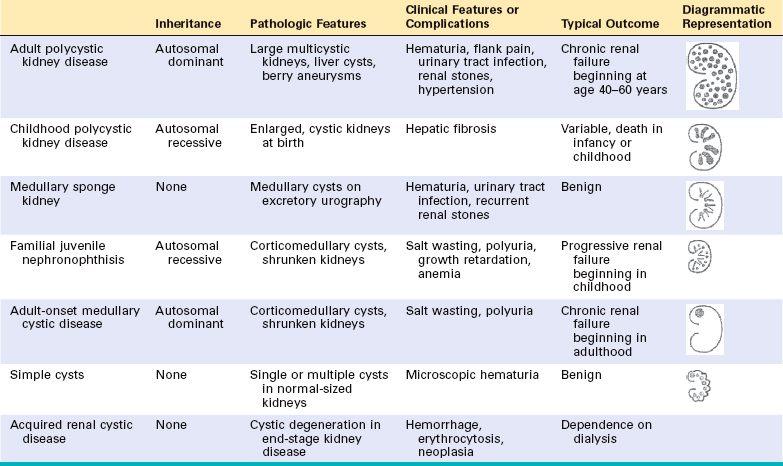
Tubular and Interstitial Diseases
Most forms of tubular injury involve the interstitium as well; therefore, diseases affecting these two components are discussed together. Under this heading we consider two major groups of processes: (1) ischemic or toxic tubular injury, leading to acute kidney injury (AKI) and acute renal failure, and (2) inflammatory reactions of the tubules and interstitium (tubulointerstitial nephritis).
ACUTE KIDNEY INJURY (AKI) (ACUTE TUBULAR NECROSIS, ATN)
AKI, a term increasingly favored over the often synonymously used terms acute tubular necrosis (ATN) and acute tubular injury, is a clinicopathologic entity characterized clinically by acute diminution of renal function and often, but not invariably, morphologic evidence of tubular injury. It is the most common cause of acute renal failure,58,59 which signifies rapid reduction of renal function and urine flow, falling within 24 hours to less than 400 mL per day. It can be caused by a variety of conditions, including
AKI accounts for some 50% of cases of acute renal failure in hospitalized patients. Other causes of acute renal failure are discussed elsewhere in this chapter.
AKI is a reversible renal lesion that arises in a variety of clinical settings. Most of these, ranging from severe trauma to acute pancreatitis, have in common a period of inadequate blood flow to the peripheral organs, usually accompanied by marked hypotension and shock. This pattern of AKI is called ischemic AKI. The second pattern, called nephrotoxic AKI, is caused by a multitude of drugs, such as gentamicin and other antibiotics; radiographic contrast agents; poisons, including heavy metals (e.g., mercury); and organic solvents (e.g., carbon tetrachloride). Combinations of ischemic and nephrotoxic AKI also can occur, exemplified by mismatched blood transfusions and other hemolytic crises causing hemoglobinuria and skeletal muscle injuries causing myoglobinuria. Such injuries result in characteristic intratubular hemoglobin or myoglobin casts, respectively; the toxic iron content of these globin molecules contributes to the AKI. In addition to its frequency, the potential reversibility of AKI adds to its clinical importance. Proper management means the difference between full recovery and death.
Pathogenesis.
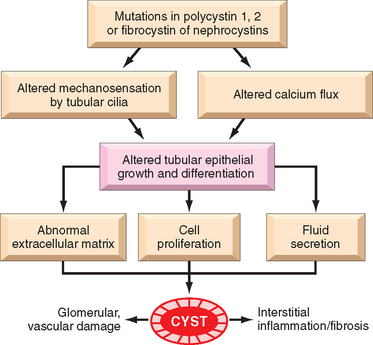
The critical events in both ischemic and nephrotoxic AKI are believed to be (1) tubular injury and (2) persistent and severe disturbances in blood flow60 (Fig. 20-23).
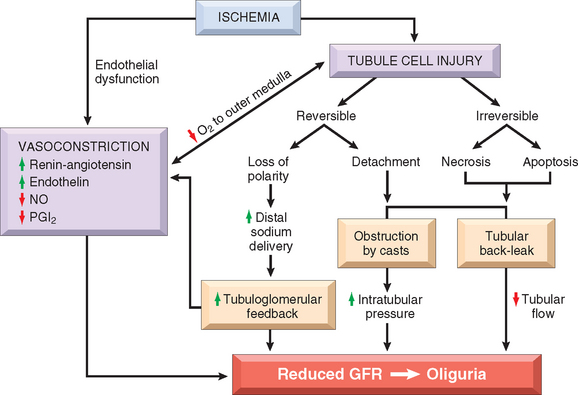
FIGURE 20-23 Postulated sequence in acute kidney injury. GFR, glomerular filtration rate; NO, nitric oxide; PGI2, prostaglandin I2 (prostacyclin).
(Modified from Brady HR et al.: Acute renal failure. In Brenner BM [ed]: Brenner and Rector’s The Kidney, 5th ed, Vol II. Philadelphia, WB Saunders, 1996, p 1210).
Ischemia causes numerous structural and functional alterations in epithelial cells, as discussed in Chapter 1. The structural changes include those of reversible injury (such as cellular swelling, loss of brush border and polarity, blebbing, and cell detachment) and those associated with lethal injury (necrosis and apoptosis). Biochemically there is depletion of ATP; accumulation of intracellular calcium; activation of proteases (e.g., calpain), which cause cytoskeletal disruption; activation of phospholipases, which damage membranes; generation of reactive oxygen species; and activation of caspases, which induce apoptotic cell death. One early reversible result of ischemia is loss of cell polarity due to redistribution of membrane proteins (e.g., the enzyme Na+, K+-ATPase) from the basolateral to the luminal surface of the tubular cells, resulting in abnormal ion transport across the cells, and increased sodium delivery to distal tubules. The latter incites vasoconstriction via tubuloglomerular feedback, which will be discussed below. In addition, ischemic tubular cells express cytokines (such as monocyte chemoattractant protein 1) and adhesion molecules (such as intercellular adhesion molecule 1), thus recruiting leukocytes that appear to participate in the subsequent injury. In time, injured cells detach from the basement membranes and cause luminal obstruction, increased intratubular pressure, and decreased GFR. In addition, fluid from the damaged tubules leaks into the interstitium, resulting in interstitial edema, increased interstitial pressure, and further damage to the tubule. All these effects, as shown in Figure 20-23, contribute to the decreased GFR.
The patchiness of tubular necrosis and maintenance of the integrity of the basement membrane along many segments allow ready repair of the necrotic foci and recovery of function if the precipitating cause is removed. This repair is dependent on the capacity of reversibly injured epithelial cells to proliferate and differentiate. Re-epithelialization is mediated by a variety of growth factors and cytokines produced locally by the tubular cells themselves (autocrine stimulation) or by inflammatory cells in the vicinity of necrotic foci (paracrine stimulation).61 Of these, epidermal growth factor, TGF-α, insulin-like growth factor type 1, and hepatocyte growth factor have been shown to be particularly important in renal tubular repair. Growth factors, indeed, are being explored as possible therapeutic agents to enhance re-epithelialization in AKI, although clinical trials to date have been disappointing.61
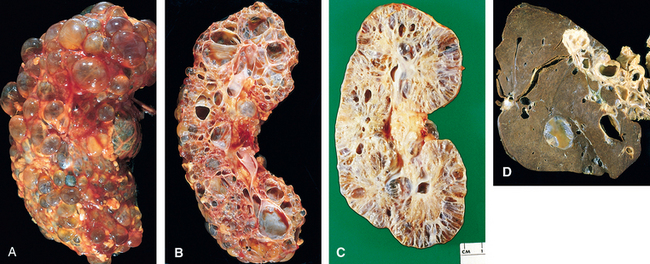
Morphology. Ischemic AKI is characterized by focal tubular epithelial necrosis at multiple points along the nephron, with large skip areas in between, often accompanied by rupture of basement membranes (tubulorrhexis) and occlusion of tubular lumens by casts62 (Figs. 20-24 and 20-25). The straight portion of the proximal tubule and the ascending thick limb in the renal medulla are especially vulnerable, but focal lesions may also occur in the distal tubule, often in conjunction with casts. Paradoxically, the clinical syndrome of AKI is often associated with lesser degrees of tubular injury. This includes attenuation or loss of proximal tubule brush borders, simplification of cell structure, cell swelling and vacuolization, and sloughing of non-necrotic tubular cells into the tubular lumina (see Fig. 20-25). The severity of the morphologic findings often does not correlate well with the severity of the clinical manifestations.
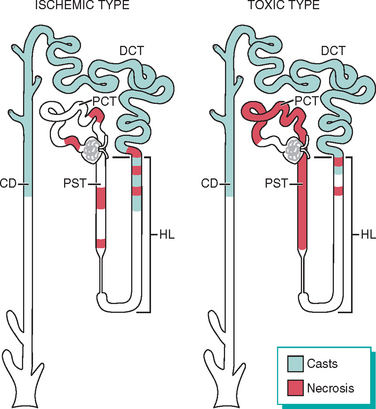
FIGURE 20-24 Patterns of tubular damage in ischemic and toxic acute kidney injury. In the ischemic type, tubular necrosis is patchy, relatively short lengths of tubules are affected, and straight segments of proximal tubules (PST) and ascending limbs of Henle’s loop (HL) are most vulnerable. In toxic acute kidney injury, extensive necrosis is present along the proximal convoluted tubule segments (PCT) with many toxins (e.g., mercury), but necrosis of the distal tubule, particularly ascending HL, also occurs. In both types, lumens of the distal convoluted tubules (DCT) and collecting ducts (CD) contain casts.
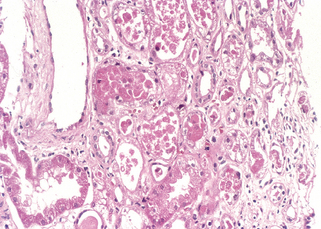
FIGURE 20-25 Acute kidney injury. Some of the tubular epithelial cells in the tubules are necrotic, and many have become detached (from their basement membranes) and been sloughed into the tubular lumens, whereas others are swollen, vacuolated, and regenerating.
(Courtesy of Dr. Agnes Fogo, Vanderbilt University, Nashville, TN.)
Eosinophilic hyaline casts, as well as pigmented granular casts, are common, particularly in distal tubules and collecting ducts. These casts consist principally of Tamm-Horsfall protein (a urinary glycoprotein normally secreted by the cells of ascending thick limb and distal tubules) in conjunction with other plasma proteins. Other findings in ischemic AKI are interstitial edema and accumulations of leukocytes within dilated vasa recta. There is also evidence of epithelial regeneration: flattened epithelial cells with hyperchromatic nuclei and mitotic figures are often present. In the course of time this regeneration repopulates the tubules so that, no residual evidence of damage is seen.
Toxic AKI is manifested by acute tubular injury, most obvious in the proximal convoluted tubules. On histologic examination the tubular necrosis may be entirely nonspecific, but it is somewhat distinctive in poisoning with certain agents. With mercuric chloride, for example, severely injured cells may contain large acidophilic inclusions. Later, these cells become totally necrotic, are desquamated into the lumen, and may undergo calcification. Carbon tetrachloride poisoning, in contrast, is characterized by the accumulation of neutral lipids in injured cells; again, such fatty change is followed by necrosis. Ethylene glycol produces marked ballooning and hydropic or vacuolar degeneration of proximal convoluted tubules. Calcium oxalate crystals are often found in the tubular lumens in such poisoning.
Clinical Course.
The clinical course of AKI is highly variable, but the classic case may be divided into initiation, maintenance, and recovery stages. The initiation phase, lasting for about 36 hours, is dominated by the inciting medical, surgical, or obstetric event in the ischemic form of AKI. The only indication of renal involvement is a slight decline in urine output with a rise in BUN. At this point, oliguria could be explained on the basis of a transient decrease in blood flow and declining GFR.
The maintenance phase is characterized by sustained decreases in urine output to between 40 and 400 mL/day (oliguria), salt and water overload, rising BUN concentrations, hyperkalemia, metabolic acidosis, and other manifestations of uremia. With appropriate attention to the balance of water and blood electrolytes, including dialysis, the patient can be supported through this oliguric crisis.
The recovery phase is ushered in by a steady increase in urine volume that may reach up to 3 L/day. The tubules are still damaged, so large amounts of water, sodium, and potassium are lost in the flood of urine. Hypokalemia, rather than hyperkalemia, becomes a clinical problem. There is a peculiar increased vulnerability to infection at this stage. Eventually, renal tubular function is restored and concentrating abilityimproves. At the same time, BUN and creatinine levels begin to return to normal. Subtle tubular functional impairment may persist for months, but most patients who reach this phase eventually recover completely.
The prognosis of AKI depends on the clinical setting. Recovery is expected with nephrotoxic AKI when the toxin has not caused serious damage to other organs, such as the liver or heart. With current supportive care, 95% of those who do not succumb to the precipitating cause recover. Conversely, in shock related to sepsis, extensive burns, or other causes of multi-organ failure, the mortality rate can rise to more than 50%.
Up to 50% of patients with AKI do not have oliguria and instead often have increased urine volumes. This so-called nonoliguric AKI occurs particularly often with nephrotoxins, and it generally tends to follow a more benign clinical course.
TUBULOINTERSTITIAL NEPHRITIS
This group of renal diseases is characterized by histologic and functional alterations that involve predominantly the tubules and interstitium. We have previously seen that chronic tubulointerstitial injury may occur in diseases that primarily affect the glomerulus (see Fig. 20-22) and that such injury may be an important cause of progression in these diseases.18 Secondary tubulointerstitial nephritis is also present in a variety of vascular, cystic (polycystic kidney disease), and metabolic (diabetes) renal disorders, in which it may also contribute to progressive damage. Here we discuss disorders in which tubulointerstitial injury seems to be a primary event. These disorders have diverse causes and different pathogenetic mechanisms (Table 20-8). Glomerular and vascular abnormalities may also be present but either are mild or occur only in advanced stages of these diseases.
TABLE 20-8 Causes of Tubulointerstitial Nephritis
| INFECTIONS |
| TOXINS |
| METABOLIC DISEASES |
| PHYSICAL FACTORS |
| Chronic urinary tract obstruction |
| NEOPLASMS |
| Multiple myeloma (light-chain cast nephropathy) |
| IMMUNOLOGIC REACTIONS |
| VASCULAR DISEASES |
| MISCELLANEOUS |
Tubulointerstitial nephritis can be acute or chronic. Acute tubulointerstitial nephritis has a rapid clinical onset and is characterized histologically by interstitial edema, often accompanied by leukocytic infiltration of the interstitium and tubules, and focal tubular necrosis. In chronic interstitial nephritis there is infiltration with predominantly mononuclear leukocytes, prominent interstitial fibrosis, and widespread tubular atrophy. Morphologic features that are helpful in separating acute from chronic tubulointerstitial nephritis include edema and, when present, eosinophils and neutrophils in the acute form, while fibrosis and tubular atrophy characterize the chronic form.
These conditions are distinguished clinically from the glomerular diseases by the absence, in early stages, of such hallmarks of glomerular injury as nephritic or nephrotic syndrome and by the presence of defects in tubular function. The latter may be subtle and include impaired ability to concentrate urine, evidenced clinically by polyuria or nocturia; salt wasting; diminished ability to excrete acids (metabolic acidosis); and isolated defects in tubular reabsorption or secretion. The advanced forms, however, may be difficult to distinguish clinically from other causes of renal insufficiency.
Some of the specific conditions listed in Table 20-8 are discussed elsewhere in this book. In this section we deal principally with pyelonephritis and interstitial diseases induced by drugs.
Pyelonephritis and Urinary Tract Infection
Pyelonephritis is a renal disorder affecting the tubules, interstitium, and renal pelvis and is one of the most common diseases of the kidney. It occurs in two forms. Acute pyelonephritis is caused by bacterial infection and is the renal lesion associated with urinary tract infection. Chronic pyelonephritis is a more complex disorder; bacterial infection plays a dominant role, but other factors (vesicoureteral reflux, obstruction) are involved in its pathogenesis. Pyelonephritis is a serious complication of urinary tract infections that affect the bladder (cystitis), the kidneys and their collecting systems (pyelonephritis), or both. Bacterial infection of the lower urinary tract may be completely asymptomatic (asymptomatic bacteriuria) and most often remains localized to the bladder without the development of renal infection. However, lower urinary tract infection always carries the potential of spread to the kidney.
Etiology and Pathogenesis.
The dominant etiologic agents, accounting for more than 85% of cases of urinary tract infection, are the gram-negative bacilli that are normal inhabitants of the intestinal tract.63 By far the most common is Escherichia coli, followed by Proteus, Klebsiella, and Enterobacter. Streptococcus faecalis, also of enteric origin, staphylococci, and virtually every other bacterial and fungal agent can also cause lower urinary tract and renal infection. In immunocompromised persons, particularly those with transplanted organs, viruses such as Polyomavirus, cytomegalovirus, and adenovirus can also be a cause of renal infection.
In most patients with urinary tract infection, the infecting organisms are derived from the patient’s own fecal flora. This is thus a form of endogenous infection. There are two routes by which bacteria can reach the kidneys: (1) through the bloodstream (hematogenous infection) and (2) from the lower urinary tract (ascending infection) (Fig. 20-26). The hematogenous route is the less common of the two and results from seeding of the kidneys by bacteria from distant foci in the course of septicemia or infective endocarditis. Hematogenous infection is more likely to occur in the presence of ureteral obstruction, in debilitated patients, in patients receiving immunosuppressive therapy, and with nonenteric organisms, such as staphylococci and certain fungi and viruses.
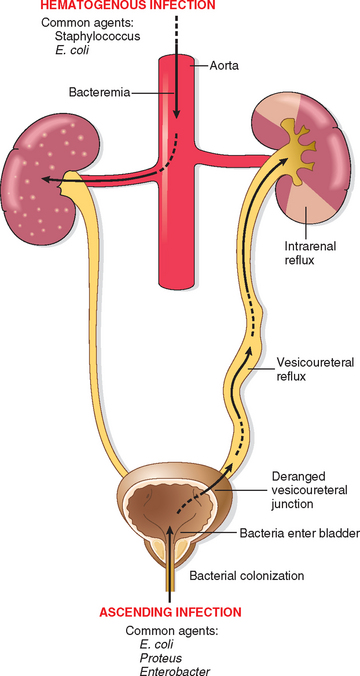
FIGURE 20-26 Schematic representation of pathways of renal infection. Hematogenous infection results from bacteremic spread. More common is ascending infection, which results from a combination of urinary bladder infection, vesicoureteral reflux, and intrarenal reflux.
Ascending infection is the most common cause of clinical pyelonephritis. Normal human bladder and bladder urine are sterile; therefore, a number of steps must occur for renal infection to occur:
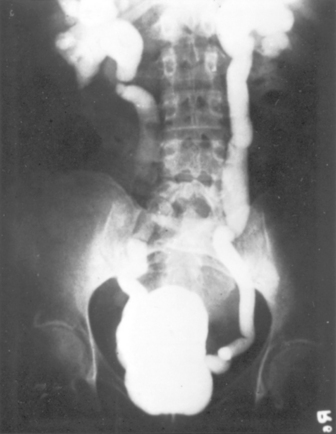
FIGURE 20-27 Vesicoureteral reflux demonstrated by a voiding cystourethrogram. Dye injected into the bladder refluxes into both dilated ureters, filling the pelvis and calyces.
In the absence of vesicoureteral reflux, infection usually remains localized in the bladder. Thus, the majority of individuals with repeated or persistent bacterial colonization of the urinary tract suffer from cystitis and urethritis (lower urinary tract infection) rather than pyelonephritis.
Acute Pyelonephritis
Acute pyelonephritis is an acute suppurative inflammation of the kidney caused by bacterial and sometimes viral (e.g., polyomavirus) infection, whether hematogenous and induced by septicemic spread or ascending and associated with vesicoureteral reflux.66
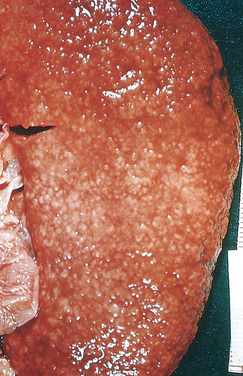
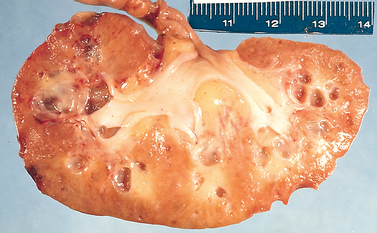
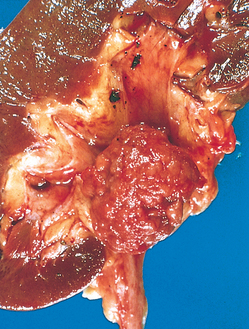
Morphology. The hallmarks of acute pyelonephritis are patchy interstitial suppurative inflammation, intratubular aggregates of neutrophils, and tubular necrosis. The suppuration may occur as discrete focal abscesses involving one or both kidneys, which can extend to large wedge-shaped areas of suppuration (Fig. 20-28). The distribution of these lesions is unpredictable and haphazard, but in pyelonephritis associated with reflux, damage occurs most commonly in the lower and upper poles.
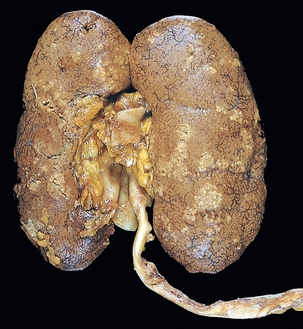
FIGURE 20-28 Acute pyelonephritis. Cortical surface shows grayish white areas of inflammation and abscess formation.
In the early stages, the neutrophilic infiltration is limited to the interstitial tissue. Soon, however, the reaction involves tubules and produces a characteristic abscess with the destruction of the engulfed tubules (Fig. 20-29). Since the tubular lumens present a ready pathway for the extension of the infection, large masses of intraluminal neutrophils frequently extend along the involved nephron into the collecting tubules. Characteristically, glomeruli seem to be relatively resistant to the infection. Large areas of severe necrosis, however, eventually destroy the glomeruli, and fungal pyelonephritis (e.g., Candida) often affects glomeruli.
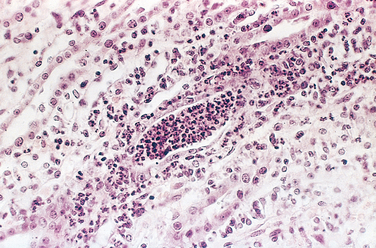
FIGURE 20-29 Acute pyelonephritis marked by an acute neutrophilic exudate within tubules and interstitial inflammation.
Three complications of acute pyelonephritis are encountered in special circumstances.
After the acute phase of pyelonephritis, healing occurs. The neutrophilic infiltrate is replaced by one that is predominantly composed of macrophages, plasma cells, and (later) lymphocytes. The inflammatory foci are eventually replaced by irregular scars that can be seen on the cortical surface as fibrous depressions. Such scars are characterized microscopically by tubular atrophy, interstitial fibrosis, and a lymphocytic infiltrate in a characteristic patchy, jigsaw pattern with intervening preserved parenchyma. The pyelonephritic scar is almost always associated with inflammation, fibrosis, and deformation of the underlying calyx and pelvis, reflecting the role of ascending infection and vesicoureteral reflux in the pathogenesis of the disease.
Clinical Features.
Acute pyelonephritis is often associated with predisposing conditions, some of which were mentioned before. These include the following:
When acute pyelonephritis is clinically apparent, the onset is usually sudden, with pain at the costovertebral angle and systemic evidence of infection, such as fever and malaise. There are usually indications of bladder and urethral irritation, such as dysuria, frequency, and urgency. The urine contains many leukocytes (pyuria) derived from the inflammatory infiltrate, but pyuria does not differentiate upper from lower urinary tract infection. The finding of leukocyte casts, typically rich in neutrophils (pus casts), indicates renal involvement, because casts are formed only in tubules. The diagnosis of infection is established by quantitative urine culture.
Uncomplicated acute pyelonephritis usually follows a benign course, and the symptoms disappear within a few days after the institution of appropriate antibiotic therapy. Bacteria, however, may persist in the urine, or there may be recurrence of infection with new serologic types of E. coli or other organisms. Such bacteriuria then either disappears or may persist, sometimes for years. In the presence of unrelieved urinary obstruction, diabetes mellitus, or immunodeficiency, acute pyelonephritis may be more serious, leading to repeated septicemic episodes. The superimposition of papillary necrosis may lead to acute renal failure.
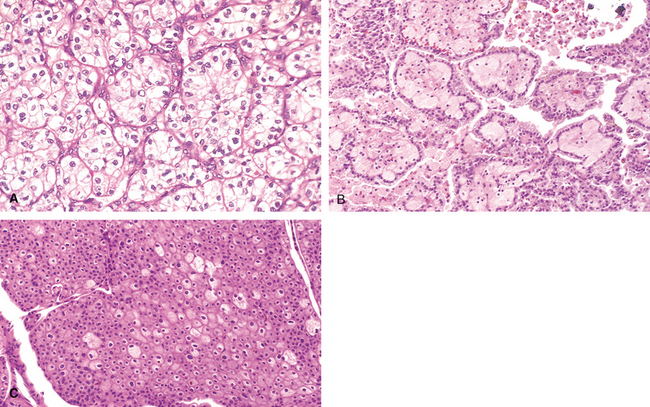
An emerging viral pathogen causing pyelonephritis in kidney allografts is polyomavirus. Latent infection with polyomavirus is widespread in the general population, but immunosuppression of the allograft recipient can lead to reactivation of latent infection and the development of a nephropathy resulting in allograft failure in as many as 1% to 5% of kidney transplant recipients.67 This form of pyelonephritis, now referred to as polyomavirus nephropathy, is characterized by viral infection of tubular epithelial cell nuclei, leading to nuclear enlargement and intranuclear inclusions visible by light microscopy (viral cytopathic effect). The inclusions are composed of virions arrayed in distinctive crystalline-like lattices when visualized by electron microscopy (Fig. 20-31). An interstitial inflammatory response is invariably present. Treatment consists of a reduction in immunosuppression.
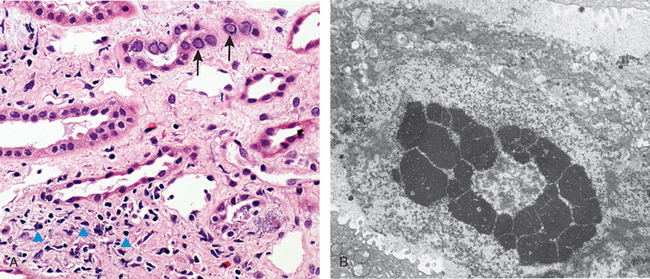
FIGURE 20-31 Polyomavirus nephropathy. A, The kidney shows enlarged tubular epithelial cells with nuclear inclusions (arrows) and interstitial inflammation (arrowheads). B, Intranuclear viral inclusions visualized by electron microscopy.
(Courtesy of Dr. Jean Olson, Department of Pathology, University of California San Francisco, San Francisco, CA.)
Chronic Pyelonephritis and Reflux Nephropathy
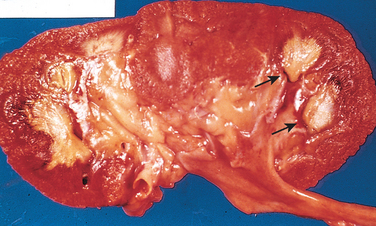
Chronic pyelonephritis is a disorder in which chronic tubulointerstitial inflammation and renal scarring are associated with pathologic involvement of the calyces and pelvis (Fig. 20-32). Pelvocalyceal damage is important in that virtually all the disease etiologies listed in Table 20-8 produce chronic tubulointerstitial alterations, but with the exception of chronic pyelonephritis and analgesic nephropathy, none affect the calyces. Chronic pyelonephritis is an important cause of end-stage kidney disease; at one time it accounted for as many as 10% to 20% of patients in renal transplant or dialysis units, until predisposing conditions such as reflux became better recognized. This condition remains an important cause of kidney destruction in children with severe lower urinary tract abnormalities.
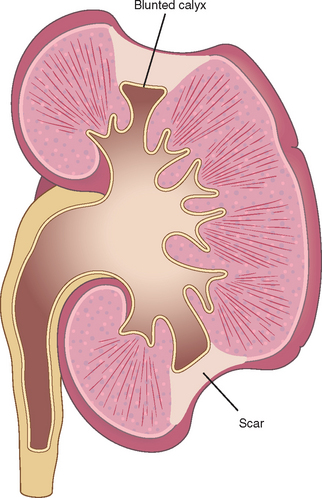
FIGURE 20-32 Typical coarse scars of chronic pyelonephritis associated with vesicoureteral reflux. The scars are usually polar and are associated with underlying blunted calyces.
Chronic pyelonephritis can be divided into two forms: chronic reflux-associated and chronic obstructive.
Reflux Nephropathy.
This is by far the more common form of chronic pyelonephritic scarring. Renal involvement in reflux nephropathy occurs early in childhood as a result of superimposition of a urinary infection on congenital vesicoureteral reflux and intrarenal reflux. Reflux may be unilateral or bilateral; thus, the resultant renal damage may cause scarring and atrophy of one kidney or involve both, leading to chronic renal insufficiency. Vesicoureteral reflux occasionally causes renal damage in the absence of infection (sterile reflux), but only when obstruction is severe.
Chronic Obstructive Pyelonephritis.
We have seen that obstruction predisposes the kidney to infection. Recurrent infections superimposed on diffuse or localized obstructive lesions lead to recurrent bouts of renal inflammation and scarring, resulting in a picture of chronic pyelonephritis. In this condition, the effects of obstruction contribute to the parenchymal atrophy; indeed, it is sometimes difficult to differentiate the effects of bacterial infection from those of obstruction alone. The disease can be bilateral, as with posterior urethral valves, resulting in renal insufficiency unless the anomaly is corrected, or unilateral, such as occurs with calculi and unilateral obstructive anomalies of the ureter.
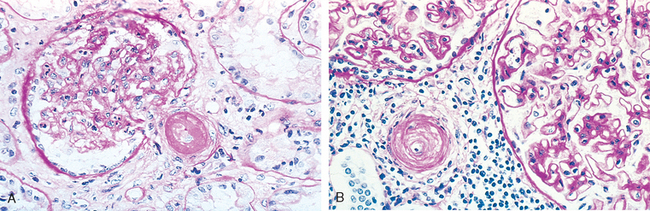
Morphology. The characteristic changes of chronic pyelonephritis are seen on gross examination (Figs. 20-32 and 20-33). The kidneys usually are irregularly scarred; if bilateral, the involvement is asymmetric. This contrasts with chronic glomerulonephritis, in which both kidneys are diffusely and symmetrically scarred. The hallmarks of chronic pyelonephritis are coarse, discrete, corticomedullary scars overlying dilated, blunted, or deformed calyces, and flattening of the papillae (see Fig. 20-33). The scars can vary from one to several in number and may affect one or both kidneys. Most are in the upper and lower poles, consistent with the frequency of reflux in these sites.
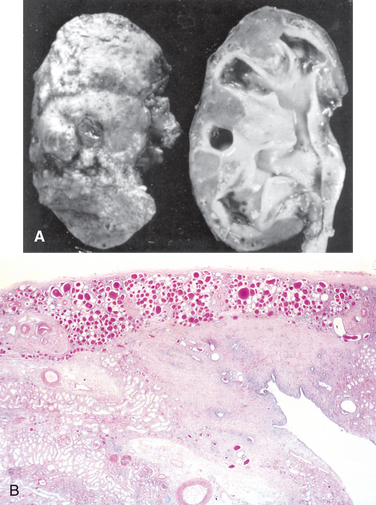
FIGURE 20-33 A, Chronic pyelonephritis. The surface (left) is irregularly scarred. The cut section (right) reveals characteristic dilation and blunting of calyces. The ureter is dilated and thickened, a finding that is consistent with chronic vesicoureteral reflux. B, Low-power view showing a corticomedullary renal scar with an underlying dilated deformed calyx. Note the thyroidization of tubules in the cortex.
The microscopic changes involve predominantly tubules and interstitium. The tubules show atrophy in some areas and hypertrophy or dilation in others. Dilated tubules with flattened epithelium may be filled with colloid casts (thyroidization). There are varying degrees of chronic interstitial inflammation and fibrosis in the cortex and medulla. In the presence of active infection there may be neutrophils in the interstitium and pus casts in the tubules. Arcuate and interlobular vessels demonstrate obliterative intimal sclerosis in the scarred areas; and in the presence of hypertension, hyaline arteriosclerosis is seen in the entire kidney. There is often fibrosis around the calyceal epithelium as well as a marked chronic inflammatory infiltrate. Glomeruli may appear normal except for periglomerular fibrosis, or exhibit a variety of changes, including ischemic fibrous obliteration and secondary changes related to hypertension. Individuals with chronic pyelonephritis and reflux nephropathy who develop proteinuria in advanced stages show secondary focal segmental glomerulosclerosis, as described later.
Xanthogranulomatous pyelonephritis is an unusual and relatively rare form of chronic pyelonephritis characterized by accumulation of foamy macrophages intermingled with plasma cells, lymphocytes, polymorphonuclear leukocytes, and occasional giant cells. Often associated with Proteus infections and obstruction, the lesions sometimes produce large, yellowish orange nodules that may be grossly confused with renal cell carcinoma.
Clinical Features.
Chronic obstructive pyelonephritis may be insidious in onset or present with clinical manifestations of acute recurrent pyelonephritis, such as back pain, fever, frequent pyuria, and bacteriuria. Chronic pyelonephritis associated with reflux may have a silent onset. These patients come to medical attention relatively late in the course of their disease because of the gradual onset of renal insufficiency and hypertension or because of the discovery of pyuria or bacteriuria on routine examination. Reflux nephropathy is often discovered when hypertension in children is investigated. Loss of tubular function—in particular of concentrating ability—gives rise to polyuria and nocturia. Radiographic studies show asymmetrically contracted kidneys with characteristic coarse scars and blunting and deformity of the calyceal system. Significant bacteriuria may be present, but it is often absent in the late stages.
Although proteinuria is usually mild, some individuals with pyelonephritic scars develop secondary focal segmental glomerulosclerosis with significant proteinuria, even in the nephrotic range, usually several years after the scarring has occurred and often in the absence of continued infection or persistent vesicoureteral reflux. The appearance of proteinuria and focal segmental glomerulosclerosis is a poor prognostic sign, and patients with these findings may proceed to chronic or end-stage renal failure. The glomerulosclerosis, as we have discussed, may be attributable to the adaptive glomerular alterations secondary to loss of renal mass caused by pyelonephritic scarring (renal ablation nephropathy).
Tubulointerstitial Nephritis Induced by Drugs and Toxins
Toxins and drugs can produce renal injury in at least three ways: (1) They may trigger an interstitial immunological reaction, exemplified by the acute hypersensitivity nephritis induced by such drugs as methicillin; (2) they may cause acute renal failure, as described earlier; and (3) they may cause subtle but cumulative injury to tubules that takes years to become manifest, resulting in chronic renal insufficiency.68 The last type of damage is especially treacherous, because it may be clinically unrecognized until significant renal damage has occurred. Such is the case with analgesic abuse nephropathy, which is usually detected only after the onset of chronic renal insufficiency.
Acute Drug-Induced Interstitial Nephritis
This is a well-recognized adverse reaction to a constantly increasing number of drugs. First reported after the use of sulfonamides, acute tubulointerstitial nephritis most frequently occurs with synthetic penicillins (methicillin, ampicillin), other synthetic antibiotics (rifampin), diuretics (thiazides), NSAIDs, and miscellaneous drugs (allopurinol, cimetidine). The disease begins about 15 days (range: 2–40) after exposure to the drug and is characterized by fever, eosinophilia (which may be transient), a rash in about 25% of patients, and renal abnormalities. The latter take the form of hematuria, mild proteinuria, and leukocyturia (often including eosinophils). A rising serum creatinine level or acute renal failure with oliguria develops in about 50% of cases, particularly in older patients.
Pathogenesis.
Many features of the disease suggest an immune mechanism. The immune response is idiosyncratic and not dose-related. Clinical evidence of hypersensitivity includes the latent period, the eosinophilia and rash, the fact that the onset of nephropathy is not dose-related, and the recurrence of hypersensitivity after re-exposure to the same or a cross-reactive drug. In some patients, serum IgE levels are increased, and IgE-containing plasma cells and basophils are present in the lesions, suggesting that the late-phase reaction of an IgE-mediated (type I) hypersensitivity may be involved in the pathogenesis (Chapter 6). In other cases, mononuclear or granulomatous infiltrate, together with positive results of skin tests to drug haptens, suggest a T cell–mediated delayedhypersensitivity reaction (type IV).
The most likely sequence of events is that the drugs act as haptens, which covalently bind to some cytoplasmic or extracellular component of tubular cells and become immunogenic. The resultant injury is then due to IgE and/or cell-mediated immune reactions to tubular cells or their basement membranes.
Morphology. On histologic examination the abnormalities are in the interstitium, which shows variable but frequently pronounced edema and infiltration by mononuclear cells, principally lymphocytes and macrophages. Eosinophils and neutrophils may be present (Fig. 20-34), often in clusters and large numbers, and plasma cells and basophils are sometimes found in small numbers. With some drugs (e.g., methicillin, thiazides), interstitial non-necrotizing granulomas containing giant cells may be seen. “Tubulitis,” the infiltration of tubules by lymphocytes, is common. Variable degrees of tubular necrosis and regeneration are present. The glomeruli are normal except in some cases caused by NSAIDs, when minimal-change disease and the nephrotic syndrome develop concurrently (see the discussion of NSAIDs later in the chapter).
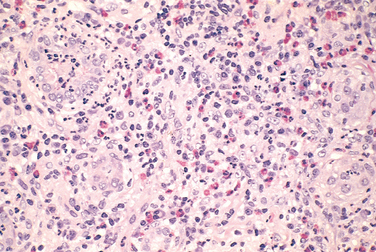
Clinical Features.
It is important to recognize drug-induced renal failure because withdrawal of the offending drug is followed by recovery, although it may take several months, and irreversible damage occurs occasionally in older subjects. It is also important to remember that while drugs are the leading identifiable cause of acute interstitial nephritis, in many affected patients (approximately 30% to 40%) an offending drug or mechanism cannot be identified.
Analgesic Nephropathy
This is a form of chronic renal disease caused by excessive intake of analgesic mixtures and characterized morphologically by chronic tubulointerstitial nephritis and renal papillary necrosis.69
The incidence of analgesic nephropathy reflects the consumption of analgesics in various populations throughout the world. In some parts of Australia, it ranked as one of the most common causes of chronic renal insufficiency until public health measures reduced its incidence. Its incidence in the United States is relatively low but varies among states, being highest in the southeast. Overall, it accounted for 9%, 3%, and 1% of patients undergoing dialysis in Australia, Europe, and the United States, respectively, before the recent surge in end-stage renal disease attributable to diabetes reduced these relative percentages. The renal damage was first ascribed to phenacetin, but the analgesic mixtures that are consumed often contain, in addition, aspirin, caffeine, acetaminophen (a metabolite of phenacetin), and codeine. Patients who develop this disease usually ingest large quantities of mixtures of at least two antipyretic analgesics. Most patients consume phenacetin-containing mixtures, and cases ascribed to ingestion of aspirin, phenacetin, or acetaminophen alone are uncommon. In most countries, restriction of over-the-counter sale of phenacetin or analgesic mixtures has reduced the incidence of the disorder but has not eradicated it, presumably because non-phenacetin-containing mixtures are available.
Pathogenesis.
Papillary necrosis is readily induced experimentally by a mixture of aspirin and phenacetin, usually combined with water depletion. It is now clear that in the sequence of events leading to renal damage, papillary necrosis occurs first, and cortical tubulointerstitial nephritis follows as a consequence of impeded urine outflow. The phenacetin metabolite acetaminophen, which can deplete cells of glutathione, then injures these cells by subsequent generation of oxidative metabolites. Aspirin induces its potentiating effect by inhibiting the vasodilatory effects of prostaglandins, predisposing the papillae to ischemia. Thus, the papillary damage may be due to a combination of direct toxic effects of phenacetin metabolites and ischemic injury to both tubular cells and vessels.
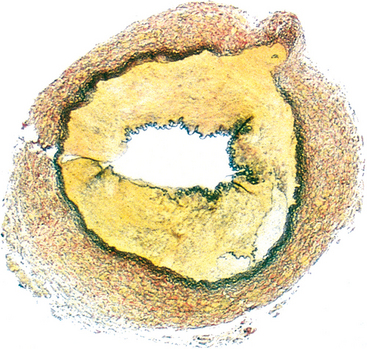
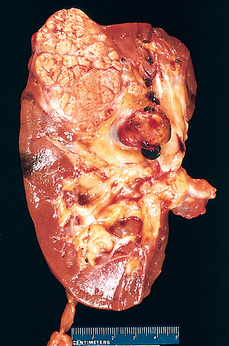
Morphology. In gross appearance the kidneys are either normal or slightly reduced in size, and the cortex shows depressed areas representing cortical atrophy overlying necrotic papillae. The papillae show various stages of necrosis, calcification, fragmentation, and sloughing. This gross appearance contrasts with the papillary necrosis seen in diabetic patients, in which all papillae are at the same stage of injury. On microscopic examination the papillary changes may take one of several forms. In early cases there is patchy necrosis, but in the advanced form the entire papilla is necrotic, often remaining in place as a structureless mass containing “ghosts” of tubules and foci of dystrophic calcification (Fig. 20-35). Segments of entire portions of the papilla may then be sloughed and excreted in the urine.
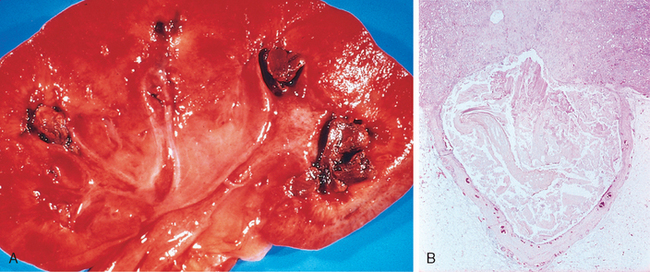
FIGURE 20-35 Analgesic nephropathy. A, The brownish necrotic papilla, transformed to a necrotic, structureless mass, fills the pelvis. B, Microscopic view. Note the fibrosis in the medulla.
(Courtesy of Dr. F.J. Gloor, Institut für Pathologie, Kantonsspital, St. Gallen, Switzerland.)
The cortical changes consist of loss and atrophy of tubules and interstitial fibrosis and inflammation. These changes are mainly due to obstructive atrophy caused by the tubular damage in the papillae. The cortical columns of Bertin are characteristically spared from this atrophy.
Clinical Features.
Analgesic nephropathy is more common in women than in men and is particularly prevalent in individuals with recurrent headaches and muscle pain, in psychoneurotic patients, and in factory workers. Early renal findings include inability to concentrate the urine (hyposthenuria), as would be expected for papillary lesions. Acquired distal renal tubular acidosis contributes to the development of renal stones. Headache, anemia, gastrointestinal symptoms, and hypertension are common accompaniments of analgesic nephropathy. Urinary tract infection complicates about 50% of cases. On occasion, entire tips of necrotic papillae are excreted, and these may cause gross hematuria or renal colic due to obstruction of the ureter by necrotic fragments. Magnetic resonance and computed tomographic imaging are helpful in detecting papillary necrosis and calcifications. Progressive impairment of renal function may lead to chronic renal failure, but with drug withdrawal, renal function may either stabilize or actually improve.
Unfortunately, a small percentage of patients with analgesic nephropathy develop transitional papillary carcinoma of the renal pelvis. Whether the carcinogenic effect is due to a metabolite of phenacetin or to some other component of the analgesic compounds is unsettled.
Papillary necrosis is not specific for analgesic nephropathy. It is also seen in diabetes mellitus, as was mentioned earlier, as well as in urinary tract obstruction, sickle cell disease or trait (described later), and focally in renal tuberculosis. Table 20-9 lists certain features of papillary necrosis in these conditions.

Nephropathy Associated with NSAIDs
NSAIDs, one of the most common classes of drugs currently in use, produce several forms of renal injury. Although these complications are uncommon, they should be kept in mind since NSAIDs are frequently administered to patients with other potential causes of renal disease. Many NSAIDs are nonselective cyclooxygenase inhibitors, and their adverse renal effects are related to their ability to inhibit cyclooxygenase-dependent prostaglandin synthesis. The selective COX-2 inhibitors, while sparing the gastrointestinal tract, do affect the kidneys because COX-2 is expressed in human kidneys.70 NSAID-associated renal syndromes include
Aristolochic Nephropathy
A syndrome of chronic tubulointerstitial nephritis caused by aristolochic acid, a supplement found in some herbal remedies, has been recognized recently. The drug forms covalent adducts with DNA and causes a distinctive picture of renal failure and interstitial fibrosis associated with a relative paucity of infiltrating leukocytes. As with analgesic nephropathy, there is an increased incidence of carcinoma in the kidney and urinary tract. Ingestion of aristolochic acid has also been identified as the cause of Balkan nephropathy, a chronic tubulointerstitial nephritis common in that part of the world.71
Other Tubulointerstitial Diseases
Urate Nephropathy
Three types of nephropathy can occur in persons with hyperuricemic disorders:
Hypercalcemia and Nephrocalcinosis
Disorders associated with hypercalcemia, such as hyperparathyroidism, multiple myeloma, vitamin D intoxication, metastatic cancer, or excess calcium intake (milk-alkali syndrome), may induce the formation of calcium stones and deposition of calcium in the kidney (nephrocalcinosis). Extensive degrees of calcinosis, under certain conditions, may lead to chronic tubulointerstitial disease and renal insufficiency. The earliest damage induced by the hypercalcemia is to the tubular epithelial cells in the form of mitochondrial distortion and other signs of cell injury. Subsequently, calcium deposits appear within the mitochondria, cytoplasm, and basement membrane. Calcified cellular debris may obstruct tubular lumens and cause obstructive atrophy of nephrons and secondary interstitial fibrosis and inflammation. Atrophy of entire cortical areas drained by calcified tubules may occur, accounting for the alternating areas of normal and scarred parenchyma seen in such kidneys.
The earliest functional defect is an inability to concentrate the urine. Other tubular defects, such as tubular acidosis and salt-losing nephritis, may also occur. With further damage, a slowly progressive renal insufficiency develops. This is usually due to nephrocalcinosis, but many of these patients also have calcium stones and secondary pyelonephritis.
Acute Phosphate Nephropathy
Extensive accumulations of calcium phosphate crystals in tubules can occur in patients consuming high doses of oral phosphate solutions in preparation for colonoscopy.72 These patients are not hypercalcemic, but excess phosphate load, perhaps complicated by dehydration, causes marked precipitation of calcium phosphate, typically presenting as renal insufficiency several weeks after the exposure. Patients typically only partially recover renal function.
Light-Chain Cast Nephropathy (“Myeloma Kidney”)
Nonrenal malignant tumors, particularly those of hematopoietic origin, affect the kidneys in several ways (Table 20-10). The most common involvements are tubulointerstitial, caused by complications of the tumor (hypercalcemia, hyperuricemia, obstruction of ureters) or therapy (irradiation, hyperuricemia, chemotherapy, infections in immunosuppressed patients). As the survival rate of persons with malignant neoplasms increases, so do these renal complications. We limit the discussion here to the tubulointerstitial lesions in multiple myeloma that sometimes dominate the clinical picture in people with this disease.
TABLE 20-10 Renal Disease Related to Nonrenal Neoplasms
| Hypercalcemia |
| Hyperuricemia |
| Amyloidosis (AL, light-chain type) |
| Excretion of abnormal proteins (multiple myeloma) |
| Effects of radiation therapy, chemotherapy, secondary infection |
Overt renal insufficiency occurs in half of those with multiple myeloma and related lymphoplasmacytic disorders. Several factors contribute to renal damage:
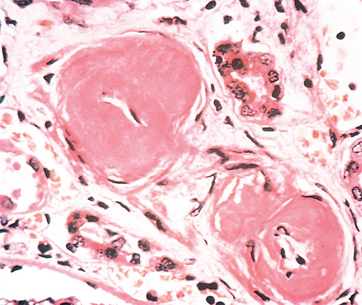
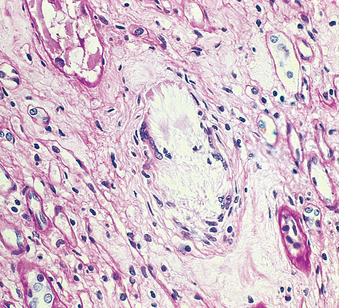
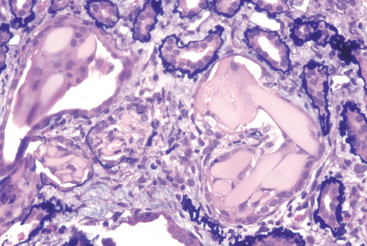
Morphology. The tubulointerstitial changes in light-chain cast nephropathy are fairly characteristic. The Bence Jones tubular casts appear as pink to blue amorphous masses, sometimes concentrically laminated and often fractured, which fill and distend the tubular lumens. Some of the casts are surrounded by multinucleate giant cells that are derived from mononuclear phagocytes (Fig. 20-37). The adjacent interstitial tissue usually shows a nonspecific inflammatory response and fibrosis. On occasion, the casts erode their way from the tubules into the interstitium and here evoke a granulomatous inflammatory reaction. Amyloidosis, light-chain deposition disease, nephrocalcinosis, and infection may also be present.
Clinical Features.
Clinically, the renal manifestations are of several types. In the most common form, chronic renal failure develops insidiously and usually progresses slowly during a period of several months to years. Another form occurs suddenly and is manifested by acute renal failure with oliguria. Precipitating factors in these patients include dehydration, hypercalcemia, acute infection, and treatment with nephrotoxic antibiotics. Bence Jones proteinuria occurs in 70% of individuals with multiple myeloma; the presence of significant non–light-chain proteinuria (e.g., albuminuria) suggests AL amyloidosis or light-chain deposition disease.