Chapter 5 Genetic Disorders
Human Genetic Architecture
The sequence of the human genome is complete, and much has been learned about the “genetic architecture” of humans.1 Some of what has been revealed was quite unexpected. For example, we now know that less than 2% of the human genome encodes proteins, whereas more than one half represents blocks of repetitive DNA sequences whose functions remain mysterious. What was totally unexpected was that humans have a mere 20,000 to 25,000 genes that code for proteins rather than the 100,000 predicted. Quite remarkably, this figure is about the same as that of the mustard plant, with 26,000 genes! However, it is also known that by alternative splicing, the 25,000 human genes can give rise to greater than 100,000 proteins. Humans are not so poor, after all. With the completion of the Human Genome Project, a new term, genomics, has been added to the medical vocabulary. Whereas genetics is the study of single or a few genes and their phenotypic effects, genomics is the study of all the genes in the genome and their interactions.2 DNA microarray analysis of tumors (Chapter 7) is an excellent example of genomics in current clinical use.
Another surprising revelation from the recent progress in genomics is that, on average, any two individuals share greater than 99.5% of their DNA sequences.3 Thus, the remarkable diversity of humans is encoded in less than 0.5% of our DNA. The secrets to disease predisposition and response to environmental agents and drugs must therefore reside within these variations. Though small when compared to the total nucleotide sequences, this 0.5% represents about 15 million base pairs. The two most common forms of DNA variations in the human genome are single-nucleotide polymorphisms (SNPs) and copy number variations (CNVs). SNPs represent variation at single isolated nucleotide positions and are almost always biallelic (i.e., one of only two choices exist at a given site within the population, such as A or T). Much effort has been devoted to making SNP maps of the human genome. These efforts have identified over 6 million SNPs in the human population, many of which show wide variation in frequency in different populations. SNPs may occur anywhere in the genome—within exons, introns, or intergenic regions, but less than 1% of SNPs occur in coding regions. These coding sequence variations are important, since they could of course alter the gene product and predispose to a phenotypic difference or to a disease. Much more commonly, however, the SNP is just a marker that is co-inherited with a disease-associated gene as a result of physical proximity. Another way of expressing this is to say that the SNP and the causative genetic factor are in linkage disequilibrium. There is optimism that groups of SNPs could serve as reliable markers of risk for multigenic complex diseases such as type II diabetes and hypertension, and that by identifying such variants strategies for disease prevention could be developed (discussed later).
CNVs are a recently identified form of genetic variation consisting of different numbers of large contiguous stretches of DNA from 1000 base pairs to millions of base pairs.4,5 In some instances these loci are, like SNPs, biallelic and simply duplicated or deleted in a subset of the population. In other instances there are complex rearrangements of genomic material, with multiple alleles in the human population. Current estimates are that CNVs are responsible for between 5 and 24 million base pairs of sequence difference between any two individuals.6 Approximately 50% of CNVs involve gene-coding sequences; thus, CNVs may underlie a large portion of human phenotypic diversity. There is a significant over-representation of certain gene families in regions affected by CNVs; these include genes involved in the immune system and in the nervous system. It is assumed that copy number diversity in such gene families has been subject to strong evolutionary selection, since they would enhance human adaptation to changing environmental factors. We currently know much less about CNVs than SNPs, therefore their influence on disease susceptibility is less established, though predicted to be substantial.
It should be pointed out that despite all these advances in the understanding of human variations, it is clear that alterations in DNA sequence cannot by themselves explain the diversity of phenotypes in human populations. Nor can classic genetics explain how monozygotic twins can have differing phenotypes.7 The answer must lie in epigenetics, which is defined as heritable changes in gene expression that are not caused by alterations in DNA sequence. Epigenetic changes are involved in tissue-specific expression of genes and genomic imprinting. The biochemical basis of epigenetic changes and its detection are discussed under “Molecular Diagnosis.”
Just as genomics involves the study of all the DNA sequences, proteomics concerns itself with the measurement of all proteins expressed in a cell or tissue. To simultaneously analyze patterns of expression involving thousands of genes and proteins has required the parallel development of computer-based techniques that can manage vast collections of data. In response to this, an exciting new discipline called bioinformatics has sprouted.8
It is worth noting that until recently the major focus of gene hunting has been discovery of structural genes whose products encode proteins. Recent studies indicate, however, that a very large number of genes do not encode proteins. Instead, their products play important regulatory functions. The most recently discovered among this class of genes are those that encode small RNA molecules, so-called microRNAs (miRNAs). miRNAs, unlike other RNAs, do not encode proteins but instead inhibit gene expression. Silencing of gene expression by miRNA is preserved in all living forms from plants to humans and therefore must be a fundamental mechanism of gene regulation. Because of their profound influence on gene regulation, miRNAs are assuming central importance in understanding normal developmental pathways, as well as pathologic conditions, such as cancer.9 Such is the importance of the discovery of gene silencing by miRNAs that Andrew Fire and Craig Mello were awarded the Nobel Prize in physiology or medicine in 2006, a mere 8 years after they published their initial work.
By current estimates there are approximately 1000 genes in humans that encode miRNAs, accounting for about 5% of the human genome. Transcription of miRNA genes produces primary miRNA transcripts, which is processed within the nucleus to form another structure, called pre-miRNA (Fig. 5-1). With the help of specific transporter proteins, pre-miRNA is exported to the cytoplasm. Additional “cutting” by an enzyme, appropriately called Dicer, generates mature miRNAs that are about 21 to 30 nucleotides in length (hence the name “micro”). At this stage the miRNA is still double-stranded. Next, the miRNA unwinds, and single strands of this duplex are incorporated into a multiprotein complex called RNA-induced silencing complex (RISC). Base-pairing between the miRNA strand and its target messenger RNA (mRNA) directs the RISC to either cause mRNA cleavage or repress its translation. In this way, the gene from which the target mRNA was derived is silenced (at a post-transcriptional level).10 Given that the numbers of miRNA genes are far fewer than those that encode proteins, it follows that a given miRNA can silence many target genes. The precise mechanism by which the target specificity of miRNA is determined remains to be fully elucidated.
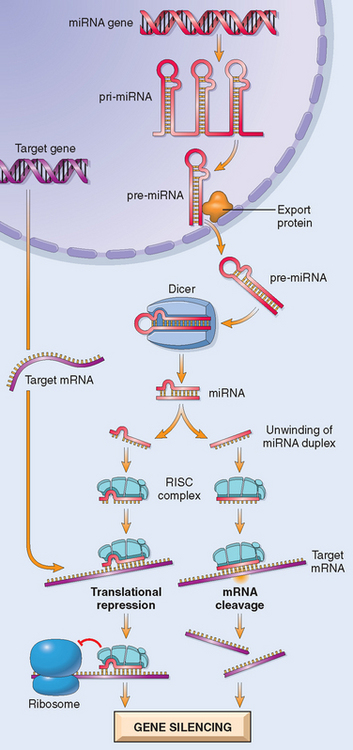
FIGURE 5-1 Generation of microRNAs and their mode of action in regulating gene function. Pri-miRNA, primary microRNA transcript; pre-miRNA, precursor microRNA; RISC, RNA-induced silencing complex.
Another species of gene-silencing RNA, called small interfering RNAs (siRNAs), works in a manner quite similar to that of miRNA. Unlike miRNA, however, siRNA precursors are introduced by investigators into the cell. Their processing by Dicer and functioning via RISC are essentially similar to that described for miRNA. siRNAs are becoming powerful tools for studying gene function and may in the future be used therapeutically to silence specific genes, such as oncogenes, whose products are involved in neoplastic transformation.
Genes and Human Diseases
Genetic disorders are far more common than is widely appreciated. The lifetime frequency of genetic diseases is estimated to be 670 per 1000.11 Furthermore, the genetic diseases encountered in medical practice represent only the tip of the iceberg, that is, those with less extreme genotypic errors permitting full embryonic development and live birth. It is estimated that 50% of spontaneous abortuses during the early months of gestation have a demonstrable chromosomal abnormality; there are, in addition, numerous smaller detectable errors and many others still beyond our range of identification. About 1% of all newborn infants possess a gross chromosomal abnormality, and approximately 5% of individuals under age 25 develop a serious disease with a significant genetic component. How many more mutations remain hidden?
Before discussing specific aberrations that may cause genetic diseases, it is useful to summarize the genetic contribution to human disease. Human genetic disorders can be broadly classified into three categories
Study of single genes and mutations with large effects has been extremely informative in medicine since a great deal of what we know about several physiologic pathways (such as cholesterol transport, chloride secretion) has been learned from analysis of single gene disorders. Although informative, these disorders are generally rare unless they are maintained in a population by strong selective forces (e.g., sickle cell anemia in areas where malaria is endemic, Chapter 14).
Since complex traits do not follow a Mendelian pattern of inheritance, the genes and polymorphisms that contribute to such diseases have been very difficult to discern. However, recent progress in genomics and high throughput sequencing technology has made possible genome wide association studies (GWAS), a systematic method of identifying disease-associated polymorphisms that is beginning to unravel the molecular basis of complex disorders. We will discuss the principle of GWAS later in the chapter.
We begin our discussion with a description of mutations that affect single genes, since they underlie Mendelian disorders. We follow with transmission patterns and selected samples of single gene disorders.
MUTATIONS
A mutation is defined as a permanent change in the DNA. Mutations that affect germ cells are transmitted to the progeny and can give rise to inherited diseases. Mutations that arise in somatic cells understandably do not cause hereditary diseases but are important in the genesis of cancers and some congenital malformations.
Mutations may result in partial or complete deletion of a gene or, more often, affect a single base. For example, a single nucleotide base may be substituted by a different base, resulting in a point mutation. Less commonly, one or two base pairs may be inserted into or deleted from the DNA, leading to alterations in the reading frame of the DNA strand; hence these are referred to as frameshift mutations (Figs. 5-2 and 5-3). Next we briefly review some general principles relating to the effects of gene mutations.
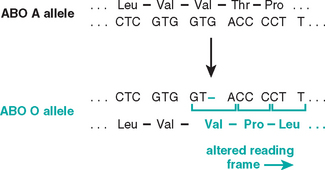
FIGURE 5-2 Single-base deletion at the ABO (glycosyltransferase) locus, leading to a frameshift mutation responsible for the O allele.
(From Thompson MW et al.: Thompson and Thompson Genetics in Medicine, 5th ed. Philadelphia, WB Saunders, 1991, p 134.)
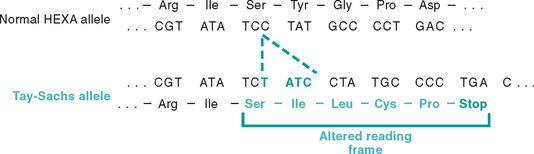
FIGURE 5-3 Four-base insertion in the hexosaminidase A gene, leading to a frameshift mutation. This mutation is the major cause of Tay-Sachs disease in Ashkenazi Jews.
(From Nussbaum RL et al.: Thompson and Thompson Genetics in Medicine, 6th ed. Philadelphia, WB Saunders, 2001, p 212.)
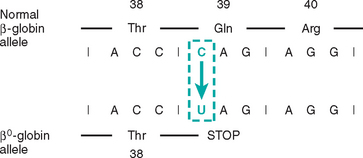
FIGURE 5-4 Point mutation leading to premature chain termination. Partial mRNA sequence of the β-globin chain of hemoglobin showing codons for amino acids 38 to 40. A point mutation (C → U) in codon 39 changes glutamine (Gln) codon to a stop codon, and hence protein synthesis stops at amino acid 38.
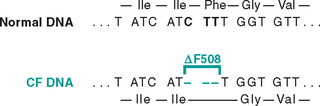
FIGURE 5-5 Three-base deletion in the common cystic fibrosis (CF) allele results in synthesis of a protein that lacks amino acid 508 (phenylalanine). Because the deletion is a multiple of three, this is not a frameshift mutation.
(From Thompson MW et al.: Thompson and Thompson Genetics in Medicine, 5th ed. Philadelphia, WB Saunders, 1991, p 135.)
To summarize, mutations can interfere with protein synthesis at various levels. Transcription may be suppressed with gene deletions and point mutations involving promoter sequences. Abnormal mRNA processing may result from mutations affecting introns or splice junctions or both. Translation is affected if a stop codon (chain termination mutation) is created within an exon. Finally, some point mutations may lead to the formation of an abnormal protein without impairing any step in protein synthesis.
In closing, it should be noted that, uncommonly, mutations are beneficial. As will be discussed in Chapter 6, the human immunodeficiency virus (HIV) uses a chemokine receptor, CCR5, to enter cells; a deletion in the CCR5 gene thus protects from HIV infection.
Against this background, we now turn our attention to the three major categories of genetic disorders: (1) disorders related to mutant genes of large effect, (2) diseases with multifactorial inheritance, and (3) chromosomal disorders. To these three well-known categories must be added a heterogeneous group of single-gene disorders with nonclassic patterns of inheritance. This group includes disorders resulting from triplet-repeat mutations, those arising from mutations in mitochondrial DNA (mtDNA), and those in which the transmission is influenced by genomic imprinting or gonadal mosaicism. Diseases within this group are caused by mutations in single genes, but they do not follow the mendelian pattern of inheritance. These are discussed later in this chapter.
It is beyond the scope of this book to review normal human genetics. It is, however, important to clarify several commonly used terms—hereditary, familial, and congenital. Hereditary disorders, by definition, are derived from one’s parents and are transmitted in the germ line through the generations and therefore are familial. The term congenital simply implies “born with.” Some congenital diseases are not genetic; for example, congenital syphilis. Not all genetic diseases are congenital; individuals with Huntington disease, for example, begin to manifest their condition only after their 20s or 30s.
Mendelian Disorders
All mendelian disorders are the result of mutations in single genes that have large effects. It is not necessary to detail Mendel’s laws here, since every student in biology, and possibly every garden pea, has learned about them at an early age. Only some comments of medical relevance are made.
It is estimated that every individual is a carrier of five to eight deleterious genes. Most of these are recessive and therefore do not have serious phenotypic effects. About 80% to 85% of these mutations are familial. The remainder represent new mutations acquired de novo by an affected individual.
Some autosomal mutations produce partial expression in the heterozygote and full expression in the homozygote. Sickle cell anemia is caused by substitution of normal hemoglobin (HbA) by hemoglobin S (HbS). When an individual is homozygous for the mutant gene, all the hemoglobin is of the abnormal, HbS, type, and even with normal saturation of oxygen the disorder is fully expressed (i.e., sickling deformity of all red cells and hemolytic anemia). In the heterozygote, only a proportion of the hemoglobin is HbS (the remainder being HbA), and therefore red cell sickling occurs only when there is exposure to lowered oxygen tension. This is referred to as the sickle cell trait to differentiate it from full-blown sickle cell anemia.
Although gene expression and mendelian traits are usually described as dominant or recessive, in some cases both of the alleles of a gene pair contribute to the phenotype—a condition called codominance. Histocompatibility and blood group antigens are good examples of codominant inheritance.
A single mutant gene may lead to many end effects, termed pleiotropism; conversely, mutations at several genetic loci may produce the same trait (genetic heterogeneity). Sickle cell anemia is an example of pleiotropism. In this hereditary disorder not only does the point mutation in the gene give rise to HbS, which predisposes the red cells to hemolysis, but also the abnormal red cells tend to cause a logjam in small vessels, inducing, for example, splenic fibrosis, organ infarcts, and bone changes. The numerous differing end-organ derangements are all related to the primary defect in hemoglobin synthesis. On the other hand, profound childhood deafness, an apparently homogeneous clinical entity, results from many different types of autosomal recessive mutations. Recognition of genetic heterogeneity not only is important in genetic counseling but also is relevant in the understanding of the pathogenesis of some common disorders, such as diabetes mellitus.
TRANSMISSION PATTERNS OF SINGLE-GENE DISORDERS
Mutations involving single genes typically follow one of three patterns of inheritance: autosomal dominant, autosomal recessive, and X-linked. The general rules that govern the transmission of single-gene disorders are well known; only a few salient features are summarized.12 Single-gene disorders with nonclassic patterns of inheritance are described later.
Autosomal Dominant Disorders
Autosomal dominant disorders are manifested in the heterozygous state, so at least one parent of an index case is usually affected; both males and females are affected, and both can transmit the condition. When an affected person marries an unaffected one, every child has one chance in two of having the disease. In addition to these basic rules, autosomal dominant conditions are characterized by the following:
The biochemical mechanisms of autosomal dominant disorders are best considered in the context of the nature of the mutation and the type of protein affected. Most mutations lead to the reduced production of a gene product or give rise to an inactive protein. The effect of such loss-of-function mutations depends on the nature of the protein affected. If the mutation affects an enzyme protein the heterozygotes are usually normal. Because up to 50% loss of enzyme activity can be compensated for, mutation in genes that encode enzymes do not manifest an autosomal dominant pattern of inheritance. By contrast, two major categories of nonenzyme proteins are affected in autosomal dominant disorders:
Less common than loss-of-function mutations are gain-of-function mutations. As the name indicates, in this type of mutation the protein product of the mutant allele acquires new properties not normally associated with the wild-type protein. The transmission of disorders produced by gain-of-function mutations is almost always autosomal dominant, as illustrated by Huntington disease (Chapter 28). In this disease the trinucleotide-repeat mutation affecting the Huntington gene (see later) gives rise to an abnormal protein, called huntingtin, that is toxic to neurons, and hence even heterozygotes develop a neurologic deficit.
To summarize, two types of mutations and two categories of proteins are involved in the pathogenesis of autosomal dominant diseases. The more common loss-of-function mutations affect regulatory proteins and subunits of multimeric proteins, the latter acting through a dominant-negative effect. Gain-of-function mutations are less common; they often endow normal proteins with toxic properties, or more rarely increase a normal activity (e.g., activating mutation in the erythropoetin receptor associated with a pathologic increase in red cell production).
Table 5-1 lists common autosomal dominant disorders. Many are discussed more logically in other chapters. A few conditions not considered elsewhere are discussed later in this chapter to illustrate important principles.
TABLE 5-1 Autosomal Dominant Disorders
| System | Disorder |
|---|---|
| Nervous | Huntington disease |
| Neurofibromatosis | |
| Myotonic dystrophy | |
| Tuberous sclerosis | |
| Urinary | Polycystic kidney disease |
| Gastrointestinal | Familial polyposis coli |
| Hematopoietic | Hereditary spherocytosis von Willebrand disease |
| Skeletal | Marfan syndrome* |
| Ehlers-Danlos syndrome (some variants)* | |
| Osteogenesis imperfecta | |
| Achondroplasia | |
| Metabolic | Familial hypercholesterolemia* |
| Acute intermittent porphyria |
* Discussed in this chapter. Other disorders listed are discussed in appropriate chapters in the book.
Autosomal Recessive Disorders
Autosomal recessive traits make up the largest category of mendelian disorders. Because autosomal recessive disorders result only when both alleles at a given gene locus are mutated, such disorders are characterized by the following features: (1) The trait does not usually affect the parents of the affected individual, but siblings may show the disease; (2) siblings have one chance in four of having the trait (i.e., the recurrence risk is 25% for each birth); and (3) if the mutant gene occurs with a low frequency in the population, there is a strong likelihood that the affected individual (proband) is the product of a consanguineous marriage. The following features generally apply to most autosomal recessive disorders and distinguish them from autosomal dominant diseases:
Autosomal recessive disorders include almost all inborn errors of metabolism. The various consequences of enzyme deficiencies are discussed later. The more common of these conditions are listed in Table 5-2. Most are presented elsewhere; a few prototypes are discussed later in this chapter.
TABLE 5-2 Autosomal Recessive Disorders
| System | Disorder |
|---|---|
| Metabolic | Cystic fibrosis |
| Phenylketonuria | |
| Galactosemia | |
| Homocystinuria | |
| Lysosomal storage diseases* | |
| α1-Antitrypsin deficiency | |
| Wilson disease | |
| Hemochromatosis | |
| Glycogen storage diseases* | |
| Hematopoietic | Sickle cell anemia |
| Thalassemias | |
| Endocrine | Congenital adrenal hyperplasia |
| Skeletal | Ehlers-Danlos syndrome (some variants)* |
| Alkaptonuria* | |
| Nervous | Neurogenic muscular atrophies |
| Friedreich ataxia | |
| Spinal muscular atrophy |
* Discussed in this chapter. Many others are discussed elsewhere in the text.
X-Linked Disorders
All sex-linked disorders are X-linked, and almost all are recessive. Several genes are located in the “male-specific region of Y”; all of these are related to spermatogenesis.13 Males with mutations affecting the Y-linked genes are usually infertile, and hence there is no Y-linked inheritance. As discussed later, a few additional genes with homologues on the X chromosome have been mapped to the Y chromosome, but no disorders resulting from mutations in such genes have been described.
X-linked recessive inheritance accounts for a small number of well-defined clinical conditions. The Y chromosome, for the most part, is not homologous to the X, and so mutant genes on the X do not have corresponding alleles on the Y. Thus, the male is said to be hemizygous for X-linked mutant genes, so these disorders are expressed in the male. Other features that characterize these disorders are as follows:
TABLE 5-3 X-Linked Recessive Disorders
| System | Disease |
|---|---|
| Musculoskeletal | Duchenne muscular dystrophy |
| Blood | Hemophilia A and B |
| Chronic granulomatous disease | |
| Glucose-6-phosphate dehydrogenase deficiency | |
| Immune | Agammaglobulinemia |
| Wiskott-Aldrich syndrome | |
| Metabolic | Diabetes insipidus |
| Lesch-Nyhan syndrome | |
| Nervous | Fragile-X syndrome* |
* Discussed in this chapter. Others are discussed in appropriate chapters in the text.
There are only a few X-linked dominant conditions. They are caused by dominant disease-associated alleles on the X chromosome. These disorders are transmitted by an affected heterozygous female to half her sons and half her daughters and by an affected male parent to all his daughters but none of his sons, if the female parent is unaffected. Vitamin D–resistant rickets is an example of this type of inheritance.
BIOCHEMICAL AND MOLECULAR BASIS OF SINGLE-GENE (MENDELIAN) DISORDERS
Mendelian disorders result from alterations involving single genes. The genetic defect may lead to the formation of an abnormal protein or a reduction in the output of the gene product. Virtually any type of protein may be affected in single-gene disorders and by a variety of mechanisms (Table 5-4). To some extent the pattern of inheritance of the disease is related to the kind of protein affected by the mutation, as was discussed earlier and is reiterated subsequently. For the purposes of this discussion, the mechanisms involved in single-gene disorders can be classified into four categories: (1) enzyme defects and their consequences; (2) defects in membrane receptors and transport systems; (3) alterations in the structure, function, or quantity of nonenzyme proteins; and (4) mutations resulting in unusual reactions to drugs.
Enzyme Defects and Their Consequences
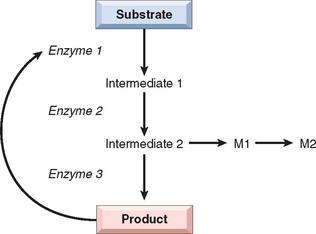
Mutations may result in the synthesis of a defective enzyme with reduced activity or in a reduced amount of a normal enzyme. In either case, the consequence is a metabolic block. Figure 5-6 provides an example of an enzyme reaction in which the substrate is converted by intracellular enzymes, denoted as 1, 2, and 3, into an end product through intermediates 1 and 2. In this model the final product exerts feedback control on enzyme 1. A minor pathway producing small quantities of M1 and M2 also exists. The biochemical consequences of an enzyme defect in such a reaction may lead to three major consequences:
Defects in Receptors and Transport Systems
Many biologically active substances have to be actively transported across the cell membrane. This transport is generally achieved by one of two mechanisms—through receptormediated endocytosis or by a transport protein. A genetic defect in a receptor-mediated transport system is exemplified by familial hypercholesterolemia, in which reduced synthesis or function of LDL receptors leads to defective transport of LDL into the cells and secondarily to excessive cholesterol synthesis by complex intermediary mechanisms. In cystic fibrosis the transport system for chloride ions in exocrine glands, sweat ducts, lungs, and pancreas is defective. By mechanisms not fully understood, impaired chloride transport leads to serious injury to the lungs and pancreas (Chapter 10).
Alterations in Structure, Function, or Quantity of Nonenzyme Proteins
Genetic defects resulting in alterations of nonenzyme proteins often have widespread secondary effects, as exemplified by sickle cell disease. The hemoglobinopathies, sickle cell disease being one, all of which are characterized by defects in the structure of the globin molecule, best exemplify this category. In contrast to the hemoglobinopathies, the thalassemias result from mutations in globin genes that affect the amount of globin chains synthesized. Thalassemias are associated with reduced amounts of structurally normal α-globin or β-globin chains (Chapter 14). Other examples of genetically defective structural proteins include collagen, spectrin, and dystrophin, giving rise to osteogenesis imperfecta (Chapter 26), heredit-ary spherocytosis (Chapter 14), and muscular dystrophies (Chapter 27), respectively.
Genetically Determined Adverse Reactions to Drugs
Certain genetically determined enzyme deficiencies are unmasked only after exposure of the affected individual to certain drugs. This special area of genetics, called pharmacogenetics, is of considerable clinical importance.14 The classic example of drug-induced injury in the genetically susceptible individual is associated with a deficiency of the enzyme G6PD. Under normal conditions glucose-6 phosphatedehydrogenase (G6PD) deficiency does not result in disease, but on administration, for example, of the antimalarial drug primaquine, a severe hemolytic anemia results (Chapter 14). In recent years an increasing number of polymorphisms of genes encoding drug-metabolizing enzymes, transporters, and receptors are being identified. In some cases these genetic factors have major impact on drug sensitivity and adverse reactions. It is expected that advances in pharmacogenetics will lead to patient-tailored therapy, or “personalized medicine.”
With this overview of the biochemical basis of single-gene disorders, we now consider selected examples grouped according to the underlying defect.
DISORDERS ASSOCIATED WITH DEFECTS IN STRUCTURAL PROTEINS
Several diseases caused by mutations in genes that encode structural proteins are listed in Table 5-4. Many are discussed elsewhere in the text. Only Marfan syndrome and Ehlers-Danlos syndromes (EDSs) are discussed here, because they affect connective tissue and hence involve multiple organ systems.
Marfan Syndrome
Marfan syndrome is a disorder of connective tissues, manifested principally by changes in the skeleton, eyes, and cardiovascular system.15 Its prevalence is estimated to be 1 in 5000. Approximately 70% to 85% of cases are familial and transmitted by autosomal dominant inheritance. The remainder are sporadic and arise from new mutations.
Pathogenesis.
Marfan syndrome results from an inherited defect in an extracellular glycoprotein called fibrillin-1. As alluded to in Chapter 3, fibrillin is the major component of microfibrils found in the extracellular matrix. These fibrils provide a scaffolding on which tropoelastin is deposited to form elastic fibers. Although microfibrils are widely distributed in the body, they are particularly abundant in the aorta, ligaments, and the ciliary zonules that support the lens; these tissues are prominently affected in Marfan syndrome.
Fibrillin occurs in two homologous forms, fibrillin-1 and fibrillin-2, encoded by two separate genes, FBN1 and FBN2, mapped on chromosomes 15q21.1 and 5q23.31, respectively. Mutations of FBN1 underlie Marfan syndrome; mutations of the related FBN2 gene are less common, and they give rise to congenital contractural arachnodactyly, an autosomal dominant disorder characterized by skeletal abnormalities. Mutational analysis has revealed more than 600 distinct mutations of the FBN1 gene in individuals with Marfan syndrome. Most of these are missense mutations that give rise to abnormal fibrillin-1. While many clinical manifestations of Marfan syndrome can be explained by changes in the mechanical properties of the extracellular matrix resulting from abnormalities of fibrillin, several others such as bone overgrowth cannot be attributed to changes in tissue elasticity. Recent studies indicate that loss of microfibrils gives rise to abnormal and excessive activation of transforming growth factor β (TGF-β), since normal microfibrils sequester TGF-β and thus control the bioavailability of this cytokine. Excessive TGF-β signaling has deleterious effects on vascular smooth muscle development and the integrity of extracellular matrix. This hypothesis is supported by two sets of observations. First, in a small number of individuals with clinical features of Marfan syndrome (MFS2) there are no mutations in FBN1 but mutations in genes that encode TGF-β receptors. Second, in mouse models of Marfan syndrome generated by mutations in Fbn1, administration of antibodies to TGF-β prevents alterations in the aorta and mitral valves.16 Human trials with a similar strategy seem to be promising.
Morphology. Skeletal abnormalities are the most striking feature of Marfan syndrome. Typically the patient is unusually tall with exceptionally long extremities and long, tapering fingers and toes. The joint ligaments in the hands and feet are lax, suggesting that the patient is double-jointed; typically the thumb can be hyperextended back to the wrist. The head is commonly dolichocephalic (long-headed) with bossing of the frontal eminences and prominent supraorbital ridges. A variety of spinal deformities may appear, including kyphosis, scoliosis, or rotation or slipping of the dorsal or lumbar vertebrae. The chest is classically deformed, presenting either pectus excavatum (deeply depressed sternum) or a pigeon-breast deformity.
The ocular changes take many forms. Most characteristic is bilateral subluxation or dislocation (usually outward and upward) of the lens, referred to as ectopia lentis. This abnormality is so uncommon in persons who do not have this genetic disease that the finding of bilateral ectopia lentis should raise the suspicion of Marfan syndrome.
Cardiovascular lesions are the most life-threatening features of this disorder. The two most common lesions are mitral valve prolapse and, of greater importance, dilation of the ascending aorta due to cystic medionecrosis. Histologically the changes in the media are virtually identical to those found in cystic medionecrosis not related to Marfan syndrome (see Chapter 12). Loss of medial support results in progressive dilation of the aortic valve ring and the root of the aorta, giving rise to severe aortic incompetence. In addition, excessive TGF-β signaling in the adventia also probably contributes to aortic dilation. Weakening of the media predisposes to an intimal tear, which may initiate an intramural hematoma that cleaves the layers of the media to produce aortic dissection. After cleaving the aortic layers for considerable distances, sometimes back to the root of the aorta or down to the iliac arteries, the hemorrhage often ruptures through the aortic wall. Such a calamity is the cause of death in 30% to 45% of these individuals.
Clinical Features
Although mitral valve lesions are more frequent, they are clinically less important than aortic lesions. Loss of connective tissue support in the mitral valve leaflets makes them soft and billowy, creating the so-called floppy valve (Chapter 12). Valvular lesions, along with lengthening of the chordae tendineae, frequently give rise to mitral regurgitation. Similar changes may affect the tricuspid and, rarely, the aortic valves. Echocardiography greatly enhances the ability to detect the cardiovascular abnormalities and is therefore extremely valuable in the diagnosis of Marfan syndrome. The great majority of deaths are caused by rupture of aortic dissections, followed in importance by cardiac failure.
While the lesions just described typify Marfan syndrome, it must be emphasized that there is great variation in the clinical expression of this genetic disorder. Patients with prominent eye or cardiovascular changes may have few skeletal abnormalities, whereas others with striking changes in body habitus have no eye changes. Although variability in clinical expression may be seen within a family, interfamilial variability is much more common and extensive. Because of such variations, the clinical diagnosis of Marfan syndrome must be based on major involvement of two of the four organ systems (skeletal, cardiovascular, ocular, and skin) and minor involvement of another organ.
To account for the variable expression of the Marfan defect, it has been hypothesized that Marfan syndrome is genetically heterogeneous. With one exception, however, all studies thus far point to mutations in the FBN1 gene, on chromosome 15q21.1, as the cause of this disease.15 Thus, variable expressivity is best explained on the basis of allelic mutations within the same locus. Because the FBN1 gene is large and many different mutations have been identified, direct diagnosis by DNA sequencing is not currently feasible, but this may change in the near future as new technologies are being developed.
Ehlers-Danlos Syndromes (EDS)
EDSs comprise a clinically and genetically heterogeneous group of disorders that result from some defect in the synthesis or structure of fibrillar collagen. Other disorders resulting from mutations affecting collagen synthesis include osteogenesis imperfecta (Chapter 26), Alport syndrome (Chapter 20), and epidermolysis bullosa (Chapter 25).
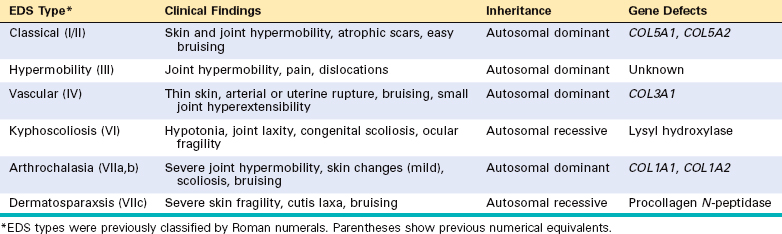
Biosynthesis of collagen is a complex process that can be disturbed by genetic errors that may affect any one of the numerous structural collagen genes or enzymes necessary for post-transcriptional modifications of collagen. Hence, the mode of inheritance of EDS encompasses all three mendelian patterns. On the basis of clinical and molecular characteristics, six variants of EDS have been recognized.17 These are listed in Table 5-5. It is beyond the scope of this book to discuss each variant individually; instead, we first summarize the important clinical features that are common to most variants and then correlate some of the clinical manifestations with the underlying molecular defects in collagen synthesis or structure.
As might be expected, tissues rich in collagen, such as skin, ligaments, and joints, are frequently involved in most variants of EDS. Because the abnormal collagen fibers lack adequate tensile strength, skin is hyperextensible, and the joints are hypermobile. These features permit grotesque contortions, such as bending the thumb backward to touch the forearm and bending the knee forward to create almost a right angle. It is believed that most contortionists have one of the EDSs. A predisposition to joint dislocation, however, is one of the prices paid for this virtuosity. The skin is extraordinarily stretchable, extremely fragile, and vulnerable to trauma. Minor injuries produce gaping defects, and surgical repair or intervention is accomplished with great difficulty because of the lack of normal tensile strength. The basic defect in connective tissue may lead to serious internal complications. These include rupture of the colon and large arteries (vascular EDS), ocular fragility with rupture of cornea and retinal detachment (kyphoscoliosis EDS), and diaphragmatic hernia (classical EDS).
The biochemical and molecular bases of these abnormalities are known in several forms of EDS. These are described briefly, because they offer some insights into the perplexing clinical heterogeneity of EDS. Perhaps the best characterized is the kyphoscoliosis type, the most common autosomal recessive form of EDS. It results from mutations in the gene encoding lysyl hydroxylase, an enzyme necessary for hydroxylation of lysine residues during collagen synthesis.18 Affected patients have markedly reduced levels of this enzyme. Because hydroxylysine is essential for the cross-linking of collagen fibers, a deficiency of lysyl hydroxylase results in the synthesis of collagen that lacks normal structural stability.
The vascular type of EDS results from abnormalities of type III collagen.19 This form is genetically heterogeneous, because at least three distinct types of mutations affecting the COL3A1 gene encoding collagen type III can give rise to this variant. Some affect the rate of synthesis of pro α1 (III) chains, others affect the secretion of type III procollagen, and still others lead to the synthesis of structurally abnormal type III collagen. Some mutant alleles behave as dominant negatives (see discussion under “Autosomal Dominant Disorders”) and thus produce severe phenotypic effects. These molecular studies provide a rational basis for the pattern of transmission and clinical features that are characteristic of this variant. First, because vascular-type EDS results from mutations involving a structural protein (rather than an enzyme protein), an autosomal dominant pattern of inheritance would be expected. Second, because blood vessels and intestines are known to be rich in collagen type III, an abnormality of this collagen is consistent with severe defects (e.g., spontaneous rupture) in these organs.
In two forms of EDS—arthrochalasia type and dermatosparaxis type—the fundamental defect is in the conversion of type I procollagen to collagen. This step in collagen synthesis involves cleavage of noncollagen peptides at the N terminus and C terminus of the procollagen molecule. This is accomplished by N terminal–specific and C terminal–specific peptidases. The defect in the conversion of procollagen to collagen in the arthrochalasia type has been traced to mutations that affect one of the two type I collagen genes, COL1A1 and COL1A2. As a result, structurally abnormal pro α1 (I) or pro α2 (I) chains that resist cleavage of N-terminal peptides are formed. In patients with a single mutant allele, only 50% of the type I collagen chains are abnormal, but because these chains interfere with the formation of normal collagen helices, heterozygotes manifest the disease. By contrast, the related dermatosparaxis type is caused by mutations in the procollagen-N-peptidase genes, essential for the cleavage of collagens. In this case the enzyme deficiency leads to an autosomal recessive form of inheritance.
Finally, the classical type of EDS is worthy of brief mention, since molecular analysis of this variant suggests that genes other than collagen genes may be involved in the pathogenesis of EDS. In 30% to 50% of these cases, mutations in the genes for type V collagen (COL5A1 and COL5A2) have been detected.20 Surprisingly, despite a phenotype typical of EDS, no other collagen gene abnormalities have been found in the remaining cases.
To summarize, the common thread in EDS is some abnormality of collagen. These disorders, however, are extremely heterogeneous. At the molecular level, a variety of defects, varying from mutations involving structural genes for collagen to those involving enzymes that are responsible for post-transcriptional modifications of mRNA, have been detected. Such molecular heterogeneity results in the expression of EDS as a clinically variable disorder with several patterns of inheritance.
DISORDERS ASSOCIATED WITH DEFECTS IN RECEPTOR PROTEINS
Familial Hypercholesterolemia
Familial hypercholesterolemia is a “receptor disease” that is the consequence of a mutation in the gene encoding the receptor for LDL, which is involved in the transport and metabolism of cholesterol. As a consequence of receptor abnormalities there is a loss of feedback control and elevated levels of cholesterol that induce premature atherosclerosis, leading to a greatly increased risk of myocardial infarction.21
Familial hypercholesterolemia is one of the most frequently occurring mendelian disorders. Heterozygotes with one mutant gene, representing about 1 in 500 individuals, have from birth a two-fold to three-fold elevation of plasma cholesterol level, leading to tendinous xanthomas and premature atherosclerosis in adult life (Chapter 11). Homozygotes, having a double dose of the mutant gene, are much more severely affected, and may have five-fold to six-fold elevations in plasma cholesterol levels. These individuals develop skin xanthomas and coronary, cerebral, and peripheral vascular atherosclerosis at an early age. Myocardial infarction may develop before age 20. Large-scale studies have found that familial hypercholesterolemia is present in 3% to 6% of survivors of myocardial infarction.
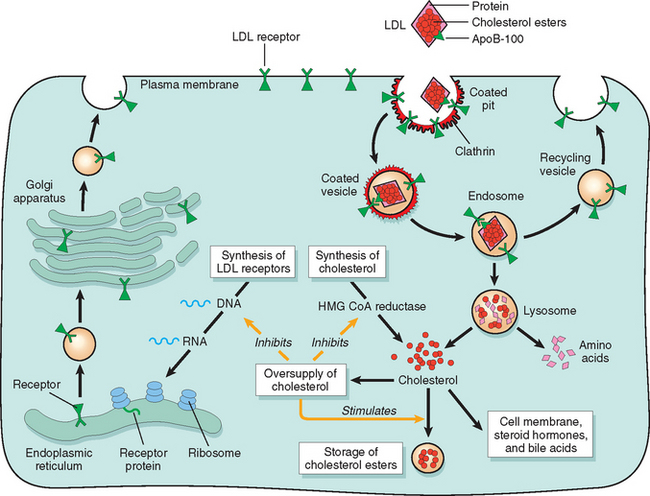
An understanding of this disorder requires that we briefly review the normal process of cholesterol metabolism and transport. Approximately 7% of the body’s cholesterol circulates in the plasma, predominantly in the form of LDL. As might be expected, the amount of plasma cholesterol is influenced by its synthesis and catabolism, and the liver plays a crucial role in both these processes (Fig. 5-7). The first step in this complex sequence is the secretion of very-low-density lipoproteins (VLDLs) by the liver into the bloodstream. VLDL particles are rich in triglycerides, but they contain lesser amounts of cholesteryl esters. When a VLDL particle reaches the capillaries of adipose tissue or muscle, it is cleaved by lipoprotein lipase, a process that extracts most of the triglycerides. The resulting molecule, called intermediate-density lipoprotein (IDL), is reduced in triglyceride content and enriched in cholesteryl esters, but it retains two of the three apoproteins (B-100 and E) present in the parent VLDL particle (see Fig. 5-7). After release from the capillary endothelium, the IDL particles have one of two fates. Approximately 50% of newly formed IDL is rapidly taken up by the liver by receptor-mediated transport. The receptor responsible for the binding of IDL to the liver cell membrane recognizes both apoprotein B-100 and apoprotein E. It is called the LDL receptor, however, because it is also involved in the hepatic clearance of LDL, as described later. In the liver cells, IDL is recycled to generate VLDL. The IDL particles not taken up by the liver are subjected to further metabolic processing that removes most of the remaining triglycerides and apoprotein E, yielding cholesterol-rich LDL particles. It should be emphasized that IDL is the immediate and major source of plasma LDL. There seem to be two mechanisms for removal of LDL from plasma, one mediated by an LDL receptor and the other by a receptor for oxidized LDL (scavenger receptor), described later. Although many cell types, including fibroblasts, lymphocytes, smooth muscle cells, hepatocytes, and adrenocortical cells, possess high-affinity LDL receptors, approximately 70% of the plasma LDL seems to be cleared by the liver, using a quite sophisticated transport process (Fig. 5-8). The first step involves binding of LDL to cell surface receptors, which are clustered in specialized regions of the plasma membrane called coated pits. After binding, the coated pits containing the receptor-bound LDL are internalized by invagination to form coated vesicles, after which they migrate within the cell to fuse with the lysosomes. Here the LDL dissociates from the receptor, which is recycled to the surface. In the lysosomes the LDL molecule is enzymatically degraded; the apoprotein part is hydrolyzed to amino acids, whereas the cholesteryl esters are broken down to free cholesterol. This free cholesterol, in turn, crosses the lysosomal membrane to enter the cytoplasm, where it is used for membrane synthesis and as a regulator of cholesterol homeostasis. The exit of cholesterol from the lysosomes requires the action of two proteins called NPC1 and NPC2 (see “Niemann-Pick Disease, Type C”). Three separate processes are affected by the released intracellular cholesterol, as follows:
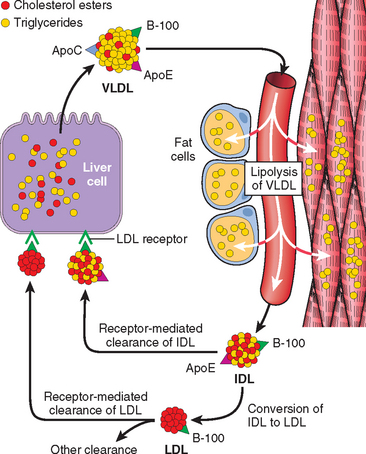
FIGURE 5-7 Low-density lipoprotein (LDL) metabolism and the role of the liver in its synthesis and clearance. Lipolysis of very-low-density lipoprotein (VLDL) by lipoprotein lipase in the capillaries releases triglycerides, which are then stored in fat cells and used as a source of energy in skeletal muscles. See text for explanation of abbreviations used.
As mentioned earlier, familial hypercholesterolemia results from mutations in the gene specifying the receptor for LDL. Heterozygotes with familial hypercholesterolemia possess only 50% of the normal number of high-affinity LDL receptors, because they have only one normal gene. As a result of this defect in transport, the catabolism of LDL by the receptor-dependent pathways is impaired, and the plasma level of LDL increases approximately two-fold. Homozygotes have virtually no normal LDL receptors in their cells and have much higher levels of circulating LDL. In addition to defective LDL clearance, both the homozygotes and heterozygotes have increased synthesis of LDL. The mechanism of increased synthesis that contributes to hypercholesterolemia also results from a lack of LDL receptors (see Fig. 5-7). Recall that IDL, the immediate precursor of plasma LDL, also uses hepatic LDL receptors (apoprotein B-100 and E receptors) for its transport into the liver. In familial hypercholesterolemia, impaired IDL transport into the liver secondarily diverts a greater proportion of plasma IDL into the precursor pool for plasma LDL.
The transport of LDL via the scavenger receptor seems to occur at least in part into the cells of the mononuclear phagocyte system. Monocytes and macrophages have receptors for chemically altered (e.g., acetylated or oxidized) LDL. Normally the amount of LDL transported along this scavenger receptor pathway is less than that mediated by the LDL receptor–dependent mechanisms. In the face of hypercholesterolemia, however, there is a marked increase in the scavenger receptor–mediated traffic of LDL cholesterol into the cells of the mononuclear phagocyte system and possibly the vascular walls (Chapter 11). This increase is responsible for the appearance of xanthomas and contributes to the pathogenesis of premature atherosclerosis.
The molecular genetics of familial hypercholesterolemia is extremely complex. More than 900 mutations, including insertions, deletions, and missense and nonsense mutations, involving the LDL receptor gene have been identified. These can be classified into five groups (Fig. 5-9): Class I mutations are relatively uncommon, and they lead to a complete failure of synthesis of the receptor protein (null allele). Class II mutations are fairly common; they encode receptor proteins that accumulate in the endoplasmic reticulum because their folding defects make it impossible for them to be transported to the Golgi complex. Class III mutations affect the LDL-binding domain of the receptor; the encoded proteins reach the cell surface but fail to bind LDL or do so poorly. Class IV mutations encode proteins that are synthesized and transported to the cell surface efficiently. They bind LDL normally, but they fail to localize in coated pits, and hence the bound LDL is not internalized. Class V mutations encode proteins that are expressed on the cell surface, can bind LDL, and can be internalized; however, the pH-dependent dissociation of the receptor and the bound LDL fails to occur. Such receptors are trapped in the endosome, where they are degraded, and hence they fail to recycle to the cell surface.
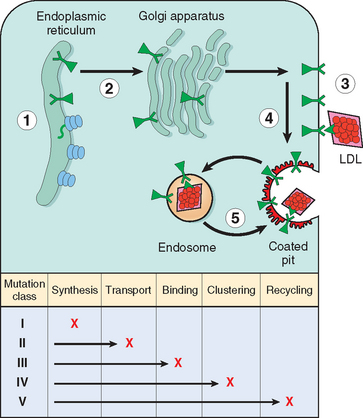
FIGURE 5-9 Classification of LDL receptor mutations based on abnormal function of the mutant protein. These mutations disrupt the receptor’s synthesis in the endoplasmic reticulum, transport to the Golgi complex, binding of apoprotein ligands, clustering in coated pits, and recycling in endosomes. Each class is heterogeneous at the DNA level.
(Modified with permission from Hobbs HH et al.: The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet 24:133–170, 1990. © 1990 by Annual Reviews.)
The discovery of the critical role of LDL receptors in cholesterol homeostasis has led to the rational design of drugs that lower plasma cholesterol by increasing the number of LDL receptors. One strategy is based on the ability of certain drugs (statins) to suppress intracellular cholesterol synthesis by inhibiting the enzyme HMG CoA reductase. This, in turn, allows greater synthesis of LDL receptors (see Fig. 5-8).
DISORDERS ASSOCIATED WITH DEFECTS IN ENZYMES
Lysosomal Storage Diseases
Lysosomes are key components of the “intracellular digestive tract.” They contain a battery of hydrolytic enzymes, which have two special properties. First, they function in the acidic milieu of the lysosomes. Second, these enzymes constitute a special category of secretory proteins that are destined not for the extracellular fluids but for intracellular organelles. This latter characteristic requires special processing within the Golgi apparatus, which is reviewed briefly. Similar to all other secretory proteins, lysosomal enzymes (or acid hydrolases, as they are sometimes called) are synthesized in the endoplasmic reticulum and transported to the Golgi apparatus. Within the Golgi complex they undergo a variety of post-translational modifications, of which one is worthy of special note. This modification involves the attachment of terminal mannose6-phosphate groups to some of the oligosaccharide side chains. The phosphorylated mannose residues serve as an “address label” that is recognized by specific receptors found on the inner surface of the Golgi membrane. Lysosomal enzymes bind these receptors and are thereby segregated from the numerous other secretory proteins within the Golgi. Subsequently, small transport vesicles containing the receptor-bound enzymes are pinched off from the Golgi and proceed to fuse with the lysosomes. Thus, the enzymes are targeted to their intracellular abode, and the vesicles are shuttled back to the Golgi (Fig. 5-10). As indicated later, genetically determined errors in this remarkable sorting mechanism may give rise to one form of lysosomal storage disease.22
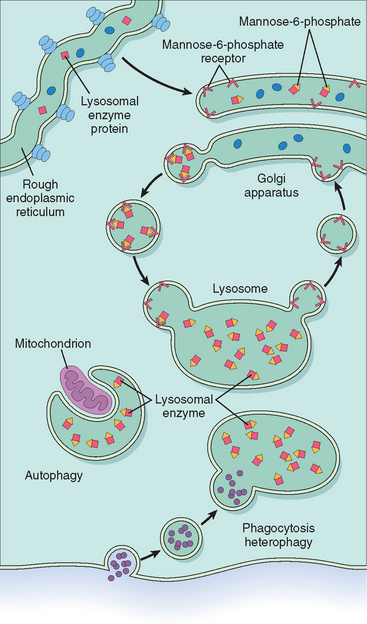
The lysosomal acid hydrolases catalyze the breakdown of a variety of complex macromolecules. These large molecules may be derived from the metabolic turnover of intracellular organelles (autophagy), or they may be acquired from outside the cells by phagocytosis (heterophagy). With an inherited deficiency of a functional lysosomal enzyme, catabolism of its substrate remains incomplete, leading to the accumulation of the partially degraded insoluble metabolite within the lysosomes. Stuffed with incompletely digested macromolecules, these organelles become large and numerous enough to interfere with normal cell functions, giving rise to the so-called lysosomal storage disorders (Fig. 5-11). In addition to “missing enzymes,” lysosomal storage diseases can result from lack of any protein essential for normal function of lysosomes. Examples are:
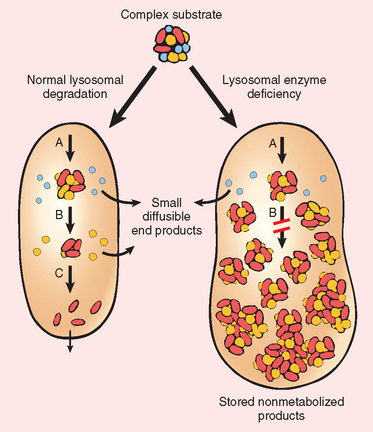
FIGURE 5-11 Pathogenesis of lysosomal storage diseases. In the example shown a complex substrate is normally degraded by a series of lysosomal enzymes (A, B, and C) into soluble end products. If there is a deficiency or malfunction of one of the enzymes (e.g., B), catabolism is incomplete and insoluble intermediates accumulate in the lysosomes.
There are three general approaches to the treatment of lysosomal storage diseases. The most obvious is enzyme replacement therapy, currently in use for several lysosomal storage diseases. Another approach, the “substrate reduction therapy,” is based on the premise that if the substrate to be degraded by the lysosomal enzyme can be reduced, the residual enzyme activity may be sufficient to catabolize it and prevent accumulation. A more recent strategy is based on the understanding of the molecular basis of enzyme deficiency. In many disorders, exemplified by Gaucher disease, the enzyme activity is low because the mutant proteins are unstable and prone to misfolding, and hence degraded in the endoplasmic reticulum. In such diseases an exogenous competitive inhibitor of the enzyme can, paradoxically, bind to the mutant enzyme and act as the “folding template” that assists proper folding of the enzyme and thus prevents its degradation. Such molecular chaperone therapy is under active investigation.23
Several distinctive and separable conditions are included among the lysosomal storage diseases (Table 5-6). In general, the distribution of the stored material, and hence the organs affected, is determined by two interrelated factors: (1) the tissue where most of the material to be degraded is found and (2) the location where most of the degradation normally occurs. For example, brain is rich in gangliosides, and hence defective hydrolysis of gangliosides, as occurs in GM1 and GM2 gangliosidoses, results primarily in accumulation within neurons and consequent neurologic symptoms. Defects in degradation of mucopolysaccharides affect virtually every organ, because mucopolysaccharides are widely distributed in the body. Because cells of the mononuclear phagocyte system are especially rich in lysosomes and are involved in the degradation of a variety of substrates, organs rich in phagocytic cells, such as the spleen and liver, are frequently enlarged in several forms of lysosomal storage disorders. The ever-expanding number of lysosomal storage diseases can be divided into rational categories based on the biochemical nature of the accumulated metabolite, thus creating such subgroups as the glycogenoses, sphingolipidoses (lipidoses), mucopolysaccharidoses (MPSs), and mucolipidoses (see Table 5-6). Only the most common disorders among the remaining groups are considered here.
TABLE 5-6 Lysosomal Storage Diseases
| Disease | Enzyme Deficiency | Major Accumulating Metabolites |
|---|---|---|
| GLYCOGENOSIS | ||
| Type 2—Pompe disease | α-1,4-Glucosidase (lysosomal glucosidase) | Glycogen |
| SPHINGOLIPIDOSES | ||
| GM1 gangliosidosis | GM1 ganglioside β-galactosidase | GM1 ganglioside, galactose-containing oligosaccharides |
| Type 1—infantile, generalized | ||
| Type 2—juvenile | ||
| GM2 gangliosidosis | ||
| Tay-Sachs disease | Hexosaminidase, α subunit | GM2 ganglioside |
| Sandhoff disease | Hexosaminidase, β subunit | GM2 ganglioside, globoside |
| GM2 gangliosidosis variant AB | Ganglioside activator protein | GM2 ganglioside |
| SULFATIDOSES | ||
| Metachromatic leukodystrophy | Arylsulfatase A | Sulfatide |
| Multiple sulfatase deficiency | Arylsulfatase A, B, C; steroid sulfatase; iduronate sulfatase; heparan N-sulfatase | Sulfatide, steroid sulfate, heparan sulfate, dermatan sulfate |
| Krabbe disease | Galactosylceramidase | Galactocerebroside |
| Fabry disease | α-Galactosidase A | Ceramide trihexoside |
| Gaucher disease | Glucocerebrosidase | Glucocerebroside |
| Niemann-Pick disease: types A and B | Sphingomyelinase | Sphingomyelin |
| MUCOPOLYSACCHARIDOSES (MPSS) | ||
| MPS I H (Hurler) | α-L-Iduronidase | Dermatan sulfate, heparan sulfate |
| MPS II (Hunter) | l-Iduronosulfate sulfatase | |
| MUCOLIPIDOSES (MLS) | ||
| I-cell disease (ML II) and pseudo-Hurler polydystrophy | Deficiency of phosphorylating enzymes essential for the formation of mannose-6-phosphate recognition marker; acid hydrolases lacking the recognition marker cannot be targeted to the lysosomes but are secreted extracellularly | Mucopolysaccharide, glycolipid |
| OTHER DISEASES OF COMPLEX CARBOHYDRATES | ||
| Fucosidosis | α-Fucosidase | Fucose-containing sphingolipids and glycoprotein fragments |
| Mannosidosis | α-Mannosidase | Mannose-containing oligosaccharides |
| Aspartylglycosaminuria | Aspartylglycosamine amide hydrolase | Aspartyl-2-deoxy-2-acetamido-glycosylamine |
| OTHER LYSOSOMAL STORAGE DISEASES | ||
| Wolman disease | Acid lipase | Cholesterol esters, triglycerides |
| Acid phosphate deficiency | Lysosomal acid phosphatase | Phosphate esters |
Tay-Sachs Disease (GM2 Gangliosidosis: Hexosaminidase α-Subunit Deficiency)
GM2 gangliosidoses are a group of three lysosomal storage diseases caused by an inability to catabolize GM2 gangliosides. Degradation of GM2 gangliosides requires three polypeptides encoded by three distinct genes. The phenotypic effects of mutations affecting these genes are fairly similar, because they result from accumulation of GM2 gangliosides.24 The underlying enzyme defect, however, is different for each. Tay-Sachs disease, the most common form of GM2 gangliosidosis, results from mutations in the α-subunit locus on chromosome 15 that cause a severe deficiency of hexosaminidase A. This disease is especially prevalent among Jews, particularly among those of Eastern European (Ashkenazic) origin, in whom a carrier rate of 1 in 30 has been reported.
Morphology. The hexosaminidase A is absent from virtually all the tissues, so GM2 ganglioside accumulates in many tissues (e.g., heart, liver, spleen), but the involvement of neurons in the central and autonomic nervous systems and retina dominates the clinical picture. On histologic examination, the neurons are ballooned with cytoplasmic vacuoles, each representing a markedly distended lysosome filled with gangliosides (Fig. 5-12A). Stains for fat such as oil red O and Sudan black B are positive. With the electron microscope, several types of cytoplasmic inclusions can be visualized, the most prominent being whorled configurations within lysosomes composed of onion-skin layers of membranes (Fig. 5-12B). In time there is progressive destruction of neurons, proliferation of microglia, and accumulation of complex lipids in phagocytes within the brain substance. A similar process occurs in the cerebellum as well as in neurons throughout the basal ganglia, brain stem, spinal cord, and dorsal root ganglia and in the neurons of the autonomic nervous system. The ganglion cells in the retina are similarly swollen with GM2 ganglioside, particularly at the margins of the macula. A cherry-red spot thus appears in the macula, representing accentuation of the normal color of the macular choroid contrasted with the pallor produced by the swollen ganglion cells in the remainder of the retina (Chapter 29). This finding is characteristic of Tay-Sachs disease and other storage disorders affecting the neurons.
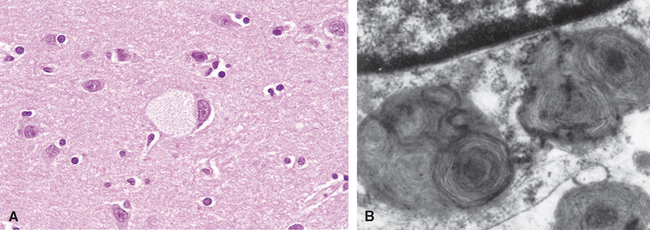
FIGURE 5-12 Ganglion cells in Tay-Sachs disease. A, Under the light microscope a large neuron has obvious lipid vacuolation. B, A portion of a neuron under the electron microscope shows prominent lysosomes with whorled configurations. Part of the nucleus is shown above.
(A, courtesy of Dr. Arthur Weinberg, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX; B, electron micrograph courtesy of Dr. Joe Rutledge, University of Texas Southwestern Medical Center, Dallas, TX.)
Clinical Features
The affected infants appear normal at birth but begin to manifest signs and symptoms at about age 6 months. There is relentless motor and mental deterioration, beginning with motor incoordination, mental obtundation leading to muscular flaccidity, blindness, and increasing dementia. Sometime during the early course of the disease, the characteristic, but not pathognomonic, cherry-red spot appears in the macula of the eye in almost all patients. Over the span of 1 or 2 years a complete vegetative state is reached, followed by death at age 2 to 3 years. More than 100 mutations have been described in the α-subunit gene; most affect protein folding. Such misfolded proteins trigger the “unfolded protein” response (Chapter 1) leading to apoptosis. These findings have given rise to the possibility of chaperone therapy of Tay-Sachs disease.
Antenatal diagnosis and carrier detection are possible by enzyme assays and DNA-based analysis. The clinical features of the two other forms of GM2 gangliosidosis, Sandhoff disease, resulting from β-subunit defect, and GM2 activator deficiency, are similar to those of Tay-Sachs disease.
Niemann-Pick Disease, Types A and B
Niemann-Pick disease types A and B refers to two related disorders that are characterized by lysosomal accumulation of sphingomyelin due to an inherited deficiency of sphingomyelinase.25 Type A is a severe infantile form with extensive neurologic involvement, marked visceral accumulations of sphingomyelin, and progressive wasting and early death within the first 3 years of life. In contrast, type B disease patients have organomegaly but generally no central nervous system involvement. They usually survive into adulthood. As with Tay-Sachs disease, Niemann-Pick disease types A and B are common in Ashkenazi Jews. The acid sphingomyelinase gene maps to chromosome 11p15.4 and is one of the imprinted genes that is preferentially expressed from the maternal chromosome as a result of epigenetic silencing of the paternal gene (discussed later). More than 100 mutations have been found in the acid sphingomyelinase gene and there seems to be a correlation between the type of mutation, the severity of enzyme deficiency, and the phenotype.
Morphology. In the classic infantile type A variant, a missense mutation causes almost complete deficiency of sphingomyelinase. Sphingomyelin is a ubiquitous component of cellular (including organellar) membranes, and so the enzyme deficiency blocks degradation of the lipid, resulting in its progressive accumulation within lysosomes, particularly within cells of the mononuclear phagocyte system. Affected cells become enlarged, sometimes to 90 μm in diameter, due to the distention of lysosomes with sphingomyelin and cholesterol. Innumerable small vacuoles of relatively uniform size are created, imparting foaminess to the cytoplasm (Fig. 5-13). In frozen sections of fresh tissue, the vacuoles stain for fat. Electron microscopy confirms that the vacuoles are engorged secondary lysosomes that often contain membranous cytoplasmic bodies resembling concentric lamellated myelin figures, sometimes called “zebra” bodies.
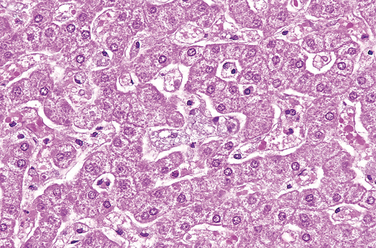
FIGURE 5-13 Niemann-Pick disease in liver. The hepatocytes and Kupffer cells have a foamy, vacuolated appearance due to deposition of lipids.
(Courtesy of Dr. Arthur Weinberg, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX.)
The lipid-laden phagocytic foam cells are widely distributed in the spleen, liver, lymph nodes, bone marrow, tonsils, gastrointestinal tract, and lungs. The involvement of the spleen generally produces massive enlargement, sometimes to ten times its normal weight, but the hepatomegaly is usually not quite so striking. The lymph nodes are generally moderately to markedly enlarged throughout the body.
Involvement of the brain and eye deserves special mention. In the brain the gyri are shrunken and the sulci widened. The neuronal involvement is diffuse, affecting all parts of the nervous system. Vacuolation and ballooning of neurons constitute the dominant histologic change, which in time leads to cell death and loss of brain substance. A retinal cherry-red spot similar to that seen in Tay-Sachs disease is present in about one third to one half of affected individuals.
Clinical manifestations in type A disease may be present at birth and almost invariably become evident by age 6 months. Infants typically have a protuberant abdomen because of the hepatosplenomegaly. Once the manifestations appear, they are followed by progressive failure to thrive, vomiting, fever, and generalized lymphadenopathy as well as progressive deterioration of psychomotor function. Death comes, usually within the first or second year of life.
The diagnosis is established by biochemical assays for sphingomyelinase activity in liver or bone marrow biopsy. Individuals affected with types A and B as well as carriers can be detected by DNA analysis.
Niemann-Pick Disease, Type C (NPC)
Although previously considered to be related to types A and B, Niemann-Pick disease type C (NPC) is quite distinct at the biochemical and molecular levels and is more common than types A and B combined. Mutations in two related genes, NPC1 and NPC2, can give rise to it, with NPC1 being responsible for 95% of cases. Unlike most other lysosomal storage diseases, NPC is due to a primary defect in lipid transport. Affected cells accumulate cholesterol as well as gangliosides such as GM1 and GM2. Both NPC1 and NPC2 are involved in the transport of free cholesterol from the lysosomes to the cytoplasm.26 NPC is clinically heterogeneous. It may present as hydrops fetalis and stillbirth, as neonatal hepatitis, or as a chronic form characterized by progressive neurologic damage. The most common form presents in childhood and is marked by ataxia, vertical supranuclear gaze palsy, dystonia, dysarthria, and psychomotor regression.
Gaucher Disease
Gaucher disease refers to a cluster of autosomal recessive disorders resulting from mutations in the gene encoding glucocerebrosidase.27 This disease is the most common lysosomal storage disorder. The affected gene encodes glucocerebrosidase, an enzyme that normally cleaves the glucose residue from ceramide. As a result of the enzyme defect, glucocerebroside accumulates principally in phagocytes but in some subtypes also in the central nervous system. Glucocerebrosides are continually formed from the catabolism of glycolipids derived mainly from the cell membranes of senescent leukocytes and erythrocytes. It is clear now that the pathologic changes in Gaucher disease are caused not just by the burden of storage material but also by activation of macrophages and the consequent secretion of cytokines such as IL-1, IL-6, and tumor necrosis factor (TNF). Three clinical subtypes of Gaucher disease have been distinguished. The most common, accounting for 99% of cases, is called type I, or the chronic non-neuronopathic form. In this type, storage of glucocerebrosides is limited to the mononuclear phagocytes throughout the body without involving the brain. Splenic and skeletal involvements dominate this pattern of the disease. It is found principally in Jews of European stock. Individuals with this disorder have reduced but detectable levels of glucocerebrosidase activity. Longevity is shortened but not markedly. Type II, or acute neuronopathic Gaucher disease, is the infantile acute cerebral pattern. This form has no predilection for Jews. In these patients there is virtually no detectable glucocerebrosidase activity in the tissues. Hepatosplenomegaly is also seen in this form of Gaucher disease, but the clinical picture is dominated by progressive central nervous system involvement, leading to death at an early age. A third pattern, type III, is intermediate between types I and II. These patients have the systemic involvement characteristic of type I but have progressive central nervous system disease that usually begins in adolescence or early adulthood.
Morphology. Glucocerebrosides accumulate in massive amounts within phagocytic cells throughout the body in all forms of Gaucher disease. The distended phagocytic cells, known as Gaucher cells, are found in the spleen, liver, bone marrow, lymph nodes, tonsils, thymus, and Peyer’s patches. Similar cells may be found in both the alveolar septa and the air spaces in the lung. In contrast to other lipid storage diseases, Gaucher cells rarely appear vacuolated but instead have a fibrillary type of cytoplasm likened to crumpled tissue paper (Fig. 5-14). Gaucher cells are often enlarged, sometimes up to 100 μm in diameter, and have one or more dark, eccentrically placed nuclei. Periodic acid–Schiff staining is usually intensely positive. With the electron microscope the fibrillary cytoplasm can be resolved as elongated, distended lysosomes, containing the stored lipid in stacks of bilayers.
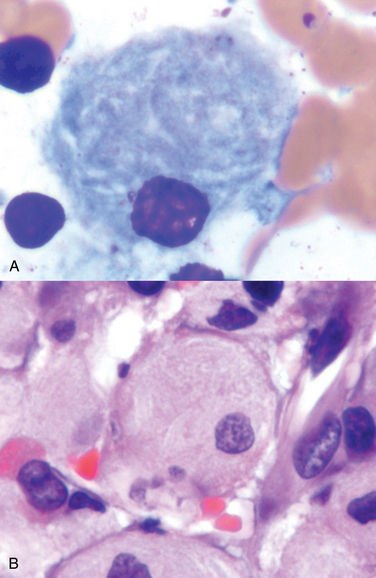
FIGURE 5-14 Gaucher disease involving the bone marrow. Gaucher cells (A, H&E; B, Wright stain) are plump macrophages that characteristically have the appearance in the cytoplasm of crumpled tissue paper (B), due to accumulation of glucocerebroside.
(Courtesy of Dr. John Anastasi, Department of Pathology, University of Chicago, Chicago, IL.)
In type I disease, the spleen is enlarged, sometimes up to 10 kg. The lymphadenopathy is mild to moderate and is body-wide. The accumulation of Gaucher cells in the bone marrow occurs in 70% to 100% of cases of type I Gaucher disease. It produces areas of bone erosion that are sometimes small but in other cases sufficiently large to give rise to pathologic fractures. Bone destruction occurs due to the secretion of cytokines by activated macrophages. In patients with cerebral involvement, Gaucher cells are seen in the Virchow-Robin spaces, and arterioles are surrounded by swollen adventitial cells. There is no storage of lipids in the neurons, yet neurons appear shriveled and are progressively destroyed. It is suspected that the lipids that accumulate in the phagocytic cells around blood vessels secrete cytokines that damage nearby neurons.
Clinical Features
The clinical course of Gaucher disease depends on the clinical subtype. In type I, symptoms and signs first appear in adult life and are related to splenomegaly or bone involvement. Most commonly there is pancytopenia or thrombocytopenia secondary to hypersplenism. Pathologic fractures and bone pain occur if there has been extensive expansion of the marrow space. Although the disease is progressive in the adult, it is compatible with long life. In types II and III, central nervous system dysfunction, convulsions, and progressive mental deterioration dominate, although organs such as the liver, spleen, and lymph nodes are also affected.
The diagnosis of homozygotes can be made by measurement of glucocerebrosidase activity in peripheral blood leukocytes or in extracts of cultured skin fibroblasts. In principle, heterozygotes can be identified by detection of mutations. However, because more than 150 mutations in the glucocerebroside gene can cause Gaucher disease, it is not possible to use a single genetic test.
Replacement therapy with recombinant enzymes is the mainstay for treatment of Gaucher disease; it is effective, and those with type I disease can expect normal life expectancy with this form of treatment. However, such therapy is extremely expensive. Because the fundamental defect resides in mononuclear phagocytic cells originating from marrow stem cells, bone marrow transplantation has been attempted. Other work is directed toward correction of the enzyme defect by transfer of the normal glucocerebrosidase gene into the patient’s bone marrow cells. Substrate reduction therapy with inhibitors of glucosylceramide synthetase is also being evaluated.
Mucopolysaccharidoses
The MPSs are a group of closely related syndromes that result from genetically determined deficiencies of lysosomal enzymes involved in the degradation of mucopolysaccharides (glycosaminoglycans). Chemically, mucopolysaccharides are long-chain complex carbohydrates that are linked with proteins to form proteoglycans. They are abundant in the ground substance of connective tissue. The glycosaminoglycans that accumulate in MPSs are dermatan sulfate, heparan sulfate, keratan sulfate, and chondroitin sulfate. The enzymes involved in the degradation of these molecules cleave terminal sugars from the polysaccharide chains disposed along a polypeptide or core protein. In the absence of enzymes, these chains accumulate within lysosomes in various tissues and organs of the body.
Several clinical variants of MPS, classified numerically from MPS I to MPS VII, have been described, each resulting from the deficiency of one specific enzyme. All the MPSs except one are inherited as autosomal recessive traits; the exception, called Hunter syndrome, is an X-linked recessive trait. Within a given group (e.g., MPS I, characterized by a deficiency of α-l-iduronidase), subgroups exist that result from different mutant alleles at the same genetic locus. Thus, the severity of enzyme deficiency and the clinical picture even within subgroups are often different.
In general, MPSs are progressive disorders, characterized by coarse facial features, clouding of the cornea, joint stiffness, and mental retardation. Urinary excretion of the accumulated mucopolysaccharides is often increased.
Morphology. The accumulated mucopolysaccharides are generally found in mononuclear phagocytic cells, endothelial cells, intimal smooth muscle cells, and fibroblasts throughout the body. Common sites of involvement are thus the spleen, liver, bone marrow, lymph nodes, blood vessels, and heart.
Microscopically, affected cells are distended and have apparent clearing of the cytoplasm to create so-called balloon cells. Under the electron microscope, the clear cytoplasm can be resolved as numerous minute vacuoles. These are swollen lysosomes containing a finely granular periodic acid–Schiff–positive material that can be identified biochemically as mucopolysaccharide. Similar lysosomal changes are found in the neurons of those syndromes characterized by central nervous system involvement. In addition, however, some of the lysosomes in neurons are replaced by lamellated zebra bodies similar to those seen in Niemann-Pick disease. Hepatosplenomegaly, skeletal deformities, valvular lesions, and subendothelial arterial deposits, particularly in the coronary arteries, and lesions in the brain, are common threads that run through all of the MPSs. In many of the more protracted syndromes, coronary subendothelial lesions lead to myocardial ischemia. Thus, myocardial infarction and cardiac decompensation are important causes of death.
Clinical Features
Of the seven recognized variants, only two well-characterized syndromes are described briefly here. Hurler syndrome, also called MPS I-H, results from a deficiency of α-l-iduronidase.28 It is one of the most severe forms of MPS. Affected children appear normal at birth but develop hepatosplenomegaly by age 6 to 24 months. Their growth is retarded, and, as in other forms of MPS, they develop coarse facial features and skeletal deformities. Death occurs by age 6 to 10 years and is often due to cardiovascular complications. Hunter syndrome, also called MPS II, differs from Hurler syndrome in mode of inheritance (X-linked), absence of corneal clouding, and milder clinical course.29
Glycogen Storage Diseases (Glycogenoses)
The glycogen storage diseases result from a hereditary deficiency of one of the enzymes involved in the synthesis or sequential degradation of glycogen. Depending on the tissue or organ distribution of the specific enzyme in the normal state, glycogen storage in these disorders may be limited to a few tissues, may be more widespread while not affecting all tissues, or may be systemic in distribution.30
The significance of a specific enzyme deficiency is best understood from the perspective of the normal metabolism of glycogen (Fig. 5-15). Glycogen is a storage form of glucose. Glycogen synthesis begins with the conversion of glucose to glucose-6-phosphate by the action of a hexokinase (glucokinase). A phosphoglucomutase then transforms the glucose-6-phosphate to glucose-1-phosphate, which, in turn, is converted to uridine diphosphoglucose. A highly branched, large polymer is then built (molecular weight as high as 100 million), containing as many as 10,000 glucose molecules linked together by α-1,4-glucoside bonds. The glycogen chain and branches continue to be elongated by the addition of glucose molecules mediated by glycogen synthetases. During degradation, distinct phosphorylases in the liver and muscle split glucose-1-phosphate from the glycogen until about four glucose residues remain on each branch, leaving a branched oligosaccharide called limit dextrin. This can be further degraded only by the debranching enzyme. In addition to these major pathways, glycogen is also degraded in the lysosomes by acid maltase. If the lysosomes are deficient in this enzyme, the glycogen contained within them is not accessible to degradation by cytoplasmic enzymes such as phosphorylases.
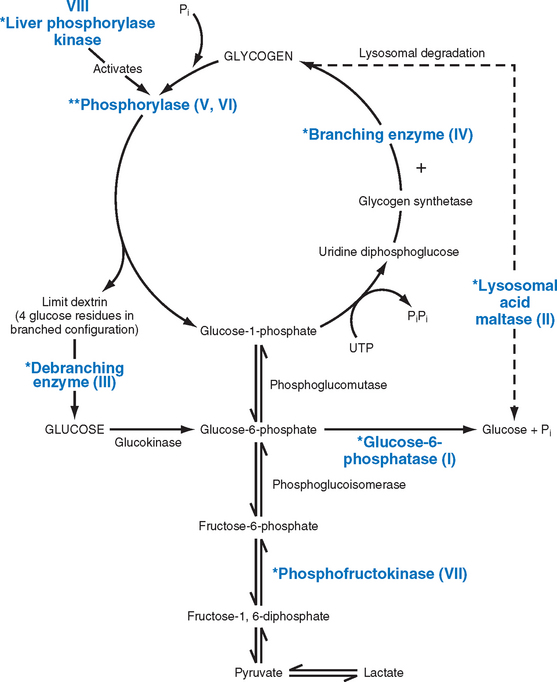
FIGURE 5-15 Pathways of glycogen metabolism. Asterisks mark the enzyme deficiencies associated with glycogen storage diseases. Roman numerals indicate the type of glycogen storage disease associated with the given enzyme deficiency. Types V and VI result from deficiencies of muscle and liver phosphorylases, respectively.
(Modified from Hers H et al.: Glycogen storage diseases. In Scriver CR et al. [eds]: The Metabolic Basis of Inherited Disease, 6th ed. New York, McGraw-Hill, 1989, p 425.)
On the basis of specific enzyme deficiencies and the resultant clinical pictures, glycogenoses have traditionally been divided into a dozen or so syndromes designated by roman numerals, and the list continues to grow.31 On the basis of pathophysiology glycogenoses can be divided into three major subgroups (Table 5-7).
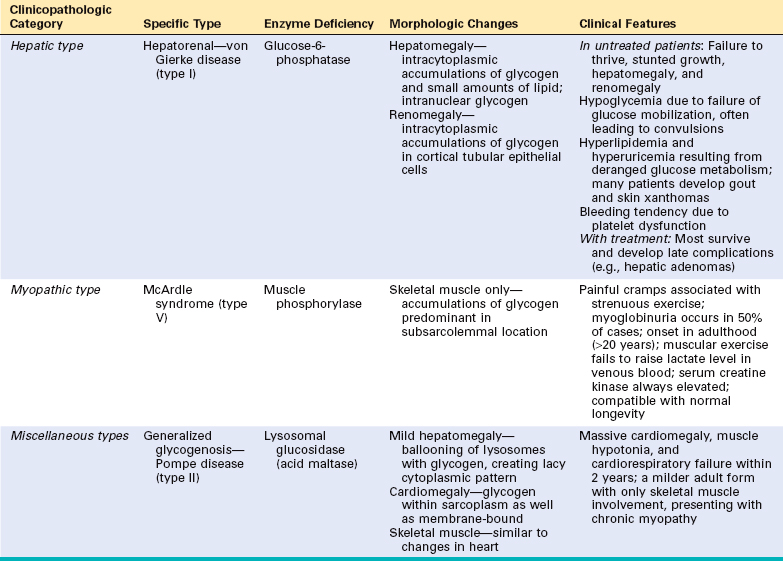
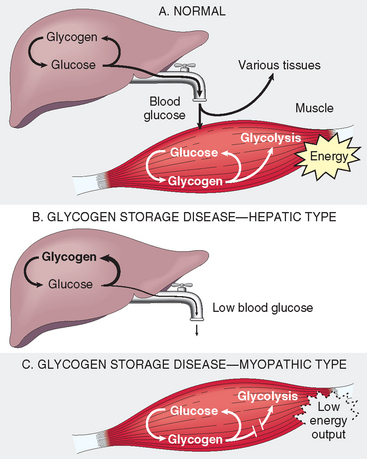
FIGURE 5-16 A, Normal glycogen metabolism in the liver and skeletal muscles. B, Effects of an inherited deficiency of hepatic enzymes involved in glycogen metabolism. C, Consequences of a genetic deficiency in the enzymes that metabolize glycogen in skeletal muscles.
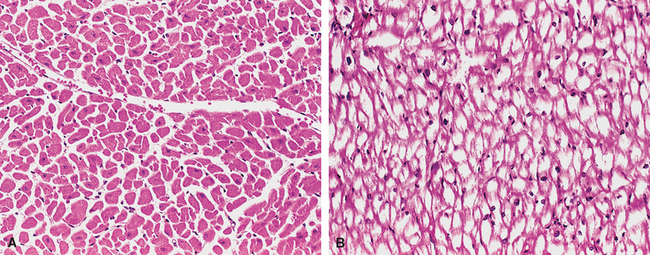
FIGURE 5-17 Pompe disease (glycogen storage disease type II). A, Normal myocardium with abundant eosinophilic cytoplasm. B, Patient with Pompe disease (same magnification) showing the myocardial fibers full of glycogen seen as clear spaces.
(Courtesy of Dr. Trace Worrell, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX.)
Alkaptonuria (Ochronosis)
Alkaptonuria, the first human inborn error of metabolism to be discovered, is an autosomal recessive disorder in which there is lack of homogentisic oxidase, an enzyme that converts homogentisic acid to methylacetoacetic acid in the tyrosine degradation pathway.35 Thus, homogentisic acid accumulates in the body. A large amount is excreted, imparting a black color to the urine if allowed to stand and undergo oxidation.
Morphology. The retained homogentisic acid binds to collagen in connective tissues, tendons, and cartilage, imparting to these tissues a blue-black pigmentation (ochronosis) most evident in the ears, nose, and cheeks. The most serious consequences of ochronosis, however, stem from deposits of the pigment in the articular cartilages of the joints. The pigment accumulation causes the cartilage to lose its normal resiliency and become brittle and fibrillated. Wear-and-tear erosion of this abnormal cartilage leads to denudation of the subchondral bone, and often tiny fragments of the fibrillated cartilage are driven into the underlying bone, worsening the damage. The vertebral column, particularly the intervertebral disc, is the prime site of attack, but later the knees, shoulders, and hips may be affected. The small joints of the hands and feet are usually spared.
Clinical Features
The metabolic defect is present from birth, but the degenerative arthropathy develops slowly and usually does not become clinically evident until the 30s. Although it is not life-threatening, it may be severely crippling. The arthropathy may be as extreme as that encountered in the severe forms of osteoarthritis (Chapter 26) of the elderly, but it occurs at a much earlier age.
DISORDERS ASSOCIATED WITH DEFECTS IN PROTEINS THAT REGULATE CELL GROWTH
Normal growth and differentiation of cells are regulated by two classes of genes; proto-oncogenes and tumor suppressor genes, whose products promote or restrain cell growth (Chapter 7). It is now well established that mutations in these two classes of genes are important in the pathogenesis of tumors. In the vast majority of cases, cancer-causing mutations affect somatic cells and hence are not passed in the germ line. In approximately 5% of all cancers, however, mutations transmitted through the germ line contribute to the development of cancer. Most familial cancers are inherited in an autosomal dominant fashion, but a few recessive disorders have also been described. This subject is discussed in Chapter 7. Specific forms of familial tumors are described in various chapters.
Complex Multigenic Disorders
As discussed previously, such disorders are caused by interactions between variant forms of genes and environmental factors. A genetic variant that has at least two alleles and occurs in at least 1% of the population is called a polymorphism. According to the common disease/common variant hypothesis, complex genetic disorders occur when many polymorphisms, each with a modest effect and low penetrance, are inherited.36 Two additional facts that have emerged from studies of common complex disorders, such as type I diabetes, are:
Several normal phenotypic characteristics are governed by multifactorial inheritance, such as hair color, eye color, skin color, height, and intelligence. These characteristics show a continuous variation in population groups, producing the standard bell-shaped curve of distribution. Environmental influences, however, significantly modify the phenotypic expression of complex traits. For example, type II diabetes mellitus has many of the features of a multifactorial disorder. It is well recognized clinically that individuals often first manifest this disease after weight gain. Thus, obesity as well as other environmental influences unmasks the diabetic genetic trait. Nutritional influences may cause even monozygous twins to achieve different heights. The culturally deprived child cannot achieve his or her full intellectual capacity.
Assigning a disease to this mode of inheritance must be done with caution. It depends on many factors but first on familial clustering and the exclusion of mendelian and chromosomal modes of transmission. A range of levels of severity of a disease is suggestive of a complex multigenic disorder, but, as pointed out earlier, variable expressivity and reduced penetrance of single mutant genes may also account for this phenomenon. Because of these problems, sometimes it is difficult to distinguish between mendelian and multifactorial disease.
Chromosomal Disorders
NORMAL KARYOTYPE
As is well known, human somatic cells contain 46 chromosomes; these comprise 22 homologous pairs of autosomes and two sex chromosomes, XX in the female and XY in the male. The study of chromosomes—karyotyping—is the basic tool of the cytogeneticist. The usual procedure to examine chromosomes is to arrest dividing cells in metaphase with mitotic spindle inhibitors (e.g., N-deacetyl-N-methylcolchicine [Colcemid]) and then to stain the chromosomes. In a metaphase spread, the individual chromosomes take the form of two chromatids connected at the centromere. A karyotype is obtained by arranging each pair of autosomes according to length, followed by sex chromosomes.
A variety of staining methods have been developed that allow identification of individual chromosomes on the basis of a distinctive and reliable pattern of alternating light and dark bands. The one most commonly used involves a Giemsa stain and is hence called G banding. A normal male karyotype with G banding is illustrated in Figure 5-18. With standard G banding, approximately 400 to 800 bands per haploid set can be detected. The resolution obtained by banding can be markedly improved by obtaining the cells in prophase. The individual chromosomes appear markedly elongated, and as many as 1500 bands per karyotype can be recognized. The use of these banding techniques permits certain identification of each chromosome and roughly delineates breakpoints and other gross alterations, to be described later.
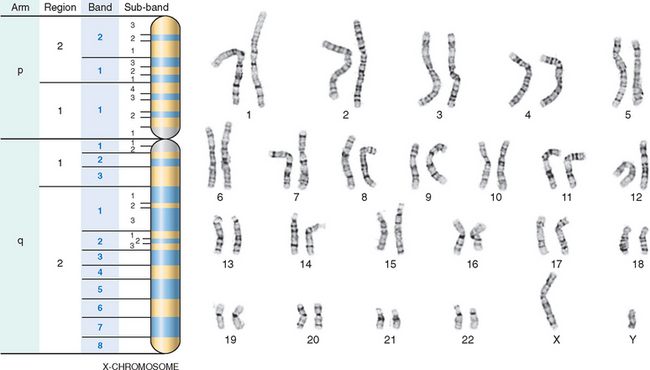
FIGURE 5-18 G-banded karyotype from a normal male (46,XY). Also shown is the banding pattern of the X-chromosome with nomenclature of arms, regions, bands, and sub-bands.
(Courtesy of Dr. Stuart Schwartz, Department of Pathology, University of Chicago, Chicago, IL.)
Before this discussion of the normal karyotype is concluded, reference must be made to commonly used cytogenetic terminology. Karyotypes are usually described using a shorthand system of notations. The following order is used: Total number of chromosomes is given first, followed by the sex chromosome complement, and finally the description of abnormalities in ascending numerical order. For example, a male with trisomy 21 is designated 47,XY,+21. Some of the notations denoting structural alterations of chromosomes are described along with the abnormalities in a later section. Here we should mention that the short arm of a chromosome is designated p (for petit), and the long arm is referred to as q (the next letter of the alphabet). In a banded karyotype, each arm of the chromosome is divided into two or more regions bordered by prominent bands. The regions are numbered (e.g., 1, 2, 3) from the centromere outward. Each region is further subdivided into bands and sub-bands, and these are ordered numerically as well (see Fig. 5-18). Thus, the notation Xp21.2 refers to a chromosomal segment located on the short arm of the X chromosome, in region 2, band 1, and sub-band 2.
STRUCTURAL ABNORMALITIES OF CHROMOSOMES
The aberrations underlying cytogenetic disorders may take the form of an abnormal number of chromosomes or alterations in the structure of one or more chromosomes. The normal chromosome complement is expressed as 46,XX for the female and 46,XY for the male. Any exact multiple of the haploid number is called euploid. If an error occurs in meiosis or mitosis and a cell acquires a chromosome complement that is not an exact multiple of 23, it is referred to as aneuploidy. The usual causes for aneuploidy are nondisjunction and anaphase lag. When nondisjunction occurs during gametogenesis, the gametes formed have either an extra chromosome (n + 1) or one less chromosome (n − 1). Fertilization of such gametes by normal gametes results in two types of zygotes—trisomic (2n + 1) or monosomic (2n − 1). In anaphase lag, one homologous chromosome in meiosis or one chromatid in mitosis lags behind and is left out of the cell nucleus. The result is one normal cell and one cell with monosomy. As seen subsequently, monosomy or trisomy involving the sex chromosomes, or even more bizarre aberrations, are compatible with life and are usually associated with variable degrees of phenotypic abnormalities. Monosomy involving an autosome generally causes loss of too much genetic information to permit live birth or even embryogenesis, but several autosomal trisomies do permit survival. With the exception of trisomy 21, all yield severely handicapped infants who almost invariably die at an early age.
Occasionally, mitotic errors in early development give rise to two or more populations of cells with different chromosomal complement, in the same individual, a condition referred to as mosaicism. Mosaicism can result from mitotic errors during the cleavage of the fertilized ovum or in somatic cells. Mosaicism affecting the sex chromosomes is relatively common. In the division of the fertilized ovum, an error may lead to one of the daughter cells receiving three sex chromosomes, whereas the other receives only one, yielding, for example, a 45,X/47,XXX mosaic. All descendent cells derived from each of these precursors thus have either a 47,XXX complement or a 45,X complement. Such a patient is a mosaic variant of Turner syndrome, with the extent of phenotypic expression dependent on the number and distribution of the 45,X cells.
Autosomal mosaicism seems to be much less common than that involving the sex chromosomes. An error in an early mitotic division affecting the autosomes usually leads to a nonviable mosaic due to autosomal monosomy. Rarely, the nonviable cell population is lost during embryogenesis, yielding a viable mosaic (e.g., 46,XY/47,XY,+21). Such a patient is a trisomy 21 mosaic with variable expression of Down syndrome, depending on the proportion of cells containing the trisomy.
A second category of chromosomal aberrations is associated with changes in the structure of chromosomes. To be visible by routine banding techniques, a fairly large amount of DNA (approximately 2–4 million base pairs), containing many genes, must be involved. The resolution is much higher with fluorescence in situ hybridization (FISH), which can detect changes as small as kilobases. Structural changes in chromosomes usually result from chromosome breakage followed by loss or rearrangement of material. Such alterations occur spontaneously at a low rate that is increased by exposure to environmental mutagens, such as chemicals and ionizing radiation. In the next section we briefly review the more common forms of alterations in chromosome structure and the notations used to signify them.
Deletion refers to loss of a portion of a chromosome (Fig. 5-19). Most deletions are interstitial, but rarely terminal deletions may occur. Interstitial deletions occur when there are two breaks within a chromosome arm, followed by loss of the chromosomal material between the breaks and fusion of the broken ends. One can specify in which region(s) and at what bands the breaks have occurred. For example, 46,XY, del(16) (p11.2p13.1) describes breakpoints in the short arm of chromosome 16 at 16p11.2 and 16p13.1 with loss of material between breaks. Terminal deletions result from a single break in a chromosome arm, producing a fragment with no centromere, which is then lost at the next cell division, and a chromosome bearing a deletion. The end of the chromosome is protected by acquiring telomeric sequences.
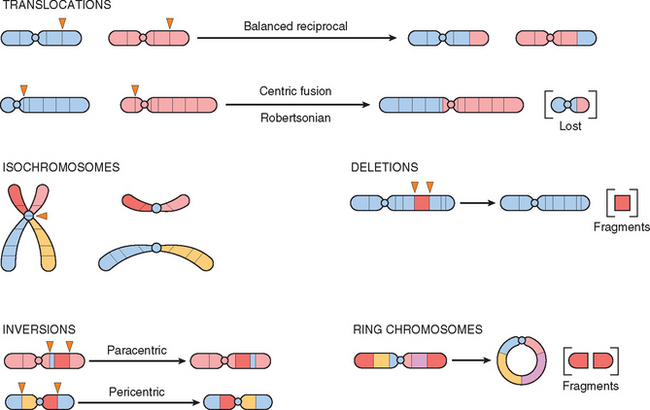
A ring chromosome is a special form of deletion. It is produced when a break occurs at both ends of a chromosome with fusion of the damaged ends (see Fig. 5-19). If significant genetic material is lost, phenotypic abnormalities result. This might be expressed as 46,XY,r(14). Ring chromosomes do not behave normally in meiosis or mitosis and usually result in serious consequences.
Inversion refers to a rearrangement that involves two breaks within a single chromosome with reincorporation of the inverted, intervening segment (see Fig. 5-19). An inversion involving only one arm of the chromosome is known as paracentric. If the breaks are on opposite sides of the centromere, it is known as pericentric. Inversions are often fully compatible with normal development.
Isochromosome formation results when one arm of a chromosome is lost and the remaining arm is duplicated, resulting in a chromosome consisting of two short arms only or of two long arms (see Fig. 5-19). An isochromosome has morphologically identical genetic information in both arms. The most common isochromosome present in live births involves the long arm of the X and is designated i(X)(q10). The Xq isochromosome is associated with monosomy for genes on the short arm of X and with trisomy for genes on the long arm of X.
In a translocation, a segment of one chromosome is transferred to another (see Fig. 5-19). In one form, called balanced reciprocal translocation, there are single breaks in each of two chromosomes, with exchange of material. A balanced reciprocal translocation between the long arm of chromosome 2 and the short arm of chromosome 5 would be written 46,XX,t(2;5)(q31;p14). This individual has 46 chromosomes with altered morphology of one of the chromosomes 2 and one of the chromosomes 5. Because there has been no loss of genetic material, the individual is likely to be phenotypically normal. A balanced translocation carrier, however, is at increased risk for producing abnormal gametes. For example, in the case cited above, a gamete containing one normal chromosome 2 and a translocated chromosome 5 may be formed. Such a gamete would be unbalanced because it would not contain the normal complement of genetic material. Subsequent fertilization by a normal gamete would lead to the formation of an abnormal (unbalanced) zygote, resulting in spontaneous abortion or birth of a malformed child. The other important pattern of translocation is called a robertsonian translocation (or centric fusion), a translocation between two acrocentric chromosomes. Typically the breaks occur close to the centromeres of each chromosome. Transfer of the segments then leads to one very large chromosome and one extremely small one. Usually the small product is lost (see Fig. 5-19); however, because it carries only highly redundant genes (e.g., ribosomal RNA genes), this loss is compatible with a normal phenotype. Robertsonian translocation between two chromosomes is encountered in 1 in 1000 apparently normal individuals. The significance of this form of translocation also lies in the production of abnormal progeny, as discussed later with Down syndrome.
Many more numerical and structural chromosomal aberrations are described in specialized texts, and more and more abnormal karyotypes are being identified in disease. As pointed out earlier, the clinically detected chromosome disorders represent only the “tip of the iceberg.” It is estimated that approximately 7.5% of all conceptions have a chromosomal abnormality, most of which are not compatible with survival or live birth. Even in live-born infants the frequency is approximately 0.5% to 1.0%. It is beyond the scope of this book to discuss most of the clinically recognizable chromosomal disorders. Hence, we focus attention on those few that are most common.
CYTOGENETIC DISORDERS INVOLVING AUTOSOMES
Trisomy 21 (Down Syndrome)
Down syndrome is the most common of the chromosomal disorders and is a major cause of mental retardation. In the United States the incidence in newborns is about 1 in 700. Approximately 95% of affected individuals have trisomy 21, so their chromosome count is 47. FISH with chromosome 21–specific probes reveals the extra copy of chromosome 21 in such cases (Fig. 5-20). Most others have normal chromosome numbers, but the extra chromosomal material is present as a translocation. As mentioned earlier, the most common cause of trisomy and therefore of Down syndrome is meiotic nondisjunction. The parents of such children have a normal karyotype and are normal in all respects.
FIGURE 5-20 FISH analysis of an interphase nucleus, using locus-specific probes to chromosome 13 (green) and chromosome 21 (red), revealing three red signals consistent with trisomy 21.
(Courtesy of Dr. Stuart Schwartz, Department of Pathology, University of Chicago, Chicago, IL.)
Maternal age has a strong influence on the incidence of trisomy 21. It occurs once in 1550 live births in women under age 20, in contrast to 1 in 25 live births for mothers over age 45. The correlation with maternal age suggests that in most cases the meiotic nondisjunction of chromosome 21 occurs in the ovum. Studies in which DNA polymorphisms were used to trace the parental origin of chromosome 21 have revealed that in 95% of the cases with trisomy 21 the extra chromosome is of maternal origin. Although many hypotheses have been advanced, the reason for the increased susceptibility of the ovum to nondisjunction remains unknown.
In about 4% of cases of Down syndrome, the extra chromosomal material derives from the presence of a robertsonian translocation of the long arm of chromosome 21 to another acrocentric chromosome (e.g., 22 or 14). Because the fertilized ovum already possesses two normal autosomes 21, the translocated material provides the same triple gene dosage as in trisomy 21. Such cases are frequently (but not always) familial, and the translocated chromosome is inherited from one of the parents (usually the mother), who is a carrier of a robertsonian translocation, for example, a mother with karyotype 45,XX,der(14;21)(q10;q10).
Approximately 1% of Down syndrome patients are mosaics, usually having a mixture of cells with 46 and 47 chromosomes. This mosaicism results from mitotic nondisjunction of chromosome 21 during an early stage of embryogenesis. Symptoms in such cases are variable and milder, depending on the proportion of abnormal cells. Clearly, in cases of translocation or mosaic Down syndrome, maternal age is of no importance.
The diagnostic clinical features of this condition—flat facial profile, oblique palpebral fissures, and epicanthic folds (Fig. 5-21)—are usually readily evident, even at birth.37 Down syndrome is a leading cause of severe mental retardation; approximately 80% of those afflicted have an IQ of 25 to 50. Ironically, these severely disadvantaged children may have a gentle, shy manner and may be more easily directed than their more fortunate normal siblings. It should be pointed out that some mosaics with Down syndrome have mild phenotypic changes and often even have normal or near-normal intelligence. In addition to the phenotypic abnormalities and the mental retardation already noted, some other clinical features are worthy of note.

Despite all these problems, improved medical care has increased the longevity of individuals with trisomy 21. Currently the median age at death is 47 years (up from 25 years in 1983).
Although the karyotype and clinical features of trisomy 21 have been known for decades, little is known about the molecular basis of Down syndrome. Chromosome 21 contains approximately 430 genes. Interestingly, there are several gene clusters, each of which is predicted to participate in the same biologic pathway. For example, there are 16 genes that are involved in the mitochondrial energy pathway, several that are likely to influence central nervous system development, and a group that is involved in folate metabolism. It is not known how each of these groups of genes is related to Down syndrome. The gene dosage hypothesis assumes that the phenotypic features of the trisomy 21 are related to overexpression of genes. In reality only about 37% of the genes on chromosomes 21 are overexpressed by 150%, whereas others have variable degrees of changes in expression. Further complexity in defining the specific genes involved in the pathogenesis of Down syndrome is related to the presence of several miRNA genes on chromosome 21 that can shut down translation of genes that map elsewhere in the genome.39 Thus, despite the availability of the gene map of chromosome 21, the progress in understanding the molecular basis of Down syndrome remains slow.40
Other Trisomies
A variety of other trisomies, involving chromosomes 8, 9, 13, 18, and 22, have been described. Only trisomy 18 (Edwards syndrome) and trisomy 13 (Patau syndrome) are common enough to merit brief mention here. As noted in Figure 5-21, they share several karyotypic and clinical features with trisomy 21. Thus, most cases result from meiotic nondisjunction and therefore carry a complete extra copy of chromosome 18 or 13. As in Down syndrome, an association with increased maternal age is also noted. In contrast to trisomy 21, however, the malformations are much more severe and wide-ranging. As a result, only rarely do these infants survive beyond the first year of life. Most succumb within a few weeks to months.
Chromosome 22q11.2 Deletion Syndrome
Chromosome 22q11.2 deletion syndrome encompasses a spectrum of disorders that result from a small deletion of band q11.2 on the long arm of chromosome 22.41 The syndrome is fairly common, occurring in as many as 1 in 4000 births, but it is often missed because of variable clinical features. These include congenital heart defects, abnormalities of the palate, facial dysmorphism, developmental delay, and variable degrees of T-cell immunodeficiency and hypocalcemia. Previously, these clinical features were considered to represent two different disorders—DiGeorge syndrome and velocardiofacial syndrome. Patients with DiGeorge syndrome have thymic hypoplasia, with resultant T-cell immunodeficiency (Chapter 6), parathyroid hypoplasia giving rise to hypocalcemia, a variety of cardiac malformations affecting the outflow tract, and mild facial anomalies. The clinical features of the so-called velocardiofacial syndrome include facial dysmorphism (prominent nose, retrognathia), cleft palate, cardiovascular anomalies, and learning disabilities. Less frequently, these patients also have immunodeficiency. Until recently the overlapping clinical features of these two conditions (e.g., cardiac malformations, facial dysmorphology) were not appreciated; it was only after these two apparently unrelated syndromes were found to be associated with a similar cytogenetic abnormality that the clinical overlap came into focus. Recent studies indicate that in addition to the numerous structural malformations, individuals with the 22q11.2 deletion syndrome are at a particularly high risk for psychotic illnesses, such as schizophrenia and bipolar disorders.42 In fact, it is estimated that approximately 25% of adults with this syndrome develop schizophrenia. Conversely, deletions of the region can be found in 2% to 3% of individuals with childhood-onset schizophrenia. In addition, attention deficit hyperactivity disorder is seen in 30% to 35% of affected children.
The diagnosis of this condition may be suspected on clinical grounds but can be established only by detection of the deletion by FISH (Fig. 5-22). By this test, approximately 90% of those previously diagnosed as having DiGeorge syndrome and 80% of those with the velocardiofacial syndrome have a deletion of 22q11.2. Thirty percent of individuals with conotruncal cardiac defects but no other features of this syndrome also reveal deletions of the same chromosomal region.
FIGURE 5-22 FISH on both metaphase chromosomes and an interphase cell from a patient with Di George syndrome demonstrating the deletion of the TUPLE1 probe (official name HIRA) localized to chromosome 22q11.2. The TUPLE1 probe is in red, and the control probe, localized to 22q, is in green. The metaphase spread shows one chromosome 22 with both a green signal (control probe) and a red signal (from the TUPLE1 probe). The other chromosome 22 shows only hybridization with the control probe (green), but no red signal since there is a deletion on this chromosome. The interphase cell shows two areas of hybridization with the control probe (in green) but also only one area of hybridization with the TUPLE1 probe (in red), illustrating a deletion of chromosome 22q11.2.
(Courtesy of Dr. Stuart Schwartz, Department of Pathology, University of Chicago, Chicago, IL.)
The molecular basis of this syndrome is not fully understood. The deleted region is large (approximately 1.5 megabases) and includes many genes. The clinical heterogeneity, with predominant immunodeficiency in some cases (DiGeorge syndrome) and predominant dysmorphology and cardiac malformations in other cases, probably reflects the variable position and size of the deleted segment from this genetic region. Approximately 30 candidate genes have been mapped to the deleted region. Among these, TBX1, a T-box transcription factor is most closely associated with the phenotypic features of this syndrome.41 This gene is expressed in the pharyngeal mesenchyme and endodermal pouch from which facial structures, thymus, and parathyroid are derived. The targets of TBX1 include PAX9, a gene that controls the development of the palate, parathyroids, and thymus. Clearly there are other genes that contribute to the behavioral and psychiatric disorders that remain to be identified.
CYTOGENETIC DISORDERS INVOLVING SEX CHROMOSOMES
Genetic diseases associated with changes involving the sex chromosomes are far more common than those related to autosomal aberrations. Furthermore, imbalances (excess or loss) of sex chromosomes are much better tolerated than are similar imbalances of autosomes. In large part, this latitude relates to two factors that are peculiar to the sex chromosomes: (1) lyonization or inactivation of all but one X chromosome and (2) the modest amount of genetic material carried by the Y chromosome.43 We discuss these features briefly to aid our understanding of sex chromosomal disorders.
In 1961, Lyon44 outlined the idea of X-inactivation, now commonly known as the Lyon hypothesis. It states that (1) only one of the X chromosomes is genetically active, (2) the other X of either maternal or paternal origin undergoes heteropyknosis and is rendered inactive, (3) inactivation of either the maternal or paternal X occurs at random among all the cells of the blastocyst on or about day 16 of embryonic life, and (4) inactivation of the same X chromosome persists in all the cells derived from each precursor cell. Thus, the great preponderance of normal females are in reality mosaics and have two populations of cells, one with an inactivated maternal X and the other with an inactivated paternal X. Herein lies the explanation of why females have the same dosage of X-linked active genes as have males. The inactive X can be seen in the interphase nucleus as a darkly staining small mass in contact with the nuclear membrane known as the Barr body, or X chromatin. The molecular basis of X inactivation involves a unique gene called XIST, whose product is a noncoding RNA that is retained in the nucleus, where it “coats” the X chromosome that it is transcribed from and initiates a gene-silencing process by chromatin modification and DNA methylation. The XIST allele is switched off in the active X.45
Although it was initially thought that all the genes on the inactive X are “switched off,” more recent studies have revealed that many genes escape X inactivation. Molecular studies suggest that 21% of genes on Xp, and a smaller number (3%) on Xq escape X inactivation. At least some of the genes that are expressed from both X chromosomes are important for normal growth and development.46 This notion is supported by the fact that patients with monosomy of the X chromosome (Turner syndrome: 45,X) have severe somatic and gonadal abnormalities. If a single dose of X-linked genes were sufficient, no detrimental effect would be expected in such cases. Furthermore, although one X chromosome is inactivated in all cells during embryogenesis, it is selectively reactivated in oogonia before the first meiotic division. Thus, it seems that both X chromosomes are required for normal oogenesis.
With respect to the Y chromosome, it is well known that this chromosome is both necessary and sufficient for male development. Regardless of the number of X chromosomes, the presence of a single Y determines the male sex. The gene that dictates testicular development (SRY: sex-determining region Y gene) has been located on its distal short arm. For quite some time this was considered to be the only gene of significance on the Y chromosome. Recent studies of the Y chromosome, however, have yielded a rich harvest of gene families in the so-called “male-specific Y,” or MSY region.47 All of these are believed to be testes-specific genes involved in spermatogenesis. With this background, we review some features that are common to all sex chromosome disorders.
The most important disorders arising in aberrations of sex chromosomes are described briefly here.
Klinefelter Syndrome
Klinefelter syndrome is best defined as male hypogonadism that occurs when there are two or more X chromosomes and one or more Y chromosomes.48 It is one of the most frequent forms of genetic disease involving the sex chromosomes as well as one of the most common causes of hypogonadism in the male. The incidence of this condition is approximately 1 in 660 live male births.49 It can rarely be diagnosed before puberty, particularly because the testicular abnormality does not develop before early puberty. Most patients have a distinctive body habitus with an increase in length between the soles and the pubic bone, which creates the appearance of an elongated body. Also characteristic are eunuchoid body habitus with abnormally long legs; small atrophic testes often associated with a small penis; and lack of such secondary male characteristics as deep voice, beard, and male distribution of pubic hair. Gynecomastia may be present. The mean IQ is somewhat lower than normal, but mental retardation is uncommon. There is increased incidence of type 2 diabetes and the metabolic syndrome; and curiously, mitral valve prolapse is seen in about 50% of adults with Klinefelter syndrome. It should be evident that the clinical features of this condition are variable, the only consistent finding being hypogonadism. Plasma gonadotropin concentrations, particularly follicle-stimulating hormone, are consistently elevated, whereas testosterone levels are variably reduced. Mean plasma estradiol levels are elevated by an as yet unknown mechanism. The ratio of estrogens and testosterone determines the degree of feminization in individual cases.
Klinefelter syndrome is an important genetic cause of reduced spermatogenesis and male infertility.50 In some patients the testicular tubules are totally atrophied and replaced by pink, hyaline, collagenous ghosts. In others, apparently normal tubules are interspersed with atrophic tubules. In some patients all tubules are primitive and appear embryonic, consisting of cords of cells that never developed a lumen or progressed to mature spermatogenesis. Leydig cells appear prominent, as a result of the atrophy and crowding of tubules and elevation of gonadotropin concentrations.
Patients with Klinefelter syndrome have a higher risk for breast cancer (20 times more common than in normal males), extragonadal germ cell tumors, and autoimmune diseases such as systemic lupus erythematosus.
The classic pattern of Klinefelter syndrome is associated with a 47,XXY karyotype (90% of cases). This complement of chromosomes results from nondisjunction during the meiotic divisions in one of the parents. Maternal and paternal nondisjunction at the first meiotic division are roughly equally involved. There is no phenotypic difference between those who receive the extra X chromosome from their father and those who receive it from their mother. Maternal age is increased in the cases associated with errors in oogenesis. In addition to this classic karyotype, approximately 15% of patients with Klinefelter syndrome have been found to have a variety of mosaic patterns, most of them being 46,XY/47,XXY. Other patterns are 47,XXY/48,XXXY and variations on this theme. As is the case with normal females, all but one X undergoes inactivation in patients with Klinefelter syndrome. Why then, do the patients with this disorder have hypogonadism and associated features? The explanation of this lies in the pattern of X inactivation. The gene encoding the androgen receptor, through which testosterone mediates its effects, maps on the X chromosome. The androgen receptor gene contains highly polymorphic CAG (trinucleotide) repeats. The functional response to androgens is dictated, in part, by the number of CAG repeats. With shorter CAG repeats, the effect of androgens is more pronounced. In persons with Klinefelter syndrome the X chromosome, bearing androgen receptor with the shortest CAG repeat, is preferentially inactivated. Such nonrandom X inactivation leaves the allele with the longest CAG repeat active, thus accounting for hypogonadism.
Turner Syndrome
Turner syndrome results from complete or partial monosomy of the X chromosome and is characterized primarily by hypogonadism in phenotypic females.51 It is the most common sex chromosome abnormality in females, affecting about 1 in 2000 live-born females.
With routine cytogenetic methods, three types of karyotypic abnormalities are seen in individuals with Turner syndrome. Approximately 57% are missing an entire X chromosome, resulting in a 45,X karyotype. Of the remaining 43%, approximately one third (approximately 14%) have structural abnormalities of the X chromosomes, and two thirds (approximately 29%) are mosaics. The common feature of the structural abnormalities is to produce partial monosomy of the X chromosome. In order of frequency, the structural abnormalities of the X chromosome include (1) an isochromosome of the long arm, 46,X,i(X)(q10) resulting in the loss of the short arm; (2) deletion of portions of both long and short arms, resulting in the formation of a ring chromosome, 46,X,r(X); and (3) deletion of portions of the short or long arm, 46X,del(Xq) or 46X,del(Xp). The mosaic patients have a 45,X cell population along with one or more karyotypically normal or abnormal cell types. Examples of karyotypes that mosaic Turner females may have are the following: (1) 45,X/46,XX; (2) 45,X/46,XY; (3) 45,X/47,XXX; or (4) 45,X/46,X,i(X)(q10). Studies suggest that the prevalence of mosaicism in Turner syndrome may be much higher than the 30% detected by conventional cytogenetic studies. With the use of more sensitive techniques, including FISH (discussed later) and polymerase chain reaction (PCR), and analysis of more than one cell type (e.g., peripheral blood and fibroblasts), the prevalence of mosaic Turner syndrome increases to 75%. Because 99% of 45,X conceptuses are nonviable, many authorities believe that there are no truly nonmosaic Turner syndrome patients. While this issue remains controversial, it is important to appreciate the karyotypic heterogeneity associated with Turner syndrome, because it is responsible for significant variations in phenotype. In patients who are truly 45,X, or in whom the proportion of 45,X cells is high, the phenotypic changes are more severe than in those who have readily detectable mosaicism. The latter may have an almost normal appearance and may present only with primary amenorrhea. Similarly, those with a Y chromosome–containing population (e.g., 45,X/46,XY karyotype) may be at risk of developing a gonadal tumor (gonadoblastoma).
The most severely affected patients generally present during infancy with edema of the dorsum of the hand and foot due to lymph stasis, and sometimes swelling of the nape of the neck. The latter is related to markedly distended lymphatic channels, producing a so-called cystic hygroma (Chapter 10). As these infants develop, the swellings subside but often leave bilateral neck webbing and persistent looseness of skin on the back of the neck. Congenital heart disease is also common, affecting 25% to 50% of patients. Left-sided cardiovascular abnormalities, particularly preductal coarctation of the aorta and bicuspid aortic valve, are seen most frequently. Cardiovascular abnormalities are the most important cause of increased mortality in children with Turner syndrome.52
The principal clinical features in the adolescent and adult are illustrated in Figure 5-23. At puberty there is failure to develop normal secondary sex characteristics. The genitalia remain infantile, breast development is inadequate, and there is little pubic hair. The mental status of these patients is usually normal, but subtle defects in nonverbal, visual-spatial information processing have been noted. Of particular importance in establishing the diagnosis in the adult is the shortness of stature (rarely exceeding 150 cm in height) and amenorrhea. Turner syndrome is the single most important cause of primary amenorrhea, accounting for approximately one third of the cases. For reasons not quite clear, approximately 50% of patients develop autoantibodies that react with the thyroid gland, and up to half of these develop clinically manifest hypothyroidism. Equally mysterious is the presence of glucose intolerance, obesity, and insulin resistance in a minority of patients. The last mentioned is significant, because therapy with growth hormone, commonly used in these patients, worsens insulin resistance.
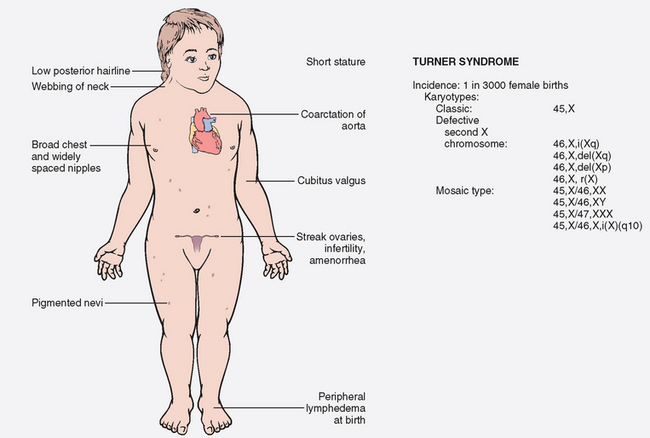
The molecular pathogenesis of Turner syndrome is not completely understood, but studies have begun to shed some light.53 As mentioned earlier, both X chromosomes are active during oogenesis and are essential for normal development of the ovaries. During normal fetal development, ovaries contain as many as 7 million oocytes. The oocytes gradually disappear so that by menarche their numbers have dwindled to a mere 400,000, and when menopause occurs fewer than 10,000 remain. In Turner syndrome, fetal ovaries develop normally early in embryogenesis, but the absence of the second X chromosome leads to an accelerated loss of oocytes, which is complete by age 2 years. In a sense, therefore, “menopause occurs before menarche,” and the ovaries are reduced to atrophic fibrous strands, devoid of ova and follicles (streak ovaries). Because patients with Turner syndrome also have other (nongonadal) abnormalities, it follows that some genes for normal growth and development of somatic tissues must also reside on the X chromosome. Among the genes involved in the Turner phenotype is the short stature homeobox (SHOX) gene at Xp22.33. This is one of several genes that remain active in both X chromosomes and has an active homologue on the short arm of the Y chromosome. Thus, both normal males and females have two copies of this gene. Haploinsufficiency of SHOX gives rise to short stature. Indeed, deletions of the SHOX gene are noted in 2% to 5% of otherwise normal children with short stature. In keeping with its role as a critical regulator of growth, the SHOX gene is expressed during fetal life in the growth plates of several long bones including the radius, ulna, tibia, and fibula. It is also expressed in the first and second pharyngeal arches. Just as the loss of SHOX is always associated with short stature, excess copies of this gene are associated with tall stature. Whereas haploinsufficiency of SHOX can explain growth deficit in Turner syndrome, it cannot explain other important clinical features such as cardiac malformations and endocrine abnormalities. Clearly several other genes located on the X chromosome are also involved.
Hermaphroditism and Pseudohermaphroditism
The problem of sexual ambiguity is exceedingly complex, and only limited observations are possible here; for more details, reference should be made to specialized sources.54 It will be no surprise to medical students that the sex of an individual can be defined on several levels. Genetic sex is determined by the presence or absence of a Y chromosome. No matter how many X chromosomes are present, a single Y chromosome dictates testicular development and the genetic male gender. The initially indifferent gonads of both the male and the female embryos have an inherent tendency to feminize, unless influenced by Y chromosome–dependent masculinizing factors. Gonadal sex is based on the histologic characteristics of the gonads. Ductal sex depends on the presence of derivatives of the müllerian or wolffian ducts. Phenotypic, or genital, sex is based on the appearance of the external genitalia. Sexual ambiguity is present whenever there is disagreement among these various criteria for determining sex.
The term true hermaphrodite implies the presence of both ovarian and testicular tissue. In contrast, a pseudohermaphrodite represents a disagreement between the phenotypic and gonadal sex (i.e., a female pseudohermaphrodite has ovaries but male external genitalia; a male pseudohermaphrodite has testicular tissue but female-type genitalia).
True hermaphroditism, implying the presence of both ovarian and testicular tissue, is an extremely rare condition. In some cases there is a testis on one side and an ovary on the other, whereas in other cases there may be combined ovarian and testicular tissue, referred to as ovotestes. The karyotype is 46,XX in 50% of patients; of the remaining, most are mosaics with a 46,XX/46,XY karyotype. Only rarely is the chromosomal constitution 46,XY. The presence of testes implies that those with the 46,XX karyotype might possess Y-chromosomal material, in particular, the SRY gene, which dictates testicular differentiation. Indeed, molecular analysis has revealed SRY gene expression in the ovotestis of 46,XX true hermaphrodites, indicating either cryptic chimerism localized to the gonads or possibly a Y-to-autosome translocation.55
Female pseudohermaphroditism is much less complex. The genetic sex in all cases is XX, and the development of the gonads (ovaries) and internal genitalia is normal. Only the external genitalia are ambiguous or virilized. The basis of female pseudohermaphroditism is excessive and inappropriate exposure to androgenic steroids during the early part of gestation. Such steroids are most commonly derived from the fetal adrenal affected by congenital adrenal hyperplasia, which is transmitted as an autosomal recessive trait. Biosynthetic defects in the pathway of cortisol synthesis are present in these patients, which lead secondarily to excessive synthesis of androgenic steroids by the fetal adrenal cortex (Chapter 24).
Male pseudohermaphroditism represents the most complex of all disorders of sexual differentiation. These individuals possess a Y chromosome, and thus their gonads are exclusively testes, but the genital ducts or the external genitalia are incompletely differentiated along the male phenotype. Their external genitalia are either ambiguous or completely female. Male pseudohermaphroditism is extremely heterogeneous, with a multiplicity of causes. Common to all is defective virilization of the male embryo, which usually results from genetically determined defects in androgen synthesis or action or both. The most common form, called complete androgen insensitivity syndrome (testicular feminization), results from mutations in the gene encoding the androgen receptor.56 This gene is located at Xq12, and hence this disorder is inherited as an X-linked recessive.
Single-Gene Disorders with Nonclassic Inheritance
It has become increasingly evident that transmission of certain single-gene disorders does not follow classic mendelian principles. This group of disorders can be classified into four categories:
Clinical and molecular features of some single-gene diseases that exemplify nonclassic patterns of inheritance are described next.
DISEASES CAUSED BY TRINUCLEOTIDE-REPEAT MUTATIONS
The discovery in 1991 of expanding trinucleotide repeats as a cause of fragile-X syndrome was a landmark in human genetics. Since then the origins of about 40 human diseases (Table 5-8) have been traced to unstable nucleotide repeats,57 and the number continues to grow. Some general principles that apply to these diseases are as follows:
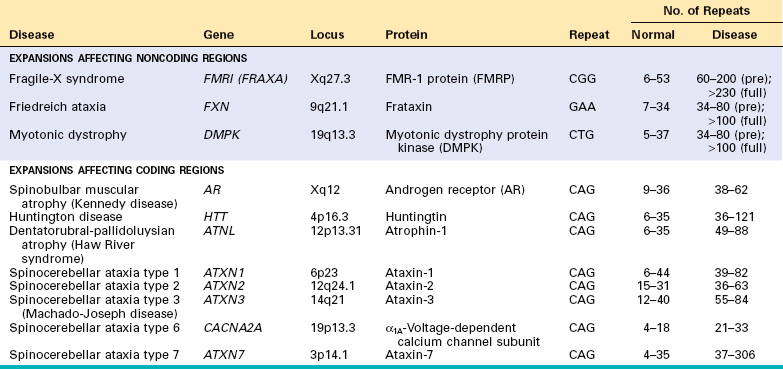
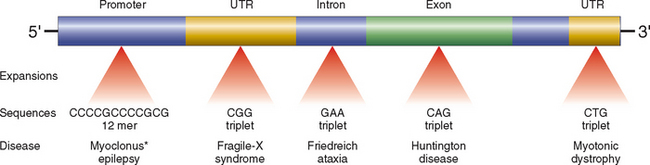
FIGURE 5-24 Sites of expansion and the affected sequence in selected diseases caused by nucleotide-repeat mutations. UTR, untranslated region.
*Though not strictly a trinucleotide-repeat disease, progressive myoclonus epilepsy is caused, like others in this group, by a heritable DNA expansion. The expanded segment is in the promoter region of the gene.
The pathogenetic mechanisms underlying disorders caused by mutations that affect coding regions seem to be distinct from those in which the expansions affect noncoding regions.58 The former usually involve CAG repeats coding for polyglutamine tracts in the corresponding proteins. Such “polyglutamine diseases” are characterized by progressive neurodegeneration, typically striking in midlife. Polyglutamine expansions lead to toxic gain of function, whereby the abnormal protein interferes with the function of the normal protein.59 The precise mechanisms by which expanded polyglutamine proteins cause disease is not fully understood. However, some general features have emerged. In most cases the proteins are misfolded and tend to aggregate; the aggregates may suppress transcription of other genes, cause mitochondrial dysfunction, or trigger the unfolded-protein stress response and apoptosis (Chapter 1). A morphologic hallmark of these diseases is the accumulation of aggregated mutant proteins in large intranuclear inclusions. By contrast, when expansions affect noncoding regions the resulting mutations are loss-of-function type, since protein synthesis (e.g., FMRP) is suppressed. Typically, such disorders affect many systems. Finally, many noncoding repeat disorders are characterized by intermediate-size expansions, or premutations, that expand to full mutations in germ cells.
Fragile-X Syndrome
Fragile-X syndrome is the prototype of diseases in which the mutation is characterized by a long repeating sequence of three nucleotides. Although the specific nucleotide sequence that undergoes amplification differs in the 20 or so disorders included in this group, in most cases the affected sequences share the nucleotides guanine (G) and cytosine (C). In the ensuing discussion we consider the clinical features and inheritance pattern of the fragile-X syndrome, to be followed by the causative molecular lesion. The remaining disorders in this group are discussed later, in this chapter and elsewhere in the book.
With a frequency of 1 in 1550 for affected males and 1 in 8000 for affected females, fragile-X syndrome is the second most common genetic cause of mental retardation, after Down syndrome. It is an X-linked disorder characterized by an inducible cytogenetic abnormality in the X chromosome and an unusual mutation within the familial mental retardation-1 (FMR1) gene. The cytogenetic alteration is seen as a discontinuity of staining or as a constriction in the long arm of the X chromosome when cells are cultured in a folate-deficient medium. Because it appears that the chromosome is “broken” at this locale, it is referred to as a fragile site (Fig. 5-25). It should be noted that more than 100 “fragile sites” have been found in the human genome.60 Many, like the one seen in fragile-X syndrome, are sensitive to lack of folate in the medium, whereas others require different culture conditions. The significance of most fragile sites is unknown, since many are present in normal individuals.
FIGURE 5-25 Fragile X, seen as discontinuity of staining.
(Courtesy of Dr. Patricia Howard-Peebles, University of Texas Southwestern Medical Center, Dallas, TX.)
In fragile-X syndrome, the affected males are mentally retarded, with an IQ in the range of 20 to 60. They express a characteristic physical phenotype that includes a long face with a large mandible, large everted ears, and large testicles (macro-orchidism). Hyperextensible joints, a high arched palate, and mitral valve prolapse noted in some patients mimic a connective tissue disorder. These and other physical abnormalities described in this condition, however, are not always present and, in some cases, are quite subtle. The most distinctive feature is macro-orchidism, which is observed in at least 90% of postpubertal males.
As with all X-linked diseases, fragile-X syndrome affects males. Analysis of several pedigrees, however, reveals some patterns of transmission not typically associated with other X-linked recessive disorders (Fig. 5-26). These include the following61:
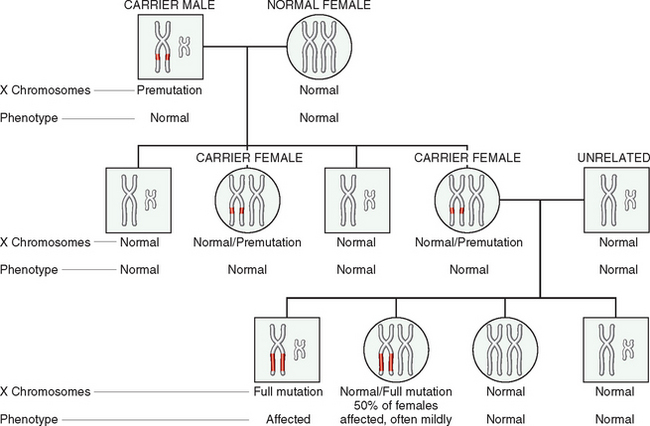
FIGURE 5-26 Fragile-X pedigree. Note that in the first generation all sons are normal and all females are carriers. During oogenesis in the carrier female, premutation expands to full mutation; hence, in the next generation all males who inherit the X with full mutation are affected. However, only 50% of females who inherit the full mutation are affected, and only mildly.
(Courtesy of Dr. Nancy Schneider, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX.)
These unusual patterns perplexed geneticists for years, but molecular studies have begun to unravel the complexities of this condition.62,63 The first breakthrough came when linkage studies localized the mutation responsible for this disease to Xq27.3, within the cytogenetically abnormal region. Within this region lies the FMR1 gene, characterized by multiple tandem repeats of the nucleotide sequence CGG in its 5′ untranslated region. In the normal population, the number of CGG repeats is small, ranging from 6 to 55 (average, 29). The presence of clinical symptoms and a cytogenetically detectable fragile site is related to the amplification of the CGG repeats. Thus, normal transmitting males and carrier females carry 55 to 200 CGG repeats. Expansions of this size are called premutations. In contrast, affected individuals have an extremely large expansion of the repeat region (200–4000 repeats, or full mutations). Full mutations are believed to arise by further amplification of the CGG repeats seen in premutations. How this process takes place is quite peculiar. Carrier males transmit the repeats to their progeny with small changes in repeat number. When the premutation is passed on by a carrier female, however, there is a high probability of a dramatic amplification of the CGG repeats, leading to mental retardation in most male offspring and 50% of female offspring. Thus, it seems that during the process of oogenesis, but not spermatogenesis, premutations can be converted to mutations by triplet-repeat amplification. This explains the unusual inheritance pattern; that is, the likelihood of mental retardation is much higher in grandsons than in brothers of transmitting males because grandsons incur the risk of inheriting a premutation from their grandfather that is amplified to a “full mutation” in their mothers’ ova. By comparison, brothers of transmitting males, being “higher up” in the pedigree, are less likely to have a full mutation. These molecular details also provide a satisfactory explanation of anticipation—a phenomenon observed by clinical geneticists but not believed by molecular geneticists until triplet-repeat mutations were identified. Why only 50% of the females with the full mutation are clinically affected is not clear. Presumably in those that are clinically affected there is unfavorable lyonization (i.e., there is a higher frequency of cells in which the X chromosome carrying the mutation is active). Recent studies indicate that premutations are not so benign after all. Approximately 30% of females carrying the premutation have premature ovarian failure (before the age of 40 years), and about one third of premutation-carrying males exhibit a progressive neurodegenerative syndrome starting in their sixth decade. This syndrome, referred to as fragile X–associated tremor/ataxia, is characterized by intention tremors and cerebellar ataxia and may progress to parkinsonism. However, it is clear that the abnormalities in premutation carriers are milder and occur later in life.
The molecular basis of mental retardation and other somatic changes is related to a loss of function of the familial mental retardation protein (FMRP). As mentioned earlier, the normal FMR1 gene contains up to 46 CGG repeats in its 5′ untranslated region. When the trinucleotide repeats in the FMR1 gene exceed approximately 230, the DNA of the entire 5′ region of the gene becomes abnormally methylated. Methylation also extends upstream into the promoter region of the gene, resulting in transcriptional suppression of FMR1. The resulting absence of FMRP is believed to cause the phenotypic changes.
FMRP is a widely expressed cytoplasmic protein, most abundant in the brain and testis, the two organs most affected in this disease. The function of FMRP in the brain is beginning to be unraveled.64 FMRP is an RNA-binding protein associated with polysomes. Unlike other cells, in neurons protein synthesis occurs both in the perinuclear cytoplasm and in dendritic spines. According to current understanding, FMRP is first transported from the cytoplasm to the nucleus, where it assembles into a complex containing specific mRNA transcripts. The assembled complex is then exported to the cytoplasm. From here the FMRP-mRNA complex is transported to the dendrites close to the synapse (Fig. 5-27). Not all species of mRNA are transported by FMRP to the dendrites. Only those that encode proteins that regulate synaptic function are so shuttled by FMRP. At the synaptic junctions, FMRP suppresses protein synthesis from the bound mRNAs in response to signaling through group I metabotropic glutamate receptors (mGlu-R). In fragile-X syndrome a reduction in FMRP results in increased translation of the bound mRNAs at the synaptic junctions. Such imbalance in turn causes permanent changes in synaptic activity and ultimately mental retardation.
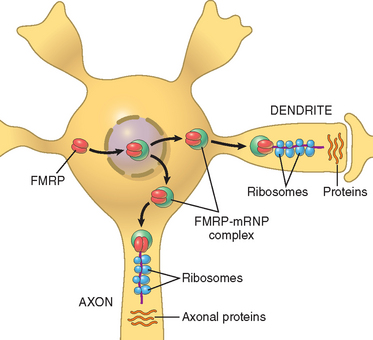
FIGURE 5-27 A model for the action of familial mental retardation protein (FMRP) in neurons.
(Adapted from Hin P, Warren ST: New insights into fragile X syndrome: from molecules to neurobehavior. Trends Biochem Sci 28:152, 2003.)
Although demonstration of an abnormal karyotype led to the identification of this disorder, PCR-based detection of the repeats is now the method of choice for diagnosis. With Southern blot analysis, distinction between premutations and mutations can be made prenatally as well as postnatally. Hence, this technique is valuable not only for establishing the diagnosis, but also for guiding genetic counseling. These techniques are described later.
MUTATIONS IN MITOCHONDRIAL GENES—LEBER HEREDITARY OPTIC NEUROPATHY
The vast majority of genes are located on chromosomes in the cell nucleus and are inherited in classical Mendelian fashion. There exist several mitochondrial genes, however, that are inherited in quite a different manner. A feature unique to mtDNA is maternal inheritance. This peculiarity exists because ova contain numerous mitochondria within their abundant cytoplasm, whereas spermatozoa contain few, if any. Hence, the mtDNA complement of the zygote is derived entirely from the ovum. Thus, mothers transmit mtDNA to all their offspring, male and female; however, daughters but not sons transmit the DNA further to their progeny (Fig. 5-28). Several other features apply to mitochondrial inheritance.65,66 They are as follows:
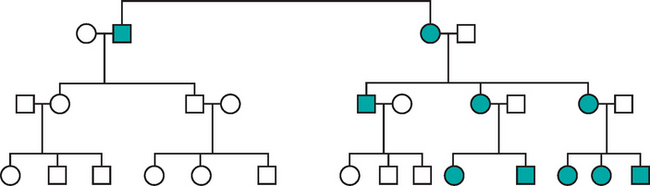
FIGURE 5-28 Pedigree of Leber hereditary optic neuropathy, a disorder caused by mutation in mitochondrial DNA. Note that all progeny of an affected male (shaded squares) are normal, but all children, male and female, of the affected female (shaded circles) manifest disease.
Diseases associated with mitochondrial inheritance are rare and, as mentioned earlier, many of them affect the neuromuscular system. Leber hereditary optic neuropathy is a prototype of this type of disorder. It is a neurodegenerative disease that manifests as a progressive bilateral loss of central vision. Visual impairment is first noted between ages 15 and 35, and it leads eventually to blindness. Cardiac conduction defects and minor neurologic manifestations have also been observed in some families.68
GENOMIC IMPRINTING
We all inherit two copies of each autosomal gene, carried on homologous maternal and paternal chromosomes. In the past, it had been assumed that there is no functional difference between the alleles derived from the mother or the father. Studies over the past two decades have provided definite evidence that, at least with respect to some genes, important functional differences exist between the paternal allele and the maternal allele. These differences result from an epigenetic process (discussed later), called imprinting. In most cases, imprinting selectively inactivates either the maternal or paternal allele. Thus, maternal imprinting refers to transcriptional silencing of the maternal allele, whereas paternal imprinting implies that the paternal allele is inactivated. Imprinting occurs in the ovum or the sperm, before fertilization, and then is stably transmitted to all somatic cells through mitosis.69 As with other instances of epigenetic regulation, imprinting is associated with differential patterns of DNA methylation at CG nucleotides. Other mechanisms include histone H4 deacetylation and methylation. Regardless of the mechanism, it is believed that such marking of paternal and maternal chromosomes occurs during gametogenesis, and thus it seems that from the moment of conception some chromosomes remember where they came from. The exact number of imprinted genes is not known; estimates range from 200 to 600. Although imprinted genes may occur in isolation, more commonly they are found in groups that are regulated by common cis-acting elements called imprinting control regions. As is often the case in medicine, genomic imprinting is best illustrated by considering two uncommon genetic disorders: Prader-Willi syndrome and Angelman syndrome.
Prader-Willi Syndrome and Angelman Syndrome
Prader-Willi syndrome is characterized by mental retardation, short stature, hypotonia, profound hyperphagia, obesity, small hands and feet, and hypogonadism.70 In 65% to 70% of cases, an interstitial deletion of band q12 in the long arm of chromosome 15, del(15)(q11.2q13), can be detected. In most cases the breakpoints are the same, causing a 5-Mb deletion. It is striking that in all cases the deletion affects the paternally derived chromosome 15. In contrast with the Prader-Willi syndrome, patients with the phenotypically distinct Angelman syndrome are born with a deletion of the same chromosomal region derived from their mothers. Patients with Angelman syndrome are also mentally retarded, but in addition they present with ataxic gait, seizures, and inappropriate laughter. Because of their laughter and ataxia they have been referred to as “happy puppets.”71 A comparison of these two syndromes clearly demonstrates the parent-of-origin effects on gene function.
The molecular basis of these two syndromes lies in the genomic imprinting (Fig. 5-29). It is known that a gene or set of genes on maternal chromosome 15q12 is imprinted (and hence silenced), and thus the only functional allele(s) are provided by the paternal chromosome. When these are lost as a result of a deletion, the person develops Prader-Willi syndrome. Conversely, a distinct gene that also maps to the same region of chromosome 15 is imprinted on the paternal chromosome. Only the maternally derived allele of this gene is normally active. Deletion of this maternal gene on chromosome 15 gives rise to the Angelman syndrome. Molecular studies of cytogenetically normal patients with the Prader-Willi syndrome (i.e., those without the deletion) have revealed that they have two maternal copies of chromosome 15. Inheritance of both chromosomes of a pair from one parent is called uniparental disomy. The net effect is the same (i.e., the person does not have a functional set of genes from the [nonimprinted] paternal chromosomes 15). Angelman syndrome, as might be expected, can also result from uniparental disomy of paternal chromosome 15.
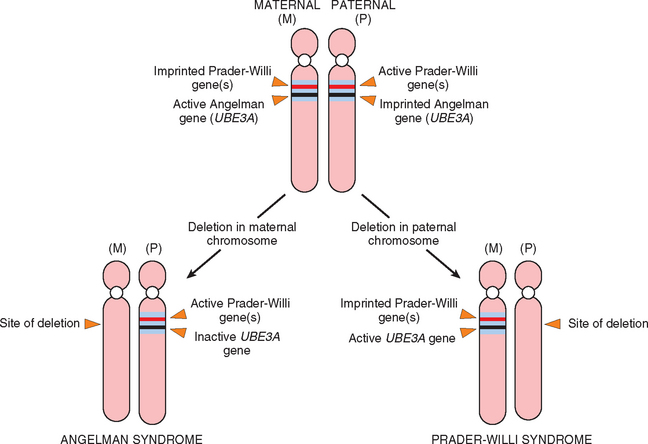
The genetic basis of these two imprinting disorders is now being unraveled. In the Angelman syndrome, the affected gene is a ubiquitin ligase that is involved in catalyzing the transfer of activated ubiquitin to target protein substrates. The gene, called UBE3A, maps within the 15q12 region, is imprinted on the paternal chromosome, and is expressed from the maternal allele primarily in specific regions of the brain.72 The imprinting is tissue-specific in that UBE3A is expressed from both alleles in most tissues. In approximately 10% of cases, Angelman syndrome occurs not as a result of imprinting but of a point mutation in the maternal allele, thus establishing a firm link between the UBE3A gene and Angelman syndrome. In contrast to Angelman syndrome, no single gene has been implicated in Prader-Willi syndrome. Instead, a series of genes located in the 15q11.2–q13 interval (which are imprinted on the maternal chromosome and expressed from the paternal chromosome) are believed to be involved. These include a gene that encodes small nuclear riboprotein N, which controls gene splicing and is expressed highly in the brain and heart. Loss of small nuclear riboprotein N function is believed to contribute to Prader-Willi syndrome. Molecular diagnosis (see later) of these syndromes is based on assessment of methylation status of marker genes and FISH.
The importance of imprinting is not restricted to rare chromosomal disorders. Parent-of-origin effects have been identified in a variety of inherited diseases, such as Huntington disease and myotonic dystrophy and in tumorigenesis.
GONADAL MOSAICISM
It was mentioned earlier that with every autosomal dominant disorder some patients do not have affected parents. In such patients the disorder results from a new mutation in the egg or the sperm from which they were derived; as such, their siblings are neither affected nor at increased risk of developing the disease. This is not always the case, however. In some autosomal dominant disorders, exemplified by osteogenesis imperfecta, phenotypically normal parents have more than one affected child. This clearly violates the laws of mendelian inheritance. Studies indicate that gonadal mosaicism may be responsible for such unusual pedigrees.73 Gonadal mosaicism results from a mutation that occurs postzygotically during early (embryonic) development. If the mutation affects only cells destined to form the gonads, the gametes carry the mutation, but the somatic cells of the individual are completely normal. Such an individual is said to exhibit germ line or gonadal mosaicism. A phenotypically normal parent who has germ line mosaicism can transmit the disease-causing mutation to the offspring through the mutant gamete. Because the progenitor cells of the gametes carry the mutation, there is a definite possibility that more than one child of such a parent would be affected. Obviously the likelihood of such an occurrence depends on the proportion of germ cells carrying the mutation.
Molecular Diagnosis of Genetic Diseases
Medical applications of recombinant DNA technology have come of age. With the completion of the Human Genome Project, DNA-based analysis has become a powerful tool for the diagnosis of human disease, both genetic and acquired. Molecular diagnostic techniques have found application in virtually all areas of medicine. In the era predating the ready availability of molecular diagnostic assays, assays for single-gene (“mendelian”) disorders depended on the identification of abnormal gene products (e.g., mutant hemoglobin or abnormal metabolites) or their clinical effects, such as mental retardation (e.g., in phenylketonuria). Now, it is possible to identify mutations at the DNA level and offer diagnostic tests for an increasing number of genetic disorders. In addition, molecular tools have become extremely important in discovery of the genetic basis of common complex disorders such as diabetes mellitus, atherosclerosis, and cancer. The molecular diagnosis of inherited diseases at the nucleic acid level has distinct advantages over other surrogate techniques:
INDICATIONS FOR ANALYSIS OF GERM LINE GENETIC ALTERATIONS
While many techniques are available today for the diagnosis of genetic diseases, to judiciously use these methods it is important to ascertain which individuals require genetic testing. In general, testing for alterations inherited in the germ line can be divided into prenatal and postnatal analysis. It may involve conventional cytogenetics, fluorescent in situ hybridization (FISH), other molecular diagnostic assays, or a combination of these techniques.
Prenatal genetic analysis should be offered to all patients who are at risk of having cytogenetically abnormal progeny. It can be performed on cells obtained by amniocentesis, on chorionic villus biopsy material, or on umbilical cord blood. Some important indications are as follows74:
Postnatal genetic analysis is usually performed on peripheral blood lymphocytes. Indications are as follows:
INDICATIONS FOR ANALYSIS OF ACQUIRED GENETIC ALTERATIONS
In this era of molecularly targeted therapies it is becoming increasingly important to identify specific molecular genetic signatures for acquired diseases (i.e., cancer and infectious disease), which were formerly diagnosed and managed with nonmolecular clinicopathologic data. The technical approaches are the same as those used for germ line mendelian disorders, and the common indications include:
Diagnosis and management of cancer (see also Chapter 7)
Diagnosis and management of infectious disease (see also Chapter 8)
PCR AND DETECTION OF DNA SEQUENCE ALTERATIONS
PCR analysis, which involves exponential amplification of DNA, has revolutionized molecular biology and is now widely used in the molecular diagnosis of human disease. By using appropriate DNA polymerases and thermal cycling, the target DNA is greatly amplified, producing millions of copies of the DNA sequence between the two primer sites. The subsequent identification of an abnormal sequence can then be performed using an ever-increasing number of assays. Direct sequence analysis of PCR products is currently the most straightforward method.
Direct Detection of DNA Sequence Alterations by DNA Sequencing
DNA can be sequenced to obtain a readout of the order of nucleotides, and by comparison with a normal (wild-type) sequence, mutations can be identified. The ready availability of Sanger di-deoxynucleotide sequencing and automated capillary electrophoresis allows thousands of base pairs of genomic DNA to be routinely sequenced in a matter of hours.75 The genes mutated in hundreds of mendelian disorders have been identified, and definitive diagnosis is possible by direct sequencing in most of them. Some disorders, most with recessive inheritance, are associated with a limited number of recurrent mutations, such as cystic fibrosis. Many others, especially those with dominant inheritance, can have mutations throughout the gene-coding region. Challenges to sole use of gene sequencing for the diagnosis of such diseases include the difficulty and high cost of analyzing large genes. For example, the gene associated with Duchenne muscular dystrophy possesses 79 exons, and the FBN1 gene mutated in Marfan syndrome possesses 65 exons; the sequencing of these genes in their entirety can be prohibitively expensive with current methodologies. Among other difficulties, it is not uncommon to detect sequence alterations of unknown significance, which cannot be definitively determined to be pathogenic in the absence of any functional data.
However, this picture is changing with remarkable speed. Rapidly advancing technology will make large-scale germ line sequencing applications feasible and may lead in the not distant future to the routine sequencing of entire individual human genomes. One high-throughput technology uses gene chips (microarrays) to sequence genes or portions of genes.76 Short sequences of DNA (oligonucleotides) that are complementary to the wild-type sequence and to known mutations are “tiled” adjacent to each other on the gene chip, and the DNA sample to be tested is hybridized to the array (Fig. 5-30). Before hybridization the sample is labeled with fluorescent dyes. The hybridization (and consequently, the fluorescent signal emitted) will be strongest at the oligonucleotide that is complementary to wild-type sequence if no mutations are present, while the presence of a mutation will cause hybridization to occur at the complementary mutant oligonucleotide. Computerized algorithms can then rapidly “decode” the DNA sequence for hundreds of thousands of base pairs of sequence from the fluorescent hybridization pattern on the chip, and identify potential mutations. Perhaps the most exciting recent advance is technology termed “next-generation” sequencing, which involves PCR performed in an oil emulsion that allows over one million individual PCR reactions at once.77 While at present very costly, over one billion nucleotides (one third of the human genome!) can be sequenced per run. Bioinformatics challenges of handling and interpreting such massive amounts of data are currently staggering, and much effort is devoted to such analysis.
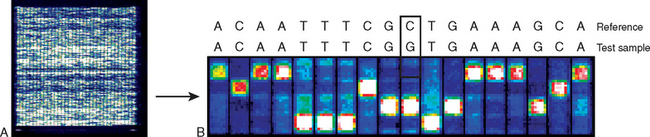
FIGURE 5-30 Microarray-based DNA sequencing. A, A low-power digitized scan of a “gene chip” that is no larger than a nickel in size but is capable of sequencing thousands of base pairs of DNA. High-throughput microarrays have been used for sequencing whole organisms (such as viruses), organelles (such as mitochondria), and entire human chromosomes. B, A high-resolution view of the gene chip illustrates hybridization patterns corresponding to a stretch of DNA sequence. Typically, a computerized algorithm is available that can convert the individual hybridization patterns across the entire chip into actual sequence data within a matter of minutes (“conventional” sequencing technologies would require days to weeks for such analysis). Here, the upper sequence is the reference (wild-type) sequence, while the lower one corresponds to the test sample sequence. As shown, the computerized algorithm has identified a C→G mutation in the test sample.
(Adapted from Maitra A et al.: The Human MitoChip: a high-throughput sequencing microarray for the mitochondrial mutation detection. Genome Res 14:812, 2004.)
Detection of DNA Mutations by Indirect Methods
There are a large number of molecular techniques that detect DNA mutations without direct sequencing. Their development is driven by lower costs and higher throughput.

FIGURE 5-31 Allele-specific PCR for mutation detection in a heterogeneous sample containing an admixture of normal and mutant DNA. Nucleotides complementary to the mutant and wild-type nucleotides at the queried base position are labeled with different fluorophores, such that incorporation into the resulting PCR product yields fluorescent signals of varying intensity based on the ratio of mutant to wild-type DNA present.
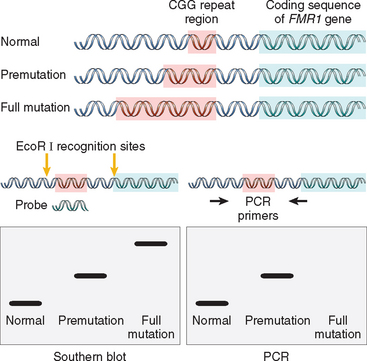
FIGURE 5-32 Diagnostic application of PCR and Southern blot analysis in fragile-X syndrome. With PCR the differences in the size of CGG repeats between normal and premutation give rise to products of different sizes and mobility. With a full mutation, the region between the primers is too large to be amplified by conventional PCR. In Southern blot analysis the DNA is cut by enzymes that flank the CGG repeat region, and is then probed with a complementary DNA that binds to the affected part of the gene. A single small band is seen in normal males, a band of higher molecular weight in males with premutation, and a very large (usually diffuse) band in those with the full mutation.
POLYMORPHIC MARKERS AND MOLECULAR DIAGNOSIS
Detection of mutations by the methods outlined above is possible only if the gene responsible for a genetic disorder is known and its sequence has been identified. In some diseases that have a genetic basis such approaches are not possible, either because the causal gene has not been identified or because the disease is multifactorial and no single gene is involved. In such cases, surrogate markers in the genome, also known as marker loci, can be used to localize the chromosomal regions of interest, on the basis of their linkage to one or more putative disease-causing genes. Linkage analysis deals with assessing these marker loci in family members having the disease or trait of interest, with the assumption that marker loci very close to the disease allele are transmitted through pedigrees (linkage disequilibrium). With time it becomes possible to define a “disease haplotype” based on a panel of marker loci, all of which co-segregate with the putative disease allele. Eventually, linkage analysis facilitates localization and cloning of the disease allele. The marker loci used in linkage studies are naturally occurring variations in DNA sequences known as polymorphisms. Two types of genetic polymorphism are most useful for linkage analysis. They are SNPs (including small insertion-deletion polymorphisms) and repeat-length polymorphisms known as minisatellite and microsatellite repeats. Each of the two types is described next.
SNPs occur at a frequency of approximately one nucleotide in every stretch of approximately 1000 base pairs and are found throughout the genome (e.g., in exons and introns and in regulatory sequences). They serve both as a physical landmark within the genome and as a genetic marker whose transmission can be followed from parent to child. Because of their prevalence throughout the genome and relative stability, SNPs can be used in linkage analysis for identifying haplotypes associated with disease.
Human DNA contains short repetitive sequences of DNA giving rise to what are called repeat-length polymorphisms. These polymorphisms are often subdivided on the basis of their length into microsatellite repeats and minisatellite repeats. Microsatellites are usually less than 1 kilobase and are characterized by a repeat size of 2 to 6 base pairs. Minisatellite repeats, by comparison, are larger (1–3 kilobases), and the repeat motif is usually 15 to 70 base pairs. It is important to note that the number of repeats, both in microsatellites and minisatellites, is extremely variable within a given population, and hence these stretches of DNA can be used quite effectively to establish genetic identity for linkage analysis. Microsatellites and the smaller minisatellites can be readily distinguished by utilizing PCR primers that flank the repeat region (Fig. 5-33A). Note that in the example given in Figure 5-33, three different alleles generate PCR products of different lengths (hence the name “length polymorphism”).
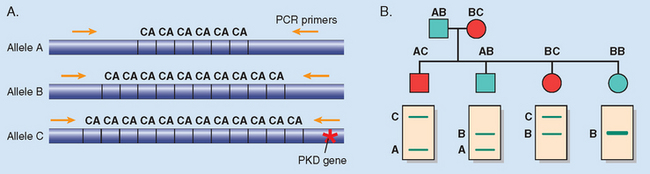
FIGURE 5-33 DNA polymorphisms resulting from a variable number of CA repeats. The three alleles produce PCR products of different sizes, thus identifying their origins from specific chromosomes. In the example depicted, allele C is linked to a mutation responsible for autosomal-dominant polycystic kidney disease (PKD). Application of this to detect progeny carrying the disease-related gene (red symbols) is illustrated in one hypothetical pedigree. Males (squares); females (circles).
Linkage analysis can be been useful in the antenatal or presymptomatic diagnosis of disorders such as Huntington disease and autosomal dominant polycystic kidney disease, even though the disease-associated gene is known in each of these conditions. In general, when the disease-associated gene is known, detection of the causative mutation by direct sequencing is the method of choice. However, if the disease originates from several different mutations in a given gene (e.g., fibrillin-1; see earlier), and gene sequencing is either not practical or negative but there is very strong clinical suspicion, linkage analysis can be useful. Figure 5-33B illustrates how microsatellite polymorphisms can be used to track the inheritance of autosomal-dominant polycystic kidney disease. In this case allele C, which produces a larger PCR product than allele A or B, carries the disease-related gene. Hence all individuals who carry the C allele are affected.
Assays to detect genetic polymorphisms are also important in many other areas of medicine, including in the determination of relatedness and identity in transplantation, cancer genetics, paternity testing, and forensic medicine. Since microsatellite markers are scattered throughout the human genome and have such a high level of polymorphism, they are ideal for differentiating two individuals and to follow transmission of the marker from parent to child. Panels of microsatellite marker PCR assays have been extensively validated and are now routinely used for determining paternity and for criminal investigations. Since PCR can be performed even with highly degraded biologic samples, DNA technology is critical in forensic identifications. The same assays have been applied to the detection and quantification of transplant chimerism in allogeneic bone marrow transplants.
Polymorphisms and Genome-Wide Analyses
As described earlier, linkage analysis utilizing DNA of affected families has been used to detect the presence of genes with large effects and high penetrance, the kind that give rise to Mendelian diseases. Similar analyses of complex (multifactorial) disorders, however, have been unsuccessful since conventional linkage studies lack the statistical power to detect variants with small effects and low penetrance, which are typical of the genes that contribute to complex disorders.
These limitations appear to have been overcome through genome-wide association studies (GWAS), a powerful method of identifying genetic variants that are associated with an increased risk of developing a particular disease.78 Such variants may themselves be causative, or may be in linkage disequilibrium with other genetic variants that are responsible for the increased risk. In GWAS large cohorts of patients with and without a disease (rather than families) are examined across the entire genome for genetic variants or polymorphisms that are over-represented in patients with the disease. This identifies regions of the genome that contain a variant gene or genes that confer disease susceptibility. The causal variant within the region is then provisionally identified using a “candidate gene” approach, in which genes are selected based on how tightly they are associated with the disease and whether their biologic function seems likely to be involved in the disease under study. For example, a variant in a gene whose product regulates vascular smooth muscle tone (e.g., angiotensinogen) is a strong candidate to influence the risk of hypertension. As might be imagined, however, some linked genes would not have been expected to be associated with particular diseases based on prior knowledge; such surprises are one of the benefits of the unbiased, systematic nature of GWAS.
GWAS have been enabled by two major technological breakthroughs. First is the completion of the so-called “HapMap” project, which has provided more complete linkage disequilibrium patterns in three major ethno-racial groups, based on genome-wide single nucleotide polymorphism (SNP) mapping. The entire human genome can now be divided into blocks known as “haplotypes,” which contain varying numbers of contiguous SNPs on the same chromosome that are in linkage disequilibrium and hence inherited together as a cluster. As a result, rather than querying every single SNP in the human genome, it is possible to glean comparable information about shared DNA by simply looking for shared haplotypes, using single or a small number of SNPs that “tag” or identify a specific haplotype. Second, it is now possible to simultaneously genotype hundreds of thousands to a million SNPs at one time, in a cost-effective way, using high-density SNP-chip technology. Figure 5-34 demonstrates how information from the publicly available “HapMap” is utilized to manufacture SNP chips that can query genome-wide haplotypes in an unbiased manner. Thereafter, DNA from a cohort of individuals with a defined trait (say, hypertension) is analyzed using SNP chips for haplotypes that are overrepresented compared to individuals without the trait (i.e., in controls). This is followed by the “candidate gene” approach described above to further localize the causal gene (and in some instances, the functional polymorphism within that gene), associated with the trait.
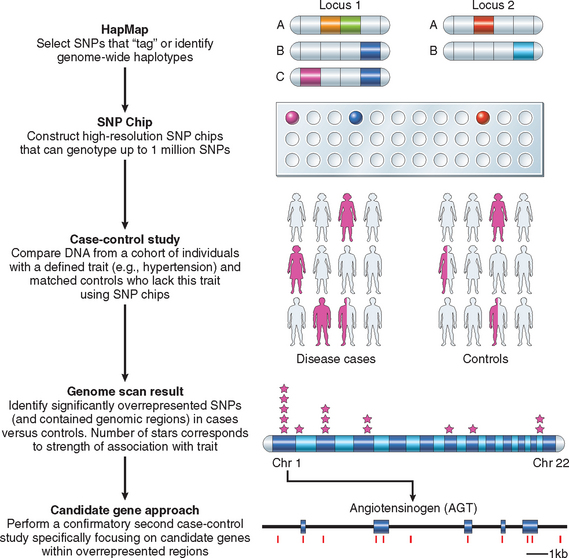
FIGURE 5-34 General scheme for conducting a genome wide association study (GWAS). Using the publicly available “HapMap” data, the human genome is divided into “haplotypes” or regions of contiguous DNA inherited as a block, each identified by one or a few “tag” SNPs that identify the haplotype. In the example shown, locus 1 contains three haplotypes defined by different combinations of SNPs, where white signifies the more common “normal” sequence and each color designates a different SNP; thus, these haplotypes can be distinguished by assaying for only the blue and purple “tag” SNPs. Thereafter, high density SNP chips are constructed that contain these “tag” SNPs, in order to enable an unbiased genome-wide assessment of shared haplotypes between disease and control populations. Of note, “disease” refers to any defined phenotype, and could pertain to an actual disease entity like hypertension, or simply a quantitative trait like hair or eye color. Next, DNA obtained from the two cohorts is analyzed for overrepresented SNPs in the disease population (“cases”) versus the control samples—this is known as a case-control study. The most significant shared genomic regions of interest are then examined for candidate genes of interest—an example shown here in a search for loci associated with hypertension is angiotensinogen, a gene on chromosome 1 whose product regulates vascular smooth muscle tone. The final step is to perform a second case control study, this time using SNPs located within the gene of interest in order to confirm or refute the association with the trait, often in an independent population from the one in which the initial GWAS was conducted. In this example, individual SNPs within angiotensinogen gene are denoted as red vertical bars, and these SNPs will be tested in the second round of case-control study.
(Modified from Mathew CG: New links to the pathogenesis of Crohn disease provided by genome-wide association scans. Nat Rev Genet 9(1):9–14, 2008.)
In addition to shedding light on some of the most frequent human ailments such as diabetes, hypertension, coronary artery disease, schizophrenia and other mental disorders, and asthma, GWAS have also led to the identification of genetic loci that modulate common quantitative traits in humans, such as height, body mass, hair and eye color, and bone density. An updated catalog of published GWAS is maintained by the National Human Genome Research Institute (www.genome.gov), with the list currently at >200 studies and growing. The power of GWAS is underscored by the fact that within a very short time, nearly a dozen genes that confer risk for type 2 diabetes have been identified, of which one in particular, TCF7L2, has emerged as a strong candidate gene (see Chapter 24 for a detailed discussion).
With the incrementally lowered costs for genotyping individual patients for SNPs that might render them “at-risk” for a variety of multifactorial disease over their lifetime, there is emerging concern in the biomedical community that such information would be utilized for discrimination in the workplace or by healthcare agencies. In the United States, a law was passed in 2008 that explicitly prohibits discrimination based on an individual’s genetic makeup.
MOLECULAR ANALYSIS OF GENOMIC ALTERATIONS
A significant number of genetic lesions involve large deletions, duplications, or more complex rearrangements that are not easily assayed using PCR methods or sequence analysis. Such “genomic” alterations can be studied using a variety of hybridization-based techniques.
Southern Blotting
Changes in the structure of specific loci can be detected by Southern blotting, which involves hybridization of radiolabeled sequence-specific probes to genomic DNA that has been first digested with a restriction enzyme and separated by gel electrophoresis. The probe usually detects one germ line band in normal individuals. Importantly, a normal DNA sample is required to compare the pattern of the DNA in question. With the advent of FISH and microarray technology, Southern blotting is rarely used but remains useful in the detection of large-trinucleotide-expansion diseases including the fragile-X syndrome (see Fig. 5-32), and in detection of clonal immunoglobulin gene rearrangements in the diagnosis of lymphoma. The latter is being replaced by PCR-based methods.
Fluorescence in Situ Hybridization
FISH uses DNA probes that recognize sequences specific to particular chromosomal regions. As part of the Human Genome Project, large libraries of bacterial artificial chromosomes that span the entire human genome were created. The human DNA insert in these clones is on the order of 100,000–200,000 base pairs, which defines the limit of resolution of FISH for identifying chromosomal changes. These DNA clones are labeled with fluorescent dyes and applied to metaphase spreads or interphase nuclei. The probe hybridizes to its homologous genomic sequence and thus labels a specific chromosomal region that can be visualized under a fluorescent microscope. The ability of FISH to circumvent the need for dividing cells is invaluable when a rapid diagnosis is warranted (e.g., when deciding to treat a patient with acute myeloid leukemia with retinoic acid, which is only effective in a particular subtype with a chromosomal translocation involving the retinoic acid receptor gene [Chapter 14]). FISH can be performed on prenatal samples (e.g., cells obtained by amniocentesis, chorionic villus biopsy, or umbilical cord blood), peripheral blood lymphocytes, touch preparations from cancer biopsies, and even archival tissue sections. FISH has been used for detection of numeric abnormalities of chromosomes (aneuploidy) (see Fig. 5-20); for the demonstration of subtle microdeletions (see Fig. 5-22) or complex translocations not detectable by routine karyotyping; for analysis of gene amplification (e.g., HER2/NEU in breast cancer or N-MYC amplification in neuroblastomas); and for mapping newly isolated genes of interest to their chromosomal loci. Chromosome painting is an extension of FISH, whereby probes are prepared for entire chromosomes. The number of chromosomes that can be detected simultaneously by chromosome painting is limited by the availability of fluorescent dyes that emit different wavelengths of visible light. This limitation has been overcome by the introduction of spectral karyotyping (also called multicolor FISH). By using a combination of five fluorochromes and appropriate computer-generated signals, the entire human genome can be visualized (Fig. 5-35). So powerful is spectral karyotyping that it might well be called “spectacular karyotyping.”
FIGURE 5-35 FISH studies using multicolor FISH in a child with an undetermined abnormality. This technique uses ratio-labeled probes labeled with 23 distinct mixtures of 5 fluorophores to create a unique “color” for each chromosome. This analysis revealed a derivative chromosome 9, with 9p containing additional material from 22q.
(Courtesy of Dr. Stuart Schwartz, Department of Pathology, University of Chicago, Chicago, IL.)
Array-Based Comparative Genomic Hybridization (Array CGH)
It is obvious from the preceding discussion that FISH requires prior knowledge of the one or few specific chromosomal regions suspected of being altered in the test sample. However, genomic abnormalities can also be detected without prior knowledge of what these aberrations may be, using a global strategy such as array CGH. In array CGH the test DNA and a reference (normal) DNA are labeled with two different fluorescent dyes (most commonly Cy5 and Cy3, which fluoresce red and green, respectively) (Fig. 5-36). The differentially labeled samples are then hybridized to a glass slide spotted with DNA probes that span the human genome at regularly spaced intervals, and usually cover all 22 autosomes and the X chromosome. If the contributions of both samples are equal for a given chromosomal region (i.e., the test sample is diploid), then all spots on the array will fluoresce yellow (the result of an equal admixture of green and red dyes). In contrast, if the test sample shows an excess of DNA at any given chromosomal region (such as resulting from an amplification), there will be a corresponding excess of signal from the dye with which this sample was labeled. The reverse will be true in the event of a deletion, with an excess of the signal used for labeling the reference sample. Amplifications and deletions in the test sample can now be significantly better localized, often down to a few thousand base pairs. Newer arrays provide even higher resolution with more than 100,000 probes per array, and are at present being used to uncover copy number abnormalities in a variety of diseases, from cancer to autism. Array CGH is regularly performed in cases of mental retardation–developmental delay of unknown etiology or in children with dysmorphic features with negative karyotypes.

FIGURE 5-36 A, Array CGH is performed by hybridization of fluorescently labeled “test” DNA and “control” DNA on a slide that contains thousands of probes corresponding to defined chromosomal regions across the human genome. The resolution of most currently available array CGH assays is in the order of about 200 to 500 kilobases. Higher power view of the array demonstrates copy number aberrations in the “test” sample (Cy5, red), including regions of amplification (spots with excess of red signal) and deletion (spots with excess of green signal); yellow spots correspond to regions of normal (diploid) copy number. B, The hybridization signals are digitized, resulting in a virtual karyotype of the genome of the “test” sample. In the illustrated example, array CGH of a cancer cell line identifies an amplification on the distal long arm of chromosome 8, which corresponds to increased number of the oncogenic MYC.
(A, From Snijders AM et al.: Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet 29:263, 2001. Web Figure A, Copyright 2001. Reprinted by permission from Macmillan Publishers Ltd.)
As discussed earlier in the chapter, CNVs are a recently discovered source of genetic polymorphism, which was uncovered using array CGH technology. While intriguing in terms of understanding the marked differences between individual genomes, they can be problematic in the clinical interpretation of array CGH data.79 There are usually many CNVs observed when comparing any two genomes encompassing millions of bases of DNA. Deciding whether a specific change is a benign polymorphism or a critical disease-causing duplication or deletion can be difficult. Databases of CNVs now exist that are very helpful guides in deciding on the relevance of questionable CNVs. Another limitation of existing array CGH platforms is that they cannot detect balanced translocations, since there is a rearrangement but no genetic material is gained or lost. Nonetheless, the vastly superior sensitivity of molecular approaches should make assays such as array CGH first-line genomic diagnostic tests that have the potential of replacing traditional karyotyping.
EPIGENETIC ALTERATIONS
Epigenetics is defined as the study of heritable chemical modification of DNA or chromatin that does not alter the DNA sequence itself. Examples of such modification include the methylation of DNA, and the methylation and acetylation of histones. Our understanding of these types of molecular alterations is rapidly growing, and it is clear that epigenetic modifications are critical for normal human development—including the regulation of tissue-specific gene expression, X chromosome inactivation, and imprinting, as well as for understanding of the cellular perturbations in the aging process and cancer.80,81
Gene expression frequently correlates with the level of methylation of DNA, usually of cytosines specifically in the CG dinucleotide-rich promoter regions known as CpG islands. As discussed earlier in the section on genomic imprinting, increased methylation of these loci is associated with decreased gene expression and is accompanied by concomitant specific patterns of histone methylation and acetylation. An ever-increasing number of disease states warrant analysis of promoter methylation—for example, in the diagnosis of fragile-X syndrome, in which hypermethylation results in FMR1 silencing. Methylation analysis is also essential in the diagnosis of Prader-Willi and Angelman syndromes.
Since traditional Sanger sequencing alone cannot detect DNA methylation, other techniques have been developed to uncover these chemical modifications. One common approach is to treat genomic DNA with sodium bisulfite, a chemical that converts unmethylated cytosines to uracil, while methylated cytosines are protected from modification. An assay termed methylation-specific PCR uses two PCR primer sets to analyze single DNA loci: one to detect a DNA sequence with unmethylated cytosines (which are converted to uracils after bisulfite treatment) and the other to detect DNA sequences with methylated cytosines (which remain cytosines after bisulfite treatment).82 Additional techniques are evolving that provide a genome-wide snapshot of epigenetically altered DNA. These techniques are based on the ability to detect histone modifications such as methylation and acetylation (which, like DNA methylation, are important regulators of gene expression) by using antibodies against specifically modified histones. Such antibodies can be used to pull down bound DNA sequences, a method termed chromatin immunoprecipitation (ChIP). These pulled-down sequences can be amplified and analyzed by hybridizing to microarrays (“ChIP on Chip”) or sequencing (“ChIP-Seq”) to map epigenetically modified genes throughout the genome.83,84
RNA ANALYSIS
Changes in DNA lead to alterations in mRNA expression; hence in principle it should be possible to use mRNA expression analysis in the diagnosis of genetic diseases. From a practical standpoint, however, DNA-based diagnosis is much preferred, since DNA is much more stable. Nonetheless, RNA analysis is critical in several areas of molecular diagnostics. The most important application is the detection and quantification of RNA viruses such as HIV and hepatitis C virus. Furthermore, mRNA expression profiling (described in Chapters 7 and 23 is rapidly becoming an important tool for molecular stratification of tumors. In some instances cancer cells bearing particular chromosomal translocations are detected with greater sensitivity by analyzing mRNA (e.g., BCR-ABL fusion in CML). The principal reason for this is that most translocations occur in scattered locations within particular introns, which can be very large, beyond the capacity of conventional PCR amplification. Since introns are removed by splicing during the formation of mRNA, PCR analysis is possible if RNA is first converted to cDNA by reverse transcriptase. PCR performed on cDNA is the method of choice for detection of minimal residual disease in patients with chronic myeloid leukemia (Chapter 13).
In closing, it should be pointed out that the progress in unraveling the genetic basis of human disease promises to be breathtaking in the coming years. An entirely new field of personalized and genomic medicine is waiting to be developed.
1 International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931.
2 Plomin R, Schalkwyk LC. Microarrays. Dev Sci. 2007;10:19.
3 Gresham D, et al. Comparing whole genomes using DNA microarrays. Nat Rev Genet. 2008;9:291.
4 Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949.
5 Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525.
6 Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444.
7 Esteller M. Epigenetics and cancer. N Engl J Med. 2008;358:1148.
8 Bayat A. Science, medicine, and the future: bioinformatics. BMJ. 2002;324:1018.
9 Jay C, et al. miRNA profiling for diagnosis and prognosis of human cancer. DNA Cell Biol. 2007;26:293.
10 Eulalio A, et al. Getting to the root of miRNA-mediated gene silencing. Cell. 2008;132:9.
11 Rimoin DL, et al. Nature and frequency of genetic disease. In: Rimoin DL, et al, editors. Emery and Rimoin’s Principles and Practice of Medical Genetics. 3rd ed. New York: Churchill Livingstone; 1997:32.
12 Ensenauer RE, et al. Primer on medical genomics. Part VIII: essentials of medical genetics for the practicing physician. Mayo Clin Proc. 2003;78:846.
13 Willard HF. Tales of the Y chromosome. Nature. 2003;423:810.
14 Gomase VS, et al. Pharmacogenomics. Curr Drug Metab. 2008;9:207.
15 Ramirez F, Dietz HC. Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr Opin Genet Dev. 2007;17:252.
16 Judge DP, Dietz HC. Therapy of Marfan syndrome. Ann Rev Med. 2008;59:43.
17 Mao JR, Bristow J. The Ehlers-Danlos syndrome: on beyond collagens. J Clin Invest. 2001;07:1063.
18 Yeowell HN, Walker LC. Mutations in the lysyl hydroxylase 1 gene that result in enzyme deficiency and the clinical phenotype of Ehlers-Danlos syndrome type VI. Mol Genet Metab. 2000;71:212.
19 Pepin MG, Byers PH. Ehler-Danlos syndrome, vascular type. Gene Rev. Available at <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=eds4>, 2006.
20 Wenstrup R, De Paepe A. Ehler-Danlos syndrome, classic type. Gene Rev. Available at <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=eds>, 2007.
21 Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2007;4:214.
22 Vellodi A. Lysosomal storage disorders. Br J Hematol. 2004;128:413.
23 Fan JQ. A counterintuitive approach to treat enzyme deficiencies: use of enzyme inhibitors for restoring mutant enzyme activity. Biol Chem. 2008;389:1.
24 Kaback MM. Hexosaminidase A deficiency. Gene Rev. Available at <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=tay-sachs>, 2006.
25 Schuchman EH. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J Inherit Metab Dis. 2007;30:654.
26 Liu B, et al. Receptor-mediated and bulk-phase endocytosis cause macrophage and cholesterol accumulation in Niemann-Pick C disease. J Lipid Res. 2007;48:1710.
27 Pastores GM, Hughes DA. Gaucher disease. Gene Rev. Available at <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=gaucher>, 2008.
28 Clarke LA. Mucopolysaccharidosis type I. Gene Rev. Available at <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=mps1>, 2007.
29 Martin RA. Mucopolysaccharidosis type II. Gene Rev. Available at <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=hunter>, 2007.
30 Ozen H. Glycogen storage diseases: new perspectives. World J Gastroenterol. 2007;13:2541.
31 Shin YS. Glycogen storage disease: clinical, biochemical, and molecular heterogeneity. Semin Pediatr Neurol. 2006;13:115.
32 Bali DS, Chen YT. Glycogen storage disease type I. Gene Rev. Available at <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=gsd1>, 2006.
33 Arenas J, et al. Glycogen storage disease type V. Gene Rev. Available at <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=gsd5>, 2007.
34 Tinkle BT, Leslie N. Glycogen storage disease type II (Pompe disease). Gene Rev. Available at <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=gsd2>, 2007.
35 Introne WJ, et al. Alkaptonuria. Gene Rev. Available at <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=alkap>, 2007.
36 Lango H, Weedon MN. What will whole genome searches for susceptibility genes for common complex disease offer to clinical practice? J Internal Med. 2007;263:16.
37 Roizen NJ, Patterson D. Down’s syndrome. Lancet. 2003;361:1281.
38 Izraeli S, et al. Trisomy of chromosome 21 in leukemogenesis. Blood Cells Mol Dis. 2007;39:156.
39 Patterson D. Genetic mechanisms involved in the phenotype of Down syndrome. Ment Retard Dev Disabil Res Rev. 2007;13:199.
40 Antonarakis SE, et al. Chromosome 21 and Down syndrome: from genomics to pathophysiology. Nat Rev Genet. 2004;5:725.
41 Sullivan KE. Chromosome 22q11.2 deletion syndrome: DiGeorge syndrome/velocardiofacial syndrome. Immunol Allergy Clin N Am. 2008;28:353.
42 Arinami T. Analyses of the associations between the genes of 22q11 deletion syndrome and schizophrenia. J Hum Genet. 2006;51:1037.
43 Ross MT, et al. The sequences of the human sex chromosomes. Curr Opin Genet Dev. 2006;16:213.
44 Lyon MF. X-chromosome inactivation and human genetic disease. Acta Paediatr. 2002;91(Suppl):107.
45 Salstrom JL. X-inactivation and the dynamic maintenance of gene silencing. Mol Genet Metab. 2007;92:56.
46 Heard E, Disteche M. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev. 2006;20:1848.
47 Hawley RS. The human Y chromosome: rumors of its death have been greatly exaggerated. Cell. 2003;113:825.
48 Visootsak J, Graham JMJr. Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet J Rare Dis. 2006;1:42.
49 Bojesen A, Gravholt CH. Klinefelter syndrome in clinical practice. Nat Clin Pract Urol. 2007;4:192.
50 Ferlin A, et al. Genetic causes of male infertility. Reprod Toxicol. 2006;22:133.
51 Hjerrild BE, et al. Turner syndrome and clinical treatment. Br Med Bull. 2008;86:77.
52 Bondy CA. Congenital cardiovascular disease in Turner syndrome. Congenit Heart Dis. 2008;3:2.
53 Marchini A, et al. SHOX at a glance: from gene to protein. Arch Physiol Biochem. 2007;113:116.
54 McLauglin DT, Donahoe PK. Sex determination and differentiation. N Engl J Med. 2004;350:367.
55 Ortenberg J, et al. SRY gene expression in the ovotestes of XX true hermaphrodites. J Urol. 2002;167:1828.
56 Brinkmann AO. Molecular basis of androgen insensitivity. Mol Cell Endocrinol. 2001;179:105.
57 Lutz RE. Trinucleotide repeat disorders. Semin Pediatr Neurol. 2007;14:26.
58 Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575.
59 Shao J, Diamond MI. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum Mol Genet. 2007;16:R115.
60 Debacker K, Kooy RF. Fragile sites and human disease. Hum Mol Genet. 2007;16:R150.
61 Penagarikano O, et al. The pathophysiology of fragile X syndrome. Annu Rev Genomics Hum Genet. 2007;8:109.
62 Venkitaramani DV, Lombroso PJ. Molecular basis of genetic neuropsychiatric disorders. Child Adolesc Psychiatr Clin N Am. 2007;16:541.
63 Bear MF, et al. Fragile X: translation in action. Neuropsychopharmacology. 2008;33:84.
64 Garber KB, et al. Fragile X syndrome. Eur J Hum Genet. 2008;16:666.
65 Haas RH, et al. Mitochondrial disease: a practical approach for primary care physicians. Pediatrics. 2007;120:1326.
66 Schapira AH. Mitochondrial disease. Lancet. 2006;368:70.
67 Mancuso M, et al. Mitochondrial DNA-related disorders. Biosci Rep. 2007;27:31.
68 Man PY, Turnbull DM, et al. Leber hereditary optic neuropathy. J Med Genet. 2002;39:162.
69 Wood AJ, Oakey RJ. Genomic imprinting in mammals: emerging themes and established theories. PLoS Genet. 2006;2:e147.
70 Chen C, et al. Prader-Willi syndrome: an update and review for the primary pediatrician. Clin Pediatr. 2007;46:580.
71 Williams CA. Neurological aspects of the Angelman syndrome. Brain Dev. 2005;27:88.
72 Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007;64:947.
73 Bernards A, Gusella JF. The importance of genetic mosaicism in human disease. N Engl J Med. 1994;331:1447.
74 American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 88, December 2007. Invasive prenatal testing for aneuploidy. Obstet Gynecol. 2007;110:1459.
75 Metzker ML. Emerging technologies in DNA sequencing. Genome Res. 2005;15:1767.
76 Gresham D, Dunham MJ, et al. Comparing whole genomes using DNA microarrays. Nat Rev Genet. 2008;9:291.
77 Shendure J, et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728.
78 Altshuler D, Daly MJ, Lander ES. Genetic mapping of human disease. Science. 2008;322:881.
79 Beaudet AL, Belmont JW. Array-based DNA diagnostics: let the revolution begin. Ann Rev Med. 2008;59:113.
80 Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148.
81 Gosden RG, Feinberg AP. Genetics and epigenetics—nature’s pen-and-pencil set. N Engl J Med. 2007;356:731.
82 Herman JG, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821.
83 Wu J, et al. ChIP—chip comes of age for genome-wide functional analysis. Cancer Res. 2006;66:6899.
84 Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823.