, appearing as irregular spikes protruding from the GBM. These spikes are best seen by silver stains, which color the basement membrane, but not the deposits, black. In time, these spikes thicken to produce domelike protrusions and eventually close over the immune deposits, burying them within a markedly thickened, irregular membrane. Immunofluorescence microscopy demonstrates that the granular deposits contain both immunoglobulins and complement (see Fig. 20-12C). As the disease advances sclerosis may occur; in the course of time glomeruli may become totally sclerosed. The epithelial cells of the proximal tubules contain protein reabsorption droplets, and there may be considerable interstitial mononuclear cell inflammation.
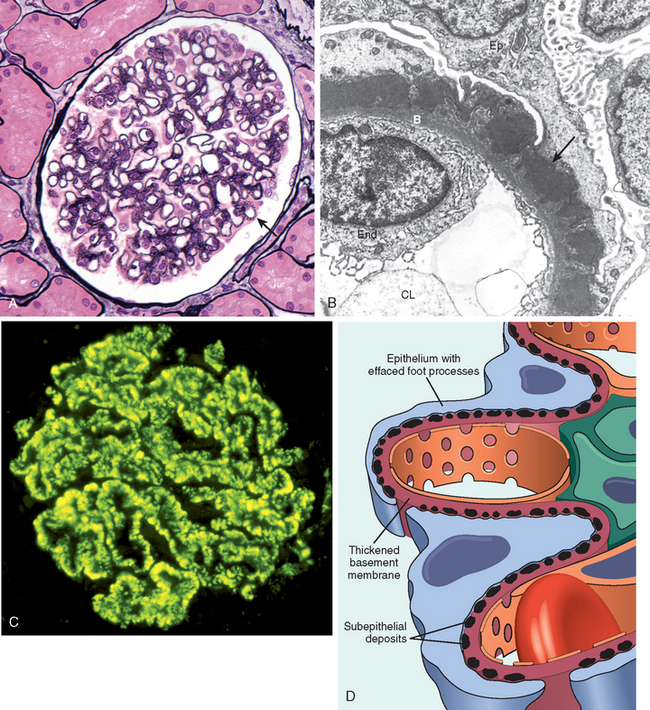
FIGURE 20-12 Membranous nephropathy. A, Silver methenamine stain. Note the marked diffuse thickening of the capillary walls without an increase in the number of cells. There are prominent “spikes” of silver-staining matrix (arrow) projecting from the basement membrane lamina densa toward the urinary space, which separate and surround deposited immune complexes that lack affinity for the silver stain. B, Electron micrograph showing electron-dense deposits (arrow) along the epithelial side of the basement membrane (B). Note the effacement of foot processes overlying deposits. CL, capillary lumen; End, endothelium; Ep, epithelium. C, Characteristic granular immunofluorescent deposits of IgG along GBM. D, Diagrammatic representation of membranous nephropathy.
(A, Courtesy of Dr. Charles Lassman, UCLA School of Medicine, Los Angeles, CA.)
Clinical Features.
In a previously healthy individual, this disorder usually begins with the insidious onset of the nephrotic syndrome or, in 15% of patients, with non-nephrotic proteinuria. Hematuria and mild hypertension are present in 15% to 35% of cases. It is necessary in any patient to first rule out the secondary causes described earlier, since treatment of the underlying condition (malignant neoplasm, infection, or SLE) or discontinuance of the offending drug can reverse the injury.
The course of the disease is variable but generally indolent. In contrast to minimal-change disease, described later, the proteinuria is nonselective and usually does not respond well to corticosteroid therapy. Progression is associated with increasing sclerosis of glomeruli, rising serum creatinine reflecting renal insufficiency, and development of hypertension. Although proteinuria persists in more than 60% of patients, only about 10% die or progress to renal failure within 10 years, and no more than 40% eventually develop renal insufficiency. Concurrent sclerosis of glomeruli in the renal biopsy at the time of diagnosis is a predictor of poor prognosis. Spontaneous remissions and a relatively benign outcome occur more commonly in women and in those with proteinuria in the non-nephrotic range. Because of the variable course of the disease, it has been difficult to evaluate the overall effectiveness of corticosteroids or other immunosuppressive therapy in controlling the proteinuria or progression.
Minimal-Change Disease
This relatively benign disorder is the most frequent cause of nephrotic syndrome in children, but it is less common in adults (see Table 20-7). It is characterized by diffuse effacement of foot processes of visceral epithelial cells (podocytes) in glomeruli that appear virtually normal by light microscopy. The peak incidence is between 2 and 6 years of age. The disease sometimes follows a respiratory infection or routine prophylactic immunization. Its most characteristic feature is its usually dramatic response to corticosteroid therapy.33
Etiology and Pathogenesis.
Although the absence of immune deposits in the glomerulus excludes classic immune complex mechanisms, several features of the disease point to an immunological basis,34 including (1) the clinical association with respiratory infections and prophylactic immunization; (2) the response to corticosteroids and/or other immunosuppressive therapy; (3) the association with other atopic disorders (e.g., eczema, rhinitis); (4) the increased prevalence of certain HLA haplotypes in patients with minimal-change disease associated with atopy (suggesting a genetic predisposition); (5) the increased incidence of minimal-change disease in patients with Hodgkin lymphoma, in whom defects in T cell–mediated immunity are well recognized; and (6) reports of proteinuria-inducing factors in the plasma or lymphocyte supernatants of patients with minimal-change disease.
The current leading hypothesis is that minimal-change disease involves some immune dysfunction, eventually resulting in the elaboration of a cytokine that damages visceral epithelial cells and causes proteinuria. The ultrastructural changes point to a primary visceral epithelial cell injury, and studies in animal models suggest the loss of glomerular polyanions. Thus, defects in the charge barrier may contribute to the proteinuria. The actual route by which protein traverses the epithelial cell portion of the capillary wall remains an enigma. Possibilities include transcellular passage through the epithelial cells, passage through residual spaces between remaining but damaged foot processes or through abnormal spaces developing underneath the portion of the foot process that directly abuts the basement membrane, or leakage through foci in which the epithelial cells have become detached from the basement membrane.
Additional insight into mechanisms by which epithelial cell injury results in proteinuria in minimal-change disease, focal segmental glomerulosclerosis, and related entities comes from the discovery of mutations in several podocyte proteins, including nephrin and podocin, discussed in the section on focal glomerulosclerosis below. These structural proteins are localized to the slit diaphragm, and nephrotic syndrome resulting from mutations in these proteins illustrates that structural defects of the podocyte are sufficient to cause marked proteinuria in the absence of an immune injury. A mutation in the nephrin gene causes a hereditary form of congenital nephrotic syndrome (Finnish type) with minimal-change glomerular morphology.8
Morphology. The glomeruli are normal by light microscopy (Fig. 20-13). By electron microscopy the GBM appears normal, and no electron-dense material is deposited. The principal lesion is in the visceral epithelial cells, which show a uniform and diffuse effacement of foot processes, these being replaced by a rim of cytoplasm often showing vacuolization, swelling, and hyperplasia of villi (see Fig. 20-13). This change, often incorrectly termed “fusion” of foot processes, actually represents simplification of the epithelial cell architecture with flattening, retraction, and swelling of foot processes. Foot process effacement is also present in other proteinuric states (e.g., membranous glomerulopathy, diabetic nephropathy); it is only when effacement is associated with normal glomeruli by light microscopy that the diagnosis of minimal-change disease can be made. The visceral epithelial changes are completely reversible after corticosteroid therapy, concomitant with remission of the proteinuria. The cells of the proximal tubules are often laden with lipid and protein, reflecting tubular reabsorption of lipoproteins passing through diseased glomeruli (thus, the historical name lipoid nephrosis for this disease). Immunofluorescence studies show no Ig or complement deposits.
Clinical Features.
Despite massive proteinuria, renal function remains good, and there is commonly no hypertension or hematuria. The proteinuria usually is highly selective, most of the protein being albumin. Most children (>90%) with minimal-change disease respond rapidly to corticosteroid therapy. However, proteinuria may recur, and some patients may become steroid-dependent or resistant. Nevertheless, the long-term prognosis for patients is excellent, and even steroid-dependent disease resolves when children reach puberty. Although adults are slower to respond, their long-term prognosis is also excellent.
As has been noted, minimal-change disease in adults can be associated with Hodgkin lymphoma and, less frequently, other lymphomas and leukemias. In addition, secondary minimal-change disease may follow NSAID therapy, usually in association with acute interstitial nephritis, to be described later in this chapter.
Focal Segmental Glomerulosclerosis
As the name implies, this lesion is characterized by sclerosis of some, but not all, glomeruli (thus, it is focal); and in the affected glomeruli, only a portion of the capillary tuft is involved (thus, it is segmental). Focal segmental glomerulosclerosis is frequently manifest clinically by the nephrotic syndrome or heavy proteinuria.
Classification and Types.
Focal segmental glomerulosclerosis (FSGS) occurs in the following settings35:
Idiopathic focal segmental glomerulosclerosis accounts for as many as 10% and 35% of cases of nephrotic syndrome in children and adults in many series, respectively. FSGS (both primary and secondary forms) has increased in incidence and is now the most common cause of nephrotic syndrome in adults in the United States,29 particularly in Hispanic and African-American patients. The clinical signs differ from those of minimal-change disease in the following respects: (1) there is a higher incidence of hematuria, reduced GFR, and hypertension; (2) proteinuria is more often nonselective; (3) there is poor response to corticosteroid therapy; and (4) there is progression to chronic kidney disease, with at least 50% developing end-stage renal disease within 10 years.
Pathogenesis.
Whether idiopathic FSGS represents a distinct disease or is simply a phase in the evolution of a subset of patients with minimal-change disease remains unresolved. The characteristic degeneration and focal disruption of visceral epithelial cells are thought to represent an accentuation of the diffuse epithelial cell change typical of minimal-change disease. It is this epithelial damage that is the hallmark of FSGS. Multiple different mechanisms can cause this epithelial damage, including circulating cytokines and genetically determined defects affecting components of the slit diaphragm complex. The hyalinosis and sclerosis stem from entrapment of plasma proteins in extremely hyperpermeable foci and increased ECM deposition. The recurrence of proteinuria, sometimes within 24 hours after transplantation, with subsequent progression to overt lesions of FSGS, suggests that a circulating factor, perhaps a cytokine, may be the cause of the epithelial damage in some patients. An approximately 50-kD non-Ig factor causing proteinuria has been isolated from sera of such patients, but more precise characterization of this factor has not been achieved.36
Morphology. By light microscopy the focal and segmental lesions may involve only a minority of the glomeruli and may be missed if the biopsy specimen contains an insufficient number of glomeruli (Fig. 20-14A). The lesions initially tend to involve the juxtamedullary glomeruli, although they subsequently become more generalized. In the sclerotic segments there is collapse of capillary loops, increase in matrix, and segmental deposition of plasma proteins along the capillary wall (hyalinosis), which may become so pronounced as to occlude capillary lumens. Lipid droplets and foam cells are often present (Fig. 20-14B). Glomeruli that do not show segmental lesions usually appear normal on light microscopy but may show increased mesangial matrix. On electron microscopy both sclerotic and nonsclerotic areas show diffuse effacement of foot processes, and there may also be focal detachment of the epithelial cells and denudation of the underlying GBM. By immunofluorescence microscopy IgM and C3 may be present in the sclerotic areas and/or in the mesangium. In addition to the focal sclerosis, there may be pronounced hyalinosis and thickening of afferent arterioles. With the progression of the disease, increased numbers of glomeruli become involved and sclerosis spreads within each glomerulus. In time, this leads to total (i.e., global) sclerosis of glomeruli, with pronounced tubular atrophy and interstitial fibrosis.
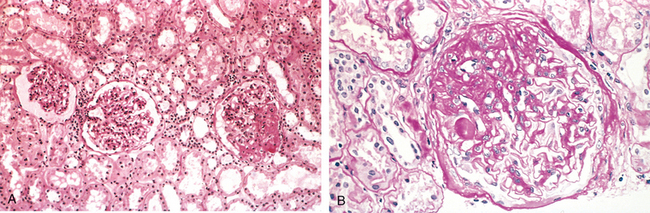
FIGURE 20-14 Focal segmental glomerulosclerosis, PAS stain. A, Low-power view showing segmental sclerosis in one of three glomeruli (at 3 o’clock). B, High-power view showing hyaline insudation and lipid (small vacuoles) in sclerotic area.
A morphologic variant of FSGS, called collapsing glomerulopathy, is characterized by retraction and/or collapse of the entire glomerular tuft, with or without additional FSGS lesions of the type described above (Fig. 20-15). A characteristic feature is proliferation and hypertrophy of glomerular visceral epithelial cells. This lesion may be idiopathic, but it is the most characteristic lesion of HIV-associated nephropathy. In both cases there is associated prominent tubular injury with formation of microcysts. It has a particularly poor prognosis.37,38

FIGURE 20-15 Collapsing glomerulopathy. Visible are retraction of the glomerular tuft, narrowing of capillary lumens, proliferation and swelling of visceral epithelial cells, and prominent accumulation of intracellular protein absorption droplets in the visceral epithelial cells. The appearance is identical in cases wherein the etiology is idiopathic to cases associated with HIV infection. Silver methenamine stain.
(Courtesy of Dr. Jolanta Kowalewska, University of Washington, Seattle, WA.)
The discovery of a genetic basis for some cases of FSGS and other causes of the nephrotic syndrome has improved the understanding of the pathogenesis of proteinuria in the nephrotic syndrome and has provided new methods for diagnosis and prognosis of affected patients. The first relevant gene to be identified, NPHS1, maps to chromosome 19q13 and encodes the protein nephrin.8 Nephrin is a key component of the slit diaphragm (see Fig. 20-3), the zipper-like structure between podocyte foot processes that might control glomerular permeability. Several mutations of the NPHS1 gene have been identified that give rise to congenital nephrotic syndrome of the Finnish type, producing a minimal-change disease–like glomerulopathy with extensive foot process effacement. Prenatal diagnosis of congenital nephrotic syndrome is possible by analysis of the NPHS1 gene.
A distinctive pattern of autosomal recessive FSGS results from mutations in the NPHS2 gene, which maps to chromosome 1q25–q31 and encodes the protein product podocin. Podocin has also been localized to the slit diaphragm. Mutations in NPHS2 result in a syndrome of steroid-resistant nephrotic syndrome of childhood onset. Affected children usually show pathologic features of FSGS. Podocin mutations may account for as many as 30% of cases of steroid-resistant nephrotic syndrome in children.8,39 A third set of mutations in the gene encoding the podocyte actin-binding protein α-actinin 4 underlies some cases of autosomal dominant FSGS, which can be insidious in onset but has a high rate of progression to renal insufficiency.39
A fourth type of mutation was found in some kindreds with adult-onset FSGS, in the gene encoding TRPC6. This protein is widely expressed, including in podocytes, and the pathogenic mutations may perturb podocyte function by increasing calcium flux in these cells.
What these proteins have in common is their localization to the slit diaphragm and to adjacent podocyte cytoskeletal structures. Their specific functions and interactions are incompletely understood, but it is clear that the integrity of each is necessary to maintain the normal glomerular filtration barrier. Additional components of the podocyte/slit diaphragm apparatus, such as CD2-associated protein (CD2AP), have been identified that may also contribute to proteinuria, as has been suggested in studies of knockout mice (but only rarely demonstrated in humans).6 While identification of these genetic defects has clarified the pathogenesis of some cases of the so-called idiopathic nephrotic syndrome, many other factors contribute to permeability defects. These include cell-cell and cell-matrix interactions, particularly those mediated by α3β1 integrins, dystroglycans, and laminins. Defects in these interactions may also cause a loss of podocyte adhesion to the GBM.
Renal ablation FSGS, a secondary form of FSGS, occurs as a complication of glomerular and nonglomerular diseases causing reduction in functioning renal tissue, particularly reflux nephropathy and unilateral agenesis. These may lead to progressive glomerulosclerosis and renal failure. The pathogenesis of FSGS in this setting has been described earlier in this chapter.
Clinical Course.
There is little tendency for spontaneous remission in idiopathic FSGS, and responses to corticosteroid therapy are variable. In general, children have a better prognosis than adults do. Progression of renal failure occurs at variable rates. About 20% of patients follow an unusually rapid course, with intractable massive proteinuria ending in renal failure within 2 years. Recurrences are seen in 25% to 50% of patients receiving allografts.
HIV-Associated Nephropathy
HIV infection can result directly or indirectly in several renal complications, including acute renal failure and/or acute interstitial nephritis induced by drugs or infection, thrombotic microangiopathies, postinfectious glomerulonephritis, and, most commonly, a severe form of the collapsing variant of FSGS.40 The latter occurs in 5% to 10% of HIV-infected individuals in some series, more frequently in blacks than in whites. In rare cases the nephrotic syndrome may precede the development of acquired immunodeficiency syndrome. The morphologic features are characterized by
FIGURE 20-16 The alternative complement pathway in MPGN. Note that C3NeF, an antibody present in the serum of individuals with membranoproliferative glomerulonephritis, acts at the same step as properdin, serving to stabilize the alternative pathway C3 convertase, thus enhancing C3 activation and consumption, causing hypocomplementemia.
The pathogenesis of HIV-related FSGS and tubular injury is probably due to infection of glomerular and tubular cells by HIV, which has been detected in a few cases by very sensitive polymerase chain reaction methods. Studies from animal models indicate that the glomerular lesion is most specifically the result of podocyte expression of the HIV gene products vpr and nef. Altered systemic or local release of cytokines may also modify and promote this particular renal injury.
Membranoproliferative Glomerulonephritis
MPGN is characterized histologically by alterations in the glomerular basement membrane, proliferation of glomerular cells, and leukocyte infiltration. Because the proliferation is predominantly in the mesangium but also may involve the capillary loops, a frequently used synonym is mesangiocapillary glomerulonephritis. MPGN accounts for 10% to 20% of cases of nephrotic syndrome in children and young adults. Some patients present only with hematuria or proteinuria in the non-nephrotic range, but many others have a combined nephrotic-nephritic picture. Like many other glomerulonephritides, MPGN either can be associated with other systemic disorders and known etiologic agents (secondary MPGN) or may be idiopathic (primary MPGN).41
Primary MPGN is divided into two major types on the basis of distinct ultrastructural, immunofluorescent, and pathologic findings: type I and type II MPGN (dense-deposit disease).
Pathogenesis.
In most cases of type I MPGN there is evidence of immune complexes in the glomerulus and activation of both classical and alternative complement pathways.42 The antigens involved in idiopathic MPGN are unknown. In many cases they are believed to be proteins derived from infectious agents such as hepatitis C and B viruses, which presumably behave either as “planted” antigens after first binding to or becoming trapped within glomerular structures or are contained in preformed immune complexes deposited from the circulation.
Most patients with dense-deposit disease (type II MPGN) have abnormalities that suggest activation of the alternative complement pathway.43 These patients have a consistently decreased serum C3 but normal C1 and C4, the immune complex–activated early components of complement. They also have diminished serum levels of factor B and properdin, components of the alternative complement pathway. In the glomeruli, C3 and properdin are deposited, but IgG is not. Recall that in the alternative complement pathway, C3 is directly cleaved to C3b (Fig. 20-16; see also Chapter 2, Fig. 2-14). The reaction depends on the initial interaction of C3 with such substances as bacterial polysaccharides, endotoxin, and aggregates of IgA in the presence of factors B and D. This leads to the generation of C3bBb, the alternative pathway C3 convertase. This C3 convertase is labile, being degraded by factors I and H, but it can be stabilized by properdin. More than 70% of patients with dense-deposit disease have a circulating antibody termed C3 nephritic factor (C3NeF), which is an autoantibody that binds to the alternative pathway C3 convertase (see Fig. 20-16). Binding of the antibody stabilizes the convertase, protecting it from enzymatic degradation and thus favoring persistent C3 activation and hypocomplementemia. There is also decreased C3 synthesis by the liver, further contributing to the profound hypocomplementemia. Precisely how C3NeF is related to glomerular injury and the nature of the dense deposits is unknown. C3NeF activity also occurs in some patients with a genetically determined disease, partial lipodystrophy, some of whom develop dense-deposit disease (type II MPGN).
Morphology. By light microscopy both types of MPGN are similar. The glomeruli are large and hypercellular. The hypercellularity is produced both by proliferation of cells in the mesangium and so-called endocapillary proliferation involving capillary endothelium and infiltrating leukocytes. Crescents are present in many cases. The glomeruli have an accentuated “lobular” appearance due to the proliferating mesangial cells and increased mesangial matrix (Fig. 20-17). The GBM is thickened, often segmentally; this is most evident in the peripheral capillary loops. The glomerular capillary wall often shows a “double-contour” or “tram-track” appearance, especially evident in silver or PAS stains. This is caused by “duplication” of the basement membrane (also commonly referred to as splitting), usually as the result of new basement membrane synthesis in response to the subendothelial deposits of immune complexes. Within the duplicated basement membranes there is inclusion or interposition of cellular elements, which can be of mesangial, endothelial, or leukocytic origin. Such interposition also gives rise to the appearance of “split” basement membranes (Fig. 20-18A).
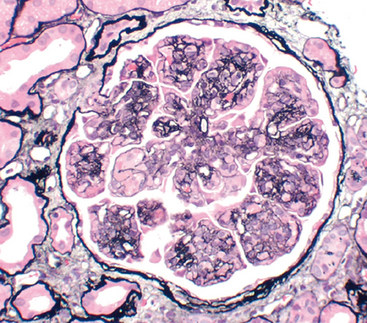
FIGURE 20-17 Membranoproliferative glomerulonephritis, showing mesangial cell proliferation, increased mesangial matrix (staining black with silver stain), basement membrane thickening with segmental splitting, accentuation of lobular architecture, swelling of cells lining peripheral capillaries, and influx of leukocytes (endocapillary proliferation).
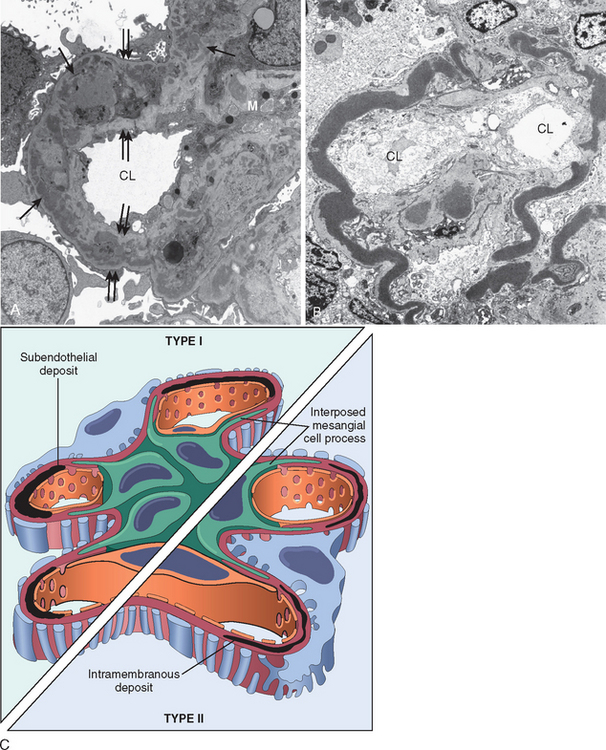
FIGURE 20-18 A, Membranoproliferative glomerulonephritis, type I. Note discrete electron-dense deposits (arrows) incorporated into the glomerular capillary wall between duplicated (split) basement membranes (double arrows), and in mesangial regions (M); CL, capillary lumen. B, Dense-deposit disease (type II membranoproliferative glomerulonephritis). There are markedly dense homogeneous deposits within the basement membrane proper. CL, capillary lumen. In both, mesangial interposition gives the appearance of split basement membranes when viewed in the light microscope. C, Schematic representation of patterns in the two types of membranoproliferative GN. In type I there are subendothelial deposits; type II is characterized by intramembranous dense deposits (dense-deposit disease). In both, mesangial interposition gives the appearance of split basement membranes when viewed in the light microscope.
(A, Courtesy of Dr. Jolanta Kowalewska, University of Washington, Seattle, WA.)
Types I and II MPGN differ in their ultrastructural and immunofluorescent features (Fig. 20-18).
Type I MPGN (the great majority of cases) is characterized by the presence of discrete subendothelial electron-dense deposits. Mesangial and occasional subepithelial deposits may also be present (see Fig. 20-18A). By immunofluorescence, C3 is deposited in a granular pattern, and IgG and early complement components (C1q and C4) are often also present, suggesting an immune complex pathogenesis.
In dense-deposit disease (type II MPGN) (Fig. 20-18B), a relatively rare entity, the lamina densa of the GBM is transformed into an irregular, ribbon-like, extremely electron-dense structure due to the deposition of dense material of unknown composition in the GBM proper. C3 is present in irregular granular or linear foci in the basement membranes on either side but not within the dense deposits. C3 is also present in the mesangium in characteristic circular aggregates (mesangial rings). IgG is usually absent, as are the early-acting complement components (C1q and C4).
Clinical Features.
Most patients present in adolescence or as young adults with nephrotic syndrome and a nephritic component manifested by hematuria or, more insidiously, as mild proteinuria. Few remissions occur spontaneously in either type, and the disease follows a slowly progressive but unremitting course. Some patients develop numerous crescents and a clinical picture of RPGN. About 50% develop chronic renal failure within 10 years. Treatments with steroids, immunosuppressive agents, and antiplatelet drugs have not been proved to be materially effective. There is a high incidence of recurrence in transplant recipients, particularly in dense-deposit disease; dense deposits may recur in 90% of such patients, although renal failure in the allograft is much less common.
Secondary MPGN
Secondary MPGN (invariably type I) is more common in adults and arises in the following settings41:
The mechanisms underlying the process of immune complex deposition in the last three categories above remain unknown.
ISOLATED URINARY ABNORMALITIES
IgA Nephropathy (Berger Disease)
This form of glomerulonephritis is characterized by the presence of prominent IgA deposits in the mesangial regions, detected by immunofluorescence microscopy. The disease can be suspected by light microscopic examination, but the diagnosis is made only by immunocytochemical techniques (Fig. 20-19). IgA nephropathy is a frequent cause of recurrent gross or microscopic hematuria and is probably the most common type of glomerulonephritis worldwide.44 Mild proteinuria is usually present, and the nephrotic syndrome may occasionally develop. Rarely, patients may present with cresentic RPGN.
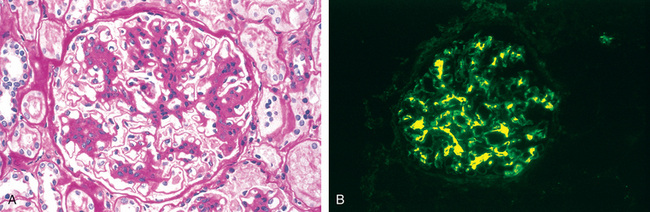
FIGURE 20-19 IgA nephropathy. A, Light microscopy showing mesangial proliferation and matrix increase. B, Characteristic deposition of IgA, principally in mesangial regions, detected by immunofluorescence.
Whereas IgA nephropathy is typically an isolated renal disease, similar IgA deposits are present in a systemic disorder of children, Henoch-Schönlein purpura, to be discussed later, which has many overlapping features with IgA nephropathy. In addition, secondary IgA nephropathy occurs in patients with liver and intestinal diseases, as discussed below.
Pathogenesis.
IgA, the main Ig in mucosal secretions, is present in plasma at low concentrations, mostly in monomeric form, the polymeric forms being catabolized in the liver. In patients with IgA nephropathy, plasma polymeric IgA is increased, and circulating IgA-containing immune complexes are present in some patients. However, it is clear that increased production of IgA cannot itself cause this disease. Although there are two subclasses of IgA molecules in humans (IgA1 and IgA2), only IgA1 forms the nephritogenic deposits of IgA nephropathy. The prominent mesangial deposition of IgA suggests entrapment of IgA immune complexes in the mesangium, and the presence of C3 combined with the absence of C1q and C4 in glomeruli points to activation of the alternative complement pathway. A genetic influence is suggested by the occurrence of this condition in families and in HLA-identical brothers and the increased frequency of certain HLA and complement genotypes in some populations.
Taken together, these clues suggest a genetic or acquired abnormality of immune regulation leading to increased IgA synthesis in response to respiratory or gastrointestinal exposure to environmental agents (e.g., viruses, bacteria, food proteins). IgA1 and IgA1-containing immune complexes are then trapped in the mesangium, where they activate the alternative complement pathway and initiate glomerular injury. In support of this scenario, IgA nephropathy occurs with increased frequency in individuals with gluten enteropathy (celiac disease), in whom intestinal mucosal defects are well defined, and in liver disease, in which there is defective hepatobiliary clearance of IgA complexes (secondary IgA nephropathy).
The nature of the initiating antigens is unknown, and several infectious agents and food products have been implicated. The deposited IgA appears to be polyclonal, and it may be that a variety of antigens are involved in the course of the disease. Alternatively, there is evidence for qualitative alterations in the IgA1 molecule itself, specifically a defect in normal galactosylation that makes it immunogenic, giving rise to autoantibodies against the IgA1 that form immune complexes that deposit in the mesangium.45
Morphology. On histologic examination the lesions vary considerably. The glomeruli may be normal or may show mesangial widening and endocapillary proliferation (mesangioproliferative glomerulonephritis), segmental proliferation confined to some glomeruli (focal proliferative glomerulonephritis), or rarely, overt crescentic glomerulonephritis. The presence of leukocytes within glomerular capillaries is a variable feature. The mesangial widening may be the result of cell proliferation, accumulation of matrix, immune deposits, or some combination of these abnormalities. Healing of the focal proliferative lesion may lead to secondary focal segmental sclerosis. The characteristic immunofluorescent picture is of mesangial deposition of IgA (Fig. 20-19B), often with C3 and properdin and lesser amounts of IgG or IgM. Early complement components are usually absent. Electron microscopy confirms the presence of electron-dense deposits in the mesangium.
Clinical Features.
The disease affects people of any age, but older children and young adults are most commonly affected. Many patients present with gross hematuria after an infection of the respiratory or, less commonly, gastrointestinal or urinary tract; 30% to 40% have only microscopic hematuria, with or without proteinuria; and 5% to 10% develop a typical acute nephritic syndrome. The hematuria typically lasts for several days and then subsides, only to return every few months. The subsequent course is highly variable. Many patients maintain normal renal function for decades. Slow progression to chronic renal failure occurs in 15% to 40% of cases over a period of 20 years. Onset in old age, heavy proteinuria, hypertension, and the extent of glomerulosclerosis on biopsy are clues to an increased risk of progression. Recurrence of IgA deposits in transplanted kidneys is frequent. In approximately 15% of those with recurrent IgA deposits, there is resulting clinical disease, which most frequently runs the same slowly progressive course as that of the primary IgA nephropathy.
Alport Syndrome
Hereditary nephritis refers to a group of heterogeneous familial renal diseases associated primarily with glomerular injury. Two deserve discussion: Alport syndrome, because the lesions and genetic defects have been well studied,46 and thin basement membrane lesion, the most common cause of benign familial hematuria.47
Alport syndrome, when fully developed, is manifest by hematuria with progression to chronic renal failure, accompanied by nerve deafness and various eye disorders, including lens dislocation, posterior cataracts, and corneal dystrophy.48 The disease is inherited as an X-linked trait in approximately 85% of cases. In this X-linked form, males express the full syndrome, and females are carriers in whom manifestations of disease are typically limited to hematuria. Autosomal recessive and autosomal dominant pedigrees also exist, in which males and females are equally susceptible to the full syndrome.
Pathogenesis.
The disease manifestations are due to abnormal α3 (COL4A3), α4 (COL4A4), or α5 (COL4A5) chain of type IV collagen. This is due to mutation of COL4A5 in the classic X-linked form and COL4A3 or COL4A4 in the autosomal forms. In all cases, the result is defective assembly of type IV collagen, which is crucial for function of the GBM, the lens of the eye, and the cochlea. Because the GBM consists of networks of trimeric collagen molecules composed of α3, α4, and α5 chains, mutations in COL4A5 also result in defective assembly of the collagen network.46 The α3 chain includes the Goodpasture antigen, and glomeruli from patients with Alport syndrome who lack the α3 chain fail to react with anti-GBM antibodies from patients with Goodpasture syndrome.
Morphology. On histologic examination the glomeruli are always involved. The early lesion is detectable only by electron microscopy and consists of diffuse GBM thinning. Interstitial foam cells stuffed with neutral fats and mucopolysaccharides are a nonspecific finding consequent to proteinuria that for unknown reasons can be unusually prominent in this disorder. As the disease progresses there is development of focal segmental and global glomerulosclerosis and other changes of progressive renal injury, including vascular sclerosis, tubular atrophy, and interstitial fibrosis.
The characteristic electron microscopic findings of fully developed disease are found in most individuals with hereditary nephritis. The GBM shows irregular foci of thickening alternating with attenuation (thinning), and pronounced splitting and lamination of the lamina densa, often producing a distinctive basket-weave appearance (Fig. 20-20). Similar alterations can be found in the tubular basement membranes.
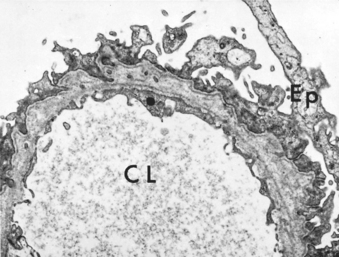
FIGURE 20-20 Hereditary nephritis (Alport syndrome). Electron micrograph of glomerulus with irregular thickening of the basement membrane, lamination of the lamina densa, and foci of rarefaction. Such changes may be present in other diseases but are most pronounced and widespread in hereditary nephritis. CL, capillary lumen; Ep, epithelium.
Immunohistochemistry can be helpful in cases with absent or borderline basement membrane lesions, because antibodies to α3, α4, and α5 collagen fail to stain both glomerular and tubular basement membranes in the classic X-linked form. There is also absence of α5 staining in skin biopsy specimens from these patients.
Clinical Features.
The most common presenting sign is gross or microscopic hematuria, frequently accompanied by red cell casts. Proteinuria may develop later, and rarely, the nephrotic syndrome develops. Symptoms appear at ages 5 to 20 years, and the onset of overt renal failure is between ages 20 and 50 years in men. The auditory defects may be subtle, requiring sensitive testing.
Thin Basement Membrane Lesion (Benign Familial Hematuria)
This is a fairly common hereditary entity manifested clinically by familial asymptomatic hematuria—usually uncovered on routine urinalysis—and morphologically by diffuse thinning of the GBM to between 150 and 250 nm (compared with 300–400 nm in normal adult individuals). Although mild or moderate proteinuria may also be present, renal function is normal and prognosis is excellent.
The disorder should be distinguished from IgA nephropathy, another common cause of hematuria, and X-linked Alport syndrome. In contrast to Alport syndrome, hearing loss, ocular abnormalities, and a family history of renal failure are absent.
The anomaly in thin basement membrane lesion has also been traced to mutations in genes encoding α3 or α4 chains of type IV collagen.46,47 Most patients are heterozygous for the defective gene and thus may be carriers. The disorder in homozygotes resembles autosomal recessive Alport syndrome. Homozygotes or compound heterozygotes may progress to renal failure. Thus, these diseases illustrate a continuum of changes resulting from mutations in collagen type IV genes.
CHRONIC GLOMERULONEPHRITIS
Chronic glomerulonephritis is best considered a pool of end-stage glomerular disease fed by several streams of specific types of glomerulonephritis (Fig. 20-21). Most of these diseases were described earlier in this chapter. Poststreptococcal glomerulonephritis is a rare antecedent of chronic glomerulonephritis, except in adults. Patients with crescentic glomerulonephritis, if they survive the acute episode, usually progress to chronic glomerulonephritis. Membranous nephropathy, MPGN, IgA nephropathy, and FSGS all may progress to chronic renal failure. Nevertheless, in any series of individuals with chronic glomerulonephritis, a variable percentage of cases arise mysteriously with no antecedent history of any of the well-recognized forms of acute glomerulonephritis. These cases must represent the end result of relatively asymptomatic forms of glomerulonephritis, either known or still unrecognized, that progress to uremia. Clearly, the proportion of such unexplained cases depends on the availability of renal biopsy material from patients early in their disease.
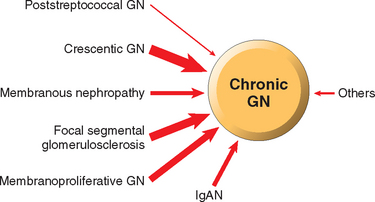
FIGURE 20-21 Primary glomerular diseases leading to chronic glomerulonephritis (GN). The thickness of the arrows reflects the approximate proportion of patients in each group who progress to chronic GN: poststreptococcal (1% to 2%); rapidly progressive (crescentic) (90%), membranous (30% to 50%), focal segmental glomerulosclerosis (50% to 80%), membranoproliferative GN (50%), IgA nephropathy (IgAN, 30% to 50%).
Morphology. The kidneys are symmetrically contracted and have diffusely granular cortical surfaces. On section, the cortex is thinned, and there is an increase in peripelvic fat. The glomerular histology depends on the stage of the disease. In early cases, the glomeruli may still show evidence of the primary disease (e.g., membranous nephropathy or MPGN). However, there eventually ensues obliteration of glomeruli, transforming them into acellular eosinophilic masses, representing a combination of trapped plasma proteins, increased mesangial matrix, basement membrane–like material, and collagen (Fig. 20-22). Because hypertension is an accompaniment of chronic glomerulonephritis, arterial and arteriolar sclerosis may be conspicuous. Marked atrophy of associated tubules, irregular interstitial fibrosis, and mononuclear leukocytic infiltration of the interstitium also occur.
FIGURE 20-22 Chronic glomerulonephritis. A Masson trichrome preparation shows complete replacement of virtually all glomeruli by blue-staining collagen.
(Courtesy of Dr. M.A. Venkatachalam, Department of Pathology, University of Texas Health Sciences Center, San Antonio, TX.)
Dialysis Changes. Kidneys from patients with end-stage disease on long-term dialysis show a variety of changes that are unrelated to the primary disease. These include arterial intimal thickening caused by accumulation of smooth muscle–like cells and a loose, proteoglycan-rich stroma; focal calcification, usually within residual tubular segments; extensive deposition of calcium oxalate crystals in tubules and interstitium; acquired cystic disease, discussed later; and increased numbers of renal adenomas and adenocarcinomas.
Uremic Complications. Individuals dying with chronic glomerulonephritis also exhibit pathologic changes outside the kidney that are related to the uremic state and are also present in other forms of chronic renal failure. Often clinically important, these include uremic pericarditis, uremic gastroenteritis, secondary hyperparathyroidism with nephrocalcinosis and renal osteodystrophy, left ventricular hypertrophy due to hypertension, and pulmonary changes of diffuse alveolar damage often ascribed to uremia (uremic pneumonitis).
Clinical Course.
In most individuals, chronic glomerulonephritis develops insidiously and slowly progresses to renal insufficiency or death from uremia during a span of years or possibly decades (see the discussion of chronic renal failure). Not infrequently, patients present with such nonspecific complaints as loss of appetite, anemia, vomiting, or weakness. In some, the renal disease is suspected with the discovery of proteinuria, hypertension, or azotemia on routine medical examination. In others, the underlying renal disorder is discovered in the course of investigation of edema. Most patients are hypertensive, and sometimes the dominant clinical manifestations are cerebral or cardiovascular. In all, the disease is relentlessly progressive, though at widely varying rates. In nephrotic patients, as glomeruli become obliterated and therefore the GFR decreases, the protein loss in the urine diminishes. If patients with chronic glomerulonephritis do not recieve dialysis or if they do not receive a renal transplant, they invariably succumb to their disease.
GLOMERULAR LESIONS ASSOCIATED WITH SYSTEMIC DISEASES
Many immunologically mediated, metabolic, or hereditary systemic disorders are associated with glomerular injury; in some (e.g., SLE and diabetes mellitus), the glomerular involvement is a major clinical manifestation. Most of these diseases are discussed elsewhere in this book. Here we briefly recall some of the lesions and discuss only those not considered in other sections.
Lupus Nephritis
The various types of lupus nephritis were described and illustrated in Chapter 6. As discussed, SLE gives rise to a heterogeneous group of lesions and clinical presentations. The clinical manifestations can include recurrent microscopic or gross hematuria, the nephritic syndrome, the nephrotic syndrome, chronic renal failure, and hypertension.
Henoch-Schönlein Purpura
This syndrome consists of purpuric skin lesions characteristically involving the extensor surfaces of arms and legs as well as buttocks; abdominal manifestations including pain, vomiting, and intestinal bleeding; nonmigratory arthralgia; and renal abnormalities. The renal manifestations occur in one third of patients and include gross or microscopic hematuria, nephritic syndrome, and nephrotic syndrome, or some combination of these. A small number of patients, mostly adults, develop a rapidly progressive form of glomerulonephritis with many crescents. Not all components of the syndrome need to be present, and individual patients may have purpura, abdominal pain, or urinary abnormalities as the dominant feature. The disease is most common in children 3 to 8 years old, but it also occurs in adults, in whom the renal manifestations are usually more severe. There is a strong background of atopy in about one third of patients, and onset often follows an upper respiratory infection. IgA is deposited in the glomerular mesangium in a distribution similar to that of IgA nephropathy. This has led to the concept that IgA nephropathy and Henoch-Schönlein purpura are manifestations of the same disease.49
Morphology. On histologic examination, the renal lesions vary from mild focal mesangial proliferation to diffuse mesangial proliferation and/or endocapillary to crescentic glomerulonephritis. Whatever the histologic lesions, the prominent feature by fluorescence microscopy is the deposition of IgA, sometimes with IgG and C3, in the mesangial region. The skin lesions consist of subepidermal hemorrhages and a necrotizing vasculitis involving the small vessels of the dermis. IgA deposits are also present in such vessels. Vasculitis also occurs in other organs, such as the gastrointestinal tract, but is rare in the kidney.
The course of the disease is variable, but recurrences of hematuria may persist for many years after onset. Most children have an excellent prognosis. Patients with the more diffuse lesions, crescents, or the nephrotic syndrome have a somewhat poorer prognosis.
Bacterial Endocarditis–Associated Glomerulonephritis
Glomerular lesions occurring in the course of bacterial endocarditis represent a type of immune complex nephritis initiated by complexes of bacterial antigen and antibody. Hematuria and proteinuria of various degrees characterize this entity clinically, but an acute nephritic presentation is not uncommon, and even RPGN may occur in rare instances. The histologic lesions, when present, generally reflect these clinical manifestations. Milder forms have a more focal and segmental necrotizing glomerulonephritis, whereas more severe ones show a diffuse proliferative glomerulonephritis, and the rapidly progressive forms show large numbers of crescents. Immunofluorescence and electron microscopy show the presence of glomerular immune deposits.
Diabetic Nephropathy
Diabetes mellitus is a major cause of renal morbidity and mortality, and diabetic nephropathy is one of the leading causes of chronic kidney failure in the United States (see Chapter 24). Advanced or end-stage kidney disease occurs in as many as 40% of both insulin-dependent type 1 diabetics and type 2 diabetics. By far the most common lesions involve the glomeruli and are associated clinically with three glomerular syndromes: non-nephrotic proteinuria, nephrotic syndrome, and chronic renal failure.50 However, diabetes also affects the arterioles (causing hyalinizing arteriolar sclerosis), increases susceptibility to the development of pyelonephritis and particularly papillary necrosis, and causes a variety of tubular lesions. The term diabetic nephropathy is applied to the conglomerate of lesions that often occur concurrently in the diabetic kidney.
The morphologic changes in the glomeruli include (1) capillary basement membrane thickening, (2) diffuse mesangial sclerosis, and (3) nodular glomerulosclerosis. The morphologic manifestations of diabetic nephropathy are identical in type 1 and type 2 diabetes and are described below as a single entity.51,52
Pathogenesis.
The pathogenesis of diabetic glomerulosclerosis is intimately linked with that of generalized diabetic microangiopathy, discussed in Chapter 24. The principal points are as follows53:
To sum up, two processes seem to play a role in the fully developed diabetic glomerular lesions: a metabolic defect, probably linked to advanced glycosylation end products, that accounts for the thickened GBM and increased mesangial matrix that occur in patients; and hemodynamic effects, associated with glomerular hypertrophy, which also contribute to the development of glomerulosclerosis. Both of these processes contribute also to the loss of podocytes, which may undergo apoptosis in response to the metabolic abnormalities and exposure to reactive oxygen species, or become detached from the basement membranes as a consequence of these metabolic changes and/or stretching induced by hemodynamic perturbations.56 The morphologic alterations and clinical features of diabetic nephropathy are described in Chapter 24.
Amyloidosis
The various forms of amyloidosis and their pathogenesis are discussed in Chapter 6. Most types of disseminated amyloidosis may be associated with deposits of amyloid within the glomeruli; most commonly renal amyloid is of light-chain (AL) or AA type. The typical Congo red–positive fibrillary amyloid deposits are present within the mesangium and capillary walls and rarely are localized to the subepithelial space. Eventually, they obliterate the glomerulus completely. Deposits of amyloid also appear in blood vessel walls and in the kidney interstitium. Patients with glomerular amyloid may present with the nephrotic syndrome and later, as a result of destruction of the glomeruli, die of uremia. Characteristically, kidney size tends to be either normal or increased.
Fibrillary Glomerulonephritis and Immunotactoid Glomerulopathy
Fibrillary glomerulonephritis is a morphologic variant of glomerulonephritis associated with characteristic fibrillar deposits in the mesangium and glomerular capillary walls that resemble amyloid fibrils superficially but differ ultrastructurally and do not stain with Congo red.60 The fibrils most often are 18 to 24 nm in diameter and hence are larger than the 10 to 12 nm fibrils characteristic of amyloid. The glomerular lesions usually show membranoproliferative or mesangioproliferative patterns by light microscopy, and by immunofluorescence microscopy, there is selective deposition of polyclonal IgG, often of the IgG4 subclass, complement C3, and Igκ and Igλ light chains. Clinically, patients develop nephrotic syndrome, hematuria, and progressive renal insufficiency. The disease recurs in kidney transplants.
In immunotactoid glomerulopathy, a much rarer condition, the deposits are microtubular in structure and 30 to 50 nm in width. Patients often have circulating paraproteins and/or monoclonal Ig deposition in glomeruli.57
Other Systemic Disorders
Goodpasture syndrome (Chapter 15), microscopic polyangiitis, and Wegener granulomatosis (Chapter 11) are commonly associated with glomerular lesions, as described in the discussion of these diseases. Suffice it to say here that the glomerular lesions in these three conditions can be histologically similar and are principally characterized by foci of glomerular necrosis and crescent formation. In the early or mild forms of involvement, there is focal and segmental, sometimes necrotizing, glomerulonephritis, and most of these patients will have hematuria with mild decline in GFR. In the more severe cases associated with RPGN, there is more extensive necrosis, fibrin deposition, and extensive formation of epithelial (cellular) crescents, which can become organized to form fibrocellular and fibrous crescents if the glomerular injury evolves into segmental or global scarring (sclerosis).
Essential mixed cryoglobulinemia is another systemic condition in which deposits of cryoglobulins composed principally of IgG-IgM complexes induce cutaneous vasculitis, synovitis, and a proliferative glomerulonephritis, typically MPGN. Most cases of essential mixed cryoglobulinemia have been associated with infection with hepatitis C virus, and this condition in particular is associated with glomerulonephritis, usually MPGN type I.
Plasma cell dyscrasias may also induce glomerular lesions. Multiple myeloma and other dyscrasias producing circulating monoclonal immunoglobulins are associated with (1) amyloidosis, in which the fibrils are usually composed of monoclonal λ light chains, (2) deposition of monoclonal immunoglobulins or light chains in the GBM, and (3) distinctive nodular glomerular lesions resulting from the deposition of nonfibrillar light chains. This so-called light-chain or monoclonal Ig deposition disease sometimes occurs in the absence of overt myeloma and is usually characterized by deposition of monoclonal Igκ or Igλ light chains in glomeruli. The glomeruli show PAS-positive mesangial nodules, lobular accentuation, and mild mesangial hypercellularity. These lesions must be differentiated from diabetic nodular glomerulosclerosis and other glomerulopathies that can cause nodular mesangial expansion, such as MPGN. These patients usually present with proteinuria or the nephrotic syndrome, hypertension, and progressive azotemia. Other renal manifestations of multiple myeloma are discussed later.